


Two major protein recycling pathways have emerged as key regulators of enduring forms of synaptic plasticity, such as long-term potentiation (LTP), yet how these pathways are recruited during plasticity is unknown. Phosphatidylinositol-3-phosphate (PI(3)P) is a key regulator of endosomal trafficking and alterations in this lipid have been linked to neurodegeneration. Here, using primary hippocampal neurons, we demonstrate dynamic PI(3)P synthesis during chemical induction of LTP (cLTP), which drives coordinate recruitment of the SNX17–Retriever and SNX27–Retromer pathways to endosomes and synaptic sites. Both pathways are necessary for the cLTP-dependent structural enlargement of dendritic spines and act in parallel by recycling distinct sets of cell surface proteins at synapses. Importantly, preventing PI(3)P synthesis blocks synaptic recruitment of SNX17 and SNX27, decreases cargo recycling, and blocks LTP in cultured neurons and hippocampal slices. These findings provide mechanistic insights into the regulation of endocytic recycling at synapses and define a role for dynamic PI(3)P synthesis in synaptic plasticity.
Introduction
Neuronal synapses have a rich complement of cell surface proteins that are dynamically regulated both by posttranslational modification (PTM) and by membrane trafficking. Protein endocytosis, endosomal sorting, and recycling back to the plasma membrane are critical for synaptic functions (Diering and Huganir, 2018; van Oostrum et al., 2020). As such, defects in these sorting mechanisms are associated with deficits in synaptic plasticity, synapse loss, and neurodegeneration (Saitoh, 2022; McDonald, 2020). Much of what we know about endocytic recycling in neurons has come from studies focused on the SNX27–Retromer pathway (Wang et al., 2013; Hussain et al., 2014; Loo et al., 2014; McMillan et al., 2021). A distinct, parallel SNX17–Retriever recycling pathway was recently discovered in non-neuronal cells (McNally et al., 2017), and in neurons, this pathway also plays a key role in regulating synapse function and plasticity (Rivero-Ríos et al., 2023). However, it is not yet known how specific patterns of neuronal activity engage these recycling pathways and to what extent upstream signals drive a coordinated or independent activation of each.
In non-neuronal cells, SNX17 defines a major endomembrane recycling pathway responsible for the recycling of over 120 cell surface proteins from endosomes to the plasma membrane. SNX17 cargoes include numerous cell adhesion proteins, signaling receptors, and solute transporters (McNally et al., 2017). SNX17-dependent recycling requires the Retriever complex (VPS35L, VPS26C, and VPS29), the CCC complex (COMMD/CCDC22/CCDC93), and the actin-regulatory WASH complex (Chen et al., 2019; McNally and Cullen, 2018; Simonetti and Cullen, 2019; Wang et al., 2018). While most neuronal SNX17 cargoes remain unidentified, our findings show that the role of SNX17 in long-term potentiation (LTP) is mediated, at least in part, by promoting the recycling of the adhesion protein β1-integrin (Rivero-Ríos et al., 2023). β1-integrin is a key regulator of synaptic function and plasticity. It localizes to postsynaptic sites in hippocampal neurons and influences cytoskeletal organization, dendritic spine maturation, and synaptic transmission (Babayan et al., 2012; Bourgin et al., 2007; Chan et al., 2006; Chun et al., 2001; Huang et al., 2006; Kramár et al., 2006; Liu et al., 2016; Mortillo et al., 2012; Ning et al., 2013; Orr et al., 2022; Warren et al., 2012; Webb et al., 2007).
Similarly, SNX27 recycles more than 200 cell surface proteins in neurons (McMillan et al., 2021). SNX27 displays high sequence homology with SNX17 but also harbors a PDZ domain for cargo binding. This unique PDZ domain directly interacts with C-terminal PDZ-binding motifs present in many neuronal surface proteins and is critical for the selective retrieval and recycling of SNX27-specific cargoes (Steinberg et al., 2013). Neuronal SNX27 cargoes include key receptors, such as α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic (AMPA)-type glutamate receptors (Loo et al., 2014; McMillan et al., 2021), β-adrenergic receptors (Lauffer et al., 2010; Temkin et al., 2011), Kir3 potassium channels (Lunn et al., 2007), the 5-hydroxytryptamine 4a receptor (Joubert et al., 2004), and the N-methyl-D-aspartate (NMDA) receptor 2C (Cai et al., 2011). SNX27 regulates cargo recycling by engaging with the Retromer complex (VPS35, VPS26A/B, and VPS29), a SNX–Bin/amphiphysin/Rvs dimer (SNX1/2 in complex with SNX5/6), and the WASH complex (Steinberg et al., 2013; Gallon et al., 2014; Harbour et al., 2012; Simonetti and Cullen, 2019; Mao et al., 2021; Jia et al., 2012; Yong et al., 2020; Kovtun et al., 2018; Yong et al., 2021). Similar to SNX17, SNX27 is also required for LTP. Alterations in this pathway prevent chemical induction of LTP (cLTP) by impairing the activity-dependent recruitment of surface AMPA receptors (Hussain et al., 2014; Loo et al., 2014; Temkin et al., 2017). AMPA receptors are rapidly inserted into the postsynaptic membrane in response to synaptic activity, a process critical for LTP (Buonarati et al., 2019). The requirement of SNX17- and SNX27-dependent recycling for LTP supports recent findings that show extensive changes in the neuronal surface proteome upon cLTP, which are mediated by the delivery of membrane proteins from intracellular compartments (van Oostrum et al., 2020). Whether these pathways coordinate to promote cargo recycling during LTP has not been directly tested.
