


Osteoporosis is caused by an imbalance of osteoclasts and osteoblasts, occurring in close proximity to hematopoietic cells in the bone marrow. Recurrent somatic mutations that lead to an expanded population of mutant blood cells is termed clonal hematopoiesis of indeterminate potential (CHIP). Analyzing exome sequencing data from the UK Biobank, we found CHIP to be associated with increased incident osteoporosis diagnoses and decreased bone mineral density. In murine models, hematopoietic-specific mutations in Dnmt3a, the most commonly mutated gene in CHIP, decreased bone mass via increased osteoclastogenesis. Dnmt3a−/− demethylation opened chromatin and altered activity of inflammatory transcription factors. Bone loss was driven by proinflammatory cytokines, including Irf3-NF-κB–mediated IL-20 expression from Dnmt3a mutant macrophages. Increased osteoclastogenesis due to the Dnmt3a mutations was ameliorated by alendronate or IL-20 neutralization. These results demonstrate a novel source of osteoporosis-inducing inflammation.
Introduction
Osteoporosis is an age-associated condition arising from an altered balance between the activity of osteoclasts that resorb bone and osteoblasts that build bone. A proinflammatory bone marrow (BM) microenvironment has been implicated in the pathophysiology of bone loss. For example, increased adipogenesis and inflammation from fatty acid excess is thought to contribute to impaired osteoblast differentiation from skeletal progenitors (Ambrosi et al., 2017; Elbaz et al., 2010; Yu et al., 2018). In addition, DNA damage induces cellular senescence in multiple cell types, including myeloid cells and osteocytes, resulting in the release of inflammatory molecules that promote osteoclastogenesis (Farr et al., 2017). Numerous immune-related cytokines critically regulate osteoclasts and osteoblasts (Ponzetti and Rucci, 2019), but mechanisms potentiating the expression and activity of these proinflammatory cytokines are less understood.
Recent studies have revealed that clonal somatic mutations in genes mutated in myeloid malignancies are detectable in hematopoietic cells at increasing prevalence with age (Busque et al., 2012; Genovese et al., 2014; Jaiswal et al., 2014; Xie et al., 2014). When such a mutation is present at a variant allele fraction (VAF) of at least 2% in the blood in an individual without a hematologic malignancy, this state has been termed clonal hematopoiesis of indeterminant potential (CHIP; Steensma et al., 2015). Individuals with CHIP have a higher incidence of hematological malignancy and coronary artery disease (Fuster et al., 2017; Genovese et al., 2014; Jaiswal et al., 2014; Jaiswal et al., 2017). CHIP mutations in macrophages increase the expression of proinflammatory cytokines (Fuster et al., 2017; Jaiswal et al., 2017). In an atherogenic murine model of Tet2-mediated CHIP, pharmacological inhibition of the NLRP3 inflammasome reduced IL-1 and reversed the effects of Tet2 loss on atherosclerosis (Fuster et al., 2017).
Mutant hematopoietic cells have the potential to alter osteoclast function via both cell-autonomous and nonautonomous mechanisms. Osteoclasts are myeloid lineage cells that are replenished from hematopoietic stem cells (HSCs; Jacome-Galarza et al., 2019), and somatic mutations in HSCs may influence osteoclast function via incorporation of nuclei bearing somatic mutations during osteoclast differentiation. Alternatively, since osteoclasts respond to cytokines released locally in the BM, CHIP may alter osteoclast function through elaboration of cytokines by mutant myeloid cells. We therefore examined whether CHIP is causally associated with osteoporosis using a combination of human genetic studies and murine models.
Results and discussion
Association between osteoporosis and somatic mutations in the UK Biobank
To examine whether CHIP is associated with osteoporosis, we analyzed the UK Biobank, a large population-based prospective cohort, for osteoporosis diagnoses. From this cohort, we included 113,641 unrelated individuals of European ancestry with available whole-exome sequencing without a diagnosis of a malignant neoplasm at the time of blood sample collection. We identified 6,097 individuals (5.7%) with somatic mutations in previously identified CHIP genes (Jaiswal et al., 2014; Fig. S1 A). Baseline characteristics were compared between individuals with and without CHIP for age, sex, body mass index (BMI), self-reported oral corticosteroid use, history of smoking, and rheumatoid arthritis as osteoporosis risk factors previously used in studies of bone mineral density and incorporated into the Fracture Risk Assessment Tool (Gourlay et al., 2012; Kanis et al., 2008). As previously reported, CHIP was associated with older age and history of smoking (Genovese et al., 2014; Jaiswal et al., 2014; Table S1).
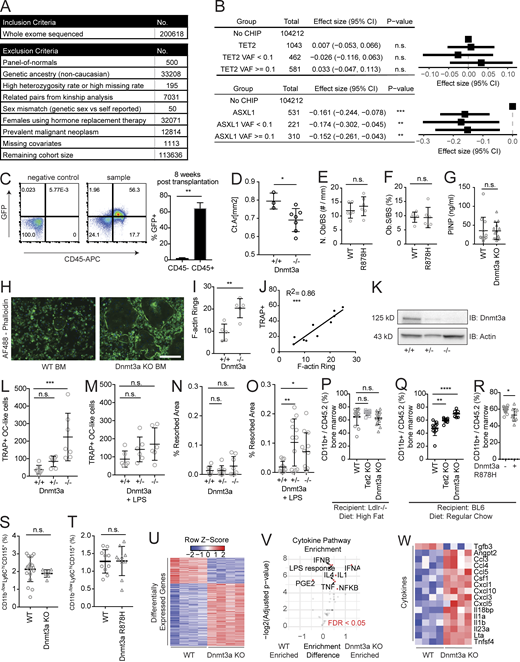
UK Biobank cohort and mouse models of CHIP. (A) Cohort selection and exclusion criteria from 200,000 individuals from the UK Biobank who had whole-exome sequencing performed. (B) Forest plot of the β-estimates for eBMD generated from multivariate linear regression model using CHIP mutations stratified by VAF ≥10% or <10%, and adjusted for age ≥65 yr, sex, BMI <18.5 kg/m2 (underweight), BMI >30 kg/m2 (obese), prior or current smoking status, self-reported history of steroid use, and rheumatoid arthritis diagnosis. The horizontal lines represent Wald 95% CIs. (C) Analysis of cells reconstituted after BM transplantation. BM from mouse with constitutive expression of GFP under a CAG promoter (JAX 024858) was transplanted into WT recipient mice. BM was analyzed at 8 wk after transplantation for CD45+ and GFP+. Most of the GFP positivity is retained in the CD45+ hematopoietic population. On the right is a quantification of GFP percentage in the hematopoietic (CD45+) and nonhematopoietic (CD45−) populations. n = 5. (D) Influence of Dnmt3a KO on bone mass in a nontransplant setting. Female WT (Vav1-Cre) or Dnmt3a KO (Dnmt3afl/flVav1-Cre) mice were aged 33–34 wk and analyzed on µCT for the femoral mid-shaft Ct Ar. n = 3–7. (E and F) Tibias of transplanted WT or Dnmt3a R878H mice were stained with Goldener’s Trichrome stain 20 wk after transplantation. n = 6. Samples were analyzed in a blinded fashion for Ob.N/BS (E) and Ob.S/BS (F) by the Center for Skeletal Research at Massachusetts General Hospital. (G) Procollagen 1 N-terminal propeptide (P1NP) serum levels in mice transplanted with WT or Dnmt3a KO BM into 8– to 10-wk-old Ldlr KO mice fed an HFD. ELISA analysis of serum obtained at 4 mo after transplantation. n = 11–14. (H) Representative images of F-actin rings marking osteoclasts shown by phalloidin immunofluorescence staining. White bar represents 100 µm. Green represents Alex Fluor 488–phalloidin. Blue represents DAPI staining. (I) Quantification of osteoclast differentiation from whole BM of 12-wk-old Dnmt3a KO and WT female mice. n = 5. (J) Correlation between TRAP quantification for osteoclasts and F-actin ring staining with simultaneous staining for TRAP and F-actin rings. (K) Immunoblot (IB) for Dnmt3a in RAW264.7 cells with heterozygous (+/−) or homozygous (−/−) frame-shift mutations in Dnmt3a. (L and M) Quantification of number of osteoclast-like cells from osteoclast differentiation of Dnmt3a +/+, +/−, −/− RAW264.7 cells. (L and M) Osteoclast differentiation was performed using vehicle treatment (L; n = 7) or LPS (M; 0.1 ng/ml; n = 6). (N and O) Quantitative hydroxyapatite resorption assay using osteoclast differentiation of Dnmt3a +/+, +/−, and −/− RAW264.7 cells. Osteoclast differentiation was performed using vehicle treatment (N; n = 7) or LPS (O; 0.1 ng/ml; n = 7). Resorbed areas were distinguished by image thresholding using ImageJ software. Each data plot represents average resorbed areas across all nonoverlapping images taken in a well on day 7 of differentiation. Error bars represent SEM. (P) Quantification of donor CD11b+ in the Dnmt3a KO and WT BM of transplants into Ldlr−/− recipient mice fed a HFD. n = 10–14. (Q) Quantification of donor CD11b+ in the Dnmt3a KO and WT BM of transplants into WT mice fed a ND. n = 7–10. (R) Quantification of donor CD11b+ in the Dnmt3a R878H and WT BM of transplants into WT mice fed a ND. n = 10–15. (S) Quantification of CD11b−/lowCD115+Ly6Chi osteoclast precursors in the Dnmt3a KO and WT BM of transplants into WT mice fed a ND. n = 7–19. (T) Quantification of CD11b−/lowCD115+Ly6Chi osteoclast precursors in the Dnmt3a R878H and WT BM of transplants into WT mice fed a ND. n = 10–15. (U–W) RNA sequencing of unstimulated BMDMs derived from WT or Dnmt3a−/− mice. n = 4. (U) Differentially expressed genes from all genes. (V) Cytokine pathway enrichment analysis using gene set variation analysis (Hänzelmann et al., 2013). Highlighted pathways indicate FDR < 0.05. (W) Significant differentially expressed cytokines/chemokines genes. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. All error bars represent SD unless specified. Source data are available for this figure: SourceData FS1.
