


We describe the discovery of an agonist of the nuclear receptor NR2F1 that specifically activates dormancy programs in malignant cells. The agonist led to a self-regulated increase in NR2F1 mRNA and protein and downstream transcription of a novel dormancy program. This program led to growth arrest of an HNSCC PDX line, human cell lines, and patient-derived organoids in 3D cultures and in vivo. This effect was lost when NR2F1 was knocked out by CRISPR-Cas9. RNA sequencing revealed that agonist treatment induces transcriptional changes associated with inhibition of cell cycle progression and mTOR signaling, metastasis suppression, and induction of a neural crest lineage program. In mice, agonist treatment resulted in inhibition of lung HNSCC metastasis, even after cessation of the treatment, where disseminated tumor cells displayed an NR2F1hi/p27hi/Ki-67lo/p-S6lo phenotype and remained in a dormant single-cell state. Our work provides proof of principle supporting the use of NR2F1 agonists to induce dormancy as a therapeutic strategy to prevent metastasis.
Introduction
Metastasis is the leading cause of cancer-related deaths and arises from disseminated tumor cells (DTCs) that seed secondary organs. These DTCs can remain dormant for years or decades before growing into symptomatic overt metastases (Aguirre-Ghiso, 2007). This period of latency occurs in multiple cancer types and is not limited to distant organs, as there can be loco-regional recurrences, a problem that is particularly significant in head and neck squamous cell carcinoma (HNSCC). More than half the patients with advanced disease develop recurrences that are usually not responsive to conventional therapies (Sacco and Cohen, 2015). This highlights the need for interventional therapies that prevent reactivation of dormant DTCs after treatment of primary tumors, a window of treatment opportunity that is currently missed.
We identified the nuclear receptor subfamily 2 group F member 1 (NR2F1; also known as COUP-TF1) as a master regulator of tumor cell dormancy (Sosa et al., 2015). NR2F1 is an orphan nuclear receptor of the steroid/thyroid hormone receptors superfamily. It has a DNA-binding domain with two conserved zinc-finger motifs and a ligand-binding domain (LBD; Tsai and Tsai, 1997). It regulates transcription either directly by binding as a dimer to direct repeats on DNA and recruiting coactivator or corepressor complexes or indirectly by acting as a cofactor to other nuclear receptors (Bertacchi et al., 2019). Depending on the context, NR2F1 can activate or repress transcription of effector genes (Bertacchi et al., 2019). NR2F1 also plays an epigenetic role in mediating global histone modifications by interacting with or recruiting chromatin-remodeling enzymes (Bertacchi et al., 2019).
We found that NR2F1 is epigenetically silenced in proliferating cancer cells, and it is up-regulated in dormant residual HNSCC cells in a patient-derived xenograft (PDX) model and prostate cancer DTCs isolated from patients (Sosa et al., 2015). In HNSCC patients, NR2F1 expression is absent or low in primary tumors, recurrent tumors, and metastases compared with benign adjacent oral mucosa (Sosa et al., 2015). When up-regulated, NR2F1 induces expression of a dormancy gene signature, including the transcription factors SOX9 and retinoic acid (RA) receptor β (RARβ). These factors activate expression of cyclin-dependent kinase (CDK) inhibitors p27 and p16, which leads to G0/G1 cell cycle arrest and cell quiescence (Sosa et al., 2015). NR2F1-associated signature is also enriched in estrogen receptor+ breast cancer tumors, and patients carrying primary lesions enriched for this signature showed delayed time to metastasis (Kim et al., 2012). More specific to DTC biology, we also reported that breast cancer patients who carried in their bone marrow DTCs positive for NR2F1 were less likely to develop and die from bone metastasis compared with those negative or low for NR2F1 (Borgen et al., 2018). NR2F1 is also associated with alterations in a breast cancer susceptibility locus (Mcs1; Smits et al., 2013). These data argue that NR2F1 is a strong negative regulator of HNSCC, breast cancer, and other cancers and that a unique function of NR2F1 is the induction of cancer cell dormancy, likely a role related to its lineage commitment function in development (Rada-Iglesias et al., 2012; Zhuang and Gudas, 2008).
shRNA-mediated down-regulation of NR2F1 results in reactivation of dormant HNSCC tumor cells, leading to locoregional relapse in surgery margins and development of lung and splenic metastasis in mice (Sosa et al., 2015). This shows that NR2F1 plays a critical role in initiating and maintaining tumor cell dormancy, a biological function that has been reproduced by independent laboratories and ours in various cancer models (Cackowski et al., 2017; Fluegen et al., 2017; Kim et al., 2012; Liu et al., 2018; Sosa et al., 2015). Epigenetic reprogramming therapy using low-dose 5-azacytidine (AZA; a DNMT1 inhibitor) and RA results in an up-regulation of NR2F1 and dormancy induction in various cancer models (Sosa et al., 2015). While NR2F1 is silenced, it is not completely absent in many tumors (Kim et al., 2012; Sosa et al., 2015). Additionally, although not directly tested, NR2F1 is predicted to regulate its own expression based on computational analysis and chromatin status data (Adam et al., 2009; Sosa et al., 2015). Hence, experimental and clinical data support that activating NR2F1 using a small-molecule agonist represents an attractive clinical strategy to induce dormancy and prevent recurrence and metastasis by restoring its expression and function in DTCs. Here, we report the discovery of an NR2F1 selective agonist that was used as a proof-of-concept tool compound to induce and activate NR2F1 leading to the powerful activation of a dormancy program in cancer cells. Importantly, use of this agonist in a preclinical neoadjuvant and adjuvant setting completely suppresses macrometastatic growth by inducing DTC dormancy. This anti-metastatic effect can persist even after cessation of the treatment, arguing for a durable reprogramming of malignant cells into dormancy. Our work reveals a proof-of-principle strategy to limit metastatic growth by activation of dormancy mechanisms.
Results
Modeling of the NR2F1 LBD identifies NR2F1 agonists in silico
There is no available crystal structure of NR2F1 or its LBD. We aimed to build a structural model based on homologous proteins of known structure, among which the most similar to NR2F1 is NR2F2 (Kruse et al., 2008). However, the available NR2F2 structure is in the autorepressed conformation, in which the agonist-binding site is occluded. Hence, we built a model of NR2F1 LBD using as template the structure of RA receptor α (RXRα) in complex with 9-cis-RA (PDB accession no. 1FM6, ∼40% sequence identify to NR2F1 over the LBD; Gampe et al., 2000). The resulting NR2F1 LBD model represents its active conformation and was used in a structure-based virtual screening approach to identify small molecules with the potential to act as NR2F1 agonists (Fig. 1 A).

NR2F1 LBD modeling and agonist screen. (A) Diagram depicting the approach used to screen for NR2F1 agonists. (B) Left: Ribbon diagram (in red) of NR2F1 LBD modeled using MODELLER v9.10 shown with the agonist C26 (sphere representation) docked in the binding site. Right: Close-up of C26 docked in the binding site; white dotted line represents the part of the helix that was removed for a better view of the binding site. (C) Stick representation showing the interaction between C26 and mostly hydrophobic residues in the binding site of the NR2F1 LBD. (D) Graph showing the fold change of luciferase/Renilla activity using the RARE-luciferase reporter system in HEK293T cells with NR2F1 overexpression and treated for 18 h with DMSO, 0.1 µM atRA, or 0.1 µM C26. Data are shown as mean ± SEM from three independent experiments. *, P < 0.05 by t test. (E) Graph showing the mean fold change of luciferase/Renilla activity using the NR2F1-luciferase reporter system in D-HEp3 cells treated for 18 h with DMSO or C26 (0.2, 0.5, or 1 µM) ± SEM from three independent experiments. *, P < 0.05; **, P < 0.01 by ANOVA. (F and G) Graphs showing the fold change of luciferase/Renilla activity using RARE-luciferase (F) or NR2F1-luciferase (G) reporter systems in D-HEp3 cells expressing NT gRNA or with NR2F1 knockout using two separate gRNAs (guide 2 and guide 4). Cells were treated for 18 h with DMSO or 0.5 µM C26. Data are mean ± SEM from three independent experiments. *, P < 0.05; ***, P < 0.001 by ANOVA. See also Fig. S1.
NR2F1 LBD modeling and agonist screen. (A) Diagram depicting the approach used to screen for NR2F1 agonists. (B) Left: Ribbon diagram (in red) of NR2F1 LBD modeled using MODELLER v9.10 shown with the agonist C26 (sphere representation) docked in the binding site. Right: Close-up of C26 docked in the binding site; white dotted line represents the part of the helix that was removed for a better view of the binding site. (C) Stick representation showing the interaction between C26 and mostly hydrophobic residues in the binding site of the NR2F1 LBD. (D) Graph showing the fold change of luciferase/Renilla activity using the RARE-luciferase reporter system in HEK293T cells with NR2F1 overexpression and treated for 18 h with DMSO, 0.1 µM atRA, or 0.1 µM C26. Data are shown as mean ± SEM from three independent experiments. *, P < 0.05 by t test. (E) Graph showing the mean fold change of luciferase/Renilla activity using the NR2F1-luciferase reporter system in D-HEp3 cells treated for 18 h with DMSO or C26 (0.2, 0.5, or 1 µM) ± SEM from three independent experiments. *, P < 0.05; **, P < 0.01 by ANOVA. (F and G) Graphs showing the fold change of luciferase/Renilla activity using RARE-luciferase (F) or NR2F1-luciferase (G) reporter systems in D-HEp3 cells expressing NT gRNA or with NR2F1 knockout using two separate gRNAs (guide 2 and guide 4). Cells were treated for 18 h with DMSO or 0.5 µM C26. Data are mean ± SEM from three independent experiments. *, P < 0.05; ***, P < 0.001 by ANOVA. See also Fig. S1.
AutoDock and eHiTS programs were used to screen a library of 110,000 drug-like compounds. The top 50 compounds identified by each program (based on docking score) were combined with the top 50 compounds ranked by the sum of their AutoDock 4 and eHiTS ranks. All selected compounds were visually inspected in the context of the target-binding site to select the most promising ones. Of the 150 initial compounds, 67 were selected for experimental validation using a RA response element (RARE) luciferase reporter system in HEK293T cells with NR2F1 overexpression (data not shown). Of the 67 compounds, one agonist herein referred to as compound C26 (C26; chemical structure shown in Fig. S1 A), consistently and significantly resulted in RARE reporter activation and was chosen for further validation. A model of NR2F1 in complex with C26, generated using the methods described above (Fig. 1 B), shows that C26 interacts with mostly hydrophobic residues in the binding pocket (Fig. 1 C). C26 was found to induce luciferase expression from RARE reporter by 2.4-fold, a level that is comparable to 100 nM all-trans retinoic acid (atRA; Fig. 1 D), which activates transcription of target genes with RARE via binding to RARs (Arisi et al., 2014). Since the RARE-luciferase bioassay reports on the combined signaling of RARs and NR2F1 as a coactivator (Lin et al., 2000), we used another reporter system where luciferase expression is driven by NR2F1 cis-regulatory element in dormant HEp3 (D-HEp3) HNSCC cells that express high endogenous levels of NR2F1 (Adam et al., 2009; Sosa et al., 2015). Results show that C26 treatment significantly induces luciferase expression by 1.7- and 1.9-fold at 0.5 and 1 µM, respectively (Fig. 1 E), further confirming that C26 acts as an NR2F1 agonist. To verify that the effect of C26 on reporter activation is specific, we generated D-HEp3 cell lines where NR2F1 is knocked out by CRISPR-Cas9 using two different guide RNAs (gRNAs), which was confirmed by Western blot (Fig. S1 B). While C26 activates RARE- and NR2F1-luciferase reporters in control cells expressing nontargeting (NT) gRNA, the effect of C26 is completely abrogated in NR2F1 knockout cell lines (Fig. 1, F and G). This indicates that the C26 effect in cells is dependent on NR2F1 and confirms the on-target effect of C26. We next assessed whether C26 has the ability to bind and activate RXRα, whose structure was used to model NR2F1 LBD, using a time-resolved fluorescence resonance energy transfer coactivator assay in vitro. Importantly, C26 does not activate RXRα even at concentrations that are 5- to 10-fold above those that we use in our assays (Fig. S1 C). These results further support that C26 is a selective agonist for NR2F1 in human cancer cells.

Chemical structure of C26, KO controls for NR2F1, and effect of C26 on RXRα activation . (A) Chemical structure of C26 compound. (B) Western blot shows NR2F1 protein expression level in nuclear extracts of D-HEp3 cell lines with NT RNA or four different NR2F1 gRNAs. Arrows indicate the two cell lines that were selected for use in experiments (guide 2 [g2] and g4). Lamin-B1 is used as a loading control. (C) Graph showing the percent activation of RXRα using different C26 concentrations. Shaded area represents the range of C26 concentration that was used in our experiments.
