


Macropinocytosis mediates the non-selective bulk uptake of extracellular fluid, enabling cells to survey the environment and obtain nutrients. A conserved set of signaling proteins orchestrates the actin dynamics that lead to membrane ruffling and macropinosome formation across various eukaryotic organisms. At the center of this signaling network are Ras GTPases, whose activation potently stimulates macropinocytosis. However, how Ras signaling is initiated and spatiotemporally regulated during macropinocytosis is not well understood. By using the model system Dictyostelium and a proteomics-based approach to identify regulators of macropinocytosis, we uncovered Leep2, consisting of Leep2A and Leep2B, as a RasGAP complex. The Leep2 complex specifically localizes to emerging macropinocytic cups and nascent macropinosomes, where it modulates macropinosome formation by regulating the activities of three Ras family small GTPases. Deletion or overexpression of the complex, as well as disruption or sustained activation of the target Ras GTPases, impairs macropinocytic activity. Our data reveal the critical role of fine-tuning Ras activity in directing macropinosome formation.
Introduction
Macropinocytosis is an evolutionarily conserved mechanism that mediates non-selective bulk uptake of extracellular fluid. It serves diverse functions in various physiological and pathological contexts. Initially recognized as a means for cells of the innate immune system, including macrophages and dendritic cells, to survey environmental antigens (Lin et al., 2020; Norbury et al., 1995; Sallusto et al., 1995), recent studies in cancer cells and the model organism Dictyostelium discoideum have uncovered an important metabolic function of macropinocytosis. Cancer cells exploit macropinocytosis to survive in nutrient-poor microenvironments by scavenging extracellular proteins and fatty acids (Commisso et al., 2013; Kamphorst et al., 2013; Kim et al., 2018; Palm et al., 2015; Wyant et al., 2017). Laboratory strains of Dictyostelium rely on macropinocytosis for the uptake of essential nutrients, such as glucose and amino acids, from liquid media (Hacker et al., 1997; Williams and Kay, 2018; Zhang et al., 2022). In addition, macropinocytosis has been implicated in pathogen infiltration, cell migration, and plasma membrane repair (Le et al., 2021; Moreau et al., 2019; Saeed et al., 2010; Sonder et al., 2021).
Macropinocytosis is initiated through local remodeling of the plasma membrane and the underlying actin cytoskeleton, leading to the formation of cup-shaped protrusions that close to generate micrometer-sized vesicles called macropinosomes. Newly formed macropinosomes traffic through the endolysosomal system, where their contents are digested and extracted (Palm, 2019; Vines and King, 2019). Studies in Dictyostelium and mammalian macrophages have revealed that the formation of macropinocytic cups is organized around intensive signaling patches of PIP3 and active small GTPases from the Ras and Rac families, which promote actin rearrangement and membrane deformation (Fujii et al., 2013; Mylvaganam et al., 2021; Parent et al., 1998; Sasaki et al., 2007; Swanson and Araki, 2022; Veltman et al., 2016; Welliver and Swanson, 2012). Actin polymerization driven by the Scar/WAVE complex and formin proteins is directed to the periphery and base of the signaling patches, respectively (Junemann et al., 2016; Veltman et al., 2014, 2016). This arrangement enables the concentration of branched actin filaments at the rim of the cup, promoting outward extension of the plasma membrane. Meanwhile, linear actin filaments align parallel to the base of the cup, providing structural support for the developing macropinosome in the absence of a physical template.
Although we have gained a basic understanding of macropinosome formation, the complete set of regulatory components and their spatiotemporal integration in driving this process remain to be fully elucidated. Earlier studies in Dictyostelium revealed that many signaling and cytoskeletal molecules involved in the formation of macropinocytic cups, such as PIP3 and active Ras, are also key regulators of pseudopodia protrusion, as indicated by their enrichment at the leading edge of motile cells (Parent et al., 1998; Sasaki et al., 2004; Yang et al., 2021). Furthermore, global chemoattractant stimulation induces transient production of these molecules at the plasma membrane or cell cortex, leading to the translocation of their effectors or other associated proteins from the cytoplasm to the cell periphery (Sobczyk et al., 2014; Swaney et al., 2010). Taking advantage of this phenomenon, we developed a proteomics-based screen to systematically identify regulators of macropinocytosis and migration by isolating proteins that exhibit the characteristic translocation behavior following chemoattractant stimulation (Yang et al., 2021). Among the candidate proteins, we previously characterized leading edge enriched protein 1 (Leep1) and demonstrated its role in coordinating PIP3 signaling and actin polymerization during macropinocytosis and migration (Yang et al., 2021). In the current study, we focus on another leading-edge protein identified from the same screen that modulates the activities of Ras family small GTPases to regulate macropinosome formation.
Results
Leep2 complex localizes to macropinocytic cups and nascent macropinosomes
In our proteomics-based screen for novel regulators of macropinocytosis and migration (Yang et al., 2021), we identified a protein with 1,640 amino acids (gene ID: DDB_G0284825). Mass spectrometry analysis revealed a translocation score of 0.61 for DDB_G0284825 (Fig. 1 A), suggesting that it may transiently accumulate in the peripheral membrane fraction following chemoattractant stimulation, a characteristic feature of leading-edge proteins (Yang et al., 2021). Therefore, we named this protein leading edge–enriched protein 2A (Leep2A). Expression of GFP-Leep2A in wild-type (WT) cells and observation of its localization dynamics confirmed the rapid translocation of a fraction of Leep2A to the plasma membrane in cells stimulated with the chemoattractant cAMP (Fig. 1, B and C). Furthermore, in cells actively engaged in macropinocytosis, GFP-Leep2A was specifically enriched at the membrane patches that invaginated to form macropinocytic cups and subsequently dispersed from the newly formed macropinosomes shortly after internalization (Fig. 1 D). In contrast to the intense concentration at macropinocytic cups, GFP-Leep2A seemed to be absent from pseudopods in randomly moving cells and exhibited a homogenous distribution in the cytosol and the actin-rich hyaline zones (Fig. 1 E).

Leep2A and Leep2B localize to macropinocytic cups and nascent macropinosomes. (A) Mass spectrometry (MS) scores of Leep2A (DDB_G0284825) at the indicated time points following cAMP stimulation. Translocation score (the sum of protein scores at 10 and 20 s divided by the sum of all four time points) of Leep2A was calculated to be 0.61. (B) Quantification of GFP-Leep2A translocation in WT cells upon cAMP stimulation (1 μM cAMP was added at time 0; mean ± SD, n represents the number of cells analyzed). (C) Time-lapse imaging of GFP-Leep2A translocation in response to cAMP stimulation in WT cells (1 μM cAMP was added at time 0). (D) Localization of GFP-Leep2A in WT cells during macropinocytosis. Arrowhead indicates an emerging macropinosome. (E) Localization of GFP-Leep2A in WT cells during random migration. Arrowhead indicates the migrating front. (F) Proteomic identification of Leep2B (DDB_G0281809) as a binding partner of Leep2A. GFP-Leep2A was immunoprecipitated from cell lysates by GFP-trap and the bound proteins analyzed by mass spectrometry. PSMs, peptide spectrum matches. (G) Co-IP of RFP-Leep2B or PHcrac-RFP with GFP or GFP-Leep2A. Fluorescent fusion proteins were expressed in WT cells. IP was performed with GFP-trap and samples were probed with GFP or RFP antibody. (H) Localization of GFP-Leep2B in WT cells during macropinocytosis. Arrowhead indicates an emerging macropinosome. (I) Colocalization of GFP-Leep2A and RFP-Leep2B in WT cells. Arrowheads indicate an emerging macropinosome. (J) Localization of GFP-Leep2A and RFP-Leep2B in WT cells migrating under agarose along a folic acid (FA) gradient. The white triangle indicates the gradient direction. Scale bars, 5 μm. Source data are available for this figure: SourceData F1.
Leep2A and Leep2B localize to macropinocytic cups and nascent macropinosomes. (A) Mass spectrometry (MS) scores of Leep2A (DDB_G0284825) at the indicated time points following cAMP stimulation. Translocation score (the sum of protein scores at 10 and 20 s divided by the sum of all four time points) of Leep2A was calculated to be 0.61. (B) Quantification of GFP-Leep2A translocation in WT cells upon cAMP stimulation (1 μM cAMP was added at time 0; mean ± SD, n represents the number of cells analyzed). (C) Time-lapse imaging of GFP-Leep2A translocation in response to cAMP stimulation in WT cells (1 μM cAMP was added at time 0). (D) Localization of GFP-Leep2A in WT cells during macropinocytosis. Arrowhead indicates an emerging macropinosome. (E) Localization of GFP-Leep2A in WT cells during random migration. Arrowhead indicates the migrating front. (F) Proteomic identification of Leep2B (DDB_G0281809) as a binding partner of Leep2A. GFP-Leep2A was immunoprecipitated from cell lysates by GFP-trap and the bound proteins analyzed by mass spectrometry. PSMs, peptide spectrum matches. (G) Co-IP of RFP-Leep2B or PHcrac-RFP with GFP or GFP-Leep2A. Fluorescent fusion proteins were expressed in WT cells. IP was performed with GFP-trap and samples were probed with GFP or RFP antibody. (H) Localization of GFP-Leep2B in WT cells during macropinocytosis. Arrowhead indicates an emerging macropinosome. (I) Colocalization of GFP-Leep2A and RFP-Leep2B in WT cells. Arrowheads indicate an emerging macropinosome. (J) Localization of GFP-Leep2A and RFP-Leep2B in WT cells migrating under agarose along a folic acid (FA) gradient. The white triangle indicates the gradient direction. Scale bars, 5 μm. Source data are available for this figure: SourceData F1.
Sequence analysis revealed that Leep2A contains a putative GTPase-activating protein (GAP) domain at the C-terminus (Fig. S1 A). It exhibits sequence homology and a domain organization similar to that of human proteins RalGAPA1 and RalGAPA2 (19.8% identity and 33.9% similarity to RalGAPA1, 20.5% identity and 35.1% similarity to human RalGAPA2), which are catalytic subunits of the heterodimeric RalGAP complex (Fig. S1, A and B) (Chen et al., 2011; Shirakawa et al., 2009). In the complex, RalGAPA1 or RalGAPA2 associates with another GAP domain-containing protein, RalGAPB. Together, they function as a GTPase activator for the Ras-like small GTPases RALA and RALB (Chen et al., 2011; Shirakawa et al., 2009). Interestingly, a Blast search of the Dictyostelium genome revealed a protein (gene ID: DDB_G0281809) homologous to human RalGAPB (17.9% identity and 29.4% similarity), which we consequently named Leep2B (Fig. S1, A and B). Immunoprecipitation and mass spectrometry showed that endogenous Leep2B was highly enriched in the immunocapture of GFP-Leep2A but not the GFP control (Fig. 1 F). A coimmunoprecipitation experiment verified this interaction (Fig. 1 G). GFP-Leep2A specifically immunoprecipitated RFP-tagged Leep2B but not RFP-tagged PHcrac, a Pleckstrin homology (PH) domain from the cytosolic regulator of adenylyl cyclase (CRAC), which shares the macropinocytic cup localization pattern with Leep2A (Parent et al., 1998; Yang et al., 2021). These results imply that, similar to their mammalian counterparts, Leep2A and Leep2B likely form a protein complex.

Leep2 complex translocates to the cell periphery in response to chemoattractant stimulation. (A) Schematic illustration of the domain organization of Leep2 and human RalGAP complexes. (B) Phylogenetic tree of Leep2 and human RalGAP complexes. (C) Quantification of the fluorescent intensity of GFP-Leep2A at macropinocytic cups in WT cells, with or without the expression of RFP-Leep2B. The scatter plot shows data points with means and SD (n represents the number of cells analyzed). Significance was determined by two-tailed unpaired t test. (D) Translocation of GFP-Leep2A and RFP-Leep2B in response to cAMP stimulation (1 μM cAMP was added at time 0). Top: Time-lapse imaging of translocation. Bottom: Quantification of translocation (mean ± SD, n represents the number of cells analyzed). (E) Translocation of GFP-Leep2A and RFP-Leep2B in response to folic acid (FA) stimulation (50 μM FA was added at time 0). Top: Time-lapse imaging of translocation. Bottom: Quantification of translocation (mean ± SD, n represents the number of cells analyzed). (F) Translocation of GFP-Leep2A and RFP-Leep2B in response to cAMP stimulation (1 μM cAMP was added at time 0) in the presence of Latrunculin A (LatA). Top: Time-lapse imaging of translocation. Bottom: Quantification of translocation (mean ± SD, n represents the number of cells analyzed). Scale bars, 5 μm.

