


The time course of signaling by peptide hormones, neural peptides, and other neuromodulators depends on their storage inside dense core vesicles (DCVs). Adaptor protein 3 (AP-3) assembles the membrane proteins that confer regulated release of DCVs and is thought to promote their trafficking from endosomes directly to maturing DCVs. We now find that regulated monoamine release from DCVs requires sorting nexin 5 (SNX5). Loss of SNX5 disrupts trafficking of the vesicular monoamine transporter (VMAT) to DCVs. The mechanism involves a role for SNX5 in retrograde transport of VMAT from endosomes to the TGN. However, this role for SNX5 conflicts with the proposed function of AP-3 in trafficking from endosomes directly to DCVs. We now identify a transient role for AP-3 at the TGN, where it associates with DCV cargo. Thus, retrograde transport from endosomes by SNX5 enables DCV assembly at the TGN by AP-3, resolving the apparent antagonism. A novel role for AP-3 at the TGN has implications for other organelles that also depend on this adaptor.
Introduction
The temporal control of signaling required for physiology, behavior, and development depends on the regulated release of signaling molecules including peptide hormones, neural peptides, neuromodulators, and classic neurotransmitters. Regulated release thus involves coordination of vesicle loading and peptide storage with the machinery for membrane fusion. Indeed, the composition of membrane proteins, including SNAREs and Ca2+-sensing synaptotagmins, dictates the properties of release (Jahn and Fasshauer, 2012; Sudhof, 2013). At the nerve terminal, these proteins assemble to form synaptic vesicles during endocytosis (Saheki and De Camilli, 2012). In the case of dense core vesicles (DCVs) that release neuromodulators such as peptides and monoamines, however, the mechanisms that assemble the required membrane proteins and even where assembly occurs have remained poorly understood.
Soluble proteins that undergo regulated secretion target to DCVs at the TGN, where they aggregate to form the dense core (Arvan and Castle, 1998; Chanat and Huttner, 1991; Orci et al., 1987). Aggregation occurs under the particular pH and redox conditions of the TGN, and metabolic labeling has confirmed that these soluble cargo separate from constitutively secreted proteins at this site (Tooze and Huttner, 1990). We know much less about the trafficking of DCV membrane proteins. After budding from the TGN, DCVs undergo remodeling in a process known as maturation that involves the removal of proteins destined for other compartments (Kuliawat et al., 1997; Tooze et al., 1991). In some cells, immature DCVs can undergo regulated exocytosis, suggesting the presence of required membrane proteins upon budding from the TGN (Tooze et al., 1991). However, the endosomal pathway may also contribute DCV components during maturation (Harrison-Lavoie et al., 2006; Kaur et al., 2017). Indeed, genetic studies in Caenorhabditis elegans and Tetrahymena have implicated endosomal trafficking machinery in DCV maturation (Ailion et al., 2014; Briguglio et al., 2013; Edwards et al., 2009; Kaur et al., 2017; Ma et al., 2020; Sasidharan et al., 2012; Shi et al., 2012; Sparvoli et al., 2018; Topalidou et al., 2020; Topalidou et al., 2016). Thus, it has generally been thought that membrane proteins take an endosomal route to maturing DCVs, after they have bud from the TGN.
For classic neurotransmitters, release by exocytosis requires storage inside secretory vesicles and hence transport from the cytoplasm, which is mediated in the case of monoamines by the vesicular monoamine transporters (VMATs). The localization of VMATs thus dictates the secretory vesicles capable of monoamine storage and release. In chromaffin cells of the adrenal medulla, the localization of VMAT2 and endocrine-specific isoform VMAT1 to DCVs (chromaffin granules; Liu et al., 1994; Weihe et al., 1994) confers the systemic release of catecholamine involved in the “fight or flight” response to stress. In previous work, we used VMAT2 to identify a role for adaptor protein 3 (AP-3) in the localization of multiple membrane proteins, including calcium sensor synaptotagmin 1 (syt 1), to DCVs (Asensio et al., 2010; Sirkis et al., 2013). In addition, AP-3 binds to vacuolar protein sorting 41 (VPS41), which also promotes DCV biogenesis and appears to act as the coat protein for AP-3 (Asensio et al., 2013). However, the site at which AP-3 and VPS41 act to promote DCV formation has remained unclear.
AP-3 has a central role in the formation of lysosomes and related organelles, targeting the membrane proteins characteristic of melanosomes, lung alveolar lamellar bodies, platelet granules, immune cell cytolytic granules, endothelial Weibel-Palade bodies, and phagosomes as well as lysosomes (Bowman et al., 2019; Dell’Angelica and Bonifacino, 2019). The perinuclear localization of AP-3 in earlier studies (Nishimura et al., 2002; Simpson et al., 1996) suggested expression at the TGN, but immuno-EM has shown localization to endosomes with very little expression at the TGN (Peden et al., 2004; Theos et al., 2005), consistent with a role for AP-3 in trafficking to lysosome-related organelles within the endocytic pathway (Bowman et al., 2019). Indeed, AP-3 contributes to the formation of synaptic-like microvesicles (SLMVs) from endosomes in endocrine cells (Faundez et al., 1998; Suckow et al., 2010) and to a subset of synaptic vesicles in neurons (Newell-Litwa et al., 2007; Silm et al., 2019). Similar to its role in formation of other lysosome-related organelles, AP-3 has been suggested to target membrane proteins from endosomes to DCVs during the process of maturation that occurs after they bud from the TGN (Harrison-Lavoie et al., 2006; Kaur et al., 2017). We now identify a protein that cooperates with AP-3 in trafficking to DCVs and find that the mechanism is not consistent with an exclusive role for AP-3 on endosomes.
Results
SNX5 confers monoamine storage and regulated release by targeting VMAT to DCVs
To identify proteins involved in membrane trafficking to DCVs, we used the cytosolic C-terminus of VMAT2, which contains multiple sorting signals (Li et al., 2005; Tan et al., 1998; Waites et al., 2001), to screen for interacting proteins by yeast two-hybrid assay. From a PC12 cell cDNA library, we identified multiple copies (5/26) of sorting nexin 5 (SNX5; Table S1), a protein associated with the retromer complex that mediates retrograde traffic from endosomes to the TGN (Bonifacino and Hurley, 2008; Elwell et al., 2017; Kvainickas et al., 2017; Simonetti et al., 2017). To assess the physiological significance of this interaction, we knocked down SNX5 in rat PC12 cells (Fig. S1 A). Loss of SNX5 greatly reduces the depolarization-dependent release of preloaded 3H-norepinephrine (3H-NE; Fig. 1 A), with no discernible effect on release of DCV polypeptide secretogranin II (SgII, Fig. 1 B). Consistent with a specific defect in monoamine storage and release, loss of SNX5 reduces the 3H-NE loaded into PC12 cells but not the cellular content of SgII (Fig. 1, A and B). In addition, SNX5 knockout (KO) mice show a major reduction in catecholamine content (Fig. 1 C), establishing the significance in vivo.
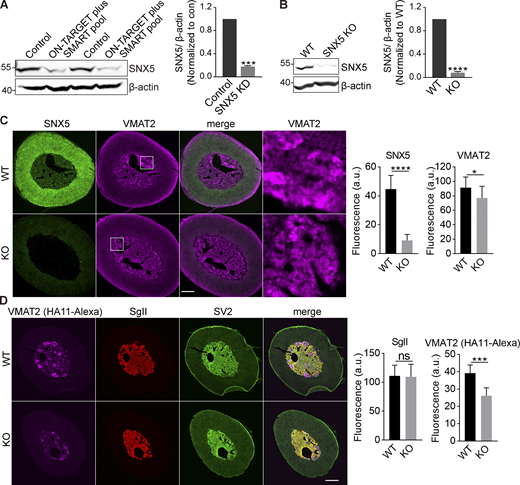
Loss of SNX5 reduces VMAT2 expression. Related to Fig. 1. (A) PC12 cells were transfected twice with control or SNX5 siRNA, and extracts were immunoblotted for SNX5. Bar graph quantifies SNX5 expression normalized to β-actin and control (n = 3). (B) Immunoblotting for SNX5 in the adrenal glands of SNX5+/+ (WT) and SNX5−/− (KO) mice. Bar graph quantifies SNX5 expression normalized to β-actin and WT (n = 3). (C and D) Slices from the adrenal medullae of WT or SNX5 KO mice expressing an HA-VMAT2 BAC transgene were immunostained for HA (magenta) and SNX5 (green) using amplification with secondary antibodies (C) or for HA11 primary antibody directly conjugated to Alexa Fluor 647 as well as for SgII (red) and SV2 (green; D). Bar graphs quantify SNX5, VMAT2, and SgII fluorescence intensity (n = 3). Scale bar, 200 μm. Bar graphs indicate mean ± SEM. *, P < 0.05; ***, P < 0.001; ****, P < 0.0001 by unpaired Student’s t test. Data S1 shows statistics source data.
Loss of SNX5 reduces VMAT2 expression. Related to Fig. 1. (A) PC12 cells were transfected twice with control or SNX5 siRNA, and extracts were immunoblotted for SNX5. Bar graph quantifies SNX5 expression normalized to β-actin and control (n = 3). (B) Immunoblotting for SNX5 in the adrenal glands of SNX5+/+ (WT) and SNX5−/− (KO) mice. Bar graph quantifies SNX5 expression normalized to β-actin and WT (n = 3). (C and D) Slices from the adrenal medullae of WT or SNX5 KO mice expressing an HA-VMAT2 BAC transgene were immunostained for HA (magenta) and SNX5 (green) using amplification with secondary antibodies (C) or for HA11 primary antibody directly conjugated to Alexa Fluor 647 as well as for SgII (red) and SV2 (green; D). Bar graphs quantify SNX5, VMAT2, and SgII fluorescence intensity (n = 3). Scale bar, 200 μm. Bar graphs indicate mean ± SEM. *, P < 0.05; ***, P < 0.001; ****, P < 0.0001 by unpaired Student’s t test. Data S1 shows statistics source data.
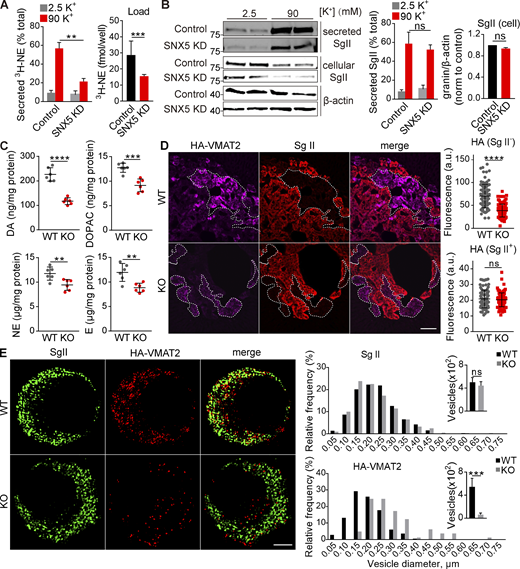
SNX5 confers monoamine storage and release by directing VMAT2 to DCVs. (A and B) PC12 cells transfected with control or SNX5 siRNA were loaded with 3H-NE and incubated in Tyrode’s solution containing 2.5 or 90 mM K+. (A) Loss of SNX5 greatly reduces the release of preloaded 3H-NE (right; n = 4). (B) Western analysis for SgII shows no change in release (normalized to total, middle, n = 4) or storage (normalized to β-actin, right, n = 6) with loss of SNX5. (C) The adrenal glands of SNX5 KO mice show reduced levels of dopamine (DA), the DA metabolite 3,4,-dihydroxyphenylacetic acid (DOPAC), NE, and epinephrine (E; n = 6 glands). (D) The adrenal medullae of WT and SNX5 KO mice both expressing an HA-VMAT2 BAC transgene were immunostained for HA with a primary antibody conjugated directly to Alexa Fluor 647 (left) and for SgII (right). Scatterplots (right) indicate total gland immunofluorescence (n = 3). Scale bar, 200 μm. (E) Chromaffin cells from WT or SNX5 KO mice crossed to the HA-VMAT2 BAC transgene were immunostained for SgII (left) and HA (right), and the images were reconstructed from a 125-nm section by structured illumination (left). Bar graphs indicate the relative frequency distribution and number of HA+ or SgII+ punctae (right; n = 6–10 cells/genotype). Scale bar, 2 μm. Bar graphs and scatterplots indicate mean ± SEM. **, P < 0.005; ***, P < 0.001; ****, P < 0.0001 by unpaired t test. Data S1 shows statistics source data. Source data are available for this figure: SourceData F1.
SNX5 confers monoamine storage and release by directing VMAT2 to DCVs. (A and B) PC12 cells transfected with control or SNX5 siRNA were loaded with 3H-NE and incubated in Tyrode’s solution containing 2.5 or 90 mM K+. (A) Loss of SNX5 greatly reduces the release of preloaded 3H-NE (right; n = 4). (B) Western analysis for SgII shows no change in release (normalized to total, middle, n = 4) or storage (normalized to β-actin, right, n = 6) with loss of SNX5. (C) The adrenal glands of SNX5 KO mice show reduced levels of dopamine (DA), the DA metabolite 3,4,-dihydroxyphenylacetic acid (DOPAC), NE, and epinephrine (E; n = 6 glands). (D) The adrenal medullae of WT and SNX5 KO mice both expressing an HA-VMAT2 BAC transgene were immunostained for HA with a primary antibody conjugated directly to Alexa Fluor 647 (left) and for SgII (right). Scatterplots (right) indicate total gland immunofluorescence (n = 3). Scale bar, 200 μm. (E) Chromaffin cells from WT or SNX5 KO mice crossed to the HA-VMAT2 BAC transgene were immunostained for SgII (left) and HA (right), and the images were reconstructed from a 125-nm section by structured illumination (left). Bar graphs indicate the relative frequency distribution and number of HA+ or SgII+ punctae (right; n = 6–10 cells/genotype). Scale bar, 2 μm. Bar graphs and scatterplots indicate mean ± SEM. **, P < 0.005; ***, P < 0.001; ****, P < 0.0001 by unpaired t test. Data S1 shows statistics source data. Source data are available for this figure: SourceData F1.
The specific defect in monoamine but not peptide storage and release suggested a role for SNX5 in the trafficking of VMATs, consistent with the observed interaction. Using HA-VMAT2 BAC transgenic mice (Silm et al., 2019; Zhang et al., 2015) and primary HA antibody directly conjugated to Alexa Fluor 647 for quantitation, we find that loss of SNX5 reduces HA immunoreactivity in the SgII− population of chromaffin cells where VMAT2 is normally expressed, with no detectable change in the low levels expressed by the SgII+ population (Figs. 1 D and S1 D; Weihe et al., 1994). The effect on monoamine stores (Fig. 1 C) suggests that loss of SNX5 has a similar effect on the closely related VMAT1 isoform expressed by SgII+ chromaffin cells. Consistent with the specificity for monoamines observed in PC12 cells, loss of SNX5 has no effect on staining for SgII in the adrenal medulla (Fig. 1 D).
We then assessed effects on subcellular localization of VMAT2. Loss of SNX5 reduces the number of VMAT2+ punctae in cultured primary chromaffin cells and increases their size, with no change in SgII+ punctae (Fig. 1 E). In PC12 cells stably expressing Flag-tagged VMAT2, loss of SNX5 disrupts the normal colocalization with SgII at the tips of processes induced by NGF (Figs. 2 A and S2). By density gradient fractionation, HA-VMAT2 comigrates with SgII in heavy fractions from control cells and redistributes to light fractions after knockdown of SNX5 (Fig. 2 B). By both immunofluorescence and gradient fractionation, the localization of VMAT2 to DCVs thus requires SNX5.
