


Epilepsy is a common neurological disease worldwide, and one of its causes is genetic abnormalities. Here, we identified a point mutation in KIF4A, a member of kinesin superfamily molecular motors, in patients with neurological disorders such as epilepsy, developmental delay, and intellectual disability. KIF4 is involved in the poly (ADP-ribose) polymerase (PARP) signaling pathway, and the mutation (R728Q) strengthened its affinity with PARP1 through elongation of the KIF4 coiled–coil domain. Behavioral tests showed that KIF4-mutant mice exhibited mild developmental delay with lower seizure threshold. Further experiments revealed that the KIF4 mutation caused aberrant morphology in dendrites and spines of hippocampal pyramidal neurons through PARP1-TrkB-KCC2 pathway. Furthermore, supplementing NAD, which activates PARP1, could modulate the TrkB-KCC2 pathway and rescue the seizure susceptibility phenotype of the mutant mice. Therefore, these findings indicate that KIF4 is engaged in a fundamental mechanism regulating seizure susceptibility and could be a potential target for epilepsy treatment.
Introduction
Epilepsy is a common chronic neurological condition that lowers quality of life, affecting more than 50 million people worldwide, and a considerable subset of patients do not respond to antiepileptic drugs. Unfortunately, the cause of this disease is not yet clear. Recent study has shown that most epilepsy cases are caused by interactions between multiple genes and environmental factors (Steinlein, 2008), while the pathogenesis from single-gene mutations or microdeletion of chromosomes are rare. However, so far, mechanistic research on single-gene-triggered genetic epilepsies has contributed to the current understanding of the primary process of epileptogenesis. Several kinds of epilepsy model mice were generated by single-gene mutations on the X chromosome, and in vivo and in vitro studies revealed that they exhibited hyper-branched dendrites and irregular dendritic spine morphologies of hippocampal pyramidal neurons with the high energy of hippocampus electroencephalogram (EEG) waves (Scheffer et al., 2002; Beguin et al., 2013). However, the detailed pathogenic mechanism is not yet known. Therefore, unraveling the regulatory mechanisms of genetic epilepsies is necessary for understanding the molecular mechanism of epilepsy to provide novel diagnosis methods and antiepileptic targets.
Kinesin superfamily proteins (KIFs) play critical roles mainly in microtubule dependent intracellular transport, which is essential for cell survival, movement, division, and maintenance of cellular morphology (Hirokawa et al., 2010). Kif4a, an X chromosome-linked kinesin-4 family gene, is critically involved in chromatin condensation in humans through stabilizing chromosome scaffold formation (Poonperm et al., 2017). The paralog of the Kif4a gene is Kif4b, an intron-less version of Kif4a localized on chromosome 5 (Ha et al., 2000). Kif4, without paralogous genes, exists on chromosome X of mice, and has 82% of the nucleic acid sequence and 85% of the amino acid sequence in common with the human’s gene Kif4a (Ha et al., 2000). KIF4 has been first identified as an anterograde transport motor enriched in the neuron cell body and the growth cones of juvenile neurons (Sekine et al., 1994). In the nervous system, cells with abolished KIF4 expression exhibit an anti-apoptotic phenotype and enhanced activity of KIF4 binding partner, poly (ADP-ribose) polymerase 1 (PARP1; Midorikawa et al., 2006). Recently, a human study revealed that four males from a single family have a disruptive mutation in Kif4a, resulting in a lack of its exon 15. This caused all four men to suffer from mild intellectual disability (ID) and epilepsy during late childhood or early adolescence (Willemsen et al., 2014). Epileptic patients and some epileptic animal models often exhibit hippocampal neuronal death, which is considered to be a result of excitotoxic neuronal injury (Teocchi and D’Souza-Li, 2016). However, considering the known functions of KIF4, we cannot explain how the mutation contributes to epileptogenesis. This indicates that the role of KIF4 in higher brain function is still poorly defined.
KIF4 contains two conserved DNA-binding motifs and therefore is abundantly present in cellular nuclei (Mazumdar et al., 2004). In fact, KIF4 binds globally to chromatin during interphase and alters the expression levels of many genes by acting as a modulator of large-scale heterochromatin architecture (Mazumdar et al., 2011). On the other hand, recent evidence has suggested that PARP1 plays crucial roles in both single and double-strand DNA repair pathways and the regulation of immune gene transcription (Ray Chaudhuri and Nussenzweig, 2017; Feng et al., 2015). Accordingly, PARP1 inhibitors are clinically used for treating malignant neoplasms and considered as potential drug candidates for diabetes complications (Feng et al., 2015).
The transcriptional role of PARP1 is mainly controlled by its physiological activation-produced poly (ADP-ribose; PAR), a post-transcriptional modifier composed of a polymer of two or more ADP-ribose units (Gibson and Kraus, 2012). This polymeric PAR takes either linear or branched forms, covalently connected with different amino acids, including aspartate, glutamate, and lysine residues or ribose group of other ADP-ribose units (Alemasova and Lavrik, 2019). In the PARP1-mediated suppression of transcription, the transcriptional initial protein complex is disrupted by the increased activity of PARP1 (Lai et al., 2012). PARP1 also mediates transcriptional facilitation, in which PAR leads to disassembly of some repressors including HSP70 (Petesch and Lis, 2012). Hydrogen peroxide (H2O2) is a well-known oxidizing agent that induces PARP1 activation resulting in PAR production (Veith et al., 2019).
Tropomyosin receptor kinase B (TrkB) has several isoforms, including the full-length and the truncated, determined by different mRNA splicing patterns. These isoforms bind with neurotrophic factors which can regulate neurite outgrowth, differentiation, survival, and synaptic plasticity through the homodimer or heterodimer model (Stoilov et al., 2002). Furthermore, previous studies have shown that the TrkB expression pattern is associated with the abnormal dendritic morphology and regulation of the potassium-chloride cotransporter 2 (KCC2). Both these processes can promote epileptic seizures in mice (Ferrini et al., 2020; Rivera et al., 2002). However, the up-stream factors of this TrkB-KCC2 pathway are still unknown.
From these previous studies, we hypothesized that KIF4-PARP1 can functionally regulate transcription in epileptic seizures. Here, we provide evidence for a novel point mutation in the coiled–coil domain of KIF4 that can trigger morphological changes of the neuronal dendrites and spines and a higher intracellular chloride concentration. These observations result from altered TrkB and KCC2 expression levels caused by impaired PARP1 activity. Experiments using in vivo mouse model exhibited phenotypes of KIF4 mutant mice (Kif4Mut/Y), including increased seizure susceptibility, mild developmental delay, and intellectual disability. These findings indicate that KIF4 has a fundamental role in epileptogenesis.
Results
A mutation in the highly conserved KIF4A coiled–coil domain was found from an intractable childhood epilepsy patient
The KIF4A mutation (R728Q) in human exon 20 was discovered in a patient who exhibited global developmental delay, severe intellectual disability, and intractable seizure (IV-3 in Fig. 1 A), and he was hospitalized at the age of one. The 1-h video-EEG recording of the child during wakening revealed frequent epileptic spasms without any focal epileptiform discharges. The magnetic resonance imaging (MRI) diagnosis of the brain revealed that each area was normal and had no focal or diffuse cerebral abnormal intensities (Fig. S1 A). Whole exome sequencing was performed, including parents III-1/2 and children IV-1/2/3, with the genotyping results as follows: III-1, XWTXMut; III-2, XWTY; IV-1/2, XWTXMut; IV-3, XMutY (Fig. 1 A). Compared with the 100 healthy controls from the population, the sequencing results indicated that the mother (III-1) and two daughters (IV-1 and IV-2) have both the wild-type and the mutant Kif4a (Fig. S1 B). Although this mother (III-1), two daughters (IV-1 and IV-2), and one son (IV-3) were all mutation carriers, only the son (IV-3) showed the severe symptoms.

The genomic background of patients and the Kif4-mutant mice exhibit a high embryonic mortality rate and developmental delay. (A) The pedigree chart of a Kif4 mutation-carrying family. Note that III-1, IV-1, and IV-2 (Kif4aWT/Mut) exhibited no symptoms, whereas IV-3 (Kif4aMut/Y) showed global developmental delay, severe intellectual disability, and intractable seizure. (B) Illustration of the mouse KIF4 protein with its point mutation identified from the patients. LCR, low complexity region; CC, coiled coil; CXC, tesmin/TSO1-like CXC domain. (C) Coiled–coil prediction of the mouse KIF4 peptide from amino acid 524–732. Note that the Mut-KIF4 has a higher coil probability, as indicated with the asterisks. (D) Mouse mating strategy and the mortality rate. The embryonic mortality rate of Kif4 mutation-carrying mice (Mut, 26%) is significantly higher than that of wild-type mice (WT, 7%) Data are presented as mean ± SEM (n = 12 mating mouse pairs). ***P < 0.001 (Student’s t tests). (E) The composition of the offspring genotypes from the Kif4-mutant background exhibited a lower Kif4Mut/Y pup population (n = 6 mating mouse pairs). (F) The photograph of Kif4WT/Y and Kif4Mut/Y mice at E16.5 and the statistical analysis results of embryo size. Data are presented as mean ± SEM (n = 8 mouse pairs). ****P < 0.0001 (Student’s t tests). (G) The postnatal weight comparison between Kif4WT/Y and Kif4Mut/Y mice from P3 to P14. Data are presented as mean ± SEM (n = 12 mouse pairs). *P < 0.05, **P < 0.01 (two-way ANOVA). (H) The assessment of Kif4WT/Y and Kif4Mut/Y mouse developmental milestones. Note that Surface Righting, Auditory, Open field, and Air Righting exhibited statistical significance. Data are presented as mean ± SEM (n = 12 mouse pairs). *P < 0.05, **P < 0.01, ***P < 0.001 (two-way ANOVA). (I) HE staining of sagittal mouse brain sections from the indicated genotypes at E16.5 and the corresponding statistical analysis of the hippocampal cell density. The red stars in the left panels indicate the hippocampus regions that are magnified in the right panels. Scale bars, 1000 μm (left panel) and 100 μm (right panel). Data are presented as mean ± SEM (n = 12 hippocampi from 6 mouse pairs). ****P < 0.0001 (Student’s t tests).
The genomic background of patients and the Kif4-mutant mice exhibit a high embryonic mortality rate and developmental delay. (A) The pedigree chart of a Kif4 mutation-carrying family. Note that III-1, IV-1, and IV-2 (Kif4aWT/Mut) exhibited no symptoms, whereas IV-3 (Kif4aMut/Y) showed global developmental delay, severe intellectual disability, and intractable seizure. (B) Illustration of the mouse KIF4 protein with its point mutation identified from the patients. LCR, low complexity region; CC, coiled coil; CXC, tesmin/TSO1-like CXC domain. (C) Coiled–coil prediction of the mouse KIF4 peptide from amino acid 524–732. Note that the Mut-KIF4 has a higher coil probability, as indicated with the asterisks. (D) Mouse mating strategy and the mortality rate. The embryonic mortality rate of Kif4 mutation-carrying mice (Mut, 26%) is significantly higher than that of wild-type mice (WT, 7%) Data are presented as mean ± SEM (n = 12 mating mouse pairs). ***P < 0.001 (Student’s t tests). (E) The composition of the offspring genotypes from the Kif4-mutant background exhibited a lower Kif4Mut/Y pup population (n = 6 mating mouse pairs). (F) The photograph of Kif4WT/Y and Kif4Mut/Y mice at E16.5 and the statistical analysis results of embryo size. Data are presented as mean ± SEM (n = 8 mouse pairs). ****P < 0.0001 (Student’s t tests). (G) The postnatal weight comparison between Kif4WT/Y and Kif4Mut/Y mice from P3 to P14. Data are presented as mean ± SEM (n = 12 mouse pairs). *P < 0.05, **P < 0.01 (two-way ANOVA). (H) The assessment of Kif4WT/Y and Kif4Mut/Y mouse developmental milestones. Note that Surface Righting, Auditory, Open field, and Air Righting exhibited statistical significance. Data are presented as mean ± SEM (n = 12 mouse pairs). *P < 0.05, **P < 0.01, ***P < 0.001 (two-way ANOVA). (I) HE staining of sagittal mouse brain sections from the indicated genotypes at E16.5 and the corresponding statistical analysis of the hippocampal cell density. The red stars in the left panels indicate the hippocampus regions that are magnified in the right panels. Scale bars, 1000 μm (left panel) and 100 μm (right panel). Data are presented as mean ± SEM (n = 12 hippocampi from 6 mouse pairs). ****P < 0.0001 (Student’s t tests).

Brain MRI, the specifications of the Kif4a mutation in patients’ family and the sequence alignment of KIF4. (A) Brain MRI in the IV-3 patient exhibited unremarkable MRI features. (B) Whole exome sequencing of the Kif4a gene in controls and patients was performed on DNA extracted from blood samples. Arrows (red) indicate the position of the mutation. Genotyping results for the parents III-1/2 and the children IV-1/2/3 were as follows: III-1, XWTXMut; III-2, XWTY; IV-1/2, XWTXMut; and IV-3, XMutY. (C) Multiple sequence alignment of the KIF4 coiled–coil region from the indicated species. R728 is highly conserved in KIF4 throughout evolution.