Leep2 complex translocates to the cell periphery in response to chemoattractant stimulation. (A) Schematic illustration of the domain organization of Leep2 and human RalGAP complexes. (B) Phylogenetic tree of Leep2 and human RalGAP complexes. (C) Quantification of the fluorescent intensity of GFP-Leep2A at macropinocytic cups in WT cells, with or without the expression of RFP-Leep2B. The scatter plot shows data points with means and SD (n represents the number of cells analyzed). Significance was determined by two-tailed unpaired t test. (D) Translocation of GFP-Leep2A and RFP-Leep2B in response to cAMP stimulation (1 μM cAMP was added at time 0). Top: Time-lapse imaging of translocation. Bottom: Quantification of translocation (mean ± SD, n represents the number of cells analyzed). (E) Translocation of GFP-Leep2A and RFP-Leep2B in response to folic acid (FA) stimulation (50 μM FA was added at time 0). Top: Time-lapse imaging of translocation. Bottom: Quantification of translocation (mean ± SD, n represents the number of cells analyzed). (F) Translocation of GFP-Leep2A and RFP-Leep2B in response to cAMP stimulation (1 μM cAMP was added at time 0) in the presence of Latrunculin A (LatA). Top: Time-lapse imaging of translocation. Bottom: Quantification of translocation (mean ± SD, n represents the number of cells analyzed). Scale bars, 5 μm.
When expressed as a GFP-fusion protein alone in WT cells, Leep2B exhibited minimal localization at the macropinocytic cups (Fig. 1 H). However, coexpression with Leep2A led to robust colocalization of the two proteins at macropinocytic cups and nascent macropinosomes, consistent with complex formation (Fig. 1 I and Video 1). The accumulation of Leep2A at macropinocytic cups was also enhanced by coexpression with Leep2B (Fig. S1 C). Similar to Leep2A (Fig. 1 C), the coexpressed Leep2A and Leep2B responded to chemoattractant stimulation by translocating from the cytoplasm to the plasma membrane (Fig. S1, D and E). The translocation occurred in cells treated with the F-actin depolymerizing agent Latrunculin A (LatA), indicating that actin polymerization is not essential for membrane association of the Leep2A–Leep2B complex (Fig. S1 F). In addition, similar to Leep2A (Fig. 1 D), coexpressed Leep2A and Leep2B did not localize to pseudopods, even in cells migrating under agarose along a chemical gradient (Fig. 1 J and Video 2), a condition previously used to reveal the pseudopodial enrichment of Leep1 (Yang et al., 2021). These results collectively demonstrate that Leep2A and Leep2B form a complex localized specifically at macropinocytic cups and newly formed macropinosomes.
Colocalization of GFP-Leep2A and RFP-Leep2B in WT cells during macropinocytosis. Corresponds to Fig. 1 I. Scale bar = 5 μm. Images were captured at 6-s intervals and played back at three frames per second.
Colocalization of GFP-Leep2A and RFP-Leep2B in WT cells during macropinocytosis. Corresponds to Fig. 1 I. Scale bar = 5 μm. Images were captured at 6-s intervals and played back at three frames per second.
Localization of GFP-Leep2A and RFP-Leep2B in WT cells migrating under agarose along a folic acid gradient. Corresponds to Fig. 1 J. Scale bar = 5 μm. Images were captured at 6-s intervals and played back at three frames per second.
Localization of GFP-Leep2A and RFP-Leep2B in WT cells migrating under agarose along a folic acid gradient. Corresponds to Fig. 1 J. Scale bar = 5 μm. Images were captured at 6-s intervals and played back at three frames per second.
Complex formation contributes to the positioning of Leep2A and Leep2B
We analyzed the spatiotemporal dynamics of the Leep2 complex. During macropinosome formation, the interior surface of the macropinocytic cup initially contains high levels of PIP3, which rapidly converts to PI(3,4)P2 during cup closure and macropinosome internalization (Maekawa et al., 2014; Yang et al., 2021). Coexpression of GFP-Leep2A, Leep2B, and sensors for different phosphoinositides revealed that the Leep2 complex colocalized with the PIP3/PI(3,4)P2 sensor PHcrac at macropinocytic cups (Fig. 2 A). As PIP3 was replaced by PI(3,4)P2 on internalized macropinosomes, which resulted in a modest increase in the PHcrac signal on macropinosomes, the Leep2 complex dissociated from the vesicles (Fig. 2, A and B). Similarly, the dissociation of the Leep2 complex from the newly formed macropinosomes was accompanied by a gradual accumulation of the PI(3,4)P2 sensor, TAPP1 (Fig. 2, C and D). Though a temporal correlation was observed between membrane association of the Leep2 complex and PIP3 accumulation, cup localization of the Leep2 complex was independent of the PIP3 signal. In cells lacking the two major PI3 kinases (pi3k1−2−) or PIP3 phosphatase Pten (pten−) (Funamoto et al., 2002; Iijima and Devreotes, 2002), the cup localization of Leep1, a validated PIP3-binding protein (Yang et al., 2021), was abolished and greatly enhanced, respectively (Fig. 2, E–G). Conversely, localization of the Leep2 complex remained unaltered (Fig. 2, H–J).

Localization dynamics of the Leep2 complex. (A) Time-lapse imaging of WT cells co-expressing GFP-Leep2A, Leep2B, and PHcrac-RFP. Arrowheads indicate an emerging macropinosome. (B) Quantification of the changes in fluorescent intensity of PHcrac-RFP and GFP-Leep2A on newly formed macropinosomes, as shown in A. The frame at which the macropinocytic cup closed was set as time 0, and the fluorescence intensity at other time points was normalized to that at time 0. (C) Time-lapse imaging of WT cells co-expressing GFP-Leep2A, Leep2B, and TAPP1-RFP. Arrowheads indicate an emerging macropinosome. (D) Quantification of the changes in fluorescent intensity of TAPP1-RFP and GFP-Leep2A on newly formed macropinosomes, as shown in C. The frame at which the macropinocytic cup closed was set as time 0, and the fluorescence intensity at other time points was normalized to that at time 0. (E–G) Localization of GFP-Leep1 in WT (E), pi3k1−2− (F), and pten− (G) cells. (H–J) Localization of RFP-Leep2A and GFP-Leep2B in WT (H), pi3k1−2− (I), and pten− (J) cells. Scale bars, 5 μm.
Localization dynamics of the Leep2 complex. (A) Time-lapse imaging of WT cells co-expressing GFP-Leep2A, Leep2B, and PHcrac-RFP. Arrowheads indicate an emerging macropinosome. (B) Quantification of the changes in fluorescent intensity of PHcrac-RFP and GFP-Leep2A on newly formed macropinosomes, as shown in A. The frame at which the macropinocytic cup closed was set as time 0, and the fluorescence intensity at other time points was normalized to that at time 0. (C) Time-lapse imaging of WT cells co-expressing GFP-Leep2A, Leep2B, and TAPP1-RFP. Arrowheads indicate an emerging macropinosome. (D) Quantification of the changes in fluorescent intensity of TAPP1-RFP and GFP-Leep2A on newly formed macropinosomes, as shown in C. The frame at which the macropinocytic cup closed was set as time 0, and the fluorescence intensity at other time points was normalized to that at time 0. (E–G) Localization of GFP-Leep1 in WT (E), pi3k1−2− (F), and pten− (G) cells. (H–J) Localization of RFP-Leep2A and GFP-Leep2B in WT (H), pi3k1−2− (I), and pten− (J) cells. Scale bars, 5 μm.
Truncations of Leep2A and Leep2B were generated to determine the regions required for complex assembly and localization. Coimmunoprecipitation experiments revealed that truncating the C-terminus, including the GAP domain, of Leep2A (Leep2A T1) did not affect the interaction with Leep2B, whereas further truncation (Leep2A T2) abolished the interaction (Fig. 3 A). The region required for interaction with Leep2B was further mapped to a fragment (Leep2A T5) adjacent to the GAP domain of Leep2A (Fig. 3 A). Similarly, a short fragment (Leep2B T5) adjacent to the GAP domain of Leep2B was found to be necessary for interaction with Leep2A (Fig. 3 B). In addition, a weak interaction was observed between the Leep2B T2 truncation and Leep2A (Fig. 3 B). In cells expressing GFP-tagged truncations of Leep2A and RFP-Leep2B, Leep2A truncations removing up to 1,194 amino acids from the C-terminus (Leep2A T1, Leep2A T2, and Leep2A T3) were still able to localize to macropinocytic cups (Fig. 3 C), but only truncations retaining the ability to interact with Leep2B (Leep2A T1) triggered the cup recruitment of coexpressed Leep2B (Fig. 3 C). In cells expressing GFP-Leep2A and RFP-tagged truncations of Leep2B, T1 and T2 truncations of Leep2B were observed at macropinocytic cups (Fig. 3 D), likely due to an interaction with Leep2A, although the Leep2B T2–Leep2A interaction was weak (Fig. 3 B). However, Leep2B T5, which exhibited a robust interaction with Leep2A, was not detected at macropinocytic cups (Fig. 3 D), suggesting that other regions of Leep2B may function to strengthen complex formation and localization. Taken together, these experiments provide further evidence for the formation of a physical complex between Leep2A and Leep2B during macropinocytosis.

Mapping the regions required for complex formation and localization. (A) Top: Schematic representation of full-length Leep2A and truncation constructs. Bottom: Co-IP of GFP, GFP-Leep2A, and GFP-tagged Leep2A truncations with RFP-Leep2B. Fluorescent fusion proteins were expressed in WT cells. IP was performed with RFP-trap, and samples were probed with GFP or RFP antibody. (B) Top: Schematic representation of full-length Leep2B and truncation constructs. Bottom: Co-IP of GFP-Leep2A with RFP, RFP-Leep2B, and RFP-tagged Leep2B truncations. Fluorescent fusion proteins were expressed in WT cells. IP was performed with RFP-trap and samples were probed with GFP or RFP antibody. (C) Localization of GFP-Leep2A or GFP-tagged Leep2A truncations and RFP-Leep2B in WT cells. (D) Localization of GFP-Leep2A and RFP-Leep2B or RFP-tagged Leep2B truncations in WT cells. Scale bars, 5 μm. Source data are available for this figure: SourceData F3.
Mapping the regions required for complex formation and localization. (A) Top: Schematic representation of full-length Leep2A and truncation constructs. Bottom: Co-IP of GFP, GFP-Leep2A, and GFP-tagged Leep2A truncations with RFP-Leep2B. Fluorescent fusion proteins were expressed in WT cells. IP was performed with RFP-trap, and samples were probed with GFP or RFP antibody. (B) Top: Schematic representation of full-length Leep2B and truncation constructs. Bottom: Co-IP of GFP-Leep2A with RFP, RFP-Leep2B, and RFP-tagged Leep2B truncations. Fluorescent fusion proteins were expressed in WT cells. IP was performed with RFP-trap and samples were probed with GFP or RFP antibody. (C) Localization of GFP-Leep2A or GFP-tagged Leep2A truncations and RFP-Leep2B in WT cells. (D) Localization of GFP-Leep2A and RFP-Leep2B or RFP-tagged Leep2B truncations in WT cells. Scale bars, 5 μm. Source data are available for this figure: SourceData F3.
Leep2 complex modulates macropinocytic activity
The distinct subcellular localization of Leep2 prompted us to investigate its role in the regulation of macropinocytic activity. To this end, we generated single (leep2A−, leep2B−) and double knockout (leep2A−leep2B−, DKO) mutants using homologous recombination (Fig. S2, A and B). Deletion of either leep2A or leep2B resulted in a 25–30% reduction in the uptake of macropinocytosis-specific fluidic tracer, 70 kDa tetramethylrhodamine isothiocyanate (TRITC)-dextran (Fig. 4, A and B). This defect was also observed by flow cytometry analysis (Fig. 4 C). Consistent with complex formation, deletion of both leep2A and leep2B led to an equivalent reduction in macropinocytosis efficiency as with the single deletion mutant (Fig. 4, A–C). In line with the critical role of macropinocytosis in axenic growth, the generation time in a liquid medium increased from ∼10 h for the WT cells to ∼13 h for the DKO cells (Fig. 4 D).
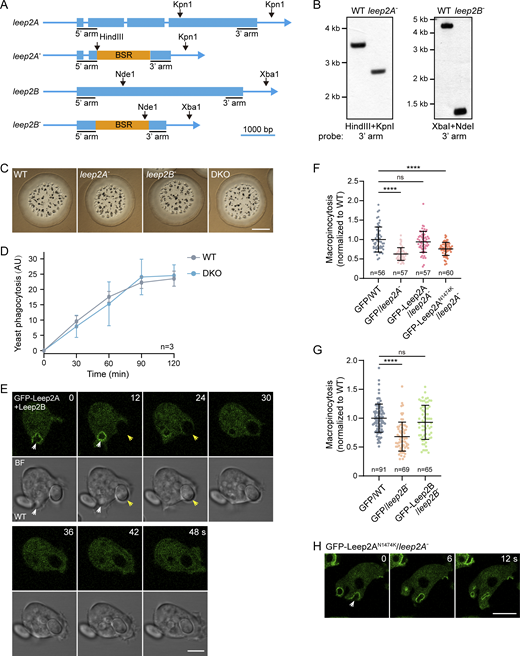
Characterization of leep2 knockout cells. (A) Design of the leep2A and leep2B knockout constructs. A blasticidin-resistant cassette (BSR) was inserted to replace part of the open reading frame of leep2A or leep2B via homologous recombination. (B) Knockout clones were confirmed by Southern blotting. Genomic DNA samples were digested with the indicated enzymes and hybridized with the indicated probes. (C) Growth of WT, leep2A−, leep2B−, and DKO cells on bacterial lawns. Cells were plated clonally with bacteria (Klebsiella aerogenes) on standard medium agar for 5 days. Scale bar, 2 mm. (D) Quantification of phagocytosis of TRITC-labeled yeast in WT and DKO cells. (E) Time-lapse images of yeast phagocytosis in WT cells expressing GFP-Leep2A and Leep2B. White arrowheads indicate macropinocytosis events and yellow arrowheads indicate phagocytosis events. Scale bar, 5 μm. (F) Quantification of TRITC-Dextran uptake in WT and leep2A− cells expressing the indicated constructs. (G) Quantification of TRITC-Dextran uptake in WT and leep2B− cells expressing the indicated constructs. (H) Localization of GFP-Leep2AN1474K in leep2A− cells. Scale bar, 5 μm. The plot in D shows data points with means and SEM; data were from three independent experiments. The scatter plots in F and G show data points with means and SD; n represents the number of cells analyzed. Significance was determined by one-way ANOVA in F and G. Source data are available for this figure: SourceData FS2.

Characterization of leep2 knockout cells. (A) Design of the leep2A and leep2B knockout constructs. A blasticidin-resistant cassette (BSR) was inserted to replace part of the open reading frame of leep2A or leep2B via homologous recombination. (B) Knockout clones were confirmed by Southern blotting. Genomic DNA samples were digested with the indicated enzymes and hybridized with the indicated probes. (C) Growth of WT, leep2A−, leep2B−, and DKO cells on bacterial lawns. Cells were plated clonally with bacteria (Klebsiella aerogenes) on standard medium agar for 5 days. Scale bar, 2 mm. (D) Quantification of phagocytosis of TRITC-labeled yeast in WT and DKO cells. (E) Time-lapse images of yeast phagocytosis in WT cells expressing GFP-Leep2A and Leep2B. White arrowheads indicate macropinocytosis events and yellow arrowheads indicate phagocytosis events. Scale bar, 5 μm. (F) Quantification of TRITC-Dextran uptake in WT and leep2A− cells expressing the indicated constructs. (G) Quantification of TRITC-Dextran uptake in WT and leep2B− cells expressing the indicated constructs. (H) Localization of GFP-Leep2AN1474K in leep2A− cells. Scale bar, 5 μm. The plot in D shows data points with means and SEM; data were from three independent experiments. The scatter plots in F and G show data points with means and SD; n represents the number of cells analyzed. Significance was determined by one-way ANOVA in F and G. Source data are available for this figure: SourceData FS2.

