


In animal cells, spindle elongation during anaphase is temporally coupled with cleavage furrow formation. Spindle elongation during anaphase is regulated by NuMA/dynein/dynactin complexes that occupy the polar region of the cell membrane and are excluded from the equatorial membrane. How NuMA/dynein/dynactin are excluded from the equatorial membrane and the biological significance of this exclusion remains unknown. Here, we show that the centralspindlin (Cyk4/Mklp1) and its interacting partner RhoGEF Ect2 are required for NuMA/dynein/dynactin exclusion from the equatorial cell membrane. The Ect2-based (Ect2/Cyk4/Mklp1) and NuMA-based (NuMA/dynein/dynactin) complexes occupy mutually exclusive membrane surfaces during anaphase. The equatorial membrane enrichment of Ect2-based complexes is essential for NuMA/dynein/dynactin exclusion and proper spindle elongation. Conversely, NuMA-based complexes at the polar region of the cell membrane ensure spatially confined localization of Ect2-based complexes and thus RhoA. Overall, our work establishes that membrane compartmentalization of NuMA-based and Ect2-based complexes at the two distinct cell surfaces restricts dynein/dynactin and RhoA for coordinating spindle elongation with cleavage furrow formation.
Introduction
Animal cells elongate their mitotic spindle and segregate sister chromatids to the opposite poles before setting up their new boundary by forming a cleavage furrow during anaphase (reviewed in Green et al., 2012; Basant and Glotzer, 2018; Pollard and O’Shaughnessy, 2019). Spindle elongation and sister chromatids’ separation are tightly coordinated with cleavage furrow formation. The proper functioning of these processes is vital for preventing the instability of chromosomes and tumorigenesis (reviewed in Ganem and Pellman, 2007; Lens and Medema, 2019).
Spindle elongation and chromosomes segregation is regulated by an evolutionarily conserved cortically anchored protein nuclear mitotic apparatus (NuMA; reviewed in di Pietro et al., 2016; Bergstralh et al., 2017; Kotak, 2019; Lechler and Mapelli, 2021). Cortically anchored NuMA serves as an adaptor for the microtubule-dependent minus-end-directed motor protein complex dynein and its associated dynactin complex (reviewed in Kotak and Gönczy, 2013; Kiyomitsu, 2019; Kotak, 2019; Lechler and Mapelli, 2021). The pulling forces generated by the cortically anchored dynein/dynactin are assumed to promote spindle elongation and chromosomes segregation. The cortical level of NuMA during mitosis is temporally controlled by a biochemical crosstalk between cyclin-dependent kinase 1 (Cdk1) and PP2A-B55γ-based phosphatase complex (Kiyomitsu and Cheeseman, 2013; Kotak et al., 2013; Seldin et al., 2013; Zheng et al., 2014; Keshri et al., 2020). During anaphase, NuMA is localized to the cell cortex by directly associating with the membrane lipids PI(4)P and PI(4,5)P2 (referred to as PIP and PIP2; Zheng et al., 2014; Kotak et al., 2014). Notably, despite the presence of PIP and PIP2 across the entire cell membrane, NuMA is excluded from the equatorial region of the cell membrane (Kotak et al., 2014). Further, it was shown the equatorial exclusion of NuMA is dependent on Rho GTPase-activating protein (RhoGAP) Cyk4 (also known as MgcRacGAP; Kotak et al., 2014). However, the mechanism and the biological relevance of Cyk4-dependent NuMA exclusion from the equatorial membrane are not known.
In metazoans, the cleavage furrow formation at the equatorial membrane is initiated by local activation of the small GTPase RhoA. RhoA directly controls actin polymerization and indirectly regulates myosin II activation at the equatorial membrane and thus helps in cleavage furrow formation. Coordinated assemblies of multiple protein complexes at the spindle midzone (also known as the central spindle) regulate the spatiotemporal activation of RhoA at the equatorial membrane (reviewed in Green et al., 2012; Basant and Glotzer, 2018; Pollard and O’Shaughnessy, 2019). The spindle midzone is a stable array of overlapping microtubules of opposite polarity that assembles during metaphase-to-anaphase transition halfway between the segregating chromosomes. One of the critical complexes that assemble at the spindle midzone and regulate RhoA activation at the equatorial membrane is centralspindlin (Somers and Saint, 2003; Yüce et al., 2005). Centralspindlin is a heterotetrametric complex consisting of a dimer of kinesin-6 family member mitotic kinesin-like protein (Mklp1) and a dimer of Cyk4 (Pavicic-Kaltenbrunner et al., 2007). The centralspindlin helps in recruiting the conserved RhoA guanine nucleotide exchange factor (RhoGEF)-epithelial cell transforming sequence 2 (Ect2) to the spindle midzone (Yüce et al., 2005; Chalamalasetty et al., 2006; Kamijo et al., 2006; Su et al., 2011; Kotýnková et al., 2016; Gómez-Cavazos et al., 2020; Schneid et al., 2021). Ect2 is essential for RhoA activation and thus for cleavage furrow ingression and cytokinesis in animal cells (Miki et al., 1993; Prokopenko et al., 1999; Kimura et al., 2000; Su et al., 2011; reviewed in Basant and Glotzer, 2018; Pollard and O’Shaughnessy, 2019). Ect2 consists of BRCT (BRCA1-C-terminal) domains at the N-terminus, and RhoGEF, pleckstrin homology (PH), and polybasic cluster (PBC) domains at the C-terminus (Chalamalasetty et al., 2006; Su et al., 2011; Kotýnková et al., 2016; Schneid et al., 2021; Fig. 5 A). Through BRCT domains, Ect2 interacts with Cyk4, and the PH and PBC domains are critical for its membrane localization (Burkard et al., 2007; Somers and Saint, 2003; Yüce et al., 2005; Wolfe et al., 2009; Su et al., 2011; Kotýnková et al., 2016; Gómez-Cavazos et al., 2020; Schneid et al., 2021). The presence of these multiple domains in Ect2 ensures Ect2 localization and RhoA-activation at the equatorial region of the membrane for cleavage furrow formation. However, how Ect2 levels at the equatorial membrane are confined and maintained to a narrow region for proper RhoA activation is poorly understood and is a crucial aspect of understanding the molecular mechanism of cleavage furrow formation.
Here, we show that the polarized distribution of Ect2/Cyk4/Mklp1 (also referred to as Ect2-based complexes) at the equatorial membrane restricts NuMA/dynein/dynactin (also referred to as NuMA-based complexes) to the polar region of the cell membrane for proper chromosome separation. Conversely, NuMA-based complexes at the polar region of the cell membrane control RhoA levels at the equatorial membrane, possibly by confining the Ect2-based complexes. Moreover, we reveal that NuMA localization at the polar membrane acts redundantly with the spindle midzone localized pool of Ect2-based complexes to ensure proper cleavage furrow formation. In summary, this work provides insights into the mechanism that limits RhoA and dynein/dynactin at two membrane surfaces by establishing mutually exclusive membrane localization of two evolutionarily conserved protein complex assemblies.
Results
Centralspindlin and Rho GEF Ect2 exclude NuMA from the equatorial membrane
During anaphase, NuMA is restricted to the polar region of the cell membrane, while the equatorial membrane is mutually exclusively occupied by RhoA (Fig. 1, A–C). We showed earlier that siRNA-mediated depletion of Cyk4, which is critical for RhoA accumulation, results in ectopic accumulation of NuMA and its associated dynein/dynactin motor protein complexes at the equatorial membrane (Kotak et al., 2014). To investigate the mechanism of NuMA/dynein/dynactin exclusion from the equatorial membrane in anaphase, we first analyzed the localization of NuMA and dynactin subunit p150Glued in cells depleted for proteins that act downstream of Cyk4 and are crucial for RhoA recruitment (Fig. 1 D; reviewed in Eggert et al., 2006; Green et al., 2012; Basant and Glotzer, 2018). siRNA-mediated depletion of Cyk4, Mklp1, Ect2, or Anillin leads to a significant accumulation of binucleated or multinucleated cells, indicating robust cytokinesis failure (Fig. 1, E–I and Fig. S1, A–D). As expected, depletion of Cyk4 and its associated kinesin Mklp1 resulted in NuMA and p150Glued localization at the equatorial membrane (Fig. 1, J–N and Q). Further, cells transfected with Ect2 siRNA showed robust enrichment of NuMA and p150Glued at the equatorial membrane (Fig. 1, O and Q).

Centralspindlin (Cyk4/Mklp1) and Ect2 are required for NuMA/p150Glued exclusion from the equatorial membrane. (A) Immunofluorescence (IF) analysis of HeLa cells in anaphase. Cells were fixed and costained using anti-RhoA (green) and anti-NuMA (red) antibodies, as indicated. In this and other IF-analysis panels, DNA is shown in blue unless specified. More than 30 cells were visually analyzed from three independent experiments, and the representative cell is shown here. Scale bar in this and for the following panels represent 10 μm. (B and C) Schematic representation of line scan analysis (B) and the outcome (C) of such analysis for RhoA and NuMA normalized intensity (in arbitrary unit [au]) for the specified membrane region (in orange). (n = 5 cells were used for this quantification as depicted; shaded region indicates SEM). (D) Schematic representation of an evolutionarily conserved pathway required for RhoA activation in metazoans. (E–I) IF analysis of HeLa cells that are either transfected with control siRNA (E) or siRNA-against Cyk4 (F), Mklp1 (G), Ect2 (H), or Anillin (I). Cells were fixed after 36 h of siRNA transfection and stained with anti-p150Glued antibodies (green). Multinucleation percentage (%) is shown for each siRNA condition (n > 500 cells each from three independent experiments). Knockdown efficiency was also determined by immunoblot analysis (see Fig. S1, A–D). (J and K) Schematic representation of line scan analysis (J) and quantification method (K) used for analyzing equatorial membrane enrichment of NuMA fluorescence intensity. bkgd., eq. mem., and cyt. represent background, equatorial membrane, and cytoplasmic intensity, respectively. (L–P) IF analysis of HeLa cells transfected with control siRNA (L) or siRNA against- Cyk4 (M), Mklp1 (N), Ect2 (O), or Anillin (P). Cells were fixed after 36 h of siRNA transfection and thereafter costained with anti-NuMA (green) and anti-p150Glued (red) antibodies. The percentage on the figure panels represents the fraction of anaphase cells that show equatorial NuMA enrichment for each condition as analyzed by visual quantification (n > 100 cells from three independent experiments). Yellow arrowheads depict NuMA localization at the equatorial membrane. Line scan analysis on the right represents NuMA fluorescence intensity as indicated in J. Red asterisk represents intensity at the equatorial membrane. (Q) Quantification of equatorial membrane NuMA intensity in cells transfected with control siRNA or siRNA against- Cyk4, Mklp1, Ect2, or Anillin as indicated in Figure panel K (n = 25 cells for all conditions, as listed; error bars: SD). ns, P > 0.05; ***, P < 0.001 as determined by two-tailed unpaired Student’s t test. (R and S) Confocal live-imaging analysis of HeLa Kyoto cells stably coexpressing AcGFP-NuMA (green) and mCherry-H2B (magenta) that are transfected with control siRNA (R) or siRNA against Ect2 (S). The recording was initiated 26 h post-transfection for control and Ect2 siRNA. “0 min” time-point represents metaphase to anaphase transition. Yellow arrowheads depict NuMA localization at the equatorial membrane. (T) Quantification of equatorial membrane AcGFP-NuMA intensity as described in K. ns, P > 0.05; **, P < 0.01; ***, P < 0.001 as determined by two-tailed unpaired Student’s t test. n = 10 cells for control siRNA, and n = 15 for Ect2 siRNA transfected cells; error bars: SD.
Centralspindlin (Cyk4/Mklp1) and Ect2 are required for NuMA/p150Glued exclusion from the equatorial membrane. (A) Immunofluorescence (IF) analysis of HeLa cells in anaphase. Cells were fixed and costained using anti-RhoA (green) and anti-NuMA (red) antibodies, as indicated. In this and other IF-analysis panels, DNA is shown in blue unless specified. More than 30 cells were visually analyzed from three independent experiments, and the representative cell is shown here. Scale bar in this and for the following panels represent 10 μm. (B and C) Schematic representation of line scan analysis (B) and the outcome (C) of such analysis for RhoA and NuMA normalized intensity (in arbitrary unit [au]) for the specified membrane region (in orange). (n = 5 cells were used for this quantification as depicted; shaded region indicates SEM). (D) Schematic representation of an evolutionarily conserved pathway required for RhoA activation in metazoans. (E–I) IF analysis of HeLa cells that are either transfected with control siRNA (E) or siRNA-against Cyk4 (F), Mklp1 (G), Ect2 (H), or Anillin (I). Cells were fixed after 36 h of siRNA transfection and stained with anti-p150Glued antibodies (green). Multinucleation percentage (%) is shown for each siRNA condition (n > 500 cells each from three independent experiments). Knockdown efficiency was also determined by immunoblot analysis (see Fig. S1, A–D). (J and K) Schematic representation of line scan analysis (J) and quantification method (K) used for analyzing equatorial membrane enrichment of NuMA fluorescence intensity. bkgd., eq. mem., and cyt. represent background, equatorial membrane, and cytoplasmic intensity, respectively. (L–P) IF analysis of HeLa cells transfected with control siRNA (L) or siRNA against- Cyk4 (M), Mklp1 (N), Ect2 (O), or Anillin (P). Cells were fixed after 36 h of siRNA transfection and thereafter costained with anti-NuMA (green) and anti-p150Glued (red) antibodies. The percentage on the figure panels represents the fraction of anaphase cells that show equatorial NuMA enrichment for each condition as analyzed by visual quantification (n > 100 cells from three independent experiments). Yellow arrowheads depict NuMA localization at the equatorial membrane. Line scan analysis on the right represents NuMA fluorescence intensity as indicated in J. Red asterisk represents intensity at the equatorial membrane. (Q) Quantification of equatorial membrane NuMA intensity in cells transfected with control siRNA or siRNA against- Cyk4, Mklp1, Ect2, or Anillin as indicated in Figure panel K (n = 25 cells for all conditions, as listed; error bars: SD). ns, P > 0.05; ***, P < 0.001 as determined by two-tailed unpaired Student’s t test. (R and S) Confocal live-imaging analysis of HeLa Kyoto cells stably coexpressing AcGFP-NuMA (green) and mCherry-H2B (magenta) that are transfected with control siRNA (R) or siRNA against Ect2 (S). The recording was initiated 26 h post-transfection for control and Ect2 siRNA. “0 min” time-point represents metaphase to anaphase transition. Yellow arrowheads depict NuMA localization at the equatorial membrane. (T) Quantification of equatorial membrane AcGFP-NuMA intensity as described in K. ns, P > 0.05; **, P < 0.01; ***, P < 0.001 as determined by two-tailed unpaired Student’s t test. n = 10 cells for control siRNA, and n = 15 for Ect2 siRNA transfected cells; error bars: SD.
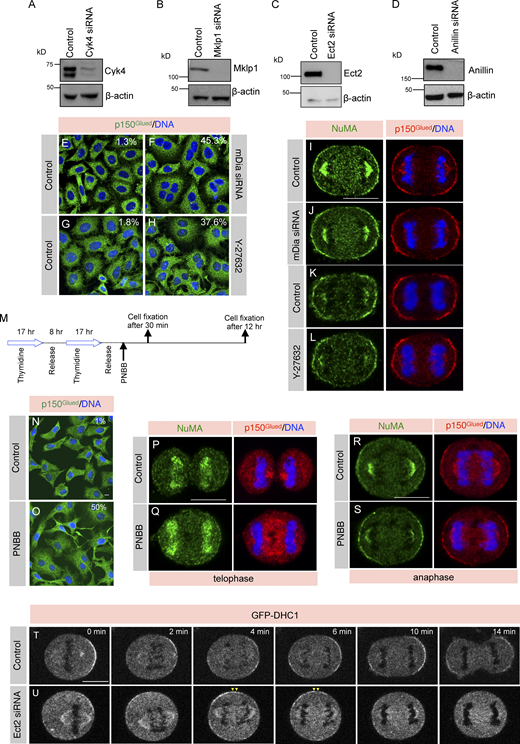
Formin, Rock, and Myosin II are not critical for NuMA exclusion from equatorial membrane. (A–D) Assessing the depletion efficiency of siRNA against Cyk4, Mklp1, Ect2, or Anillin by immunoblot analysis. Mitotically synchronized protein extracts made from HeLa Kyoto cells were either transfected with control siRNA or siRNA-against above-mentioned genes for 60 h. The resulting blot was probed with antibodies directed against Cyk4 (A), Mklp1 (B), Ect2 (C), or Anillin (D). Antibodies against β-actin were used for loading control. In this and other Figure panels for immunoblot analysis, the molecular mass is indicated in kilodaltons (kD) and is shown on the left. (E–H) Immunofluorescence (IF) analysis of HeLa cells that were either transfected with control siRNA (E) siRNA-against mDia (Formin; F) for 60 h, treated with DMSO (Control; G), or with Y-27632 (H) for 12 h to inactivate Rho-associated protein kinase (Rock). Cells were fixed thereafter and stained with anti-p150Glued antibodies (green). In this, and other IF-analysis panels DNA is shown in blue. Multinucleation percentage (%) is shown in the figure panel. (n > 500 cells were analyzed from two independent experiments). The scale bar in this panel and the following panels represent 10 μm. (I–L) HeLa cells transfected with control siRNA (I), siRNA against mDia (J) for 60 h, treated with DMSO (Control; K), or treated with Y-27632 (L) for 1 h to inactivate Rock in anaphase. Cells were fixed and thereafter costained with anti-NuMA (green) and anti-p150Glued (red) antibodies. More than 50 cells from two independent experiments were analyzed, and the representative cells are shown here. (M) Synchronization scheme for myosin II inactivation using PNBB to analyze cells in anaphase for NuMA and p150Glued localization, and after 12 h for assessing cytokinesis failure. (N and O) IF analysis of mitotically synchronized populations of HeLa cells that were either treated with DMSO (Control; N) or PNBB (O) for 12 h. Thereafter, cells were fixed and stained with anti-p150Glued antibodies (green). Multinucleation percentage (%) is shown in the figure panel. (n > 500 cells were analyzed from two independent experiments). (P–S) HeLa cells in telophase (P and Q) or during early anaphase (R and S) that were either treated with DMSO (Control) or PNBB for 30 min. Cells were fixed and costained with anti-NuMA (green) and anti-p150Glued (red) antibodies. Please note that during telophase (P and Q), NuMA has localized to the nucleus, but the cleavage furrow has not formed because of myosin II inactivation. n > 50 cells, from two independent experiments, and the representative cells are shown here. (T and U) Confocal live-imaging analysis of HeLa Kyoto cells stably expressing GFP-DHC1 that were transfected with control siRNA (T) or siRNA against Ect2 (U). The recording was started 26 h post-transfection for control and Ect2 siRNA. “0 min” time-point represents metaphase to anaphase transition. Yellow arrowheads depict GFP-DHC1 localization at the equatorial membrane. Representative images from the time-lapse recording are shown here (n > 10 cells). Source data are available for this figure: SourceData FS1.