Brain MRI, the specifications of the Kif4a mutation in patients’ family and the sequence alignment of KIF4. (A) Brain MRI in the IV-3 patient exhibited unremarkable MRI features. (B) Whole exome sequencing of the Kif4a gene in controls and patients was performed on DNA extracted from blood samples. Arrows (red) indicate the position of the mutation. Genotyping results for the parents III-1/2 and the children IV-1/2/3 were as follows: III-1, XWTXMut; III-2, XWTY; IV-1/2, XWTXMut; and IV-3, XMutY. (C) Multiple sequence alignment of the KIF4 coiled–coil region from the indicated species. R728 is highly conserved in KIF4 throughout evolution.
Protein function is intimately related to its three-dimensional (3D) structure which is determined by its amino acid sequence (Dietzen, 2018). Protein evolution analysis has shown that the functional region or catalytic pocket of a protein is typically located within the conserved sequence (Sievers and Higgins, 2014). Because of these knowledges, we performed multisequence alignments of KIF4 in several species. We compared the region of interest in the KIF4 coiled–coil domain from amino acids 524–732 and found that amino acid 728 (the mutation site) is highly conserved in the compared five species (Fig. S1 C). Crystallographic research has indicated that the KIF4 protein has an N-terminal motor domain spanning amino acids 1–336, with a coiled–coil connected region from 350 to 1,000 and a flexible C-terminal tail domain from 1001 to 1231 (Fig. 1 B). Using COILS, one of the online bioinformatics tools, we performed the coiled–coil prediction from amino acids 524–732 of wild-type KIF4 (WT-KIF4) and mutant-carrying KIF4 (Mut-KIF4). The resulting prediction score, which represents probability of forming a coiled–coil in the range of 0–1, at site 728 was ∼0.45 in the WT-KIF4, but increased to 0.92 in Mut-KIF4 because of the single amino acid change (Fig. 1 C). Considering these results, we believed that this mutation within the highly conserved portion of the KIF4 protein may elongate the coiled–coil domain and subsequently influence its binding partner and downstream cascades.
Kif4 mutant mouse exhibited a developmental delay phenotype
Kif4 transgenic C57BL/6J mouse was established using the clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR associated protein 9 (Cas9) system through single-stranded oligodeoxynucleotides (ssODNs)-mediated homology-directed repair (HDR; Kumita et al., 2019). This transgenic mouse model had several nucleotides substituted in the relative site in exon 19, the corresponding positions in the human Kif4a exon 20. The amino acid at position 728 was arginine converted to glutamine (Fig. S2, A and B). The first generation of Kif4 mutant mice was confirmed by Sanger sequencing (Fig. S2 C). The genotypes of the offspring of the transgenic mice were confirmed through the enzymatic digestion method using the XhoI restriction enzyme. After conducting PCRs with the genomic DNA, both the wild-type and mutant mice produced 494 base pair (bp) fragments, while only the mutant PCR product could be digested with XhoI, thereby exhibiting smaller bands seen by gel electrophoresis (Fig. S2, D and E).

Construction of a R728Q KIF4 transgenic mouse. (A–D) Red (functional mutation) and dark-blue (XhoI site) letters indicate the substitution target site. (A) Schematic illustration showing the locations of the gRNAs and ssODNs. Black bar indicates the position of the gRNA target with the orange bar showing the PAM sequence. ssODNs are shown in green letters (intron area) and blue letters (exon area). (B) The amino acid sequences from wild type (WT) and substituted sequences are shown, with red letters indicating the target and the substitution. (C) WT and donor DNA sequences are displayed in the top panel. The lower panel shows the electropherogram of the direct sanger-sequencing result, in which one of the first-generation male mice received the specific substitution. (D) Schematic illustration showing the locations of the PCR primers, along with the mouse Kif4 locus. Black bars on both sides indicate the position of the PCR primer sequences. (E) Representative image of the genotyping results. The products from genomic PCR of mouse tail DNAs were the same length in the WT and the substituted. Subsequently, XhoI digestion was performed to distinguish the individual genotypes. The blue arrow indicates WT Kif4 and the pink arrow indicates Mut Kif4.

Construction of a R728Q KIF4 transgenic mouse. (A–D) Red (functional mutation) and dark-blue (XhoI site) letters indicate the substitution target site. (A) Schematic illustration showing the locations of the gRNAs and ssODNs. Black bar indicates the position of the gRNA target with the orange bar showing the PAM sequence. ssODNs are shown in green letters (intron area) and blue letters (exon area). (B) The amino acid sequences from wild type (WT) and substituted sequences are shown, with red letters indicating the target and the substitution. (C) WT and donor DNA sequences are displayed in the top panel. The lower panel shows the electropherogram of the direct sanger-sequencing result, in which one of the first-generation male mice received the specific substitution. (D) Schematic illustration showing the locations of the PCR primers, along with the mouse Kif4 locus. Black bars on both sides indicate the position of the PCR primer sequences. (E) Representative image of the genotyping results. The products from genomic PCR of mouse tail DNAs were the same length in the WT and the substituted. Subsequently, XhoI digestion was performed to distinguish the individual genotypes. The blue arrow indicates WT Kif4 and the pink arrow indicates Mut Kif4.
The patient information made us investigate the survival rate, growth, and development of the Kif4-mutant mouse. We initially mated the Kif4+/Mut female mouse with the Kif4Mut/Y male mouse and compared the embryonic mortality rate and the embryo size with embryos from a Kif4+/+ female and Kif4WT/Y male mouse pair. The fetal demise rate of the mutant mating group was significantly higher than that of the wild-type mating group (Fig. 1 D). The genotypes of the offspring of these two groups were detected, with male pups accounting for 42% of the total embryos, yet the Kif4Mut/Y genotype only covered 31% of the males (Fig. 1 E).
We then recorded the mouse embryo sizes at embryonic days 16.5 (E16.5), which were quantified by the perimeter and vertical axis lengths. These two measurements were both shorter in Kif4Mut/Y mice (Fig. 1 F). Subsequently, the postnatal weight (P3–P14) examinations showed lighter body weights in Kif4Mut/Y mice (Fig. 1 G). We found a global delay trend in the Kif4Mut/Y mice, especially in the surface righting, auditory, open field, and air righting projects, through the assessment of developmental milestones experiment (Fig. 1 H). Furthermore, the apparent smaller brains in the Kif4Mut/Y mice were confirmed by hematoxylin and eosin (HE) staining. The hippocampus area was especially smaller in the Kif4Mut/Y mice (Fig. 1 I). From the test results of delayed surface righting, we speculated that Kif4Mut/Y mice had a weaker locomotive ability. The delayed phenotype in the opening field and air righting could be caused by aberrant complex locomotive skills in Kif4Mut/Y pups, like motor coordination. Furthermore, the delayed response to sound suggested abnormal nerve development in the mutant mice. Collectively, these results indicate a delayed neurodevelopmental trend in Kif4Mut/Y mice.
Mouse behavioral tests showed Kif4 mutation-induced anxiety, intellectual disability, and epileptic phenotype
Next, to determine whether the Kif4Mut/Y mouse exhibited the behavior associated with the human patients’ symptoms, we performed a series of behavioral tests which were often used to measure anxiety-like behaviors. In the open field test (OFT), we found that, compared with the Kif4WT/Y mice, Kif4Mut/Y mice preferred to stay in the edge zone rather than explore the central area (Fig. 2 A). Both the in-zone frequency and time spent in the central zone were significantly reduced in Kif4Mut/Y mice. The total distance that the mice traveled at the OFT showed no significant difference between Kif4+/Y and Kif4Mut/Y mice (Fig. 2, B–E). In the elevated plus maze test (EPM), Kif4Mut/Y mice spent less time on the open arm and entered the open arm fewer times compared with Kif4WT/Y mice (Fig. 2, F and G). Collectively, these results indicated that Kif4Mut/Y mice exhibited abnormal levels of anxiety phenotype similar to the other seizure models, such as the cyclin-dependent kinase-like 5 (Cdkl5), X chromosome localized gene, defected mouse model (Okuda et al., 2018). In the novel object recognition test, there is an object location task (OLT) and a novel object recognition task (NORT), which are two effective methods that can examine the hippocampus-dependent spatial memory and general memory function (Denninger et al., 2018). Kif4WT/Y mice spent more time with the object in a novel location or took longer time with the new item. As a result, they exhibited significant increases in discrimination indexes in both the OLT and NORT. On the other hand, Kif4Mut/Y mice spent the same time in all objects and locations that showed no significant changes in the discrimination indexes between the training session and the probe test for either the OLT or NORT, suggesting an impaired learning and memory phenotype in Kif4Mut/Y mice (Fig. 2, H and I). The fear conditioning test (FCT) is often used to examine whether a rodent has hippocampus-dependent learning and memory impairments (Izquierdo et al., 2016). Compared with Kif4WT/Y mice, Kif4Mut/Y mice exhibited a less freezing time on both Day 1 and Day 7 after foot-shock stimulation on Day 0 (Fig. 2 J). These FCT results support our speculation that the KIF4 mutation can negatively impact learning and memory.

Kif4 Mut/Y mice exhibit anxiety, anti-depression and intellectual disability. (A) The heatmap of the occupancy of individual mice in OFT. A warmer color represents more time spent (red > yellow > green > blue), and red rectangles are put at the central zones in the fields. (B–E) The results of OFT. Total distances that mice ran during tests (B), cumulative durations in the edge zone (C) or the central zone (D), and in-zone frequencies to the central zone (E) are represented. Data are presented as mean ± SEM (n = 12 mouse pairs). ns, P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001 (Student’s t tests). (F and G) The results of the EPM test. The number of entries into the open arm (F) and the cumulative durations mice spent in the open arm (G) were decreased in the Kif4Mut/Y mice. Data are presented as mean ± SEM (n = 12 mouse pairs). **P < 0.01, (Student’s t tests). (H) Schematic illustration showing the schedule of novel objective recognition test (NORT). (I) The results of OLT and NORT. The discrimination index is calculated from the time difference between two events divided by the sum of that time. Positive values indicate that the mouse spent more time investigating the novel object or location. Note that Kif4Mut/Y mice had a value of ∼0, indicating a memory dysfunction phenotype. Data are presented as mean ± SEM (n = 12 mouse pairs). ns, P > 0.05, ***P < 0.001, ****P < 0.0001 (two-way ANOVA). (J) The results of FCT. Total freezing durations were recorded. Kif4Mut/Y mice exhibited shorter freezing time at both Day 1 and 7. Data are presented as mean ± SEM (n = 12 mouse pairs). ****P < 0.0001 (two-way ANOVA).
Kif4 Mut/Y mice exhibit anxiety, anti-depression and intellectual disability. (A) The heatmap of the occupancy of individual mice in OFT. A warmer color represents more time spent (red > yellow > green > blue), and red rectangles are put at the central zones in the fields. (B–E) The results of OFT. Total distances that mice ran during tests (B), cumulative durations in the edge zone (C) or the central zone (D), and in-zone frequencies to the central zone (E) are represented. Data are presented as mean ± SEM (n = 12 mouse pairs). ns, P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001 (Student’s t tests). (F and G) The results of the EPM test. The number of entries into the open arm (F) and the cumulative durations mice spent in the open arm (G) were decreased in the Kif4Mut/Y mice. Data are presented as mean ± SEM (n = 12 mouse pairs). **P < 0.01, (Student’s t tests). (H) Schematic illustration showing the schedule of novel objective recognition test (NORT). (I) The results of OLT and NORT. The discrimination index is calculated from the time difference between two events divided by the sum of that time. Positive values indicate that the mouse spent more time investigating the novel object or location. Note that Kif4Mut/Y mice had a value of ∼0, indicating a memory dysfunction phenotype. Data are presented as mean ± SEM (n = 12 mouse pairs). ns, P > 0.05, ***P < 0.001, ****P < 0.0001 (two-way ANOVA). (J) The results of FCT. Total freezing durations were recorded. Kif4Mut/Y mice exhibited shorter freezing time at both Day 1 and 7. Data are presented as mean ± SEM (n = 12 mouse pairs). ****P < 0.0001 (two-way ANOVA).
Furthermore, we used the pentylenetetrazole (PTZ), a non-competitive GABAA receptor (GABAAR) antagonist, to validate mouse epileptic susceptibility in the different genotype backgrounds (Shimada and Yamagata, 2018). Immediately after the PTZ (38 mg/kg) was intraperitoneally injected, we observed an immobilization (Stage 1) in Kif4WT/Y mice but a severe seizure with continuous rearing and falling (Stage 4) in Kif4Mut/Y mice (Fig. 3 A and Video 1). Then, the behavioral seizures were evaluated every 60 min according to a modified Racine’s score (Lüttjohann et al., 2009; Fig. 3 A). The statistical data revealed that the cumulative seizure score and the maximum seizure stage in Kif4Mut/Y mice were significantly higher than those in Kif4WT/Y mice. Additionally, the percentage of stage 4 occurrence was elevated in Kif4Mut/Y mice (Fig. 3, B–D). Eleven of fifteen Kif4Mut/Y mice exhibited stage 5 seizures and three died, while none of the Kif4WTY mice died and only seven mice experienced stage 3 seizures under the same conditions (Fig. 3 A). These behavioral results suggested that Kif4Mut/Y mice were more susceptible to PTZ-induced seizures.