Leep2 complex regulates macropinocytosis. (A) TRITC-Dextran uptake in WT, leep2A−, leep2B−, and leep2A−leep2B− (DKO) cells. (B) Quantification of TRITC-Dextran uptake, as shown in A. (C) Quantification of TRITC-Dextran uptake by flow cytometry analysis. (D) Generation time of WT and DKO cells. (E) Time-lapse imaging of macropinosome formation in WT and DKO cells. PHcrac-GFP was expressed to indicate the macropinocytic structures. Arrowheads point to macropinocytic cups that closed. (F) Quantification of the size of membrane ruffles. (G) Quantification of the rate of membrane ruffle formation. (H) Quantification of the rate of macropinosome formation. (I) Quantification of the size of nascent macropinosomes. (J) Quantification of random motility. Top: Cell trajectories. Bottom: Summary of motility parameters. (K) Quantification of chemotaxis along the folic acid gradient. Top: Cell trajectories. Bottom: Summary of chemotaxis parameters; FMI, forward migration index. The scatter plots show data points with means and SD; n represents the number of cells or events analyzed. The plot in D shows means and SEM; data were from three independent experiments. For J and K, the cell trajectories at the top were from one representative experiment, n represents the number of cells analyzed; data shown in the table were from three independent experiments, and the average of each biological replicate was used to calculate the means and SEM. Significance was determined by one-way ANOVA in B and by two-tailed unpaired t test in D, F, G, H, and I. Scale bars, 5 μm.
Leep2 complex regulates macropinocytosis. (A) TRITC-Dextran uptake in WT, leep2A−, leep2B−, and leep2A−leep2B− (DKO) cells. (B) Quantification of TRITC-Dextran uptake, as shown in A. (C) Quantification of TRITC-Dextran uptake by flow cytometry analysis. (D) Generation time of WT and DKO cells. (E) Time-lapse imaging of macropinosome formation in WT and DKO cells. PHcrac-GFP was expressed to indicate the macropinocytic structures. Arrowheads point to macropinocytic cups that closed. (F) Quantification of the size of membrane ruffles. (G) Quantification of the rate of membrane ruffle formation. (H) Quantification of the rate of macropinosome formation. (I) Quantification of the size of nascent macropinosomes. (J) Quantification of random motility. Top: Cell trajectories. Bottom: Summary of motility parameters. (K) Quantification of chemotaxis along the folic acid gradient. Top: Cell trajectories. Bottom: Summary of chemotaxis parameters; FMI, forward migration index. The scatter plots show data points with means and SD; n represents the number of cells or events analyzed. The plot in D shows means and SEM; data were from three independent experiments. For J and K, the cell trajectories at the top were from one representative experiment, n represents the number of cells analyzed; data shown in the table were from three independent experiments, and the average of each biological replicate was used to calculate the means and SEM. Significance was determined by one-way ANOVA in B and by two-tailed unpaired t test in D, F, G, H, and I. Scale bars, 5 μm.
We expressed PHcrac-GFP in WT and DKO cells to visualize the dynamics of macropinosome formation. WT cells constantly formed membrane ruffles labeled by PHcrac-GFP, which expanded and then closed to generate large macropinosomes (Fig. 4 E and Video 3). Although membrane ruffles were produced at similar frequencies and in similar sizes, the progression to negatively curved cups frequently stalled in the DKO cells, causing PHcrac-labeled patches to remain flat for extended periods of time or abort without vesicle formation (Fig. 4, F and G; and Video 3). Consequently, the number of successful cup-closure events per cell per minute significantly decreased in the DKO cells (Fig. 4 H). In addition, the size of newly formed macropinosomes was slightly reduced (Fig. 4 I), supporting the role of the Leep2 complex in regulating macropinosome formation.
PHcrac-GFP dynamics in WT and DKO cells during macropinocytosis. Corresponds to Fig. 4 E. Scale bar = 5 μm. Images were captured at 3-s intervals and played back at six frames per second.
PHcrac-GFP dynamics in WT and DKO cells during macropinocytosis. Corresponds to Fig. 4 E. Scale bar = 5 μm. Images were captured at 3-s intervals and played back at six frames per second.
To further investigate the function of the Leep2 complex, we examined other prominent leading-edge activities, including cell motility and phagocytosis. In random motility assays, the deletion of Leep2 did not lead to a noticeable deviation in the motile behavior of DKO cells compared with WT cells (Fig. 4 J). Similarly, in under-agarose chemotaxis assays, DKO cells migrated up the gradient with speed, persistence, and directness comparable with WT cells (Fig. 4 K). When assessed for phagocytosis on bacterial lawns, the plaque growth of DKO cells was indistinguishable from that of WT cells, indicating that bacterial phagocytosis and digestion were not significantly affected (Fig. S2 C). In addition, the deletion of Leep2 had no significant impact on the phagocytosis of large yeast particles (Fig. S2 D). Consistent with this observation, the Leep2 complex was only faintly visible in phagocytic cups during yeast particle engulfment (Fig. S2 E and Video 4). Collectively, these experiments demonstrate that the Leep2 complex plays a primary and specific role in regulating macropinocytosis.
Localization of GFP-Leep2A in WT cells expressing GFP-Leep2 and Leep2B during yeast phagocytosis. Corresponds to Fig. S2 E. Scale bar = 5 μm. Images were captured at 6-s intervals and played back at three frames per second.
Localization of GFP-Leep2A in WT cells expressing GFP-Leep2 and Leep2B during yeast phagocytosis. Corresponds to Fig. S2 E. Scale bar = 5 μm. Images were captured at 6-s intervals and played back at three frames per second.
Co-expression of GFP-Leep2A and Leep2B fully restored the macropinocytosis defect in DKO cells (Fig. 5, A–C). In contrast, expression of either GFP-Leep2A or GFP-Leep2B failed to rescue the defect (Fig. 5, A–C), despite their ability to rescue the respective single knockout mutant (Fig. S2, F and G). The impaired cup localization of GFP-Leep2A in the absence of Leep2B likely contributed to its inability to restore macropinocytosis in DKO cells (Fig. 5 D). This finding verified the importance of complex formation to functional integrity. Furthermore, we observed that overexpression of the Leep2 complex also led to a substantial decrease in macropinocytic activity (Fig. 5, E and F), indicating that the cellular level of the complex needs to be properly adjusted for efficient macropinocytosis.

Leep2A and Leep2B form a complex to regulate macropinocytosis. (A) TRITC-Dextran uptake in WT cells expressing GFP or DKO cells expressing GFP, GFP-Leep2A, GFP-Leep2B, or GFP-Leep2A and Leep2B. (B) Quantification of TRITC-Dextran uptake, as shown in A. (C) Quantification of TRITC-Dextran uptake by flow cytometry analysis. (D) Localization of GFP-Leep2A in DKO cells with (top) or without (bottom) the expression of Leep2B. (E) Quantification of TRITC-Dextran uptake in WT cells overexpressing Flag and GFP or Flag-Leep2A and GFP-Leep2B. (F) Quantification of the uptake of Alexa647-Dextran in WT cells overexpressing GFP and RFP or GFP-Leep2A and RFP-Leep2B by flow cytometry analysis. The scatter plots show data points with means and SD; n represents the number of cells analyzed. Significance was determined by one-way ANOVA in B and by two-tailed unpaired t test in E. Scale bars, 5 μm.
Leep2A and Leep2B form a complex to regulate macropinocytosis. (A) TRITC-Dextran uptake in WT cells expressing GFP or DKO cells expressing GFP, GFP-Leep2A, GFP-Leep2B, or GFP-Leep2A and Leep2B. (B) Quantification of TRITC-Dextran uptake, as shown in A. (C) Quantification of TRITC-Dextran uptake by flow cytometry analysis. (D) Localization of GFP-Leep2A in DKO cells with (top) or without (bottom) the expression of Leep2B. (E) Quantification of TRITC-Dextran uptake in WT cells overexpressing Flag and GFP or Flag-Leep2A and GFP-Leep2B. (F) Quantification of the uptake of Alexa647-Dextran in WT cells overexpressing GFP and RFP or GFP-Leep2A and RFP-Leep2B by flow cytometry analysis. The scatter plots show data points with means and SD; n represents the number of cells analyzed. Significance was determined by one-way ANOVA in B and by two-tailed unpaired t test in E. Scale bars, 5 μm.
Leep2 complex exhibits GAP activity toward Ras GTPases
To investigate how the Leep2 complex regulates macropinosome formation, we examined whether the putative GAP domains in Leep2A and Leep2B are required for their function. Deletion of either GAP domain impaired the complex’s ability to rescue the macropinocytosis defect in DKO cells (Fig. 6, A–C), without affecting the complex’s localization (Fig. 6, D and E) or the interaction between Leep2A and Leep2B (Fig. 6 F). Unlike most GAPs that supply an arginine residue into the active site of the small GTPase to catalyze GTP hydrolysis (Bos et al., 2007; Scheffzek et al., 1997), RalGAPA1 and RalGAPA2, with which Leep2A shares sequence homology, do not contain a catalytic arginine. Instead, they utilize an asparagine residue, referred to as the “asparagine thumb,” as in Rap1GAP, for catalysis (Chen et al., 2011; Daumke et al., 2004; Shirakawa et al., 2009). Sequence comparison suggested that Leep2A likely employs the asparagine thumb mechanism (Fig. S3 A). When the conserved asparagine residue was mutated to lysine (Leep2AN1474K), the mutant Leep2A exhibited a reduced capacity to rescue the macropinocytosis defect in leep2A− cells (Fig. S2 F), though it was still able to localize to macropinocytic cups (Fig. S2 H). Conversely, the GAP domain of Leep2B, similar to that of RalGAPB, lacks the conserved catalytic motif and appears non-functional (Fig. S3 A).

The GAP domains of Leep2A and Leep2B are required for complex function. (A) TRITC-Dextran uptake in WT expressing GFP or DKO cells expressing GFP, GFP-Leep2A and Leep2B, GFP-Leep2ΔGAP and Leep2B, or GFP-Leep2A and Leep2BΔGAP. (B) Quantification of TRITC-Dextran uptake, as shown in A. The scatter plot shows data points with means and SD; n represents the number of cells analyzed. Significance was determined by one-way ANOVA. (C) Quantification of TRITC-Dextran uptake by flow cytometry analysis. (D) Localization of GFP-Leep2ΔGAP and RFP-Leep2B in DKO cells. (E) Localization of GFP-Leep2A and RFP-Leep2BΔGAP in DKO cells. (F) Co-IP of GFP, GFP-Leep2A, or GFP-Leep2AΔGAP with RFP-Leep2B or RFP-Leep2BΔGAP. Fluorescent fusion proteins were expressed in DKO cells. IP was performed with RFP-trap and samples were probed with GFP or RFP antibody. Scale bars, 5 μm. Source data are available for this figure: SourceData F6.
The GAP domains of Leep2A and Leep2B are required for complex function. (A) TRITC-Dextran uptake in WT expressing GFP or DKO cells expressing GFP, GFP-Leep2A and Leep2B, GFP-Leep2ΔGAP and Leep2B, or GFP-Leep2A and Leep2BΔGAP. (B) Quantification of TRITC-Dextran uptake, as shown in A. The scatter plot shows data points with means and SD; n represents the number of cells analyzed. Significance was determined by one-way ANOVA. (C) Quantification of TRITC-Dextran uptake by flow cytometry analysis. (D) Localization of GFP-Leep2ΔGAP and RFP-Leep2B in DKO cells. (E) Localization of GFP-Leep2A and RFP-Leep2BΔGAP in DKO cells. (F) Co-IP of GFP, GFP-Leep2A, or GFP-Leep2AΔGAP with RFP-Leep2B or RFP-Leep2BΔGAP. Fluorescent fusion proteins were expressed in DKO cells. IP was performed with RFP-trap and samples were probed with GFP or RFP antibody. Scale bars, 5 μm. Source data are available for this figure: SourceData F6.

GAP domain alignment and Y2H analysis. (A) Sequence alignment of the GAP domains of Leep2A, Leep2B, human Rap1GAP, human RalGAPA1, and human RalGAPB. Red box indicates the catalytic motif. The catalytic asparagine residues conserved in Leep2A, human Rap1GAP, and human RalGAPA1 are highlighted in red. (B) Yeast two-hybrid screen of the active forms of Ras, Rac, and Rab subfamily GTPases for interaction with the GAP domain of Leep2A. The GAP domain was fused to the Gal4 activation domain (AD) and small GTPases to the Gal4 binding domain (BD). Yeast was transformed with the indicated constructs and selected for the presence of prey and bait plasmids by growth on double-dropout agar plate lacking leucine and tryptophan (-LW). Interactions were assayed by growth on quadruple-dropout agar plate additionally lacking histidine and adenine (-LWHA). The GAP-RacM interaction was non-specific since yeast expressing RacM alone also grew on the quadruple-dropout plate.

GAP domain alignment and Y2H analysis. (A) Sequence alignment of the GAP domains of Leep2A, Leep2B, human Rap1GAP, human RalGAPA1, and human RalGAPB. Red box indicates the catalytic motif. The catalytic asparagine residues conserved in Leep2A, human Rap1GAP, and human RalGAPA1 are highlighted in red. (B) Yeast two-hybrid screen of the active forms of Ras, Rac, and Rab subfamily GTPases for interaction with the GAP domain of Leep2A. The GAP domain was fused to the Gal4 activation domain (AD) and small GTPases to the Gal4 binding domain (BD). Yeast was transformed with the indicated constructs and selected for the presence of prey and bait plasmids by growth on double-dropout agar plate lacking leucine and tryptophan (-LW). Interactions were assayed by growth on quadruple-dropout agar plate additionally lacking histidine and adenine (-LWHA). The GAP-RacM interaction was non-specific since yeast expressing RacM alone also grew on the quadruple-dropout plate.
The above findings support the role of the Leep2 complex in GTPase activation. As Dictyostelium cells lack genuine homologs of RalA and RalB, the Leep2 complex, despite its overall similarity to the RalGAP complex, is unlikely to function through Ral proteins. To identify the substrates of the Leep2 complex, we initially employed the yeast two-hybrid (Y2H) approach. Earlier studies indicated that small GTPases, when stabilized in their active conformation, may trap interactions with GAPs (Chotard et al., 2010; Haas et al., 2005). We screened 13 Ras, 19 Rac, and 21 Rab GTPases, which are potentially involved in endocytosis in Dictyostelium cells, in their constitutively activated forms for interaction with the GAP domain of Leep2A (Fig. S3 B). These active forms were generated through amino acid substitution at positions corresponding to either position 12 within the P-loop or position 61 within switch II of human HRAS. However, no specific interactions were detected (Fig. S3 B).
Previous studies also indicated that GAPs preferentially interact with their target GTPases during the transition state of GTP hydrolysis (Bos et al., 2007; Scheffzek et al., 1997). The binding of GTPases to GDP in complex with aluminum fluoride (GDP/AlFx) mimics the transition state (Fig. 7 A), thereby facilitating a stabilized GTPase–GAP interaction (Scheffzek et al., 1997; Wittinghofer, 1997). We exploited this feature in a proteomics approach to identify potential substrates of the Leep2 complex. GFP-Leep2A/RFP-Leep2B was immunoprecipitated from cell lysates supplemented with or without GDP/AlFx, followed by mass spectrometry analysis of bound proteins (Fig. 7 B). Three Ras family GTPases (RasB, RasD, and RasG) showed a preferential association with the Leep2 complex in the presence of GDP/AlFx, which is characteristic of the GAP–substrate GTPase interaction (Fig. 7 B). The interactions were confirmed in glutathione-S-transferase (GST) pull-down assays, which demonstrated specific interactions of the Leep2 complex with recombinant GST-RasB, -RasD, and -RasG loaded with GDP/AlFx, but not with unloaded GST-Ras proteins or GST alone (Fig. 7, C and D). Furthermore, we observed that an intact complex was required for the interaction, as the lack of either Leep2A or Leep2B abolished the interaction (Fig. 7 E).