Formin, Rock, and Myosin II are not critical for NuMA exclusion from equatorial membrane. (A–D) Assessing the depletion efficiency of siRNA against Cyk4, Mklp1, Ect2, or Anillin by immunoblot analysis. Mitotically synchronized protein extracts made from HeLa Kyoto cells were either transfected with control siRNA or siRNA-against above-mentioned genes for 60 h. The resulting blot was probed with antibodies directed against Cyk4 (A), Mklp1 (B), Ect2 (C), or Anillin (D). Antibodies against β-actin were used for loading control. In this and other Figure panels for immunoblot analysis, the molecular mass is indicated in kilodaltons (kD) and is shown on the left. (E–H) Immunofluorescence (IF) analysis of HeLa cells that were either transfected with control siRNA (E) siRNA-against mDia (Formin; F) for 60 h, treated with DMSO (Control; G), or with Y-27632 (H) for 12 h to inactivate Rho-associated protein kinase (Rock). Cells were fixed thereafter and stained with anti-p150Glued antibodies (green). In this, and other IF-analysis panels DNA is shown in blue. Multinucleation percentage (%) is shown in the figure panel. (n > 500 cells were analyzed from two independent experiments). The scale bar in this panel and the following panels represent 10 μm. (I–L) HeLa cells transfected with control siRNA (I), siRNA against mDia (J) for 60 h, treated with DMSO (Control; K), or treated with Y-27632 (L) for 1 h to inactivate Rock in anaphase. Cells were fixed and thereafter costained with anti-NuMA (green) and anti-p150Glued (red) antibodies. More than 50 cells from two independent experiments were analyzed, and the representative cells are shown here. (M) Synchronization scheme for myosin II inactivation using PNBB to analyze cells in anaphase for NuMA and p150Glued localization, and after 12 h for assessing cytokinesis failure. (N and O) IF analysis of mitotically synchronized populations of HeLa cells that were either treated with DMSO (Control; N) or PNBB (O) for 12 h. Thereafter, cells were fixed and stained with anti-p150Glued antibodies (green). Multinucleation percentage (%) is shown in the figure panel. (n > 500 cells were analyzed from two independent experiments). (P–S) HeLa cells in telophase (P and Q) or during early anaphase (R and S) that were either treated with DMSO (Control) or PNBB for 30 min. Cells were fixed and costained with anti-NuMA (green) and anti-p150Glued (red) antibodies. Please note that during telophase (P and Q), NuMA has localized to the nucleus, but the cleavage furrow has not formed because of myosin II inactivation. n > 50 cells, from two independent experiments, and the representative cells are shown here. (T and U) Confocal live-imaging analysis of HeLa Kyoto cells stably expressing GFP-DHC1 that were transfected with control siRNA (T) or siRNA against Ect2 (U). The recording was started 26 h post-transfection for control and Ect2 siRNA. “0 min” time-point represents metaphase to anaphase transition. Yellow arrowheads depict GFP-DHC1 localization at the equatorial membrane. Representative images from the time-lapse recording are shown here (n > 10 cells). Source data are available for this figure: SourceData FS1.
We next assessed the importance of other proteins that act downstream of RhoA for NuMA and p150Glued exclusion from the equatorial membrane. Depletion of proteins, such as Anillin, a structural component of the cytokinetic contractile ring and mDia (also known as Diaphanous-related formin-1), a protein required for actin polymerization at the cytokinetic ring, or the inhibition of Rho-associated protein kinase (Rock) using a specific inhibitor Y-27632 did not cause NuMA and p150Glued mislocalization to the equatorial membrane (Fig. 1, P and Q; and Fig. S1, E–L). As RhoA is essential for myosin II activation (Fig. 1 D; reviewed in Green et al., 2012; Basant and Glotzer, 2018; Pollard and O’Shaughnessy, 2019), we next investigated if myosin II activity is crucial for excluding NuMA from the equatorial membrane during anaphase. For this purpose, we utilized a non-photo toxic myosin II inhibitor para-nitroblebbistatin (PNBB; Képiró et al., 2014) and treated mitotically synchronized HeLa cell population with PNBB (Fig. S1 M). Despite a strong impact of PNBB that resulted in a significant increase in binucleated cells and impairment of the cleavage furrow formation (Fig. S1, N–Q), NuMA remained excluded from the equatorial membrane in cells treated with PNBB during anaphase (Fig. S1, R and S).
To track the spatiotemporal localization of NuMA, we performed live-cell imaging in monoclonal stable HeLa Kyoto cells coexpressing AcGFP (Aequorea coerulescens GFP) and a mono FLAG epitope-tagged NuMA and mCherry-H2B (Rajeevan et al., 2020). Analogous to the endogenous protein, AcGFP-NuMA localizes to the equatorial membrane in cells transfected with Ect2 siRNA compared with control cells (Fig. 1, R–T). Similar results were observed in Cyk4 or Mklp1 siRNA-transfected cells (data not shown). Since NuMA is a cortical adaptor for dynein, we further analyzed dynein localization in cells stably expressing GFP-tagged dynein heavy chain (DHC1; Poser et al., 2008). We found that, similar to NuMA localization, GFP-DHC1 is no longer restricted to the polar region of the cell cortex in cells transfected with Ect2 siRNA (Fig. S1, T and U). Altogether, these results suggest that the centralspindlin complex (Cyk4/Mklp1) and Ect2 are necessary for excluding NuMA/dynein/dynactin from the equatorial membrane. However, the contractile ring proteins that act downstream of RhoA and that we tested in our study do not appear to be essential for NuMA exclusion from the equatorial membrane.
Failure in cell elongation is not responsible for equatorial membrane localization of NuMA
Cells depleted either for the centralspindlin complex or Ect2 fail to elongate significantly during anaphase. Therefore, an alternative possibility could be that the compromised cell elongation in cells transfected with siRNA against Ect2 (or Cyk4/Mklp1) is the cause of NuMA/dynein/dynactin localization at the equatorial membrane. To determine whether it is (1) cells’ inability to elongate upon centralspindlin/Ect2 depletion or (2) a direct role of these proteins in NuMA exclusion, we monitored the equatorial localization of NuMA in control and Ect2 siRNA transfected cells in the early stages of anaphase before cell elongation begins. We noticed no significant difference in the cell length in cells transfected with Ect2 siRNA versus control siRNA at 1–4 min post anaphase entry (Fig. 2, A–D). However, cells transfected with Ect2 siRNA significantly localized AcGFP-NuMA at the equatorial membrane as early as 3 min post anaphase entry (Fig. 2, A–D).

NuMA/p150Glued localization at the equatorial membrane is not due to failure in cell elongation. (A and B) Confocal live-imaging analysis of HeLa Kyoto cells stably coexpressing AcGFP-NuMA (green) and mCherry-H2B (magenta) that are transfected with control siRNA (A) or siRNA against Ect2 (B). The recording was started 26 h post-transfection for control and Ect2 siRNA transfected cells. “0 min” time-point represents metaphase to anaphase transition, and the images were acquired every minute. Yellow arrowheads depict NuMA localization at the equatorial membrane. The scale bar in this and for the following panels represents 10 μm. (C and D) Schematic representation of quantification method used for analyzing equatorial membrane enrichment of AcGFP-NuMA fluorescence intensity and for computing cell length (d [μm]) (C). Quantification of equatorial membrane enrichment of NuMA, along with the cell length (D) as described pictorially in C. Bar graph represents the cell length for cells that are either transfected with control siRNA (blue), or siRNA against Ect2 (green). The line plot graph represents the equatorial NuMA membrane intensity in cells transfected with control siRNA (violet) or Ect2 siRNA (pink). Time (in min) is shown on the x-axis, and NuMA equatorial intensity together with the cell length is plotted on the y-axis. ns, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001 as determined by two-tailed unpaired Student’s t test. n > 10 cells as listed; error bars: SEM. (E and F) IF analysis of HeLa cells that were either control siRNA transfected and treated with Cdk1 inhibitor RO-3306 (E) or transfected with siRNA-against Ect2 and treated with RO-3306 (F) for 5 min. Cells were fixed and costained using anti-NuMA (green) and anti-p150Glued (red) antibodies. Yellow arrowheads depict NuMA localization at the equatorial membrane. More than 30 cells were visually analyzed, and the representative cells are shown here. The experiments were repeated three times. (G) Visual quantification of the cortical NuMA distribution in RO-3306 or in Ect2 siRNA plus RO-3306 treated cells. The stacked blue bar, and brown bar represent the percentage of the cells that show cortical NuMA distribution at the polar cortical region and equatorial and polar cortical region, respectively. ***, P < 0.001 as determined by two-tailed unpaired Student’s t test. n > 30 cells/experiment, from three independent experiments; error bars: SD.
NuMA/p150Glued localization at the equatorial membrane is not due to failure in cell elongation. (A and B) Confocal live-imaging analysis of HeLa Kyoto cells stably coexpressing AcGFP-NuMA (green) and mCherry-H2B (magenta) that are transfected with control siRNA (A) or siRNA against Ect2 (B). The recording was started 26 h post-transfection for control and Ect2 siRNA transfected cells. “0 min” time-point represents metaphase to anaphase transition, and the images were acquired every minute. Yellow arrowheads depict NuMA localization at the equatorial membrane. The scale bar in this and for the following panels represents 10 μm. (C and D) Schematic representation of quantification method used for analyzing equatorial membrane enrichment of AcGFP-NuMA fluorescence intensity and for computing cell length (d [μm]) (C). Quantification of equatorial membrane enrichment of NuMA, along with the cell length (D) as described pictorially in C. Bar graph represents the cell length for cells that are either transfected with control siRNA (blue), or siRNA against Ect2 (green). The line plot graph represents the equatorial NuMA membrane intensity in cells transfected with control siRNA (violet) or Ect2 siRNA (pink). Time (in min) is shown on the x-axis, and NuMA equatorial intensity together with the cell length is plotted on the y-axis. ns, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001 as determined by two-tailed unpaired Student’s t test. n > 10 cells as listed; error bars: SEM. (E and F) IF analysis of HeLa cells that were either control siRNA transfected and treated with Cdk1 inhibitor RO-3306 (E) or transfected with siRNA-against Ect2 and treated with RO-3306 (F) for 5 min. Cells were fixed and costained using anti-NuMA (green) and anti-p150Glued (red) antibodies. Yellow arrowheads depict NuMA localization at the equatorial membrane. More than 30 cells were visually analyzed, and the representative cells are shown here. The experiments were repeated three times. (G) Visual quantification of the cortical NuMA distribution in RO-3306 or in Ect2 siRNA plus RO-3306 treated cells. The stacked blue bar, and brown bar represent the percentage of the cells that show cortical NuMA distribution at the polar cortical region and equatorial and polar cortical region, respectively. ***, P < 0.001 as determined by two-tailed unpaired Student’s t test. n > 30 cells/experiment, from three independent experiments; error bars: SD.
To further corroborate that the equatorial membrane localization of NuMA is possibly not because of the altered cell size in Ect2 depleted cells, we analyzed the localization of NuMA and p150Glued in roundish metaphase cells that are acutely treated with Cdk1 inhibitor RO-3306 (Vassilev et al., 2006) to program them in anaphase like state. Brief incubation (5 min) of metaphase cells with RO-3306 facilitates centralspindlin accumulation at the spindle midzone as observed by analyzing Cyk4 localization (Fig. S2, A and B; Keshri et al., 2020), and in the majority (75%) of these cells, NuMA is excluded from the equatorial membrane (Fig. 2, E and G). However, a significant fraction of metaphase cells transfected with Ect2 siRNA and treated with RO-3306 robustly localize NuMA at the equatorial membrane (compare Fig. 2 F with Fig. 2 E; Fig. 2 G). These results strongly suggest that NuMA localization at the equatorial membrane in Ect2 depleted cells is not simply because of failure in proper cell elongation.

Centralspindlin component Cyk4 robustly localizes to the spindle midzone in cells treated with Cdk1 inhibitor RO-3306. (A and B) IF analysis of HeLa cells that were either control siRNA transfected and treated with RO-3306 (A) or transfected with siRNA-against Ect2 and treated with RO-3306 (B) for 5 min. Cells were fixed and costained using anti-NuMA (green) and anti-Cyk4 (red) antibodies. Yellow arrowheads depict NuMA localization at the equatorial membrane. The scale bar represents 10 µm. More than 30 cells were analyzed from three independent experiments, and the representative cells are shown here.