The signaling lipid phosphatidylinositol-3-phosphate (PI(3)P) is a key regulator of endosomal sorting and trafficking (Di Paolo and De Camilli, 2006; Raiborg et al., 2013) that can potentially interact with both SNX17 and SNX27 (Chandra et al., 2019). PI(3)P is predominantly localized on early endosomes and functions by recruiting a variety of effectors that contain PI(3)P-binding domains, such as phox homology (PX) and FYVE (Fab1, YOTB, Vac1, and EEA1) domains (Marat and Haucke, 2016); both SNX17 and SNX27 can interact with PI(3)P through their PX domains. PI(3)P is synthesized from phosphatidylinositol by phosphorylation at the third position of the inositol ring. Approximately 60–70% of the PI(3)P pool is produced by the lipid kinase VPS34 (Devereaux et al., 2013; Ikonomov et al., 2015). In addition, class II phosphatidylinositol 3 kinases and INPP4 phosphatases also contribute to PI(3)P levels (Heng and Maffucci, 2022; Gozzelino et al., 2020; Burke et al., 2022).
In neurons, PI(3)P is widely distributed in both dendrites and axons, with an enrichment at synapses, where it colocalizes with the postsynaptic scaffold PSD95 (Wang et al., 2011). PI(3)P plays key roles in neurotransmission by regulating synaptic vesicle cycling (Liu et al., 2022) and GABA receptor clustering at inhibitory synapses (Papadopoulos et al., 2017). Chronic defects in PI(3)P synthesis are linked to reductions in dendritic spine density, reactive gliosis, and progressive neurodegeneration (Wang et al., 2011; Zhou et al., 2010). However, whether dynamic changes in PI(3)P synthesis impact synaptic function and plasticity remains unclear.
Here, we demonstrate a key role for PI(3)P in coordinately driving the recycling of cell surface proteins during LTP. We show that PI(3)P levels increase upon cLTP and are necessary for the structural changes in dendritic spines. We find that the role of PI(3)P in synaptic plasticity is mediated, at least in part, by regulation of the SNX17 and SNX27 pathways. Specifically, cLTP-dependent PI(3)P synthesis promotes the recruitment of SNX17 and SNX27 to endosomes and to dendritic spines. Moreover, we show that SNX17 and SNX27 define two distinct pathways that act in parallel to recycle different sets of cell surface receptors necessary for LTP. Preventing dynamic PI(3)P synthesis blocks the activity-dependent recycling of SNX17 and SNX27 cargoes and prevents LTP in hippocampal slices as well as in cultured hippocampal neurons. Together, these findings provide mechanistic insights into the regulation of endocytic recycling at synapses and define a key role for dynamic PI(3)P synthesis in enduring forms of synaptic plasticity.
Results
cLTP results in an increase in PI(3)P-containing puncta in dendrites
LTP is characterized by extensive synapse remodeling and changes in cell surface proteins, which occur through the controlled delivery of membrane proteins from intracellular compartments (van Oostrum et al., 2020). Previous studies have identified an important role for the SNX27–Retromer pathway in recycling AMPARs during LTP (Loo et al., 2014; Hussain et al., 2014), and we recently identified the SNX17–Retriever endomembrane recycling pathway as a critical regulator of LTP (Rivero-Ríos et al., 2023). The mechanism by which these parallel recycling pathways are coordinated during plasticity, however, is unknown.
One possibility is that a common upstream effector recruits both pathways. Notably, SNX17 and SNX27 both interact with the signaling lipid PI(3)P via their PX domains (Knauth et al., 2005; Lunn et al., 2007; Gillooly et al., 2000). To explore this idea, we examined dynamic changes in PI(3)P with a specific PI(3)P reporter, dsRed-EEA1-FYVE (Singla et al., 2019), which consists of the fusion between dsRed and the FYVE domain from EEA1. Using this probe in primary cultured DIV17 hippocampal neurons, we found that PI(3)P exhibited a punctate distribution pattern, with puncta distributed throughout dendrites (Fig. 1 A). A similar PI(3)P distribution in neurons was observed using the recombinant 2xFYVE domain of Hrs (Liu et al., 2022). In addition, we found that PI(3)P formation in dendrites is dependent on the lipid kinase VPS34. A 30-min treatment with either VPS34-IN1 or SAR405, chemically distinct VPS34 inhibitors, resulted in ∼72% and 85% decreases, respectively, in the numbers of dendritic PI(3)P-positive puncta (Fig. 1, A and B). To avoid a potential competition with endogenous PI(3)P, we also used a recently developed recombinant biosensor for PI(3)P, SNAP-FYVE-Hrs x 2 on fixed cells (Maib et al., 2024), which confirmed the punctate distribution of PI(3)P along dendrites and its disappearance upon VPS34-IN1 treatment (Fig. S1, A and B).