Leep2 complex promotes GTP hydrolysis of RasB, RasD, and RasG. (A) GAPs interact preferentially with their target GTPases during the transition state of GTP hydrolysis. Top: Schematic view of GAP-stimulated GTP hydrolysis. Bottom: GAP-GTPase interaction during the transition state of GTP hydrolysis (left) or in the presence of GDP and AlFx (right). (B) Proteomic identification of the substrates of Leep2. The Leep2 complex was immunoprecipitated from cell lysates of DKO cells expressing GFP-Leep2A and RFP-Leep2B, supplemented with or without GDP and AlFx, using GFP-trap. The bound proteins were analyzed by mass spectrometry. The abundance ratio was determined by dividing the abundance from AlFx-containing samples by that from AlFx-free samples. (C) Pull-down of Leep2 complex from cell lysates using GST or GST-fused RasB, RasD, and RasG immobilized on beads. The cell lysates were obtained from WT cells expressing Flag-Leep2A and GFP-Leep2B, supplemented with or without GDP and AlFx. Samples were probed with Flag antibody. Coomassie bright blue-stained gel at the bottom shows purified GST and GST-fusion proteins. (D) Pull-down of Leep2 complex from cell lysates using GST or GST-fused Ras subfamily GTPases immobilized on beads. The cell lysates were obtained from WT cells expressing GFP-Leep2A and RFP-Leep2B, supplemented with GDP and AlFx. Samples were probed with GFP antibody. Coomassie bright blue-stained gel at the bottom shows purified GST and GST-fusion proteins. (E) Pull-down of Leep2 components from cell lysates using GST, GST-RasB, GST-RasD, or GST-RasG. The cell lysates were obtained from DKO cells coexpressing GFP-Leep2A and RFP-Leep2B or DKO cells expressing GFP-Leep2A or GFP-Leep2B, supplemented with GDP and AlFx. Samples were probed with GFP or RFP antibody. Coomassie bright blue-stained gel at the bottom shows purified GST and GST-fusion proteins. (F) Luminescence-based GAP assay: Purified recombinant GST, GST-RasB, GST-RasD, or GST-RasG immobilized on beads were incubated with immunopurified complexes of Flag-Leep2A and GFP-Leep2B, Flag-Leep2AN1474K and GFP-Leep2B, as well as Flag-Leep2A, or Flag control. The luminescence of GST incubated with the Flag control was set to 1, and other values were normalized correspondingly (means ± SD, n = 3). AU, arbitrary unit. Source data are available for this figure: SourceData F7.
Leep2 complex promotes GTP hydrolysis of RasB, RasD, and RasG. (A) GAPs interact preferentially with their target GTPases during the transition state of GTP hydrolysis. Top: Schematic view of GAP-stimulated GTP hydrolysis. Bottom: GAP-GTPase interaction during the transition state of GTP hydrolysis (left) or in the presence of GDP and AlFx (right). (B) Proteomic identification of the substrates of Leep2. The Leep2 complex was immunoprecipitated from cell lysates of DKO cells expressing GFP-Leep2A and RFP-Leep2B, supplemented with or without GDP and AlFx, using GFP-trap. The bound proteins were analyzed by mass spectrometry. The abundance ratio was determined by dividing the abundance from AlFx-containing samples by that from AlFx-free samples. (C) Pull-down of Leep2 complex from cell lysates using GST or GST-fused RasB, RasD, and RasG immobilized on beads. The cell lysates were obtained from WT cells expressing Flag-Leep2A and GFP-Leep2B, supplemented with or without GDP and AlFx. Samples were probed with Flag antibody. Coomassie bright blue-stained gel at the bottom shows purified GST and GST-fusion proteins. (D) Pull-down of Leep2 complex from cell lysates using GST or GST-fused Ras subfamily GTPases immobilized on beads. The cell lysates were obtained from WT cells expressing GFP-Leep2A and RFP-Leep2B, supplemented with GDP and AlFx. Samples were probed with GFP antibody. Coomassie bright blue-stained gel at the bottom shows purified GST and GST-fusion proteins. (E) Pull-down of Leep2 components from cell lysates using GST, GST-RasB, GST-RasD, or GST-RasG. The cell lysates were obtained from DKO cells coexpressing GFP-Leep2A and RFP-Leep2B or DKO cells expressing GFP-Leep2A or GFP-Leep2B, supplemented with GDP and AlFx. Samples were probed with GFP or RFP antibody. Coomassie bright blue-stained gel at the bottom shows purified GST and GST-fusion proteins. (F) Luminescence-based GAP assay: Purified recombinant GST, GST-RasB, GST-RasD, or GST-RasG immobilized on beads were incubated with immunopurified complexes of Flag-Leep2A and GFP-Leep2B, Flag-Leep2AN1474K and GFP-Leep2B, as well as Flag-Leep2A, or Flag control. The luminescence of GST incubated with the Flag control was set to 1, and other values were normalized correspondingly (means ± SD, n = 3). AU, arbitrary unit. Source data are available for this figure: SourceData F7.
Using immunopurified complex and a luminescence-based GAP assay, we assessed the ability of the Leep2 complex to promote GTP hydrolysis by the three Ras proteins. In this assay, the GTP remaining after completion of the GTPase reaction is converted to ATP, which is then used in a luciferase reaction to produce light. Thus, the greater the GTP consumption, the lower the luminescence output. We observed different intrinsic GTPase activities for RasB, RasD, and RasG, with RasG displaying the highest activity (Fig. 7 F). The addition of purified Flag-Leep2A/GFP-Leep2B complex accelerated GTP hydrolysis of all three Ras proteins (Fig. 7 F). A mutation in the conserved catalytic asparagine residue in Leep2A abrogated the GAP activity (Fig. 7 F), consistent with the compromised ability of Leep2AN1474K to rescue the leep2A knockout (Fig. S2 F). Moreover, even though Leep2B lacks a functional GAP domain (Fig. S3 A), its removal from the complex rendered the complex inactive against the tested Ras GTPases (Fig. 7 F). This observation aligns with the findings that Leep2A failed to complement the DKO cells (Fig. 5, A–C) or exhibit interaction with Ras GTPases (Fig. 7 E) when expressed alone without Leep2B. Thus, Leep2A and Leep2B likely function as a RasGAP complex, and the presence of both proteins is essential for GAP activity and the regulation of macropinocytosis.
Leep2 complex regulates macropinocytosis through Ras GTPases
We performed cell experiments to investigate whether the Leep2 complex regulates macropinocytosis by modulating the activity of RasB, RasD, and RasG. In Dictyostelium, Ras proteins are proposed to act upstream of a number of signaling and cytoskeletal molecules, including class I PI3Ks and the actin polymerization factor formin G, thereby facilitating macropinosome formation (Hoeller et al., 2013; Junemann et al., 2016). Although previous studies have implicated RasB and RasG in the regulation of macropinocytosis, there have been inconsistencies regarding the effects of Ras deletions. Deletion mutants of rasG from different backgrounds all exhibited poor growth in shaken suspension, initially attributed to a cytokinesis failure, but subsequent research linked this phenotype to a defect in macropinocytosis (Tuxworth et al., 1997; Williams et al., 2019). The original rasB mutant displayed markedly reduced growth in a liquid medium, which was assumed to be associated with impaired macropinocytosis, but later research demonstrated that rasB deletion only weakly affects macropinocytosis (Junemann et al., 2016; Sutherland et al., 2001). The differences in genetic backgrounds and growth conditions may contribute to these discrepancies. RasD has received less attention as it was assumed to be expressed only during development and not be functional in growing cells (Wilkins et al., 2000). Nevertheless, it is known that rasD is upregulated when rasG is deleted, indicating compensatory effects between Ras genes (Khosla et al., 2000). Our analysis of the Leep2-interacting proteins also revealed a substantial level of RasD in growing cells (Fig. 7 B), promoting a reassessment of its function. Based on this information, we decided to systematically evaluate the role of these Ras proteins in macropinocytosis.
We examined the localization and dynamics of fluorescently tagged RasB, RasD, and RasG. All three proteins localized prominently to the plasma membrane and membrane invaginations that evolved into macropinosomes (Fig. S4 A). To visualize their activation status, we expressed the Ras-binding domain (RBD) of yeast Byr2 kinase fused with GFP in cells. This domain interacts with the active forms of several Dictyostelium Ras GTPases, including RasB, RasD, and RasG (Junemann et al., 2016). Byr2RBD-GFP was strongly recruited to macropinocytic cups and nascent macropinosomes, with the signal disappearing from the macropinosomes shortly after their internalization (Fig. S4 B). Although we are currently unable to determine if the sensor’s localization is specifically mediated by RasB, RasD, and RasG, this observation aligns with the presence of active RasB, RasD, and RasG at macropinocytic cups.

Characterization of ras knockout cells. (A) Localization of GFP-tagged RasB, RasD, and RasG in WT cells. Scale bar, 5 μm. (B) Time-lapse imaging of Byr2RBD-GFP in WT cells during macropinocytosis. Scale bar, 5 μm. (C) Top: Design of ras knockout construct. A blasticidin-resistant cassette (BSR) or a Hygromycin-resistant cassette (HygR) was inserted to replace part of the open reading frame of rasB, rasD, or rasG via homologous recombination. Bottom: Knockout clones were confirmed by PCR using the indicated primers. (D) Growth of WT and ras knockout cells on bacterial lawns. Cells were plated clonally with bacteria (Klebsiella aerogenes) on standard medium agar for 5 days. Scale bar, 2 mm. (E) Generation time of WT and ras knockout cells. The plot shows means and SEM; data were from three independent experiments. (F) Localization of GFP-Leep2A and RFP-Leep2B in WT and ras knockout cells. Scale bar, 5 μm.

Characterization of ras knockout cells. (A) Localization of GFP-tagged RasB, RasD, and RasG in WT cells. Scale bar, 5 μm. (B) Time-lapse imaging of Byr2RBD-GFP in WT cells during macropinocytosis. Scale bar, 5 μm. (C) Top: Design of ras knockout construct. A blasticidin-resistant cassette (BSR) or a Hygromycin-resistant cassette (HygR) was inserted to replace part of the open reading frame of rasB, rasD, or rasG via homologous recombination. Bottom: Knockout clones were confirmed by PCR using the indicated primers. (D) Growth of WT and ras knockout cells on bacterial lawns. Cells were plated clonally with bacteria (Klebsiella aerogenes) on standard medium agar for 5 days. Scale bar, 2 mm. (E) Generation time of WT and ras knockout cells. The plot shows means and SEM; data were from three independent experiments. (F) Localization of GFP-Leep2A and RFP-Leep2B in WT and ras knockout cells. Scale bar, 5 μm.
The reduction in macropinocytic activity observed with either deletion or overexpression of the Leep2 complex (Fig. 4, A–C; and Fig. 5, E and F) suggests the need for precise control of Ras activities. Thus, both disruption and overactivation of Ras could have a negative impact on macropinocytosis. To further analyze the role of these Ras proteins, cell lines lacking rasB, rasD, or rasG were generated in a uniform genetic background (Fig. S4, C and D). Moreover, cells lacking both rasD and rasG were generated to investigate potential compensatory effects (Fig. S4, C and D). The deletion of rasB or rasG resulted in a modest decrease in macropinocytic efficiency, whereas the deletion of rasD alone had no effect (Fig. 8, A and B). Consistent with a potential functional overlap between RasD and RasG (Khosla et al., 2000), the simultaneous deletion of both genes severely impaired macropinocytosis, reducing the activity by >80%, as quantified by microscopic examination of TRITC-dextran uptake (Fig. 8, A and B). The generation time of the rasD and rasG double knockout mutant was extended to ∼40 h (Fig. S4 E). To assess the effects of overactivation of Ras and avoid potential complications from prolonged expression, an inducible system was utilized in which the constitutively activated forms of RasB, RasD, or RasG were expressed under a doxycycline-inducible promoter. The expression of active Ras proteins also inhibited macropinocytosis (Fig. 8, C and D), and intensely fluorescent cells tended to accumulate less TRITC-dextran than weakly fluorescent cells and cells without expression (Fig. 8 E). The above results collectively demonstrate the key role of RasB, RasD, and RasG in regulating macropinocytosis.