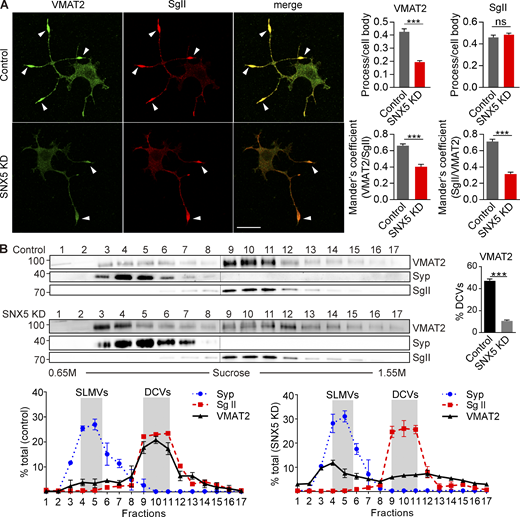
Loss of SNX5 disrupts localization of VMAT2 to DCVs. (A) PC12 cells stably expressing 3Flag-VMAT2 were transfected with control or SNX5 siRNA and immunostained for Flag (green) and SgII (red) after treatment with 50 ng/ml NGF for 48 h. Bar graphs (right) show reduced localization of VMAT2 but not SgII in processes relative to the cell body (upper) and Mander’s overlap coefficient of the proportion of pixels labeled strongly for VMAT2 that also stain for SgII (VMAT2/SgII) and vice versa (SgII/VMAT2; n = 3). (B) Equilibrium sucrose density gradient fractionation of PC12 cells stably expressing 3HA-VMAT2 shows that SNX5 knockdown (KD) redistributes VMAT2 but not SgII or synaptophysin (Syp) to light fractions (n = 3). The gradient fractions were loaded onto two gels (separated here by vertical lines) and then transferred to one membrane for detection by ECL. Bar graphs indicate mean ± SEM. ****, P < 0.0001 by unpaired t test. Data S1 shows statistics source data. Source data are available for this figure: SourceData F2.
Loss of SNX5 disrupts localization of VMAT2 to DCVs. (A) PC12 cells stably expressing 3Flag-VMAT2 were transfected with control or SNX5 siRNA and immunostained for Flag (green) and SgII (red) after treatment with 50 ng/ml NGF for 48 h. Bar graphs (right) show reduced localization of VMAT2 but not SgII in processes relative to the cell body (upper) and Mander’s overlap coefficient of the proportion of pixels labeled strongly for VMAT2 that also stain for SgII (VMAT2/SgII) and vice versa (SgII/VMAT2; n = 3). (B) Equilibrium sucrose density gradient fractionation of PC12 cells stably expressing 3HA-VMAT2 shows that SNX5 knockdown (KD) redistributes VMAT2 but not SgII or synaptophysin (Syp) to light fractions (n = 3). The gradient fractions were loaded onto two gels (separated here by vertical lines) and then transferred to one membrane for detection by ECL. Bar graphs indicate mean ± SEM. ****, P < 0.0001 by unpaired t test. Data S1 shows statistics source data. Source data are available for this figure: SourceData F2.
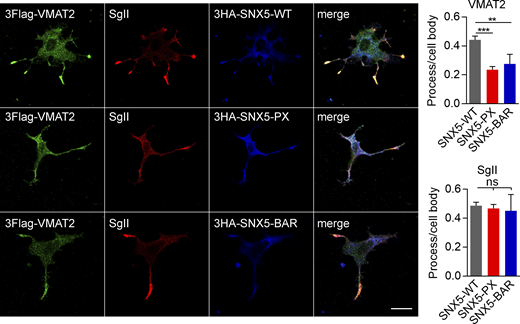
Isolated SNX5 PX and BAR domains disrupt the localization of VMAT2 to DCVs. Related to Fig. 2. PC12 cells stably expressing 3Flag-VMAT2 were transfected with HA-tagged WT, PX, or BAR domains of SNX5; treated with 50 ng/ml NGF for 48 h; and immunostained for Flag (green), SgII (red), and HA (blue). Bar graphs show reduced localization of VMAT2 but not SgII in processes relative to the cell body (n = 10 images from three independent experiments). Bar graphs indicate mean ± SEM. **, P < 0.005; ***, P < 0.001 by one-way ANOVA with Tukey’s multiple comparisons test. Data S1 contains statistics source data.
Isolated SNX5 PX and BAR domains disrupt the localization of VMAT2 to DCVs. Related to Fig. 2. PC12 cells stably expressing 3Flag-VMAT2 were transfected with HA-tagged WT, PX, or BAR domains of SNX5; treated with 50 ng/ml NGF for 48 h; and immunostained for Flag (green), SgII (red), and HA (blue). Bar graphs show reduced localization of VMAT2 but not SgII in processes relative to the cell body (n = 10 images from three independent experiments). Bar graphs indicate mean ± SEM. **, P < 0.005; ***, P < 0.001 by one-way ANOVA with Tukey’s multiple comparisons test. Data S1 contains statistics source data.
SNX5 interacts with VMAT2 at a site also recognized by adaptor proteins
To understand how SNX5 influences the trafficking of VMAT2, we characterized the interaction between them. In transfected cells, HA-tagged VMAT2 coimmunoprecipitates (co-IPs) Flag-tagged SNX5 and vice versa (Fig. 3, A and B). SNX5 also co-IPs with VMAT2 from brain extracts, indicating interaction of the endogenous proteins (Fig. 3 C). Previous work has suggested that a dimer of sorting nexins (either SNX1 or 2 and either SNX5 or 6) associates with the retromer complex (VPS26, VPS29, and VPS35) that mediates retrograde transport of the cation-independent mannose-6-phosphate receptor (CI-MPR) to the TGN (Bonifacino and Hurley, 2008; Gallon and Cullen, 2015). However, recent work has shown that SNX5 can recognize cargo directly, independently of retromer (Kvainickas et al., 2017; Simonetti et al., 2019; Yong et al., 2020). We also find that the C-terminus of VMAT2 recognizes his-tagged SNX5 in vitro (Fig. 3 D), indicating a direct interaction. Previous work has suggested an interaction of VMAT2 with retromer component VPS35 (Wu et al., 2016), but we now find that knockdown of SNX5 reduces the interaction with VPS35, not vice versa (Fig. S3, A and B), consistent with SNX5 mediating the interaction with VPS35. Knockdown of VPS35 has an effect on regulated release of 3H-NE, but this is much smaller than that due to loss of SNX5 (Fig. S3 C). Testing the specificity for other SNX proteins that contain a Bin-Amphiphysin-Rvs (BAR) domain implicated in membrane curvature (Simunovic et al., 2019), VMAT2 does not co-IP SNX1 or 6 (Fig. S3 D).
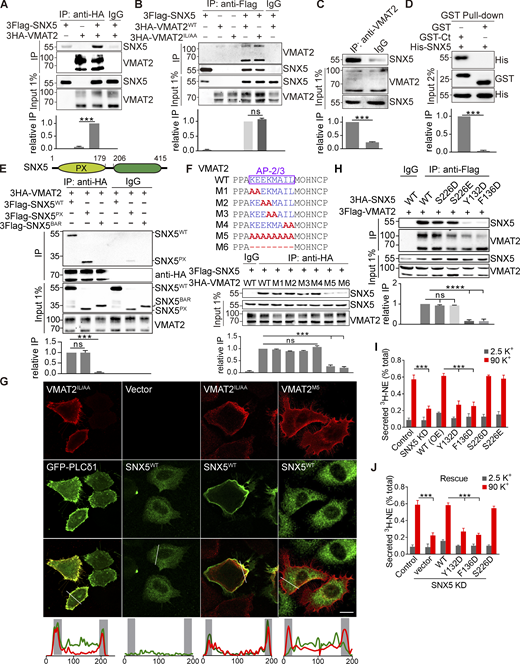
SNX5 recognizes upstream residues in an adaptor binding site. (A and B) Extracts from COS7 cells transfected with 3Flag-SNX5 and 3HA-VMAT2 were immunoprecipitated for HA (A) or Flag (B), and the precipitates were immunoblotted for Flag (A) and HA (B). (C) Rat brain extract was immunoprecipitated for VMAT2 and immunoblotted for SNX5. (D) A GST fusion to the C-terminus of VMAT2 pulls down bacterially expressed 6His-SNX5 (full length). (E) Top: Diagram of SNX5 protein containing PX and BAR domains. Bottom: Extracts from cells transfected with 3Flag-tagged WT or mutant SNX5 and 3HA-VMAT2 were immunoprecipitated for HA (n = 3). (F) Top: Sequence alignment of the extended dileucine-like motif and alanine-scanning mutants. Bottom: Extracts from cells transfected with 3Flag-SNX5 and 3HA-tagged WT or mutant VMAT2 were immunoprecipitated for HA (n = 3). (G) Cotransfected 3Flag-SNX5 (green) redistributes to the plasma membrane with IL/AA VMAT2 but not the M5 mutant (red). Line scans of fluorescence intensity are shown to the right. Scale bars, 10 µm. (H) Extracts from cells transfected with 3Flag-VMAT2 and 3Flag-SNX5 (WT and mutants) were immunoprecipitated for Flag (n = 3) and immunoblotted for HA. (I and J) Secretion assays as described in Fig. 1 (n = 3–6). Input (1%) is shown below the immunoprecipitation. Bar graphs indicate mean ± SEM of the band intensities normalized to maximum co-IP for each experiment (n = 3). ***, P < 0.001; ****, P < 0.0001 by unpaired t test (A–D) or one-way ANOVA with Tukey’s multiple comparisons test (E, F, and H–J). Data S1 contains statistics source data. Source data are available for this figure: SourceData F3.
SNX5 recognizes upstream residues in an adaptor binding site. (A and B) Extracts from COS7 cells transfected with 3Flag-SNX5 and 3HA-VMAT2 were immunoprecipitated for HA (A) or Flag (B), and the precipitates were immunoblotted for Flag (A) and HA (B). (C) Rat brain extract was immunoprecipitated for VMAT2 and immunoblotted for SNX5. (D) A GST fusion to the C-terminus of VMAT2 pulls down bacterially expressed 6His-SNX5 (full length). (E) Top: Diagram of SNX5 protein containing PX and BAR domains. Bottom: Extracts from cells transfected with 3Flag-tagged WT or mutant SNX5 and 3HA-VMAT2 were immunoprecipitated for HA (n = 3). (F) Top: Sequence alignment of the extended dileucine-like motif and alanine-scanning mutants. Bottom: Extracts from cells transfected with 3Flag-SNX5 and 3HA-tagged WT or mutant VMAT2 were immunoprecipitated for HA (n = 3). (G) Cotransfected 3Flag-SNX5 (green) redistributes to the plasma membrane with IL/AA VMAT2 but not the M5 mutant (red). Line scans of fluorescence intensity are shown to the right. Scale bars, 10 µm. (H) Extracts from cells transfected with 3Flag-VMAT2 and 3Flag-SNX5 (WT and mutants) were immunoprecipitated for Flag (n = 3) and immunoblotted for HA. (I and J) Secretion assays as described in Fig. 1 (n = 3–6). Input (1%) is shown below the immunoprecipitation. Bar graphs indicate mean ± SEM of the band intensities normalized to maximum co-IP for each experiment (n = 3). ***, P < 0.001; ****, P < 0.0001 by unpaired t test (A–D) or one-way ANOVA with Tukey’s multiple comparisons test (E, F, and H–J). Data S1 contains statistics source data. Source data are available for this figure: SourceData F3.
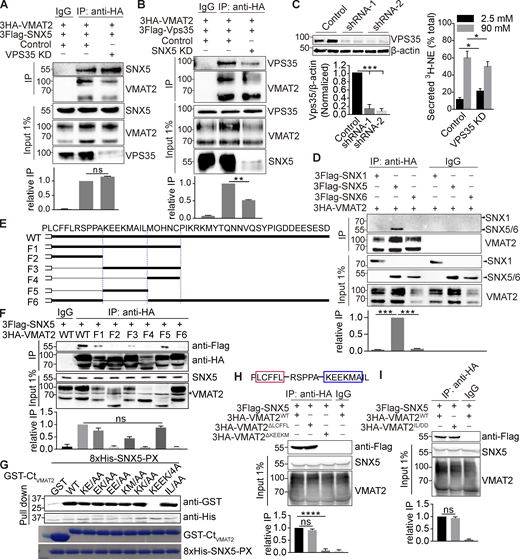
SNX5 recognizes the extended dileucine-like motif of VMAT2. Related to Fig. 3. (A and B) 3HA-VMAT2 and 3Flag-SNX5 (A) or 3Flag-Vps35 (B) were expressed in COS7 cells with Vps35 (A), SNX5 (B), or control siRNA, and extracts were immunoprecipitated for HA. (C) Vps35 and control shRNA were transduced into PC12 cells by lentivirus, and the extracts were immunoblotted for VPS35. Knockdown with each of the constructs slightly impairs regulated release of preloaded 3H-NE (n = 6). (D) Extracts from COS7 cells transiently transfected with 3HA-VMAT2 and 3Flag-SNX1, -SNX5, or -SNX6 were immunoprecipitated for HA. (E) Sequence of the VMAT2 C-terminus and deletions F1–F6. (F) 3Flag-SNX5 was cotransfected into COS7 cells with WT and mutant 3HA-VMAT2, and extracts were immunoprecipitated for HA. The input shows variable expression of the deletions, but F1, 3, and 5 still recognize SNX5. (G) Bacterially expressed 8His-SNX5-PX pulls down all of the GST-VMAT2 fusions except the replacement of KEEK by alanine. Below, 0.2-µg input stained with Coomassie Brilliant Blue (n = 3). (H) Sequence of VMAT2 C-terminus with potential β hairpin structure. Extracts from HEK293T cells transfected with 3Flag-SNX5 and 3HA-VMAT2 WT, LCFFL, or KEEKM deletion mutants were immunoprecipitated for HA, and the precipitates were immunoblotted for Flag (lower panel). (I) Extracts from HEK293T cells transfected with 3Flag-SNX5 and 3HA-VMAT2 WT or IL/DD mutant were immunoprecipitated for HA, and the precipitates were immunoblotted for Flag. 25% of the immunoprecipitates were used for immunoblotting. Inputs (1%) are shown below the immunoprecipitations. Bar graphs indicate mean ± SEM of the band intensities normalized to maximum co-IP for each experiment (n = 3). **, P < 0.005; ***, P < 0.001 by one-way ANOVA with Tukey’s multiple comparisons test. Data S1 contains statistics source data. Source data are available for this figure: SourceData FS3.
