


Juvenile myelomonocytic leukemia (JMML) is a poor-prognosis childhood leukemia usually caused by RAS-pathway mutations. The cellular hierarchy in JMML is poorly characterized, including the identity of leukemia stem cells (LSCs). FACS and single-cell RNA sequencing reveal marked heterogeneity of JMML hematopoietic stem/progenitor cells (HSPCs), including an aberrant Lin−CD34+CD38−CD90+CD45RA+ population. Single-cell HSPC index-sorting and clonogenic assays show that (1) all somatic mutations can be backtracked to the phenotypic HSC compartment, with RAS-pathway mutations as a “first hit,” (2) mutations are acquired with both linear and branching patterns of clonal evolution, and (3) mutant HSPCs are present after allogeneic HSC transplant before molecular/clinical evidence of relapse. Stem cell assays reveal interpatient heterogeneity of JMML LSCs, which are present in, but not confined to, the phenotypic HSC compartment. RNA sequencing of JMML LSC reveals up-regulation of stem cell and fetal genes (HLF, MEIS1, CNN3, VNN2, and HMGA2) and candidate therapeutic targets/biomarkers (MTOR, SLC2A1, and CD96), paving the way for LSC-directed disease monitoring and therapy in this disease.
Introduction
Juvenile myelomonocytic leukemia (JMML) is an aggressive subtype of childhood myelodysplastic syndrome (MDS), usually presenting in the first 5 yr of life, that is characterized by abnormal proliferation of dysplastic cells of the monocytic and granulocytic lineages (Locatelli and Niemeyer, 2015). Although JMML is a rare childhood cancer, it has several key features that make it an important paradigm. In particular, it is definitively caused by RAS-pathway mutations, typically in PTPN11, KRAS, NRAS, CBL, and NF1, in >90% of cases, and the molecular landscape appears otherwise relatively simple with a low number of somatic mutations in comparison with other malignancies (Caye et al., 2015; Stieglitz et al., 2015a). Some patients show evidence of clonal evolution, with acquisition of monosomy 7 (Locatelli and Niemeyer, 2015) or secondary somatic mutations of SETBP1, ASXL1, EZH2, and other genes that are associated with a worse prognosis (Caye et al., 2015; Stieglitz et al., 2015a, b). The only curative therapy for JMML is allogeneic hematopoietic stem cell transplantation (HSCT); however, relapse rates are high (Locatelli and Niemeyer, 2015), indicating a failure to eradicate the disease-propagating cells in this condition.
The presence of distinct populations of rare disease-propagating cancer stem cells (CSCs) has been demonstrated in some cancers, and this is a crucial step toward understanding cellular pathways of disease relapse (Magee et al., 2012). In adults with chronic myeloid neoplasms, including myeloproliferative neoplasms (MPNs; Mead and Mullally, 2017), chronic myeloid leukemia (CML; Gallipoli et al., 2011), and MDS (Woll et al., 2014), rare and distinct CSCs have been identified that share phenotypic features with normal hematopoietic stem cell (HSCs). However, in acute myeloid leukemia (AML), leukemia stem cells (LSCs) are more heterogeneous, and although the disease may originate in HSCs, various different progenitor cell populations are transformed and are responsible for disease propagation (Thomas and Majeti, 2017). Furthermore, in childhood acute lymphoblastic leukemia (ALL), blast cells across all stages of differentiation have LSC properties (le Viseur et al., 2008). In view of this marked heterogeneity of CSC/LSC between different liquid tumors, it is crucial to properly characterize the CSCs in specific disease entities, particularly when the prognosis is poor and targeted therapy remains elusive. Indeed, for JMML, while the genetic basis is now well described, very little is known about the cellular hierarchy, including the identity of cells that propagate the disease and cause relapse.
Results
Single-cell phenotypic, functional, and molecular analyses reveal heterogeneity of hematopoietic stem/progenitor cells (HSPCs) in JMML
To characterize LSCs in JMML, we established a national prospective study and collected bone marrow (BM) and peripheral blood (PB) samples from a cohort of 26 patients with JMML for phenotypic, functional, and molecular analysis of HSPCs before and after HSCT (Fig. 1 A and Table S1). At diagnosis, the phenotype of early (CD38-negative) HSPCs was markedly disrupted in JMML (Fig. 1, B and C). Although numbers of phenotypic HSCs (Lineage [Lin]−CD34+CD38−CD90+CD45RA−) and lymphoid primed multipotent progenitors (LMPPs) (Lin−CD34+CD38−CD90−CD45RA+) were normal, MPPs (Lin−CD34+CD38−CD90−CD45RA−) were frequently reduced in JMML patients in comparison with patients with normal pediatric BM (P = 0.047), although with considerable patient-to-patient variation (Fig. 1, B and C). A number of patients (10/17 analyzed) also showed the presence of an aberrant Lin−CD34+CD38−CD90+CD45RA+ (+/+) population of cells (P = 0.008). The CD38+ myeloid progenitor compartment showed a normal frequency of phenotypically defined common myeloid progenitors (CMPs; Lin−CD34+CD38+CD123+CD45RA−) and megakaryocyte erythroid progenitors (MEPs; Lin−CD34+CD38+CD123−CD45RA−) with increased numbers of granulocyte monocyte progenitors (GMPs; Lin−CD34+CD38+CD123+CD45RA+; P = 0.05) (Fig. 1, B and C).

Single-cell phenotypic and functional analysis reveals heterogeneity of HSPCs in JMML. (A) Outline of experimental design used in this study. (B) Top panels: Representative FACS profiles of samples of normal pediatric BM (age 14 yr); middle and bottom panels: BM samples from two JMML patients (ID5 and ID3) at diagnosis. Samples gated on singlets, live, and Lin− cells (left column); CD34+CD38− cells (middle); CD34+CD38+ cells (right). Percentages on figure represent percentage of Lin−CD34+ cells. Red boxes indicate the aberrant Lin-CD34+CD38−CD90+CD45RA+ population. (C) Frequency of immunophenotypically defined HSCs and progenitors in total Lin−CD34+ cells from normal pediatric BM (NBM; biological replicates, n = 12; black dots) and JMML BM (biological replicates, n = 17; red dots); HSC P = 0.52, MPP P = 0.0476 (*), LMPP P = 0.13, +/+ P = 0.0082 (**), CMP P = 0.57, MEP P = 0.45, and GMP P = 0.05 (*). Data acquired over four independent experiments. Error bars represent mean ± SEM. Statistical analysis performed by Mann-Whitney test and Welch t test for data that were normally distributed. (D) Clonogenic output of single HSC and progenitor cells sorted into methylcellulose; expressed as the percentage of sorted cells that generated a colony and the type of colony generated. Results for seven different biological replicates from JMML BM samples (ID1, ID2, ID5, ID6, ID7, ID15, and ID18) compared with five different biological replicate normal (CB) controls. Data acquired over 10 independent experiments. Error bars represent mean ± SEM. (E) Bar chart showing the percentage of HSPCs at different time points from diagnosis, day +30 after BMT, and at relapse (biological replicates, n = 3: ID1, ID5, and ID17). P value indicated on figure represents comparison of +/+ at diagnosis and relapse (P = 0.0047 [**]). HSC comparison at diagnosis versus relapse (P = 0.0175). Data acquired over six independent experiments. Statistical analysis performed by two-way ANOVA. Error bars represent mean ± SEM. GEMM, granulocyte/erythroid/megakaryocyte/monocyte; LMPP, lymphoid primed multipotent progenitor.
Single-cell phenotypic and functional analysis reveals heterogeneity of HSPCs in JMML. (A) Outline of experimental design used in this study. (B) Top panels: Representative FACS profiles of samples of normal pediatric BM (age 14 yr); middle and bottom panels: BM samples from two JMML patients (ID5 and ID3) at diagnosis. Samples gated on singlets, live, and Lin− cells (left column); CD34+CD38− cells (middle); CD34+CD38+ cells (right). Percentages on figure represent percentage of Lin−CD34+ cells. Red boxes indicate the aberrant Lin-CD34+CD38−CD90+CD45RA+ population. (C) Frequency of immunophenotypically defined HSCs and progenitors in total Lin−CD34+ cells from normal pediatric BM (NBM; biological replicates, n = 12; black dots) and JMML BM (biological replicates, n = 17; red dots); HSC P = 0.52, MPP P = 0.0476 (*), LMPP P = 0.13, +/+ P = 0.0082 (**), CMP P = 0.57, MEP P = 0.45, and GMP P = 0.05 (*). Data acquired over four independent experiments. Error bars represent mean ± SEM. Statistical analysis performed by Mann-Whitney test and Welch t test for data that were normally distributed. (D) Clonogenic output of single HSC and progenitor cells sorted into methylcellulose; expressed as the percentage of sorted cells that generated a colony and the type of colony generated. Results for seven different biological replicates from JMML BM samples (ID1, ID2, ID5, ID6, ID7, ID15, and ID18) compared with five different biological replicate normal (CB) controls. Data acquired over 10 independent experiments. Error bars represent mean ± SEM. (E) Bar chart showing the percentage of HSPCs at different time points from diagnosis, day +30 after BMT, and at relapse (biological replicates, n = 3: ID1, ID5, and ID17). P value indicated on figure represents comparison of +/+ at diagnosis and relapse (P = 0.0047 [**]). HSC comparison at diagnosis versus relapse (P = 0.0175). Data acquired over six independent experiments. Statistical analysis performed by two-way ANOVA. Error bars represent mean ± SEM. GEMM, granulocyte/erythroid/megakaryocyte/monocyte; LMPP, lymphoid primed multipotent progenitor.
Clonogenic assays of bulk BM (Fig. S1 A) or purified single HSPCs showed apparently preserved myeloid output but aberrant erythroid potential of CMP and MEP from JMML patients in comparison with cord blood (CB; Fig. 1 D) and pediatric BM (Fig. S1 B). Erythroid colonies derived from JMML patients were frequently dysplastic (Fig. S1 C), in keeping with anemia seen in all patients (Table S1). The aberrant +/+ population showed exclusively myeloid output (Fig. 1 D and Fig. S1 B). Lymphoid potential of JMML HSCs was reduced (Fig. S1 D) and megakaryocytic potential severely reduced (Fig. S1 E) in keeping with patients’ thrombocytopenia (Table S1).

Single-cell phenotypic and functional analysis reveals heterogeneity of HSPCs in JMML (related to Fig. 1 ). (A) Bulk clonogenicity in methylcellulose, 100 cells sorted per assay (CB control, n = 2 different biological replicates; JMML, n = 2 different biological replicates); one experiment. CFU-M, CFU-macrophage; CFU-GM, CFU–granulocyte macrophage. (B) Clonogenic output of purified HSCs and progenitor populations from normal pediatric BM (PBM; n = 3 different biological replicates) and JMML (n = 7 different biological replicates) in single-cell methylcellulose assays. 11 independent experiments performed. (C) Representative examples of colonies (left column) and MGG-stained cells from individual plucked colonies (right column) from clonogenic assays in methylcellulose. (Ci) Macrophage colonies from the JMML Lin−CD34+CD38−CD90+CD45RA+ population; scale bars, 1,000 µm (left), 50 µm (right). (Cii) “Normal-appearing” erythroid colonies; scale bar, 200 µm (left), 50 µm (right). (Ciii) Abnormal erythroid colonies derived from JMML MEP; scale bar, 400 µm (left), 50 µm (right). (D) Lymphoid output of sorted HSCs (100 cells) from four different biological replicates from JMML BM samples (red bar) compared with four different biological replicates from normal controls (CB; blue bars). Cells cultured on MS-5 stromal lines, and output measured as percent CD19+ cells on gated hCD45+ cells (mean ± SEM) after 4 wk. Mann Whitney test, *** P = 0.0006. Data acquired over two independent experiments. (E) Percentage of wells with CD42+CD41+ megakaryocytes (Mk) in single-cell liquid cultures of normal CB (n = 2) and JMML (n = 2). Two independent experiments performed. (F) Representative FACS plots of serial patient samples from diagnosis, after BMT, and at clinical relapse: ID1 (Fi); ID5 (Fii); and ID17 (Fiii). Top panels for each patient represent Lin−CD34+CD38− compartment and bottom panel Lin−CD34+CD38+ compartments. Percentage of each population shown in the figure represents the percentage of Lin−CD34+. Green boxes indicate the aberrant Lin-CD34+CD38-CD90+CD45RA+ population. Data acquired over six independent experiments. BFU-E, burst-forming unit–erythroid; GEMM, granulocyte/erythroid/megakaryocyte/monocyte; LMPP, lymphoid primed multipotent progenitor.