Leep2 complex regulates macropinocytosis through RasB, RasD, and RasG. (A) TRITC-Dextran uptake in WT and the indicated ras mutants. Independent clones of rasD and rasG double knockout cells were generated by deleting rasG in a rasD knockout background (rasD−rasG−) or deleting rasD in a rasG knockout background (rasG−rasD−). (B) Quantification of TRITC-Dextran uptake, as shown in A. (C) TRITC-Dextran uptake in WT cells expressing constitutively active RasB, RasD, or RasG under a doxycycline-inducible promoter. (D) Quantification of TRITC-Dextran uptake, as shown in C. (E) Scatter plots showing a negative correlation between fluorescence intensity (I) corresponding to GFP-fusion proteins and TRITC-dextran. The correlation coefficients are shown on the right. (F) Localization of Byr2RBD and Leep2B in WT cells co-expressing Byr2RBD-GFP, Flag-Leep2A, and RFP-Leep2B. (G) Quantification of the changes in fluorescent intensity of RFP-Leep2B and Byr2RBD-GFP on newly formed macropinosomes, as shown in F. The frame at which the macropinocytic cup closed was set as time 0, and the fluorescence intensity at other time points was normalized to that at time 0. (H) Localization of Byr2RBD-GFP in WT and DKO cells during macropinocytosis. (I) Quantification of the fluorescent intensity of Byr2RBD-GFP at the macropinocytic cups in WT and DKO cells. (J) Quantification of the changes in fluorescent intensity of Byr2RBD-GFP on newly formed macropinosomes. The frame at which the cup closed was set as time 0. The scatter plots in B, D, and I show data points with means and SD; n represents the number of cells analyzed. The plots in G and J show means and SD; n represents the number of cells analyzed. Significance was determined by one-way ANOVA in B and D and by two-tailed unpaired t test in I. Scale bars, 5 μm.
Leep2 complex regulates macropinocytosis through RasB, RasD, and RasG. (A) TRITC-Dextran uptake in WT and the indicated ras mutants. Independent clones of rasD and rasG double knockout cells were generated by deleting rasG in a rasD knockout background (rasD−rasG−) or deleting rasD in a rasG knockout background (rasG−rasD−). (B) Quantification of TRITC-Dextran uptake, as shown in A. (C) TRITC-Dextran uptake in WT cells expressing constitutively active RasB, RasD, or RasG under a doxycycline-inducible promoter. (D) Quantification of TRITC-Dextran uptake, as shown in C. (E) Scatter plots showing a negative correlation between fluorescence intensity (I) corresponding to GFP-fusion proteins and TRITC-dextran. The correlation coefficients are shown on the right. (F) Localization of Byr2RBD and Leep2B in WT cells co-expressing Byr2RBD-GFP, Flag-Leep2A, and RFP-Leep2B. (G) Quantification of the changes in fluorescent intensity of RFP-Leep2B and Byr2RBD-GFP on newly formed macropinosomes, as shown in F. The frame at which the macropinocytic cup closed was set as time 0, and the fluorescence intensity at other time points was normalized to that at time 0. (H) Localization of Byr2RBD-GFP in WT and DKO cells during macropinocytosis. (I) Quantification of the fluorescent intensity of Byr2RBD-GFP at the macropinocytic cups in WT and DKO cells. (J) Quantification of the changes in fluorescent intensity of Byr2RBD-GFP on newly formed macropinosomes. The frame at which the cup closed was set as time 0. The scatter plots in B, D, and I show data points with means and SD; n represents the number of cells analyzed. The plots in G and J show means and SD; n represents the number of cells analyzed. Significance was determined by one-way ANOVA in B and D and by two-tailed unpaired t test in I. Scale bars, 5 μm.
Using the Byr2RBD probe, we compared the dynamics of Leep2 and active Ras, as well as the levels of Ras activation in WT and DKO cells, during macropinosome formation. The Leep2 complex colocalized with Byr2RBD-GFP on both macropinocytic cups and newly formed macropinosomes. Notably, the signal of Byr2RBD-GFP disappeared slightly before the Leep2 complex from the internalized macropinosomes (Fig. 8, F and G), consistent with the proposed role of Leep2 in terminating Ras signaling. The unsynchronized nature of macropinocytosis made it difficult to determine the degree of activation of individual Ras proteins in WT versus DKO cells using the RBD pull-down method, previously employed to assess Ras activation following chemoattractant stimulation (Cai et al., 2012; Xu et al., 2017). Thus, we expressed Byr2RBD-GFP in WT and DKO cells to monitor Ras activity at the sites of macropinocytosis. Compared with WT cells, there was a small but significant increase in the fluorescent intensity of Byr2RBD-GFP at macropinocytic cups in DKO cells, indicating elevated Ras activation (Fig. 8, H and I). However, there were no significant differences in the proportion of the cell periphery labeled with the RBD probe or the duration of the probe on macropinosomes (Fig. 8 J). The lack of specificity of the Byr2RBD probe (Junemann et al., 2016) and the potential redundancy among RasGAP proteins in Dictyostelium cells (Buckley et al., 2020; Marinovic et al., 2019; Xu et al., 2022) may account for the limited Ras activation observed in the absence of the Leep2 complex.
We speculate that elevated Ras activation in DKO cells may alter the normal dynamics of downstream effectors, thereby interfering with macropinocytosis. To investigate this hypothesis, we focused on PI3K signaling. Several lines of evidence indicate that the macropinocytosis defect observed in DKO cells may, at least partially, be due to perturbed PIP3 dynamics. First, stimulation-induced PI3K activation, which leads to the transient production of PIP3 at the plasma membrane as evidenced by the membrane translocation and cytoplasmic depletion of the PIP3-sensor GFP-PHPkgE (Lutton et al., 2023), was markedly reduced in rasD−rasG− cells. This verified the role of Leep2-targeted Ras proteins in PI3K activation (Fig. S5 A). Second, the levels and dynamics of PIP3 production were affected in DKO cells. We observed that membrane ruffles often failed to close properly in DKO cells, which correlated with a small yet significant increase in PIP3 levels at these membrane ruffles (Fig. S5 B). Furthermore, upon stimulation to produce PIP3, the PIP3 signal persisted for a slightly longer duration in DKO cells (Fig. S5 C). Third, we found that sustained PIP3 generation impaired macropinocytosis. Deletion of the PIP3 phosphatase Pten (Iijima and Devreotes, 2002) or Dd5P4 (Loovers et al., 2007), which converts PIP3 to PI(4,5)P2 or PI(3,4)P2, respectively, inhibited macropinocytosis (Fig. S5 D). Last, we examined whether partial inhibition of PIP3 production could alleviate the macropinocytosis defect in DKO cells. Treating WT and DKO cells with the PI3K inhibitor LY294002 revealed that although this treatment did not restore the macropinocytic activity of DKO cells to WT levels, low doses of LY294002 inhibited macropinocytosis in WT but not DKO cells, suggesting an increased tolerance of DKO cells to PI3K inhibition (Fig. S5 E). Together, these results substantiate the importance of precise regulation of Ras activities and downstream signaling in macropinocytosis.

PIP 3 signaling and the effect of its perturbation on macropinocytosis. (A) Quantification of GFP-PHPkgE translocation in WT and rasD−rasG− cells in response to FA stimulation (250 μM FA was added at time 0) in the presence of LatA. (B) Quantification of the fluorescent intensity of GFP-PHPkgE at the macropinocytic cups in WT and DKO cells. (C) GFP-PHPkgE translocation in WT and DKO cells in response to FA stimulation (250 μM FA was added at time 0) in the presence of LatA. Top: Time-lapse imaging of translocation. Bottom: Quantification of translocation. (D) Quantification of TRITC-Dextran uptake in WT, pten−, and Dd5P4− cells. (E) Quantification of TRITC-Dextran uptake in WT and DKO cells treated with DMSO or LY294002 (12.5 μM). The plots in A and C show mean and SD; n represents the number of cells analyzed. The plots in B, D, and E show data points with means and SD; n represents the number of cells analyzed. Significance was determined by one-way ANOVA in D and by two-tailed unpaired t test in B, C, and E. Scale bars, 5 μm.

PIP 3 signaling and the effect of its perturbation on macropinocytosis. (A) Quantification of GFP-PHPkgE translocation in WT and rasD−rasG− cells in response to FA stimulation (250 μM FA was added at time 0) in the presence of LatA. (B) Quantification of the fluorescent intensity of GFP-PHPkgE at the macropinocytic cups in WT and DKO cells. (C) GFP-PHPkgE translocation in WT and DKO cells in response to FA stimulation (250 μM FA was added at time 0) in the presence of LatA. Top: Time-lapse imaging of translocation. Bottom: Quantification of translocation. (D) Quantification of TRITC-Dextran uptake in WT, pten−, and Dd5P4− cells. (E) Quantification of TRITC-Dextran uptake in WT and DKO cells treated with DMSO or LY294002 (12.5 μM). The plots in A and C show mean and SD; n represents the number of cells analyzed. The plots in B, D, and E show data points with means and SD; n represents the number of cells analyzed. Significance was determined by one-way ANOVA in D and by two-tailed unpaired t test in B, C, and E. Scale bars, 5 μm.
Discussion
Ras GTPases play a central role in the signaling network that orchestrates macropinosome formation (Kay et al., 2022; Mylvaganam et al., 2021; Palm, 2022). However, the precise spatiotemporal regulation of Ras activity during macropinocytosis remains incompletely understood. In this study, we identified Leep2 as a RasGAP complex that specifically localizes to macropinocytic cups and nascent macropinosomes, where it regulates macropinosome formation by modulating the activities of three Ras GTPases. Both the deletion and overexpression of this complex compromise macropinocytic activity, highlighting the importance of proper Ras activation for efficient macropinocytosis. This finding is supported by previous studies showing that Ras proteins undergo transient activation during macropinocytosis in both Dictyostelium cells and mammalian macrophages (Hoeller et al., 2013; Welliver and Swanson, 2012). Furthermore, we linked the macropinocytosis defect caused by leep2 deletion to perturbed PIP3 signaling. This result aligns with our previous observation that PIP3 rapidly converts to PI(3,4)P2 during macropinosome formation in Dictyostelium (Tu et al., 2022; Yang et al., 2021). The need for timely degradation of PIP3 during macropinocytosis has also been noted in various mammalian cells, suggesting it to be a general phenomenon (Maekawa et al., 2014; Schink et al., 2021; Welliver and Swanson, 2012). However, the precise mechanism by which PIP3 turnover modulates macropinosome closure remains to be elucidated.
In contrast to classical single-protein RasGAPs that typically utilize an arginine residue for catalysis, the GAP activity of Leep2 depends on the assembly of a protein complex and employs the asparagine thumb mechanism, similar to RalGAPs and Rap1GAP. We used AlphaFold2.3 to predict the structure of the Leep2 complex (Evans et al., 2022, Preprint; Jumper et al., 2021), which revealed the formation of an elongated tail-to-tail heterodimer between Leep2A and Leep2B (Fig. 9 A). In line with our co-immunoprecipitation experiments demonstrating a critical role of the T5 fragments in Leep2A and Leep2B for complex formation (Fig. 3, A and B), both T5 fragments contain extensive interacting interfaces with each other and with additional sites in Leep2A and Leep2B (Fig. 9 B). Importantly, the interacting face residues exhibit a high pLDDT (predicted local distance difference test) score (Fig. 9 C). We further predicted the complex structure of Leep2A-Leep2B-Ras with a 1:1:1 stoichiometry (Fig. 9, D and E). The Ras protein preferentially binds to the GAP domain of Leep2A, where the interface exhibits a high pLDDT score (Fig. 9, F and G), consistent with the presence of a conserved RHIGND catalytic motif solely in the GAP domain of Leep2A and not in Leep2B (Fig. S3 A). Our observation that Leep2A alone could not bind to Ras protein or catalyze GTP hydrolysis (Fig. 7, E and F) suggests that the Leep2A GAP domain may either be an inactive conformation or, more likely, in a dynamic state requiring interaction with Leep2B to assume a functional conformation. Thus, although Leep2B does not directly interact with Ras in the predicted trimer complex, we speculate that it may provide a scaffold for the Leep2B-Leep2A interface, leading to reorganization or stabilization of the interacting modules around the GAP domain of Leep2A, ultimately positioning it for Ras activation.