PI(3)P is present at dendrites and its synthesis increases upon cLTP. (A) Rat hippocampal neurons were transfected at DIV16 with dsRed-EEA1-FYVE and eGFP as a filler. 24 h later, neurons were treated with either DMSO, 1 μM VPS34-IN1, or 1 μM SAR405 for 30 min before fixation. Scale bar, 5 µm. (B) The number of dsRed-EEA1-FYVE–positive puncta in the first 30 μm of secondary dendrites was quantified. DMSO: 0.247 ± 0.016, N = 30 neurons; VPS34-IN1: 0.070 ± 0.011, N = 30 neurons; SAR405: 0.037 ± 0.007, N = 30 neurons. Three independent experiments. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (C) Example confocal images taken from DIV17 neurons expressing dsRed-EEA1-FYVE and eGFP (filler) under conditions of cLTP or HBS control (mock). Images of live neurons in an environmental chamber were captured before cLTP (baseline); during cLTP; and at 0, 5, 10, 15, 20, 25, and 30 min after the cLTP stimulus. Representative images of baseline, t = 10, and t = 30 are shown. Intensity presented in the “fire” LUT color scheme. Scale bar, 5 µm. (D) The number of dsRed-EEA1-FYVE–positive puncta in 30 µm of dendritic spines at the different time points following cLTP (or mock) was quantified and normalized to the baseline for each dendrite. Mock: N = 15 dendrites (baseline: 1.00 ± 0.071, cLTP: 1.04 ± 0.077, 0: 1.06 ± 0.076, 5: 1.05 ± 0.072, 10: 1.10 ± 0.069, 15: 1.08 ± 0.080, 20: 1.09 ± 0.083, 25: 1.14 ± 0.079, 30: 1.15 ± 0.079); cLTP: N = 15 dendrites (baseline: 1.00 ± 0.051, cLTP: 1.20 ± 0.070, 0: 1.38 ± 0.072, 5: 1.46 ± 0.076, 10: 1.51 ± 0.075, 15: 1.63 ± 0.089, 20: 1.69 ± 0.082, 25: 1.72 ± 0.072, 30: 1.75 ± 0.076). Three independent experiments. Statistical significance was determined using two-way ANOVA with Sidak’s multiple comparison test, *P < 0.05, **P < 0.01, ***P < 0.005, and ****P < 0.001. Error bars are SEM. (E) The size of dsRed-EEA1-FYVE–positive puncta in 30 µm of dendritic spines at the indicated time points following cLTP (or mock) was quantified. Mock: N = 15 dendrites (baseline: 0.184 ± 0.013, 10: 0.187 ± 0.011, 30: 0.186 ± 0.014); cLTP: N = 15 dendrites (baseline: 0.184 ± 0.023, 10: 0.188 ± 0.023, 30: 0.181 ± 0.021). Three independent experiments. Data were analyzed using one-way ANOVA with Tukey’s post hoc test. Error bars are SEM. (F) Hippocampal neurons were co-transfected at DIV16 with dsRed-EEA1-FYVE and either an empty pRK5 vector or pRK5-HA-CaMKII-T286D. 24 h later, neurons were fixed, permeabilized, and incubated with an anti-HA antibody. The number of dsRed-EEA1-FYVE–positive puncta in the first 30 μm of secondary dendrites was quantified. Ctrl: 0.201 ± 0.010, N = 30 neurons; CaMKII-T286D: 0.311 ± 0.017, N = 30 neurons. Statistical significance was determined using unpaired two-tailed Student’s t test, ****P < 0.001. Error bars are SEM. DIV, days in vitro.
