


Adaptive immunity is a fundamental component in controlling COVID-19. In this process, follicular helper T (Tfh) cells are a subset of CD4+ T cells that mediate the production of protective antibodies; however, the SARS-CoV-2 epitopes activating Tfh cells are not well characterized. Here, we identified and crystallized TCRs of public circulating Tfh (cTfh) clonotypes that are expanded in patients who have recovered from mild symptoms. These public clonotypes recognized the SARS-CoV-2 spike (S) epitopes conserved across emerging variants. The epitope of the most prevalent cTfh clonotype, S864–882, was presented by multiple HLAs and activated T cells in most healthy donors, suggesting that this S region is a universal T cell epitope useful for booster antigen. SARS-CoV-2–specific public cTfh clonotypes also cross-reacted with specific commensal bacteria. In this study, we identified conserved SARS-CoV-2 S epitopes that activate public cTfh clonotypes associated with mild symptoms.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has caused a worldwide outbreak of coronavirus disease 2019 (COVID-19). B cells (antibodies) and cytotoxic (CD8+) and helper (CD4+) T cells are the fundamental components of adaptive immunity, the process by which the body attempts to control COVID-19 (Ni et al., 2020; Rydyznski Moderbacher et al., 2020). B cells produce neutralizing antibodies, and their critical epitopes are determined within the spike (S) protein (Liu et al., 2020); these epitopes have contributed to the development of current vaccines that employ S protein as an antigen (Baden et al., 2021;Walsh et al., 2020; Sadoff et al., 2021; Voysey et al., 2021; Shinde et al., 2021). In contrast, key T cell epitopes that contribute to protective immunity against COVID-19 have not been well characterized. In addition to the importance of CD8+ T cells (Sette and Crotty, 2021), rapid induction of CD4+ T cells is associated with mild COVID-19 symptoms (Peng et al., 2020; Tan et al., 2021;Sette and Crotty, 2021). Among CD4+ T cells, follicular helper T (Tfh) cells help B cells to produce protective antibodies (Liu et al., 2013; Vinuesa et al., 2016;Crotty, 2019), and inefficient induction of Tfh cells is correlated with more severe and fatal COVID-19 (Kaneko et al., 2020; Zhang et al., 2021; Gong et al., 2020). Although TCR clonotypes for SARS-CoV-2 are being extensively examined (Shomuradova et al., 2020; Dykema et al., 2021), public Tfh clonotypes associated with COVID-19 recovery have not yet been reported. The identification of such public clonotypes and their common epitopes would elucidate the S regions essential for protective T cell immunity and thereby provide invaluable information for future vaccine design against emerging variants. Furthermore, any cross-reactive antigens of such clonotypes may contribute to the severity of COVID-19.
In this study, using single-cell–based paired and bulk TCR sequencing (TCR-seq) of COVID-19 patient T cells and the worldwide TCR database, we identified public circulating Tfh (cTfh) clonotypes associated with mild symptoms and their epitopes in the S regions. These epitopes are conserved among the emerging SARS-CoV-2 variants.
Results
Identification of SARS-CoV-2 S protein–specific cTfh clones in convalescent COVID-19 patients
We first analyzed SARS-CoV-2–specific T cell subsets and their clonotypes using a single-cell–based RNA-seq platform. Peripheral blood mononuclear cells (PBMCs) isolated from healthy donors and convalescent COVID-19 patients (Table S1 andTable S2) were stimulated with antigens derived from SARS-CoV-2, including inactivated virus, recombinant S protein, overlapped peptide pools derived from S protein (S peptide pool) or membrane (M) and nucleocapsid (N) proteins (MN peptide pool). Activation marker–positive T cells were sorted and analyzed for their TCR sequences together with RNA expression by single-cell TCR-seq and RNA-seq analyses (Fig. S1 A). Uniform manifold approximation and projection (UMAP) embedding and clustering allowed identification of a cluster (#8) consisting of CD4+ cells expressing Tfh-related genes, such as CD200,PDCD1, ICOS, CXCL13,CD40LG, and CXCR5 (Fig. 1, A and B; Crotty, 2019; Schmitt et al., 2014), and activation signature genes (Fig. S1 B). Indeed, cluster #8 also exhibited a high Tfh score based on the reported gene sets (Meckiff et al., 2020), suggesting that it includes cTfh cells (Fig. 1, C and D). Within this cluster, we could identify 1,735 TCRs (Table S3). Among 147 TCRs from the first batch of analyses, which included five patients (Ts-002, -016, -017, -018, and -020) and one healthy donor (Oo-001), we detected S-reactive clones bearing TCRαβ pairs shared between patients Ts-017 and Ts-018, who exhibited anti-S and neutralizing antibodies (Table S1). Considering the cell number of single-cell TCR-seq, these are likely to be frequent TCRs, and we designated them as TCR-017 and TCR-018, respectively. TCR-017 and -018 had the same Vα, Jα, Vβ, and Jβ usages, and their complementarity-determining region 3 (CDR3) sequences were identical except for one amino acid in CDR3β (Fig. 1, E and F). TCR-018 was detected in patient Ts-018 as three different barcoded clones (Table S3).

Single-cell–based analyses of SARS-CoV-2–responsive T cells. PBMCs from healthy donors and convalescent COVID-19 patients were stimulated with SARS-CoV-2–derived antigens as indicated for 20 h before antigen-responding T cells were sorted and analyzed by single-cell TCR- and RNA-seq. (A) A workflow of single-cell–based analyses. Antigen-stimulated PBMCs were stained with eight different hashtag antibodies (#1–8), anti-CD3, and the indicated activation marker antibodies. Hashtagged cells from two patients were pooled. Antigen-responding (CD69+ or CD137+ or CD154+) CD3+ T cells were sorted and immediately subjected to single-cell TCR- and RNA-seq. Each collection day after diagnosis is shown after the donor identifier (ID). (B) Activation score of each single cell was calculated based on the expression of activation genes (CD69, TNFRSF9,TNFRSF4, and CD40LG).
Single-cell–based analyses of SARS-CoV-2–responsive T cells. PBMCs from healthy donors and convalescent COVID-19 patients were stimulated with SARS-CoV-2–derived antigens as indicated for 20 h before antigen-responding T cells were sorted and analyzed by single-cell TCR- and RNA-seq. (A) A workflow of single-cell–based analyses. Antigen-stimulated PBMCs were stained with eight different hashtag antibodies (#1–8), anti-CD3, and the indicated activation marker antibodies. Hashtagged cells from two patients were pooled. Antigen-responding (CD69+ or CD137+ or CD154+) CD3+ T cells were sorted and immediately subjected to single-cell TCR- and RNA-seq. Each collection day after diagnosis is shown after the donor identifier (ID). (B) Activation score of each single cell was calculated based on the expression of activation genes (CD69, TNFRSF9,TNFRSF4, and CD40LG).

TCRs shared by SARS-CoV-2–reactive Tfh cells from different patients. (A) UMAP projection of T cells in single-cell analysis of PBMCs identified a cluster representing Tfh cells (cluster #8). Each dot corresponds to a single cell and is colored according to the cluster.(B) The expression levels of the canonical Tfh cell markers CD200, PDCD1,ICOS, CXCL13,CD40LG, and CXCR5 are shown as heatmaps in the UMAP plot. (C) Tfh scores of T cells in single-cell analysis of PBMCs are shown as heatmaps in the UMAP plot.(D) Tfh score of each cluster determined in A is shown in a violin plot. (E) Usages of V and J gene segments and CDR3 sequences of the α (TRAV, TRAJ, and CDR3α, respectively) and β chains of TCR-017 and TCR-018. For TCR-018, three different barcoded clones having the same nucleotide sequences detected in Ts-018 are shown. (F) Alignment of CDR3α and CDR3β nucleotide and amino acid sequences from TCR-017 and TCR-018. Nucleotide sequences within the white boxes indicate N-nucleotides. (G) Surface expression of paired TCRs on reporter cells are assessed by surface anti-CD3 staining. Filled histogram, anti-mouse CD3 mAb; open histogram, isotype control antibody. Data are representative of three independent experiments (G).
TCRs shared by SARS-CoV-2–reactive Tfh cells from different patients. (A) UMAP projection of T cells in single-cell analysis of PBMCs identified a cluster representing Tfh cells (cluster #8). Each dot corresponds to a single cell and is colored according to the cluster.(B) The expression levels of the canonical Tfh cell markers CD200, PDCD1,ICOS, CXCL13,CD40LG, and CXCR5 are shown as heatmaps in the UMAP plot. (C) Tfh scores of T cells in single-cell analysis of PBMCs are shown as heatmaps in the UMAP plot.(D) Tfh score of each cluster determined in A is shown in a violin plot. (E) Usages of V and J gene segments and CDR3 sequences of the α (TRAV, TRAJ, and CDR3α, respectively) and β chains of TCR-017 and TCR-018. For TCR-018, three different barcoded clones having the same nucleotide sequences detected in Ts-018 are shown. (F) Alignment of CDR3α and CDR3β nucleotide and amino acid sequences from TCR-017 and TCR-018. Nucleotide sequences within the white boxes indicate N-nucleotides. (G) Surface expression of paired TCRs on reporter cells are assessed by surface anti-CD3 staining. Filled histogram, anti-mouse CD3 mAb; open histogram, isotype control antibody. Data are representative of three independent experiments (G).
