


Membrane integrity at the endoplasmic reticulum (ER) is tightly regulated, and its disturbance is implicated in metabolic diseases. Using an engineered sensor that activates the unfolded protein response (UPR) exclusively when normal ER membrane lipid composition is compromised, we identified pathways beyond lipid metabolism that are necessary to maintain ER integrity in yeast and in C. elegans. To systematically validate yeast mutants that disrupt ER membrane homeostasis, we identified a lipid bilayer stress (LBS) sensor in the UPR transducer protein Ire1, located at the interface of the amphipathic and transmembrane helices. Furthermore, transcriptome and chromatin immunoprecipitation analyses pinpoint the UPR as a broad-spectrum compensatory response wherein LBS and proteotoxic stress deploy divergent transcriptional UPR programs. Together, these findings reveal the UPR program as the sum of two independent stress responses, an insight that could be exploited for future therapeutic intervention.

Introduction
The unfolded protein response (UPR) is a stress response that is canonically activated by the accumulation of misfolded proteins in the ER lumen. Recent studies showed that the UPR is similarly triggered by lipid bilayer stress (LBS; Halbleib et al., 2017; Hou et al., 2014; Koh et al., 2018; Promlek et al., 2011; Tam et al., 2018; Volmer et al., 2013), especially stress to the ER membrane (Covino et al., 2018). In mammals, the UPR regulates gene expression by signaling through the ER stress sensors IRE1, ATF6, and PKR-like endoplasmic reticulum kinase (PERK). These UPR components have the potential to alleviate cellular stress or to promote apoptosis.
First identified as the sole ER stress transducer in Saccharomyces cerevisiae, Ire1 is essential for cell viability during ER stress (Cox et al., 1993). Active Ire1 cleaves the intron of the HAC1 mRNA precursor, which is translated into the transcription factor Hac1 (Mori et al., 1996; Sidrauski et al., 1996). Hac1 up-regulates the expression of hundreds of genes, including lipid biosynthetic genes (Travers et al., 2000). Conversely, the deletion of lipid regulatory genes such as modulators of sphingolipid synthesis genes ORM1 and ORM2 in yeast or changes in sphingolipid levels in mammalian cells lead to lipid imbalance-induced UPR (Han et al., 2010; Jonikas et al., 2009; Tam et al., 2018). Similarly, increasing cellular saturated fatty acids through genetic manipulation or by exogenous supplementation strongly activates the UPR, likely triggered by a change in lipid bilayer properties (Halbleib et al., 2017; Hou et al., 2014; Kitai et al., 2013; Pineau et al., 2009; Volmer et al., 2013). Moreover, in obese mice and humans, altered membrane lipids composition in the liver and in adipose tissues is associated with UPR induction (Fu et al., 2011; Gregor et al., 2009). Likewise, perturbing membrane phospholipids, including too high or too low phosphatidylcholine (PC) to phosphatidylethanolamine (PE) ratios, can lead to ER stress and UPR activation (Fu et al., 2011; Hou et al., 2014; Koh et al., 2018; Shyu et al., 2019; Thibault et al., 2012). Collectively, these studies suggest that UPR sensors can detect changes in membrane lipids and/or membrane properties.
Despite the intimate relationship between lipid dysregulation and the UPR, our insight into how Ire1 senses changes in the ER membrane lipids is incomplete. A yeast Ire1 mutant (ΔIII Ire1) bearing a truncation in the luminal domain (LD) remains capable of activating the UPR upon inositol depletion (Promlek et al., 2011). Similarly, a mammalian LD-deleted Ire1 variant remains sensitive to LBS and induces the UPR in cells (Volmer et al., 2013). A conserved amphipathic helix near the transmembrane helix of Ire1 drives Ire1 oligomerization during LBS (Halbleib et al., 2017). However, mutating this amphipathic helix of Ire1 severely diminished UPR activation during proteotoxic stress, suggesting that this helix is universally required to activate Ire1. These results point to a conserved UPR sensing and activation mechanism of LBS that is independent of proteotoxicity-induced ER stress (Robblee et al., 2016).
Here, we identified cellular perturbations inducing the UPR through LBS (termed UPRLBS). These were found by monitoring UPR activation in a genome-wide genetic screen in a yeast strain lacking the Ire1 LD, i.e., this domain was dispensable to induce the UPR. Several identified genes are conserved in metazoans, and their inactivation in Caenorhabditis elegans similarly led to UPRLBS. In yeast, OPI3 deletion was one of the strongest hits inducing the UPR. Since the mammalian homologue of OPI3, Pemt, is required to maintain membrane lipid homeostasis (Gao et al., 2015; Koh et al., 2018; Li et al., 2006), we further characterized how OPI3 deletion activates Ire1. We found that mutation of residue R537 renders Ire1 insensitive to LBS while permitting UPR activation by proteotoxic stress (termed UPRPT). Transcriptomic and chromatin immunoprecipitation (ChIP)–quantitative real-time PCR (qPCR) data revealed that the UPR program differs when activated by proteotoxic stress or LBS. For example, Hac1 was found to additionally associate to the promoters of the genes PIR3 and PUT1 during LBS. Together, our data support a model whereby the UPR is a broad-spectrum compensatory pathway in which UPRLBS and UPRPT deploy divergent transcriptional programs.
Results
Various cellular perturbations activate Ire1, independently of its LD
LBS-induced UPR has been demonstrated by either depleting culture media of the phospholipid precursor inositol or by supplementing the media with an excess of saturated fatty acids (Halbleib et al., 2017; Promlek et al., 2011; Volmer et al., 2013). Additionally, 17 yeast deletion strains were tested to identify perturbations that are sensed by Ire1 containing a truncated LD (ΔIII Ire1; Promlek et al., 2011). This study identified nine genes with ER and metabolism-related functions but failed to provide a global view of perturbations that activate the UPR through Ire1’s transmembrane domain. To systematically identify cellular perturbations inducing LBS, we generated a yeast Ire1 mutant lacking its LD (Ire1ΔLD; Fig. S1 A). We exploited Ire1ΔLD’s inability to bind misfolded proteins and used it to monitor gene deletions that activate the UPR to identify cellular processes necessary for ER membrane integrity. Using this tool, we performed a genome-wide genetic screen to measure the in vivo UPR activation with two query strains expressing either WT Ire1 or Ire1ΔLD (Fig. 1 A). The two query strains also express a GFP gene driven by the UPR element (UPRE)–containing promoter and a mCherry gene driven by a constitutive promoter (Breker et al., 2013). The output was measured as the median of single-cell fluorescence ratio (GFP/mCherry) using high-throughput flow cytometry.

The UPR is activated in IRE1 and IRE1ΔLD cells. (A) Schematic representation of Ire1 and Ire1ΔLD domain boundaries. (B) Query strains IRE1 and IRE1ΔLD with genomically integrated UPREpr-GFP and TEF2pr-mCherry were incubated 1h with 10 mM DTT, when indicated, before visualization by confocal microscopy. Scale bar, 5 µm. (C) Flow-cytometric histograms of strains and conditions as in A. Numbers represent the maximum relative GFP fluorescence. (D) Query strains with mutation scj1Δ or spc2Δ were visualized as in A. (E) Flow-cytometric histograms of IRE1 or IRE1ΔLD query strains with mutation get1Δ, spc2Δ, scj1Δ, and ste24Δ. Numbers represent the maximum relative GFP fluorescence. Images shown are representatives of three independent experiments. Dot plots indicate the relative mRNA levels of hsp-4 (F) and of Y41C4A.11 (G) at the larval 4 stage (i.e., after 44 h; n = 3 or 4 per RNAi treatment). Error bars represent SEM. Statistical analysis: Kruskal–Wallis test; all comparisons are to vector RNAi (negative control). Note, only the RNAi clones that evoked significant changes in hsp-4 or Y41C4A.11 are shown, out of 15 total tested clones.
The UPR is activated in IRE1 and IRE1ΔLD cells. (A) Schematic representation of Ire1 and Ire1ΔLD domain boundaries. (B) Query strains IRE1 and IRE1ΔLD with genomically integrated UPREpr-GFP and TEF2pr-mCherry were incubated 1h with 10 mM DTT, when indicated, before visualization by confocal microscopy. Scale bar, 5 µm. (C) Flow-cytometric histograms of strains and conditions as in A. Numbers represent the maximum relative GFP fluorescence. (D) Query strains with mutation scj1Δ or spc2Δ were visualized as in A. (E) Flow-cytometric histograms of IRE1 or IRE1ΔLD query strains with mutation get1Δ, spc2Δ, scj1Δ, and ste24Δ. Numbers represent the maximum relative GFP fluorescence. Images shown are representatives of three independent experiments. Dot plots indicate the relative mRNA levels of hsp-4 (F) and of Y41C4A.11 (G) at the larval 4 stage (i.e., after 44 h; n = 3 or 4 per RNAi treatment). Error bars represent SEM. Statistical analysis: Kruskal–Wallis test; all comparisons are to vector RNAi (negative control). Note, only the RNAi clones that evoked significant changes in hsp-4 or Y41C4A.11 are shown, out of 15 total tested clones.

A high-throughput screen reveals gene clusters that mount a UPR response in IRE1ΔLD cells. (A) Strategy of the genome-wide high-throughput screen adapted from the synthetic genetic array methodology. Query strains were mated to the deletion strains. Selected haploid strains were analyzed by flow cytometry to measure the UPR activation (GFP) normalized to cytosolic mCherry. (B) Venn diagram depicting number of deletion mutants expressing IRE1 or IRE1ΔLD. Shown are the number of strains giving fold changes that were ≥1.5 compared with the median GFP/mCherry signal. (C) UPR reporter levels of up-regulated hits in both IRE1 and IRE1ΔLD query strains. Black lined circles are previously reported deletion mutants that activated the UPR in IRE1 query strain. (D and E) UPR reporter levels of up-regulated hits in either IRE1 (D) or IRE1ΔLD (E) query strains.
A high-throughput screen reveals gene clusters that mount a UPR response in IRE1ΔLD cells. (A) Strategy of the genome-wide high-throughput screen adapted from the synthetic genetic array methodology. Query strains were mated to the deletion strains. Selected haploid strains were analyzed by flow cytometry to measure the UPR activation (GFP) normalized to cytosolic mCherry. (B) Venn diagram depicting number of deletion mutants expressing IRE1 or IRE1ΔLD. Shown are the number of strains giving fold changes that were ≥1.5 compared with the median GFP/mCherry signal. (C) UPR reporter levels of up-regulated hits in both IRE1 and IRE1ΔLD query strains. Black lined circles are previously reported deletion mutants that activated the UPR in IRE1 query strain. (D and E) UPR reporter levels of up-regulated hits in either IRE1 (D) or IRE1ΔLD (E) query strains.
