


Homeostatic maintenance and repair of lymphatic vessels are essential for health. We investigated the dynamics and the molecular mechanisms of lymphatic endothelial cell (LEC) renewal in adult mesenteric quiescent lymphatic vasculature using label-retention, lineage tracing, and cell ablation strategies. Unlike during development, adult LEC turnover and proliferation was confined to the valve regions of collecting vessels, with valve cells displaying the shortest lifespan. Proliferating valve sinus LECs were the main source for maintenance and repair of lymphatic valves. We identified mechanistic target of rapamycin complex 1 (mTORC1) as a mechanoresponsive pathway activated by fluid shear stress in LECs. Depending on the shear stress level, mTORC1 activity drives division of valve cells or dictates their mechanic resilience through increased protein synthesis. Overactivation of lymphatic mTORC1 in vivo promoted supernumerary valve formation. Our work provides insights into the molecular mechanisms of maintenance of healthy lymphatic vascular system.
Introduction
Lymphatic vessels perform essential roles in maintenance of fluid homeostasis and regulation of immune responses in virtually every organ of the body (Oliver et al., 2020; Petrova and Koh, 2020). The mature lymphatic vascular network is composed of blind-ended capillaries that uptake interstitial fluid and cells and collecting vessels that return lymph into blood circulation. Lymphatic capillaries and collecting vessels display distinct morphological features that support their specialized functions. Intraluminal bi-leaflet valves are a hallmark of collecting vessels and ensure unidirectional lymph flow (Zawieja, 2009).
During development and tissue regeneration, active and complex signaling is required for lymphangiogenesis and formation of the lymphatic vascular tree (Tammela and Alitalo, 2010). VEGF-C is a key ligand that binds to the receptor tyrosine kinase VEGFR-3 to drive lymphatic endothelial cell (LEC) survival, proliferation, and migration (Karkkainen et al., 2004; Mäkinen et al., 2001). Adult lymphatic vasculature is considered to be essentially quiescent, with the exception of intestinal lacteals and meningeal lymphatic vessels, which require continuous VEGFR-3 signaling for their maintenance (Antila et al., 2017; Bernier-Latmani et al., 2015; Nurmi et al., 2015). Nevertheless, lymphatic vasculature must be maintained throughout life to ensure its proper functioning. Little is known about the pathways involved in lymphatic vessel maintenance and repair in adulthood. Acquiring better knowledge of such processes is important for understanding and treatment of pathologies in which lymphatic vessels have been shown to be involved, such as cancer, inflammation, neurodegeneration, and cardiovascular diseases (Oliver et al., 2020; Petrova and Koh, 2020).
Here, we assessed the proliferation pattern of the LECs in adult mice under physiological conditions and the molecular pathways involved in lymphatic vessel maintenance. We report that under homeostatic conditions lymphatic collecting vessels represent the main site of LEC turnover and proliferating cells are mostly concentrated in valve sinuses. We further demonstrate that shear stress-responsive mechanistic target of rapamycin complex 1 (mTORC1) signaling is active in quiescent collecting vessels and valves, and is crucial for LEC proliferation and deposition of valve ECM components, like fibronectin EIIIA (FN-EIIIA). In vivo activation of mTORC1 signaling induces supernumerary lymphatic valve formation, further confirming a key role of this pathway in collecting lymphatics.
Results
Collecting LECs proliferate more than capillary LECs
The mature lymphatic vascular network comprises blind-ended lymphatic capillaries, which uptake the interstitial fluid and immune cells, and collecting vessels, which transport lymph to lymph nodes (González-Loyola and Petrova, 2021). During developmental and pathological lymphangiogenesis, capillary LECs proliferate at higher rates compared to collecting vessel LECs (Baluk et al., 2017; Sabine et al., 2015). However, the dynamics of LEC turnover in normal adult lymphatic vasculature has not been characterized.
We analyzed the mouse mesentery, a model that allows parallel study of lymphatic capillaries and collecting vessels in three dimensions at single-cell resolution (Fig. 1 A and Sabine et al., 2018). Blind-ended lymphatic capillaries are distinct from collecting vessels that are longer conduits composed of lymphangion segments intercalated by valves (Fig. 1, B and C, and González-Loyola and Petrova, 2021). Moreover, capillary and collecting vessel LECs are molecularly distinct: while both cell types express PROX1, a transcription factor, capillary, but not collecting vessel LECs express the hyaluronan receptor LYVE-1 (Fig. S1 A and Mäkinen et al., 2005). To visualize proliferating LECs, we stained mesenteries with KI67, the pan-endothelial marker PECAM1 and PROX1 (Fig. 1, B and C). In contrast to embryonic and early postnatal mesenteric lymphatic vessels (Bernier-Latmani et al., 2015; Sabine et al., 2015), we detected a higher percentage of proliferating LECs in adult PROX1+ collecting vessels compared to PROX1+ capillaries (Fig. 1 D). While during embryonic development up to 30% of LECs were KI67+ (Bernier-Latmani et al., 2015), the proportion of proliferating LECs in adult collecting vessels was only 0.5%, indicating that, as expected, the majority of adult LECs are quiescent (Fig. 1 D).

Proliferating LECs are confined to the valve areas of adult lymphatic vessels. (A) Schematic organization of the mesenteric vasculature. B and C insets indicate localization of images shown in B and C. LN, lymph node. (B and C) Proliferating LECs were detected in whole-mount lymphatic collecting vessel (B) and capillary (C) stained for KI67 (red), PROX1 (green), and PECAM1 (white). Coll, collecting vessel; Cap, capillary; Pre-col, pre-collector. Bracket, lymphatic valve area. Scale bar, 50 µm. (D) Proliferating LECs are more frequent in collecting vessels versus capillaries. Quantification of percentage of KI67+/PROX1+ among PROX1+ cells in whole-mount mesenteric vessels. n = 7 mice of 12–22 wk old (17 collecting vessels and 10 capillaries; 200–2,000 PROX1+ cells per mesentery). Two-tailed unpaired Mann–Whitney test, P = 0.0455. (E) Higher proliferation rate of collecting vessel LECs in valve area versus lymphangion. Quantification of percentage of KI67+/PROX1+ among PROX1+ cells in whole-mount mesenteric vessels. n = 9 mice of 10–12 wk old (25 collecting vessels; 200–2,000 PROX1+ cells per mesentery). Two-tailed unpaired Mann–Whitney test, P = 0.0434. (F) Definition of the valve area in whole-mount collecting vessel from a 12-wk-old mouse stained for PROX1 (green) and PECAM1 (white). Scale bar, 50 µm. (G) Quantification from data presented in E of percentage of KI67+/PROX1+ among PROX1+ cells in regions defined in F. Kruskal–Wallis test with Dunn’s correction for multiple comparison, P = 0.0071 between categories “0” and “50–100,” and P = 0.0272 between categories “0” and “>100.” (H) Workflow of flow cytometry analysis of LEC proliferation. w, week. (I) Quantification of percentage of KI67+ LECs among total LECs in collecting vessels and capillaries. n = 3 samples, each sample is a pool of three mesenteries isolated from 10-wk-old mice (2,700–10,000 cells analyzed per sample; 5–7% of PROX1+ cells detected as LYVE1neg collecting vessel LECs. Ratio paired t test, P = 0.0343. Data are shown as mean ± SD. *P < 0.05, **P < 0.01. H is created with help from BioRender. See also Fig. S1.
Proliferating LECs are confined to the valve areas of adult lymphatic vessels. (A) Schematic organization of the mesenteric vasculature. B and C insets indicate localization of images shown in B and C. LN, lymph node. (B and C) Proliferating LECs were detected in whole-mount lymphatic collecting vessel (B) and capillary (C) stained for KI67 (red), PROX1 (green), and PECAM1 (white). Coll, collecting vessel; Cap, capillary; Pre-col, pre-collector. Bracket, lymphatic valve area. Scale bar, 50 µm. (D) Proliferating LECs are more frequent in collecting vessels versus capillaries. Quantification of percentage of KI67+/PROX1+ among PROX1+ cells in whole-mount mesenteric vessels. n = 7 mice of 12–22 wk old (17 collecting vessels and 10 capillaries; 200–2,000 PROX1+ cells per mesentery). Two-tailed unpaired Mann–Whitney test, P = 0.0455. (E) Higher proliferation rate of collecting vessel LECs in valve area versus lymphangion. Quantification of percentage of KI67+/PROX1+ among PROX1+ cells in whole-mount mesenteric vessels. n = 9 mice of 10–12 wk old (25 collecting vessels; 200–2,000 PROX1+ cells per mesentery). Two-tailed unpaired Mann–Whitney test, P = 0.0434. (F) Definition of the valve area in whole-mount collecting vessel from a 12-wk-old mouse stained for PROX1 (green) and PECAM1 (white). Scale bar, 50 µm. (G) Quantification from data presented in E of percentage of KI67+/PROX1+ among PROX1+ cells in regions defined in F. Kruskal–Wallis test with Dunn’s correction for multiple comparison, P = 0.0071 between categories “0” and “50–100,” and P = 0.0272 between categories “0” and “>100.” (H) Workflow of flow cytometry analysis of LEC proliferation. w, week. (I) Quantification of percentage of KI67+ LECs among total LECs in collecting vessels and capillaries. n = 3 samples, each sample is a pool of three mesenteries isolated from 10-wk-old mice (2,700–10,000 cells analyzed per sample; 5–7% of PROX1+ cells detected as LYVE1neg collecting vessel LECs. Ratio paired t test, P = 0.0343. Data are shown as mean ± SD. *P < 0.05, **P < 0.01. H is created with help from BioRender. See also Fig. S1.

Representative FACS plots and gating strategy for sorting LECs from mouse mesentery (related to Fig. 1, H and I ). (A) Representative image of whole-mount lymphatic collecting vessel and capillary stained for PROX1 (green), LYVE1 (magenta), and PECAM1 (white). Asterisk, lymphatic valve. Scale bar, 50 µm. (B) LECs (CD45neg CD31+ Gp38+), LECs of collecting vessels (CD45neg CD31+ Gp38+ LYVE1neg), LECs from capillaries (CD45neg CD31+ Gp38+ LYVE1+).
Representative FACS plots and gating strategy for sorting LECs from mouse mesentery (related to Fig. 1, H and I ). (A) Representative image of whole-mount lymphatic collecting vessel and capillary stained for PROX1 (green), LYVE1 (magenta), and PECAM1 (white). Asterisk, lymphatic valve. Scale bar, 50 µm. (B) LECs (CD45neg CD31+ Gp38+), LECs of collecting vessels (CD45neg CD31+ Gp38+ LYVE1neg), LECs from capillaries (CD45neg CD31+ Gp38+ LYVE1+).
We next analyzed the position of KI67+ LECs along the collecting vessel. We observed that proliferating LECs were mostly located in the valve region (Fig. 1 E), defined by PROX1high valve LECs (Fig. 1 F and Sabine et al., 2012; Sabine et al., 2015). Accordingly, quantification confirmed that proliferating LECs are concentrated in the valve area (Fig. 1, F and G). To independently confirm these results, we further analyzed mesenteric adult LECs by flow cytometry (Fig. 1 H and Fig. S1 B). We found that the proportion of KI67+ cells was higher in CD31+LYVE1neg collecting LECs compared with CD31+LYVE1+ capillary LECs (Fig. 1 I). The percentage of KI67+ LECs was higher in flow cytometry experiments, likely due to superior sensitivity of signal detection as compared to visual inspection of immunofluorescent staining (Jenson et al., 1998).
In summary, these results confirm low proliferation rates of LECs in adult lymphatic vessels, identify collecting vessels as the main location of LEC proliferation in the adult quiescent vasculature, and lymphatic valve areas as hot spots of adult LEC renewal.