TCR-017 and -018 recognize an identical S epitope restricted by the same HLAs
To examine the antigen specificity of TCR-017 and -018, the respective TCRα and β chains were reconstituted in a TCR-deficient T cell hybridoma bearing an NFAT-GFP reporter (Fig. 1 G). TCR transfectants were stimulated with SARS-CoV-2 antigens in the presence of transformed B cells derived from the same patients as autologous APCs. Cells expressing TCR-017 and -018 responded to recombinant S protein and the S peptide pool but not to the MN peptide pool (Fig. 2 A), which is consistent with the initial antigen specificity revealed in single-cell analysis (Table S3). Among two halves of pooled S peptides, #1 (S1–643) and #2 (S633–1273), only #2 pool reacted with both TCRs (Fig. 2 B). However, another SARS-CoV-2 S peptide pool (S304–338, 421–475, 492–519, 683–707, 741–770, 785–802, 885–1273; termed selected S pool) did not activate either clone (Fig. S2 A and Fig. 2 B). These results suggest that the antigen epitopes for TCR-017 and -018 are located in regions 633–682, 708–740, 771–784, or 803–884 of the S protein (Fig. S2 A).

TCR-017 and -018 recognize the same core epitope, S870–878, presented on DRB1*15:01. (A) Cells expressing TCR-017 or TCR-018 were left unstimulated or stimulated with 10 μg/ml recombinant S protein, 1 μg/ml S protein peptide pool, or 1 μg/ml MN peptide pool in the presence of autologous APCs and analyzed for GFP and CD69 expression. (B–D) Cells expressing TCR-017 or TCR-018 were stimulated with 0.3 μg/ml S peptide pool #1, #2, or selected S pool (S304–338, 421–475, 492–519, 683–707, 741–770, 785–802, 885–1273; B), 1 μg/ml S peptide pool #2 in the presence of HEK293T cells expressing the indicated HLAs (C), or 1 μg/ml single peptides S828–846 or S864–882 (D). (E) Sensitivity of TCR-017 and TCR-018 to the S864–882 epitope. TCR transfectants were stimulated with the indicated concentrations of S864–882 peptide and APCs from donor Ts-018. Activities are shown as percentages of the maximum responses induced by plate-coated anti-CD3 mAb. Data are shown as mean ± SD of triplicates. (F) Determination of the core epitope recognized by TCR-017 and TCR-018 (shown in red). Cells were stimulated with 1 μg/ml of serial overlapping 15-mer peptides covering the S861–887 region. Data are shown as mean ± SD of triplicates. (G and H) Occurrence of TCRα-017/018 (CDR3, CVVNRGSSYKLIF) and TCRβ-017/018 (CDR3, CASS[Q or P]TYEQYF) in a prepandemic healthy cohort from Japan (n = 27; G) and in cohorts of prepandemic healthy donors (n = 786) and convalescent COVID-19 patients (n = 1,413) from multiple ethnicities (H). Numbers of individuals possessing the TCRs among cohorts are indicated. Data are representative of two independent experiments (A–D and F), or four independent experiments (E). pep, peptide; Unstim, unstimulated.
TCR-017 and -018 recognize the same core epitope, S870–878, presented on DRB1*15:01. (A) Cells expressing TCR-017 or TCR-018 were left unstimulated or stimulated with 10 μg/ml recombinant S protein, 1 μg/ml S protein peptide pool, or 1 μg/ml MN peptide pool in the presence of autologous APCs and analyzed for GFP and CD69 expression. (B–D) Cells expressing TCR-017 or TCR-018 were stimulated with 0.3 μg/ml S peptide pool #1, #2, or selected S pool (S304–338, 421–475, 492–519, 683–707, 741–770, 785–802, 885–1273; B), 1 μg/ml S peptide pool #2 in the presence of HEK293T cells expressing the indicated HLAs (C), or 1 μg/ml single peptides S828–846 or S864–882 (D). (E) Sensitivity of TCR-017 and TCR-018 to the S864–882 epitope. TCR transfectants were stimulated with the indicated concentrations of S864–882 peptide and APCs from donor Ts-018. Activities are shown as percentages of the maximum responses induced by plate-coated anti-CD3 mAb. Data are shown as mean ± SD of triplicates. (F) Determination of the core epitope recognized by TCR-017 and TCR-018 (shown in red). Cells were stimulated with 1 μg/ml of serial overlapping 15-mer peptides covering the S861–887 region. Data are shown as mean ± SD of triplicates. (G and H) Occurrence of TCRα-017/018 (CDR3, CVVNRGSSYKLIF) and TCRβ-017/018 (CDR3, CASS[Q or P]TYEQYF) in a prepandemic healthy cohort from Japan (n = 27; G) and in cohorts of prepandemic healthy donors (n = 786) and convalescent COVID-19 patients (n = 1,413) from multiple ethnicities (H). Numbers of individuals possessing the TCRs among cohorts are indicated. Data are representative of two independent experiments (A–D and F), or four independent experiments (E). pep, peptide; Unstim, unstimulated.

Epitope peptide and restricting HLAs of clonotype-017/018. (A) Relative positions within the S protein that are covered by each S peptide pool are shown. Strong binding peptides (%RANK ≤2) presented by DRA-DRB1*15:01 in regions 633–682, 708–740, 771–784, and 803–884 (pink regions) were predicted using NetMHC 4.0 Server. 19-mer peptides (red bars) that cover the strong binding peptides were synthesized and used for further epitope identification. (B) Cells expressing TCR-017 or -018 were stimulated with 1 μg/ml of S peptide pool #2 in the presence of heterologous APCs from exchanged donors (Ts-018 and Ts-017, respectively). (C) Cells expressing TCR-017 or -018 were stimulated with 1 μg/ml of peptide S864–882 in the presence of HEK293T cells expressing the indicated HLAs. Data are representative of two independent experiments (B and C). NTD, N-terminal domain; FP, fusion peptide; TM, transmembrane domain.
Epitope peptide and restricting HLAs of clonotype-017/018. (A) Relative positions within the S protein that are covered by each S peptide pool are shown. Strong binding peptides (%RANK ≤2) presented by DRA-DRB1*15:01 in regions 633–682, 708–740, 771–784, and 803–884 (pink regions) were predicted using NetMHC 4.0 Server. 19-mer peptides (red bars) that cover the strong binding peptides were synthesized and used for further epitope identification. (B) Cells expressing TCR-017 or -018 were stimulated with 1 μg/ml of S peptide pool #2 in the presence of heterologous APCs from exchanged donors (Ts-018 and Ts-017, respectively). (C) Cells expressing TCR-017 or -018 were stimulated with 1 μg/ml of peptide S864–882 in the presence of HEK293T cells expressing the indicated HLAs. Data are representative of two independent experiments (B and C). NTD, N-terminal domain; FP, fusion peptide; TM, transmembrane domain.
To further reduce the number of epitope candidates, we determined the HLA alleles that restrict TCR-017 and -018. Heterologous APCs from patient Ts-017 could activate TCR-018, and vice versa, demonstrating that these APCs are interchangeable (Fig. S2 B). Thus, the shared HLA class II alleles, DRA-DRB1*15:01, DQA1*01:02-DQB1*06:02, or DPA1*02:02-DPB1*05:01 (shown in red in Table S2), are considered to restrict this recognition. Since DRB1*15:01 and DQB1*06:02 are in linkage disequilibrium and would not be segregated from each other (Begovich et al., 1992), these HLA alleles were separately transfected into HEK293T cells to examine their ability to present S peptides to TCR-017 and -018. Only cells transfected with DRA and DRB1*15:01 could activate these TCRs in the presence of S peptides (Fig. 2 C). Using NetMHC Server software (Reynisson et al., 2020), two epitopes, S828–846 and S864–882, were predicted as strong binders to DRA-DRB1*15:01 within the candidate S regions described above (Fig. S2 A). Among them, S864–882 was ultimately identified as the epitope for both TCR-017 and -018 (Fig. 2 D). Judging from their dose–response curves, the relative affinities of TCR-017 and -018 to this peptide appeared comparable (Fig. 2 E). Furthermore, serial overlapping peptides determined S870–878 as a minimum epitope for both TCRs (Fig. 2 F). Given the same epitope on the same HLA with similar affinities, TCR-017 and -018 are considered as one clonotype, clonotype-017/018, shared by different patients. As would be expected from their similarity to DRB1*15:01, DRB1*15:02/*15:03/*15:04 also present this epitope and activate clonotype-017/018 (Fig. S2 C).
Public clonotypes expanded in recovered COVID-19 patients preferentially recognize conserved S epitopes
Consistent with the high frequency of DRB1*15, which is shared by 33% of the Japanese population (http://hla.or.jp), bulk TCR-seq of prepandemic healthy Japanese donors revealed that TCRα and β chains of clonotype-017/018 are prevalent (Fig. 2 G). As DRB1*15 is not a minor allele worldwide (Gonzalez-Galarza et al., 2020), we analyzed TCR databases of healthy and convalescent COVID-19 cohorts containing donors of various ethnicities (Emerson et al., 2017; Nolan et al., 2020 Preprint) and found that approximately one-fifth of donors possessed TCRβ-017/018 (Fig. 2 H). These results suggest that clonotype-017/018 is a public clonotype.