To test our reporter system, the query strains were treated with the proteotoxic stress-inducing reagent DTT. The fluorescence signal ratio was increased significantly in WT Ire1-expressing cells but not Ire1ΔLD-expressing cells (Fig. S1, B and C). Thus, DTT-induced UPR requires the LD of Ire1. To uncouple UPR activation by UPRLBS from UPRPT, we deleted STE24, SPC2, SCJ1, and GET1, genes known to induce LBS (Promlek et al., 2011). As expected, deletion of these genes activated the UPR independently of the LD of Ire1 (Fig. S1, D and E). Interestingly, these four genes have roles in protein folding and translocation (Antonin et al., 2000; Ast et al., 2016; Schuldiner et al., 2008; Silberstein et al., 1998), suggesting that lipid composition can modulate protein folding and/or protein trafficking through the ER. These genes are therefore required for proper ER quality control as a result of UPRLBS. Together, these data demonstrate that the query strains selectively report UPR activation through UPRLBS and UPRPT.
We mated query strains expressing WT Ire1 or Ire1ΔLD with 4,847 strains from a yeast deletion library (Giaever et al., 2002) using synthetic genetic array methodology (Tong et al., 2001; Fig. 1 A and Table S1). We identified 629 and 958 gene deletions that activate the UPR in an Ire1 LD-dependent and -independent manner, respectively (Fig. 1 B). We identified known pathways that are required for protein homeostasis, which specifically activates UPRPT in the IRE1 strain but not the IRE1ΔLD strain. For example, the deletion of genes involved in the synthesis and transfer of N-linked glycosylation induced the UPR in the IRE1 strain, whereas no significant UPR activation was observed in the IRE1ΔLD strain. Dolichol-linked oligosaccharide synthesis genes ALG3, ALG6, and ALG8 and oligosaccharyltransferase complex gene OST3 also specifically induced UPRPT (Fig. 1 D and Table S1). Identifying these known UPRPT-associated genes validates our screen.
We found 181 gene deletions that activated the UPR in both query strains, strongly suggesting that the lack of these genes specifically activates the UPR independently of Ire1 LD and thus presumably through LBS (Fig. 1 C and Table S1). As reported (Promlek et al., 2011), we found that loss of ARV1, GET1, PMT2, OPI3, SCJ1, SPC2, and STE24 activated the UPR independently of Ire1 LD, validating our approach (Fig. 1 D). Additionally, we identified the lack of TLG2 activates the UPR, a gene known to cause LBS (Mousley et al., 2008). Vacuolar protein sorting genes VPS8, VPS29, VPS61, VPS63, and VPS72 are also required for ER membrane integrity (Markgraf et al., 2009). Another important process associated with the UPRLBS is the ER-associated protein degradation (ERAD) machinery. The ERAD component Hrd1 forms a ubiquitin-gated protein conducting channel for the retro-translocation of misfolded ER luminal protein across the ER lipid bilayer. Given that Hrd1-Hrd3 contacts the ER membrane bilayer (Schoebel et al., 2017), there is a potential for this complex to regulate changes to the membrane bilayer to buffer UPRLBS. This adds to the protective role of Hrd1 during UPRPT through the degradation of misfolded proteins accumulated in the ER. Together, our findings argue that maintenance of vesicular trafficking and the ERAD pathways are necessary to maintain ER membrane integrity.
Conserved cellular functions are necessary to maintain ER membrane integrity in C. elegans
To identify evolutionarily conserved cellular perturbations linked to UPRLBS, we performed a reverse genetic RNAi screen in the animal model C. elegans. We focused on identifying genes whose inactivation caused UPR activation through metabolic changes. Specifically, we depleted 1,247 predicted metabolic genes (Fig. 2 A and Table S2; Yilmaz and Walhout, 2016; Zhang et al., 2013); empty vector and mediator subunit 15 (mdt-15) RNAi clones served as negative and positive controls, respectively (Hou et al., 2014). To carry out the screen, synchronized stage 1 larvae bearing the IRE-1– and XBP-1– (C. elegans homologue of yeast Hac1) dependent reporter hsp-4p::gfp were subjected to RNAi, and GFP fluorescence was scored after 48 and 72 h (Fig. 2 A). The screen was completed in duplicate, and hits were subsequently confirmed in three independent validation experiments, yielding 34 RNAi clones that reproducibly induced hsp-4p::gfp fluorescence (Fig. 2, B and C; and Table S2). As published, we identified roles for the fatty acid desaturation enzyme fat-6, PC synthesis enzymes (pcyt-1, sams-1), the 3-hydroxy-3-methylglutaryl CoA synthase hmgs-1, and the sarco-endoplasmic reticulum calcium ATPase sca-1, validating our screen (Hou et al., 2014; Koh et al., 2018; Mörck et al., 2009).
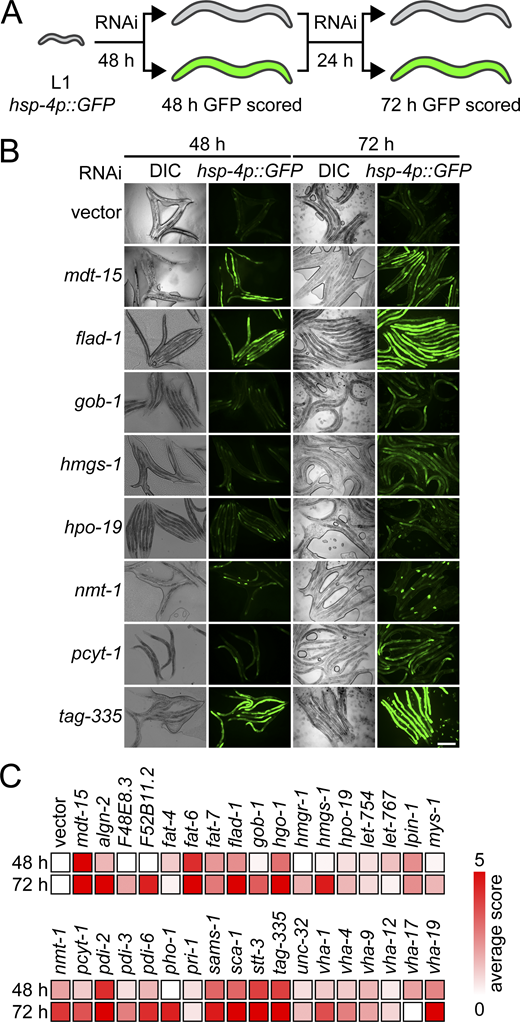
Membrane aberration activating the UPR is conserved in C. elegans. (A) Schematic of the C. elegans screen. Hits were scored as positive when above-background fluorescence was detected at either 48 h or 72 h. (B) Representative fluorescence and DIC micrographs show hsp-4p::gfp worms at 48 h and 72 h on RNAi bacteria. Vector refers to the empty RNAi vector negative control, mdt-15 serves as positive control. Scale bar, 200 µm. DIC, differential interference contrast. (C) Heat map of average scores reflecting hsp-4p::gfp fluorescence intensity. Average score is aggregated from two screen and three validation experiments (see Materials and methods and Table S2).
Membrane aberration activating the UPR is conserved in C. elegans. (A) Schematic of the C. elegans screen. Hits were scored as positive when above-background fluorescence was detected at either 48 h or 72 h. (B) Representative fluorescence and DIC micrographs show hsp-4p::gfp worms at 48 h and 72 h on RNAi bacteria. Vector refers to the empty RNAi vector negative control, mdt-15 serves as positive control. Scale bar, 200 µm. DIC, differential interference contrast. (C) Heat map of average scores reflecting hsp-4p::gfp fluorescence intensity. Average score is aggregated from two screen and three validation experiments (see Materials and methods and Table S2).
We confirmed reliance on canonical IRE-1 signaling by monitoring fluorescence after RNAi in a strain lacking XBP-1 (xbp-1; hsp-4p::gfp), and found that all 34 clones required xbp-1 for induction (Table S2). Next, we confirmed induction of the endogenous UPR in WT worms using real-time PCR, quantifying the mRNA levels of hsp-4 and Y41C4A.11 (a coatomer protein complex subunit), both targets of the IRE-1 branch (Fig. S1, F and G). Testing 15 of 34 RNAi clones, we found that 11 induced at least either gene at 44 h post-RNAi treatment (note that in our screen, the other four were classified as nonhits at this time point; Table S2). Last, we tested whether choline supplementation, which suppresses UPRLBS activation in worms defective for PC synthesis through the cytidine diphosphate diacylglycerol (CDP-DAG) pathway (Hou et al., 2014; Koh et al., 2018), is sufficient to suppress UPR activation. We observed partial rescue of RNAi clones hmgs-1, pdi-2, and vha-4 as well as the expected complete rescue of sams-1 RNAi-treated animals, which are unable to synthesize PC through this pathway (Table S2). Thus, 30 of 34 hits likely induce the UPR without dramatically altering PC levels.
We next tested whether the genes identified in the yeast screen are linked to UPRLBS activation in C. elegans. Of 181 genes whose inactivation induced the UPR in both IRE1 WT and IRE1ΔLD yeast strains, we tested 38 and found that RNAi inactivation of one, the signal peptidase complex subunit homologue spcs-2, activated the hsp-4p::gfp reporter (Table S3). We also compared the 181 candidates from the yeast screen to the 34 candidates from our C. elegans screen to identify evolutionarily conserved processes or pathways whose impairment activates the UPR in both species. Some genes whose inactivation induced the UPR in C. elegans are essential in yeast (e.g., fatty acid desaturation genes OLE1, protein disulfide isomerase PDI1), thus preventing us from assessing their conservation. However, the impairment of Ole1 activation by Ubx2 loss induces the UPR via increased saturated membrane lipids (Surma et al., 2013). Notably, inactivation of genes in several other pathways resulted in robust UPRLBS induction across species, for example, genes involved in PC synthesis, genes encoding the vacuolar H+-ATPase, and several related metabolic genes (Table S4).
Phospholipid perturbation activates Ire1 independently of its LD
As decrease in PC levels activated the UPR in both yeast and C. elegans, we studied how Ire1 is activated in yeast cells lacking the PC biosynthesis gene OPI3, which display a severe PC imbalance (Shyu et al., 2019; Thibault et al., 2012). Increased or decreased PC/PE ratios in membranes are linked to ER stress and UPR activation in several organisms (Fu et al., 2011; Hou et al., 2014; Koh et al., 2018; Li et al., 2006; Thibault et al., 2012). Therefore, we used opi3Δ cells to delineate the mechanisms that sense PC depletion and to identify pertinent downstream pathways activated by Ire1.