Single-cell phenotypic and functional analysis reveals heterogeneity of HSPCs in JMML (related to Fig. 1 ). (A) Bulk clonogenicity in methylcellulose, 100 cells sorted per assay (CB control, n = 2 different biological replicates; JMML, n = 2 different biological replicates); one experiment. CFU-M, CFU-macrophage; CFU-GM, CFU–granulocyte macrophage. (B) Clonogenic output of purified HSCs and progenitor populations from normal pediatric BM (PBM; n = 3 different biological replicates) and JMML (n = 7 different biological replicates) in single-cell methylcellulose assays. 11 independent experiments performed. (C) Representative examples of colonies (left column) and MGG-stained cells from individual plucked colonies (right column) from clonogenic assays in methylcellulose. (Ci) Macrophage colonies from the JMML Lin−CD34+CD38−CD90+CD45RA+ population; scale bars, 1,000 µm (left), 50 µm (right). (Cii) “Normal-appearing” erythroid colonies; scale bar, 200 µm (left), 50 µm (right). (Ciii) Abnormal erythroid colonies derived from JMML MEP; scale bar, 400 µm (left), 50 µm (right). (D) Lymphoid output of sorted HSCs (100 cells) from four different biological replicates from JMML BM samples (red bar) compared with four different biological replicates from normal controls (CB; blue bars). Cells cultured on MS-5 stromal lines, and output measured as percent CD19+ cells on gated hCD45+ cells (mean ± SEM) after 4 wk. Mann Whitney test, *** P = 0.0006. Data acquired over two independent experiments. (E) Percentage of wells with CD42+CD41+ megakaryocytes (Mk) in single-cell liquid cultures of normal CB (n = 2) and JMML (n = 2). Two independent experiments performed. (F) Representative FACS plots of serial patient samples from diagnosis, after BMT, and at clinical relapse: ID1 (Fi); ID5 (Fii); and ID17 (Fiii). Top panels for each patient represent Lin−CD34+CD38− compartment and bottom panel Lin−CD34+CD38+ compartments. Percentage of each population shown in the figure represents the percentage of Lin−CD34+. Green boxes indicate the aberrant Lin-CD34+CD38-CD90+CD45RA+ population. Data acquired over six independent experiments. BFU-E, burst-forming unit–erythroid; GEMM, granulocyte/erythroid/megakaryocyte/monocyte; LMPP, lymphoid primed multipotent progenitor.
Analysis of the HSPC compartment in patients with serial samples available after HSCT and at relapse indicated that the aberrant Lin−CD34+CD38−CD90+CD45RA+ (+/+) population, which was present in all three cases at diagnosis, was absent early after transplant (Fig. 1 E and Fig. S1 F). However, at relapse, there was a striking recurrence of +/+ HSPCs, which represented the predominant Lin−CD34+CD38− cell population at the time of disease recurrence (Fig. 1 E and Fig. S1 F).
Although FACS analysis supported relative preservation of a number of HSPC subpopulations in JMML, bulk phenotypic analysis may mask the underlying heterogeneity of HSPCs in JMML in comparison with normal hematopoiesis. We therefore next assessed the global cellular architecture of JMML Lin−CD34+ HSPCs in an unbiased manner by high-throughput single-cell RNA sequencing (RNA-seq) of 17,547 single HSPCs from JMML (n = 2) and CB (n = 2). Using 593 selected genes showing a high level of dispersion, t-distributed stochastic neighbor embedding (tSNE) showed highly distinct clustering of JMML HSPCs (Fig. 2 A), which were not equally distributed between the identified HSPC subpopulations in comparison with CB HSPCs (Fig. 2, B and C). JMML-specific clusters of HSPCs showed up-regulation of myeloid genes (MS4A3 and MPO), stem cell and fetal genes (THY1, ZFP36L1, HMGA2), and proliferation markers (MKI67) and aberrant expression of leukemia (HOPX and FOS) and erythroid differentiation-associated (GATA1) genes (Fig. 2, D and E). Taken together, these findings support that the HSPC compartment is heterogeneous in JMML, with an aberrant phenotype, myeloid bias, and distinct molecular signatures in comparison with normal controls.

Single-cell RNA-seq of JMML HSPCs reveals distinct clustering. (A) tSNE analysis of Lin−CD34+ single-cell RNA-seq dataset based on 593 variable genes. 17,547 single cells that passed filtering from two CB controls (light and dark blue) and two JMML BM samples (orange and red) are shown. (B) Single-cell RNA-seq clusters identified by the k-SNN method and projected by tSNE plot. Colors correspond to computationally identified clusters of single cells. (C) Number of single cells within each cluster from CB (blue) and JMML (red). (D) Selected highly variable genes from top principal components plotted on tSNE analysis from A. Color scale represents normalized UMI counts (gray, not detected [ND]; red, maximum count [Max]). (E) Heatmap depicting the top five positively identifying genes that distinguish each cluster of single cells shown in B. Data acquired in one experiment.
Single-cell RNA-seq of JMML HSPCs reveals distinct clustering. (A) tSNE analysis of Lin−CD34+ single-cell RNA-seq dataset based on 593 variable genes. 17,547 single cells that passed filtering from two CB controls (light and dark blue) and two JMML BM samples (orange and red) are shown. (B) Single-cell RNA-seq clusters identified by the k-SNN method and projected by tSNE plot. Colors correspond to computationally identified clusters of single cells. (C) Number of single cells within each cluster from CB (blue) and JMML (red). (D) Selected highly variable genes from top principal components plotted on tSNE analysis from A. Color scale represents normalized UMI counts (gray, not detected [ND]; red, maximum count [Max]). (E) Heatmap depicting the top five positively identifying genes that distinguish each cluster of single cells shown in B. Data acquired in one experiment.
Somatic mutations are present with a high clonal burden in the phenotypic HSC compartment in JMML
As driver mutations must be present in cells with self-renewal capability in order to propagate the disease, we next set out to track RAS-pathway somatic mutations to the HSPC cellular hierarchy in JMML in order to gain insights into the identity of JMML LSCs. In adult MDS, this approach has been used to identify phenotypic HSCs as the population with stem cell properties (Woll et al., 2014). In contrast, in AML, some mutations can be tracked to progenitor cell populations but are absent in HSCs, supporting the presence of aberrant stem cell properties in progenitor cells in AML (Jan et al., 2012). We first performed a targeted mutation analysis of JMML-associated mutations in our cohort of 26 JMML cases. RAS pathway–activating mutations were present in all patients, while secondary spliceosome and epigenetic mutations and/or monosomy 7 was identified in 8/26 (31%) of patients, including the majority of patients with NF1 mutations and 2/26 patients who had two RAS pathway mutations, as previously reported (Fig. 3 A; Caye et al., 2015).
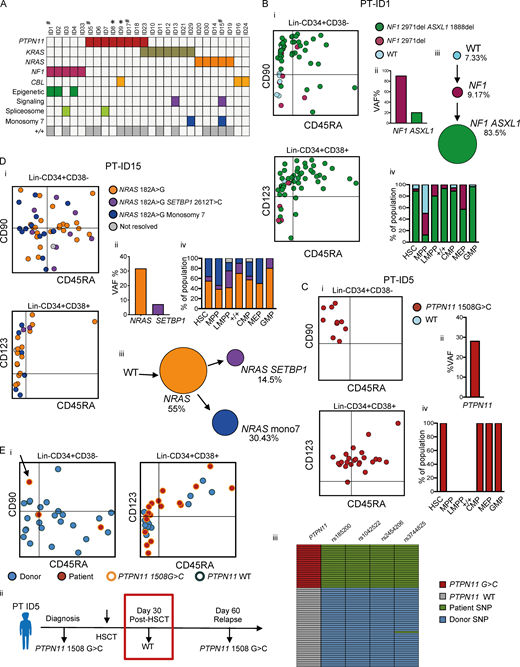
Single-cell mutation tracing to phenotypic HSCs in JMML.(A) Mutations detected through a customized MDS/JMML targeted sequencing panel in whole JMML BM and/or PB samples (n = 26) at diagnosis except where indicated; each column represents an individual patient. All patients had RAS gene pathway mutations, in two cases involving two different RAS-pathway genes (ID9 and ID23). Secondary mutations were detected in 7/26 in epigenetic regulators (ASXL1, TET2), signaling (SETBP1), and spliceosome (SRSF2) genes; also two patients with monosomy 7 (see Table S1). Patients where the aberrant +/+ population was detected by FACS are shown in the bottom row; #, samples for single-cell genotyping; *, relapse or early posttreatment samples. (B–D) Single-cell mutational profiling for three JMML BM samples (ID1 [B], ID5 [C], and ID15 [D]). For each panel: (i) FACS indexing of CD34+ single cells with points colored by mutational status; top CD38−; bottom CD38+; (ii) variant allele frequency (VAF) at diagnosis for each mutation detected in bulk BM; (iii) inferred mutation hierarchy and proportion of each subclone (for ID1 and ID15 only); and (iv) proportion of each subclone in the indicated HSPC subpopulation. (Ei) FACS indexing of posttransplant (day +30) BM CD34+ cells from patient ID5 with single cells colored by donor/patient and mutational status; left: Lin−CD34+CD38−; right: Lin−CD34+CD38+. Arrow indicates a mutant HSC detected in the posttransplant sample. (Eii) Timeline of pre- and posttransplant PTPN11 mutation detection by targeted sequencing of bulk BM cells showing that the mutation was not detected by NGS at day +30. Marked day 30 sample used for single-cell genotyping. (Eiii) Heatmap of the 65 single cell–derived colonies analyzed showing PTPN11 mutational status and patient- and donor-derived SNPs. Details for number of cells sorted, processed, and analyzed are outlined in the relevant Materials and methods section.
Single-cell mutation tracing to phenotypic HSCs in JMML.(A) Mutations detected through a customized MDS/JMML targeted sequencing panel in whole JMML BM and/or PB samples (n = 26) at diagnosis except where indicated; each column represents an individual patient. All patients had RAS gene pathway mutations, in two cases involving two different RAS-pathway genes (ID9 and ID23). Secondary mutations were detected in 7/26 in epigenetic regulators (ASXL1, TET2), signaling (SETBP1), and spliceosome (SRSF2) genes; also two patients with monosomy 7 (see Table S1). Patients where the aberrant +/+ population was detected by FACS are shown in the bottom row; #, samples for single-cell genotyping; *, relapse or early posttreatment samples. (B–D) Single-cell mutational profiling for three JMML BM samples (ID1 [B], ID5 [C], and ID15 [D]). For each panel: (i) FACS indexing of CD34+ single cells with points colored by mutational status; top CD38−; bottom CD38+; (ii) variant allele frequency (VAF) at diagnosis for each mutation detected in bulk BM; (iii) inferred mutation hierarchy and proportion of each subclone (for ID1 and ID15 only); and (iv) proportion of each subclone in the indicated HSPC subpopulation. (Ei) FACS indexing of posttransplant (day +30) BM CD34+ cells from patient ID5 with single cells colored by donor/patient and mutational status; left: Lin−CD34+CD38−; right: Lin−CD34+CD38+. Arrow indicates a mutant HSC detected in the posttransplant sample. (Eii) Timeline of pre- and posttransplant PTPN11 mutation detection by targeted sequencing of bulk BM cells showing that the mutation was not detected by NGS at day +30. Marked day 30 sample used for single-cell genotyping. (Eiii) Heatmap of the 65 single cell–derived colonies analyzed showing PTPN11 mutational status and patient- and donor-derived SNPs. Details for number of cells sorted, processed, and analyzed are outlined in the relevant Materials and methods section.
To track these disease-causing mutations to the HSPC hierarchy, single Lin−CD34+ cells from three patients were index-sorted into methylcellulose colony-forming assays and individual colonies picked for targeted genotyping (n = 299); the index-sorting data allowed us to derive the FACS phenotype, and hence the HSPC population of origin, of each colony (Fig. S2, A and B). Genotyping of single-nucleotide polymorphisms (SNPs) demonstrated the low allelic dropout of this assay, with no false-positive mutations seen in control samples (Fig. S2, C–H). Patient ID1 showed evidence of linear evolution with acquisition of an NF1 followed by an ASXL1 mutation, both of which could be tracked to all HSPC subpopulations, including HSCs (Fig. 3 B). The few residual wild-type cells were present in the HSC compartment and enriched in MPPs, whereas mutation-positive cells were infrequent in MPPs in keeping with phenotypic data (Fig. 1 C). ID5 was characterized by a PTPN11 mutation alone that was again traceable to HSCs as well as CMPs and GMPs (Fig. 3 C). Patient ID15 showed evidence of branching clonal evolution within the HSC compartment, with a first hit NRAS mutation followed by acquisition of monosomy 7 and a SETBP1 mutation in separate subclones (Fig. 3 D). All subclones were present in the HSC compartment. We also analyzed a sample from patient ID5 taken after HSCT, at a time when the patient was in clinical remission but subsequently suffered overt evidence of relapse of JMML (Fig. 3 E). Parallel genotyping of the PTPN11 mutation together with donor- and recipient-specific SNPs demonstrated the presence of PTPN11 mutation–positive HSPCs (including a single HSC), which predate (and potentially might have helped to predict) subsequent relapse. Notably, mutant-positive progenitors (CD38+) were more prominent than HSCs after HSCT, in keeping with phenotypic data (Fig. 1 E), raising the possibility that these cells may predominantly drive relapse.

Colony genotyping mutation tracing to phenotypic HSCs in JMML–QC (related to Fig. 3 ). (A) Diagram of the workflow for single-cell genotyping. (B) Pathway for samples analysis. (C–E) Variant allele frequency (VAF) and coverage of amplicons sequenced and that passed QC from patient-derived colonies and CB controls. (ID1, n = 129 cells used for analysis; ID15, n = 71 cells used for analysis; and ID5, n = 34 cells used for analysis). (F) VAF and coverage of amplicons sequenced from patient-derived colonies (ID5) after BMT and CB controls (ID5 after BMT, n = 65 cells used for analysis). (G) Examples of coverage of amplicons from heterozygous SNPs used to calculate the allelic dropout of the method. (H) Summary of amplicons from heterozygous SNPs for each sequencing run; coverage applied and allelic dropout for each amplicon. Six independent experiments; details for number of colonies are outlined in the Materials and methods section.