Centralspindlin component Cyk4 robustly localizes to the spindle midzone in cells treated with Cdk1 inhibitor RO-3306. (A and B) IF analysis of HeLa cells that were either control siRNA transfected and treated with RO-3306 (A) or transfected with siRNA-against Ect2 and treated with RO-3306 (B) for 5 min. Cells were fixed and costained using anti-NuMA (green) and anti-Cyk4 (red) antibodies. Yellow arrowheads depict NuMA localization at the equatorial membrane. The scale bar represents 10 µm. More than 30 cells were analyzed from three independent experiments, and the representative cells are shown here.
Chromosome passenger complex (CPC) is dispensable for NuMA exclusion from the equatorial membrane
Centrosome-localized Aurora A kinase phosphorylates NuMA and regulates its cortical distribution in metaphase (Gallini et al., 2016; Kotak et al., 2016). Therefore, we wondered if Aurora B, an essential part of chromosome passenger complex (CPC), which localizes to the spindle midzone and regulates cytokinesis, is involved in excluding NuMA from the equatorial membrane (Gruneberg et al., 2004; Hümmer and Mayer, 2009; Kitagawa et al., 2013; Kitagawa et al., 2014; Basant et al., 2015). To this end, we combined Aurora B inactivation with cell synchronization using a specific Aurora B inhibitor, ZM447439 (Fig. 3 A; Ditchfield et al., 2003). Despite significant cytokinesis failure seen in cells that are treated with ZM447439, NuMA/p150Glued remained restricted to the polar region of the membrane identical to the control cells during anaphase (Fig. 3, B–E). To strengthen these findings, we further depleted kinesin-6 Mklp2, which is necessary to localize Aurora B at the spindle midzone in anaphase (Fig. 3, F–I; Gruneberg et al., 2004; Hümmer and Mayer, 2009; Kitagawa et al., 2013). siRNA-mediated depletion of Mklp2 did not impact cortical NuMA/p150Glued distribution (compare Fig. 3 K with Fig. 3 J). These results indicate that the CPC component Aurora B, which localizes to the central spindle similar to the centralspindlin complex, is not involved in NuMA/dynactin exclusion from the equatorial membrane.

Central spindle localized Aurora B is not essential for NuMA exclusion from the equatorial membrane. (A) Synchronization method for inactivation of Aurora B using ZM447439 to analyze cells in anaphase for NuMA and p150Glued localization, and after 12 h for assessing cytokinesis failure. (B and C) IF analysis of mitotically synchronized populations of HeLa cells as mentioned above either treated with DMSO (Control; B) or ZM447439 (C) for 12 h. Thereafter, cells were fixed and stained with anti-p150Glued antibodies (green). The scale bar in the panel and following panels represent 10 µm. Multinucleation percentage (%) is shown in the figure panel. (n > 500 cells were analyzed each from two independent experiments). (D and E) IF analysis of mitotically synchronized populations of HeLa cells that were either treated with DMSO (Control; D) or ZM447439 (E) for 30 min. Cells were fixed and costained with anti-NuMA (green) and anti-p150Glued (red) antibodies. More than 50 anaphase cells were analyzed from two independent experiments, and the representative cells are shown here. (F and G) IF analysis of HeLa cells in interphase that were either transfected with control siRNA (F) or siRNA-against Mklp2 (G). Cells were fixed after 40 h of siRNA transfection and stained with anti-p150Glued antibodies (green). Multinucleation percentage (%) is shown for each siRNA condition. (n > 500 cells from two independent experiments). (H and I) IF analysis of HeLa cells during anaphase that were either transfected with control siRNA (H) or siRNA-against Mklp2 (I). Cells were fixed after 40 h of siRNAs transfection and thereafter stained with anti-Aurora B antibodies (red). More than 30 anaphase cells were analyzed from two independent experiments, and the representative cells are shown here. (J and K) IF analysis of HeLa cells in anaphase that were either transfected with control siRNA (Control; J) or siRNA-against Mklp2 (K). Cells were fixed after 40 h of siRNA transfection and costained with anti-NuMA (green) and anti-p150Glued (red) antibodies. More than 50 anaphase cells were analyzed from two independent experiments, and the representative cells are shown here.
Central spindle localized Aurora B is not essential for NuMA exclusion from the equatorial membrane. (A) Synchronization method for inactivation of Aurora B using ZM447439 to analyze cells in anaphase for NuMA and p150Glued localization, and after 12 h for assessing cytokinesis failure. (B and C) IF analysis of mitotically synchronized populations of HeLa cells as mentioned above either treated with DMSO (Control; B) or ZM447439 (C) for 12 h. Thereafter, cells were fixed and stained with anti-p150Glued antibodies (green). The scale bar in the panel and following panels represent 10 µm. Multinucleation percentage (%) is shown in the figure panel. (n > 500 cells were analyzed each from two independent experiments). (D and E) IF analysis of mitotically synchronized populations of HeLa cells that were either treated with DMSO (Control; D) or ZM447439 (E) for 30 min. Cells were fixed and costained with anti-NuMA (green) and anti-p150Glued (red) antibodies. More than 50 anaphase cells were analyzed from two independent experiments, and the representative cells are shown here. (F and G) IF analysis of HeLa cells in interphase that were either transfected with control siRNA (F) or siRNA-against Mklp2 (G). Cells were fixed after 40 h of siRNA transfection and stained with anti-p150Glued antibodies (green). Multinucleation percentage (%) is shown for each siRNA condition. (n > 500 cells from two independent experiments). (H and I) IF analysis of HeLa cells during anaphase that were either transfected with control siRNA (H) or siRNA-against Mklp2 (I). Cells were fixed after 40 h of siRNAs transfection and thereafter stained with anti-Aurora B antibodies (red). More than 30 anaphase cells were analyzed from two independent experiments, and the representative cells are shown here. (J and K) IF analysis of HeLa cells in anaphase that were either transfected with control siRNA (Control; J) or siRNA-against Mklp2 (K). Cells were fixed after 40 h of siRNA transfection and costained with anti-NuMA (green) and anti-p150Glued (red) antibodies. More than 50 anaphase cells were analyzed from two independent experiments, and the representative cells are shown here.
Chromosome separation is significantly affected in cells depleted for Ect2 or Cyk4
NuMA/dynein/dynactin localization at the polar cortical region during anaphase is critical for proper spindle elongation, possibly by generating cortical pulling forces (Collins et al., 2012; Kotak et al., 2013; Zheng et al., 2014; Keshri et al., 2020; reviewed in Kotak, 2019; Kiyomitsu and Boerner, 2021). Thus, we set out to determine whether the presence of NuMA/dynein/dynactin at the equatorial membrane in cells depleted for Ect2 or centralspindlin component Cyk4 perturbs proper spindle elongation due to exerting a counteracting force from the equatorial cortical region. To this end, we performed the live-imaging in AcGFP-NuMA and mCherry-H2B coexpressing cells that are transfected with either control, Ect2 siRNA, or Cyk4 siRNA. Since spindle elongation is coupled with chromosome separation in human cells (Roostalu et al., 2010), we measured the distance between separating sister chromatids in cells undergoing metaphase-to-anaphase transition. We noticed a modest but significant impact on the chromosome separation kinetics in cells depleted either for Ect2 or Cyk4 (Fig. 4, A and B). These data indicate that the ectopic accumulation of NuMA/dynein/dynactin at the equatorial membrane may result in an imbalance of pulling forces that hamper proper chromosomes segregation kinetics and possibly spindle elongation during anaphase.

Equatorial localization of NuMA-based complexes impacts proper chromosomes separation during anaphase. (A and B) Schematic representation for the calculation of the distance (d [μm]) between inter chromatids in cells undergoing metaphase to anaphase transition at various time intervals (A). Quantification of inter chromatids distance in HeLa Kyoto cells that are stably coexpressing AcGFP-NuMA and mCherry-H2B and are transfected with either control siRNA, siRNA-against Ect2, or siRNA-against Cyk4 (B). Note that Ect2 or Cyk4 siRNA transfected cells that show no cleavage furrow, and thus 100% penetrance of the siRNA were analyzed for quantification of inter chromatids distance. ns, P > 0.05; **, P < 0.01; ***, P < 0.001 as determined by two-tailed unpaired Student’s t test. n = 10 cells from two independent experiments as depicted in the figure panel; error bars: SD shown by shaded region-pink: control; green: Ect2 depletion; red: Cyk4 depletion. (C–E) Confocal live-imaging analysis of HeLa Kyoto cells expressing AcGFP-Ect2r (C), or AcGFP-Ect2rΔmem (D), with mCherry-H2B, and the quantification of inter chromatids distance in these cells (E). In these cells, endogenous Ect2 was depleted by siRNA targeting endogenous copy of the mRNA, but not ectopically expressed transgene. The recording was initiated 26 h post-transfection of Ect2 siRNA. “0 min” time-point represents metaphase to anaphase transition. ns, P > 0.05; *, P < 0.05; **, P < 0.01 as determined by two-tailed unpaired Student’s t test. n = 10 cells as depicted in the figure panel; error bars: SD represented by the shaded region-pink: cells coexpressing AcGFP-Ect2r and mCherry-H2B; green: cells coexpressing AcGFP-Ect2rΔmem and mCherry-H2B.
Equatorial localization of NuMA-based complexes impacts proper chromosomes separation during anaphase. (A and B) Schematic representation for the calculation of the distance (d [μm]) between inter chromatids in cells undergoing metaphase to anaphase transition at various time intervals (A). Quantification of inter chromatids distance in HeLa Kyoto cells that are stably coexpressing AcGFP-NuMA and mCherry-H2B and are transfected with either control siRNA, siRNA-against Ect2, or siRNA-against Cyk4 (B). Note that Ect2 or Cyk4 siRNA transfected cells that show no cleavage furrow, and thus 100% penetrance of the siRNA were analyzed for quantification of inter chromatids distance. ns, P > 0.05; **, P < 0.01; ***, P < 0.001 as determined by two-tailed unpaired Student’s t test. n = 10 cells from two independent experiments as depicted in the figure panel; error bars: SD shown by shaded region-pink: control; green: Ect2 depletion; red: Cyk4 depletion. (C–E) Confocal live-imaging analysis of HeLa Kyoto cells expressing AcGFP-Ect2r (C), or AcGFP-Ect2rΔmem (D), with mCherry-H2B, and the quantification of inter chromatids distance in these cells (E). In these cells, endogenous Ect2 was depleted by siRNA targeting endogenous copy of the mRNA, but not ectopically expressed transgene. The recording was initiated 26 h post-transfection of Ect2 siRNA. “0 min” time-point represents metaphase to anaphase transition. ns, P > 0.05; *, P < 0.05; **, P < 0.01 as determined by two-tailed unpaired Student’s t test. n = 10 cells as depicted in the figure panel; error bars: SD represented by the shaded region-pink: cells coexpressing AcGFP-Ect2r and mCherry-H2B; green: cells coexpressing AcGFP-Ect2rΔmem and mCherry-H2B.
NuMA exclusion from the equatorial membrane is not merely due to NuMA and Ect2 competition for the membrane phosphoinositides
Ect2 localizes at the spindle midzone with the help of Cyk4/Mklp1 and accumulates at the equatorial membrane during anaphase (Fig. 5 O; reviewed in Green et al., 2012; Basant and Glotzer, 2018). As Ect2 interacts with similar PIP and PIP2 lipid species at the cell membrane to which NuMA binds (Su et al., 2011; Kotak et al., 2014; Zheng et al., 2014), we asked if Ect2 prevents NuMA localization at the equatorial membrane purely by competing for these lipids. We reasoned if Ect2 solely leads to NuMA exclusion via its ability to associate PIP/PIP2 at the equatorial membrane, then targeting the PIP/PIP2-binding module present at the C-terminus of Ect2 to the polar region of the cell membrane might delocalize NuMA from that membrane surface (Fig. S3 A; Chalamalasetty et al., 2006; Su et al., 2011). Importantly, overexpression of the lipid-interacting C-terminus fragment of Ect2 consisting of pleckstrin homology (PH) and polybasic cluster (PBC) caused no significant changes to the NuMA localization at the polar region of the cell membrane (Fig. S3, B–E; Su et al., 2011). Therefore, this finding suggests that the membrane compartmentalization between Ect2 and NuMA is not based on a simple competition-based model between Ect2 and NuMA for the identical pool of membrane lipids (see Discussion).

Ect2/Cyk4/Mklp1-based complexes are localized to the equatorial membrane during anaphase. (A) Schematic representation of siRNA-resistant Ect2 construct with AcGFP (Aequorea coerulescens GFP)-tag and mono FLAG-tag at the N-terminus (referred to as AcGFP-Ect2r). (B) Immunoblot analysis of mitotically synchronized protein extracts made from HeLa Kyoto cells, or Kyoto cells that were stably expressing AcGFP-Ect2r. These extracts were prepared 60 h post-transfection with control siRNA (−) or siRNA against Ect2 (+). The resulting blot was probed with antibodies directed against Ect2 and β-actin. As mentioned, transgenic AcGFP-Ect2r and endogenous (Endo.) Ect2 were detected on this immunoblot. In this and other panels, the molecular mass is indicated in kilodaltons (kD) and is shown on the left. (C–E) IF analysis of the Kyoto cells that are transfected with control siRNA (C), siRNA against Ect2 (D), or Kyoto cells stably expressing AcGFP-Ect2r after transfection with siRNA against Ect2 (E) for 60 h. Cells were fixed and stained using anti-α-tubulin (green) antibodies. DNA is shown in gray. Multinucleation percentage (%) is shown for each siRNA condition. (n > 500 cells each from three independent experiments). The scale bar in the panel and the following panels represent 10 μm. (F) Experimental protocol for chemically induced anaphase onset of mitotically synchronized HeLa Kyoto (Control), or HeLa Kyoto cells that are stably expressing AcGFP-Ect2r by acute Cdk1 inactivation using RO-3306. (G–L) IF analysis of HeLa Kyoto cells (Control) that are either treated with proteasome inhibitor MG132 (G and I) or MG132 and Cdk1 inhibitor RO-3306 (H and J) to synchronize them in the anaphase-like state as explained in F. Cells were fixed and stained using either anti-Plk1 (G and H; green) or anti-Mklp1 (I and J; red) antibodies. The experiment was repeated twice, and the representative cells are shown here. Quantification on the right represents midzone enrichment for Plk1 (K) and Mklp1 (L) in cells treated with MG132 or MG132+RO-3306. ***, P < 0.001 as determined by a two-tailed unpaired Student’s t test. n ≥ 12 cells, error bars: SD. (M) Co-immunoprecipitation (IP) by GFP-Trap in lysates from the AcGFP-Ect2r expressing cells that are chemically induced in the anaphase-like state. The resulting blots were probed for Cyk4, Mklp1, p150Glued, and GFP as indicated. IN (1% of total), IP: 20% of the total. Please note that for GFP detection in the IP fraction, only 3% of IP fraction was loaded. Note that AcGFP-Ect2r interacts with Cyk4, and Mklp1 (centralspindlin complex), but not with dynein interacting dynactin subunit p150Glued. (N) Schematic representation of quantification method used for analyzing equatorial membrane enrichment of AcGFP-Ect2r fluorescence intensity. (O–Q) Images from confocal live-imaging analysis of HeLa Kyoto cells stably expressing AcGFP-Ect2r (green) that are transfected with control siRNA (O) or siRNA against Cyk4 (P) or Mklp1 (Q). The signal intensity of AcGFP-Ect2r is also specified by the pseudocolor gradient on the right side of the image. (R) Equatorial membrane quantification for AcGFP-Ect2r for control siRNA, Cyk4 siRNA, or Mklp1 siRNA was performed as described in N. ***, P < 0.001 as determined by a two-tailed unpaired Student’s t test. n > 12 cells, error bars: SD. (S–U) The membrane localization of AcGFP (green) in HeLa Kyoto cells that are either expressing AcGFP-Ect2r (S) or AcGFP-NuMA (T). The signal intensity of AcGFP-Ect2r or AcGFP-NuMA is also specified by the pseudocolor gradient on the right side of the image. The linescan analysis of the membrane AcGFP intensity for the region (in orange) is depicted in (U). For such measurements, one quadrant of an anaphase cell from the equatorial cell membrane to the polar region of the cell membrane was analyzed to calculate the normalized intensity (U). Source data are available for this figure: SourceData F5.
Ect2/Cyk4/Mklp1-based complexes are localized to the equatorial membrane during anaphase. (A) Schematic representation of siRNA-resistant Ect2 construct with AcGFP (Aequorea coerulescens GFP)-tag and mono FLAG-tag at the N-terminus (referred to as AcGFP-Ect2r). (B) Immunoblot analysis of mitotically synchronized protein extracts made from HeLa Kyoto cells, or Kyoto cells that were stably expressing AcGFP-Ect2r. These extracts were prepared 60 h post-transfection with control siRNA (−) or siRNA against Ect2 (+). The resulting blot was probed with antibodies directed against Ect2 and β-actin. As mentioned, transgenic AcGFP-Ect2r and endogenous (Endo.) Ect2 were detected on this immunoblot. In this and other panels, the molecular mass is indicated in kilodaltons (kD) and is shown on the left. (C–E) IF analysis of the Kyoto cells that are transfected with control siRNA (C), siRNA against Ect2 (D), or Kyoto cells stably expressing AcGFP-Ect2r after transfection with siRNA against Ect2 (E) for 60 h. Cells were fixed and stained using anti-α-tubulin (green) antibodies. DNA is shown in gray. Multinucleation percentage (%) is shown for each siRNA condition. (n > 500 cells each from three independent experiments). The scale bar in the panel and the following panels represent 10 μm. (F) Experimental protocol for chemically induced anaphase onset of mitotically synchronized HeLa Kyoto (Control), or HeLa Kyoto cells that are stably expressing AcGFP-Ect2r by acute Cdk1 inactivation using RO-3306. (G–L) IF analysis of HeLa Kyoto cells (Control) that are either treated with proteasome inhibitor MG132 (G and I) or MG132 and Cdk1 inhibitor RO-3306 (H and J) to synchronize them in the anaphase-like state as explained in F. Cells were fixed and stained using either anti-Plk1 (G and H; green) or anti-Mklp1 (I and J; red) antibodies. The experiment was repeated twice, and the representative cells are shown here. Quantification on the right represents midzone enrichment for Plk1 (K) and Mklp1 (L) in cells treated with MG132 or MG132+RO-3306. ***, P < 0.001 as determined by a two-tailed unpaired Student’s t test. n ≥ 12 cells, error bars: SD. (M) Co-immunoprecipitation (IP) by GFP-Trap in lysates from the AcGFP-Ect2r expressing cells that are chemically induced in the anaphase-like state. The resulting blots were probed for Cyk4, Mklp1, p150Glued, and GFP as indicated. IN (1% of total), IP: 20% of the total. Please note that for GFP detection in the IP fraction, only 3% of IP fraction was loaded. Note that AcGFP-Ect2r interacts with Cyk4, and Mklp1 (centralspindlin complex), but not with dynein interacting dynactin subunit p150Glued. (N) Schematic representation of quantification method used for analyzing equatorial membrane enrichment of AcGFP-Ect2r fluorescence intensity. (O–Q) Images from confocal live-imaging analysis of HeLa Kyoto cells stably expressing AcGFP-Ect2r (green) that are transfected with control siRNA (O) or siRNA against Cyk4 (P) or Mklp1 (Q). The signal intensity of AcGFP-Ect2r is also specified by the pseudocolor gradient on the right side of the image. (R) Equatorial membrane quantification for AcGFP-Ect2r for control siRNA, Cyk4 siRNA, or Mklp1 siRNA was performed as described in N. ***, P < 0.001 as determined by a two-tailed unpaired Student’s t test. n > 12 cells, error bars: SD. (S–U) The membrane localization of AcGFP (green) in HeLa Kyoto cells that are either expressing AcGFP-Ect2r (S) or AcGFP-NuMA (T). The signal intensity of AcGFP-Ect2r or AcGFP-NuMA is also specified by the pseudocolor gradient on the right side of the image. The linescan analysis of the membrane AcGFP intensity for the region (in orange) is depicted in (U). For such measurements, one quadrant of an anaphase cell from the equatorial cell membrane to the polar region of the cell membrane was analyzed to calculate the normalized intensity (U). Source data are available for this figure: SourceData F5.