PI(3)P is present at dendrites and its synthesis increases upon cLTP. (A) Rat hippocampal neurons were transfected at DIV16 with dsRed-EEA1-FYVE and eGFP as a filler. 24 h later, neurons were treated with either DMSO, 1 μM VPS34-IN1, or 1 μM SAR405 for 30 min before fixation. Scale bar, 5 µm. (B) The number of dsRed-EEA1-FYVE–positive puncta in the first 30 μm of secondary dendrites was quantified. DMSO: 0.247 ± 0.016, N = 30 neurons; VPS34-IN1: 0.070 ± 0.011, N = 30 neurons; SAR405: 0.037 ± 0.007, N = 30 neurons. Three independent experiments. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (C) Example confocal images taken from DIV17 neurons expressing dsRed-EEA1-FYVE and eGFP (filler) under conditions of cLTP or HBS control (mock). Images of live neurons in an environmental chamber were captured before cLTP (baseline); during cLTP; and at 0, 5, 10, 15, 20, 25, and 30 min after the cLTP stimulus. Representative images of baseline, t = 10, and t = 30 are shown. Intensity presented in the “fire” LUT color scheme. Scale bar, 5 µm. (D) The number of dsRed-EEA1-FYVE–positive puncta in 30 µm of dendritic spines at the different time points following cLTP (or mock) was quantified and normalized to the baseline for each dendrite. Mock: N = 15 dendrites (baseline: 1.00 ± 0.071, cLTP: 1.04 ± 0.077, 0: 1.06 ± 0.076, 5: 1.05 ± 0.072, 10: 1.10 ± 0.069, 15: 1.08 ± 0.080, 20: 1.09 ± 0.083, 25: 1.14 ± 0.079, 30: 1.15 ± 0.079); cLTP: N = 15 dendrites (baseline: 1.00 ± 0.051, cLTP: 1.20 ± 0.070, 0: 1.38 ± 0.072, 5: 1.46 ± 0.076, 10: 1.51 ± 0.075, 15: 1.63 ± 0.089, 20: 1.69 ± 0.082, 25: 1.72 ± 0.072, 30: 1.75 ± 0.076). Three independent experiments. Statistical significance was determined using two-way ANOVA with Sidak’s multiple comparison test, *P < 0.05, **P < 0.01, ***P < 0.005, and ****P < 0.001. Error bars are SEM. (E) The size of dsRed-EEA1-FYVE–positive puncta in 30 µm of dendritic spines at the indicated time points following cLTP (or mock) was quantified. Mock: N = 15 dendrites (baseline: 0.184 ± 0.013, 10: 0.187 ± 0.011, 30: 0.186 ± 0.014); cLTP: N = 15 dendrites (baseline: 0.184 ± 0.023, 10: 0.188 ± 0.023, 30: 0.181 ± 0.021). Three independent experiments. Data were analyzed using one-way ANOVA with Tukey’s post hoc test. Error bars are SEM. (F) Hippocampal neurons were co-transfected at DIV16 with dsRed-EEA1-FYVE and either an empty pRK5 vector or pRK5-HA-CaMKII-T286D. 24 h later, neurons were fixed, permeabilized, and incubated with an anti-HA antibody. The number of dsRed-EEA1-FYVE–positive puncta in the first 30 μm of secondary dendrites was quantified. Ctrl: 0.201 ± 0.010, N = 30 neurons; CaMKII-T286D: 0.311 ± 0.017, N = 30 neurons. Statistical significance was determined using unpaired two-tailed Student’s t test, ****P < 0.001. Error bars are SEM. DIV, days in vitro.
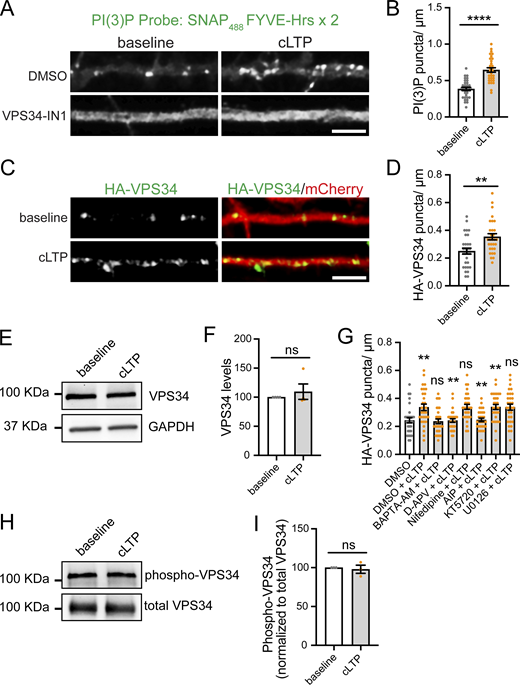
cLTP promotes the formation of PI(3)P- and VPS34-positive puncta, which is dependent on the NMDAR–calcium–CaMKII pathway. (A) Rat hippocampal neurons were transfected at DIV16 with mCherry as a filler. 24 h later, neurons were treated with either DMSO or 1 μM VPS34-IN1 for 30 min. Neurons were either fixed in the absence of cLTP (baseline) or treated with cLTP for 5 min and fixed 10 min after the cLTP stimulus. DMSO or VPS34-INH were maintained during the course of the experiment. After fixation, cells were immunostained with the PI(3)P recombinant biosensor conjugated to Alexa488 (SNAP488). Scale bar, 5 µm. (B) The number of PI(3)P-positive puncta in the first 30 μm of secondary dendrites was quantified for DMSO-treated cells. Baseline: 0.385 ± 0.023, N = 31 neurons; cLTP: 0.645 ± 0.031, N = 31 neurons. Three independent experiments. Statistical significance was determined using Student’s unpaired t test, ****P < 0.001. Error bars are SEM. (C) DIV16 hippocampal neurons were co-transfected at DIV16 with HA-VPS34 and mCherry as a filler. 24 h later, neurons were either fixed in the absence of cLTP (baseline) or treated with cLTP for 5 min and fixed 10 min after the cLTP stimulus, followed by immunostaining with an anti-HA antibody. Scale bar, 5 µm. (D) The number of HA-VPS34–positive puncta in the first 30 μm of secondary dendrites was quantified. Baseline: 0.250 ± 0.021, N = 30 neurons; cLTP: 0.353 ± 0.022, N = 30 neurons. Three independent experiments. Statistical significance was determined using Student’s unpaired t test, **P < 0.01. Error bars are SEM. (E) DIV17 rat cortical neurons were either left untreated (baseline) or treated with cLTP for 5 min. Extracts were collected 10 min after cLTP and analyzed by western blot for the levels of VPS34, and GAPDH was used as a loading control. (F) VPS34 levels were quantified and normalized to the baseline protein levels. Baseline: 100; cLTP: 109.50 ± 13.24. Four independent experiments. Statistical significance was determined using Student’s unpaired t test. Error bars are SEM. (G) DIV16 hippocampal neurons were co-transfected at DIV16 with HA-VPS34 and mCherry as a filler. 