UK Biobank cohort and mouse models of CHIP. (A) Cohort selection and exclusion criteria from 200,000 individuals from the UK Biobank who had whole-exome sequencing performed. (B) Forest plot of the β-estimates for eBMD generated from multivariate linear regression model using CHIP mutations stratified by VAF ≥10% or <10%, and adjusted for age ≥65 yr, sex, BMI <18.5 kg/m2 (underweight), BMI >30 kg/m2 (obese), prior or current smoking status, self-reported history of steroid use, and rheumatoid arthritis diagnosis. The horizontal lines represent Wald 95% CIs. (C) Analysis of cells reconstituted after BM transplantation. BM from mouse with constitutive expression of GFP under a CAG promoter (JAX 024858) was transplanted into WT recipient mice. BM was analyzed at 8 wk after transplantation for CD45+ and GFP+. Most of the GFP positivity is retained in the CD45+ hematopoietic population. On the right is a quantification of GFP percentage in the hematopoietic (CD45+) and nonhematopoietic (CD45−) populations. n = 5. (D) Influence of Dnmt3a KO on bone mass in a nontransplant setting. Female WT (Vav1-Cre) or Dnmt3a KO (Dnmt3afl/flVav1-Cre) mice were aged 33–34 wk and analyzed on µCT for the femoral mid-shaft Ct Ar. n = 3–7. (E and F) Tibias of transplanted WT or Dnmt3a R878H mice were stained with Goldener’s Trichrome stain 20 wk after transplantation. n = 6. Samples were analyzed in a blinded fashion for Ob.N/BS (E) and Ob.S/BS (F) by the Center for Skeletal Research at Massachusetts General Hospital. (G) Procollagen 1 N-terminal propeptide (P1NP) serum levels in mice transplanted with WT or Dnmt3a KO BM into 8– to 10-wk-old Ldlr KO mice fed an HFD. ELISA analysis of serum obtained at 4 mo after transplantation. n = 11–14. (H) Representative images of F-actin rings marking osteoclasts shown by phalloidin immunofluorescence staining. White bar represents 100 µm. Green represents Alex Fluor 488–phalloidin. Blue represents DAPI staining. (I) Quantification of osteoclast differentiation from whole BM of 12-wk-old Dnmt3a KO and WT female mice. n = 5. (J) Correlation between TRAP quantification for osteoclasts and F-actin ring staining with simultaneous staining for TRAP and F-actin rings. (K) Immunoblot (IB) for Dnmt3a in RAW264.7 cells with heterozygous (+/−) or homozygous (−/−) frame-shift mutations in Dnmt3a. (L and M) Quantification of number of osteoclast-like cells from osteoclast differentiation of Dnmt3a +/+, +/−, −/− RAW264.7 cells. (L and M) Osteoclast differentiation was performed using vehicle treatment (L; n = 7) or LPS (M; 0.1 ng/ml; n = 6). (N and O) Quantitative hydroxyapatite resorption assay using osteoclast differentiation of Dnmt3a +/+, +/−, and −/− RAW264.7 cells. Osteoclast differentiation was performed using vehicle treatment (N; n = 7) or LPS (O; 0.1 ng/ml; n = 7). Resorbed areas were distinguished by image thresholding using ImageJ software. Each data plot represents average resorbed areas across all nonoverlapping images taken in a well on day 7 of differentiation. Error bars represent SEM. (P) Quantification of donor CD11b+ in the Dnmt3a KO and WT BM of transplants into Ldlr−/− recipient mice fed a HFD. n = 10–14. (Q) Quantification of donor CD11b+ in the Dnmt3a KO and WT BM of transplants into WT mice fed a ND. n = 7–10. (R) Quantification of donor CD11b+ in the Dnmt3a R878H and WT BM of transplants into WT mice fed a ND. n = 10–15. (S) Quantification of CD11b−/lowCD115+Ly6Chi osteoclast precursors in the Dnmt3a KO and WT BM of transplants into WT mice fed a ND. n = 7–19. (T) Quantification of CD11b−/lowCD115+Ly6Chi osteoclast precursors in the Dnmt3a R878H and WT BM of transplants into WT mice fed a ND. n = 10–15. (U–W) RNA sequencing of unstimulated BMDMs derived from WT or Dnmt3a−/− mice. n = 4. (U) Differentially expressed genes from all genes. (V) Cytokine pathway enrichment analysis using gene set variation analysis (Hänzelmann et al., 2013). Highlighted pathways indicate FDR < 0.05. (W) Significant differentially expressed cytokines/chemokines genes. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. All error bars represent SD unless specified. Source data are available for this figure: SourceData FS1.
In a univariate analysis, CHIP was associated with an increased incidence of osteoporosis diagnoses, with a median follow up of 10.7 yr (Fig. 1 A). After adjusting for osteoporosis risk factors, the risk of incident osteoporosis in individuals with CHIP was 1.44 times (95% confidence interval [CI], 1.22–1.72; P = 3e-05) that of non-CHIP (Fig. 1 B).

Increased risk for osteoporosis in humans and murine models with CHIP. (A) Cumulative incidence of osteoporosis diagnoses by ICD based on CHIP status. Individuals were censored at the time of osteoporosis diagnosis, the end of follow up, or at the time of death or malignant neoplasm diagnosis as competing risks. (B) Forest plot of hazard ratios (HR) for the association between incident ICD osteoporosis diagnoses and the presence of CHIP stratified by VAF ≥10% or <10% in the Cox proportional hazard model, adjusted for age ≥65 yr, sex, BMI <18.5 kg/m2 (underweight), BMI >30 kg/m2 (obesity), history of smoking, oral corticosteroid use, and rheumatoid arthritis. The horizontal lines represent Wald 95% CIs. (C) Forest plot of the β-estimates for eBMD generated from multivariate linear regression model using CHIP or DNMT3A mutations stratified by VAF ≥10% or <10% and osteoporosis risk covariates as above. The horizontal lines represent Wald 95% CIs. (D–G)Tet2fl/flVav1-Cre (Tet2−/−), Dnmt3afl/flVav1-Cre (Dnmt3a−/−), and Vav1-Cre (WT) BM transplants into WT male mice and sacrificed at 20 wk after transplantation for blinded µCT analysis. To assess statistical significance, one-way ANOVA was used against WT, and P values were adjusted for multiple comparisons using the two-stage linear step-up procedure of BY. Error bars represent SD. (D and E) BM transplants into WT recipient mice fed a ND. µCT analysis for Tb BV/TV (D) and femoral mid-shaft Ct Ar (E). n = 10–14. (F and G) BM transplants into Ldlr−/− recipient mice fed HFD 4-wk after transplantation. µCT analysis for F) Tb BV/TV and G) Ct Ar. n = 9–19. (H) Representative sagittal µCT images of the distal femur in Tet2−/−, Dnmt3a−/−, and WT BM transplants into Ldlr−/− recipients fed a HFD (top row) or WT recipients fed a ND (bottom row). Scale bar (bottom right), 1 mm. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Increased risk for osteoporosis in humans and murine models with CHIP. (A) Cumulative incidence of osteoporosis diagnoses by ICD based on CHIP status. Individuals were censored at the time of osteoporosis diagnosis, the end of follow up, or at the time of death or malignant neoplasm diagnosis as competing risks. (B) Forest plot of hazard ratios (HR) for the association between incident ICD osteoporosis diagnoses and the presence of CHIP stratified by VAF ≥10% or <10% in the Cox proportional hazard model, adjusted for age ≥65 yr, sex, BMI <18.5 kg/m2 (underweight), BMI >30 kg/m2 (obesity), history of smoking, oral corticosteroid use, and rheumatoid arthritis. The horizontal lines represent Wald 95% CIs. (C) Forest plot of the β-estimates for eBMD generated from multivariate linear regression model using CHIP or DNMT3A mutations stratified by VAF ≥10% or <10% and osteoporosis risk covariates as above. The horizontal lines represent Wald 95% CIs. (D–G)Tet2fl/flVav1-Cre (Tet2−/−), Dnmt3afl/flVav1-Cre (Dnmt3a−/−), and Vav1-Cre (WT) BM transplants into WT male mice and sacrificed at 20 wk after transplantation for blinded µCT analysis. To assess statistical significance, one-way ANOVA was used against WT, and P values were adjusted for multiple comparisons using the two-stage linear step-up procedure of BY. Error bars represent SD. (D and E) BM transplants into WT recipient mice fed a ND. µCT analysis for Tb BV/TV (D) and femoral mid-shaft Ct Ar (E). n = 10–14. (F and G) BM transplants into Ldlr−/− recipient mice fed HFD 4-wk after transplantation. µCT analysis for F) Tb BV/TV and G) Ct Ar. n = 9–19. (H) Representative sagittal µCT images of the distal femur in Tet2−/−, Dnmt3a−/−, and WT BM transplants into Ldlr−/− recipients fed a HFD (top row) or WT recipients fed a ND (bottom row). Scale bar (bottom right), 1 mm. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
To investigate the effect of CHIP on bone mineral density, we examined data from quantitative heel ultrasounds, which were performed on individuals in the UK Biobank cohort. Estimated bone mineral density (eBMD) was derived from the heel ultrasound data, as established previously as a reliable predictor of osteoporosis (Kemp et al., 2017). Larger CHIP clones, especially in DNMT3A, with VAF ≥10% were significant correlated with lower eBMD, consistent with accelerated bone loss (Fig. 1 C and Fig. S1 B). These data indicate that CHIP mutations are associated with decreased bone mineral density and an increased risk of osteoporosis.
Murine models of CHIP have decreased trabecular and cortical bone mass
To examine whether the association of CHIP and osteoporosis is causal, we assessed the impact of mutated blood cells on bone morphology using murine models with hematopoietic-specific inactivation of Dnmt3a or Tet2, the most commonly mutated genes in CHIP. We performed BM transplantations from mice with hematopoietic-specific Dnmt3a KO (Dnmt3afl/flVav1-Cre, referred to as Dnmt3a−/−), Tet2 KO (Tet2fl/flVav1-Cre, referred to as Tet2−/−), or WT (Vav1-Cre) mice into WT recipient mice and assessed the effect on bone mass of the recipients (Fig. S1 C). Mice were aged for 20 wk after transplantation before analysis to allow for the replacement of host osteoclasts (Jacome-Galarza et al., 2019).
To assess mutation-associated changes in bone mass, the femurs of transplanted mice were isolated, imaged using micro–computed tomography (µCT), and analyzed for bone density by a blinded reviewer. Mice with either Dnmt3a−/− or Tet2−/− BM transplants had significant reductions in trabecular bone volume relative to total volume (Tb BV/TV) of the femoral metaphysis, as well as cortical area (Ct Ar) of the femoral mid-shaft, consistent with an overall reduction in bone mass (Fig. 1, D and E; and Table S2).
Studies of the effect of hematopoietic-specific Tet2 inactivation on cardiovascular disease have demonstrated that atherosclerosis-susceptible Ldlr−/− mice fed a high-fat diet (HFD) increases inflammatory cytokine levels released from Tet2-deficient macrophages (Fuster et al., 2017; Jaiswal et al., 2017). Since the elevated cholesterol levels in the Ldlr−/− mice increase systemic inflammation (Bieghs et al., 2012), and inflammatory cytokines influence osteoclast and osteoblast activity (Ponzetti and Rucci, 2019), we used this model to study the effect of CHIP mutations on bone density in a proinflammatory context. Similar to our studies in WT recipient mice fed a normal diet (ND), Dnmt3a−/− BM transplants into Ldlr−/− mice fed a HFD had significant reductions in Tb BV/TV and Ct Ar compared with WT and Tet2−/− BM transplants, which trended lower on both measures (Fig. 1, F–H).
Given the strong effects on bone phenotype in mice with hematopoietic-specific inactivation of Dnmt3a, we focused further studies on this model. To exclude the effects of gamma irradiation used in the BM transplant model, we assessed bone mass in mice with specific inactivation of Dnmt3a in hematopoietic cells using Vav1-Cre. Dnmt3a−/− decreased Ct Ar compared with WT (Fig. S1 D). Taken together, Dnmt3a inactivation in blood cells is sufficient to lower bone mass in mice.
Increased osteoclast activity in vivo mediated by Dnmt3a mutations
Our results from human genetic studies and murine models suggest that somatic hematopoietic DNMT3A mutations are associated with lower bone mass and can contribute causally to the pathogenesis of osteoporosis. Mechanistically, these findings could be due to increased osteoclast activity or decreased osteoblast activity. To evaluate osteoclast numbers, we examined histological sections from the distal femoral metaphysis of mice with Dnmt3a−/− or WT BM transplants. We observed an increased number of osteoclasts on bone surfaces (N. Oc/BS) by tartrate-resistant acid phosphatase (TRAP)–positive stain in Dnmt3a−/− compared with WT BM transplants mice in both Ldlr−/− and WT recipient mice (Fig. 2, A–C). Similar results were obtained using Dnmt3afl-R878H/+ crossed to Vav1-Cre (referred to here as Dnmt3aR878H; Loberg et al., 2019), which models the most common heterozygous CHIP mutation in DNMT3A, occurring in residue R882 (Fig. 2 D).