Lymphatic collecting vessels display higher turnover rate compared to capillaries
The occurrence of KI67+ adult LECs was a very rare event. Therefore, we used an alternative approach to analyze adult LEC proliferation by cumulatively labeling proliferating cells with the thymidine analog EdU administered in drinking water for 2 wk (Fig. 2 A). In agreement with the expression pattern of KI67, we observed that the proportion of EdU+ LECs was significantly higher in collecting vessels compared to capillaries (Fig. 2, B and C), further confirming that under homeostatic conditions collecting vessels represent the main site of LEC proliferation.

Valve LECs are short-lived. (A) Scheme of EdU administration in adult mice. (B) Detection of EdU+ LECs in lymphatic vessels. Paraffin sections of mesenteries were stained for PROX1 (green), EdU (red), LYVE1 (magenta). Arrowhead, EdU+/PROX1+ cell; arrow, EdU+/PROX1neg cell; asterisk, valve. Inset, zoom of the box. Scale bar, 50 µm. (C) Quantification of EdU+/PROX1+ among PROX1+ cells in collecting vessels and capillaries. n = 6 mice of 10–15 wk old (32 collecting vessels and 21 capillaries). Two-tailed unpaired Mann–Whitney test, P = 0.0022. (D) Scheme of EdU washout experiment in adult mice. (E) Preferential localization of EdU+ cells in the valve area. Whole-mount staining of mesenteric collecting lymphatic vessels for PROX1 (green), EdU (red), and PECAM1 (white) at the indicated time points. Asterisk, valve area; purple arrowhead, valve area EdU+/PROX1+ cell; blue arrow, lymphangion EdU+/PROX1+ cell. Scale bar, 50 µm. (F) Quantification of percentage of EdU+/PROX1+ among PROX1+ cells in valve area and lymphangion. n = 10 mice of 11 wk old (2–3 mice and 12–18 images per time point). (G) Half-life (t1/2) curve for LECs in valve area from data in E and F. Half-life for valve area cells is 9–10 d. (H) Half-life (t1/2) curve for LECs in lymphangion from data in E and F. Half-life for lymphangion cells is 30–60 d. Data are shown as mean ± SD. ***P < 0.005. A and D are created with help from BioRender. w, week.
Valve LECs are short-lived. (A) Scheme of EdU administration in adult mice. (B) Detection of EdU+ LECs in lymphatic vessels. Paraffin sections of mesenteries were stained for PROX1 (green), EdU (red), LYVE1 (magenta). Arrowhead, EdU+/PROX1+ cell; arrow, EdU+/PROX1neg cell; asterisk, valve. Inset, zoom of the box. Scale bar, 50 µm. (C) Quantification of EdU+/PROX1+ among PROX1+ cells in collecting vessels and capillaries. n = 6 mice of 10–15 wk old (32 collecting vessels and 21 capillaries). Two-tailed unpaired Mann–Whitney test, P = 0.0022. (D) Scheme of EdU washout experiment in adult mice. (E) Preferential localization of EdU+ cells in the valve area. Whole-mount staining of mesenteric collecting lymphatic vessels for PROX1 (green), EdU (red), and PECAM1 (white) at the indicated time points. Asterisk, valve area; purple arrowhead, valve area EdU+/PROX1+ cell; blue arrow, lymphangion EdU+/PROX1+ cell. Scale bar, 50 µm. (F) Quantification of percentage of EdU+/PROX1+ among PROX1+ cells in valve area and lymphangion. n = 10 mice of 11 wk old (2–3 mice and 12–18 images per time point). (G) Half-life (t1/2) curve for LECs in valve area from data in E and F. Half-life for valve area cells is 9–10 d. (H) Half-life (t1/2) curve for LECs in lymphangion from data in E and F. Half-life for lymphangion cells is 30–60 d. Data are shown as mean ± SD. ***P < 0.005. A and D are created with help from BioRender. w, week.
We next analyzed the life span of LECs in collecting vessels using a label washout approach (Vibert and Thomas-Vaslin, 2017). We continuously administered EdU for 2 wk and analyzed EdU incorporation after 0, 2, 4, and 8 wk of washout (Fig. 2 D). In collecting vessels, we detected a higher proportion of EdU in valve areas than in lymphangions, a difference that was lost after 2 wk of washout (Fig. 2, E and F). In agreement with this observation, the estimated half-life of valve LECs was about 9–10 d whereas it was 3–6 times longer for lymphangion LECs (Fig. 2, G and H). Collectively, these data indicate that valve areas are the main place of LEC turnover in adult lymphatic vessels.
Valve sinus LECs proliferate and migrate to valve leaflets
We next investigated in further detail the location of KI67+ LECs in collecting vessels. The majority of LECs have low PROX1 levels in collecting vessels, with the exception of valve leaflet LECs that are characterized by high levels of PROX1 (Norrmén et al., 2009; Sabine et al., 2012; Sabine et al., 2015). As we never observed PROX1highKI67+ LECs in the valve leaflet of adult mice (Fig. 1), maybe due to the extremely low occurrence of KI67+ adult LECs (Fig. 1 C), we also studied younger 3-wk-old mice, which overall had many more KI67+ LECs in collecting vessels in comparison with adult animals (Fig. 3 A). However, as in adult mice, the overwhelming majority of KI67+ LECs in collecting vessels displayed low PROX1 levels (Fig. 3 B and Fig. S2, A–C) and only a few cells PROX1highKI67+ cells were observed in valves (Fig. 3 B and Fig. S2, D–F). This result identifies the proliferating LECs as valve sinus or lymphangion cells and indicates that proliferation in the valve region mostly takes place in the valve sinus rather than on the leaflets, the latter being subjected to the highest levels of shear stress, which may preclude cell division (Pujari et al., 2020).

Proliferating LECs reside in the valve sinus. (A) Whole-mount staining of 3-wk-old mouse mesenteric lymphatic vessels for PROX1 (green), KI67 (red), and PECAM1 (white). Arrowhead, PROX1low/KI67+ cell in valve sinus; arrow, PROX1high/KI67neg cell in valve leaflet. Scale bar, 20 µm. (B) Quantification of PROX1low/KI67+ and PROX1high/KI67+ cells. n = 6 valves from two mice (50–90 PROX1+ cells per vessels); each color represents a valve; each dot represents a valve cell. Most of valve KI67+ cells are PROX1low. Two-tailed unpaired Mann–Whitney test, P = 0.0065. **P < 0.01. (C)MKi67-CreERT2/mTmG mouse model and tamoxifen administration schedule. (D) GFP+ LECs are found mostly in valve sinus early after tamoxifen administration (upper panels), but in valve leaflet later after tamoxifen delivery (lower panels). Whole-mount mesenteric collecting vessels of 3-wk-old mice presented as confocal z-stack (left) or 1.2-µm z-slice (right) for Tomato (red) and GFP (green). Schemes are presented under each microscopy image, with the corresponding legend. Asterisk, valve leaflet; #, valve sinus; arrowhead, valve GFP+ LEC (a and b in valve sinus; c in valve leaflet). Scale bar, 50 µm. (E) Quantification from D of GFP+ valve LECs in valve sinus or leaflet. n = 3 mice per time point (5–6 valves per mouse). Data are shown as mean ± SD. (F) Representative images of GFP+ LECs in valve sinuses (upper panels) or at the edge of the leaflet (lower panels) 2 d or 1 wk after tamoxifen administration, respectively. Legends correspond to D. Scale bar, 50 µm. C is created with help from BioRender. See also Fig. S2.
Proliferating LECs reside in the valve sinus. (A) Whole-mount staining of 3-wk-old mouse mesenteric lymphatic vessels for PROX1 (green), KI67 (red), and PECAM1 (white). Arrowhead, PROX1low/KI67+ cell in valve sinus; arrow, PROX1high/KI67neg cell in valve leaflet. Scale bar, 20 µm. (B) Quantification of PROX1low/KI67+ and PROX1high/KI67+ cells. n = 6 valves from two mice (50–90 PROX1+ cells per vessels); each color represents a valve; each dot represents a valve cell. Most of valve KI67+ cells are PROX1low. Two-tailed unpaired Mann–Whitney test, P = 0.0065. **P < 0.01. (C)MKi67-CreERT2/mTmG mouse model and tamoxifen administration schedule. (D) GFP+ LECs are found mostly in valve sinus early after tamoxifen administration (upper panels), but in valve leaflet later after tamoxifen delivery (lower panels). Whole-mount mesenteric collecting vessels of 3-wk-old mice presented as confocal z-stack (left) or 1.2-µm z-slice (right) for Tomato (red) and GFP (green). Schemes are presented under each microscopy image, with the corresponding legend. Asterisk, valve leaflet; #, valve sinus; arrowhead, valve GFP+ LEC (a and b in valve sinus; c in valve leaflet). Scale bar, 50 µm. (E) Quantification from D of GFP+ valve LECs in valve sinus or leaflet. n = 3 mice per time point (5–6 valves per mouse). Data are shown as mean ± SD. (F) Representative images of GFP+ LECs in valve sinuses (upper panels) or at the edge of the leaflet (lower panels) 2 d or 1 wk after tamoxifen administration, respectively. Legends correspond to D. Scale bar, 50 µm. C is created with help from BioRender. See also Fig. S2.

Representative lymphatic valve confocal analysis showing most of PROX1 high cells are KI67 neg (related to Fig. 3, A and B ). (A–F) Whole-mount staining of 3-wk-old mouse mesenteric lymphatic vessels for PROX1 (green), KI67 (red), and PECAM1 (white). Confocal z-stack images are shown on the left for each panel (50–100 µm in thickness). Scale bar, 30 µm. Confocal 0.8-µm thick z-slices are shown as close-ups of the dashed cyan box on the right. Scale bars, 30 µm for A–C and E, 20 µm for D and F. Arrowheads, valve LECs; their location within the valve is indicated under the confocal z-slice. Arrowhead color depicts the valve LEC identity, KI67+/PROX1high (yellow), KI67+/PROX1low (cyan), KI67low/PROX1high (white), and KI67neg/PROX1high (magenta). Note that the highest proportion of PROX1high cells are KI67neg and most of KI67+ valve LECs are PROX1low. Only a few cells PROX1highKI67+ were observed in valves, either with very low levels of KI67 (D, white arrowhead), with high levels of PROX1 due to mitosis (E, white arrow) or localized at the bottom of the leaflet at the junction with the sinus (F, yellow arrowhead).
Representative lymphatic valve confocal analysis showing most of PROX1 high cells are KI67 neg (related to Fig. 3, A and B ). (A–F) Whole-mount staining of 3-wk-old mouse mesenteric lymphatic vessels for PROX1 (green), KI67 (red), and PECAM1 (white). Confocal z-stack images are shown on the left for each panel (50–100 µm in thickness). Scale bar, 30 µm. Confocal 0.8-µm thick z-slices are shown as close-ups of the dashed cyan box on the right. Scale bars, 30 µm for A–C and E, 20 µm for D and F. Arrowheads, valve LECs; their location within the valve is indicated under the confocal z-slice. Arrowhead color depicts the valve LEC identity, KI67+/PROX1high (yellow), KI67+/PROX1low (cyan), KI67low/PROX1high (white), and KI67neg/PROX1high (magenta). Note that the highest proportion of PROX1high cells are KI67neg and most of KI67+ valve LECs are PROX1low. Only a few cells PROX1highKI67+ were observed in valves, either with very low levels of KI67 (D, white arrowhead), with high levels of PROX1 due to mitosis (E, white arrow) or localized at the bottom of the leaflet at the junction with the sinus (F, yellow arrowhead).
