


The human placenta is a dynamic organ that modulates physiological adaptations to pregnancy. To define the immunological signature of the human placenta, we performed unbiased profiling of secreted immune factors from human chorionic villi isolated from placentas at mid and late stages of pregnancy. We show that placental trophoblasts constitutively secrete the inflammasome-associated cytokines IL-1β and IL-18, which is blocked by NLRP3 inflammasome inhibitors and occurs without detectable gasdermin D cleavage. We further show that placenta-derived IL-1β primes monocytes for inflammasome induction to protect against Listeria monocytogenes infection. Last, we show that the human placenta responds to L. monocytogenes infection through additional inflammasome activation and that inhibition of this pathway sensitizes villi to infection. Our results thus identify the inflammasome as an important mechanism by which the human placenta regulates systemic and local immunity during pregnancy to defend against L. monocytogenes infection.

Introduction
Infections are one of the most common threats to human reproductive health. Infections during pregnancy can cause prematurity and stillbirth and can be vertically transmitted to the fetus, causing congenital infection or disease (Silasi et al., 2015; Bicker et al., 2008; Kourtis et al., 2014). In addition to fetal risks, infections and the systemic inflammatory responses to these infections remain the second leading known cause of maternal death in the United States (Petersen et al., 2019). Despite advances in technology and implementation of rapid response systems, the rates of morbidity and mortality from infections in pregnancy have remained unchanged since maternal surveillance data were collected in 1986 (Davis et al., 2019; Petersen et al., 2019). A basic understanding of many of the mechanisms by which the immune system changes and is regulated throughout pregnancy remains unclear.
The placenta forms the sole interface between the maternal and fetal compartments throughout pregnancy, and teratogenic microbes must traverse this barrier to cause fetal disease. In humans, the placenta is composed of chorionic villi that form during the first trimester and are covered by a single layer of contiguous, multinucleated syncytiotrophoblast. This layer forms a physical barrier to vertical transmission and is the primary source of communication from the fetus-derived placenta to the maternal systemic circulation. These cells possess substantial exocrine and endocrine activity. Syncytiotrophoblasts are highly immunologically active, even in the absence of microbial infections (reviewed in Ander et al., 2019; Arora et al., 2017). For example, trophoblasts constitutively secrete substrates such as type III IFNs that restrict viral infections in both fetus- and mother-derived tissues (Bayer et al., 2016; Corry et al., 2017; Jagger et al., 2017). In addition to factors that are constitutively released, syncytiotrophoblasts also respond to infections through the release of specific immunoregulatory factors such as chemokines and cytokines (Ander et al., 2018). However, the full repertoire of immunological factors secreted from the placenta and whether these factors differ at different points in gestation or in response to different infectious pathogens remain largely unknown.
Given that placental villi are in direct contact with the maternal circulation, the placenta has the capacity to regulate inflammatory processes systemically through contact with circulating maternal blood and immune cells and locally at the maternal–fetal interface. Although pregnancy has classically been described as an immunosuppressed state in order to prevent rejection of the semiallogeneic fetus (Billingham et al., 1953), pregnant women are able to mount robust immune responses, as evidenced by their effective responses to vaccines (Kay et al., 2015; Muňoz et al., 2019; Vilajeliu et al., 2015). In fact, many infections commonly associated with higher rates of morbidity and mortality during pregnancy may arise from enhanced inflammatory responses to infection rather than from direct pathogen-induced damage or immunosuppression (Periolo et al., 2015; Rasmussen et al., 2012; Raj et al., 2014; Le Gars et al., 2019; Kourtis et al., 2014). Likewise, adverse fetal outcomes can be correlated with maternal-derived immunological responses that alter maternal tolerance or dysregulate placental function (Koga et al., 2009; Cardenas et al., 2010; Yockey et al., 2018).
In this study, we sought to define the immunological secretome of human placental chorionic villi under basal states and in response to microbial infection. Using multianalyte Luminex-based profiling of 80 cytokines, chemokines, and growth factors released from human chorionic villi isolated from >25 human placentas, we found that placental trophoblasts constitutively release the inflammasome-associated cytokines IL-1β and IL-18. The inflammasome is a protein complex initially identified in the myeloid cell lineage whose signaling is initiated through a two-step process (Man and Kanneganti, 2015; He et al., 2016). Inflammasome priming involves the transcriptional up-regulation of sensors such as NLRP3 and the inactive forms of IL-1β, IL-18, and caspase-1 through NF-κB, along with ubiquitination and phosphorylation of apoptosis-related speck-like protein (ASC; Budai et al., 2017; Man and Kanneganti, 2015). The activation step, most commonly through pattern- or damage-associated molecular signaling, induces inflammasome complex aggregation to activate caspase-1, which cleaves the precursor forms of IL-1β and IL-18 into their active proinflammatory forms. Caspase-1 also cleaves gasdermin D, which induces membrane pore formation that facilitates secretion of IL-1β and cell death by pyroptosis (Liu et al., 2016). In cases of sustained inflammasome activation, IL-1β release may occur independent of gasdermin D cleavage and pyroptosis, as has been described in myeloid cells (Monteleone et al., 2018). In addition to the constitutive release of IL-1β and IL-18, we found that release of these cytokines was enhanced in response to infection of the human placenta by Listeria monocytogenes. We also found that inflammasome signaling was involved in the defense of the human placenta from L. monocytogenes infection and that inhibition of this pathway enhanced infection. Our study thus identifies the inflammasome as a key driver of innate immunity at the maternal–fetal interface.
Results
Chorionic villi constitutively release IL-1β and IL-18
To define the immunological profile of the human placenta under basal conditions, we generated conditioned media (CM) containing secreted products from placental chorionic villi isolated from 29 human placentas (donor characteristics listed in Table S1). To compare differences over advancing gestational age, we collected placentas from midgestation elective terminations, preterm deliveries in the third trimester, and full-term deliveries, and we performed immune profiling of ∼80 cytokines, chemokines, and growth factors using multianalyte arrays on a Luminex platform. Of note, all placentas were gathered in the absence of labor, other proinflammatory conditions (e.g., infection, premature preterm rupture of membranes, abruption, diabetes), or genetic anomalies. Tissue function was confirmed by measuring the levels of β–human chorionic gonadotropin (βhCG) and pregnancy-specific glycoprotein 1 (PSG-1), which were released at high levels from chorionic villi isolated at both mid and late stages of pregnancy (Fig. S1, A and B). We performed similar analyses in CM isolated from the choriocarcinoma cell lines BeWo and JEG-3 to identify factors that were abundant in CM from primary placental villous explants compared with cell line–derived models.

βhCG and PSG-1 secretion in CM from chorionic villi and FM preparations and the difference in cytokine abundance after sPLS-DA. (A and B) Luminex assays for β-hCG (A) or ELISA for PSG-1 (B) in CM derived from midgestation (MG) or full-term (Term) chorionic villi (Villi) or FM. Data are shown as mU/ml (A) or ng/ml (B). (C) The variable importance plot derived from sPLS-DA represents the contribution of each cytokine selected on the first component, with length of the bar representing the importance of each cytokine to the component (importance from bottom to top). Colors indicate the samples from villi- and FM-derived CM in which the cytokine is most abundant. (D and E) Correlation analyses for IL-1β levels in villi-derived CM relative to the levels of IL-1α (D) or IL-18 (E). Correlation coefficients are shown in red at top left. (F) IL-1β levels in CM isolated from chorionic villi in the second or third trimesters and at term. Each symbol represents villi from unique placental preparations. (G) Secreted alkaline phosphatase activity (shown as OD655) in the medium of HEK293 reporter cells stably expressing IL-1R (in red) or the receptor for IFN-λs (in blue) and treated with recombinant IFN-λ3 (100 µg/ml) or IL-1β (100 µg/ml) or with CM from choriocarcinoma cell lines (BeWo, JEG3, and JAR) or with CM from three placental preparations of villi-derived CM. In A and B, individual villi or FM preparations are indicated by symbols. Data in G are shown as mean ± SD of three independent experiments. Significance in F was determined with the Kruskal-Wallis test. **, P < 0.01. Significance in G was determined with one-way ANOVA with Dunnett’s test for multiple comparisons compared with NCM-treated controls. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
βhCG and PSG-1 secretion in CM from chorionic villi and FM preparations and the difference in cytokine abundance after sPLS-DA. (A and B) Luminex assays for β-hCG (A) or ELISA for PSG-1 (B) in CM derived from midgestation (MG) or full-term (Term) chorionic villi (Villi) or FM. Data are shown as mU/ml (A) or ng/ml (B). (C) The variable importance plot derived from sPLS-DA represents the contribution of each cytokine selected on the first component, with length of the bar representing the importance of each cytokine to the component (importance from bottom to top). Colors indicate the samples from villi- and FM-derived CM in which the cytokine is most abundant. (D and E) Correlation analyses for IL-1β levels in villi-derived CM relative to the levels of IL-1α (D) or IL-18 (E). Correlation coefficients are shown in red at top left. (F) IL-1β levels in CM isolated from chorionic villi in the second or third trimesters and at term. Each symbol represents villi from unique placental preparations. (G) Secreted alkaline phosphatase activity (shown as OD655) in the medium of HEK293 reporter cells stably expressing IL-1R (in red) or the receptor for IFN-λs (in blue) and treated with recombinant IFN-λ3 (100 µg/ml) or IL-1β (100 µg/ml) or with CM from choriocarcinoma cell lines (BeWo, JEG3, and JAR) or with CM from three placental preparations of villi-derived CM. In A and B, individual villi or FM preparations are indicated by symbols. Data in G are shown as mean ± SD of three independent experiments. Significance in F was determined with the Kruskal-Wallis test. **, P < 0.01. Significance in G was determined with one-way ANOVA with Dunnett’s test for multiple comparisons compared with NCM-treated controls. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Using this approach, we identified 27 factors that were released from midgestation chorionic villi at very low (<100 pg/ml) to undetectable levels, 28 that were released at mid to high levels (>100 to <1,000 pg/ml), and 23 that were released at high to very high (>1,000 pg/ml) levels (Fig. 1 A). The two most abundant molecules present were soluble CD163 and IL-8, which were present in CM with average levels of ∼162 ng/ml and ∼30 ng/ml, respectively. Cytokines known to be constitutively released from chorionic villi, including the type III IFN-λ2, were present at values consistent with those reported previously (∼165 pg/ml; Fig. 1 A; Bayer et al., 2016; Corry et al., 2017). Of these cytokines, very few were retained in placenta-derived BeWo and JEG-3 cells (Fig. 1 A). To identify factors that could directly reach the maternal circulation, we compared secreted factors from chorionic villi and fetal membrane (FM) specimens from matched midgestation placentas (Fig. 1 B). FM purity was confirmed by comparative analysis of βhCG and PSG-1 in CM from matched villi and FM tissue explants (Fig. S1, A and B). Luminex-based profiling was performed on matched villi- and FM-derived CM, and sparse partial least squares discriminant analysis (sPLS-DA) was used to identify factors that contributed maximally to the differentiation of these two groups. The variable importance in the projection for the multiplex cytokine profiles identified a number of cytokines that were specifically released from chorionic villi compared with FM. Among the top villi-enriched cytokines was IL-1β, with chorionic villi containing high levels (∼145 pg/ml, on average) and CM from matched FMs containing low to undetectable levels (<10 to 15 pg/ml, on average; Fig. 1 C and Fig. S1 C). Related cytokines in the IL-1 family, such as IL-18 and IL-1α, exhibited similar trends, with elevated levels in CM isolated from chorionic villi compared with CM from FM (Fig. 1, D and E). IL-1β abundance in CM correlated with IL-1α and IL-18 levels, suggesting that a common molecular pathway might be responsible for the secretion of these cytokines (r2 = 0.92 and 0.66, respectively; Fig. S1, D and E). We also found that the levels of IL-1β were significantly lower in CM isolated from villi obtained from full-term placentas than in those isolated from the second trimester or from preterm placentas, suggesting that its secretion might decline with advanced gestation or before parturition (Fig. 1 F and Fig. S1 F). In contrast, there were no differences in the levels of other cytokines, such as IL-8 or IFN-λs, between midgestation and full-term villi (Fig. 1 A). This suggests that the differential amounts of IL-1β with advancing gestation are physiological and that these findings are not secondary to differences in tissue processing, tissue viability, or acute inflammation. We confirmed that IL-1β present in villi-derived CM was active by measuring the induction of IL-1β–mediated signaling using an HEK293 reporter cell line that expresses the IL-1 receptor (IL-1R) and a luciferase reporter to monitor downstream signaling. We found that villi-derived CM induced IL-1β–mediated signaling in these cells, which was comparable to signaling induced in similar reporter cells expressing the receptor for IFN-λs (Fig. S1 G). Together, these data identify IL-1β and IL-18 as being constitutively released from chorionic villi from normal placentas.