TCR-017/018 is not an exceptional public clonotype because 855 of 1,735 TCRs identified in our samples were also detected in these cohorts (Table S3). Among them, we identified 10 public cTfh clonotypes that were significantly expanded in recovered COVID-19 patients compared with the healthy cohort (Fig. 3 A, column J, asterisks). Of note, one-half of such clonotypes were indeed detected in multiple patients in our sample pool by single-cell TCR-seq (Fig. 3 A, column A, #), and TCR-017/018 was the fifth most expanded clonotype (Fig. 3 A, columns A and J).
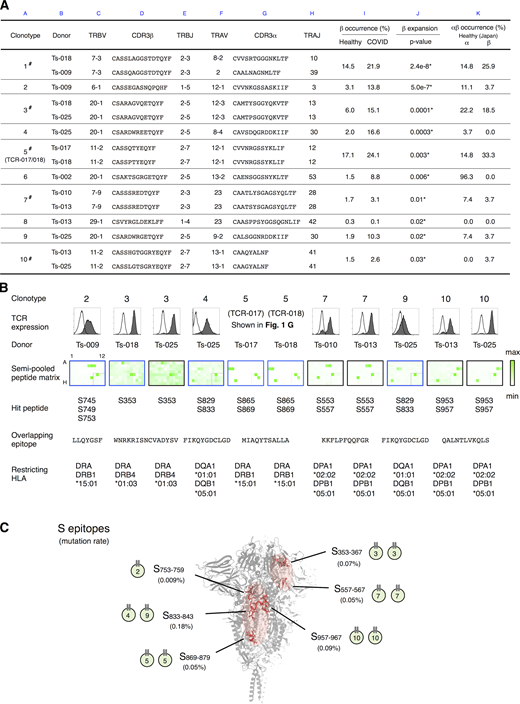
Determination of epitopes of public Tfh clonotypes expanded in COVID-19 patients within conserved S regions exhibiting low mutation rates.(A) Characterization of public clonotypes. Clonotypes detected in multiple donors from our sample pool are indicated by # (column A). Clonotype occurrences were calculated based on a prepandemic healthy cohort from Japan (n = 27, column K) and cohorts of prepandemic healthy donors (n = 786) and convalescent COVID-19 patients (n = 1,413) from multiple ethnicities (column I). For clonotypes that contain different TCRα or TCRβ sequences, occurrences were calculated using pooled sequences (columns K and I). Clonotypes are listed following the order of significance of TCRβ expansion. Clonotypes expanded significantly in recovered patients are indicated by * (P < 0.05, column J). (B) The TCR pair of each clonotype listed in A was reconstituted into reporter cells, and their epitopes and restricting HLAs were identified. TCR expression levels of paired TCRs on reporter cells are indicated by surface CD3 expression as shown in the histograms. Filled histogram, TCR-reconstituted reporter cells; open histogram, parental cells. Semi-pooled peptide matrix described in Fig. S3 A was used for epitope determination, in which reporter cells were stimulated in the presence of HEK293T expressing shared HLAs or autologous APCs and GFP reporter activities are shown as heatmaps. Unless noted otherwise, a peptide name that starts with an S followed by a number in normal size refers to a 15-mer S peptide, and the number indicates the position of its N-terminal amino acid in S protein. Restricting HLAs were determined as described in Fig. S3, B–G; Table S2, and Materials and methods. Clonotypes 1, 6, and 8 did not respond to S peptide pools. (C) Locations of epitopes described in B are highlighted in red in the three-dimensional structure of the SARS-CoV-2 S protein (Protein Data Bank under accession no. 6XR8). Frequencies of mutation (mutation rate) defined relative to Wuhan-Hu-1 were calculated based on data from CoV-GLUE and GISAID. Data are representative of two independent experiments (B).
Determination of epitopes of public Tfh clonotypes expanded in COVID-19 patients within conserved S regions exhibiting low mutation rates.(A) Characterization of public clonotypes. Clonotypes detected in multiple donors from our sample pool are indicated by # (column A). Clonotype occurrences were calculated based on a prepandemic healthy cohort from Japan (n = 27, column K) and cohorts of prepandemic healthy donors (n = 786) and convalescent COVID-19 patients (n = 1,413) from multiple ethnicities (column I). For clonotypes that contain different TCRα or TCRβ sequences, occurrences were calculated using pooled sequences (columns K and I). Clonotypes are listed following the order of significance of TCRβ expansion. Clonotypes expanded significantly in recovered patients are indicated by * (P < 0.05, column J). (B) The TCR pair of each clonotype listed in A was reconstituted into reporter cells, and their epitopes and restricting HLAs were identified. TCR expression levels of paired TCRs on reporter cells are indicated by surface CD3 expression as shown in the histograms. Filled histogram, TCR-reconstituted reporter cells; open histogram, parental cells. Semi-pooled peptide matrix described in Fig. S3 A was used for epitope determination, in which reporter cells were stimulated in the presence of HEK293T expressing shared HLAs or autologous APCs and GFP reporter activities are shown as heatmaps. Unless noted otherwise, a peptide name that starts with an S followed by a number in normal size refers to a 15-mer S peptide, and the number indicates the position of its N-terminal amino acid in S protein. Restricting HLAs were determined as described in Fig. S3, B–G; Table S2, and Materials and methods. Clonotypes 1, 6, and 8 did not respond to S peptide pools. (C) Locations of epitopes described in B are highlighted in red in the three-dimensional structure of the SARS-CoV-2 S protein (Protein Data Bank under accession no. 6XR8). Frequencies of mutation (mutation rate) defined relative to Wuhan-Hu-1 were calculated based on data from CoV-GLUE and GISAID. Data are representative of two independent experiments (B).
To determine the epitopes of these clonotypes more efficiently, we established a rapid epitope determination platform (Fig. S3 A) and validated it using TCR-017/018 (Fig. 3 B, clonotype 5). Using this platform, we could determine the epitopes of additional public cTfh clonotypes expanded in patients (Fig. 3 B). The restricting HLAs of these epitopes were determined using transformed B cells and HLA transfectants (Table S2 andFig. S3, B–G). All identified epitopes were located in two major regions within the conserved trimer-forming interface of the S protein and exhibited low mutation rates (Fig. 3 C; Singer et al., 2020,Preprint; Elbe and Buckland-Merrett, 2017). These results reveal that public cTfh clonotypes expanded during SARS-CoV-2 infection preferentially recognize epitopes located within the S region, and these regions are conserved across the emerging variants (Emerson et al., 2017; Nolan et al., 2020 Preprint). Of note, sera from donors who possessed these clonotypes were positive for anti–receptor binding domain (RBD) and neutralizing antibodies (Table S1).

Identification of the epitopes of public clonotypes. (A) A scheme of the rapid epitope determination platform used for identification of the epitopes for T cell clonotypes. 15-mer peptides with 11-amino acid overlap that cover the full length of the S protein of SARS-CoV-2 were synthesized and pooled as described in Materials and methods. Each peptide is present in a unique combination of two different pools; thus, the epitope can be revealed by the common peptides in the pools that activate the clonotype. Peptide pools of S, M, and N proteins of SARS-CoV-2 or HCoV S proteins were also added to the matrix as controls. S864 indicates S864-882 peptide.(B) Identification of DRB1*15:01 as a restricting HLA of S753–759 for clonotype 2. Reporter cells expressing clonotype 2 were activated by S749–763 in the presence of HEK293T cells transfected with DRA*01:01-DRB1*15:01. Data are shown as mean ± SD of triplicates. (C) Exclusion of DPA1*02:02-DPB1*05:01 as a restricting HLA of S353–367 for clonotype 3. Reporter cells expressing clonotype 3 were not activated by S353-including peptide pools, such as A5, E8, and E9 (S pool #1) in A, in the presence of HEK293T cells transfected with DPA1*02:02-DPB1*05:01.(D) DRB4*01:03 is a restricting HLA of S353–367 for clonotype 3. Reporter cells expressing clonotype 3 were activated by S353 in the presence of B cells from Ts-009. (E) DQA1*01:01-DQB1*05:01 is a restricting HLA of S833–843 for clonotypes 4 and 9. Reporter cells expressing clonotype 4 or 9 were activated by S828–846 in the presence of B cells from donors Ts-025 (autologous), Ts-013, and Ts-003 but not activated in the presence of B cells from donors Ts-001 and HIWA. (F) DPA1*02:02-DPB1*05:01 is a restricting HLA of S557–567 for clonotype 7. Reporter cells expressing clonotype 7 were activated by S553- or S557-including peptide pools, such as B7, B8, F4, and E9 (S pool #1) in A, in the presence of HEK293T cells transfected with DPA1*02:02-DPB1*05:01.(G) DPA1*02:02-DPB1*05:01 is a restricting HLA of S957–967 for clonotype 10. Reporter cells expressing clonotype 10 were activated by S953- or S957-including peptide pools, such as C11, C12, G4, and E10 (S pool #2) in A, in the presence of HEK293T cells transfected with DPA1*02:02-DPB1*05:01. Data are representative of two independent experiments (B–G).