Dnmt3a-mutated myeloid cells increase osteoclastogenesis. (A) Representative sections of the distal femur in WT or Dnmt3a−/− BM transplants into Ldlr−/− recipients fed an HFD. For A–D, mice were sacrificed at 20 wk after transplantation, and sections were stained for TRAP (red). Scale bar (bottom right), 200 µm. (B and C) Quantification of N. Oc/BS in the distal femur in BM transplants with Ldlr−/− recipients fed a HFD (B; n = 13–14) and WT recipients fed an ND (C; n = 9–19). (D) Quantification of N. Oc/BS in the tibias of WT recipients fed an ND with WT or Dnmt3aR878H BM transplants (n = 6). (E and F) Rescue of decline in cortical bone mass in Dnmt3aR878H mice with alendronate. Dnmt3aR878H or WT BM transplants into WT recipients fed an ND. Mice were treated with subcutaneous injections of alendronate or vehicle twice a week for 7 wk starting 14 wk after transplantation. Mice were sacrificed 20 wk after transplantation for µCT analysis of the femur for Ct Ar (E) and ultimate moment using mechanical testing (F). Changes in Tb BV/TV of the femoral metaphysis were not significant between WT and Dnmt3aR878H transplants. n = 10–15. P values were determined via comparison to vehicle-treated Dnmt3aR878H and adjusted for multiple comparisons using BY. P = 0.08 for comparison between WT and Dnmt3a R878H on mechanical testing. (G) Osteoclast differentiation ex vivo using whole BM cells from 12–to 15-wk-old male mice from Dnmt3a+/+Vav1-Cre (WT), Dnmt3afl/+Vav1-Cre (+/−), and Dnmt3afl/flVav1-Cre (−/−) backgrounds (n = 7). Osteoclast differentiation was performed in the presence of vehicle or LPS (0.1 ng/ml). Statistical significance was determined via two-way ANOVA (P < 0.0001 for genotype-specific effect), and P values were adjusted for multiple comparisons using BY. Error bars represent SEM. (H) BM-sorted 500 CD11b−/lowCD115+Ly6Chi osteoclast precursors differentiated from WT and Dnmt3a−/− BM were quantified via TRAP staining on day 6 of culture. Osteoclast precursors were plated in 10 ng/ml M-CSF and increasing concentrations of RANKL (n = 7), 10 ng/ml RANKL and increasing concentrations of M-CSF (n = 8), or RANKL and M-CSF at 10 ng/ml with increasing concentrations of LPS. Statistical significance was assessed by two-way ANOVA. No significance differences are observed between WT and Dnmt3a−/−. (I) Osteoclast differentiation from 200 WT osteoclast precursors in the Transwell assays in the presence of 20,000 WT or Dnmt3a KO cells in the upper compartment. Osteoclasts are assessed by phalloidin-FITC for F-actin rings. Statistical significance was determined via two-way ANOVA (P < 0.0001 for genotype-specific effect), and P values were adjusted for multiple comparisons using BY. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. All error bars represent SD unless specified.
Dnmt3a-mutated myeloid cells increase osteoclastogenesis. (A) Representative sections of the distal femur in WT or Dnmt3a−/− BM transplants into Ldlr−/− recipients fed an HFD. For A–D, mice were sacrificed at 20 wk after transplantation, and sections were stained for TRAP (red). Scale bar (bottom right), 200 µm. (B and C) Quantification of N. Oc/BS in the distal femur in BM transplants with Ldlr−/− recipients fed a HFD (B; n = 13–14) and WT recipients fed an ND (C; n = 9–19). (D) Quantification of N. Oc/BS in the tibias of WT recipients fed an ND with WT or Dnmt3aR878H BM transplants (n = 6). (E and F) Rescue of decline in cortical bone mass in Dnmt3aR878H mice with alendronate. Dnmt3aR878H or WT BM transplants into WT recipients fed an ND. Mice were treated with subcutaneous injections of alendronate or vehicle twice a week for 7 wk starting 14 wk after transplantation. Mice were sacrificed 20 wk after transplantation for µCT analysis of the femur for Ct Ar (E) and ultimate moment using mechanical testing (F). Changes in Tb BV/TV of the femoral metaphysis were not significant between WT and Dnmt3aR878H transplants. n = 10–15. P values were determined via comparison to vehicle-treated Dnmt3aR878H and adjusted for multiple comparisons using BY. P = 0.08 for comparison between WT and Dnmt3a R878H on mechanical testing. (G) Osteoclast differentiation ex vivo using whole BM cells from 12–to 15-wk-old male mice from Dnmt3a+/+Vav1-Cre (WT), Dnmt3afl/+Vav1-Cre (+/−), and Dnmt3afl/flVav1-Cre (−/−) backgrounds (n = 7). Osteoclast differentiation was performed in the presence of vehicle or LPS (0.1 ng/ml). Statistical significance was determined via two-way ANOVA (P < 0.0001 for genotype-specific effect), and P values were adjusted for multiple comparisons using BY. Error bars represent SEM. (H) BM-sorted 500 CD11b−/lowCD115+Ly6Chi osteoclast precursors differentiated from WT and Dnmt3a−/− BM were quantified via TRAP staining on day 6 of culture. Osteoclast precursors were plated in 10 ng/ml M-CSF and increasing concentrations of RANKL (n = 7), 10 ng/ml RANKL and increasing concentrations of M-CSF (n = 8), or RANKL and M-CSF at 10 ng/ml with increasing concentrations of LPS. Statistical significance was assessed by two-way ANOVA. No significance differences are observed between WT and Dnmt3a−/−. (I) Osteoclast differentiation from 200 WT osteoclast precursors in the Transwell assays in the presence of 20,000 WT or Dnmt3a KO cells in the upper compartment. Osteoclasts are assessed by phalloidin-FITC for F-actin rings. Statistical significance was determined via two-way ANOVA (P < 0.0001 for genotype-specific effect), and P values were adjusted for multiple comparisons using BY. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. All error bars represent SD unless specified.
We next examined the effect of Dnmt3a mutations on osteoblast number and activity. On histology, osteoblast number and surface per bone surface (Ob.N/BS and Ob.S/BS) were not significantly different between WT and Dnmt3aR878H mice (Fig. S1, E and F). To evaluate changes in osteoblast activity, we tested the serum for bone formation byproduct P1NP and found no differences between WT and Dnmt3a−/− mice (Fig. S1 G). The results suggest that the predominant effect of hematopoietic Dnmt3a mutations on bone mass is mediated through increased osteoclasts.
Alendronate reverses lower bone mass caused by Dnmt3a mutations
If Dnmt3a mutations cause bone loss through increased osteoclasts, then pharmacologic inhibition of osteoclasts should rescue the effect of Dnmt3a mutations. Bisphosphonates such as alendronate incorporate into areas of active bone remodeling and inhibit osteoclasts (Drake et al., 2008). We transplanted Dnmt3aR878H or WT hematopoietic cells into WT recipient mice and treated with alendronate for 7 wk. Alendronate administration fully rescued the decrease in Ct Ar observed in Dnmt3aR878H mice (Fig. 2 E). We tested the functional effect of the alendronate intervention via destructive biomechanical testing. We found that femoral bending strength was significantly improved in alendronate-treated Dnmt3aR878H mice compared with vehicle-treated Dnmt3aR878H mice (Fig. 2 F). The histological analyses and alendronate experiments highlight the importance of osteoclasts in Dnmt3a-mediated decline in bone mass in vivo.
Inflammatory Dnmt3a-mutated myeloid cells increase osteoclast production in vitro
Mutations in the hematopoietic cells could influence osteoclasts through two potential mechanisms: Dnmt3a−/− monocytes could incorporate into multinucleated osteoclasts and alter function in a cell-autonomous manner (Jacome-Galarza et al., 2019), or Dnmt3a−/− hematopoietic cells may express molecules that influence osteoclast function in a non–cell-autonomous fashion. We sought to address these possibilities using ex vivo differentiation of WT or Dnmt3a-mutant whole BM cells into osteoclasts. Hematopoietic-specific Dnmt3a KO increased the number of osteoclasts (Fig. 2 G; and Fig. S1, H and I). This effect was magnified by inflammatory molecules such as LPS (Fig. 2 G).
To confirm the effect of Dnmt3a in in vitro models, we used immortalized RAW264.7 macrophages, generating isogenic Dnmt3a+/− and Dnmt3a−/− lines via CRISPR-Cas9 (Fig. S1 K). Differentiation of Dnmt3a−/− cell line via RANKL resulted in an increase in the number of TRAP+ osteoclast-like cells compared with WT (Fig. S1, L and M). To confirm hydroxyapatite resorption by osteoclast-like cells, we repeated prior differentiation experiments and assessed the resorbed areas. Similar to ex vivo experiments (Fig. 2 G), the Dnmt3a−/− cells increased hydroxyapatite resorption in the presence of LPS relative to WT cells (Fig. S1, N and O). Overall, these results confirm that Dnmt3a mutations increase osteoclast number and function, particularly in the presence of inflammatory stimuli.
During osteoclast differentiation of whole BM cells or RAW264.7 cells in vitro, a portion of cells are myeloid hematopoietic cells that do not become osteoclasts, and these cells can influence the differentiation of osteoclasts. Our in vitro findings could therefore be explained either by mutations causing intrinsic differences in differentiation potential of osteoclast precursors or by exogenous factors from mutant myeloid cells that alter osteoclast differentiation and function. We first examined whether Dnmt3a−/− causes intrinsic differences in differentiation of CD11b−/lowLy6ChiCD115+ BM osteoclast precursors (Charles et al., 2012), but the differentiating potential was equivalent (Fig. 2 H), and the percentage of osteoclast precursors was unchanged (Fig. S1, P–T). Taken together, these findings indicate that the cell-intrinsic differentiation potential and response to inflammatory stimuli of Dnmt3a−/− osteoclast precursors are not significantly different than that of WT osteoclast precursors.
We then examined whether mutant myeloid cells influence osteoclast differentiation. We purified WT osteoclast precursors and induced differentiation into osteoclasts in the presence of sorted WT or Dnmt3a−/− CD11b+ myeloid cells, which include macrophages, monocytes, neutrophils, and natural killer cells. Using Transwell assays, which allow for the diffusion of molecules, but not cells, between two chambers, we found that the presence of Dnmt3a−/− CD11b+ myeloid cells increased the number of osteoclasts (F-actin ring+) compared with WT (Fig. 2 I). In contrast, sorted lymphoid cells (CD3+/B220+) or nonhematopoietic cells (CD45−) did not alter differentiation of osteoclast precursors (Fig. 2 I). These co-culture experiments indicate that Dnmt3a−/− myeloid cells, especially macrophages, alter osteoclast differentiation non–cell autonomously via secreting molecules that increase osteoclast differentiation.
To identify soluble factors released from Dnmt3a−/− macrophages that might drive differences in osteoclast differentiation, we performed RNA sequencing (RNA-seq) on unstimulated BM-derived macrophages (BMDMs) from WT and Dnmt3a−/− mice. Similar to prior studies on Tet2 (Fuster et al., 2017; Jaiswal et al., 2017), we observed increased expression of proinflammatory cytokines in Dnmt3a−/− BMDMs compared with WT BMDMs (Fig. S1, U–W; and Table S3). Overall, these results indicate that proinflammatory Dnmt3a−/− macrophages provide a mechanistic basis for increased osteoclast differentiation in Dnmt3a−/− mice.
CRISPR screen for regulators of Dnmt3a-mediated osteoclast differentiation
To gain insight into pathways in osteoclasts that may participate in sensing inflammatory signals from Dnmt3a-mutated myeloid cells, we performed a genome-wide CRISPR-Cas9 screen using Cas9-expressing RAW264.7 cells. First, to identify osteoclast-like cells during differentiation, we generated a reporter for cathepsin K (Ctsk), a protease used by osteoclasts for bone resorption, via CRISPR-Cas9 (Fig. 3 A). From a single-cell clone, we generated an isogenic Dnmt3a−/− cell line and verified that Dnmt3a−/− cells treated with RANKL increased Ctsk reporter expression compared with WT cells (Fig. 3 A, and Fig. S2 A).

CRISPR screen and cytokine array reveal increased IL-20 expression. (A) Generation of RAW264.7 Ctsk-T2A-R647 reporter cell line stably expressing SpCas9-P2A-GFP, and analysis of Ctsk expression in the APC channel by flow cytometry on day 5 of osteoclast differentiation. The percentage of the APC+ population is calculated from the total GFP+ population and shown as boxplots. n = 16. (B) CRISPR screen for genes regulating osteoclast differentiation in the Dnmt3a KO Ctsk reporter. sgRNA-targeted genes ranked (x axis) by log2-fold change of read counts (y axis). The top right indicates genes enriched in the population with lower Ctsk reporter expression (bottom APC 5%) over higher Ctsk reporter expression (top APC 5%). These genes potentially enhance or are required for osteoclast differentiation. FDR < 0.05 (red). Cytokine receptors identified in the higher APC-expressing populations (bottom left) are not shown to due to cell fusion occurring during osteoclast differentiation. (C) Cytokine array of Dnmt3a−/− versus WT serum. n = 13–14. Statistical significance via Wilcoxon rank sum test. FDR < 0.05 is denoted by a horizontal dotted line. (D) ELISA quantification of IL-20 from WT or Dnmt3a−/− BMDMs (n = 10) or RAW264.7 cells (n = 12) cultured at a density of 50,000/cm2 for 4 d. Error bars represent SD. *, P < 0.05; ***, P < 0.001.
