


NF-κB2/p100 (p100) is an inhibitor of κB (IκB) protein that is partially degraded to produce the NF-κB2/p52 (p52) transcription factor. Heterozygous NFKB2 mutations cause a human syndrome of immunodeficiency and autoimmunity, but whether autoimmunity arises from insufficiency of p52 or IκB function of mutated p100 is unclear. Here, we studied mice bearing mutations in the p100 degron, a domain that harbors most of the clinically recognized mutations and is required for signal-dependent p100 degradation. Distinct mutations caused graded increases in p100-degradation resistance. Severe p100-degradation resistance, due to inheritance of one highly degradation-resistant allele or two subclinical alleles, caused thymic medullary hypoplasia and autoimmune disease, whereas the absence of p100 and p52 did not. We inferred a similar mechanism occurs in humans, as the T cell receptor repertoires of affected humans and mice contained a hydrophobic signature of increased self-reactivity. Autoimmunity in autosomal dominant NFKB2 syndrome arises largely from defects in nonhematopoietic cells caused by the IκB function of degradation-resistant p100.

Introduction
Rare mutations in NFKB2 cause an autosomal dominant human syndrome of hypogammaglobulinemia and increased susceptibility to infections, often accompanied by organ-specific autoimmunity (Chen et al., 2013; Klemann et al., 2019; Lee et al., 2014). Key insights into immunological mechanisms of health and disease have come from analysis of rare Mendelian human syndromes (Cheng and Anderson, 2012). Accurate mouse models of these syndromes have been informative and provide a means to elucidate cell types in which the mutations act to cause pathology, with implications for therapy in humans. In the human NFKB2 syndrome, autoimmune manifestations are frequent but heterogenous (Klemann et al., 2019), and the determinants of susceptibility to autoimmunity are unknown.
The NF-κB family comprises five proteins: RelA, RelB, c-Rel, NF-κB1 (p105/p50), and NF-κB2 (p100/p52). Full-length NF-κB2/p100 (p100) dimerizes with other NF-κB family members and binds preformed NF-κB dimers, inhibiting their function until the signal-dependent degradation of p100 releases active NF-κB dimers (Basak et al., 2007; Scheinman et al., 1993; Sun et al., 1994). In resting cells, p100 serves as the core of high-molecular-weight, cytoplasmic complexes called kappaBsomes, which contain all NF-κB members (Savinova et al., 2009; Tao et al., 2014). Degradation of p100 requires noncanonical NF-κB activation induced by engagement of cell-surface receptors, predominantly of the tumor necrosis factor receptor superfamily (Sun, 2017). This leads to intracellular accumulation of NF-κB–inducing kinase (NIK), which cooperates with IκB kinase α (IKKα) to phosphorylate the p100 degron at specific sites (Ser866, Ser870, and possibly Ser872) before ubiquitination of Lys855, tagging the C-terminal portion of p100 for proteasomal removal (Liang et al., 2006; Sun, 2012; Xiao et al., 2001). Heterozygous mutations that truncate the NF-κB2 precursor protein proximal to the degron, enabling signal-independent production of NF-κB2/p52 (p52), have been found in patients with immunodeficiency or cytopenias (Klemann et al., 2019; Kotlinowski et al., 2019; Kuehn et al., 2017). Most of the clinically recognized NFKB2 mutations, including all those variants identified in patients with alopecia areata, hypopituitarism, and/or serum autoantibodies, cluster in the p100 degron (Klemann et al., 2019). All tested p100 degron mutations confer a decrease in p52 abundance (Chen et al., 2013; Kuehn et al., 2017; Lee et al., 2014; Lindsley et al., 2014; Liu et al., 2014; Maccari et al., 2017; Ramakrishnan et al., 2018; Slade et al., 2019), and pathology has been proposed to arise from p52 insufficiency (Chen et al., 2013; Klemann et al., 2019). However, some effects of NFKB2 degron mutations, such as cytoplasmic retention of RelA (Lee et al., 2014) and reduced cytokine release in response to LPS stimulation (Kuehn et al., 2017), cannot be explained by p52 insufficiency and are consistent with exaggerated IκB function of mutated p100. The contributions of these two mechanisms to autoimmune susceptibility are unclear.
Studies of mice with Nfkb2 mutations indicate that p52 insufficiency and IκB function of mutated p100 both cause autoimmune manifestations, albeit differing in severity. Nfkb2−/− mice, which lack p100 and p52, exhibit inflammatory infiltrates in multiple organs (Zhu et al., 2006) but have a near-normal lifespan (O’Reilly et al., 2015). Nfkb2Lym1 encodes a mutant p100 protein (p.Y868*) that is truncated in the p100 degron (Tucker et al., 2007). In Nfkb2Lym1/Lym1 cells, p100 is detectable, but p52 is not (Tucker et al., 2007). During noncanonical NF-κB activation, newly synthesized p100 is predominantly processed into p52, whereas most of the preexisting p100 undergoes complete degradation (Yilmaz et al., 2014). In contrast, noncanonical NF-κB activation causes p100 to accumulate in Nfkb2Lym1/Lym1 cells, demonstrating that the mutation inhibits the partial and complete degradation of p100 (Tucker et al., 2007). On the BALB/c genetic background, Nfkb2Lym1/Lym1 mice developed extensive inflammatory infiltrates in the lung and liver and had a shortened lifespan, while Nfkb2+/Lym1 mice had milder inflammation and no increase in mortality up to 250 d of age (Tucker et al., 2007). AIRE-expressing medullary thymic epithelial cells (mTECs) are present but reduced in Nfkb2−/− mice (O’Reilly et al., 2015; Zhu et al., 2006), while thymic expression of Aire mRNA is markedly reduced in Nfkb2Lym1/Lym1 mice (Tucker et al., 2007). AIRE is required for the expression and presentation of tissue-specific antigens in the thymus and mutations in AIRE cause autoimmune polyendocrine syndrome type I (Proekt et al., 2017). NFKB2 mutations have been proposed to confer susceptibility to autoimmunity by causing a deficiency of AIRE (Chen et al., 2013; Klemann et al., 2019; Lee et al., 2014; Liu et al., 2014; Ramakrishnan et al., 2018; Shi et al., 2016). Whether and how NFKB2/Nfkb2 mutations predispose to autoimmunity by mechanisms distinct from diminishing AIRE expression is unclear.
Here, we analyzed a murine Nfkb2 allelic series, including a strain carrying an allele orthologous to the variant identified in our index patient (Lee et al., 2014). We found that some Nfkb2 degron mutations conferred moderate p100-degradation resistance, which did not trigger spontaneous autoimmunity in heterozygous mice. However, homozygosity for subclinical alleles, or one copy of the Nfkb2Lym1 allele, caused thymic medullary hypoplasia and fully penetrant, fatal, multiorgan autoimmunity in C57BL/6 (B6) mice. These effects were distinct from those caused by the absence of p100 and p52 or AIRE. The TCR repertoires of autoimmunity-prone strains were enriched in hydrophobic motifs, a biomarker of self-reactivity (Stadinski et al., 2016), and a similar abnormality was present in patients with NFKB2 mutations.
Results
Decreased lifespan of mice with mutations in the p100 degron
The N-terminal portion of p100 is p52 (aa ∼1–405; Betts and Nabel, 1996), which contains a Rel homology domain, a feature of all NF-κB members (Fig. 1 A). The C-terminal portion of p100 contains a helix-turn-helix domain (aa 435–481) and an ankyrin repeat domain (aa 490–752) that mediate IκB activity (Savinova et al., 2009; Sun et al., 1994; Tao et al., 2014) and a processing inhibitory domain (aa 753–849) that inhibits signal-independent (aberrant) processing of p100 into p52 (Xiao et al., 2001). On the other hand, signal-dependent processing of p100 into p52 requires the p100 degron (aa 850–900; Xiao et al., 2001). 16 of the 21 pathogenic NFKB2 mutations identified thus far cluster in the p100 degron (Fig. 1 A; Klemann et al., 2019).

Decreased lifespan in mice with Nfkb2 genotypes that result in above-threshold p100 accumulation relative to p52 (see also Fig. S1 ). (A) Cartoon of human NF-κB2, showing its domains and known pathological variants. (B and C) Lifespans of WT mice compared with mice that were heterozygous (B) or homozygous (C) for the indicated Nfkb2 or Aire genotypes. Small vertical lines indicate observations censored for reasons unrelated to illness. See Fig. S1 B for statistical analyses. (D)Nfkb2 alleles studied in this paper. See Fig. S1 A for DNA sequence chromatograms. (E)Nfkb2 genotype affects p100 and p52 expression. Spleen lysates were subjected to SDS-PAGE followed by immunoblotting with an antibody reactive with p100 and p52 or GAPDH as a loading control. Graphs (right) show the density of p100 or p52 normalized to GAPDH for each sample and then divided by the mean of Nfkb2+/+ control samples on the same gel. Far right graph shows the p100/p52 ratio, excluding Nfkb2xdr/xdr samples, in which the p100 and p52 densities were below the limit of detection. Each symbol on a graph represents an individual mouse, determined as the mean of one to five technical replicates. Vertical dashed lines mark the mean of the Nfkb2+/+ group. Data from female and male mice were comparable and were pooled from 26 experiments. Nfkb2S866fs/S866fs mice (mean ± SD, 372 ± 289 d; range, 61–614 d) were older than Nfkb2+/+ mice (mean ± SD, 163 ± 99 d; range, 60–571 d); otherwise, ages were not significantly different from the Nfkb2+/+ group. Each genotype was compared with the Nfkb2+/+ group using one-way ANOVA with Dunnett’s post-test; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. (F) For the indicated mouse strains (far right), the graphs show the time to 20% mortality plotted against the mean p100 density (left), mean p52 density (middle), or mean p100/p52 ratio (right), annotated with P and r values calculated using Pearson’s test for correlation.
Decreased lifespan in mice with Nfkb2 genotypes that result in above-threshold p100 accumulation relative to p52 (see also Fig. S1 ). (A) Cartoon of human NF-κB2, showing its domains and known pathological variants. (B and C) Lifespans of WT mice compared with mice that were heterozygous (B) or homozygous (C) for the indicated Nfkb2 or Aire genotypes. Small vertical lines indicate observations censored for reasons unrelated to illness. See Fig. S1 B for statistical analyses. (D)Nfkb2 alleles studied in this paper. See Fig. S1 A for DNA sequence chromatograms. (E)Nfkb2 genotype affects p100 and p52 expression. Spleen lysates were subjected to SDS-PAGE followed by immunoblotting with an antibody reactive with p100 and p52 or GAPDH as a loading control. Graphs (right) show the density of p100 or p52 normalized to GAPDH for each sample and then divided by the mean of Nfkb2+/+ control samples on the same gel. Far right graph shows the p100/p52 ratio, excluding Nfkb2xdr/xdr samples, in which the p100 and p52 densities were below the limit of detection. Each symbol on a graph represents an individual mouse, determined as the mean of one to five technical replicates. Vertical dashed lines mark the mean of the Nfkb2+/+ group. Data from female and male mice were comparable and were pooled from 26 experiments. Nfkb2S866fs/S866fs mice (mean ± SD, 372 ± 289 d; range, 61–614 d) were older than Nfkb2+/+ mice (mean ± SD, 163 ± 99 d; range, 60–571 d); otherwise, ages were not significantly different from the Nfkb2+/+ group. Each genotype was compared with the Nfkb2+/+ group using one-way ANOVA with Dunnett’s post-test; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. (F) For the indicated mouse strains (far right), the graphs show the time to 20% mortality plotted against the mean p100 density (left), mean p52 density (middle), or mean p100/p52 ratio (right), annotated with P and r values calculated using Pearson’s test for correlation.
To investigate immunological effects of p100 degron mutations, we used CRISPR/Cas9 gene editing to generate mice with an allele (Nfkb2D865G) orthologous to the variant identified in our index patient (Lee et al., 2014; Fig. 1 D and Fig. S1 A). The gene editing procedure also generated other variants. The Nfkb2Ser866fs allele is predicted to encode a lengthened p100 protein in which the 49 C-terminal amino acids are altered. The Nfkb2Y868indel variant is predicted to cause a net deletion of one residue between Ser866 and Ser870. To enable comparison with established models, we studied mice bearing the Nfkb2+/Lym1 allele (Tucker et al., 2007) or the Nfkb2xdr allele, in which an intronic substitution disrupts splicing of Nfkb2 mRNA and prevents expression of p100 and p52 (Miosge et al., 2002). All strains had a B6 genetic background.