Identification of the epitopes of public clonotypes. (A) A scheme of the rapid epitope determination platform used for identification of the epitopes for T cell clonotypes. 15-mer peptides with 11-amino acid overlap that cover the full length of the S protein of SARS-CoV-2 were synthesized and pooled as described in Materials and methods. Each peptide is present in a unique combination of two different pools; thus, the epitope can be revealed by the common peptides in the pools that activate the clonotype. Peptide pools of S, M, and N proteins of SARS-CoV-2 or HCoV S proteins were also added to the matrix as controls. S864 indicates S864-882 peptide.(B) Identification of DRB1*15:01 as a restricting HLA of S753–759 for clonotype 2. Reporter cells expressing clonotype 2 were activated by S749–763 in the presence of HEK293T cells transfected with DRA*01:01-DRB1*15:01. Data are shown as mean ± SD of triplicates. (C) Exclusion of DPA1*02:02-DPB1*05:01 as a restricting HLA of S353–367 for clonotype 3. Reporter cells expressing clonotype 3 were not activated by S353-including peptide pools, such as A5, E8, and E9 (S pool #1) in A, in the presence of HEK293T cells transfected with DPA1*02:02-DPB1*05:01.(D) DRB4*01:03 is a restricting HLA of S353–367 for clonotype 3. Reporter cells expressing clonotype 3 were activated by S353 in the presence of B cells from Ts-009. (E) DQA1*01:01-DQB1*05:01 is a restricting HLA of S833–843 for clonotypes 4 and 9. Reporter cells expressing clonotype 4 or 9 were activated by S828–846 in the presence of B cells from donors Ts-025 (autologous), Ts-013, and Ts-003 but not activated in the presence of B cells from donors Ts-001 and HIWA. (F) DPA1*02:02-DPB1*05:01 is a restricting HLA of S557–567 for clonotype 7. Reporter cells expressing clonotype 7 were activated by S553- or S557-including peptide pools, such as B7, B8, F4, and E9 (S pool #1) in A, in the presence of HEK293T cells transfected with DPA1*02:02-DPB1*05:01.(G) DPA1*02:02-DPB1*05:01 is a restricting HLA of S957–967 for clonotype 10. Reporter cells expressing clonotype 10 were activated by S953- or S957-including peptide pools, such as C11, C12, G4, and E10 (S pool #2) in A, in the presence of HEK293T cells transfected with DPA1*02:02-DPB1*05:01. Data are representative of two independent experiments (B–G).
Crystal structure of TCR-017 reveals sequence flexibility in CDR3 extending the publicity of SARS-CoV-2–specific clonotypes
The only difference between TCR-017 and -018 is the amino acid at position 94 (Q94 and P94, respectively). Since these two residues have distinct characteristics, we suspected that any amino acid at this position allows epitope recognition. Indeed, any of 17 amino acid substitutions, except for D94, retained reactivity to the S864–882 epitope on DRA-DRB1*15:01 (Fig. 4 A). We thus solved the crystal structure of TCR-017 and found that the residue at position 94 in CDR3β is located away from CDR3α and thus unlikely to contribute to antigen recognition (Fig. 4, B and C). C90, A91, S92, and S93 were also distant from CDR3α (Fig. 4 C), suggesting that this TCRβ may mainly use a C-terminal half of CDR3β for recognition and allow diverse TCR sequence variants or even other Vβ genes.

Antigen recognition and clonal expansion of public clonotype 5 (TCR-017/018). (A) TCRβ-017, -018, or mutants at position 94 were coexpressed with TCRα-017/018 and human CD4 in reporter cells. Cells were kept unstimulated or stimulated with peptide S864–882 or anti-CD3 antibody in the presence of APCs from donor Ts-018. Percentages of GFP+ cells in the CD3+ populations are shown. Data are shown as mean ± SD of duplicates. (B and C) Crystal structure of TCRαβ heterodimer of clonotype 5 (TCR-017). The entire view (B) and a close-up of the CDR3 domains (C) are shown. Q94 and adjacent amino acids, C90, A91, S92, and S93 within CDR3β are indicated. (D) Occurrences of extended antigen-reactive TCRβ chains harboring the 17 acceptable substitutions at position 94 (shown as X; except for D94 of TCRβ-017/018) as determined in A within individuals in a prepandemic healthy cohort from Japan (n = 27) and within cohorts of prepandemic healthy donors (n = 786) and convalescent COVID-19 patients (n = 1,413) from multiple ethnicities. Numbers of individuals possessing this TCRβ among cohorts are indicated. (E) Frequencies of extended antigen-reactive TCRβ chains harboring the 17 acceptable substitutions at position 94 (except for D94 of TCRβ-017/018) as determined in A within the prepandemic healthy donors, all convalescent COVID-19 patients, and patients admitted to the ICU and non-ICU setting from multiple ethnicities. Horizontal line, median; bottom/top, first/third quartile; whiskers, 1.5 times the interquartile range. Data are representative of two independent experiments (A). *, P < 0.05; ***, P < 0.00001.
Antigen recognition and clonal expansion of public clonotype 5 (TCR-017/018). (A) TCRβ-017, -018, or mutants at position 94 were coexpressed with TCRα-017/018 and human CD4 in reporter cells. Cells were kept unstimulated or stimulated with peptide S864–882 or anti-CD3 antibody in the presence of APCs from donor Ts-018. Percentages of GFP+ cells in the CD3+ populations are shown. Data are shown as mean ± SD of duplicates. (B and C) Crystal structure of TCRαβ heterodimer of clonotype 5 (TCR-017). The entire view (B) and a close-up of the CDR3 domains (C) are shown. Q94 and adjacent amino acids, C90, A91, S92, and S93 within CDR3β are indicated. (D) Occurrences of extended antigen-reactive TCRβ chains harboring the 17 acceptable substitutions at position 94 (shown as X; except for D94 of TCRβ-017/018) as determined in A within individuals in a prepandemic healthy cohort from Japan (n = 27) and within cohorts of prepandemic healthy donors (n = 786) and convalescent COVID-19 patients (n = 1,413) from multiple ethnicities. Numbers of individuals possessing this TCRβ among cohorts are indicated. (E) Frequencies of extended antigen-reactive TCRβ chains harboring the 17 acceptable substitutions at position 94 (except for D94 of TCRβ-017/018) as determined in A within the prepandemic healthy donors, all convalescent COVID-19 patients, and patients admitted to the ICU and non-ICU setting from multiple ethnicities. Horizontal line, median; bottom/top, first/third quartile; whiskers, 1.5 times the interquartile range. Data are representative of two independent experiments (A). *, P < 0.05; ***, P < 0.00001.
Considering this flexibility within the CDR3β sequence, the occurrence of extended clonotype 5 was increased (Fig. 4 D), which gives us a more precise expansion ratio in recovered patients. Indeed, increase in the frequencies of this extended clonotype in individual COVID-19 patients compared with the healthy individuals in Fig. 3 A became more significant (Fig. 4 E). In the COVID-19 cohort, the frequency was also significantly higher in patients not admitted to the intensive care unit (non-ICU), but not in ICU patients, than that in the healthy control cohort. Furthermore, it was significantly higher in non-ICU than in ICU patients (Fig. 4 E). These results suggest that expansion of this clonotype during COVID-19 is associated with mild symptoms and, therefore, its epitope may serve as an antigen for inducing protective immunity.
Public cTfh clonotypes did not cross-react with HCoVs but with symbiotic bacteria
We next examined the cross-reactive antigens of these clonotypes. “Common cold” HCoVs have been reported as cross-reactive examples for SARS-CoV-2 (Grifoni et al., 2020; Mateus et al., 2020; Braun et al., 2020). However, TCR-017/018 did not react with S proteins derived from HCoV-OC43 (Fig. S4, A and B). Furthermore, none of the SARS-CoV-2–specific public clonotypes characterized in Fig. 3 responded to HCoV peptides (Fig. 3 B; and Fig. S4, C and D). Thus, these clonotypes are unlikely to cross-react with reported HCoVs present before the outbreak of COVID-19. Instead, a homology search of core epitopes activating the most prevalent clonotype 5, S870–878, revealed that oral commensal bacteria,Selenomonas noxia, possesses the same epitope in a protein called multidrug and toxic compound extrusion (MATE) family efflux transporter (Fig. 5 A). Indeed, the corresponding peptides (MATE241–260 and MATE242–256) that include this epitope were recognized by both TCR-017 and -018 on DRB1*15:01 (Fig. 5 B).

Cross-reactivity of public Tfh clonotypes. (A) Cells expressing TCR-017 or -018 were cocultured with HEK293T cells expressing the indicated HLAs and S proteins derived from the indicated CoVs. (B) Cells expressing TCR-017 or -018 were stimulated with 1 μg/ml of peptide pool derived from HCoV-OC43 S protein. (C) Sequences of peptides from HCoVs corresponding to S867–881 of SARS-CoV-2. Amino acids that are the same as those in SARS-CoV-2 in the core epitope are shown in black. (D) Cells expressing TCR-017 or -018 were stimulated with 1 μg/ml of the peptides shown in C. Data are representative of two independent experiments (A, B, and D). pep, peptide; Unstim, unstimulated.