Chemical structure of C26, KO controls for NR2F1, and effect of C26 on RXRα activation . (A) Chemical structure of C26 compound. (B) Western blot shows NR2F1 protein expression level in nuclear extracts of D-HEp3 cell lines with NT RNA or four different NR2F1 gRNAs. Arrows indicate the two cell lines that were selected for use in experiments (guide 2 [g2] and g4). Lamin-B1 is used as a loading control. (C) Graph showing the percent activation of RXRα using different C26 concentrations. Shaded area represents the range of C26 concentration that was used in our experiments.
C26 up-regulates NR2F1 and downstream target genes that regulate dormancy
We previously showed that NR2F1 knockdown reduces repressive chromatin marks on its own promoter allowing an active chromatin state (Sosa et al., 2015). Additionally, computational identification of a transcription factor network active in dormant HNSCC cells revealed that NR2F1 is a central node, and it was predicted to regulate its own expression (Adam et al., 2009). These observations suggest that upon its activation, NR2F1 might up-regulate its own expression and that even NR2F1lo tumor cells could be activated to reexpress this nuclear receptor. This has not been tested before due to lack of experimental tools to activate NR2F1. To test this, we used tumorigenic HEp3 (T-HEp3) cells, a highly proliferative HNSCC PDX line that was obtained from a lymph node metastasis with primary carcinoma in the buccal mucosa (Moore et al., 1955) and maintained in vivo. These cells express low but detectable levels of NR2F1 (Fluegen et al., 2017; Sosa et al., 2015). T-HEp3 cells were pretreated with DMSO or C26 and inoculated in vivo on chicken embryo chorioallantoic membrane (CAM) with continuous daily treatments. After 7 d, tumors were excised, and mRNA was extracted. Quantitative PCR (qPCR) analysis revealed that C26 treatment results in 2.3-fold increase in NR2F1 mRNA levels in tumors (Fig. 2 A). To evaluate if C26 treatment also leads to up-regulation of NR2F1 protein, we performed a similar experiment, but this time, tumors were dissociated, and cytospins of cells were immunostained for NR2F1. T-HEp3 cells are distinguished from avian cells by immunostaining for vimentin, an intermediate filament that is abundantly expressed by HEp3 cells and has been previously used to detect these cells (Bragado et al., 2013; Fluegen et al., 2017; Sosa et al., 2015). The percentage of NR2F1+ cells is dramatically increased from ∼4% in DMSO control tumors, which is consistent with what was previously reported (Sosa et al., 2015), to 42% in tumors treated with C26 (Fig. 2, B and C). This was determined using a stringent mask for strong nuclear NR2F1 signal, which appears as prominent clusters that we had reported previously in HNSCC and breast cancer DTCs (Borgen et al., 2018; Fluegen et al., 2017; Sosa et al., 2015). To further confirm the effect of C26 on NR2F1 expression, we used an in vitro 3D assay, where T-HEp3 cells were plated at low density in Matrigel and treated with DMSO or C26 for 4 d. Immunostaining for NR2F1 showed a significant increase in the mean fluorescence intensity (MFI) of nuclear NR2F1 in C26-treated cells compared with control (Fig. 2, D and E). These data confirm that NR2F1 activation by C26 treatment results in up-regulation of NR2F1 mRNA and nuclear protein levels.

C26 up-regulates NR2F1 expression. (A) T-HEp3 cells pretreated for 6 d with DMSO or 0.5 µM C26 were inoculated on CAM and treated daily. After 7 d, tumors were collected and RNA extracted. Graph shows mean fold change in NR2F1 mRNA levels over DMSO obtained by qPCR ± SEM from four tumors per group. *, P < 0.05 by t test. (B and C) CAM tumors treated as in A were dissociated and cell cytospins were generated and immunostained for NR2F1 and nuclei counterstained with DAPI. Graph shows the mean percentage of NR2F1+ cells ± SEM from four tumors per group (DMSO, 87 cells; C26, 128 cells). Scale bar, 10 µm; arrowheads indicate nuclear NR2F1. *, P < 0.05 by t test. (D and E) T-HEp3 cells were plated in Matrigel and treated with DMSO or 0.5 µM C26. After 4 d, cells were fixed and immunostained for NR2F1. Scale bar, 50 µm; arrowheads indicate nuclear NR2F1. Graph shows box (25th to 75th percentile) and whiskers (minimum to maximum values) of nuclear NR2F1 MFI per cell (DMSO, 64 cells; 0.2 µM, 26 cells; 0.5 µM, 29 cells; 1 µM, 23 cells from two independent experiments). ****, P < 0.0001. (F) SOX9, RARβ, p27, and DEC2 mRNA levels were measured using qPCR in CAM tumors described in A. Graph shows fold change in mRNA levels over DMSO ± SEM from four tumors per group. *, P < 0.05 by t test.
C26 up-regulates NR2F1 expression. (A) T-HEp3 cells pretreated for 6 d with DMSO or 0.5 µM C26 were inoculated on CAM and treated daily. After 7 d, tumors were collected and RNA extracted. Graph shows mean fold change in NR2F1 mRNA levels over DMSO obtained by qPCR ± SEM from four tumors per group. *, P < 0.05 by t test. (B and C) CAM tumors treated as in A were dissociated and cell cytospins were generated and immunostained for NR2F1 and nuclei counterstained with DAPI. Graph shows the mean percentage of NR2F1+ cells ± SEM from four tumors per group (DMSO, 87 cells; C26, 128 cells). Scale bar, 10 µm; arrowheads indicate nuclear NR2F1. *, P < 0.05 by t test. (D and E) T-HEp3 cells were plated in Matrigel and treated with DMSO or 0.5 µM C26. After 4 d, cells were fixed and immunostained for NR2F1. Scale bar, 50 µm; arrowheads indicate nuclear NR2F1. Graph shows box (25th to 75th percentile) and whiskers (minimum to maximum values) of nuclear NR2F1 MFI per cell (DMSO, 64 cells; 0.2 µM, 26 cells; 0.5 µM, 29 cells; 1 µM, 23 cells from two independent experiments). ****, P < 0.0001. (F) SOX9, RARβ, p27, and DEC2 mRNA levels were measured using qPCR in CAM tumors described in A. Graph shows fold change in mRNA levels over DMSO ± SEM from four tumors per group. *, P < 0.05 by t test.
NR2F1 induces tumor cell quiescence by binding to the promoters of SOX9, RARβ, and p27 and inducing their expression (Sosa et al., 2015). To determine if activation of NR2F1 by C26 and its nuclear accumulation is accompanied by an increase in NR2F1 target gene expression, we measured the effect of C26 treatment on mRNA levels of these factors in CAM tumors. mRNA levels of SOX9, RARβ, and p27 are all significantly up-regulated in C26-treated tumors compared with control (Fig. 2 F). Interestingly, C26 treatment has no effect on mRNA levels of DEC2 (Fig. 2 F), a transcription factor that in dormant HNSCC and breast cancer cells is primarily regulated by TGF-β2 signaling (Bragado et al., 2013). Hence, C26 does not affect the TGF-β2–DEC2 pathway, which has been shown to mediate dormancy signals in a manner that is parallel to and independent from NR2F1 (Sosa et al., 2015). Collectively, these results show that C26 treatment induces NR2F1 expression and nuclear accumulation and selectively activates canonical NR2F1-driven dormancy pathway genes.
RNA-sequencing (RNA-seq) analysis reveals that NR2F1 activation induces neural crest–like and growth suppression programs in HNSCC cells
To gain an unbiased insight on transcriptional changes that are induced by C26-mediated activation of NR2F1, we performed RNA-seq of T-HEp3 tumors treated with C26 in vivo. T-HEp3-GFP cells were pretreated in culture with C26 or DMSO for 6 d and then inoculated on CAM with continuous daily treatment. After 7 d, tumors were recovered and dissociated, GFP+ cells were isolated using FACS (Fig. S2 A), and mRNA was sequenced using next-generation sequencing. Principal-component analysis (PCA) of the RNA-seq data using the top 500 genes with the highest variation in gene expression across samples revealed that replicate samples from each condition cluster together and away from the other condition (Fig. S2 B). Bioinformatics analysis showed that 1,324 genes were down-regulated and 456 genes were up-regulated by C26 treatment compared with DMSO control (Fig. 3 A). Gene set enrichment analysis (GSEA) showed that the Hallmark gene sets with the highest normalized enrichment score in DMSO control (hence negatively regulated by C26 treatment) included epithelial-to-mesenchymal transition (EMT), down-regulation of UV light response, mTORC1 pathway, protein secretion, and oxidative phosphorylation (Fig. S2 C). EMT is also one of the most significantly down-regulated pathways in C26 in the WikiPathways database (Fig. S2 D; left panel). Interestingly, C26 treatment also significantly down-regulates the phosphoinositide 3-kinase (PI3K) signaling pathway, which is activated in more than 90% of HNSCC cases and plays a critical role in the pathogenesis of HNSCC (Marquard and Jücker, 2020). This is evidenced by the focal adhesion_PI3K_AKT_mTOR and the PI3K_AKT signaling pathways being the two most significantly down-regulated pathways from the WikiPathways database in C26-treated samples (Fig. S2 D; left panel) and the significant enrichment of the Hallmark PI3K_AKT_mTOR pathway in DMSO control (Fig. 3 B). Since NR2F1 is known to regulate neural crest gene expression during development (Rada-Iglesias et al., 2012), we explored if the neural crest differentiation pathway was up-regulated upon C26 treatment. Results show that this pathway is significantly enriched in C26-treated samples (Fig. 3 C) and is among the top up-regulated pathways from the WikiPathways database (Fig. S2 D; right panel). In agreement, 27 genes involved in cranial neural crest cell development (Betancur et al., 2010) are significantly up-regulated upon C26 treatment (Fig. 3 D). Among these, C26 treatment up-regulated NOTCH1, which is found mutated in a fraction of HNSCC tumors and is thought to restrict progression (Stransky et al., 2011), and ZIC1, a negative regulator of sonic hedgehog signaling that is also commonly methylated in HNSCC (Paluszczak et al., 2017).

Sorting and RNA - seq controls and description and additional gene expression analysis. (A) Representative FACS plots showing gating strategy used to sort GFP+ cells from T-HEp3-GFP tumors. Negative control, T-HEp3 cells with no GFP; positive control, T-HEp3-GFP cells grown in culture; DMSO, cells from DMSO-treated T-HEp3-GFP tumors; C26, cells from C26-treated T-HEp3-GFP tumors; SSC, side scatter. (B) PCA plot generated using the top 500 genes with the highest variation (Var) in gene expression across samples. (C) Gene set enrichment profiles of the top 5 enriched gene sets in DMSO control. (D) Top 15 most down-regulated or up-regulated pathways from the WikiPathways database in C26 treatment. Arrows indicate the pathways that were discussed in text. (E) Venn diagrams showing DEGs down-regulated in D-HEp3 versus T-HEp3 and up-regulated in C26 versus DMSO (top) and DEGs up-regulated in D-HEp3 versus T-HEp3 and down-regulated in C26 versus DMSO (bottom). Statistical analysis was performed using a hypergeometric probability test.
Sorting and RNA - seq controls and description and additional gene expression analysis. (A) Representative FACS plots showing gating strategy used to sort GFP+ cells from T-HEp3-GFP tumors. Negative control, T-HEp3 cells with no GFP; positive control, T-HEp3-GFP cells grown in culture; DMSO, cells from DMSO-treated T-HEp3-GFP tumors; C26, cells from C26-treated T-HEp3-GFP tumors; SSC, side scatter. (B) PCA plot generated using the top 500 genes with the highest variation (Var) in gene expression across samples. (C) Gene set enrichment profiles of the top 5 enriched gene sets in DMSO control. (D) Top 15 most down-regulated or up-regulated pathways from the WikiPathways database in C26 treatment. Arrows indicate the pathways that were discussed in text. (E) Venn diagrams showing DEGs down-regulated in D-HEp3 versus T-HEp3 and up-regulated in C26 versus DMSO (top) and DEGs up-regulated in D-HEp3 versus T-HEp3 and down-regulated in C26 versus DMSO (bottom). Statistical analysis was performed using a hypergeometric probability test.