To find out whether the Ire1ΔLD remains an integral ER membrane protein during PC depletion, we assessed its subcellular localization by indirect immunofluorescence. Ire1-HA and Ire1ΔLD-HA colocalized with the ER marker Kar2 in both mutants (Fig. S2 A). We further validated the integration of Ire1ΔLD into the ER membrane by alkaline carbonate extraction from the membrane (Wang et al., 2011). Ire1 and Ire1ΔLD were both present in the pellet fraction, together with the ER-localized transmembrane protein Emc4, indicating proper integration (Fig. S2 B).

Ire1 and Ire1ΔLD are properly integrated into the ER membrane. (A) Cells were grown to early log phase in selective synthetic complete media before being fixed in formaldehyde and permeabilized. Staining was performed using anti-HA and anti-Kar2 primary antibodies. Scale bar, 5 µm. (B) Membrane prepared from the indicated cells were treated with 0.1 M sodium carbonate, pH 11, for 30 min on ice. A portion was kept as the total fraction (T), and the remaining was subjected to centrifugation at 100,000 g. Supernatant (S) and membrane pellet (P) fractions were collected and analyzed by immunoblotting. Proteins were detected using antibodies against HA. Kar2 and Sec61 serve as soluble and integral membrane protein controls, respectively. The asterisk indicates unspecific bands. (C) Cells were grown and treated as in A. (D) Ire1-HA protein levels upon 4-PBA treatment. Proteins were separated by SDS-PAGE and detected by immunoblotting with antibodies against the HA tag and Tub1 as loading control. (E) Cells expressing Ire1-mNG or Ire1ΔLD-mNG were depleted of inositol (-inositol). Cells also expressed the dsRed-HDEL ER marker. (F) Mutant strains get1Δ, scj1Δ, and ste24Δ treated as in E. Scale bars, 5 µm. Images shown are representative of three independent experiments.
Ire1 and Ire1ΔLD are properly integrated into the ER membrane. (A) Cells were grown to early log phase in selective synthetic complete media before being fixed in formaldehyde and permeabilized. Staining was performed using anti-HA and anti-Kar2 primary antibodies. Scale bar, 5 µm. (B) Membrane prepared from the indicated cells were treated with 0.1 M sodium carbonate, pH 11, for 30 min on ice. A portion was kept as the total fraction (T), and the remaining was subjected to centrifugation at 100,000 g. Supernatant (S) and membrane pellet (P) fractions were collected and analyzed by immunoblotting. Proteins were detected using antibodies against HA. Kar2 and Sec61 serve as soluble and integral membrane protein controls, respectively. The asterisk indicates unspecific bands. (C) Cells were grown and treated as in A. (D) Ire1-HA protein levels upon 4-PBA treatment. Proteins were separated by SDS-PAGE and detected by immunoblotting with antibodies against the HA tag and Tub1 as loading control. (E) Cells expressing Ire1-mNG or Ire1ΔLD-mNG were depleted of inositol (-inositol). Cells also expressed the dsRed-HDEL ER marker. (F) Mutant strains get1Δ, scj1Δ, and ste24Δ treated as in E. Scale bars, 5 µm. Images shown are representative of three independent experiments.
To determine if Ire1ΔLD is sufficient to rescue the synthetic lethality of an ire1Δopi3Δ double mutant, we performed growth assays. As expected, both ire1Δ and ire1Δopi3Δ strains expressing WT IRE1 grew in the presence of the proteotoxic stress inducer tunicamycin (Tm; Fig. 3 A). Similar to ire1Δ cells, cells lacking the LD of IRE1 failed to survive during Tm-induced proteotoxic stress. In contrast, the expression of IRE1ΔLD was sufficient to rescue the synthetic lethality of ire1Δopi3Δ. opi3ΔIRE1ΔLD cells displayed an exacerbated growth rate during proteotoxic stress, potentially because these cells failed to further up-regulate the UPR by the additional source of ER stress. We conclude that Ire1 LD is dispensable for restoring ER homeostasis during LBS.

Ire1LD is sufficient to uncouple the UPR activation triggered by LBS and proteotoxic stress. (A) ire1Δ and ire1Δopi3Δ strains expressing IRE1 or IRE1ΔLD (IΔLD) were grown at 30°C and serial dilutions of the culture were spotted onto synthetic complete selective medium supplemented with 0.25 µg/ml Tm, when indicated, and incubated until the appearance of colonies. (B) RT-PCR of unspliced (HAC1U) and spliced (HAC1S) HAC1 mRNA. Media was supplemented with 1 mM choline (cho) or incubated 1 h with 1 mM DTT, when indicated. Actin (ACT1) was used as loading control. Percentages (%) of HAC1S indicated below the image are of three independent experiments. (C–E) UPR induction was measured using a β-galactosidase reporter assay of indicated strains (C) with the overexpression of IRE1 luminal domain (OE IRE1LD; D), or preincubated with 1 mM 4-PBA (E). (F) qPCR results comparing percentages of HAC1S of samples treated as in E. Data shown are the mean ± SEM (n = 3). Statistical analysis was subjected to paired two-tailed Student’s t test; ns, not significant.
Ire1LD is sufficient to uncouple the UPR activation triggered by LBS and proteotoxic stress. (A) ire1Δ and ire1Δopi3Δ strains expressing IRE1 or IRE1ΔLD (IΔLD) were grown at 30°C and serial dilutions of the culture were spotted onto synthetic complete selective medium supplemented with 0.25 µg/ml Tm, when indicated, and incubated until the appearance of colonies. (B) RT-PCR of unspliced (HAC1U) and spliced (HAC1S) HAC1 mRNA. Media was supplemented with 1 mM choline (cho) or incubated 1 h with 1 mM DTT, when indicated. Actin (ACT1) was used as loading control. Percentages (%) of HAC1S indicated below the image are of three independent experiments. (C–E) UPR induction was measured using a β-galactosidase reporter assay of indicated strains (C) with the overexpression of IRE1 luminal domain (OE IRE1LD; D), or preincubated with 1 mM 4-PBA (E). (F) qPCR results comparing percentages of HAC1S of samples treated as in E. Data shown are the mean ± SEM (n = 3). Statistical analysis was subjected to paired two-tailed Student’s t test; ns, not significant.
To monitor the UPR, we assayed HAC1 mRNA splicing (HAC1S), which is mediated by activated Ire1. Under DTT-induced proteotoxic stress, HAC1 mRNA was only spliced in ire1Δ cells expressing IRE1, whereas the expression of IRE1ΔLD failed to generate HAC1S (Fig. 3 B). Corroborating our growth assays, IRE1ΔLD was sufficient for HAC1 mRNA splicing in ire1Δopi3Δ cells during LBS. Choline supplementation inhibited HAC1 splicing, presumably by alleviating LBS through the PC synthesis CDP-choline pathway, suggesting that the UPR is specifically activated by a decrease in PC. To further validate our UPRLBS model, we monitored UPR activation using the UPRE-LacZ reporter (Cox and Walter, 1996). As expected, Ire1 LD was necessary to induce UPRPT but dispensable during UPRLBS (Fig. 3 C). We note that UPR activation in opi3ΔIRE1ΔLD was about half of that seen in opi3ΔIRE1, suggesting the strong UPR activation in opi3Δ cells is a combination of proteotoxic- and LBS-induced ER stress, consistent with our growth assay (Fig. 3 A). In sum, our data show that LBS directly activates Ire1 independently of its LD to support cell survival.
To further uncouple the contribution of UPRLBS and UPRPT to the overall UPR program, we titrated the accumulation of misfolded proteins in the ER lumen by overexpressing (OE) IRE1 (IRE1LD) in the ER (Fig. S2 A; Korennykh et al., 2009; Liu et al., 2000, 2002). In WT cells, OE IRE1LD was sufficient to attenuate UPR activation upon Tm treatment (Fig. 3 D). This indicates that OE IRE1LD prevents UPRPT. Similarly, in opi3Δ cells, OE IRE1LD reduced UPR activation by 39.1% compared with empty vector. This level of UPR attenuation is comparable to the 48.2% reduction observed from IRE1 to IRE1ΔLD expression in opi3Δ cells (Fig. 3 C). These findings suggest that Ire1 senses both the accumulation of misfolded proteins and LBS, resulting in a strong activation of the UPR in opi3Δ cells. Together, our data suggest that IRE1LD binds unfolded proteins in the ER lumen to partially inhibit UPR activation.
Next, we asked if 4-phenylbutyric acid (4-PBA) attenuates the UPR activation in opi3Δ cells. 4-PBA inhibits the aggregation of proteins and consequently suppresses the UPR (Kubota et al., 2006; Le et al., 2016; Ozcan et al., 2006; Pineau et al., 2009). More recently, 4-PBA was shown to attenuate ER retention via COPII and consequently clearing the ER of misfolded proteins (Ma et al., 2017). We monitored UPR activation using the UPRE-LacZ reporter and the HAC1 mRNA splicing assay. As expected, 4-PBA attenuated Tm-induced UPR in IRE1 cells (Fig. 3, E and F). In opi3ΔIRE1 cells, 4-PBA failed to reduce the UPR. In contrast to a previous report that 4-PBA accelerates Ire1 degradation (Mai et al., 2018), Ire1 protein levels were unchanged in IRE1 and opi3ΔIRE1 cells upon 4-PBA treatment (Fig. S2 D). These findings reinforce the notion that LBS is the main driver of the UPR in opi3Δ.