Predicted structures of the Leep2 complex. (A) Ribbon representation of the predicted structure of the Leep2 complex in two different views. (B) Ribbon representation of the predicted structure of the Leep2 complex. The T5 fragments and GAP domains in Leep2A and Leep2B are highlighted. (C) Mapping of pLDDT score per residue onto the predicted Leep2 complex structure. pLDDT stands for predicted local distance difference test, which indicates the confidence level of the prediction. (D) Ribbon representation of the predicted heterodimeric structure of Leep2A, Leep2B, and RasD. Comparable structures were predicted with RasG or RasB. (E) Surface representation of the predicted structure in D, except that the GAP domain in Leep2A and Leep2B is colored in cyan and pink, respectively. (F) Left: Ribbon representation of the predicted structure of the GAP domain of Leep2A and RasD. Right: Mapping of pLDDT score per residue onto the predicted complex structure of Leep2A GAP domain and RasD. (G) Left: Ribbon representation of the predicted structure of the GAP domain of Leep2B and RasD. Right: Mapping of pLDDT score per residue onto the predicted complex structure of Leep2B GAP domain and RasD.
Predicted structures of the Leep2 complex. (A) Ribbon representation of the predicted structure of the Leep2 complex in two different views. (B) Ribbon representation of the predicted structure of the Leep2 complex. The T5 fragments and GAP domains in Leep2A and Leep2B are highlighted. (C) Mapping of pLDDT score per residue onto the predicted Leep2 complex structure. pLDDT stands for predicted local distance difference test, which indicates the confidence level of the prediction. (D) Ribbon representation of the predicted heterodimeric structure of Leep2A, Leep2B, and RasD. Comparable structures were predicted with RasG or RasB. (E) Surface representation of the predicted structure in D, except that the GAP domain in Leep2A and Leep2B is colored in cyan and pink, respectively. (F) Left: Ribbon representation of the predicted structure of the GAP domain of Leep2A and RasD. Right: Mapping of pLDDT score per residue onto the predicted complex structure of Leep2A GAP domain and RasD. (G) Left: Ribbon representation of the predicted structure of the GAP domain of Leep2B and RasD. Right: Mapping of pLDDT score per residue onto the predicted complex structure of Leep2B GAP domain and RasD.
Our analysis of the function of Leep2 and its target Ras GTPases offered additional insights into the role of Ras signaling in macropinocytosis. In Dictyostelium, Ras proteins are proposed to act upstream of class I PI3Ks and formin G to facilitate the expansion and subsequent closure of macropinocytic cups (Hoeller et al., 2013; Junemann et al., 2016). We demonstrated that a marked reduction in macropinocytosis (>80%) could be achieved via simultaneous deletion of rasD and rasG, two of the three targets of Leep2 (a triple knockout has not been successfully generated, possibly due to severe growth deficiency). Thus, Ras signaling seems to play an essential role in the process of macropinocytosis in Dictyostelium. Conversely, Ras signaling appears to be dispensable for macropinocytosis in certain contexts in mammalian cells, despite functioning through similar effectors, such as PI3Ks (Charpentier et al., 2020; Hobbs et al., 2020; King et al., 2020; Palm et al., 2017). For example, fibroblasts lacking all major Ras isoforms exhibit no defect in macropinocytosis induction upon growth factor stimulation (Palm et al., 2017). This discrepancy may arise from different mechanisms by which PI3Ks are recruited and activated. Mammalian PI3Ks are recruited to the plasma membrane by interactions with receptor tyrosine kinases (RTKs), potentially enabling their activation independent of Ras (Cantley, 2002; King et al., 2020). In Dictyostelium cells, which lack RTKs (as do other unicellular eukaryotes), Ras may become indispensable for PI3K activation and initiation of macropinocytosis. The presence of additional regulatory mechanisms in mammalian cells may also explain why constitutively active Ras variants negatively affect macropinocytosis in axenic Dictyostelium cells (Fig. 8, C and D) (Bloomfield et al., 2015) yet promote it in mammalian cells (Bar-Sagi and Feramisco, 1986; Commisso et al., 2013). It is yet to be determined whether macropinocytosis can proceed in mammalian cells despite the continuous activation of Ras proteins and downstream signaling, or if the downstream signaling can adapt effectively to persistent Ras activation. Therefore, further research is needed not only to identify the shared core machinery essential for macropinocytosis but also to elucidate the distinct regulations present in different organisms to reveal the fundamental principles underlying macropinocytosis.
How the Leep2 complex is selectively targeted in cells requires further investigation. The localization of the complex closely resembles the pattern observed with reporters for F-actin, PIP3, and active Ras (Veltman et al., 2016; Yang et al., 2021). Nevertheless, the membrane recruitment of Leep2 seems to be independent of these factors. Leep2 can still translocate to the plasma membrane upon chemoattractant stimulation, even in the absence of an intact actin network (Fig. S1 F). Its accumulation at macropinocytic cups is unaffected by changes in PIP3 levels on the plasma membrane (Fig. 2, H–J). In rasD−rasG− cells, which lack two primary targets of Leep2 and exhibit infrequent macropinocytosis, Leep2 remains associated with membrane patches resembling flattened cups at the leading edge of motile cells (Fig. S4 F). Thus, the membrane targeting of Leep2 may be facilitated by its interaction with the remaining Ras proteins or through other yet-to-be-identified mechanisms. In line with the latter possibility, the N-terminal region of Leep2A, which lacks the GAP domain or the region required for complex formation, exhibits a similar localization pattern as the full-length protein (Fig. 3 C).
Another interesting yet unresolved question relates to the coordination of Leep2 with other regulators in controlling Ras activation. The axenic laboratory strains of Dictyostelium, which were derived from the selection of wild-type amoeba capable of growth in liquid medium, express at least 14 proteins containing RasGAP-related domains (Xu et al., 2017). Among these proteins, the deletion of RGBARG results in slightly more but significantly smaller macropinosomes and an ∼50% reduction in macropinocytosis, whereas the deletion of IqgC or C2GAP2 leads to minor increases in macropinocytosis (Buckley et al., 2020; Marinovic et al., 2019; Xu et al., 2022). The wild-type amoeba, on the other hand, encodes another RasGAP known as NF1, a homolog of the tumor suppressor Neurofibromin (Bloomfield et al., 2015). The disruption of NF1 leads to a marked elevation of macropinocytic activity, serving as one of the key genetic determinants for axenic growth (Bloomfield et al., 2015; Clarke and Kayman, 1987). These various RasGAPs share localization and substrates to some extent (Buckley et al., 2020; Marinović et al., 2019; Xu et al., 2022). Nevertheless, the varying impacts of their disruption on the scale and kinetics of macropinocytosis imply that they do not perform entirely redundant functions. The development of improved methods for manipulating and monitoring the activities of these RasGAPs is crucial for clarifying their relative contributions and coordination in regulating macropinocytosis and other cellular activities.
In summary, our study identified a RasGAP complex that plays a specific role in macropinocytosis in Dictyostelium. Similar to Dictyostelium, mammalian cells express multiple RasGAP domain-containing proteins. Given the pivotal roles played by Ras signaling and macropinocytosis in tumorigenesis, it would be of great interest to investigate whether any of these RasGAP proteins contribute to the tumorigenic effects of Ras signaling by modulating macropinocytic activity of the cell.
Materials and methods
Cell culture
WT cells were derived from the Ax2 (Ka) axenic strain provided by the Robert Kay laboratory. Cells were cultured at 22°C in HL5 medium (cat#HLF2; Formedium) supplemented with glucose (cat#GLU03; Formedium) and antibiotics. Plasmids for gene expression or disruption were introduced into the cells by electroporation, and the resulting cell lines were selected and maintained in HL5 medium containing G418 (10–20 μg/ml), Hygromycin (50 μg/ml), or Blasticidin S (10 μg/ml). Growth on bacterial lawns was performed as previously described (Cai et al., 2014). To induce the expression of GFP-tagged constitutively active Ras GTPases, cells were incubated in an HL5 medium containing 25 μg/ml doxycycline for 14–16 h. The cell growth rate was assessed by seeding 2 × 104 cells in triplicate wells of a 6-well plate in the HL5 medium and then counting the cell numbers every 24 h for a total of 72 h.
Plasmid construction
The plasmids and primers used in this study are listed in Table 1. To generate constructs expressing GFP-, RFP-, or Flag-fusion proteins, DNA fragments were PCR-amplified from genomic DNA or cDNA and then inserted into pDM vectors (Paschke et al., 2018; Veltman et al., 2009) containing an added multiple cloning site. To generate pDM317-HygR, the G418-resistant cassette in pDM317 was replaced with a Hygromycin-resistant cassette. The Leep2A and Leep2B dual-expression plasmids were constructed by first cloning Leep2B or Leep2BΔGAP into pDM344, followed by subsequent release of the expression cassette through NgoMIV digestion and cloning into pDM317-Leep2A. For inducible expression of constitutively active Ras, a DNA fragment encoding RasBG15V, RasDQ61L, or RasGG12V was inserted into pDM371. To generate constructs expressing GST-fusion proteins, cDNA fragments encoding small GTPases were cloned into pGEX-6P-1 vector. For the yeast two-hybrid assay, Leep2A1359-1560 was cloned into pGADT7 vector as prey, while the constitutively active forms of Ras GTPases were cloned into pGBKT7 vector. GFP-Leep1, TAPP1-RFP, GFP-PHPkgE, and yeast two-hybrid constructs expressing constitutively active forms of Rab and Rac GTPases were generated previously (Li et al., 2023; Tu et al., 2022; Yang et al., 2021).
Plasmids and primers used in this study
| Plasmid | Plasmid backbone | Sequence, 5′-3′ |
| Expression in Dictyostelium cells | ||
| GFP-Leep2A | pDM317 | F: 5′-GGAAGATCTATGAGCTCAACAGCGAATAATTCAC-3′ |
| R: 5′-GCTCTAGAAATTGGTGAAGGTATACCAATTGTTG-3′ | ||
| Flag-Leep2A | pDM320 | F: 5′-AGATGATGATGATAAAAGATCTATGAGCTCAACAGCGAATAATTCAC-3′ |
| R: 5′-TAAATAATTTATTTATTTAACTAGTTTAAATTGGTGAAGGTATACC-3′ | ||
| RFP-Leep2A | pDM449 | F: 5′-GGAAGATCTATGAGCTCAACAGCGAATAATTCAC-3′ |
| R: 5′-GCTCTAGAAATTGGTGAAGGTATACCAATTGTTG-3′ | ||
| GFP-Leep2AΔGAP | pDM317 | F: 5′-CTATCTAGAATGAGCTCAACAGCGAATAATTCAC-3′ |
| F: 5′-CTAGTCGACACCACCTTGACTATTGTATTGTTTG-3′ | ||
| GFP-Leep2A1-1344 (T1) | pDM317 | F: 5′-GGAAGATCTATGAGCTCAACAGCGAATAATTCAC-3′ |
| R: 5′-GTCGACTTATTCCAATTGACGTAGAGTGTTGGTATAG-3′ | ||
| GFP-Leep2A1-1100 (T2) | pDM317 | F: 5′-GGAAGATCTATGAGCTCAACAGCGAATAATTCAC-3′ |
| R: 5′-GTCGACTTAAATGAATTCTGCACCTTGTTTTGATG-3′ | ||
| GFP-Leep2A1-446 (T3) | pDM317 | F: 5′-GGAAGATCTATGAGCTCAACAGCGAATAATTCAC-3′ |
| R: 5′-ACGCGTCGACTGATTCTGCTTCGAATACATGTAAC-3′ | ||
| GFP-Leep2A447-1100 (T4) | pDM317 | F: 5′-CTATCTAGAATGCAAAATTTTGAAAAAGAGAGAGAAGC-3′ |
| R: 5′-GTCGACTTAAATGAATTCTGCACCTTGTTTTGATG-3′ | ||
| GFP-Leep2A1101-1344 (T5) | pDM317 | F: 5′-TCTAGAAAAGGTACCAAAAGATCTATGAGTTCAATGATTGATGAAGAAG-3′ |
| R: 5′-TTAAATAATTTATTTATTTAACTAGTTTCCAATTGACGTAGAGTGTTGG-3′ | ||
| GFP-Leep2A1345-1640 (T6) | pDM317 | F: 5′-GAAGATCTATGTCAACAAATAAATTAAGTCGTGCTC-3′ |
| R: 5′-GCTCTAGAAATTGGTGAAGGTATACCAATTGTTG-3′ | ||
| GFP-Leep2AN1474K | pDM317 | F: 5′-TCATGTGGGTAAAGATATTGTAAACATCATTTG-3′ |
| R: 5′-ACAATATCTTTACCCACATGACGTTTCTTATGAATTTG-3′ | ||
| GFP-Leep2B | pDM317 | F: 5′-CTATCTAGAATGATTACAAACTCATTTACTAATAATTTTC-3′ |
| F: 5′-CTAGTCGACTTTATGAGATTGATCTTCAGATTG-3′ | ||
| GFP-Leep2B | pDM317-HygR | F: 5′-CTATCTAGAATGATTACAAACTCATTTACTAATAATTTTC-3′ |
| F: 5′-CTAGTCGACTTTATGAGATTGATCTTCAGATTG-3′ | ||
| RFP-Leep2B | pDM449 | F: 5′-CTATCTAGAATGATTACAAACTCATTTACTAATAATTTTC-3′ |
| F: 5′-CTAGTCGACTTTATGAGATTGATCTTCAGATTG-3′ | ||
| RFP-Leep2BΔGAP | pDM449 | F: 5′-CCGGATCCATGATTACAAACTCATTTACTAATA-3′ |
| R: 5′-CTATCTAGAATCGTCTTCGGTTGAGTCACCCTT-3′ | ||
| RFP-Leep2B1-1264 (T1) | pDM449 | F: 5′-CTATCTAGAATGATTACAAACTCATTTACTAATAATTTTC-3′ |
| R:5′-GTCGACTTACTCCAATGGTGTCAATTTATTTCTATTC-3′ | ||
| RFP-Leep2B1-1010 (T2) | pDM449 | F: 5′-CTATCTAGAATGATTACAAACTCATTTACTAATAATTTTC-3′ |
| R:5′-GTCGACTTAGGTATTGGTTGGTGTAGCATTATATGG-3′ | ||