SNX5 recognizes the extended dileucine-like motif of VMAT2. Related to Fig. 3. (A and B) 3HA-VMAT2 and 3Flag-SNX5 (A) or 3Flag-Vps35 (B) were expressed in COS7 cells with Vps35 (A), SNX5 (B), or control siRNA, and extracts were immunoprecipitated for HA. (C) Vps35 and control shRNA were transduced into PC12 cells by lentivirus, and the extracts were immunoblotted for VPS35. Knockdown with each of the constructs slightly impairs regulated release of preloaded 3H-NE (n = 6). (D) Extracts from COS7 cells transiently transfected with 3HA-VMAT2 and 3Flag-SNX1, -SNX5, or -SNX6 were immunoprecipitated for HA. (E) Sequence of the VMAT2 C-terminus and deletions F1–F6. (F) 3Flag-SNX5 was cotransfected into COS7 cells with WT and mutant 3HA-VMAT2, and extracts were immunoprecipitated for HA. The input shows variable expression of the deletions, but F1, 3, and 5 still recognize SNX5. (G) Bacterially expressed 8His-SNX5-PX pulls down all of the GST-VMAT2 fusions except the replacement of KEEK by alanine. Below, 0.2-µg input stained with Coomassie Brilliant Blue (n = 3). (H) Sequence of VMAT2 C-terminus with potential β hairpin structure. Extracts from HEK293T cells transfected with 3Flag-SNX5 and 3HA-VMAT2 WT, LCFFL, or KEEKM deletion mutants were immunoprecipitated for HA, and the precipitates were immunoblotted for Flag (lower panel). (I) Extracts from HEK293T cells transfected with 3Flag-SNX5 and 3HA-VMAT2 WT or IL/DD mutant were immunoprecipitated for HA, and the precipitates were immunoblotted for Flag. 25% of the immunoprecipitates were used for immunoblotting. Inputs (1%) are shown below the immunoprecipitations. Bar graphs indicate mean ± SEM of the band intensities normalized to maximum co-IP for each experiment (n = 3). **, P < 0.005; ***, P < 0.001 by one-way ANOVA with Tukey’s multiple comparisons test. Data S1 contains statistics source data. Source data are available for this figure: SourceData FS3.
SNX5 contains a C-terminal BAR domain and an N-terminal Phox (PX) domain originally presumed to interact with phosphoinositides but more recently found to recognize proteins (Chandra and Collins, 2019; Fig. 3 E). Transfected HA-VMAT2 co-IPs only the PX domain (Fig. 3 E), consistent with its role in cargo recognition (Elwell et al., 2017; Kvainickas et al., 2017; Simonetti et al., 2017; Simonetti et al., 2019; Yong et al., 2020). However, expression of isolated PX or BAR domains both impair the localization of VMAT2 to DCVs (Fig. S2), suggesting the importance of both domains.
The analysis of C-terminal deletions in VMAT2 shows that the extended dileucine-like motif KEEKMAIL (F5) is both required and sufficient to recognize SNX5 (Fig. S3, E and F; bold values denote the central, conserved core of the motif; values in italics are somewhat less conserved). Replacement of all eight residues with alanine eliminates SNX5 recognition (Fig. 3 F), and the analysis of purified proteins confirms the requirement for KEEK to interact directly (Fig. S3 G). This sequence does not conform to the β hairpin consensus recently reported for recognition by SNX5 (Simonetti et al., 2019; Yong et al., 2020), and Fig. 3 F raises the possibility of residual binding in the KEEKMAIL deletion mutant. Indeed, the upstream sequence LCFFL conforms to the consensus ΦXΩXΦ (where Φ are hydrophobic and Ω are aromatic residues) and may contribute to the first part of the bipartite binding site for SNX5 (Simonetti et al., 2019; Yong et al., 2020). However, this sequence apparently resides within transmembrane domain 12 of the VMATs, (Jumper et al., 2021) and deletion of LCFFL does not impair the interaction with SNX5 (Fig. S3 H). Thus, the extended dileucine-like motif KEEKMAIL appears both necessary and sufficient for interaction with SNX5. Interestingly, this is the same sequence recognized by adaptor proteins (Asensio et al., 2010; Mattera et al., 2011). In contrast to the adaptors, however, replacement of the IL core with either alanine or aspartate does not impair the interaction with SNX5 in cells (Fig. 3, B and F; and Fig. S3 I) or in vitro (Fig. S3 G). SNX5 thus recognizes a block of residues three to six positions upstream within the dileucine-like motif, not the core dileucine itself. Acidic residues upstream of a dileucine motif are known to interact with adaptor proteins, particularly AP-3 (Kelly et al., 2008; Mattera et al., 2011), and two glutamates upstream of the dileucine-like motif in VMAT2 have been shown to interact functionally with AP-3 in targeting to DCVs (Asensio et al., 2010; Sirkis et al., 2013). Thus, SNX5 binds to the same sequence upstream of the dileucine that is recognized by AP-3. Recent work has indeed observed that recognition sites for SNX5 and multiple adaptors frequently overlap (Simonetti et al., 2019), but the significance has remained unclear.
Interactions important for trafficking are often transient, so we assessed the strength of interaction between VMAT2 and SNX5 in cells. Alanine replacement of the core dileucine-like motif (IL) disrupts internalization of the transporter, as previously shown (Fig. 3 G; Tan et al., 1998), and cotransfected SNX5 redistributes to the plasma membrane with internalization-defective IL/AA VMAT2 (Fig. 3 G), indicating a persistent interaction between the two proteins. With replacement of the extended dileucine motif, VMAT2 remains at the plasma membrane, but SNX5 redistributes back to the cytoplasm (Fig. 3 G), because residues upstream of the dileucine are required for the interaction with SNX5 (Fig. 3). Although highly overlapping, the motifs in VMAT2 recognized by SNX5 and adaptor proteins are thus distinct.
Since the sequence in VMAT2 recognized by SNX5 diverges from the recently established bipartite consensus motif (Elwell et al., 2017; Simonetti et al., 2019; Yong et al., 2020), we determined whether SNX5 recognizes VMAT2 using the same residues shown to recognize other cargo. By both overexpression and rescue of the SNX5 knockdown, we find that SNX5 mutants Y132D and F136D defective in recognition of VMAT2 as well as other cargo (Simonetti et al., 2019; Fig. 3 H) impair the function of SNX5 in regulated catecholamine release (Fig. 3, I and J). Although the site in VMAT2 does not conform to the established consensus, SNX5 uses the same residues previously described, consistent with a hydrophobic binding site on the surface of the SNX5 PX domain (Elwell et al., 2017). Extended dileucine-like motifs may therefore recruit sorting nexins as well as adaptor proteins. We also used mutations S226D and S226E to assess a requirement for dimerization of SNX-BAR proteins (Itai et al., 2018). S226D and S226E SNX5 do not act as dominant negatives on regulated release of 3H-NE (Fig. 3 I), and S226D SNX5 rescues the loss of SNX5 (Fig. 3 J). The function of SNX5 in trafficking of VMAT2 thus does not require dimerization with other SNX-BAR proteins.
SNX5 directs VMAT2 from the endosome to the TGN
How does SNX5 target VMAT2 to DCVs? SNX5 mediates retrograde transport of CI-MPR from endosomes to the Golgi complex (Kvainickas et al., 2017; Simonetti et al., 2017; Simonetti et al., 2019). In PC12 cells, VMAT2 does not localize strongly to the TGN (Fig. 2 A), presumably due to rapid exit in DCVs or other membranes. In HeLa cells, however, VMAT2 colocalizes with TGN46 (Figs. 3 G, 4 A, and S4 A), and knocking down SNX5 (Fig. S4 B) redistributes VMAT2 from the TGN (Fig. 4 A) to endosomes labeling for EEA1 (Fig. 4 B). The accumulation of VMAT2 on endosomes supports a role for SNX5 in retrograde trafficking to the TGN, and coexpression of RNAi-resistant SNX5 rescues both the reduced Golgi and increased endosome localization (Fig. 4, A and B). Overexpression of isolated PX and BAR domains but not full-length SNX5 also redistributes VMAT2 to endosomes (Fig. S4, C and D). SNX5 thus promotes the trafficking of VMAT2 from endosomes to the TGN.
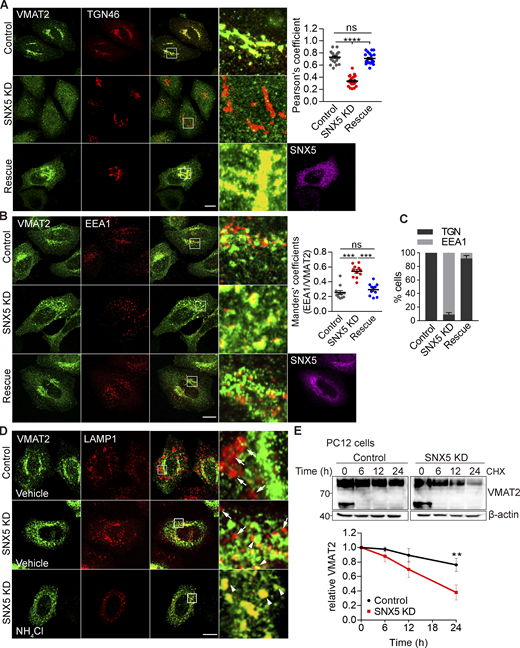
Loss of SNX5 redistributes VMAT2 from TGN to endosomes and promotes its lysosomal degradation. (A and B) Loss of SNX5 redistributes VMAT2 away from the TGN (A) to endosomes (B). HeLa cells transfected with 3HA-VMAT2 and control siRNA, SNX5 siRNA, or SNX5 siRNA with RNAi-resistant SNX5 were immunostained for HA (green), SNX5 (magenta), and either TGN46 (red; A) or EEA1 (red; B). The colocalization of VMAT2 and TGN46 was quantified by Pearson’s correlation coefficient (n = 20 images from three independent experiments). The VMAT2 colocalizing with EEA1 was quantified using the Mander’s overlap coefficient analyzing the proportion of pixels labeled strongly for EEA1 that also stain for VMAT2 (n = 10 images from three independent experiments). (C) The bar graph shows the percentage of cells with overall colocalization of VMAT2 to either TGN46 or EEA1 (n = 25 images from three independent experiments). (D) HeLa cells transfected with 3HA-VMAT2 and either control or SNX5 siRNA, without or with pretreatment for 1 h in NH4Cl (10 mM), were immunostained for HA (green) and LAMP1 (red) and the colocalization quantified by Pearson’s correlation coefficient shown in Fig. S5 D (n = 15 images from three independent experiments). Scale bars, 10 µm. (E) PC12 cells stably expressing 3HA-VMAT2 were treated with cycloheximide (CHX) at the times indicated, and VMAT2 was quantified by Western analysis, normalizing to β-actin and the amount at time 0 (n = 3). The lower band seen only at 0 min is an immature form of VMAT2. Scatterplots show mean ± SEM (n = 3). **, P < 0.005; ***, P < 0.001 by one-way ANOVA with Tukey’s multiple comparisons test (A and B) or unpaired t test (E). Data S1 contains statistics source data. Source data are available for this figure: SourceData F4.
Loss of SNX5 redistributes VMAT2 from TGN to endosomes and promotes its lysosomal degradation. (A and B) Loss of SNX5 redistributes VMAT2 away from the TGN (A) to endosomes (B). HeLa cells transfected with 3HA-VMAT2 and control siRNA, SNX5 siRNA, or SNX5 siRNA with RNAi-resistant SNX5 were immunostained for HA (green), SNX5 (magenta), and either TGN46 (red; A) or EEA1 (red; B). The colocalization of VMAT2 and TGN46 was quantified by Pearson’s correlation coefficient (n = 20 images from three independent experiments). The VMAT2 colocalizing with EEA1 was quantified using the Mander’s overlap coefficient analyzing the proportion of pixels labeled strongly for EEA1 that also stain for VMAT2 (n = 10 images from three independent experiments). (C) The bar graph shows the percentage of cells with overall colocalization of VMAT2 to either TGN46 or EEA1 (n = 25 images from three independent experiments). (D) HeLa cells transfected with 3HA-VMAT2 and either control or SNX5 siRNA, without or with pretreatment for 1 h in NH4Cl (10 mM), were immunostained for HA (green) and LAMP1 (red) and the colocalization quantified by Pearson’s correlation coefficient shown in Fig. S5 D (n = 15 images from three independent experiments). Scale bars, 10 µm. (E) PC12 cells stably expressing 3HA-VMAT2 were treated with cycloheximide (CHX) at the times indicated, and VMAT2 was quantified by Western analysis, normalizing to β-actin and the amount at time 0 (n = 3). The lower band seen only at 0 min is an immature form of VMAT2. Scatterplots show mean ± SEM (n = 3). **, P < 0.005; ***, P < 0.001 by one-way ANOVA with Tukey’s multiple comparisons test (A and B) or unpaired t test (E). Data S1 contains statistics source data. Source data are available for this figure: SourceData F4.
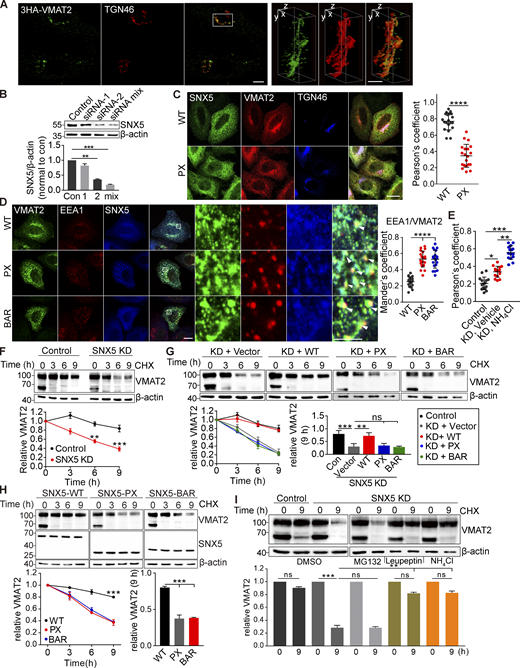
Isolated SNX5 PX and BAR domains redistribute VMAT2 from TGN to endosomes and increase its lysosomal degradation. Related to Fig. 4. (A) HeLa cells transfected with 3HA-VMAT2 were fixed and immunostained for TGN46 (red) and VMAT2 (green). The images were obtained by SIM, shown here as reconstructions of a 125-nm slice. Representative 3D SIM volume rendering of VMAT2 and TGN46 shows that VMAT2 colocalizes with TGN46. Scale bars, 10 µm (left) and 1 µm (right). (B) SNX5 siRNA reduces SNX5 expression in HeLa cells by Western analysis, normalizing to β-actin and control (n = 3). (C) HeLa cells were transfected with either WT or PX-SNX5, immunostained for Flag (green), HA (red), and TGN46 (blue), and the colocalization of VMAT2 with TGN46 was quantified by Pearson’s correlation coefficient (n = 15 images from three independent experiments). Scale bars, 10 µm. (D) HeLa cells were transfected with WT, PX-SNX5, or BAR-SNX5 and immunostained for HA (green), EEA1 (red), and Flag (blue), and the colocalization was quantified by Mander’s overlap coefficient of the proportion of pixels labeled strongly for EEA1 that also stained for VMAT2 (n = 15 images from three independent experiments). Scale bar, 10 µm. (E) The scatterplot shows the Pearson’s correlation coefficient from Fig. 4 D. (F–H) CHO cells stably expressing 3HA-VMAT2 were transfected with control or SNX5 siRNA (F) or siRNA-resistant WT, PX-SNX5, or BAR-SNX5 (G and H) and treated with cycloheximide (CHX) for the times indicated, and extracts were immunoblotted for VMAT2, normalizing to β-actin and the amount at time 0 (n = 3). (I) CHO cells stably expressing 3HA-VMAT2 were transfected with control or SNX5 siRNA and treated with CHX, and the remaining VMAT was determined after 9 h in DMSO, proteasome inhibitor MG132, lysosomal protease inhibitor leupeptin, or NH4Cl (n = 3). Bar graph and scatterplots show mean ± SEM (n = 3). *, P < 0.05; **, P < 0.005; ***, P < 0.001; ****, P < 0.0001 by one-way ANOVA with Tukey’s multiple comparisons test (B and D–I) or unpaired t test (C). Data S1 contains statistics source data. Source data are available for this figure: SourceData FS4.