NFKB2/Nfkb2 variants and statistical analysis of lifespan in the murine Nfkb2 allelic series (related to Fig. 1 ). (A) Sanger sequencing chromatograms for DNA encoding the p100 degron of a control B6 mouse (Nfkb2+/+) and mice of the indicated Nfkb2 genotypes generated in this study. (B) For pairs of survival curves shown in Fig. 1, B and C, the grid shows the P values of log-rank tests. (C) NFKB2 variants identified in patients, including effects of variants on expression and phosphorylation of the mutant precursor protein and on p52 expression, as well as the reference. NA, not available.
NFKB2/Nfkb2 variants and statistical analysis of lifespan in the murine Nfkb2 allelic series (related to Fig. 1 ). (A) Sanger sequencing chromatograms for DNA encoding the p100 degron of a control B6 mouse (Nfkb2+/+) and mice of the indicated Nfkb2 genotypes generated in this study. (B) For pairs of survival curves shown in Fig. 1, B and C, the grid shows the P values of log-rank tests. (C) NFKB2 variants identified in patients, including effects of variants on expression and phosphorylation of the mutant precursor protein and on p52 expression, as well as the reference. NA, not available.
As the human NFKB2 syndrome is autosomal dominant, we studied mice with heterozygous Nfkb2 mutations. While most strains had a normal lifespan, Nfkb2+/D865G mice had a slight reduction in lifespan compared with Nfkb2+/+ control mice (Fig. 1 B and Fig. S1 B). B6.Nfkb2+/Lym1 mice had a markedly shortened lifespan with a median survival of 215 d (Fig. 1 B), shorter than previously observed in BALB/c.Nfkb2+/Lym1 mice (Tucker et al., 2007). This is remarkable because autoimmune manifestations in B6.Aire−/− mice are milder than in BALB/c.Aire−/− mice (Jiang et al., 2005).
To study effects of Nfkb2 mutations in the absence of WT p100 and p52, homozygous mutant mice were examined. Attempts to study Nfkb2Lym1/Lym1 mice were unsuccessful, because all offspring of Nfkb2+/Lym1 females died before weaning (data not shown), possibly due to a lactation defect akin to that observed in mice with hypomorphic mutations in IKKα (Cao et al., 2001). Nfkb2D865G/D865G and Nfkb2Y868indel/Y868indel mice had markedly shortened lifespans, with a median survival of 170 d and 128 d, respectively (Fig. 1 C). Compared with WT mice, lifespan was also shortened in Nfkb2xdr/xdr mice, but not in Nfkb2S866fs/S866fs or Aire−/− mice (Fig. 1 C and Fig. S1 B).
To assess effects of Nfkb2 mutations on the abundance of p100 and p52, spleen lysates were analyzed by immunoblotting. In humans, recognized p100 degron mutations result in normal or increased abundance of p100, whereas the abundance of p52 is decreased (Fig. S1 C). This combination of changes was observed in four genotypes: Nfkb2+/D865G, Nfkb2D865G/D865G, Nfkb2+/Lym1, and Nfkb2Y868indel/Y868indel (Fig. 1 E). Similar results were obtained for the Nfkb2+/Y868indel genotype, but the trend toward a decrease in p52 abundance was not statistically significant. Those findings suggest that the p100D865G, p100Lym1, and p100Y868indel proteins resist degradation. Notably, the p100 band in Nfkb2+/Lym1 samples migrated faster than other samples, consistent with a truncation in the p100Lym1 protein (Fig. 1 E). While no changes were detected in the Nfkb2+/S866fs group, both p100 and p52 were decreased in samples from Nfkb2S866fs/S866fs mice. Interestingly, in Nfkb2+/xdr mice, the abundance of p100 was halved, whereas the p52 abundance was not significantly different from WT. As expected, neither p100 nor p52 was detectable in Nfkb2xdr/xdr samples.
We hypothesized that the p100/p52 ratio may indicate the extent to which the p100 protein pool in each strain resists processing into p52. Consistent with this hypothesis, in mice bearing the Nfkb2D865G or Nfkb2Y868indel alleles, the absence of WT p100 protein in homozygous mutant mice resulted in a higher p100/p52 ratio than in heterozygous mutant mice (Fig. 1 E). The p100/p52 ratio in heterozygous Nfkb2+/Lym1 samples was comparable to that in homozygous Nfkb2D865G/D865G and Nfkb2Y868indel/Y868indel samples, suggesting that p100Lym1 is more resistant to degradation than the p100D865G and p100Y868indel proteins. Combining the data thus far, lifespan was not correlated with the abundance of either p100 or p52 but was negatively correlated with the p100/p52 ratio (Fig. 1 F).
T cell–dependent multiorgan autoimmunity caused by p100 degron mutations
The reduction in lifespan described above was T cell dependent, as Tcra−/− Nfkb2+/Lym1 mice remained healthy (Fig. 2 A). At necropsy, Nfkb2+/Lym1 mice had dilated intestines and small pancreases, consistent with exocrine pancreatitis (Fig. 2, B and C). Nfkb2+/Lym1 mice commonly developed dermatitis on the face and ears, accompanied occasionally by vitiligo (Fig. 2 D). Histology revealed severe exocrine pancreatitis in all Nfkb2D865G/D865G, Nfkb2+/Lym1, and Nfkb2Y868indel/Y868indel mice, but not in Nfkb2xdr/xdr mice (Fig. 2, E and F; and data not shown). Previous studies revealed mild to moderate lymphocytic infiltrates in liver and lung, but not exocrine pancreatitis, in B6.Nfkb2−/− mice (O’Reilly et al., 2015; Zhu et al., 2006). Inflammatory infiltrates were also common in the lacrimal glands, salivary glands, liver, and lung of Nfkb2D865G/D865G and Nfkb2+/Lym1 mice (Fig. 2, E and F). Nfkb2+/Lym1 mice also had inflammatory infiltrates in small exocrine glands that line the ear canal and prepuce (data not shown). Some humans with NFKB2 mutations have central adrenal insufficiency (Brue et al., 2014), but pituitary gland histology in Nfkb2+/Lym1 mice was normal (data not shown). Pancreatitis was uncommon in B6.Aire−/− mice, whereas prostate and retina were affected, as described previously (Jiang et al., 2005; Leonard et al., 2017; Taniguchi et al., 2012; Fig. 2, E and F). Mice with mutations in the p100 degron thus developed T cell–mediated autoimmunity, which affected a set of organs distinct from those affected by the absence of p100 and p52 or AIRE.

T cell–dependent multiorgan autoimmunity caused by p100 degron mutations . (A) Lifespans of cohoused Tcra+/− Nfkb2+/Lym1 and Tcra−/− Nfkb2+/Lym1 sibling mice (P value, log-rank test). Small vertical lines indicate observations censored for reasons unrelated to illness. (B) Abdominal viscera of Nfkb2+/+ and Nfkb2+/Lym1 littermates aged 150 d. Yellow arrows indicate the pancreas, which is small in the Nfkb2+/Lym1 mouse; scale bar, 1 cm. (C) Pancreas mass in Nfkb2+/+ and Nfkb2+/Lym1 mice stratified by sex. (D) A male Nfkb2+/Lym1 mouse aged 225 d with dermatitis on the face and ears and vitiligo; scale bar, 1 cm. (E) Histology of organs (denoted above) showing normal (top row) and affected (bottom row) tissue sections; scale bars, 100 µm. (F) Pathology score for each organ colored by genotype, with bars showing the group mean. The number of mice per group is shown below the x axis. In F, the Aire−/− mice (mean ± SD, 182 ± 60 d; range, 92–272 d) were significantly older than the Nfkb2+/+ mice (mean ± SD, 118 ± 43 d; range, 84–211 d). The ages of all other groups in F and the Nfkb2+/Lym1 mice in C were not significantly different from the Nfkb2+/+ group. In A and F, data from female and male mice were comparable and therefore pooled. For each sex in C or organ in F, each genotype was compared with the Nfkb2+/+ group using two-way ANOVA with Sidak’s post-test (C) or Kruskal-Wallis tests with Dunn’s post-test (F); *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
T cell–dependent multiorgan autoimmunity caused by p100 degron mutations . (A) Lifespans of cohoused Tcra+/− Nfkb2+/Lym1 and Tcra−/− Nfkb2+/Lym1 sibling mice (P value, log-rank test). Small vertical lines indicate observations censored for reasons unrelated to illness. (B) Abdominal viscera of Nfkb2+/+ and Nfkb2+/Lym1 littermates aged 150 d. Yellow arrows indicate the pancreas, which is small in the Nfkb2+/Lym1 mouse; scale bar, 1 cm. (C) Pancreas mass in Nfkb2+/+ and Nfkb2+/Lym1 mice stratified by sex. (D) A male Nfkb2+/Lym1 mouse aged 225 d with dermatitis on the face and ears and vitiligo; scale bar, 1 cm. (E) Histology of organs (denoted above) showing normal (top row) and affected (bottom row) tissue sections; scale bars, 100 µm. (F) Pathology score for each organ colored by genotype, with bars showing the group mean. The number of mice per group is shown below the x axis. In F, the Aire−/− mice (mean ± SD, 182 ± 60 d; range, 92–272 d) were significantly older than the Nfkb2+/+ mice (mean ± SD, 118 ± 43 d; range, 84–211 d). The ages of all other groups in F and the Nfkb2+/Lym1 mice in C were not significantly different from the Nfkb2+/+ group. In A and F, data from female and male mice were comparable and therefore pooled. For each sex in C or organ in F, each genotype was compared with the Nfkb2+/+ group using two-way ANOVA with Sidak’s post-test (C) or Kruskal-Wallis tests with Dunn’s post-test (F); *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Thymic tolerance defects in mice with p100 degron mutations
In a WT mouse, more than half of the nascent thymic lymphocytes (thymocytes) that receive a detectable αβ TCR signal undergo apoptosis at the CD4+ CD8+ double-positive (DP) stage, or before up-regulation of the chemokine receptor, CCR7 (wave 1 deletion; Daley et al., 2013; Sinclair et al., 2013; Stritesky et al., 2013). To test whether Nfkb2 mutations affect wave 1 deletion, we quantified nascent thymocytes that received a strong TCR signal in mice carrying an antiapoptotic, B cell lymphoma 2 transgene (BCL2-tg; Ogilvy et al., 1999; Wirasinha et al., 2019; Fig. 3 A). To identify nascent thymocytes, mice were injected once with 5-ethynyl-2′-deoxyuridine (EdU), which is taken up by thymocytes that are proliferating just before the onset of αβ TCR expression (Lucas et al., 1993). At 3 d after EdU injection, Nfkb2+/D865G mice had normal frequencies of EdU+ thymocytes and TCR-signaled CD5+ TCRβ+ cells (Saini et al., 2010; Fig. S2, A and B). In Nfkb2+/D865G mice with normal apoptosis, the frequencies of strongly TCR-signaled Helios+ CCR7− cells and weakly TCR-signaled Helios− CCR7+ cells were normal (Fig. 3 B). However, when apoptosis was inhibited by BCL2-tg expression, Nfkb2+/D865G mice had decreased Helios+ CCR7− cell induction and increased Helios− CCR7+ cell induction (Fig. 3 B). Similar results were obtained in Nfkb2+/Lym1 mice (Fig. 3 C and Fig. S2, C and D). Thus, only the Nfkb2+/Lym1 genotype caused autoimmunity, but the Nfkb2+/D865G and Nfkb2+/Lym1 genotypes both decreased the number of thymocytes that received a strong TCR signal and increased the number of thymocytes that received a weak TCR signal at the wave 1 checkpoint.