LBS-activated Ire1 induces the UPR independently of its oligomeric state
An Ire1 variant missing the misfolded protein binding region in the LD, ΔIII Ire1, clusters into puncta upon inositol depletion in yeast (Halbleib et al., 2017; Promlek et al., 2011), but with noticeable fewer puncta compared with proteotoxic stress, suggesting lower levels of dimerization in LBS. However, whether Ire1 lacking its entire LD retains its oligomerization property remains unknown (Kimata et al., 2007). To monitor Ire1 clusters, we inserted HA tag with ymNeonGreen between the Ire1 kinase domain residues I571 and G572 (Ire1-mNG), as previously reported (Aragón et al., 2009; Botman et al., 2019). To validate Ire1 variants, we assayed HAC1 mRNA splicing. Ire1-mNG was sufficient to induce HAC1S upon DTT treatment, upon inositol depletion, and in opi3Δ cells, while ire1Δopi3Δ cells expressing Ire1ΔLD-mNG displayed HAC1S accumulation (Fig. 4 A). Ire1-mNG was uniformly distributed and colocalized with the ER marker dsRed-HDEL in ire1Δ cells (Fig. 4 B). As expected, we detected Ire1-mNG clusters as bright punctate structures at the ER upon DTT treatment. Unexpectedly, in ire1Δopi3Δ cells, puncta were absent with both Ire1-mNG and Ire1ΔLD-mNG, while Ire1-mNG formed fewer puncta compared with ire1Δ cells in response to DTT. To assess this discrepancy with previous reports, LBS was induced with a 4-h inositol depletion in ire1Δ cells. Few puncta were detected with Ire1-mNG reporter but not with Ire1ΔLD-mNG (Figs. 4 C and,S2 E). Consistent with the lack of clusters observed in opi3Δ cells, get1Δ, scj1Δ, and ste24Δ mutants showed the absence of fluorescent puncta (Fig. 4 D). As expected, clusters of Ire1 were evident in these mutants upon DTT treatment (Fig. S2 F). To further validate our findings, we used a pair of split Venus fragments to monitor Ire1 clusters by bimolecular fluorescence complementation assay (BiFC; Robida and Kerppola, 2009). Clusters of Ire1 were observed only upon DTT treatment (Fig. S3). Together, these findings suggest that the formation of large Ire1 clusters is mostly driven by proteotoxic stress and that Ire1ΔLD is unable to form oligomers.

Ire1 forms clusters during proteotoxic stress that are absent during LBS. (A) RT-PCR of unspliced (HAC1U) and spliced (HAC1S) HAC1 mRNA. Media was depleted of inositol, supplemented with 1 mM choline (cho) or incubated 1 h with 1 mM DTT, when indicated. Actin (ACT1) was used as loading control. Percentages (%) of HAC1S indicated below the image are of three independent experiments. (B and C) Cells expressing Ire1-mNG or Ire1ΔLD-mNG (IΔLD-mNG) were treated 1 h with 10 mM DTT (B) or depleted of inositol (-inositol; C), when indicated. dsRed-HDEL was used as ER marker. (D)get1Δ, scj1Δ, and ste24Δ mutants expressing Ire1-mNG were treated as in B. Scale bar, 5 µm. Images shown are representatives of three independent experiments. Statistical analysis was subjected to paired two-tailed Student’s t test.
Ire1 forms clusters during proteotoxic stress that are absent during LBS. (A) RT-PCR of unspliced (HAC1U) and spliced (HAC1S) HAC1 mRNA. Media was depleted of inositol, supplemented with 1 mM choline (cho) or incubated 1 h with 1 mM DTT, when indicated. Actin (ACT1) was used as loading control. Percentages (%) of HAC1S indicated below the image are of three independent experiments. (B and C) Cells expressing Ire1-mNG or Ire1ΔLD-mNG (IΔLD-mNG) were treated 1 h with 10 mM DTT (B) or depleted of inositol (-inositol; C), when indicated. dsRed-HDEL was used as ER marker. (D)get1Δ, scj1Δ, and ste24Δ mutants expressing Ire1-mNG were treated as in B. Scale bar, 5 µm. Images shown are representatives of three independent experiments. Statistical analysis was subjected to paired two-tailed Student’s t test.

Ire1 clusters are mostly driven by DTT-induced proteotoxic stress. (A and B) Cells were grown to early log phase in selective synthetic complete media before being fixed in formaldehyde and permeabilized. Staining was performed using anti-HA (A) or anti-FLAG (B) and anti-Kar2 primary antibodies. Scale bar, 5 µm. (C) RT-PCR of unspliced HAC1 (HAC1U) and spliced (HAC1S) mRNA. Media was depleted of inositol (-inositol), supplemented with 1 mM choline (cho) or incubated 1 h with 1 mM DTT, when indicated. Actin (ACT1) was used as loading control. (D and E) Cells coexpressing the pair of split Venus fragments to monitor IRE1-HA-VN173 and IRE1-FLAG-VC155 or IRE1ΔLD-HA-VN173 and IRE1ΔLD-FLAG-VC155 to dimerization in vivo by BiFC in ire1Δ and ire1Δopi3Δ. Cells were treated 1 h with 10 mM DTT (D) or depleted of inositol (-inositol; E), when indicated. CFP-HDEL was used as ER marker. Red arrowheads indicate Ire1 puncta. (F) Mutant strains get1Δ, scj1Δ, and ste24Δ treated as in D.
Ire1 clusters are mostly driven by DTT-induced proteotoxic stress. (A and B) Cells were grown to early log phase in selective synthetic complete media before being fixed in formaldehyde and permeabilized. Staining was performed using anti-HA (A) or anti-FLAG (B) and anti-Kar2 primary antibodies. Scale bar, 5 µm. (C) RT-PCR of unspliced HAC1 (HAC1U) and spliced (HAC1S) mRNA. Media was depleted of inositol (-inositol), supplemented with 1 mM choline (cho) or incubated 1 h with 1 mM DTT, when indicated. Actin (ACT1) was used as loading control. (D and E) Cells coexpressing the pair of split Venus fragments to monitor IRE1-HA-VN173 and IRE1-FLAG-VC155 or IRE1ΔLD-HA-VN173 and IRE1ΔLD-FLAG-VC155 to dimerization in vivo by BiFC in ire1Δ and ire1Δopi3Δ. Cells were treated 1 h with 10 mM DTT (D) or depleted of inositol (-inositol; E), when indicated. CFP-HDEL was used as ER marker. Red arrowheads indicate Ire1 puncta. (F) Mutant strains get1Δ, scj1Δ, and ste24Δ treated as in D.
Ire1 arginine 537 (R537) is necessary for LBS-activated UPR
Two residues in the amphipathic helix of Ire1 near the transmembrane helix are necessary to activate the UPR (Halbleib et al., 2017) but likely disrupt Ire1’s secondary structure (Fig. 5 A). Therefore, we used a different approach to assess the role of Ire1’s transmembrane helical domain in sensing LBS (van Anken et al., 2014). Most of the Ire1 transmembrane α-helix amino acid sequence is hydrophobic, while two residues in the core are charged. We hypothesized that the arginine residue at the interface of the amphipathic and transmembrane helices (R537) may act as an LBS sensor and thus mutated it to glutamine [Ire1(R537Q)]. As expected, Ire1(R537Q) localized to the ER in both WT and opi3Δ cells (Fig. S4). To validate the functionality of Ire1(R537Q) during proteotoxic stress, we performed a spotting assay (Fig. 5 B). Both IRE1(R537Q) and IRE1 exhibited similar growth on synthetic complete media supplemented with Tm. Next, we measured the splicing of HAC1 mRNA by RT-PCR (Fig. 5 C). During LBS in opi3Δ cells, we found no significant HAC1 mRNA splicing in opi3ΔIRE1(R537Q) cells compared with WT, while Ire1 WT and Ire1(R537Q) spliced HAC1 mRNA at similar levels upon Tm treatment. We detected no HAC1 mRNA splicing by Ire1ΔLD(R537Q) during proteotoxic stress or LBS.

A key Ire1 arginine residue is critical to sense LBS. (A) Ire1-predicted amphipathic and transmembrane helices are highlighted in gray and orange, respectively. Point mutations highlighted in green were previously reported to be important in sensing both proteotoxic stress and LBS (Halbleib et al., 2017), and the point mutation in purple is part of this study. (B) The strains were grown, and serial dilutions of the culture were spotted onto synthetic complete selective medium supplemented with 0.25 µg/ml Tm or 1 mM choline (cho), when indicated, and incubated until the appearance of colonies. (C) RT-PCR of unspliced (HAC1U) and spliced (HAC1S) HAC1 mRNA. Media was supplemented with 1 mM choline or incubated 1 h with 2.5 µg/ml Tm, when indicated. Actin (ACT1) was used as loading control. Percentages of HAC1S indicated below the image are of three independent experiments. (D) qPCR results comparing KAR2. Media were supplemented with 1 mM choline or incubated 1 h with 2.5 µg/ml Tm, when indicated. Images shown are representatives of three independent experiments. (E) UPR induction was measured using a β-galactosidase reporter assay of indicated strains. Data shown are the mean ± SEM (n = 3). Statistical analysis was subjected to paired two-tailed Student’s t test.
A key Ire1 arginine residue is critical to sense LBS. (A) Ire1-predicted amphipathic and transmembrane helices are highlighted in gray and orange, respectively. Point mutations highlighted in green were previously reported to be important in sensing both proteotoxic stress and LBS (Halbleib et al., 2017), and the point mutation in purple is part of this study. (B) The strains were grown, and serial dilutions of the culture were spotted onto synthetic complete selective medium supplemented with 0.25 µg/ml Tm or 1 mM choline (cho), when indicated, and incubated until the appearance of colonies. (C) RT-PCR of unspliced (HAC1U) and spliced (HAC1S) HAC1 mRNA. Media was supplemented with 1 mM choline or incubated 1 h with 2.5 µg/ml Tm, when indicated. Actin (ACT1) was used as loading control. Percentages of HAC1S indicated below the image are of three independent experiments. (D) qPCR results comparing KAR2. Media were supplemented with 1 mM choline or incubated 1 h with 2.5 µg/ml Tm, when indicated. Images shown are representatives of three independent experiments. (E) UPR induction was measured using a β-galactosidase reporter assay of indicated strains. Data shown are the mean ± SEM (n = 3). Statistical analysis was subjected to paired two-tailed Student’s t test.

Ire1 and Ire1ΔLD containing the transmembrane mutation R537Q are properly localized into the ER membrane. Cells were grown to early log phase in selective synthetic complete media before being fixed in formaldehyde and permeabilized. Staining was performed using anti-FLAG and anti-Kar2 primary antibodies. Scale bar, 5 µm. Images shown are representative of three independent experiments.
Ire1 and Ire1ΔLD containing the transmembrane mutation R537Q are properly localized into the ER membrane. Cells were grown to early log phase in selective synthetic complete media before being fixed in formaldehyde and permeabilized. Staining was performed using anti-FLAG and anti-Kar2 primary antibodies. Scale bar, 5 µm. Images shown are representative of three independent experiments.