Colony genotyping mutation tracing to phenotypic HSCs in JMML–QC (related to Fig. 3 ). (A) Diagram of the workflow for single-cell genotyping. (B) Pathway for samples analysis. (C–E) Variant allele frequency (VAF) and coverage of amplicons sequenced and that passed QC from patient-derived colonies and CB controls. (ID1, n = 129 cells used for analysis; ID15, n = 71 cells used for analysis; and ID5, n = 34 cells used for analysis). (F) VAF and coverage of amplicons sequenced from patient-derived colonies (ID5) after BMT and CB controls (ID5 after BMT, n = 65 cells used for analysis). (G) Examples of coverage of amplicons from heterozygous SNPs used to calculate the allelic dropout of the method. (H) Summary of amplicons from heterozygous SNPs for each sequencing run; coverage applied and allelic dropout for each amplicon. Six independent experiments; details for number of colonies are outlined in the Materials and methods section.
Single-cell colony genotyping might fail to identify single cells unable to generate colonies in vitro, which may have led us to underestimate the number of wild-type cells present. We therefore also performed a direct single-cell genotyping analysis (n = 645 single cells that passed quality control [QC]) of two patients with PTPN11 mutation (ID5 and ID17). These data (Fig. S3, A–F) support the findings based on colony genotyping that disease-associated driver mutations can be tracked back to the phenotypic HSC compartment and that wild-type HSPCs are rare in JMML patients, indicating that JMML HSPCs are highly clonally dominant. Taken together, these data support the conclusion that all somatic mutations driving JMML disease initiation and evolution are present in the phenotypic HSC compartment, with a high degree of clonal dominance over wild-type HSCs in all cases analyzed, with RAS-pathway mutations as a “first hit.”

Single-cell genotyping using TARGET-seq (related to Fig. 3 ). (A) Variant allele frequency (VAF) and coverage of PTPN11 c.226G>A amplicon sequenced from patient ID17 (total of 384 single cells sorted, 365 cells passed QC, coverage cutoff 100). (B) VAF and coverage of PTPN11 c.1508G>C amplicon sequenced from patient ID5 (total of 384 cells single sorted, 280 cells passed QC, coverage cutoff 30). (C) Single-cell mutation profiling: cells plotted as per FACS index data and colored according to their mutational status. Plots showing Lin−CD34+CD38− cells from patient ID17. (D) Single-cell mutation profiling: cells plotted as per FACS index data and colored according to their mutational status. Plots showing Lin−CD34+CD38− cells from patient ID5. (E) Single-cell mutation profiling: cells plotted as per FACS index data and colored according to their mutational status. Plots showing Lin−CD34+CD38+ cells from patient ID17. (F) Single-cell mutation profiling: cells plotted as per FACS index data and colored according to their mutational status. Plots showing Lin−CD34+CD38+ cells from patient ID5. Data acquired with two independent experiments (one per biological replicate).
Single-cell genotyping using TARGET-seq (related to Fig. 3 ). (A) Variant allele frequency (VAF) and coverage of PTPN11 c.226G>A amplicon sequenced from patient ID17 (total of 384 single cells sorted, 365 cells passed QC, coverage cutoff 100). (B) VAF and coverage of PTPN11 c.1508G>C amplicon sequenced from patient ID5 (total of 384 cells single sorted, 280 cells passed QC, coverage cutoff 30). (C) Single-cell mutation profiling: cells plotted as per FACS index data and colored according to their mutational status. Plots showing Lin−CD34+CD38− cells from patient ID17. (D) Single-cell mutation profiling: cells plotted as per FACS index data and colored according to their mutational status. Plots showing Lin−CD34+CD38− cells from patient ID5. (E) Single-cell mutation profiling: cells plotted as per FACS index data and colored according to their mutational status. Plots showing Lin−CD34+CD38+ cells from patient ID17. (F) Single-cell mutation profiling: cells plotted as per FACS index data and colored according to their mutational status. Plots showing Lin−CD34+CD38+ cells from patient ID5. Data acquired with two independent experiments (one per biological replicate).
Heterogeneity of LSCs in JMML
To characterize JMML LSCs, we next performed in vitro and in vivo stem cell assays to assess the self-renewal potential of JMML HSPC subpopulations. Long-term culture–initiating cell (LTC-IC) assays showed LTC-IC potential was present in both HSC and +/+ populations in JMML but was absent in GMP, while in CB controls LTC-IC potential was restricted to HSCs, as expected (Fig. 4 A). We then performed xenotransplantation of purified HSC, +/+, and GMP or total CD34+ cells from four JMML patients as shown in Fig. 4 B, with results of serial readout of engraftment shown in Fig. 4, C and D. JMML CD34+ HSPCs were highly efficient at supporting reconstitution in this NOD scid gamma (NSG) xenograft model, unlike adult MDS (Woll et al., 2014). Lineage analysis (Fig. 4 D and Fig. S4 A) showed that transplantation from JMML donors resulted in highly myeloid-biased reconstitution in comparison with CB. In all cases, mice showed reconstitution following transplantation of purified HSCs from CB or JMML. Engraftment potential of specific JMML HSPC subpopulations (+/+ and GMP), however, showed marked inter-patient heterogeneity of JMML LSCs (Fig. 4, C and D). In patient ID1, JMML LSC potential was restricted to the phenotypic HSC compartment. Patient ID3, who died from refractory disease following HSCT, showed similar repopulating potential in all populations (HSC, +/+, and GMP). Patient ID5, who also relapsed after HSCT, showed the most robust reconstitution with HSCs, but late reconstitution was also observed with +/+ and total CD34+CD38+ cells. It is also noteworthy that at relapse after HSCT, the mutant-positive JMML HSPC compartment in patient ID5 was dominated by mature progenitor cells (Fig. 3 E), unlike at diagnosis, when HSCs were frequent (Fig. 3 C). This is consistent with relapse being primarily propagated by progenitors rather than HSCs in this patient, in keeping with phenotypic (Fig. S1 F) and single-colony genotyping (Fig. 3 E) data. Finally, patient ID15 showed reconstitution following transplantation of HSCs and +/+, but not GMP.

Heterogeneity of LSCs in JMML. (A) LTC-IC assay. 100 cells from each population from JMML BM samples (five different biological replicates) and CB controls (five different biological replicates) sorted and cultured for 8 wk. Results shown as the percentage of cells from each sample with clonogenic activity. Data acquired over three independent LTC-IC experiments. Error bars represent mean ± SEM. (B) Schematic of in vivo transplantation experiments with purified HSPC populations into NSG mice. (C) Percentage of human (h)CD45+ cells in PB from xenograft CB and JMML HSPCs. Below: Key for each patient for population, number of cells transplanted, and number of replicates. Student's t test, * P < 0.05. ID1 HSC versus ID1 +/+, * P = 0.041; ID1 HSC versus ID1 GMP, * P = 0.040. ID5 HSC (n = 250) versus ID5 +/+ (n = 250), * P = 0.044; ID5 HSC (n = 250) versus ID5 GMP (n = 250), * P = 0.02. Four different JMML BM samples were used (ID1, ID3, ID5, and ID15) and four different CB controls. Number of animals per group is shown in Fig. 4 C. (D) Lineage of PB reconstitution of mice with >1% human engraftment at terminal time point. Color bars (top) correspond to population and cell number groups as in C. Two-way ANOVA, compared with CB controls. CB lymphoid versus ID1 lymphoid, **** P < 0.0001; CB lymphoid versus ID3 lymphoid, **** P < 0.0001; CB lymphoid versus ID5 lymphoid, **** P < 0.0001; CB lymphoid versus ID15 lymphoid, ** P < 0.0024; CB myeloid (My) versus ID3 myeloid, ** P = 0.0038; CB myeloid versus ID5 myeloid **** P < 0.0001, and CB myeloid versus ID15 myeloid, * P = 0.016. Rep., representative. (E) BM analysis of mice transplanted with HSCs from CB (n = 12) or JMML BM (n = 4 samples; 16 mice). Mean percent human engraftment at terminal time point (top); percent engraftment (of total hCD45+ cells) for each HSPC population shown below each population; and log fold-change (FC) for JMML compared with CB controls indicated by the color chart. (F) Comparison of percent hCD45+ cells in PB versus BM at terminal time point with best fit (black) and a 1:1 ratio (red). (G) PB hCD45+ cell percentage in secondary transplantation of CB and JMML HSCs. (H) Photomicrograph of Giemsa-stained BM sections from mice transplanted with purified CB (left) or JMML (right) HSCs. Scale bars, 100 µm. (I) Left panel: Terminal spleen weight from mice transplanted with CB versus JMML HSCs, ** P = 0.0087, Mann-Whitney test. Right panel: Representative images of increased spleen size in JMML versus CB HSC-transplanted mice. (J) Kaplan-Meier leukemia-free survival curve of mice transplanted with purified JMML HSCs or CB HSCs. Significance by log-rank test, **** P < 0.0001. LMPP, lymphoid primed multipotent progenitor.
Heterogeneity of LSCs in JMML. (A) LTC-IC assay. 100 cells from each population from JMML BM samples (five different biological replicates) and CB controls (five different biological replicates) sorted and cultured for 8 wk. Results shown as the percentage of cells from each sample with clonogenic activity. Data acquired over three independent LTC-IC experiments. Error bars represent mean ± SEM. (B) Schematic of in vivo transplantation experiments with purified HSPC populations into NSG mice. (C) Percentage of human (h)CD45+ cells in PB from xenograft CB and JMML HSPCs. Below: Key for each patient for population, number of cells transplanted, and number of replicates. Student's t test, * P < 0.05. ID1 HSC versus ID1 +/+, * P = 0.041; ID1 HSC versus ID1 GMP, * P = 0.040. ID5 HSC (n = 250) versus ID5 +/+ (n = 250), * P = 0.044; ID5 HSC (n = 250) versus ID5 GMP (n = 250), * P = 0.02. Four different JMML BM samples were used (ID1, ID3, ID5, and ID15) and four different CB controls. Number of animals per group is shown in Fig. 4 C. (D) Lineage of PB reconstitution of mice with >1% human engraftment at terminal time point. Color bars (top) correspond to population and cell number groups as in C. Two-way ANOVA, compared with CB controls. CB lymphoid versus ID1 lymphoid, **** P < 0.0001; CB lymphoid versus ID3 lymphoid, **** P < 0.0001; CB lymphoid versus ID5 lymphoid, **** P < 0.0001; CB lymphoid versus ID15 lymphoid, ** P < 0.0024; CB myeloid (My) versus ID3 myeloid, ** P = 0.0038; CB myeloid versus ID5 myeloid **** P < 0.0001, and CB myeloid versus ID15 myeloid, * P = 0.016. Rep., representative. (E) BM analysis of mice transplanted with HSCs from CB (n = 12) or JMML BM (n = 4 samples; 16 mice). Mean percent human engraftment at terminal time point (top); percent engraftment (of total hCD45+ cells) for each HSPC population shown below each population; and log fold-change (FC) for JMML compared with CB controls indicated by the color chart. (F) Comparison of percent hCD45+ cells in PB versus BM at terminal time point with best fit (black) and a 1:1 ratio (red). (G) PB hCD45+ cell percentage in secondary transplantation of CB and JMML HSCs. (H) Photomicrograph of Giemsa-stained BM sections from mice transplanted with purified CB (left) or JMML (right) HSCs. Scale bars, 100 µm. (I) Left panel: Terminal spleen weight from mice transplanted with CB versus JMML HSCs, ** P = 0.0087, Mann-Whitney test. Right panel: Representative images of increased spleen size in JMML versus CB HSC-transplanted mice. (J) Kaplan-Meier leukemia-free survival curve of mice transplanted with purified JMML HSCs or CB HSCs. Significance by log-rank test, **** P < 0.0001. LMPP, lymphoid primed multipotent progenitor.

Functional characterization of JMML HSPCs (related to Fig. 4 ). Number of animals per group as outlined in index of Fig. 1 C. (A) Representative FACS analysis of PB from JMML xenograft showing gating strategy. FSC, forward scatter. (B) Representative FACS analysis of BM from JMML xenograft overall showing gating strategy. (C) Terminal PB analysis from mice transplanted with CB or JMML HSCs; * P < 0.05, Student’s t test. t test hemoglobin CB versus JMML, P = 0.018; platelets CB versus JMML, P = 0.0311. Error bars represent mean ± SEM. HGB, hemoglobin; PLT, platelets. (D) Representative histology photographs of spleen CB HSC control (left) and JMML HSC (right). Scale bars, 100 µm. (E) Kaplan-Meier survival curves for JMML +/+ and JMML GMP compared with CB control; log-rank test, * P = 0.024. (F) Spleen size of NSG mice at termination from CB HSC controls; JMML HSC, JMML +/+, and JMML GMP; ** P = 0.0087; Student’s t test. Error bars represent mean ± SEM. ns, not significant.