Diminished thymic tolerance mechanisms in mice with p100 degron mutations (see also Fig. S2 ). (A) Quantification of wave 1 deletion. At 3 d after proliferating thymocytes incorporate EdU, nascent (EdU+) TCR-signaled (CD5+ TCRβ+) thymocytes were analyzed to resolve Helios+ CCR7− and Helios− CCR7+ subsets, which have received a strong or weak TCR signal, respectively. BCL2-tg expression inhibits apoptotic deletion, enabling measurement of the scale of wave 1 deletion. (B) Helios/CCR7 phenotypes of EdU+ CD5+ TCRβ+ thymocytes from Nfkb2+/+ or Nfkb2+/D865G mice, which were negative or positive for BCL2-tg (top), 3 d after EdU injection, with summaries (right) showing the frequencies of gated populations among EdU+ thymocytes. (C)Nfkb2+/+ and Nfkb2+/Lym1 mice were examined as in B. The age ranges of mice were 68–94 d (B) and 40–70 d (C). (D) Quantification of thymocytes that received a weak or strong TCR signal at the wave 2 checkpoint. At 5 d after EdU injection, the EdU+ CD5+ TCRβ+ CD4SP population was analyzed to resolve Helios+ CCR7+ (strongly TCR-signaled) and Helios− CCR7+ (weakly TCR-signaled) subsets. (E) Plots show the Helios/CCR7 phenotype of EdU+ CD5+ TCRβ+ CD4SP thymocytes from mice of the indicated genotypes 5 d after EdU injection, with summaries showing the frequencies of gated populations among EdU+ thymocytes. The Nfkb2xdr/xdr mice (45 d old) were significantly younger than the Nfkb2+/+ mice (mean ± SD, 86 ± 22 d; range 53–105 d). The ages of all other groups were not significantly different from the Nfkb2+/+ group. (F) For the genotypes examined in E, plots show Foxp3/CCR7 phenotype of CD4SP thymocytes, with a graph showing the frequency and number of Foxp3+ cells in the CD4SP population. Age of mice did not differ significantly between groups (mean ± SD, 84 ± 24 d; range, 50–158 d). (G)Nfkb2+/+, Nfkb2+/D865G, Nfkb2xdr/xdr, Aire+/+, or Aire−/− mice aged 42–98 d were irradiated then reconstituted with WT BM. Graphs show the frequencies of Helios+ Foxp3− and Foxp3+ cells among CD4SP CCR7+ thymocytes in the chimeric mice 5–14 wk after transplantation. (H) WT CD451/1 mice aged 57–145 d were irradiated then reconstituted with WT CD451/2 BM mixed with Nfkb2+/+, Nfkb2+/Lym1, or Nfkb2xdr/xdr CD452/2 BM. All donors and recipients were male. 8–27 wk later, flow cytometry was used to determine the frequency of CD452/2 cells among DP thymocytes and among three subsets of CD4SP CCR7+ thymocytes defined by Helios and Foxp3 expression as gated in the plots. The graph shows the frequency of CD452/2 cells in the subsets indicated on the x axis, divided by the frequency of CD452/2 cells among DP thymocytes in the same sample. To enable comparisons between CD4SP CCR7+ thymocyte subsets, data were then divided by the mean of the Nfkb2+/+ group for each subset. (I)Nfkb2+/+, Nfkb2+/Lym1, Aire+/+, or Aire−/− mice aged 42–96 d were irradiated then reconstituted with BCL2-tg+ BM. Graphs show the frequencies of Helios+ Foxp3− and Foxp3+ cells among CD4SP CCR7+ thymocytes in the chimeric mice 7–10 wk after transplantation. Each symbol in a summary graph represents an individual mouse, and horizontal bars show the group means. Each graph shows data compiled from one (G and I), two (B, C, and E), three (H), or eight (F) separate experiments. Unless otherwise stated, graphs show data from female and male mice. Statistical comparisons used one-way (E and F) or two-way (B, C, and H) ANOVA with Sidak’s multiple comparisons tests or Student’s t tests (G and I); *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Diminished thymic tolerance mechanisms in mice with p100 degron mutations (see also Fig. S2 ). (A) Quantification of wave 1 deletion. At 3 d after proliferating thymocytes incorporate EdU, nascent (EdU+) TCR-signaled (CD5+ TCRβ+) thymocytes were analyzed to resolve Helios+ CCR7− and Helios− CCR7+ subsets, which have received a strong or weak TCR signal, respectively. BCL2-tg expression inhibits apoptotic deletion, enabling measurement of the scale of wave 1 deletion. (B) Helios/CCR7 phenotypes of EdU+ CD5+ TCRβ+ thymocytes from Nfkb2+/+ or Nfkb2+/D865G mice, which were negative or positive for BCL2-tg (top), 3 d after EdU injection, with summaries (right) showing the frequencies of gated populations among EdU+ thymocytes. (C)Nfkb2+/+ and Nfkb2+/Lym1 mice were examined as in B. The age ranges of mice were 68–94 d (B) and 40–70 d (C). (D) Quantification of thymocytes that received a weak or strong TCR signal at the wave 2 checkpoint. At 5 d after EdU injection, the EdU+ CD5+ TCRβ+ CD4SP population was analyzed to resolve Helios+ CCR7+ (strongly TCR-signaled) and Helios− CCR7+ (weakly TCR-signaled) subsets. (E) Plots show the Helios/CCR7 phenotype of EdU+ CD5+ TCRβ+ CD4SP thymocytes from mice of the indicated genotypes 5 d after EdU injection, with summaries showing the frequencies of gated populations among EdU+ thymocytes. The Nfkb2xdr/xdr mice (45 d old) were significantly younger than the Nfkb2+/+ mice (mean ± SD, 86 ± 22 d; range 53–105 d). The ages of all other groups were not significantly different from the Nfkb2+/+ group. (F) For the genotypes examined in E, plots show Foxp3/CCR7 phenotype of CD4SP thymocytes, with a graph showing the frequency and number of Foxp3+ cells in the CD4SP population. Age of mice did not differ significantly between groups (mean ± SD, 84 ± 24 d; range, 50–158 d). (G)Nfkb2+/+, Nfkb2+/D865G, Nfkb2xdr/xdr, Aire+/+, or Aire−/− mice aged 42–98 d were irradiated then reconstituted with WT BM. Graphs show the frequencies of Helios+ Foxp3− and Foxp3+ cells among CD4SP CCR7+ thymocytes in the chimeric mice 5–14 wk after transplantation. (H) WT CD451/1 mice aged 57–145 d were irradiated then reconstituted with WT CD451/2 BM mixed with Nfkb2+/+, Nfkb2+/Lym1, or Nfkb2xdr/xdr CD452/2 BM. All donors and recipients were male. 8–27 wk later, flow cytometry was used to determine the frequency of CD452/2 cells among DP thymocytes and among three subsets of CD4SP CCR7+ thymocytes defined by Helios and Foxp3 expression as gated in the plots. The graph shows the frequency of CD452/2 cells in the subsets indicated on the x axis, divided by the frequency of CD452/2 cells among DP thymocytes in the same sample. To enable comparisons between CD4SP CCR7+ thymocyte subsets, data were then divided by the mean of the Nfkb2+/+ group for each subset. (I)Nfkb2+/+, Nfkb2+/Lym1, Aire+/+, or Aire−/− mice aged 42–96 d were irradiated then reconstituted with BCL2-tg+ BM. Graphs show the frequencies of Helios+ Foxp3− and Foxp3+ cells among CD4SP CCR7+ thymocytes in the chimeric mice 7–10 wk after transplantation. Each symbol in a summary graph represents an individual mouse, and horizontal bars show the group means. Each graph shows data compiled from one (G and I), two (B, C, and E), three (H), or eight (F) separate experiments. Unless otherwise stated, graphs show data from female and male mice. Statistical comparisons used one-way (E and F) or two-way (B, C, and H) ANOVA with Sidak’s multiple comparisons tests or Student’s t tests (G and I); *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.

Quantification of thymic deletion at waves 1 and 2 (related to Fig. 3 ). (A and B) Thymocyte phenotypes of Nfkb2+/+ and Nfkb2+/D865G mice, which were negative or positive for BCL2-tg (top), 3 d after a single dose of EdU. (A) Forward scatter (FSC) versus EdU on all thymocytes with a gate for EdU+ cells, including a negative control sample from an uninjected mouse (column 6). (B) CD5/TCRβ phenotype of EdU+ thymocytes with a gate to identify CD5+ TCRβ+ (TCR-signaled) cells, including a negative control sample from a Tcra–/– mouse (column 5). (C and D) Nfkb2+/+ and Nfkb2+/Lym1 mice, which were negative or positive for BCL2-tg (top), were examined as above in A and B. A BCL2-tg+ B2m–/– H2-Aa–/– mouse was used as a negative control for the gating of CD5+ TCRβ+ cells (column 5). (E–G) Mice of the indicated genotypes were injected with a single dose of EdU and thymocytes were analysed 5 d later. (E) FSC versus EdU on all thymocytes with a gate for EdU+ cells. (F and G) Plots show gating of CD5+ TCRβ+ cells among EdU+ thymocytes (F) and CD4+ CD8α− cells among EdU+ CD5+ TCRβ+ thymocytes (G). Each symbol in a graph (right) represents a measurement from one mouse and horizontal bars show the group means. Data in A and B, C and D, and E–G were compiled from two separate experiments each. Statistical comparisons used one-way ANOVA with Sidak’s multiple comparisons tests; *, P < 0.05; **, P < 0.01.
Quantification of thymic deletion at waves 1 and 2 (related to Fig. 3 ). (A and B) Thymocyte phenotypes of Nfkb2+/+ and Nfkb2+/D865G mice, which were negative or positive for BCL2-tg (top), 3 d after a single dose of EdU. (A) Forward scatter (FSC) versus EdU on all thymocytes with a gate for EdU+ cells, including a negative control sample from an uninjected mouse (column 6). (B) CD5/TCRβ phenotype of EdU+ thymocytes with a gate to identify CD5+ TCRβ+ (TCR-signaled) cells, including a negative control sample from a Tcra–/– mouse (column 5). (C and D) Nfkb2+/+ and Nfkb2+/Lym1 mice, which were negative or positive for BCL2-tg (top), were examined as above in A and B. A BCL2-tg+ B2m–/– H2-Aa–/– mouse was used as a negative control for the gating of CD5+ TCRβ+ cells (column 5). (E–G) Mice of the indicated genotypes were injected with a single dose of EdU and thymocytes were analysed 5 d later. (E) FSC versus EdU on all thymocytes with a gate for EdU+ cells. (F and G) Plots show gating of CD5+ TCRβ+ cells among EdU+ thymocytes (F) and CD4+ CD8α− cells among EdU+ CD5+ TCRβ+ thymocytes (G). Each symbol in a graph (right) represents a measurement from one mouse and horizontal bars show the group means. Data in A and B, C and D, and E–G were compiled from two separate experiments each. Statistical comparisons used one-way ANOVA with Sidak’s multiple comparisons tests; *, P < 0.05; **, P < 0.01.
At the subsequent stage of development, strong TCR signaling induces some CD4+ CD8− CCR7+ thymocytes to up-regulate Helios as they commit to undergo deletion (wave 2 deletion) or up-regulate Foxp3 (Hu et al., 2016, 2017). To quantify strongly TCR-signaled cells at the wave 2 checkpoint, we analyzed thymocytes 5 d after EdU injection, when wave 2 deletion peaks in EdU+ thymocytes (Hu et al., 2016; Fig. 3 D). Sequential gating of thymocytes that were EdU+, CD5+ TCRβ+, CD4+ CD8− (CD4 single positive [CD4SP]), and Helios− CCR7+ revealed increased induction of weakly TCR-signaled cells in Nfkb2+/Lym1 and Aire−/− mice (Fig. 3 E and Fig. S2, E–G). The induction of Helios+ CCR7+ cells was decreased in Nfkb2+/D865G and Nfkb2+/Lym1 mice, but not in Nfkb2xdr/xdr mice (Fig. 3 E). A caveat with this result is that the Nfkb2xdr/xdr mice were significantly younger than the mice in other groups, which may have contributed to the greater Helios+ CCR7+ cell frequencies detected in Nfkb2xdr/xdr mice compared with control mice (Fig. 3 E). Analysis of bulk CD4SP thymocytes revealed a decreased frequency and number of Foxp3+ regulatory T cells (T reg cells) in Nfkb2+/D865G, Nfkb2+/Lym1 and Aire−/− mice, but not in Nfkb2xdr/xdr mice (Fig. 3 F). A single copy of the Nfkb2D865G or Nfkb2Lym1 allele was thus sufficient to strongly decrease TCR-signaled thymocyte populations at the wave 1 and 2 checkpoints and decrease the thymic T reg cell population.
T cell–extrinsic and intrinsic defects in T reg cell differentiation
During Foxp3+ T reg cell differentiation in CD4SP CCR7+ thymocytes, the up-regulation of Helios and Foxp3 mark the onsets of requirements for thymocyte-intrinsic c-Rel function (Daley et al., 2013) and IL-2 signaling (Hu et al., 2017), respectively. To test for T cell–extrinsic defects in T reg cell differentiation, irradiated WT or mutant mice were reconstituted with WT bone marrow (BM) and thymocytes from the chimeric mice were examined 5–14 wk later. Among CD4SP CCR7+ thymocytes, the frequencies of Helios+ Foxp3− and Foxp3+ cells were decreased in Nfkb2+/D865G recipients, but not in Nfkb2xdr/xdr recipients (Fig. 3 G). The frequency of Foxp3+ cells was decreased in Aire−/− recipients of WT BM (Fig. 3 G). In a similar experiment designed to “capture” cells that would otherwise undergo apoptosis, irradiated WT or mutant mice were reconstituted with BCL2-tg+ BM. At 7–10 wk after transplantation, both Helios+ Foxp3− and Foxp3+ cells were decreased in Nfkb2+/Lym1 recipients, whereas only Foxp3+ cells were decreased in Aire−/− recipients (Fig. 3 I). These results establish that the capacity of nonhematopoietic cells to induce strongly TCR-signaled Helios+ Foxp3− thymocytes at the CD4SP CCR7+ stage is decreased in Nfkb2+/D865G and Nfkb2+/Lym1 mice in comparison to WT, Nfkb2xdr/xdr and Aire−/− mice.