In addition to KI67 analysis by immunostaining, we also crossed Mki67-CreERT2 mice (Basak et al., 2018) with reporter mTmG reporter mice (Muzumdar et al., 2007), which express membrane-targeted red fluorescent Tomato (mTomato) protein prior to Cre-mediated excision, and membrane-targeted GFP (mGFP) after excision (Fig. 3 C). Using this model, we found that 2 d after injection of a single dose of tamoxifen, the majority of adult LECs remained mTomato+, while the rare mGFP+ LECs detected were mostly located in the lymphatic valve sinus (Fig. 3, D–F). In contrast, 1 wk after tamoxifen injection, the mGFP+ LECs were observed in a significantly higher proportion in valve leaflets, which suggests that LECs proliferate in the valve sinus and further populate the leaflet over longer periods (Fig. 3, D–F). Interestingly, most of the 1-wk labeled cells were located at the tip of the leaflet, lining the edge of the valve opening, perpendicular to the direction of flow (Fig. 3 F).
LECs proliferate to maintain valve integrity
We next sought to establish the contribution of valve sinus LEC proliferation to collecting vessel development and maintenance. Esco2 encodes a cohesin acetyltransferase, which is necessary for sister chromatid cohesion and separation during mitosis and meiosis (Whelan et al., 2012); its ablation thus eliminates proliferating cells (Hou and Zou, 2005; Vega et al., 2005). We generated Esco2fl/fl/Prox1-CreERT2 mice for conditional ablation of proliferating LECs (Esco2lecKO). We deleted Esco2 in LECs of postnatal mice and analyzed mesentery and diaphragm lymphatic vessels (Fig. 4 A). Staining for KI67 revealed decreased proliferation of collecting LECs as early as 4 d after tamoxifen administration (Fig. 4, B and C), indicating efficient Esco2 ablation. Further quantification showed a significant decrease in the number of mesenteric lymphatic valves of Esco2lecKO mice in comparison with the controls (Fig. 4, D and E). Reduced valve number was also found in the diaphragm, where smaller PROX1high valve-like structures were observed on one side of the vessel, suggesting shortened or ablated valve leaflets (Fig. 4, F and G). These data thus establish proliferating valve sinus LECs as a major cellular source for lymphatic valve leaflet maintenance.

Depletion of dividing cells reduces the number of valves. (A) Scheme of Esco2lecKO/Prox1-CreERT2 mouse model and tamoxifen administration schedule. (B)Esco2 ablation reduces LEC proliferation. Whole-mount staining of mesenteric collecting vessels of post-natal P10 wild-type and Esco2lecKO mice for PROX1 (green), KI67 (red), and PECAM1 (white). KI67 was masked to PROX1 channel and shown in black in a separate window. Scale bar, 20 µm. (C) Quantification of percentage of KI67+/PROX1+ among PROX1+ cells from data shown in B from control wild-type (n = 4) and Esco2lecKO mice (n = 6; 4–5 vessels per mouse, 50–150 PROX1+ cells per vessel). Two-tailed unpaired Welch’s t test, P = 0.0381. (D) Decreased number of lymphatic valves in Esco2lecKO mice. Whole-mount staining of mesenteric collecting vessels of wild-type and Esco2lecKO mice for PROX1 (black). Arrow, valve. Scale bar, 50 µm. (E) Quantification of mesenteric lymphatic valve number in wild-type (n = 4) and Esco2lecKO (n = 5) mice (4–5 mesenteric lymphatic vessels per mouse). Two-tailed unpaired Welch’s t test, P = 0.0299. (F) Decreased number of lymphatic valves in Esco2lecKO mice. Whole-mount staining of diaphragm collecting vessels of wild-type and Esco2lecKO mice for PROX1 (black). Arrowhead, mature valve; arrow, residual valve site. Scale bar, 25 µm. (G) Quantification of diaphragm lymphatic valve number in wild-type (n = 4) and Esco2lecKO (n = 5) mice (40–60 mm diaphragm lymphatic vessels per mouse). Two-tailed unpaired Welch’s t test, P = 0.0123. A is created with help from BioRender. Data are shown as mean ± SD. *P < 0.05.
Depletion of dividing cells reduces the number of valves. (A) Scheme of Esco2lecKO/Prox1-CreERT2 mouse model and tamoxifen administration schedule. (B)Esco2 ablation reduces LEC proliferation. Whole-mount staining of mesenteric collecting vessels of post-natal P10 wild-type and Esco2lecKO mice for PROX1 (green), KI67 (red), and PECAM1 (white). KI67 was masked to PROX1 channel and shown in black in a separate window. Scale bar, 20 µm. (C) Quantification of percentage of KI67+/PROX1+ among PROX1+ cells from data shown in B from control wild-type (n = 4) and Esco2lecKO mice (n = 6; 4–5 vessels per mouse, 50–150 PROX1+ cells per vessel). Two-tailed unpaired Welch’s t test, P = 0.0381. (D) Decreased number of lymphatic valves in Esco2lecKO mice. Whole-mount staining of mesenteric collecting vessels of wild-type and Esco2lecKO mice for PROX1 (black). Arrow, valve. Scale bar, 50 µm. (E) Quantification of mesenteric lymphatic valve number in wild-type (n = 4) and Esco2lecKO (n = 5) mice (4–5 mesenteric lymphatic vessels per mouse). Two-tailed unpaired Welch’s t test, P = 0.0299. (F) Decreased number of lymphatic valves in Esco2lecKO mice. Whole-mount staining of diaphragm collecting vessels of wild-type and Esco2lecKO mice for PROX1 (black). Arrowhead, mature valve; arrow, residual valve site. Scale bar, 25 µm. (G) Quantification of diaphragm lymphatic valve number in wild-type (n = 4) and Esco2lecKO (n = 5) mice (40–60 mm diaphragm lymphatic vessels per mouse). Two-tailed unpaired Welch’s t test, P = 0.0123. A is created with help from BioRender. Data are shown as mean ± SD. *P < 0.05.
Collecting vessel LEC proliferation is VEGFR-2 and VEGFR-3 independent
We next sought to investigate the molecular pathway(s) that promote valve LEC renewal. VEGF-A/VEGFR-2 and VEGF-C/VEGFR-3 signaling are key pathways regulating blood and LEC proliferation (Bahram and Claesson-Welsh, 2010; Secker and Harvey, 2015), and we sought to determine whether these pathways are required for proliferation of adult collecting vessel LECs under physiological conditions. We treated adult mice with VEGFR-2 or VEGFR-3 blocking antibodies, DC101 (Prewett et al., 1999) and mF4-31C1 (Pytowski et al., 2005), respectively, and assessed the cumulative EdU labeling index in lymphatic vessels after 2 wk of treatment (Fig. S3 A). Previously, we and others have shown that, while most adult endothelial cells are quiescent, the small intestinal blood and lymphatic ECs undergo VEGF-A– and VEGF-C–dependent renewal (Bernier-Latmani et al., 2015; Nurmi et al., 2015). As expected, VEGFR-2 and VEGFR-3 blockade in adult mice efficiently reduced proliferation of intestinal blood and lymphatic capillary ECs (Fig. S3, B and C), respectively. However, mesentery collecting vessel LEC proliferation was not affected (Fig. S3, D and E), indicating that valve LEC renewal occurs via a VEGFR-2/3-ligand independent mechanism.

Proliferation of mature LECs is VEGF-A/VEGFR-2 and VEGF- C/VEGFR-3 signaling independent. (A) Scheme of blocking antibody injections and EdU administration. w, week. (B) Decreased proliferation of blood endothelial cells (BECs) upon anti-VEGFR2 treatment in intestine. n = 8 for mice control and n = 9 for anti-R2 (4–6 images per sample). Two-tailed unpaired Welch’s t test, P = 0.0082. (C) Decreased proliferation of LECs upon anti-VEGFR3 treatment in intestine. n = 5 mice for control and n = 4 for anti-R3 (4–6 images per sample). (D) Immunofluorescent staining of 8-μm-thick paraffin mesentery sections for PROX1 (green), EdU (red), LYVE1 (magenta). Arrowheads, proliferating PROX1+/EdU+ LECs. Scale bar, 20 µm. (E) Blocking VEGFR-2 or -3 does not alter LEC proliferation in mesentery. Quantification of EdU+ LECs in mesentery. n = 3 mice for control, n = 4 for anti-R2 and anti-R3 (4–6 images per sample). Kruskal–Wallis test with Dunn’s correction for multiple comparison, P = 0.0257. (F) Higher mTORC1 activity in valve LECs. Quantification of pS6 intensity in embryo and adult valve and lymphangion LECs (related to Fig. 5, C and G). n = 3 embryo (left) and n = 3 adult (right) mesenteries. Two-tailed unpaired Welch’s t test, P = 0.0021 for embryos. Data are shown as mean ± SD. *P < 0.05, **P < 0.001, and ***P < 0.005. A is created with help from BioRender.
Proliferation of mature LECs is VEGF-A/VEGFR-2 and VEGF- C/VEGFR-3 signaling independent. (A) Scheme of blocking antibody injections and EdU administration. w, week. (B) Decreased proliferation of blood endothelial cells (BECs) upon anti-VEGFR2 treatment in intestine. n = 8 for mice control and n = 9 for anti-R2 (4–6 images per sample). Two-tailed unpaired Welch’s t test, P = 0.0082. (C) Decreased proliferation of LECs upon anti-VEGFR3 treatment in intestine. n = 5 mice for control and n = 4 for anti-R3 (4–6 images per sample). (D) Immunofluorescent staining of 8-μm-thick paraffin mesentery sections for PROX1 (green), EdU (red), LYVE1 (magenta). Arrowheads, proliferating PROX1+/EdU+ LECs. Scale bar, 20 µm. (E) Blocking VEGFR-2 or -3 does not alter LEC proliferation in mesentery. Quantification of EdU+ LECs in mesentery. n = 3 mice for control, n = 4 for anti-R2 and anti-R3 (4–6 images per sample). Kruskal–Wallis test with Dunn’s correction for multiple comparison, P = 0.0257. (F) Higher mTORC1 activity in valve LECs. Quantification of pS6 intensity in embryo and adult valve and lymphangion LECs (related to Fig. 5, C and G). n = 3 embryo (left) and n = 3 adult (right) mesenteries. Two-tailed unpaired Welch’s t test, P = 0.0021 for embryos. Data are shown as mean ± SD. *P < 0.05, **P < 0.001, and ***P < 0.005. A is created with help from BioRender.
mTORC1 signaling is active in lymphatic collecting vessels
As mTOR is a ubiquitous pathway controlling cell growth and proliferation (Laplante and Sabatini, 2013), we hypothesized that endothelial mTOR signaling may promote valve LEC renewal. In line with this hypothesis, adult organ transplant patients treated with the pharmacological inhibitor of mTORC1, rapamycin, but not with unrelated immunosuppressive drugs, display impaired lymph drainage and increased incidence of limb lymphedema (Al-Otaibi et al., 2007; Baliu et al., 2014; Desai et al., 2009; Romagnoli et al., 2005), indicating the continuous need of mTORC1 signaling for postnatal lymphatic homeostasis.
We analyzed the mTORC1 pathway in vivo by staining developing and adult lymphatic vessels for the phosphorylated form of ribosomal protein S6 (pS6; Ser240/Ser244) as a readout of mTORC1 activity (Isotani et al., 1999). In the embryonic mesenteric vasculature (Fig. 5 A), we observed activated mTORC1 signaling in growing lymphatic capillaries (Fig. 5 B and Martinez-Corral et al., 2020) and in developing collecting vessels (Fig. 5 C), with a higher percentage of pS6+ LECs in the valve area compared to lymphangion, with more than 15% of valve cells being pS6+ (Fig. 5, C and D). In the adult mouse mesentery (Fig. 5 E), pS6+ LECs were observed in collecting vessels and valves (Fig. 5, G and H), but were mostly absent from resting capillaries (Fig. 5 F). While LECs with active mTORC1 signaling were observed less frequently in adult lymphatic vessels than in embryos (Fig. 5, D and H), in agreement with the low proliferation and renewal rate of adult LECs, pS6 intensity was similar between embryonic and adult LECs (Fig. 5, C and G; and Fig. S3 F), suggesting that the mTORC1 signaling pathway remains active throughout life. Taken together, these observations indicate that mTORC1 signaling is active during lymphatic valve formation and persists in the adult collecting lymphatic vessels to maintain valve homeostasis.