Luminex-based profiling of human midgestation and full-term placentas. (A) Heatmap of CM generated from chorionic villi isolated from 22 midgestation placentas and six third-trimester or term placentas analyzed by Luminex-based multianalyte profiling for the indicated cytokines, chemokines, and growth factors. Top: CM isolated from choriocarcinoma JEG-3 or BeWo cells. Middle: CM isolated from chorionic villi in the second trimester (17–23 wk). Bottom: CM isolated from chorionic villi in the third trimester (24–41 wk). Red indicates high levels of expression (>1,000 pg/ml), and blue indicates low (<100 pg/ml) to undetectable levels. Scale is shown at right. Luminex assays were performed in duplicate from at least three to five independent villi per placenta. (B) Heatmap of CM generated from FMs (isolated from 11 midgestation placentas matched to chorionic villi in A). Luminex assays were performed in duplicate from at least two or three independent explants per placenta. (C–E) Concentrations of IL-1β (C), IL-18 (D), and IL-1α (E) from chorionic villi (in red) or matched FM (in blue) samples shown in A and B. (F) Comparison of the level of IL-1β in CM isolated from chorionic villi (red, orange) or FM (dark blue, light blue) from midgestation (MG; red, dark blue) or full-term (orange, light blue) placentas. In C–F, each symbol represents villi from unique placental preparations. In C–F, each symbol represents a unique donor. Significance was determined by Mann-Whitney U test. ***, P < 0.001. In A and B, the red ** indicate cytokines described in the text.
Luminex-based profiling of human midgestation and full-term placentas. (A) Heatmap of CM generated from chorionic villi isolated from 22 midgestation placentas and six third-trimester or term placentas analyzed by Luminex-based multianalyte profiling for the indicated cytokines, chemokines, and growth factors. Top: CM isolated from choriocarcinoma JEG-3 or BeWo cells. Middle: CM isolated from chorionic villi in the second trimester (17–23 wk). Bottom: CM isolated from chorionic villi in the third trimester (24–41 wk). Red indicates high levels of expression (>1,000 pg/ml), and blue indicates low (<100 pg/ml) to undetectable levels. Scale is shown at right. Luminex assays were performed in duplicate from at least three to five independent villi per placenta. (B) Heatmap of CM generated from FMs (isolated from 11 midgestation placentas matched to chorionic villi in A). Luminex assays were performed in duplicate from at least two or three independent explants per placenta. (C–E) Concentrations of IL-1β (C), IL-18 (D), and IL-1α (E) from chorionic villi (in red) or matched FM (in blue) samples shown in A and B. (F) Comparison of the level of IL-1β in CM isolated from chorionic villi (red, orange) or FM (dark blue, light blue) from midgestation (MG; red, dark blue) or full-term (orange, light blue) placentas. In C–F, each symbol represents villi from unique placental preparations. In C–F, each symbol represents a unique donor. Significance was determined by Mann-Whitney U test. ***, P < 0.001. In A and B, the red ** indicate cytokines described in the text.
Trophoblasts secrete IL-1β through constitutive NLRP3 inflammasome activity
To determine the cell type(s) responsible for the secretion of IL-1β, we generated CM from primary human trophoblasts isolated from human midgestation chorionic villi and compared it with CM from matched villous explants. Levels of IL-1β in CM isolated from primary trophoblasts was comparable to those in matched villous explants, suggesting that these cells are the primary producers of IL-1β (Fig. 2 A). Consistent with our findings from full-term villous explants, we found that the levels of IL-1β were significantly lower in trophoblasts isolated from full-term placentas than in those isolated from midgestation placentas (Fig. S2 A). In contrast, levels of other cytokines, such as IL-8 and IFN-λs, were similar in CM from trophoblasts isolated from either midgestation or full-term placentas (Fig. S2 A). In addition to IL-1β in CM isolated from chorionic villi, we found that this medium also contained high levels of active caspase-1 (Fig. S2 B), which can be secreted following inflammasome activation (Shamaa et al., 2015; Laliberte et al., 1999).

Expression and localization of inflammasome-associated components in placental tissue. (A) Levels of IL-1β secreted by JEG-3 choriocarcinoma cells (gray), chorionic villi isolated from midgestation placentas (dark blue), or primary human trophoblasts (light blue) isolated from these midgestation villi. Each symbol represents a unique placental preparation. Luminex assays were performed in duplicate from three to five independent villi per placenta or from three independent trophoblast isolations. (B) Heatmap of inflammasome-associated transcripts in JEG-3 cells, primary human trophoblasts (PHT cells), and chorionic villi based on log2 reads per kilobase million values. Transcripts with no reads are shown in gray. Hierarchical clustering is shown at top. (C) Immunoblot from total protein isolated from chorionic villi (from five villi isolated from two unique midgestation placentas). Shown are immunoblots for AIM2 (top), gasdermin D (GasD), NLRP3 (third row), and GAPDH (bottom row) as a loading control. Molecular weights are shown at right. Immunoblots were performed at least three times. (D) Confocal microscopy of chorionic villi immunostained for total gasdermin D (top, green), pro–caspase-1 (second row, green), NLRP3 (third row, green), or cleaved IL-1β (bottom row, green) and counterstained for actin (red in all). DAPI-stained nuclei are shown in blue. At far right are zoomed images from the white boxes shown at left. White arrows denote staining in syncytiotrophoblasts (SYN), and clear arrows denote staining in Hofbauer cells (HC). Scale bar is 10 µm. Images are representative of at least five villi immunostained from at least two or three unique placentas. (E) Immunohistochemistry demonstrating the location of gasdermin D, cleaved gasdermin D, or pro–IL-1β in midgestation chorionic villi. At right are zoomed images of red boxes at left. Arrows demonstrate the multinuclear SYN layer. Scale bar is 10 µm. Images are representative of at least five villi from at least two or three unique placentas. (F) Immunoblots from total protein isolated from chorionic villi (from five villi isolated from two unique midgestation placentas) or from THP-1 cells treated with LPS overnight and then exposed to nigericin for 60 min (+Nig) or to mock control (−Nig). Shown are immunoblots for ASC (top), gasdermin D (GasD; second row), two exposures (short, top; long, bottom) of the N-terminal cleavage product of GasD (third and fourth rows), cleaved IL-1β (fifth row), and GAPDH (bottom row) as a loading control. Molecular weights are shown at right. Immunoblots were performed at least three times.
Expression and localization of inflammasome-associated components in placental tissue. (A) Levels of IL-1β secreted by JEG-3 choriocarcinoma cells (gray), chorionic villi isolated from midgestation placentas (dark blue), or primary human trophoblasts (light blue) isolated from these midgestation villi. Each symbol represents a unique placental preparation. Luminex assays were performed in duplicate from three to five independent villi per placenta or from three independent trophoblast isolations. (B) Heatmap of inflammasome-associated transcripts in JEG-3 cells, primary human trophoblasts (PHT cells), and chorionic villi based on log2 reads per kilobase million values. Transcripts with no reads are shown in gray. Hierarchical clustering is shown at top. (C) Immunoblot from total protein isolated from chorionic villi (from five villi isolated from two unique midgestation placentas). Shown are immunoblots for AIM2 (top), gasdermin D (GasD), NLRP3 (third row), and GAPDH (bottom row) as a loading control. Molecular weights are shown at right. Immunoblots were performed at least three times. (D) Confocal microscopy of chorionic villi immunostained for total gasdermin D (top, green), pro–caspase-1 (second row, green), NLRP3 (third row, green), or cleaved IL-1β (bottom row, green) and counterstained for actin (red in all). DAPI-stained nuclei are shown in blue. At far right are zoomed images from the white boxes shown at left. White arrows denote staining in syncytiotrophoblasts (SYN), and clear arrows denote staining in Hofbauer cells (HC). Scale bar is 10 µm. Images are representative of at least five villi immunostained from at least two or three unique placentas. (E) Immunohistochemistry demonstrating the location of gasdermin D, cleaved gasdermin D, or pro–IL-1β in midgestation chorionic villi. At right are zoomed images of red boxes at left. Arrows demonstrate the multinuclear SYN layer. Scale bar is 10 µm. Images are representative of at least five villi from at least two or three unique placentas. (F) Immunoblots from total protein isolated from chorionic villi (from five villi isolated from two unique midgestation placentas) or from THP-1 cells treated with LPS overnight and then exposed to nigericin for 60 min (+Nig) or to mock control (−Nig). Shown are immunoblots for ASC (top), gasdermin D (GasD; second row), two exposures (short, top; long, bottom) of the N-terminal cleavage product of GasD (third and fourth rows), cleaved IL-1β (fifth row), and GAPDH (bottom row) as a loading control. Molecular weights are shown at right. Immunoblots were performed at least three times.

Differences in cytokine secretion between midgestation and term villous preparations. (A) Levels of IL-8, IFN-λ2, IFNλ-1, and IL-1β in CM isolated from primary human trophoblasts from full-term or midgestation placentas as determined by Luminex-based profiling and ELISA. (B) Caspase-1 activity in CM isolated from non-CM (NCM) or from CM isolated from BeWo and JEG-3 cells or from chorionic villi isolated from midgestation or full-term placentas. (C) Confocal microscopy for pro–caspase-1 (in green) in chorionic villi isolated from midgestation placentas and costained with the Hofbauer cell marker CD163 (in red). (D) Confocal microscopy for cleaved IL-1β (in green) from FM isolated from the placenta used in Fig. 2 D. Actin staining is shown in red and DAPI-stained nuclei in blue. At top, focal plane is the amnion, and at bottom, it is the chorion. x–z cross-section is shown at bottom. DAPI-stained nuclei are shown in blue. (E) Caspase-1 activity in the media of villi treated with ILG or MCC950 or mock-treated controls. (F) Levels of LDH in media collected from placental chorionic villi (villi) or matched FM. As controls, media from THP-1 cells treated with LPS overnight and then exposed to nigericin for 2 h (+Nig) or mock-treated controls were also determined. In A, B, E, and F, symbols represent individual placental preparations. In C and D, scale bar is 10 µm. Significance was determined with the Kruskal-Wallis test or one-way ANOVA with Dunnett’s test for multiple comparisons (F). *, P < 0.05; **, P < 0.01.
Differences in cytokine secretion between midgestation and term villous preparations. (A) Levels of IL-8, IFN-λ2, IFNλ-1, and IL-1β in CM isolated from primary human trophoblasts from full-term or midgestation placentas as determined by Luminex-based profiling and ELISA. (B) Caspase-1 activity in CM isolated from non-CM (NCM) or from CM isolated from BeWo and JEG-3 cells or from chorionic villi isolated from midgestation or full-term placentas. (C) Confocal microscopy for pro–caspase-1 (in green) in chorionic villi isolated from midgestation placentas and costained with the Hofbauer cell marker CD163 (in red). (D) Confocal microscopy for cleaved IL-1β (in green) from FM isolated from the placenta used in Fig. 2 D. Actin staining is shown in red and DAPI-stained nuclei in blue. At top, focal plane is the amnion, and at bottom, it is the chorion. x–z cross-section is shown at bottom. DAPI-stained nuclei are shown in blue. (E) Caspase-1 activity in the media of villi treated with ILG or MCC950 or mock-treated controls. (F) Levels of LDH in media collected from placental chorionic villi (villi) or matched FM. As controls, media from THP-1 cells treated with LPS overnight and then exposed to nigericin for 2 h (+Nig) or mock-treated controls were also determined. In A, B, E, and F, symbols represent individual placental preparations. In C and D, scale bar is 10 µm. Significance was determined with the Kruskal-Wallis test or one-way ANOVA with Dunnett’s test for multiple comparisons (F). *, P < 0.05; **, P < 0.01.