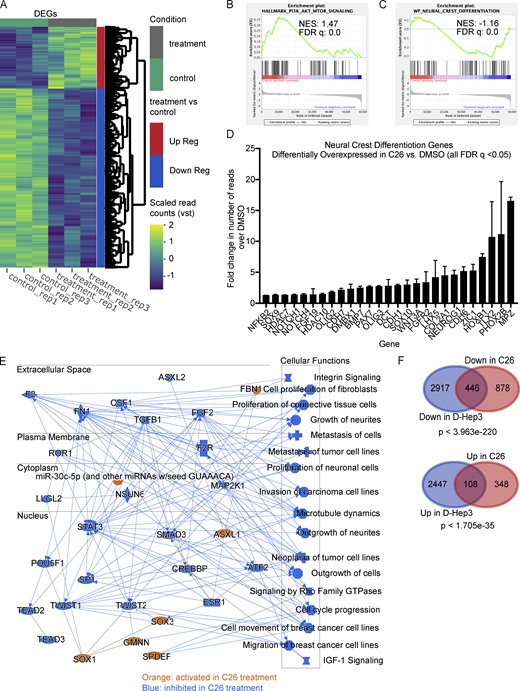
RNA-seq analysis showing transcriptional changes induced by C26 treatment. T-HEp3-GFP cells pretreated for 6 d with DMSO or 0.5 µM C26 were inoculated on CAM and treated daily. After 7 d, GFP+ T-HEp3 cells were sorted by FACS from dissociated tumors, and mRNA was isolated and sequenced using next-generation sequencing as described in Materials and methods. (A) Heatmap showing differentially expressed genes (DEGs) between DMSO control and C26-treated samples (three replicates per condition). Up Reg, up-regulated; Down Reg, down-regulated; vst, variable stabilizing transformation. (B and C) Gene set enrichment profile showing enrichment of the PI3K_AKT_mTOR signaling pathway in DMSO (B) and the neural crest differentiation pathway in C26 (C). FDR, false discovery rate; NES, normalized enrichment score. (D) Graph showing fold change in the number of RNA-seq reads of 27 neural crest differentiation pathway genes that were significantly (q value < 0.05) up-regulated in C26 treatment; error bars show the standard deviation for mRNA levels for each gene. (E) Graphical summary of the most significant entities predicted in the core analysis using IPA software (Qiagen). Entities include canonical pathways, upstream regulators, transcription factors, and biological functions. Orange color, activated entities; blue color, inhibited entities in C26 treatment. (F) Venn diagrams showing DEGs down-regulated (Down) in D-HEp3 versus T-HEp3 and in C26 versus DMSO (top) and DEGs up-regulated (Up) in D-HEp3 versus T-HEp3 and in C26 versus DMSO (bottom). Statistical analysis was performed using a hypergeometric probability test. See also Figs. S2 and S3 and Table S1.
RNA-seq analysis showing transcriptional changes induced by C26 treatment. T-HEp3-GFP cells pretreated for 6 d with DMSO or 0.5 µM C26 were inoculated on CAM and treated daily. After 7 d, GFP+ T-HEp3 cells were sorted by FACS from dissociated tumors, and mRNA was isolated and sequenced using next-generation sequencing as described in Materials and methods. (A) Heatmap showing differentially expressed genes (DEGs) between DMSO control and C26-treated samples (three replicates per condition). Up Reg, up-regulated; Down Reg, down-regulated; vst, variable stabilizing transformation. (B and C) Gene set enrichment profile showing enrichment of the PI3K_AKT_mTOR signaling pathway in DMSO (B) and the neural crest differentiation pathway in C26 (C). FDR, false discovery rate; NES, normalized enrichment score. (D) Graph showing fold change in the number of RNA-seq reads of 27 neural crest differentiation pathway genes that were significantly (q value < 0.05) up-regulated in C26 treatment; error bars show the standard deviation for mRNA levels for each gene. (E) Graphical summary of the most significant entities predicted in the core analysis using IPA software (Qiagen). Entities include canonical pathways, upstream regulators, transcription factors, and biological functions. Orange color, activated entities; blue color, inhibited entities in C26 treatment. (F) Venn diagrams showing DEGs down-regulated (Down) in D-HEp3 versus T-HEp3 and in C26 versus DMSO (top) and DEGs up-regulated (Up) in D-HEp3 versus T-HEp3 and in C26 versus DMSO (bottom). Statistical analysis was performed using a hypergeometric probability test. See also Figs. S2 and S3 and Table S1.
To further narrow the effect of C26 on transcriptional changes, we performed a pathway enrichment analysis using Ingenuity Pathway Analysis (IPA; Fig. 3 E), which revealed that transcriptional changes induced by C26 are predicted to cause inhibition of cell cycle progression and proliferation as well as suppression of invasion and metastasis. Interestingly, one of the signaling nodes that are inhibited upon C26 treatment is TGF-β1, which we have previously shown to induce reactivation of dormant HNSCC cells (Bragado et al., 2013). IPA analysis also revealed Sox1, Sox3, and GMNN, which are transcription factors linked to neural crest stem cell lineage function, as significant activity nodes. While the function of these factors remains unknown in the context of NR2F1 activation, they are involved in one or more processes that regulate neural crest cell development and differentiation and stem cell fate decisions (Ekonomou et al., 2005; Kan et al., 2007; Rizzoti et al., 2004; Spella et al., 2011). We conclude that C26 induces a program of growth and metastasis suppression associated with a reduction in the PI3K–mTOR pathway and induction of components of a developmental program found in differentiating neural crest cells.
We next aimed to determine to what extent the transcriptional changes that are induced in C26 treatment are found in spontaneously dormant HNSCC cells, as described previously (Sosa et al., 2015). We compared RNA-seq data from our current C26 treatment study with RNA-seq data comparing dormant D-HEp3 cells to proliferative T-HEp3 cells. This analysis showed that the genes regulated by C26 activation of NR2F1 represent only a fraction of the genes regulated in D-HEp3 cells. Of these, 446 genes that are down-regulated by C26 treatment are also down-regulated in dormant cells, and 108 genes that are up-regulated by C26 treatment are also up-regulated in dormant cells (Fig. 3 F). Comparing GSEA of both datasets revealed that a subset of the Hallmark pathways that are significantly down-regulated in C26, including the EMT signature, are also down-regulated in spontaneously dormant cells (Table S1; left column). However, there were differences between these programs as well (Table S1; right column). For example, the mTORC1 signaling pathway is significantly down-regulated after C26 treatment, but not in D-HEp3 cells. Of note, although far less statistically significant, a subset of genes was modulated in the opposite direction in D-HEp3 cells (i.e., down-regulated in D-HEp3/up-regulated in C26 and up-regulated in D-HEp3/down-regulated in C26; Fig. S2 E). We conclude that C26-mediated activation of NR2F1 in malignant cells controls a subprogram of gene expression found in spontaneously dormant D-HEp3 cells but that C26 also modulates a distinct subset of unique gene expression programs.
Finally, to corroborate the RNA-seq gene expression findings and assess the specificity of the C26 effect on gene expression, we used CRISPR-Cas9 to knock out NR2F1 in T-HEp3 cells and confirmed the knockout by Western blot (Fig. S3 A). We then performed qPCR analysis of multiple markers from the neural crest cell development pathway (neurogenin 1, cadherin 6, SOX10, and PHOX2B), as well as NR2F1 downstream effectors (p27, RARβ, and SOX9, which is also part of the neural crest cell development pathway). The results show that C26 treatment significantly increases expression of neurogenin 1, cadherin 6, SOX10, and PHOX2B (Fig. S3 B; left panel), which validates the RNA-seq results. Importantly, the effect of C26 on gene expression of these markers, as well as p27, RAR, and SOX9, was completely abrogated in the R2F1 knockout cell line (Fig. S3 B; right panel), indicating that this effect is specifically mediated by NR2F1.

Control for NR2F1 KO in T-HEp3 cells and on neural crest cell marker gene expression. (A) Western blot shows NR2F1 protein expression level in nuclear extracts of T-HEp3 cell lines with NT RNA or four different NR2F1 gRNAs. Arrows indicate the two cell lines that were selected for use in experiments (g2 and g4). Lamin-B1 was used as a loading control. (B) mRNA levels of indicated transcripts were measured using qPCR in DMSO- or C26-treated CAM tumors from T-HEp3 cells with NT or NR2F1 gRNA (g2). Graph shows fold difference in mRNA levels obtained by qPCR and normalized to DMSO. Data represent mean ± SEM from six to eight tumors per group. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 by t test.
Control for NR2F1 KO in T-HEp3 cells and on neural crest cell marker gene expression. (A) Western blot shows NR2F1 protein expression level in nuclear extracts of T-HEp3 cell lines with NT RNA or four different NR2F1 gRNAs. Arrows indicate the two cell lines that were selected for use in experiments (g2 and g4). Lamin-B1 was used as a loading control. (B) mRNA levels of indicated transcripts were measured using qPCR in DMSO- or C26-treated CAM tumors from T-HEp3 cells with NT or NR2F1 gRNA (g2). Graph shows fold difference in mRNA levels obtained by qPCR and normalized to DMSO. Data represent mean ± SEM from six to eight tumors per group. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 by t test.
C26 induces growth arrest via NR2F1
Our RNA-seq data showing that C26 treatment induces transcriptional changes associated with growth suppression and lineage commitment, and the fact that we had shown that NR2F1 controls a cancer cell quiescence program in HNSCC and other cancer cells (Sosa et al., 2015), led us to test its effect on malignant tumor cell growth. To this end, results showed that C26 treatment of T-HEp3 cells plated in Matrigel kept them in a single-cell stage, as evidenced by the higher percentage of single cells (Fig. 4 A) and lower percentage of clusters (Fig. 4 B) compared with DMSO. Similar results were obtained in two separate HNSCC cell lines, FaDu and SQ20B (Fig. 4 C). Accordingly, staining for the proliferation marker Ki-67 in the Matrigel assay showed that C26 significantly decreased the percentage of Ki-67+ cells in T-HEp3, SQ20B, and FaDu cells compared with DMSO (Figs. 4 D and S4 A). These data collectively indicate that C26 treatments arrest HNSCC cells in a nonproliferative single-cell state. We then evaluated the effect of C26 on tumor growth in vivo in the CAM model. T-HEp3 cells were pretreated for 6 d in culture and then inoculated on CAM, with or without continuous treatment. Results show that C26 inhibits tumor growth on CAM both with pretreatment only (Fig. 4 E) and, to a higher extent, with continuous treatment (Fig. 4 F). Staining for cleaved caspase-3 revealed that there is no difference in the percentage of cells that are positive for this apoptotic marker between control and treated groups, which indicates that C26 treatment does not induce apoptotic cell death (Fig. S4 B).

C26 induces growth arrest via NR2F1. (A–C) T-HEp3, SQ20B, or FaDu cells were plated in Matrigel and treated with DMSO or C26 (0.2, 0.5, or 1 µM for T-HEp3; 0.5 µM for SQ20B and FaDu). After 4 d, cells were counted manually under a microscope. Graphs show percentage of single cells (A and C) or colonies of three or more cells (B). Data are shown as mean ± SEM from three independent experiments. *, P < 0.05; **, P < 0.01 by ANOVA (A and B). *, P < 0.05 by t test (C). (D) T-HEp3, SQ20B, or FaDu cells from A–C were fixed and immunostained for Ki-67. Graph shows the percentage of Ki-67+ cells. Data are shown as mean ± SEM from three (T-HEp3 and FaDu) or two (SQ20B) independent experiments. The total number of cells analyzed is 81 (DMSO) and 68 cells (C26) for T-HEp3; 125 (DMSO) and 117 cells (C26) for FaDu; and 49 (DMSO) and 34 cells (C26) for SQ20B. *, P < 0.05; **, P < 0.01 by t test. (E and F) T-HEp3 cells were pretreated in culture for 6 d with DMSO or 0.5 µM C26 and inoculated on CAM without continuous treatment (E) or with daily treatment (F). After 7 d, tumors were collected and dissociated. Graph shows the mean number of cells per tumor ± SEM from 20 tumors (DMSO) and 21 tumors (C26; E) or 7 tumors (DMSO) and 8 tumors (C26; F). *, P < 0.05; **, P < 0.01 by t test. (G–I) Single cells from patient-derived organoids were plated in Matrigel and treated with DMSO or 0.5 µM C26. After 7 d, 5–10 random widefield images per sample were acquired on a confocal microscope. Images in G are representative z-stack projections (scale bar, 100 µm). Graph in H shows the percentage of single cells or organoids per well. Data are mean ± SEM from three independent experiments. *, P < 0.05 by t test. Graph in I shows the colony area of 112 colonies (DMSO) or 33 colonies (C26) from three independent experiments. ****, P < 0.0001 by t test. (J) T-HEp3 cells expressing CDK2 biosensor were plated on Matrigel and treated with DMSO or 0.5 µM C26 for 48 h. Cells in different phases of the cell cycle were manually counted under a microscope. Graph shows the percentage of cells in G0/G1, S, or G2/M phases of the cell cycle. Data are shown as mean ± SEM from three independent experiments. *, P < 0.05; **, P < 0.01. (K and L) T-HEp3 cells with NT gRNA or two different NR2F1 gRNAs (guide 2 and guide 4) were plated in Matrigel and treated with DMSO or 0.5 µM C26. After 4 d, cells were counted manually under a microscope. Graphs show the mean number of single cells (J) or colonies with more three or more cells (K). Data represent mean ± SEM from two independent experiments. *, P < 0.05; **, P < 0.01 by ANOVA. See also Fig. S4.