Cross-reactivity of public Tfh clonotypes. (A) Cells expressing TCR-017 or -018 were cocultured with HEK293T cells expressing the indicated HLAs and S proteins derived from the indicated CoVs. (B) Cells expressing TCR-017 or -018 were stimulated with 1 μg/ml of peptide pool derived from HCoV-OC43 S protein. (C) Sequences of peptides from HCoVs corresponding to S867–881 of SARS-CoV-2. Amino acids that are the same as those in SARS-CoV-2 in the core epitope are shown in black. (D) Cells expressing TCR-017 or -018 were stimulated with 1 μg/ml of the peptides shown in C. Data are representative of two independent experiments (A, B, and D). pep, peptide; Unstim, unstimulated.

Cross-reactivity of SARS-CoV-2–specific public Tfh clonotypes. (A) Alignment of corresponding peptides. Amino acids identical to SARS-CoV-2 epitopes are shown in black. (B) Cells expressing clonotype 5 (TCR-017 or TCR-018) were stimulated with 1 μg/ml of peptides derived from the MATE family efflux transporter of S. noxia. (C) Cells expressing the TCR from clonotype 2 were stimulated with 1 μg/ml of peptides derived from K. pneumoniae yersiniabactin ABC transporter ATP-binding/permease protein (YbtP) or Bacteroidales bacterium TonB-dependent receptor (TBDR). Accession numbers of TBDRs from different Bacteroidales bacteria in GenBank are shown. (D) Cells expressing TCRs from clonotype 5 (TCR-017) or clonotype 2 were stimulated with heat-killed E. coli transformed with expression vectors for the indicated microbial proteins in the presence of monocyte-derived dendritic cells (Mo-DCs) expressing DRB1*15:01. Data are representative of two independent experiments (B–D).
Cross-reactivity of SARS-CoV-2–specific public Tfh clonotypes. (A) Alignment of corresponding peptides. Amino acids identical to SARS-CoV-2 epitopes are shown in black. (B) Cells expressing clonotype 5 (TCR-017 or TCR-018) were stimulated with 1 μg/ml of peptides derived from the MATE family efflux transporter of S. noxia. (C) Cells expressing the TCR from clonotype 2 were stimulated with 1 μg/ml of peptides derived from K. pneumoniae yersiniabactin ABC transporter ATP-binding/permease protein (YbtP) or Bacteroidales bacterium TonB-dependent receptor (TBDR). Accession numbers of TBDRs from different Bacteroidales bacteria in GenBank are shown. (D) Cells expressing TCRs from clonotype 5 (TCR-017) or clonotype 2 were stimulated with heat-killed E. coli transformed with expression vectors for the indicated microbial proteins in the presence of monocyte-derived dendritic cells (Mo-DCs) expressing DRB1*15:01. Data are representative of two independent experiments (B–D).
Among other epitopes, the epitope of the most expanded clonotype 2 (5.9-fold;Table S3), S753–759, was contained in gut symbiotic microbes, such as Bacteroidales and Klebsiella pneumoniae (Fig. 5 A; Atarashi et al., 2017; Sefik et al., 2015). We confirmed that corresponding synthetic peptides activated T cells expressing the clonotype 2 TCR (Fig. 5 C). Furthermore, Escherichia coli transformed with expression vectors for these proteins could activate public clonotypes in the presence of HLA-matched dendritic cells (Fig. 5 D), confirming that cross-reactive antigenic epitopes are processed and presented. These observations imply that these bacteria might affect protective immunity against SARS-CoV-2 infection.
S864–882 could activate multiple public cTfh clonotypes
S864–882 is an epitope activating the most prevalent clonotypes (Fig. 4 D). To examine the ability of this epitope to activate multiple cTfh clonotypes, T cells from DRB1*15 donors were subjected to single-cell TCR- and RNA-seq (Fig. 6 A and Table S4). We identified 104 public cTfh clonotypes that responded to S864–882 after filtering by two criteria: (1) expanded during stimulation based on single-cell sequencing (>0.1% of total CD4+ T cells) and (2) detected in cohort databases (Table S4, columns P–S). Bulk TCR-seq confirmed that both α and β chains of all 104 clonotypes were indeed expanded after S864–882 stimulation (Table S4, columns J and K). These results indicate that the S864–882 peptide is capable of expanding multiple public cTfh clones in DRB1*15 donors. Consistent with this, we indeed identified another clonotype within the Tfh cluster (Table S3) that recognizes the identical epitope S864–882 on the same HLA through TCR distinct from clonotype 5 (Fig. 6, B–D); this clonotype was also public (Table S3, Seurat barcode, DB2_CAAGAAAGTGCGGTAA).
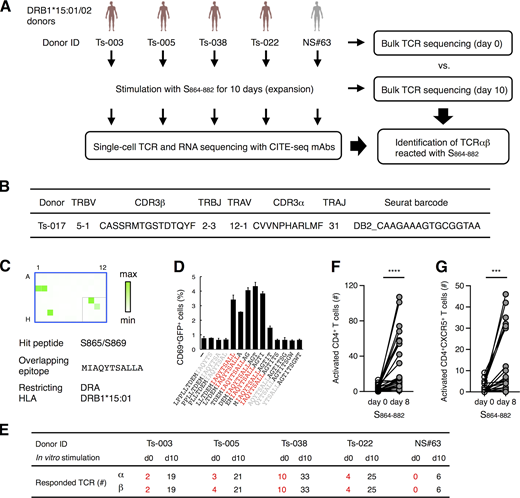
Characterization of S864–882-reactive T cells.(A) Workflow of the analysis for the S864–882-induced T cell expansion. PBMCs from convalescent COVID-19 patients (red) and healthy (gray) donors possessing DRB1*15:01 or *15:02 were analyzed by bulk TCR-seq or stimulated with 1 μg/ml S864–882 peptide for 10 d before CD4+ T cells were sorted and analyzed by single-cell TCR- and RNA-seq as well as bulk TCR-seq. (B) V and J usages and CDR3 sequences of the newly identified clonotype that recognizes S870–878 epitope. (C) Reporter cells expressing TCRαβ in B were stimulated with pooled peptide matrix described in Fig. S3 A in the presence of DRA-DRB1*15:01–expressing APCs. Wells containing S865 or S869 (C1, C2, and G3), S pool #2 (E10), and S864–882 (F11) were positive. (D) 1 μg/ml of serial overlapped 15-mer peptides covering the S861–887 region were tested for reactivity to the clonotype in B to determine the minimum epitope (shown in red). Data are shown as mean ± SD of triplicates. (E) Numbers of TCRα and β of expanded clonotypes (proportion >0.1% in single-cell TCR-seq) detected in bulk TCR-seq described in Table S4 are shown. (F and G) 105 of PBMCs from healthy donors were left unstimulated (day 0) or stimulated with S864–882 for 8 d (day 8). Numbers of CD4+CD69+CD137+ T cells (F) and CD4+CD69+CD137+CXCR5+ Tfh cells (G) are shown. Data are representative of two independent experiments (C and D). ***, P < 0.005; ****, P < 0.001. ID, identifier.
Characterization of S864–882-reactive T cells.(A) Workflow of the analysis for the S864–882-induced T cell expansion. PBMCs from convalescent COVID-19 patients (red) and healthy (gray) donors possessing DRB1*15:01 or *15:02 were analyzed by bulk TCR-seq or stimulated with 1 μg/ml S864–882 peptide for 10 d before CD4+ T cells were sorted and analyzed by single-cell TCR- and RNA-seq as well as bulk TCR-seq. (B) V and J usages and CDR3 sequences of the newly identified clonotype that recognizes S870–878 epitope. (C) Reporter cells expressing TCRαβ in B were stimulated with pooled peptide matrix described in Fig. S3 A in the presence of DRA-DRB1*15:01–expressing APCs. Wells containing S865 or S869 (C1, C2, and G3), S pool #2 (E10), and S864–882 (F11) were positive. (D) 1 μg/ml of serial overlapped 15-mer peptides covering the S861–887 region were tested for reactivity to the clonotype in B to determine the minimum epitope (shown in red). Data are shown as mean ± SD of triplicates. (E) Numbers of TCRα and β of expanded clonotypes (proportion >0.1% in single-cell TCR-seq) detected in bulk TCR-seq described in Table S4 are shown. (F and G) 105 of PBMCs from healthy donors were left unstimulated (day 0) or stimulated with S864–882 for 8 d (day 8). Numbers of CD4+CD69+CD137+ T cells (F) and CD4+CD69+CD137+CXCR5+ Tfh cells (G) are shown. Data are representative of two independent experiments (C and D). ***, P < 0.005; ****, P < 0.001. ID, identifier.
To examine the presence and longevity of these S864–882-reactive clones in convalescent patients, we assembled all 104 clonotypes with bulk TCR-seq of unstimulated peripheral T cells (Fig. 6 A, day 0). 19 of these 104 clonotypes were present in the periphery of convalescent patients (Fig. 6 E). These data suggest that a substantial frequency of S864–882-reactive public cTfh clones was maintained as a memory pool for at least 3 mo after infection (Table S1).