NuMA exclusion from the equatorial membrane is not merely dependent on Ect2 interaction with the membrane phosphoinositides. (A) Schematic representation of AcGFP (Aequorea coerulescens GFP), and mono FLAG -tagged siRNA-resistant Ect2 full-length (referred to as AcGFP-Ect2r), Ect2 without its membrane binding (PH and PBC) domain (referred to as AcGFP-Ect2rΔmem), and mCherry-tagged C-terminus of Ect2 with membrane binding (PH and PBC) domains together with mutations in the DH domain (referred to as mCherry-Ect2CTGEF4A) that impair GDP/GTP exchange activity but not its membrane binding (see panel C, and Materials and methods, and Su et al., 2011). (B and C) Confocal live-imaging analysis of HeLa Kyoto cells stably expressing AcGFP-NuMA (B) or AcGFP-NuMA cells that are transiently transfected with phosphoinositides binding C-terminus of Ect2 fragment (mCherry-Ect2CTGEF4A). “0 min” time-point represents metaphase to anaphase transition. The scale bar in this panel and the following panels represent 10 µm. (D) Schematic representation of quantification method used for analyzing polar membrane enrichment of AcGFP-NuMA fluorescence intensity. (E) Polar membrane quantification for AcGFP-NuMA in untransfected control cells or in cells expressing mCherry-Ect2CTGEF4A. ns, P > 0.05 as determined by two-tailed unpaired Student’s t test. n = 10 cells; error bars: SD. (F and G) Confocal live-imaging analysis of HeLa Kyoto cells stably expressing AcGFP- Ect2r (F) or AcGFP-Ect2rΔmem (G). The recording was started 26 h post-transfection for Ect2 siRNA targeting endogenous Ect2. “0 min” time-point represents metaphase to anaphase transition. Maximum intensity projected images are shown for each time point. (H) Schematic representation of quantification method used for analyzing midzone enrichment of AcGFP fluorescence intensity in cells stably expressing AcGFP-Ect2r, or AcGFP-Ect2rΔmem and are depleted for endogenous Ect2. (I) Quantification of midzone enrichment of AcGFP signal in cells expressing either AcGFP-Ect2r, or AcGFP-Ect2rΔmem and are depleted for endogenous Ect2. “0 min” time-point represents metaphase to anaphase transition. ns, P > 0.05 as determined by two-tailed unpaired Student’s t test. n = 15 cells; error bars: SD.

NuMA exclusion from the equatorial membrane is not merely dependent on Ect2 interaction with the membrane phosphoinositides. (A) Schematic representation of AcGFP (Aequorea coerulescens GFP), and mono FLAG -tagged siRNA-resistant Ect2 full-length (referred to as AcGFP-Ect2r), Ect2 without its membrane binding (PH and PBC) domain (referred to as AcGFP-Ect2rΔmem), and mCherry-tagged C-terminus of Ect2 with membrane binding (PH and PBC) domains together with mutations in the DH domain (referred to as mCherry-Ect2CTGEF4A) that impair GDP/GTP exchange activity but not its membrane binding (see panel C, and Materials and methods, and Su et al., 2011). (B and C) Confocal live-imaging analysis of HeLa Kyoto cells stably expressing AcGFP-NuMA (B) or AcGFP-NuMA cells that are transiently transfected with phosphoinositides binding C-terminus of Ect2 fragment (mCherry-Ect2CTGEF4A). “0 min” time-point represents metaphase to anaphase transition. The scale bar in this panel and the following panels represent 10 µm. (D) Schematic representation of quantification method used for analyzing polar membrane enrichment of AcGFP-NuMA fluorescence intensity. (E) Polar membrane quantification for AcGFP-NuMA in untransfected control cells or in cells expressing mCherry-Ect2CTGEF4A. ns, P > 0.05 as determined by two-tailed unpaired Student’s t test. n = 10 cells; error bars: SD. (F and G) Confocal live-imaging analysis of HeLa Kyoto cells stably expressing AcGFP- Ect2r (F) or AcGFP-Ect2rΔmem (G). The recording was started 26 h post-transfection for Ect2 siRNA targeting endogenous Ect2. “0 min” time-point represents metaphase to anaphase transition. Maximum intensity projected images are shown for each time point. (H) Schematic representation of quantification method used for analyzing midzone enrichment of AcGFP fluorescence intensity in cells stably expressing AcGFP-Ect2r, or AcGFP-Ect2rΔmem and are depleted for endogenous Ect2. (I) Quantification of midzone enrichment of AcGFP signal in cells expressing either AcGFP-Ect2r, or AcGFP-Ect2rΔmem and are depleted for endogenous Ect2. “0 min” time-point represents metaphase to anaphase transition. ns, P > 0.05 as determined by two-tailed unpaired Student’s t test. n = 15 cells; error bars: SD.
Ect2/Cyk4/Mklp1 tripartite complex excludes NuMA from the equatorial membrane
To get further mechanistic insights on how Ect2, Cyk4, and Mklp1 can exclude NuMA, we generated monoclonal stable HeLa Kyoto cells expressing siRNA-resistant allele of Ect2 that is N-terminally tagged with AcGFP and a FLAG-tag (referred to as AcGFP-Ect2r), similar to what has been reported previously (Fig. 5 A; Su et al., 2011). This transgenic line expresses ectopic AcGFP-Ect2r protein in amounts comparable with the endogenous copy of the gene (Fig. 5 B). The robust cytokinesis failure seen upon the depletion of endogenous Ect2 is fully rescued in this cell line (Fig. 5, C–E), suggesting that the AcGFP-Ect2r protein is functional. Next, we sought to investigate the interaction of AcGFP-Ect2r with the centralspindlin (Cyk4/Mklp1) in anaphase-synchronized cells. To set up anaphase synchronization, we sequentially treated HeLa Kyoto cells with microtubule poison Nocodazole and proteasome inhibitor MG132 to obtain cells in metaphase. These cells were then acutely treated with the Cdk1 inhibitor RO-3306 to synchronize them in the anaphase-like state (Fig. 5 F). Acute treatment with RO-3306 led to a robust localization of the centralspindlin component (Mklp1) and Polo-like kinase 1 (Plk1) at the spindle midzone (Fig. 5, G–L), indicating an anaphase-like state. Importantly, immunoprecipitates (IP) from AcGFP-Ect2r expressing cells synchronized in an anaphase-like state revealed that AcGFP-Ect2r interacts with endogenous Cyk4 and Mklp1, as reported previously (Fig. 5 M; Somers and Saint, 2003; Yüce et al., 2005). This data suggests that Cyk4, Mklp1, and Ect2 make a tripartite complex during anaphase. AcGFP-Ect2r, which is in complex with Cyk4 and Mklp1 in anaphase, is confined to the equatorial membrane, and this restricted localization of AcGFP-Ect2r is significantly perturbed in cells depleted for Cyk4 or Mklp1 (Fig. 5, N–R). Crucially, AcGFP-Ect2r localizes to the equatorial membrane surfaces devoid of AcGFP-NuMA (Fig. 5, S–U). These data indicate that Ect2 enriches at the equatorial membrane in a complex with centralspindlin (Cyk4/Mklp1) and raise a possibility that the confined localization of Ect2/Cyk4/Mklp1-based tripartite complex, instead of Ect2 alone, at the equatorial membrane may restrict NuMA/dynein/dynactin to the polar membrane.
How might Ect2/Cyk4/Mklp1-based tripartite complex exclude NuMA/dynein/dynactin from the equatorial cell membrane? As mentioned previously, the carboxy-terminal of Ect2 (Ect2CT) possesses a conserved PH and PBC domain capable of directly interacting with membrane phosphoinositides chiefly PIP/PIP2 (Chalamalasetty et al., 2006; Su et al., 2011). Therefore, we sought to investigate if the localization of Ect2/Cyk4/Mklp1-based tripartite complex at the equatorial membrane via membrane-binding potential of Ect2 is responsible for NuMA/dynein/dynactin exclusion. To this end, we generated a monoclonal HeLa Kyoto cell line expressing a siRNA-resistant allele of Ect2 lacking its C-terminal PH and PBC domain and tagged with AcGFP and a FLAG-tag (Fig. 6 A; and referred to as AcGFP-Ect2rΔmem). The transgenic protein made by AcGFP-Ect2rΔmem expressing cell line is analogous to AcGFP-Ect2r expressing line (Fig. 6 B). Depletion of endogenous protein in cells expressing the wild-type allele of Ect2 (AcGFP-Ect2r), but not AcGFP-Ect2rΔmem, rescues cytokinesis failure as reported earlier (Fig. 6, C–F; Su et al., 2011). Furthermore, as expected, AcGFP-Ect2rΔmem fails to localize at the equatorial membrane; however, the midzone localization of AcGFP-Ect2rΔmem is not significantly affected compared to AcGFP-Ect2r (Fig. 6, G and H; and Fig. S3, F–I; Su et al., 2011). Next, we examined the localization of NuMA in these lines upon endogenous Ect2 depletion. Transiently expressed mCherry-NuMA is excluded from the equatorial membrane in cells stably expressing AcGFP-Ect2r (Fig. 6, G, I, and J). Notably, mCherry-NuMA localizes to the equatorial membrane surface in cells stably expressing AcGFP-Ect2rΔmem (Fig. 6, H, I, and J).