Behavior and EEG recordings of the Kif4 Mut/Y mouse exhibit high susceptibility to seizure induction. (A–D) The results of the PTZ kindling test. The chart representing the seizure stage classification (A) of the individual scores from Kif4WT/Y and Kif4Mut/Y mice, before and after the PTZ injection. Statistical analysis results of the cumulative number of maximum (B) and all (C) seizure stage, and the ratio of the occurrence of stage 4 (D). Data are presented as mean ± SEM (n = 15 mouse pairs). ****P < 0.0001 (Student’s t tests). (E–H) The representative EEG recordings (E) and the results of the related time-frequency analysis using Morlet-Wavelet’s method (F). Red arrows and star marks indicate the timepoints of PTZ injections and the more energetic brain waves representing seizure onsets, respectively; M1, primary motor cortex; Hippo, hippocampus. (G) 45-second-long brain waves were picked up from hippocampal data in (E). The waveform the Kif4Mut/Y mice exhibited was a spike-and-slow-wave pattern with a high electric current. (H) Spectrum analysis of post-PTZ injected individual mice using the FFT method. Note that the energy levels from 0.5 to 1.25 and 2 Hz in Kif4Mut/Y mice were substantially higher than the control; PSD, power spectral density. Data are presented in (H) as mean ± SEM (n = 5 mouse pairs). *P < 0.05, ****P < 0.0001 (two-way ANOVA).
Behavior and EEG recordings of the Kif4 Mut/Y mouse exhibit high susceptibility to seizure induction. (A–D) The results of the PTZ kindling test. The chart representing the seizure stage classification (A) of the individual scores from Kif4WT/Y and Kif4Mut/Y mice, before and after the PTZ injection. Statistical analysis results of the cumulative number of maximum (B) and all (C) seizure stage, and the ratio of the occurrence of stage 4 (D). Data are presented as mean ± SEM (n = 15 mouse pairs). ****P < 0.0001 (Student’s t tests). (E–H) The representative EEG recordings (E) and the results of the related time-frequency analysis using Morlet-Wavelet’s method (F). Red arrows and star marks indicate the timepoints of PTZ injections and the more energetic brain waves representing seizure onsets, respectively; M1, primary motor cortex; Hippo, hippocampus. (G) 45-second-long brain waves were picked up from hippocampal data in (E). The waveform the Kif4Mut/Y mice exhibited was a spike-and-slow-wave pattern with a high electric current. (H) Spectrum analysis of post-PTZ injected individual mice using the FFT method. Note that the energy levels from 0.5 to 1.25 and 2 Hz in Kif4Mut/Y mice were substantially higher than the control; PSD, power spectral density. Data are presented in (H) as mean ± SEM (n = 5 mouse pairs). *P < 0.05, ****P < 0.0001 (two-way ANOVA).
Related toFig. 3 A . Pentylenetetrazol (PTZ) kindling test showing that Kif4Mut/Y mice are highly sensitive to epilepsy.
Related toFig. 3 A . Pentylenetetrazol (PTZ) kindling test showing that Kif4Mut/Y mice are highly sensitive to epilepsy.
Kif4Mut/Y mice exhibited epileptic patterns in EEG recordings
To further understand the mechanism of Kif4 mutation-induced susceptibility to seizures, we simultaneously recorded the EEGs and behavior of Kif4WT/Y and Kif4Mut/Y mice at postnatal week 3. The electrodes were stereotaxically inserted into the primary motor cortex (M1) and hippocampus (Hippo). At the baseline stage (pre-PTZ), neither epileptic pattern nor EEG power exhibited a significant difference in the M1 and Hippo region between Kif4WT/Y and Kif4Mut/Y mice (Fig. 3 E). In response to an intraperitoneal injection of PTZ (38 mg/kg), a spike-wave pattern was recorded on the EEG in the Kif4Mut/Y mice (Fig. 3 E). The total time-frequency analyses derived the two high energy peaks at 500 and 1,000 s, through the Morlet wavelet transforms, suggesting that Kif4Mut/Y neurons fired twice after PTZ injection (Star marks in Fig. 3 F).
We selected the 45 s corresponding waveforms that contained seizure events. Higher energy and classic seizure waveforms including the irregular spike-and-slow-wave complex pattern were clearly visible in the Kif4Mut/Y mouse group (Fig. 3 G). To further assess the waveforms, we performed power analysis of the picked-up 45 s long waveforms with the fast Fourier transform (FFT). The EEGs of the Kif4Mut/Y mice showed a more than 10–fold increase in power, suggesting that these mice were more susceptible to seizure inductions (Fig. 3 H).
Mut-KIF4 strongly bound to PARP1, leading to a high probability of cell death
Our previous study has shown that PARP1 is a binding partner of KIF4 in neurons, and underlies neuronal apoptosis through its enzymatic activity (Midorikawa et al., 2006). Thus, from the bioinformatics results of the elongated coiled–coil domain (Fig. 1 C), we wondered whether the mutation in KIF4 had any effect on its PARP1-binding strength. Therefore, we performed co-immunoprecipitation (co-IP) and proximity-dependent biotin identification (BioID) experiments to examine the binding strength between KIF4 and PARP1. In the co-IP experiment, the primary culture neuron lysates were separated into nuclear and other cytosol components using ultra-centrifugation and individually immunoprecipitated (IPed) using an anti-KIF4 antibody or control IgG. Immunoblots against PARP1 identified a stronger PARP1 signal in the mutant nucleus than in wild-type nucleus after normalized by the input PARP1 volumes. The results indicated that PARP1 had higher affinity for Mut-KIF4 in the nucleus, compared to WT-KIF4 (Fig. 4, A and B). Next, in the BioID experiments, the WT-KIF4 and Mut-KIF4 were individually overexpressed in the Neuro 2A (N2A) cells, and the biotinylated proteins were purified by the streptavidin magnetic bead-based pulldown. While the immunoblots of the input samples showed no changes in PARP1 and endogenous KIF4 expression levels (Fig. S3 A), higher amounts of PARP1 were detected in the purified lysates expressing Mut-KIF4 (Fig. 4, C and D). These data suggested a dramatically higher affinity between PARP1 and Mut-KIF4 compared with WT-KIF4.

Mut-KIF4 strongly binds to PARP1, whose activity influences cell survival rate. (A and B) Co-immunoprecipitation (Co-IP) immunoblotting results of the primary culture neuron lysates (A), and quantification of amounts of 110 kD PARP1 bands (B). The statistical values were normalized to relevant bands in the input lanes. The input samples and immunoprecipitated complexes including KIF4 were analyzed by immunoblotting using an antibody specific to PARP1; IP, immunoprecipitation; IB, immunoblotting; IgG, immunoglobulin G; Cyt., Cytosol; Nuc., Nuclear. Data are presented as mean ± SEM (n = 3 independent experiments). ***P < 0.001 (Student’s t tests). (C and D) Immunoblotting results of BioID pulldown (C), and quantification of amounts of PARP1 protein bands (D). The statistical values were normalized to overexpressed KIF4 bands. The biotinylated PARP1 was clearly detected in a lysate of N2A cells expressing Mut-KIF4. Data are presented as mean ± SEM (n = 3 independent experiments). ***P < 0.001 (Student’s t tests). (E and F) The survival assay using CMFDA with or without indicated reagents. Representative images of hippocampal culture neurons (E), and quantification of survival rates for each condition (F). Scale bar, 50 µm. Data are presented as mean ± SEM (n = 3 independent experiments). ns, P > 0.05, ****P < 0.0001 (two-way ANOVA). (G and H) Hippocampal neurons immunostained with anti-PAR and anti-MAP2 antibodies (G), and quantification of the normalized fluorescent intensity for each condition (H). Scale bar, 10 µm. Data are presented as mean ± SEM (n = 60, 3 independent experiments). ns, P > 0.05, ****P < 0.0001 (Student’s t tests). Source data are available for this figure: SourceData F4.
Mut-KIF4 strongly binds to PARP1, whose activity influences cell survival rate. (A and B) Co-immunoprecipitation (Co-IP) immunoblotting results of the primary culture neuron lysates (A), and quantification of amounts of 110 kD PARP1 bands (B). The statistical values were normalized to relevant bands in the input lanes. The input samples and immunoprecipitated complexes including KIF4 were analyzed by immunoblotting using an antibody specific to PARP1; IP, immunoprecipitation; IB, immunoblotting; IgG, immunoglobulin G; Cyt., Cytosol; Nuc., Nuclear. Data are presented as mean ± SEM (n = 3 independent experiments). ***P < 0.001 (Student’s t tests). (C and D) Immunoblotting results of BioID pulldown (C), and quantification of amounts of PARP1 protein bands (D). The statistical values were normalized to overexpressed KIF4 bands. The biotinylated PARP1 was clearly detected in a lysate of N2A cells expressing Mut-KIF4. Data are presented as mean ± SEM (n = 3 independent experiments). ***P < 0.001 (Student’s t tests). (E and F) The survival assay using CMFDA with or without indicated reagents. Representative images of hippocampal culture neurons (E), and quantification of survival rates for each condition (F). Scale bar, 50 µm. Data are presented as mean ± SEM (n = 3 independent experiments). ns, P > 0.05, ****P < 0.0001 (two-way ANOVA). (G and H) Hippocampal neurons immunostained with anti-PAR and anti-MAP2 antibodies (G), and quantification of the normalized fluorescent intensity for each condition (H). Scale bar, 10 µm. Data are presented as mean ± SEM (n = 60, 3 independent experiments). ns, P > 0.05, ****P < 0.0001 (Student’s t tests). Source data are available for this figure: SourceData F4.
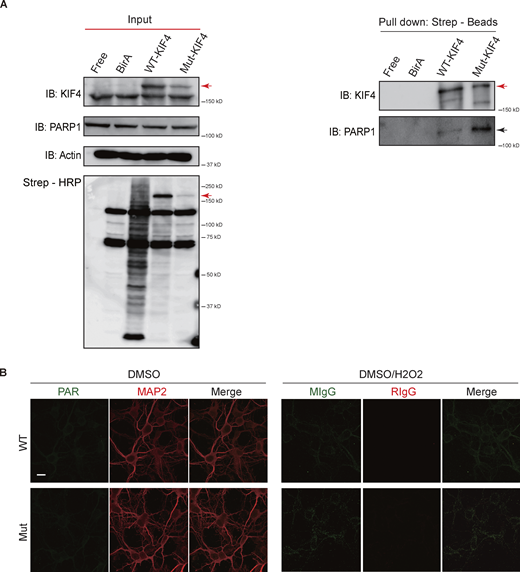
Binding strength between KIF4 and PARP1 was stronger in mutant KIF4. (A) Immunoblotting results of BioID. BirA indicated the mock vector transfected cells. Input or pull-down lysates were blotted with the indicated antibodies. Total biotinylated proteins were confirmed by streptavidin-conjugated horse radish peroxidase (Strep-HRP). Red arrows indicate the overexpressed KIF4 band and the black arrow indicates endogenous PARP1. (B) Immunocytochemistry of primary neurons using the indicated antibodies, mouse (MIgG), and rabbit IgG (RIgG); Scale bar, 10 μm. Source data are available for this figure: SourceData FS3.

Binding strength between KIF4 and PARP1 was stronger in mutant KIF4. (A) Immunoblotting results of BioID. BirA indicated the mock vector transfected cells. Input or pull-down lysates were blotted with the indicated antibodies. Total biotinylated proteins were confirmed by streptavidin-conjugated horse radish peroxidase (Strep-HRP). Red arrows indicate the overexpressed KIF4 band and the black arrow indicates endogenous PARP1. (B) Immunocytochemistry of primary neurons using the indicated antibodies, mouse (MIgG), and rabbit IgG (RIgG); Scale bar, 10 μm. Source data are available for this figure: SourceData FS3.
The PARP1 activity has been shown to underlie a protection of neuron from apoptosis (Nicoletti and Stella, 2003). To compare the PARP1 activity between the genotypes, we performed hippocampal culture neuron survival assay using the fluorescence probe, 5-chloromethylfluorescein diacetate (CMFDA), which accumulates only in live cells (Sebastià et al., 2003). We first compared cell survival rates for each genotype of culture neurons kept in a medium containing B27 serum-free supplement with or without PARP1 inhibitor, 1,5-dihydroxyisoquinoline (DHIQ). There was no difference in survival rate between the individual KIF4 variants when cultured in DHIQ-free conditions, while, in the presence of DHIQ, the cell survival rate of mutant neurons was substantially decreased compared with that of wild-type neurons (Fig. 4, E and F). On the other hand, after 6 d of B27 deprivation, the cell survival rate of mutant neurons was much lower than that of wild-type neurons (Fig. 4, E and F). Accordingly, we examined whether PARP1 activity could prevent the neurons deprived of B27 supplementation from inducing apoptosis. In order to activate PARP1, we supplied a high-KCl medium or a treatment with nicotinamide adenine dinucleotide (NAD), one of the specific substrates of PARP1 (Midorikawa et al., 2006; Murata et al., 2019), to the culture neurons as additional chemical components. In wild-type neurons, there was no significant difference in survival rates with KCl and NAD treatments, which indicated that PARP1 activity did not affect apoptotic induction pathway in these conditions. However, KCl and NAD treatments recovered the survival rates in mutant neurons without B27 supplementation, demonstrating that the PARP1 activity facilitated the cell survival of the mutant neurons in the B27-free conditions (Fig. 4, E and F). Furthermore, additional DHIQ treatment was enough to eliminate the rescue effects of KCl and NAD in mutant neurons (Fig. 4, E and F). Together, these data suggested that Mut-KIF4 can suppress PARP1 activity through a stronger binding affinity, which affects the antiapoptotic pathway. Additionally, to confirm this suggestion, we performed immunocytochemistry using an antibody against PAR which is a PARP1 catalytic product and measured the PAR fluorescence intensity in nuclei of MAP2-positive cells under different culture conditions. Within 5 min after 1 mM H2O2-induced stimulation, wild-type neurons exhibited significantly higher PAR levels than mutant neurons (Fig. 4, G and H). There was no significant difference in PAR levels between the genotypes when the neurons were treated with DHIQ, indicating that DHIQ abrogated the PAR production (Fig. 4, G and H). PAR-positive fluorescent signals could not be detected under H2O2 free condition (Fig. S3 B). These results strengthened our hypothesis that Mut-KIF4 strictly suppresses PARP1 activity.
Histological analysis revealed a hyper-branched phenotype of CA3 pyramidal neurons from Kif4Mut/Y mice
To investigate the possible irregular branches of Kif4Mut/Y neurons, we initially visualized the morphology of a single neuron in the hippocampus ex vivo using the Golgi staining technique. A total of 20 cells from five pairs of Kif4WT/Y and Kif4Mut/Y mouse hippocampal CA3 pyramidal neurons were selected and subsequently analyzed with the Sholl method. Hyper-branched phenotype was observed in the Kif4Mut/Y neurons (Fig. 5 A). To further verify the specification of CA3 pyramidal neuron morphology, we crossed the Kif4Mut/Y mice with Thy1-EGFP transgenic mice (M-line) in which EGFP is genetically encoded downstream of the Thy1 promoter and selectively expressed in the hippocampal pyramidal neurons (Feng et al., 2000). A total 20 EGFP-expressing cells from five pairs of Kif4WT/Y-Thy1+ and Kif4Mut/Y-Thy1+ mouse hippocampal CA3 pyramidal neurons were selected. The analysis showed that the mutant group exhibited a significant hyper-branched phenotype (Fig. 5 B).