S864–882 is a promiscuous epitope presented by multiple HLA alleles
DRB1*15 alleles are frequent, being shared by 25% of the population worldwide (Gonzalez-Galarza et al., 2020). However, it is also important to examine whether S864–882 can be presented by individuals lacking DRB1*15. In silico analysis predicted 77 major HLA alleles as potential binders to S864–882, implying that most of the world population is capable of presenting peptides that include this epitope (Table S5; Vita et al., 2019). In line with this, most randomly collected healthy donors possessed some of these predicted binding alleles (Table S6); this was confirmed by assessing the direct binding of biotinylated epitope (Fig. S5). Indeed, peripheral T cells from these donors responded to S864–882 to express activation markers (Fig. 6 F). The number of activated CD4+CXCR5+ cells was also increased upon epitope stimulation (Fig. 6 G). These results suggest that S864–882 is a universal epitope activating multiple CD4+ T cells upon presentation on multiple HLAs and, therefore, is a critical region of the S protein serving as a T cell antigen in SARS-CoV-2 infection.

Presentation of S868–880 epitope on multiple HLA alleles. Transformed B cells from healthy donors described inFig. 6, F and G, were stained with (filled histogram) or without (open histogram) 10 μg/ml biotinylated S868–880 peptide with a GSGSGS linker at the N-terminus at 37°C for 12 h followed by adding streptavidin-phycoerythrin. DRA-DRB1*15:01 transfectant was used as a control. Data are representative of two independent experiments.
Presentation of S868–880 epitope on multiple HLA alleles. Transformed B cells from healthy donors described inFig. 6, F and G, were stained with (filled histogram) or without (open histogram) 10 μg/ml biotinylated S868–880 peptide with a GSGSGS linker at the N-terminus at 37°C for 12 h followed by adding streptavidin-phycoerythrin. DRA-DRB1*15:01 transfectant was used as a control. Data are representative of two independent experiments.
Discussion
This study reports the identification of cTfh epitopes within the SARS-CoV-2 S protein that may contribute to the protective T cell responses against COVID-19. In the wake of the COVID-19 outbreak, some public TCRs recognizing SARS-CoV-2 have recently been reported (Shomuradova et al., 2020; Dykema et al., 2021). In the present study, we first identified and determined the crystal structure of SARS-CoV-2–specific public TCRs of CD4+ T cells associated with mild COVID-19 symptoms. Individuals possessing such public cTfh clonotypes are expected to recognize S protein and to induce the production of antibodies against this cognate antigen for immune protection (Kaneko et al., 2020; Zhang et al., 2021; Gong et al., 2020). Consistent with this view, convalescent patients who expressed the public cTfh clonotypes in our sample pool had anti–S-neutralizing antibodies and recovered from COVID-19 (Table S1).
As these public clonotypes were widely detected in multiple ethnicities, precise monitoring of the frequencies of public Tfh clonotypes by developing specific probes could become a novel option for prognosis prediction. Given that their restricting HLAs cover a large global population (Gonzalez-Galarza et al., 2020) and, in particular, S864–882 is a universal epitope presented by multiple HLAs, such common S epitopes are promising antigens for promoting protective Tfh responses against SARS-CoV-2 worldwide.
Since most of the identified epitopes are located in the non-RBD region and included in the current full-length S vaccines, this study provides a molecular affirmation of and additional rationale for the vaccines being administered worldwide (Baden et al., 2021; Walsh et al., 2020; Sadoff et al., 2021; Voysey et al., 2021; Shinde et al., 2021). As these epitopes are conserved among SARS-CoV-2 variants (Fig. 3 C; Singer et al., 2020,Preprint; Elbe and Buckland-Merrett, 2017), these peptides are expected to generate T cell memory against Alpha: B.1.1.7 (UK), Beta: B.1.351 (South Africa), Gamma: P.1 (Brazil), Delta: B.1.617.2 (India), and future mutants that escape from neutralizing antibodies (Wang et al., 2021; Collier et al., 2021; Cele et al., 2021). The universal T epitope S864–882 could be a candidate for peptide vaccines upon coupling with appropriate linear B cell epitopes (Sauer et al., 2021); in fact such a combined peptide may provide an “adjustable” booster for variants in the postvaccine era. Furthermore, identification of additional public clonotypes and their epitopes using the platform established in this study would be an intriguing next step in extending the number of candidate T cell antigens. In addition, cross-reacting symbiotic bacteria may contribute to the priming of SARS-CoV-2–specific T cells, thus providing a novel perspective on prognosis and prevention. The search for antigenic cross-reactive epitopes having acceptable substitutions will expand the pool of environmental “priming” antigens. Further global metagenomic analysis might clarify the relationship between the presence of cross-reactive commensals and resistance to COVID-19, offering a potential explanation of local/ethnic variation in disease severity and another route to fighting this virus.
The current study has a limitation in the definition of Tfh cells, as we collected T cells from PBMCs. Although circulating Tfh cells in the periphery reflect germinal center Tfh cells (Locci et al., 2013; Hill et al., 2019; Heit et al., 2017; Schmitt et al., 2014; Crotty, 2019), the analysis of tissue-resident Tfh cells using biopsy samples is ideal. Different TCR-seq methods also have advantages and limitations. Single-cell TCR-seq can determine αβ pairs, but throughput is limited. Bulk sequencing can detect more diverse clonotypes, but pair information cannot be obtained. Taken together, the current study shows that a combination of single-cell and bulk TCR-seq, global TCR databases, and the TCR reconstitution/epitope determination platform is an efficient workflow to identify beneficial public helper/cytotoxic clonotypes and epitopes. This methodology could provide a powerful tool for future outbreaks of other infectious diseases.
Materials and methods
Human subjects
The institutional review boards of Osaka University (approval number 898-4) and National Institute of Infectious Diseases (approval number 1237) approved blood draw protocols for convalescent COVID-19 and healthy individuals. The institutional review board of the nonprofit organization MINS (approval number 190210) approved the analyses in KOTAI Biotechnologies using blood samples from healthy individuals. The research was performed in accordance with all relevant guidelines and regulations. Written informed consent was obtained from all participants or designated health care surrogates if participants were unable to provide informed consent. Study enrollment criteria included subjects >20 yr old, regardless of disease severity and genders (Table S1). Prior to enrollment in this study, all COVID-19 donors were confirmed to be positive for SARS-CoV-2 by PCR using nasopharyngeal swab specimens. Blood from convalescent COVID-19 donors and healthy donors was obtained at Tokyo Shinagawa Hospital and Osaka University. Samples were deidentified before analyses. For PBMC preparation, whole blood was collected in heparin-coated tubes and centrifuged to separate the cellular fraction and plasma followed by density-gradient sedimentation. For neutralizing antibody assay, plasma samples from patients were heat inactivated for 30 min at 56°C. Plasma diluted at 1:5 followed by twofold serial dilutions was incubated with equal volume of solution containing 100 median tissue culture infectious dose of SARS-CoV-2 for 1 h at 37°C and added to VeroE6/TMPRSS2 cells. After incubation for 5 d, the highest plasma dilution that protected 100% of cells from cytopathic effect was taken as the neutralization titer. Disease severity was defined as mild, moderate I, moderate II, or severe in accordance with the Japanese COVID-19 Clinical Practice Guideline Version 2.2 (Ministry of Health, Labour and Welfare, 2020). The study using samples from healthy donors was approved by the ethical committee of Osaka University Hospital (approval number 20483). Comprehensive informed consent has been previously obtained and approved by the institutional review board of Osaka Universal Hospital (approval number 209008-B). Waiver of informed consent was approved for projects involving the secondary analysis of the residual samples, and an opt-out method was applied to obtain consent in this study by using the announcement online.
SARS-CoV-2
For the preparation of inactivated SARS-CoV-2, SARS-CoV-2 (KNG19-020) was kindly supplied by Dr. Tomohiko Takasaki (Kanagawa Prefectural Institute of Public Health, Kanagawa, Japan). The virus was propagated in VeroE6/TMPRSS2 cells (JCRB1819) and purified by sucrose gradient centrifugation (Dent and Neuman, 2015). Concentrated virus was then inactivated by ultraviolet light (0.6 J/cm2).
Antigens
Inactivated virus was used at 2.9 × 106 viral particles/ml (∼1 μg S protein/ml) for stimulation. For the preparation of recombinant SARS-CoV-2 S protein, the ectodomain of S protein was cloned into a mammalian expression vector pCMV, with a foldon sequence followed by 9× His-tag and Strep-tag at the C-terminus. Polybasic cleavage site (RRAR) of S protein was replaced by a single alanine, and K986P and V987P mutations were introduced to stabilize the conformation as previously described (Amanat et al., 2021). After transient transfection into Expi293F cells (Gibco), secreted protein was purified from the culture supernatant 4 d after transfection using TALON Metal Affinity Resin (Clontech) and an Amicon Ultra 10K Centrifugal Filter Device (Millipore). For the expression of S proteins of SARS-CoV-2 and HCoV-OC43 for antigen presentation, pME18S expression vectors for each protein were transfected into HEK293T cells. For the expression of bacterial proteins in E. coli, cDNA for MATE family efflux transporter, TonB-dependent receptor, and TonB-dependent receptor plug domain–containing protein was cloned into pCold expression vector and transformed into E. coli BL21 (DE3) competent cells, Champion21 (SMBIO Technology). The protein expressions were induced by the addition of 0.5 or 1.0 mM isopropyl-β-D-thiogalactopyranoside at 37°C. Bacterial pellets were washed and resuspended with PBS and incubated at 95°C for 5 min for inactivation. The expressions of the proteins were confirmed by SDS-PAGE and Coomassie brilliant blue staining. PepMix SARS-CoV-2 (Spike Glycoprotein), which contains pool #1 and #2, was purchased from JPT Peptide Technologies. PepTivator SARS-CoV-2 for S, M, and N protein were purchased from Miltenyi Biotec. Domains of SARS-CoV-2 S protein shown in Fig. S2 A were previously described (Wrapp et al., 2020). All of other peptides were from GenScript. Three-dimensional structure of SARS-CoV-2 S protein (Protein Data Bank accession no. 6XR8) was depicted with program PyMOL (The PyMOL Molecular Graphics System, Version 2.0; Schrödinger, LLC).