C26 induces growth arrest via NR2F1. (A–C) T-HEp3, SQ20B, or FaDu cells were plated in Matrigel and treated with DMSO or C26 (0.2, 0.5, or 1 µM for T-HEp3; 0.5 µM for SQ20B and FaDu). After 4 d, cells were counted manually under a microscope. Graphs show percentage of single cells (A and C) or colonies of three or more cells (B). Data are shown as mean ± SEM from three independent experiments. *, P < 0.05; **, P < 0.01 by ANOVA (A and B). *, P < 0.05 by t test (C). (D) T-HEp3, SQ20B, or FaDu cells from A–C were fixed and immunostained for Ki-67. Graph shows the percentage of Ki-67+ cells. Data are shown as mean ± SEM from three (T-HEp3 and FaDu) or two (SQ20B) independent experiments. The total number of cells analyzed is 81 (DMSO) and 68 cells (C26) for T-HEp3; 125 (DMSO) and 117 cells (C26) for FaDu; and 49 (DMSO) and 34 cells (C26) for SQ20B. *, P < 0.05; **, P < 0.01 by t test. (E and F) T-HEp3 cells were pretreated in culture for 6 d with DMSO or 0.5 µM C26 and inoculated on CAM without continuous treatment (E) or with daily treatment (F). After 7 d, tumors were collected and dissociated. Graph shows the mean number of cells per tumor ± SEM from 20 tumors (DMSO) and 21 tumors (C26; E) or 7 tumors (DMSO) and 8 tumors (C26; F). *, P < 0.05; **, P < 0.01 by t test. (G–I) Single cells from patient-derived organoids were plated in Matrigel and treated with DMSO or 0.5 µM C26. After 7 d, 5–10 random widefield images per sample were acquired on a confocal microscope. Images in G are representative z-stack projections (scale bar, 100 µm). Graph in H shows the percentage of single cells or organoids per well. Data are mean ± SEM from three independent experiments. *, P < 0.05 by t test. Graph in I shows the colony area of 112 colonies (DMSO) or 33 colonies (C26) from three independent experiments. ****, P < 0.0001 by t test. (J) T-HEp3 cells expressing CDK2 biosensor were plated on Matrigel and treated with DMSO or 0.5 µM C26 for 48 h. Cells in different phases of the cell cycle were manually counted under a microscope. Graph shows the percentage of cells in G0/G1, S, or G2/M phases of the cell cycle. Data are shown as mean ± SEM from three independent experiments. *, P < 0.05; **, P < 0.01. (K and L) T-HEp3 cells with NT gRNA or two different NR2F1 gRNAs (guide 2 and guide 4) were plated in Matrigel and treated with DMSO or 0.5 µM C26. After 4 d, cells were counted manually under a microscope. Graphs show the mean number of single cells (J) or colonies with more three or more cells (K). Data represent mean ± SEM from two independent experiments. *, P < 0.05; **, P < 0.01 by ANOVA. See also Fig. S4.

Effect of C26 on HNSCC cell lines, apoptosis, cell cycle, and knockdown controls and C26 effect on single cell and colony frequencies in 3D cultures. ( A) Representative images of THEp3, FaDu, and SQ20B cells plated in Matrigel and treated with DMSO or C26 (0.5 µM) for 4 d then fixed and immunostained for Ki-67. Scale bar, 25 µm. (B) DMSO or C26 (0.5 µM) treated CAM tumors were dissociated and cell cytospins were generated. Cytospins were immunostained for cleaved caspase-3 and nuclei counterstained with DAPI. Graph shows the percentage of cleaved caspase-3+ cells from four tumors per group. (C) Representative images of T-HEp3 cells expressing CDK2 biosensor in G0/G1, S, or G2/M phases of the cell cycle. Scale bar, 25 µm. (D) NR2F2 mRNA levels in T-HEp3 cells transfected with control siRNA or two different NR2F2 siRNAs. (E and F) T-HEp3 cells transfected with control siRNA or two different NR2F2 siRNAs were plated in Matrigel and treated with DMSO or C26 (0.5 µM). After 4 d, cells were counted manually under a microscope. Graphs show the percentage of single cells (E) or colonies of three or more cells (F). Data are mean ± SEM from two independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001 by ANOVA.
Effect of C26 on HNSCC cell lines, apoptosis, cell cycle, and knockdown controls and C26 effect on single cell and colony frequencies in 3D cultures. ( A) Representative images of THEp3, FaDu, and SQ20B cells plated in Matrigel and treated with DMSO or C26 (0.5 µM) for 4 d then fixed and immunostained for Ki-67. Scale bar, 25 µm. (B) DMSO or C26 (0.5 µM) treated CAM tumors were dissociated and cell cytospins were generated. Cytospins were immunostained for cleaved caspase-3 and nuclei counterstained with DAPI. Graph shows the percentage of cleaved caspase-3+ cells from four tumors per group. (C) Representative images of T-HEp3 cells expressing CDK2 biosensor in G0/G1, S, or G2/M phases of the cell cycle. Scale bar, 25 µm. (D) NR2F2 mRNA levels in T-HEp3 cells transfected with control siRNA or two different NR2F2 siRNAs. (E and F) T-HEp3 cells transfected with control siRNA or two different NR2F2 siRNAs were plated in Matrigel and treated with DMSO or C26 (0.5 µM). After 4 d, cells were counted manually under a microscope. Graphs show the percentage of single cells (E) or colonies of three or more cells (F). Data are mean ± SEM from two independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001 by ANOVA.
To assess if C26 treatment has an effect on tumor cells directly derived from patients with HNSCC, we established a patient-derived organoid line following previously established methods (Driehuis et al., 2019). After establishing the propagatable organoid line, organoids were dissociated into single cells and plated in Matrigel with DMSO or C26. By determining the percentage of single cells and colonies after 7 d of culture, we found that C26 significantly inhibits the organoid formation efficiency, where cells are arrested in a single-cell state (Fig. 4, G and H). Additionally, organoids that form have a markedly smaller area in C26 treatment compared with DMSO (Fig. 4 I). These results indicate that C26 treatment has the ability to induce quiescence of cells directly derived from patient tumors.
Our RNA-seq and growth arrest data suggest that C26-mediated activation of NR2F1 induces cell cycle arrest. To further support this conclusion, we used T-HEp3 cells expressing a previously described DNA Helicase B (DHB)-Venus, a CDK2 biosensor that translocates from the nucleus to the cytoplasm upon phosphorylation by CDK2 (Di Martino et al., 2021,Preprint; Spencer et al., 2013). Hence, this biosensor enables the identification of the different phases of the cell cycle using fluorescent microscopy, where cells in G0/G1 phase show nuclear accumulation, cells in S phase show both nuclear and cytoplasmic localization, and cells in G2 phase show cytoplasmic and no nuclear localization (Fig. S4 C). Cells were pretreated with C26 for 48 h, plated on Matrigel, and treated for an additional 48 h. Cells were then fixed, stained with DAPI, and imaged. Using this approach, we show that C26 treatment significantly increases the percentage of cells in G0/G1 from 7% to 41% while decreasing the percentage of cells in S and G2/M phases (Fig. 4 J). These results indicate that NR2F1 activation by C26 induces cell cycle arrest at G0/G1.
To confirm that the effect of C26 on cell growth is mediated by NR2F1, we used T-HEp3 cell lines in which NR2F1 is knocked out by CRISPR-Cas9 using two different gRNAs (g2 and g4 in Fig. S3 A). C26 effect on these cells lines was evaluated in the previously described 3D Matrigel assay. While C26 increases the percentage of single cells and decreases the number of clusters in cells with NT gRNA in a manner similar to the parental cell line, the effect of C26 was completely abrogated in the two NR2F1 knockout cell lines (Fig. 4, K and L). However, knockdown of NR2F2 using two different siRNAs (see Fig. S4 D for qPCR verification) did not alter the effect of C26. C26 treatment of cells with NR2F2 knockdown increased the percentage of single cells (Fig. S4 E) and decreased the percentage of clusters (Fig. S4 F) compared with DMSO similar to cells with control siRNA (compare to Fig. 4, A, B, K, and L). These results, along with the in vivo detection of dormancy genes detected in Fig. 2 F and the selectivity controls showed in Fig. 1, indicate that the growth-suppressive effects of C26 in cancer cells is tightly dependent on an NR2F1-driven dormancy program.
C26 inhibits primary tumor growth and metastatic growth in lungs
The above RNA-seq data and growth experiments establish that C26 activates NR2F1 and induces dormancy of HNSCC cells in in vitro 3D models and in vivo on CAM. However, the CAM system does not allow monitoring long-term phenotypes in target organs, since tumors can be grown for a maximum of 7 d. Thus, we assessed whether C26-mediated activation of NR2F1 induces tumor cell dormancy and inhibits metastatic growth in mice. GFP-tagged T-HEp3 PDX cells were injected subcutaneously in BALB/c nu/nu mice. Tumors were allowed to develop and reach 300 mm3 in volume, after which neoadjuvant treatment was administered i.p. for 5 d. After 2 d of rest, tumors were surgically removed, and four cycles of adjuvant treatment (5 d of treatment and 2 d of rest) were administered (Fig. 5 A). Excised tumors were weighed at the time of surgery, and results unexpectedly showed that C26 treatment inhibited primary tumor growth in 8 out 12 mice (Fig. 5 B). Analysis of resected tumors for NR2F1 expression showed that as early as 1 wk after neoadjuvant treatment, C26 induced up-regulation of NR2F1 expression in primary tumors (Fig. S5 A). This data supports that C26 dosed in mice with the current schedule engages the target and activates NR2F1 function and expression. Importantly, C26 treatment did not induce apoptosis of proliferating primary tumor cells as evident by the absence of change in percentage of cleaved caspase-3+ cells compared with DMSO (Fig. S5 B).

C26 suppresses primary tumor growth and lung metastasis in mice. (A) Schematic depicting xenograft establishment as well as neoadjuvant and adjuvant treatment with DMSO or C26 (0.5 mg/kg/day) in mice. (B) Graph showing the weight of primary tumors surgically resected after the neoadjuvant phase. Data are mean ± SEM from 12 mice per group. ***, P < 0.001 by t test. (C) Graph showing the number of T-HEp3-GFP+ cells in collagenase-digested lung lobules counted under a fluorescence microscope. Data are mean ± SEM from 11 mice (DMSO) and 10 mice (C26). ****, P < 0.0001 by t test. (D) Lung sections from A were immunostained for vimentin to detect single DTCs, micrometastases (<50 cells), or macrometastases (>50 cells). Scale bar, 50 µm. (E) Graph showing the percentage of DMSO- or C26-treated mice with single DTCs only, micrometastases only, or micrometastases and macrometastases. **, P < 0.01 by Fisher’s exact test. (F–H) Experimental metastasis assay was performed by injecting T-HEp3 cells into tail veins of BALB/c nu/nu mice, which were treated by i.p. injection of DMSO for 3 wk, C26 (0.5 mg/kg/day) for 3 wk, or C26 (0.5 mg/kg/day) for 1 wk and then DMSO for 2 wk. Two representative images of lungs stained with H&E are shown in F. Arrowheads indicate metastatic lesions. Scale bar, 10 mm. Graphs show the number of metastases per lung section (G) and percentage of lung area section with metastasis (H). *, P < 0.05; **, P < 0.01 by t test. See also Fig. S5.
C26 suppresses primary tumor growth and lung metastasis in mice. (A) Schematic depicting xenograft establishment as well as neoadjuvant and adjuvant treatment with DMSO or C26 (0.5 mg/kg/day) in mice. (B) Graph showing the weight of primary tumors surgically resected after the neoadjuvant phase. Data are mean ± SEM from 12 mice per group. ***, P < 0.001 by t test. (C) Graph showing the number of T-HEp3-GFP+ cells in collagenase-digested lung lobules counted under a fluorescence microscope. Data are mean ± SEM from 11 mice (DMSO) and 10 mice (C26). ****, P < 0.0001 by t test. (D) Lung sections from A were immunostained for vimentin to detect single DTCs, micrometastases (<50 cells), or macrometastases (>50 cells). Scale bar, 50 µm. (E) Graph showing the percentage of DMSO- or C26-treated mice with single DTCs only, micrometastases only, or micrometastases and macrometastases. **, P < 0.01 by Fisher’s exact test. (F–H) Experimental metastasis assay was performed by injecting T-HEp3 cells into tail veins of BALB/c nu/nu mice, which were treated by i.p. injection of DMSO for 3 wk, C26 (0.5 mg/kg/day) for 3 wk, or C26 (0.5 mg/kg/day) for 1 wk and then DMSO for 2 wk. Two representative images of lungs stained with H&E are shown in F. Arrowheads indicate metastatic lesions. Scale bar, 10 mm. Graphs show the number of metastases per lung section (G) and percentage of lung area section with metastasis (H). *, P < 0.05; **, P < 0.01 by t test. See also Fig. S5.