CRISPR screen and cytokine array reveal increased IL-20 expression. (A) Generation of RAW264.7 Ctsk-T2A-R647 reporter cell line stably expressing SpCas9-P2A-GFP, and analysis of Ctsk expression in the APC channel by flow cytometry on day 5 of osteoclast differentiation. The percentage of the APC+ population is calculated from the total GFP+ population and shown as boxplots. n = 16. (B) CRISPR screen for genes regulating osteoclast differentiation in the Dnmt3a KO Ctsk reporter. sgRNA-targeted genes ranked (x axis) by log2-fold change of read counts (y axis). The top right indicates genes enriched in the population with lower Ctsk reporter expression (bottom APC 5%) over higher Ctsk reporter expression (top APC 5%). These genes potentially enhance or are required for osteoclast differentiation. FDR < 0.05 (red). Cytokine receptors identified in the higher APC-expressing populations (bottom left) are not shown to due to cell fusion occurring during osteoclast differentiation. (C) Cytokine array of Dnmt3a−/− versus WT serum. n = 13–14. Statistical significance via Wilcoxon rank sum test. FDR < 0.05 is denoted by a horizontal dotted line. (D) ELISA quantification of IL-20 from WT or Dnmt3a−/− BMDMs (n = 10) or RAW264.7 cells (n = 12) cultured at a density of 50,000/cm2 for 4 d. Error bars represent SD. *, P < 0.05; ***, P < 0.001.

CRISPR screen and identification of IL-20 as a target. (A) Immunoblot confirmation of Dnmt3a KO in the Ctsk-T2A-R647 reporter line expressing Cas9. (B) Experimental schema for the genome-wide CRISPR screen using the Brie library. (C) Normalized read counts for Tnfrsf11a and Il20ra. (D and E) Limited receptor-based screen in Dnmt3a KO (D) or WT (E) in the Ctsk-T2A-R647 reporter line. Red indicates genes with FDR < 0.05. The screen could not be performed with WT cells due to the low rate of baseline differentiation toward osteoclasts in the WT. (F and G) Serum levels of IL-20 (F) and fractalkine (G) in mice transplanted with WT, Dnmt3a KO, and Tet2 KO cells. (H–J) Effect of increasing concentrations of IL-11 (H), Lif (I), or fractalkine (J) on osteoclast differentiation from 200 CD11b−/lowCD115+Ly6Chi osteoclast precursors sorted from the BM of 12-wk-old Dnmt3a KO (n = 11), shown as boxplots. Data are normalized by the number of TRAP+ cells from the control arm. (K and L) ELISA quantification of IL-11 (K) or Lif (L) from unstimulated WT or Dnmt3a KO BMDMs or RAW264.7 cells cultured at a density of 50,000/cm2 for 4 d. n = 8 for BMDMs and n = 12 for RAW264.7 cells. (M) ELISA quantification of IL-1β from WT or Dnmt3a KO BMDMs cultured at a density of 50,000/cm2 for 4 d. A positive control using LPS and uric acid treatment is shown. n = 6–8. (O and P) Il20 expression in the context of Nlrp3fl/flVav1-Cre (referred to as Nlrp3 KO). (N)Il20 expression quantified by quantitative PCR. BMDMs were stimulated with 10 ng/ml LPS for 10 h. n = 4. Error bars represent SEM. (O) ELISA quantification of IL-20 of unstimulated BMDMs cultured for 5 d. n = 4. Error bars represent SD unless otherwise noted. Source data are available for this figure: SourceData FS2.
CRISPR screen and identification of IL-20 as a target. (A) Immunoblot confirmation of Dnmt3a KO in the Ctsk-T2A-R647 reporter line expressing Cas9. (B) Experimental schema for the genome-wide CRISPR screen using the Brie library. (C) Normalized read counts for Tnfrsf11a and Il20ra. (D and E) Limited receptor-based screen in Dnmt3a KO (D) or WT (E) in the Ctsk-T2A-R647 reporter line. Red indicates genes with FDR < 0.05. The screen could not be performed with WT cells due to the low rate of baseline differentiation toward osteoclasts in the WT. (F and G) Serum levels of IL-20 (F) and fractalkine (G) in mice transplanted with WT, Dnmt3a KO, and Tet2 KO cells. (H–J) Effect of increasing concentrations of IL-11 (H), Lif (I), or fractalkine (J) on osteoclast differentiation from 200 CD11b−/lowCD115+Ly6Chi osteoclast precursors sorted from the BM of 12-wk-old Dnmt3a KO (n = 11), shown as boxplots. Data are normalized by the number of TRAP+ cells from the control arm. (K and L) ELISA quantification of IL-11 (K) or Lif (L) from unstimulated WT or Dnmt3a KO BMDMs or RAW264.7 cells cultured at a density of 50,000/cm2 for 4 d. n = 8 for BMDMs and n = 12 for RAW264.7 cells. (M) ELISA quantification of IL-1β from WT or Dnmt3a KO BMDMs cultured at a density of 50,000/cm2 for 4 d. A positive control using LPS and uric acid treatment is shown. n = 6–8. (O and P) Il20 expression in the context of Nlrp3fl/flVav1-Cre (referred to as Nlrp3 KO). (N)Il20 expression quantified by quantitative PCR. BMDMs were stimulated with 10 ng/ml LPS for 10 h. n = 4. Error bars represent SEM. (O) ELISA quantification of IL-20 of unstimulated BMDMs cultured for 5 d. n = 4. Error bars represent SD unless otherwise noted. Source data are available for this figure: SourceData FS2.
To identify genes required for enhanced osteoclast differentiation in Dnmt3a−/− cells, we performed a genome-wide CRISPR screen during osteoclast differentiation of our Dnmt3a−/− reporter line (Fig. S2, B–E). Based on our findings implicating secreted inflammatory molecules in informing the Dnmt3a-mediated phenotype, we focused our analysis on the receptors in osteoclast-like cells that would reveal effects from surrounding cells, as osteoclast differentiation leaves a portion of the culture as undifferentiated myeloid cells capable of exerting paracrine effects. Single guide RNAs (sgRNAs) that reduce Ctsk reporter expression highlight genes that may promote osteoclast differentiation. As a positive control, we noted that Tnfrsf11a, the receptor for RANKL required for osteoclast differentiation, was significantly enriched in the population with lower Ctsk expression (Fig. 3 B). Other hits included Il20ra (IL-20 receptor), Cx3cr1 (fractalkine receptor), and Ltbr (lymphotoxin-β receptor). These results suggest that proinflammatory molecules IL-20, fractalkine and lymphotoxin-β may play a local role in osteoclast differentiation in the context of hematopoietic inactivation of Dnmt3a−/−.
Increased IL-20 expression in Dnmt3a KO
To discover whether circulating cytokines contribute to the Dnmt3a-mediated phenotype, we assayed 44 cytokines in the serum of Dnmt3a−/− and WT mice. The expression of IL-20 and fractalkine was increased specifically in Dnmt3a−/− mice compared with WT (Fig. 3 C; and Fig. S2, F and G), complementary to our CRISPR screen that highlighted the importance of these cytokine receptors on osteoclast-lineage cells (Fig. 3 B). Additional cytokines increased in Dnmt3a−/− serum relative to WT include IL-11 and leukemia inhibitory factor (Lif; Fig. 3 C). These cytokines have known roles in promoting osteoclast formation and migration (Bozec et al., 2008; Girasole et al., 1994; Hsu et al., 2011; Koizumi et al., 2009), which we confirmed using the differentiation of sorted osteoclast precursors (Fig. S2, H–J).
To determine whether the secretion of these cytokines promoting osteoclastogenesis is increased in Dnmt3a-mutated myeloid cells, we performed enzyme-linked immunosorbent assays from BMDM and RAW264.7 cultured medium. IL-20 levels were increased in Dnmt3a−/− compared with WT cultures in contrast to IL-11, Lif, or IL-1β (Fig. 3 D; and Fig. S2, K–M) and did not depend on the NLRP3 inflammasome (Fig. S2, N and O).
Dnmt3a loss causes differential Irf3-NF-κB signaling to increase Il20 expression
To investigate the mechanistic basis for the inflammatory phenotype of myeloid cells with loss of the DNA methyltransferase Dnmt3a, we performed reduced representation bisulfite sequencing (RRBS) on Dnmt3a−/− and WT BMDMs to assess methylation changes (Fig. S3, A–C). Concurrently, we performed assay for transposase-accessible chromatin (ATAC) sequencing to assess differences in chromatin accessibility between WT and Dnmt3a−/− BMDMs in the context of changes in methylation (Fig. S3, D–G). Consistent with prior studies of Dnmt3a−/− in other cell types (Yang et al., 2016), Dnmt3a−/− resulted in significant global hypomethylation of cytosine-guanine residues (CpGs) without a preference for promoters, CpG islands, or open chromatin regions globally (Fig. 4 A). Hypomethylation was enriched in large undermethylated regions termed “canyons,” as described previously for Dnmt3a−/− HSCs, progenitors, and leukemic cells (Jeong et al., 2014; Yang et al., 2016), but the most hypomethylated regions occurred in Dnmt3a−/−-specific open chromatin regions (Fig. 4, A and B). Open chromatin regions were increased in Dnmt3a−/− BMDMs compared with WT, primarily in intergenic and intronic regions (Fig. 4 C).

Chromatin analysis and investigation of DNMT3A rs6722613 SNP. (A) RRBS analysis of Dnmt3a−/− and WT BMDMs using cutoffs of FDR < 0.05 and differential methylation >20%. n = 5. Volcano density plot showing changes in CpG methylation (x axis) across all detected CpGs versus statistical significance (y axis). (B) Volcano plot showing changes in CpG methylation (x axis) in the gene body, promoter, and associated 5-kb flanking regions in all cytokine and chemokine ligands with statistical significance (y axis). CpG distribution across regions (right bar graph). (C) Methylation and gene expression changes. For each cytokine or chemokine gene, methylation analysis was performed over all CpGs in the gene body and 1-kb promoter region around transcriptional start sites (TSS). The top panel shows the percentage of significant hypomethylated gene-associated CpGs, the middle panel shows average changes in methylation across all gene-associated CpGs, and the bottom panel shows gene expression changes on mRNA sequencing. Filled bars show significant gene expression changes based on FDR < 0.05 regardless of fold change difference. Of all the cytokines/chemokines, only ones with CpG changes are shown. (D) KEGG pathway analysis for genes within 20 kb of LPS-treated Dnmt3a KO–specific ATAC peaks. This could not be performed for vehicle-treated Dnmt3a KO due to a lack of genes. (E) Venn diagram of peaks specific to LPS-treated Dnmt3a KO or vehicle-treated Dnmt3a, and peaks with genes annotated in Gene Ontology biological process or KEGG pathways are shown. Bold highlighted genes in Dnmt3a KO LPS are genes part of the TNF signaling pathway group in KEGG. P value indicates P value of overlap, as assessed by hypergeometric test. (F) ATAC peaks specific to Dnmt3a KO plus LPS were ranked by fold change relative to WT plus LPS. Peaks with proximal genes (within 20 kb) and log2(fold change [FC]) > 1 were included. Corresponding genes from RNA-seq analysis were included on the right, with significant genes FDR < 0.05 highlighted in red and labeled. (G) ATAC peaks specific to Dnmt3a KO plus vehicle versus WT plus vehicle were ranked in the same order as F. Corresponding genes from RNA-seq analysis were included on the right, with significant genes FDR < 0.05 highlighted in red and labeled. (H and I) ChIP-seq for Irf3 and Rela. (H) Venn diagrams of Irf3 and Rela ChIP-seq peaks with and without LPS administration (10 ng/ml). (I) Peak occupancy plots, sorted by Irf3 occupancy on the left. Rela peaks are displayed in the same order on the right. (J) KEGG pathway enrichment analysis for genes within 10 kb of Dnmt3a KO–specific peaks. Rela binding accounted for all inflammatory signaling pathway enrichment. (K) RNA-seq differential expression analysis of genes identified in J. Genes with FDR < 0.05 are highlighted with an asterisk. (L) ChIP PCR of Irf3 binding in the Il20 locus. n = 4–6. Statistical significance via Wilcoxon rank sum test. Error bars represent SEM. (M) Analysis of rs6722613 SNP across three large population studies. B, β; EA, effect allele; EAF, effect allele frequency; NEA, noneffect allele. *, P < 0.05; ****, P < 0.0001.