| RFP-Leep2B1-400 (T3) | pDM449 | F: 5′-CTATCTAGAATGATTACAAACTCATTTACTAATAATTTTC-3′ |
| R: 5′-CTAGTCGACACTATTGGAATGAATACCAAGTATAC-3′ | ||
| RFP-Leep2B401-1010 (T4) | pDM449 | F: 5′-TCTAGAAAAGGTACCAAAAGATCTATGGAAATGTTGGCACGTAATTTATG-3′ |
| R: 5′-TAAATAATTTATTTATTTAACTAGTTTAGGTATTGGTTGGTGTAGCATTATATG-3′ | ||
| RFP-Leep2B1011-1264 (T5) | pDM449 | F: 5′-TCTAGAAAAGGTACCAAAAGATCTATGTCATCAAAAGTAACCGAATCTG-3′ |
| R: 5′-TTAAATAATTTATTTATTTAACTAGTTTACTCCAATGGTGTCAATTTATTTCTATTC-3′ | ||
| RFP-Leep2B1265-1619 (T6) | pDM449 | F: 5′-TGCTCTAGAATGAATAGTGTTCAATTTTTCCAATCTTTAAATATG-3′ |
| R: 5′-CTAGTCGACTTTATGAGATTGATCTTCAGATTG-3′ | ||
| Leep2B | pDM344 | F: 5′-CCGGATCCTACCCATACGATGTTCCAGATTACGCTATGATTACAAACTCATTTACTAATAATTTTC-3′ |
| R: 5′-CTATCTAGATTTATGAGATTGATCTTCAGATTG-3′ | ||
| Leep2BΔGAP | pDM344 | F: 5′-CCGGATCCTACCCATACGATGTTCCAGATTACGCTATGATTACAAACTCATTTACTAATAATTTTC-3′ |
| R: 5′-CTATCTAGAATCGTCTTCGGTTGAGTCACCCTT-3′ | ||
| GFP-RasB | pDM317 | F: 5′-GGAAGATCTATGTCAGTTTCAAATGAATATAAATTAG-3′ |
| R: 5′-GGACTAGTCTAAAGGATTAAACAATCACCACC-3′ | ||
| GFP-RasD | pDM317 | F: 5′-GGGGATCCATGACAGAATATAAATTAGTTATTGTAGGTGG-3′ |
| R: 5′-GGAACTAGTTTATAAAATTAAACATTGTTTTTTCTTTTTTTGAGC-3′ | ||
| GFP-RasG | pDM317 | F: 5′-GGGGATCCATGACAGAATACAAATTAGTTATTGTTGG-3′ |
| R: 5′-GGAACTAGTTTATAAAAGAGTACAAGCTTTTAATGGTC-3′ | ||
| GFP-RasBG15V | pDM371 | F: 5′-GGAAGATCTATGTCAGTTTCAAATGAATATAAATTAG-3′ |
| R: 5′-GGACTAGTCTAAAGGATTAAACAATCACCACC-3′ | ||
| F: 5′-TTGTTATGGGTGGTGTTGGTGTTGGTAAGAGTG-3′ | ||
| R: 5′-TCTTACCAACACCAACACCACCCATAACAACTAATTTATATTC-3′ | ||
| GFP-RasDQ61L | pDM371 | F: 5′-GGGGATCCATGACAGAATATAAATTAGTTATTGTAGGTGG-3′ |
| R: 5′-GGAACTAGTTTATAAAATTAAACATTGTTTTTTCTTTTTTTGAGC-3′ | ||
| F: 5′-ACTGCAGGTTTAGAGGAATATAGTGCAATG-3′ | ||
| R: 5′-TATTCCTCTAAACCTGCAGTATCTAAAATATCTAATAAAC-3′ | ||
| GFP-RasGG12V | pDM371 | F: 5′-GGGGATCCATGACAGAATACAAATTAGTTATTGTTGG-3′ |
| R: 5′-GGAACTAGTTTATAAAAGAGTACAAGCTTTTAATGGTC-3′ | ||
| F: 5′-TGTTGGTGGTGTTGGTGTCGGTAAAAGTGCCTTAAC-3′ | ||
| R: 5′-TTTACCGACACCAACACCACCAACAATAACTAATTTGTATTC-3′ | ||
| Gene distruption | ||
| leep2A KO | pBluescript-BSR | ARM1-F: 5′-CGGGGTACCCAATCCTCACAATCACCAGTTGCG-3′ |
| ARM1-R: 5′-CCCAAGCTTGTACATTTTCAATACCTAACATTTTCT-3′ | ||
| ARM2-F: 5′-CGGGGATCCGGTTGAGGCCATCTTTCACGATATC-3′ | ||
| ARM2-R: 5′-CCGGCGGCCGCAATTGTTGATGGTGGAGTTGTTGAAG-3′ | ||
| P1: 5′-GGAAGATCTATGAGCTCAACAGCGAATAATTCAC-3′ | ||
| P2: 5′-GTAGCGACAGAGAAGATTACAATGC-3′ | ||
| P3: 5′-TCGGGTATATTTGAGTGGAATGAG-3′ | ||
| P4: 5′-GCTCTAGAAATTGGTGAAGGTATACCAATTGTTG-3′ | ||
| leep2B KO | pBluescript-BSR | ARM1-F: 5′-CCGGTACCATGATTACAAACTCATTTACTAA-3′ |
| ARM1-R: 5′-CAAGCTTTGGTGAATTATTATTACTTGATTC-3′ | ||
| ARM2-F: 5′-CCGGATCCATTAGTAAACATATACTTGCC-3′ | ||
| ARM2-R: 5′-ATAAGGCGGCCGCTTTATGAGATTGATCTTCAGATTG-3′ | ||
| P1: 5′-ATGTTTCTTGACTGGGCCTCATCC-3′ | ||
| P2: 5′-GTAGCGACAGAGAAGATTACAATGC-3′ | ||
| P3: 5′-TCGGGTATATTTGAGTGGAATGAG-3′ | ||
| P4: 5′-CCCAGTCATTGCACCAGCCAAGCC-3′ | ||
| rasB KO | pBluescript-BSR | ARM1-F: 5′-CGGGGTACCCAATTCCAATAGTAAAAAGTC-3′ |
| ARM1-R: 5′-CTGGTCGACGGATAGTAAGTGCACTCTTACTG-3′ | ||
| ARM2-F: 5′-CGGGATCCGTCAAGATGATTACAGTGCTA-3′ | ||
| ARM2-R: 5′-CCGGCGGCCGCCTTTGATCTGCCTGGCTCTTTG-3′ | ||
| P1: 5′-AAAAAGTATGAGATTTTTAAAGTTGG-3′ | ||
| P2: 5′-GTAGCGACAGAGAAGATTACAATGC-3′ | ||
| P4: 5′-ATCATCTTGACCTGCAGTATC-3′ | ||
| rasD KO | pDM1801 | ARM1-F: 5′-ATTGCCGGCTTGAATAGATATAAGAGGTCATATTC-3′ |
| ARM1-R: 5′-CCCAAGCTTACCACCTACAATAACTAATTTATATTCTGTC-3′ | ||
| ARM2-F: 5′-GGAAGATCTATCAATCAAGTGGCAAAGCTC-3′ | ||
| ARM2-R: 5′-GGAACTAGTACTACAAGCACTTTTCCAGATACC-3′ | ||
| P1: 5′-ATCTATTTGTCAGTGAATTTATACC-3′ | ||
| P2: 5′-TTTATCGGCACTTTGCATCGG-3′ | ||
| P3: 5′-AAACTTCTCGACAGACGTCG-3′ | ||
| P4: 5′-TAAATCCTGCCAATGTAATTTCAGTAC-3′ | ||
| P5: 5′-TATGATCCAACAATTGAAGATAGTTATCG-3′ | ||
| P6: 5′-AATTCTACTTTTAGCAGATGACTCC-3′ | ||
| rasG KO | pLPBLP | ARM1-F: 5′-GAATTCATCTACACATGCGATTAACTGCCAC-3′ |
| ARM1-R: 5′-CATTGATGAATACGATCCAACTATCG-3′ | ||
| ARM2-F: 5′-GCGGCCGCGTGTCTACTCTATCACTTCAAG-3′ | ||
| ARM2-R: 5′-GTAAATTAGAAATAATAATTCAACCACCGCGG-3′ | ||
| P1: 5′-AAATCTACACATGCGATTAAC-3′ | ||
| P2: 5′-GTAGCGACAGAGAAGATTACAATGC-3′ | ||
| P3: 5′-TCGGGTATATTTGAGTGGAATGAG-3′ | ||
| P4: 5′-TTTTAATTGATCGGATTGGA-3′ | ||
| Expression in bacteria | ||
| GST-RapA | pGEX-6p-1 | F: 5′-GCGGATCCATGCCTCTTAGAGAATTCAAAATCG-3′ |
| R: 5′-ACGCGTCGACTTACAATAAAGCACATTTTGATTTAGC-3′ | ||
| GST-RapB | pGEX-6p-1 | F: 5′-GCGGATCCATGGGTAAGGGAAATGGTAAATC-3′ |
| R: 5′-ACGCGTCGACTTACATAATGATACATTTTTCTTTT-3′ | ||
| GST-RapC | pGEX-6p-1 | F: 5′-GCGGATCCATGCAAACCTATAAAGTAGTTGTTTTG-3′ |
| R: 5′-ACGCGTCGACTTACATGATTAAACATTTTCCTTTTG-3′ | ||
| GST-RanA | pGEX-6p-1 | F: 5′-GCGGATCCATGGCAGAAAAAGAACAAATTAAATTAG-3′ |
| R: 5′-ACGCGTCGACTTACAAGTCATCATTGTCTTCTGG-3′ | ||
| GST-RasB | pGEX-6p-1 | F: 5′-GGGGATCCATGTCAGTTTCAAATGAATATAAATTAG-3′ |
| R: 5′-CGGAATTCCTAAAGGATTAAACAATCACCAC-3′ | ||
| GST-RasC | pGEX-6p-1 | F: 5′-GGGGATCCATGTCAAAATTATTAAAATTAGTTATCGTTGG-3′ |
| R: 5′-CGGAATTCTTACAATATAATACATCCCCTTTTCTTTGG-3′ | ||
| GST-RasD | pGEX-6p-1 | F: 5′-GGGGATCCATGACAGAATATAAATTAGTTATTGTAGGTGG-3′ |
| R: 5′-CGGAATTCTTATAAAATTAAACATTGTTTTTTCTTTTTTTGAGC-3′ | ||
| GST-RasG | pGEX-6p-1 | F: 5′-GGGGATCCATGACAGAATACAAATTAGTTATTGTTGG-3′ |
| R: 5′-CGGAATTCTTATAAAAGAGTACAAGCTTTTAATGGTC-3′ | ||
| GST-RasS | pGEX-6p-1 | F: 5′-GGGGATCCATGTTTAATTTTAAATTAGTATTAGTTGG-3′ |
| R: 5′-CGGAATTCTTATAATAAATTACAAGATTTCTTTTTTTTAATTGGTTG-3′ | ||
| GST-RasU | pGEX-6p-1 | F: 5′-CGGGATCCATGTCAGCCTTTATATATAATAACAG-3′ |
| R: 5′-GAGTCGACTTATATCATTTTACAAATAGAATGAAATG-3′ | ||
| GST-RasV | pGEX-6p-1 | F: 5′-CGGGATCCATGTCAATTAAAATTAAAAATTTTAAAAAAAATA-3′ |
| R: 5′-GAGTCGACTTACATAACTTTACAAATTTCATTAATTTTTTTTTTTAATG-3′ | ||
| GST-RasW | pGEX-6p-1 | F: 5′-CGGGATCCATGACTTCCTATAAAAATAATAATG-3′ |
| R: 5′-GAGTCGACTTACATCATTTTACAAATTGAAGTTTTAG-3′ | ||
| GST-RasX | pGEX-6p-1 | F: 5′-CGGGATCCATGTCAGGATATAAAAATAATAATTTAG-3′ |
| R: 5′-GAGTCGACTTACATCATTTTACAAATTGAATTTTTAG-3′ | ||
| GST-RasY | pGEX-6p-1 | F: 5′-CGGGATCCATGACAACAAATAAAAGTAATGGTAATTTAG-3′ |
| R: 5′-GAGTCGACTTACATCATTTTACAAATTGATATTTTGGC-3′ | ||
| GST-RasZ | pGEX-6p-1 | F: 5′-CGGGATCCATGGCATCATATAAAAATAATAATTTAG-3′ |
| R: 5′-GAGTCGACTTACATCATTTTACAAATTGAGGTTTTAG-3′ | ||
| Yeast two-hybrid | ||
| Leep2A1359-1560 | pGADT7 | F: 5′-CGCCATATGACTCCTGGTCGTGAAATCTTAAAG-3′ |
| R: 5′-CGGGATCCTTATGGAGTATTATGACGAACATAACG-3′ | ||
| RasBQ64L ΔCAAX | pGBKT7 | F: 5′-CGGAATTCATGTCAGTTTCAAATGAATATAAATTAG-3′ |
| R: 5′-CGGGATCCTTAATCACCACCTTTAAACTTTTTAAGG-3′ | ||
| RasCQ62L ΔCAAX | pGBKT7 | F: 5′-CGGAATTCATGTCAAAATTATTAAAATTAGTTATC-3′ |
| R: 5′-CGGGATCCTTATCCCCTTTTCTTTGGTGGG-3′ | ||
| RasDQ61L ΔCAAX | pGBKT7 | F: 5′-CGGAATTCATGACAGAATATAAATTAGTTATTG-3′ |
| R: 5′-CGGGATCCTTATTGTTTTTTCTTTTTTTGAGC-3′ | ||
| RasGQ61L ΔCAAX | pGBKT7 | F: 5′-CGGAATTCATGACAGAATACAAATTAGTTATTG-3′ |
| R: 5′-CGGGATCCTTAAGCTTTTAATGGTCTCTTC-3′ | ||
| RasSQ61L ΔCAAX | pGBKT7 | F: 5′-CGGAATTCATGTTTAATTTTAAATTAGTATTAGTTG-3′ |
| R: 5′-CGGGATCCTTAAGATTTCTTTTTTTTAATTGG-3′ | ||
| RasUQ72L ΔCAAX | pGBKT7 | F: 5′-GGCATATGTCAGCCTTTATATATAATAACAG-3′ |
| R: 5′-CGGGATCCTTAAATAGAATGAAATGTATTATTTAATTTTTG-3′ | ||
| RasVQ91L ΔCAAX | pGBKT7 | F: 5′-CGGAATTCATGTCAATTAAAATTAAAAATTTTA-3′ |
| R: 5′-CGGGATCCTTAAATTTCATTAATTTTTTTTTTTAATGG-3′ | ||
| RasWQ67L ΔCAAX | pGBKT7 | F: 5′-CGGAATTCATGACTTCCTATAAAAATAATAATG-3′ |
| R: 5′-CGGGATCCTTAAATTGAAGTTTTAGTTTG-3′ | ||
| RasXQ67L ΔCAAX | pGBKT7 | F: 5′-CGGAATTCATGTCAGGATATAAAAATAATAAT-3′ |
| R: 5′-CGGGATCCTTAAATTGAATTTTTAGTATTATTTAC-3′ | ||
| RasYQ68L ΔCAAX | pGBKT7 | F: 5′-CGGAATTCATGACAACAAATAAAAGTAATGG-3′ |
| R: 5′-CGGGATCCTTAAATTGATATTTTGGCACCATTTACTTTTTG-3′ | ||
| RasZQ67L ΔCAAX | pGBKT7 | F: 5′-CGGAATTCATGGCATCATATAAAAATAATAAT-3′ |
| R: 5′-CGGGATCCTTAAATTGAGGTTTTAGTATTATTTAC-3′ | ||
| RanAQ65L | pGBKT7 | F: 5′-GGCATATGGCAGAAAAAGAACAAATTAAATTAG-3′ |
| R: 5′-GCGGATCCTTACAAGTCATCATTGTCTTCTGG-3′ | ||
| RapAQ65E ΔCAAX | pGBKT7 | F: 5′-GGCATATGCCTCTTAGAGAATTCAAAATCG-3′ |
| R: 5′-CGGGATCCTTATTTTGATTTAGCTTTGCTTGG-3′ | ||
To create leep2A and leep2B knockout constructs, a blasticidin S resistance (BSR) cassette (Kimmel and Faix, 2006) was inserted into pBlueScript II SK+ to generate pBlueScript-BSR. Subsequently, 5′- and -3′-arms were PCR-amplified from genomic DNA and inserted upstream and downstream of the BSR cassette, respectively. The resulting disruption cassette was then amplified by PCR and electroporated into cells. Gene disruption was validated by resistance to blasticidin (10 μg/ml), PCR, Southern blotting, and rescue experiment. To generate leep2A and leep2B double knockout cells, the BSR cassette was first removed from leep2A− cells by transformation with a Cre recombinase expression plasmid, pDEX-NLS-Cre (Faix et al., 2004), followed by selection with 20 μg/ml G418. The leep2B gene was then disrupted. Similar approaches were used to generate rasB and rasG knockout cells, while a Hygromycin-resistant cassette from pDM1081 (Paschke et al., 2018) was used to generate rasD knockout cells and rasD and rasG double knockout cells. Dd5P4− cell line was generated previously (Li et al., 2022).
Translocation assays
To induce responsiveness to cAMP, cells grown in HL5 medium were washed with development buffer (DB; 5 mM Na2HPO4, 5 mM KH2PO4, 2 mM MgSO4, 0.2 mM CaCl2, pH 6.5), starved in DB for 1 h, and pulsed with 100 nM cAMP every 6 min for 3–5 h. To image protein translocation in response to cAMP stimulation, 105 cells were plated in an eight-well coverslip chamber (cat#C8-1.5H-N; Cellvis) and allowed to adhere for at least 15 min. 1 μM cAMP was added for stimulation. To analyze the requirement of an intact actin cytoskeleton for translocation, cells were pretreated with 5 μM LatA (cat#BML-T119-0100; Enzo Life Sciences) for 5 min before the addition of cAMP. To image protein translocation in response to folic acid stimulation, cells were plated in a coverslip chamber in HL5. After the cells settled, HL5 was replaced with DB. After 30 min, 50 μM folic acid was added for stimulation. For the folic acid stimulation assay in the presence of LatA, cells were incubated with DB for 15 min, followed by treatment with 5 μM LatA for 5–10 min. Subsequently, 250 μM folic acid was added to the cells to initiate stimulation. Images were taken every 3 or 6 s on a Zeiss 880 inverted microscope equipped with a 63×/1.4 oil-immersion objective. To quantify translocation, the cytosol-to-membrane fluorescent intensity ratio was quantified using Fiji ImageJ (National Institutes of Health).
Macropinocytosis assays
For microscopy imaging, 0.6-1 × 105 cells seeded in an eight-well coverslip chamber were incubated with HL5 containing 0.5 mg/ml TRITC-Dextran (cat#T1162; Sigma-Aldrich) for 30–45 min. Images were taken on a Zeiss 880 inverted microscope equipped with a 63×/1.4 oil-immersion objective. Fluorescent intensity within the cells was quantified using Fiji ImageJ and normalized to the cell area. For the LY294002 treatment, cells were preincubated with either 0.1% DMSO or 12.5 μM LY249002 in HL5 medium for 30 min before the addition of TRITC-Dextran. For analysis of macropinocytosis by flow cytometry, 1.5 × 106 cells were seeded in a six-well plate. After the cells settled, the HL5 medium was replaced by an HL5 medium containing 0.5 mg/ml TRITC-Dextran or 20 μg/ml Alexa647-Dextran (cat#D22914; Invitrogen) for 30–45 min. Following the incubation, the cells were resuspended in ice-cold HL5, washed once with ice-cold KK2 buffer (6.5 mM KH2PO4 and 3.8 mM K2HPO4, pH 6.2) containing 5 mM EDTA, and then resuspended for analysis. The total fluorescence intensity per cell was determined by a BD Biosciences Influx flow cytometer. Data were analyzed by FlowJo.
Macropinocytosis dynamics was recorded in cells expressing PHcrac-GFP. Time-lapse images were taken every 3 s for 5 min on a Zeiss 880 inverted microscope equipped with a 63×/1.4 oil-immersion objective. The rate of macropinocytosis was quantified by tracking the number of membrane ruffles or enclosed macropinosomes formed in 5 min. The size of the membrane ruffle was determined by measuring the length of the membrane ruffle with the freehand line tool in ImageJ. The size of the macropinosome was determined by measuring the area of newly formed macropinosomes immediately after ruffle closure with the same tool. For Fig. 2, B and D and Fig. 8 G, the time evolution of the fluorescent intensity during macropinosome formation was determined by recording the medial optical section. After background subtraction, the average intensity at the macropinosomal membrane was determined for each channel at three positions. For Fig. 8 E, the average GFP fluorescent intensity at the plasma membrane was measured. For Fig. 8, I and J; and Fig. S5 B, the average fluorescence intensity in the cytosol was subtracted from that at the macropinosomal membrane, and the resulting value was then divided by the average fluorescence intensity in the cytosol.