24 h later, neuron cultures were treated with DMSO, 10 μM BAPTA-AM, 100 μM D-APV, 10 μM nifedipine, 10 uM AIP, 2 μM KT5720, or 10 μM U0126 for 30 min, followed by a 5-min cLTP stimulus in the presence of compounds where indicated. Neurons were further incubated in the presence of the indicated compounds for 10 min before fixation, followed by permeabilization and incubation with an anti-HA antibody. The number of HA-VPS34–positive puncta in the first 30 μm of secondary dendrites was quantified. DMSO: 0.243 ± 0.019, N = 30 neurons; DMSO + cLTP: 0.337 ± 0.023, N = 30 neurons; BAPTA-AM + cLTP: 0.237 ± 0.017, N = 30 neurons; D-APV + cLTP: 0.242 ± 0.013, N = 30 neurons; nifedipine + cLTP: 0.340 ± 0.018, N = 30 neurons; AIP + cLTP: 0.247 ± 0.012, N = 30 neurons; KT5720 + cLTP: 0.339 ± 0.018, N = 30 neurons; U0126 + cLTP: 0.338 ± 0.021, N = 30 neurons. Three independent experiments. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, **P < 0.01. Error bars are SEM. (H) DIV16 rat hippocampal neurons were either left untreated (baseline) or treated with cLTP for 5 min. Extracts were collected 10 min after cLTP and used for immunoprecipitation with an anti-VPS34 antibody, followed by immunoblotting with an antibody against pan-phospho-Ser/Thr. (I) The ratios of pan-phospho-Ser/Thr to VPS34 were quantified and normalized to the baseline group. Baseline: 100; cLTP: 97.95 ± 5.22. Three independent experiments. Statistical significance was determined using Student’s unpaired t test. Error bars are SEM. DIV, days in vitro. Source data are available for this figure: SourceData FS1.
cLTP promotes the formation of PI(3)P- and VPS34-positive puncta, which is dependent on the NMDAR–calcium–CaMKII pathway. (A) Rat hippocampal neurons were transfected at DIV16 with mCherry as a filler. 24 h later, neurons were treated with either DMSO or 1 μM VPS34-IN1 for 30 min. Neurons were either fixed in the absence of cLTP (baseline) or treated with cLTP for 5 min and fixed 10 min after the cLTP stimulus. DMSO or VPS34-INH were maintained during the course of the experiment. After fixation, cells were immunostained with the PI(3)P recombinant biosensor conjugated to Alexa488 (SNAP488). Scale bar, 5 µm. (B) The number of PI(3)P-positive puncta in the first 30 μm of secondary dendrites was quantified for DMSO-treated cells. Baseline: 0.385 ± 0.023, N = 31 neurons; cLTP: 0.645 ± 0.031, N = 31 neurons. Three independent experiments. Statistical significance was determined using Student’s unpaired t test, ****P < 0.001. Error bars are SEM. (C) DIV16 hippocampal neurons were co-transfected at DIV16 with HA-VPS34 and mCherry as a filler. 24 h later, neurons were either fixed in the absence of cLTP (baseline) or treated with cLTP for 5 min and fixed 10 min after the cLTP stimulus, followed by immunostaining with an anti-HA antibody. Scale bar, 5 µm. (D) The number of HA-VPS34–positive puncta in the first 30 μm of secondary dendrites was quantified. Baseline: 0.250 ± 0.021, N = 30 neurons; cLTP: 0.353 ± 0.022, N = 30 neurons. Three independent experiments. Statistical significance was determined using Student’s unpaired t test, **P < 0.01. Error bars are SEM. (E) DIV17 rat cortical neurons were either left untreated (baseline) or treated with cLTP for 5 min. Extracts were collected 10 min after cLTP and analyzed by western blot for the levels of VPS34, and GAPDH was used as a loading control. (F) VPS34 levels were quantified and normalized to the baseline protein levels. Baseline: 100; cLTP: 109.50 ± 13.24. Four independent experiments. Statistical significance was determined using Student’s unpaired t test. Error bars are SEM. (G) DIV16 hippocampal neurons were co-transfected at DIV16 with HA-VPS34 and mCherry as a filler. 