Functional characterization of JMML HSPCs (related to Fig. 4 ). Number of animals per group as outlined in index of Fig. 1 C. (A) Representative FACS analysis of PB from JMML xenograft showing gating strategy. FSC, forward scatter. (B) Representative FACS analysis of BM from JMML xenograft overall showing gating strategy. (C) Terminal PB analysis from mice transplanted with CB or JMML HSCs; * P < 0.05, Student’s t test. t test hemoglobin CB versus JMML, P = 0.018; platelets CB versus JMML, P = 0.0311. Error bars represent mean ± SEM. HGB, hemoglobin; PLT, platelets. (D) Representative histology photographs of spleen CB HSC control (left) and JMML HSC (right). Scale bars, 100 µm. (E) Kaplan-Meier survival curves for JMML +/+ and JMML GMP compared with CB control; log-rank test, * P = 0.024. (F) Spleen size of NSG mice at termination from CB HSC controls; JMML HSC, JMML +/+, and JMML GMP; ** P = 0.0087; Student’s t test. Error bars represent mean ± SEM. ns, not significant.
Terminal analysis of reconstituted mice showed relative enrichment of HSC and +/+ cells relative to CB-engrafted animals in three of four cases analyzed (Fig. 4 E and Fig. S4 B), with higher engraftment in BM than in PB (Fig. 4 F). JMML HSCs also strongly supported robust reconstitution in secondary transplantations (Fig. 4 G). Mice transplanted with JMML HSCs developed a JMML-like disease with cytopenias (Fig. S4 C), characteristic histology (Fig. 4 H and Fig. S4 D), marked splenomegaly (Fig. 4 I), and reduced leukemia-free survival (Fig. 4 J). Mice also developed JMML-like disease after transplantation of +/+ HSPCs but not GMP (Fig. S4, E and F). Taken together, these findings demonstrate that JMML LSC activity is present in, but not confined to, the phenotypic HSC compartment. JMML HSCs nevertheless not only showed the most robust reconstituting potential but were also the key population that induced JMML-like disease in vivo.
Conservation of molecular hierarchy of JMML HSPCs
To characterize transcriptomic signatures of JMML LSCs, we next performed RNA-seq analysis of three different JMML LSC populations (HSC, +/+, and GMP) from six JMML patients. JMML HSCs showed a higher number of differentially expressed genes in comparison with CB HSCs (n = 5) than in comparison with different JMML HSPC subpopulations (Fig. 5 A), and, notably, +/+ cells from JMML shared molecular signatures with both HSC and GMP (Fig. 5 B). Expression of known HSC genes was higher in HSCs than other populations in both JMML and CB, with expression in JMML +/+ cells intermediate between HSC and GMP, suggesting that a hierarchy may be preserved in JMML (Fig. 5 C). In keeping with this, myeloid-associated genes were more highly expressed in GMP in both CB and JMML, with +/+ cells again showing intermediate expression (Fig. 5 D). Analysis of cell cycle genes supported that quiescence-associated gene expression was higher in CB HSCs with proliferation-associated transcriptional changes in all JMML HSPC populations, including HSCs, but more so in JMML GMPs (Fig. 5 E). Interestingly, the fetal HSC genes HMGA2, CNN3, and VNN2 were highly expressed in JMML HSCs and to a lesser extent in JMML +/+ and GMP cells (Fig. 5 F). Gene set enrichment analysis (GSEA) further supported that JMML HSCs showed more HSC- and quiescence-associated gene expression than JMML GMPs (Fig. 5 G). In contrast, JMML GMPs showed up-regulation of proliferation and pediatric cancer signatures in comparison with JMML HSCs (Fig. 5 G). Direct comparison of CB versus JMML HSCs showed a number of clusters of aberrantly expressed genes, clearly distinguishing JMML HSCs (Fig. 5 H). Hallmark GSEA revealed enrichment of a number of proliferation-associated (G2M checkpoint, MYC, and E2F) and DNA repair gene sets in JMML HSCs versus CB HSCs (Fig. 5 I). We identified a core set of 24 genes that distinguished JMML HSCs from JMML GMPs and CB HSCs (Fig. 5 J) and were overexpressed in JMML-specific HSPC subpopulations identified by single-cell RNA-seq (Fig. 5 K). This set of genes included the non–DNA-binding homeodomain protein HOPX, a regulator of primitive hematopoiesis (Palpant et al., 2017) and the serine/threonine kinase STK24, a target of the kinase inhibitor bosutinib (Fig. 5 L; Remsing Rix et al., 2009).

Conservation of molecular hierarchy of JMML HSPCs and novel therapeutic targets for JMML HSCs. (A) Nearest neighbor analysis of purified HSPC populations from CB and JMML. Numbers represent significant differentially expressed genes <0.1 false discovery rate (FDR). JMML BM, n = 6 different biological replicates, and normal CB control, n = 5 different biological replicates. (B) Heatmap of top differentially expressed genes between JMML HSC, JMML +/+, and JMML GMP. (C–F) Gene expression for CB HSC (black), CB GMP (gray), JMML HSC (blue), JMML CD90+/CD45RA+ (+/+; orange), and JMML GMP (red) for known stem cell genes (C); myeloid genes up-regulated in JMML and CB GMP (D); cell cycle–related genes (E); and expression in JMML HSPCs of known fetal HSC genes (F). (G) GSEA of JMML HSCs versus JMML GMPs with up-regulation of gene sets for HSC markers and CML quiescence in HSCs (top) and up-regulation of gene sets for pediatric cancer and CML proliferation gene sets in GMPs (bottom). NES, normalized enrichment score. (H) Heatmap depicting k-means clustering of top differentially expressed genes between JMML HSCs and CB HSCs with an FDR cutoff <0.1. Cluster size corresponds to the number of genes within each significant cluster. (I) Hallmark GSEA between JMML HSCs and CB HSCs depicting up-regulated gene sets in JMML. Dashed line represents an FDR cutoff of <0.1; numbers next to the bars represent NES. (J) Identification of a JMML LSC–specific gene signature from bulk RNA-seq. (K) Single-cell gene expression of the JMML stem cell genes identified in Fig. 5 J. Clusters are as identified in Fig. 2 B. Expression plotted as mean UMI. (L) Heatmap of top differentially expressed genes between JMML HSC, JMML +/+, and JMML GMP. Data acquired over two independent experiments. TPM, transcripts per million.
Conservation of molecular hierarchy of JMML HSPCs and novel therapeutic targets for JMML HSCs. (A) Nearest neighbor analysis of purified HSPC populations from CB and JMML. Numbers represent significant differentially expressed genes <0.1 false discovery rate (FDR). JMML BM, n = 6 different biological replicates, and normal CB control, n = 5 different biological replicates. (B) Heatmap of top differentially expressed genes between JMML HSC, JMML +/+, and JMML GMP. (C–F) Gene expression for CB HSC (black), CB GMP (gray), JMML HSC (blue), JMML CD90+/CD45RA+ (+/+; orange), and JMML GMP (red) for known stem cell genes (C); myeloid genes up-regulated in JMML and CB GMP (D); cell cycle–related genes (E); and expression in JMML HSPCs of known fetal HSC genes (F). (G) GSEA of JMML HSCs versus JMML GMPs with up-regulation of gene sets for HSC markers and CML quiescence in HSCs (top) and up-regulation of gene sets for pediatric cancer and CML proliferation gene sets in GMPs (bottom). NES, normalized enrichment score. (H) Heatmap depicting k-means clustering of top differentially expressed genes between JMML HSCs and CB HSCs with an FDR cutoff <0.1. Cluster size corresponds to the number of genes within each significant cluster. (I) Hallmark GSEA between JMML HSCs and CB HSCs depicting up-regulated gene sets in JMML. Dashed line represents an FDR cutoff of <0.1; numbers next to the bars represent NES. (J) Identification of a JMML LSC–specific gene signature from bulk RNA-seq. (K) Single-cell gene expression of the JMML stem cell genes identified in Fig. 5 J. Clusters are as identified in Fig. 2 B. Expression plotted as mean UMI. (L) Heatmap of top differentially expressed genes between JMML HSC, JMML +/+, and JMML GMP. Data acquired over two independent experiments. TPM, transcripts per million.
Identification of new disease-specific biomarkers
One of the top differentially expressed genes (overexpressed in JMML HSCs) was the cell surface receptor CD96 (Fig. 6 A), also confirmed to be more frequently expressed in JMML-specific subpopulations of HSPCs in our single-cell RNA-seq dataset (Figs. 6, B and C). CD96 has previously been proposed as a biomarker and therapeutic target in AML (Hosen et al., 2007). We confirmed aberrant surface expression of CD96 protein on JMML LSCs in the majority of patients when analyzing either total Lin−CD34+90+ (Fig. 6 D) or Lin−CD34+CD38−CD90+CD45RA+ (Fig. S5 A) cells. Furthermore, we showed that this aberrant population of CD96-expressing cells could be detected in two patients after HSCT who subsequently relapsed (Fig. 6 E and Fig. S5 B) at a time point when there was no evidence of disease relapse, raising the possibility that CD96 could be a useful biomarker for JMML LSCs. CD96 expression was also positively correlated with the level of fetal hemoglobin at diagnosis (Fig. 6 F; Niemeyer and Flotho, 2019). By contrast, CD96 expression on mature myeloid or B cells in JMML was low (Fig. S5 C), although expression was higher on CD3+ T cells from JMML patients as reported for healthy individuals (Wang et al., 1992). Interestingly, CD96 expression was clearly higher on Lin−CD34+CD90+ samples from JMML patients compared with the same immunophenotypic population in patients with chronic or transforming MPNs (Fig. 6 G and Fig. S5 D), while in pediatric AML, expression of CD96 was extremely heterogeneous.

CD96 expression in JMML HSPCs. (A) CD96 expression by bulk RNA-seq in JMML and CB HSCs, P < 0.05 Benjamini-Hochberg–corrected false discovery rate. Data acquired through bulk RNA-seq. (B) CD96 expression by single-cell RNA-seq (left) on tSNE analysis from CB and JMML (right). (C) Violin plots of single-cell CD96 expression in JMML and CB CD34+ HSPCs. (D) CD96 expression by FACS in CB and JMML Lin−CD34+CD90+ cells showing percentage of Lin−CD34+90+ cells expressing surface CD96 (JMML, n = 19 different biological replicates; CB controls, n = 11 different biological replicates). Data acquired over five independent experiments. Statistical analysis performed by Mann-Whitney test, **** P < 0.0001. Error bars represent mean ± SEM. (E) Presence of Lin−CD34+CD90+CD96+ cells in paired diagnostic and posttransplant samples in patient ID5 detected by flow cytometry. Data acquired over two independent experiments; representative plots from one experiment shown. Dashed line represents the CD96 gate. (F) Dot plot showing correlation between CD96 expression on Lin−CD34+CD90+ cells from JMML patients (age > 6 mo) at diagnosis and fetal hemoglobin (HbF) percentage at the same time point. Pearson's correlation and P value generated by ggplot2 and ggExtra R package (P = 0.0043). JMML, n = 17 different biological replicates. Data acquired over five independent experiments. (G) CD96 expression by FACS in Lin−CD34+CD90+ cells in JMML patient samples (n = 19 different biological replicates), CB controls (n = 11 different biological replicates), and different pediatric and adult myeloid malignancies (MPN: essential thrombocytopenia, n = 3 different biological replicates; myelofibrosis, n = 3 different biological replicates; CML, n = 3 different biological replicates; polycythemia vera, n = 3 different biological replicates; MPN transformed to AML (tMPN AML), n = 5 different biological replicates; and pediatric AML cases, n = 3 different biological replicates. **** P < 0.0001. Error bars represent mean ± SEM. Data acquired over six independent experiments. (Hi) Percent human CD45+ cells in PB from mice transplanted with CD96+ (red line) or CD96− (black dashed line) Lin−CD34+CD38− cells. Data acquired from one independent experiment. Error bars represent mean ± SEM. (Hii) Kaplan-Meier leukemia-free survival curve of mice transplanted with purified JMML Lin−CD34+CD38− CD96+ (n = 1,802 cells per recipient) or JMML Lin−CD34+CD38− CD96− cells (n = 382 cells per recipient). Three animals transplanted per group. Significance by log-rank test, * P = 0.025. TPM, transcripts per million.
CD96 expression in JMML HSPCs. (A) CD96 expression by bulk RNA-seq in JMML and CB HSCs, P < 0.05 Benjamini-Hochberg–corrected false discovery rate. Data acquired through bulk RNA-seq. (B) CD96 expression by single-cell RNA-seq (left) on tSNE analysis from CB and JMML (right). (C) Violin plots of single-cell CD96 expression in JMML and CB CD34+ HSPCs. (D) CD96 expression by FACS in CB and JMML Lin−CD34+CD90+ cells showing percentage of Lin−CD34+90+ cells expressing surface CD96 (JMML, n = 19 different biological replicates; CB controls, n = 11 different biological replicates). Data acquired over five independent experiments. Statistical analysis performed by Mann-Whitney test, **** P < 0.0001. Error bars represent mean ± SEM. (E) Presence of Lin−CD34+CD90+CD96+ cells in paired diagnostic and posttransplant samples in patient ID5 detected by flow cytometry. Data acquired over two independent experiments; representative plots from one experiment shown. Dashed line represents the CD96 gate. (F) Dot plot showing correlation between CD96 expression on Lin−CD34+CD90+ cells from JMML patients (age > 6 mo) at diagnosis and fetal hemoglobin (HbF) percentage at the same time point. Pearson's correlation and P value generated by ggplot2 and ggExtra R package (P = 0.0043). JMML, n = 17 different biological replicates. Data acquired over five independent experiments. (G) CD96 expression by FACS in Lin−CD34+CD90+ cells in JMML patient samples (n = 19 different biological replicates), CB controls (n = 11 different biological replicates), and different pediatric and adult myeloid malignancies (MPN: essential thrombocytopenia, n = 3 different biological replicates; myelofibrosis, n = 3 different biological replicates; CML, n = 3 different biological replicates; polycythemia vera, n = 3 different biological replicates; MPN transformed to AML (tMPN AML), n = 5 different biological replicates; and pediatric AML cases, n = 3 different biological replicates. **** P < 0.0001. Error bars represent mean ± SEM. Data acquired over six independent experiments. (Hi) Percent human CD45+ cells in PB from mice transplanted with CD96+ (red line) or CD96− (black dashed line) Lin−CD34+CD38− cells. Data acquired from one independent experiment. Error bars represent mean ± SEM. (Hii) Kaplan-Meier leukemia-free survival curve of mice transplanted with purified JMML Lin−CD34+CD38− CD96+ (n = 1,802 cells per recipient) or JMML Lin−CD34+CD38− CD96− cells (n = 382 cells per recipient). Three animals transplanted per group. Significance by log-rank test, * P = 0.025. TPM, transcripts per million.