To test for T cell–intrinsic defects in T reg cell differentiation, we reconstituted irradiated CD451/1 mice with WT CD451/2 BM mixed with WT, Nfkb2+/Lym1, or Nfkb2xdr/xdr CD452/2 BM. In all groups, the frequencies of CD452/2 cells among weakly TCR-signaled CD4SP CCR7+ Helios− Foxp3− thymocytes were similar to the frequencies of CD452/2 cells among their DP precursors (Fig. 3 H), indicating that naive CD4+ T cell differentiation was not impaired. However, chimeras bearing CD452/2 Nfkb2+/Lym1 BM had a lower CD452/2 cell frequency in the Helios+ Foxp3− subset and a further decrease in the Foxp3+ subset (Fig. 3 H), indicative of defects at both c-Rel–dependent and IL-2–dependent stages of thymic T reg cell differentiation. In contrast, T reg cell differentiation was not impaired in Nfkb2xdr/xdr cells, which actually showed enhanced differentiation to the c-Rel–dependent Helios+ Foxp3− stage (Fig. 3 H). Together, the results in Fig. 3 show that the Nfkb2+/D865G and Nfkb2+/Lym1 genotypes conferred T cell–extrinsic and intrinsic defects in thymic tolerance that were not recapitulated by the Nfkb2xdr/xdr genotype, suggesting that these defects arise predominantly from an exaggerated IκB function of mutant p100 proteins and not from insufficiency of p52.
Quantitative and functional T reg cell deficiencies conferred by p100 degron mutations
Decreased T reg cell populations have been described in patients with heterozygous NFKB2 mutations and homozygous AIRE mutations (Klemann et al., 2019; Sng et al., 2019). We observed that heterozygous NFKB2 mutations confer a greater T reg cell defect than homozygous AIRE mutations (Fig. 4 A and Table S1). Except for the Nfkb2+/S866fs mice, all mouse strains with p100 degron mutations had a decreased Foxp3+ T reg cell frequency in the spleen (Fig. 4 B). In contrast, Foxp3+ T reg cell frequency was normal in Nfkb2xdr/xdr mice (Fig. 4 B), as observed in Nfkb2−/− mice (O’Reilly et al., 2015; Zhu et al., 2006). Despite the T reg cell deficiency, antigen-experienced CD44hi Foxp3− cells were not increased in any mutant mouse strain; in fact, they were significantly decreased in several strains (Fig. 4 B). Analysis of mixed chimeras showed that the Nfkb2+/Lym1 genotype impaired differentiation of splenic T reg cells and antigen-experienced CD44hi Foxp3− cells in a cell-intrinsic manner, whereas the Nfkb2xdr/xdr genotype did not (Fig. 4 C). Capacity for T reg cell differentiation was increased in Nfkb2xdr/xdr cells, as observed when the Nfkb2 gene is deleted specifically in T reg cells or all T cells (Grinberg-Bleyer et al., 2018).
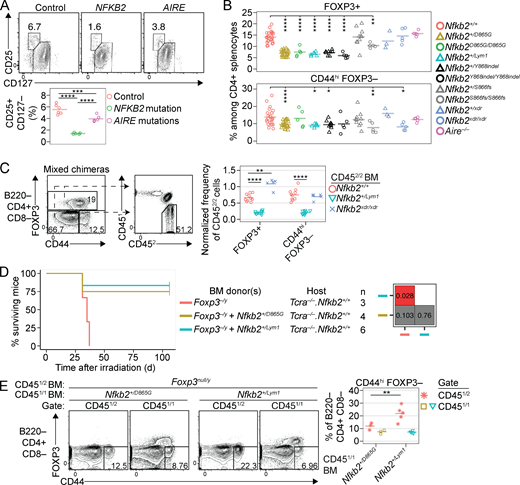
Quantitative and functional T reg cell deficiencies conferred by p100 degron mutations. (A) Plots show CD25/CD127 phenotypes of human peripheral blood CD19− CD4+ CD8− cells, with a graph showing the CD25+ CD127− T reg cell frequency from healthy control subjects or patients with mutations in NFKB2 or AIRE (see Table S1 for details). (B) Frequencies of Foxp3+ and CD44hi Foxp3− cells among CD4+ splenocytes in mice of the indicated genotypes. Age of mice did not differ significantly between groups (mean ± SD, 119 ± 48 d; range, 51–219 d). (C) From mixed chimeras (described in Fig. 3 H) harboring WT CD451/2 BM and WT, Nfkb2+/Lym1, or Nfkb2xdr/xdr CD452/2 BM; plots show the gates used to resolve three subsets of B220− CD4+ CD8− splenocytes based on CD44/Foxp3 phenotype. The graph shows the frequency of CD452/2 cells in the subsets indicated on the x axis, divided by the frequency of CD452/2 cells among CD44lo Foxp3− cells in the same sample. (D) Survival curves of Tcra−/− Nfkb2+/+ female mice after irradiation at 62–90 d of age followed by reconstitution with CD451/2Foxp3−/y BM alone or mixed 1:1 with CD451/1Nfkb2+/D865G BM or CD451/1Nfkb2+/Lym1 BM (see key, middle). All BM donors were male. Grid (right) shows the P values of log-rank tests comparing each pair of experimental groups. (E) For the chimeras described in D at 106 d after transplantation, plots show the gates used to determine the frequency of Foxp3+ and CD44+ Foxp3− cells among the CD451/2 and CD451/1 subsets of B220− CD4+ CD8− splenocytes, enumerated for multiple mice in the graph (right). Unless otherwise stated, graphs show data from female and male mice compiled from 1 (D and E), 3 (C), 5 (A), or 11 (B) separate experiments. Statistical comparisons used one-way (A and B) or two-way (C and E) ANOVA with Sidak’s multiple comparisons tests; *, P < 0.05; **, P < 0.01; ****, P < 0.0001.
Quantitative and functional T reg cell deficiencies conferred by p100 degron mutations. (A) Plots show CD25/CD127 phenotypes of human peripheral blood CD19− CD4+ CD8− cells, with a graph showing the CD25+ CD127− T reg cell frequency from healthy control subjects or patients with mutations in NFKB2 or AIRE (see Table S1 for details). (B) Frequencies of Foxp3+ and CD44hi Foxp3− cells among CD4+ splenocytes in mice of the indicated genotypes. Age of mice did not differ significantly between groups (mean ± SD, 119 ± 48 d; range, 51–219 d). (C) From mixed chimeras (described in Fig. 3 H) harboring WT CD451/2 BM and WT, Nfkb2+/Lym1, or Nfkb2xdr/xdr CD452/2 BM; plots show the gates used to resolve three subsets of B220− CD4+ CD8− splenocytes based on CD44/Foxp3 phenotype. The graph shows the frequency of CD452/2 cells in the subsets indicated on the x axis, divided by the frequency of CD452/2 cells among CD44lo Foxp3− cells in the same sample. (D) Survival curves of Tcra−/− Nfkb2+/+ female mice after irradiation at 62–90 d of age followed by reconstitution with CD451/2Foxp3−/y BM alone or mixed 1:1 with CD451/1Nfkb2+/D865G BM or CD451/1Nfkb2+/Lym1 BM (see key, middle). All BM donors were male. Grid (right) shows the P values of log-rank tests comparing each pair of experimental groups. (E) For the chimeras described in D at 106 d after transplantation, plots show the gates used to determine the frequency of Foxp3+ and CD44+ Foxp3− cells among the CD451/2 and CD451/1 subsets of B220− CD4+ CD8− splenocytes, enumerated for multiple mice in the graph (right). Unless otherwise stated, graphs show data from female and male mice compiled from 1 (D and E), 3 (C), 5 (A), or 11 (B) separate experiments. Statistical comparisons used one-way (A and B) or two-way (C and E) ANOVA with Sidak’s multiple comparisons tests; *, P < 0.05; **, P < 0.01; ****, P < 0.0001.
To test for cell-intrinsic defects in T reg cell function, WT hosts were irradiated and reconstituted with Foxp3null/y BM alone or Foxp3null/y BM mixed 1:1 with Nfkb2+/D865G BM or Nfkb2+/Lym1 BM. All recipients of Foxp3null/y BM alone developed disease due to the absence of Foxp3+ T reg cells (Fontenot et al., 2003), whereas most of the chimeras that also received Nfkb2+/D865G or Nfkb2+/Lym1 BM remained healthy for >100 d after BM transplantation (Fig. 4 D). However, the antigen-experienced CD44hi Foxp3− cell frequency in the Foxp3null/y CD4+ compartment was increased in mice with Nfkb2+/Lym1 T reg cells (Fig. 4 E). Thus, while both Nfkb2+/D865G and Nfkb2+/Lym1 T reg cells can suppress disease, Nfkb2+/Lym1 T reg cells are defective in the trans-acting control of spontaneous CD4+ T cell activation.
Autoimmune susceptibility arises from effects within nonhematopoietic cells
Susceptibility to autoimmunity arising from NFKB2/Nfkb2 mutations could be due to effects within hematopoietic cells, including T cells, and/or nonhematopoietic cells, which play a crucial role in antigen presentation to developing thymocytes. To distinguish between these possibilities, chimeras were made in which T cell–depleted BM from Nfkb2+/+ or Nfkb2+/Lym1 donors was transferred into irradiated Nfkb2+/+, Nfkb2+/D865G, or Nfkb2+/Lym1 hosts. To prevent the onset of autoimmune disease before the BM transplant, all hosts were αβ T cell–deficient (Tcra−/−). Transplantation of Nfkb2+/Lym1 BM into WT recipients did not cause disease (Fig. 5 A), indicating that the cell-intrinsic defects in T reg cell development and function do not cause autoimmunity when the nonhematopoietic compartment is normal. In contrast, most of the Nfkb2+/Lym1 hosts that received Nfkb2+/+ BM developed a fatal disease with a median survival after BM transplantation of 49 d, significantly shorter than all other groups (Fig. 5 A). This disease could be inhibited by exogenous T reg cells, as purified T reg cells injected 8 d after BM transplantation rescued half of the Nfkb2+/Lym1 hosts bearing Nfkb2+/+ BM (Fig. 5 B). No disease occurred in unirradiated Tcra−/− Nfkb2+/Lym1 hosts that received Nfkb2+/+ splenocytes, suggesting that T cell development in the Nfkb2+/Lym1 host thymus was required to induce pathology. These data establish that susceptibility to autoimmunity in Nfkb2+/Lym1 mice stems from abnormalities in nonhematopoietic cells.

Autoimmune susceptibility arises from effects within nonhematopoietic cells . (A) Posttransplant survival curves of Nfkb2+/+, Nfkb2+/Lym1 or Nfkb2+/D865G mice, which were αβ T cell–deficient (Tcra−/−) and aged 63–85 d at irradiation and reconstitution with T cell–depleted BM from Nfkb2+/+ or Nfkb2+/Lym1 donors (see key, middle). (B) Survival curves of Nfkb2+/Lym1 mice after irradiation and reconstitution with Nfkb2+/+ BM on day 0 without further treatment (blue trace) with some Nfkb2+/Lym1 mice subsequently receiving FACS-sorted Foxp3GFP+ spleen and lymph node cells from WT Foxp3GFP mice at day 8 (red trace). In the group receiving T reg cells, some recipients were Tcra−/− and aged 78–122 d, whereas others were Tcra+/+ and aged 54–88 d, at irradiation. Since T reg cell transfer prevented disease in three out of four Tcra−/− recipients and two out of six Tcra+/+ recipients, these groups were combined. Other Tcra−/− Nfkb2+/Lym1 mice received whole Nfkb2+/+ splenocytes on day 0 in the absence of any other treatment (gray trace). Data in A and B were combined from two separate experiments each. Grids show the P values of log-rank tests comparing each pair of experimental groups.
Autoimmune susceptibility arises from effects within nonhematopoietic cells . (A) Posttransplant survival curves of Nfkb2+/+, Nfkb2+/Lym1 or Nfkb2+/D865G mice, which were αβ T cell–deficient (Tcra−/−) and aged 63–85 d at irradiation and reconstitution with T cell–depleted BM from Nfkb2+/+ or Nfkb2+/Lym1 donors (see key, middle). (B) Survival curves of Nfkb2+/Lym1 mice after irradiation and reconstitution with Nfkb2+/+ BM on day 0 without further treatment (blue trace) with some Nfkb2+/Lym1 mice subsequently receiving FACS-sorted Foxp3GFP+ spleen and lymph node cells from WT Foxp3GFP mice at day 8 (red trace). In the group receiving T reg cells, some recipients were Tcra−/− and aged 78–122 d, whereas others were Tcra+/+ and aged 54–88 d, at irradiation. Since T reg cell transfer prevented disease in three out of four Tcra−/− recipients and two out of six Tcra+/+ recipients, these groups were combined. Other Tcra−/− Nfkb2+/Lym1 mice received whole Nfkb2+/+ splenocytes on day 0 in the absence of any other treatment (gray trace). Data in A and B were combined from two separate experiments each. Grids show the P values of log-rank tests comparing each pair of experimental groups.