The membrane tethering of Ect2-based complexes is essential for NuMA exclusion from the equatorial membrane. (A) Schematic representation of AcGFP (Aequorea coerulescens GFP), and mono FLAG -tagged siRNA-resistant Ect2 full-length (referred to as AcGFP-Ect2r), and Ect2 without its membrane binding (PH and PBC) domain (referred to as AcGFP-Ect2rΔmem). (B) Immunoblot analysis of mitotically synchronized protein extracts made from HeLa Kyoto cells, Kyoto cells that were stably expressing either AcGFP-Ect2r or AcGFP- Ect2rΔmem. The resulting blot was probed with anti-GFP or anti-β-actin antibodies. (C–F) IF analysis of the HeLa Kyoto cells transfected with either control siRNA (C), siRNA against Ect2 (D), HeLa Kyoto cells stably expressing AcGFP-Ect2r and transfected with Ect2 siRNA (E), or HeLa Kyoto cells stably expressing AcGFP- Ect2rΔmem and transfected with Ect2 siRNA (F) for 48 h. Cells were fixed and stained with anti-α-tubulin (green) antibodies. DNA is shown in gray. % on each panel marks the cytokinesis failure as calculated by analyzing the occurrence of binucleated and multinucleated cells (n > 500 cells each from three independent experiments). The scale bar in the panel and following panels represent 10 μm. (G and H) Confocal live-imaging analysis of HeLa Kyoto cells stably expressing AcGFP-Ect2r (G) or AcGFP-Ect2rΔmem (H), which are transiently transfected with mCherry-NuMA and are depleted for endogenous Ect2 by siRNA. The recording was started 26 h post-transfection for control and Ect2 siRNA targeting endogenous Ect2. “0 min” time-point represents metaphase to anaphase transition. Yellow arrowheads depict NuMA localization at the equatorial membrane. (I) Schematic representation of quantification method used for analyzing equatorial membrane enrichment of mCherry-NuMA fluorescence intensity. (J) Equatorial membrane quantification for mCherry-NuMA in cells expressing either AcGFP-Ect2r or AcGFP-Ect2rΔmem and are depleted for endogenous Ect2 as described in I. ns, P > 0.05; **, P < 0.01; ***, P < 0.001 as determined by two-tailed unpaired Student’s t test. n = 10 cells; error bars: SD. Source data are available for this figure: SourceData F6.
The membrane tethering of Ect2-based complexes is essential for NuMA exclusion from the equatorial membrane. (A) Schematic representation of AcGFP (Aequorea coerulescens GFP), and mono FLAG -tagged siRNA-resistant Ect2 full-length (referred to as AcGFP-Ect2r), and Ect2 without its membrane binding (PH and PBC) domain (referred to as AcGFP-Ect2rΔmem). (B) Immunoblot analysis of mitotically synchronized protein extracts made from HeLa Kyoto cells, Kyoto cells that were stably expressing either AcGFP-Ect2r or AcGFP- Ect2rΔmem. The resulting blot was probed with anti-GFP or anti-β-actin antibodies. (C–F) IF analysis of the HeLa Kyoto cells transfected with either control siRNA (C), siRNA against Ect2 (D), HeLa Kyoto cells stably expressing AcGFP-Ect2r and transfected with Ect2 siRNA (E), or HeLa Kyoto cells stably expressing AcGFP- Ect2rΔmem and transfected with Ect2 siRNA (F) for 48 h. Cells were fixed and stained with anti-α-tubulin (green) antibodies. DNA is shown in gray. % on each panel marks the cytokinesis failure as calculated by analyzing the occurrence of binucleated and multinucleated cells (n > 500 cells each from three independent experiments). The scale bar in the panel and following panels represent 10 μm. (G and H) Confocal live-imaging analysis of HeLa Kyoto cells stably expressing AcGFP-Ect2r (G) or AcGFP-Ect2rΔmem (H), which are transiently transfected with mCherry-NuMA and are depleted for endogenous Ect2 by siRNA. The recording was started 26 h post-transfection for control and Ect2 siRNA targeting endogenous Ect2. “0 min” time-point represents metaphase to anaphase transition. Yellow arrowheads depict NuMA localization at the equatorial membrane. (I) Schematic representation of quantification method used for analyzing equatorial membrane enrichment of mCherry-NuMA fluorescence intensity. (J) Equatorial membrane quantification for mCherry-NuMA in cells expressing either AcGFP-Ect2r or AcGFP-Ect2rΔmem and are depleted for endogenous Ect2 as described in I. ns, P > 0.05; **, P < 0.01; ***, P < 0.001 as determined by two-tailed unpaired Student’s t test. n = 10 cells; error bars: SD. Source data are available for this figure: SourceData F6.
siRNA-mediated depletion of Ect2 or Cyk4 significantly impairs chromosome separation during metaphase to anaphase transition (Fig. 4 B). Since cells expressing AcGFP-Ect2rΔmem fail o exclude NuMA from the equatorial membrane, we sought to measure chromosome separation kinetics in cells expressing either AcGFP-Ect2r or AcGFP-Ect2rΔmem with a DNA marker mCherry-H2B upon endogenous Ect2 depletion. Consistent with the previous results (Fig. 4 B), cells expressing AcGFP-Ect2rΔmem show significantly slow kinetics of chromosome separation compared with cells expressing full-length AcGFP-Ect2r (Fig. 4, C–E).
Altogether, these results indicate that the membrane-binding ability of Ect2 in complex with Cyk4/Mklp1 ensures compartmentalization of Ect2/Cyk4/Mklp1 at the equatorial membrane, which is critical for NuMA/dynein/dynactin exclusion and proper chromosomes separation.
NuMA ensures proper RhoA levels at the equatorial membrane
As NuMA-based and Ect2-based complexes piggyback on identical lipid species (PIP/PIP2) for their accumulation at two regions of the cell membrane (polar and equatorial), we asked what if the accumulation of NuMA-based complexes restrict Ect2/Cyk4/Mklp1 to the equatorial membrane by causing molecular crowding at the polar membrane. If this hypothesis is correct, then NuMA depletion should allow Ect2-based complexes to spread, and concomitantly, this should lead to a reduction in the levels of these complexes at the equatorial membrane. To assess the localization of Ect2-based tripartite complexes in cells depleted of NuMA, we analyzed the localization of RhoA since Ect2-based complexes are critical for regulating and confining RhoA at the equatorial membrane (Yüce et al., 2005). Notably, cells transfected with NuMA siRNA show modest but significantly reduced levels of RhoA at the equatorial membrane during anaphase (compare Fig. 7 B with Fig. 7 A, quantification in Fig. 7, E–J). These reductions in RhoA intensity at the equatorial membrane in NuMA depleted cells appear not because of the impact of NuMA depletion on the organization of spindle midzone or spindle midzone localized key regulators of RhoA such as Cyk4 or Ect2 (Fig. S4, A–K). Nevertheless, irrespective of this reduction in the RhoA levels at the equatorial membrane, NuMA siRNA transfected cells establish and ingress cytokinetic furrow at a time comparable with control cells (Fig. 8 B and Video 2). We reasoned that no visible impact of NuMA depletion on cytokinetic furrow formation might be due to robust enrichment of Ect2/Cyk4/Mklp1 complexes at the spindle midzone that could rapidly exchange with the proximal equatorial membrane and will help in maintaining a critical RhoA level necessary for cytokinetic furrow formation, even when NuMA is depleted. What would happen to cleavage furrow if the localization of Ect2/Cyk4/Mklp1 complexes at the spindle midzone is compromised along with NuMA depletion? To investigate this, we analyzed the localization of RhoA in cells co-depleted for NuMA and protein regulator of cytokinesis 1 (Prc1). Prc1 crosslinks antiparallel microtubules and is vital for the assembly of the spindle midzone (Fig. S4, L–N; Mollinari et al., 2002; Mollinari et al., 2005; Zhu et al., 2006; Kellogg et al., 2016). Prc1 depletion drastically diminishes Cyk4, Mklp1, and Ect2 localization at the spindle midzone (Fig. 7, K–R). However, Prc1 depletion does not significantly impact RhoA levels at the equatorial membrane, as reported previously (compare Fig. 7 C with Fig. 7 A; quantification in Fig. 7, E–J; Verbrugghe and White, 2004; Mollinari et al., 2005). Crucially, in cells that are co-depleted for NuMA and Prc1, we observed either significantly diminished (referred to as weak) or no RhoA (referred to as absent) enrichment at the equatorial membrane (Fig. 7, DCatA and DCatB, and related quantification in Fig. 7, E–J). We think that the RhoA zone, which usually occupies ∼10–16% of the cell perimeter during mid-anaphase in control cells (Fig. S4, O and P), had possibly spread to a non-RhoA membrane (∼84–90% of the cell perimeter) in cells that are either depleted for NuMA or co-depleted for NuMA and Prc1 (Fig. 7, G and H), which likely resulted in the reduced intensity of RhoA at the equatorial membrane. Indeed, we noted a significant increase (P = 0.0018) in the pixel intensity of RhoA at the polar membrane upon NuMA depletion, which further increases in conditions that destabilize the spindle midzone (Cat. B, NuMA and Prc1 siRNA; P < 0.0001; Fig. 7, G and H). Furthermore, the cytoplasmic levels or total levels of RhoA remain unchanged in cells depleted for NuMA or NuMA and Prc1 (Fig. 7 S; and Fig. S4, Q and R). This observation indicates that the weak levels of RhoA in NuMA or NuMA and Prc1 depleted cells cannot be attributed to a change in the cytoplasmic or total amount of RhoA. Also, we confirmed that this diminished RhoA zone in cells co-depleted for NuMA and Prc1 is not because of any direct impact of NuMA and Prc1 depletion on the total protein levels of centralspindlin component Cyk4 or Ect2 during mitosis (Fig. 7 S).

NuMA and Prc1 co-depletion impact RhoA accumulation at the equatorial membrane. (A–D) IF analysis of HeLa Kyoto cells that are either transfected with control siRNA (A) or siRNA-against NuMA (B), Prc1 (C), or Prc1 and NuMA (DCat.A and DCat.B). Cells were fixed 60 h post siRNA transfection and stained with anti-RhoA (gray) and anti-NuMA (red) antibodies. A minimum of 60 cells from three independent experiments were visually analyzed for RhoA based on their interchromatid distance during anaphase and representative images are shown in the Figure panel. The relative frequency of these category are mentioned in F. Note that NuMA and Prc1 co-depleted cells either show weak (Cat. A) or no RhoA (Cat. B) enrichment at the equatorial membrane. Also, note that Cat. B cells do not show cytokinetic furrow despite significant inter chromatid distance. In Prc1 depleted cells, in addition to its usual membrane localization, NuMA mislocalizes next to the DNA, possibly because of the absence of an intact spindle midzone. The scale bar in the panel and following panels represent 10 μm. (E and F) Schematic representation of the visual quantification of equatorial RhoA membrane intensity (E), and the % of cells based on such visual quantification in various siRNA transfected conditions (F). For such analysis, cells were grouped into four categories: strong (depicted in blue), as in control siRNA transfected cells, reduced (depicted in pink), as in NuMA siRNA, weak (depicted in orange) as in NuMA and Prc1 siRNA Cat. A, and absent (depicted in brown) as in NuMA and Prc1 siRNA Cat. B. (n = 109 cells for control siRNA; n = 81 cells for NuMA siRNA; n = 96 cells for Prc1 siRNA, and n = 60 cells for NuMA and Prc1 siRNA transfected cells from three independent experiments). (G and H) Schematic representation of line scan analysis (G) and the outcome (H) of this analysis for Rho A intensity at the equatorial membrane (depicted in blue) and polar membrane (shown in orange) in different siRNA transfected conditions. Please note a decrease in RhoA equatorial intensity in NuMA siRNA (P < 0.0001) as well as NuMA and Prc1 siRNA (P < 0.0001) transfected cells. Also, note a marginal increase in RhoA intensity at the polar membrane surface in cells transfected with either NuMA siRNA (P = 0.0018) or NuMA and Prc1 siRNA (Cat A: P < 0.04; Cat B: P < 0.0001). The RhoA intensity in Prc1 depleted cells at the polar membrane is non-significant (P = 0.108). n = 15 representative cells from all siRNA conditions were used for this quantification; the shaded region indicates SEM; the intensity value at the peak of each line scan curve has been compared to that of control to measure the P value by two-tailed unpaired Student’s t test. (I and J) Schematic representation of the quantification method (I), and the outcome of such analysis for equatorial membrane enrichment of RhoA fluorescence intensity (J). Please note a significant decrease in equatorial membrane RhoA intensity in cells depleted for NuMA, or NuMA and Prc1. The distribution for the RhoA intensity for all the cells depleted for NuMA and Prc1 is shown in the inset. Note that ∼30% of cells show the ratio of a selected region at the equatorial membrane to that of a similar area at the polar membrane is ∼1 (shown as red dots in the inset). ns, P > 0.05; ***, P < 0.001 as determined by two-tailed unpaired Student’s t test. n = 25 cells for control siRNA, NuMA siRNA, or Prc1 siRNA, and n = 27 cells for NuMA and Prc1 siRNA transfected cells were taken from three independent experiments; error bars: SD. (K–R) IF analysis of the central spindle localization of Prc1 (K and O), Cyk4 (L and P), Mklp1 (M and Q), or Ect2 (N and R) in HeLa Kyoto cells that are transfected with either control siRNA or siRNA against Prc1. Yellow-line represents the area that was used for computing the line scan intensity (in the arbitrary unit, au) on the right of each panel. n = 5 cells in each condition, as depicted; error bars: SD. (S) Immunoblot analysis of mitotically synchronized protein extracts made from HeLa Kyoto cells that were either transfected with control siRNA or siRNA against Prc1, NuMA, or Prc1 and NuMA for 60 h. The resulting blot was probed with anti-NuMA, Anti-Prc1, anti-Ect2, anti-Cyk4, and anti-RhoA antibodies. Anti-β-actin antibodies were used to analyze the equal loading of the samples. The values below the Ect2, Cyk4, and RhoA represent the band intensity, normalized to the intensity value from the respective β-actin control. Please note that we have run various % of SDS-PAGE to detect proteins with distinct molecular weight (MW). For instance, NuMA (MW ∼238kD) is detected by running a 6% gel, and for RhoA (MW ∼22 kD) detection, we have run the samples on 12% gel. Source data are available for this figure: SourceData F7.
NuMA and Prc1 co-depletion impact RhoA accumulation at the equatorial membrane. (A–D) IF analysis of HeLa Kyoto cells that are either transfected with control siRNA (A) or siRNA-against NuMA (B), Prc1 (C), or Prc1 and NuMA (DCat.A and DCat.B). Cells were fixed 60 h post siRNA transfection and stained with anti-RhoA (gray) and anti-NuMA (red) antibodies. A minimum of 60 cells from three independent experiments were visually analyzed for RhoA based on their interchromatid distance during anaphase and representative images are shown in the Figure panel. The relative frequency of these category are mentioned in F. Note that NuMA and Prc1 co-depleted cells either show weak (Cat. A) or no RhoA (Cat. B) enrichment at the equatorial membrane. Also, note that Cat. B cells do not show cytokinetic furrow despite significant inter chromatid distance. In Prc1 depleted cells, in addition to its usual membrane localization, NuMA mislocalizes next to the DNA, possibly because of the absence of an intact spindle midzone. The scale bar in the panel and following panels represent 10 μm. (E and F) Schematic representation of the visual quantification of equatorial RhoA membrane intensity (E), and the % of cells based on such visual quantification in various siRNA transfected conditions (F). For such analysis, cells were grouped into four categories: strong (depicted in blue), as in control siRNA transfected cells, reduced (depicted in pink), as in NuMA siRNA, weak (depicted in orange) as in NuMA and Prc1 siRNA Cat. A, and absent (depicted in brown) as in NuMA and Prc1 siRNA Cat. B. (n = 109 cells for control siRNA; n = 81 cells for NuMA siRNA; n = 96 cells for Prc1 siRNA, and n = 60 cells for NuMA and Prc1 siRNA transfected cells from three independent experiments). (G and H) Schematic representation of line scan analysis (G) and the outcome (H) of this analysis for Rho A intensity at the equatorial membrane (depicted in blue) and polar membrane (shown in orange) in different siRNA transfected conditions. Please note a decrease in RhoA equatorial intensity in NuMA siRNA (P < 0.0001) as well as NuMA and Prc1 siRNA (P < 0.0001) transfected cells. Also, note a marginal increase in RhoA intensity at the polar membrane surface in cells transfected with either NuMA siRNA (P = 0.0018) or NuMA and Prc1 siRNA (Cat A: P < 0.04; Cat B: P < 0.0001). The RhoA intensity in Prc1 depleted cells at the polar membrane is non-significant (P = 0.108). n = 15 representative cells from all siRNA conditions were used for this quantification; the shaded region indicates SEM; the intensity value at the peak of each line scan curve has been compared to that of control to measure the P value by two-tailed unpaired Student’s t test. (I and J) Schematic representation of the quantification method (I), and the outcome of such analysis for equatorial membrane enrichment of RhoA fluorescence intensity (J). Please note a significant decrease in equatorial membrane RhoA intensity in cells depleted for NuMA, or NuMA and Prc1. The distribution for the RhoA intensity for all the cells depleted for NuMA and Prc1 is shown in the inset. Note that ∼30% of cells show the ratio of a selected region at the equatorial membrane to that of a similar area at the polar membrane is ∼1 (shown as red dots in the inset). ns, P > 0.05; ***, P < 0.001 as determined by two-tailed unpaired Student’s t test. n = 25 cells for control siRNA, NuMA siRNA, or Prc1 siRNA, and n = 27 cells for NuMA and Prc1 siRNA transfected cells were taken from three independent experiments; error bars: SD. (K–R) IF analysis of the central spindle localization of Prc1 (K and O), Cyk4 (L and P), Mklp1 (M and Q), or Ect2 (N and R) in HeLa Kyoto cells that are transfected with either control siRNA or siRNA against Prc1. Yellow-line represents the area that was used for computing the line scan intensity (in the arbitrary unit, au) on the right of each panel. n = 5 cells in each condition, as depicted; error bars: SD. (S) Immunoblot analysis of mitotically synchronized protein extracts made from HeLa Kyoto cells that were either transfected with control siRNA or siRNA against Prc1, NuMA, or Prc1 and NuMA for 60 h. The resulting blot was probed with anti-NuMA, Anti-Prc1, anti-Ect2, anti-Cyk4, and anti-RhoA antibodies. Anti-β-actin antibodies were used to analyze the equal loading of the samples. The values below the Ect2, Cyk4, and RhoA represent the band intensity, normalized to the intensity value from the respective β-actin control. Please note that we have run various % of SDS-PAGE to detect proteins with distinct molecular weight (MW). For instance, NuMA (MW ∼238kD) is detected by running a 6% gel, and for RhoA (MW ∼22 kD) detection, we have run the samples on 12% gel. Source data are available for this figure: SourceData F7.