Aberrant CD96 expression in JMML (related to Fig. 6 ). (Ai) Histograms of CD96 expression on the aberrant Lin−CD34+CD38−CD90+CD45RA+ cells from patient samples (ID1, ID3, ID5, ID6, ID14, and ID15) and Lin−CD34+CD38−CD90+ cells from a normal CB sample. Dashed line represents the CD96 gate. Six biological replicates; representative data shown from one experiment. (Aii) CD96 expression by FACS in CB Lin−CD34+CD38−CD90+ and JMML Lin−CD34+CD38−CD90+CD45RA+ samples showing percentage of cells expressing CD96 (JMML, n = 12; CB, n = 4). Mann-Whitney test; ** P = 0.0049. Error bars represent mean ± SEM. Data shown from two independent experiments. (Aiii) Representative FACS plots of CD96 expression on the aberrant Lin−CD34+CD38−CD90+CD45RA+ cells from patient samples (ID3 and ID5) and CD96 expression of Lin−CD34+CD90+ cells from a normal CB sample. Representative data shown from one experiment. (B) Histogram showing CD96 expression on Lin−CD34+CD90+ cells of a JMML sample (ID19) compared with pediatric BM at diagnosis and 1 mo after transplantation. Dashed line represents the CD96 gate. Patient subsequently relapsed, requiring a second BMT 6 mo after the first BMT. Representative data from one experiment. (C) Representative FACS profile of CD96 expression on mature hematopoietic populations in three JMML patients (ID2, ID5, and ID6). Dashed line represents the CD96 gate. Representative data from one experiment. (D) Representative FACS plots of CD96 expression on Lin−CD34+CD90+ cells in JMML (ID13), chronic phase MPN (CML), and transformed MPN (tMPN AML). Representative data from one experiment. FSCA, forward scatter area.
Aberrant CD96 expression in JMML (related to Fig. 6 ). (Ai) Histograms of CD96 expression on the aberrant Lin−CD34+CD38−CD90+CD45RA+ cells from patient samples (ID1, ID3, ID5, ID6, ID14, and ID15) and Lin−CD34+CD38−CD90+ cells from a normal CB sample. Dashed line represents the CD96 gate. Six biological replicates; representative data shown from one experiment. (Aii) CD96 expression by FACS in CB Lin−CD34+CD38−CD90+ and JMML Lin−CD34+CD38−CD90+CD45RA+ samples showing percentage of cells expressing CD96 (JMML, n = 12; CB, n = 4). Mann-Whitney test; ** P = 0.0049. Error bars represent mean ± SEM. Data shown from two independent experiments. (Aiii) Representative FACS plots of CD96 expression on the aberrant Lin−CD34+CD38−CD90+CD45RA+ cells from patient samples (ID3 and ID5) and CD96 expression of Lin−CD34+CD90+ cells from a normal CB sample. Representative data shown from one experiment. (B) Histogram showing CD96 expression on Lin−CD34+CD90+ cells of a JMML sample (ID19) compared with pediatric BM at diagnosis and 1 mo after transplantation. Dashed line represents the CD96 gate. Patient subsequently relapsed, requiring a second BMT 6 mo after the first BMT. Representative data from one experiment. (C) Representative FACS profile of CD96 expression on mature hematopoietic populations in three JMML patients (ID2, ID5, and ID6). Dashed line represents the CD96 gate. Representative data from one experiment. (D) Representative FACS plots of CD96 expression on Lin−CD34+CD90+ cells in JMML (ID13), chronic phase MPN (CML), and transformed MPN (tMPN AML). Representative data from one experiment. FSCA, forward scatter area.
To investigate whether CD96 was differentially expressed on JMML LSCs, we FACS-sorted Lin−CD34+CD38−CD96+ and Lin−CD34+CD38−CD96− cells from patient ID5 and assessed the LSC potential of these populations by xenotransplantation. Recipients of Lin−CD34+CD38−CD96+ cells showed rapid engraftment (Fig. 6 Hi), with all mice succumbing to JMML-like disease 11 wk after transplantation (Fig. 6 Hii). While recipients of the CD34+CD38−CD96− fraction also showed engraftment, this was at a lower level than for the mice receiving CD96+ cells, and all mice remained alive up to 19 wk after transplantation (Fig. 6 Hii).
GLUT1: A novel therapeutic target for JMML HSCs
A number of putative therapeutic targets were up-regulated in JMML HSCs, including overexpression of SLC2A1, which encodes the glucose transporter protein GLUT1 (Fig. 7 A). Small-molecule inhibition of GLUT1 is well known to induce apoptosis in cancer cells (Wood et al., 2008). To explore whether this target is relevant for JMML, we initially performed apoptosis assays on the RAS-mutated myeloid leukemia cell lines (THP1, TF1, and GDM1) and demonstrated a dose-related cytotoxic effect of the GLUT1 inhibitors WZB117 and fasentin on all three cell lines (Fig. 7 B). We next showed that both GLUT1 inhibitors induced apoptosis in primary JMML HSPCs in vitro in comparison with pediatric BM (Fig. 7 C). GLUT1 inhibition selectively eradicated proliferating cells and enriched for quiescent HSPCs (Fig. 7 D).

GLUT1: A n ovel therapeutic target for JMML HSCs. (A) Gene expression of SLC2A1 by bulk RNA-seq in JMML HSCs and CB HSCs. (B) Percentage of nonviable cells (Annexin-V+ and propidium iodide–positive) from THP1 cell line (left panels), GDM-1 cell line (middle panels), and TF1 cell line (right panels) treated with GLUT1 inhibitors (WZB117: top three panels; fasentin: bottom three panels) and analyzed 24 h after treatment. THP1, four independent experiments with at least two replicates per dose; GMD1, three independent experiments with at least two replicates per dose; TF1, four independent experiments with at least two replicates per dose. Error bars represent mean ± SEM. (C) Percent Annexin-V+ CD34+ cells in vehicle control–treated normal pediatric BM (PBM) controls (n = 4) and JMML CD34+ cells (n = 4) 24 h after treatment with WZB117 50 μM, ** P = 0.0028 (top panel), or fasentin 200 μM, * P = 0.047 (bottom panel). Two independent experiments. Error bars represent mean ± SEM. Statistical analysis performed by Mann-Whitney test. (D) Cell cycle analysis of JMML CD34+ (n = 3) and CB CD34+ controls (n = 4) 7 d after treatment with GLUT1 inhibitors. Two independent experiments. Error bars represent mean ± SEM. Two-way ANOVA: JMML DMSO versus JMML WZB117 (G0-G1), ** P = 0.0083; JMML DMSO versus JMML fasentin (G0-G1), ** P = 0.0041; and JMML DMSO versus JMML WZB117 (S/G2/M), ** P = 0.0021. (E) In vitro single-cell clonogenicity of CB (n = 3) and JMML CD34+CD38− HSPCs (n = 5) with selected targeted inhibitors. Error bars represent mean ± SEM. Three independent experiments, 30 cells from each biological replicate plated per condition. Mann-Whitney test, * P = 0.0411; ** P = 0.0079. FC indicates fold change. (F) Matrix illustrating drug synergy between GLUT1 inhibitor (fasentin) and MEK-inhibitor (PD901), representative result of two independent experiments on THP1 cell lines measuring percentage of nonviable cells after 24 h after treatment, synergy z-score 27.488. (G) Percent Annexin-V+ over vehicle control–treated CB CD34+ (n = 4) and JMML CD34+ (n = 4) 24 h after treatment with WZB117 50 μM, fasentin 200 μM, PD901 50 μM, and combinations of WZB117 with PD901 and fasentin with PD901 at the same doses. Three independent experiments; error bars represent mean ± SEM. CB WZB117 versus JMML WZB117, ** P = 0.002; CB WZB117 + PD901 versus JMML WZB117 + PD901, ** P = 0.0019; CB fasentin versus JMML fasentin, * P = 0.029; CB fasentin + PD901 versus JMML fasentin + PD901, * P = 0.028; and JMML WZB117 versus JMML WZB117 + PD901, * P = 0.013; Mann-Whitney test. TPM, transcripts per million.
GLUT1: A n ovel therapeutic target for JMML HSCs. (A) Gene expression of SLC2A1 by bulk RNA-seq in JMML HSCs and CB HSCs. (B) Percentage of nonviable cells (Annexin-V+ and propidium iodide–positive) from THP1 cell line (left panels), GDM-1 cell line (middle panels), and TF1 cell line (right panels) treated with GLUT1 inhibitors (WZB117: top three panels; fasentin: bottom three panels) and analyzed 24 h after treatment. THP1, four independent experiments with at least two replicates per dose; GMD1, three independent experiments with at least two replicates per dose; TF1, four independent experiments with at least two replicates per dose. Error bars represent mean ± SEM. (C) Percent Annexin-V+ CD34+ cells in vehicle control–treated normal pediatric BM (PBM) controls (n = 4) and JMML CD34+ cells (n = 4) 24 h after treatment with WZB117 50 μM, ** P = 0.0028 (top panel), or fasentin 200 μM, * P = 0.047 (bottom panel). Two independent experiments. Error bars represent mean ± SEM. Statistical analysis performed by Mann-Whitney test. (D) Cell cycle analysis of JMML CD34+ (n = 3) and CB CD34+ controls (n = 4) 7 d after treatment with GLUT1 inhibitors. Two independent experiments. Error bars represent mean ± SEM. Two-way ANOVA: JMML DMSO versus JMML WZB117 (G0-G1), ** P = 0.0083; JMML DMSO versus JMML fasentin (G0-G1), ** P = 0.0041; and JMML DMSO versus JMML WZB117 (S/G2/M), ** P = 0.0021. (E) In vitro single-cell clonogenicity of CB (n = 3) and JMML CD34+CD38− HSPCs (n = 5) with selected targeted inhibitors. Error bars represent mean ± SEM. Three independent experiments, 30 cells from each biological replicate plated per condition. Mann-Whitney test, * P = 0.0411; ** P = 0.0079. FC indicates fold change. (F) Matrix illustrating drug synergy between GLUT1 inhibitor (fasentin) and MEK-inhibitor (PD901), representative result of two independent experiments on THP1 cell lines measuring percentage of nonviable cells after 24 h after treatment, synergy z-score 27.488. (G) Percent Annexin-V+ over vehicle control–treated CB CD34+ (n = 4) and JMML CD34+ (n = 4) 24 h after treatment with WZB117 50 μM, fasentin 200 μM, PD901 50 μM, and combinations of WZB117 with PD901 and fasentin with PD901 at the same doses. Three independent experiments; error bars represent mean ± SEM. CB WZB117 versus JMML WZB117, ** P = 0.002; CB WZB117 + PD901 versus JMML WZB117 + PD901, ** P = 0.0019; CB fasentin versus JMML fasentin, * P = 0.029; CB fasentin + PD901 versus JMML fasentin + PD901, * P = 0.028; and JMML WZB117 versus JMML WZB117 + PD901, * P = 0.013; Mann-Whitney test. TPM, transcripts per million.
We next explored whether the cytotoxic effect of GLUT1 inhibition was synergistic with the MEK inhibitor PD901, as MEK inhibitors are in clinical development in JMML. We first demonstrated that both agents independently reduced clonogenicity of JMML HSPCs (Fig. 7 E). We then demonstrated clear synergy between PD901 and fasentin when these agents were used in combination in THP1 cells (Fig. 7 F). Finally, we demonstrated that the addition of PD901 to single-agent GLUT1 inhibition with either fasentin or WZB117 resulted in markedly enhanced cytotoxicity in JMML (Fig. 7 G). These data support further investigation of GLUT1 inhibitors as a potential novel therapeutic approach in JMML.