24 h later, neuron cultures were treated with DMSO, 10 μM BAPTA-AM, 100 μM D-APV, 10 μM nifedipine, 10 uM AIP, 2 μM KT5720, or 10 μM U0126 for 30 min, followed by a 5-min cLTP stimulus in the presence of compounds where indicated. Neurons were further incubated in the presence of the indicated compounds for 10 min before fixation, followed by permeabilization and incubation with an anti-HA antibody. The number of HA-VPS34–positive puncta in the first 30 μm of secondary dendrites was quantified. DMSO: 0.243 ± 0.019, N = 30 neurons; DMSO + cLTP: 0.337 ± 0.023, N = 30 neurons; BAPTA-AM + cLTP: 0.237 ± 0.017, N = 30 neurons; D-APV + cLTP: 0.242 ± 0.013, N = 30 neurons; nifedipine + cLTP: 0.340 ± 0.018, N = 30 neurons; AIP + cLTP: 0.247 ± 0.012, N = 30 neurons; KT5720 + cLTP: 0.339 ± 0.018, N = 30 neurons; U0126 + cLTP: 0.338 ± 0.021, N = 30 neurons. Three independent experiments. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, **P < 0.01. Error bars are SEM. (H) DIV16 rat hippocampal neurons were either left untreated (baseline) or treated with cLTP for 5 min. Extracts were collected 10 min after cLTP and used for immunoprecipitation with an anti-VPS34 antibody, followed by immunoblotting with an antibody against pan-phospho-Ser/Thr. (I) The ratios of pan-phospho-Ser/Thr to VPS34 were quantified and normalized to the baseline group. Baseline: 100; cLTP: 97.95 ± 5.22. Three independent experiments. Statistical significance was determined using Student’s unpaired t test. Error bars are SEM. DIV, days in vitro. Source data are available for this figure: SourceData FS1.
To determine whether synaptic activity regulates PI(3)P synthesis, we performed live-cell imaging with dsRed-EEA1-FYVE and monitored PI(3)P dynamics over time in the same dendrite. We used a well-established cLTP protocol (400 µM glycine and 0 Mg2+; 5 min), which promotes synaptic NMDAR activation to induce a long-lasting increase in postsynaptic strength (Lu et al., 2001; Park et al., 2004; Rivero-Ríos et al., 2023) and an enduring enlargement of dendritic spines (Henry et al., 2017). In mock-treated cells, which did not receive the cLTP stimulus, PI(3)P puncta size and number were stable over the course of the experiment. By contrast, the numbers of dsRed-FYVE-–positive puncta began increasing immediately after the cLTP stimulus and remained significantly elevated throughout the remaining time points analyzed, up to 30 min after cLTP. (Fig. 1, C and D). The size of the PI(3)P-positive compartments remained constant across all time points analyzed (Fig. 1, C and E). Note that we did not detect trafficking of the PI(3)P-positive compartments to dendritic spines, which suggests that PI(3)P-enriched endosomes may primarily localize to the dendritic shaft. Alternatively, the levels of PI(3)P moving into spines may be substantially lower than in the shaft, limiting detection by the probe. Using the recombinant PI(3)P bioprobe, we observed a similar increase in endogenous PI(3)P-positive puncta upon cLTP (Fig. S1, A and B).
Since PI(3)P formation in dendrites depends on VPS34, we hypothesized that the cLTP-induced increase in PI(3)P-positive endosomes may result from enhanced VPS34 activity. Because no suitable antibody is available for immunolocalizing endogenous rat VPS34, we transfected neurons with HA-tagged VPS34 (Park et al., 2016). We found that the increase in PI(3)P-positive puncta correlates with an increase in the number of VPS34-positive endosomes 10 min after cLTP treatment (Fig. S1, C and D). Notably, this increase in VPS34-positive endosomes was not due to elevated VPS34 expression levels (Fig. S1, E and F), suggesting that it reflects changes in VPS34 localization. The protein is likely being recruited from the cytosol to endosomal membranes.
cLTP depends on NMDA receptor activation, which increases postsynaptic calcium levels and triggers downstream signaling cascades, including calcium/CaM-dependent kinase II (CaMKII), protein kinase A, and Ras extracellular signal-regulated kinase (Musleh et al., 1997; Lisman et al., 2012). To determine whether cLTP-induced VPS34 recruitment to endosomes is mediated by NMDA receptor activation and calcium-dependent signaling, we pharmacologically disrupted key LTP pathways. These manipulations included intracellular calcium chelation with membrane-permeable BAPTA-AM, NMDA receptor blockade with D-APV, inhibition of L-type voltage-dependent calcium channels with nifedipine, inhibition of CaMKII with autocamtide-2–related inhibitory peptide (AIP), inhibition of protein kinase A with KT5720, and blockade of extracellular signal-regulated kinase signaling with U0126. Among these treatments, BAPTA-AM, D-APV, and AIP fully blocked the cLTP-dependent increase in VPS34 endosomes, while the other inhibitors had no effect (Fig. S1 G). These findings suggest that activity-dependent NMDAR activation, intracellular calcium elevation, and CaMKII signaling are required for recruiting VPS34 to endosomes to generate PI(3)P upon cLTP.