Spindle midzone and astral microtubules are not affected by NuMA depletion. (A) Schematic representation of quantification method used for analyzing midzone microtubule fluorescence intensity. (B and C) IF analysis of HeLa cells that were either transfected with control siRNA (B), or siRNA directed against NuMA (C) for 60 h, were fixed and stained with antibodies directed against ⍺-tubulin (in green). The scale bar in the panel and following panels represent 10 μm. More than 30 anaphase cells were analyzed from two independent experiments, and the representative cells are shown here. (D) Midzone microtubule fluorescence intensity quantification for control siRNA, or NuMA siRNA was performed as described in A. ns- P > 0.05 as determined by two-tailed unpaired Student’s t test. n = 15 cells, error bars: SD. (E) Schematic representation of quantification method used for analyzing midzone enrichment for Cyk4 (F and G) or Ect2 (I and J). (F–K) IF analysis of HeLa cells that were either transfected with control siRNA (F and I), or siRNA directed against NuMA (G and J) for 60 h, were fixed and stained with antibodies directed against Cyk4 or Ect2 (in green) as indicated. More than 30 anaphase cells were analyzed from two independent experiments, and the representative cells are shown here. Quantification on the right represent midzone enrichment for Cyk4 (H) or Ect2 (K) for control siRNA, or NuMA siRNA as described in E. ns, P > 0.05 as determined by two-tailed unpaired Student’s t test. n = 15 cells, error bars: SD. (L) A schematic representation of anaphase cell and highlighting the midzone microtubules bundles created by Prc1 (Protein Regulator of Cytokinesis 1) homodimer. (M and N) IF analysis of HeLa cells that were either transfected with control siRNA (M), or siRNA directed against Prc1 (N) for 36 h were fixed and stained with antibodies directed against ⍺-tubulin (in green). More than 30 anaphase cells were analyzed from two independent experiments, and the representative cells are shown here. (O and P) Schematic representation of quantification method (O) used to determine the RhoA zone (in gray) and outcome of such analysis in cells transfected with control siRNA (P). n = 25 cells; error bars: SD. (Q and R) Schematic representation of quantification method (Q) used to determine the cytoplasmic RhoA intensity in arbitrary unit (au), and outcome of such analysis in cells transfected with control siRNA, NuMA siRNA, Prc1 siRNA, or NuMA and Prc1 siRNA (R). ns, P > 0.05 as determined by two-tailed unpaired Student’s t test. n = 10 cells in each condition; error bars: SD. (S and T) Representative IF image of HeLa Kyoto cell in anaphase fixed and stained with α-tubulin antibodies (gray). The areas used to analyze relative astral microtubules intensity is shown (S). Quantification of relative astral microtubule intensity in cells transfected with control siRNA (control), Prc1 siRNA, NuMA siRNA or NuMA and Prc1 siRNA (T). The intensities of the spindle (Ispindle) and the total cell (Itotal) were determined with ImageJ software. Relative astral MT intensity was calculated by ([Itotal−Ispindle]/Ispindle). ns, P > 0.05 as determined by two-tailed unpaired Student’s t test. n = 11 cells in each condition; error bars: SD.

Spindle midzone and astral microtubules are not affected by NuMA depletion. (A) Schematic representation of quantification method used for analyzing midzone microtubule fluorescence intensity. (B and C) IF analysis of HeLa cells that were either transfected with control siRNA (B), or siRNA directed against NuMA (C) for 60 h, were fixed and stained with antibodies directed against ⍺-tubulin (in green). The scale bar in the panel and following panels represent 10 μm. More than 30 anaphase cells were analyzed from two independent experiments, and the representative cells are shown here. (D) Midzone microtubule fluorescence intensity quantification for control siRNA, or NuMA siRNA was performed as described in A. ns- P > 0.05 as determined by two-tailed unpaired Student’s t test. n = 15 cells, error bars: SD. (E) Schematic representation of quantification method used for analyzing midzone enrichment for Cyk4 (F and G) or Ect2 (I and J). (F–K) IF analysis of HeLa cells that were either transfected with control siRNA (F and I), or siRNA directed against NuMA (G and J) for 60 h, were fixed and stained with antibodies directed against Cyk4 or Ect2 (in green) as indicated. More than 30 anaphase cells were analyzed from two independent experiments, and the representative cells are shown here. Quantification on the right represent midzone enrichment for Cyk4 (H) or Ect2 (K) for control siRNA, or NuMA siRNA as described in E. ns, P > 0.05 as determined by two-tailed unpaired Student’s t test. n = 15 cells, error bars: SD. (L) A schematic representation of anaphase cell and highlighting the midzone microtubules bundles created by Prc1 (Protein Regulator of Cytokinesis 1) homodimer. (M and N) IF analysis of HeLa cells that were either transfected with control siRNA (M), or siRNA directed against Prc1 (N) for 36 h were fixed and stained with antibodies directed against ⍺-tubulin (in green). More than 30 anaphase cells were analyzed from two independent experiments, and the representative cells are shown here. (O and P) Schematic representation of quantification method (O) used to determine the RhoA zone (in gray) and outcome of such analysis in cells transfected with control siRNA (P). n = 25 cells; error bars: SD. (Q and R) Schematic representation of quantification method (Q) used to determine the cytoplasmic RhoA intensity in arbitrary unit (au), and outcome of such analysis in cells transfected with control siRNA, NuMA siRNA, Prc1 siRNA, or NuMA and Prc1 siRNA (R). ns, P > 0.05 as determined by two-tailed unpaired Student’s t test. n = 10 cells in each condition; error bars: SD. (S and T) Representative IF image of HeLa Kyoto cell in anaphase fixed and stained with α-tubulin antibodies (gray). The areas used to analyze relative astral microtubules intensity is shown (S). Quantification of relative astral microtubule intensity in cells transfected with control siRNA (control), Prc1 siRNA, NuMA siRNA or NuMA and Prc1 siRNA (T). The intensities of the spindle (Ispindle) and the total cell (Itotal) were determined with ImageJ software. Relative astral MT intensity was calculated by ([Itotal−Ispindle]/Ispindle). ns, P > 0.05 as determined by two-tailed unpaired Student’s t test. n = 11 cells in each condition; error bars: SD.

Co-depletion of NuMA and Prc1 affect cytokinetic furrow initiation. (A–D) Confocal live-imaging analysis in combination with differential interphase contrast (DIC) microscopy images of HeLa Kyoto cells stably expressing mCherry-H2B, and were either transfected with control siRNA (A; Video 1), siRNA against NuMA (B; Video 2), siRNA against Prc1 (C; Video 3), or siRNA against Prc1 and NuMA (D(i); Video 4 and D(ii); Video 5). D(i) represents cells where the cytokinetic furrow was initiated but did not fully ingress and regress back before complete ingression (∼20% of the cells). In contrast, D(ii) represents cells where no cytokinetic furrow initiation was observed (∼10% of the cells). The recording was started 50 h post-transfection. “0 min” time-point represents metaphase to anaphase transition. The white arrowhead represents appearance of cytokinetic furrow. The yellow asterisk depicts cytokinesis failure (binucleated cell) in Prc1 siRNA transfected cell after complete furrow ingression, followed by regression. The scale bar in these panels represents 10 μm. (E) Quantification of various cytokinetic phenotypes as indicated in HeLa Kyoto cells stably expressing mCherry-H2B that were transfected with Prc1 siRNA or Prc1 and NuMA siRNA (n > 50 cells from three independent experiments). The recording was started 50 h post-transfection. “0 min” time-point represents metaphase to anaphase transition, and for every cell time-lapse, movies were recorded till 70 min from time 0 min. Since control siRNA transfected cells, and NuMA siRNA transfected cells do not reveal any furrow regression and cytokinesis failure within 70 min of time-lapse movies acquisition, their data are not included in the graph. (Total cells analyzed for Prc1 siRNA and for NuMA and Prc1 siRNA transfected cells were 51 and 87, respectively). (F–J) Schematic representation of anaphase cell depicting the method of measurements of the cytokinetic furrow width (d [µm]) from time-lapse confocal live imaging obtained for A–D and F. Furrow width measurements in cells stably expressing mCherry-H2B that were transfected with control siRNA (G), siRNA against NuMA (H), siRNA against Prc1 (I), and siRNA against Prc1 and NuMA (J). Please note that in contrast to Prc1 siRNA transfected cells, 10% of cells transfected with NuMA and Prc1 siRNA do not initiate cytokinetic furrow ingression, while 20% of these cells do not fully ingress cytokinetic furrow and proceed with furrow regression in time <35 min from time “0 min” that indicates metaphase to anaphase transition. (a minimum of 16 cells were used for such analysis for each siRNA condition, as depicted on the figure panels).
Co-depletion of NuMA and Prc1 affect cytokinetic furrow initiation. (A–D) Confocal live-imaging analysis in combination with differential interphase contrast (DIC) microscopy images of HeLa Kyoto cells stably expressing mCherry-H2B, and were either transfected with control siRNA (A; Video 1), siRNA against NuMA (B; Video 2), siRNA against Prc1 (C; Video 3), or siRNA against Prc1 and NuMA (D(i); Video 4 and D(ii); Video 5). D(i) represents cells where the cytokinetic furrow was initiated but did not fully ingress and regress back before complete ingression (∼20% of the cells). In contrast, D(ii) represents cells where no cytokinetic furrow initiation was observed (∼10% of the cells). The recording was started 50 h post-transfection. “0 min” time-point represents metaphase to anaphase transition. The white arrowhead represents appearance of cytokinetic furrow. The yellow asterisk depicts cytokinesis failure (binucleated cell) in Prc1 siRNA transfected cell after complete furrow ingression, followed by regression. The scale bar in these panels represents 10 μm. (E) Quantification of various cytokinetic phenotypes as indicated in HeLa Kyoto cells stably expressing mCherry-H2B that were transfected with Prc1 siRNA or Prc1 and NuMA siRNA (n > 50 cells from three independent experiments). The recording was started 50 h post-transfection. “0 min” time-point represents metaphase to anaphase transition, and for every cell time-lapse, movies were recorded till 70 min from time 0 min. Since control siRNA transfected cells, and NuMA siRNA transfected cells do not reveal any furrow regression and cytokinesis failure within 70 min of time-lapse movies acquisition, their data are not included in the graph. (Total cells analyzed for Prc1 siRNA and for NuMA and Prc1 siRNA transfected cells were 51 and 87, respectively). (F–J) Schematic representation of anaphase cell depicting the method of measurements of the cytokinetic furrow width (d [µm]) from time-lapse confocal live imaging obtained for A–D and F. Furrow width measurements in cells stably expressing mCherry-H2B that were transfected with control siRNA (G), siRNA against NuMA (H), siRNA against Prc1 (I), and siRNA against Prc1 and NuMA (J). Please note that in contrast to Prc1 siRNA transfected cells, 10% of cells transfected with NuMA and Prc1 siRNA do not initiate cytokinetic furrow ingression, while 20% of these cells do not fully ingress cytokinetic furrow and proceed with furrow regression in time <35 min from time “0 min” that indicates metaphase to anaphase transition. (a minimum of 16 cells were used for such analysis for each siRNA condition, as depicted on the figure panels).
In sea urchin embryos, astral microtubules are linked to maintaining a localized RhoA zone at the equatorial membrane (Bement et al., 2005; Foe and von Dassow, 2008; Von Dassow et al., 2009). Thus, we wondered if the impact of Prc1 and NuMA codepletion on RhoA localization could be via their influence on the astral microtubules. However, we found no significant difference in the intensity profile of astral microtubules in cells transfected with siRNA against Prc1 and NuMA in comparison with the control cells (Fig. S4, S and T). Altogether these data indicate that NuMA’s polarized membrane distribution acts in concert with the spindle midzone to ensure proper RhoA accumulation at the equatorial membrane (see Discussion).
NuMA-based complexes at the polar membrane act redundantly with the spindle midzone to control cleavage furrow ingression
To scrutinize the relevance of the diminished or weak RhoA zone for cytokinesis in cells depleted for NuMA and Prc1, we conducted live-imaging experiments in HeLa Kyoto cells stably expressing mCherry-H2B. Control cells initiate cytokinetic furrow at ∼12 min and completely ingress furrow within ∼20 min (Fig. 8, A, F, and G; and Video 1). Despite reduced levels of RhoA at the equatorial membrane in cells transfected with NuMA siRNA, these cells do not show any visible sign of cytokinesis defects (Fig. 8, B, F, and H; and Video 2). As expected from the RhoA equatorial levels, Prc1 depletion in cells does not impact the timing of furrow initiation, but invariably these cells fail cytokinesis because of the instability of the abscission zone (Fig. 8, C, E, F, and I; and Video 3). Notably, ∼20% of cells codepleted for NuMA and Prc1 start furrow ingression at a time comparable with the control cells (∼12 min); however, the cleavage furrow does not fully ingress and undergoes regression, leading to cytokinesis failure (Fig. 8, D(i), E, F, and J; and Video 4). Around 10% of NuMA and Prc1 codepleted cells do not initiate furrow and completely fail cytokinesis (Fig. 8, D(ii), E, F, and J; and Video 5). Taken together, these results suggest that the compartmentalized distribution of NuMA at the polar region of the cell membrane act redundantly with the Ect2/Cyk4/Mklp1 accumulation at the spindle midzone to maintain a confined zone of RhoA for proper assembly and ingression of the cleavage furrow.
Confocal live-imaging analysis in combination with DIC microscopy images of HeLa Kyoto cells stably expressing mCherry-H2B and are transfected with control siRNA (related toFig. 8 A ). The mCherry signal is shown in red. Time-lapse videos were acquired using a laser-scanning confocal microscope (Olympus FV 3000). Time is in minutes (min); Images were acquired every 2 min; Playback 3 frames/s.
Confocal live-imaging analysis in combination with DIC microscopy images of HeLa Kyoto cells stably expressing mCherry-H2B and are transfected with control siRNA (related toFig. 8 A ). The mCherry signal is shown in red. Time-lapse videos were acquired using a laser-scanning confocal microscope (Olympus FV 3000). Time is in minutes (min); Images were acquired every 2 min; Playback 3 frames/s.
Confocal live-imaging analysis in combination with DIC microscopy images of HeLa Kyoto cells stably expressing mCherry-H2B and are transfected with siRNA against NuMA (related toFig. 8 B ). The mCherry signal is shown in red. Time-lapse videos were acquired using a laser-scanning confocal microscope (Olympus FV 3000). Time is in minutes (min); Images were acquired every 2 min; Playback 3 frames/s.
Confocal live-imaging analysis in combination with DIC microscopy images of HeLa Kyoto cells stably expressing mCherry-H2B and are transfected with siRNA against NuMA (related toFig. 8 B ). The mCherry signal is shown in red. Time-lapse videos were acquired using a laser-scanning confocal microscope (Olympus FV 3000). Time is in minutes (min); Images were acquired every 2 min; Playback 3 frames/s.
Confocal live-imaging analysis in combination with DIC microscopy images of HeLa Kyoto cells stably expressing mCherry-H2B and are transfected with siRNA against Prc1 (related toFig. 8 C ). The mCherry signal is shown in red. Time-lapse videos were acquired using a laser-scanning confocal microscope (Olympus FV 3000). Time is in minutes (min); Images were acquired every 2 min; Playback 3 frames/s.
Confocal live-imaging analysis in combination with DIC microscopy images of HeLa Kyoto cells stably expressing mCherry-H2B and are transfected with siRNA against Prc1 (related toFig. 8 C ). The mCherry signal is shown in red. Time-lapse videos were acquired using a laser-scanning confocal microscope (Olympus FV 3000). Time is in minutes (min); Images were acquired every 2 min; Playback 3 frames/s.
Confocal live-imaging analysis in combination with DIC microscopy images of HeLa Kyoto cells stably expressing mCherry-H2B and are transfected with siRNA against Prc1 and NuMA (related toFig. 8 D(i) ). In ∼20% of the cells depleted for NuMA and Prc1, the cytokinetic furrow was initiated, but it did not fully ingress and regress before complete ingression. The mCherry signal is shown in red. Time-lapse videos were acquired using a laser-scanning confocal microscope (Olympus FV 3000). Time is in minutes (min); Images were acquired every 2 min; Playback 3 frames/s.
Confocal live-imaging analysis in combination with DIC microscopy images of HeLa Kyoto cells stably expressing mCherry-H2B and are transfected with siRNA against Prc1 and NuMA (related toFig. 8 D(i) ). In ∼20% of the cells depleted for NuMA and Prc1, the cytokinetic furrow was initiated, but it did not fully ingress and regress before complete ingression. The mCherry signal is shown in red. Time-lapse videos were acquired using a laser-scanning confocal microscope (Olympus FV 3000). Time is in minutes (min); Images were acquired every 2 min; Playback 3 frames/s.
Confocal live-imaging analysis in combination with DIC microscopy images of HeLa Kyoto cells stably expressing mCherry-H2B and are transfected with siRNA against Prc1 and NuMA (related toFig. 8 D(ii) ). In ∼10% of the cells depleted for NuMA and Prc1, no cytokinetic furrow initiation was observed. The mCherry signal is shown in red. Time-lapse videos were acquired using a laser-scanning confocal microscope (Olympus FV 3000). Time is in minutes (min); Images were acquired every 2 min; Playback 3 frames/s.
Confocal live-imaging analysis in combination with DIC microscopy images of HeLa Kyoto cells stably expressing mCherry-H2B and are transfected with siRNA against Prc1 and NuMA (related toFig. 8 D(ii) ). In ∼10% of the cells depleted for NuMA and Prc1, no cytokinetic furrow initiation was observed. The mCherry signal is shown in red. Time-lapse videos were acquired using a laser-scanning confocal microscope (Olympus FV 3000). Time is in minutes (min); Images were acquired every 2 min; Playback 3 frames/s.
Discussion
The polarized distribution of protein complexes at the cell membrane is critical for polarity establishment, spindle positioning, and cytokinesis. How protein complexes regulating these processes are localized and maintained at different membrane regions remains incompletely understood. During anaphase, antiparallel microtubules emanated from the two opposite centrosomes are dynamically reorganized between the segregated sister chromatids and form a spindle midzone (reviewed in Green et al., 2012; D’Avino et al., 2015). The assembly of multiple-protein complexes at the spindle midzone and the proximal membrane establishes a narrow RhoA zone at the equatorial membrane for cleavage furrow formation (reviewed in Bement et al., 2006; Green et al., 2012; Basant and Glotzer, 2018). Yet, how membrane RhoA levels are confined and maintained to the narrow equatorial zone of the cell membrane to promote cytokinesis is not well understood. It is hypothesized that a crosstalk between a stimulatory cue at the equatorial membrane and an inhibitory signal at the polar region of the cell cortex maintains RhoA to a restricted membrane zone (reviewed in Green et al., 2012; Basant and Glotzer, 2018). However, the molecular nature of inhibitory signals and the mechanism that prevents RhoA accumulation at the polar region of the cell membrane remains largely unknown. Our results suggest that the polarized distribution of NuMA-based complexes (NuMA/dynein/dynactin) at the polar region of the cell membrane could be involved in restricting the localization of RhoA, possibly via antagonizing with Ect2-based complexes (Ect2/Cyk4/Mklp1) at the equatorial membrane. This is crucial for establishing a confined RhoA zone. Conversely, the enrichment of Ect2-based complexes at the equatorial membrane limits NuMA/dynein/dynactin to the polar region of the cell membrane to ensure proper chromosome separation during anaphase.
Ect2-based complexes at the equatorial membrane restrict NuMA localization to the polar membrane
We uncovered that Ect2 and centralspindlin restrict NuMA/dynein/dynactin to the polar membrane. How do Ect2, Cyk4, and Mklp1 limit NuMA to the polar membrane surface? Ect2 interacts with similar phosphoinositide species (PIP and PIP2) to which NuMA binds (Su et al., 2011; Kotak et al., 2014; Zheng et al., 2014). Therefore, one plausibility was that the enriched population of Ect2 at the spindle midzone would interact with the identical pools of PIP and PIP2 at the proximal equatorial membrane, which will compete out NuMA from the equatorial membrane. This model predicted that ectopic targeting of the lipid-binding domain of Ect2 at the polar region of the cell membrane should reduce NuMA from those membrane surfaces. However, overexpression of the membrane-anchoring C-terminus fragment of Ect2 that robustly localizes to the polar region of the cell membrane (Chalamalasetty et al., 2006; Su et al., 2011) does not significantly impact NuMA localization. Likewise, the depletion of Cyk4 that leads to Ect2 accumulation in the polar membrane does not perturb NuMA localization at the polar membrane (data not shown). These data indicate that ectopic localization of the Ect2 alone at the polar membrane surface is insufficient in outcompeting NuMA by binding to similar lipid species. Therefore, our results cannot be explained by a simple competition-based model between Ect2 and NuMA for the identical pool of PIP/PIP2. An alternative hypothesis that we favor is that the competition between membrane complexes comprising Ect2/Cyk4/Mklp1 (instead of single-molecule of Ect2) creates compartmentalized membrane domains of Ect2-based complexes (Fig. 9). As established previously, we show that Ect2 is in a complex with Cyk4 and Mklp1 during anaphase (Somers and Saint, 2003; Yüce et al., 2005; Fig. 5). This association ensures robust accumulation of Ect2-based complexes at the spindle midzone and the proximal equatorial membrane because of the membrane-binding potential of Ect2 (Fig. 9 i). We propose that enrichment of these Ect2-based complexes at the equatorial membrane creates spatial crowding that blocks NuMA/dynein/dynactin localization (Fig. 9 i). Indeed, cells expressing Ect2Δmem, which can localize to the spindle midzone because of its interaction with centralspindlin but are unable to tether the complex at the membrane, fail to restrict NuMA at the polar region of the membrane.