The hyper-branched phenotype induced by the KIF4 mutation is accompanied by altered TrkB-KCC2 pathway. (A) Golgi staining of the hippocampal CA3 pyramidal neurons of individual mice at P21; Scale bar, 25 µm. Sholl analysis for the dendritic arborization of each genotypic neuron. Data are presented as mean ± SEM (n = 20, from 5 mouse pairs). *P < 0.05, **P < 0.01, ***P < 0.001 (two-way ANOVA). (B) Representative images of GFP-expressing CA3 pyramidal neurons from individual mice at P90; Scale bar, 20 µm. Sholl analysis for the dendritic arborization of each genotypic neuron. Data are presented as mean ± SEM (n = 20, from 5 mouse pairs). **P < 0.01, ***P < 0.001, ****P < 0.0001 (two-way ANOVA). (C and D) Immunoblotting of M1, Hippo (C) and primary neuron (D) lysates from individual samples using the indicated antibodies and the statistics of the expression ratio. Quantification of individual protein bands with reference to tubulin and actin control bands. Data are presented as mean ± SEM (n = 6 independent experiments). ns, P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (two-way ANOVA). (E) Immunocytochemistry images of hippocampal culture neurons (DIV 18). Both the surface (top panel) and internal (bottom panel) TrkB were detected; Scale bars, 10 µm in the images of WT and Mut, and 30 µm in the images of IgG. Data are presented as mean ± SEM (n = 20, four independent experiments). ****P < 0.0001 (Student’s t tests). (F) The RT-qPCR assay revealed the markedly increased mRNA expression of trkb fl, trkb t1, and decreased kcc2 in the mutant hippocampal and primary culture neurons. Primary neurons cultured under 5 μm AraC condition. Quantification of individual mRNA level with reference to act control. Data are presented as mean ± SEM (n = 6 independent experiments). ns, P > 0.05, **P < 0.01, ****P < 0.0001 (two-way ANOVA). Source data are available for this figure: SourceData F5.
The hyper-branched phenotype induced by the KIF4 mutation is accompanied by altered TrkB-KCC2 pathway. (A) Golgi staining of the hippocampal CA3 pyramidal neurons of individual mice at P21; Scale bar, 25 µm. Sholl analysis for the dendritic arborization of each genotypic neuron. Data are presented as mean ± SEM (n = 20, from 5 mouse pairs). *P < 0.05, **P < 0.01, ***P < 0.001 (two-way ANOVA). (B) Representative images of GFP-expressing CA3 pyramidal neurons from individual mice at P90; Scale bar, 20 µm. Sholl analysis for the dendritic arborization of each genotypic neuron. Data are presented as mean ± SEM (n = 20, from 5 mouse pairs). **P < 0.01, ***P < 0.001, ****P < 0.0001 (two-way ANOVA). (C and D) Immunoblotting of M1, Hippo (C) and primary neuron (D) lysates from individual samples using the indicated antibodies and the statistics of the expression ratio. Quantification of individual protein bands with reference to tubulin and actin control bands. Data are presented as mean ± SEM (n = 6 independent experiments). ns, P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (two-way ANOVA). (E) Immunocytochemistry images of hippocampal culture neurons (DIV 18). Both the surface (top panel) and internal (bottom panel) TrkB were detected; Scale bars, 10 µm in the images of WT and Mut, and 30 µm in the images of IgG. Data are presented as mean ± SEM (n = 20, four independent experiments). ****P < 0.0001 (Student’s t tests). (F) The RT-qPCR assay revealed the markedly increased mRNA expression of trkb fl, trkb t1, and decreased kcc2 in the mutant hippocampal and primary culture neurons. Primary neurons cultured under 5 μm AraC condition. Quantification of individual mRNA level with reference to act control. Data are presented as mean ± SEM (n = 6 independent experiments). ns, P > 0.05, **P < 0.01, ****P < 0.0001 (two-way ANOVA). Source data are available for this figure: SourceData F5.
The suppression of PARP1 activity driven by Mut-KIF4 affected the expression of TrkB-KCC2 epilepsy-related pathway
PARP1 is a well-known epigenetic factor that induces critical physiological changes (Ciccarone et al., 2017). To investigate whether PARP1 affects the expression levels of epilepsy-related proteins, we performed immunoblotting with homogenates of the M1 and Hippo regions from Kif4WT/Y and Kif4Mut/Y brains. Intriguingly, full-length and truncated TrkB protein expression levels were substantially higher in both regions from Kif4Mut/Y brains, while PARP1 expression levels remained the same (Fig. 5 C). On the other hand, the expression levels of KCC2, which is regulated by TrkB activity and maintains chloride homeostasis (Rivera et al., 2002), and PAR were significantly lower in both regions from Kif4Mut/Y brains. The expression levels of other epilepsy-related proteins including GluR1 and GABAA receptor (GABAAR), a PTZ-targeted chloride channel, in the brain regions of Kif4Mut/Y mice did not significantly change compared with those of Kif4WT/Y mice (Fig. 5 C).
To further understand the TrkB expression profile between wild-type and mutant neurons, we performed immunoblotting and immunocytochemistry using the dissociated hippocampal culture neurons. To exclude the glia effects, we extracted total proteins from the neurons 18 d in vitro (DIV 18) cultured under cytarabine (AraC, 5 μM), then lysates were analyzed by immunoblotting, which showed that full-length and truncated TrkB protein levels were significantly higher in the mutant neurons (Fig. 5 D). This upregulation triggered the activation of the brain-derived neurotrophic factor (BDNF)-TrkB pathway, by which phosphorylation of the protein kinase B (AKT) and the extracellular signal-regulated protein kinase 1/2 (ERK1/2) was facilitated in the mutant neurons, while the expression levels of PAR and KCC2 were lower (Fig. 5 D). The protein levels of GABAAR, AKT, ERK1/2, and KIF4 did not exhibit significant difference (Fig. 5 D). To identify the internal/surface expression levels of TrkB (full-length and truncated isoforms), we treated culture neurons with or without 0.5% TritonX-100 for 5 min to permeabilize the cell membrane. Immunocytochemistry of the hippocampal culture neurons (DIV 18) showed a consistent significant increase in TrkB levels in the mutant neurons both internally and on the surface (Fig. 5 E). To further evaluate the effects of mutant kif4 upon the TrkB-KCC2 pathway, we determined the mRNA levels of the full-length trkb (trkb-fl), truncated trkb (trkb-t1), kcc2, kif4, and parp1 genes. The hippocampal and primary culture samples revealed that the two isoforms of trkb were significantly higher and kcc2 was significantly lower in the mutant group (Fig. 5 F).
ANA-12 exhibited anti-epileptic effect and recovery of KCC2 expression in the ischemia induced seizure mouse model (Carter et al., 2018). Immunoblotting results revealed that the treatment of wild-type neurons with DHIQ (10 μM) dramatically increased TrkB protein expression at nearly the same levels as mutant neurons. The decrease in KCC2 expression was accompanied by an increase in TrkB activity. Further TrkB inhibition by ANA-12 (20 μM) rescued the KCC2 expression levels (Fig. 6, A–D). These results indicated that the suppression of PARP1 activity driven by Mut-KIF4 increased the expression of TrkB and decreased the expression of KCC2, one of the downstream chloride ion channels, through the TrkB activity.

The KIF4 mutation affects TrkB signaling through suppression of PARP1 activity. (A–D) Hippocampal culture neurons are cultured under 5 μm Arac condition. Immunoblotting of the hippocampal culture neuron lysates (DIV 18) using the indicated antibodies (A) and the statistics of the expression ratios (B–D). The expression ratios of TrkB FL (B), TrkB p705 (C), and KCC2 (D) are represented. Quantification of individual protein bands with reference to actin control bands. Data are presented as mean ± SEM (n = 6, three independent experiments). n.s., P > 0.5, ***P < 0.001, ****P < 0.0001 (two-way ANOVA). (E–I) Immunoblotting of the hippocampal culture neuron lysates (DIV 18) using the indicated antibodies (E) and the statistics of the expression ratio (F–I). The expression ratios of TrkB FL (F), TrkB T1 (G), PAR (H), and KCC2 (I) are represented. Quantification of individual protein bands with reference to tubulin and actin control bands. Data are presented as mean ± SEM (n = 6, three independent experiments). n.s., P > 0.5, **P < 0.01, ***P < 0.001, ****P < 0.0001 (two-way ANOVA). (J–K) Immunocytochemistry of primary neurons using the indicated antibodies, and IgG (J) and the statistics of the fluorescence density (K); Scale bar, 10 μm. Data are presented as mean ± SEM (n = 20, 4 independent experiments). **P < 0.01, ****P < 0.0001 (two-way ANOVA). Source data are available for this figure: SourceData F6.
The KIF4 mutation affects TrkB signaling through suppression of PARP1 activity. (A–D) Hippocampal culture neurons are cultured under 5 μm Arac condition. Immunoblotting of the hippocampal culture neuron lysates (DIV 18) using the indicated antibodies (A) and the statistics of the expression ratios (B–D). The expression ratios of TrkB FL (B), TrkB p705 (C), and KCC2 (D) are represented. Quantification of individual protein bands with reference to actin control bands. Data are presented as mean ± SEM (n = 6, three independent experiments). n.s., P > 0.5, ***P < 0.001, ****P < 0.0001 (two-way ANOVA). (E–I) Immunoblotting of the hippocampal culture neuron lysates (DIV 18) using the indicated antibodies (E) and the statistics of the expression ratio (F–I). The expression ratios of TrkB FL (F), TrkB T1 (G), PAR (H), and KCC2 (I) are represented. Quantification of individual protein bands with reference to tubulin and actin control bands. Data are presented as mean ± SEM (n = 6, three independent experiments). n.s., P > 0.5, **P < 0.01, ***P < 0.001, ****P < 0.0001 (two-way ANOVA). (J–K) Immunocytochemistry of primary neurons using the indicated antibodies, and IgG (J) and the statistics of the fluorescence density (K); Scale bar, 10 μm. Data are presented as mean ± SEM (n = 20, 4 independent experiments). **P < 0.01, ****P < 0.0001 (two-way ANOVA). Source data are available for this figure: SourceData F6.
Altered TrkB signaling and higher intracellular chloride concentration in the mutant neuron were rescued by PARP1 activation
Afterward, we performed immunoblotting to confirm pharmacological effects on the KIF4-PARP1-TrkB-KCC2 pathway using total protein lysates from the culture neurons (DIV 18) pretreated with NAD and DHIQ individually or together. NAD treatment to mutant neurons up-regulated the PARP1 activity and production of PAR, and rescued TrkB and KCC2 expression levels (Fig. 6, E–I). The increased TrkB expression in wild-type neurons with DHIQ application was rescued by a further addition of 20 μM NAD. Collectively, these results demonstrated that PARP1 activity regulated the TrkB-KCC2 expression. To further understand the alteration of KCC2 expression level, we performed immunocytochemistry of the hippocampal culture neurons (DIV 18). The result showed a consistent significant decrease in KCC2 level in the mutant neurons on the cell surface, and the additional NAD rescued the KCC2 protein expression (Fig. 6, J and K).
We next visualized the intracellular chloride concentration using a live cell ratio metric chloride sensor, ClopHensorN (Werley et al., 2020), in order to examine how chloride homeostasis was regulated in mutant neurons. Intracellular chloride calibration was conducted by adding an ionophore cocktail to the gradient chloride buffer. We confirmed using wild-type primary neurons that ClopHensorN reduced the green/red fluorescent intensity ratio with higher chloride concentration, as expected (Fig. 7 A). We then applied ClopHensorN to the wild-type and mutant culture neurons (DIV 16). The green/red fluorescence ratio in the mutant neurons was significantly lower than the wild-type neurons, indicating a higher intracellular chloride concentration (Fig. 7 B). This low chloride ion phenotype in the mutant neurons was rescued by a further addition of 20 μM NAD or 10 μM ANA-12. These results suggested that the suppression of PARP1 activity induced by Mut-KIF4 altered TrkB-KCC2 pathway, leading to the impaired intracellular chloride homeostasis.