Given that IL-1β and active caspase-1 secretion is associated with inflammasome activation, we next analyzed the transcriptional levels of inflammasome-associated genes in primary human trophoblasts and midgestation chorionic villous explants and compared these levels with those in JEG-3 cells, which do not produce IL-1β (Fig. 2 A). Using previously generated RNA sequencing (RNA-seq) datasets (Bayer et al., 2016; Corry et al., 2017), we found that primary trophoblasts and chorionic villi express high levels of many transcripts in the inflammasome pathway, including caspase-1, IL-1β, IL-18, NLRP3, and gasdermin D, among others (Fig. 2 B). Consistent with this, chorionic villi also express absent in melanoma 2 (AIM2), NLRP3, and gasdermin D at the protein level (Fig. 2 C). Immunofluorescence and immunohistochemistry confirmed the presence of NLRP3, gasdermin D, and cleaved IL-1β in the syncytiotrophoblast layer of placental villi (Fig. 2, D and E). Although NLRP3 and pro–IL-1β were largely restricted in localization to the syncytial layer, pro–caspase-1 was also localized within the villous core within fetus-derived M2 Hofbauer cell macrophages (Fig. 2 D and Fig. S2 C). Consistent with our Luminex findings supporting a low to undetectable level of IL-1β secretion from FM, we found that matched FM samples had undetectable cleaved IL-1β immunostaining (Fig. S2 D).
We next determined whether the constitutive release of IL-1β from chorionic villi was associated with cleavage of gasdermin D, which, after cleavage, can oligomerize to function as a pore for secretion of IL-1β and initiates pyroptosis-mediated cell death. We were unable to detect the N-terminal cleavage product of gasdermin D by immunohistochemistry (Fig. 2 E). Consistent with this, we were also unable to detect this product by immunoblotting, despite its abundance in lysates of LPS-treated THP-1 monocytes treated with nigericin, which functions to regulate K+ flux and is a well-known activator of the NLRP3 inflammasome (Fig. 2 F). In contrast to the differential abundance of cleaved gasdermin D, lysates of chorionic villi contained levels of cleaved IL-1β and ASC similar to those of THP-1 cells (Fig. 2 F). Consistent with this, the levels of lactate dehydrogenase (LDH) in villi-derived CM were comparable to those in LPS-treated THP-1 monocytes and significantly lower than when LPS-treated THP-1 cells underwent inflammasome-mediated cytolytic cell death following treatment with nigericin (Fig. S2 F). Together, these data suggest that placental trophoblasts possess high levels of basal inflammasome activity and that release of inflammasome-associated cytokines occurs independent from gasdermin D cleavage and pyroptosis.
To confirm that inflammasome activity is responsible for the constitutive release of IL-1β, we treated human chorionic villi with the inflammasome inhibitor isoliquiritigenin (ILG; Liu et al., 2017; Honda et al., 2014) and the specific NLRP3 inflammasome inhibitor MCC950 (Coll et al., 2015) and measured cytokine levels in CM by Luminex multianalyte profiling. Treatment of placental villous explants with ILG reduced the levels of 14 cytokines by more than twofold, but it had no effect on 67 other cytokines or chemokines tested (Fig. 3, A and C–E). The most reduced cytokine levels with ILG treatment were for IL-1β (13-fold down-regulation) and thymic stromal lymphopoietin, which is regulated by IL-1β (23-fold down-regulation; Fig. 3 A; Elder et al., 2016). MCC950 reduced levels of only three cytokines: IL-1β, IL-18, and IL-1α (∼6.5-, 3-, and 2.5-fold reductions, respectively; Fig. 3, B–E). Both ILG and MCC950 treatment also reduced the levels of active caspase-1 in villi CM (Fig. S2 E). Importantly, ILG and MCC950 treatment did not change the production of many other cytokines, such as IL-8 (Fig. 3, A, B, and F). These data suggest that the constitutive release of IL-1β and IL-18 is mediated primarily by NLRP3 inflammasome activity in trophoblasts.

Inhibition of the NLRP3 inflammasome reduces the constitutive release of IL-1β and IL-18. (A and B) Heatmaps demonstrating the reduction (shown as fold change from mock-treated controls) in matched chorionic villi specimens treated with or without ILG (A) or MCC950 (MCC; B) by multiplex Luminex-based profiling. AVG denotes the average change in concentration of cytokines over villi obtained from unique placentas. Blue denotes significantly decreased cytokines compared with untreated. Gray or white denotes little to no change (scale at top left). The red arrow demonstrates cytokines with greater than twofold down-regulation observed in the average of separate experiments. Data are from two or three unique placental preparations with three to five villi used per placenta. Luminex assays were performed in duplicate. (C–F) IL-1β (C), IL-18 (D), IL-1α (E), and IL-8 (F) cytokine levels present in CM from chorionic villi samples with and without treatment with ILG or MCC. Symbols represent individual villi from two or three different placenta preparations. In C–F, significance was determined with the Kruskal-Wallis test. *, P < 0.05; **, P < 0.01; ***, P < 0.0001; ns, not significant.
Inhibition of the NLRP3 inflammasome reduces the constitutive release of IL-1β and IL-18. (A and B) Heatmaps demonstrating the reduction (shown as fold change from mock-treated controls) in matched chorionic villi specimens treated with or without ILG (A) or MCC950 (MCC; B) by multiplex Luminex-based profiling. AVG denotes the average change in concentration of cytokines over villi obtained from unique placentas. Blue denotes significantly decreased cytokines compared with untreated. Gray or white denotes little to no change (scale at top left). The red arrow demonstrates cytokines with greater than twofold down-regulation observed in the average of separate experiments. Data are from two or three unique placental preparations with three to five villi used per placenta. Luminex assays were performed in duplicate. (C–F) IL-1β (C), IL-18 (D), IL-1α (E), and IL-8 (F) cytokine levels present in CM from chorionic villi samples with and without treatment with ILG or MCC. Symbols represent individual villi from two or three different placenta preparations. In C–F, significance was determined with the Kruskal-Wallis test. *, P < 0.05; **, P < 0.01; ***, P < 0.0001; ns, not significant.
Trophoblast-derived IL-1β primes monocytes for inflammasome activation
Factors released from placental villi have the capacity to act in a paracrine manner to directly impact the function of maternal-derived cells, particularly if these factors reach the systemic circulation. To determine whether placenta-derived IL-1β has the potential to impact maternal immunity, we measured IL-1β levels in serum isolated from pregnant and nonpregnant women. We detected IL-1β in serum isolated from pregnant women but not in serum isolated from nonpregnant women, suggesting that IL-1β is increased in the systemic circulation during pregnancy (Fig. 4 A). We also found that monocytes isolated from pregnant women in the second trimester of pregnancy (20–27 wk gestation; Table S2) exhibited elevated expression of caspase-1 and IL-1β compared with nonpregnant control subjects (Fig. 4, B and C), suggesting that inflammasome priming of monocytes occurs in normal pregnancy. To determine if placenta-secreted factors could be responsible for inflammasome priming of monocytes, we exposed primary monocytes and THP-1 cells with CM from villous explants. We found that treatment of THP-1 cells with CM from placental villous explants, but not matched FMs, significantly induced the expression of IL-1β, caspase-1, and NLRP3, which was dependent on gestational age (Fig. S3, A–C). Similar results were obtained in primary monocytes from nonpregnant female donors, in which exposure to villous CM increased RNA levels of IL-1β, caspase-1, and NLRP3 (Fig. 4 D).
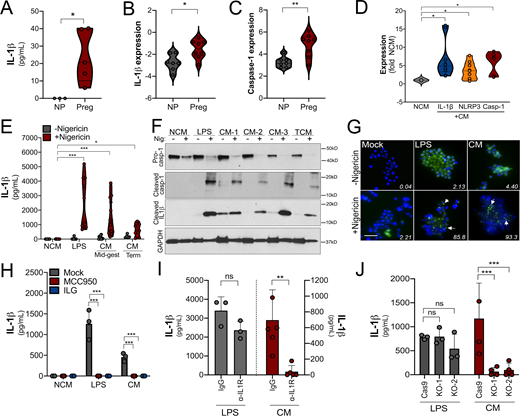
Trophoblast-derived IL-1β primes monocytes for inflammasome activation.(A) Levels of IL-1β as determined by ELISA in the plasma from pregnant female donors (20–27 wk; n = 5) compared with nonpregnant control subjects. Donor characteristics are shown in Table S2. (B and C) IL-1β (B) and caspase-1 (C) transcripts are increased in primary human monocytes isolated from pregnant (Preg) women compared with monocytes from nonpregnant women (NP) of reproductive age as assessed by RT-qPCR. Samples from five nonpregnant and five pregnant donors were used, and RT-qPCR was performed in duplicate. Each symbol is an average of the qPCR replicates. (D) IL-1β, caspase-1, and NLRP3 transcripts as assessed by RT-qPCR in primary human monocytes (from nonpregnant donors) after exposure to chorionic villi–derived CM. Fold change in transcript compared with exposure to non-CM (NCM) is shown. Samples from three nonpregnant donors were exposed to villi-derived CM from three unique preparations. RT-qPCR was performed in duplicate. (E) Nigericin treatment leads to increased IL-1β secretion as assessed by ELISA from THP-1 monocytes exposed to chorionic villi CM isolated from midgestation (Mid-gest) or term placentas. LPS was used as a positive control. Each symbol represents a unique CM preparation. (F) Immunoblot from THP-1 cells treated with villi-derived CM from three midgestation placentas (CM 1–3) or one full-term placenta (TCM) for pro–caspase-1 (top row), cleaved caspase-1 (second row), cleaved IL-1β (third row), or GAPDH (bottom row) as a loading control. Molecular weights are shown at right. Immunoblots were performed at least three times. (G) THP-1 cells stably expressing ASC-GFP were treated with villi-derived CM, with LPS as a positive control, or with NCM, and GFP expression and localization were assessed by fluorescence microscopy with (bottom row) or without (top row) nigericin treatment for 2 h. DAPI-stained nuclei are shown in blue. Scale bar is 5 µm. White text denotes the average percentage of cells containing punctae averaged from three independent experiments. Images are representative of imaging of at least three to five independent fields from experiments performed in triplicate. (H) IL-1β present in the media of THP-1 cells treated with LPS or CM and then exposed to nigericin (or mock treated as a control) in the absence (mock) or presence of ILG or MCC950 as determined by ELISA. (I) THP-1 cells incubated with anti–IL-1R blocking antibody (10 µg/ml) or with isotype control antibody (IgG) were treated with villi-derived CM (red, right) or with LPS (gray, left) and then treated with nigericin for 2 h. Levels of IL-1β were assessed by ELISA. (J) THP-1 cells lacking expression of IL-1R (two clones, KO-1 and KO-2) or Cas9 control cells were treated with villi-derived CM or LPS and then exposed to nigericin for 2 h. Levels of IL-1β were assessed by ELISA (in pg/ml). Immunoblotting for IL-1R and GAPDH from the indicated cells is shown at bottom. In H–J, experiments were performed a minimum or three times with technical duplicates or triplicates, with symbols representing experimental replicates. In H–J, data are presented as mean ± SD. Significance was determined with a t test (A, C, and D) or one-way ANOVA with Dunnett’s test for multiple comparisons (D, E, and H–J). *, P < 0.05; **, P < 0.01; ***, P < 0.001. Symbols represent unique donor cells, CM isolated from individual placental preparations, or experimental replicates.