Isolated SNX5 PX and BAR domains redistribute VMAT2 from TGN to endosomes and increase its lysosomal degradation. Related to Fig. 4. (A) HeLa cells transfected with 3HA-VMAT2 were fixed and immunostained for TGN46 (red) and VMAT2 (green). The images were obtained by SIM, shown here as reconstructions of a 125-nm slice. Representative 3D SIM volume rendering of VMAT2 and TGN46 shows that VMAT2 colocalizes with TGN46. Scale bars, 10 µm (left) and 1 µm (right). (B) SNX5 siRNA reduces SNX5 expression in HeLa cells by Western analysis, normalizing to β-actin and control (n = 3). (C) HeLa cells were transfected with either WT or PX-SNX5, immunostained for Flag (green), HA (red), and TGN46 (blue), and the colocalization of VMAT2 with TGN46 was quantified by Pearson’s correlation coefficient (n = 15 images from three independent experiments). Scale bars, 10 µm. (D) HeLa cells were transfected with WT, PX-SNX5, or BAR-SNX5 and immunostained for HA (green), EEA1 (red), and Flag (blue), and the colocalization was quantified by Mander’s overlap coefficient of the proportion of pixels labeled strongly for EEA1 that also stained for VMAT2 (n = 15 images from three independent experiments). Scale bar, 10 µm. (E) The scatterplot shows the Pearson’s correlation coefficient from Fig. 4 D. (F–H) CHO cells stably expressing 3HA-VMAT2 were transfected with control or SNX5 siRNA (F) or siRNA-resistant WT, PX-SNX5, or BAR-SNX5 (G and H) and treated with cycloheximide (CHX) for the times indicated, and extracts were immunoblotted for VMAT2, normalizing to β-actin and the amount at time 0 (n = 3). (I) CHO cells stably expressing 3HA-VMAT2 were transfected with control or SNX5 siRNA and treated with CHX, and the remaining VMAT was determined after 9 h in DMSO, proteasome inhibitor MG132, lysosomal protease inhibitor leupeptin, or NH4Cl (n = 3). Bar graph and scatterplots show mean ± SEM (n = 3). *, P < 0.05; **, P < 0.005; ***, P < 0.001; ****, P < 0.0001 by one-way ANOVA with Tukey’s multiple comparisons test (B and D–I) or unpaired t test (C). Data S1 contains statistics source data. Source data are available for this figure: SourceData FS4.
The SNX5 KO also reduces the expression of VMAT2 (Fig. 1 D), raising the possibility that a defect in retrograde trafficking to the Golgi complex might increase delivery of VMAT2 to the lysosome, increasing degradation. Indeed, loss of SNX5 increases colocalization of VMAT2 with lysosomal protein LAMP1 (Figs. 4 D and S4 E). To determine whether SNX5 reduces the degradation of VMAT2, we used cycloheximide to block protein synthesis in PC12 transformants. In control cells, VMAT2 is stable for >24 h, but loss of SNX5 reduces the half-life to ∼18 h (Fig. 4 E). Loss of SNX5 also reduces the stability of VMAT2 in CHO cells (Fig. S4 F). Expression of the isolated PX and BAR domains has a similar effect, and only full-length SNX5 rescues the stability of VMAT2 after SNX5 knockdown (Fig. S4, G and H). Both the lysosomal protease inhibitor leupeptin and NH4Cl but not the proteasome inhibitor MG132 stabilize VMAT2 in the absence of SNX5 (Fig. S4 I). Thus, retrograde transport to the TGN by SNX5 also prevents the degradation of VMAT2 in lysosomes. In this case, loss of VPS35 has a similar effect (Wu et al., 2016), implicating the entire retromer complex in this effect on VMAT2.
How does retrograde transport from endosomes to the TGN promote trafficking to DCVs? AP-3 is thought to direct membrane proteins to DCVs within the endolysosomal pathway (Harrison-Lavoie et al., 2006; Kaur et al., 2017; Peden et al., 2004; Theos et al., 2005). The role of SNX5 in retrograde transport from endosomes to the TGN would thus divert VMAT2 away from the endosomal pathway where AP-3 is presumed to function. The recognition of an overlapping sequence in VMAT2 is also difficult to reconcile with a role for both SNX5 and AP-3 in sorting from endosomes to DCVs.
AP-3 and SNX5 have distinct roles in DCV formation
To understand the relationship between SNX5 and AP-3 in DCV biogenesis, we determined whether they have additive effects on regulated exocytosis. Similar to SNX5, loss of AP-3 impairs the regulated release of preloaded 3H-NE, but also affects peptide release (Fig. 5, A and B; and Fig. S5 A). However, loss of SNX5 greatly reduces the regulated release of 3H-NE (Fig. 1 A), making it difficult to determine whether loss of AP-3 has an additional effect. Thus, we used an assay which shows some residual activity in the absence of SNX5. Fusion of a pH-sensitive form of GFP (pHluorin; Miesenböck et al., 1998) to a lumenal loop of VMAT2 shows quenching due to the low pH of DCVs, and exposure to the higher external pH at exocytosis relieves this quenching, increasing fluorescence (Onoa et al., 2010). In PC12 cells, loss of either SNX5 or AP-3 reduces the rate and extent of regulated VMAT2-pHluorin exocytosis, and the combined knockdown of SNX5 and AP-3 has an additive effect (Fig. 5 C; and Fig. S5, C and D). Importantly, the partial effect of AP-3 does not reflect incomplete knockdown because the complete loss of AP-3 by gene inactivation with CRISPR shows no difference from the knockdown in regulated monoamine release or VMAT2 exocytosis, although it has a bigger effect on monoamine loading (Fig. S5, B–E). Thus, SNX5 and AP-3 both contribute to DCV formation but appear to have distinct roles.
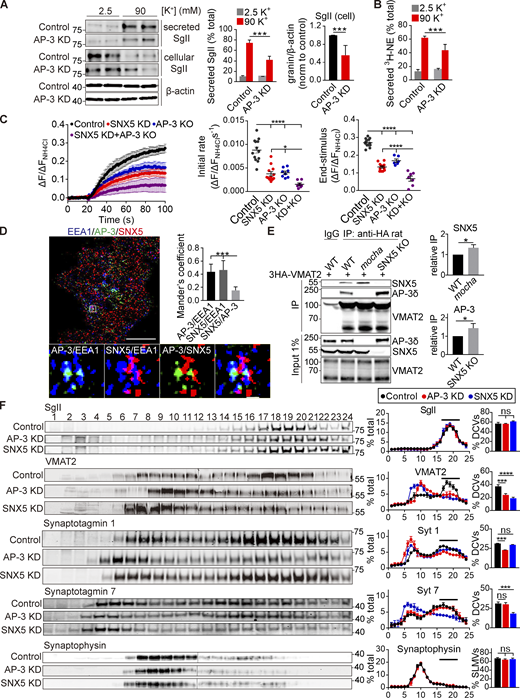
AP-3 and SNX5 have distinct roles in DCV formation. (A and B) Release of SgII (A) and preloaded 3H-NE (B) from PC12 cells transfected with control or AP-3 siRNA and stimulated as in Fig. 1 C. WT or AP-3 KO PC12 cells transfected with VMAT2-pHluorin and siRNA to SNX5 were stimulated with 90 mM K+, and the fluorescence was normalized to that observed in 50 mM NH4Cl (n = 3). (D) PC12 cells were immunostained for EEA1 (blue), AP-3δ (green), and SNX5 (red), and the images were acquired using SIM and reconstructed as in Fig. 1. Scale bar, 5 μm. (E) Glial cells from WT, SNX5 KO, and mocha mice were transduced with lentivirus encoding 3HA-VMAT2 and immunoprecipitated for HA. (F) PC12 cells stably expressing 3HA-VMAT2 were transfected with control, SNX5 siRNA, or AP-3 siRNA and extracts separated by equilibrium density gradient fractionation. The fractions were loaded onto one or two gels (in the case of VMAT2 and synaptophysin) and then transferred onto one membrane for fluorescent detection. Bar graphs indicate the percentage of protein in DCV fractions 16–21 (n = 3 independent experiments). Bar graphs and scatterplots indicate mean ± SEM. **, P < 0.005; ***, P < 0.001 by unpaired t test (A, B, and E) or one-way ANOVA with Tukey’s multiple comparisons test (C, D, and F). Data S1 contains statistics source data. Source data are available for this figure: SourceData F5.
AP-3 and SNX5 have distinct roles in DCV formation. (A and B) Release of SgII (A) and preloaded 3H-NE (B) from PC12 cells transfected with control or AP-3 siRNA and stimulated as in Fig. 1 C. WT or AP-3 KO PC12 cells transfected with VMAT2-pHluorin and siRNA to SNX5 were stimulated with 90 mM K+, and the fluorescence was normalized to that observed in 50 mM NH4Cl (n = 3). (D) PC12 cells were immunostained for EEA1 (blue), AP-3δ (green), and SNX5 (red), and the images were acquired using SIM and reconstructed as in Fig. 1. Scale bar, 5 μm. (E) Glial cells from WT, SNX5 KO, and mocha mice were transduced with lentivirus encoding 3HA-VMAT2 and immunoprecipitated for HA. (F) PC12 cells stably expressing 3HA-VMAT2 were transfected with control, SNX5 siRNA, or AP-3 siRNA and extracts separated by equilibrium density gradient fractionation. The fractions were loaded onto one or two gels (in the case of VMAT2 and synaptophysin) and then transferred onto one membrane for fluorescent detection. Bar graphs indicate the percentage of protein in DCV fractions 16–21 (n = 3 independent experiments). Bar graphs and scatterplots indicate mean ± SEM. **, P < 0.005; ***, P < 0.001 by unpaired t test (A, B, and E) or one-way ANOVA with Tukey’s multiple comparisons test (C, D, and F). Data S1 contains statistics source data. Source data are available for this figure: SourceData F5.
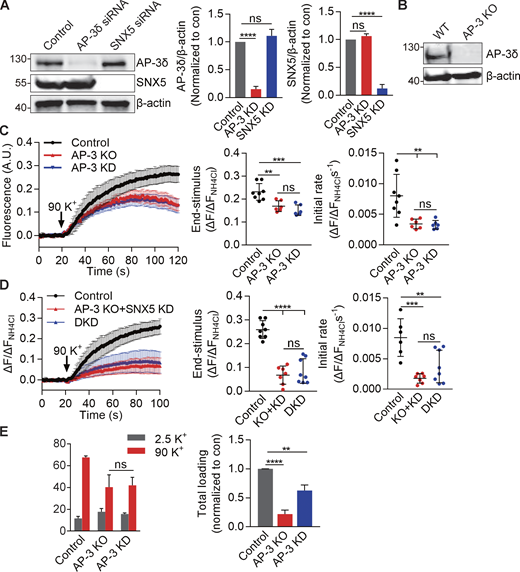
AP-3 knockdown impairs VMAT2 exocytosis and monoamine loading similarly to KO of the AP-3δ gene by CRISPR. Related to Fig. 5. (A) PC12 cells were transfected twice with control, AP-3, or SNX5 siRNA, and extracts were immunoblotted for AP-3 and SNX5, respectively. Bar graph quantifies AP-3 (middle) and SNX5 (right) expression normalized to β-actin and control (n = 3). (B) Western blot of PC12 cell lines confirms the absence of AP-3 protein from KO cells. (C) WT or AP-3 KO PC12 cells transfected with VMAT2-pHluorin and siRNA to AP-3 (in WT only) were stimulated in 90 mM K+, and the fluorescence was normalized to that observed in 50 mM NH4Cl (n = 3 independent experiments). (D) WT or AP-3 KO PC12 cells transfected with VMAT2-pHluorin and siRNA to SNX5 and AP-3 in WT or siRNA to SNX5 in KO cells were stimulated in 90 mM K+, and the fluorescence was normalized to that observed in 50 mM NH4Cl (n = 3 independent experiments). (E) Left: WT or AP-3 KO PC12 cells transfected with control or AP-3 siRNA (in WT cells only) were preloaded with 3H-NE and stimulated as in Fig. 1. Right: Total loaded 3H-NE in WT, AP-3 KO, or knockdown (KD) cells. Bar graphs indicate mean ± SEM. **, P < 0.005; ***, P < 0.001; ****, P < 0.0001 by one-way ANOVA with Tukey’s multiple comparisons test. Statistics source data can be found in Data S1.
AP-3 knockdown impairs VMAT2 exocytosis and monoamine loading similarly to KO of the AP-3δ gene by CRISPR. Related to Fig. 5. (A) PC12 cells were transfected twice with control, AP-3, or SNX5 siRNA, and extracts were immunoblotted for AP-3 and SNX5, respectively. Bar graph quantifies AP-3 (middle) and SNX5 (right) expression normalized to β-actin and control (n = 3). (B) Western blot of PC12 cell lines confirms the absence of AP-3 protein from KO cells. (C) WT or AP-3 KO PC12 cells transfected with VMAT2-pHluorin and siRNA to AP-3 (in WT only) were stimulated in 90 mM K+, and the fluorescence was normalized to that observed in 50 mM NH4Cl (n = 3 independent experiments). (D) WT or AP-3 KO PC12 cells transfected with VMAT2-pHluorin and siRNA to SNX5 and AP-3 in WT or siRNA to SNX5 in KO cells were stimulated in 90 mM K+, and the fluorescence was normalized to that observed in 50 mM NH4Cl (n = 3 independent experiments). (E) Left: WT or AP-3 KO PC12 cells transfected with control or AP-3 siRNA (in WT cells only) were preloaded with 3H-NE and stimulated as in Fig. 1. Right: Total loaded 3H-NE in WT, AP-3 KO, or knockdown (KD) cells. Bar graphs indicate mean ± SEM. **, P < 0.005; ***, P < 0.001; ****, P < 0.0001 by one-way ANOVA with Tukey’s multiple comparisons test. Statistics source data can be found in Data S1.
SNX5 and AP-3 differ in their localization on endosomes. SNX5 and AP-3 both colocalize with EEA1 but do not colocalize with each other by structured illumination microscopy (SIM; Fig. 5 D), segregating to different endosomal domains. In addition, we observe competition for binding to VMAT2: VMAT2 co-IPs more SNX5 in glial cells from AP-3–deficient mocha mice than from WT, and more AP-3 from the SNX5 KO (Fig. 5 E). Gradient fractionation supports different roles for the two proteins, with redistribution of VMAT2 and syt 1 but not syt 7 by AP-3 knockdown and of VMAT2 and syt 7 but not syt1 by knockdown of SNX5 (Fig. 5 F). The effect on syt 7 shows that the role of SNX5 in DCV formation extends beyond VMAT, and the difference from AP-3 suggests mechanisms that may contribute to the observed diversity of DCVs (Kreutzberger et al., 2020; Rao et al., 2014).
SNX5 and AP-3 also differ in their role on endosomes. In contrast to retrograde transport by SNX5, AP-3 on endosomes contributes to the formation of SLMVs in neuroendocrine cells and to a subset of synaptic vesicles in neurons (Blumstein et al., 2001; Faundez et al., 1998; Newell-Litwa et al., 2010; Suckow et al., 2010). Loss of AP-3 and SNX5 both impair localization of VMAT2 to the processes of PC12 cells where SgII+ DCVs accumulate (Fig. 6 A), and as anticipated, SNX5 has less effect than AP-3 on SgII. However, loss of AP-3 also increases the localization of VMAT2 to endosomes (Fig. 6 A), perhaps because the transporter is inaccessible to SNX5 in this segregated endosomal domain (Fig. 5 D). In contrast to HeLa cells (Fig. 4, A and B), loss of SNX5 does not increase the endosomal localization of VMAT2 in PC12 cells (Fig. 6 A). AP-3 and SNX5 thus have divergent effects on the trafficking of VMAT2 at endosomes. To determine whether AP-3 targets a subset of VMAT2 to SLMVs, we separated SLMVs from other cell membranes including endosomes by velocity gradient sedimentation. Loss of AP-3 redistributes VMAT2 and other SLMV proteins away from SLMVs (Fig. 6 B). In contrast, loss of SNX5 does not affect the migration of VMAT2 and syt 1, apparently because AP-3 targets these proteins from endosomes to SLMVs. Thus, AP-3 and SNX5 have divergent effects on the membrane trafficking of VMAT2 from endosomes but both promote trafficking to DCVs.