Effect of C26 on NR2F1 expression and apoptosis in primary tumors, association of DTC numbers and tumor weight, effect of AZA+ at RA on primary tumor weight and DTC burden, effect of C26 on apoptosis of DTCs and quantification, and quantification of NR2F1 signal intensity in DTCs. (A) Representative images of primary tumors from the spontaneous metastasis experiment immunostained for NR2F1. Scale bar, 75 µm. Graph shows the percentage of NR2F1+ cells. Data are mean ± SEM from eight mice per group. *, P < 0.05 by t test. Arrowheads indicate examples of NR2F1+ cells. (B) Representative images of primary tumors from the spontaneous metastasis experiment immunostained for cleaved caspase-3 (cc-3). Scale bar, 100 µm. Graph shows the percentage of cc-3+ cells. Data are mean ± SEM from eight mice per group. Arrowheads indicate examples of cc-3+ cells. (C) Supplementary data for Fig. 5. Graphs show correlation between primary tumor weight and number of DTC per lung lobule of DMSO- or C26-treated mice. (D) Schematic depicting treatment protocol used for the combination of AZA and then ATRA/C26 in the neoadjuvant setting. (E) Graph shows weight of primary tumors surgically resected after the neoadjuvant phase. Data are mean ± SEM from 12 mice per group. *, P < 0.05; ***, P < 0.001 by t test. (F) Graph shows number of T-HEp3-GFP+ cells in collagenase-digested lung lobules counted under a fluorescence microscope. Data are mean ± SEM from 11 mice (DMSO), 10 mice (C26), and 10 mice (AZA+atRA/C26). *, P < 0.05; ****, P < 0.0001 by t test. Data for DMSO and C26 are the same as those presented in Fig. 5, B and C, as the AZA+atRA/C26 treatment was done in parallel. (G) Graph shows weight of mice (in grams) measured weekly in the experimental metastasis experiment. (H) Lungs from DMSO- or C26-treated mice described in Fig. 4 were immunostained for vimentin and cleaved caspase-3. Scale bar, 50 µm. (I) Graph shows percentage of cleaved caspase-3−/vimentin+ tumor cells in lungs. (J) Graph shows box (25th to 75th percentile) and whiskers (minimum to maximum values) of nuclear NR2F1 MFI in single DTCs only in lungs from DMSO- and C26-treated mice. ****, P < 0.0001 by t test.
Effect of C26 on NR2F1 expression and apoptosis in primary tumors, association of DTC numbers and tumor weight, effect of AZA+ at RA on primary tumor weight and DTC burden, effect of C26 on apoptosis of DTCs and quantification, and quantification of NR2F1 signal intensity in DTCs. (A) Representative images of primary tumors from the spontaneous metastasis experiment immunostained for NR2F1. Scale bar, 75 µm. Graph shows the percentage of NR2F1+ cells. Data are mean ± SEM from eight mice per group. *, P < 0.05 by t test. Arrowheads indicate examples of NR2F1+ cells. (B) Representative images of primary tumors from the spontaneous metastasis experiment immunostained for cleaved caspase-3 (cc-3). Scale bar, 100 µm. Graph shows the percentage of cc-3+ cells. Data are mean ± SEM from eight mice per group. Arrowheads indicate examples of cc-3+ cells. (C) Supplementary data for Fig. 5. Graphs show correlation between primary tumor weight and number of DTC per lung lobule of DMSO- or C26-treated mice. (D) Schematic depicting treatment protocol used for the combination of AZA and then ATRA/C26 in the neoadjuvant setting. (E) Graph shows weight of primary tumors surgically resected after the neoadjuvant phase. Data are mean ± SEM from 12 mice per group. *, P < 0.05; ***, P < 0.001 by t test. (F) Graph shows number of T-HEp3-GFP+ cells in collagenase-digested lung lobules counted under a fluorescence microscope. Data are mean ± SEM from 11 mice (DMSO), 10 mice (C26), and 10 mice (AZA+atRA/C26). *, P < 0.05; ****, P < 0.0001 by t test. Data for DMSO and C26 are the same as those presented in Fig. 5, B and C, as the AZA+atRA/C26 treatment was done in parallel. (G) Graph shows weight of mice (in grams) measured weekly in the experimental metastasis experiment. (H) Lungs from DMSO- or C26-treated mice described in Fig. 4 were immunostained for vimentin and cleaved caspase-3. Scale bar, 50 µm. (I) Graph shows percentage of cleaved caspase-3−/vimentin+ tumor cells in lungs. (J) Graph shows box (25th to 75th percentile) and whiskers (minimum to maximum values) of nuclear NR2F1 MFI in single DTCs only in lungs from DMSO- and C26-treated mice. ****, P < 0.0001 by t test.
Lungs were collected when the mice were sacrificed at the end of the adjuvant treatment period. One lung lobe was processed for collagenase dissociation, while other lobes were prepared for formalin fixation and paraffin embedding. Analysis of the total number of GFP+ tumor cells per lung lobe revealed that mice treated with C26 had almost half the number of tumor cells compared with control mice (Fig. 5 C). This metastasis inhibitory effect of C26 is independent of the effect on primary tumor growth, since primary tumor weight and number of lung DTCs are not correlated (Fig. S5 C). Since the above analysis does not distinguish between solitary DTCs and large or small metastatic lesion frequency, we immunostained lungs using an antibody for vimentin, which has been previously used to detect T-HEp3 cells in lungs (Bragado et al., 2013; Fluegen et al., 2017; Sosa et al., 2015), and determined the number of mice with solitary cells only, micrometastases (<50 cells), and macrometastases (>50 cells; Fig. 5 D). Results showed that 33% and 50% of DMSO-treated mice have micrometastases only or micrometastases and macrometastases in their lungs, respectively. However, 0% of mice treated with C26 showed micro- or macrometastases, and only single DTCs persisted in the lungs of these mice (Fig. 5 E). We have previously reported that in this aggressive model of HNSCC and in the absence of NR2F1, solitary DTCs in lungs transition out of a short-term quiescence and form metastasis (Sosa et al., 2015). Our current data indicate that while tumor cells were able to disseminate to the lungs and lodge in a solitary cell state as we showed previously (Bragado et al., 2013), C26 treatment prevents single cells from dividing and growing into overt metastases. This is consistent with what we found in vitro in the 3D Matrigel cultures, where C26 treatment arrested cells in a single-cell state.
In a parallel group to the two groups presented above, we combined in the neoadjuvant phase a pretreatment with AZA, atRA, and C26 (Fig. S5 D). This treatment shortened the time of C26 treatment to accommodate the combination. The rationale for this treatment is that low NR2F1 expression in T-HEp3 cells may not allow for a full effect of the C26 agonist. As shown previously, the AZA+atRA treatment may restore NR2F1 expression (among other gene expression changes) and allow for more receptor to be available for the C26 treatment. However, the AZA+atRA pretreatment did not improve the effect of C26 on primary tumor or metastatic growth (Fig. S5, E and F). In the primary tumors the lack of effect could be due to the shorter exposure to C26 or an interfering effect from the AZA+atRA treatment. Nevertheless, the results show that C26 as a single agent can engage the target systemically and suppress metastasis development.
To assess the reversibility and stability of the metastasis suppression phenotype caused by C26 treatment, we performed an experimental metastasis experiment by injecting T-HEp3 cells into the tail vein of BALB/c nu/nu mice. These cells were treated in culture with DMSO or C26 for 1 wk before injection similar to Fig. 4, E and F. Mice injected with DMSO-pretreated cells were treated with DMSO in vehicle for 3 wk via i.p. delivery. Mice injected with C26 pretreated cells were divided into two groups. The first group was treated continuously with C26 for 3 wk, and the second group was treated with C26 for 1 wk, then the treatment was stopped and followed by 2 wk of DMSO. At the end of treatment, mice were euthanized, and the resected lungs were stained with H&E and analyzed for metastasis frequency and burden. Our results show that significantly, both continuous and interrupted treatments with C26 inhibited metastasis to the same extent as evidenced by a comparable decrease in number of metastases (frequency) as well as the percentage of lung area covered with metastases (burden) compared with DMSO (Fig. 5, F–H).
To determine if C26 treatment causes adverse toxic effects in mice, we monitored the weight of the mice weekly, and at the end of the treatment, we performed autopsy, complete blood count, and liver and kidney function analysis. Neither continuous nor interrupted C26 treatment induced weight loss (Fig. S5 G). Additionally, as determined by a veterinary pathologist, there were no signs of gross, blood, kidney, or liver toxicity upon C26 treatment. These results indicated that at least for 3 wk, C26 induces a potent and, as predicted, self-sustained long-lasting metastasis inhibitory effect, with no evidence of toxicity.
C26 suppresses lung metastasis by inducing an NR2F1hi/p27hi/Ki67lo/p-S6lo dormancy profile in solitary DTCs
We next sought to determine if the inability of lung DTCs to proliferate and grow into overt metastases in C26-treated mice is due to induction of NR2F1 and a dormancy phenotype. To verify that single DTCs in C26-treated mice are nonproliferative dormant cells, we immunostained lungs, which were collected from the previously described spontaneous dissemination experiment (Fig. 5 A), for Ki-67 and determined the percentage of proliferative Ki-67+/vimentin+ tumor cells in lungs. While 45% of human-specific vimentin+ tumor cells in control are Ki-67+, this is significantly reduced to 7% in C26-treated mice (Fig. 6, A and B). Importantly, more than 99% tumor cells in the lungs of both DMSO and C26-treated mice were negative for cleaved caspase-3 (Fig. S5, H and I), indicating that solitary DTCs in C26-treated lungs are growth arrested, but not apoptotic. These data show that single DTCs in lungs of C26-treated mice are in a quiescent nonproliferative state. To determine if these single DTCs are in a dormant state, we immunostained for NR2F1, which not only is the target of C26, but also serves as a dormancy marker and is more informative on time to metastasis than proliferation markers alone, as we showed in breast cancer DTCs from patients (Borgen et al., 2018). Results showed that lung DTCs in C26-treated had significantly higher NR2F1 expression, as evidenced by an increase in the percentage of NR2F1hi cells (Fig. 6, A and C) and MFI per nucleus (Fig. 6, A and D). Interestingly, NR2F1 expression was significantly higher in DTCs of C26-treated mice compared with solitary DTCs in DMSO-treated mice (Fig. S5 J). We also performed a similar analysis for the CDK inhibitor p27, which is a target of NR2F1 and also serves as a complementary dormancy marker (Sosa et al., 2014). The percentage of cells with nuclear accumulation of p27 (Fig. 6, E and F) and nuclear p27 MFI (Fig. 6, E and G) are significantly higher in lung DTCs of C26-treated mice (Fig. 6, E and F). Altogether, these results confirm that C26 treatment prevents outgrowth of lung metastases by activating NR2F1, which up-regulates its own expression as well as downstream targets, including p27, and keeps DTCs in a dormant state.

C26 suppresses metastatic growth in lungs by inducing dormancy. (A) Lungs from DMSO- or C26-treated mice described in Fig. 4 were immunostained for vimentin, Ki-67, and NR2F1. Scale bar, 50 µm. (B and C) Graphs showing the percentage of Ki-67+/vimentin+ (B) or NR2F1+/vimentin+ (C) tumor cells in lungs. Data are mean ± SEM. (D) Graph showing box (25th to 75th percentile) and whiskers (minimum to maximum values) of nuclear NR2F1 MFI per cell. Data in B–D are from five mice per group (DMSO, 250 cells; C26, 150 cells). *, P < 0.05; **, P < 0.01; ****, P < 0.0001 by t test. (E) Lungs from DMSO- or C26-treated mice described in Fig. 4 were immunostained for vimentin and p27. Scale bar, 50 µm. (F) Graph showing the percentage of vimentin+ tumor cells with nuclear accumulation of p27. Data are mean ± SEM. (G) Graph shows box (25th to 75th percentile) and whiskers (minimum to maximum values) of nuclear p27 MFI per cell in vimentin+ tumor cells. Data in F and G are from five mice per group (DMSO, 174 cells; C26, 85 cells). **, P < 0.01; ****, P < 0.0001 by t test. (H) Lungs from DMSO- or C26-treated mice described in Fig. 4 were immunostained for vimentin and p-S6. Scale bar, 50 µm. Arrowheads in C26 indicate solitary DTCs that are negative for p-S6. (I) Graph showing the percentage of vimentin+ tumor cells with a positive p-S6 signal. Data are mean ± SEM from five mice per group. **, P < 0.01 by t test. See also Fig. S5.