To test whether CaMKII activation is sufficient to increase PI(3)P-positive puncta, we utilized a hyperactive CaMKII mutant, T286D, which has been shown to induce LTP when introduced with a viral expression system (Pettit et al., 1994; Hayashi et al., 2000) or by direct injection into postsynaptic cells (Lledo et al., 1995). We co-transfected rat hippocampal neurons with CaMKII-T286D (or an empty vector) together with dsRed-EEA1-FYVE. Expression of CaMKII-T286D for 24 h led to an increase in PI(3)P-positive endosomes in the absence of an external cLTP stimulus (Fig. 1 F). Taken together, these observations suggest that increased dendritic PI(3)P synthesis accompanies strong synaptic NMDAR activity and CaMKII activation.
CaMKII activation promotes the phosphorylation of a wide array of synaptic proteins, which modulates their activity and/or trafficking to drive synaptic plasticity (Coultrap and Bayer, 2012; Lisman et al., 2012). Therefore, we hypothesized that the cLTP-induced recruitment of VPS34 to endosomes might result from direct CaMKII-dependent phosphorylation. To test this, we performed immunoprecipitation of endogenous VPS34 followed by immunoblotting with a pan phospho-serine/threonine antibody. We observed no significant change in VPS34 phosphorylation levels 10 min after cLTP (Fig. S1, H and I). These results suggest that VPS34 endosomal recruitment is not regulated by direct CaMKII-mediated phosphorylation. Instead, CaMKII may act indirectly, potentially by phosphorylating VPS34-interacting partners or by modulating upstream regulators of endosomal trafficking.
Increased PI(3)P levels upon cLTP regulate functional and structural plasticity
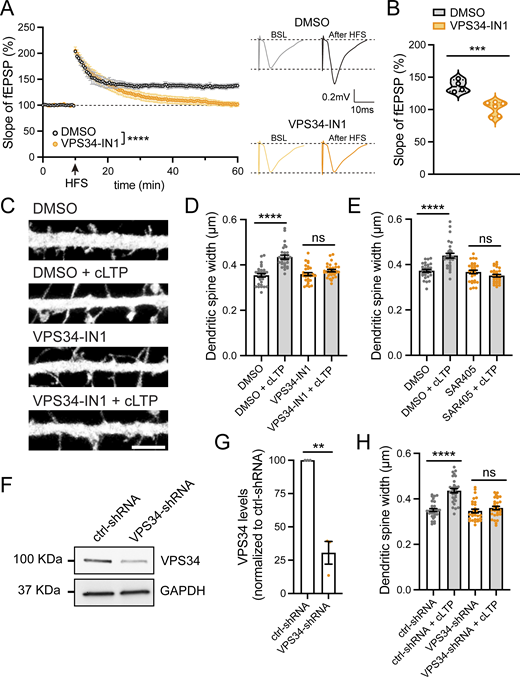
To analyze the functional consequences of reduced PI(3)P synthesis at synapses, we tested the effect of pharmacological inhibition of VPS34 on LTP induction using electrophysiological recordings in acute hippocampal slices from wild-type mice. Slices were incubated with either DMSO or 5 μM VPS34-IN1 for 30 min prior to recording. Following a stable 10-min baseline, LTP was induced by two high-frequency stimulations (100 Hz, 1 s) separated by 30 s (Temkin et al., 2017). VPS34-IN1 treatment impaired LTP induction compared with DMSO-treated controls (Fig. 2, A and B), indicating that PI(3)P synthesis is essential for LTP.
A decrease in PI(3)P levels blocks functional and structural plasticity during LTP. (A) Field excitatory postsynaptic potentials (fEPSPs) were recorded in the CA1 stratum radiatum using ACSF-filled glass pipettes (3–5 MΩ). Schaffer collateral fibers were stimulated every 30 s with 0.1-ms pulses (50–250 μA), and slices were pretreated with DMSO (n = 4 slices, 4 mice) or 5 μM VPS34-IN1 (n = 6 slices, 4 mice) for 30 min. After a 10-min stable baseline, LTP was induced with two high-frequency stimulations (HFS; 100 Hz, 1 s) separated by 30 s. Data were analyzed by two-way ANOVA with Sidak’s post hoc test, ****P < 0.0001. Error bars are SEM. (B) Summary data (55–60 min after HFS) corresponding to (A). DMSO: 136.441 ± 4.657; VPS34-IN1: 102.142 ± 4.388. Data were analyzed by Student’s unpaired t test, ***P < 0.001. (C) Representative confocal images of dendritic spines in DIV16 hippocampal neurons transfected with eGFP (filler) at DIV12. Neurons were treated with either DMSO or 1 μM VPS34-IN1 for 30 min, followed by a 5-min cLTP stimulus in the presence of the compounds where indicated. Neurons were further incubated in the presence of DMSO or VPS34-IN1 for 50 min before fixation. Scale bar, 5 µm. (D) The maximum width for each spine was quantified, and the average size of the dendritic spines in the first 30 μm of secondary dendrites was calculated. DMSO: 0.354 ± 0.007, N = 30 neurons; DMSO with cLTP: 0.435 ± 0.009, N = 30 neurons; VPS34-IN1: 0.359 ± 0.007, N = 30 neurons; VPS34-IN1 with cLTP: 0.375 ± 0.006, N = 30 neurons. Three independent experiments. Data were analyzed by one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (E) Neurons were transfected at DIV12 with eGFP (filler). At DIV16, neurons were treated with either DMSO or 1 μM SAR405 for 30 min, followed by a 5-min cLTP stimulus in the presence of compounds where indicated. Neurons were further