mTORC1 signaling is active in both growing and adult lymphatic valves. (A) Analysis of E18.5 embryo mesenteric lymphatic vasculature. (B and C) Detection of mTORC1 target pS6 in embryonic lymphatic growing vessels. Whole-mount staining of E18.5 mesenteric lymphatic capillaries (B) and collecting vessels (C) for PROX1 (green), pS6 (red), and PECAM1 (white). Confocal z-stack (left) and confocal thick z-section (right) are presented. Arrowhead, PROX1+pS6+ cell. Scale bar, 50 µm for z-stack, 10 µm (B) or 20 µm (C) for zoomed z-sections. (D) Higher mTORC1 activity in valve LECs. Quantification of pS6 intensity in valve and lymphangion LECs. n = 3 embryos (35 collecting vessels). (E) Analysis of adult mesenteric lymphatic vasculature. (F and G) Detection of mTORC1 target pS6 in adult resting lymphatic vessels. Whole-mount staining of adult mesenteric lymphatic capillary (F) and collecting vessel (G) for PROX1 (green), pS6 (red), and PECAM1 (white). Confocal z-stack (left) and confocal thick z-section (right) are presented. Arrowhead, PROX1+pS6+ cell; arrows, pS6+ non-LEC. Scale bar, 50 µm for z-stack, 20 µm (F and G) for zoomed z-sections. (H) mTOR activity is detected in collecting vessel LECs. Quantification of pS6 intensity in valve and lymphangion LECs. n = 3 mice (15 collecting vessels). Data are shown as mean ± SD. A and E are created with help from BioRender. See also Fig. S3 F.
mTORC1 signaling is active in both growing and adult lymphatic valves. (A) Analysis of E18.5 embryo mesenteric lymphatic vasculature. (B and C) Detection of mTORC1 target pS6 in embryonic lymphatic growing vessels. Whole-mount staining of E18.5 mesenteric lymphatic capillaries (B) and collecting vessels (C) for PROX1 (green), pS6 (red), and PECAM1 (white). Confocal z-stack (left) and confocal thick z-section (right) are presented. Arrowhead, PROX1+pS6+ cell. Scale bar, 50 µm for z-stack, 10 µm (B) or 20 µm (C) for zoomed z-sections. (D) Higher mTORC1 activity in valve LECs. Quantification of pS6 intensity in valve and lymphangion LECs. n = 3 embryos (35 collecting vessels). (E) Analysis of adult mesenteric lymphatic vasculature. (F and G) Detection of mTORC1 target pS6 in adult resting lymphatic vessels. Whole-mount staining of adult mesenteric lymphatic capillary (F) and collecting vessel (G) for PROX1 (green), pS6 (red), and PECAM1 (white). Confocal z-stack (left) and confocal thick z-section (right) are presented. Arrowhead, PROX1+pS6+ cell; arrows, pS6+ non-LEC. Scale bar, 50 µm for z-stack, 20 µm (F and G) for zoomed z-sections. (H) mTOR activity is detected in collecting vessel LECs. Quantification of pS6 intensity in valve and lymphangion LECs. n = 3 mice (15 collecting vessels). Data are shown as mean ± SD. A and E are created with help from BioRender. See also Fig. S3 F.
Lymphatic mTORC1 signaling induces LEC proliferation, PROX1 expression, FN-EIIIA deposition, and valve formation
As in vivo ablation of proliferating LECs in Esco2lecKO mice prevented formation of lymphatic valves (Fig. 4), we next tested whether enforced mTORC1 signaling is sufficient to promote endothelial proliferation. The heterodimeric TSC1/2 protein complex negatively regulates mTORC1 signaling by exerting its GTPase-activating activity on RHEB, which is a direct activator of mTORC1 (Huang and Manning, 2008; Liu and Sabatini, 2020). Therefore, loss of TSC1 is a strong activator of mTORC1 signaling. To test the role of mTORC1 signaling in vivo we generated Tsc1/Flt4-CreERT2 (Tsc1lecKO) mice, in which administration of tamoxifen inactivates Tsc1 in VEGFR-3–expressing LECs (Fig. 6 A). We activated mTORC1 at early postnatal stages (Fig. 6 B) and Tsc1 ablation was validated by quantitative RT-PCR (RT-qPCR) in sorted mesenteric LECs from wild-type and Tsc1lecKO animals (Fig. S4, A and B). Staining of postnatal P10 mouse mesenteries for pS6 confirmed efficient activation of mTORC1 pathway in collecting lymphatic vessels of Tsc1lecKO mice (Fig. 6 C). We observed a heterogenous pattern of pS6 induction in Tsc1-deficient collecting lymphatics; in some vessels all LECs were pS6+ (Fig. 6 C, left), while other vessels displayed pS6 exclusively in the valve region (Fig. 6 C, right), which may reflect the degree of collecting vessel maturity. Although further longitudinal analyses are necessary, we propose that valve region LECs are primed for mTORC1 activation and poised to respond to loss of Tsc1 or activation of the mTORC1 pathway.

mTORC1 activation promotes formation of lymphatic valves . (A) Tsc1 lecKO mouse model. (B) Experimental setup and tamoxifen administration schedule. (C) Whole-mount staining of mesenteric lymphatic vessels of control or Tsc1lecKO mice for PROX1 (green), pS6 (red), PECAM1 (white). Blue arrowhead, valve region with increased pS6. Scale bar, 50 µm. (D) Increased LEC proliferation in Tsc1lecKO mice. Whole-mount staining of mesenteric lymphatic vessels of post-natal P9 control or Tsc1lecKO mice for PROX1 (green) and EdU (red, masked to Prox1 staining). Scale bar, 40 µm. (E) Quantification of proliferating PROX1+/EdU+ LECs in control (n = 2) and Tsc1lecKO (n = 2) mice. (F) Increased number of lymphatic valves in Tsc1lecKO mice. Whole-mount staining of P10 mesenteric lymphatic vessels for PROX1 (black). Arrowhead, valve site. Scale bar, 100 µm. (G) Quantification of lymphatic valves per vessel length in control (n = 6) or Tsc1lecKO (n = 5) mice. Two-tailed unpaired Welch’s t test, P = 0.0404. (H) mTORC1 activation promotes FN-EIIIA deposition in lymphatic valves. Whole-mount staining of mesenteric lymphatic vessels of P10 control or Tsc1lecKO mice for PROX1 (green), FNEIIIA (red), and PECAM1 (white). Scale bar, 50 µm. (I) Quantification of FN-EIIIA staining in control (n = 6) or Tsc1lecKO (n = 5) mice. Two-tailed unpaired Welch’s t test, P = 0.0671. Data are shown as mean ± SD. *P < 0.05. A and B are created with help from BioRender. See also Fig. S4, A and B.
mTORC1 activation promotes formation of lymphatic valves . (A) Tsc1 lecKO mouse model. (B) Experimental setup and tamoxifen administration schedule. (C) Whole-mount staining of mesenteric lymphatic vessels of control or Tsc1lecKO mice for PROX1 (green), pS6 (red), PECAM1 (white). Blue arrowhead, valve region with increased pS6. Scale bar, 50 µm. (D) Increased LEC proliferation in Tsc1lecKO mice. Whole-mount staining of mesenteric lymphatic vessels of post-natal P9 control or Tsc1lecKO mice for PROX1 (green) and EdU (red, masked to Prox1 staining). Scale bar, 40 µm. (E) Quantification of proliferating PROX1+/EdU+ LECs in control (n = 2) and Tsc1lecKO (n = 2) mice. (F) Increased number of lymphatic valves in Tsc1lecKO mice. Whole-mount staining of P10 mesenteric lymphatic vessels for PROX1 (black). Arrowhead, valve site. Scale bar, 100 µm. (G) Quantification of lymphatic valves per vessel length in control (n = 6) or Tsc1lecKO (n = 5) mice. Two-tailed unpaired Welch’s t test, P = 0.0404. (H) mTORC1 activation promotes FN-EIIIA deposition in lymphatic valves. Whole-mount staining of mesenteric lymphatic vessels of P10 control or Tsc1lecKO mice for PROX1 (green), FNEIIIA (red), and PECAM1 (white). Scale bar, 50 µm. (I) Quantification of FN-EIIIA staining in control (n = 6) or Tsc1lecKO (n = 5) mice. Two-tailed unpaired Welch’s t test, P = 0.0671. Data are shown as mean ± SD. *P < 0.05. A and B are created with help from BioRender. See also Fig. S4, A and B.
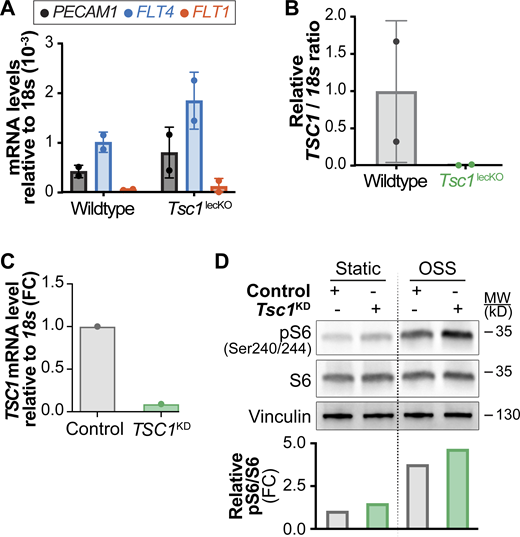
Ablation of Tsc1 in vivo or in vitro promotes mTORC1 signaling. A and B related to Fig. 6; C and D related to Fig. 9. (A) CD45neg CD31+ Gp38+ LECs were sorted from mesenteries of 3-wk-old wild-type or Tsc1lecKO mice treated at birth with tamoxifen according to Fig. 6 B. Identity of sorted cells was validated by RT-qPCR for the indicated markers. (B) Efficient deletion of Tsc1 in Tsc1lecKO animals. Analysis of Tsc1 mRNA levels by RT-qPCR in sorted LECs shown in A. (C) Efficient reduction of TSC1 in LECs transfected with TSC1 siRNA shown by RT-qPCR analysis. (D) Knockdown of TSC1 in LECs promotes mTORC1 signaling. Western blot analysis of LECs under static and OSS conditions for the indicated proteins. Quantification of pS6 activation is shown in the graph below the Western blot. Data normalized to total S6 and STAT control conditions. FC, fold change. Source data are available for this figure: SourceData FS4.
Ablation of Tsc1 in vivo or in vitro promotes mTORC1 signaling. A and B related to Fig. 6; C and D related to Fig. 9. (A) CD45neg CD31+ Gp38+ LECs were sorted from mesenteries of 3-wk-old wild-type or Tsc1lecKO mice treated at birth with tamoxifen according to Fig. 6 B. Identity of sorted cells was validated by RT-qPCR for the indicated markers. (B) Efficient deletion of Tsc1 in Tsc1lecKO animals. Analysis of Tsc1 mRNA levels by RT-qPCR in sorted LECs shown in A. (C) Efficient reduction of TSC1 in LECs transfected with TSC1 siRNA shown by RT-qPCR analysis. (D) Knockdown of TSC1 in LECs promotes mTORC1 signaling. Western blot analysis of LECs under static and OSS conditions for the indicated proteins. Quantification of pS6 activation is shown in the graph below the Western blot. Data normalized to total S6 and STAT control conditions. FC, fold change. Source data are available for this figure: SourceData FS4.