Phagocytosis assay
Yeast phagocytosis assay was performed following a standard protocol (Junemann et al., 2016). Briefly, cells were adjusted to a density of 2 × 106 cells/ml and cultured in a shaken suspension in HL5 medium at 150 rpm. TRITC-labeled yeast was added at a sixfold excess. Subsequently, aliquots of 1 ml were taken at each time point and incubated on ice for 3 min with 100 μl of Trypan Blue solution (2 mg/ml in 20 mM citrate and 150 mM NaCl, pH 4.5). Cells were then pelleted, washed once with ice-cold Sorensen buffer (14.6 mM KH2PO4 and 2.0 mM Na2HPO4, pH 6.1), and resuspended in 1 ml of Sorensen buffer for immediate measurement using a Tecan Spark fluorescence spectrophotometer (544-nm excitation and 574-nm emission).
Migration assays
To assess random motility, 2 × 105 cells were cultured in a six-well plate in HL5 medium for 4 h, and the cells were imaged following medium replacement. Under-agarose folic acid chemotaxis assay was performed following previously described methods (Li et al., 2023). In brief, 5 ml of 0.5% agarose (cat#50071; Lonza) melted in LoFlo medium (cat#LFG0501; Formedium) was poured into a 50 mm glass-bottom dish (cat#P50G-1.5-30-F; MatTek). Once the agarose solidified, two troughs 5 mm apart were cut; one was filled with 1 mM folic acid and the other with cells resuspended in LoFlo medium. Cells were allowed to migrate for 4–5 h. For both random motility and chemotaxis assays, time-lapse images were captured every 15 s using a Zeiss 880 inverted microscope equipped with a 10×/0.45 objective. Cell tracking was performed using the manual tracking plugin of Fiji ImageJ, and the data were analyzed using Ibidi chemotaxis tool software.
Yeast two-hybrid assay
To screen for Leep2-interacting GTPases, yeast two-hybrid analyses were performed using the Matchmaker GAL4 Two-Hybrid System3 (Clontech Laboratories). The GAP domain of Leep2A was cloned into pGADT7 as prey, while the active forms of small GTPases from the Ras, Rab, and Rac subfamilies were cloned into pGBDT7 as bait. S. cerevisiae strain AH109 was cotransfected with both bait and prey plasmids and grown on double-dropout (DD, lacking leucine and tryptophan) agar plates following the manufacturer’s instructions. Clones were collected, resuspended in 100 μl H2O, and 2 μl of the suspension was spotted on quadruple-dropout (QD, deficiency in leucine, tryptophan, histidine, and adenine) agar plates. The interactions between tested proteins were analyzed according to the yeast growth on QD agar plates.
Protein purification
To purify GST-tagged small GTPases, expression plasmids were introduced into Escherichia coli BL21 cells. Cells were cultured in LB supplemented with ampicillin at 37°C until reaching an OD600 of 0.6–0.8. Protein expression was induced with 0.4 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 20 h at 16–20°C. The bacteria were centrifuged at 4,000 g, and the pellet was resuspended in suspension buffer (50 mM Hepes pH 7.4, 500 mM NaCl, 1 mM EDTA, 1 mM DTT, 1 mM PSMF) and lysed using sonication or a high-pressure homogenizer. The resulting cell suspension was centrifuged at 20,000 g for 20 min to pellet debris. The supernatant was then incubated with prewashed Glutathione Sepharose 4B beads (cat#17075605; Cytiva) for 1.5 h at 4°C. The beads were washed three times with suspension buffer and once with washing buffer (50 mM Hepes pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM DTT). The proteins bound to the beads were stored at −20°C in a buffer containing 50 mM Hepes, 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, and 50% glycerol.
For the GAP assay, the Leep2 complex or control was purified from DKO cells expressing Flag-Leep2A and GFP-Leep2B, Flag-Leep2AN1474K and GFP-Leep2B, Flag-Leep2A, or the Flag control. 2 × 108 cells were pelleted, washed twice with DB, and then lysed in 4 ml lysis buffer (20 mM Hepes pH 7.4, 300 mM NaCl, 1 mM EDTA, 1% Triton X-100) supplemented with complete EDTA-free protease inhibitor cocktail (cat#11873580001; Roche) and 1 mM NaF. After a 10-min incubation on ice, the cells were centrifuged at 15,000 g at 4°C. The supernatant was incubated with EZview red anti-Flag M2 affinity gel (cat#F2426; Sigma-Aldrich) for 2 h at 4°C. Subsequently, the beads were washed three times with wash buffer (20 mM Hepes pH 7.4, 150 mM NaCl) and once with storage buffer (20 mM Hepes pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM DTT). The proteins bound to the beads were stored at 4°C. The amount of purified proteins was quantified by comparing the intensity of the protein bands to a known concentration of BSA run on the same gel and stained with Coomassie blue.
Immunoprecipitation and GST pull-down assays
For coimmunoprecipitation experiments, cells expressing GFP- or RFP-fusion proteins were lysed in ice-cold lysis buffer (50 mM Hepes pH7.4, 0.5% NP-40, 150 mM NaCl, 10% glycerol, complete EDTA-free protease inhibitor cocktail, 1 mM NaF) and incubated on ice for 10 min. Cell lysates were centrifuged at 15,000 g for 10 min at 4°C. The supernatants were incubated with anti-GFP affinity beads (cat#SA070005; Smart Lifesciences) or anti-RFP affinity beads (cat#SA072005; Smart Lifesciences) for 1 h at 4°C. Beads were washed four times in lysis buffer without protease inhibitor. Samples were eluted with SDS loading buffer and subjected to SDS-PAGE and Western blotting. Anti-GFP antibody (1:5,000; WB) was purchased from Roche (cat#11814460001, RRID: AB_390913). Anti-DsRed antibody (1:5,000; WB), which was used to detect RFP-fusion proteins, was purchased from Takara (cat#632496, RRID: AB_10013483).
To identify Leep2-interacting proteins, cells expressing GFP-Leep2A and RFP-Leep2B were starved in DB for 3 h and lysed in buffer A (20 mM Hepes pH7.4, 0.5% NP-40, 150 mM NaCl, 10 μM GDP, 10% glycerol, 5 mM MgCl2, 1 mM DTT) or buffer B (20 mM Hepes pH 7.4, 0.5% NP-40, 150 mM NaCl, 10 μM GDP, 10 mM NaF, 30 μM AlCl3, 10% glycerol, 5 mM MgCl2, 1 mM DTT) supplemented with complete EDTA-free protease inhibitor cocktail. After a 10-min incubation on ice, cell lysates were centrifuged at 15,000 g at 4°C. The supernatants were incubated with anti-GFP affinity beads at 4°C for 1 h. Subsequently, the beads were washed four times with buffer A or buffer B. Samples were eluted with SDS loading buffer and subjected to SDS-PAGE. Protein bands were visualized by Coomassie blue staining and subjected to in-gel trypsin digestion and mass spectrometry analysis.
To verify the interaction between the Leep2 complex and RasB, RasD, and RasG as shown in Fig. 7 D, beads containing 30 μg purified GST-fused Ras GTPases were preincubated in buffer C (20 mM Hepes pH 7.4, 5 mM MgCl2, 1 mM DTT, 20 μM GDP) for 1 h at room temperature. Cells expressing GFP-Leep2A and RFP-Leep2B were starved in DB for 3 h and lysed in buffer B supplemented with a complete EDTA-free protease inhibitor cocktail. Following a 10-min incubation on ice, the cell lysates were centrifuged at 15,000 g at 4°C. The resulting supernatants were incubated with the pretreated beads for 2 h at 4°C. Subsequently, the beads were washed four times with buffer B. Samples were eluted with SDS loading buffer and subjected to SDS-PAGE. Purified GST-fusion proteins were visualized by Coomassie blue staining, while the Leep2 complex pulled down in the assay was detected by Western blotting with a GFP antibody.
To confirm the interaction between the Leep2 complex and GST-Ras proteins under the GDP-AlFx condition, cells expressing Flag-Leep2A and GFP-Leep2B were lysed in buffer A or buffer B supplemented with a complete EDTA-free protease inhibitor cocktail. Beads containing 30 μg GST-fusion proteins were preincubated in buffer C for 1 h at room temperature and then mixed with the cell lysates for 2 h at 4°C. The beads were washed four times with buffer A or buffer B. Samples were eluted with SDS loading buffer and subjected to SDS-PAGE. The purified GST-fusion proteins were visualized by Coomassie blue staining, while the Leep2 complex pulled down in the assay was detected by Western blotting with a Flag antibody. Anti-Flag antibody (1:2,000; WB) was purchased from Sigma-Aldrich (cat#F1804, RRID: AB_262044).
GAP assay
GAP assay was performed with the GTPase-Glo assay kit (cat#TM452; Promega) following the manufacturers protocol. GST-RasB, GST-RasD, or GST-RasG bound to glutathione-agarose were washed with buffer containing 20 mM Hepes pH 7.4, 150 mM NaCl, 1 mM EDTA, and 1 mM DTT. They were then titrated in a GAP buffer and mixed with Leep2-bound agarose in the same buffer. Each reaction contained ∼0.16 μM of the Leep2 complex or Leep2A and 1 μM of GST-fusion proteins. GTP and DTT were added to the reaction mixer at final concentrations of 10 µM and 1 mM, respectively. The reaction was carried out for 2 h at room temperature. Relative luminescence was measured using a Tecan Spark fluorescence spectrophotometer.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 9.0. Statistical significance was determined by one-way ANOVA with Dunnett or Tukey post-test or two-tailed unpaired t test with Welch’s correction. Data distribution was assumed to be normal, but this was not formally tested. In all figures, **** indicates P < 0.0001, *** indicates P < 0.001, ** P < 0.01, * P < 0.05, and ns, not significant. The statistical details are mentioned in the figure legends.
Online supplemental material
Fig. S1 shows the quantification of fluorescent intensity of GFP-Leep2A at macropinocytic cups and the translocation of the Leep2 complex in response to chemoattractant stimulation. Fig. S2 shows the characterization of leep2 knockout cells. Fig. S3 shows sequence alignment of the GAP domains and yeast two-hybrid screen of small GTPases for interaction with the GAP domain of Leep2A. Fig. S4 shows the characterization of ras knockout cells. Fig. S5 shows the impact of leep2 or ras deletion on PIP3 signaling, as well as the effect of perturbed PIP3 signaling on macropinocytosis. Video 1 shows the colocalization of GFP-Leep2A and RFP-Leep2B in WT cells during macropinocytosis. Video 2 shows the localization of GFP-Leep2A and RFP-Leep2B in WT cells migrating under agarose along a folic acid gradient. Video 3 shows PHcrac-GFP dynamics in WT and DKO cells during macropinocytosis. Video 4 shows the localization of GFP-Leep2A in WT cells expressing GFP-Leep2 and Leep2B during yeast phagocytosis.
Data availability
All data needed to evaluate the conclusions in the paper are available in the published article and/or its online supplementary materials. Raw data are available from the corresponding author upon reasonable request. All unique materials reported in this study will be provided upon request with no restrictions.
Acknowledgments
The authors thank Dr. Peter N. Devreotes (Johns Hopkins University, Baltimore, MD, USA) for PHcrac-GFP and PHcrac-RFP plasmids and pten− cells; Dr. Robert R. Kay (MRC Laboratory of Molecular Biology, London, UK) for pDM vectors and Ax2 cells; Dr. Richard Firtel (University of California, San Diego, CA, USA) for pi3k1−2− cells; Dr. Arjan Kortholt (University of Groningen, Groningen, Netherlands) for Byr2RBD-GFP plasmid; the proteomics core facility in the Institute of Biophysics (CAS) for mass spectrometry analysis; Junying Jia (Core Facility, Institute of Biophysics, CAS) for technical support in flow cytometry experiments; Yan Teng (Center for Biological Imaging, Institute of Biophysics, CAS) for help with confocal imaging.
This work was supported by grants from the Ministry of Science and Technology of China (2021YFA1300301 to H. Cai), the National Natural Science Foundation of China (92254303 and 32170701 to H. Cai and 32270743 to Y. Yang), the Strategic Priority Research Program of CAS (XDB37020304 to H. Cai), the Beijing Natural Science Foundation (5234029 to H. Tu), and the National Laboratory of Biomacromolecules.
Author contributions: X. Chao: Conceptualization, Data curation, Formal analysis, Methodology, Visualization, Y. Yang: Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Resources, Validation, Visualization, Writing - original draft, W. Gong: Data curation, Formal analysis, Investigation, Methodology, Resources, Visualization, Writing - original draft, Writing - review & editing, S. Zou: Validation, H. Tu: Resources, Validation, D. Li: Investigation, Resources, Visualization, W. Feng: Project administration, Supervision, H. Cai: Conceptualization, Formal analysis, Funding acquisition, Methodology, Project administration, Supervision, Visualization, Writing - original draft, Writing - review & editing.