Trophoblast-derived IL-1β primes monocytes for inflammasome activation.(A) Levels of IL-1β as determined by ELISA in the plasma from pregnant female donors (20–27 wk; n = 5) compared with nonpregnant control subjects. Donor characteristics are shown in Table S2. (B and C) IL-1β (B) and caspase-1 (C) transcripts are increased in primary human monocytes isolated from pregnant (Preg) women compared with monocytes from nonpregnant women (NP) of reproductive age as assessed by RT-qPCR. Samples from five nonpregnant and five pregnant donors were used, and RT-qPCR was performed in duplicate. Each symbol is an average of the qPCR replicates. (D) IL-1β, caspase-1, and NLRP3 transcripts as assessed by RT-qPCR in primary human monocytes (from nonpregnant donors) after exposure to chorionic villi–derived CM. Fold change in transcript compared with exposure to non-CM (NCM) is shown. Samples from three nonpregnant donors were exposed to villi-derived CM from three unique preparations. RT-qPCR was performed in duplicate. (E) Nigericin treatment leads to increased IL-1β secretion as assessed by ELISA from THP-1 monocytes exposed to chorionic villi CM isolated from midgestation (Mid-gest) or term placentas. LPS was used as a positive control. Each symbol represents a unique CM preparation. (F) Immunoblot from THP-1 cells treated with villi-derived CM from three midgestation placentas (CM 1–3) or one full-term placenta (TCM) for pro–caspase-1 (top row), cleaved caspase-1 (second row), cleaved IL-1β (third row), or GAPDH (bottom row) as a loading control. Molecular weights are shown at right. Immunoblots were performed at least three times. (G) THP-1 cells stably expressing ASC-GFP were treated with villi-derived CM, with LPS as a positive control, or with NCM, and GFP expression and localization were assessed by fluorescence microscopy with (bottom row) or without (top row) nigericin treatment for 2 h. DAPI-stained nuclei are shown in blue. Scale bar is 5 µm. White text denotes the average percentage of cells containing punctae averaged from three independent experiments. Images are representative of imaging of at least three to five independent fields from experiments performed in triplicate. (H) IL-1β present in the media of THP-1 cells treated with LPS or CM and then exposed to nigericin (or mock treated as a control) in the absence (mock) or presence of ILG or MCC950 as determined by ELISA. (I) THP-1 cells incubated with anti–IL-1R blocking antibody (10 µg/ml) or with isotype control antibody (IgG) were treated with villi-derived CM (red, right) or with LPS (gray, left) and then treated with nigericin for 2 h. Levels of IL-1β were assessed by ELISA. (J) THP-1 cells lacking expression of IL-1R (two clones, KO-1 and KO-2) or Cas9 control cells were treated with villi-derived CM or LPS and then exposed to nigericin for 2 h. Levels of IL-1β were assessed by ELISA (in pg/ml). Immunoblotting for IL-1R and GAPDH from the indicated cells is shown at bottom. In H–J, experiments were performed a minimum or three times with technical duplicates or triplicates, with symbols representing experimental replicates. In H–J, data are presented as mean ± SD. Significance was determined with a t test (A, C, and D) or one-way ANOVA with Dunnett’s test for multiple comparisons (D, E, and H–J). *, P < 0.05; **, P < 0.01; ***, P < 0.001. Symbols represent unique donor cells, CM isolated from individual placental preparations, or experimental replicates.

Response of monocytes to exposure to CM. (A) IL-1β transcript as assessed by RT-qPCR in THP-1 monocytes following exposure to CM from placental villi or matched FM samples. Fold change in transcript by qPCR compared with control non-CM (NCM) is shown. THP-1 cells were exposed to five separate CM preparations from midgestation villi and two from term villi. Experiments were performed in triplicate, and RT-qPCR was performed in duplicate. Each symbol is an average of the qPCR replicates. (B and C) Caspase-1 (B) and NLRP3 (C) transcripts as assessed by RT-qPCR in THP-1 monocytes exposed to LPS or CM from midgestation or term placental chorionic villi preparations. Each symbol is CM from a separate preparation. (D) Quantification of ASC-GFP punctae formation in THP-1 cells treated with LPS or CM and then exposed to nigericin for 2 h. Data are matched to representative images shown in Fig. 4 G. (E) Caspase-1 activity in the media of THP-1 cells treated with LPS or CM (or NCM treated control) and then exposed to nigericin for 2 h in the absence (−MCC) or presence of MCC950 (+MCC). (F) Quantification of ASC-GFP punctae formation in THP-1 cells treated with LPS or CM and then exposed to nigericin for 2 h in the absence (Mock) or presence of MCC950. (G) Immunoblots for IL-1R in control THP-1 cells (Cas9 control) or two clones of THP-1 cells lacking expression of IL-1R (IL1RKO). GAPDH is included as a loading control. (H) THP-1 monocytes incubated with anti–IL-1R blocking antibody (10 µg/ml; red) or with isotype control antibody (IgG; gray) were treated with villi-derived CM (PCM) or with LPS and then treated with nigericin for 2 h. Caspase-1 activity in culture supernatants was assessed using the Caspase-1 Glo Inflammasome Activity kit. (I) Caspase-1 activity in culture supernatants from THP-1 monocytes with IL-1R deletion (IL-1R KO; red) or from Cas9 control (Cas9 Con; gray) treated with villi-derived CM (PCM) or LPS followed by exposure to nigericin. Data in E and F are shown as mean ± SD, and significance was determined with a t test or one-way ANOVA with Dunnett’s test for multiple comparisons. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Symbols represent CM from individual placental preparations or experimental replicates (NCM, LPS).
Response of monocytes to exposure to CM. (A) IL-1β transcript as assessed by RT-qPCR in THP-1 monocytes following exposure to CM from placental villi or matched FM samples. Fold change in transcript by qPCR compared with control non-CM (NCM) is shown. THP-1 cells were exposed to five separate CM preparations from midgestation villi and two from term villi. Experiments were performed in triplicate, and RT-qPCR was performed in duplicate. Each symbol is an average of the qPCR replicates. (B and C) Caspase-1 (B) and NLRP3 (C) transcripts as assessed by RT-qPCR in THP-1 monocytes exposed to LPS or CM from midgestation or term placental chorionic villi preparations. Each symbol is CM from a separate preparation. (D) Quantification of ASC-GFP punctae formation in THP-1 cells treated with LPS or CM and then exposed to nigericin for 2 h. Data are matched to representative images shown in Fig. 4 G. (E) Caspase-1 activity in the media of THP-1 cells treated with LPS or CM (or NCM treated control) and then exposed to nigericin for 2 h in the absence (−MCC) or presence of MCC950 (+MCC). (F) Quantification of ASC-GFP punctae formation in THP-1 cells treated with LPS or CM and then exposed to nigericin for 2 h in the absence (Mock) or presence of MCC950. (G) Immunoblots for IL-1R in control THP-1 cells (Cas9 control) or two clones of THP-1 cells lacking expression of IL-1R (IL1RKO). GAPDH is included as a loading control. (H) THP-1 monocytes incubated with anti–IL-1R blocking antibody (10 µg/ml; red) or with isotype control antibody (IgG; gray) were treated with villi-derived CM (PCM) or with LPS and then treated with nigericin for 2 h. Caspase-1 activity in culture supernatants was assessed using the Caspase-1 Glo Inflammasome Activity kit. (I) Caspase-1 activity in culture supernatants from THP-1 monocytes with IL-1R deletion (IL-1R KO; red) or from Cas9 control (Cas9 Con; gray) treated with villi-derived CM (PCM) or LPS followed by exposure to nigericin. Data in E and F are shown as mean ± SD, and significance was determined with a t test or one-way ANOVA with Dunnett’s test for multiple comparisons. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Symbols represent CM from individual placental preparations or experimental replicates (NCM, LPS).
The data described above suggest that villi-derived factors can prime monocytes for inflammasome activation. To test this, we used nigericin, which is a well-known activator of the NLRP3 inflammasome in primed monocytes. As has been shown previously, nigericin treatment of THP-1 cells alone did not induce inflammasome activation, as assessed by an ELISA for IL-1β production (Fig. 4 E). However, exposure of THP-1 cells to villi-derived CM (or LPS as a positive control) sensitized cells to nigericin-induced IL-1β secretion (Fig. 4 E), consistent with inflammasome priming. Of note, we found that this response was less robust when cells were exposed to CM from full-term placental explants (Fig. 4 E). Consistent with this, immunoblotting for inflammasome-associated components in THP-1 cells exposed to villi-derived CM isolated from midgestation or full-term placentas revealed significant cleavage of pro–caspase-1 into active caspase-1, which correlated with the enhanced expression of cleaved IL-1β (Fig. 4 F). As an additional measure of inflammasome activation, we used fluorescence microscopy for ASC punctae formation in THP-1 ASC-GFP reporter cells that stably express ASC-GFP under the control of an NF-κB–inducible promoter and a secreted caspase-1 luciferase-based assay. In ASC-GFP–expressing cells, NF-κB activation induces ASC-GFP expression; subsequent inflammasome activation leads to aggregation of the inflammasome complex; and the ASC-GFP fluorescence signal coalesces into subcellular punctae. Consistent with our ELISA results, we found that LPS- and villi-derived CM up-regulated the expression of ASC-GFP (Fig. 4 G and Fig. S3 D). When these cells were subsequently exposed to nigericin, those treated with CM or LPS developed ASC-GFP punctae (Fig. 4 G and Fig. S3 D). In addition, CM treatment significantly increased the levels of activated caspase-1 in cell culture supernatants (Fig. S3 E). Consistent with the role of inflammasome priming and activation, treatment of cells exposed to LPS or CM with ILG or MCC950 inhibited nigericin-mediated IL-1β secretion (Fig. 4 H), caspase-1 activation (Fig. S3 E), and ASC-GFP punctae formation (Fig. S3 F). Collectively, these data show that a factor or factors released into the media of human placental explants prime monocytes for inflammasome induction.
To determine whether IL-1β constitutively secreted by placental trophoblasts is responsible for the CM-mediated inflammasome priming in monocytes, we used an antibody that blocks binding of IL-1β to IL-1R and also generated THP-1 cells lacking expression of IL-1R through CRISPR-mediated gene editing (Fig. S3 G). We found that a blocking antibody against IL-1R or depletion of IL-1R expression significantly inhibited CM-mediated priming of monocytes for nigericin-induced IL-1β and active caspase-1 release while having no effect on LPS-mediated priming (Fig. 4, I and J; and Fig. S4, H and I). Together, these data show that IL-1β released by trophoblasts participates in priming of monocytes for inflammasome activation.

ASC-GFP punctae with LPS or CM priming. (A) THP-1 ASC-GFP cells were exposed to non-CM (NCM) control (top panel), LPS (middle panel), or chorionic villi CM from midgestation placentas (CMMG; bottom panel) and then infected with L. monocytogenes (red). ASC-GFP expression (diffuse green staining), punctae, and L. monocytogenes were visualized. Scale bar is 5 µm. (B)L. monocytogenes growth in NCM or in villi-derived CM. CFU/ml was measured 6 h after cultures were initiated. Data are shown as percentage of NCM. (C) Caspase-1 activity in the media of THP-1 cells treated with LPS (blue) or placental CM (red; or NCM control, gray) and then infected with L. monocytogenes for 2 h in the absence (solid bars) or presence (hatched bars) of MCC950. In B and C, data are shown as mean ± SD. Symbols represent CM from individual placental preparations or experimental replicates (NCM, LPS). Data were analyzed with a t test or one-way ANOVA with Dunnett’s test for multiple comparisons. *, P < 0.05; **, P < 0.01.