To further validate the role of IRE1(R537Q), we monitored KAR2 mRNA expression, which is induced through the Ire1-Hac1 axis upon ER stress (Fig. 5 D). As expected, KAR2 was significantly up-regulated upon Tm treatment in IRE1 WT and IRE1(R537Q) cells. In contrast, Ire1ΔLD(R537Q) failed to up-regulate KAR2 in opi3Δ cells compared with opi3Δ cells treated with choline, validating the requirement of Ire1 R537 in sensing LBS. This is the first demonstration that LBS sensing by Ire1 can be disrupted while retaining Ire1 activation by proteotoxic stress. Interestingly, HAC1S was undetected in IRE1ΔLD(R537Q) upon inositol depletion, suggesting that the lack of inositol induces proteotoxic stress and LBS (Fig. 5 E). Although the role of R537 in sensing LBS is unlikely to be conserved in higher organisms, positively charged lysine residues are usually found at the edge of the transmembrane helical domain of IRE1 in metazoans, suggesting similar mechanistic uncoupling of stresses in these proteins.
A novel gene subset is specifically up-regulated by LBS-induced UPR
The UPR activates a broad compensatory response during ER stress. To restore ER homeostasis, UPR-activated genes may vary depending on the source of stress (Fun and Thibault, 2019; Thibault et al., 2011). To explore the LBS-induced UPR transcriptional program, we performed a DNA microarray analysis in ire1Δ and ire1Δopi3Δ cells expressing either IRE1 or IRE1ΔLD. We induced proteotoxic stress with DTT and found 264 genes that were up-regulated compared with unstressed WT IRE1 and IRE1ΔLD (Fig. 6 A, Fig. S5, and Table S5). Therefore, these genes are modulated independently of Ire1LD.

A subset of genes is up-regulated by Hac1 specifically during LBS. (A) Diagram representing up-regulated transcriptional targets of IRE1 and IRE1ΔLD cells treated 1 h with 1 mM DTT, opi3ΔIRE1, and opi3ΔIRE1ΔLD cells. UpSet plot highlights intersections of selected group of genes differentially regulated during proteotoxic stress or LBS. Shown are number of genes giving fold changes that were >1.5 and at P < 0.05 (one-way ANOVA). (B) Bar plot of the GO analysis of genes up-regulated in color-coded conditions as in A. Genes are highlighted in yellow in Table S6. TPP, thiamine pyrophosphate. (C) Heat maps of selected LBS-induced genes. Based on log2 fold changes in gene expression normalized to untreated IRE1 strain. Highlighted in green are genes containing a predicted UPRE within the promoter region. (D) Potential Hac1 binding sites of different UPRE motifs within the promoter region of highlighted genes in C. (E–K) ChIP-qPCR validation of predicted HAC1 binding sites within the promoter regions of KAR2 (E), ACA1 (F), GAC1 (G), GPH1 (H), PBI2 (I), PIR3 (J), and PUT1 (K). IRE1 cells were treated with 2.5 µg/ml of Tm, and opi3ΔIRE1 cells were supplemented with 1 mM choline (cho), when indicated. (L and M) Time-course qPCR comparing relative mRNA levels of spliced HAC1 (HAC1S) mRNA (L) or UPR target genes (M) in IRE1 cells treated 4 h with 1 mM DTT or 24 h depletion of choline in opi3ΔIRE1 cells. Data shown are the mean ± SEM (n = 3). Statistical analysis was subjected to paired one-tailed Student’s t test.
A subset of genes is up-regulated by Hac1 specifically during LBS. (A) Diagram representing up-regulated transcriptional targets of IRE1 and IRE1ΔLD cells treated 1 h with 1 mM DTT, opi3ΔIRE1, and opi3ΔIRE1ΔLD cells. UpSet plot highlights intersections of selected group of genes differentially regulated during proteotoxic stress or LBS. Shown are number of genes giving fold changes that were >1.5 and at P < 0.05 (one-way ANOVA). (B) Bar plot of the GO analysis of genes up-regulated in color-coded conditions as in A. Genes are highlighted in yellow in Table S6. TPP, thiamine pyrophosphate. (C) Heat maps of selected LBS-induced genes. Based on log2 fold changes in gene expression normalized to untreated IRE1 strain. Highlighted in green are genes containing a predicted UPRE within the promoter region. (D) Potential Hac1 binding sites of different UPRE motifs within the promoter region of highlighted genes in C. (E–K) ChIP-qPCR validation of predicted HAC1 binding sites within the promoter regions of KAR2 (E), ACA1 (F), GAC1 (G), GPH1 (H), PBI2 (I), PIR3 (J), and PUT1 (K). IRE1 cells were treated with 2.5 µg/ml of Tm, and opi3ΔIRE1 cells were supplemented with 1 mM choline (cho), when indicated. (L and M) Time-course qPCR comparing relative mRNA levels of spliced HAC1 (HAC1S) mRNA (L) or UPR target genes (M) in IRE1 cells treated 4 h with 1 mM DTT or 24 h depletion of choline in opi3ΔIRE1 cells. Data shown are the mean ± SEM (n = 3). Statistical analysis was subjected to paired one-tailed Student’s t test.

Transcriptional changes reveal LD-specific strategies in coping with different stresses. (A) Pearson correlation of relative RNA abundance in IRE1 and IRE1ΔLD treated with 2 mM DTT. (B) Pearson correlation of relative RNA abundance in opi3ΔIRE1 and opi3ΔIRE1ΔLD. (C) Pearson correlation of relative RNA abundance in IRE1 treated with 2 mM DTT and opi3ΔIRE1. (D) Pearson correlation of relative RNA abundance in IRE1ΔLD treated with 2 mM DTT and opi3ΔIRE1ΔLD. (E) Pearson correlation of relative RNA abundance ratio of IRE1/IRE1ΔLD treated with 2 mM DTT and opi3ΔIRE1/opi3ΔIRE1ΔLD. (F) Pearson correlation of relative RNA abundance in opi3ΔIRE1 and opi3ΔIRE1ΔLD supplemented with 1 mM choline.
Transcriptional changes reveal LD-specific strategies in coping with different stresses. (A) Pearson correlation of relative RNA abundance in IRE1 and IRE1ΔLD treated with 2 mM DTT. (B) Pearson correlation of relative RNA abundance in opi3ΔIRE1 and opi3ΔIRE1ΔLD. (C) Pearson correlation of relative RNA abundance in IRE1 treated with 2 mM DTT and opi3ΔIRE1. (D) Pearson correlation of relative RNA abundance in IRE1ΔLD treated with 2 mM DTT and opi3ΔIRE1ΔLD. (E) Pearson correlation of relative RNA abundance ratio of IRE1/IRE1ΔLD treated with 2 mM DTT and opi3ΔIRE1/opi3ΔIRE1ΔLD. (F) Pearson correlation of relative RNA abundance in opi3ΔIRE1 and opi3ΔIRE1ΔLD supplemented with 1 mM choline.
In opi3Δ cells, 214 genes were exclusively up-regulated during LBS (Fig. 6 A, red). These genes are involved in transmembrane and glucose transport, aerobic respiration, and mitophagy (Fig. 6 B). Interestingly, ATG11 and ATG32, required for autophagy and ER-phagy, were up-regulated, suggesting a role of ER-phagy in buffering UPRLBS (Fregno and Molinari, 2018). However, a gene subset is likely up-regulated independently of the UPR, as we previously reported during PC depletion in yeast and C. elegans (Koh et al., 2018; Thibault et al., 2012).
To identify genes specifically regulated via the UPRLBS, cells lacking IRE1 or HAC1 could not be studied because of the synthetic lethality with opi3Δ (Costanzo et al., 2010; Thibault et al., 2011). Instead, we further analyzed the data to get around this hurdle. First, we used the gene ontology (GO) tool DAVID to functionally annotate up-regulated genes in opi3ΔIRE1 and/or opi3ΔIRE1ΔLD, and identified genes also induced in Ire1 cells treated with DTT (Table S6). These 77 genes, in part, mount a response to stress pathways, including UPR target gene DER1 (Fig. 6 B; Leber et al., 2004). Remodeling of the proteome and UPR activation occurs in opi3Δ mutants, and we confirmed the up-regulation of known target genes such as SEC62 and SEC72 in ire1Δopi3Δ cells expressing Ire1 and Ire1ΔLD (Table S6). Manual inspection of our microarray data showed that known UPR target genes are strongly up-regulated in opi3ΔIRE1ΔLD, as in our previous studies (Koh et al., 2018; Thibault et al., 2012; Travers et al., 2000). The exclusion of known UPR target genes within this cluster confirmed 139 genes were activated by LBS. Genes involved in ER stress and cellular stress resistance were strongly elevated in response to LBS. These include SRX1 and HSP33, required for oxidative stress resistance (Dahl et al., 2015), and PDH1, activated by diauxic shift (Fig. 6 C; Ohlmeier et al., 2004). Other genes involved in DNA replicative stress were up-regulated, including glycogen degradation gene GDB1, carbohydrate metabolism gene CAR2, endopeptidase inhibitor activity gene PBI2, and transporter gene CYC7.
Next, we analyzed genes that are differentially regulated between opi3ΔIRE1ΔLD and opi3ΔIRE1 strains. This group of genes is potentially up-regulated in response to UPRPT. For instance, the gene encoding the transporter SEC24 was up-regulated in a LD-dependent manner, suggesting its up-regulation is only during UPRPT (Iwasaki et al., 2015). CIS1 encodes a protein required for autophagosome formation and is of interest because autophagy is required for cellular homeostasis during the UPR (Koh et al., 2018).
To better understand how LBS affects the UPR, we further examined Hac1-specific target genes. To induce gene expression, Hac1 binds to three known UPRE consensus sequences (Fordyce et al., 2012). We performed a bioinformatics analysis to identify putative UPRE consensus sequences in the promoters of the differentially regulated genes. We identified six genes containing the predicted UPRE motif (Fig. 6, C and D). These genes include PBI2, carbohydrate metabolism genes ACA1, GAC1, and GPH1, amino acid metabolism gene PUT1, and cell wall synthesis gene PIR3. This suggests that Hac1 promotes metabolic processes to restore cellular homeostasis to compensate for disturbed lipid biosynthesis during LBS. Similarly, genes maintaining cell wall integrity were up-regulated, arguing that cell wall stress occurs during LBS-induced ER stress and pinpointing to the importance of coordinating cell wall biogenesis and the UPR response.
Next, we performed ChIP in ire1Δ and ire1Δopi3Δ mutants expressing HA-Hac1 and Ire1 to confirm Hac1 binding to UPRE motifs. We studied the KAR2 promoter as positive control as it contains a well-characterized UPRE (Fig. 6 E; Fordyce et al., 2012). Association of Hac1 to KAR2 promoter was indeed increased during Tm treatment. During Tm-induced protein stress, qPCR showed the absence of fold enrichment of the five gene promoters GAC1, GPH1, PBI2, PIR3, and PUT1 (Fig. 6, G-K). The ACA1 promoter was significantly bound by Hac1 during protein stress (Fig. 6 F). ACA1 belongs to the family of bZIP proteins (including Hac1) and acts as ATF/CREB activators (Garcia-Gimeno and Struhl, 2000). Our findings, however, suggest that ACA1 is a UPR target gene, possibly through carbon source regulation (Garcia-Gimeno and Struhl, 2000). In contrast, we detected enrichment of the three genes, PBI2, PIR3, and PUT1, that are up-regulated during LBS.