Impaired thymic medullary development in mice with p100 degron mutations
As autoimmunity arises when the Nfkb2+/Lym1 genotype is confined to the nonhematopoeitic compartment, which includes thymic epithelial cells (TECs), we characterized thymic structure. The histological demarcation between cortex and medulla was unclear in Nfkb2D865G/D865G and Nfkb2Y868indel/Y868indel mice (Fig. 6 A). Except for Nfkb2+/S866fs mice, which were similar to WT (data not shown), the medullary area tended to be smaller in all strains bearing mutations in the Nfkb2 degron (Fig. 6 B). In contrast, thymus sections from Nfkb2xdr/xdr mice had clearly demarcated regions of cortex and medulla with normal medullary area (Fig. 6, A and B), as observed in Nfkb2−/− mice (Zhu et al., 2006). In all strains examined by immunofluorescence, we found distinct regions of expression of cytokeratin-8 (K8) and cytokeratin-14 (K14), which characterize the cortex and medulla, respectively (Fig. 6 C). The lectin Ulex Europaeus agglutinin 1 (UEA-1) binds to mature mTECs (Farr and Anderson, 1985). UEA-1 staining was reduced in Nfkb2+/Lym1 and Nfkb2xdr/xdr mice (Fig. 6 D). While AIRE was readily detected in Nfkb2+/+, Nfkb2+/D865G, and Nfkb2xdr/xdr mice, AIRE+ cells were rare in thymic sections from Nfkb2+/Lym1 mice (Fig. 6 E).

Impaired thymic medullary development in mice with p100 degron mutations . (A and B) Thymus sections stained with hematoxylin and eosin (A; scale bars, 500 µm) with a graph showing the percentage area occupied by the medulla (B). Age did not differ significantly between genotypes (mean ± SD, 110 ± 35 d; range, 55–181 d). (C–E) Immunofluorescence microscopy on thymus sections from mice at 61–86 d of age detecting K8 (cyan) and cytokeratin-14 (K14; magenta; C), UEA-1 (yellow; D), or cytokeratin-5 (K5; magenta) and AIRE (cyan; E); c, cortex; m, medulla; dashed line, corticomedullary border (n = 2 mice per genotype in a single experiment; scale bars, 50 µm). The detection of different proteins in the same set of sections is shown in C and D. (F and G) From mice aged 84–133 d, thymic cells released by enzymatic digestion were analyzed by flow cytometry for CD45/EpCAM (F) to identify CD45− EpCAM+ TECs that were analyzed for binding of UEA-1 and expression of AIRE (G). (H) Plots show the CD19/CD11c phenotype of thymic CD45+ cells with gates for B cells (CD19+) and DCs (CD11c+). For each subset gated in the plots, graphs show the number of cells per thymus (right). Each symbol in a graph represents an individual mouse and horizontal bars mark the group mean compiled from two separate experiments. Graphs show data from female and male mice. Each genotype was compared with the Nfkb2+/+ group using one-way ANOVA with Sidak’s multiple comparisons test; log10-transformed values were used in F–H; *, P < 0.05; ***, P < 0.001; ****, P < 0.0001.
Impaired thymic medullary development in mice with p100 degron mutations . (A and B) Thymus sections stained with hematoxylin and eosin (A; scale bars, 500 µm) with a graph showing the percentage area occupied by the medulla (B). Age did not differ significantly between genotypes (mean ± SD, 110 ± 35 d; range, 55–181 d). (C–E) Immunofluorescence microscopy on thymus sections from mice at 61–86 d of age detecting K8 (cyan) and cytokeratin-14 (K14; magenta; C), UEA-1 (yellow; D), or cytokeratin-5 (K5; magenta) and AIRE (cyan; E); c, cortex; m, medulla; dashed line, corticomedullary border (n = 2 mice per genotype in a single experiment; scale bars, 50 µm). The detection of different proteins in the same set of sections is shown in C and D. (F and G) From mice aged 84–133 d, thymic cells released by enzymatic digestion were analyzed by flow cytometry for CD45/EpCAM (F) to identify CD45− EpCAM+ TECs that were analyzed for binding of UEA-1 and expression of AIRE (G). (H) Plots show the CD19/CD11c phenotype of thymic CD45+ cells with gates for B cells (CD19+) and DCs (CD11c+). For each subset gated in the plots, graphs show the number of cells per thymus (right). Each symbol in a graph represents an individual mouse and horizontal bars mark the group mean compiled from two separate experiments. Graphs show data from female and male mice. Each genotype was compared with the Nfkb2+/+ group using one-way ANOVA with Sidak’s multiple comparisons test; log10-transformed values were used in F–H; *, P < 0.05; ***, P < 0.001; ****, P < 0.0001.
Flow cytometry revealed that the Nfkb2D865G allele caused a dose-dependent reduction in the number of TECs, identified as CD45− EpCAM+ cells, with the magnitude of the defect in Nfkb2D865G/D865G mice similar to that observed in Nfkb2+/Lym1 mice (Fig. 6 F). Aire+ TECs were present but reduced in Nfkb2+/D865G mice, while these cells were rare in Nfkb2D865G/D865G and Nfkb2+/Lym1 mice (Fig. 6 G). A single Nfkb2−/− mouse, which was obtained for this experiment, had a clearly identifiable Aire+ mTEC population, albeit reduced in frequency compared with Nfkb2+/+ controls (Fig. 6 G). Consistent with the immunohistology, mature-phenotype UEA-1+ AIRE− mTECs were decreased in Nfkb2+/Lym1 mice, as they were in Nfkb2D865G/D865G mice (Fig. 6 G). Thymic B cells were decreased in Nfkb2+/D865G, Nfkb2D865G/D865G, and Nfkb2+/Lym1 mice, and thymic dendritic cells (DCs) were decreased in Nfkb2D865G/D865G and Nfkb2+/Lym1 mice (Fig. 6 H). Overall, while UEA-1+ and Aire+ mTECs were reduced in Nfkb2xdr/xdr and Nfkb2−/− mice, consistent with previous reports (O’Reilly et al., 2015; Zhu et al., 2006), medullary size and haemopoietic APCs were normal in these strains lacking p100 and p52. In contrast, the Nfkb2+/Lym1, Nfkb2D865G/D865G, and Nfkb2Y868indel/Y868indel genotypes caused a severe block in thymic medullary development that affected both epithelial and hematopoietic APCs.
Accumulation of pancreas-specific CD4+ T cells
In models of autoimmune exocrine pancreatitis, protein disulfide isomerase family A member 2 (PDIA2) is a self-antigen that is targeted by B and T cells in NOD.Aire−/− mice and BALB/c.Ctla4−/− mice, respectively (Ise et al., 2010; Niki et al., 2006). To test whether Nfkb2 mutations affect T cell tolerance to PDIA2, we analyzed CD4+ T cells bound by tetramers of the MHC class II molecule IAb, presenting a peptide corresponding to PDIA2 residues 83–93 (IAb-PDIA2; Malhotra et al., 2016). Compared with Nfkb2+/+ controls, IAb-PDIA2–specific CD4+ T cell populations were larger in Nfkb2+/Lym1 mice, but not in Nfkb2+/D865G mice (Fig. 7 A). The IAb-PDIA2–specific CD4+ T cell population in B6.Aire−/− mice was normal in size (Fig. 7 A); this was expected, because B6.Aire−/− mice do not develop autoimmune exocrine pancreatitis (Jiang et al., 2005). Tetramer staining intensity was increased in Nfkb2+/Lym1 mice (Fig. 7 A), suggesting that many of the expanded CD4+ T cells had a high TCR affinity for IAb-PDIA2. CD44 expression was also increased (Fig. 7 A), indicating some self-antigen–specific CD4+ T cells in Nfkb2+/Lym1 mice had acquired an antigen-experienced phenotype. In contrast, the size and phenotype of CD4+ T cell populations specific for a foreign peptide corresponding to residues 81–95 of GFP (Malhotra et al., 2016) were comparable in Nfkb2+/+ and Nfkb2+/Lym1 mice (Fig. 7 B). These data show that CD4+ T cell tolerance to the pancreatic autoantigen, PDIA2, is impaired in Nfkb2+/Lym1 mice.

Increased self-reactivity of T cells selected in the presence of pathogenic NFKB2/Nfkb2 genotypes (see also Fig. S3 ). (A) After magnetic bead-based enrichment of IAb-PDIA2 tetramer-binding cells from pooled spleen and lymph nodes, plots show IAb-PDIA2 tetramer staining versus CD44 expression on CD4+ T cells, with summaries (right) showing the total number of tetramer-binding CD4+ cells detected per mouse, tetramer relative fluorescence intensity (RFI), and CD44 RFI of the tetramer-binding cells. To calculate RFI, mean fluorescence intensities were divided by the mean of Nfkb2+/+ samples analyzed on the same day. (B) For mice of the indicated genotypes (top), some of which had been immunized with GFP81–95 emulsified in CFA, plots show IAb-GFP81-95 tetramer staining versus CD44 expression on CD4+ T cells, with summaries presented as in A (right). In A and B, numbers on plots indicate the number of cells in the gate shown, and each symbol in a graph represents one mouse while horizontal bars show group means. Age did not differ significantly between genotypes or peptides (mean ± SD, 135 ± 48 d; range, 63–312 d). Graphs show data from female and male mice compiled from seven (A) or two (B) separate experiments. (C) For T cell subsets sorted from the thymus or spleen (top) of female mice of the indicated genotypes (color coded, right), graphs show the percentage of unique TCRα (squares) or TCRβ (circles) sequences with a self-reactivity-promoting amino acid doublet at CDR3 P6-P7 (hydrophobic index; Stadinski et al., 2016). Age did not differ significantly between genotypes (mean ± SD, 105 ± 12 d; range, 84–120 d). IELp, precursors of CD8αα intestinal intraepithelial lymphocytes. (D) Hydrophobic index of the TCRβ repertoire of T cell subsets (top) sorted from the blood of healthy control subjects or individuals with mutations in NFKB2 or AIRE (see key, right). Each symbol represents an individual sample. Statistical comparisons used one-way (A, B, and D) or two-way (C) ANOVA to compare each group with the control group, followed by Sidak’s multiple comparisons test, using log10-transformed values for tetramer+ cell counts (A and B) and TCRα and TCRβ values matched by mouse (C); *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Increased self-reactivity of T cells selected in the presence of pathogenic NFKB2/Nfkb2 genotypes (see also Fig. S3 ). (A) After magnetic bead-based enrichment of IAb-PDIA2 tetramer-binding cells from pooled spleen and lymph nodes, plots show IAb-PDIA2 tetramer staining versus CD44 expression on CD4+ T cells, with summaries (right) showing the total number of tetramer-binding CD4+ cells detected per mouse, tetramer relative fluorescence intensity (RFI), and CD44 RFI of the tetramer-binding cells. To calculate RFI, mean fluorescence intensities were divided by the mean of Nfkb2+/+ samples analyzed on the same day. (B) For mice of the indicated genotypes (top), some of which had been immunized with GFP81–95 emulsified in CFA, plots show IAb-GFP81-95 tetramer staining versus CD44 expression on CD4+ T cells, with summaries presented as in A (right). In A and B, numbers on plots indicate the number of cells in the gate shown, and each symbol in a graph represents one mouse while horizontal bars show group means. Age did not differ significantly between genotypes or peptides (mean ± SD, 135 ± 48 d; range, 63–312 d). Graphs show data from female and male mice compiled from seven (A) or two (B) separate experiments. (C) For T cell subsets sorted from the thymus or spleen (top) of female mice of the indicated genotypes (color coded, right), graphs show the percentage of unique TCRα (squares) or TCRβ (circles) sequences with a self-reactivity-promoting amino acid doublet at CDR3 P6-P7 (hydrophobic index; Stadinski et al., 2016). Age did not differ significantly between genotypes (mean ± SD, 105 ± 12 d; range, 84–120 d). IELp, precursors of CD8αα intestinal intraepithelial lymphocytes. (D) Hydrophobic index of the TCRβ repertoire of T cell subsets (top) sorted from the blood of healthy control subjects or individuals with mutations in NFKB2 or AIRE (see key, right). Each symbol represents an individual sample. Statistical comparisons used one-way (A, B, and D) or two-way (C) ANOVA to compare each group with the control group, followed by Sidak’s multiple comparisons test, using log10-transformed values for tetramer+ cell counts (A and B) and TCRα and TCRβ values matched by mouse (C); *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Increased self-reactivity of T cells selected in the presence of pathogenic NFKB2/Nfkb2 genotypes
The presence of cysteine and hydrophobic residues at specific sites in complementarity-determining region 3 (CDR3) of αβ TCRs promotes T cell self-reactivity (Stadinski et al., 2016; Wirasinha et al., 2018). As the frequency of these TCR-intrinsic motifs varies predictably across T cell subsets in healthy mice and humans, they serve as biomarkers to diagnose and classify T cell tolerance defects (Daley et al., 2019). To test whether Nfkb2 or Aire mutations affect the self-reactivity of mature T cell populations in mice, we sequenced the TCRα and TCRβ repertoires of six T cell subsets sorted from the thymus and spleen (Fig. S3, A and B). In all mouse strains, the percentage of unique CDR3 sequences with cysteine within two positions of the CDR3 apex (cysteine index) was highest in thymic type A precursors of CD8αα intestinal intraepithelial lymphocytes (Fig. S3 C), which are induced by strong TCR signaling in the thymic cortex (Ruscher et al., 2017). No strain had an increased cysteine index in the T reg cell, CD4+ conventional T cell (T conv cell), or CD8+ T conv cell TCR repertoires (Fig. S3 C). Likewise, the cysteine index was normal in samples from patients with NFKB2 or AIRE mutations (Fig. S3, D and E). These genetic lesions do not perturb tolerance induction in T cells with cysteine at the CDR3 apex.