Chromatin analysis and investigation of DNMT3A rs6722613 SNP. (A) RRBS analysis of Dnmt3a−/− and WT BMDMs using cutoffs of FDR < 0.05 and differential methylation >20%. n = 5. Volcano density plot showing changes in CpG methylation (x axis) across all detected CpGs versus statistical significance (y axis). (B) Volcano plot showing changes in CpG methylation (x axis) in the gene body, promoter, and associated 5-kb flanking regions in all cytokine and chemokine ligands with statistical significance (y axis). CpG distribution across regions (right bar graph). (C) Methylation and gene expression changes. For each cytokine or chemokine gene, methylation analysis was performed over all CpGs in the gene body and 1-kb promoter region around transcriptional start sites (TSS). The top panel shows the percentage of significant hypomethylated gene-associated CpGs, the middle panel shows average changes in methylation across all gene-associated CpGs, and the bottom panel shows gene expression changes on mRNA sequencing. Filled bars show significant gene expression changes based on FDR < 0.05 regardless of fold change difference. Of all the cytokines/chemokines, only ones with CpG changes are shown. (D) KEGG pathway analysis for genes within 20 kb of LPS-treated Dnmt3a KO–specific ATAC peaks. This could not be performed for vehicle-treated Dnmt3a KO due to a lack of genes. (E) Venn diagram of peaks specific to LPS-treated Dnmt3a KO or vehicle-treated Dnmt3a, and peaks with genes annotated in Gene Ontology biological process or KEGG pathways are shown. Bold highlighted genes in Dnmt3a KO LPS are genes part of the TNF signaling pathway group in KEGG. P value indicates P value of overlap, as assessed by hypergeometric test. (F) ATAC peaks specific to Dnmt3a KO plus LPS were ranked by fold change relative to WT plus LPS. Peaks with proximal genes (within 20 kb) and log2(fold change [FC]) > 1 were included. Corresponding genes from RNA-seq analysis were included on the right, with significant genes FDR < 0.05 highlighted in red and labeled. (G) ATAC peaks specific to Dnmt3a KO plus vehicle versus WT plus vehicle were ranked in the same order as F. Corresponding genes from RNA-seq analysis were included on the right, with significant genes FDR < 0.05 highlighted in red and labeled. (H and I) ChIP-seq for Irf3 and Rela. (H) Venn diagrams of Irf3 and Rela ChIP-seq peaks with and without LPS administration (10 ng/ml). (I) Peak occupancy plots, sorted by Irf3 occupancy on the left. Rela peaks are displayed in the same order on the right. (J) KEGG pathway enrichment analysis for genes within 10 kb of Dnmt3a KO–specific peaks. Rela binding accounted for all inflammatory signaling pathway enrichment. (K) RNA-seq differential expression analysis of genes identified in J. Genes with FDR < 0.05 are highlighted with an asterisk. (L) ChIP PCR of Irf3 binding in the Il20 locus. n = 4–6. Statistical significance via Wilcoxon rank sum test. Error bars represent SEM. (M) Analysis of rs6722613 SNP across three large population studies. B, β; EA, effect allele; EAF, effect allele frequency; NEA, noneffect allele. *, P < 0.05; ****, P < 0.0001.

Dnmt3a-mediated increase in Irf3-Rela binding regulates Il20 expression. (A) RRBS analysis of Dnmt3a−/− and WT BMDMs. n = 5. Heatmap on top reveals CpG regions proportionally affected by hypo- or hypermethylation. Bar graph quantifies change in CpG methylation (y axis) at the defined loci (x axis). To assess statistical significance, one-way ANOVA was used against Dnmt3a ATAC or Dnmt3a ATAC with LPS, and P values were adjusted for multiple comparisons using BY. Error bars represent SD. (B) CpG methylation density plot at the ATAC sites increased in Dnmt3a−/− compared with WT BMDMs in vehicle-treated (top) or LPS-treated (bottom) samples. (C) ATAC-sequencing analysis of Dnmt3a−/− and WT BMDMs. n = 2. (D) TF binding site motif analysis. Statistical significance was assessed using hypergeometric test of overlap using all ATAC sites as the atlas. Log2(FDR) of LPS-treated samples (y axis) is plotted against that of vehicle-treated samples (x axis). (E) ChIP-seq of Irf3 and Rela, an NF-κB subunit. Bar graph of binding sites overlapped with ATAC sites globally. (F) Venn diagram of Irf3 and Rela-specific peaks and their overlap. Significance of overlap was assessed by a hypergeometric test. (G) Irf3 and Rela binding at the open chromatin regions in the Il20 locus. Shaded areas represent significant TF peaks detected. n = 3. Read densities are normalized to a million reads. (H)Il20 expression in RAW264.7 cells with Dnmt3a−/−, Irf3−/−, or both assessed 2 d after LPS treatment (10 ng/ml). n = 4. One-way ANOVA was used, and P values were adjusted for multiple comparisons using BY. *, P < 0.05; ****, P < 0.0001.
Dnmt3a-mediated increase in Irf3-Rela binding regulates Il20 expression. (A) RRBS analysis of Dnmt3a−/− and WT BMDMs. n = 5. Heatmap on top reveals CpG regions proportionally affected by hypo- or hypermethylation. Bar graph quantifies change in CpG methylation (y axis) at the defined loci (x axis). To assess statistical significance, one-way ANOVA was used against Dnmt3a ATAC or Dnmt3a ATAC with LPS, and P values were adjusted for multiple comparisons using BY. Error bars represent SD. (B) CpG methylation density plot at the ATAC sites increased in Dnmt3a−/− compared with WT BMDMs in vehicle-treated (top) or LPS-treated (bottom) samples. (C) ATAC-sequencing analysis of Dnmt3a−/− and WT BMDMs. n = 2. (D) TF binding site motif analysis. Statistical significance was assessed using hypergeometric test of overlap using all ATAC sites as the atlas. Log2(FDR) of LPS-treated samples (y axis) is plotted against that of vehicle-treated samples (x axis). (E) ChIP-seq of Irf3 and Rela, an NF-κB subunit. Bar graph of binding sites overlapped with ATAC sites globally. (F) Venn diagram of Irf3 and Rela-specific peaks and their overlap. Significance of overlap was assessed by a hypergeometric test. (G) Irf3 and Rela binding at the open chromatin regions in the Il20 locus. Shaded areas represent significant TF peaks detected. n = 3. Read densities are normalized to a million reads. (H)Il20 expression in RAW264.7 cells with Dnmt3a−/−, Irf3−/−, or both assessed 2 d after LPS treatment (10 ng/ml). n = 4. One-way ANOVA was used, and P values were adjusted for multiple comparisons using BY. *, P < 0.05; ****, P < 0.0001.
To determine which transcription factors (TFs) have increased binding in open chromatin of Dnmt3a−/− BMDMs, we quantified TF binding motif enrichment in Dnmt3a−/− specific open chromatin. The top TF binding site enriched in both LPS and vehicle-treated Dnmt3a−/− BMDMs was that of Irf3 (Fig. 4 D), which activates and cooperates with NF-κB signaling to modulate inflammation (Csumita et al., 2020; Yan et al., 2020). To confirm increased DNA binding of these two TFs in the context of Dnmt3a−/−, we performed chromatin immunoprecipitation sequencing (ChIP-seq) for Irf3 and Rela, a subunit of the NF-κB complex, using WT and Dnmt3a−/− BMDMs (Fig. S3, H–K). At open chromatin regions globally, Irf3 and Rela ChIP-seq peaks increased in Dnmt3a−/− BMDMs compared with WT (Fig. 4 E and Fig. S3 H) and had significant overlap (Fig. 4 F). Dnmt3a−/− specific Rela peaks were associated with increased expression of several inflammatory genes, such as Il1b, Lcn2, Nfkbiz, and Tlr2 (Fig. S3 K).
Having identified IL-20 as a mediator of increased osteoclast activity in mice with hematopoietic KO of Dnmt3a, we examined the Il20 locus for Irf3 and Rela ChIP-seq peaks. We identified Irf3 and Rela binding sites at the Il20 locus with significantly increased Irf3 binding especially in the context of LPS administration (Fig. 4 H and Fig. S3 L). As Irf3 activates NF-κB signaling (Csumita et al., 2020; Yan et al., 2020) and is cobound with Rela (Fig. 4 F), we tested whether Irf3 is required for Il20 expression. In Dnmt3a−/− cells, Irf3−/− resulted in a significant reduction in Il20 expression to levels comparable to WT (Fig. 4 H), suggesting that Irf3 in Dnmt3a−/− is required for Il20 expression.
IL-20 as a regulator of Dnmt3a-mediated osteoclast differentiation in vitro
Based on the CRISPR screen, cytokine array, and in vitro models of Dnmt3a−/− implicating IL-20 secretion as a primary mechanism driving Dnmt3a-mediated osteoporosis, we investigated whether IL-20 is sufficient to increase osteoclast differentiation. Administration of IL-20 to BM osteoclast precursors resulted in a significant dose-dependent increase in osteoclast differentiation (Fig. 5 A). Conversely, a neutralizing IL-20 monoclonal antibody 7E (Hsu et al., 2011) abrogated the effect of Dnmt3a−/− on osteoclast differentiation (Fig. 5, B and C). In vivo, administration of the 7E antibody for 4 wk decreased serum markers of bone turnover in mice transplanted with Dnmt3a−/− BM (Fig. 5 D). Overall, these data indicate that IL-20 secretion by Dnmt3a mutant myeloid cells is sufficient to drive osteoclast differentiation, and blocking IL-20 inhibits Dnmt3a-mediated osteoclastogenesis.

IL-20 regulates Dnmt3a-mediated osteoclastogenesis. (A) Effect of increasing concentrations of IL-20 on osteoclast differentiation from 200 CD11b−/lowCD115+Ly6Chi osteoclast precursors sorted from the BM of 12-wk-old Dnmt3a−/− mice, shown as boxplots. n = 11. (B) Co-culture experiments of 200 CD11b−/lowCD115+Ly6Chi osteoclast precursors from WT or Dnmt3a−/− mice and 2,000 BMDMs from respective sources in the presence of IL-20 neutralizing antibody 7E or isotype IgG during osteoclast differentiation. Boxplot representation of TRAP+ cells at day 6 of differentiation. n = 8. (C) Expression of the Ctsk reporter (APC+) as a percentage of GFP+ cells using Ctsk-T2A-R647 reporter cell line expressing Cas9-GFP in the presence WT or Dnmt3a−/−, and IL-20 neutralizing antibody 7E or isotype antibody during osteoclast differentiation. n = 8. (D) WT or Dnmt3a−/− BM transplants into WT male mice treated with isotype antibody or IL-20 neutralizing antibody 7E at a dose of 6 μg/kg every 3 d intraperitoneally starting at 8 wk after transplantation for 4 wk. Serum was collected at 12 wk and analyzed for CTX-I, a marker of bone resorption. Statistical significance via Wilcoxon rank sum test and adjusted by FDR. n = 7–14. For A–C, one-way ANOVA was used, and P values were adjusted for multiple comparisons using BY. *, P < 0.05; **, P < 0.01. Error bars otherwise note SD.