Membrane compartmentalization of Ect2 and NuMA-based complexes ensure proper chromosome separation and cytokinetic furrow formation. Model for localization of Ect2/Cyk4/Mklp1 and NuMA/dynein/dynactin at the equatorial and polar membrane regions, respectively. During anaphase, the spindle midzone assembly ensures efficient localization of centralspindlin (2Cyk4:2Mklp1) together with its interacting component Ect2 enrichment at the spindle midzone and at the equatorial membrane region. On the contrary, NuMA/dynein/dynactin complexes are enriched at the spindle poles and at the polar membrane surface. Inset (i) highlights the equatorial membrane region where Ect2, because of its ability to directly associate with the membrane phosphoinositides (shown in violet), ensures constant flux of Ect2/Cyk4/Mklp1-based complexes at the equatorial membrane in the proximity of spindle midzone. This regulates sufficient levels of RhoA for proper cytokinesis. In the absence of such robust flux (for instance, in Prc1 [siRNA]), RhoA localization and cleavage furrow ingression is not affected because of the occupancy of NuMA/dynein/dynactin complexes at the polar region of the membrane (inset ii). This polarization between Ect2/Cyk4/Mklp1 and NuMA/dynein/dynactin ensures proper chromosome separation and the formation of the cytokinetic furrow. Violet and orange lipids represent phosphoinositides and other phospholipids, respectively.
Membrane compartmentalization of Ect2 and NuMA-based complexes ensure proper chromosome separation and cytokinetic furrow formation. Model for localization of Ect2/Cyk4/Mklp1 and NuMA/dynein/dynactin at the equatorial and polar membrane regions, respectively. During anaphase, the spindle midzone assembly ensures efficient localization of centralspindlin (2Cyk4:2Mklp1) together with its interacting component Ect2 enrichment at the spindle midzone and at the equatorial membrane region. On the contrary, NuMA/dynein/dynactin complexes are enriched at the spindle poles and at the polar membrane surface. Inset (i) highlights the equatorial membrane region where Ect2, because of its ability to directly associate with the membrane phosphoinositides (shown in violet), ensures constant flux of Ect2/Cyk4/Mklp1-based complexes at the equatorial membrane in the proximity of spindle midzone. This regulates sufficient levels of RhoA for proper cytokinesis. In the absence of such robust flux (for instance, in Prc1 [siRNA]), RhoA localization and cleavage furrow ingression is not affected because of the occupancy of NuMA/dynein/dynactin complexes at the polar region of the membrane (inset ii). This polarization between Ect2/Cyk4/Mklp1 and NuMA/dynein/dynactin ensures proper chromosome separation and the formation of the cytokinetic furrow. Violet and orange lipids represent phosphoinositides and other phospholipids, respectively.
Since Ect2-based complexes play a vital role in RhoA activation, and Ect2 and RhoA are engaged in a positive feedback loop (Chen et al., 2020), our results do not exclude the possibility that RhoA, either alone or in combination with Ect2/Cyk4/Mklp1 is involved in NuMA/dynein/dynactin exclusion from the equatorial membrane. Also, we do not entirely rule out an additional possibility that the contractile ring protein/s other than Anillin or mDia (formin) could function either with Ect2-based complexes or by yet unknown means to exclude NuMA/dynein/dynactin. Moreover, our data support that RhoA downstream effectors Rock and myosin II are not engaged in NuMA exclusion from the equatorial membrane in anaphase.
NuMA controls RhoA levels at the equatorial membrane
NuMA-based and Ect2-based complexes accumulate in mutually exclusive membrane surfaces during anaphase (Fig. 9). Since these complexes rely on identical lipids for their membrane localization, we assumed that the robust accumulation of NuMA/dynein/dynactin at the polar region of the cell membrane might suppress the lateral movement of Ect2-based complexes by creating spatial crowding on the polar membrane (Fig. 9 ii). This would ensure a confined localization of Ect2-based complexes and thus RhoA at the equatorial membrane for proper cleavage furrow formation. Notably, cells depleted of NuMA alone had reduced RhoA intensity at the equatorial membrane. However, such reduced RhoA levels did not impact cytokinetic furrow formation. This data aligns with the observations that furrow initiation requires significantly less RhoA activity (Loria et al., 2012; Tse et al., 2012). We rationalized that cells with NuMA depletion would still maintain sufficient RhoA levels at the equatorial membrane for furrow initiation, possibly because of the enrichment of Ect2-based complexes at the spindle midzone, which would rapidly exchange with the proximal equatorial membrane. We hypothesized that if we compromise the enrichment of Ect2-based complexes at the spindle midzone in NuMA-depleted condition, then we expect to see a further reduction in the RhoA levels at the equatorial membrane. Indeed, affecting the enrichment of Ect2-based complexes at the spindle midzone (by Prc1 siRNA) together with NuMA depletion further decreases RhoA intensity. Importantly, we detected no RhoA enrichment at the equatorial membrane in a significant number of cells (∼27%) that were co-depleted for NuMA and Prc1. Consistently, ∼30% of cells co-depleted for NuMA and Prc1 showed either a complete failure in furrow ingression or ingression followed by early regression. These phenotypes were not observed in cells depleted for either NuMA or Prc1. The reduced levels of RhoA at the membrane in NuMA or NuMA and Prc1 depleted cells are not due to any change in the total RhoA levels. Therefore, we are of the opinion that the reduction in RhoA intensity at the equatorial membrane in either NuMA or NuMA and Prc1 depleted cells is likely due to the spreading of RhoA regulators Ect2/Cyk4/Mklp1 to a larger surface area which leads to an overall decrease in RhoA intensity at the equatorial membrane. In line with this, we noted a significant increase in the pixel intensity of RhoA at the polar membrane in cells that are depleted for NuMA or NuMA and Prc1.
Our model of NuMA-dependent physical exclusion of Ect2-based complexes from the polar cortex can also explain why the depletion of Prc1 alone does not affect RhoA levels at the equatorial membrane and therefore has no impact on cytokinetic furrow formation. We reasoned that despite the fact that depletion of Prc1 results in loss of Ect2-based complexes at the spindle midzone, their critical threshold at the equatorial membrane is constantly maintained owing to the presence of NuMA-based complexes at the polar membrane, which prevents them from diffusing out of the equatorial zone. This restricted mobility of Ect2-based complexes ensures timely cleavage furrow formation in Prc1-depleted cells (Fig. 8; Verbrugghe and White, 2004; Mollinari et al., 2005). Therefore, we propose that NuMA-based complexes act redundantly with the spindle midzone localized Ect2-based complexes to establish a confined zone of RhoA for cleavage furrow formation. In summary, this work has characterized an essential but unrecognized mechanism that coordinates chromosome segregation with cleavage furrow formation by polarizing two evolutionarily conserved protein complexes at different cell membrane regions.
Materials and methods
Cell culture, transfections, and stable cell line generation
All the cell lines used in this study were cultured in high-glucose DMEM supplemented with 10% fetal bovine serum (10270-106; Gibco) at 37°C in a humidified 5% CO2 incubator. All the stable cell lines generated during this study were made in HeLa Kyoto cells (a gift from Daniel Gerlich, Institute of Molecular Biotechnology, Vienna, Austria). HeLa Kyoto cells stably expressing GFP-tagged dynein heavy chain (DHC1) were kindly provided by Anthony Hyman (Max Plank Institute of Molecular Cell Biology and Genetics, Dresden, Germany; Poser et al., 2008).
For plasmid transfections, 4 µg of plasmid DNA was suspended in 400 μl of serum-free DMEM. After 5 min incubation, 6 μl of either lipofectamine 2000 (11668019; Life Technologies) or 6 μl turbofect (R0531; ThermoFisher Scientific) was added to the mixture and incubated for 15 min. This mixture was added to the cells plated at 80% confluency on coverslips in six-well plates or on an imaging dish (0030740017; Eppendorf). Cells were fixed for immunostaining or imaged 30–36 h after plasmid transfection.
For the siRNA transfection, 6 or 9 μl of 20 μM siRNA was suspended in 100 μl of water (W4502; Sigma-Aldrich) and incubated for 5 min. Parallelly, 4 μl of lipofectamine RNAi MAX (13778150; Invitrogen) suspended in water was incubated for 5 min. These solutions were mixed and incubated for an additional 15 min, following which this mixture was added to the cells plated on coverslips in 6-well plates or on an imaging dish. Cells were then grown for specified hours before fixation and immunostaining or live-imaging analysis.
For the generation of HeLa Kyoto cell lines stably expressing AcGFP-Ect2r or AcGFP-Ect2rΔmem, cells were cultured at 80% confluency in a 10-cm dish. These cells were then transfected with 6 μg of pIRES-AcGFP-FLAG-Ect2r or pIRES-AcGFP-FLAG-Ect2rΔmem plasmid using 12 μl of lipofectamine 2000 (11668019; Life Technologies). After 36 h of transfection, 400 ng/μl puromycin media was added for the selection. Isolated clones were confirmed by immunostaining and immunoblot analysis. Similarly, for the generation of HeLa Kyoto cells stably expressing mCherry-H2B, cells were transfected with pIRES-H2B-mCherry plasmid (plasmid # 21044; Addgene), and stably expressing cells were selected by adding 500 μg/ml G418 (10131035; Life Technologies).
Plasmids and siRNAs
All human Ect2 clones were generated using cDNA from the human cells as a template via PCR amplification. siRNA resistant Ect2 full-length was cloned into a pIRES-AcGFP-FLAG plasmid (a gift from Mark Petronczki, Boehringer Ingelheim Regional Center, Vienna, Austria; Su et al., 2011) between Age1 and EcoR1 sites using the forward primer 5′-GCACCGGTATGGCTGAAAATAGTGTATTAACATCC-3′ and reverse primer 5′-GCGAATTCCTATATCAAATGAGTTGTAGATCTACTTAACG-3′. siRNA resistance was generated using the primer 5′-CTATATTCTCTTCTACTACCAGGTGGGTACATCTTTCATCTCC-3′. Similarly, Ect2rΔmem was cloned into pIRES-AcGFP-FLAG using the forward primer 5′-GCACCGGTATGGCTGAAAATAGTGTATTAACATCC-3′ and the reverse primer 5′-GCGAATTCCTAGCATCCATCTACTTCATAAACAACATC-3′. To create pIRES-mCherry plasmid, mCherry sequence was amplified from pIRES-H2B-mCherry (plasmid # 21044; Addgene) using the forward primer 5′-GCGAATTCATGGTGAGCAAGGGCGAGGAGG-3′ and the reverse primer 5′-GCGCGGCCGCCTACTTGTACAGCTCGTCCATGCC-3′. The amplified mCherry sequence was cloned into the pIRES-AcGFP plasmid (a from Mark Petronczki, Boehringer Ingelheim Regional Center, Vienna, Austria; Su et al., 2011), replacing the AcGFP using EcoR1 and Not1 enzymes.
Ect2CTGEF4A (amino acids 445–914) with a mutation in the conserved PVQR residues within the CR3 helix of the DH domain (generated by the primer 5′-GCAACACTGGGTAACGCCGCTGCTGCTCGGATAAGAAGTTC-3′) was cloned into the pIRES-mCherry plasmid using the forward primer 5′-GCGCTAGCATGCCAGTTCCTTCAAAGCAGTCAGCAAGG-3′ and the reverse primer 5′-GCGAATTCTATCAAATGAGTTGTAGATCTACTTAACG-3′. NuMA full-length was amplified from previously existing plasmid using the forward primer 5′-GCACCGGTATGACACTCCACGCCACCCGGGGG-3′ and the reverse primer 5′-CGGAATTCTTAGTGCTTTGCCTTGCCCTTGGC-3′, and was cloned into the pIRES-AcGFP-FLAG plasmid using Age1 and EcoR1 sites, as reported in Rajeevan et al. (2020). All the clones were confirmed by sequencing analysis.
Double-stranded siRNA oligonucleotides used were 5′-GCACUCACCUUGUAGUUGA-3′ (Ect2 siRNA; Eurogentec), 5′-AAUGGAACCAGAUUCAUCAAUGGUU-3′ (Cyk4 siRNA; Invitrogen), 5′-CAGGAUGUACAGAAGUUGAAGUGAA-3′ (Mklp1 siRNA; Eurogentec), 5′-CGAUGCCUCUUUGAAUAAA-3′ (Anillin siRNA; Eurogentec), 5′-AGAACGAUGAGUUGGCGCUGUCUGG-3′ (NuMA siRNA; Invitrogen), 5′-GAGAGACGACCAUCUUGCAACUAGA-3′ (Prc1 siRNA; Eurogentec), 5′-CUCCGGCACAAUUCAGUUCAA-3′ (mDia siRNA; Eurogentec), and 5′-AACGAACUGCUUUAUGACCUA-3′ (Mklp2 siRNA; Eurogentec).
Drug treatments