ASC-GFP punctae with LPS or CM priming. (A) THP-1 ASC-GFP cells were exposed to non-CM (NCM) control (top panel), LPS (middle panel), or chorionic villi CM from midgestation placentas (CMMG; bottom panel) and then infected with L. monocytogenes (red). ASC-GFP expression (diffuse green staining), punctae, and L. monocytogenes were visualized. Scale bar is 5 µm. (B)L. monocytogenes growth in NCM or in villi-derived CM. CFU/ml was measured 6 h after cultures were initiated. Data are shown as percentage of NCM. (C) Caspase-1 activity in the media of THP-1 cells treated with LPS (blue) or placental CM (red; or NCM control, gray) and then infected with L. monocytogenes for 2 h in the absence (solid bars) or presence (hatched bars) of MCC950. In B and C, data are shown as mean ± SD. Symbols represent CM from individual placental preparations or experimental replicates (NCM, LPS). Data were analyzed with a t test or one-way ANOVA with Dunnett’s test for multiple comparisons. *, P < 0.05; **, P < 0.01.
Trophoblast-derived IL-1β protects monocytes from L. monocytogenes infection
L. monocytogenes can traverse the maternal–fetal barrier and cause fetal infection with associated miscarriage and stillbirth. Inflammasome signaling is important in the defense against bacterial infections and is a primary sentinel of the innate immune defense against L. monocytogenes (reviewed in von Moltke et al., 2013). Therefore, we next determined whether placenta-derived IL-1β sensitized peripheral cells to inflammasome activation as a mechanism of host defense against this bacterium. We found that, similar to cells primed with LPS, placental CM-primed ASC-GFP–expressing THP-1 cells exhibited robust inflammasome activation in response to L. monocytogenes compared with control-treated cells (Fig. 5, A and B). Consistent with this, IL-1β secretion in response to infection was significantly higher with LPS and CM priming than with controls (Fig. 5 C). Also, we found that cells primed for inflammasome activation by exposure to CM or LPS were less susceptible to L. monocytogenes infection as assessed by bacterial cell count– and fluorescence-based assays (Fig. 5 D and Fig. S4 A). Importantly, we found that L. monocytogenes growth was not reduced when cultured in villi-derived CM or nonconditioned control media, demonstrating that bacterial growth was not inhibited by a component present in this media (Fig. S4 B). L. monocytogenes–induced IL-1β and caspase-1 release in cells exposed to LPS or CM was reversed by treatment with MCC950, supporting the role of the inflammasome in this process (Fig. 5 E and Fig. S5 C). In addition, the reduction of L. monocytogenes infection in CM-treated cells was significantly reversed when cells were treated with MCC950, supporting a direct role for the NLPR3 inflammasome in this effect (Fig. 5 F). Last, we found that deletion of the IL-1R in THP-1 cells by CRISPR-mediated gene editing significantly reduced the placental CM-induced enhancement of IL-1β (Fig. 5 G) and also its anti–L. monocytogenes effects (Fig. 5 H), suggesting that the IL-1β in CM is responsible for mediating the priming of monocytes to respond to L. monocytogenes infection. Collectively, these data demonstrate that inflammasome priming of monocytes by placenta-derived IL-1β restricts L. monocytogenes infection.

Trophoblast-derived IL-1β primes monocytes for inflammasome activation in response to L. monocytogenes infection. (A and B) THP-1 cells stably expressing ASC-GFP were treated with villi-derived CM, with LPS as a positive control, or with non-CM (NCM). GFP expression and localization were assessed by fluorescence microscopy following infection with L. monocytogenes at an MOI of 5 (in gray; A) or 10 (in blue; A) for 2 h. Cells were fixed, and fluorescence microscopy was performed to determine the extent of ASC-GFP punctae formation (B). In B, representative images are shown, with mCherry-L. monocytogenes shown in red. Bright-field images are shown at left. Scale bar is 10 µm. In A, quantification of ASC-GFP punctae is shown from three independent experiments. Images are representative of imaging of at least three to five independent fields from experiments performed in triplicate. (C) ELISA of IL-1β from THP-1 cells treated with villi-derived CM, LPS, or NCM and then infected with L. monocytogenes at MOI of 1 (in gray) or 5 (in blue) for 2–4 h. (D) Levels of L. monocytogenes infection as determined by CFU in THP-1 cells treated with villi-derived CM, LPS, or NCM for 24 h followed by bacterial inoculation (MOI, 1). Data are shown as a percentage of growth in NCM-treated controls. (E) IL-1β concentration from supernatants of THP-1 cells after treatment with NCM, LPS, or CM followed by infection with L. monocytogenes (for 2 h) in the absence (mock) or presence of MCC950. (F) Levels of L. monocytogenes infection as determined by CFU in THP-1 cells treated with villi-derived CM, LPS, or NCM for 24 h followed by bacterial inoculation (MOI, 1) in the absence (−MCC) or presence (+MCC) of MCC950. Data are shown as a percentage of growth in NCM-treated controls. (G) IL-1β concentration from supernatants of THP-1 cells lacking IL-1R or Cas9 control cells after treatment with villi-derived CM, LPS, or NCM followed by infection with L. monocytogenes (MOI, 5). Data are mean ± SD, with individual villi preparations (and replicates) shown by symbols. Levels of IL-1R are shown in Fig. S3 G. (H) Levels of L. monocytogenes infection as determined by CFU in control (Cas9) THP-1 cells or THP-1 cells lacking expression of IL-1R (ΔIL1R) treated with villi-derived CM (+CM) or NCM for 24 h followed by bacterial inoculation (MOI, 1). Data are shown as a percentage of growth in NCM-treated controls. Experiments were performed a minimum of three times with technical duplicates or triplicates, with symbols representing experimental replicates. Significance was determined with one-way ANOVA with Dunnett’s test for multiple comparisons. *, P < 0.05; **, P < 0.01; ***, P < 0.001. In A and C–H, symbols represent CM isolated from individual placental preparations or experimental replicates.
Trophoblast-derived IL-1β primes monocytes for inflammasome activation in response to L. monocytogenes infection. (A and B) THP-1 cells stably expressing ASC-GFP were treated with villi-derived CM, with LPS as a positive control, or with non-CM (NCM). GFP expression and localization were assessed by fluorescence microscopy following infection with L. monocytogenes at an MOI of 5 (in gray; A) or 10 (in blue; A) for 2 h. Cells were fixed, and fluorescence microscopy was performed to determine the extent of ASC-GFP punctae formation (B). In B, representative images are shown, with mCherry-L. monocytogenes shown in red. Bright-field images are shown at left. Scale bar is 10 µm. In A, quantification of ASC-GFP punctae is shown from three independent experiments. Images are representative of imaging of at least three to five independent fields from experiments performed in triplicate. (C) ELISA of IL-1β from THP-1 cells treated with villi-derived CM, LPS, or NCM and then infected with L. monocytogenes at MOI of 1 (in gray) or 5 (in blue) for 2–4 h. (D) Levels of L. monocytogenes infection as determined by CFU in THP-1 cells treated with villi-derived CM, LPS, or NCM for 24 h followed by bacterial inoculation (MOI, 1). Data are shown as a percentage of growth in NCM-treated controls. (E) IL-1β concentration from supernatants of THP-1 cells after treatment with NCM, LPS, or CM followed by infection with L. monocytogenes (for 2 h) in the absence (mock) or presence of MCC950. (F) Levels of L. monocytogenes infection as determined by CFU in THP-1 cells treated with villi-derived CM, LPS, or NCM for 24 h followed by bacterial inoculation (MOI, 1) in the absence (−MCC) or presence (+MCC) of MCC950. Data are shown as a percentage of growth in NCM-treated controls. (G) IL-1β concentration from supernatants of THP-1 cells lacking IL-1R or Cas9 control cells after treatment with villi-derived CM, LPS, or NCM followed by infection with L. monocytogenes (MOI, 5). Data are mean ± SD, with individual villi preparations (and replicates) shown by symbols. Levels of IL-1R are shown in Fig. S3 G. (H) Levels of L. monocytogenes infection as determined by CFU in control (Cas9) THP-1 cells or THP-1 cells lacking expression of IL-1R (ΔIL1R) treated with villi-derived CM (+CM) or NCM for 24 h followed by bacterial inoculation (MOI, 1). Data are shown as a percentage of growth in NCM-treated controls. Experiments were performed a minimum of three times with technical duplicates or triplicates, with symbols representing experimental replicates. Significance was determined with one-way ANOVA with Dunnett’s test for multiple comparisons. *, P < 0.05; **, P < 0.01; ***, P < 0.001. In A and C–H, symbols represent CM isolated from individual placental preparations or experimental replicates.

Lack of visualization of bacteria in placental trophoblast explants after L. monocytogenes infection. (A) Infection of chorionic villi with mCherry-expressing L. monocytogenes (104 CFU/ml; right panel) or mock-infected controls (left panel). DAPI-stained nuclei are shown in blue and actin in green. Scale bar is 10 µm. (B and C) TNF-α (B) and RANTES (C) levels as determined by Luminex assays in placental villi infected with L. monocytogenes (104 CFU/ml) for 24 h (or uninfected controls [CON]) treated without (Mock) or with ILG or MCC950 (MCC). Data are from two unique placental preparations, with three unique villi used per placenta. Significance was determined with the Kruskal-Wallis test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Symbols represent CM from individual villi from unique placental preparations.
Lack of visualization of bacteria in placental trophoblast explants after L. monocytogenes infection. (A) Infection of chorionic villi with mCherry-expressing L. monocytogenes (104 CFU/ml; right panel) or mock-infected controls (left panel). DAPI-stained nuclei are shown in blue and actin in green. Scale bar is 10 µm. (B and C) TNF-α (B) and RANTES (C) levels as determined by Luminex assays in placental villi infected with L. monocytogenes (104 CFU/ml) for 24 h (or uninfected controls [CON]) treated without (Mock) or with ILG or MCC950 (MCC). Data are from two unique placental preparations, with three unique villi used per placenta. Significance was determined with the Kruskal-Wallis test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Symbols represent CM from individual villi from unique placental preparations.
L. monocytogenes infection of placental villi activates the inflammasome
Because we found that placenta-derived IL-1β worked in a paracrine manner to protect monocytes from L. monocytogenes infection, we next determined whether inflammasome signaling protects the placenta itself from infection. Previous work has shown that placental syncytiotrophoblasts resist infection by L. monocytogenes (Robbins et al., 2010). However, little is known regarding the mechanisms that confer this protection and the pathways by which these cells sense and respond to infection. To define the placental response to L. monocytogenes, we isolated chorionic villi from second-trimester human placentas, infected them with L. monocytogenes for 24 h, and then performed multianalyte Luminex-based cytokine profiling to identify the innate pathways induced. Consistent with previous work in first-trimester placentas (Robbins et al., 2010), we found that midgestation chorionic villi were highly resistant to L. monocytogenes infection, and there was little to no bacterial growth detected in villi by confocal microscopy (Fig. S5 A). Despite the very low levels of infection, we identified nine cytokines whose levels were induced greater than fivefold in villi infected with L. monocytogenes (Fig. 6 A). The most induced cytokine was IL-1β, which was up-regulated an average of 20-fold compared with mock-infected controls (Fig. 6, A and B). In addition, IL-18 was induced approximately fourfold (Fig. 6, A and C). Based on immunofluorescence microscopy, induced IL-1β was restricted to the syncytial layer and not to other cellular compartments (Fig. 6 D). In contrast to infection with L. monocytogenes, treatment of villi with polyinosinic-polycytidylic acid (poly I:C), a synthetic mimic of viral double-stranded RNA, robustly induced IFN-λ2 but had no effect on IL-1β (Fig. 6, E and F), suggesting that this is a pathogen-specific effect. Unlike poly I:C, the TLR7/8 ligand R848/resiquimod significantly increased the levels of IL-1β without having an effect on IFN-λs (Fig. 6, E and F). The structural relative of resiquimod, imiquimod, has been shown to mediate inflammasome activation through K+ efflux (Groß et al., 2016). These data suggest that the inflammasome hyperactivation is further enhanced as a response of the human placenta to L. monocytogenes infection and other inflammasome activators.