The concentration of intracellular chloride is altered by KIF4 mutation in a PARP1-TrkB dependent manner in hippocampal primary neurons. (A) Calibration of ClopHensorN fluorescence in the hippocampal neurons. Scale bar, 20 µm. Statistical results exhibited green/red fluorescence ratio decreasing with intracellular chloride concentration gradient. Data are presented as mean ± SEM (n = 30, 3 independent experiments). (B) ClopHensorN fluorescence images of hippocampal culture neurons (DIV 18) demonstrating a higher intracellular chloride concentration in the mutant neurons and the NAD or ANA-12 could rescue this phenotype. Scale bar, 20 µm. Data are presented as mean ± SEM (n = 30, 3 independent experiments). ***P < 0.001, ****P < 0.0001 (two-way ANOVA).
The concentration of intracellular chloride is altered by KIF4 mutation in a PARP1-TrkB dependent manner in hippocampal primary neurons. (A) Calibration of ClopHensorN fluorescence in the hippocampal neurons. Scale bar, 20 µm. Statistical results exhibited green/red fluorescence ratio decreasing with intracellular chloride concentration gradient. Data are presented as mean ± SEM (n = 30, 3 independent experiments). (B) ClopHensorN fluorescence images of hippocampal culture neurons (DIV 18) demonstrating a higher intracellular chloride concentration in the mutant neurons and the NAD or ANA-12 could rescue this phenotype. Scale bar, 20 µm. Data are presented as mean ± SEM (n = 30, 3 independent experiments). ***P < 0.001, ****P < 0.0001 (two-way ANOVA).
A Kif4 mutation-induced hyper-branched phenotype was regulated by TrkB signaling and was rescued by enhancement of PARP1 activity
To further understand the cellular characteristics of KIF4-mutant neurons, we examined the morphology by a Sholl analysis to quantify dendritic branching in hippocampal culture neurons expressing EGFP protein. First, we found that mutant neurons exhibited the substantially increased number of dendritic branches compared with the wild type (Fig. 8, A and B). Then we treated neurons with DHIQ and TrkB-specific inhibitor, ANA-12 (10 μM), at DIV 16 for 2 d combinedly or respectively and found that the hyper-branched phenotype caused by PARP1 inhibition was rescued by reduced TrkB activity (Fig. 8, A and B). These results indicated that KIF4 mutation induced neuronal branching through the TrkB activation. To further understand whether PARP1 activity influences the neuronal morphology, culture neurons at DIV 16 were treated with 10 μM DHIQ or 10 μM NAD for 2 d. Both wild-type and mutant neurons exhibited hyper-branched phenotype with DHIQ treatment, but the mutant neurons were found to be more branched. However, treatment with NAD restored in both neuron phenotypes. Furthermore, overexpression of Mut-KIF4 in the wild-type neurons also induced a dendritic hyper-branched phenotype as in the mutant neurons (Fig. 8, C and E). To determine whether the enhancement of PARP1 activity could rescue the mutant-induced hyper-branched phenotype, we overexpressed PARP1 in both the wild-type and mutant neurons, resulting in cell death and suppression for the hyper-branched phenotype, respectively (Fig. 8, D and F). Additionally, this morphological alteration of mutant neurons was reversed by addition of 10 μM DHIQ to the medium. The hyper-branched phenotype in mutant neurons with both PARP1 overexpression and DHIQ application was rescued by a further addition of 20 μM NAD (Fig. 8, D and F). These results suggested that KIF4 had a critical role in neuronal branching through the regulation of PARP1 enzymatic activity.

Aberrant dendritic morphology in mutant neurons is regulated by TrkB and is rescued by enhancement of PARP1 activity. (A) Fluorescence microscopic images of hippocampal culture neurons (DIV 18) from the indicated genotypes. DHIQ or ANA treatments were performed. (B) Quantification of Sholl analysis for the dendritic arborization of neurons in A. (C and D) Fluorescence microscopic images of hippocampal culture neurons (DIV 18) from the indicated genotypes. DHIQ or NAD treatments (both C and D) and transfection with Mut-KIF4-EGFP in C or PARP1-tagRFP in D were performed. Scale bars, 50 µm. D/N, both DHIQ and NAD. (E and F) Quantification of Sholl analysis for the dendritic arborization of neurons in A and B. Data are presented as mean ± SEM (n = 18, 3 independent experiments). ns, P > 0.05, ****P < 0.0001 (two-way ANOVA).
Aberrant dendritic morphology in mutant neurons is regulated by TrkB and is rescued by enhancement of PARP1 activity. (A) Fluorescence microscopic images of hippocampal culture neurons (DIV 18) from the indicated genotypes. DHIQ or ANA treatments were performed. (B) Quantification of Sholl analysis for the dendritic arborization of neurons in A. (C and D) Fluorescence microscopic images of hippocampal culture neurons (DIV 18) from the indicated genotypes. DHIQ or NAD treatments (both C and D) and transfection with Mut-KIF4-EGFP in C or PARP1-tagRFP in D were performed. Scale bars, 50 µm. D/N, both DHIQ and NAD. (E and F) Quantification of Sholl analysis for the dendritic arborization of neurons in A and B. Data are presented as mean ± SEM (n = 18, 3 independent experiments). ns, P > 0.05, ****P < 0.0001 (two-way ANOVA).
KIF4-regulated PARP1 activity altered dendritic spine morphology
To determine whether the KIF4 mutation and PARP1 activity are sufficient to alter spine density and morphology, we alternated PARP1 activation with inhibition in hippocampal culture neurons (DIV 18). Both endogenous and exogenous expression of mutant KIF4 induced elongated dendritic spines, which was rescued by overexpression of PARP1 or additional NAD treatment (Fig. S4, A–C). Notably, the density of mushroom and stubby dendritic spines positively correlated with PARP1 activity. The overall density of dendritic protrusions did not vary among individual treatments (Fig. S4, A–C).

Aberrant dendritic spine morphology in mutant neurons is regulated by TrkB and is rescued by enhancement of PARP1 activity. (A–C) Fluorescence microscopic images of dendritic spines. (A) From the indicated genotypes. Pharmacological treatments and transfections were performed as indicated; scale bar, 2 µm. Quantification of the average length of the spine (B) and the spine density (C) along a 720 µm-long dendrite. Mus., mushroom; Stu., stubby. D, DHIQ; D/N, both DHIQ and NAD. Data are presented as mean ± SEM (n = 18 neurons, three independent experiments, total 540 spines). ns, P > 0.05, ***P < 0.001, ****P < 0.0001 (two-way ANOVA).

Aberrant dendritic spine morphology in mutant neurons is regulated by TrkB and is rescued by enhancement of PARP1 activity. (A–C) Fluorescence microscopic images of dendritic spines. (A) From the indicated genotypes. Pharmacological treatments and transfections were performed as indicated; scale bar, 2 µm. Quantification of the average length of the spine (B) and the spine density (C) along a 720 µm-long dendrite. Mus., mushroom; Stu., stubby. D, DHIQ; D/N, both DHIQ and NAD. Data are presented as mean ± SEM (n = 18 neurons, three independent experiments, total 540 spines). ns, P > 0.05, ***P < 0.001, ****P < 0.0001 (two-way ANOVA).
Pharmacological manipulation of PARP1 activity influenced mouse anxiety-like behavior and seizure susceptibility phenotype
To examine effects of pharmacological manipulations that regulate PARP1 activity on mouse behavior and seizure susceptibility phenotype, we performed intraperitoneal injections of DHIQ or NAD into the Kif4+/Y and Kif4Mut/Y mice. The optimal concentrations of injections were adjusted to 6 mg/kg DHIQ or 300 mg/kg NAD per day for successive 10 and 15 d, respectively (Fig. S5, A–H). DHIQ injection triggered anxiety-like behaviors of Kif4+/Y mice in the OFT, while NAD injection inhibited those of Kif4Mut/Y mice (Fig. S5, I–M). Furthermore, PTZ-induced epilepsy experiments demonstrated that DHIQ injection was enough to trigger the susceptible phenotype with severe seizure stages in Kif4+/Y mice, while the NAD injection successfully rescued the epileptic phenotype in Kif4Mut/Y mice (Fig. 9, A–D and Video 2). These results indicated that the pharmacological manipulation could rescue the phenotypes of Kif4Mut/Y mice.

The optimized activation of PARP1 regulated mice anxiety and epileptic behavior. (A–H) The results of the PTZ kindling test. The chart representing the seizure stage classification of the scores from mice with indicated genotypes and treatments, before and after the PTZ injection (A and E). Statistical analysis results of the cumulative number of maximum (B and F) and all (C and G) seizure stage, and the ratio of the occurrence of stage 4 (D and H). Data are presented as mean ± SEM (n = 10 mouse pairs). **P < 0.01, ****P < 0.0001 (Student’s t tests). (I–M) The heatmap of the occupancy of individual mice with indicated pharmacological treatments in OFT. A warmer color represents more time spent (red > yellow > green > blue), and red rectangles are put at the central zones in the fields (I). The OFT results of individual mice with the indicated treatments. Total distances that mice ran during tests (J), cumulative durations in the edge zone (K) or the central zone (L), and in-zone frequencies to the central zone (M) are represented. Data are presented as mean ± SEM (n = 10 mouse pairs). n.s., P > 0.5, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (two-way ANOVA).

The optimized activation of PARP1 regulated mice anxiety and epileptic behavior. (A–H) The results of the PTZ kindling test. The chart representing the seizure stage classification of the scores from mice with indicated genotypes and treatments, before and after the PTZ injection (A and E). Statistical analysis results of the cumulative number of maximum (B and F) and all (C and G) seizure stage, and the ratio of the occurrence of stage 4 (D and H). Data are presented as mean ± SEM (n = 10 mouse pairs). **P < 0.01, ****P < 0.0001 (Student’s t tests). (I–M) The heatmap of the occupancy of individual mice with indicated pharmacological treatments in OFT. A warmer color represents more time spent (red > yellow > green > blue), and red rectangles are put at the central zones in the fields (I). The OFT results of individual mice with the indicated treatments. Total distances that mice ran during tests (J), cumulative durations in the edge zone (K) or the central zone (L), and in-zone frequencies to the central zone (M) are represented. Data are presented as mean ± SEM (n = 10 mouse pairs). n.s., P > 0.5, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (two-way ANOVA).