C26 suppresses metastatic growth in lungs by inducing dormancy. (A) Lungs from DMSO- or C26-treated mice described in Fig. 4 were immunostained for vimentin, Ki-67, and NR2F1. Scale bar, 50 µm. (B and C) Graphs showing the percentage of Ki-67+/vimentin+ (B) or NR2F1+/vimentin+ (C) tumor cells in lungs. Data are mean ± SEM. (D) Graph showing box (25th to 75th percentile) and whiskers (minimum to maximum values) of nuclear NR2F1 MFI per cell. Data in B–D are from five mice per group (DMSO, 250 cells; C26, 150 cells). *, P < 0.05; **, P < 0.01; ****, P < 0.0001 by t test. (E) Lungs from DMSO- or C26-treated mice described in Fig. 4 were immunostained for vimentin and p27. Scale bar, 50 µm. (F) Graph showing the percentage of vimentin+ tumor cells with nuclear accumulation of p27. Data are mean ± SEM. (G) Graph shows box (25th to 75th percentile) and whiskers (minimum to maximum values) of nuclear p27 MFI per cell in vimentin+ tumor cells. Data in F and G are from five mice per group (DMSO, 174 cells; C26, 85 cells). **, P < 0.01; ****, P < 0.0001 by t test. (H) Lungs from DMSO- or C26-treated mice described in Fig. 4 were immunostained for vimentin and p-S6. Scale bar, 50 µm. Arrowheads in C26 indicate solitary DTCs that are negative for p-S6. (I) Graph showing the percentage of vimentin+ tumor cells with a positive p-S6 signal. Data are mean ± SEM from five mice per group. **, P < 0.01 by t test. See also Fig. S5.
Our RNA-seq analysis revealed a suppression of mTOR signaling pathway upon C26 treatment (Fig. 3 B). This was a surprising finding, as we had not previously connected NR2F1 signaling to mTOR signaling. To assess if this pathway is down-regulated in solitary DTCs in C26-treated mice, we immunostained for phosphorylated ribosomal protein S6 (p-S6), which is a downstream readout for mTOR pathway activation (Liu and Sabatini, 2020). This analysis showed that while ∼50% of tumor cells in lungs of DMSO-treated mice are positive for p-S6, none of the solitary DTCs in lungs of C26-treated mice were positive for this marker (Fig. 6, H and I). These data corroborate the findings from the RNA-seq analysis and further indicate that C26 treatment and NR2F1 activation function as novel suppressors of mTOR signaling coupled with a gain of neural crest differentiation markers in HNSCC cells.
NR2F1 levels and activation inform on solitary human DTCs state and disease progression in HNSCC
We previously reported that NR2F1 expression is silenced in primary tumors and overt HNSCC lymph node metastasis (Sosa et al., 2015). However, we did not have access to lymph nodes with solitary residual DTCs to investigate whether NR2F1 is up-regulated when DTCs are in a solitary state and presumably dormant (Sproll et al., 2018). To address this specific question, we stained patient samples for NR2F1 in normal adjacent epithelium, primary tumors, and lymph nodes that were confirmed to have solitary HNSCC DTCs, but not overt metastasis (Sproll et al., 2018). Pan-cytokeratin (pan-CK) was used as a marker to identify cells of epithelial origin in all tissues (Sproll et al., 2017). Quantitative image analysis was used to score pan-CK+ DTCs as NR2F1hi, NR2F1med, or NR2F1−/low. This analysis revealed that the percentage of NR2F1hi cells drops from 28% in normal epithelium to 2.4% in primary tumors, while 20% of solitary DTCs are NR2F1hi. Conversely, the percentage of NR2F1−/low is 39% in normal epithelium, 84% in primary tumors, and 63% in solitary DTCs (Fig. 7, A and B). These results indicate that a subpopulation of solitary DTCs are more frequently positive for NR2F1 than cells in primary tumors, supporting that NR2F1 is dynamically regulated in cancer cells in primary versus secondary organs and could also mark dormant solitary DTCs in HNSCC patients.

NR2F1 levels in solitary human DTCs and C26 signature in disease progression in HNSCC. (A) HNSCC patient samples of normal epithelium, primary tumors, or lymph nodes with solitary DTCs were immunostained for NR2F1 and pan-CK and nuclei counterstained with DAPI. Scale bar, 50 µm (normal epithelium and primary tumor) or 10 µm (solitary DTC). The total number of cells analyzed is 1,814 cells for normal epithelium and 3,841 cells for primary tumors from two patients and 271 solitary DTCs from three patients. (B) Graph showing the percentage of NR2F1hi, NR2F1med, or NR2F1−/low cells in normal epithelium (n = 1,814 cells from two patients), primary tumor (PT; n = 3,841 cells from two patients), or solitary DTCs (n = 271 cells from three patients). ***, P < 0.001 by Fisher’s exact test adjusted with false discovery rate correction by Benjamini–Hochberg. (C) Kaplan–Meier plots of overall survival (left panel) or relapse-free survival (right panel) generated from 500 HNSCC patients using the Kaplan–Meier plotter database. Hazard ratio (HR) with 95% confidence intervals and log-rank P value are shown.
NR2F1 levels in solitary human DTCs and C26 signature in disease progression in HNSCC. (A) HNSCC patient samples of normal epithelium, primary tumors, or lymph nodes with solitary DTCs were immunostained for NR2F1 and pan-CK and nuclei counterstained with DAPI. Scale bar, 50 µm (normal epithelium and primary tumor) or 10 µm (solitary DTC). The total number of cells analyzed is 1,814 cells for normal epithelium and 3,841 cells for primary tumors from two patients and 271 solitary DTCs from three patients. (B) Graph showing the percentage of NR2F1hi, NR2F1med, or NR2F1−/low cells in normal epithelium (n = 1,814 cells from two patients), primary tumor (PT; n = 3,841 cells from two patients), or solitary DTCs (n = 271 cells from three patients). ***, P < 0.001 by Fisher’s exact test adjusted with false discovery rate correction by Benjamini–Hochberg. (C) Kaplan–Meier plots of overall survival (left panel) or relapse-free survival (right panel) generated from 500 HNSCC patients using the Kaplan–Meier plotter database. Hazard ratio (HR) with 95% confidence intervals and log-rank P value are shown.
To further explore the relevance of the transcriptional changes induced by C26 treatment, we explored how the C26-induced dormancy signature in T-HEp3 cells is associated with outcomes of HNSCC patients. To this end, we generated a C26 gene signature (Table S2) and assessed whether the presence of this signature in primary tumors from HNSCC patients revealed different patterns of overall survival or relapse-free survival using Kaplan–Meier plot analysis. The C26 gene signature includes the top 25 up-regulated and top 25 down-regulated genes that had the largest fold changes in the RNA-seq data we obtained, including neural crest cell differentiation genes (Table S2). We also included NR2F1, as we had prior data showing that under certain conditions, such as hypoxia, it can be turned on in HNSCC primary tumors and spawn dormant DTCs (Fluegen et al., 2017). Kaplan–Meier plots showed that the probability of both overall survival and relapse-free survival is significantly higher in patients carrying tumors with the C26 gene signature compared with patients without the signature (Fig. 7 C). These data support that a subpopulation of HNSCC patients carry primary tumors enriched for a unique NR2F1 activation dormancy signature that occurs spontaneously. Importantly, patients carrying these tumors had improved relapse-free survival, suggesting that cancer cells that disseminated locoregionally or distantly from these tumors may have taken longer to produce recurrences due to higher NR2F1 activity. Since the signature has no enrichment in proliferation versus growth arrest cell cycle genes, it may not be simply interpreted as a difference in proliferative potential in patients’ tumors.
Discussion
We previously discovered that NR2F1 is an important regulator of cancer cell dormancy (Sosa et al., 2015), a finding that was independently confirmed (Cackowski et al., 2017; Fluegen et al., 2017; Kim et al., 2012; Liu et al., 2018; Sosa et al., 2015). We sought to determine if this dormancy marker could be drugged to take advantage of its stable quiescence-inducing function. Our results using a structure-based in silico screening approach leveraged a new agonist (C26) that could accomplish our objective of turning “on” an NR2F1 pathway in malignant cancer cells. The inability of C26 to activate RXRα as well as the NR2F1 knockout and NR2F2 knockdown controls support the selectivity of C26. It is possible that a crystal structure of NR2F1 LBD, which as of now has not been solved yet, may yield more potent and selective agonists. NR2F2 also does not appear to be a target of C26 as the sgRNAs were designed to target only NR2F1, and KO of NR2F1, but not NR2F2 knockdown, eliminated the C26 effect. A significant finding was that C26 treatment resulted in autoinduction of NR2F1 expression in HNSCC T-HEp3 cells, which show silencing (albeit not complete) of NR2F1 through repressive histone modifications (Sosa et al., 2015). Induction of endogenous NR2F1 by C26 suggests that this silencing is not as tight as previously expected and that, although not tested directly, engaging NR2F1 with C26 may allow it to rapidly remodel the repressive chromatin at promoters, turn on its own expression, and drive downstream signals for dormancy onset. The lack of additional effect of the AZA+atRA treatment on metastasis further supports that C26 can alone restore high NR2F1 expression and activity. Whether other epigenetic drugs such as histone deacetylase or bromodomain inhibitors (Manzotti et al., 2019) might have additive or synergistic effects with C26 is unknown and will be tested in future studies. This is interesting, as activating this orphan nuclear receptor may be useful in treating not only tumors with abundant expression of NR2F1 but also those that express low levels of NR2F1. NR2F1 agonists might be particularly useful in breast cancer, where NR2F1 expression is highly enriched in estrogen receptor+ tumors. Interestingly, these are the patients that commonly show late relapse (Kim et al., 2012). Thus, agonists for NR2F1 may be used as a therapeutic approach to suppress reawakening of dormant cells kept in that state by anti-estrogen therapies or even in triple-negative and other breast cancer subtypes.
We also wondered whether in vivo treatment with an NR2F1 agonist would be sufficient to prevent awakening of DTCs, and if the effect is long-lived. Our in vivo experiments in the CAM avian PDX system and in mice support that C26-mediated activation of NR2F1 induces a strong and long-lived growth suppressive signal that can persist even if the treatment is removed for at least 2 wk (equivalent to ∼1.6 yr in humans). While pretreatment of T-HEp3 cells in culture with C26 did not significantly inhibit cell proliferation in 2D culture in vitro (data not shown), it did suppress growth in 3D cultures, which was corroborated using a CDK2-biosensor of cell cycle progression, and 1 wk later in vivo on CAM, an effect that was stronger with continuous treatment in vivo. These data support that perhaps a treatment schedule that activates NR2F1 might not need to be continuous, as it may operate as a growth arrest program that is self-sustained, similar to what we previously reported for AZA and atRA (Sosa et al., 2015). A unique set of findings was that the C26-regulated program might be linked to a differentiation-like program in the neural crest lineage that may stabilize the growth arrest phenotype induced by C26 in DTCs. This program may be reinforced by the strong inhibition of S6-kinase, a canonical mTOR target. In the mouse PDX experiments, the neoadjuvant plus adjuvant strategy was chosen based on prior data showing that the very aggressive T-HEp3 PDX model spreads soon after subcutaneous implantation and that metastases can grow almost simultaneously with the primary tumor (Bragado et al., 2013). Nevertheless, our results strongly support that C26 was able to hold these DTCs, which are highly efficient at initiating metastasis (Aguirre-Ghiso et al., 2004; Ossowski et al., 1987), in a dormant state. This result, and the stability of the C26 effect without any detectable toxicity, was further corroborated in the experimental metastasis assay that as performed here exclusively targeted freshly seeded DTCs in lungs. The dormancy phenotype caused by C26 was supported by the NR2F1hi/p27hi/Ki67lo/p-S6lo profile that these DTCs displayed. Furthermore, none of the animals treated with C26 displayed micro- or macrometastasis, strengthening the significance of activating NR2F1 to prevent metastatic relapse. Even when analyzing only NR2F1 levels specifically in solitary DTCs, which are more frequently spontaneously high for NR2F1 (Sosa et al., 2015), we found that C26-treated animals displayed DTCs with higher NR2F1 expression than control animals. This corroborates that C26 might be preventing metastasis initiation by maintaining a dormant solitary cancer cell state. Surprisingly, C26 inhibited growth of some primary tumors and engaged the target in all tested tumors regardless of size. This further highlights the ability to use NR2F1 detection as a marker of target engagement and the power of using NR2F1 agonists as a treatment strategy not only in an adjuvant setting (M0 patients in the clinic) but also to cotarget proliferative and dormant DTCs coexisting in stage IV (M1) patients.