Infection of chorionic villi with L. monocytogenes enhances IL-1β and IL-18 secretion. (A) Heatmap demonstrating the induction (shown as fold change from mock-infected controls) in chorionic villi specimens infected with L. monocytogenes (104 CFU/ml) for 24 h. At least three independent villi from four unique placental preparations were included, and Luminex assay was performed in duplicate. AVG denotes the average change in concentration of cytokines over villi obtained from four placentas. Blue denotes significantly up-regulated factors, and gray or white denotes little to no change (scale at top left). The red arrow demonstrates cytokines with greater than fivefold difference observed in the average of all experiments. (B and C) IL-1β (B) and IL-18 (C) cytokine levels present in CM from chorionic villi samples with or without infection. Symbols represent individual villi from four different placental preparations. (D) Confocal microscopy of cleaved IL-1β (in green) in chorionic villi infected with L. monocytogenes (104 CFU/ml) for 24 h. DAPI-stained nuclei are shown in blue. y–z and x–z cross-sections are shown at right and below. White arrows denote staining in syncytiotrophoblasts. Scale bar is 10 µm. Images are representative of imaging of at least three to five independent fields from three unique placental preparations. (E and F) Levels of IL-1β (E) or IFN-λ2 (F) following exposure of chorionic villi to poly I:C (10 µg), resiquimod (R848; 10 µg), or mock treated as a control for 24 h. Data are from three unique placental preparations with three to five villi used per placenta, with Luminex assay performed in duplicate. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Symbols represent villi (averaged) from individual placental preparations.
Infection of chorionic villi with L. monocytogenes enhances IL-1β and IL-18 secretion. (A) Heatmap demonstrating the induction (shown as fold change from mock-infected controls) in chorionic villi specimens infected with L. monocytogenes (104 CFU/ml) for 24 h. At least three independent villi from four unique placental preparations were included, and Luminex assay was performed in duplicate. AVG denotes the average change in concentration of cytokines over villi obtained from four placentas. Blue denotes significantly up-regulated factors, and gray or white denotes little to no change (scale at top left). The red arrow demonstrates cytokines with greater than fivefold difference observed in the average of all experiments. (B and C) IL-1β (B) and IL-18 (C) cytokine levels present in CM from chorionic villi samples with or without infection. Symbols represent individual villi from four different placental preparations. (D) Confocal microscopy of cleaved IL-1β (in green) in chorionic villi infected with L. monocytogenes (104 CFU/ml) for 24 h. DAPI-stained nuclei are shown in blue. y–z and x–z cross-sections are shown at right and below. White arrows denote staining in syncytiotrophoblasts. Scale bar is 10 µm. Images are representative of imaging of at least three to five independent fields from three unique placental preparations. (E and F) Levels of IL-1β (E) or IFN-λ2 (F) following exposure of chorionic villi to poly I:C (10 µg), resiquimod (R848; 10 µg), or mock treated as a control for 24 h. Data are from three unique placental preparations with three to five villi used per placenta, with Luminex assay performed in duplicate. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Symbols represent villi (averaged) from individual placental preparations.
Inhibition of inflammasome signaling sensitizes the human placenta to L. monocytogenes infection
To confirm that the inflammasome was required for L. monocytogenes–mediated IL-1β and IL-18 secretion, we infected chorionic villi in the presence of ILG and MCC950 and performed Luminex-based cytokine profiling. We found that both ILG and MCC950 treatment suppressed the induction of IL-1β and IL-18 (Fig. 7, A–D). MCC950 treatment inhibited only IL-1β, IL-18, and IL-1α by greater than twofold, whereas ILG treatment also inhibited the induction of noninflammasome cytokines including IFN-λ2, consistent with its possible pleiotropic effects on other pathways including NF-κB (Fig. 7 A; Liu et al., 2017; Kumar et al., 2007). Importantly, our Luminex-based multianalyte assays allowed the profiling of ∼80 growth factors, cytokines, and chemokines, which demonstrated that other cytokines induced by L. monocytogenes infection, such as RANTES, were unaffected by this treatment (Fig. 7, A and B; and Fig. S5, B and C). In fact, both ILG and MCC950 treatment led to enhanced TNF-α secretion from L. monocytogenes–infected villi (Fig. S5 B).

Inflammasome inhibition sensitizes chorionic villi to L. monocytogenes infection. (A and B) Heatmap demonstrating the induction (shown as fold change from mock-treated controls) in chorionic villi specimens infected with L. monocytogenes (104 CFU/ml) for 24 h in the presence of ILG (A) or MCC950 (MCC; B). At least three independent villi from two unique placental preparations were included in each, and Luminex assay was performed in duplicate. AVG denotes the average change in concentration of cytokines over villi obtained from two placentas. Blue denotes significantly up-regulated factors, and gray or white denotes little to no change (scale at top left). The red line demonstrates cytokines with greater than twofold reduction observed following ILG or MCC treatment. (C and D) IL-1β (C) and IL-18 (D) levels as determined by Luminex assay in placental villi infected with L. monocytogenes (104 CFU/ml) for 24 h (or uninfected controls [CON]) treated without (Mock) or with ILG or MCC. Data are from two unique placental preparations, with three unique villi used per placenta. (E)L. monocytogenes infection (shown as log10 CFU) in mock- or ILG-treated chorionic villi infected for 24 h. As a control, Caco-2 cells were included to compare growth from permissive cells. Experiments were performed a minimum of three times with technical duplicates or triplicates, with symbols representing experimental replicates from unique placentas. (F) Mean fluorescence intensity (MFI) in placental villi infected with L. monocytogenes (104 CFU/ml) for 24 h (or uninfected CON) treated without (Mock) or with ILG or MCC. Data are shown as fold changes in MFI from CON. Individual symbols represent unique villi. Data are from two unique placental preparations, with three to five villi used per placenta. Experiments were performed on three or four independent villi isolated from two unique placental preparations. Symbols represent individual fields quantified from these villi. (G) Confocal microscopy of placental villi infected with L. monocytogenes (104 CFU/ml) for 24 h (or uninfected CON) treated without (Mock) or with ILG or MCC. Actin-stained nuclei are shown in green and DAPI-stained nuclei in blue. Scale bar is 10 µm. Images at right are zoomed areas of white boxes in right panels. Data are from two unique placental preparations, with three villi used per placenta. *, P < 0.05; **, P < 0.01; ***, P < 0.001. In C and D, symbols represent villi from individual placental preparations.
Inflammasome inhibition sensitizes chorionic villi to L. monocytogenes infection. (A and B) Heatmap demonstrating the induction (shown as fold change from mock-treated controls) in chorionic villi specimens infected with L. monocytogenes (104 CFU/ml) for 24 h in the presence of ILG (A) or MCC950 (MCC; B). At least three independent villi from two unique placental preparations were included in each, and Luminex assay was performed in duplicate. AVG denotes the average change in concentration of cytokines over villi obtained from two placentas. Blue denotes significantly up-regulated factors, and gray or white denotes little to no change (scale at top left). The red line demonstrates cytokines with greater than twofold reduction observed following ILG or MCC treatment. (C and D) IL-1β (C) and IL-18 (D) levels as determined by Luminex assay in placental villi infected with L. monocytogenes (104 CFU/ml) for 24 h (or uninfected controls [CON]) treated without (Mock) or with ILG or MCC. Data are from two unique placental preparations, with three unique villi used per placenta. (E)L. monocytogenes infection (shown as log10 CFU) in mock- or ILG-treated chorionic villi infected for 24 h. As a control, Caco-2 cells were included to compare growth from permissive cells. Experiments were performed a minimum of three times with technical duplicates or triplicates, with symbols representing experimental replicates from unique placentas. (F) Mean fluorescence intensity (MFI) in placental villi infected with L. monocytogenes (104 CFU/ml) for 24 h (or uninfected CON) treated without (Mock) or with ILG or MCC. Data are shown as fold changes in MFI from CON. Individual symbols represent unique villi. Data are from two unique placental preparations, with three to five villi used per placenta. Experiments were performed on three or four independent villi isolated from two unique placental preparations. Symbols represent individual fields quantified from these villi. (G) Confocal microscopy of placental villi infected with L. monocytogenes (104 CFU/ml) for 24 h (or uninfected CON) treated without (Mock) or with ILG or MCC. Actin-stained nuclei are shown in green and DAPI-stained nuclei in blue. Scale bar is 10 µm. Images at right are zoomed areas of white boxes in right panels. Data are from two unique placental preparations, with three villi used per placenta. *, P < 0.05; **, P < 0.01; ***, P < 0.001. In C and D, symbols represent villi from individual placental preparations.
Given that we observed that ILG and MCC950 inhibited L. monocytogenes–induced inflammasome activation, we next determined whether this signaling restricted bacterial infection. We found that treatment of villi with ILG or MCC950 significantly increased the susceptibility of placental villi to L. monocytogenes infection, whereas control villi were resistant to infection. ILG or MCC950 treatment of villous explants promoted bacterial growth or invasion of villi, as supported by the presence of bacteria in villi, possibly in cytotrophoblasts subjacent to the syncytium (Fig. 7, E–G). Taken together, these data show that the NLRP3 inflammasome is a primary component in the placental innate response and defense against L. monocytogenes infection.
Discussion
Our work presented in this article identifies the NLRP3 inflammasome as a regulator of immunity at the maternal–fetal interface. We show that placental trophoblasts constitutively release IL-1β and IL-18 through the activity of the inflammasome and that this is further enhanced by L. monocytogenes infection. Moreover, we demonstrate that inflammasome hyperactivation and IL-1β secretion occur in trophoblasts in the absence of detectable gasdermin D cleavage and without detectable cell death. Our work also suggests that inflammasome activation controls L. monocytogenes infection and that inhibition of this pathway sensitizes the human placenta to infection. We also demonstrate that placenta-derived IL-1β may play a role in sensitizing maternal-derived immune or other cells to subsequent infection or inflammatory stimuli. Our study thus defines a pathway by which the placenta regulates local and systemic innate immunity during human pregnancy.
Although previous work on the inflammasome in pregnancy associated this signaling with pathogenic conditions such as spontaneous preterm delivery and preeclampsia, the role of this pathway in normal pregnancies matched to gestational age at delivery was not fully investigated (Gomez-Lopez et al., 2017; Matias et al., 2015). Within this context, our data implicate placenta-derived inflammasome activity as being associated with maintenance of healthy pregnancy because the placentas used in our study were not associated with any known pathologies. Consistent with this, we detected IL-1β in the systemic circulation of healthy women in the second trimester of pregnancy, suggesting that placenta-derived IL-1β is secreted in nonpathological states. However, it is possible that the elevated levels of IL-1β circulating during pregnancy also arise from additional sources. Although the biological function of IL-1β in healthy pregnancy is unknown, it is likely involved in mediating the complex immunological crosstalk between maternal and fetal compartments during pregnancy, and alterations in its levels may send a danger signal to the maternal host.
Interestingly, we found that villi isolated from full-term placentas exhibited reduced release of IL-1β and other inflammasome-associated cytokines compared with midgestation placentas. This finding further suggests the role of the inflammasome in maintenance of normal pregnancy. The mechanistic basis for these differences remains unclear, but several key changes in the placenta between these gestational stages may be responsible. For example, placental villi undergo rapid expansion and growth throughout the second trimester until term, which corresponds with changes in nuclear morphology and abundance of heterochromatin, among other changes (Jackson et al., 1992). Despite these differences, syncytiotrophoblasts remain transcriptionally active across all stages of gestation (Ellery et al., 2009). Thus, gestation-associated differences associated with changes in placental structure or cellular activity may impact the secretion of placenta-derived immunological factors. Previous studies using cell-signaling elastic network analysis have defined that peripheral cellular programming changes with gestational age (Aghaeepour et al., 2017). Future studies focused on these networks and corresponding alteration of cytokines secreted by trophoblasts may provide additional insight into the regulation of the immune secretome of the placenta.