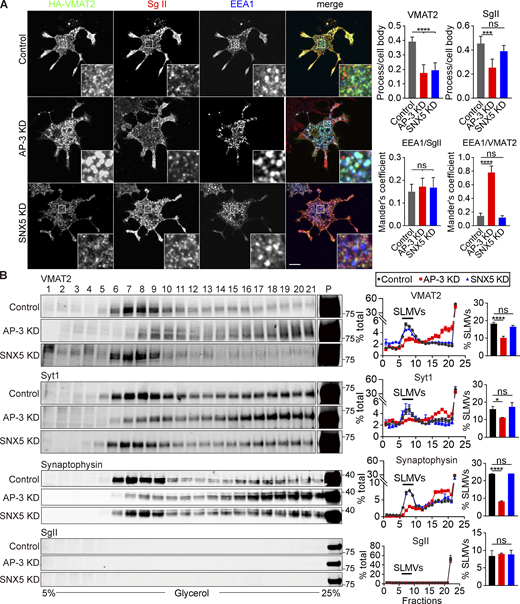
Loss of AP-3 but not SNX5 impairs trafficking to SLMVs. (A) PC12 cells stably expressing 3HA-VMAT2 and transfected with control, SNX5 siRNA, or AP-3 siRNA were immunostained for VMAT2 (green), SgII (red), and EEA1 (blue) after NGF treatment. Bar graphs (right) show that AP-3 or SNX5 knockdown (KD) both reduced localization of VMAT2, but only AP-3 KD reduced localization of SgII in processes relative to the cell body (upper). Mander’s overlap coefficient analyzing the proportion of pixels labeled strongly for EEA1 that also stain for SgII or VMAT2 suggested that AP-3 KD increased colocalization of VMAT2 with EEA1 (lower; n = 3). (B) PC12 cells stably expressing 3HA-VMAT2 were transfected with control, SNX5 siRNA, or AP-3 siRNA, and extracts were separated by glycerol velocity gradient fractionation. Bar graphs indicate the percentage of protein in SLMV fractions 6–9 (n = 3 independent experiments). Bar graphs indicate mean ± SEM. *, P < 0.05; ***, P < 0.001; ****, P < 0.0001 by one-way ANOVA with Tukey’s multiple comparisons test. Data S1 contains statistics source data. Source data are available for this figure: SourceData F6.
Loss of AP-3 but not SNX5 impairs trafficking to SLMVs. (A) PC12 cells stably expressing 3HA-VMAT2 and transfected with control, SNX5 siRNA, or AP-3 siRNA were immunostained for VMAT2 (green), SgII (red), and EEA1 (blue) after NGF treatment. Bar graphs (right) show that AP-3 or SNX5 knockdown (KD) both reduced localization of VMAT2, but only AP-3 KD reduced localization of SgII in processes relative to the cell body (upper). Mander’s overlap coefficient analyzing the proportion of pixels labeled strongly for EEA1 that also stain for SgII or VMAT2 suggested that AP-3 KD increased colocalization of VMAT2 with EEA1 (lower; n = 3). (B) PC12 cells stably expressing 3HA-VMAT2 were transfected with control, SNX5 siRNA, or AP-3 siRNA, and extracts were separated by glycerol velocity gradient fractionation. Bar graphs indicate the percentage of protein in SLMV fractions 6–9 (n = 3 independent experiments). Bar graphs indicate mean ± SEM. *, P < 0.05; ***, P < 0.001; ****, P < 0.0001 by one-way ANOVA with Tukey’s multiple comparisons test. Data S1 contains statistics source data. Source data are available for this figure: SourceData F6.
Retrograde transport by SNX5 enables DCV assembly at the TGN by AP-3
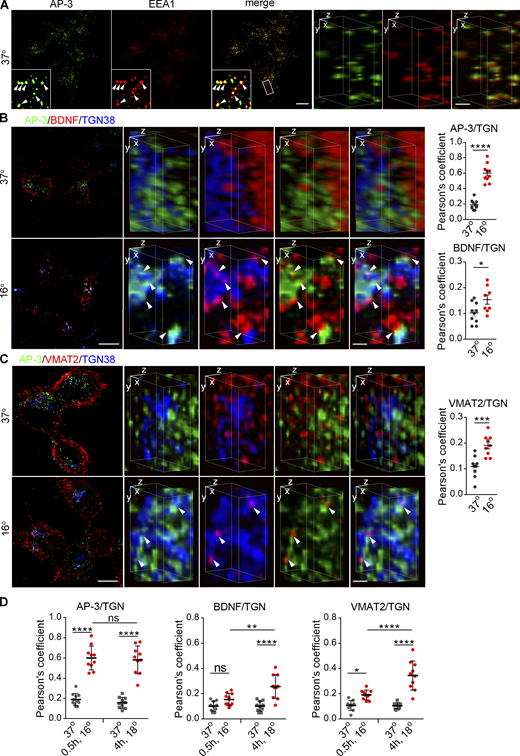
To reconcile the different, apparently antagonistic, roles of SNX5 and AP-3 with the importance of both proteins for DCV formation, we hypothesized that their role in DCV biogenesis does not involve function on the same, endosomal membranes. In yeast, AP-3 appears to act at the Golgi (Cowles et al., 1997; Darsow et al., 2001). In mammals, however, AP-3 localizes at steady state to endosomes, not the Golgi complex (Peden et al., 2004; Theos et al., 2005), although one study inferred a role at the TGN from the effect of temperature shift on proteins destined for lysosomes and melanosomes (Chapuy et al., 2008). To test the possibility that AP-3 localizes transiently to other membranes such as the Golgi, we thus slowed exit from the TGN by lowering the temperature (Chapuy et al., 2008; Saraste et al., 1986). In control cells maintained at physiological temperature, AP-3 colocalizes with EEA1 by SIM (Fig. S8 A), consistent with its role in endolysosomal trafficking. Under these conditions, very little AP-3 colocalizes with TGN38 by confocal (Fig. 7 A) or 3D reconstruction (Fig. 7 B). However, temperature shift to 18°C for 4 h dramatically redistributes AP-3 from peripheral membranes to a perinuclear compartment staining for TGN38 (Fig. 7, A and B). Temperature shift does not affect the localization of endosomes labeled using EEA1 (Fig. 7, C and D). Previous work has suggested Golgi complex tubulation and changes in the localization of multiple Golgi proteins on temperature shift (Gilbert et al., 2018; Martinez-Alonso et al., 2005), and we do observe weak cytoplasmic staining for TGN38 with a rabbit but not mouse antibody after temperature shift to 18°C for 4 h (Fig. 7, A, D, and E). However, the strong immunoreactivity for TGN38 does not change in extent with this temperature shift (Fig. S6 A), and when selecting this strong immunoreactivity, the Mander’s overlap coefficients show a robust increase in colocalization with AP-3 (Fig. S6, B and C). Further, we do not observe the cytoplasmic labeling for TGN38 after temperature shift to 16°C for 30 min, yet the colocalization with AP-3 increases to the same extent as at 18°C for 4 h (Fig. S8 D). Thus, AP-3 redistributes to the TGN independent of any change in the localization of TGN proteins. In contrast to AP-3, SNX5 does not redistribute to the TGN on temperature shift by confocal (Fig. 7 E) or 3D reconstruction (Fig. 7 F), consistent with a different role in trafficking to DCVs. We further tested the TGN association of AP-3 by velocity sedimentation and find that temperature shift redistributes AP-3 from small to large membranes containing Golgin 58 (Fig. 7 G). In mammalian cells, AP-3 thus localizes transiently to the TGN.
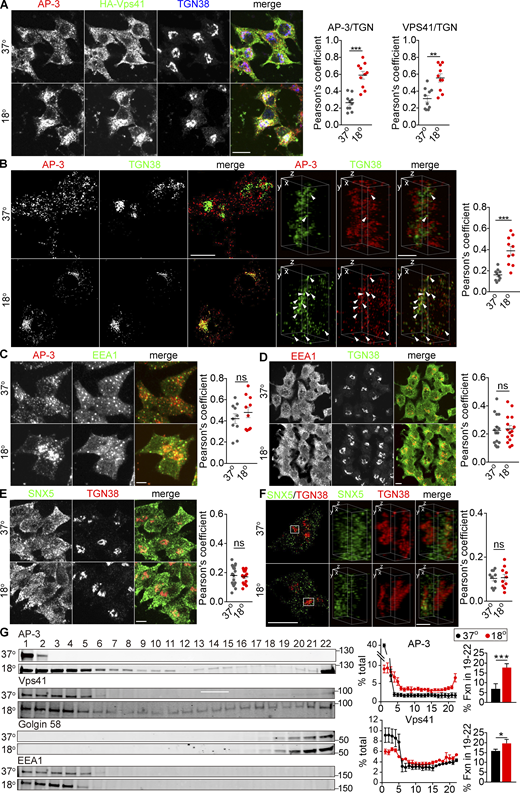
Temperature shift redistributes AP-3 to the TGN. (A) PC12 cells infected with HA-Vps41 virus were incubated for 4 h at either 37 or 18°C, immunostained for AP-3δ (red), HA-Vps41 (green), and TGN38 (blue). Scatterplot (right) indicates the Pearson’s correlation coefficient of AP-3 or VPS41 with TGN (n = 10 cells from three independent experiments). Scale bar, 5 µm. (B) PC12 cells were incubated for 4 h at either 37 or 18°C, immunostained for AP-3δ (red) and TGN38 (green), and the images were reconstructed as in Fig. 1 (left scale bar, 5 μm). Representative 3D SIM volume rendering shows colocalization of AP-3 with TGN38 only at 18°C (right scale bar, 0.5 µm). Arrowheads indicate AP-3 colocalizing with TGN38. Scatterplot (right) indicates the Pearson’s correlation coefficient of AP-3 with TGN (n = 10 cells from three independent experiments). (C and D) PC12 cells were incubated for 4 h at either 37 or 18°C and immunostained either for AP-3δ (red) and EEA1 (green; C) or for EEA1 (red) and TGN38 (green; D). Representative micrographs show that EEA1 does not accumulate during temperature shift. Scatterplot (right) indicates the Pearson’s correlation coefficient of each channel (n = 10 cells from three independent experiments). Scale bar, 5 µm. (E and F) PC12 cells were incubated for 4 h at either 37 or 18°C and immunostained for endogenous SNX5 (green) and TGN38 (red), and images were acquired by confocal (E) or SIM (F) reconstructed as in Fig. 1 (left scale bar, 5 μm; right scale bar, 0.5 µm). Scatterplot (right) indicates the Pearson’s correlation coefficient of each channel (n = 10 cells from three independent experiments). Representative images show that SNX5 does not accumulate at the TGN with temperature shift. Scale bar, 5 μm. (G) PC12 cells infected with HA-Vps41 were incubated for 4 h at either 37 or 18°C, the postnuclear supernatant separated by velocity sedimentation through 0.3–1.2 M sucrose, the fractions immunoblotted for AP-3, HA-Vps41, golgin-58, and EEA1. AP-3 and HA-Vps41 migrate to heavier fractions after incubation at 18°C (n = 3 independent experiments). Bar graphs indicate the percentage of protein in TGN fractions (19–22). Error bars indicate mean ± SEM. *, P < 0.05; **, P < 0.05; ***, P < 0.001 relative to control by two-tailed Student’s t test. Data S1 contains statistics source data. Source data are available for this figure: SourceData F7.
Temperature shift redistributes AP-3 to the TGN. (A) PC12 cells infected with HA-Vps41 virus were incubated for 4 h at either 37 or 18°C, immunostained for AP-3δ (red), HA-Vps41 (green), and TGN38 (blue). Scatterplot (right) indicates the Pearson’s correlation coefficient of AP-3 or VPS41 with TGN (n = 10 cells from three independent experiments). Scale bar, 5 µm. (B) PC12 cells were incubated for 4 h at either 37 or 18°C, immunostained for AP-3δ (red) and TGN38 (green), and the images were reconstructed as in Fig. 1 (left scale bar, 5 μm). Representative 3D SIM volume rendering shows colocalization of AP-3 with TGN38 only at 18°C (right scale bar, 0.5 µm). Arrowheads indicate AP-3 colocalizing with TGN38. Scatterplot (right) indicates the Pearson’s correlation coefficient of AP-3 with TGN (n = 10 cells from three independent experiments). (C and D) PC12 cells were incubated for 4 h at either 37 or 18°C and immunostained either for AP-3δ (red) and EEA1 (green; C) or for EEA1 (red) and TGN38 (green; D). Representative micrographs show that EEA1 does not accumulate during temperature shift. Scatterplot (right) indicates the Pearson’s correlation coefficient of each channel (n = 10 cells from three independent experiments). Scale bar, 5 µm. (E and F) PC12 cells were incubated for 4 h at either 37 or 18°C and immunostained for endogenous SNX5 (green) and TGN38 (red), and images were acquired by confocal (E) or SIM (F) reconstructed as in Fig. 1 (left scale bar, 5 μm; right scale bar, 0.5 µm). Scatterplot (right) indicates the Pearson’s correlation coefficient of each channel (n = 10 cells from three independent experiments). Representative images show that SNX5 does not accumulate at the TGN with temperature shift. Scale bar, 5 μm. (G) PC12 cells infected with HA-Vps41 were incubated for 4 h at either 37 or 18°C, the postnuclear supernatant separated by velocity sedimentation through 0.3–1.2 M sucrose, the fractions immunoblotted for AP-3, HA-Vps41, golgin-58, and EEA1. AP-3 and HA-Vps41 migrate to heavier fractions after incubation at 18°C (n = 3 independent experiments). Bar graphs indicate the percentage of protein in TGN fractions (19–22). Error bars indicate mean ± SEM. *, P < 0.05; **, P < 0.05; ***, P < 0.001 relative to control by two-tailed Student’s t test. Data S1 contains statistics source data. Source data are available for this figure: SourceData F7.
Temperature shift redistributes AP-3 to the TGN independent of any change in the localization of TGN proteins. Related to Fig. 7. (A) PC12 cells were incubated either at 16°C for 30 min or 18°C for 4 h, fixed immediately, and immunostained. The images were acquired by confocal (left) or SIM (right) and reconstructed as in Fig. 1. Representative 3D SIM volume rendering shows distribution of TGN38 at the indicated temperature. Scatterplot indicates the area occupied by TGN38+ structures (n = 20 cells from three independent experiments). Scale bars, 5 µm (left) and 1 µm (right). (B) PC12 cells were incubated at 37°C, 16°C for 30 min, or 18°C for 4 h and immunostained for AP-3δ (green) and TGN38 (magenta). Magnified images show the colocalization of AP-3 with TGN38. Scatterplots show the Pearson’s correlation coefficient of AP-3 with TGN (top) and the Mander’s overlap coefficient for the proportion of pixels labeled strongly for TGN that also stained for AP-3 (TGN/AP-3, middle) and vice versa (AP-3/TGN, bottom) using autothreshold (n = 15 cells from three independent experiments). Scale bar, 5 µm. (C) PC12 cells were incubated at 37°C, 16°C for 30 min, or 18°C for 4 h and immunostained for AP-3δ (green) and TGN38 (red). The Mander’s overlap coefficient was used to quantify AP-3/TGN using autothreshold (above) or manual threshold (below). White puncta indicated the colocalization of AP-3 and TGN38 by auto- or manual threshold. Scatterplot indicates the Mander’s overlap coefficient for AP-3/TGN using a manual threshold. Scale bar, 5 µm. Error bars indicate mean ± SEM. ****, P < 0.0001 relative to control by one-way ANOVA with Tukey’s multiple comparisons test. Data S1 contains statistics source data.