As the UPR is constitutively activated in opi3Δ cells (chronic ER stress), we grew opi3ΔIRE1 cells under unstressed condition by supplementing the media with choline to maintain PC synthesis followed by 24 h choline depletion to induce acute ER stress, which is comparable to 4-h DTT treatment in IRE1 cells. Around 40% HAC1S was detected at 0.5 h and 12 h in IRE1 and opi3ΔIRE1 cells, respectively (Fig. 6 L). Concurrently, only the mRNA level of thiol oxidase ERO1 was significantly up-regulated at 0.5 h compared with 0 h in DTT-treated IRE1 cells, while the mRNA levels of ERO1, PBI2, and PIR3 were increased by 2.8-, 3.8-, and 14.5-fold, respectively, at 12 h compared with 0 h in opi3ΔIRE1 cells (Fig. 6 M). Furthermore, only ERO1 levels correlate with HAC1S levels in IRE1 cells, whereas ERO1, PBI2, and PIR3 levels resemble the increase of HAC1S in opi3ΔIRE1 cells. Together with the ChIP data, these findings strongly argue for the existence of a specific UPRLBS transcriptional program.
Discussion
Recently, yeast Ire1 was shown to contain a sensing domain that monitors LBS at the ER (Halbleib et al., 2017; Promlek et al., 2011). Conserved in higher eukaryotes, IRE1 is similarly activated by LBS together with PERK (Volmer et al., 2013), while ATF6 is activated by an increased level of sphingolipid species (Tam et al., 2018). A detailed LBS-sensing mechanism of yeast Ire1 has revealed that the rotational orientation of its amphipathic helix stabilizes its activation during proteostatic and lipostatic ER stress (Halbleib et al., 2017). To date, LBS has only been demonstrated by few manipulations. As such, the extent of cellular perturbations activating the UPR through LBS was unknown. To address this knowledge gap, we performed large-scale genetic screens and identified a set of genes necessary to maintain ER membrane integrity in yeast and C. elegans (Figs. 1, 2, 3, and 4). Furthermore, we identified a residue at the interface of the amphipathic and transmembrane helices of Ire1 that senses ER membrane integrity while being dispensible for sensing proteotoxic stress (Fig. 5). By uncoupling LBS- and proteotoxic-induced UPR, we demonstrated that the UPR program is a broad-spectrum compensatory pathway with divergent transcriptomes (Fig. 6).
Conical PE and cylindrical PC promote negative intrinsic curvature and minimal membrane curvature, respectively (Ho et al., 2018; Szule et al., 2002; Vance and Tasseva, 2013). The phospholipid intermediate N-monomethyl phosphatidylethanolamine, generated during de novo PC synthesis, exhibits physical properties similar to PE and is highly abundant in opi3Δ cells (Shyu et al., 2019; Thibault et al., 2012). Together, the absence of sterol and the replacement of PC with N-monomethyl phosphatidylethanolamine contribute to membrane stiffening (Dawaliby et al., 2016; Shyu et al., 2019; Subczynski et al., 2017; Weete et al., 2010; Zinser et al., 1993). Conceivably, during LBS, membrane stiffening might prevent the formation of large Ire1 clusters. In contrast, the large accumulation of unfolded proteins in the ER, by Tm or DTT, might be necessary to promote Ire1 clustering through the binding of Ire1 luminal domain during LBS (Credle et al., 2005; Gardner and Walter, 2011; Kimata et al., 2004, 2007). However, no Ire1α homodimers or oligomers were detected by palmitic acid–induced LBS, which is in agreement with our findings (Kitai et al., 2013). Accordingly, nonoligomerized Ire1 spliced HAC1 mRNA during PC depletion (Fig. 4, C and D). Although a dimerization-dependent conformational switch is required to activate the Ire1 RNase domain, Ire1 RNase activity is preserved in the monomer (Rubio et al., 2011; Tam et al., 2014). Kinase-inactive Ire1 splices HAC1 mRNA while displaying defects in Ire1 deactivation (Rubio et al., 2011), leading to chronic ER stress. On the other hand, ER stress–induced clustering might be undetectable by conventional methods during LBS due to low abundance of endogenous Ire1 (Ho et al., 2018). For instance, LBS-induced Ire1 puncta are weaker and less abundant compared with proteotoxic stress–induced puncta, suggesting higher Ire1 oligomerization states during proteotoxic stress (Halbleib et al., 2017; Promlek et al., 2011). Interestingly, mammalian Ire1α transmembrane domain residues W457 and S450 are required for dimerization during membrane saturation by palmitic acid (Cho et al., 2019). Together with our results, this suggests that the Ire1 activation mechanism differs between proteotoxic stress and LBS. These two activation routes might work synergistically or independently to transduce the UPR signal (Fig. 7).

Ire1 signaling pathway induces differential UPR programs to restore ER homeostasis. Upon the accumulation of unfolded proteins or during membrane perturbation, ER stress sensor Ire1 activates downstream transcription factors, resulting in the up-regulation of UPR target genes. Initially a linear stress response pathway, the UPR transcriptional program diverges in response to unresolved ER stress, UPRPT and UPRLBS. The source of stress, acute or chronic, may direct the differential UPR program to restore ER homeostasis by catering to an organism’s specific needs.
Ire1 signaling pathway induces differential UPR programs to restore ER homeostasis. Upon the accumulation of unfolded proteins or during membrane perturbation, ER stress sensor Ire1 activates downstream transcription factors, resulting in the up-regulation of UPR target genes. Initially a linear stress response pathway, the UPR transcriptional program diverges in response to unresolved ER stress, UPRPT and UPRLBS. The source of stress, acute or chronic, may direct the differential UPR program to restore ER homeostasis by catering to an organism’s specific needs.
In C. elegans, we demonstrated that the transcriptome diverges dramatically between UPRPT and UPRLBS, including hundreds of genes up-regulated in an IRE-1–dependent manner during LBS (Koh et al., 2018). Similarly, the mammalian ATF6-modulated UPRLBS program diverged from the UPRPT program (Tam et al., 2018). During ER stress, the Hac1-mediated response is assumed to be linear until ER homeostasis is restored. However, some evidence suggests the contrary in yeast and mammals, particularly during LBS. Previously, we demonstrated that the UPR transcriptome is modulated through differential target gene expression depending on the source of stress, including SCJ1-deficient cells (Thibault et al., 2011). The ablation of Hsp70 cochaperone SCJ1 was identified to induce UPRLBS in our genetic screen (Figs. 1 and S1). In agreement with these findings, the HAC1 mRNA level was enhanced by a bipartite signal, misfolded proteins, and either inositol depletion or temperature shift (Leber et al., 2004). In the presence of one of the latter signals in ire1Δ cells, the levels of HAC1 mRNA more than doubled, indicating an Ire1-independent mechanism maintaining protein quality control, mounting an alternative transcriptional program. This higher magnitude of UPR, termed the “Super-UPR,” could be classified with its own transcriptional program. Additionally, autoregulation of HAC1 occurs during periods of extreme and prolonged ER stress via a positive feedback loop of Hac1 binding to its own promoter (Ogawa and Mori, 2004). Additionally, Gcn4, a transcriptional activator of amino acid biosynthetic genes, works synergistically with Hac1 at the promoter of UPR target genes (Patil et al., 2004). In accordance with these findings, we identified and validated endopeptidase inhibitor activity PBI2 and cell wall synthesis PIR3 as genes that are up-regulated only during UPRLBS, which supports a mechanism of integration of multiple stimuli to mount a divergent transcriptional response by Hac1. Additional unidentified trans-acting factors might regulate Hac1 transcriptional factor and should be explored in future studies. Ire1 faces a delicate balance in response to the stress it encounters and induces a fine-tuned response, i.e., the activation of specific genes to adapt to specific cellular changes (Leber et al., 2004).
The UPR is linked to many diseases including diabetes, nonalcoholic fatty liver disease, liver failure, cystic fibrosis, neurodegenerative diseases, and cancer (Cubillos-Ruiz et al., 2017; Hetz et al., 2013; Wu et al., 2014). Despite mounting evidence on the role of the UPR in metabolic diseases, the contribution of the ER membrane composition in activating the UPR is poorly understood. Our genome-wide genetic screen revealed a wide variety of cellular processes that are necessary to maintain ER membrane integrity (Fig. 1). For instance, UPRLBS was induced by disrupting cellular pathways related to fatty acid and phospholipid biosynthesis, vesicle trafficking, and ER-phagy. Intuitively, low PC- and palmitic acid–induced LBS disrupt ER structure and integrity of which the UPR transcriptional program is essential for cell survival (Borradaile et al., 2006; Thibault et al., 2012). In accordance with our findings, the selective autophagy pathway ER-phagy is required to maintain cellular homeostasis by recycling ER membrane during ER stress (Grumati et al., 2018; Schuck et al., 2014). Similarly, vesicular transport from the ER might be necessary to remove excessive ER membrane. Although lipid synthesis is tightly regulated according to cellular needs, the overall buffering of ER membrane integrity is undoubtedly a coordinated effort of multiple regulatory pathways.
In this report, we show evidence linking the UPR to ER membrane integrity, implicating pathways beyond lipid metabolism. To overcome LBS, the activation mechanism of Ire1, which senses fluctuation at the ER membrane, diverges from the activation mechanism by proteotoxic stress. In addition, through a mechanism that remains unclear, transcription factor Hac1 deploys a curated UPR transcriptional program to restore cellular homeostasis during LBS. Taken together, our data demonstrate the remarkably diverse cellular pathways working in concert with the UPR to maintain ER membrane integrity. How each of the regulatory pathways contribute to UPR-associated metabolic diseases will be the challenge of future studies.
Materials and methods
Strains and antibodies
S. cerevisiae strains used in this study are listed in Table S7. Strains were prepared using standard transformation protocols. Anti-HA mouse monoclonal antibodies HA.11 (MMS-101R-1000, Covance), anti-FLAG mouse M2 monoclonal antibody (F-1804, Sigma-Aldrich), and anti-Tub1 mouse monoclonal antibody (12G10, Developmental Studies Hybridoma Bank) were commercially purchased. Anti-Kar2 rabbit polyclonal was a gift from D. Ng (Temasek Life Sciences Laboratories, Singapore). Secondary antibodies goat anti-mouse IgG-DyLight 488 (35503, Thermo Fisher Scientific), goat anti-rabbit IgG DyLight 550 (84541, Thermo Fisher Scientific), goat anti-mouse IgG-IRDye 800 (926–32210, LI-COR Biosciences), and goat anti-rabbit IgG-IRDye 680 (926–68021, LI-COR Biosciences) were commercially purchased.