The in vivo PARP1 activity regulates epileptic behavior (A-D) and graphical model summarizing the molecular mechanism of the KIF4 mutation-induced epilepsy (E). (A–D) The results of the PTZ kindling test. The chart representing the seizure stage classification (A) of the individual scores from mice with indicated genotypes and treatments, before and after the PTZ injection. Statistical analysis results of the cumulative number of maximum (B) and all (C) seizure stage, and the ratio of the occurrence of stage 4 (D). Data are presented as mean ± SEM (n = 10 mouse pairs). ****P < 0.0001 (two-way ANOVA). (E) Schematic model of the involvement of KIF4 in epileptogenesis. (a) The WT-KIF4 moderately binds to PARP1, and the cell exhibits a baseline level of PAR in the nucleus. (a′) Mut-KIF4 excessively binds to PARP1, and the cell exhibits a dramatically inhibited PARP1 activity. (b and b′) The suppressed PARP1 activity can alter the TrkB-KCC2 expression in the mutant group. Meanwhile, the aberrant protein expression triggers a higher intracellular chloride concentration. (c–d′) Subsequently, the hyper-branched dendrites and immature dendritic spines are dominantly observed in the mutant neuron.
The in vivo PARP1 activity regulates epileptic behavior (A-D) and graphical model summarizing the molecular mechanism of the KIF4 mutation-induced epilepsy (E). (A–D) The results of the PTZ kindling test. The chart representing the seizure stage classification (A) of the individual scores from mice with indicated genotypes and treatments, before and after the PTZ injection. Statistical analysis results of the cumulative number of maximum (B) and all (C) seizure stage, and the ratio of the occurrence of stage 4 (D). Data are presented as mean ± SEM (n = 10 mouse pairs). ****P < 0.0001 (two-way ANOVA). (E) Schematic model of the involvement of KIF4 in epileptogenesis. (a) The WT-KIF4 moderately binds to PARP1, and the cell exhibits a baseline level of PAR in the nucleus. (a′) Mut-KIF4 excessively binds to PARP1, and the cell exhibits a dramatically inhibited PARP1 activity. (b and b′) The suppressed PARP1 activity can alter the TrkB-KCC2 expression in the mutant group. Meanwhile, the aberrant protein expression triggers a higher intracellular chloride concentration. (c–d′) Subsequently, the hyper-branched dendrites and immature dendritic spines are dominantly observed in the mutant neuron.
Related toFig. 9 A . Pentylenetetrazol (PTZ) kindling test of representative mouse behavior injected with the indicated compound. Note that DHIQ induced epilepsy in Kif4WT/Y mouse and NAD + rescued the epileptic sensitivity of Kif4Mut/Y mice.
Related toFig. 9 A . Pentylenetetrazol (PTZ) kindling test of representative mouse behavior injected with the indicated compound. Note that DHIQ induced epilepsy in Kif4WT/Y mouse and NAD + rescued the epileptic sensitivity of Kif4Mut/Y mice.
Discussion
In this study, we demonstrate that the KIF4 mutation (R728Q) strikingly affects higher brain functions. Kif4Mut/Y mice exhibit a seizure-susceptibility phenotype with an irregular spike-and-slow-wave complex pattern in the EEG recording. Furthermore, our data indicate that the KIF4 mutation strengthens the interaction with PARP1 and influences cell survival, neuron branching, and dendritic spine morphology. Concomitant with these findings, we show that the KIF4 mutant neuron exhibits dysregulation of TrkB-KCC2 signaling pathway, leading to the impairment in intracellular chloride homeostasis. As PARP1 activation induced by the pharmacological treatment rescues the phenotypes in vitro and in vivo, our data demonstrate the importance of KIF4 and PARP1 in preventing epilepsy (Fig. 9 E).
KIF4 mutant mouse serves as a novel model for understanding the molecular mechanism of seizure susceptibility
Although the roles of KIF4 in cell division, cell death, and chromosome configuration have been extensively studied, the lethality by full knocking out the Kif4 gene has dampened the development of research for a relevant mouse model (Hirokawa and Tanaka, 2015). Previous studies have used Kif4 knockout embryonic stem (ES) cells to investigate the function of KIF4 (Midorikawa et al., 2006), causing that the in vivo fundamental aspects remain poorly understood. The embryonic mortality rate in the KIF4 mutant group studied here is ∼27%, and the surviving offspring exhibited a smaller population of Kif4Mut/Y male mice than the female mutant carriers including homozygous (Fig. 1). These results indicate that the Kif4 mutation is a fundamental semi-lethal allele, which is much more dominant in male. Mutant mouse exhibits weight loss, size reduction, and developmental delay phenotypes that are likely caused by multiple mechanisms including losing interactions with the protein regulator of cytokinesis (PRC) protein, malfunctioning spindle formation, or dysregulated Aurora B-mediated phosphorylation of KIF4 (Bastos et al., 2013).
An epileptic mouse model can be established through several different tools, including neurochemical reagents, electoral stimulation, hypoxic, injury, and genetic disorder-induced seizure (Kandratavicius et al., 2014), although these tools have each limitation. For example, a famous chemoconvulsant, kainic acid, shows a high mortality rate and unspecified neural toxicity (Sharma et al., 2007). In the hypoxia model, age and strain are common seizure susceptibility factors (Jensen et al., 1991). Some KIFs are already reported to contribute to various epileptogenesis by our group. For instance, loss of KIF2A triggers hippocampal epilepsy through many aberrantly elongated dendrites in the dentate granule cells (Homma et al., 2018), whereas KIF5A knockout mice shows epilepsy through reduced cell surface expression of GABAAR and impaired GABAAR-mediated synaptic transmission (Nakajima et al., 2012). In our present study, the Kif4Mut/Y mice displayed an epileptic phenotype following PTZ compound injection (Fig. 3). The specific mechanism causing this observation needs to be more elucidated to accelerate the development of specific epilepsy therapeutics (Löscher and Schmidt, 1988). KIF4 mutation induces weight loss, mild developmental delay, intellectual disability, and decreased epileptic threshold in mice (Figs. 1, 2, and 3). Since the Kif4 contains differences in the genome between humans and mice, this might explain why the patient with the Kif4a mutation shows a more severe phenotype. Furthermore, we believe that the KIF4 mutant mouse is a novel tool for investigating unknown functions of KIF4 both in vitro and in vivo, providing a better way to save expenditures and time.
A coiled–coil domain of Mut-KIF4 exhibits a gain-of-function phenotype
In recent years, next-generation sequencing analyses have revealed many critical single nucleotide polymorphisms (SNPs) involved in epileptogenesis (Ellis et al., 2020). However, it is difficult to understand the effects of a SNP in the coiled–coil domain using crystallography analysis because of this region’s limited flexibility and stability. Even though many bioinformatics tools have been developed to explore SNP-induced changes in coiled–coil domains, understanding its effects on the whole protein’s function also remains difficult (Kalman et al., 2020). The patient exhibiting epileptic phenotypes in our study has a mutation in the X chromosome gene Kif4a, which inspired us to investigate the detailed mechanisms of KIF4 in neurons using transgenic mouse model techniques (Fig. 1). The KIF4 mutation studied here causes the elongation of the coiled–coil domain, which triggers strengthened affinity for PARP1 and dramatically suppresses its activation (Fig. 4). Furthermore, neuron survival assays suggest that KIF4 is critical in PARP1 activity-regulated cell death (Fig. 4). Contrarily, a disease-causing SNP in a DNA repair protein 50 (RAD50), which dropped the length of coiled–coil domain, exhibits a loss-of-function phenotype (Chansel-Da Cruz et al., 2020). These pieces of evidence imply that the length of the coiled–coil domain could be directly related to protein function. Hyperactivated PARP1 induces mitochondria to release apoptosis-inducing factor (AIF) or triggers intracellular ATP over consumption which could cause cell death (Yu et al., 2002; Chiarugi, 2002). On the opposite, suppression of PARP1 also triggers cell death through disrupted DNA homeostasis (Zhang et al., 2014). Our pharmacological and genetic data support the view that the optimal activity of PARP1 is critical for cell survival (Fig. 4 and Fig. 8 D). DHIQ treatment was continued for five consecutive days in the neuron survival assay. However, we observed a hyper-branched phenotype in neurons that were treated with DHIQ only for 2 d. These data indicated that different levels of PARP1 suppression could cause different cell fates (Fig. 4 and Fig. 8 D).
Interestingly, a different genomic modification in Kif4a, the loss of exon 15, triggers a similar phenotype in human with mild intellectual disability and epilepsy (Willemsen et al., 2014). Both exon 15 and 19 in Kif4a code for the coiled–coil region. This subdomain widely exists in KIFs including the KIF5, KIF13, and KIF21A, and plays critical roles in autoinhibition to regulate kinesin activity (Bianchi et al., 2016). KIF21A, which belongs to the same kinesin-4 subfamily as KIF4, exhibits disrupted autoinhibition by affecting the structural integrity of the coiled–coil domain through a point mutation, which causes decreased microtubules affinity and congenital fibrosis of the extraocular muscles type 1 (CFEOM1; Bianchi et al., 2016). Another kinesin-4 subfamily member, KIF7, strongly binds to microtubules following dysfunctional autoinhibition resulting from a missense mutation or deletion in the coiled–coil domain (Lynne Blasius et al., 2021). Willemsen et al. (2014) reported that PARP1 binds to the intact tail-domain of KIF4A (Willemsen et al., 2014). The lack of exon 15 may induce disrupted autoinhibitory regulation and result in an altered binding capacity between KIF4A and PARP1, consequently affecting PARP1 activity, which should be further elucidated.
The KIF4-PARP1 axis contributes to seizure susceptibility working as a potential antiepileptic agent target
So far, in vivo and in vitro studies have demonstrated that disruption of proper development of neuronal morphology is involved in epileptogenesis (Llorens-Martín et al., 2016). The reduction of Kif3b can induce a hyper-branched phenotype in cortical pyramidal cells (Yoshihara et al., 2021). Additionally, Kif26a knockout mouse with allodynia exhibits the dorsal root ganglion (DRG) sensory neuron to exuberant growth its neurites (Wang et al., 2018). In this study, we confirmed both in vitro and in vivo that hippocampal pyramidal cells in Kif4Mut/Y mouse display hyper-branched morphology (Figs. 5 and 8). Some studies have revealed that overexpression of TrkB, both the full-length and the truncated versions, causes an increased number of dendrites (Yacoubian and Lo, 2000). Here, we manipulated PARP1 activity using genetic and pharmacological tools and repeatedly confirmed that KIF4-PARP1 is involved in dendritic growth (Fig. 8). Furthermore, we observed that TrkB signaling and dendritic spine morphology are affected by the KIF4 mutation. Abnormal up-regulation of truncated-TrkB level can induce long-thin and filopodia spine formation (Hartmann et al., 2004). An additional possible mechanism is that attenuated expression of KCC2 can also force dendritic spine morphological change (Li et al., 2007). We observed that KIF4-PARP1 is the upstream factor that controls TrkB-KCC2 expression and involves an induction of a deviant intracellular chloride concentration in mutant neurons (Figs. 5, 6, and 7). Treatment of the wild-type neuron with a PARP1 inhibitor triggered a TrkB-KCC2 alteration similar to that seen in the mutant group. Down-regulation of KCC2 expression by TrkB hyperactivity triggered epilepsy through the impairment of intracellular chloride concentration, which could be rescued by the TrkB inhibitor that restores transcription of kcc2 (Rivera et al., 2002, 2004; Carter et al., 2018). Our protein expression and mRNA transcription data revealed that KIF4-PARP1 controls the TrkB-KCC2 pathway by regulating transcription (Figs. 5 and 7). Furthermore, the NAD-induced rescue experiment supports our hypothesis that KIF4 regulates PARP1 activity and contributes to seizure susceptibility through the TrkB-KCC2 axis (Figs. 5, 6, and 7). Various transcription factors, including c-Fos, Arc, CBP/P300, and CREB, can regulate TrkB expression in neurons. However, PARP1-associated proteins like NF-κB or RNA Polymerase II are also involved in transcription, making it difficult to fully evaluate the transcriptional role of KIF4-PARP1 (Vuong et al., 2015). Therefore, the cascade of KIF4 with PARP1-regulated transcription should be further studied.
Over the past decades, PARP1 inhibitors, such as Olaparib, Rucaparib, and Niraparib, have emerged as antitumor agents that have been approved by the U.S. Food and Drug Administration (FDA) and by the European Medicines Agency (EMA). The intraperitoneal injection of these molecules can also attenuate diabetes complications including nerve damage, cardiovascular disease, or kidney damage through suppression of the NF-κB transcription (Szabó et al., 2006). An adult-onset mitochondrial myopathy clinical research reveals that a high dose oral supplement of niacin can raise NAD levels with a simultaneous enhancement of PARP1 activity (Pirinen et al., 2020). In the present study, administration of a PARP1 inhibitor could sufficiently make mouse susceptible to PTZ-induced epilepsy (Figs. 9 and S5). Additional intraperitoneal injection of NAD could rescue this seizure susceptibility phenotype in Kif4Mut/Y mutant mice (Figs. 9 and S5). Oral PARP1 inhibitors are widely used to treat advanced or recurrent ovarian cancers, with 600–800 mg/d commonly used. The side effects such as headaches caused by these medications (Lheureux et al., 2015) indicate the risk and possibility that PARP1 inhibitors may lower the threshold of epilepsy. Another report introduced a specific TrkB inhibitor peptide that can prevent epilepsy (Gu et al., 2015). Overall, our data reveal that KIF4-PARP1 is a potential therapeutic target for the prevention of seizure susceptibility.