Temperature shift redistributes AP-3 to the TGN independent of any change in the localization of TGN proteins. Related to Fig. 7. (A) PC12 cells were incubated either at 16°C for 30 min or 18°C for 4 h, fixed immediately, and immunostained. The images were acquired by confocal (left) or SIM (right) and reconstructed as in Fig. 1. Representative 3D SIM volume rendering shows distribution of TGN38 at the indicated temperature. Scatterplot indicates the area occupied by TGN38+ structures (n = 20 cells from three independent experiments). Scale bars, 5 µm (left) and 1 µm (right). (B) PC12 cells were incubated at 37°C, 16°C for 30 min, or 18°C for 4 h and immunostained for AP-3δ (green) and TGN38 (magenta). Magnified images show the colocalization of AP-3 with TGN38. Scatterplots show the Pearson’s correlation coefficient of AP-3 with TGN (top) and the Mander’s overlap coefficient for the proportion of pixels labeled strongly for TGN that also stained for AP-3 (TGN/AP-3, middle) and vice versa (AP-3/TGN, bottom) using autothreshold (n = 15 cells from three independent experiments). Scale bar, 5 µm. (C) PC12 cells were incubated at 37°C, 16°C for 30 min, or 18°C for 4 h and immunostained for AP-3δ (green) and TGN38 (red). The Mander’s overlap coefficient was used to quantify AP-3/TGN using autothreshold (above) or manual threshold (below). White puncta indicated the colocalization of AP-3 and TGN38 by auto- or manual threshold. Scatterplot indicates the Mander’s overlap coefficient for AP-3/TGN using a manual threshold. Scale bar, 5 µm. Error bars indicate mean ± SEM. ****, P < 0.0001 relative to control by one-way ANOVA with Tukey’s multiple comparisons test. Data S1 contains statistics source data.
To test the role of AP-3 at the TGN using an orthogonal approach that does not rely on temperature shift, we knocked down the putative coat protein VPS41 (Fig. S7). If AP-3 works with VPS41 at the TGN, knockdown of VPS41 might also be expected to trap AP-3 at this site, and like AP-3, VPS41 also redistributes to larger membranes on temperature shift (Fig. 7, A and G). Loss of VPS41 increases the colocalization of AP-3 with TGN38 by both standard confocal and SIM (Fig. 8), further supporting a role for AP-3 at this site.
Localization of VPS41. Related to Fig. 8. (A) PC12 cells were transiently cotransfected with 1 or 0.2 µg HA-Vps41 and either control or Vps41 siRNA, and extracts were immunoblotted for HA. Bar graph quantifies HA-Vps41 expression normalized to β-actin and control (n = 3). (B) PC12 cells infected with HA-Vps41 were stained for HA and organelle markers (TGN38 for TGN, EEA1 for endosomes, and LAMP1 for lysosomes). Arrowheads indicate VPS41. Representative micrographs show that VPS41 does not colocalize with EEA1 and partially colocalizes with LAMP1. Scale bar, 5 µm. Error bars indicate mean ± SEM. ****, P < 0.0001 relative to control by two tailed Student’s t test. Data S1 contains statistics source data. Source data are available for this figure: SourceData FS7.
Localization of VPS41. Related to Fig. 8. (A) PC12 cells were transiently cotransfected with 1 or 0.2 µg HA-Vps41 and either control or Vps41 siRNA, and extracts were immunoblotted for HA. Bar graph quantifies HA-Vps41 expression normalized to β-actin and control (n = 3). (B) PC12 cells infected with HA-Vps41 were stained for HA and organelle markers (TGN38 for TGN, EEA1 for endosomes, and LAMP1 for lysosomes). Arrowheads indicate VPS41. Representative micrographs show that VPS41 does not colocalize with EEA1 and partially colocalizes with LAMP1. Scale bar, 5 µm. Error bars indicate mean ± SEM. ****, P < 0.0001 relative to control by two tailed Student’s t test. Data S1 contains statistics source data. Source data are available for this figure: SourceData FS7.
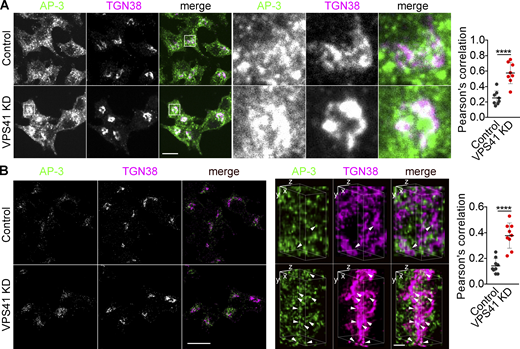
Loss of VPS41 traps AP-3 at the TGN. (A and B) PC12 cells were transfected twice with control or Vps41 siRNA and immunostained for AP-3δ (green) and TGN38 (red), and images were acquired by confocal (A) or SIM (B) and reconstructed as in Fig. 1 (left scale bar, 5 μm). Representative 3D SIM volume rendering shows colocalization of AP-3 with TGN38 (arrowheads; right scale bar, 0.5 µm). Scatterplot (right) indicates the Pearson’s correlation coefficient of AP-3 with TGN (n = 3 independent experiments). Error bars indicate mean ± SEM. ****, P < 0.0001 relative to control by two-tailed Student’s t test. Data S1 contains statistics source data.
Loss of VPS41 traps AP-3 at the TGN. (A and B) PC12 cells were transfected twice with control or Vps41 siRNA and immunostained for AP-3δ (green) and TGN38 (red), and images were acquired by confocal (A) or SIM (B) and reconstructed as in Fig. 1 (left scale bar, 5 μm). Representative 3D SIM volume rendering shows colocalization of AP-3 with TGN38 (arrowheads; right scale bar, 0.5 µm). Scatterplot (right) indicates the Pearson’s correlation coefficient of AP-3 with TGN (n = 3 independent experiments). Error bars indicate mean ± SEM. ****, P < 0.0001 relative to control by two-tailed Student’s t test. Data S1 contains statistics source data.
If AP-3 assembles DCVs at the TGN, it should colocalize there with both soluble and membrane DCV cargo. To test this, we increased the flux of soluble cargo through the Golgi by transfecting BDNF-mScarlet. Due to the accumulation in DCVs, mScarlet+ punctae do not generally colocalize with TGN38 at 37°C (Fig. 9 A). On temperature shift, however, the colocalization increases and most of the TGN38+ mScarlet+ punctae colocalize with AP-3 (Fig. 9 A). With regard to membrane protein cargo, VMAT2 localizes primarily to DCVs but colocalizes more with TGN38 after temperature shift, and these punctae also colocalize with AP-3 (Fig. 9 B). Velocity sedimentation confirms the shift of VMAT2 and SV2 to larger membranes containing golgin 58 (Fig. 9 C). It is possible that the accumulation of membrane proteins traps AP-3 nonspecifically at the TGN, but temperature shift to 16°C for only 30 min similarly redistributes AP-3 with less accumulation of DCV cargo than at 18°C for 4 h (Fig. S8, B and D). Thus, temperature block rapidly redistributes AP-3 to the TGN, whereas DCV soluble and membrane cargo accumulate more slowly due to transit through the secretory pathway.
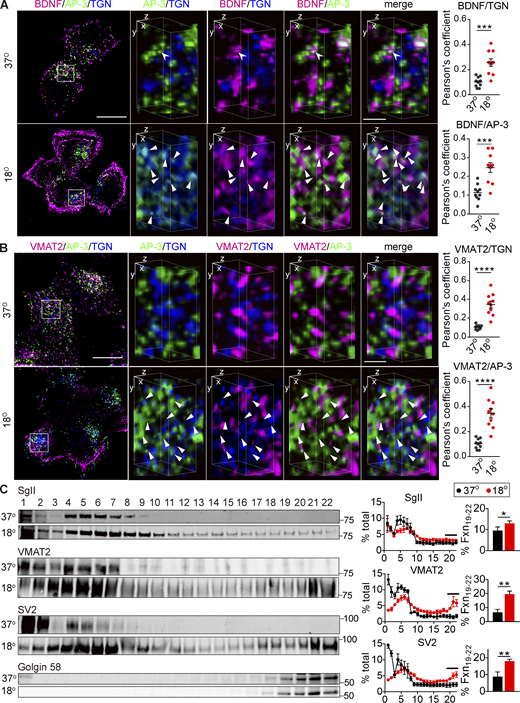
AP-3 trapped at the TGN colocalizes with soluble and membrane DCV cargo. (A and B) PC12 cells transduced with lentivirus encoding BDNF-mScarlet (A) or 3HA-VMAT2 (B) were incubated at 18°C for 4 h and immunostained for AP-3δ (green), BDNF (red), and TGN38 (blue; A); or AP-3δ (green), VMAT2 (red), and TGN38 (blue; B). The images were obtained using SIM. Left scale bars, 5 μm. Representative 3D SIM volume rendering (lower panels) shows that BDNF and VMAT2 colocalize with AP-3 after temperature shift. Right scale bars, 0.5 µm. Arrowheads indicate cargo colocalizing with both AP-3 and TGN38. Scatterplots indicate the Pearson’s correlation coefficients for colocalization of the indicated channels (n = 10 images from three independent experiments). (C) PC12 cells were incubated for 4 h at either 37 or 18°C, the postnuclear supernatant was separated by velocity sedimentation and fractions were immunoblotted for SgII, VMAT2, SV2, and Golgin 58 as described in Fig. 7. SgII, VMAT2, and SV2 migrate to heavier fractions after incubation at 18°C (n = 3 independent experiments). Bar graphs indicate the percentage of protein in TGN fractions (19–22). Error bars indicate mean ± SEM. *, P < 0.05; **, P < 0.005; ***, P < 0.001; and ****, P < 0.0001 by two-tailed Student’s t test. Table S1 contains statistics source data. Source data are available for this figure: SourceData F9.
AP-3 trapped at the TGN colocalizes with soluble and membrane DCV cargo. (A and B) PC12 cells transduced with lentivirus encoding BDNF-mScarlet (A) or 3HA-VMAT2 (B) were incubated at 18°C for 4 h and immunostained for AP-3δ (green), BDNF (red), and TGN38 (blue; A); or AP-3δ (green), VMAT2 (red), and TGN38 (blue; B). The images were obtained using SIM. Left scale bars, 5 μm. Representative 3D SIM volume rendering (lower panels) shows that BDNF and VMAT2 colocalize with AP-3 after temperature shift. Right scale bars, 0.5 µm. Arrowheads indicate cargo colocalizing with both AP-3 and TGN38. Scatterplots indicate the Pearson’s correlation coefficients for colocalization of the indicated channels (n = 10 images from three independent experiments). (C) PC12 cells were incubated for 4 h at either 37 or 18°C, the postnuclear supernatant was separated by velocity sedimentation and fractions were immunoblotted for SgII, VMAT2, SV2, and Golgin 58 as described in Fig. 7. SgII, VMAT2, and SV2 migrate to heavier fractions after incubation at 18°C (n = 3 independent experiments). Bar graphs indicate the percentage of protein in TGN fractions (19–22). Error bars indicate mean ± SEM. *, P < 0.05; **, P < 0.005; ***, P < 0.001; and ****, P < 0.0001 by two-tailed Student’s t test. Table S1 contains statistics source data. Source data are available for this figure: SourceData F9.
Temperature block rapidly redistributes AP-3 to the TGN, whereas DCV cargo accumulate more slowly. Related to Fig. 9. (A) PC12 cells were immunostained for AP-3δ (green) and EEA1 (red), and the images were acquired by SIM and reconstructed as in Fig. 1. Arrowheads indicate punctate labeling for both AP-3 and EEA1 (left scale bar, 5 μm). Representative 3D SIM volume rendering shows AP-3 colocalizing with EEA1 (right scale bar, 0.5 µm). (B and C) PC12 cells transduced with lentivirus encoding BDNF-mScarlet (B) or 3HA-VMAT2 (C) were incubated at either 37 or 16°C for 30 min and immunostained for AP-3δ (green), BDNF (red), and TGN38 (blue; B) or AP-3δ (green), VMAT2 (red), and TGN38 (blue; C). The images were acquired using SIM (n = 8–10 images from three independent experiments). Arrowheads indicate cargo colocalizing with AP-3 and TGN38. Scatterplots indicate Pearson’s correlation coefficients for the colocalization of cargo with AP-3 in the region surrounding the TGN. (D) Scatterplots indicate Pearson’s correlation coefficients for the colocalization of AP-3, BDNF, or VMAT2 with TGN at 16°C for 30 min or 18°C for 4 h. Error bars indicate mean ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 by two tailed Student’s t test (B) or one-way ANOVA with Tukey’s multiple comparisons test (C). Table S1 contains statistics source data.
Temperature block rapidly redistributes AP-3 to the TGN, whereas DCV cargo accumulate more slowly. Related to Fig. 9. (A) PC12 cells were immunostained for AP-3δ (green) and EEA1 (red), and the images were acquired by SIM and reconstructed as in Fig. 1. Arrowheads indicate punctate labeling for both AP-3 and EEA1 (left scale bar, 5 μm). Representative 3D SIM volume rendering shows AP-3 colocalizing with EEA1 (right scale bar, 0.5 µm). (B and C) PC12 cells transduced with lentivirus encoding BDNF-mScarlet (B) or 3HA-VMAT2 (C) were incubated at either 37 or 16°C for 30 min and immunostained for AP-3δ (green), BDNF (red), and TGN38 (blue; B) or AP-3δ (green), VMAT2 (red), and TGN38 (blue; C). The images were acquired using SIM (n = 8–10 images from three independent experiments). Arrowheads indicate cargo colocalizing with AP-3 and TGN38. Scatterplots indicate Pearson’s correlation coefficients for the colocalization of cargo with AP-3 in the region surrounding the TGN. (D) Scatterplots indicate Pearson’s correlation coefficients for the colocalization of AP-3, BDNF, or VMAT2 with TGN at 16°C for 30 min or 18°C for 4 h. Error bars indicate mean ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 by two tailed Student’s t test (B) or one-way ANOVA with Tukey’s multiple comparisons test (C). Table S1 contains statistics source data.
Discussion
In this work, we identify a requirement for SNX5 in the trafficking of VMAT2 that enables the regulated release of catecholamine from DCVs. In the absence of SNX5, monoamine storage is greatly reduced and release severely impaired, reflecting the absence of VMAT on DCVs. SNX5 may thus resemble the chaperones (stonin and SV2 for synaptotagmin 1 and AP180 for v-SNARE VAMP2) responsible for the trafficking of other membrane proteins important for neurotransmitter release (Diril et al., 2006; Dittman and Kaplan, 2006; Kaempf et al., 2015; Koo et al., 2015; Martina et al., 2001; Nonet et al., 1999; Schivell et al., 1996). However, loss of SNX5 also affects the distribution of syt 7, indicating a role beyond the VMATs that may contribute to the differences in release between subpopulations of DCVs (Kreutzberger et al., 2020; Rao et al., 2014).
We also identify the mechanism by which SNX5 promotes VMAT trafficking to DCVs: retrograde transport from endosomes to the TGN, similar to its role with the CI-MPR (Kvainickas et al., 2017; Simonetti et al., 2017; Simonetti et al., 2019). In the process, SNX5 stabilizes VMAT2 by preventing its degradation in the lysosome. However, retrograde traffic to the TGN seemed inconsistent with the role of AP-3 in endolysosomal trafficking (Bowman et al., 2019). Retrograde transport to the TGN would compete with this role for AP-3. In addition, SNX5 and AP-3 compete for binding to closely overlapping sequences in VMAT2.