Mechanistically, there are some open questions. For example, one specific NR2F1 antibody that was used in the Matrigel and CAM assays revealed a specific speckled nuclear localization of NR2F1 in large areas of the nucleus of the size of nucleoli. This signal was highly induced upon C26 treatment. Another NR2F1-specific antibody, which was used for paraffin-embedded lung tissues, reveals a more homogeneous specific nuclear signal. These differences may indicate that there are different nuclear NR2F1 pools (and epitopes) that might exhibit differential spatial localization and function, which requires further analysis. For example, it will be important to determine if C26 is inducing the same promoter and enhancer occupancy that NR2F1 shows during development (Rada-Iglesias et al., 2012) or in cells that have endogenously high NR2F1 levels. To this end our RNA-seq data and confirmatory experiments using quantitative RT-PCR revealed that signatures associated with neural crest differentiation and repression of EMT programs were regulated by C26, arguing that strong activation and up-regulation of NR2F1 via C26 may direct this nuclear receptor to activate these developmental programs while also suppressing mTOR signaling and proliferation programs, the latter commonly linked to a differentiated state. The inhibition of mTOR signaling strongly argues that C26 may also induce via NR2F1 a metabolic reprogramming that will be explored in separate studies. In HNSCC, mTOR inhibition has been tested as a potential therapy and showed mainly disease stabilization and partial response (Patel et al., 2020). It is tempting to speculate that agonists of NR2F1 plus mTOR inhibitors may be a potential effective treatment to manage residual but also overt recurrent disease in HNSCC. It was intriguing to find that patients with HNSCC primary tumors that expressed a C26-NR2F1 activity signature had better relapse-free and overall survival. This suggests that NR2F1-positive tumors may turn on a C26-like program that affects the fate of DTCs after dissemination. It also suggests that the signature, along with NR2F1 detection, may identify a priori patients who could benefit from an NR2F1 agonist. We have shown that primary tumor hypoxia can induce and NR2F1-dependent program that manifests after dissemination (Fluegen et al., 2017). However, it is unknown if the hypoxia-induced NR2F1 program overlaps with the C26 activity. We have not tested if C26 stabilizes the protein and whether this adds to the long-term effects we observed in vitro and in vivo. Finally, studies on reprogramming strategies, which were first designed in a cancer cell–centric way, revealed that they were also modulating the immune system to favor tumor control (Lu et al., 2020). We have so far not explored the contribution of non-T cell–dependent responses to the effect of C26 on metastasis in nude mouse PDX models.
A long-term objective of our work is to provide proof-of-principle data that dormancy modulation can be exploited to find alternative strategies to prevent metastatic disease. Our experience using a combination of AZA and RA to activate global dormancy programs (Sosa et al., 2015) was a first step. This knowledge led to the development of an ongoing clinical trial that has as a goal to treat prostate cancer patients with biochemical relapse (NCT03572387). We have now advanced a step further by rationally designing a strategy based on the selective activation of NR2F1. Our data revealed that a selective NR2F1 agonist stably suppresses aggressive metastasis in a HNSCC PDX model, showing that it is possible to exploit dormancy mechanisms as therapeutic strategies to prevent metastasis. There is skepticism in the industry setting about advancing dormancy-inducing strategies, because there is a paralyzing misconception that trials need to last for many years, which would be costly (Aguirre-Ghiso, 2021). Our clinical trial in prostate cancer and other trials (Aguirre-Ghiso, 2021) are clear examples that it is not accurate to assume that readouts of dormancy therapies would take long to be obtained. One could envision many scenarios in which an agonist such as C26 could be tested within months. For example, in HNSCC, window-of-opportunity trials testing the biological response for further patient selection for treatment in the adjuvant setting could be designed by randomizing treatment between biopsy-based diagnosis and surgery. Overall, our study reveals a mechanism-based and rationally designed strategy to exploit NR2F1-activated dormancy as a therapeutic option to prevent metastatic relapse.
Materials and methods
Cell lines
T-HEp3 cells were derived from a lymph node metastasis from a HNSCC patient as described previously (Ossowski and Reich, 1983) and kept as PDXs on chick CAM. D-HEp3 cells were obtained by passing T-HEp3 cells for more than 40 generations in vitro (Ossowski and Reich, 1983). In vitro, these cells were cultured in DMEM with 10% of FBS and 100 U penicillin/0.1 mg/ml streptomycin. SQ20B, FaDu, and HEK293T cells were obtained from ATCC and grown in the same medium as HEp3 cells. All cells were grown at 5% CO2 and 37°C and regularly tested for mycoplasma. Cell transfection was performed using Lipofectamine 3000 (Invitrogen) following manufacturer’s instructions. siRNA transfection was performed using Lipofectamine RNAiMAX (Invitrogen) following manufacturer’s instructions.
Reagents and antibodies
C26 (molecular formula C28H30N4OS) was purchased from ChemBridge (6596020). atRA was purchased from Sigma-Aldrich (R2625). Human NR2F1 plasmid was a kind gift from Dr. Gilles Salbert (Rennes University, Rennes, France). Primary antibodies used for Western blot and immunofluorescence (IF) are listed in Table S3. Human NR2F2 siRNA was purchased from Horizon Discovery (LQ-003422-00-0002).
In silico NR2F1 LBD modeling and agonist screen
The 3D structure of NR2F1 LBD domain was predicted by comparative protein structure modeling based on its alignment with the structure of RXRα LBD, whose crystal structure in complex with 9-cis-RA has been solved (PDB accession no. 1FM6; Gampe et al., 2000). This analysis was performed using the software MODELLER v9.10 (Webb and Sali, 2014). The resulting NR2F1 LBD was used to screen for potential agonists from a library of 110,000 drug-like small molecules (ChemBridge) using computational docking. Docking scores were generated using two different software packages, AutoDock 4 release 4.2.3 (Goodsell et al., 1996) and eHiTS (Zsoldos et al., 2007). In both cases, the docking grid was centered on the position of the 9-cis-RA in the RXRα structure. The target and the ligands were prepared using AutoDock Tools.
Luciferase reporter assay
HEK293T or D-HEp3 cells were transfected with a firefly luciferase expression reporter driven by RARE (Farias et al., 2005) or NR2F1 cis element (Signosis; LR-2029). Cells were cotransfected with Renilla luciferase as an internal control. H3K293T cells were also transfected with wild-type NR2F1 cDNA. Cells were treated with DMSO or C26 for 36 h, after which they were lysed. Both Renilla and firefly luciferase activities were measured using dual-luciferase reporter assay kit (Promega; E1910) following manufacturer’s instructions. Data were reported as firefly/Renilla luciferase activity.
RXRα activation assay
RXRα activation by C26 was assessed using LanthaScreen time-resolved fluorescence resonance energy transfer RXR α coactivator assay (Thermo Fisher Scientific; PV4797). Briefly, a mixture of fluorescein-coregulator peptide and terbium anti-glutathione-S transferase (anti-GST) antibody was added to GST-conjugated RXRα LBD with varying concentrations of C26. DMSO and 9-cis-RA were used as 0% activation control and 100% activation control, respectively. The terbium label on the anti-GST antibody was then excited at 340 nm, and the energy transferred to the fluorescein label on the coactivator peptide was detected as emission at 520 nm. The emission ratio (ER) was then calculated using the following formula: fluorescein emission (520 nm)/terbium emission (495 nm). Percent activation was then calculated using the following formula: (ERsample − ER0% activation control/ER100% activation control − ER0% activation control) × 100.
Generation of knockout cell lines
NR2F1-knockout cell lines were generated using CRISPR-Cas9–targeted genome editing. Briefly, two separate NR2F1 gRNAs (guide 2, 5′-GATCCGCAGGACGACGTGGC-3′; and guide 4, 5′-GGCTGCCGTAGCGCGACGTG-3′) were cloned into pLentiCRISPRv2 (Addgene; 52961). An NT gRNA (5′-GTATTACTGATATTGGTGGG-3′) was used as a control. Lentiviral vectors were produced in HEK293T cells and viral supernatants were used to infect T-HEp3 or D-HEp3 cells. Cells were then selected using 2 µg/ml puromycin (Sigma-Aldrich; P8833). NR2F1 knockout was confirmed by Western blot.
Generation of patient-derived organoids
Fresh HNSCC tissue was provided to our laboratory via the Cancer Biorepository at Icahn School of Medicine at Mount Sinai (New York, NY). The sample was deidentified, obtained with institutional review board approval, and deemed nonhuman subject research.
The tissue was then processed, and organoids were generated following previously described methods, with some modifications (Driehuis et al., 2019). Briefly, tumor tissue was digested with collagenase-1A (Sigma-Aldrich; C9891) for 30 min at 37°C, after which the reaction was stopped with cold medium and the suspension was strained over a 100-µm filter and centrifuged at 1,000 rpm at 4°C. The resulting pellet was resuspended in ice-cold Matrigel (Corning; 356231), and 40-µl droplets were plated on a preheated 24-well plate and incubated at 37°C for 30 min, after which growth medium was added. Organoid growth medium consisted of DMEM/F12 (Life Technologies; 12634-034), supplemented with 1x GlutaMAX (Life Technologies; 12634-034), penicillin–streptomycin (Life Technologies; 15140-122), and 10 mmol/liter Hepes (Life Technologies; 15630-056). Medium also contained 1 × B27 supplement (Life Technologies; 17504-044), 1.25 mmol/liter N-acetyl-l-cysteine (Sigma-Aldrich; A9165), 10 mmol/liter Nicotinamide (Sigma-Aldrich; N0636), 50 ng/ml human EGF (Peprotech; AF-100-15), 500 nmol/liter A83-01, 10 ng/ml human FGF10 (Peprotech; 100-26), 5 ng/ml human FGF2 (Peprotech; 100-18B), 1 μmol/liter Prostaglandin E2 (Tocris Bioscience; 2296), 0.3 μmol/liter CHIR 99021 (Sigma-Aldrich; SML1046), 1 μmol/liter Forskolin (R&D Systems; 1099), 0.5 µg/ml R-spondin (R&D Systems; 4645-RS-025), and 100 ng/ml Noggin (Peprotech; 120-10C). 10 μmol/liter Y-27632 (Sigma-Aldrich; Y0503) was added when single cells were plated. The medium was changed every 3 d. The organoids were passaged every 2 wk.
3D Matrigel assay
1,000 cells (T-HEp3, FaDu, SQ20B, or cells from patient-derived organoids) were seeded in 50 µl growth factor–reduced Matrigel (Corning; 356231) in 8-well chamber slides (Falcon). T-HEp3, FaDu, and SQ20B cells were grown in their growth medium with reduced FBS content (2–5%), while cells from patient-derived organoids were grown in organoid growth medium. Cultures were treated every 24 h starting at day 0 with DMSO or 0.5 µM C26. Single cells and clusters were counted after 4 d (T-HEp3, FaDu, and SQ20B) or 7 d (cells from patient-derived organoids) using a light microscope, and the cultures were then fixed with 2% paraformaldehyde for further analysis.
Chick CAM assay
T-HEp3 tumor growth on CAM was previously described (Aguirre Ghiso et al., 1999). Briefly, 150 × 103 T-HEp3 cells were inoculated on CAM and allowed to grow. Tumors were treated every 24 h with 50 µl DMSO or 0.5 µM C26 starting at day 0. After 7 d, tumors were harvested and digested with collagenase-1A (Sigma-Aldrich; C9891) for 30 min at 37°C. T-HEp3 tumor cells (recognized by their very large size compared with chicken cells) were counted using a hemacytometer.
RNA-seq from in vivo CAM tumors
T-HEp3-GFP cells were pretreated with DMSO or 0.5 µM C26 for 6 d then inoculated on CAM and treated every 24 h. After 7 d, tumors were collected and dissociated into single cells. FACS was used to isolate T-Hep3 tumor cells using GFP. RNA extraction, library preparations, and sequencing were conducted at Genewiz as follows: total RNA was extracted from fresh frozen cell pellets using the Qiagen RNeasy Plus Universal mini kit following the manufacturer’s instructions (Qiagen). RNA samples received were quantified using Qubit 2.0 Fluorometer (Life Technologies) and RNA integrity was checked using Agilent TapeStation 4200 (Agilent Technologies). RNA-seq libraries were prepared using the NEBNext Ultra RNA Library Prep Kit from Illumina following the manufacturer’s instructions (New England Biolabs). Briefly, mRNA was first enriched with oligo(dT) beads. Enriched mRNA was fragmented for 15 min at 94°C. First-strand and second-strand cDNA was subsequently synthesized. cDNA fragments were end repaired and adenylated at 3′ ends, and universal adapters were ligated to cDNA fragments, followed by index addition and library enrichment by limited-cycle PCR. The sequencing libraries were validated on the Agilent TapeStation (Agilent Technologies) and quantified using a Qubit 2.0 Fluorometer (Invitrogen) and qPCR (KAPA Biosystems). The sequencing libraries were clustered on one lane of a flow cell. After clustering, the flow cell was loaded on the Illumina HiSeq instrument (4,000 or equivalent) according to manufacturer’s instructions. The samples were sequenced using a 2 × 150-bp paired-end configuration. Image analysis and base calling were conducted by the HiSeq Control Software. Raw sequence data generated from Illumina HiSeq were converted into FASTQ files and demultiplexed using Illumina bcl2fastq 2.17 software. One mismatch was allowed for index sequence identification.