In vitro stimulation of PBMCs
Cryopreserved PBMCs were thawed and washed with RPMI 1640 medium (Sigma) supplemented with 5% human AB serum (Gemini Bio), penicillin (Sigma), streptomycin (MP Biomedicals), and 2-mercaptoethanol (Nacalai Tesque). 5 × 105 PBMCs were stimulated in the same medium with inactivated SARS-CoV-2, 1 or 10 μg/ml of recombinant S protein, 1 μg/ml of S peptide pool, or 1 μg/ml of MN peptide pool for 20 h, followed by staining with anti-human CD3, CD69, CD137, CD154, and TotalSeq-C Hashtag antibodies. CD69+, CD137+, or CD154+ cells within a CD3+-gated population were sorted by SH800S Cell Sorter (Sony Biotechnology) and used for single-cell TCR- and RNA-seq analyses. For epitope-specific clonal expansion, 1–5 × 105 PBMCs were stimulated with 1 μg/ml of S864–882 for 10 d, and recombinant human IL-2 (1 ng/ml, PeproTech) was added at day 4 and day 7. CD4+ T cells were sorted and analyzed by single-cell TCR- and RNA-seq.
Single-cell–based transcriptome and TCR repertoire analysis
Single-cell capturing and library preparation were performed using the following reagents: Chromium Next GEM Single Cell 5′ Library & Gel Bead Kit v1.1, 16 rxns, PN-1000165; Chromium Next GEM Chip G Single Cell Kit, 48 rxns, PN-1000120; Chromium Single Cell V(D)J Enrichment Kit, Human T Cell, 96 rxns, PN-1000005; Single Index Kit T Set A, 96 rxns, PN-1000213; Chromium Single Cell 5′ Feature Barcode Library Kit, 16 rxns, PN-1000080; and Single Index Kit N Set A, 96 rxns, PN-1000212. Single-cell suspension containing ∼2 × 104 cells were loaded into Chromium microfluidic chips to generate single-cell gel bead-in-emulsion using Chromium Controller (10x Genomics) according to the manufacturer’s instructions. RNA from the barcoded cells for each sample was subsequently reverse-transcribed inside gel bead-in-emulsion using a Veriti Thermal Cycler (Thermo Fisher Scientific), and all subsequent steps to generate single-cell libraries were performed according to the manufacturer’s protocol, with 14 cycles used for cDNA amplification. Then, ∼50 ng of cDNA was used for gene expression library amplification by 14 cycles in parallel with cDNA enrichment and library construction for TCR libraries. Fragment size of the libraries was confirmed with an Agilent 2100 Bioanalyzer. Libraries were sequenced on Illumina NovaSeq 6000 as paired-end mode (read 1, 28 bp; read 2, 91 bp). The raw reads were processed by Cell Ranger 3.1.0 (10x Genomics). Gene expression–based clustering was performed using the Seurat R package (v3.1; Hafemeister and Satija, 2019). Briefly, cells with a mitochondrial content >10% and cells with <200 or >4,000 genes detected were considered outliers (dying cells and empty droplets and doublets, respectively) and filtered out. The Seurat SCTransform function was used for normalization, and data were integrated without performing batch-effect correction as all samples were processed simultaneously. Hashtag oligo demultiplexing was performed on centered log ratio–normalized hashtag unique molecular identifier counts, and clonotypes were matched to the gene expression data through their droplet barcodes, using Python scripts. Only cells assigned a single hashtag and a β-chain clonotype were retained for downstream analyses. Tfh scores and activation scores were generated using a published list of Tfh-enriched transcripts (Meckiff et al., 2020) and four well-known activation genes (CD69, TNFRSF9, TNFRSF4, and CD40LG), respectively. Calculations were performed in R with the AddmoduleScore function of Seurat, using the default parameters. Briefly, the average expression levels of all genes of the gene list were computed and subtracted by the average expression of control genes randomly selected from similar aggregate expression level bins.
Bulk TCR-seq and analysis
1–3 × 105 PBMCs were lysed in QIAzol (QIAGEN). Full-length cDNA was then synthesized using SMARTer technology (Takara Bio), and the variable regions of TCRα and β genes were amplified using TRAC/TRBC-specific primers. After sequencing of the variable region amplicons, each pair of reads was assigned a clonotype (defined as TR(A/B)V and TR(A/B)J genes and CDR3) using MiXCR software (Bolotin et al., 2015). For each α/β clonotype, expansion was defined as the fraction of reads for that clone divided by the total number of reads for the α/β chain, respectively, and fractions were converted to log10 for plotting and statistical analyses.
HLA class II typing
For HLA class II typing, genomic DNA samples were isolated from PBMCs using QIA DNA Mini Kit (QIAGEN). AllType FASTplex NGS 11 Loci Flex Kit (One Lambda) was used to prepare DNA sequence libraries (DPA1, DPB1, DQA1, DQB1, DRB1, and DRB3/4/5) according to the manufacturer’s protocol. Sequencing was performed on a MiSeq System (Illumina). TypeStream Visual version 2.0 (One Lambda) was used to analyze the DNA sequences. For DRB1*15:01/02 typing, genomic DNA was amplified with a DRB1*15/16-specific forward primer (5′-CGTTTCCTGTGGCAGCCTAAGAGG-3′) and a DRB1*15-specific reverse primer (5′-CCGCGCCTGCTCCAGGAT-3′) followed by DNA sequencing.
APCs
Recombinant EBV was produced as previously reported (Kanda et al., 2015). Viral stocks were obtained by concentrating virus-containing culture supernatants by ultracentrifugation at 32,000 rpm for 1 h. 3 × 105 PBMCs were incubated with an aliquot of the viral stock for 1 h at 37°C. The infected cells were cultured with RPMI 1640 medium supplemented with 20% FBS (Capricorn Scientific GmbH) containing 0.1 μg/ml cyclosporine A (Cayman Chemical). EBV-immortalized B lymphoblastoid cell lines were obtained after 3-wk culture and used as APCs. To generate APCs expressing specific HLA, HEK293T cells were transfected with plasmids encoding HLA class II alleles as previously described (Jiang et al., 2013). EBV experiments were approved by the Ministry of Education, Culture, Sports, Science and Technology (approval number 539) and the institutional review board of Osaka University (approval number 04658). To generate monocyte-derived dendritic cells, CD14+ cells were isolated from PBMCs using CD14 MicroBeads, human (Miltenyi Biotec), and cultured in RPMI 1640 medium supplemented with 10% FBS, 0.1 mM Non-Essential Amino Acids Solution (Gibco), 1 mM sodium pyruvate (Gibco), 10 ng/ml human GM-CSF (PeproTech), and IL-4 (PeproTech) for 6 d.
TCR reconstitution and stimulation
TCRα and β chain cDNA sequences were synthesized and cloned into retroviral vectors pMX-IRES-rat CD2 and pMX-IRES-human CD8, respectively. Two vectors containing paired TCRα and β chains were transfected together into Phoenix packaging cells using PEI MAX (Polysciences). Supernatant containing retroviruses was used for infection into mouse T cell hybridoma with an NFAT-GFP reporter gene (Matsumoto et al., 2021) to reconstitute TCRαβ pairs. TCRβ mutants were constructed by site-directed mutagenesis. For antigen stimulation, TCR-reconstituted cells were cocultured with stimulants in the presence of immortalized autologous B cells unless indicated otherwise. After 20 h, T cell activation was assessed by GFP or CD69 expression.
Rapid epitope determination platform
For the preparation of pooled peptide matrix, 15-mer peptides with 11-amino acid overlap that cover the full length of S protein of SARS-CoV-2 were individually synthesized (GenScript). Peptides were dissolved in DMSO at 12 mg each peptide/ml, and 2–12 peptides were mixed to create 75 different semipools, as indicated in Fig. S3 A, so that the responsible epitopes could be determined from the cell reactivities to horizontal and vertical pools. Pooled peptides were adjusted to 1 mg each peptide/ml in DMSO, followed by 10× dilution with water, and 1 μl of the solution was added into each corresponding well. Commercial S peptide pools and S864–882 peptide were also loaded as controls. S peptide pools of four common cold HCoVs (229E, NL63, OC43, and HKU1) were loaded to assess cross-reactivities. For reporter cell assay, 100 μl of media containing T cells and APCs were added to the wells so that T cells were stimulated with each peptide at 1 μg/ml. After 20 h, GFP reporter induction was assessed in a T cell–gated population using an Attune NxT flow cytometer (Thermo Fisher Scientific).