Although this role for SNX5 and competition for binding might be expected to antagonize the endosomal role of AP-3, we find that the adaptor associates transiently with the TGN, where it colocalizes with soluble and membrane DCV cargo. Thus, retrograde delivery enables AP-3 to assemble membrane proteins into DCVs at the TGN rather than on endosomes. Because AP-3 promotes the trafficking of many membrane proteins to DCVs, we hypothesize that different SNX-BAR or related proteins may contribute to the retrograde transport of other DCV membrane proteins required for assembly by AP-3 at the TGN. In addition, overlap between SNX5 and adaptor binding sequences restricts the function of these trafficking proteins to different membranes. SNX5 recognition of adaptor binding sites in other proteins (Simonetti et al., 2019) suggests this may be a general mechanism to coordinate membrane traffic. The requirement for SNX5 also indicates the importance of recycling from endosomes for DCV biogenesis. Efficient sorting in the biosynthetic pathway would require SNX5 only for endocytic recycling after DCV exocytosis. The importance of retrograde transport for localization to DCVs suggests inefficient sorting at the TGN and a requirement for iterative recycling in DCV formation.
If SNX5 and AP-3 operate in tandem to promote the trafficking of VMAT2 to DCVs, why do they have additive effects on the regulated exocytosis of VMAT2? This does not reflect a partial loss of function, because inactivation of AP-3 using CRISPR has the same effect as the knockdown. Rather, the additivity reflects distinct biological roles for these proteins, each of which is required for different features of DCV formation, but without which DCV formation still proceeds. In particular, loss of SNX5 dramatically affects the trafficking of VMAT but has little effect on most other DCV membrane proteins. In contrast, loss of AP-3 impairs the trafficking of multiple DCV membrane proteins but only partially. Thus, AP-3 inactivation would be expected to disrupt the regulated exocytosis of DCVs with marginal amounts of VMAT2 due to loss of SNX5, and SNX5 inactivation would be expected to reduce further the VMAT2 on DCVs with impaired exocytosis due to the reduction in other membrane proteins due to loss of AP-3.
The results further indicate important differences between the roles of AP-3 on endosomes and at the TGN. On endosomes, AP-3 contributes to the formation of SLMVs (Blumstein et al., 2001; Faundez et al., 1998; Newell-Litwa et al., 2010; Suckow et al., 2010). Consistent with this, loss of AP-3 increases the VMAT2 on endosomes and impairs targeting to SLMVs. Surprisingly, loss of SNX5 does not trap VMAT2 on endosomes in PC12 cells as it does in HeLa cells. This presumably reflects diversion to SLMVs by AP-3 and to lysosomes for degradation. The steady-state location of AP-3 to endosomes also suggests that its role there involves a more stable association. In contrast, AP-3 appears to have a transient role in the formation of DCVs at the TGN, and loss of AP-3 does not trap VMAT2 at the TGN as it does on endosomes. Indeed, previous work showing constitutive exocytosis of DCV cargo (SgII and VMAT2) in the absence of AP-3 (Asensio et al., 2010) suggests alternative, constitutive pathways out of the Golgi complex. Loss of AP-3 thus traps cargo on endosomes but not the TGN.
The roles of SNX5 and AP-3 at distinct steps on the pathway to DCV formation require mechanisms to regulate their interaction with cargo. Since SNX5 can remain associated with VMAT2 even at the plasma membrane, this interaction does not apparently require localization at a particular site. SNX5 might thus be expected to outcompete AP-3 on endosomes. However, the two proteins occupy different endosomal domains, and VMAT2 accumulates on endosomes in the absence of AP-3, suggesting this fraction may be inaccessible to retrograde transport by SNX5. At the TGN, AP-3 must displace SNX5 to bind VMAT2, and this may require additional signals such as GTPases or lipid modification specific to this site. ADP ribosylation factor (ARF) 1, phosphatidylinositol 4-kinase type IIα, and other protein complexes such as biogenesis of lysosome organelle complex 1 (BLOC-1) have indeed been implicated in the function of AP-3 (Faundez et al., 1997; Salazar et al., 2005; Salazar et al., 2006). The transient role of AP-3 at the TGN may also reflect preassembly of the membrane proteins dependent on AP-3 or simply more efficient budding.
In summary, the results show that retrograde transport by SNX5 enables assembly of VMAT2 into DCVs by AP-3 at the TGN. AP-3 also promotes trafficking of VMAT and other proteins to SLMVs, but this does not require SNX5. This selectivity suggests a mechanism for SNX5 to control the composition of DCVs independently from SLMVs, with implications for the properties of release. SNX5 and other proteins involved in retrograde transport to the TGN may similarly affect the targeting of other proteins involved in regulated secretion. Localization of AP-3 to the TGN as well as endosomes also raises the possibility of other noncanonical roles for the adaptor in the formation of lysosomes and related organelles.
Materials and methods
Animals
The SNX5 KO mouse line (RIKEN accession no. CDB0840K) was obtained from the RIKEN Center for Biosystems Dynamics Research. All mouse lines were congenic with the C57BL/6 background. All rodent procedures were performed according to guidelines established by the University of California San Francisco (UCSF) Institutional Animal Care and Use Committee.
Antibodies
The HA.11 mouse monoclonal antibody (MMS-101R) was obtained from Covance; the Alexa Fluor 647–conjugated anti-HA antibody (26183-A647) from Thermo Fisher Scientific; the HA rabbit polyclonal (ab9110), SNX5 rabbit polyclonal (ab180520), and LAMP1 mouse monoclonal (ab25630) antibodies from Abcam; the M2-Flag mouse monoclonal (F1804), Flag rabbit polyclonal (F2555), and TGN38 rabbit polyclonal (T9826) antibodies from Sigma-Aldrich; the HA rat monoclonal antibody (11867423001) from Roche; the EEA1 mouse monoclonal (610457) and TGN38 monoclonal (610899) antibodies from BD Transduction Labs; the EEA1 rabbit polyclonal (237002), synaptophysin mouse monoclonal (101011), synaptotagmin 1 mouse monoclonal (105011), and synaptotagmin 7 rabbit polyclonal (105173) antibodies from Synaptic Systems; the SgII rabbit antibody from Meridian Life Science (K55101R) and Proteintech (20357-1-AP); the β-actin mouse monoclonal (A2228) and VMAT2 polyclonal (V9014) antibodies from Millipore; and the AP-3δ (anti-delta SA4, AB_2056641) mouse monoclonal and SV2 mouse monoclonal (AB_2315387) antibodies from the Developmental Studies Hybridoma Bank. The following secondary antibodies were obtained from Invitrogen: Alexa Fluor 488 or 568 conjugated to donkey anti-rabbit or anti-mouse antibodies, and Alexa Fluor 674–conjugated donkey anti-rat antibody.
Yeast two-hybrid screen
The LexA yeast two-hybrid system (Clontech) was used to identify proteins interacting with the C-terminus of VMAT2, following the manufacturer’s instructions. Briefly, the C-terminus of rat VMAT2 extending from Pro-466 to the termination codon was inserted into bait vector pLexA, this construct introduced into yeast along with a PC12 cDNA library cloned into the activation domain vector pB42AD, expression of the activation domain library induced with galactose, and clones exhibiting an interaction selected in Leu− media.
Molecular biology
The plasmids encoding 3Flag- or 3HA-tagged rat SNX5 were amplified by PCR to add 5′ KpnI and 3′ XbaI sites and then subcloned into the pcDNA3.1-3xFlag or -3xHA expression vector, respectively. 3Flag-SNX5-PX1–180 and -BAR206–415 were amplified by PCR from the 3Flag-rat SNX5 plasmid and cloned into pcDNA3.1-3xFlag or -3xHA vectors. RNAi-resistant constructs were generated by making point mutations in the site of recognition by siRNA. Construction of plasmids encoding 3Flag- or 3HA-tagged rat VMAT2 involved the same strategy as for SNX5. Stop codons required for the 3HA-VMAT2 deletion mutants were introduced by overlap extension PCR. 3HA-VMAT2 point mutations were generated by site-directed mutagenesis. For expression in a lentiviral vector, 3HA-VMAT2, VMAT2-pHluorin, BDNF-mScarlet, and HA-Vps41 (Asensio et al., 2013) were amplified by PCR to add 5′ XbaI and 3′ HpaI sites and subcloned into the modified FUGW lentiviral expression vector. 3Flag human SNX1 and rat SNX6 constructs were subcloned into pcDNA3.1-3xFlag using KpnI and NotI. The 3Flag-Vps35 construct was amplified by PCR to add 5′ KpnI and 3′ XhoI sites, then subcloned into the pcDNA3.1-3xFlag vector. Full-length 6xHis-SNX5 was amplified by PCR to add 5′ HindIII and 3′ BamHI sites and subcloned into pET28 (a). 8xHis-SNX5-PX20–180 was kindly provided by O.S. Rosenberg (UCSF). The cytoplasmic C-terminus of VMAT2 was amplified by PCR to add 5′ BamHI and 3′ EcoRI sites and subcloned into pGEX-6P-1 to produce GST-VMAT2-Ct. All constructs were sequenced to confirm the desired and exclude off-target mutations.
siRNA and shRNA
siRNA to human SNX5 (sense 1, 5′-GCUGCAUUGAUUUAUUCAATT-3′; and sense 2, 5′-GGCAACAUGAAGACUUUGUTT-3′) and corresponding, nontargeting control siRNA were obtained from GenePharma Company. siRNA On-target SMARTpool to rat SNX5 (sense 1, 5′-UGACAGAGCUCCUCCGAUA′-3′; sense 2, 5′-GAGUGGGCAGCAUUUCGAAA-3′; sense 3, 5′-UGAUAAACUUCAAACGGAA-3′; and sense 4, 5′-GAAAUAGCUUUGACUAGUU-3′), siRNAs to rat AP-3δ (antisense, 5′-UUCUUGGUCAUGAUCCAUGTG-3′) and siRNA On-Target SMARTpool to rat Vps41 (sense 1, 5′-ACGACAUGGCCGCACGAAA-3′; sense 2, 5′-UGAUAUAAGUCUUCGCCCA-3′; sense 3, 5′-GGGAACAUAAGGAGCGUGA-3′; and sense 4, 5′-CGUAAAGGAGGUUUCGGAA-3′) were obtained from Dharmacon. siRNA to human Vps35 (sense, 5′-CCAGACTATCAGTGCTTTGAT-3′) from GenePharma. Lentiviral constructs expressing shRNA to rat Vps35 (shVps35-1, 5′-TCAGAGGACGTTGTATCTTTA-3′; shVps35-2, 5′-GCTTCACACTGCCACCTTTGG-3′; and scramble shRNA, 5′-TTCTCCGAACGTGTCACGT-3′) were made in a pFH1UChW vector, a variant of pFH1UGW containing mCherry in place of GFP.
Cell culture and transfection
All cells were maintained in 5% CO2 at 37°C. HeLa and COS-7 cells were maintained in DMEM (Invitrogen) with 10% cosmic calf serum (Hyclone). HEK293T cells were maintained in DMEH-21 medium with 10% FBS (Hyclone) and 1% GlutaMAX. CHO cells were maintained in Ham’s F12 medium (Invitrogen) with 5% cosmic calf serum, and PC12 cells in DMEM (Invitrogen) with 10% equine serum, 5% FCS (both Hyclone), and 1% GlutaMAX. Stable transformants were produced as previously described (Liu and Edwards, 1997b). Briefly, CHO cells were transfected with 3Flag-VMAT2 and selected in 500 µg/ml G418 for 1 wk. PC12 cells were transfected with 3Flag- or 3HA-VMAT2 and selected in 800 µg/ml G418 for 1 wk. Single-cell clones were obtained by serial dilution and screened by immunofluorescence and Western analysis. Stable transformants were maintained in 50 µg/ml G418.
For immunofluorescence, the cells were seeded onto poly-D-lysine– and Matrigel-coated glass coverslips, transfected 24 h later using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions, and differentiated 12 h after transfection using 100 nM NGF for 24–36 h before fixation in 4% PFA/0.1 M NaPO4 buffer, pH 7.2. For biochemical assays, transfections were also done using Lipofectamine 2000, and the cells were incubated at 37°C for 36–48 h before harvesting.
For temperature shift to 16°C, PC12 cells in DMEM with 10% equine serum, 5% FCS (both Hyclone), and 1% GlutaMAX were placed in a 16°C refrigerator for 30 min. For temperature shift to 18°C, PC12 cells in the same medium were incubated in 5% CO2 at 18°C for 4 h. In both cases, the cells were then immediately placed on ice before immunostaining or biochemical fractionation.
Mouse adrenal chromaffin cells were isolated and cultured as previously described (Larsen et al., 2006). Briefly, adrenal glands were dissected and placed in cold Ca2+-, Mg2+-free (CMF) HBSS, the surrounding fat and cortex were removed, and the medullae were transferred to CMF-HBSS containing 20 U/ml papain and dissociated by shaking for 40 min at 37°C. Papain solution was then replaced by CMF-HBSS containing 200 µg/ml M1 DNAse I (Sigma-Aldrich) and 1% heat-inactivated FBS (Gibco). The tissue was triturated first with a P200 pipette tip, and then with a 23-gauge needle. The cells were pelleted at 300 g for 2 min at room temperature and resuspended in prewarmed culture medium. Cells were maintained in DMEH-21 medium supplemented with 10% FBS and antibiotics. For lentiviral transduction, freshly isolated chromaffin cells were plated in viral supernatant, and fresh medium was added the next morning.
AP-3 δ CRISPR/CAS9-mediated KO in PC12 cells
AP-3 δ subunit KO PC12 cells were generated using the indel strategy. A pair of annealed oligonucleotides (20 bp) was ligated into the single gRNA scaffold of a modified pLentiCRISPR_v2 vector (AddGene; Zhang Lab, Massachusetts Institute of Technology) with mCherry replacing puromycin. The following gRNA sequences were used: AP-3 δ forward, 5′-CACCGCTTGATCTCGTCAATGCAC-3′; and AP-3 δ reverse, 5′-AAACGTGCATTGACGAGATCAAGC-3′. All gRNA sequences were determined using the CRISPR design tool. Individual PC12 cells expressing mCherry were selected by cell sorting and expanded into clonal cell lines. A BD FACS Aria machine was used for cell sorting in conjunction with the Diva software suite.
Lentivirus production
Low-passage HEK293T cells were seeded onto six-well plates coated with poly-L-lysine and transfected overnight with a mixture of third-generation lentiviral vector pFUGW encoding the gene of interest as well as accessory plasmids pMDLG/pRRE, pIVS-VSVG, and pRSV-Rev, using FuGene HD transfection reagent (Promega) according to the manufacturer’s instructions. Cells were switched into PC12 cell medium the next morning and 36 h later, the culture medium was collected and cell debris was sedimented at 1,000 g for 2 min. The viral supernatant was either used immediately or aliquoted and frozen at −80°C.
Immunoprecipitation
Cells were washed with PBS and disrupted for 10 min on ice in NP-40 lysis buffer (0.5% NP-40, 150 mM NaCl, 50 mM Tris-HCl, pH 7.0, and 5 mM EDTA) supplemented with 1 mM PMSF and 10 µM leupeptin. The cell lysate was sedimented at 1,600 g, and the supernatant was incubated for 2 h at 4°C with 30 μl protein A/G-agarose beads (Thermo Scientific Pierce) bound to polyclonal antibody. After immunoprecipitation, the beads were washed four times with lysis buffer, and the bound proteins were separated by SDS-PAGE and immunoblotted or stained directly with Coomassie Blue.