IL-20 regulates Dnmt3a-mediated osteoclastogenesis. (A) Effect of increasing concentrations of IL-20 on osteoclast differentiation from 200 CD11b−/lowCD115+Ly6Chi osteoclast precursors sorted from the BM of 12-wk-old Dnmt3a−/− mice, shown as boxplots. n = 11. (B) Co-culture experiments of 200 CD11b−/lowCD115+Ly6Chi osteoclast precursors from WT or Dnmt3a−/− mice and 2,000 BMDMs from respective sources in the presence of IL-20 neutralizing antibody 7E or isotype IgG during osteoclast differentiation. Boxplot representation of TRAP+ cells at day 6 of differentiation. n = 8. (C) Expression of the Ctsk reporter (APC+) as a percentage of GFP+ cells using Ctsk-T2A-R647 reporter cell line expressing Cas9-GFP in the presence WT or Dnmt3a−/−, and IL-20 neutralizing antibody 7E or isotype antibody during osteoclast differentiation. n = 8. (D) WT or Dnmt3a−/− BM transplants into WT male mice treated with isotype antibody or IL-20 neutralizing antibody 7E at a dose of 6 μg/kg every 3 d intraperitoneally starting at 8 wk after transplantation for 4 wk. Serum was collected at 12 wk and analyzed for CTX-I, a marker of bone resorption. Statistical significance via Wilcoxon rank sum test and adjusted by FDR. n = 7–14. For A–C, one-way ANOVA was used, and P values were adjusted for multiple comparisons using BY. *, P < 0.05; **, P < 0.01. Error bars otherwise note SD.
Concluding remarks
Bone health depends on the balance between resorption and formation, and osteoclasts are responsible for bone resorption. Numerous studies have examined the role of inflammatory cytokines on osteoclast function (Ponzetti and Rucci, 2019), but little is known about how these cytokines might arise in aging individuals to modulate osteoclast activity. In this study, we demonstrate that CHIP is associated with osteoporosis and heel ultrasound–based eBMD in a prospective human cohort and that Dnmt3a mutations in mice cause an osteoclast-mediated reduction in bone mass. While Dnmt3a−/− osteoclast precursor cells differentiate normally into osteoclasts, Dnmt3a−/− myeloid cells elaborate proinflammatory cytokines, including IL-20, that promote osteoclastogenesis.
Prior studies indicate that macrophages in CHIP autonomously contribute to atherosclerotic plaques (Fuster et al., 2017; Jaiswal et al., 2017). Our studies demonstrate that mutations in myeloid cells promote osteoclast differentiation in a non–cell-autonomous manner. In ex vivo experiments using BM-derived cells, Dnmt3a mutations increased osteoclast differentiation from precursors. This effect on osteoclast differentiation required the presence of whole BM, sorted CD11b+ myeloid cells, or BMDMs, demonstrating that Dnmt3a−/− macrophages contribute to this phenotype. In contrast, WT and Dnmt3a mutant osteoclast precursors cultured in isolation had equivalent differentiation potential. Experiments allowing for the diffusion of molecules, but not cells, revealed that secreted factors from macrophages are responsible for this phenotype.
Dnmt3a-mutated macrophages elaborated increased levels of inflammatory mediators, and Dnmt3a−/− mice had higher levels of proinflammatory cytokines. Gene expression analysis of Dnmt3a−/− BMDMs revealed increased expression of inflammatory cytokines, suggesting a proinflammatory polarization. In Dnmt3a−/− BMDMs, methylation erosion is associated with increased open chromatin susceptible to increased inflammatory TF binding. We identified increased TF binding of Irf3 at open chromatin regions, which cooperates with NF-κB signaling. Irf3 is required for LPS-mediated inflammatory signaling (Sakaguchi et al., 2003) and could explain the potentiation of Dnmt3a loss on osteoclast formation with the addition of LPS.
We identified the IL-20 receptor in an in vitro CRISPR screen for genes required for Dnmt3a-mediated osteoclastogenesis, and cytokine analysis revealed IL-20 among the most enriched cytokines in the serum of Dnmt3a−/− compared with WT mice. The neutralization of IL-20 secreted from Dnmt3a−/− BMDMs or RAW264.7 cells by the neutralizing antibody reversed Dnmt3a-mediated osteoclast differentiation. IL-20 is expressed primarily in monocytes/macrophages and epithelial cells (Blumberg et al., 2001; Wolk et al., 2002); is controlled by Irf3-NF-κB−mediated signaling; and has been implicated in psoriasis, rheumatoid arthritis, and osteoporosis (Blumberg et al., 2001; Hsu et al., 2011; Hsu et al., 2006).
Prior bone studies have investigated the role of Dnmt3a or Tet2 but did not use pan-hematopoietic deletions, as we used, that model CHIP including the effect of myeloid cells, and thus, the association with osteoporosis remains elusive. In one study, the examination of the cell-intrinsic function of Dnmt3a in osteoclasts via osteoclast-specific (e.g., RANK-Cre or Ctsk-Cre) loss of Dnmt3a early in development increased bone mass via decreased osteoclasts (Nishikawa et al., 2015). However, the other hematopoietic lineages, which represent the majority of the BM cells influencing osteoclasts, did not bear the Dnmt3a mutation in this model. In agreement, we also did not observe an increase in osteoclasts when Dnmt3a-mutated osteoclast precursors are differentiated in isolation from other BM cells. Additionally, germline Tet2−/− mice have been shown to have increased bone mass through impaired osteoclast differentiation (Chu et al., 2018), but this effect is confounded by increased osteoblast differentiation from Tet2−/− mesenchymal cells (Chu et al., 2018). Transcriptomic analysis of osteoblasts has linked a DNMT3A single-nucleotide polymorphism (SNP) to osteoporosis (Panach et al., 2020), but we could not find support for the conclusions in larger association studies (Fig. S3 M).
In summary, human genetic studies and murine models demonstrate that CHIP mutations increase osteoporosis risk. In the UK Biobank, individuals with CHIP have an increased risk of osteoporosis after adjusting for osteoporosis risk factors. In murine studies, CHIP mutations cause a reduction in bone mass. Individuals with CHIP may benefit from treatment with bisphosphonates or IL-20–specific therapies.
Materials and methods
UK Biobank cohort
The UK Biobank project comprises >500,000 adult individuals from across the UK recruited between 2006 and 2010 and followed prospectively (Bycroft et al., 2018; Van Hout et al., 2019,Preprint). At their baseline visit, the participants had a physical examination, gave samples, and provided detailed information regarding medical history and medications. We did not have human participants in the study, and we used anonymized data from the UK Biobank through the proposal ID 50834 approved by the UK Biobank Ethics Advisory Committee. In this study, the UK Biobank cohort includes individuals of European ancestry aged 40–70 yr at the time of blood sample collection for sequencing and available whole-exome sequences. A summary of the inclusion and exclusion criteria is shown in Fig. S1 A. Individuals with a history of any malignant neoplasm at baseline were excluded. The KING (Kinship-Based Inference for Genome-Wide Association Studies) tool was used to derive unrelated kinship pairs in a third-degree kinship analysis (Manichaikul et al., 2010). Samples were also filtered for a mismatch between genetic sex and reported sex, missing BMI, and missing smoking history. Samples were filtered for history of hormone replacement therapy given history of surgical and premature menopause in this population, and incomplete information on the nature of hormone replacement therapy. For incident analysis, samples with a diagnosis of osteoporosis by the International Classification of Diseases (ICD) codes before sequencing were excluded and for heel ultrasound data, samples with missing heel ultrasound were excluded. Participant characteristics at baseline were compared among individuals with and without CHIP using the Wilcoxon rank sum test for continuous variables and Fisher’s exact test for categorical variables.
The statistical models were performed using the R statistical software. The time-to-event analysis was performed in R using the survival package. The association between incident osteoporosis and CHIP was analyzed by Cox proportional hazard model using coxph function adjusting for established covariates at baseline, including age ≥65 yr, sex, BMI <18.5 kg/m2 (underweight), BMI >30 kg/m2 (obese), ever or never smoking status, rheumatoid arthritis diagnosis, and self-reported oral corticosteroid use. An age cutoff of 65 yr was used, as it is the age at which the U.S. Preventive Services Task Force in 2011 and the National Institute for Health and Clinical Excellence in 2012 recommends bone densitometry screening for women. The diagnosis of osteoporosis with or without related fractures was ascertained based on the ICD10 codes M80.* and M81.* and the ICD9 code 733.* in any of the primary or secondary diagnosis fields. Asterisks indicate wild cards. The ICD code for osteopenia (ICD10 M85.8) included other disorders of bone and was not analyzed. Osteoporosis is a markedly underdiagnosed condition, but the osteoporosis incidence reported here for age 40–70 yr is consistent with that seen in a large Danish cohort using similar ICD codes (Vestergaard et al., 2005). The diagnosis of rheumatoid arthritis was ascertained based on ICD10 codes M05.* and M06.* and the ICD9 code 714.*. Oral corticosteroid use was determined from self-reported fields. The event of interest was osteoporosis diagnosis, and individuals were censored at death, malignant neoplasm diagnosis, or the end of the follow-up period (March 2020), whichever came first. Death and malignant neoplasm diagnosis were included as competing risks in cumulative incidence graphs. The horizontal lines represent Wald 95% CIs in the forest plots.
Curation of CHIP calls
The UK Biobank exomes were sequenced from blood samples at the Regeneron Genetics Center (Van Hout et al., 2019,Preprint). The alignment cram files processed through the SPB pipeline were obtained from the UK Biobank in Jan-Feb 2021. CHIP calls were curated as described previously (Gibson et al., 2017; Jaiswal et al., 2014; Jaiswal et al., 2017). Briefly, aligned exome sequences were analyzed using Mutect2 in the top 58 genes known to be commonly mutated in healthy individuals and myeloid malignancies (Benjamin et al., 2019,Preprint; Gibson et al., 2017; Jaiswal et al., 2014). Initial studies have examined a larger panel of genes (n = 160) recurrently mutated in hematological malignancies, but subsequent studies have demonstrated that most of the mutations are in DNMT3A, TET2, and ASXL1. Our 58-gene analysis is sufficient to capture previously described CHIP mutations. Putative somatic variants were further curated to exclude frequent germline polymorphisms and common sequencing artifacts using a “panel of normal” and manual inspection of sequencing reads at the loci of the mutation, and this approach was verified independently by two molecular pathologists.
Animal maintenance
Animal experiments were performed with the approval of Institutional Animal Care and Use Committee at Brigham and Woman’s Hospital and at Dana Farber Cancer Institute. The following floxed strains were used: Tet2-floxed line B6;129S-Tet2tm1.1laai/J (JAX 017573), Dnmt3a-floxed line B6;129S4-Dnmt3atm3.1Enl/J (Okano et al., 1999), B6(Cg)-Dnmt3atm1Trow/J line (JAX 032289), and Nlrp3-floxed line B6.129S6-Nlrp3tm1Bhk/J (JAX 021302). Floxed strains were crossed with mice with constitutive expression of Cre recombinase under the control of the Vav1 promoter to generate hematopoietic-specific KOs (Georgiades et al., 2002), which avoids targeting Commd10. For constitutive GFP expression, Gt(ROSA)26Sortm1.1(CAG-cas9*,-EGFP)Fezh/J (JAX 024858) was used. For transplant recipients, the Ldlr KO strain B6;129S7-Ldlrtm1Her/J and WT B6.SJL-Ptprca Pepcb/BoyJ (JAX 002014) strain were used. Ldlr KO strain was partially backcrossed to B6.SJL-Ptprca Pepcb/BoyJ (JAX 002014) mice to generate recipient mice homozygous for CD45.1. Genotyping was performed by Transnetyx.
BM transplantation
8- to 10-wk-old recipient mice were lethally irradiated with two doses of λ-irradiation (475 cGy) separated by 3 h. CD45.2+ donor marrow was obtained from respective floxed mice with Vav1-Cre controls, and 1–2 million cells were injected retro-orbitally. Following transplantation, recipient mice were provided with sterilized cages, food, and water for a period of 2 wk. For transplant into Ldlr KO mice, mice were started on a high-fat, high-cholesterol diet (TD.96121; 21% milk fat, 1.25% cholesterol diet; Harlan-Teklad) 4 wk after transplantation. We primarily studied male recipient mice to exclude the effect of variable estrogen levels in female mice following irradiation conditioning for transplantation. BM chimerism was assessed by flow cytometry after transplantation by analyzing the CD45.2/(CD45.1+CD45.2) ratio.
Isolation of BM and peripheral blood
Peripheral blood was obtained retro-orbitally. BM cells were obtained by crushing femurs with a mortar and pestle and strained through a 100-µm strainer. Red blood cells were lysed using lysis buffer (BD Pharm Lyse) for 15 min at room temperature before flow cytometry analysis.