Our data suggest that placenta-derived IL-1β, and likely other cytokines, alters the function of maternal-derived immune cells. In the case of IL-1β, we show that this cytokine released by trophoblasts primes maternal monocytes for enhanced inflammasome signaling. This concept is supported by our data showing that the transcripts of inflammasome-associated components such as caspase-1 and IL-1β are elevated in monocytes isolated from pregnant women. These findings are consistent with the work of others who have also identified increased expression of inflammasome components in maternal-derived monocytes, which are more responsive to inflammasome activators, although the mechanistic basis for these differences was not explored (Matias et al., 2015). Our study thus describes a direct mechanism by which the placental immune secretome could modulate circulating innate immune cells in this manner. This has substantial clinical relevance because it has been appreciated for some time that immune cells derived from pregnant women exhibit alterations in phenotype and function (Mor and Cardenas, 2010; Le Gars et al., 2019). Our data suggest that the placenta is a highly reactive immunological organ that provides signals that directly alter maternal immunity. In addition, our data support a model whereby infection during pregnancy is subject to robust, placenta-derived immune modulation that alters maternal susceptibility to infection and disease.
The maternal–fetal interface has been demonstrated to exhibit increased inflammation during the events of implantation and with parturition (Racicot et al., 2014; Dekel et al., 2014). However, our study identifies proinflammatory cytokines, including IL-1β, in healthy pregnancies even in the early stages of the second trimester. We show that the inflammasome is constitutively active in placental syncytiotrophoblasts and that these cells secrete high levels of the inflammasome-associated cytokines IL-1β and IL-18. Importantly, although inflammasome induction is associated with pyroptosis, a highly inflammatory means of programmed cell death, there is no evidence that syncytiotrophoblasts themselves undergo this form of cell death. This is supported by our findings that the integrity of this layer is intact, that syncytiotrophoblasts remain active and secrete known pregnancy-associated hormones and other factors, that they are responsive to various infection-associated stimuli (e.g., poly I:C and R848), and that they retain their barrier properties against microbial infections. In addition, we were unable to detect the N-terminal cleavage product of gasdermin D in chorionic villi by immunohistochemistry or immunoblotting, suggesting that the release of IL-1β occurs through a gasdermin D–independent pathway in trophoblasts. This form of IL-1β secretion has been proposed to occur in myeloid cells undergoing hyperactivated states of prolonged inflammation (Monteleone et al., 2018) but has not been described in tissue. Of note, placental villi are constitutively in a hyperactivated state that is associated with the constitutive release of many proinflammatory factors and may have a unique shared mechanism for secretion that has yet to be characterized.
Previous work has demonstrated that L. monocytogenes can activate the NLRP3 and AIM2 inflammasomes (Li et al., 2016; Kim et al., 2010; Sauer et al., 2010). Our data implicate the NLRP3 inflammasome as an important mechanism by which the placenta responds to and controls L. monocytogenes infection, both at the maternal fetal interface and systemically. We show that L. monocytogenes infection of chorionic villi robustly induces the release of IL-1β and IL-18. In mice, IL-1RA (which competes with IL-1β to antagonize the IL-1R) has been shown to be an important determinant of listeriosis, and its overexpression increases susceptibility to infection and fetal loss (Irikura et al., 1999). Our data also suggest that inflammasome activation in response to L. monocytogenes infection controls infection in the placenta itself. Consistent with previous work in the first trimester (Robbins et al., 2010), our data show that syncytiotrophoblasts potently resist infection by L. monocytogenes in the mid to late stages of gestation. Although the mechanism for this resistance has remained largely unknown, others have shown that the physical properties of this layer, including a dense cortical actin cytoskeleton, might be involved (Robbins et al., 2010). Our data support a model by which syncytiotrophoblasts use physical and innate immunological barriers in the defense against L. monocytogenes, given that inhibition of inflammasome activation increased infection in the syncytium. However, it should be noted that the levels of overall infection remain low, supporting parallel pathways of resistance, as exist for nonbacterial pathogens such as Toxoplasma gondii (Ander et al., 2018).
Taken together, our results show that placental inflammasome activity plays an important role in antimicrobial defenses at the maternal–fetal interface. In addition, our work also defines the immunological landscape of the placenta from early to middle stages of pregnancy and highlights its potential impacts on maternal immunity. Last, our work supports a role for inflammasome-mediated cytokine secretion in the altered responses of pregnant women to infections or other inflammatory stimuli, which has implications for understanding the enhanced inflammatory responses to pathogenic stimuli during pregnancy.
Materials and methods
Placenta sample collection and processing
Human placental tissue from <24-wk gestation that resulted from elective terminations was obtained from the University of Pittsburgh Health Sciences Tissue Bank through an honest broker system after approval was obtained from the University of Pittsburgh Institutional Review Board (IRB) and in accordance with the University of Pittsburgh’s tissue procurement guidelines. Tissue was from healthy normal donors, and tissue was excluded in cases of fetal anomalies or aneuploidy. Third-trimester and full-term human placentas were obtained through the Magee Obstetric Maternal & Infant Database and Biobank after approval was received from the University of Pittsburgh IRB. Women who had previously consented to tissue donation and had undergone cesarean delivery were included. Women with labor, chorioamnionitis, preterm premature rupture of membranes, maternal complications such as systemic infection, diabetic ketoacidosis, or sepsis or fetal anomalies were excluded. After delivery, a full-thickness biopsy was obtained and immediately processed for dissection (<2 h from collection). Placental tissue was dissected to isolate chorionic villi and FM (∼0.5 cm × 0.5 cm in size).
Primary human trophoblasts
Cells from midgestation or full-term placentas were isolated using the trypsin-DNase-dispase/Percoll method as described elsewhere (Kliman et al., 1986; Tang et al., 2011; Stromberg et al., 1978). Cells were maintained in MEM (10-010-CV; MilliporeSigma) containing 10% FBS (HyClone Laboratories) and penicillin/streptomycin. Media were changed every 24 h, and cells were maintained at 37°C in 5% CO2 for 72–96 h after plating.
Cell culture
THP-1 (TIB-202), JEG-3 (HTB-36), BeWo (CCL-98), and Caco-2 (HTB-37) cells were purchased from the American Type Culture Collection. ASC-GFP–expressing THP-1 cells (thp-ascgfp) and reporter HEK-Blue cells for IL-1R (hkb-il1r) and IFNLR (hkb-ifnl) were purchased from InvivoGen. THP-1 cells were cultured in RPMI-1640 medium (SH30027.01; HyClone Laboratories) supplemented with 10% Gibco FBS (26140079; Life Technologies) with Gibco L-glutamine (25030; Life Technologies) and 1% penicillin/streptomycin (17-602E; Lonza) and were maintained at 37°C in an incubator at densities of 1 × 104 to 106 cells/ml. BeWo cells were cultured in Gibco Ham’s F-12K media with Kaighn’s modification (21127-022; Life Technologies) and containing 10% FBS with 1% penicillin/streptomycin. JEG-3 cells were cultured in Gibco Eagle's MEM (12492-013; Life Technologies) supplemented with 10% FBS and 1% penicillin/streptomycin. Caco-2 cells were cultured in Gibco DMEM (10-017-CV; Life Technologies) supplemented with 10% FBS and 1% penicillin/streptomycin. Cells were enumerated using a TC20 cell counter (catalog no. 1450102; Bio-Rad Laboratories).
Generation of CM
Chorionic villi were dissected and cultured in 1 ml (per villus) of DMEM/F-12 (1:1) supplemented with 10% FBS and penicillin/streptomycin for 24 h. Supernatants were collected and centrifuged to remove debris. Media from at least five individual villi were collected per placental preparation and pooled to generate CM. BeWo and JEG3 cells were grown to confluence and incubated overnight in the appropriate media to collect CM. Primary human trophoblasts were cultured for 48 h, and then media were collected as CM.
Maternal blood sample processing
Samples from pregnant women were obtained through the Magee Obstetric Maternal & Infant Database and Biobank following approval from the University of Pittsburgh IRB. Women with any preexisting pregnancy complications (e.g., diabetes, hypertension, preterm labor) were excluded, and they were also excluded if they experienced any symptoms within 7 d of an infection including but not limited to rhinorrhea, cough, systemic malaise, odynophagia, or fatigue. Women who met study criteria donated at the time of routine Glucola challenge testing. Blood was collected in a Na-heparin tube and immediately processed (within ∼1 h). Samples from nonpregnant women were obtained through venipuncture from healthy volunteers in the absence of symptoms of an infection within 7 d, as noted above. Samples were diluted 1:1 in PBS 2 mM EDTA, layered over Ficoll-Percoll PLUS (17-1440-02; GE Healthcare Life Sciences), and centrifuged at 400 ×g at room temperature for 30 min. Plasma was collected, aliquoted, and stored at −80°C. The peripheral blood mononuclear cell (PBMC) layer was removed, resuspended in PBS 2 mM EDTA, and centrifuged at 800 ×g for 5 min to pellet the cells. The PBMCs were resuspended and centrifuged at 800 ×g for 5 min to remove any remaining Ficoll before use. PBMCs from nonpregnant women of reproductive age (25–45 yr old) were also purchased from the American Type Culture Collection (PCS-800-011). PBMCs were then used to isolate CD14+ monocytes using the Pan Monocyte Isolation Kit (130-096-537; Miltenyi Biotec) according to the manufacturer’s instructions. Monocytes were plated at a density of 5 × 105 in DMEM/F-12 with 2.5% human serum (H3667; MilliporeSigma) and penicillin/streptomycin and allowed to rest for 1–2 h at 37°C before collection or use. Pregnant and nonpregnant donor characteristics are described in Table S2.
Immunofluorescence microscopy
Cells or tissues were fixed in 4% paraformaldehyde (in 1× PBS), followed by permeabilization in 0.25% Triton X-100 for 15–30 min, then they were washed in PBS and incubated with primary antibody for at least 1 h at room temperature. Following washing in PBS, cells/tissues were incubated with Alexa Fluor–conjugated secondary antibodies. Alexa Fluor–conjugated phalloidin was purchased from Invitrogen (A12381). Rabbit anti–IL-1β (ab9722) and anti–caspase-1 (ab62698) antibodies were purchased from Abcam. Rabbit anti–gasdermin D (96458), cleaved gasdermin D (36425), and cleaved IL-1β (83186) were purchased from Cell Signaling Technology. Cells or tissues were mounted with VECTASHIELD mounting medium (H-1200; Vector Laboratories) containing DAPI, and images were captured using an Olympus IX83 inverted fluorescence microscope or a Zeiss LSM confocal microscope. Images were adjusted for brightness/contrast using Adobe Photoshop (Adobe). For three-dimensional sections, confocal images were acquired at 0.35–0.5-µm intervals.
Immunohistochemistry
Tissue sections were deparaffinized with xylene and rehydrated with decreasing concentrations of ethanol (100%, 95%, and 80%), then washed with double-distilled water. Antigen retrieval was performed with slides submerged in 10 mM citrate buffer (pH 6.0) and heated in a steamer at 90°C for 20 min. Slides were cooled to room temperature and incubated with 6% H2O2 in methanol for 30 min. Following washing in 0.1% PBS-T (PBS with 0.1% Tween-20), slides were incubated in avidin blocking solution for 15 min, followed by subsequent blocking in biotin blocking solution for 15 min (SP-2001; Vector Laboratories). Following washing in 0.1% PBS-T, slides were incubated with serum-free protein block (ab156024; Abcam) for 10 min. Sections were incubated with primary antibodies diluted 1:100 or 1:250 in PBS-T overnight in a humidified chamber at 4°C. Next, slides were washed with PBS-T and incubated with secondary antibody (biotinylated goat antirabbit IgG, BA-1000; Vector BioLabs) for 30 min, washed, and then incubated with avidin/biotin-based peroxidase (VECTASTAIN Elite ABC HRP Kit, PK-6100; Vector Laboratories) for an additional 30 min. Following washes in PBS-T, sections were incubated with 3,3′-diaminobenzidine substrate (SK-4100; Vector Laboratories) for ∼5 min. Slides were washed with double-distilled H2O and then counterstained with hematoxylin for 1 min, thoroughly rinsed with H2O, and incubated in 0.1% sodium bicarbonate in H2O for 5 min. Slides were then dehydrated with increasing concentrations of ethanol, cleared with xylene, and mounted with VectaMount Permanent Mounting Medium (H-5000; Vector Laboratories). Images were captured on an IX83 inverted microscope (Olympus) using a UC90 color charge-coupled device camera (Olympus).