C. elegans strains and RNAi constructs
All strains were grown at 20°C using standard C. elegans methods, as previously described (Brenner, 1974; Stiernagle, 2006). Nematode growth medium (NGM) agar plates were seeded with Escherichia coli strain OP50 for normal growth or with HT115 bacteria for RNAi feeding, as indicated. The WT N2 Bristol, SJ4005 (hsp-4p::gfp), and SJ17 (xbp-1(zc12) III;zcIs4 [hsp-4p::GFP] V) strains were obtained from Caenorhabditis Genetic Center. RNAi was performed using solid NGM-RNAi media, i.e., NGM containing 25 µg/ml carbenicillin, 2 mM IPTG, and 12.5 µg/ml tetracycline, seeded with the appropriate HT115 RNAi bacteria. RNAi clones including positive controls mdt-15 and fat-6 were from the Ahringer library and were sequenced to verify insert identity.
Plasmids used in this study
Plasmids and oligonucleotides used in this study are listed in Table S8 and Table S9, respectively. Plasmids were constructed by either restriction or Gibson cloning. All coding sequences of plasmid constructs used in this study were fully sequenced. The plasmid pGT0421 containing HA-tagged HAC1 in pMR366 was constructed as previously described (Thibault et al., 2011). The plasmid pGT0330 containing endogenously expressed IRE1 was generated as previously described (Ng et al., 2000). The plasmid pGT0201 containing IRE1ΔLD was generated by amplifying the endogenous promoter, signal sequence (fragment 1), and transmembrane cytosolic domain (fragment 2) with primer pairs GTO275-276 and GTO277-GTO278, respectively, from template genomic DNA of WT cells. The fragments were further digested with XhoI and PstI (fragment 1) and Pstl and Notl (fragment 2) before ligation into a pRS313 XhoI and NotI linearized plasmid. The plasmids pGT0223 and pGT0225 were generated by genomic DNA of WT cells amplification using primer pair HWO15-16 and ligated into pSW177 (Wang et al., 2011). pGT0285 was generated by amplifying the luminal domain with primers B29 containing a BamHI cut site and B30 that contain HA-HDEL-NcoI overhang sequences followed by ligation into BamHI/NcoI linearized pGT0223. The PGK promoter was amplified from pGT0121 with primer pair B36-37 and subsequently digested with NotI and BamHI and ligated with BamHI/Not1 linearized pGT0285 to make pGT0289. The plasmids BGT0261 and BGT0262 were generated by digesting pGT0223 and pGT0225 with NcoI/NotI and ligation into NcoI/NotI digested pGT101. The plasmid pGT0334 was generated by Phusion site-directed mutagenesis from pGT0289 with GTO311-312 and GTO313-314 as previously described (Nelson et al., 1993). pGT0442 and pGT0443 were generated from pGT0261 and pGT0262, respectively, by Phusion site-directed mutagenesis using the primer pair HN107-108 as previously described (Nelson et al., 1993). pGT0448 was a gift from Madhusudan Dey (University of Wisconsin-Milwaukee, Milwaukee, WI) and constructed as previously described (Sathe et al., 2015). pGT0557 and pGT0558 were generated from pGT0330 and pGT0201, respectively, by Phusion site-directed mutagenesis using the primer pair HN107-108. The split Venus constructs pGT0544 and pGT0546 were generated by Gibson assembly to join HA-VN173 (synthesized by Gblock) with HN177-178 linearized pGT0330 and pGT0201. The split Venus constructs pGT0545 and pGT0547 were generated by Gibson assembly to join FLAG-VC155 (synthesized) with HN177-178 linearized pGT0059 and pGT0435. pGT0584 and pGT585 were generated by Gibson assembly to join synthesized ymNeonGreen-HA (mNG) with WS45 primer pairs linearized pGT0330 and WS46 primer pairs linearized pGT0201, respectively. pGT0586 was generated by Gibson assembly to join PCR-amplified dsRed-Express2-HDEL from template pGT0370 with WS47 primer pairs and XbaI-digested pGT0383. pGT0370 (21770, Addgene) was a gift from Benjamin Glick (The University of Chicago, Chicago, IL). Yeast knockout strains in ire1Δ BY4741 background (Brachmann et al., 1998) were constructed by homologous recombination with the following primers: scj1Δ with HN199 and HN200, opi3Δ with HN205 and HN206, ste24Δ with HN207 and HN208, and get1Δ with HN209 and HN210.
Yeast genetic screen
The yeast deletion library was used to conduct the genetic screen (Giaever et al., 2002). Using the Synthetic Genetic Array methodology (Tong and Boone, 2006), the reporter strains YGT1228 and YGT1202 were cloned from YMS612 strain (Cohen et al., 2017) and were mated to the MATa yeast deletion library containing a single gene deleted with KanR. In short, following mating and sporulation on nitrogen-starved medium plates for 7 d, the MATα cells were ultimately passaged onto synthetic defined (SD) plates containing geneticin sulfate (200 µg/ml), hygromycin B (200 µg/ml), and the toxic amino acid derivatives canavanine (100 µg/ml) and thialysine (100 µg/ml) to select for strains carrying either Ire1/Ire1ΔLD and Kan-marked gene deletions. The genetic screen was condensed with the 384 Solid Pin Multi-Blot Replicator (V&P Scientific) and performed in 384-format until analysis. Cells were subsequently pinned from 384 spots on agar to four 96-well plates using the 96 Solid Pin Multi-Blot Replicator (V&P Scientific) and inoculated in 200 µl yeast extract–peptone–dextrose (YPD) medium per well and grown overnight at 30°C. An automated high-throughput sampler connected to the LSRFortessa X-20 (BD) was used to measure the relative levels of GFP and mCherry. The program FACSDiVA v 8.0 (BD) was used to acquire data in .fcs file format. Files were read with the program FlowJo X 10.0.7r2 (FlowJo, LLC). GFP and mCherry were excited at 488 and 561 nm, collected through a 505- and 595-nm long-pass filter and a 530/30 and 610/20 band pass filter, respectively. Reporter GFP fluorescence levels were normalized to the constitutive translation elongation factor (TEF2) promoter–driven mCherry expression to correct for nonspecific GFP expression. The median readout from 10,000 cells was obtained. The log2 GFP/mCherry ratio of each mutant (m) was normalized to WT levels from each plate and used as the final sample’s reporter level using Eq. 1.
C. elegans RNAi screen
To identify genes whose inactivation induced the UPR in C. elegans, we used a strain with a stably integrated hsp-4p::gfp reporter [strain SJ4005 zcIs4 (hsp-4p::gfp) V], which is widely used as a reporter for UPR induction (Calfon et al., 2002; Hou et al., 2014; Koh et al., 2018). We compiled a list of 1,695 metabolic genes from two published datasets (Yilmaz and Walhout, 2016; Zhang et al., 2013), and obtained RNAi clones for 1,247 of these from the Ahringer RNAi library (Source BioScience). We performed the RNAi screen in duplicate in 12-well format in nematode growth medium (NGM)-RNAi media seeded with appropriate HT115 RNAi bacteria. RNAi clones were tested in batches of 30–40 clones, and each batch included negative (empty vector) and positive (mdt-15 and/or fat-6) RNAi clones. Synchronized (by standard bleaching) stage 1 larvae were placed on RNAi bacteria lawns and allowed to develop into stage 4 larvae (∼44–48 h) and subsequently young adults (∼70–72 h); at both stages, hsp-4p::gfp levels were scored visually in a Leica M205FA upright fluorescent microscope. Clones causing visual developmental or growth delay were noted. Fluorescence was visually classified into three categories, low, medium, or high. The number of worms in each category was counted, and converted into a semi-quantitative rubric as follows: a hit was classified as a “strong” hit if >70% of worms displayed high or medium fluorescence; as a “moderate” hit if 10–69% of worms displayed strong or medium fluorescence; and as a “weak” hit if any worms displayed fluorescence above that seen in the negative control (in screen #1, fluorescence at 72 h was only assessed for 398 clones, and only on a qualitative basis). Thus, we identified 107 RNAi clones that were scored as initial hits at either time point in either screen (Table S2, tabs “Screen #1” and “Screen #2”). 106 of these (one subsequently failed to grow) were then subjected to three validation experiments, using the same readout and scoring approach (Table S2, tabs “Valid. #1” to “Valid. #3”). We assigned values of 5, 3, and 1 in the individual experiments to the rubrics “strong,” “moderate,” and “weak,” and then aggregated values to generate a summative score; a hit was considered validated if it obtained summative score of at least 3 (equivalent to 3 weak or 1 moderate outcome; Table S2, tab “Valid. summary”). This yielded 38 hits. Sanger sequencing of the contained RNAi vectors revealed three clones with an insert other than the one identified in the library, and these were removed (Table S2, tab “Sequencing”). The remaining 35 clones were tested for their ability to induce the hsp-4p::gfp reporter in strain SJ17 (xbp-1(zc12) III; zcIs4 [hsp-4p::gfp] V); one clone failed to reliably induce hsp-4p::gfp and was removed (Table S2, tab “xbp-1 dep.”). For the remaining 34 bona fide hits, the values of 5, 3, and 1 (for “strong,” “moderate,” and “weak”) from both screens and all three validation experiments were averaged into an overall score; this score was then visualized as the heat map in Fig. 2 (Table S2, tab “Hits Fig. 2;” and Fig. 2). Choline supplementation analysis was performed identically except that plates were additionally supplemented with 30 mM choline chloride. C. elegans homologues of yeast hits were tested as above; of the 181 hits, 54 had C. elegans homologues, of which 38 had corresponding RNAi clones in the Ahringer RNAi library; of these, Sanger sequencing revealed that 35 had the correct insert (Table S3).
Indirect immunofluorescence
Indirect immunofluorescence was performed as previously described (Spear and Ng, 2005). In brief, cells were grown to early log phase in selective media, fixed by 3.7% formaldehyde treatment, and permeabilized. Monoclonal mouse anti-HA (1:500), anti-FLAG tag mouse M2 monoclonal antibody (1:500), and rabbit anti-Kar2 (1:1,000) were used as primary antibodies. Mouse anti-Dylight 488 (1:500) and rabbit anti-Dylight 550 (1:500) were used as secondary antibodies. Samples were imaged with a Zeiss LSM 710 confocal microscope with a 100× 1.4 NA oil plan-Apochromat objective (Carl Zeiss MicroImaging). Images were analyzed using ImageJ 1.48v.