Increased lymphatic mTORC1 signaling promoted proliferation of collecting vessel LECs (Fig. 6, D and E) and increased expression of PROX1 in lymphangion cells (Fig. 6, C and D). These results agree with previous observations of decreased LEC proliferation and PROX1 expression upon rapamycin treatment in a mouse model of lymphangiectasia (Baluk et al., 2017). Most importantly, we observed the formation of supernumerary valves in collecting vessels upon mTORC1 signaling induction (Fig. 6, F and G). Formation of specialized valve matrix is a pre-requisite for lymphatic valve development (Bazigou et al., 2009; Danussi et al., 2013); we therefore analyzed deposition of the alternatively spliced form of fibronectin, FN-EIIIA, required for lymphatic valve leaflet elongation during development (Bazigou et al., 2009). We observed increased accumulation of FN-EIIIA in the valves of Tsc1lecKO mice (Fig. 6, H and I), suggesting that FN-EIIIA production is mTORC1 dependent. Collectively, these data indicate that, in addition to its well-established role in lymphangiogenesis (Baluk et al., 2017), mTORC1 signaling contributes to lymphatic valve development and maintenance.
Flow shear stress activates mTOR signaling in LECs
We next sought to investigate how mTORC1 signaling is activated in the lymphatic collecting vessel endothelium. Collecting vessel LECs are subjected to varying amplitudes and patterns of fluid shear stress (Sabine et al., 2016; Zawieja, 2009; Zolla et al., 2015). Unidirectional, laminar shear stress, present in lymphangion, promotes LEC proliferation in vitro (Choi et al., 2017b; Choi et al., 2017a; Sabine et al., 2015), while reversing flow, modeled by oscillatory shear stress in vitro (OSS), enforces LEC quiescence (Sabine et al., 2015). In the valve, leaflet LECs are subjected to high fluid shear stress, whereas valve sinus LECs experience low shear stress (Fig. 7 A and Pujari et al., 2020). Based on these results, we compared the impact of low (0.7 dyn/cm2) and high (4 dyn/cm2) OSS on mTORC1 activation in cultured LECs (Fig. 7 B). We observed similar levels of activation of mTORC1 signaling in both conditions (Fig. 7, C and D), suggesting that mTORC1 could be equally active in different valve compartments. We next analyzed the impact of high or low OSS on LEC proliferation by staining for KI67. As observed previously (Sabine et al., 2015), high OSS suppressed LEC proliferation, but surprisingly, low OSS strongly promoted proliferation of LECs (Fig. 7, E and F).

Flow shear stress activates mTORC1 signaling . (A) Scheme of flow patterns in lymphatic valves. (B) Workflow of shear stress experiments. (C) Both low and high shear stress activate mTORC1. Staining of LECs subjected to 48-h low (0.7 dyn/cm2) or high (4 dyn/cm2) flow shear stress for pS6 (green) and VE-cadherin (white). Scale bar, 50 µm. (D) Quantification of pS6 signal intensity. Data normalized to static conditions. n = 3 independent flow experiments (five images and 300–450 cells per condition for each experiment). Two-tailed ratio paired t test, P = 0.0088 for 0.7 dyn/cm2 and 0.0132 for 4 dyn/cm2). (E) Low shear stress promotes, and high shear stress inhibits LEC proliferation. Staining of LECs subjected to 48-h low (0.7 dyn/cm2) or high (4 dyn/cm2) flow shear stress for KI67 (magenta) and β-catenin (white). Scale bar, 50 µm. (F) Quantification of the percentage of KI67+ LECs. Data normalized to static conditions. n = 3 independent flow experiments (five images and 300–450 cells per condition for each experiment). Two-tailed ratio paired t test, P = 0.0474 for 0.7 dyn/cm2 and 0.0607 for 4 dyn/cm2. (G) Shear stress activates mTORC1 in the absence of glucose, glutamine, and growth factors. LECs were subjected to 1-h flow shear stress (4 dyn/cm2) or kept in static conditions in medium without glutamine, amino acids, and growth factors. Staining for pS6 (green) and VE-cadherin (white). Scale bar, 50 µm. (H) Quantification of pS6 signal intensity. Data normalized to static conditions. n = 3 independent flow experiments (four images and 350–450 cells per condition for each experiment). Two-tailed ratio paired t test, P = 0.0089. Data are shown as mean ± SD. *P < 0.05, **P < 0.01. B is created with help from BioRender. See also Fig. S5, A and B.
Flow shear stress activates mTORC1 signaling . (A) Scheme of flow patterns in lymphatic valves. (B) Workflow of shear stress experiments. (C) Both low and high shear stress activate mTORC1. Staining of LECs subjected to 48-h low (0.7 dyn/cm2) or high (4 dyn/cm2) flow shear stress for pS6 (green) and VE-cadherin (white). Scale bar, 50 µm. (D) Quantification of pS6 signal intensity. Data normalized to static conditions. n = 3 independent flow experiments (five images and 300–450 cells per condition for each experiment). Two-tailed ratio paired t test, P = 0.0088 for 0.7 dyn/cm2 and 0.0132 for 4 dyn/cm2). (E) Low shear stress promotes, and high shear stress inhibits LEC proliferation. Staining of LECs subjected to 48-h low (0.7 dyn/cm2) or high (4 dyn/cm2) flow shear stress for KI67 (magenta) and β-catenin (white). Scale bar, 50 µm. (F) Quantification of the percentage of KI67+ LECs. Data normalized to static conditions. n = 3 independent flow experiments (five images and 300–450 cells per condition for each experiment). Two-tailed ratio paired t test, P = 0.0474 for 0.7 dyn/cm2 and 0.0607 for 4 dyn/cm2. (G) Shear stress activates mTORC1 in the absence of glucose, glutamine, and growth factors. LECs were subjected to 1-h flow shear stress (4 dyn/cm2) or kept in static conditions in medium without glutamine, amino acids, and growth factors. Staining for pS6 (green) and VE-cadherin (white). Scale bar, 50 µm. (H) Quantification of pS6 signal intensity. Data normalized to static conditions. n = 3 independent flow experiments (four images and 350–450 cells per condition for each experiment). Two-tailed ratio paired t test, P = 0.0089. Data are shown as mean ± SD. *P < 0.05, **P < 0.01. B is created with help from BioRender. See also Fig. S5, A and B.
Among the major stimuli for mTORC1 activation are nutrients (Kim et al., 2008; Sancak et al., 2008) and growth factors (Efeyan and Sabatini, 2013; Inoki et al., 2002; Manning et al., 2002). Interestingly, mTORC1 signaling was still activated by OSS when LECs were cultured in a basal medium devoid of growth factors, glucose and glutamine, while it was barely detectable under static conditions (Fig. 7, G and H). The magnitude of short-term mTORC1 activation by shear stress after starvation was comparable to re-feeding with glucose, glutamine, and leucine (Fig. 7, G and H). Moreover, the extent of long-term mTORC1 activation by flow was also similar to that of starved LECs acutely re-fed with nutrients (Fig. S5 A) or treated with the lymphangiogenic growth factor VEGF-C (Fig. S5 B). Thus, our results identify mTORC1 as a mechanosensitive signaling pathway in LECs that is independent of nutrient/growth factor signaling.
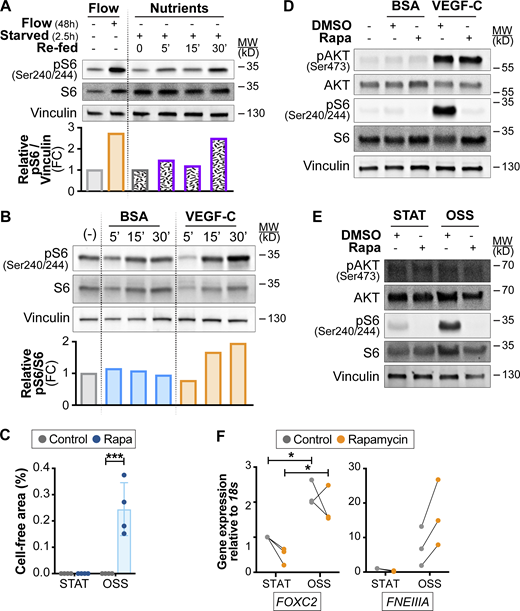
Activation of mTOR by nutrients and growth factor signaling in LECs. A and B related to Fig. 7, G and H; C related to Fig. 8, A and D; F related to Fig. 8, G and H. (A) Activation of mTORC1 by flow or by nutrients. Western blot analysis of: (left) LECs cultured under 4 dyn/cm2 OSS or kept under static conditions for 48 h; (right) LECs starved and treated with glucose/glutamine/leucine cocktail for the indicated times (0–30 min). Quantification of pS6 activation is shown in the graph below the Western blot. Data normalized to total Vinculin and static (left) or saline-treated control conditions (right). (B) Induction of mTORC1 signaling by VEGF-C growth factor. Western blot analysis of LECs starved and treated with VEGFC for the indicated times (0–30 min). Quantification of pS6 activation is shown in the graph below the Western blot. Data normalized to total S6 and BSA-treated control conditions. (C) Quantification of cell-free area in control and rapamycin-treated LECs subjected to 4 dyn/cm2 OSS (related to Fig. 8 D). n = 4 independent flow experiments. Two-way ANOVA, P = 0.0028 for OSS effects under rapamycin conditions. (D) Rapamycin inhibits mTORC1, but not mTORC2 activation upon acute stimulation of LECs with VEGF-C. Western blot analysis of LECs starved and treated with VEGF-C for 30 min in the presence or absence of rapamycin to assess its effects on S6 and AKT (Ser 473) phosphorylation. (E) mTORC2 is not activated by flow shear stress on the long term. Western blot analysis of LECs cultured under 4 dyn/cm2 OSS or kept under static conditions for 48 h in the presence or absence of rapamycin to assess its effects on S6 and AKT (Ser 473) phosphorylation. (F)FNEIIIA mRNA expression is not affected by rapamycin treatment. RT-qPCR for the indicated transcripts. Induction of FOXC2 shows the 48-h effect of high-flow shear stress. n = 3 independent flow experiments. Two-way ANOVA, P = 0.0207 for OSS effects on FOXC2 expression. Data are shown as mean ± SD. *P < 0.05, ***P < 0.005. Source data are available for this figure: SourceData FS5.
Activation of mTOR by nutrients and growth factor signaling in LECs. A and B related to Fig. 7, G and H; C related to Fig. 8, A and D; F related to Fig. 8, G and H. (A) Activation of mTORC1 by flow or by nutrients. Western blot analysis of: (left) LECs cultured under 4 dyn/cm2 OSS or kept under static conditions for 48 h; (right) LECs starved and treated with glucose/glutamine/leucine cocktail for the indicated times (0–30 min). Quantification of pS6 activation is shown in the graph below the Western blot. Data normalized to total Vinculin and static (left) or saline-treated control conditions (right). (B) Induction of mTORC1 signaling by VEGF-C growth factor. Western blot analysis of LECs starved and treated with VEGFC for the indicated times (0–30 min). Quantification of pS6 activation is shown in the graph below the Western blot. Data normalized to total S6 and BSA-treated control conditions. (C) Quantification of cell-free area in control and rapamycin-treated LECs subjected to 4 dyn/cm2 OSS (related to Fig. 8 D). n = 4 independent flow experiments. Two-way ANOVA, P = 0.0028 for OSS effects under rapamycin conditions. (D) Rapamycin inhibits mTORC1, but not mTORC2 activation upon acute stimulation of LECs with VEGF-C. Western blot analysis of LECs starved and treated with VEGF-C for 30 min in the presence or absence of rapamycin to assess its effects on S6 and AKT (Ser 473) phosphorylation. (E) mTORC2 is not activated by flow shear stress on the long term. Western blot analysis of LECs cultured under 4 dyn/cm2 OSS or kept under static conditions for 48 h in the presence or absence of rapamycin to assess its effects on S6 and AKT (Ser 473) phosphorylation. (F)FNEIIIA mRNA expression is not affected by rapamycin treatment. RT-qPCR for the indicated transcripts. Induction of FOXC2 shows the 48-h effect of high-flow shear stress. n = 3 independent flow experiments. Two-way ANOVA, P = 0.0207 for OSS effects on FOXC2 expression. Data are shown as mean ± SD. *P < 0.05, ***P < 0.005. Source data are available for this figure: SourceData FS5.