Discussion
Disease relapse after achievement of clinical/morphological remission is a major cause of treatment failure across many different human cancers. Characterization of distinct CSCs, specific to each type of malignancy, is a crucial step toward improved approaches for disease monitoring in order to predict disease relapse and the development of CSC-directed therapy. This task is difficult in rare pediatric diseases such as JMML, and yet the need for new disease biomarkers in this disease is particularly acute because HSCT, the only curative therapy, carries a high rate of relapse that is often difficult to diagnose promptly. In hematopoietic cancers such as MDS, MPN, ALL, and AML (le Viseur et al., 2008; Magee et al., 2012; Mead and Mullally, 2017; Thomas and Majeti, 2017; Woll et al., 2014), LSCs are diverse and vary considerably in their frequency and phenotype, with each providing important insights into LSC biology. However, for the rare and poor prognosis childhood leukemia JMML, very little is known about the cellular hierarchy and identity of LSCs, including whether progenitor cells are transformed in this condition. We have performed a comprehensive phenotypic, functional, and molecular analysis of a cohort of JMML patients and describe significant disruption of the HSPC hierarchy in JMML, including the presence of an aberrant early progenitor cell (Lin−CD34+CD38−CD90+CD45RA+) that may reflect an underlying aberrant HSC myeloid differentiation pathway in JMML. Interestingly, this population has also been observed in adult patients with myeloid malignancies associated with monosomy 7 (Dimitriou et al., 2016). Consistent with this, we also observed impaired megakaryocyte and erythroid output from JMML HSCs.
Backtracking of somatic genetic lesions to distinct HSPC subsets is a powerful method to help identify CSCs, as mutations acquired by short-lived progenitors that lack self-renewal ability are not able to propagate the disease (Woll et al., 2014). Using a similar approach combining single-cell index sorting and colony genotyping, we demonstrate that all JMML driver mutations could be backtracked to the phenotypic HSC compartment with RAS-pathway mutations as a “first hit.” Despite being relatively genetically simple in comparison with other tumors, we observed JMML patients with both linear and branching patterns of clonal evolution in JMML HSCs. Importantly, we were able to detect mutant HSPCs in a post-HSCT patient a month before molecular/clinical evidence of relapse. At the same time point, a population of CD96+ cells within the Lin−CD34+CD90+ gate could be detected by FACS on patient cells but not on normal controls. Taken together, this strongly suggests that the presence of JMML HSPCs with aberrant phenotype could be used to predict impending relapse in JMML patients, thus providing a much-needed biomarker of residual disease after HSCT, although this will need to be validated in larger clinical cohorts before it could be applied clinically.
Although our colony genotyping data supported that phenotypic HSCs might be the cell of origin in JMML, it remains possible that transformation occurs at the progenitor cell level with subsequent aberrant up-regulation of HSC surface markers. Analysis of patient samples cannot discriminate between these two possibilities. Relating to this point, it is noteworthy that RNA-seq also supported hierarchical organization of JMML HSPCs, with HSCs displaying higher expression of HSC-associated transcriptional programs in comparison with JMML progenitors, albeit with aberrant expression of proliferation and fetal-associated genes. Functional analysis (xenotransplantation and LTC-IC assays) revealed marked inter-patient heterogeneity of JMML LSCs. Thus, while JMML HSCs were consistently able to support LTC-IC potential and engraftment in xenograft models, some patients also showed evidence of self-renewal of progenitor cells, including GMPs and the novel +/+ cell population that we describe. Interestingly, this suggests that JMML displays a distinct biology, sharing some features with MDS/MPN, which are propagated by the counterparts of HSCs (Mead and Mullally, 2017; Woll et al., 2014), and other features with acute leukemias, which show transformation of progenitor populations but cannot usually be propagated by HSCs lacking the late driver mutations acquired by transformed progenitor/precursors (Jan et al., 2012; le Viseur et al., 2008; Thomas and Majeti, 2017). Importantly, we demonstrate that the +/+-aberrant HSPC population was particularly prominent in serial samples taken from JMML patients that relapsed. Furthermore, JMML HSPCs engrafted robustly in NSG mice, rather than exhibiting the poor in vitro and in vivo proliferative capacity characteristic of adult MDS stem cells (Woll et al., 2014). These biological features of pediatric versus adult MDS may reflect the impact of RAS driver mutations on JMML HSCs and/or coexpression of a proliferative fetal gene program, such as we observed here in the JMML HSCs but not reported in adult MDS (Woll et al., 2014). Of specific note, the fetal HSC–specific gene VNN2, encoding the glycophosphatidylinositol-anchored surface protein GPI-80, was markedly overexpressed by JMML HSCs (Prashad et al., 2015). This is of particular interest, as disease-associated mutations in JMML can often be tracked back to neonatal blood spots and fetal hemoglobin is often increased in JMML cases (Locatelli and Niemeyer, 2015), together suggesting a fetal origin of JMML. However, a limitation of this current analysis is that earlier neonatal samples taken at birth (CB or neonatal blood spots) were not available. Consequently, while our data support a fetal origin of the JMML cases studied, this cannot be definitively established.
The above findings are in line with a recent report (Caye et al., 2020) that also demonstrated the presence and heterogeneity of JMML LSCs but concluded that the HSPC compartments (phenotypically and molecularly) were globally maintained in JMML. However, although phenotypic analysis of HSPCs showed considerable overlap with normal pediatric BM HSPCs, such analyses, based on a small number of canonical surface markers, often fail to reveal underlying cellular heterogeneity. In contrast, using a combination of phenotypic analyses, functional assays, single-cell molecular lineage tracing, and RNA-seq, we show marked disruption of the HSPC compartment in JMML at diagnosis and relapse and thereby identify therapeutic vulnerabilities of JMML LSCs. Single-cell RNA-seq analysis is a powerful method to resolve such heterogeneity and has been widely used to analyze HSPCs (Povinelli et al., 2018). We have shown that in CML and MPN, such an approach can help to resolve normal and malignant stem cells (Giustacchini et al., 2017; Psaila et al., 2020; Rodriguez-Meira et al., 2019). We therefore used a similar approach to analyze over 17,000 HSPCs from CB and JMML patients, revealing that JMML HSPCs are molecularly highly distinct. Bulk RNA-seq analysis of JMML LSCs revealed evidence of a hierarchical organization of HSPCs in JMML, with HSCs residing at the apex of this hierarchy and +/+ cells sharing features of both HSCs and GMPs.
Importantly, this analysis also allowed us to identify a number of up-regulated or aberrantly expressed putative therapeutic targets in the JMML HSC. We demonstrate aberrant high-level expression of CD96 on the cell surface of JMML LSCs, which represents a potential biomarker of JMML LSCs and could be developed as an immunotherapeutic target, as also previously proposed in AML (Staudinger et al., 2013; Chávez-González et al., 2014), adult MDS (Zhang et al., 2015), and ALL (Wang et al., 2013). Furthermore, we validate GLUT1 inhibition as a novel therapeutic approach to target JMML HSPCs either as a single agent or in combination with MEK inhibition. As eradication of CSCs/LSCs is not only necessary but also potentially sufficient to achieve disease eradication (Magee et al., 2012), further evaluation of these targets is warranted.
In summary, we describe an integrated phenotypic, functional, and molecular analysis of JMML LSCs, illustrating marked intra- and inter-patient heterogeneity of LSCs. We identified a number of candidate biomarkers and therapeutic targets, paving the way for LSC-directed disease monitoring and therapy in JMML.
Materials and methods
Contact for reagent and resources
Further information and requests for resources and reagents should be directed to and will be fulfilled by Adam Mead ([email protected]).
Patient sample collection
Patient samples and normal controls were prospectively collected following informed consent, in accordance with the Declaration of Helsinki for sample collection and use in research under the UK National Institute of Health Research, National Health Service (NHS), Research Ethics Committee (REC)–approved Pediatric MDS/JMML study (NHS REC reference number 15/LO/0961) or normal pediatric BM (NHS REC reference number 12/LO/0426). An additional JMML patient sample was provided by the Bloodwise Childhood Leukemia Cell Bank (NHS REC reference number 16/SW/0219). CB samples were commercially sourced (Zenbio US; Cat# SER-CD34-F). Adult MPN samples used were collected under the INForMeD study (NHS REC number 16/LO/1376). Detailed phenotypic and clinical characteristics (Table S1) were captured on a secure online platform using the Human Ontology Database tool. Clinical details for the sample provided by the Leukemia Cell Bank were not available. Mononuclear cells (MNCs) from PB and BM samples were isolated on Ficoll density gradients and cryopreserved in 90% FBS and 10% DMSO. Cells were thawed and processed for downstream analysis as previously described (Woll et al., 2014). Additional information on FACS sorting of samples is detailed in supplemental materials.
In vitro assays
Cell lines
The M210B4 cell line was maintained in RPMI (Gibco) 10% FBS (HyClone), the Sl/Sl cell line was maintained in DMEM (Gibco) 15% FBS (HyClone), and the MS5 cell line was maintained in AlphaMEM (Invitrogen) 10% FBS (HyClone). The THP1 cell line was maintained in RPMI (Gibco) + 10% FBS + 1% penicillin-streptomycin (Sigma-Aldrich), the GDM-1 cell lines were maintained in RPMI (Gibco) + 20% FBS + 1% penicillin-streptomycin (Sigma-Aldrich), and the TF1 cell lines were maintained in RPMI (Gibco) + 20% FBS + 5 ng/ml GM-CSF (Sigma-Aldrich) + 1% penicillin-streptomycin.
FACS
FACS sorting of MNCs was performed using the FACS Aria II, FACS Aria III, and FACS Aria Fusion (Becton Dickinson). All FACS experiments included single color–stained controls (CompBeads; BD Biosciences) and fluorescence minus one controls. Bulk and single-cell HSPCs were isolated from JMML patient samples and normal controls. For all single-cell experiments, sorting was performed using the single-cell purity setting and tested by the deposition of single beads into the bottom of 96-well plates and checking by fluorescence microscopy. Index-sort data for single cells of the mean fluorescence intensities of CD34, CD38, CD90, CD45RA, and CD123 were also recorded for each individual cell isolated. To check the correct alignment of the sorter, BD FACS Accudrop Beads (BD Biosciences) were sorted into a 96-well plate or Eppendorf tube in order to ensure that all beads were delivered to the bottom of the well/tube. Purity of a sorted population was performed by reanalysis of sorted populations and was typically >95%. Flow cytometry profiles of the human HSPC compartments were analyzed using FlowJo software (version 10.1).
Antibodies for HSPC sorting and analysis were CD34 APC–eFluor780 (eBioscience; Clone: 4H11, Cat# 47–0349-42, RRID: AB_2573956), CD38 PE–Texas Red (Invitrogen; Clone: HIT2, Cat# MHCD3817, RRID: AB_10392545), CD90 BV421 (Biolegend; Clone: 5 E 10, Cat# 328122, RRID: AB_2561420), CD45RA PE (eBioscience; Clone: HI100, Cat# 12–0458-41, RRID: AB_10717397), CD123 PE Cy7 (Biolegend; Clone: 6H6, Cat# 306010, RRID: AB_493576), CD8 (Biolegend; Clone: RPA-T8, Cat# 301006, RRID: AB_314124), CD20 (Biolegend; Clone: 2H7, Cat# 302304, RRID: AB_314252), CD2 (BD Biosciences; Clone: RPA-2.10, Cat# 555326), CD3 (BD Biosciences; Clone: SK7, Cat# 345763), CD16 (eBioscience; Clone: eBioCB16, Cat# 11–0168-41, RRID: AB_10804882), CD19 (eBioscience; Clone: HIB19, Cat# 11–0199-42, RRID: AB_10669461), CD235a (eBioscience; Clone: HIR2, Cat# 11–9987-82, RRID: AB_465477), CD66b (Biolegend; Clone: G10F5, Cat# 305104, RRID: AB_314496), CD10 (Biolegend; Clone: HI10a, Cat# 312208, RRID: AB_314919), and CD127 (eBioscience; Clone: RDR5, Cat# 11–1278-42, RRID: AB_1907343), each FITC. JMML patient samples were also stained for CD96 expression (laboratory of Martin Gramatzki (Division of Stem Cell Transplantation and Immunotherapy, Department of Medicine, University of Kiel, Kiel, Germany); Clone: TH-111, donkey anti-mouse PE [eBioscience; Cat# 12–4010-82, RRID: AB_11063706] using the same panel described above with CD45RA BV650 [Biolegend; Clone: HI100, Cat# 304136, RRID: AB_2563653]).
Single-cell clonogenic assays
Single HSPCs were sorted based on their immunophenotypic definitions into 96-well plates with 50 µl of MethoCult H4435 (StemCell Technologies, Inc.). Lineage output was assessed on day 7, day 10, and day 14 by evaluating the morphology of the colonies under direct light microscopy, and individual colonies were picked for cytospins and flow cytometry or resuspended in 10 µl of PBS, flash frozen, and stored at −80° for downstream mutational analysis. For targeting candidate JMML targets, PD-0325901 (Cambridge Bioscience; Cat# HY-10254; final concentrations 1 μM or 5 μM), fasentin (Sigma-Aldrich; Cat# F5557-5MG, final concentrations 5 nM or 20 nM), or equivalent DMSO vehicle control were added to the MethoCult before FACS sorting.