T cell sorting gates for TCR-sequencing and cysteine index results (related to Fig. 7 ). (A and B) Flow cytometry gates used to sort T cell subsets from (A) thymus and (B) spleen of female Nfkb2+/+, Nfkb2+/D865G, Nfkb2D865G/D865G, Nfkb2+/Lym1, Nfkb2xdr/xdr, and Aire−/− mice aged 84–120 d (n = 3/genotype). (C) For T cell subsets sorted from the thymus or spleen (top) of mice of the indicated genotypes (color coded, right), graphs show the percentage of unique TCRα (squares) or TCRβ (circles) sequences with cysteine within two positions of the CDR3 apex (cysteine index). (D) Gating strategy to sort T cell subsets from human PBMC for TCR sequencing. (E) Cysteine index of T cell subsets (bottom) sorted from PBMC of healthy control subjects or individuals with mutations in NFKB2 or AIRE (see key, right). In C and E, filled symbols indicate samples that had zero sequences with cysteine within two positions of the CDR3 apex; in these cases, the symbols represent the reciprocal of the number of unique sequences in the sample, expressed as a percentage; *, P < 0.05; one-way ANOVA with Sidak’s multiple comparisons test.
T cell sorting gates for TCR-sequencing and cysteine index results (related to Fig. 7 ). (A and B) Flow cytometry gates used to sort T cell subsets from (A) thymus and (B) spleen of female Nfkb2+/+, Nfkb2+/D865G, Nfkb2D865G/D865G, Nfkb2+/Lym1, Nfkb2xdr/xdr, and Aire−/− mice aged 84–120 d (n = 3/genotype). (C) For T cell subsets sorted from the thymus or spleen (top) of mice of the indicated genotypes (color coded, right), graphs show the percentage of unique TCRα (squares) or TCRβ (circles) sequences with cysteine within two positions of the CDR3 apex (cysteine index). (D) Gating strategy to sort T cell subsets from human PBMC for TCR sequencing. (E) Cysteine index of T cell subsets (bottom) sorted from PBMC of healthy control subjects or individuals with mutations in NFKB2 or AIRE (see key, right). In C and E, filled symbols indicate samples that had zero sequences with cysteine within two positions of the CDR3 apex; in these cases, the symbols represent the reciprocal of the number of unique sequences in the sample, expressed as a percentage; *, P < 0.05; one-way ANOVA with Sidak’s multiple comparisons test.
Hydrophobic amino acid doublets at positions 6 and 7 (P6-P7) of the CDR3 in TCRβ (CDR3β) can promote T cell self-reactivity (Stadinski et al., 2016). In all mouse strains, the percentage of unique sequences with a hydrophobic doublet at P6-P7 (hydrophobic index) was highest in type A precursor of CD8αα intestinal intraepithelial lymphocytes (Fig. 7 C). While the hydrophobic indices in Nfkb2+/D865G, Nfkb2xdr/xdr, and Aire−/− mice were normal, Nfkb2D865G/D865G and Nfkb2+/Lym1 mice had increased hydrophobic indices in thymic and splenic CD8+ T conv cells, as well as in splenic CD4+ T conv cells (Fig. 7 C).
After establishing this important signature of defective T cell self-tolerance in mice with p100 degron mutations, we tested for a similar change in the peripheral T cell repertoire of patients. Remarkably, the hydrophobic index was also increased in the CD4+ T conv cell TCRβ repertoires of patients with heterozygous NFKB2 mutations, but not in patients with homozygous AIRE mutations (Fig. 7 D). Thus, in humans and mice, the presence of pathogenic mutations in the p100 degron results in increased T cell self-reactivity.
Discussion
Nfkb2 mutations in the p100 degron cause T cell–dependent autoimmune exocrinopathy, including severe exocrine pancreatitis. Pathology involves mTEC and T reg cell developmental defects that are distinct from those caused by the absence of p100 and p52 or AIRE. Distinct Nfkb2 degron mutations confer different extents of degradation resistance on the mutated p100 proteins in a manner that is reflected in the p100/p52 ratio. Nfkb2 mutations that cause moderate T cell tolerance defects remain subclinical in B6 mice, but fully penetrant autoimmune disease develops above a threshold of p100-degradation resistance. T cell central tolerance appears to be exquisitely sensitive to the IκB function of mutated p100 proteins.
While the defect in thymic tolerance is largely T cell extrinsic and mediated by changes in thymic epithelium, we also observed T cell–intrinsic defects. Defects in p100 degradation would be expected to have pleiotropic effects, because NF-κB members are required for the development of multiple cell types that establish immune self-tolerance. For example, T cell–specific deletion of RelA or c-Rel causes graded T reg cell deficiencies, while deletion of both RelA and c-Rel abolishes T reg cells (Oh et al., 2017). In contrast, T cell–specific or T reg cell–specific deletion of NF-κB2 causes T reg cell expansion (Grinberg-Bleyer et al., 2018). Consistent with this, Nfkb2xdr/xdr cells, which lack NF-κB2, had an enhanced capacity for T reg cell development in mixed BM chimeras. T reg cell deficiency in mice bearing the Nfkb2D865G, Nfkb2Lym1, or Nfkb2Y868indel mutations is not consistent with p52 insufficiency; instead, it is consistent with an exaggerated IκB function of the mutated p100 proteins, which may sequester RelA and c-Rel in the cytoplasm of T cells (Basak et al., 2007; Lee et al., 2014; Savinova et al., 2009; Scheinman et al., 1993; Sun et al., 1994; Tao et al., 2014). We interpret the mTEC results similarly. Mice with TEC-specific deletion of RelA, RelA and c-Rel, or RelB exhibit progressively more severe blocks in the development of AIRE− and AIRE+ mTECs (Riemann et al., 2017). Nfkb2+/Lym1 and Nfkb2D865G/D865G mice have a smaller thymic medulla and fewer mature (UEA-1hi AIRE− or AIRE+) mTECs than Nfkb2xdr/xdr and Nfkb2−/− mice. We postulate that mTEC development in Nfkb2+/Lym1 and Nfkb2D865G/D865G mice is blocked because the mutated p100 proteins inhibit RelA, c-Rel, and/or RelB.
AIRE-dependent T reg cell selection in early life is critical to establish self-tolerance in NOD.Aire−/− mice (Yang et al., 2015). Autoimmunity caused by AIRE deficiency involves aberrant thymic selection, into the CD4+ T conv cell lineage, of TCR specificities normally found in the T reg cell lineage (Malchow et al., 2016; Perry et al., 2014). A similar misdirection may occur in Nfkb2D865G/D865G and Nfkb2+/Lym1 mice in which few AIRE+ mTECs are present. However, Nfkb2 degron mutations caused defects that were not observed in Aire−/− mice, such as thymic medullary hypoplasia and decreases in the number of UEA1+ Aire− mTECs, B cells, and DCs in the thymus. When transplanted with WT or BCL2-tg BM, Nfkb2+/D865G and Nfkb2+/Lym1 hosts had fewer Helios+ Foxp3− cells among CD4SP CCR7+ thymocytes than WT and Aire−/− hosts, suggesting that the size of the thymic medulla limits the magnitude of wave 2 deletion. Spontaneous expansion of high-affinity, IAb-PDIA2–specific, CD4+ T cells in Nfkb2+/Lym1 mice is consistent with increased escape of self-reactive T cells from thymic deletion or T reg cell differentiation (Malhotra et al., 2016). As mixed chimeras provided no evidence that the Nfkb2+/Lym1 genotype confers a thymocyte-intrinsic defect in deletion, the T cell deletion defect is attributable to defects in thymic APCs. T cell–extrinsic defects in Nfkb2+/Lym1 mice are compounded by T cell–intrinsic defects. Even when selected in the presence of WT thymic APCs, Nfkb2+/Lym1 T reg cells exhibited a deficit in trans-acting control of CD4+ CD44high Foxp3− T cell differentiation. TCR sequencing failed to reveal an effect of AIRE/Aire mutations on the hydrophobic index, revealing that this index fails to detect certain changes in the TCR repertoire (Malchow et al., 2016; Perry et al., 2014). By contrast, we observed increased hydrophobic indices in mice with pathogenic Nfkb2 mutations and then confirmed that this signature could be detected within the TCR repertoire of humans, indicating that frequent escape of self-reactive T cells from tolerance mechanisms is a robust marker of thymic dysfunction conferred by severe p100-degradation resistance. Thus, in mice on the B6 genetic background, the Nfkb2+/Lym1 and Nfkb2D865G/D865G genotypes confer T cell–extrinsic and T cell–intrinsic self-tolerance defects that are not present in Aire−/− mice, providing an explanation for the greater severity of autoimmune manifestations.
Why might the extent of the developmental defect caused by degradation-resistant p100 differ between cell types? The fate of p100 is influenced by the relative concentrations of RelB and the NIK:IKKα complex, which compete with each other for binding to p100 (Fusco et al., 2016). RelB diverts p100 into kappaBsomes, whereas the NIK–IKKα complex phosphorylates p100 to trigger its degradation (Fusco et al., 2016). The p100 degron is required for the binding of p100 to IKKα (Xiao et al., 2004) but not for the binding of p100 to RelB (Fusco et al., 2008). By inhibiting p100 degradation, p100 degron mutations may promote p100 diversion into kappaBsomes in a RelB-regulated manner. Compared with splenic T reg cells, RelB mRNA expression is 1.7 times higher in follicular B cells and 7 times higher in mature mTECs and CD8+ DCs (Heng et al., 2008). Thus, while T reg cell population size was decreased by a similar magnitude in all mice with the Nfkb2D865G, Nfkb2Lym1, or Nfkb2Y868indel alleles, the greater sensitivity of mTECs, B cells, and DCs in the thymus to severe p100-degradation resistance may be due to RelB-mediated stabilization of the mutated p100 proteins.
In Nfkb2Ser866fs/S866fs mice, the T reg cell deficiency and trend toward a reduced thymic medullary area are consistent with exaggerated IκB function, suggesting that p100S866fs may be partially resistant to signal-dependent degradation. As these phenotypes were milder or similar to those of Nfkb2+/D865G and Nfkb2+/Y868indel mice, it is not surprising that Nfkb2Ser866fs/S866fs mice did not develop spontaneous autoimmunity. The decreased abundance of p100 and p52 in Nfkb2Ser866fs/S866fs mice suggests that the mutated and lengthened C-terminal domain may also perturb the structural integrity of p100. This is plausible because the unphosphorylated degron contributes to the interaction between the N- and C-terminal domains of p100 (Qing et al., 2005). To date, no human NFKB2 mutations that lengthen the p100 protein have been recognized as pathogenic.
Heterozygous NFKB1 mutations cause clinical manifestations similar to heterozygous NFKB2 mutations, but distinctions are emerging. Pathogenic NFKB1 mutations are distributed throughout the protein and reduce the abundance of p50, suggesting that haploinsufficiency of p50 is pathogenic, although with incomplete penetrance (Fliegauf et al., 2015; Tuijnenburg et al., 2018). In contrast, clinically recognized NFKB2 mutations cluster in the p100 degron (Klemann et al., 2019), consistent with a distinct underlying mechanism, such as IκB function of the mutated p100 proteins. Our mouse models have revealed that autoimmunity caused by p100 degron mutations is T cell dependent but arises largely due to defects in nonhematopoietic cells. Thus, while successful hematopoietic stem cell transplantation is likely to reduce susceptibility to recurrent infections in patients with NFKB2 mutations, this treatment may not reduce susceptibility to autoimmunity. Our findings suggest that suppressing the IκB activity of degradation-resistant p100 may mitigate autoimmunity.