Shear stress amplitude dictates mTORC1 signaling responses between LEC proliferation and protein production
To decipher the functional significance of mechanosensitive mTORC1 activation, we treated LECs subjected to low or high OSS with rapamycin and assessed cell proliferation. Rapamycin treatment prevented LEC proliferation in low OSS conditions, confirming the key role for mTORC1 signaling (Fig. 8, A and B). Surprisingly, during high OSS, when LECs become quiescent, rapamycin treatment increased the number of inter-endothelial gaps in the cell monolayer (Fig. 8 A). We also found decreased intensity of VE-cadherin staining in LEC junctions (Fig. 8, A and C), reduced junctional β-catenin (Fig. 8 D), and cell loss (Fig. 8 D and Fig. S5 C), indicating diminished cell–cell or cell–matrix adhesion.

Mechanosensitive mTORC1 signaling controls cell proliferation and protein synthesis. (A) mTORC1 is required for proliferation and junctional integrity. LECs were subjected to 0.7 dyn/cm2 or 4 dyn/cm2 flow shear stress for 48 h and treated with 10 nM rapamycin (Rapa) or DMSO (Control). Staining for pS6 (green), KI67 (magenta), and VE-cadherin (white). Arrowhead, cell-free area in LEC monolayer. Scale bar, 50 µm. (B) Quantification of the percentage of KI67+ LECs. Data normalized to control and static conditions. n = 3 independent flow experiments (five images and 500–800 cells per condition for each experiment). Two-way ANOVA, P = 0.0448 for 0.7 dyn/cm2 OSS effects under control conditions, 0.0386 for rapamycin effects under 0.7 dyn/cm2 OSS conditions, and 0.0256 for 4 dyn/cm2 OSS effects under control conditions. (C) Quantification of VE-cadherin intensity from A. Data normalized to control and static conditions. n = 3 independent flow experiments (five images and 500–800 cells per condition for each experiment). Two-way ANOVA, P = 0.0646 for rapamycin effects under 0.7 dyn/cm2 OSS, and 0.0445 for rapamycin effects under 4 dyn/cm2 OSS. (D) mTORC1 is required for junctional integrity. LECs were subjected to 0.7 dyn/cm2 or 4 dyn/cm2 flow shear stress for 48 h and treated with 10 nM rapamycin or DMSO. Staining for β-catenin (black). Arrowhead, cell-free area in LEC monolayer; arrow, weak cell–cell junction. Scale bar, 50 µm. (E) Shear stress promotes protein synthesis in a rapamycin-sensitive manner. Protein synthesis is detected by OPP labeling. Staining of control or rapamycin-treated LECs subjected to 4 dyn/cm2 OSS for 48 h for OPP (red) and β-catenin (white). Data normalized to control and static conditions. Arrow, accumulation of OPP signal; arrowhead, loss of monolayer integrity. Scale bar, 50 µm. (F) Quantification of cytoplasmic OPP intensity. Data normalized to control and static conditions. n = 3 independent flow experiments (2 in the presence of rapamycin; five images and 80–120 cells per condition for each experiment). Two-way ANOVA, P = 0.0298 for OSS effects under control conditions, and 0.0196 for rapamycin effects under OSS. (G) Increased FN-EIIIA matrix deposition by flow shear stress in a rapamycin-sensitive manner. Staining of decellularized ECM from control or rapamycin-treated LECs subjected to 4 dyn/cm2 OSS for 72 h. FN-EIIIA (red), DNA (blue, decellularization control). Scale bar, 50 µm. (H) Quantification of matrix FN-EIIIA signal intensity. Data normalized to control and static conditions. n = 4 independent flow experiments (five images per condition for each experiment). Two-way ANOVA, P = 0.0053 for OSS effects under control conditions, and 0.0494 for rapamycin effects under OSS. Data are shown as mean ± SD. *P < 0.05; **P < 0.01. See also Fig. S5, C and F.
Mechanosensitive mTORC1 signaling controls cell proliferation and protein synthesis. (A) mTORC1 is required for proliferation and junctional integrity. LECs were subjected to 0.7 dyn/cm2 or 4 dyn/cm2 flow shear stress for 48 h and treated with 10 nM rapamycin (Rapa) or DMSO (Control). Staining for pS6 (green), KI67 (magenta), and VE-cadherin (white). Arrowhead, cell-free area in LEC monolayer. Scale bar, 50 µm. (B) Quantification of the percentage of KI67+ LECs. Data normalized to control and static conditions. n = 3 independent flow experiments (five images and 500–800 cells per condition for each experiment). Two-way ANOVA, P = 0.0448 for 0.7 dyn/cm2 OSS effects under control conditions, 0.0386 for rapamycin effects under 0.7 dyn/cm2 OSS conditions, and 0.0256 for 4 dyn/cm2 OSS effects under control conditions. (C) Quantification of VE-cadherin intensity from A. Data normalized to control and static conditions. n = 3 independent flow experiments (five images and 500–800 cells per condition for each experiment). Two-way ANOVA, P = 0.0646 for rapamycin effects under 0.7 dyn/cm2 OSS, and 0.0445 for rapamycin effects under 4 dyn/cm2 OSS. (D) mTORC1 is required for junctional integrity. LECs were subjected to 0.7 dyn/cm2 or 4 dyn/cm2 flow shear stress for 48 h and treated with 10 nM rapamycin or DMSO. Staining for β-catenin (black). Arrowhead, cell-free area in LEC monolayer; arrow, weak cell–cell junction. Scale bar, 50 µm. (E) Shear stress promotes protein synthesis in a rapamycin-sensitive manner. Protein synthesis is detected by OPP labeling. Staining of control or rapamycin-treated LECs subjected to 4 dyn/cm2 OSS for 48 h for OPP (red) and β-catenin (white). Data normalized to control and static conditions. Arrow, accumulation of OPP signal; arrowhead, loss of monolayer integrity. Scale bar, 50 µm. (F) Quantification of cytoplasmic OPP intensity. Data normalized to control and static conditions. n = 3 independent flow experiments (2 in the presence of rapamycin; five images and 80–120 cells per condition for each experiment). Two-way ANOVA, P = 0.0298 for OSS effects under control conditions, and 0.0196 for rapamycin effects under OSS. (G) Increased FN-EIIIA matrix deposition by flow shear stress in a rapamycin-sensitive manner. Staining of decellularized ECM from control or rapamycin-treated LECs subjected to 4 dyn/cm2 OSS for 72 h. FN-EIIIA (red), DNA (blue, decellularization control). Scale bar, 50 µm. (H) Quantification of matrix FN-EIIIA signal intensity. Data normalized to control and static conditions. n = 4 independent flow experiments (five images per condition for each experiment). Two-way ANOVA, P = 0.0053 for OSS effects under control conditions, and 0.0494 for rapamycin effects under OSS. Data are shown as mean ± SD. *P < 0.05; **P < 0.01. See also Fig. S5, C and F.
Rapamycin treatment inhibits mTORC2 in some settings (Sarbassov et al., 2005; Schreiber et al., 2015), therefore we analyzed AKT phosphorylation at Ser473 as direct mTORC2 readout (Sarbassov et al., 2005) in LECs subjected to shear stress or treated with VEGF-C as positive control. As expected, VEGF-C stimulation induced phosphorylation of both S6 and Ser473 of AKT. Rapamycin treatment abolished activation of mTORC1 by VEGF-C while it had no effect on mTORC2 activity (Fig. S5 D). In contrast, we did not observe striking changes in activation levels of mTORC2-dependent phospho-AKT (Ser473) in LECs cultured under OSS, while there was pronounced activation of mTORC1, which was abolished by rapamycin (Fig. S5 E). Low level mTORC2 activation was mostly unaffected by rapamycin as shown by unaltered levels of phospho-AKT (Ser473; Fig. S5 E). In all, these results show that the proliferative effect of low flow on LECs in culture is mediated mostly by mTORC1.
Regulation of protein synthesis is another important function of mTORC1, and protein synthesis is uncoupled from cell proliferation and growth in many specialized cell types (Blanco et al., 2016; Laplante and Sabatini, 2013). We assessed protein synthesis in LECs subjected to high OSS using an O-propargyl-puromycin (OPP) incorporation assay. OPP is a puromycin analog and incorporates into nascent polypeptide chains during translation. We found that high OSS significantly increased total protein synthesis in quiescent LECs and this process was mTORC1 dependent, as determined by comparing OPP incorporation in LECs in the presence or absence of rapamycin (Fig. 8, E and F).
As we observed increased deposition of FN-EIIIA protein in the valves of Tsc1lecKO mice (Fig. 6, H and I), we examined whether mTORC1 inhibition affects FN-EIIIA production in vitro. We found that high OSS promoted FN-EIIIA deposition in the LEC extracellular matrix, and inhibition of mTORC1 signaling with rapamycin reduced it (Fig. 8, G and H). There was a tendency towards increased FN-EIIIA transcription by OSS, which was not inhibited by rapamycin treatment, indicating that mTORC1 signaling stimulates FN-EIIIA production on the level of protein synthesis (Fig. S5 F). In summary, our in vitro analyses indicate that mechanosensitive mTORC1 is important for cell proliferation under low shear stress conditions, while in quiescent LECs it is important for protein synthesis, in particular of the valve ECM component FN-EIIIA, and maintenance of the endothelial monolayer integrity.
mTORC1 activation under high-flow shear stress is sufficient to promote LEC proliferation
To test whether mTORC1 signaling induction overrides LEC quiescence imposed to LECs by high OSS, we transfected cultured LECs with siRNA targeting TSC1 and subjected them to high OSS. TSC1 knockdown further enhanced S6 phosphorylation by OSS (Fig. 9, A and B; and Fig. S4, C and D). Interestingly, TSC1-deficient LECs readily proliferated under high OSS conditions (Fig. 9, A and C), indicating that supra-physiological mTORC1 activation overcomes LEC quiescence dictated by high OSS.

Mechanosensitive mTORC1 signaling controls cell proliferation and protein synthesis. (A) mTORC1 activation overcomes the quiescent phenotype of LECs under high OSS. Control or TSC1KD LECs were subjected to 4 dyn/cm2 OSS for 48 h. Staining for pS6 (green), KI67 (magenta), and VE-cadherin (white). Scale bar, 50 µm. (B) Quantification of pS6 signal intensity. Data normalized to control and static conditions. n = 4 independent flow experiments (five images per condition for each experiment, 300–450 cells per condition). Two-way ANOVA, P = 0.0174 for OSS effects under control conditions, and 0.0296 for TSC1 knockdown effects under OSS. (C) Higher proliferation of LECs upon TSC1 knockdown. Quantification of the percentage of KI67+ LECs. n = 4 independent flow experiments (five images per condition for each experiment, 1,200–2,000 cells per condition). Two-way ANOVA, P = 0.0306 for TSC1 knockdown effects under OSS. Data are shown as mean ± SD. *P < 0.05. See also Fig. S4, C and D.
Mechanosensitive mTORC1 signaling controls cell proliferation and protein synthesis. (A) mTORC1 activation overcomes the quiescent phenotype of LECs under high OSS. Control or TSC1KD LECs were subjected to 4 dyn/cm2 OSS for 48 h. Staining for pS6 (green), KI67 (magenta), and VE-cadherin (white). Scale bar, 50 µm. (B) Quantification of pS6 signal intensity. Data normalized to control and static conditions. n = 4 independent flow experiments (five images per condition for each experiment, 300–450 cells per condition). Two-way ANOVA, P = 0.0174 for OSS effects under control conditions, and 0.0296 for TSC1 knockdown effects under OSS. (C) Higher proliferation of LECs upon TSC1 knockdown. Quantification of the percentage of KI67+ LECs. n = 4 independent flow experiments (five images per condition for each experiment, 1,200–2,000 cells per condition). Two-way ANOVA, P = 0.0306 for TSC1 knockdown effects under OSS. Data are shown as mean ± SD. *P < 0.05. See also Fig. S4, C and D.