In conclusion, our findings demonstrate that the mutation in the KIF4 coiled–coil domain strengthens the binding affinity to its cargo protein, PARP1, leading to the suppression of its activity (Fig. 9 E). This mutation also modifies the TrkB-KCC2 axis, neuronal branching, dendritic spine morphology, and intracellular chloride homeostasis (Fig. 9 E), thereby onsetting the various patient’s symptoms with the same mutation. This study provides a potential antiepileptic target helpful for development of novel therapeutics.
Materials and methods
KIF4 mutant transgenic mouse generation and genotyping
A modified CRISPR transgene method was adopted to generate Kif4 mutation-carrying C57BL/6J mouse line (Kumita et al., 2019). By microinjecting gRNA, hCas9 mRNA, and ssODNs into mouse zygotes, the Kif4 transgenic mouse was established in an efficient HDR way. The sgRNA was designed by searching for ‘GG’ sequences near the point mutation target site and was defined as N(22)GG. The sgRNA used in this study was as follows; 5′-CGCAAAGAGACTCAGAGCCGTGGG-3′.
The synthesized ssODNs which works as the HDR template was described as follows; 5′-ATAACCACTCGGCCTTTTGTTTTTAGGCAGCAGCTGCCAACAAACGTCTTAAGGATGCTCTGCAAAAGCAAAAGGAGGTTGCAGAAAAACAGAAAGAGACTCAATCTCGAGGGATGGAAAGCACTGCAGCTAGAATGAAGGTATGAACACCTCGAACTGCTATTCTCCTTCCATGCACTGGGAATAGGAAATAACTTTCC-3′. The sequence contains segments of the intron 18–19 and intron 19–20 with the total exon 19. The target mutation site followed with several nuclear exchanges could bring the silent mutation on the amino acid sequence (Fig. S2, A and B). This silent mutation site contained a restriction enzyme site of XhoI, which is convenient for the mouse genotyping and limits the post-recombination cleavage (Fig. S2, A and B).
The genotype of the first generation Kif4 transgenic mouse was validated by Sanger sequencing (Fig. S2 C). For the subsequent offspring, genomic PCR and agarose gel electrophoresis were used to identify the genotypes. Briefly, the genomic DNA was extracted from the tail tips of the 3-wk-old mice, and PCR was performed using the following primers: 5′-TGCGCATTCTGAAGCAGTAG -3′ (forward) and 5′-GCTGCAGGACTAGGCTCATC-3′ (reverse). The obtained PCR products were treated by ethanol precipitation and digested by the restrictive enzyme XhoI. Finally, the digested products were loaded on the agarose gel to identify the mouse genotype (Fig. S2, D and E).
Animals and behavioral assessments
All animal experiments were conducted under the University of Tokyo’s restrictions regarding animal experimentation and had been approved by the University of Tokyo Graduate School of Medicine. Mice were housed in a specific-pathogen-free environment. All behavioral tests were performed in a blinded manner, and separate cohorts of mature male mice were used for the following behavioral tests. Postnatal days (P) 1–15 male mice were used for the assessment of developmental milestones test, and 10 to 12-wk-old males were used for the other tests. All the equipment in behavior tests were cleaned with a 75% ethanol solution before and in between testing. 5 d before the test, animals were put in the experimenter room and handled daily for 1 min to reduce the stress.
Open field test (OFT)
Spontaneous locomotor activity and anxious behavior were evaluated using OFT. Mice were placed at the center of a novel rectangular box (55 × 38 cm). All movements were recorded using a digital camera for 15 min and quantified with the EthoVision XT 11.0 software (Noldus Information Technology B.V.). Total distance, central in-zone frequency, and central/edge time spent were measured (Morikawa et al., 2018).
Elevated plus maze (EPM)
Mice were tested for anxiety-related behavior in the EPM test. The mice were placed at the center of the plus-shaped apparatus containing 4 arms (10 cm wide, 50 cm long; two of them have walls) which was elevated 55 cm above the floor. Four arms were similarly illuminated by indirect lighting. All movements were recorded using a digital camera for 15 min and quantified the number of entries and time spent in the open arms (Lueptow, 2017).
Novel object recognition test
Mice were tested for learning and memory-related behavior in the novel object recognition test. Mice were put in a rectangular box (55 × 38 cm) for 10 min per day for three consecutive days with two different objects located in its corners (5 cm from the walls).
On Day 1, we placed the mouse in the box with no objects and allowed it to explore the arenas freely for 6 min. On Day 2, first in the training trail, we put two different objects at the corners and let the mouse investigate the arena and objects freely for 10 min. Then, in the OLT, one of the objects used in the training trail was moved to a new non-release corner, and let the mouse investigate for 10 min. On Day 3, in the NORT, we replaced the object that did not move during the OLT on Day 2 with a novel object and recorded the movements of the mouse using a digital camera for 10 min. The total time of nose dips to the objects was manually counted. The discrimination index was calculated as below; Discrimination index = (time with novel location or object − time with familiar location or object)/(time with novel location or object + time with familiar location or object). If the value approximates to 0, there was no difference in the amount of time that the mouse spent investigating the two objects. And a positive value indicates that the mouse spent more time investigating the novel one (Denninger et al., 2018; Lueptow, 2017).
Fear conditioning test (FCT)
Mice were tested for learning and memory-related behavior in the fear conditioning test. Each mouse was placed in a novel chamber for a total of 10 min, at the middle of the time given a foot shock (1 s, 0.25 mA; unconditioned stimulus), and returned to the home cage 5 min after the foot shock (Day 0). At 1 and 7 d after the conditioning, the mice were placed back in the same chamber for 8 min without any stimulations. All mouse movements were videotaped, and the freezing time during the test was manually calculated.
Assessment of neonatal development milestones
Mice were tested for developmental situation in the assessment of neonatal development milestones test. Pups were labeled at P2 in a well-established two-color tail tattooing method to guarantee identification. This assessment was performed from P3 to P18, including surface righting, negative geotaxis, grasping reflex, cliff aversion, front- and hindlimb suspension, eye/fur/incisor development, eye open, pinnae detachment, auditory, walking, open field, and air righting. These milestones were completed when the mouse was considered to have reached for two consecutive days, and the previous date number was recorded as the finished day (Hill et al., 2008).
Pentylenetetrazole (PTZ) injection
PTZ was obtained from Sigma-Aldrich and prepared by dissolving in sterile saline to a final 2 mg/ml. Animals are given a single intraperitoneal injection of PTZ at a dose of 38 mg/kg. During the next 60 min, all mice were observed for seizures activity according to Shimada’s study (Shimada and Yamagata, 2018).
Histological staining
Mouse brains at E16.5 were dissected and fixed with formalin solution (20% formalin solution; Wako) for 24 h at room temperature (RT). The fixed brains were dehydrated in ethanol series, embedded in paraplast (Sigma-Aldrich), cut into 4 µm thick sections using a rotary microtome (STS10514; MICROM), and stained by the HE method, as previously described (Takei et al., 2000). The HE staining was performed on paraffin sections (4 µm) using standard methodology and stained with hematoxylin (75290; Wako) for 40 s and with eosin (17372; Wako) for 30 s. The tissue sections were examined under a light microscope (1.0 NA 20× objective Olympus) after mounting with Permount mounting medium-malinol 750 cps (No.2009-2, Muto pure chemicals).
Golgi staining
For Golgi-Cox staining, 12-wk-old mice were anesthetized and perfused with Rat Ringer’s solution (156 mM NaCl, 5.4 mM KCl, 5 mM HEPES, 10 mM glucose, 4 mM EGTA, 6 mM MgCl2, and 0.5 mM NaH2PO4). The brain was dissected and subjected to Golgi-Cox staining using a superGolgi Kit (Bioenno Lifesciences) according to the manufacturer’s protocol. The brain was then coronally sectioned using a DTK-1000 micro-slicer (Dosaka-EM) at a thickness of 150 μm. Pyramidal cells from the CA3 of the hippocampus sections from the Kif4WT/Y and Kif4Mut/Y mice were compared and photographed with a DM 3000 microscope (Leica) using a 40×/0.75 Plan Apochromat (Leica) objective lens.
Thy1-EGFP mouse histology
Thy1-EGFP transgenic mice were crossed respectively with Kif4WT/Y and Kif4Mut/Y mice. The Kif4WT/Y-Thy1+ and Kif4Mut/Y-Thy1+ mice were anaesthetized and perfused transcardially with 4% (w/v) paraformaldehyde (PFA) in phosphate-buffered saline (PBS) at 12 wk of age. Then the brains were dissected, fixed, dehydrated, frozen, and coronally sectioned using a cryostat (Leica CM1950) at a thickness of 40 μm. Pyramidal cells from the CA3 layer of the hippocampus sections were compared in the individual mice and photographed with Zeiss LSM780 confocal microscope (63×/1.49 oil Zeiss objective) equipped with an Airyscan module (ZEISS).
Sholl analysis
The number of branches was evaluated through Sholl analysis. An interval of 10 μm concentric rings was drawn on neurons selected using the same criteria. Branch intersections represent the Sholl output every 10 μm, 10–200 μm away from the soma. The above processes were automatically produced by the function of Sholl-analysis of the ImageJ software (National Institutes of Health [NIH]).
Electroencephalogram (EEG) recording
Kif4WT/Y and Kif4Mut/Y siblings were anesthetized at the postnatal week 3 using isoflurane and were surgically implanted with a set of electrodes. Two silver wires with a diameter of 0.1 mm were bonded, including a reference electrode and a working electrode with a hard epoxy resin coat which served to electrically insulate the probe from the reference electrode. The length of the electrode was optimized by ordering Dental cement (GC Dental) was used to fix the electrode set to the skull. The electrode positions in the right hemisphere and the CA1 of the left hippocampus were stereotactically determined as 2.0–2.5 or 1.9–2.4 mm anterior to the bregma and 2.0–2.2 or 1.9–2.1 mm lateral to the midline at a depth of 0.9–1.5 or 0.8–1.4 mm for the Kif4WT/Y and Kif4Mut/Y mice, respectively. These differences were due to the differences in brain size between the two genotypes (Nakajima et al., 2012).
EEG records were obtained using mice 3 d after the surgery was completed. The electrodes, measurement system, and software were all purchased from Unique Medical. The EEG data were arranged and visualized by MATLAB (MathWorks). EEG records were obtained from five pairs of mice from each genotype (Dong et al., 2017).
PTZ post-injected EEG recording
PTZ injection and EEG monitoring were started 73 h after surgery. After the initial monitoring period without any treatments, all animals were subjected to a single intraperitoneal dose of 38 mg/kg PTZ. Continuous digital EEG recording was started 60 min before the PTZ injection and continued for up to 2 h for seizure monitoring.
Immunoblotting
Sample preparation and immunoblotting were performed. For mouse tissue samples, male mice were euthanized by cervical dislocation. The Hippo and M1 were surgically isolated and then homogenized in ice-cold RIPA buffer (50 mM Tris [pH 8.0], 1% Triton X-100 [Sigma-Aldrich], 0.1% SDS [Wako], and 150 mM NaCl) containing a protease inhibitor cocktail (Pis; cOmplete mini EDTA-free, Roche Diagnostics) and a phosphatase inhibitor cocktail (PHOSIs; PhosSTOP EASYpack, Roche Diagnostics). After sonication for 1 min with a duty cycle of 30% and output level of 3 (Sonifer-250; Branson), lysates were centrifugated at 16,500 g for 10 min to remove cell fragments. The supernatants were mixed with one-third volume of 4× Laemmli’s sample buffer, and boiled at 98°C for 5 min. The proteins were separated by SDS-PAGE, transferred onto a polyvinylidene difluoride (PVDF) immobilon transfer membrane (EMD Millipore) using a semi-dry device, and then probed with the appropriate primary and secondary antibodies. Detection was performed with electrochemiluminescent using Amersham ECL prime Western blotting detection reagent (RPN2232; GE Healthcare). Imaging acquisition used ImageQuant LAS 4000 imaging system (GE Healthcare). The band intensity of each protein was measured and normalized with its relevant β-actin band (Figs. 6 and 7). PARP1 band intensitie was measured and normalized with its relevant input PARP1 band (Fig. 4 A) or over-expressed KIF4 band (Fig. 4 C and Fig. S4).
Immunocytochemistry
Stable cells were fixed in 4% PFA in PBS at 37°C for 15 min. Cells were permeabilized by 0.2% (w/v) Triton X-100 in PBS at RT for 10 min. Cells were labeled with the following primary antibodies in various experiments: a chicken anti-TrkB polyclonal antibody (1:250), a mouse anti-pADPr polyclonal antibody (1:250), and a rabbit anti-MAP2 polyclonal antibody (1:2,000). Primary antibodies were detected using Alexa Fluor 568-conjugated goat anti-chicken IgG, Alexa Fluor 488-conjugated goat anti-mouse IgG, Alexa Fluor 568-conjugated goat anti-rabbit IgG (1:1,000). Images were obtained using a Zeiss LSM780 confocal microscope (63×/1.49 oil Zeiss objective) equipped with an Airyscan module (ZEISS).
Antibodies
An anti-KIF4 antibody was obtained from rabbits injected with the KIF4 protein purified from Sf9 cells as the antigen. For the antigen protein constructed by KIF4 (residues 78–1071), 20 residues (RLLTKPERKLSWLLPPLSNN) were added to C-terminus. The detail of rabbit anti-KIF4 antibody (1:1,000) was previously described (Sekine et al., 1994).