Protein purification and pull-down assays
The recombinant proteins were induced for 18 h at 16°C in Escherichia coli BL21 (DE3) using 0.2 mM IPTG. The bacteria were harvested by centrifugation, resuspended in binding buffer (50 mM Tris, 500 mM NaCl, 1 mM EDTA, and 7.5 mM imidazole, pH 8.0) containing 1× Complete protease inhibitor mix (pi; 11697498001; Roche), and lysed by sonication. The debris was pelleted for 1 h at 15,000 g in a 70Ti rotor. Supernatants were passed through a 0.2-μm filter and incubated with either Ni-NTA agarose (Qiagen) or glutathione-Sepharose beads (GE Healthcare Life Sciences) for 1 h at 4°C. The Ni-NTA agarose was then washed five times in binding buffer with 40 mM imidazole and His-SNX5-PX eluted with 250 mM imidazole. The eluates were immediately loaded onto a Superdex size exclusion column and fractions with pure His-SNX5-PX concentrated using an Amicon filter (10 kD cutoff). The glutathione-Sepharose was washed five times in binding buffer and GST-VMAT2-Ct eluted with 10 mM glutathione in 50 mM Tris, pH 8.0. Pull-down assays were performed by incubating 100 ng GST or GST fusions prebound to glutathione-Sepharose with 200 ng His fusions for 1 h at 4°C, washing five times, and elution with 2× SDS sample buffer for Western analysis.
Brain extract
Whole brain from one to two rats was homogenized in 4 ml ice-cold homogenization buffer (0.32 M sucrose and 4 mM Hepes, pH 7.4) with pi by nine strokes with a 5-ml glass Teflon homogenizer, as previously described (Goh et al., 2011). The homogenizer was rinsed with an additional 4 ml homogenization buffer, and the combined 8 ml of homogenate (H) was sedimented at 1,000 g for 10 min at 4°C in an SS34 rotor (Sorvall). The supernatant (S1) was removed from the pellet (P1) and centrifuged at 12,500 g for 15 min at 4°C in an SS34. The supernatant (S2) was removed completely, and the synaptosome-enriched pellet (P2) was resuspended in 1 ml homogenization buffer. A Bradford assay was used to determine the protein concentration of P2, and all membrane extracts were adjusted to the same final concentration of 1–2 mg/ml using homogenization buffer with protease inhibitors. For immunoprecipitation, 1 mg synaptosomes (P2) were sedimented at 12,000 g for 40 min at 4°C, resuspended in NP-40 lysis buffer with protease inhibitors, disrupted using a polytron, rotated at 4°C for 40 min, and sedimented at 12,000 g at 4°C for 40 min. The supernatant was used for immunoprecipitation as described above.
Immunofluorescence and image acquisition
Immunofluorescence of cultured cells was performed as previously described (Larsen et al., 2006). Briefly, cells were plated onto glass coverslips coated with poly-D-lysine and Matrigel (Collaborative Research), fixed with 4% PFA/0.1 M phosphate buffer, pH 7.2, permeabilized, and blocked for 1 h in PBS containing 2% BSA, 1% fish skin gelatin, and 0.02% saponin. The cells were incubated with primary antibody in the same buffer at room temperature for 1.5 h, washed three times for 10 min each in the same buffer, incubated for an additional 1 h at room temperature with the appropriate secondary antibody diluted 1:400 in the same buffer, and washed again three times for 5 min each.
To immunostain the adrenal gland, mice were perfused with 4% PFA/PBS, and the adrenal gland dissected, postfixed overnight in 4% PFA, and cryoprotected in PBS containing 30% sucrose. Floating sections (25 μm) cut using a Leica CM3050S cryostat were blocked and permeabilized in PBS containing 4% normal goat serum and 0.2% Triton X-100. Primary and secondary antibodies were diluted in PBS containing 1% normal goat serum and 0.2% Triton X-100. Primary antibodies were incubated overnight at 4°C and secondary antibodies for 2 h at room temperature in the dark. Slices were mounted in Fluoromount-G (Southern Biotech).
The stained HeLa cells were visualized with a confocal laser microscope (LSM710; Zeiss) at the imaging facility of Nanjing Medical University. Adrenal gland slices were acquired using a Nikon Ti Microscope equipped with CSU-W1 spinning disk confocal, Andor Zyla sCMOS camera, and Plan Apo 10× or Plan Apo 40× oil objective. PC12 and chromaffin cells were imaged and reconstructed with the DeltaVision OMX SR microscope at the UCSF Nikon Imaging Center. SIM images were captured on a DeltaVision OMX SR microscope (GE Healthcare) with Plan ApoN 60×/1.42 objective (Olympus), 1.516 index immersion oil, a three PCO 15-bit CMOS camera, 10–50 ms exposures, 1 pre-amp gain, and variable EM gain. Alexa Fluor 488 fluorescence was excited with a 488-nm laser and collected with a 528/488 emission filter, Alexa Fluor 555 with 568-nm laser and a 609/37 emission filter, and Alexa Fluor 647 with a 642-nm laser and 683/40 emission filter. Acquisition and all mechanics were controlled by the SR controller software. To obtain 3D images, the sample was translated vertically in steps of 125 nm. The system was controlled with OMX AquireSR software (GE Healthcare), and alignment of two channel images was performed with SoftWoRx (GE Healthcare).
Adrenal gland SNX5 and monoamine content
Adrenal glands from WT and SNX5 KO mice were homogenized in 150 mM NaCl, 50 mM Tris-HCl, pH 8.0, 1% NP-40, and 1 mM EDTA, supplemented with 1 mM PMSF. After sedimentation at 14,000 g to remove nuclei, 10 µg protein was separated by electrophoresis through polyacrylamide and transferred to nitrocellulose membrane, and the membranes were immunoblotted for SNX5 with β-actin as loading control and the appropriate secondary antibodies conjugated to IRDye800 (Rockland Immunochemicals). Immunoreactivity was quantified by imaging with an Odyssey system (LI-COR Biosciences) and ImageJ (National Institutes of Health), and results were normalized to β-actin.
To measure monoamine levels in WT and SNX5 KO mice, sex-matched littermates were euthanized by CO2 inhalation and decapitated, and the adrenal gland was dissected in HBSS and immediately frozen on dry ice. Tissue catecholamine levels were measured by HPLC with coupled electrochemical detection at the Vanderbilt University Neurochemistry Core. Briefly, tissues were homogenized in 500 μl of 0.1 M TCA with 10 mM sodium acetate, 0.1 mM EDTA, and 10.5% methanol, pH 3.8, using a handheld sonic tissue dismembrator and sedimented at 10,000 g for 20 min, and the supernatant was removed for HPLC/coupled electrochemical detection. HPLC was performed using a Kinetix 2.6 µm C18 column (4.6 × 100 mm; Phenomenex). The same buffer used for tissue homogenization was used for the HPLC mobile phase. The Waters Xevo TQ-XS mass spectrometer was equipped with an electrospray ionization source and operated in the positive ion mode. The capillary voltage was 3.1 kV, and the cone voltage was optimized at 25 V. The source and desolvation temperatures were 150 and 500°C, respectively. The desolvation gas flow was 1,000 liter/h, and the cone gas was operated at 150 liter/h.
Protein turnover
2 d after the second transfection of SNX5 siRNA, CHO cells stably expressing 3HA-VMAT2 were incubated for ∼9 h in complete medium supplemented with vehicle, the proteasome inhibitor MG132 (40 µM), the lysosomal protease inhibitor leupeptin (10 µM), or 10 mM NH4Cl. The cells were washed on ice with PBS, lysed in 50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 10 mM EDTA, 1% Triton X-100 with pi plus1 mM PMSF, and the samples were analyzed by Western blotting.
To measure VMAT2 turnover, stable CHO or PC12 transformants were transiently transfected with SNX5-specific or control siRNA. After 24 h, the cells were treated with 100 μg/ml cycloheximide and collected at 0, 3, 6, and 9 h. The amount of VMAT2 was then determined by immunoblotting, and the enhanced chemiluminescent signal was analyzed using a Tanon 5200 Multi Auto-imaging System (Tanon).
Gradient fractionation and Western analysis
For equilibrium density gradient fractionation, stably transformed or transiently transfected PC12 cells were harvested in buffer A (in mM: 150 NaCl, 1 EGTA, 0.1 MgCl2, and 10 Hepes, pH 7.4) with pi. Cells were homogenized by eight passes through a ball-bearing device (clearance 12 µm). Postnuclear supernatants were then loaded onto continuous density gradients prepared with a gradient mixer using 0.65 and 1.55 M sucrose in buffer B (in mM: 1 EGTA, 1 MgCl2, and 10 Hepes, pH 7.4) and centrifuged in an SW41 rotor (Beckman Instruments) at 153,900 g for 16 h at 4°C.
For velocity sedimentation, PC12 cells were washed twice with TBS plus sulfate (TBSS; 4.5 mM KCI, 137 mM NaCl, 0.7 mM Na2HPO4, 1.6 mM Na2SO4, and 25 mM Tris-HCI, pH 7.4) and once with TBSS plus pi, collected in 10 ml TBSS/pi, sedimented for 5 min at 700 g, washed once in homogenization buffer (0.25 M sucrose, 1 mM EDTA, 1 mM Mg acetate, 1.6 mM Na2SO4, and 10 mM Hepes-KOH, pH 7.2) and pi, sedimented again for 5 min at 1,700 g, resuspended in 1 ml homogenization buffer and pi, and passed through a 22-gauge needle to produce a single-cell suspension. The cell suspension was then homogenized in a ball-bearing device at 18-μm clearance, the nuclei and unbroken cells were sedimented at 1,000 g for 10 min, and the supernatant was loaded onto prepared 0.3–1.2 M continuous sucrose gradients and separated by velocity centrifugation at 106,900 g for 19 min at 4°C in an SW41 rotor. Fractions (0.5 ml) were collected from top to bottom, and equal amounts of each were denatured in 6× SDS sample buffer and separated by electrophoresis through 10% SDS–polyacrylamide. For glycerol velocity gradient sedimentation, postnuclear supernatants from stably transformed PC12 cells were loaded onto continuous glycerol gradients prepared using 5 and 25% glycerol and sedimented in an SW41 rotor at 270,000 g for 1 h at 4°C.
Western blotting was performed according to published procedures (Wu et al., 2016). Proteins were separated by electrophoresis through discontinuous 10% SDS–polyacrylamide before electrotransfer to nitrocellulose (Tanon). After blocking with 10% nonfat dry milk and incubation with primary and secondary antibodies, the blots were developed by enhanced chemiluminescence (ECL; Tanon) or quantitative fluorescent Western blotting with an Odyssey system (LI-COR Biosciences).
Secretion assay
Cells were transfected twice 2 d apart with siRNA (20 nM); washed 2 d after the second transfection in basal Tyrode’s solution (in mM: 119 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 30 glucose, and 25 Hepes, pH 7.4); incubated in culture medium containing 0.7 μCi/ml 3H-NE, 0.1 mM pargyline, and 1 mM Na ascorbate for 90 min; washed twice with basal Tyrode’s solution; and incubated in culture medium again for 30 min. After washing twice, cells were incubated in Tyrode’s solution containing 2.5 mM K+ (basal) or 90 mM K+ (stimulated) for 15 min at 37°C. The media were collected, and cell lysates were prepared as above. 3H-NE was measured by scintillation counting, and the proteins by quantitative fluorescent immunoblotting. In both cases, equal proportions of medium (M) and cell lysate (C) were analyzed, and the amount secreted was normalized to total using Mstim / (Mstim + Cstim).
Live-cell imaging
PC12 cells were transfected twice with siRNA and with VMAT2-pHluorin, as well on day 1 before imaging, as previously described (Asensio et al., 2010; Onoa et al., 2010). Glass coverslips containing the transfected PC12 cells were mounted in the perfusion chamber (Warner Instruments) of an inverted Nikon TE3000 microscope fitted with a 60× water objective, and the cells were imaged in modified Tyrode’s solution at room temperature with stimulation by 90 mM K+ and alkalinization of the quenched intracellular fluorophore with 10 mM NH4Cl. Cells were illuminated using a Xenon lamp (Sutter Instruments) with 470/40-nm excitation and 525/50-nm emission filters. Images were acquired every second using a QuantEM charge coupled device camera (Photometrics). MetaMorph software was used to control data collection and perform analysis offline.
Image analysis
Images were analyzed using Fiji/ImageJ software. Pearson’s correlation coefficient was quantified using the Coloc2 plug-in in ImageJ with the Costes automatic threshold function. The Mander’s overlap coefficient was calculated using the automatic threshold strategy of the measure correlation module in CellProfiler. Plotting and statistical analysis were performed using Prism (v7.0; GraphPad).
Statistical analysis
The data from multiple, independent experiments are presented as mean ± SEM. The significance of the difference between groups was determined by two-tailed, unpaired Student’s t test or one-way ANOVA followed by Tukey’s multiple comparisons test using GraphPad Prism v7.0 software. The raw data from quantitative Western blotting were first normalized to percentage of control (comparative Western blots) or to percentage of time point 0 (protein turnover assays) for each condition. Colocalization data from at least three independent experiments were averaged, and SEM was calculated across the three experiments. For all statistical tests, P < 0.05 was considered significant and indicated by an asterisk. The individual data for all experiments with statistics are shown in Data S1.
Online supplemental material
Fig. S1 shows that loss of SNX5 reduces VMAT2 expression, related to Fig. 1. Fig. S2 shows that SNX5 PX and BAR domains disrupt the localization of VMAT2 to DCVs, related to Fig. 2. Fig. S3 shows that SNX5 recognizes the extended dileucine-like motif of VMAT2, related to Fig. 3. Fig. S4 shows that isolated SNX5 PX and BAR domains redistribute VMAT2 from TGN to endosomes and increase its lysosomal degradation, related to Fig. 4. Fig. S5 shows that AP-3 knockdown impairs VMAT2 exocytosis and monoamine loading similar to KO of the AP-3δ gene by CRISPR, related to Fig. 5. Fig. S6 shows that temperature shift redistributes AP-3 to the TGN independently of any change in the localization of TGN proteins, related to Fig. 7. Fig. S7 shows localization of VPS41, related to Fig. 8. Fig. S8 shows that temperature block rapidly redistributes AP-3 to the TGN, whereas DCV cargo accumulate more slowly, related to Fig. 9. Table S1 lists candidate binding partners from yeast two-hybrid screen. Data S1 contains the statistics source file.
Acknowledgments
We thank P. Zhang, J. Eriksen, and other members of the Edwards lab for helpful discussion; DeLaine Larsen and Kari Herrington for assistance with the SIM; Mark von Zastrow for suggestion of the temperature shift experiment; and Ginger Milne from the Neurochemistry Core at Vanderbilt University Medical Center for the catecholamine measurements.
This work was supported by the National Nature Science Foundation of China (grants 31371436 and 8157051134 to Y. Liu) and by the John and Helen Cahill Family Endowed Chair, R01 MH50712 and R01 NS103938 (to R.H. Edwards).
The authors declare no competing financial interests.
Author contributions: The work was conceptualized by Y. Liu and R.H. Edwards and initiated by Y. Liu in the lab of R.H. Edwards, with most of the experiments performed subsequently by H. Xu under supervision by first Y. Liu and then R.H. Edwards. F. Chang performed the initial coimmunoprecipitation of VMAT2 and SNX5. S. Jain and B.A. Heller made the AP-3 knockout cell line. X. Han assisted in sucrose gradient fractionation. The writing was done by H. Xu and R.H. Edwards.