Flow cytometry and peripheral blood analysis
Cell populations were analyzed using FACSCANTO II (BD). Cell sorting was performed on Sony MA900 using a 100-µm sorting chip. Single-cell suspensions were blocked with Fc-Block (BD) for 15 min at room temperature. The following antibodies were used to stain cells: FITC anti-mouse Ly6C (AL-21; BD), PE/Cy7 anti-mouse CD115 (AFS98; BD), Pacific Blue anti-mouse CD45.1 (A20; BioLegend), allophycocyanin (APC) anti-mouse CD45.2 (104; BioLegend), PerCP/Cyanine5.5 anti-mouse CD11b (M1/70; BioLegend), APC-Cy7 anti-mouse B220 (RA3-6B2; BD), APC-Cy7 anti-mouse Ter-119 (TER-119; BD), and APC-Cy7 anti-mouse CD3 (17A2; BioLegend). Cells were stained for 40 min using 1:100 concentration of antibodies at 4°C in 2% FBS/PBS (vol/vol) and washed with 2% FBS/PBS (vol/vol) before analysis. Complete blood counts were measured on peripheral blood samples using a Hemavet (Drew Scientific).
µCT and mechanical testing
Trabecular bone architecture in the distal femoral metaphysis and cortical bone morphology of the femoral mid-diaphysis was analyzed using high-resolution µCT (μCT40; Scanco Medical) in a blinded manner at the Beth Israel Deaconess Medical Center for Advanced Orthopedic Studies as described previously (Wein et al., 2016). Briefly, trabecular and cortical bone was segmented using mineral density thresholds of 400 and 700 mg hydroxyapatite/cm2, respectively, and then analyzed using the standard Scanco trabecular and cortical bone morphology scripts. Mechanical testing of the femur was also performed at the same core facility. Briefly, three-point bend tests were performed using electrical force materials testing machine (Electroforce 3230; Bose Corporation) with the following parameters: bending fixture bottom span length of 8 mm, load point in displacement control moving at a rate of 0.1 mm/s with force, and displacement data collected at 60 Hz. Bending rigidity (N-mm2), ultimate moment (N-mm), and work to fracture (mJ) values were calculated using beam-bending equations.
Histological analysis
Femurs were fixed in 4% paraformaldehyde (PFA) overnight at 4°C and decalcified in 10% EDTA (pH 7.1) for variable periods of time depending on the developmental stage before paraffin embedding. Sections were de-paraffinized and rehydrated, and antigen retrieval was performed using standard protocols. Osteoclasts were stained using the TRAP staining kit (Sigma). Slides were scanned via Panoramic MIDI II (3DHistech), and the number of TRAP+ cells per bone surface was calculated using the TrapHisto analysis software (van 't Hof et al., 2017). For analysis of the tibias, samples were stained for TRAP for N. Oc/BS or Goldener’s Trichrome stain for Ob.N/BS and Ob.S/BS and analyzed in a blinded fashion at the Endocrine Histology Core at Massachusetts General Hospital.
Alendronate and IL-20 neutralizing antibody treatment
For alendronate, at 14 wk after BM transplantation, mice were subcutaneously injected twice per week with alendronate sodium (A4978; Sigma) or vehicle control at a dose of 100 μg/kg capped at 3 μg/mouse for 7 wk. For IL-20 neutralization, 7E blocking antibodies or control IgGs (Creative Biolabs) were injected intraperitoneally at 6 µg/kg per mouse every 3 d for 4 wk (lot CBL2007Z11 or CB2006YC08, respectively; Creative Biolabs).
Cell culture
Cells were maintained in 10% FBS/DMEM (Genesee Scientific) supplemented with 1% penicillin-streptomycin-glutamine (PSG) and grown in 37°C incubators at 5% CO2 unless otherwise specified. BMDMs were generated as previously described using macrophage CSF (M-CSF) at 30 ng/ml (Weischenfeldt and Porse, 2008). For in vitro osteoclast differentiation, whole BM cells from crushed femurs were plated at a density of 50,000 cells/cm2 in a 96-well tissue-culture treated plate in α-MEM (Gibco) supplemented with 10% FBS, 1% PSG, 30 ng/ml M-CSF, 30 ng/ml human sRANKL (ThermoFisher), or 10 ng/ml mouse sRANKL (R&D) for 6 d. Sorted osteoclast precursors were obtained from the BM of 12- to 14-wk-old female mice unless otherwise indicated because of the larger osteoclast pool in females. For Transwell assays, 0.4-µm-size membrane plates were used (Corning). For osteoclast differentiation from RAW264.7 cells (ATCC), single cells were plated evenly at a density of 4,000 cells/cm2 in DMEM supplemented with 10% FBS, 1% PSG, and 50 ng/ml human sRANKL (Gibco) or 10 ng/ml murine RANKL (R&D) for 5–6 d. LPS (0.1 ng/ml, L4391; Sigma) or IL-1β (0.5 ng/ml; R&D) were added during initial plating unless otherwise specified. For TRAP staining, cells were then fixed with 4% PFA for 15 min, washed with distilled water, and permeabilized with ice-cold 50%/50% acetone/methanol for 1 min. After washing with distilled water, osteoclasts were detected via TRAP staining kit (387A; Sigma), and multinucleated TRAP+ cells were counted. For staining for F-actin rings, fixed cells were stained with Alexa Fluor 488-phalloidin at 1:100 for 40 min at room temperature (ThermoFisher). For IL-20 blockade, 7E blocking antibodies or control IgGs were used at 2 µg/ml. Lif, IL-11, and fractalkine were obtained from R&D.
CRISPR-Cas9–mediated KO
Dnmt3a KO from RAW 264.7 cells was generated as previously described (Boettcher et al., 2019). Initially, a single-cell clone of RAW264.7 was obtained via limited dilution. Recombinant Cas9 protein, synthetic locus-specific CRISPR RNAs (crRNA), negative control crRNA, and transactivating crRNAs (tracrRNA) were obtained from Integrated DNA Technologies. The Dnmt3a-specific crRNAs were 5′-AGGAAGTTTACACCGACATG-3′ and 5′-GCTGGCACGTTGGAAAAGGG-3′. For DNMT3A, crRNA 5′-CCTCTTGTCACTAACGCCCA-3′ was used. The Irf3-specific crRNA was 5′-CCAGTGGTGCCTACACCCCG-3′. crRNAs and tracrRNAs (120 pmol each) were mixed in Cas9 working buffer (Hepes 20 mM, 150 mM KCl, 1 mM MgCl2, 10% glycerol, and 1 mM TCEP) in a total volume of 5 µl. crRNA–tracrRNA duplexes were formed by heating the mixtures to 98°C for 5 min and then cooled to room temperature. Recombinant Cas9-3NLS (100 pmol) in 5 µl Cas9 working buffer was added slowly to the duplex solution and incubated for 20 min at room temperature. Cells were resuspended in 10 µl of SF nucleofection solution with 2 µl Supplement 1 (Lonza), combined with duplex/Cas9 solution in a 20 µl nucleocuvette, and electroporated using the Amaxa 4D-Nucleofector using the RAW264.7 program (Lonza). After subcloning, genome editing was confirmed by either amplicon sequencing of the PCR products of 5′-ACTGTGGGTGCTATGCTAGT-3′/5′-GCGCTCATCAATGATCTCCT-3′ or 5′-AGTCCAGCCTCCGTCAG-3′/5′-CTCAGCTACAGCCCAACAAA-3′ primer pairs, respectively, or via immunoblotting. To generate virally mediated KO of target genes, sgRNA sequences were cloned into the LentiGuide-Puro backbone (52963; Addgene). For Irf3, validated crRNAs from the Brie genome-wide library was used and include 5′-GGCTGGACGAGAGCCGAACG-3′ and 5′-CTGGCGGCCTCGGTAGAAGG-3′. As a control, the backbone without crRNA was used. Amplicon sequencing was performed at the Massachusetts General Hospital Center for Computational and Integrative Biology DNA core facility (Cambridge, MA).
Immunoblotting
Protein lysates were obtained using Pierce IP lysis buffer (#87787; ThermoFisher) freshly supplemented with Halt Protease and Phosphatase Inhibitor Cocktail (#78440; ThermoFisher). Protein concentration was determined using Pierce BCA Protein Assay Kit (#23225; ThermoFisher). Equal protein amounts were run on Tris-HCl Criterion Precast gels (Bio-Rad) and transferred onto Immobilon-P membranes (Millipore). Blots were blocked in 5% nonfat dry milk (Santa Cruz Biotechnology) or 5% BSA in TBS-T 0.1% for 1 h. The antibodies used were anti-Dnmt3a (ab188470, 1:1,000; Abcam), anti-actin HRP (ab20272, 1:2,000; Abcam), anti-Irf3 (sc-33641, 1:1,000; Santa Cruz Biotechnology), and anti-Rela (S8242S, 1:1,000; Cell Signaling). Secondary stains include goat anti-rabbit HRP (1:5,000; Prometheus Labs) and rabbit anti-mouse HRP (1:5,000; Abcam).
Bone resorption assay
To assay hydroxyapatite resorption, RAW264.7 cells were differentiated into osteoclasts on OsteoAssay plates (Corning) using the abovementioned conditions for 7 d. Plates were stripped of cells using 1% bleach, rinsed with distilled water, and air dried. Modified von Kossa staining was performed by immersing wells with 5% (wt/vol) silver nitrate for 10 min and placing them under the UV light for 30 min. Images of the well surfaces were obtained in a nonoverlapping fashion on a Leica inverted microscope, and resorbed areas were quantified via image thresholding using ImageJ. Each data point represents mean resorbed area per image per well.
RNA-seq and analysis
RNA was collected on day 8 of BMDM culture after replating at a cell density of 50,000/cm2. Total RNA was extracted using the RNeasy micro kit (Qiagen), and 10 ng RNA was used to prepare sequencing libraries using the NEBNext Ultra RNA Library Prep Kit (E7530) with Poly(A) mRNA Magnetic Isolation Module (E7490) and multiplexed using NEBNext Multiplex Oligos (E6609S). NextSeq 550 sequencing (Illumina) was used to obtain single-end reads of 75 bp. Raw Illumina output was converted to FASTQ using bcl2fastq (v2.20). Trimmomatic (v0.39) was used for quality trimming with a minimum trimmed length of 36 bp. Sequencing data were aligned with STAR v2.7.0a to mm10 genome database (Dobin et al., 2013), and counts were obtained by featureCounts excluding multimapping reads (Liao et al., 2014). Differential expression was determined using DESeq2 (Love et al., 2014) using a q-value of 0.05 and log2 fold change of 0.5 unless noted otherwise. Gene set variation analysis was performed as previously described (Hänzelmann et al., 2013). Data has been deposited to the Gene Expression Omnibus under accession no. GSE183480.
RRBS and analysis
RRBS libraries were made from 300 ng of purified DNA using Zymo-Seq RRBS Kit according to manufacturer instructions (Zymo Research; Meissner et al., 2008). Libraries was amplified for 9 cycles. Indexed libraries were sequenced on Nextseq 550 using paired-end reads of 43 bp, or Novaseq SP using paired-end reads of 50 bp. Quality control was obtained using Trim Galore v0.4.5 (Krueger; Babraham Bioinformatics). Trimmed reads were aligned to the mm10 genome database via Bismark v0.22.3 using paired-end settings (Krueger and Andrews, 2011), and reads from the same biological sample were merged at the time of methylation calling. The average bisulfite conversion rate was at least 96%. Differential methylation analysis was performed using MethylKit (Akalin et al., 2012). Average coverage per CpG was at least 22×. Undermethylated regions (called canyons) and 2-kb edges are defined previously (Jeong et al., 2014). CpG islands are obtained from UCSC mm10 genome tables and shores defined as 2-kb flanking regions. Promoters are defined as ±1 kb of transcriptional start sites. For volcano density plots, display of –log10(q-values) > 100 is capped at 100. RRBS and subsequent chromatin-related data has been deposited to the Gene Expression Omnibus under accession no. GSE184446.