Based on the above observations, we propose an “escalator” model of LEC renewal in collecting lymphatic vessels. Valve leaflet LECs are subjected to continuous high mechanical stress and are damaged and eliminated more frequently in comparison to other collecting vessel LECs. Valve leaflet LECs do not proliferate; instead, in the low-flow valve sinus zone, LECs divide and may thereby promote sequential, collective migration toward the damaged valve leaflet to maintain the valve architecture (Fig. 10). Mechanistically, mTORC1 signaling is activated by flow shear stress in lymphatic collecting vessels and valves, promoting cell proliferation in low-flow valve sinus and cell adhesion in high-flow valve leaflet (Fig. 10). This model of valve LEC renewal suggests proliferation of valve sinus LECs and mTORC1 signaling maintains valve integrity throughout adult life.

Escalator model of valve LEC renewal. Left side: Valve homeostasis. Lymphatic collecting vessel is organized as sequential units, the lymphangions, separated by valves that are composed of two main compartments, sinuses and leaflets. Valve leaflets open and close to allow lymph flow and prevent backflow, respectively. Lymph flow through the valve generates shear stress of different types and magnitudes; laminar shear stress at the center of the valve is generated by unidirectional pulsatile lymph flow through the leaflets; in valve sinuses recirculation of lymph, which is caused by shape of the leaflets and is amplified by valve closure, generates OSS that is of low intensity at the bottom of valvular sinuses (Pujari et al., 2020) and of higher intensity at the leaflet. Right side: Escalator model of valve LEC renewal. Half of the valve area is shown at different stages, which are described on the figure under each panel. The proposed implication of mTORC1 downstream of shear stress is also indicated.
Escalator model of valve LEC renewal. Left side: Valve homeostasis. Lymphatic collecting vessel is organized as sequential units, the lymphangions, separated by valves that are composed of two main compartments, sinuses and leaflets. Valve leaflets open and close to allow lymph flow and prevent backflow, respectively. Lymph flow through the valve generates shear stress of different types and magnitudes; laminar shear stress at the center of the valve is generated by unidirectional pulsatile lymph flow through the leaflets; in valve sinuses recirculation of lymph, which is caused by shape of the leaflets and is amplified by valve closure, generates OSS that is of low intensity at the bottom of valvular sinuses (Pujari et al., 2020) and of higher intensity at the leaflet. Right side: Escalator model of valve LEC renewal. Half of the valve area is shown at different stages, which are described on the figure under each panel. The proposed implication of mTORC1 downstream of shear stress is also indicated.
Discussion
Aging is associated with blood and lymphatic vessel rarefication in many tissues, and activation of VEGF signaling can restore age-dependent loss of organ function (González-Loyola and Petrova, 2021; Grunewald et al., 2021). These observations underscore the importance of better understanding of mechanisms underpinning homeostatic maintenance and repair of the vasculature. Here, we investigated this process in postnatal and adult lymphatic vessels. Our data provide insights into the dynamics of lymphatic endothelium maintenance and repair and identify a link between biomechanical forces and mTORC1 signaling in this process. Differently from actively growing embryonic or early postnatal lymphatics, adult LEC renewal was mainly observed in collecting vessels. LECs proliferate in the low-flow area of valve sinuses, where they fulfill a “progenitor-like” function for replacing short-lived endothelial cells in the high-flow area of valve leaflets. We further identified mechanoresponsive mTORC1 signaling as a pathway required both for the proliferation and durability of valve sinus and leaflet LECs, respectively.
Ectopic mTORC1 signaling activation in lymphatic vessels was sufficient to promote the formation of supernumerary valves. The phenotype of Tsc1lecKO mice is thus reminiscent of that of mice with lymphatic endothelial-specific loss of Foxo1, which also form additional lymphatic valves in postnatal and adult lymphatic vessels (Niimi et al., 2021; Scallan et al., 2021). Along the same lines, OSS was shown to activate AKT signaling in vitro, which in turn prevented FOXO1 nuclear translocation and FOXO1-driven transcription (Niimi et al., 2021; Scallan et al., 2021). Shear stress activates mTORC1 as soon as 5 min after flow onset; therefore, we believe that the initial response of LECs to flow via mTORC1 is unlikely to be mediated by transcriptional changes and by FOXO1 down-regulation. As FOXO1 represses mTORC1 activity in blood endothelial cells (Chen et al., 2010), further mechanistic studies in LECs are warranted. Taken together with previous results (Niimi et al., 2021; Scallan et al., 2021), our data delineate a mechanosensitive AKT–FOXO1–mTORC1 pathway as a major regulator of postnatal lymphatic valve maintenance and repair, acting on one hand to initiate the proliferation of sinus LECs and on the other hand to sustain protein synthesis and ECM production by cells of the valve leaflet.
Our observations raise further questions, such as the exact identity of the cell surface mechanosensory complex triggering mTORC1 activation. VE-cadherin and PECAM1 are part of a general endothelial mechanosensing complex (Tzima et al., 2005). Loss of VE-cadherin impaired AKT activation in LECs subjected to shear stress and inhibited valve development (Scallan et al., 2021). Similarly, lymphatic valve morphogenesis was compromised in Pecam1−/− mice (Wang et al., 2016). However, it remains possible that in addition to VE-cadherin and PECAM1, which contribute to the overall shear stress sensing by LECs, other cell surface molecules are more directly implicated in flow-dependent activation of PI3K/AKT/mTOR pathway, most notably receptor tyrosine kinases and integrins. Using blocking antibodies, we have ruled out the ligand-dependent functions of VEGFR-2 and VEGFR-3 in maintenance of lymphatic valves. Previous studies have shown that shear stress promotes ligand-independent phosphorylation of VEGFR-2 and VEGFR-3 signaling and activation of AKT (Coon et al., 2015). However, lymphatic valves were not affected following postnatal deletion of Vegfr2 (Kdr) or Vegfr3 (Flt4) (Zarkada et al., 2015).
Another fascinating unanswered question is how loss of valve leaflet LECs is communicated to valve sinus LECs for induction of proliferation. Studies in endothelial cell monolayers demonstrated that mechanical perturbations of VE-cadherin complexes resulted in disruption of inter-endothelial contacts distant from the applied force (Barry et al., 2015). Furthermore, gap junction protein CX37 (GJA4) is necessary for collective migration of valve-forming endothelial cells (Sabine et al., 2012). The VE-cadherin– and CX37-mediated long-distance intercellular communication may therefore be part of a mechanism activating migration of sinus LECs to restore integrity of the valve leaflet.
Our data also provide a possible link with long-standing clinical observations of increased incidence of lower leg lymphedema due to collecting vessel dysfunction in solid transplant recipients treated with rapamycin (Al-Otaibi et al., 2007; Baliu et al., 2014; Desai et al., 2009; Romagnoli et al., 2005). Given the likely absence of active lymphangiogenesis in the legs of adult patients, this suggests a function for mTORC1 in human quiescent lymphatic vessels, but at present the effects of rapamycin cannot be unequivocally attributed to valve degeneration. In the future, it will be interesting to investigate whether rapamycin-induced lymphedema is a result of lymph valve insufficiency as previously shown in lymphedema-distichiasis syndrome patients (Mellor et al., 2011; Petrova et al., 2004). In addition, as tuberous sclerosis patients harbor loss-of-function mutations in TSC2 and TSC1, it will be interesting to study whether in addition to the main effect of mutation on smooth muscle cells (Carsillo et al., 2000) such patients demonstrate lymphatic anomalies due to lymphatic endothelial defects.
Materials and methods
Animal models
Animal experiments were approved by the Animal Ethics Committee of Canton Vaud. Tsc1fl/fl (Tsc1tm1Djk; Uhlmann et al., 2002), Flt4-CreERT2 (Flt4tm2.1(cre/ERT2)Sgo; Stanczuk et al., 2015; Martinez-Corral et al., 2016), Mki67-CreERT2 (Mki67tm2.1(cre/ERT2)Cle; Basak et al., 2018), mTmG (Gt(ROSA)26Sortm4(ACTB- tdTomato,-EGFP)Luo; Muzumdar et al., 2007), Prox1-CreERT2 (Tg(Prox1-cre/ERT2)1Tmak; Bazigou et al., 2011), and Esco2fl/fl (Esco2tm1.1Ge; Whelan et al., 2012) mice were described previously.
For long-term EdU labeling experiments, 10–15-wk-old C57BL6/J mice were used. 200 μg/ml of EdU (Santa Cruz) was administered in drinking water with 5% sucrose for 2 wk. For washout experiments, EdU was administered for 2 wk and mice were sacrificed immediately after 0, 2, 4, or 8 wk after EdU administration. For short-term EdU labeling, mice were injected with EdU at 5 μg/g 3 h prior to sacrifice.
DC101 and mF4-31C1 blocking antibodies, which block ligand interaction with VEGFR-2 (R2) and VEGFR-3 (R3), respectively, were described previously (Prewett et al., 1999; Pytowski et al., 2005). Anti-R2, anti-R3, and control rat anti-HRP IgGs (BioXCell) were injected i.p. at 40 mg/kg of mouse every 3 d for 2 wk.
Mouse tissue collection, staining procedures, and image acquisition
Prior to sacrifice, adult mice were anesthetized and perfused intracardially with 4% PFA. Tissues were fixed overnight at 4°C in 4% PFA, washed with ice-cold PBS and stored in PBS with 0.1% sodium azide.
For whole-mount staining, mesenteries were prepared as described (Sabine et al., 2018) and small intestines were prepared as described (Bernier-Latmani and Petrova, 2016). Briefly, tissues were blocked with PBS, 5% donkey serum, 0.5% Triton X-100, and incubated with primary antibodies for 2 d in blocking buffer at 4°C. After several washes with 0.5% Triton X-100 in PBS, tissues were incubated with secondary antibodies for 2 d at 4°C, extensively washed and cleared with Histodenz (Sigma-Aldrich).
Deparaffinized 8-µm-thick tissue sections were subjected to heat-induced epitope retrieval (High/Low pH Retrieval solution, DAKO). Sections were stained with primary antibodies overnight at 4°C, followed by secondary antibody incubation for 1 h at RT. For EdU detection Alexa Fluor 555-azide (Life Technologies) was used. Antibodies are listed in Table S1.
Imaging was performed using confocal Zeiss LSM800 or Zeiss LSM880 microscopes. Additional details about the microscopes are provided in Table S1. Images were acquired using Zen (Zeiss) software, processed using Imaris (Bitplane, v9.5) and Photoshop CC (Adobe, v2014 or v2020) softwares. Confocal images represent maximum intensity projection of Z-stacks of single tile scan images.
Flow cytometry analysis
Following removal of mesenteric lymph nodes mesenteries from three C57BL/6 mice were pooled in ice-cold Hank’s buffer containing 2% BSA and 1% penicillin-streptomycin. Tissues were cut into small pieces and digested in 8% Liberase TL (Sigma-Aldrich) solution for 1 h at 37°C with shaking at 150 rpm. Digestion was terminated with the addition of Hank’s buffer containing 2% BSA and 1% penicillin-streptomycin. Digest was filtered through 40-µm strainer and resulting single-cell suspension was centrifuged for 15 min at 1,200 rpm at 4°C. Pellet was treated with red blood cell lysis buffer (Invitrogen) for 5 min on ice. Cells were incubated with Zombie Violet dye (BioLegend) on ice for 30 min and stained with conjugated primary antibodies in FACS blocking buffer for 30 min on ice. For KI67 staining, cells were fixed with Cytofix/CytoPerm (Invitrogen) on ice for 20 min and incubated with KI67 antibody in permeabilization buffer overnight at 4°C on a rotating wheel. Cells were resuspended in FACS buffer and analyzed using LSRII flow cytometer (BD). Based on single-stained beads (BD) and fluorescence minus one controls, compensations were adjusted during analysis on a common acquisition matrix, which was generated automatically. The analysis was performed using FlowJo version 10.5.3 (FlowJo LLC). The gating strategy is shown in Fig. 1 H and Fig. S1 B.