HeLa cells were synchronized in anaphase by double thymidine release. Briefly, the cells were treated with 2 mM thymidine (T1895; Sigma-Aldrich) for 17 h, released for 8 h, followed by another round of thymidine treatment for 17 h. After 10.5 h of second thymidine release, the drug of choice was added onto the cells, as mentioned in the respective figure panels. To inactivate Aurora B kinase, cells were treated with 2 μM ZM447439 (S1103; Selleckchem). For initiating premature anaphase entry, cells were treated with 10 µM RO-3306 (S7747; Selleckchem) for 5 min. To inhibit Rock, cells were treated with 30 µM Y-27632 (Y0503; Sigma-Aldrich) for 1 or 12 h. To inhibit myosin II, cells were treated with 100 µM PNBB (Optopharma Ltd., DR-A-081) for 30 min or 12 h. Following drug treatments, the cells were fixed and immunostained.
For synchronization of cells in an anaphase-like state for immunofluorescence and immunoprecipitation analysis, HeLa Kyoto or HeLa Kyoto stably expressing AcGFP-Ect2r were synchronized in prometaphase with 100 nM Nocodazole for 17 h and released in 10 μM MG132 (S2619; Selleckchem) for 4 h to obtain metaphase-synchronized cell population. After that, cells were treated with 10 µM of Cdk1 inhibitor RO-3306 (S7747; Selleckchem) for 10 min to synchronize them in the anaphase-like state, as reported previously in Keshri et al. (2020).
Indirect immunofluorescence and live imaging
For immunofluorescence analysis, cells were fixed with cold methanol at −20°C for 10 min and washed in PBST (PBS containing 0.05% Triton X-100) or fixed with ice-cold 10% trichloroacetic acid (TCA, T0699; Sigma-Aldrich) for 15 min at 4°C and washed in PBST. Cells were blocked in 1% bovine serum albumin (MB083; Himedia) at RT for 1 h, and further incubated with primary antibody for 4 h. The primary antibodies used were 1:200 rabbit anti-NuMA (sc-48773; Santa Cruz), 1:300 mouse anti-NuMA (sc-365532; Santa Cruz), 1:200 mouse anti-p150Glued (612709; Transduction Laboratories), 1:1,000 mouse anti-α-tubulin (T6199; Sigma-Aldrich), 1:2,000 mouse anti-α-tubulin (TU-01 13-8000; Thermo Fisher Scientific), 1:20,000 rabbit anti-GFP (A11122; Invitrogen), 1:1,000 mouse anti-GFP (8H11; DSHB), 1:200 rabbit anti-Ect2 (07-1364; Merck), 1:200 rabbit anti-Cyk4 (A302-797A; Bethyl), 1:200 rabbit anti-Mklp1 (sc-867; Santacruz), 1:200 mouse anti-RhoA (sc-418; Santacruz), 1:200 rabbit anti-Prc1 (sc-8356; Santacruz), 1:200 mouse anti-Aurora B (AB_398396; BD Bioscience), and 1:200 mouse anti-Plk1 (sc-17783; Santacruz). After incubating with primary antibody, washes were done three times, 5 min each with PBST, following which the cells were incubated with secondary antibody for an hour. After three washes with PBST, cells were incubated with 1 μg/ml Hoechst 33342 (B2261; Sigma-Aldrich) for 5 min. Following this, the cells were washed three times with PBST and the coverslips were mounted using Fluoromount (0100-01; SouthernBiotech). Secondary antibodies used were 1:500 Alexa Fluor 488 goat anti-rabbit (A11008; Invitrogen), 1:500 Alexa Fluor 488 goat anti-mouse (A11004; Invitrogen), 1:500 Alexa Fluor 568 goat anti-mouse (A11004; Invitrogen), and 1:500 Alexa Fluor 568 goat anti-rabbit (A11011; Invitrogen). Confocal images were acquired on an Olympus FV 3000 confocal laser scanning microscope using a 60X (NA 1.4) oil immersion objective. All the images were processed using ImageJ (https://imagej.nih.gov/ij/download.html) maintaining relative image intensities.
Time-lapse recording of cells expressing AcGFP or mCherry-tagged proteins was performed on an Olympus FV 3000 confocal laser scanning microscope using a 40X (NA 1.3) oil immersion objective (Olympus Corporation) using an imaging dish (0030740017; Eppendorf) at 5% CO2, 37°C, and 90% humidity maintained by a Tokai Hit STR Stage Top incubator. Images were acquired on the inbuilt FV3000 software at an interval of 1, 2, or 3 min with 9–11 optical sections (3 μm apart). All the images were processed using ImageJ maintaining relative image intensities.
Immunoblotting and immunoprecipitation
For immunoblotting analysis, HeLa cells synchronized in prometaphase with 100 nM Nocodazole for 17–20 h were lysed in lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 0.1 mM sodium orthovanadate, 25 mM sodium fluoride, 0.1 mM phenylmethylsulfonyl fluoride, 0.2% Triton X-100, 0.3% NP-40, 100 nM Okadaic acid, and complete EDTA-free protease inhibitor [539134; Merck]) for 2 h on ice. The lysate was centrifuged at 14,000 rpm, and the proteins in the supernatant fractions were quantified using the Bradford reagent (500-0001; Biorad). Following this, the supernatant was denatured at 99°C in 2X SDS-PAGE buffer and analyzed by 6–12% of SDS-PAGE depending on the molecular weight of the protein of interest. After transfer to nitrocellulose membrane (1620115; Biorad), blocking was done for an hour using 5% skimmed milk (GRM1254; Himedia) in PBST. The membrane was then incubated with primary antibodies overnight at 4°C. The primary antibodies used were 1:1,000 rabbit anti-Ect2 (07-1364; Merck), 1:1,000 rabbit anti-Cyk4 (A302-797A; Bethyl), 1:1,000 rabbit anti-Mklp1 (sc-867; Santacruz), 1:1,000 mouse anti-Anillin (sc-271814; Santacruz), 1:5,000 mouse anti-β-actin (sc-58673; Santacruz), 1:1,000 rabbit anti-Prc1 (sc-8356; Santacruz), 1:1,000 mouse anti-p150Glued (612709; BD Bioscience); 1:20,000 rabbit anti-GFP (A11122; Invitrogen), 1:1,000 rabbit anti-NuMA (sc-48773; Santa Cruz), and 1:1,000 mouse anti-RhoA (sc-418; Santa Cruz). After three washes in PBST (0.05% Tween-20; P1379; Sigma-Aldrich) for 5 min each, the membrane was incubated with HRP-conjugated goat anti-rabbit IgG (111-035-045; Jackson ImmunoResearch Laboratories Inc.) or HRP-conjugated goat anti-mouse IgG (A90-116P; Bethyl) for an hour at room temperature. The membrane was further washed three times for 5 min each in PBST and developed using Luminsol (WBLUF0500; Merck).
For immunoprecipitation, the harvested cells were lysed in lysis buffer (10 mM Tris, pH 7.5, 150 mM NaCl, 0.5 mM EDTA, and 1 mM PMSF [7110; Calbiochem], 0.5% NP-40, and complete EDTA-free protease inhibitor [539134; Merck]). Then 2 mg equivalent of cell lysate was incubated with 30 μl of GFP-Trap agarose beads (ACT-CM-GFA0050; Chromotek) at 4°C for 2 hr. After binding, the beads were washed two times with wash buffer (Lysis buffer without 0.5% NP-40) at 4°C. The bead-bound complex was denatured at 99°C in 2X SDS buffer and analyzed by immunoblotting.
Quantifications and statistical analysis
All quantifications were done in ImageJ. Briefly, in Image J, the “integrated density,” which is defined as “(sum of pixel values in selection) * (area of one pixel)” was taken as intensity measurements in arbitrary units (au). A constant ROI area was used for all the quantifications in a given experiment.
Quantification of membrane signal of NuMA or GFP intensity (Fig. 1, Q and T; and Fig. 2 D and Fig. 6 J) was measured by calculating the ratio of the mean intensity of equatorial membrane signal (of a rectangular region of interest of area 3.5 µm2) divided by the mean intensity value in the cytoplasm (similar area) and correcting for the background signal (an analogous area outside the cell).
Similarly, to calculate the polar enrichment of AcGFP-NuMA (Fig. S3 E), the mean intensity of polar membrane signal (of a rectangular region of interest of area 3.5 µm2) was divided by the mean cytoplasmic intensity (similar area) and corrected for background signal. Quantification of membrane AcGFP-Ect2r (Fig. 5 R) and membrane RhoA (Fig. 7 J) intensity was measured by calculating the ratio of the mean intensity of equatorial membrane signal (of a rectangular region of interest of area 3.5 µm2) divided by the mean intensity of polar membrane signal (similar area) and corrected for background signal. Quantification of midzone enrichment of Plk1, Mklp1 (Fig. 5, K and L), Cyk4, Ect2 (Fig. S4, H and K), and AcGFP signal in Fig. S3 I was measured by calculating the ratio of the mean intensity of the midzone signal (of a rectangular region of interest of area 6.9 µm2) divided by the mean intensity of the cytoplasmic signal (similar area) and corrected for the background signal. Midzone microtubules (Fig. S4 D) were measured by subtracting the mean intensity of the midzone signal (of a rectangular region of interest of 24 µm2) from that of the background signal (similar area). The same method was used to quantify cytoplasmic RhoA intensity in Fig. S4 R (a rectangular region of interest of 3.5 µm2 was used).
The line scan intensity profile was measured using ImageJ. In ImageJ, the line scan plot shows the gray value which is the sum of the gray values of all the pixels divided by the total number of pixels in the selection. The “straight line” tool was used to draw a line of 2.5 µm through the cortex of the anaphase cells to obtain the line scan plot intensity values for NuMA at the equatorial membrane (Fig. 1, L–P), and equatorial and polar membrane for RhoA (Fig. 7, G and H).
Cell length (Fig. 2, C and D) and cytokinetic furrow width (Fig. 8, F–J) were measured at the midplane of the DIC live imaging movies using the line tool in ImageJ. The freehand tool on ImageJ was used for accurately outlining the cell cortex to get the line scan plot of membrane NuMA and RhoA (Fig. 1, A–C), and AcGFP-NuMA and AcGFP-Ect2r at t = 12 minutes after anaphase onset (Fig. 5, S–U). Similarly, the freehand tool (ImageJ) was used to determine the RhoA zone in anaphase cells by outlining the RhoA enriched equatorial membrane and the whole-cell perimeter (Fig. S4, O and P). RhoA zone (%) was determined by the equation mentioned in Fig. S4 O.
The interchromatid distance (Fig. 4) was quantified in maximum intensity–projected live imaging movies of cells expressing AcGFP-NuMA; mCherry-H2B, or AcGFP-Ect2r; mCherry-H2B, or AcGFP-Ect2rΔmem; and mCherry-H2B every 1 min after anaphase onset using ImageJ.
The relative astral microtubule intensity of the anaphase cell was measured using ImageJ. Briefly, maximum intensity projected images were used to calculate the total (astral and spindle) microtubule intensity (Intotal) and the spindle microtubule intensity (Inspindle) of the cell by using the selected area shown in Fig. S4 S. The relative astral microtubule intensity was further calculated using the equation shown in Fig. S4 S.
To calculate the significance of the differences between two mean values, two-tailed unpaired Student’s t tests were performed. The P value was considered to be significant if P < 0.05 using GraphPad Prism 8 (https://www.graphpad.com/scientific-software/prism/). The significances are mentioned as ns, P ≥ 0.05; *, -P < 0.05; **, -P < 0.01; ***, -P < 0.001. Data distribution was assumed to be normal but was not formally tested.
Online supplemental material
Fig. S1 shows that Formin, Rock, and Myosin II are not critical for NuMA exclusion from the equatorial membrane. Fig. S2 shows the accumulation of Cyk4 at the spindle midzone upon Cdk1 inhibitor RO-3306 treatment. Fig. S3 shows that NuMA exclusion from the equatorial membrane is not simply because of the competition between Ect2 and NuMA for the analogous membrane phosphoinositides (PIP/PIP2) species. Fig. S4 shows that NuMA depletion does not affect spindle midzone localization of Ect2/Cyk4 and astral microtubules. Videos 1, 2, 3, 4, and 5 show the confocal live imaging analysis of cells treated with control siRNA and siRNA against NuMA, Prc1, or NuMA and Prc1, respectively.
Acknowledgments
We thank Daniel Gerlich, Mark Petronczki, and Anthony Hyman for providing plasmids and cell lines. We thank Arshad Desai, Karen Oegema, Andrew Goryachev, and Alissa Schlientz for the fruitful discussions during this study and their critical comments on the manuscript. We further thank Iain Hagan, Emanuelle Derivery, Umesh Varshney, and the members of the Kotak Laboratory for their suggestions on the manuscript. We thank Sukriti Kapoor for help with the working model. We thank DST-FIST, UGC Centre for the Advanced Study, Department of Biotechnology-Indian Institute of Science (DBT-IISc) Partnership Program, and IISc for the infrastructure support.
This work is supported by the DBT grant (BT/PR36084/BRB/10/1857/2020) to S. Kotak, and by grants from the DBT/Wellcome Trust India Alliance Fellowship (IA/I/15/2/502077) to S. Kotak. S. Kotak is a Wellcome Trust DBT-India Alliance Intermediate Fellow.
The authors declare no competing financial interests.
Author contributions: Conceptualization: S. Kotak; Methodology: S. Sana, A. Rajeevan, and S. Kotak; Validation: S. Sana, A. Rajeevan, and S. Kotak; Formal analysis: S. Sana, A. Rajeevan, and S. Kotak; Investigation: S. Sana, A. Rajeevan, and S. Kotak; Resources: S. Kotak; Data curation: S. Sana, A. Rajeevan, and S. Kotak; Writing—original draft: S. Kotak; Writing—review & editing: A. Rajeevan, and S. Kotak; Supervision: S. Kotak; Project administration: S. Kotak; Funding acquisition: S. Kotak.