Anti-α-tubulin antibody (mouse, 1:3,000 for immunoblotting, clone: #T9026; DM1A, RRID: AB_477593) and anti-β-actin antibody (mouse, 1:3,000, #A5441, RRID: AB_476744) were obtained from Sigma-Aldrich. Anti-pADPr(10H) antibody (mouse, 1:1,000, #sc-56198, RRID: AB_212695), anti-GABA AR antibody (goat, 1:500, #sc-7363, RRID: AB_640785), anti-BDNF antibody (rabbit, 1:200, #sc-546, RRID: AB_630940), and anti-PARP1 antibody (rabbit, 1:1,000, #sc-7150, RRID: AB_2160738) were obtained from Santa Cruz Biotechnology. Anti-MAP2 antibody (rabbit, 1:1,000, #AB5622, RRID: AB_91939) and anti-KCC2 antibody (mouse, 1:1,000, #MABN88, RRID: AB_11213323) were obtained from Millipore. Anti-TrkB antibody (mouse, 1:1,000, #T161020, RRID: AB_397508) was obtained from BD Pharmingen. Anti-phospho-705 TrkB antibody (rabbit, 1:200, #ab229908, RRID: AB_2892153) was obtained from BD Pharmingen. Anti-ERK1/2 antibody (rat, 1:1,000, #686902, RRID: AB_2629535) was obtained from BioLegend. Anti-p44/42 Erk1/2 antibody (rabbit, 1:1,000, #9101, RRID: AB_331646), anti-phosphor-AKT Thr308 antibody (rabbit, 1:1,000, #2965, RRID: AB_2255933), anti-AKT antibody (rabbit, 1:1,000, #9272, RRID: AB_329827), and anti-GluR1 antibody (rabbit 1:1,000, #8850, RRID: AB_10950223) were obtained from Cell Signaling Technology. Normal rabbit IgG (#55944, RRID: AB_2334717) was obtained from Cappel/ICN/MP. Horseradish-peroxidase-linked sheep anti-mouse antibody (1:3,000, #NA931V, RRID: AB_772210) and horseradish-peroxidase-linked donkey anti-rabbit antibody (1:3,000, #NA934V, RRID: AB_772206) were obtained from GE Healthcare.
Alexa Fluor 488-conjugated goat anti-mouse and donkey anti-mouse IgG (1:1,000, #A11029, RRID: AB_2534088 and #A-11001, RRID: AB_2534069) and Alexa Fluor 568-conjugated goat anti-mouse, goat anti-chicken and goat anti-rabbit IgG (1:1,000; #A-21043, RRID: AB_2535712, #A-11041, RRID: AB_2534098, and #A-11036 RRID: AB_10563566) were obtained from Invitrogen/Thermo Fisher Scientific.
Proximity-dependent biotin identification (BioID) pulldowns
Large-scale BioID pulldowns were performed. 4 of 10 cm dishes at 80% confluency were incubated with 50 µM biotin for 16 h. After two times of PBS wash, cells were lysed in 2.4 ml of lysis buffer containing 50 mM Tris (pH 7.4), 500 mM NaCl, 0.4% SDS, 1 mM DTT, and 1× Complete protease inhibitor (Halt Phosphatase Inhibitor Cocktail; Life Technologies). After that, lysates were additionally supplied with a final 2% (w/v) of Triton X-100 and sonicated to further shear DNA for 1 min at 30% duty cycle and an output level of 3 (Sonifer-250; Branson). Lysates were centrifugated at 16,500 g for 10 min to remove the cell fragment and then incubated with Streptavidin magnetic beads (Thermo Fisher Scientific) for 12–16 h. Streptavidin beads were collected using a magnetic stand, by washing twice with 2% (w/v) SDS, once with 0.1% deoxycholate, 1% Triton X-100, 500 mM NaCl, 1 mM EDTA, and 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (pH 7.5), once with 250 mM LiCl, 0.5% NP-40, 0.5% deoxycholate, 1 mM EDTA, and 10 mM Tris (pH 8.0), and finally once with 50 mM Tris. After centrifugation at 1,600 g for 5 min, beads were collected and resuspended in a 2× sample buffer for immunoblotting analysis (Roux et al., 2018).
Nuclear and cytoplasmic co-immunoprecipitation (co-IP)
Cells were lysed with cytoplasmic lysis buffer (10 mM HEPES [pH 7.9], 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 0.05% Nonidet P-40) containing 1× proteinase inhibitor. The cell mixture was passed through a syringe with a 26.5 gauge needle more than 15 times to destroy the cell membrane and release the nuclei from the cells. The nuclei were separated from lysate by centrifugation at 3,000 rpm for 10 min, and these nuclei containing pellet was lysed with a high-salt buffer (5 mM HEPES [pH 7.9], 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 26% [v/v] glycerol, and 300 mM NaCl) containing 1× proteinase inhibitor (11836153001 Roche) and 1× phosphatase inhibitor (4906845001 Roche). Both cytoplasmic and nuclear lysates were sonicated for 1 min at 30% duty cycle and an output level of 3 (Sonifer-250; Branson) and centrifuged at 16,500 g for 10 min. The supernatants were collected and incubated with 2 µg of antibody and protein A or protein G Sepharose beads (ab193259; Abcam) for 12–16 h. Sepharose beads had precleared in cytoplasmic lysis buffer, and those procedures above were performed at 4°C. The immunocomplexes were then washed with cytoplasmic lysis buffer three times and separated by SDS-PAGE. Immunoblotting was performed as described above.
Cell culture
Primary culture of E16.5 hippocampal neurons was performed using Minimal Essential Medium (MEM), 1 mM pyruvate (Gibco), 0.6% glucose, 2 mM GlutaMAX (#35050; Gibco/Thermo Fisher Scientific), and 2% B27 (Gibco; Iwata et al., 2020). Cortical neurons from E16.5 embryos were plated at 6 × 106 cells per 6 cm culture plate coated with 1% polyethyleneimine. To exclude the glia affections, primary neurons were cultured under 5 μM cytarabine (AraC). The N2A cell line was maintained with 10% FBS (Gibco), 1% Penicillin-Streptomycin (Gibco) in DMEM (Wako) in a 5% CO2 atmosphere at 37°C and routinely passaged every 2 d.
Survival assay of hippocampal primary neurons
On day 1, hippocampal neurons were plated at 7.5 × 104 cells/cm2, and after 3 h, the medium was replaced to MEM medium with or without B27 (17504044; Thermo Fisher Scientific). From day 2, cells were maintained in the medium in the presence or absence of KCl (25 mM), DHIQ (5 μM), or NAD (20 μM). On day 6, cells were labeled with 2.5 μM CMFDA (Molecular Probes) for 30 min at 37°C, and the number and density of CMFDA-positive cells were calculated (Midorikawa et al., 2006).
Live cell imaging
Cultured hippocampal neurons at DIV 14 were transfected with the expression vectors using the calcium phosphate method on the day before observation. The cells were observed at DIV 18 using an inverted confocal laser-scanning microscope (LSM780) equipped with a ZEISS 63× lens (NA 1.46) and with an Airyscan module (ZEISS). a CSU-W1 spinning disk confocal scanning unit (Yokogawa), and a cooled iXon EM-CCD camera (Andor) used in this study. Cells were maintained at 37°C in a 5% CO2 in the MEM culture medium in the presence or absence of DMSO, DHIQ (10 μM), or NAD (20 μM), respectively or combinedly. Imaging acquisition and analysis used Zen software (ZEISS).
Chloride imaging
ClopHensorN, the ratio-metric chloride fluorescent indicator, was used to detect intracellular chloride ion concentration (Werley et al., 2020). We cotransfected into LX293T cells (#632180; TaKaRa) with the transfer vector (ClopHensorN [#163048; Addgene]), the packaging plasmid (psPAX2 [#12260; Addgene]), and the envelope plasmid (pMD2.G [#12259; Addgene]) to produce lentivirus. The hippocampal primary neuron was infected at DIV 16 and incubated in lentivirus-containing media for 6 h. After infection, lentivirus-containing media were replaced with culture media that had been used, and live cells were imaged after DIV 18. Hippocampal primary neurons were incubated in a Kreb HEPES buffer containing 128 mM NaCl, 2.5 mM KCl, 2.7 mM CaCl2, 1 mM MgSO4, 16 mM Glucose, and 20 mM HEPES (pH 7.3) under 100% O2 for 1 h at 37°C. The medium pH changes were excluded, so we can measure [Cl−] by collecting E2GFP fluorescence under just 488 nm excitation. Subsequently, cells were excited at 488 nm (for E-GFP, “green”) and 594 nm (for the red fluorophores, “red”). Emission was collected between 500 and 550 nm for the green channel and between 650 and 700 nm for the red channel by the Zeiss LSM780. Images were analyzed with the ImageJ software (NIH). To quantitatively evaluate the intracellular fluorescence intensity, 8 hippocampal neurons were calculated, and this process was repeated three times in each condition (a total of 24 cells). For calibration of intracellular chloride iron concentrations, the fluorescence of cells was measured in the presence of different known extracellular [Cl–]I (0, 20, 40, or 80 mM) in the bath. Final concentrations of 10 µM tributyltin (Cl–-OH– exchanger), 5 µM nigericin (K+-H+ exchanger), and 10 µM valinomycin (K+/Cl– ionophore) were added to the calibration buffer containing KCl and K gluconate (135 mM K+ in total with desired amount of Cl2), and 20 mM HEPES, pH adjusted to 7.3 using KOH.
Multiple sequence alignment
Sequence alignments and visualization were done using Clustal Omega (Sievers and Higgins, 2014) and ESPript (Gouet et al., 1999). The conserved sequence was marked with a quadrilateral frame and the highly conserved amino acids were marked with red values. The protein sequences used for analysis were obtained through the NCBI protein database under the accession numbers NP_036442.3 (Homo sapiens), NP_032472.2 (Mus musculus), NP_990306 (Gallus gallus), NP_957117.1 (Danio rerio), and NP_525053.1 (Drosophila melanogaster).
Coiled–coil prediction
Coiled–coil prediction was performed using COILS (Li et al., 2016), with a 21-window width, and MTIDK scoring matrices. The protein sequences used for analysis were picked up from 524 to 732 of the KIF4 amino acid sequence. Wildtype and mutant were respectively performed.
Plasmid construction and primers
For the KIF4WT-EGFP and PARP1-tagRFP expression vectors, mouse cDNAs were cloned from sequence-verified individual ORF clones ID: 6720401H10 (DNAFORM) and 5830444G22 (DNAFORM) and ligated with pEGFP-N3 and ptagRFP-N1 (Clontech), respectively.
For the mutant KIF4-EGFP expression vector, the megaprimer PCR was used to generate the mutant Kif4 cDNA and ligated with pEGFP-N3 (Midorikawa et al., 2006). Briefly, two-step PCR was performed. Initially, point mutation-containing primer and the reverse primer were used for PCR, then this product was used in the second reaction with the forward primer. The insertion of a specific mutation was confirmed by sequencing (Eurofins). For the BioID vectors, the BioID2-pcDNA3.1 backbone was used. Wildtype and mutant Kif4 cDNAs were cloned into this vector as similar to the procedure of the pEGFP vector. Primers were designed as follow: 5′-ATGAAAGAAGAGGTGAAGGG-3′ (forward), 5′-TCAGTGGGACTCTTCTTGGA-3′ (reverse), and 5′-GAGGTTGCAGAAAAACAGAAAGAGACTCAGAGC-3′ (mutation).
RNA isolation and qPCR
Total RNA was isolated using Agilent Total RNA Isolation Mini kit to the manufacturer’s instructions. Total 500 ng of RNA was reverse transcribed into cDNA using ReverTra Ace qPCR RT Master Mix kit (FSQ-201; TOYOBO). The relative mRNA expression levels were determined using real-time quantitative PCR by THUNDERBRID SYBR qPCR Mix (QPS-201; TOYOBO). Relative mRNA expression levels were determined by the ddCt method. Beta-actin was used as reference internal standard. The qPCR primers were picked up from published papers (Plotkin et al., 2014; Luo et al., 2017; Jerónimo-Santos et al., 2015; Kursan et al., 2017), the sequence described as follow: TrkB FL: 5′-AAGGAGCCTTCGGGAAAGTT-3′ (forward), 5′-GAAAGTCCTTGCGTGCATTG-3′ (reverse), TrkB T1: 5′-CACTGGATGGGTAGCTGAGATA-3′ (forward), 5′-TGCAGACATCCTCGGAGATT-3′ (reverse), KCC2: 5′-TGCCCAGAAGTCTATCCCTAC-3′ (forward), 5′-CACCAAGTTGCCATTCACAG-3′ (reverse), PARP1: 5′-TGGTTTCAAGTCCCTTGTCC-3′ (forward), 5′-TGCTGTCTATGGAGCTGTGG-3′ (reverse), KIF4: 5′-GAGCACACTAAAATGTCAGGAGG-3′ (forward), 5′-CTGTTTGCTGGCTACTTGGAG-3′ (reverse), Act: 5′-AGCCATGTACGTAGCCATCC-3′ (forward), 5′-CTCTCAGCTGTGGTGGTGAA-3′ (reverse).
Statistics
All data are presented as the mean ± SEM. Distribution of the data was assessed for normality using the Shapiro–Wilk test. Data were analyzed by an unpaired Student’s t test or a paired t test. Two-way ANOVA with Tukey’s multiple comparisons test or Sidak’s multiple comparisons tests were used for calculations of significance for multiple comparisons.
Online supplemental material
Fig. S1 shows the KIF4A mutant patient's clinic background and the protein sequence conservation analysis. Fig. S2 shows the mutant KIF4 mouse transgenic strategy and the genotyping PCR method. Fig. S3 A shows the enhanced binding affinity of mutant KIF4 with PARP1 in the BioID test. Fig. S3 B shows the IgG immunocytochemistry data in hippocampal primary culture neurons. Fig. S4 shows that the mutant KIF4 regulates dendritic spine morphology through PARP1 activity. Fig. S5 shows that the activation of PARP1 regulates mice anxiety and seizure susceptibility behavior. Video 1 shows the enhanced seizure susceptibility of KIF4 mutant mice under pentylenetetrazol. Video 2 shows that the activation of PARP1 modifies KIF4 mutant mice seizure susceptibility under pentylenetetrazol.
Data availability
All data are available in the main text or the supplementary materials.
Acknowledgments
We thank Y. Kanai, T. Ogawa, N. Homma, H.Sato, H. Fukuda, N. Onouchi, and T. Akamatsu for their advice, technical supports and secretarial help; and all the members of the N. Hirokawa laboratory for technical help and valuable discussions; S. Mizuno and S. Takahashi (University of Tsukuba) for production of KIF4 mutant mouse; and all my friends and families, especially D. Wang, L. Feng, X. Jiang, Z. Lai, J. An, W. Yi, X.T. Wang, F. Li, and X.M. Wang for all their encouragement, support, and advice. We thank J. Iacona, Ph.D., from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
This work was supported by grants from the Japan Society for the Promotion of Science (JSPS) KAKENHI (grant numbers JP23000013 and JP16H06372) to N. Hirokawa and from the Uehara Memorial Foundation to N. Hirokawa.
The authors declare no competing financial interests.
Author contributions: Y. Wan: Conceptualization, Data curation, Methodology, Software, Investigation, Visualization, Writing—original draft, Writing—review & editing. N. Hirokawa: Conceptualization, Supervision, Project administration, Writing—review & editing, Funding acquisition. M. Morikawa: Methodology, Investigation, Visualization, Writing—review & editing. M. Morikawa: Methodology, Writing—review & editing. S. Iwata: Methodology, Writing—review & editing. Y. Tanaka: Methodology, Writing—review & editing. M.I. Naseer: Resources (Clinic Data). A.G. Ahmed Chaudhary: Resources (Clinic Data).