Microscopy
For live-cell imaging, yeast cells were grown to an exponential phase at 30°C in 3 ml of selective media. Samples were treated with 10 mM DTT for 1 h or inositol depleted for 4 h. Cells undergoing inositol depletion were washed six times before transferring to inositol-free media. 500 μl of cells in selective media were placed on slides coated with 10 mg/ml Concanavalin A (Sigma-Aldrich) mounted onto Attofluor cell chambers (Thermo Fisher Scientific) and imaged at room temperature. Images were captured using Zeiss Axio Observer.Z1 fluorescence microscope (ymNeonGreen) or Zeiss LSM 710 confocal microscope (split Venus) with a 100× 1.4 NA oil plan-Apochromat objective (Carl Zeiss MicroImaging). Images were analyzed using ImageJ 1.48v. Images were deconvoluted using the ImageJ plugin DeconvolutionLab.
Spotting growth assay
Cells were grown overnight in 3 ml of selective media at 30°C and diluted to 0.2 OD600/ml, from which three 10-fold serial dilutions were prepared and spotted on selective plates (0.25 µg/ml Tm or 1 mM choline were added to the plates when indicated). Plates were incubated at 30°C until the appearance of colonies.
Alkaline carbonate extraction
Alkaline carbonate extraction was performed as previously described (Fujiki et al., 1982). In brief, cells were grown to early log phase, and the equivalent of 50 OD600 of cells were harvested. Cells were resuspended in 10 mM sodium phosphate buffer, pH 7.0, 1 mM PMSF, and protease inhibitor cocktail (Roche). An equal volume of 0.2 M sodium carbonate (pH 11.5) was added to cell lysates and incubated 30 min at 4°C and spun down at 100,000 g for 30 min, 4°C. The pellet (membrane fraction) was solubilized in 3% SDS, 100 mM Tris Cl, pH 7.4, and 3 mM DTT and incubated at 95°C for 10 min. Proteins from total cell lysate and supernatant fractions (collected from centrifuged lysate) were precipitated with 10% TCA and spun down 30 min at 18,400 g, 4°C. Proteins were resuspended in TCA resuspension buffer (1 mM Tris, pH 11, and 3% SDS) and incubated 10 min at 95°C. Solubilized proteins were separated by SGS-PAGE and transferred to nitrocellulose for immunoblot analysis. Protein loading buffer was added to each fraction and separated by SDS-PAGE followed by immunoblot analysis.
Immunoblot
Cells were grown to an early log phase overnight at 30°C. Tm was added to a final concentration of 2.5 µg/ml and incubated at 30°C for 1 h, when indicated. Harvested cells were resuspended in 10% TCA followed by the addition of 0.5 mm zirconium beads. Cells were disrupted by two 30-s cycles. The lysate was transferred to a new tube and combined with a 10% TCA bead wash. The precipitate was pelleted by centrifugation and vortexed in TCA resuspension buffer (100 mM Tris, pH 11, 3% SDS, 1 mM PMSF). The samples were incubated 10 min at 95°C and spun down 15 min at 18,400 g, 4°C. A portion of the extract was separated by SDS-PAGE using a 15% gel and transferred to nitrocellulose. The blots were probed with primary antibodies followed by secondary goat anti-mouse IgG-IRDye 800 (LI-COR) and goat anti-rabbit IgG-IRDye 680 (LI-COR) antibodies. Membranes were washed in TBS and visualized with the Odyssey CLx imaging system (Li-COR).
qRT-PCR
Cells were grown to an early log phase overnight at 30°C. Tm was added to a final concentration of 2.5 µg/ml and incubated at 30°C for 1 h, when indicated. 4-PBA was added to a final concentration of 1 mM 15 min before Tm, when indicated, as previously reported (Kubota et al., 2006; Le et al., 2016; Ozcan et al., 2006; Pineau et al., 2009). Total RNA was extracted using RNeasy Mini Kit (Qiagen) following the manufacturer’s protocol. DNase treatment in columns was performed with RNase-free DNase (Qiagen) following the manufacturer’s protocol. For C. elegans, ∼1,000–2,000 worms were grown on appropriate RNAi clones, harvested, and washed in M9 buffer, and RNA was extracted with Trizol and BCP following purification with the RNeasy Mini Kit and RNase-free DNase, as described (Hou et al., 2014). cDNA was synthesized from 2 µg of total RNA using RevertAid reverse transcription (Thermo Fisher Scientific) following the manufacturer’s protocol. SYBR Green qPCR experiments were performed following the manufacturer’s protocol using a QuantStudio 6 Flex or a StepOnePlus Real-time PCR system (Applied Biosystems). cDNA (30 ng) and 50 nM of paired-primer mix were used for each reaction. Relative mRNA was determined with the comparative Ct method (ΔΔCt) normalized to housekeeping gene ACT1. C. elegans samples were normalized to the three reference genes, tba-1, ubc-2, and act-1, as described (Hou et al., 2014). Oligonucleotide primers used are listed in Table S9.
β-Galactosidase assay
ChIP
DNA microarray
Cultures were grown to an OD600/ml of 0.25 at 30°C in selective synthetic complete media. The UPR was induced in WT cells by 1 h incubation of 2 mM DTT when indicated. Cells were harvested from cultures at cell density of less than 0.5 OD600/ml. Total RNA was extracted by the hot acid phenol method as previously described (Spellman et al., 1998). Total RNA was subsequently cleaned up using the RNeasy Mini Kit (Qiagen). RNA quality control was performed using the Agilent RNA Nano 6000 Chip (Agilent Technologies). RNA was prepared from independent triplicate samples. Probe preparation and microarray construction and analysis were performed as previously described (DeRisi et al., 1997; Shalon et al., 1996; Spellman et al., 1998). Probes were prepared using the Low Input Quick Amp Labeling System with 100 ng of Total RNA as starting material following the manufacturer’s instructions, which included One-Color Microarray-Based Gene Expression Analysis Protocol Version 6.5 (Agilent Technologies), and were hybridized on a Custom Microarray Agilent GE 8 × 60K array. Arrays were scanned using a high-resolution DNA Microarray Scanner, model G2505C (Agilent Technologies). Data were analyzed using GeneSpring GX software (Agilent Technologies). Differentially expressed genes were deemed significant with fold-change >1.5 and ANOVA P values <0.05. UpSetR was used to compare and visualize set intersections of significantly up-regulated genes in a matrix-style layout (Conway et al., 2017). GO terms analysis from gene lists acquired from the intersections was performed with DAVID (Huang et al., 2007). Heat map in the figure was generated using R Studio. The DNA microarray data discussed in this publication have been deposited in GEO under accession no. GSE131146.
Statistics
The error bars denote SEM, derived from at least three biological replicates, unless otherwise indicated. P values were calculated using two-tailed Student’s t test unless otherwise indicated in the figure legends and reported as values in figures. Scatter plots were plotted using GraphPad Prism 8.
Online supplemental material
Fig. S1 shows the validation of IRE1 and IRE1ΔLD as UPR reporter query strains and the qRT-PCR validation of the screen in C. elegans, using IRE-1 and XBP-1 regulated genes in a select set of RNAi treatments. Fig. S2 shows the localization and integration of Ire1 variants to the ER and the clustering of Ire1-mNG and Ire1ΔLD-mNG. Fig. S3 shows the localization and integration of Ire1 variants to the ER and Ire1 clustering by BiFC. Fig. S4 shows the localization of Ire1(R537Q) and Ire1ΔLD(R537Q) to the ER. Fig. S5 shows the Pearson correlations of relative RNA abundance related to Fig. 6. Table S1 shows the relative fluorescence of strains in UPRE-GFP screen. Table S2 shows the results of the RNAi screen results, validation, sequence validation, xbp-1 dependence, and choline response in C. elegans. Table S3 shows the RNAi and sequence validation of 38 yeast screen hit orthologues in C. elegans. Table S4 shows the summary of yeast and C. elegans screen hit orthologues. Table S5 shows gene list from different diagram categories related to Fig. 6 A. Table S6 shows predominant GO terms of each cluster related to Fig. 6 B. Table S7 contains the list of yeast strains used in this study. Table S8 contains the list of plasmids used in this study. Table S9 contains the list of oligonucleotide primers used in this study.
Acknowledgments
We are grateful to Dr. Maya Schuldiner (Weizmann Institute of Science, Rehovot, Israel) for providing reagents to carry out our genetic screen. We thank members of the Thibault laboratory for critical reading of the manuscript. We thank Angel Chu for help with qPCR validation in C. elegans.
This work was supported by the Nanyang Assistant Professorship program from Nanyang Technological University (G. Thibault), the National Research Foundation Singapore under its National Research Foundation of Singapore together with the National Natural Science Foundation of China (NRF-NSFC) joint research grant call (NRF2018NRFNSFC003SB-006 to G. Thibault), the Nanyang Technological University Research Scholarship to N. Ho and J.H. Koh (predoctoral fellowship), the Natural Sciences and Engineering Research Council of Canada Discovery grant (RGPIN-2018-05133 to S. Taubert), the Canadian Institutes of Health Research (PJT-153199), and British Columbia Children's Hospital Research Institute (BCCHR) Canucks for Kids Graduate and University of British Columbia (UBC) Affiliate Studentships to J. Xu. Some strains were provided by the Caenorhabditis Genetic Center, which is funded by the National Institutes of Health Office of Research Infrastructure Programs (P40 OD010440).
The authors declare no competing financial interests.
Author contributions: Conceptualization: G. Thibault; Methodology: N. Ho, W.S. Yap, J. Xu, H. Wu, S. Taubert, G. Thibault; Formal analysis: N. Ho, W.S. Yap, J. Xu, H. Wu, J.H. Koh, W.W.B. Goh; Investigation: N. Ho, W.S. Yap, J. Xu, H. Wu, J.H. Koh, B. George, S.C. Chong; Resources: N. Ho, W.S. Yap, J. Xu, H. Wu; Writing - original draft: N. Ho, J. Xu, H. Wu, S. Taubert, G. Thibault; Writing - review & editing: N. Ho, W.S. Yap, J. Xu, H. Wu, S. Taubert, G. Thibault; Supervision: S. Taubert, G. Thibault; Project administration: G. Thibault; Funding acquisition: N. Ho, J. Xu, J.H. Koh, S. Taubert, G. Thibault.