Flow cytometry cell sorting
LECs were sorted from 3-wk-old mouse mesenteries after removal of the mesenteric lymph node, following staining with conjugated primary antibodies, as described previously (González-Loyola et al., 2021). Briefly, samples were digested for 30 min in digestion buffer containing Hanks’ balanced salt solution (Gibco), 200 µg/ml Liberase TL (Roche), 50 µg/ml DNase I (Invitrogen), 2% BSA, and 1% penicillin-streptomycin at 37°C with gentle stirring. Cell suspension was incubated for 5 min at RT with RBC lysis buffer (eBioscience). Cells were blocked with anti-CD16/32 antibodies, incubated with conjugated antibodies specific for CD45, CD31, and PDPN. Cells were sorted using MoFlo Astrios EQ cell sorter (Beckman Coulter) with FACSDiva software (BD Biosciences). Following exclusion of dead cells using DAPI and RedDot1 Far-Red Nuclear Stain reagent (Biotium) and gating on single cells, single CD45−CD31+PDPN+ cells were sorted directly in RLT lysis buffer containing 1% β-mercaptoethanol (Qiagen, RNeasy Micro Kit). Antibodies used for flow cytometry are listed in Table S1.
Cell culture, transfection, flow shear stress, and immunostaining
Human primary LECs were cultured as described previously (Norrmén et al., 2010) on fibronectin-coated dishes in EBM2 complete medium (Lonza) supplemented with EGM-2 SingleQuots (Lonza) for a maximum of 12 passages.
For knockdown experiments, cells were transfected with 30 nM siRNA using Lipofectamine RNAiMAX (Invitrogen). siRNA list is provided in Table S1. LECs were used 24 h after transfection for flow experiments.
For flow shear stress experiments, LECs were seeded at confluence on fibronectin-coated slides (μ-slide I 0.8 Luer, ibidi), cultured for 24 h and subjected to oscillatory flow (0.7 dynes/cm2 or 4 dynes/cm2; 0.25 Hz, flow changes direction every 4 s) in a parallel plate flow chamber system (ibidi Pump System, ibidi) or kept under static conditions for 48 h. When applicable, cells were treated with 10 nM rapamycin (LC Laboratories) during the entire duration of flow. At the end of experiment, cells were fixed with 4% PFA containing 1 mM CaCl2 and 0.5 mM MgCl2, permeabilized with 0.1% PBS Triton X-100 and kept at 4°C in PBS. For immunostaining, cells were blocked with 5% donkey serum, permeabilized with 0.1% PBS Triton X-100, and incubated with primary antibodies for 1 h at RT. After washes with 0.1% PBS Triton X-100, cells were incubated with secondary antibodies for 1 h at RT. Following washes with 0.1% PBS Triton X-100, cells were mounted using ibidi mounting medium (ibidi).
The decellularization procedure in cultured LECs was performed as described previously (Harris et al., 2018). Briefly, LECs were exposed to flow shear stress, culture medium was removed, and cells were rinsed twice with PBS, then washed three times with 100 mM Na2HPO4, 2 mM MgCl2, 2 mM EGTA, pH 9.6, and incubated for 15 min at 37°C in lysis buffer 8 mM Na2HPO4, 1% NP-40, pH 9.6. Lysis buffer was next replaced with fresh lysis buffer and incubated for 45 min at 37°C. Remaining matrix was rinsed with 10 mM Na2HPO4, 300 mM KCl, pH 7.5, five times and four times with deionized water and stored in PBS at 4°C. Immunofluorescence staining was performed after decellularization in the presence of DAPI to confirm cell removal.
For induction of mTOR signaling with nutrients, cells were starved in DME 5030 basal medium (Sigma-Aldrich) for 2.5 h and stimulated with a mix of 2 mM L-glutamine (Sigma-Aldrich), 0.8 mM L-leucine (Sigma-Aldrich), and 5 mM glucose (Sigma-Aldrich) or control saline.
For induction of mTOR signaling with VEGF-C, cells were starved in EBM2 basal medium (Lonza) overnight and induced with 100 ng/ml human VEGF-C (kind gift from Kari Alitalo) or control BSA.
For analysis of mTOR signaling in the absence of nutrients or growth factors, cells were starved in DME 5030 basal medium (Sigma-Aldrich) for 2.5 h and subjected to 4 dyn/cm2 OSS or kept under static conditions without medium change.
For the analysis of total protein synthesis, we used Click-iT-based OPP (Jena Bioscience) protein synthesis detection method. OPP was added to fluidic units 45 min prior to stopping the flow experiment at a concentration of 10 μM. At the end of the experiment, cells were fixed with 4% PFA containing 1 mM CaCl2 and 0.5 mM MgCl2, permeabilized with 0.1% PBS Triton X-100, and kept at 4°C in PBS. Proteins were detected by imaging after the Click-iT reaction with Alexa Fluor 555 conjugated azide.
RT-qPCR
RNA was isolated using QIAGEN RNeasy Micro kit and analyzed by Fragment Analyzer (Advanced Analytical Technologies). mRNA was amplified by Ovation RNA-seq system V2 (Nugen). For RT-qPCR, Quant Studio 3 (Applied Biosystems) machine with SensiFast Sybr Lo-Rox Mix (Bioline) was used. Data were normalized to 18s using the comparative Ct (ΔΔCt) method. Sequences of primers are listed in Table S1.
Western blotting
Cells were lysed in buffer containing 1% NP40, 0.1 M PMSF, 0.5 M EDTA, 20% SDS, 10% sodium deoxycholate, PhosSTOP (Roche), and complete mini protease inhibitor (Roche). Protein concentration was measured using BCA kit (Thermo Fisher Scientific). Samples were run on Mini-PROTEAN TGX Precast gels and transferred on Immobilon-P membrane (Milipore). After blocking with 5% BSA, membranes were incubated with primary antibodies at 4°C overnight. Membranes were washed with 0.1% PBS-Tween and incubated with HRP-conjugated secondary antibody (Dako) for 1 h at RT. Western blots were developed using SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific).
Quantifications
For proliferation analysis on paraffin sections of mesentery, staining with LYVE1, PROX1, and EdU or KI67 was performed. At least five images were taken per mouse and total count of LECs was performed using PROX1 staining. Double-positive cells PROX1+/KI67+ of PROX1+/EdU+ were counted, and percentage from the total LEC population was calculated.
For in vitro pS6 intensity quantifications, images from five independent fields per slide were taken and all PROX1+ cells per image were analyzed. Analysis was done using Fiji software, and mean gray value measurement was performed.
For quantification of in vitro FN-EIIIA deposition (Fig. 8, G and H), images from five independent fields per slide were taken. Analysis was done using Fiji software, and mean gray value measurement was performed.
For quantification of in vitro cell proliferation, images from five independent fields per slide were taken. Analysis was performed using Fiji software with “Analyze Particles” function or CellProfiler software with “IdentifyPrimaryObjects” module to detect the percentage of KI67+ nuclei from the total number of DAPI+ or HOECHST+ nuclei.
For Western blot analysis, band intensities were measured using Fiji “Gels Analysis” tool. Intensities of protein of interest was normalized to loading control and set to 1, as indicated in the corresponding figure legends.
The range of samples or cells analyzed is indicated in the corresponding figure legends.
Statistics
Graphs were prepared and statistical analyses were performed using GraphPad Prism software version 9. Data are shown as mean ± SD. N indicates the sample size and is described for each quantification in the corresponding figure legends.
To assess Gaussian distribution of the data, we used Shapiro–Wilk test in Graphpad Prism; normal distribution was assumed if P value was higher than 0.05.
For in vivo experiments, to determine statistical significance between two groups, we used a two-tailed unpaired Welch’s t test when data were normal and a two-tailed unpaired Mann–Whitney test when data normality could not be confirmed; difference between the two groups was considered statistically significant when P value was <0.05. To compare multiple groups, we used a Kruskal–Wallis test with Dunn’s correction for multiple comparison when data normality could not be confirmed.
For in vitro flow experiments, to compare static and OSS conditions, we used a two-tailed ratio paired t test when data were normal. To assess the effect of rapamycin treatment in flow experiments, we used two-way ANOVA test.
For in vitro experiments with blocking antibodies, to compare control vs. blocking antibodies, we used a two-tailed unpaired Welch’s t test.
Online supplemental material
Fig. S1, related to Fig. 1, shows representative FACS plots and gating strategy for sorting LECs from mouse mesentery. Fig. S2, related to Fig. 3, shows representative lymphatic valve confocal images showing that most of PROX1high cells are KI67neg. Fig. S3 shows representative images and quantification data highlighting that proliferation of mature mesenteric LECs is independent of VEGF-A/VEGFR-2 and VEGF-C/VEGFR-3 signaling. Fig. S4, related to Fig. 6 (for panels A and B) and Fig. 9 (for panels C and D), shows that ablation of Tsc1 in vivo or in vitro promotes mTORC1 signaling. Fig. S5, related to Fig. 7 (for panels A and B) and Fig. 8 (for panels C and F), shows activation of mTOR by nutrients and growth factor signaling in LECs. Table S1, related to Materials and methods, lists materials used in this study (primary and secondary antibodies, chemicals and kits, mouse models, cell culture, primers, silencing RNAs, instruments, consumables, and softwares).
Data availability
Data that support the findings of this study are available within the article and its supplementary information, or on request from the corresponding author.
Acknowledgments
We thank E. Bovay for participating at the initial stages of the project, S. Arroz-Madeira for kind help with flow cytometry analysis, T. Mäkinen (Uppsala University, Uppsala, Sweden) for Prox1-CreERT2 mice, S. Ortega (Spanish National Cancer Research Center, Madrid, Spain) for Flt4-CreERT2 mice, A. Keller (University Hospital Zürich, Zürich, Switzerland) for Esco2fl/fl mice, M. Jelstch and K. Alitalo (University of Helsinki, Helsinki, Finland) for VEGF-C protein, C. Beauverd for mouse genotyping and colony maintenance, and S. Davanture and M. Favre for lab managing and kind help with the project. Animal, Cellular Imaging, Flow Cytometry, Mouse Pathology, and Genomic Technology facilities of the University of Lausanne are gratefully acknowledged.
This work was supported by grants from the European Union’s Horizon 2020 research and innovation program Theralymph (grant agreement no. 847939) to T.V. Petrova, the Swiss National Science Foundation (CR32I3_166326) to T.V. Petrova, Erna Hamburger Foundation (2018) to C. Saygili Demir, and Société Académique Vaudoise (2018) to C. Saygili Demir.
Author contributions: Original investigation was performed mostly by C. Saygili Demir; investigation for revision was performed by A. Sabine and M. Gong. Formal analysis was performed by A. Sabine and C. Saygili Demir. Validation of the data was performed by A. Sabine, T.V. Petrova, and C. Saygili Demir. Visualization of the data was performed by A. Sabine, T.V. Petrova, and C. Saygili Demir. Writing of the original manuscript was written by C. Saygili Demir, A. Sabine, and T.V. Petrova. Revision of the manuscript was performed by A. Sabine and T.V. Petrova. Review and editing of the manuscript were performed by A. Sabine, M. Gong, C. Saygili Demir, O. Dormond, and T.V. Petrova. Project was administered by T.V. Petrova. Funding for the project was acquired by T.V. Petrova and C. Saygili Demir.