Restricting HLA determination
For the clonotypes detected in more than one donor from our sample pool, reporter cells were stimulated in the presence of HEK293T cells expressing individual pairs of alleles shared by the donors. Otherwise, reporter cells were stimulated in the presence of transformed B cells from various donors that have partial overlapped HLAs with the original donor of the clonotype.
Database analyses
Public repertoire datasets of prepandemic healthy donors (n = 786, Adaptive ImmuneACCESS; Emerson et al., 2017) or COVID-19 convalescent donors (n = 1,413, Adaptive ImmuneRACE; Nolan et al., 2020,Preprint) of various ethnicities from the US, Italy, and Spain were downloaded from the Adaptive Biotechnologies website, and V-GENE names were renamed to match the IMGT nomenclature. Finally, clonotypes and their expansions were defined in the same way as in-house bulk TCR-seq. TCR occurrence in a dataset was defined as the fraction of patients whose repertoire contained the given TCR, regardless of its expansion. A two-sided Student’st test was used to compare expansion values. Considering the extremely high diversity of observed TCR clonotype sequences, conventional P value adjustment for multiple testing has not been performed in the field (Emerson et al., 2017), and P values are therefore nonadjusted unless stated otherwise.
Expression and purification of soluble TCRαβ heterodimer
Expression constructs encoding the extracellular domains of TCRα-017 (Q1-D207 in mature protein) and TCRβ-017 (G1-G241) subunits were incorporated into pCold vector including 6× His-tag and a tobacco etch virus protease cleavage site. For crystallization, the point mutations (T159C in α and S166C/C184A in β) were introduced to form an artificial disulfide bond as described previously (Boulter et al., 2003). The plasmids were transformed into E. coli BL21 competent cells Champion21 and Rosetta2 (DE3; Novagen), respectively. The protein expression was induced by the addition of 0.5 mM isopropyl-β-D-thiogalactopyranoside at 18°C. Cells were suspended with 500 mM NaCl-containing Tris-HCl buffer (pH 8.0) and disrupted with sonication. The inclusion bodies, including target proteins, were collected by centrifugation. The inclusion body was then solubilized by 50 mM Tris-HCl buffer (pH 8.0) containing 6 M guanidine HCl, 10 mM EDTA, and 2 mM dithiothreitol at room temperature. The equal amount of solubilized TCRα and TCRβ (35 mg each) was mixed and rapidly diluted with 1 liter of 100 mM Tris-HCl buffer (pH 8.0) containing 5 M urea, 0.4 M L-arginine, 5 mM reduced glutathione, and 0.5 mM oxidized glutathione at 4°C. The diluted solution was further dialyzed against 10 mM Tris-HCl buffer (pH 8.0) at 4°C for 2 d. The dialyzed solution was applied onto 5 ml nickel-nitrilotriacetic acid agarose (FUJIFILM Wako), and His-tagged TCRαβ were eluted with elution buffer (50 mM Tris-HCl [pH 8.0], 300 mM NaCl, and 250 mM imidazole). After removal of His-tag by tobacco etch virus protease, the eluted protein was concentrated and further applied to Superdex 75 (GE Healthcare) equilibrated with 20 mM Tris-HCl (pH 8.0) buffer containing 100 mM NaCl. The TCRαβ heterodimer fractions were concentrated up to 1.2 mg/ml by Amicon Ultra (molecular weight cutoff, 10 kD). The purity of the proteins was assessed by SDS-PAGE and Coomassie brilliant blue staining. The numbering of amino acids in CDR3 sequences of TCRs was based on mature protein.
Crystallization, data collection, and structure determination of TCR-017 ectodomain
All crystallization trials were performed by sitting drop vapor diffusion method. Initial crystallization conditions were screened using Index (Hampton Research), SG1 Screen, and SG2 Screen (Molecular Dimensions). The best diffracted crystal was obtained under the condition of 0.1 M Hepes-Na (pH 7.0) and 1.1 M sodium malonate at 20°C. Prior to x-ray diffraction experiments, crystals were soaked in the reservoir containing 20% ethylene glycol and flash cooled in liquid nitrogen. X-ray diffraction datasets were collected at the beamline BL-1A in the Photon Factory. Diffraction data were integrated with program XDS (Kabsch, 2010) and scaled with program SCALA (Evans, 2006). The phases of datasets were determined by molecular replacement method using program MOLREP (Vagin and Teplyakov, 2010) with the coordinate of 1E6 TCR (Protein Data Bank accession no. 5C0B; Cole et al., 2016). After initial phase determination, the model buildings were manually performed using program COOT (Emsley et al., 2010). Refinement was performed using program REFMAC5 (Vagin et al., 2004) at the initial step and Phenix.refine for the final model (Afonine et al., 2012). The stereochemical quality of the final model was assessed by program MolProbity (Williams et al., 2018). Data collection and refinement statistics are summarized in Table S7. Structural factors and the atomic coordinates of TCRαβ-017 ectodomain have been deposited in Protein Data Bank under accession no. 7EA6. All figures of 3D structure were depicted with program PyMOL (The PyMOL Molecular Graphics System, Version 2.0; Schrödinger, LLC).
Antibodies
Anti-human CD3 (HIT3a), anti-human CD8 (RPA-T8), anti-human CD69 (FN50), anti-human CD137 (4B4-1), anti-human CD154 (24-31), anti-human CD4 (OKT4), anti-human CXCR5 (J252D4), TotalSeq-C Hashtags (LNH-94; 2M2), anti-mouse CD3 (17A2), anti-mouse CD69 (H1.2F3), and anti-rat CD2 (OX-34) antibodies were purchased from BioLegend. Rat IgG2b κ Isotype Control (eB149/10H5) was purchased from eBioscience.
Statistical analysis
Statistical analysis in Fig. 3 A and Fig. 4 E was performed with a two-sided Student’s t test to compare expansion values, and P values are indicated. Analyses in Fig. 6, F and G, were performed with paired t test, and P values are indicated.
Online supplemental material
Fig. S1 shows our workflow for single-cell–based analyses of SARS-CoV-2–responsive T cells. Fig. S2 shows epitope peptides and restricting HLAs of clonotype-017/018. Fig. S3 shows identification of the epitopes and restricting HLAs of public clonotypes. Fig. S4 shows no cross-reactivity of clonotype-017/018 to other HCoVs. Fig. S5 shows presentation of S868–880 epitope on multiple HLA alleles. Table S1 shows donor characteristics. Table S2 shows HLA class II types of involved blood donors. Table S3 shows cTfh clonotypes identified in single-cell analysis. Table S4 shows S864–882-reactive public cTfh clonotypes. Table S5 shows frequencies of HLAs in the Japanese population that are predicted to present epitopes of public clonotypes. Table S6 shows predicted binding alleles to S864–882 in healthy donors. Table S7 shows data collection and refinement statistics of the crystallographic analysis of TCRαβ-017.
Data availability
Recombinant SARS-CoV-2 S protein, TCR-reconstituted cells, and soluble TCRαβ heterodimers are available from the corresponding author on request under a standard material transfer agreement. Single-cell–based transcriptome data have been deposited in Gene Expression Omnibus datasets under accession no. GSE184806). Structural factors and the atomic coordinates of the TCRαβ-017 ectodomain have been deposited in Protein Data Bank under accession no. 7EA6). Other data needed to support the study conclusions are included in the main text and online supplemental material.
Acknowledgments
We thank T. Ito, K. Toyonaga, Y. Adachi, S. Moriyama, Y. Harima, D. Azizah, H. Hayashi, and J. Sun for experimental support and C. Schutt, W. Ise, J. B. Wing, D. Okuzaki, D. Standley, S. Teraguchi, S. Futami, T. Sasazuki, and T. Kobayashi for discussion.
This research was supported by Japan Agency for Medical Research and Development (20fk0108542, 20fk0108403 [H. Arase], 20fk0108265, 20nk0101602, 20fk0108454, 21nf0101623, 21gm0910010, 21ak0101070, 20fk0108075, 20fk0108104, 21fk0108608, and 21fk0108534 [S. Yamasaki]), Japan Society for the Promotion of Science Grants-in-Aid for Scientific Research JP18H05279 (H. Arase) and JP20H00505 (S. Yamasaki), the Kansai Economic Federation, and the Mitsubishi Foundation (S. Yamasaki). The Department of Health Development and Medicine is an endowed department supported by AnGes, Daicel, and FunPep.
Author contributions: S. Yamasaki conceptualized research; X. Lu, Y. Hosono, S. Ishizuka, E. Ishikawa, M. Nagae, and Y. Ozaki did investigation; R. Shinnakasu, T. Inoue, T. Onodera, T. Matsumura, M. Shinkai, T. Sato, S. Mori, T. Kanda, E.E. Nakayama, T. Shioda, T. Kurosaki, H. Arase, and Y. Takahashi provided resources; D. Motooka, N. Sax, Y. Maeda, Y. Kato, T. Morita, S. Nakamura, M. Nagae, K. Takeda, A. Kumanogoh, H. Nakagami, and K. Yamashita did data curation; S. Yamasaki supervised the research; and X. Lu, Y. Hosono, and S. Yamasaki wrote the manuscript.