RNA isolation, quantitative RT-PCR (RT-qPCR), and RNA-seq
RNA was extracted and isolated using the GenElute Mammalian Total RNA Miniprep Kit and associated DNase digestion kit (RTN 350; MilliporeSigma). A NanoDrop One spectrophotometer (Thermo Fisher Scientific) was used to validate RNA quality and quantity. Total RNA was reverse transcribed using the iScript cDNA synthesis kit (1708891; Bio-Rad Laboratories). RT-qPCR was performed using iQSYBR Green Supermix (1725121; Bio-Rad Laboratories) in a Bio-Rad CFX96 Touch real-time PCR machine according to the manufacturer's instructions. Gene expression was calculated using the comparative cycle threshold method and normalized to human β-actin. Primers used were as follows: actin (5′-ACTGGGACGACATGGAGAAAA-3′; 5′-GCCACACGCAGCTC-3′), caspase-1 (5′-CAACTACAGAAGAGTTGAGG-3′; 5′-AACATTATCTGGTGTGGAAG-3′), IL-1β (5′-CTAAACAGATGAAGTGCT-3′; 5′-GGTCATTCTCCTGGAAG-3′), and NLRP3 (5′-AGGTGTTGGAATTAGACAAC-3′; 5′-AATACATTTCAGACAACCCC-3′). All sample processing was performed in duplicate, and each experiment was performed with at least three biological replicates. Heatmaps based on RNA-seq log2 reads per kilobase million values were generated using previously published datasets available in the Sequence Read Archive (SRP067137, SRP109039).
Immunoblotting
Cells lysates were prepared on ice in cold radioimmunoprecipitation assay buffer (50 mM Tris-HCl, pH 7.4, 1% Nonidet P-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA; MilliporeSigma) with 1× protease inhibitor cocktail (4-[2-aminoethyl]benzenesulfonyl fluoride hydrochloride, aprotinin, bestatin, E64, leupeptin, pepstatin A; Thermo Fisher Scientific). Lysates were separated on 4–20% Tris-HCl gels (4561093 or 3450033; Bio-Rad Laboratories) and transferred to nitrocellulose membranes. Membranes were blocked with 5% nonfat milk before being probed with primary antibodies overnight. Following this incubation, membranes were incubated with HRP-conjugated secondary antibodies. SuperSignal West Femto (Thermo Fisher Scientific) or SuperSignal West Dura (Thermo Fisher Scientific) chemiluminescence substrate was applied, and the immunoblots were imaged by autoradiography.
ELISA and Luminex assays
IL-1β and PSG-1 ELISAs were performed using the IL-1β DuoSet Kit (DY201; R&D Systems) or the PSG1 Quantikine Kit (DPSG10; R&D Systems) according to the manufacturer’s instructions. Plates were read at 450 and 520 mm using a Synergy H1 microplate reader. Results were normalized to a standard curve, and concentration per well was extrapolated. All sample processing was performed in duplicate, and each experiment was performed with at least three biological replicates. Luminex assays were performed using the following kits according to the manufacturers’ instructions: the Bio-Plex Pro Human Inflammation Panel 1, 37-Plex kit (171AL001M; Bio-Rad Laboratories); the Bio-Plex Pro Human Cytokine Screening Panel, 48-Plex kit (12007283; Bio-Rad Laboratories); the Bio-Plex Pro Human Chemokine IL-1β Set (171BK26MR2; Bio-Rad Laboratories); or the MILLIPLEX MAP Human Circulating Cancer Biomarker Magnetic Bead Panel–Cancer Multiplex Assay, β-hCG beads (HCCBP1MAG-58K; MilliporeSigma). All samples were completed in duplicate, read on a MAGPIX (EMD Millipore) Luminex machine, and analyzed using xPONENT software (EMD Millipore). Multivariate sPLS-DA was performed to identify the cytokines characterizing villi- and FM-derived CM. sPLS-DA was performed using the mixOmics package in R. Briefly, sPLS-DA was run in the classification mode, and the model was evaluated using fivefold cross-validation repeated 500 times. The variable selection and classification are performed as a one-step procedure. We visually selected the components that showed maximum separation between the classes and extracted the features that contributed to these components.
L. monocytogenes growth, maintenance, and infections
mCherry-expressing L. monocytogenes (10403S) was provided by Daniel Portnoy, University of California, Berkeley (Berkeley, CA). Bacterial colonies were resuspended in brain heart infusion broth and grown at 37°C in a shaking incubator until stationary phase was reached as measured by OD600. Bacteria were diluted and grown an additional 2–4 h at 37°C until OD600 was 0.4–0.6 nm. Bacteria were pelleted and then resuspended in 1× PBS before cells were infected at a multiplicity of infection (MOI) of 1–10 as indicated in the figure legends. After 2 h at 37°C, gentamicin (100 µg/ml) was added to the media, and cells were infected for ∼24 h. At this time, cells and supernatants were collected and pelleted at 10,000 ×g for 10 min and then resuspended in PBS. Viable bacterial counts of intracellular bacteria were determined by plating serial dilutions onto brain heart infusion agar and calculating CFU per milliliter.
Inflammasome priming and activation
Monocytes were exposed to LPS (100 ng/ml) or placenta-derived CM for at least 16 h. Cells were collected and pelleted at 1,200 rpm for 5 min; media were removed; and cell pellets were resuspended in RPMI complete media. Nigericin (6.7 µM; InvivoGen) was added, and cells were incubated for 2 h at 37°C. Cells were then centrifuged; supernatants were collected for ELISA and Luminex assays; and cell pellets were lysed for RNA extraction or protein as described.
Caspase-1 activity
Caspase-1 activity was measured in culture medium using the Caspase-Glo 1 Inflammasome Assay Kit (G9951; Promega) according to the manufacturer’s instructions.
Generation of THP-1–IL-1β–knockout cells
IL-1R was deleted from THP-1 cells with the single guide RNA sequence 5′-TTCAGGACATTACTATTGC-3′, using the LentiArray lentiviral vector (A32042; Invitrogen). THP-1 cells stably expressing mCherry-Cas9 (THP-1 Cas9-mCherrytg) were provided by Seth Masters (Walter and Eliza Hall Institute of Medical Research, Parkville, Australia) and have been described elsewhere (Baker and Masters, 2018). THP-1 Cas9 cells were transduced with LentiArray lentiviral particles expressing the IL-1R gRNA at an MOI of 0.3 in the presence of 5 µg/ml polybrene (TR-1003-G; MilliporeSigma) while centrifuging at 800 ×g for 60 min. Approximately 3 d following transduction, cells were placed in selection (puromycin, 3 µg/ml) for 7 d, and individual clones were isolated by limiting dilution. IL-1R deletion was verified by immunoblotting and Sanger sequencing.
Pharmacologic inhibitors and blocking antibody
Chorionic villi were treated with ILG (InvivoGen) at 40 µM for ∼18 h before harvesting medium. MCC950 (InvivoGen) was used at 15 µM. For blocking IL-1R activity, cells were incubated with anti–IL-1R blocking antibody (100 µg/ml; R&D Systems) for 1 h at 16°C before the addition of medium containing anti–IL-1R (100 µg/ml).
Antibodies
Pro–IL-1β (12703), cleaved IL-1β (83186), anti–gasdermin D (96458), cleaved gasdermin D (36425), and ASC (13833) rabbit monoclonal antibodies were purchased from Cell Signaling Technology. Rabbit anti-AIM2 (ab93015), cleaved IL-1β (ab2105), and NLRP3 (ab214185) were purchased from Abcam. Rabbit anti-GAPDH (sc-25778) and mouse anti–IL-1R (sc-393998) were obtained from Santa Cruz Biotechnology. Antirabbit HRP secondary antibody was purchased from Thermo Fisher Scientific (31460).
Statistics
All statistical analyses were performed using Prism software (GraphPad Software). Data are presented as mean ± SD, unless otherwise stated. Statistical significance was determined as described in the figure legends. Parametric tests were applied when data were distributed normally on the basis of D’Agostino-Pearson analyses; otherwise, nonparametric tests were applied. P values <0.05 were considered statistically significant, with specific P values noted in the figure legends.
Online supplemental material
Fig. S1 demonstrates βhCG and PSG-1 secretion in CM from chorionic villi and FM preparations, shows the differences in cytokine abundance after sPLS-DA, compares IL-1β with IL-1α, IL-18, and IL-1β levels across gestational ages, and demonstrates the differences in IL-1β in CM of various placental cell lines and compares IL-1β with IFN-λ. Fig. S2 demonstrates differences in cytokine secretion between midgestation and term villous preparations, shows caspase-1 activity in CM, and shows pro–caspase-1 localization in Hofbauer cells and lack of IL-1β in FMs and suppression of caspase-1 activity in CM with treatment with ILG and MCC950. Fig. S2 F demonstrates that minimal LDH is present in CM. Fig. S3 shows the response of monocytes to exposure to CM in IL-1β, caspase-1, and NLRP3 transcripts; in addition, it shows ASC-GFP punctae formation with nigericin treatment, the change in caspase-1 activity, and punctae formation with treatment with MCC950, and with the presence of IL-1R antibody or in the IL-1R–knockout THP-1 monocytes. Fig. S4 demonstrates ASC-GFP punctae with LPS or CM priming, provides bacterial quantification when grown in CM, and demonstrates caspase-1 activity with L. monocytogenes infection and treatment with MCC950. Fig. S5 demonstrates the lack of visualization of bacteria in placental trophoblast explants after L. monocytogenes infection and the response of TNF-α and RANTES secretion with L. monocytogenes infection with or without ILG and MCC950. Table S1 lists characteristics of placental tissue used in this study. Table S2 lists characteristics of human pregnant and nonpregnant donors.
Acknowledgments
We thank Scott Canna and William DePas (University of Pittsburgh Medical Center Children’s Hospital of Pittsburgh) for helpful suggestions; Azia Evans (University of Pittsburgh) and Ira Blader (Jacobs School of Medicine, University at Buffalo) for critical review of the manuscript; Seth Masters for providing Cas9-expressing THP-1 cells, and Daniel Portnoy for providing mCherry L. monocytogenes. We also thank the Magee Obstetric Maternal & Infant Database and Biobank at the Magee-Womens Research Institute for providing placental tissue and serum from maternal donors.
This project was supported by National Institutes of Health grants AI145828 and HD097400 (to C.B. Coyne), Magee-Womens Research Institute Clinical Trainee Research Award 4032 (to C. Megli), and the Children’s Hospital of Pittsburgh of the University of Pittsburgh Medical Center Health System (to C.B. Coyne). This project also used the University of Pittsburgh Medical Center Hillman Cancer Center and Tissue and Research Pathology/Pittsburgh Biospecimen Core and Animal Facility shared resources, which are supported in part by award P30CA047904.
Author contributions: C. Megli designed and performed experiments and participated in manuscript review and writing. S. Morosky performed experiments. D. Rajasundaram performed biostatistical analysis of cytokine profiles. C.B. Coyne designed and performed experiments, participated in manuscript review and writing, and secured funding.