Materials and methods
Mice
In each experiment, most or all of the mice described as Nfkb2+/+ were siblings of mice with Nfkb2 mutations. As results obtained from Nfkb2+/+ mice were comparable with B6 mice, which were used in some experiments, we refer to them collectively as Nfkb2+/+ (or WT) mice. The Nfkb2Lym1 line was rederived at the Australian Phenomics Facility, Canberra, using in vitro fertilization with cryopreserved sperm from a Nfkb2+/Lym1 mouse on the BALB/c background (Tucker et al., 2007). Data were obtained from Nfkb2+/Lym1 mice that were the offspring of ≥6, and mostly >10, generations of backcrossing to B6. Mice carrying BCL2-tg (Tg(Vav-BCL2)1Jad), Nfkb2xdr, Aire− (Airetm1Pltn), CD451 (Ptprca), Foxp3GFP (Foxp3tm2Ayr), Foxp3null (Foxp3tm1.1Ayr), H2-Aa− (H2-Aatm1Blt), B2m− (B2mtm1Jae), or Tcra− (Tcratm1Mom) on the B6 genetic background were bred, intercrossed in some cases, and housed in specific-pathogen–free environments at 18–24°C and 40–70% humidity with a lighting cycle of 7 a.m. to 7 p.m. light (below 350 lux) and 7 p.m. to 7 a.m. darkness, at the Australian Phenomics Facility, Canberra, or at Monash University, Melbourne. Mice were genotyped using PCR assays on genomic DNA extracted from ear or tail biopsies. All procedures were performed in accordance with protocols approved by The Animal Experimentation Ethics Committees of the Australian National University (A2014/62 and A2018/06) or Monash University (MARP/2015/64). Thymus samples from a single Nfkb2−/− (Nfkb2tm1Sbn) mouse and its Nfkb2+/+ sibling were provided by Vanessa Bryant (The Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia).
The Nfkb2D865G, Nfkb2Y868indel, and Nfkb2S866fs alleles were generated at the Australian Phenomics Facility, Canberra, using CRISPR/Cas9 gene editing (Yang et al., 2014). To synthesize Cas9 mRNA, a plasmid encoding Cas9 with a 3′ 95-nt poly(A) tail (plasmid 48625:pCAG-T3-hCAS-pA; Addgene) was linearized with Sph1 (New England Biolabs) and transcribed in vitro using mMessage mMachine T3 kit (Life Technologies). RNA was precipitated with absolute ethanol, suspended in RNase-free water, and stored at −80°C until use. The single guide RNA (sgRNA) included a sequence corresponding to the target site in exon 22 of Nfkb2 (5′-CTCCACTGACTGGCTCCCAT-3′; protospacer-associated motif italicized and underlined). To synthesize sgRNA, two complementary oligos (Integrated DNA Technologies) were mixed in the annealing buffer (1 M Tris, pH 8, 1 M MgCl2, 5 M NaCl, and 0.5 M EDTA, pH 8), gently spun down, heated to 95°C for 5 min, and then cooled by 5°C/min to a final temperature of 25°C. sgRNA was transcribed using 5–8 µl annealed oligos with the MEGAshort transcription kit (Life Technologies) followed by alcohol precipitation. The quality and quantity of Cas9 mRNA and sgRNA were analyzed using Nano drop and bioanalyzer. The homology-directed repair template was a 103-base single-stranded sense oligonucleotide with 50-nt arms corresponding to genomic DNA on either side of the codon with the desired mutation (GAC>GGC) and harboring a silent mutation in the protospacer-associated motif sequence (GGG>GGA; underlining indicates nucleotides targeted by CRISPR and the desired change; Integrated DNA Technologies).
Following established protocols (Yang et al., 2014), superovulated B6 (28-d-old) female mice were mated overnight with B6 male mice. Zygotes were collected at 20 h, and pronuclei-formed zygotes were put into the M2 medium (Sigma-Aldrich). After four to six washes in M2 medium at room temperature, embryos were transferred in to M16 medium that was precalibrated overnight at 37°C. Cas9 mRNA, sgRNA, and the homology-directed repair template (100 ng/µl) were mixed, and ∼4 pl was injected into the cytoplasm of each zygote with well-recognized pronuclei using an Eppendorf micromanipulator. After injection, all zygotes were cultured overnight in M16 medium at 37°C and 5% CO2. 15–20 fertilized one- or two-cell embryos were transferred into the oviduct of 8-wk-old female Swiss albino mice mated with vasectomized males the previous night. For genotyping, ear punches were obtained from 15-d-old pups and suspended in TE-Tween Lysis Buffer (50 mM Tris HCl, 0.125 mM EDTA, and 2% Tween 20, pH 8.0). 1.0 µl Proteinase K (20 mg/ml) was added to the mix and incubated at 56°C for 1 h followed by 99°C for 10 min to denature Proteinase K. 5 µl was used as a PCR template with the following primers: forward, 5′-CCCTGAAGCCTGAAACCTTGG-3′; and reverse, 5′-CAGCCTCCACCCTCATTAAA-3′. PCR products were purified using ExoSAP-IT (Affymetrix), sequenced using the forward primer and results analyzed with Sequencher software (Gene Codes Corporation). A founder mouse with the desired Nfkb2D865G substitution, along with founder mice bearing other substitutions (Nfkb2Y868indel and Nfkb2S866fs), were backcrossed to B6 to propagate the alleles and a further two generations of backcrossing were performed before mice were used for experiments. Effects of variant alleles on protein sequences were determined using Mutalyzer (https://mutalyzer.nl).
BM transplantation and adoptive transfers
To make chimeras, recipient mice were irradiated with x rays (two doses of 4.5 Gy given 4 h apart) and then injected i.v. with at least 2 × 106 BM cells that had been depleted of T cells using magnetic beads (Mouse CD3ε Microbead Kit; Miltenyi) and the “Depl05” program on an autoMACS machine (Miltenyi). To purify T reg cell cells for adoptive transfer, pooled spleen and lymph node cells from Foxp3GFP mice were incubated with anti-CD45R(B220)-biotin (catalog no. 130–101-998; Miltenyi) and anti-CD8α-biotin (catalog no. 130–118-074; Miltenyi), followed by incubation with anti-biotin MicroBeads (catalog no. 130–090-485; Miltenyi) to allow removal of B cells and CD8+ T cells using the “Deplete” program on an autoMACS machine, before 1 × 105–2 × 105 FACS-sorted viable CD4+ GFP+ cells were injected i.v. per recipient.
Immunoblotting
Splenocytes were incubated in lysis buffer (radioimmunoprecipitation assay buffer supplemented with 0.5% Protease Inhibitor [Pierce Net] and 1% Halt Phosphatase Inhibitor Cocktail [Thermo Fisher Scientific]) for 30 min on ice and then centrifuged at 16,000 g for 10 min at 4°C. Supernatant was harvested and protein concentration estimated using a DC protein assay (Bio-Rad). 50 µg of total protein was combined with LDS Sample Buffer and Sample Reducing Agent (Thermo Fisher Scientific) and boiled at 100°C for 10 min. Samples were then subjected to SDS-PAGE followed by wet transfer to polyvinylidene difluoride membrane. Membranes were blocked for 1 h at room temperature with 2.5% BSA in Tris-buffered saline with 0.1% Tween-20 (TBS-T) before overnight incubation with rabbit anti-p100/p52 (catalog no. 4882; Cell Signaling Technology) diluted 1:1,000 in 5% BSA in TBS-T, followed by 1 h in HRP-conjugated anti-rabbit IgG (catalog no. ab6795; Abcam) diluted 1:9,000 in 5% BSA in TBS-T, mouse anti-GAPDH (catalog no. ab8245; Abcam), and HRP-conjugated goat anti-mouse IgG (catalog no. sc-2005; Santa Cruz), diluted 1:3,000 in 5% skim milk in TBS-T, and incubated sequentially for 1 h each. Membranes were imaged using Amersham ECL detection agents (GE Lifesciences) and chemiluminescence detected on a FujiFilm LAS-4000 camera. Protein density was analyzed with ImageJ analysis software (Schneider et al., 2012). Density histograms were plotted for each lane. Histogram peaks for p100, p52, and GAPDH were identified at 98 kD, 49 kD, and 38 kD, respectively. The areas for the p100 and p52 peaks were divided by the area of GAPDH to control for differences in quantity of sample loaded. Each value was then divided by the (mean of) WT lane(s) to obtain a normalized density; p100/p52 ratios were calculated directly, ignoring GAPDH.
Histology
Organs were fixed in 10% neutral buffered formalin and embedded in paraffin, and 4-µm sections were stained with hematoxylin and eosin. Pathology was scored as the average of two or three observers, blinded to animal identifiers, with the following rubric: 0, section shows normal tissue free of inflammatory infiltrates; 1, section shows one or two inflammatory foci, typically with a perivascular distribution; 2, section shows more than two inflammatory foci, with no significant lesions in tissue parenchyma; 3, section shows <50% tissue occupied by lesions including inflammatory infiltrates; 4, section shows >50% lesions with or without inflammatory infiltrates. Lesions in the pancreas/lacrimal gland/salivary gland included acinar cell effacement with preservation of ducts with or without replacement with fat or fibroblasts and in lung included collapse, congestion, emphysema, consolidation, or fibrosis. Thymic medullary areas were determined in a genotype-blinded manner using the Lasso tool and Measurement function in Adobe Photoshop. In addition, 11 live mice were submitted to the Australian Phenomics Network Histopathology and Organ Pathology Service, University of Melbourne, Australia, for comprehensive pre- and postmortem examination.
Flow cytometry including EdU labeling
For CCR7 staining, single-cell thymocyte suspensions were incubated for 60 min at 37°C in prewarmed FACS buffer (PBS containing 2% vol/vol heat-inactivated bovine serum and 0.01% mass/vol sodium azide) containing fluorochrome- or biotin-conjugated anti-CCR7 (catalog no. 120104 or 120105; BioLegend). Cells were pelleted by centrifugation and incubated for 30 min in FACS buffer at 4°C containing assortments of antibodies against TCRβ (catalog no. 109233 or 109234; BioLegend), CD5 (Miltenyi, catalog no. 130–103-796), CD4 (catalog no. 100430; BioLegend), CD8α (catalog no. 100766; BioLegend), CD44 (catalog no. 103039; BioLegend), and B220 (catalog no. 103222; BioLegend). After washing in FACS buffer, cells were fixed and permeabilized using the Foxp3/Transcription Factor Staining Buffer Set (catalog no. 00–5523-00; Thermo Fisher Scientific) and then incubated with antibodies specific for Helios (catalog no. 137220 or 137222; BioLegend) or Foxp3 (catalog no. 11–5773-80; Thermo Fisher Scientific). In some experiments, 0.25 mg EdU in PBS was injected i.v. either 3 d or 5 d before analysis as described in the figure legends, in which case thymocyte samples were then processed using the Click-iT EdU Flow Cytometry Assay Kit (catalog no. C10420 or C10635; Thermo Fisher Scientific) following the manufacturer’s instructions except that Click-iT EdU buffer additive (Component G) was used at one-fifth of the concentration recommended. Samples were then washed in FACS buffer and incubated with streptavidin-PE or -BV650 (catalog no. 405204 or 405232; BioLegend). Human peripheral blood mononucleated cells were isolated by density gradient centrifugation on Ficoll, washed with FACS buffer, and maintained at 4°C. The following antibodies were used: CD19 (SJ25C1), CD3 (HIT3A), CD4 (SK3), CD8 (SK1), and CD25 (2A3; all from BD Biosciences) and CD127 (A7R34; eBioscience). After washing in FACS buffer, data were acquired with LSR II flow cytometers (Becton Dickinson) and analyzed using FlowJo software (FlowJo).