Single-cell erythroid/megakaryocytic liquid culture assay
Single cells were directly sorted into 96-well plates with erythroid/megakaryocyte bipotential medium for erythroid and megakaryocytic output (Psaila et al., 2016). StemSpan (StemCell Technologies, Inc.) was supplemented with thrombopoietin (TPO; 100 ng/ml), erythropoietin (EPO; 1 IU/ml), stem cell factor (SCF; 100 ng/ml), IL-3 (10 ng/ml), IL-6 (10 ng/ml; all from Peprotech), and human low-density lipoprotein (40 mg/ml; StemCell Technologies, Inc.). Single cells were cultured at 37°C in 5% CO2 for 2 wk, and colony readout was performed by direct light microscopy and flow cytometry using CD41a APC (eBioscience; Clone: HIP8, Cat# 17–0419-42, RRID: AB_2573144), CD42b PE (eBioscience; Clone: HIP1, Cat# 12–0429042, RRID: AB_10852864), CD11b FITC (eBioscience; Clone: ICRF44, Cat# 11–0118-42, RRID: AB_1582242), CD14 FITC (eBioscience; Clone: 61D3, Cat# 11–0149-41, RRID: AB_10597445), CD34 APC–eFluor 780 (Clone: 4H11, Cat# 47–0349-42, RRID: AB_2573956), and CD235a PerCp Cy5.5 (Biolegend; Clone: HIR2, Cat# 306614, RRID: AB_10683170). CD41a and CD42b double-positive cells were used to identify the megakaryocytic output.
B cell–differentiation MS5 co-cultures
100 cells from relevant HSPC populations were sorted into cytokine-containing medium (AlphaMEM), 10% heat-inactivated (batch-tested) FBS, FLT3 (10 ng/ml), SCF (20 ng/ml), IL-2 (10 ng/ml), and IL-7 (5 ng/ml; all from Peprotech). Sorted cells were cultured on MS5 stroma cell lines (DSMZ; Cat# ACC 441) for 4 wk. Co-cultures were disaggregated by vigorous pipetting, passed through a 70-µm filter, and replated on fresh MS5 cells every 4 d. Colony readout to assess the lymphoid output was performed by flow cytometry at weeks 2, 3, and 4 of the assay with CD11b FITC (eBioscience; Clone: ICRF44, Cat# 11–0118-42, RRID: AB_1582242), CD14 FITC (eBioscience; Clone: 61D3, Cat# 11–0149-41, RRID: AB_10597445), CD34 APC–eFluor 780 (eBioscience; Clone: 4H11, Cat# 47–0349-42, RRID: AB_2573956), CD235a PerCp Cy5.5 (Biolegend; Clone: HIR2, Cat# 306614, RRID: AB_10683170), human CD45 Alexa Fluor 700 (Biolegend; Clone: HI130, Cat# 304024, RRID: AB_493761), CD19 PE (BD Biosciences; Clone: SJ25C1, Cat# 345789), CD56 FITC (eBioscience; Clone: TULY56, Cat# 11–0566-42, RRID: AB_2572458), and CD33 FITC (BD Biosciences; Clone: HIM3-4, Cat# 555626).
Lymphoid output for two control samples was assessed with human CD45 Alexa Fluor 700 (BD; Clone: 2D1, Cat# 56–9459-42), CD19 APC (Biolegend; Clone: HIB19, Cat# 302212), CD56 PE (eBioscience; Clone: CMSSB, Cat# 12–0567-41, RRID: AB_10598372), CD10 PE Cy7 (eBioscience, Clone: ebioCB-CALLA, Cat# 25–0106-42), CD34 PerCPCy5.5 (Biolegend; Clone: 581, Cat# 343522), CD73 BV421 (Biolegend; Clone: AD2, Cat# 344008), CD11b (eBioscience; Clone: ICRF44, Cat# 11–0118-42, RRID: AB_1582242), CD14 (eBioscience; Clone: 61D3, Cat# 11–0149-41, RRID: AB_10597445), and CD33 (BD Biosciences; Clone: HIM3-4, Cat# 555626), all FITC.
Annexin V in vitro apoptosis assays
Apoptosis assays with cell lines
THP1, GDM1, and TF1 cell lines were cultured in vitro in the media conditions outlined above. Typically, 1,000 cells were harvested and treated with either WZB117 (Cambridge-Bioscience; Cat# HY-10254), fasentin (Sigma-Aldrich; Cat# F5557) alone or in combination with PD-0325901 (Cambridge Bioscience; Cat# HY-10254) at indicated concentrations. Cells were incubated at 37°C in 5% CO2 for 24 h and then analyzed by flow cytometry using the Annexin kit (eBioscience; Cat# 88-8007) per manufacturer’s instructions. The drug synergy score for the drug combination matrix was calculated by using the SynergyFinder R-package available through the SynergyFinder web application v2: visual analytics of multi-drug combination synergies (https://github.com/IanevskiAleksandr/SynergyFinder; Ianevski et al., 2017, 2020). Synergy scores indicate the percentage of response beyond the expected drug effect when each drug is used in isolation.
Apoptosis assays with primary cells
500–1,000 primary Lin−CD34+ cells from either JMML samples or normal CB or pediatric BM controls were FACS sorted into culture media (StemSpan with 1× BIT9500; StemCell Technologies, Inc.), 10 ng/ml SCF (Peprotech), 10 ng/ml FLT3L (Peprotech), 10 ng/ml TPO, 5 ng/ml IL-3 (Peprotech), 10 ng/ml G-CSF (Thermo Fisher), and 1 IU/ml EPO (Roche) and treated with either WZB117 (Cambridge-Bioscience; Cat# HY-10254) or fasentin (Sigma-Aldrich; Cat# F5557) at indicated concentrations. Cells were incubated at 37°C in 5% CO2 for 24 h and then analyzed by flow cytometry using the Annexin kit as per the manufacturer’s instructions.
Cell cycle in vitro assays
500–2,000 primary Lin−CD34+ cells from either JMML patients or normal CB or pediatric BM controls were FACS sorted into culture media (StemSpan with 1× BIT9500, 10 ng/ml SCF, 10 ng/ml FLT3L, 10 ng/ml TPO, 5 ng/ml IL-3, 10 ng/ml G-CSF (Thermo Fisher), and 1 IU/ml EPO (Roche) and treated with either WZB117 (Cambridge-Bioscience) or fasentin (Sigma-Aldrich) at indicated concentrations. Cells were incubated at 37°C in 5% CO2 for 7 d, then fixed with 70% ethanol and incubated for 1 h at −20°C. Cells were washed in PBS, stained with propidium iodide (eBioscience; Cat# 00–6990) at 4°C and analyzed by flow cytometry 24 h after incubation.
LTC-IC
Stem cell potential was assessed in vitro using the LTC-IC assay as previously described (Woll et al., 2014). Mouse stromal cell lines (M210B4 and SL/SL, sourced from Dr. Donna Hogge, Terry Fox Laboratory, University of British Columbia, Vancouver, Canada) were cultured for 7 d in RPMI supplemented with 10% FBS and DMEM supplemented with 15% FBS, respectively. Antibiotic selection was performed for an extra 7 d with hygromycin and G418, followed by irradiation 24 h before FACS sorting. HSPCs from JMML patient samples and CB controls were isolated by FACS as described above, and 100 cells/well were seeded in H5100 MyeloCult medium (StemCell Technologies, Inc.) supplemented with 10−6 hydrocortisone 21-hemisuccinate (StemCell Technologies, Inc.) and co-cultured with SL/SL and M210B4 stroma cells. Sorted populations were cultured for 6 wk, with half of the medium replaced once weekly. Individual wells were harvested after 6 wk and transferred to MethoCult H4435. Colony output was evaluated after 2 wk under direct light microscopy.
Electron microscopy
Electron microscopy photographs were obtained with the Olympus microscope BX60.
NSG xenotransplantation experiments
All procedures involving animals were approved by the UK Home Office Project license (PPL 30–3103). NSG mice were obtained through The Jackson Laboratory and maintained in individually ventilated cages in a specific pathogen–free facility. Male and female mice were randomly assigned to groups. NSG xenografts were performed in 10–14-wk-old mice. FACS-purified HSPC populations were transplanted into NSG (NOD.Cg-Prkdcscid II2rgtm1Wjl/SzJ) mice (The Jackson Laboratory; Cat# 005557) 10–14 wk of age, following sublethal irradiation (split dose of 2.5 Gy delivered 4 h apart). Cells were injected through tail vein injection 6–8 h after the last radiation dose. Human cell reconstitution and lineage distribution were monitored by flow cytometry in the PB at 5, 10, 16, or 22 wk after transplant. For terminal readouts, mice were sacrificed, and flow cytometry analysis of the BM, spleen, and PB was performed in order to assess human cell reconstitution and immunophenotype of the engrafting population. BM MNCs from primary transplants were cryopreserved and used for secondary transplantation. PB was stained with Human CD45 Alexa Fluor 700 (Biolegend; Clone: HI130, Cat# 304024, RRID: AB_493761), Mouse CD45 PE–Texas Red (Invitrogen; Clone: 30-F11, Cat# MCD4517, RRID: AB_10392557), CD19 PE (BD; Clone: SJ25C1, Cat# 345789), CD34 APC–eFluor 780 (eBioscience; Clone: 4H11, Cat# 47–0349-42, RRID: AB_2573956), CD235a PerCP Cy5.5 (Biolegend; Clone: HIR2, Cat# 306614, RRID: AB_10683170), CD15 (BD Biosciences, Clone: MMA, Cat# 347423), CD33 (BD Biosciences; Clone: HIM3-4, Cat# 555626), CD66b (Biolegend; Clone: G10F5, Cat# 305104, RRID: AB_314496), CD11b (eBioscience; Clone: ICRF44, Cat# 11–0118-42, RRID: AB_1582242), and CD14 (eBioscience; Clone: 61D3, Cat# 11–0149-41, RRID: AB_10597445), each FITC.
BM was stained with CD34 APC–eFluor 780 (Clone: 4H11, Cat# 47–0349-42, RRID: AB_2573956), CD38 PE–Texas Red (Invitrogen; Clone: HIT2, Cat# MHCD3817, RRID: AB_10392545), CD90 BV421 (Biolegend; Clone: 5 E 10, Cat# 328122, RRID: AB_2561420), CD45RA PE (eBioscience; Clone: HI100, Cat# 12–0458-41, RRID: AB_10717397), CD123 PE Cy7 (Biolegend; Clone: 6H6, Cat# 306010, RRID: AB_493576), Mouse CD45 BV510 (Biolegend; Clone: 30-F11, Cat# 103138, RRID: AB_2563061), Human CD45 AF700 (Biolegend; Clone: HI130, Cat# 304024, RRID: AB_493761; and BD Biosciences; Clone: 2D1, Cat# 56–9459-42), CD8 (Biolegend; Clone: RPA-T8, Cat# 301006, RRID: AB_314124), CD20 (Biolegend; Clone: 2H7, Cat# 302304, RRID: AB_314252), CD2 (BD Biosciences; Clone: RPA-2.10, Cat# 555326), CD3 (BD Biosciences; Clone: SK7, Cat# 345763), CD16 (eBioscience; Clone: eBioCB16, Cat# 11–0168-41, RRID: AB_10804882), CD19 (eBioscience; Clone: HIB19, Cat# 11–0199-42, RRID: AB_10669461), CD235a (eBioscience; Clone: HIR2, Cat# 11–9987-82, RRID: AB_465477), CD66b (Biolegend; Clone: G10F5, Cat# 305104, RRID: AB_314496), CD10 (Biolegend; Clone: HI10a, Cat# 312208, RRID: AB_314919), and CD127 (eBioscience; Clone: RDR5, Cat# 11–1278-42, RRID: AB_1907343), each FITC. Further details of the antibodies used can be found in the key resource Table S4.
NSG mice were bred at Oxford Biomedical Services. All animal protocols were approved by the UK Home Office. For leukemia-free survival, mice were monitored for health and weight loss and euthanized if presenting with health concerns. Leukemia incidence was confirmed by the presence of leukemic engraftment in PB >50%. Femurs preserved were fixed in neutral buffered formalin and embedded in paraffin for tissue sectioning and staining according to standard techniques.
Bulk targeted next-generation sequencing (NGS)
DNA was extracted from whole PB or BM using the Qiagen DNeasy blood and tissue kit. DNA was also extracted from hair and/or skin fibroblasts to screen for germline mutations. A customized targeted NGS panel was designed using the Illumina design studio (http://www.illumina.com/informatics/research/experimental-design/designstudio.html) to screen for the most common mutations identified in this patient group. The NGS panel contained 32 genes with a total of 916 amplicons (covering 301 exons; Table S2). Coverage of each nucleotide within a target region was assessed for every sample on each sequencing run using the in-house customized software Covermi. Library preparation and sequencing was performed using the Illumina Truseq Custom Amplicon panel v1.5 kit (Illumina; Cat# WG-311-8007), and sequencing was performed using the Illumina MiSeq with the v2 300 cycle reagent kit.
Data analysis of the targeted NGS dataset
Data analysis was performed using in-house bioinformatic pipelines at the Oxford Molecular Diagnostic Center. In short, FASTQ files were processed by the MiSeq Reporter, and sequences were aligned to the reference genome (hg19) by the Smith-Waterman algorithm and Illumina Somatic Variant caller (1.1.0). Variants were annotated using Illumina VariantStudio (2.2) and were filtered using the following passing filters: quality >100 and consequence: missense, frameshift, stop gained, stop lost, initiator codon, in-frame insertion, in-frame deletion, and splice filters. Variants were first verified by visual check by using the Interactive Genomics Viewer (IGV, 2.3.40) and confirmed by either Sanger sequencing or PCR-based mutational analysis performed in the Great Ormond Street Hospital diagnostic laboratory.