ATAC sequencing and analysis
ATAC-sequencing libraries were made according to manufacturer instructions (Active Motif). Briefly, 50,000 BMDM cells were subject to lysis, tagmentation, and DNA purification. DNA was amplified using Nextera i7/i5 indexed primers for 10 cycles. Indexed libraries were sequenced on Nextseq 550 using paired-end reads of 42 bp. Quality control was obtained using Trim Galore v0.4.5 (Krueger; Babraham Bioinformatics). Trimmed reads were aligned to the mm10 genome via bowtie2.2.9 using --very-sensitive -X2000 --no-mixed --no-discordant. Duplicates were removed using MarkDuplicates in Picard/2.8.0. ATAC shifts were performed using alignmentSieve in deepTools/3.5.0. Peaks were called using Macs2/2.1.1 using -f BAMPE --keep-dup all -B --SPMR --nomodel -p 1e-2. For biological duplicates, peaks were filtered using an irreproducible discovery rate < 0.05. Analysis of differential chromatin accessibility was performed using DESeq2 using the cutoff false discovery rate (FDR) < 0.05. Peaks were annotated using HOMER/4.9. Motif discovery was performed using Find Individual Motif Occurrences in MEME Suite 4.12.0.
ChIP-seq and analysis
ChIP-seq libraries were made according to manufacturer instructions (SimpleChIP; Cell Signaling). The duration of LPS treatment was 6 h with 10 ng/ml. Briefly, BMDMs were cross-linked using 37% formaldehyde for 10 min and then quenched with glycine. Cells were washed with PBS and scraped in the presence of a protease inhibitor cocktail. Cell nuclei were pelleted and partially digested with micrococcal nuclease for 20 min, and then the remaining DNA was sonicated to 100–300 bp using Covaris. Immunoprecipitation was performed in the ChIP buffer with protease inhibitor cocktail overnight with rotation. Antibodies used include anti-Irf3 (sc-33641; Santa Cruz Biotechnology) an anti-Rela (S8242S; Cell Signaling) at 3 μg each per sample. Protein G Magnetic beads were added to each sample, isolated with a magnetic separation rack, and washed according to instructions. Samples were eluted and treated with RNase H and Proteinase K. Isolated DNA was subject to end preparation and adaptor ligation and amplified for 10–12 cycles, and indexed libraries were subject to sequencing on the Nextseq 550 using paired-end reads of 42 bp. Trimmed reads were aligned to the mm10 genome via bowtie2.2.9 using --very-sensitive -X2000 --no-mixed --no-discordant. Duplicates were marked using MarkDuplicates in Picard/2.8.0. Peaks were called using Macs2/2.1.1 using -f BAMPE -B --SPMR --nomodel -p 1e-5. For each biological triplicate, peaks were filtered using ChIP-R (Newell et al., 2021) at the default α of 0.05. Peaks were annotated using HOMER/4.9. Tracks for ATAC-seq and ChIP-seq are visualized after applying toTDF function in igvtools to Macs pileups.
Generation of reporter cell lines via CRISPR homology-directed repair
To generate the Ctsk-T2A-R647 reporter cell line from RAW264.7 cells, a Ctsk-T2A-R647-P2A-neoR construct with 100-bp homologous flanking sites was synthesized (Integrated DNA Technologies). The construct was designed to target several nucleotides upstream of the stop codon of the Ctsk gene via crRNA 5′-CGCCTGCGGCATTACCAACA-3′. In the construct, nucleotides for Thr321 and Asn322 were changed to have synonymous mutations to prevent cleavage of the construct (5′-CGCCTGCGGCATTACGAATA-3′). 500 ng of the construct was electroporated with cRNA–tracrRNA duplex and Cas9 as mentioned. After 5 days of growth, reporter cell line was selected using neomycin (1 μg/μl; Sigma) for 10 d. Clones were verified via flanking PCR using a 5′-CTCGACCACCTTGATTCTCTG-3′ and 5′-GCACACACATACACACACATATAC-3′ primer pair, and ∼406-bp product was sequenced via next-generation sequencing for confirmation. Cells were then transduced with SpCas9-P2A-GFP expressing lentivirus (pL-CRISPR.SFFV.GFP, 57827; Addgene) and single-cell cloned via cell sorting and plating at limiting dilutions.
Lentiviral production
Lentiviral particles were produced by transient transfection of 293T cells using the FuGENE 6 (Promega) transfection method. Viral constructs were cotransfected with pMD2.G (plasmid 12259; Addgene) and psPAX2 (plasmid 12260; Addgene), and lentiviral particles were harvested on day 3 of transfection.
CRISPR-Cas9–mediated screen
RAW264.7 cells expressing Ctsk-T2A-R647 in the endogenous Ctsk locus and stably expressing Cas9-GFP were used to generate Dnmt3a KO cell lines. 150 million Dnmt3a KO cells were infected with the mouse Brie LentiGuide-Puro genome-wide CRISPR library (Doench et al., 2016) with 2 μg/ml polybrene (Sigma). The Brie library targets 19,674 genes with four guide RNAs (gRNAs) per gene. 24 h after infection, cells harboring the construct were selected for 2 d using 2 μg/ml puromycin (Sigma). After 3–5 d of puromycin withdrawal, cells were replated as single cells at a density of 4,000 cells/cm2 in the presence of sRANKL for 5 d. GFP+ cells with top 5% and bottom 5% in the APC channel were sorted (MA900; Sony). “APC high” represents the top 5% of Ctsk-R647–expressing cells, and “APC low” represents the bottom 5% of Ctsk-R647–expressing cells. DNA was purified from sorted cell pellets, and the guide RNA (gRNA) library was PCR amplified and then sequenced on either Illumina Nextseq 550 or Novaseq SP to obtain read counts for each gRNA. gRNAs were ranked based on fold change in read counts (bottom 5%/top 5%), and rank sum–based statistics were used to calculate statistical significance as previously described (Slabicki et al., 2020).
Quantitative RT-PCR
Total RNA isolated from the RNeasy micro kit (Qiagen) was reverse transcribed into cDNAs using the SuperScript II reverse transcription kit (Life Technologies). Real-time quantitative PCR was performed using SYBR Green Master Mix (Life Technologies) using Applied Biosystems QuantStudio 6. Primer sequences include 5′-GTCTTGCCTTTGGACTGTTCT-3′ and 5′-AGGTTTGCAGTAATCACACAGC-3′ (Il20; PrimerBank 50845426c1) and 5′-TCCTCAGACCGCTTTTTGCC-3′ and 5′-CTAATCACGACGCTGGGACT-3′ (Hprt). Quantitative PCR data were normalized to the housekeeping gene and the untreated sample using the 2−ΔΔCTmethod.
ELISA
P1NP levels in mouse serum were determined using the Rat/Mouse P1NP EIA kit (Immunodiagnostic Systems). C-terminal telopeptide of type I collagen (CTX-I) levels in fasting mouse serum were determined using the RatLaps CTX-I EIA kit (Immunodiagnostic Systems). Serum cytokine arrays were performed using 44-Plex technology (MD44) from Eve Technologies. IL-20, IL-11, Lif, and IL-1β levels were determined using mouse IL-20 (EMIL20; ThermoFisher), IL-11 (EMIL11; ThermoFisher), Lif (MLF00; ThermoFisher), and IL-1β ELISA kits (5018265; ThermoFisher). Absorbance was measured at 450 nm using Clariostar Plate Reader or Molecular Devices FilterMax.
Immunofluorescence staining
For F-actin ring staining, cells were fixed with 4% PFA for 15 min at room temperature and blocked with 10% FCS for 1 h. Cells were then stained with 1:100 phalloidin-FITC (A12379; Life Technologies) for 1 h at room temperature, washed three times using PBS, and counterstained with DAPI before imaging.
Statistical analysis
Analyses were performed in R 3.4.1/3.6.1 statistical environment or Prism 7.0. A two-sided Welch’s t test was used to assess significant differences between the two samples. For comparing one sample against all other samples, one-way ANOVA was used, and P values were adjusted for multiple comparisons using the two-stage linear step-up procedure of Benjamini, Krieger, and Yekutieli (BY). For grouped comparisons, significance was assessed by two-way ANOVA adjusted for multiple comparisons with the two-stage linear step-up procedure of BY. Otherwise, multiple comparisons after two-sided Welch’s t test were adjusted by the method of BY unless otherwise specified. The error bars represent SD, and biological replicates were used.
Online supplemental material
Fig. S1 shows additional data relevant to the UK Biobank cohort, validation of the transplant models for reconstitution of predominantly hematopoietic cells, osteoblast-related data, validation of TRAP staining, isogenic in vitro models of Dnmt3a mutants, osteoclast precursor numbers in vivo, and RNA-seq data of BMDMs. Fig. S2 shows supporting data for CRISPR screens, and validation of IL-20 as a potential target that is inflammasome independent. Fig. S3 shows data related to methylation sequencing, ATAC sequencing, ChIP-seq, and investigation of DNMT3A SNP rs6722613. Table S1 lists UK Biobank cohort characteristics and shows a comparison of baseline characteristics and CHIP calls. Table S2 shows a summary of μCT and biomechanical testing. Table S3 lists differentially expressed genes in RNA-seq data from BMDMs.
Acknowledgments
This work is supported by the National Institutes of Health (grants R01HL082945, P01CA108631, and P50CA206963), the Howard Hughes Medical Institute, and the Edward P. Evans Foundation (B.L. Ebert). P.G. Kim was supported by the National Heart, Lung, and Blood Institute Training Program in Molecular Hematology (5T32HL116324); is a Damon Runyon Physician-Scientist supported (in part) by the Damon Runyon Cancer Research Foundation (PST-35-21); and is supported by the Edward P. Evans Foundation Evans Young Investigator Award. A. Niroula is supported by the Knut and Alice Wallenberg Foundation (KAW 2017.0436). C.J. Gibson is supported by a Damon Runyon Cancer Research Foundation Physician-Scientist Training Award. G. Griffin is supported by the Damon Runyon Cancer Research Foundation. D.P. Kiel is supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (grant R01AR041398). M. Agrawal is supported by Deutsche Forschungsgemeinschaft (grant AG252/1-1). J.B. Richards is supported by the Canadian Institutes of Health Research (grants 365825, 409511, and 100558), the McGill Interdisciplinary Initiative in Infection and Immunity (MI4), the Lady Davis Institute of the Jewish General Hospital, the Jewish General Hospital Foundation, the Canadian Foundation for Innovation, the National Institutes of Health Foundation, Cancer Research UK, Genome Québec, the Public Health Agency of Canada, McGill University, Cancer Research UK (grant C18281/A29019), and the Fonds de Recherche Québec Santé. J.B. Richards is also supported by a Fonds de Recherche Québec Santé Mérite Clinical Research Scholarship, Calcul Québec, and Compute Canada. M.N. Wein is supported by the National Institutes of Health (grant R01DK116716). J.F. Charles is supported by National Institute of Arthritis and Musculoskeletal and Skin Diseases (grant R21AR077768) and the Brigham Research Institute Fund to Sustain Research. S. Jaiswal is supported by the Burroughs Wellcome Foundation Career Award for Medical Scientists, Foundation Leducq, Ludwig Center for Cancer Stem Cell Research, the American Society of Hematology Scholar Award, and the National Institutes of Health Director's New Innovator Award (DP2-HL157540).
Author contributions: P.G. Kim initiated the project; designed, performed and analyzed experiments; and wrote the manuscript. A. Niroula, A. Bick, C.J. Gibson, G. Griffin, M.M. Uddin, A. Sekar, S. Jaiswal, and P. Natarajan made CHIP calls. A. Niroula, J.P. Kemp, D.P. Kiel, J.B. Richards, D.M. Evans, and P. Natarajan helped analyze the UK Biobank data. V. Shkolnik, M. McConkey, A.E. Lin, W.J. Wong, D.N. Cohen, W.J. Shin, and M. Agrawal performed in vivo and in vitro experiments. M. Słabicki and J.M. Tsai assisted with CRISPR screen experiments. D.J. Brooks and M.L. Bouxsein performed and analyzed bone densitometry and mechanical testing. M.N. Wein and J.F. Charles provided guidance on bone biology and provided reagents. B.L. Ebert initiated the project, designed experiments, provided supervision, and wrote the manuscript. All authors contributed to manuscript editing.