Immunofluorescence
Thymi were dissected, embedded in Tissue-Tek O.C.T compound (Sakura Finetek), snap frozen in a liquid nitrogen/isopentane slurry, and stored at −80°C before use. Samples were cut into 8-µm cryosections using a Micron HM550 Cryostat (Thermo Fisher Scientific), air dried, fixed in ice-cold acetone for 1 min, air dried, and blocked with 5% (vol/vol) goat serum in PBS with 0.5% Tween-20 (vol/vol) for 30 min at room temperature. Sections were incubated for 30 min at room temperature with primary antibodies, including anti-K5 (catalog no. 905504; BioLegend), anti-K14 (catalog no. 90530; BioLegend), biotinylated UEA-1 lectin (catalog no. B-1065; Vector Laboratories), biotinylated anti-mouse AIRE (clone 5H12; Walter and Eliza Hall Institute), and anti-K8 (clone TROMA-I; Developmental Studies Hybridoma Bank). Following three 5-min washes in PBS, sections were incubated with secondary reagents including Donkey anti-rabbit IgG Alexa Fluor 555 (catalog no. A-31572; Invitrogen); Goat anti-rat IgG Alexa Fluor 555 (catalog no. A-21434; Invitrogen); Donkey anti-rabbit IgG Alexa Fluor 647 (catalog no. A-21244; Invitrogen), and Alexa Fluor 488 conjugated streptavidin (catalog no. S32354; Invitrogen) for 30 min at room temperature. Sections were then washed with PBS, counterstained with 300 nM DAPI (Sigma-Aldrich), and mounted with Vectashield (Vector Laboratories). Images were collected using an LSM780 or LSM880 confocal microscope with Zen 2012 SP2 (black) software v11.0 (Zeiss). Single optical sections were processed for presentation using OMERO (Allan et al., 2012) and FIJI (Schindelin et al., 2012).
Thymus digestion and flow cytometry
This procedure is described in detail elsewhere (Jain and Gray, 2014). Briefly, the thymic lobes were separated and cleaned of connective tissue. Snips were made in lobes with scissors, and the fragments were triturated in 5 ml RPMI-1640 medium with 25.96 mM Hepes with a wide-bore pipette tip. The supernatant was recovered and replaced by 1 ml digestion buffer (RPMI-Hepes supplemented with 0.5 Wunsch units of Liberase TM [Roche] and 0.1% wt/vol DNase I [Sigma-Aldrich]). Thymic tissue was digested at 37°C for 15 min with periodic gentle trituration. At the end of the first digestion, the supernatant was recovered, stored on ice (fraction 1), and replaced with 500 µl fresh digestion buffer. Digestion was repeated at 37°C with gentle agitation after every 5 min until a single-cell suspension was obtained (fraction 2). Cells in all fractions were counted before staining for flow cytometry. Surface staining of TECs was performed using the following antibodies for 20 min on ice: anti-mouse CD45 PerCP/Cy5.5 (catalog no. 103132; BioLegend), anti-mouse CD326 (EpCAM) APC/Cy7 (catalog no. 118218; BioLegend), and biotinylated UEA-1 lectin (catalog no. B-1065; Vector Laboratories). Intracellular staining with anti-mouse AIRE-A647 (clone 5H12; Walter and Eliza Hall Institute) was performed after fixation and permeabilization with the eBioscience Foxp3/Transcription Factor Staining Buffer Set (catalog no. 00–5523-00; Thermo Fisher Scientific). Secondary detection of biotinylated UEA-1 was performed with Alexa Fluor 488–conjugated streptavidin (catalog no. S32354; Thermo Fisher Scientific). To quantify thymic B cells and DCs, supernatants recovered from the first trituration step and fraction 1 were stained separately with anti-mouse CD45 PerCP/Cy5.5 (BioLegend), anti-mouse CD19 PE (catalog no. 115508; BioLegend), and anti-mouse CD11c PE/Cy7 (catalog no. 117318; BioLegend) before flow cytometry data were acquired and analyzed as above.
Peptide/MHC class II tetramer synthesis
The regions encoding the extracellular domains of the IAb α- and β-chains were cloned into a modified pFastBac Dual vector (Invitrogen). The IAb α-chain, driven by the polyhedrin promoter, was cloned in frame between a sequence encoding the baculovirus gp67 signal peptide and an enterokinase-cleavable fos-leucine zipper region. The IAb β-chain encoded a factor Xa–cleavable N-terminal glycine-serine linker. This modified IAb β-chain was cloned, in front of the p10 promoter, in frame between a sequence encoding the baculovirus gp67 signal peptide and an enterokinase-cleavable jun leucine zipper region, followed by a BirA recognition sequence and a poly-histidine tag. Sequences encoding peptides for mouse PDIA2 residues 83–93, EYSKAAALLAA (reference sequence NP_001074539.1) or GFP residues 81–95, HDFFKSAMPEGYVQE (reference sequence AMQ45836.1) were cloned in frame between the region encoding the gp67 signal peptide and the N-terminal glycine-serine linker of the IAb β-chain. This construct was used in the Bac-to-Bac system (Invitrogen) to produce recombinant virus as described by the manufacturer, with the exception that a DH10B strain harboring a chitinase/vcathepsin-negative AcMNPV bacmid (AcBACΔCC; Kaba et al., 2004) was used in place of DH10Bac cells. The resultant virus was used to infect High Five insect cells (Trichoplusia ni BTI-TN-5B1-4 cells; Invitrogen) for the production of IAb-PDIA283–93 and IAb-GFP81–95 proteins. Supernatants were harvested 48 h after infection, concentrated, and diafiltered using a tangential flow filtration system (Cogent M1; Merck-Millipore). Recombinant IAb was purified from the diafiltered supernatant using Ni NTA Agarose (Qiagen) and further purified by gel filtration and ion exchange chromatography on Superdex 200 16/60 and HiTrap Q HP columns (GE Biosciences), respectively. Proteins were buffer exchanged into 10 mM Tris, pH 8.0, biotinylated using BirA ligase, desalted in PBS to remove excess biotin, and tetramerized by addition of Streptavidin-PE or Streptavidin-APC (BD Biosciences) at a 4:1 molar ratio.
Detection of peptide/MHC class II–specific T cells
Peptide/MHC class II–specific CD4+ T cell populations were enumerated using tetramer-based magnetic enrichment (Moon et al., 2007). Briefly, the spleen and major lymph nodes (auxiliary, brachial, cervical, inguinal, and mesenteric) from individual mice were pooled and stained with PE-labeled or APC-labeled peptide/MHC class II tetramers. Cells were washed and incubated with anti-PE or anti-APC microbeads (Miltenyi), and tetramer-bound cells were enriched using a magnetic LS column (Miltenyi). Enriched cells were stained with antibodies specific for CD4, CD8α, TCRβ, CD44, CD11b, CD11c, B220, F4/80, and NK1.1 and analyzed using a BD LSRFortessa X-20 flow cytometer. To elicit IAb-GFP81–95 specific T cells, mice were anaesthetized with isoflurane and immunized subcutaneously at two sites with a total of 100 µg GFP81–95 peptide emulsified in a 1:1 ratio with complete Freund’s adjuvant 14 d before analysis.
Human TCR sequencing
Peripheral blood mononuclear cells were obtained from patients and healthy donors upon informed consent, and the study was approved by the Institutional Review Board of the National Institutes of Health (protocols 18-I-0041 and 16-I-N139) or the Human Research Ethics Committee of the Canberra Hospital (ETH1.15.015). CD4− CD8+ (CD8+ T conv), CD4+ CD8− CD127+ CD25− (CD4+ T conv), and CD4+ CD8− CD127− CD25+ (T reg) cells were sorted using the gating strategy shown in Fig. S3 D. For samples from two patients with AIRE mutations (identified as PTCID02 and PTCID03), methods used to amplify and sequence TCRβ mRNA transcripts were described previously (Daley et al., 2019). For all other human T cell samples, TCRβ genes were amplified using proprietary, multiplex PCR protocols with either genomic DNA (Adaptive Biotechnologies) or mRNA (iRepertoire) as template. The PCR products were sequenced using the Illumina HiSeq platform. Custom algorithms (Adaptive Biotechnologies or iRepertoire) were used to filter the raw sequences for errors and to align the sequences to reference genome sequences.
Mouse TCR sequence acquisition and filtering
T cells were sorted from the thymus and spleen according to the gating strategy shown in Fig. S3, A and B. Methods used for RNA isolation, cDNA synthesis, PCR amplification of TCRα and TCRβ transcripts, addition of sequencing adapters and sample indices, amplicon concentration, purification, sequencing, and alignment to mouse genome using molecular identifier groups–based error correction software (Shugay et al., 2014) were described previously (Wirasinha et al., 2018). Primers are listed in Table S2. Sequences with a CDR3 that was out of frame or contained a stop codon were excluded. To avoid overestimating TCR diversity due to PCR or sequencing errors, sequences detected only once in any given sample were excluded. A unique sequence was defined as a unique combination of Trav or Trbv gene and CDR3 nucleotide sequence.
TCR sequence analyses
For hydrophobic and cysteine index analyses, CDR3 sequences <8 aa were excluded because a conserved Phe or Try is present at position 6 or 7 of CDR3 sequences that are 6 or 7 aa long. The hydrophobic index equals the percentage of unique clonotypes with a CDR3 P6-P7 doublet corresponding to any of the 175 aa doublets identified as promoting self-reactivity when present at CDR3β P6-P7 (Stadinski et al., 2016). The cysteine index equals the percentage of unique clonotypes with cysteine within two positions of the CDR3 apex. Briefly, for a CDR3 sequence of n amino acids, the amino acid at the largest position not greater than (n/2 + 1) was defined as the CDR3 apex (Wirasinha et al., 2018).
Data visualization and statistical analyses
The “tidyverse,” “stringr,” “survival,” and “reshape2” packages were used to perform TCR sequence analyses, conduct Pearson’s tests for correlation, and produce graphs in RStudio. Statistical analyses were performed using GraphPad Prism version 7.0a (GraphPad Software) with multiple comparisons tests recommended by GraphPad Prism. Figures were made using Adobe Illustrator (Adobe Systems).
Materials availability
Mouse strains generated in this study are available from the Australian Phenomics Facility with a completed Materials Transfer Agreement.
Data and code availability
TCR sequencing data have been deposited in the NCBI Short Read Archive under BioProject numbers PRJNA606989 (mouse) and PRJNA606976 (human subjects PTCID02 and PTCID03). Refer to the Monash University data repository digital object identifier 10.26180/5e4a78acb70ef for the code for calculating cysteine and hydrophobic indices and the digital object identifier 10.26180/5da7d4b26478f for the filtered list of mouse TCR clonotypes plus a summary of the TCR-sequencing results at the individual-sample level.
Online supplemental material
Fig. S1 shows NFKB2/Nfkb2 variants and statistical analysis of lifespan in the murine Nfkb2 allelic series. Fig. S2 shows gating strategies to quantify thymic deletion at waves 1 and 2. Fig. S3 shows the T cell sorting gates for TCR-sequencing and cysteine index results. Table S1 shows the NFKB2 and AIRE mutations and clinical manifestations observed in patients examined. Table S2 lists primers used to amplify mouse Tcra and Tcrb transcripts.
Acknowledgments
We thank Vanessa Bryant for Nfkb2−/− thymus tissue, the Australian Phenomics Facility staff for animal husbandry and genotyping, and the Monash University platforms for Animal Research, Histology, FlowCore, and Micromon for their services.
This work was funded by National Health and Medical Research Council grants 1107464 (M.C. Cook and S.R. Daley), 1108800 (C.C. Goodnow and S.R. Daley), 1079648 (C.G. Vinuesa and M.C. Cook), 1113577 (C.G. Vinuesa and M.C. Cook), 1145888 (D.H.D. Gray), 1158024 (D.H.D. Gray), and 1121325 (D.H.D. Gray); the Monash Biomedicine Discovery Institute; and the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health. This study utilized the Australian Phenomics Network Histopathology and Organ Pathology Service of the University of Melbourne.
Author contributions: Conceptualization, M.C. Cook and S.R. Daley. Methodology, R.C. Wirasinha, A.R. Davies, M. Srivastava, C.C. Goodnow, M.C. Cook, and S.R. Daley. Software and data curation, S.R. Daley. Formal analysis, R.C. Wirasinha, A.R. Davies, M.C. Cook, and S.R. Daley. Investigation, R.C. Wirasinha, A.R. Davies, M. Srivastava, X.Y.X. Sng, K.L. Loh, O.M. Delmonte, K. Dobbs, L.A. Miosge, C.E. Lee, R. Chand, A. Chan, J.Y. Yap, H.H. Reid, C.C. Goodnow, M.C. Cook, and S.R. Daley. Resources, M.D. Keller, K. Chen, H.H. Reid, J. Rossjohn, N.L. La Gruta, C.G. Vinuesa, L.D. Notarangelo, D.H.D. Gray, C.C. Goodnow, M.C. Cook, and S.R. Daley. Writing (original draft), M.C. Cook and S.R. Daley. Writing (review and editing), A.R. Davies, M.D. Keller, J. Rossjohn, M.S. Lionakis, D.H.D. Gray, C.C. Goodnow, M.C. Cook, and S.R. Daley. Visualization, A.R. Davies and S.R. Daley. Supervision, J. Rossjohn, N.L. La Gruta, C.G. Vinuesa, H.H. Reid, L.D. Notarangelo, D.H.D. Gray, C.C. Goodnow, M.C. Cook, and S.R. Daley. Funding acquisition, C.G. Vinuesa, L.D. Notarangelo, D.H.D. Gray, C.C. Goodnow, M.C. Cook, and S.R. Daley.