Three replicate samples per group were used (in order to get enough cells, each replicate included cells combined from two tumors). RNA-seq libraries from all six samples were processed by the BiNGS bioinformatics core at Mount Sinai School of Medicine using the same pipeline for compatibility. R (v.4.0.3) was used to perform bioinformatics analysis. Quality control was performed using FastQC (v0.11.8). Trim Galore! (v0.6.5) was used to trim the adapter sequences with a quality threshold of 20. The human genome reference used was GRCh38.p13 and GENCODE release 36. Alignment was performed using STAR aligner (v2.7.5b; Dobin et al., 2013). Gene-level read counts were obtained by using Salmon (v1.2.1) for all libraries (Patro et al., 2017). All samples passed the quality control requirements, with >60% of reads uniquely mapping (>20 million uniquely mapped reads for each library) using STAR. Differential expression analysis was performed using the gene-level read counts and the DESeq2 (v1.28.1) R package (Love et al., 2014). Heatmaps show the z-scores of gene-level read counts and were generated using heatmaply (v1.1.0; Galili et al., 2018). Genes with adjusted P value < 0.05 were considered differentially expressed. Enrichment of Hallmark gene sets was done using GSEA (v4.1.0). WikiPathways gene set analysis was performed using the clusterProfiler R package (v3.16.0; Yu et al., 2012). Gene sets were obtained from the Molecular Signatures Database (Liberzon et al., 2015; Liberzon et al., 2011; Subramanian et al., 2005). Core pathway analysis was performed, and a graphical summary was generated using IPA (Qiagen; v60467501).
Mouse xenograft and experimental metastasis studies
750 × 103 cells were injected subcutaneously in 8-wk-old female BALB/c nu/nu mice (Jackson Laboratories) in the interscapular region. Mice were inspected every 48 h, and arising tumors were measured with calipers in two perpendicular diameters. When tumors reached ∼300 mm3, mice were treated i.p. with DMSO or C26 (0.5 mg/kg/day) for 5 consecutive days. Following 2 d of rest, mice were injected with the anesthetics ketamine (80–120 mg) and xylazine (5 mg), and tumors were surgically removed and weighed. Sutures were then performed using a wound clipper. After surgery, mice were subjected to four cycles of adjuvant therapy (5 d of treatment followed by 2 d of rest) with DMSO or C26 (0.5 mg/kg/day). At the end of the last cycle, mice were sacrificed, and the lungs were collected and fixed in 10% formalin. All experimental procedures in mice were approved by the Institutional Animal Care and Use Committee of Icahn School of Medicine at Mount Sinai. Mouse husbandry care was provided by The Center for Comparative Medicine and Surgery at Icahn School of Medicine in accordance with the US Department of Agriculture and/or recommendations of the Public Health Service and/or Guide for the Care and Use of Laboratory Animals.
For the experimental metastasis experiment, T-HEp3 cells were pretreated with DMSO or C26 for 1 wk, and then 2.5 × 105 cells were injected into the tail vein of 8-wk-old female BALB/c nu/nu mice (Jackson Laboratories). Mice injected with DMSO-pretreated cells were treated i.p. with DMSO for 3 wk (5 consecutive days a week). Mice injected with C26-pretreated cells were divided into two groups. The first group was treated with C26 (0.5 mg/kg/day) for 3 wk, and the second group was treated with C26 for 1 wk, after which C26 treatment was stopped, and mice were treated with DMSO for 2 wk. At the end of treatments, mice from all three groups were euthanized. Gross autopsy was performed, and blood was collected for biochemical analysis of liver and kidney functions. Lungs were collected and stained with H&E for metastasis analysis.
IF
Matrigel cultures were fixed in 2% paraformaldehyde for 20 min and permeabilized in 0.5% TritonX-100 for 10 min. Paraffin-embedded sections were incubated in xylene followed by graded ethanol rehydration. Antigen retrieval for mouse lung tissues was performed in 10 mM citrate (pH 6) for 40 min using a steamer. Antigen retrieval for human tissue samples was performed in a steamer for 60 min in 1 mM EDTA buffer. All paraffin-embedded tissues were permeabilized in 0.1% TritonX-100 for 10 min. Cells dissociated from CAM tumors were fixed in 4% paraformaldehyde, and cytospins were prepared by centrifugation onto glass slides. Cytospins were then permeabilized in 0.5% TritonX-100 for 10 min. Sections, cytospins, and Matrigel cultures were blocked with 3% BSA (Fisher Bioreagents) and 5% normal goat serum (Gibco; PCN5000) in PBS for 1 h at room temperature. Primary antibodies were incubated overnight at 4°C followed by washing and incubation with Alexa-conjugated secondary antibodies (Invitrogen; 1:1,000 dilution) at room temperature for 1–3 h in the dark. Slides were mounted with ProLong Gold Antifade reagent with DAPI (Invitrogen; P36931). Images were obtained using Leica Software on a Leica SPE confocal microscope and analyzed using ImageJ software.
Western blot
Nuclear fractions were extraction using the NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Fisher Scientific; 78835) and boiled for 5 min in sample buffer. Samples were run on SDS–PAGE gels and transferred to polyvinylidene fluoride membranes. Membranes were then blocked in 5% milk in TBS-T. Primary antibodies (Table S3) were incubated overnight at 4°C. HRP-conjugated secondary antibodies were used at room temperature for 1 h. Western blot development was done using ECL (GE RPN 2106) and GE ImageQuant LAS 4010.
qPCR
RNA was extracted using RNeasy mini kit (Qiagen; 74104), and cDNA was produced using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems; 4368814) following the manufacturer’s instructions. Real-time PCR was performed using the PowerUP SYBR Green Master Mix (Applied Biosystems; A25741). Primers used are listed in Table S4.
Human samples
Paraffin-embedded sections from HNSCC primary tumors were obtained from the Cancer Biorepository at Icahn School of Medicine at Mount Sinai, New York, NY. Paraffin-embedded tissue sections from lymph nodes biopsied from clinically diagnosed metastasis-free HNSCC patients were obtained from Dr. Karl Christoph Sproll at the Heinrich Heine University Düsseldorf, Düsseldorf, Germany. These patients presented small primary tumors, had no previously diagnosed malignancy in the head and neck region, and had not been subject to previous treatment. Samples were deidentified and obtained with institutional review board approval.
Kaplan–Meier analysis
Survival analysis using the C26 expression signature including the top 25 up-regulated and down-regulated genes from our RNA-seq data in addition to NR2F1 expression was performed using the Kaplan–Meier plotter database (https://kmplot.com/analysis/). Overall survival and relapse-free survival Kaplan–Meier plots of the two patient cohorts (patients with or without C26 signature) were generated using data from 500 HNSCC patients, and the hazard ratio with 95% confidence intervals and log-rank P value were calculated.
Data availability
Statistical analysis
To ensure reproducibility, in vitro experiments were repeated at least three times unless otherwise indicated. For CAM tumor growth analysis, a minimum of seven tumors were analyzed per group. For CAM qPCR and immunostaining experiments, a minimum of four tumors were analyzed. For mouse experiments, a minimum of 10 mice per group were used for tumor growth and number of lung DTCs, while a minimum of 5 mice per group were used for immunostaining analysis. Statistical analysis was performed on Prism software using an unpaired t test, two-way ANOVA with Holm–Sidak’s multiple comparison test, or Fisher’s exact test with false discovery rate correction using Benjamini–Hochberg where applicable. A P value < 0.05 was considered significant.
Online supplemental material
Fig. S1 shows the chemical structure of C26, Western blots for NR2F1 protein expression level in nuclear extracts of D-HEp3 cell lines with NT RNA or four different NR2F1 gRNAs, and a graph of the percent activation of RXRα using different C26 concentrations. Fig. S2 shows representative FACS plots of gating strategy used to sort GFP+ cells from T-HEp3-GFP tumors, the PCA plot generated using the top 500 genes with the highest variation in gene expression across samples, gene set enrichment profiles of the top five enriched gene sets in DMSO control, the top 15 most down-regulated or up-regulated pathways from the WikiPathways database in C26 treatment, and the number of genes that were up-regulated in opposite direction in C26 versus D-HEp3. Fig. S3 shows Western blot of NR2F1 protein expression level in nuclear extracts of T-HEp3 cell lines with NT RNA or four different NR2F1 gRNAs and mRNA levels in T-HEp3 CAM tumors with NT or NR2F1 gRNA treated with DMSO or C26. Fig. S4 shows representative images of T-HEp3, FaDu, and SQ20B cells plated in Matrigel and treated with DMSO or C26 (0.5 µM) for 4 d then fixed and immunostained for Ki-67, a graph with the percentage cleaved caspase-3+ cells, representative images of T-HEp3 cells with CDK2 biosensor in different stages of the cell cycle, mRNA levels of cells with control siRNA or two different NR2F2 siRNAs, and the percentage of single cells or colonies in T-HEp3 cells with control siRNA or two different NR2F2 siRNAs treated with DMSO or C26. Fig. S5 shows representative images of primary tumors stained for NR2F1 and the percentage of NR2F1+ cells; representative images of primary tumors stained for cleaved caspase-3 and the percentage of cleaved caspase-3+ cells; the correlation between primary tumor weight and the number of DTCs per lung lobule of DMSO- or C26-treated mice; the schematic of treatment regimen used for AZA in combination with atRA/C26 in the spontaneous metastasis experiment; primary tumor weight in DMSO, C26, or AZA+atRA/C26 from the spontaneous metastasis experiment; the number of lung DTCs in DMSO, C26, or AZA+atRA/C26 from the spontaneous metastasis experiment, the weights of mice in the experimental metastasis experiment monitored over 3 wk; representative images of lungs from DMSO- or C26-treated mice with immunostaining for vimentin and cleaved caspase-3; the percentage of cleaved caspase-3−/vimentin+ tumor cells in lungs; and graphs of nuclear NR2F1 MFI in single DTCs in lungs from DMSO- and C26-treated mice. Table S1 shows comparisons of Hallmark GSEA in C26 versus DMSO and D-HEp3 versus T-HEp3. Table S2 lists genes included in the C26 gene signature. Table S3 lists the primary antibodies used for IF and Western blot. Table S4 lists the primers used for qPCR.
Acknowledgments
We thank the Aguirre-Ghiso and Sosa laboratories for helpful discussions and Anna Freund and Marianne Hölbling for technical assistance with formalin-fixed paraffin-embedded sections. T-HEp3 cells expressing CDK2 biosensor were a generous gift from the Bravo-Cordero laboratory at Mount Sinai. RNA-seq bioinformatic analysis was performed by the BiNGS core at Mount Sinai.
This work was supported by National Institutes of Health/National Cancer Institute (grants CA109182, CA216248, CA218024, and CA196521 to J.A. Aguirre-Ghiso), the Samuel Waxman Cancer Research Foundation Tumor Dormancy Program (J.A. Aguirre-Ghiso), BioAccelerate NYC–NYC Partnership Fund (J.A. Aguirre-Ghiso and M. Soledad Sosa), and HiberCell (to J.A. Aguirre-Ghiso). M. Soledad Sosa was funded by the National Cancer Institute (K22 grant CA201054), Susan G. Komen (CCR17483357), and Melanoma Research Alliance (401181). B.D. Khalil was funded the National Cancer Institute (T32 grant CA078207). A. Rodriguez Martinez was funded by the Portuguese Foundation for Science and Technology (SFRH/BD/100380/2014). K.C. Sproll was funded by the Deutsche Krebshilfe (109600). Computational work was supported by the National Institutes of Health Office of Research Infrastructure (awards S10OD018522 and S10OD026880). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author contributions: B.D. Khalil designed, planned, and conducted experiments; analyzed data; and wrote the manuscript. R. Sanchez and M. Mezei conducted computational screen for agonists. M. Soledad Sosa, T. Rahman, S. Moritsch, and A. Rodriguez Martinez conducted the in vitro agonist screen and phenotype validation experiments. B. Miles and K.C. Sproll contributed tissue specimens and HNSCC expertise. E. Farias, A.R. Nobre, and N. Kale planned and conducted in vivo mouse experiments and processing of tissues. D. Singh performed RNA-seq studies on D-HEp3 versus T-HEp3 cells. C. Rodriguez-Tirado conducted experiments with patient samples. M. Soledad Sosa and J.A. Aguirre-Ghiso jointly conceived the project, designed experiments, analyzed data, secured funding, and wrote the manuscript.