Mutational analysis of colonies derived from single cells
Mutations were traced back to HSPCs by the mutational analysis of colonies that arise from single cells sorted from each population. Patient-specific primers were designed for downstream amplification of the region of interest using the Primer blast tool website (https://www.ncbi.nlm.nih.gov/tools/primer-blast/). The sequences of primers used can be found in Table S3. For every individual cell sorted for genotyping, all index-sort data of the mean fluorescence intensities for CD34, CD38, CD90, CD45RA, and CD123 were recorded. This allowed the immunophenotypic identification of every cell processed and mapping of the JMML mutations onto the hematopoietic hierarchy.
Colonies derived from sorted single cells from each population were picked and resuspended in 10 µl PBS and frozen at −80°C for downstream analysis. Samples were thawed and lysed using 15 µl of a custom lysis buffer containing 10% Proteinase K (Qiagen) and 0.4% Triton X. Samples were incubated at 56°C for 10 min and 75°C for 20 min to release genomic DNA and inactivate the Proteinase K, respectively. Regions of interest were amplified using a three-step nested PCR method with FastStart High Fidelity enzyme (Roche). For the first PCR, 3.6 µl of the lysate was amplified with patient-specific primers covering the regions of interest with a targeted length of ∼300-bp amplicons. For the second PCR step, 1 µl of the PCR1 product was amplified with nested target-specific primers generating ∼200-bp amplicons. Primers for PCR 2 were attached to Access Array adaptors (Fluidigm; forward adaptor: 5′-ACACTGACGACATGGTTCTACA-3′; reverse adaptor: 5′-TACGGTAGCAGAGACTTGGTCT-3′). Lastly, for the third PCR step, Illumina-compatible adaptors containing single-direction indexes (Access Array Barcode Library for Illumina Sequencers-384, Single Direction; Fluidigm) were attached to preamplified amplicons from PCR2. This final PCR product was purified with Ampure XP beads (0.8:1 ratio, beads to product), quantified using Quant-iT Picogreen (Thermo Fisher), pooled, and sequenced on the Illumina MiSeq with 150-bp paired-end reads.
Numbers of cell were analyzed for each patient as follows: Patient ID1: of 360 single cells sorted and 199 colonies derived from these cells, 136 were sequenced alongside 20 CB control colonies (total, 156 colonies sequenced); 129 colonies passed QC and were used for downstream analysis. Patient ID5: of 240 single cells sorted and 81 colonies derived from these cells, 67 were sequenced alongside 26 CB control colonies (total, 93 colonies sequenced); 34 colonies passed QC and were used for downstream analysis. Patient ID15: of 420 cells sorted from single cells and 106 colonies derived from these cells, 100 were sequenced alongside 57 CB control colonies (total, 157 colonies sequenced); 71 colonies passed QC and were used for downstream analysis. Patient ID5 after BM transplantation (BMT): of 420 cells sorted and 85 colonies derived from these cells, 81 colonies were sequenced from the posttransplant sample alongside 38 CB control colonies (total, 123 colonies sequenced); 65 patient-derived colonies after transplant passed QC and were used for downstream analysis.
Single-cell genotyping
FACS-sorted single cells were processed using an optimized method for the detection of genomic DNA from single cells and a combined approach for genomic and cDNA amplification, as previously described (Rodriguez-Meira et al., 2019). In short, single cells were sorted into 4 µl lysis buffer containing 2.5% Protease (Qiagen) and 0.2% Triton X. Cells were lysed at 56°C for 20 min and for 10 min at 95°C. The entire lysate was used for downstream multiplex PCR, with custom patient-specific primers. For the first PCR step, targeted regions were amplified with SeqAMP enzyme (Clontech), and 1 µl of the first PCR product was further amplified using nested, patient-specific primers with KAPA 2G Robust PCR (KAPA Biosystems; KK5702). This product was further barcoded using the Access Array barcoded system (Access Array Barcode Library for Illumina Sequencers-384; Fluidigm; Cat# 100–4876). PCR product was then purified with Ampure XP beads and sequenced on a MiSeq as described above.
Colony genotyping analysis
Reads were aligned to the human genome (GRCh37/hg19) using STAR with default settings (Cold Spring Harbor Laboratory; version 2.4.2a, RRID: SCR_004463). Variant calling was performed using mpileup (samtools, Wellcome Trust Sanger Institute; version 1.1, RRID: SCR_002105, options–minBQ 30,–count-orphans,–ignore overlaps), and the results were summarized with a custom Python script. Downstream analysis of the variants and annotation of the mutant cells were performed with a custom R script. Each individual amplicon was analyzed, and those with coverage of <30 reads were excluded from the downstream analysis. Samples in which one of the amplicons was not detected and samples with allelic dropout (assessed using SNPs) in three or more amplicons were also excluded from further analysis. For each sequencing run, normal controls were also screened for the variants of interest in order to ensure no false-positive samples were present. None of the mutations were detected in any of the control samples in any of the sequencing runs. Amplicons covering common heterozygous SNPs for each patient were used to calculate the allelic dropout rate of the assay, as described in Fig. S3.
Single-cell genotyping analysis
Sequencing reads were demultiplexed and aligned to the human genome; downstream analysis and variant annotation were performed as previously described (Rodriguez-Meira et al., 2019). Amplicons with >30 reads (for the analysis of patient ID5) and amplicons with >100 reads (for the analysis of patient ID17) were used for downstream variant annotation and mapping of the patient’s mutation on the hematopoietic hierarchy by integrating the index-sort data with the mutational analysis. The number of WT and homozygous cells detected was within the expected level for the assay, and allelic dropout was calculated as previously described (Rodriguez-Meira et al., 2019; Psaila et al., 2020).
Single-cell RNA-seq
18,333–24,000 single cells from the Lin−CD34+ population from two JMML patient samples (patients ID1 and ID5) and two normal CB controls were sorted and processed for RNA-seq using the 10X chromium platform per the manufacturer’s instructions (10X Genomics; Cat# 120237). In short, cells were sorted into a total volume of 40 µl, and 33.8 µl of the sample was loaded onto the Single Cell 3′ Chromium 10X Chip with 66.2 µl of the 10X master mix. cDNA was generated on the Chromium 10X controller. After RT, cleanup was performed per the manufacturer’s instructions, and the product was amplified with eight PCR cycles. After amplification, cleanup and library construction were performed per the manufacturer’s instructions, and samples were sequenced on the Illumina HiSeq4000, with read1 being 26 bp and read2 98 bp.
Single-cell RNA-seq analysis
Reads from 10X Genomics single-cell RNA-seq were aligned and counted using Cell Ranger (2.0.0) using the hg19 human genome. Unique molecular identifier (UMI) counts where analyzed using Seurat (Satija Lab; v 2.0.1). Genes were filtered by minimum number of cells expressing = 10, >0.15 mean UMI counts, <4 mean UMI counts, and >0.5 dispersion. Cells were filtered by >1,000 genes detected, <4,000 genes detected, and <5% mitochondrial reads. After filtering, 593 variable genes and 17,547 cells were used for analysis. tSNE analysis was performed after selecting the most variable principal components by use of the jackStrawPlot feature in Seurat. Clusters were identified by the k-shared nearest neighbor (k-SNN) approach FindClusters. Figures for single-cell analysis were generated in R (v 3.4.1). Top differentially expressed genes from k-SNN–identified clusters were calculated using the FindAllMarkers function in Seurat.
Bulk RNA-seq
FACS-purified populations were processed for RNA-seq using the Smart Seq2 protocol as previously described (Picelli et al., 2014). Briefly, 50 purified cells were sorted directly into 4 µl of lysis buffer containing 0.4% Triton X-100 (Sigma-Aldrich), RNase inhibitor (Clontech), 2.5 mM deoxyribonucleotide triphosphates (Thermo Fisher), and 2.5 µM oligo-dT30VN primer (Biomers.net). cDNA was generated using SuperScript II (Invitrogen), preamplified using KAPA HiFi HotStart ReadyMix using 19 cycles of amplification. After PCR amplification, the cDNA libraries were purified with AMPure XP beads (Beckman Coulter) using a ratio of 0.8:1 beads to cDNA, according to the manufacturer’s instructions. After purification, libraries were resuspended in elution buffer buffer (Qiagen). The quality of cDNA traces was assessed by using the High-Sensitivity DNA Kit in a Bioanalyzer instrument (Agilent Technologies). Tagmentation and library preparation were performed using the Nextera XT DNA Library Preparation Kit (Illumina; Cat# FC-131) according to the manufacturer’s instructions. Samples were sequenced using the Illumina NextSeq 500 platform, generating 75-bp single-end reads.
Bulk RNA-seq analysis
Data analysis was performed with in-house bioinformatics pipelines. Briefly, RNA-seq reads from bulk populations were aligned to the human genome (GRCh38) using STAR. Aligned reads were counted using featureCounts (Walter and Eliza Hall Institute of Medical Research; version 1.4.5-p1, RRID: SCR_012919) summarized to gene identifiers from the GRCh38 Ensembl transcriptome. Differential gene expression was done using the DESeq2 package (Bioconductor; 1.16.1, RRID: SCR_015687) in R (CRAN; 3.6.1). Plotting of expression values of genes was done by calculating transcripts per million and plotted in R (3.6.1). GSEA was performed using software (Broad Institute; v 2.20, RRID: SCR_003199) against Hallmark, and curated HSC and cell cycle gene sets were obtained through MSigDB (Broad Institute; RRID: SCR_003199) using the default settings. Heatmaps were generated using the package pheatmap (1.0.8).
Quantification and statistical analysis
Welch t tests or the Mann-Whitney test for comparisons of individual groups was used for parametric or nonparametric data, respectively. Two-way ANOVA and multiple t tests were used to compare experimental groups as indicated in the figure legends. Statistical analyses were performed using GraphPad Prism v7.00 and v8.00 or R v3.4.1 and 3.6.1. Survival curves were compared using the log-rank test.
Data and software availability
Single-cell RNA-seq and bulk RNA-seq are available through the GEO database (accession no. GSE111895). The single-cell colony genotyping sequencing is available at the SRA database (accession no. PRJNA438158). R and Python scripts used for the analysis are available upon request.
Online supplemental material
Fig. S1 describes further bulk and single-cell clonogenic assays with JMML HSPCs compared with normal controls, including lymphoid and megakaryocytic output of JMML HSPCs, and includes flow cytometry analysis of patients’ serial samples from diagnosis and after BMT and relapse. Fig. S2 describes the QC steps and analysis of the colony genotyping assays. Fig. S3 demonstrates single-cell genotyping of JMML HSPCs showing that the JMML mutations are highly clonal and can be traced back to the HSC compartment. Fig. S4 shows representative flow cytometry examples of the PB and BM analysis in NSG mice transplanted with JMML LSCs, the hematological parameters of mice transplanted with HSCs, histopathology sections of the spleen of JMML and CB HSC recipients, survival curves for mice that received a JMML +/+ population, and JMML GMP and spleen size of all recipients. Fig. S5 contains flow cytometry analysis of CD96 expression on Lin-CD34+CD38-CD90+CD45RA+ (+/+), mature hematopoietic compartments, and other MPNs. Table S1 contains the clinical characteristics of the patients included in the study. Table S2 shows the primer details used to design the targeted NGS panel. Table S3 includes the primer details used for colony and single-cell genotyping. Table S4 includes all key resources and reagents used.
Acknowledgments
The authors thank all patients, their families, and the UK National Institute of Health Research (NIHR) Pediatric MDS/JMML recruiting centers for contributing samples for this research and the Department of Biological and Medical Services Oxford University, for technical assistance. The authors thank Professor Matthias Peipp, University of Kiel, Kiel, Germany, for providing the CD96 antibody.
This work was funded by a Medical Research Council (MRC) Senior Clinical Fellowship (MR/L006340/1) and a Cancer Research UK Senior Cancer Research Fellowship to A.J. Mead; an NIHR Fellowship to E. Louka; an MRC-funded Oxford Consortium for Single-cell Biology grant (MR/M00919X/1); and the NIHR Oxford Biomedical Research Centre based at Oxford University Hospitals NHS Trust (I. Roberts and A.J. Mead) and University of Oxford. A. Roy is supported by a Bloodwise Clinician Scientist Fellowship (17001), N. Fordham by a Kay Kendall Leukaemia Fund Fellowship (KKL1124), and D. Iskander by a Bloodwise Clinical Research Fellowship. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health. The authors acknowledge the contributions of the Weatherall Institute of Molecular Medicine Flow Cytometry and Single Cell Facilities, supported by the MRC Human Immunology and Molecular Haematology Units; MRC Molecular Haematology Unit (MC_UU_12009); John Fell Fund (131/030 and 101/517); the E.P. Abraham fund (CF182 and CF170); and Weatherall Institute of Molecular Medicine Strategic Alliance awards (G0902418 and MC_UU_12025).
Author contributions: E. Louka and B. Povinelli performed experiments, analyzed the data, and helped to write the manuscript. A. Rodriguez-Meira performed experiments and analyzed data. A. Hamblin, R. Norfo, and A. Roy analyzed and interpreted data. G. Buck, N. Ashley, C.A.G. Booth, N. Elliott, D. Iskander, N. Sousos, N. Fordham, M. Salio, and S. O’Bryne conducted experiments and analyzed data. G. Wang, S. Thongjuea, and W.X. Wen performed bioinformatic analysis. J. de la Fuente and S. Inglott provided patient samples. A. Rao conducted the clinical study, helped to advise on experimental design, and provided patient samples. I. Roberts and A.J. Mead conceived, designed, and supervised the research, analyzed the data, and wrote the manuscript.