incubated in the presence of DMSO or SAR405 for 50 min before fixation. The maximum width for each spine was quantified, and the average size of the dendritic spines in the first 30 μm of secondary dendrites was calculated. DMSO: 0.373 ± 0.006, N = 30 neurons; DMSO with cLTP: 0.439 ± 0.011, N = 30 neurons; SAR405: 0.367 ± 0.008, N = 30 neurons; SAR405 with cLTP: 0.351 ± 0.006, N = 30 neurons. Three independent experiments. Data were analyzed by one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (F) Validation of an shRNA clone to knockdown rat VPS34. Rat cortical neurons were infected with lentiviruses carrying either VPS34-shRNA (TRCN0000025373; Millipore Sigma) or control-shRNA (pLKO.1 scrambled nontarget shRNA SHC002, Millipore Sigma) at an MOI of 2. At 6 days after infection, cell extracts were collected and analyzed by western blot. GAPDH was used as a loading control. (G) The levels of VPS34 protein were quantified in neurons infected with VPS34-shRNA and normalized to VPS34 levels in control-shRNA–infected neurons. ctrl-shRNA: 100%; VPS34-shRNA: 30.5 ± 8.50%. N = 3 independent experiments. Statistical significance was determined using unpaired two-tailed Student’s t test, **P < 0.01. Error bars are SEM. (H) DIV12 neurons were co-transfected with eGFP (filler) and either ctrl-shRNA or VPS34-shRNA. At DIV16, neurons were either treated with cLTP or left untreated and fixed 50 min after cLTP. The maximum width for each spine was quantified and the average size of the dendritic spines in the first 30 μm of secondary dendrites was calculated. ctrl-shRNA: 0.351 ± 0.007, N = 30 neurons; ctrl-shRNA with cLTP: 0.436 ± 0.010, N = 30 neurons; VPS34-shRNA: 0.347 ± 0.008, N = 30 neurons; VPS34-shRNA with cLTP: 0.360 ± 0.007, N = 30 neurons. Three independent experiments. Data were analyzed by one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. DIV, days in vitro. Source data are available for this figure: SourceData F2.
A decrease in PI(3)P levels blocks functional and structural plasticity during LTP. (A) Field excitatory postsynaptic potentials (fEPSPs) were recorded in the CA1 stratum radiatum using ACSF-filled glass pipettes (3–5 MΩ). Schaffer collateral fibers were stimulated every 30 s with 0.1-ms pulses (50–250 μA), and slices were pretreated with DMSO (n = 4 slices, 4 mice) or 5 μM VPS34-IN1 (n = 6 slices, 4 mice) for 30 min. After a 10-min stable baseline, LTP was induced with two high-frequency stimulations (HFS; 100 Hz, 1 s) separated by 30 s. Data were analyzed by two-way ANOVA with Sidak’s post hoc test, ****P < 0.0001. Error bars are SEM. (B) Summary data (55–60 min after HFS) corresponding to (A). DMSO: 136.441 ± 4.657; VPS34-IN1: 102.142 ± 4.388. Data were analyzed by Student’s unpaired t test, ***P < 0.001. (C) Representative confocal images of dendritic spines in DIV16 hippocampal neurons transfected with eGFP (filler) at DIV12. Neurons were treated with either DMSO or 1 μM VPS34-IN1 for 30 min, followed by a 5-min cLTP stimulus in the presence of the compounds where indicated. Neurons were further incubated in the presence of DMSO or VPS34-IN1 for 50 min before fixation. Scale bar, 5 µm. (D) The maximum width for each spine was quantified, and the average size of the dendritic spines in the first 30 μm of secondary dendrites was calculated. DMSO: 0.354 ± 0.007, N = 30 neurons; DMSO with cLTP: 0.435 ± 0.009, N = 30 neurons; VPS34-IN1: 0.359 ± 0.007, N = 30 neurons; VPS34-IN1 with cLTP: 0.375 ± 0.006, N = 30 neurons. Three independent experiments. Data were analyzed by one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (E) Neurons were transfected at DIV12 with eGFP (filler). At DIV16, neurons were treated with either DMSO or 1 μM SAR405 for 30 min, followed by a 5-min cLTP stimulus in the presence of compounds where indicated. Neurons were further incubated in the presence of DMSO or SAR405 for 50 min before fixation. The maximum width for each spine was quantified, and the average size of the dendritic spines in the first 30 μm of secondary dendrites was calculated. DMSO: 0.373 ± 0.006, N = 30 neurons; DMSO with cLTP: 0.439 ± 0.011, N = 30 neurons; SAR405: 0.367 ± 0.008, N = 30 neurons; SAR405 with cLTP: 0.351 ± 0.006, N = 30 neurons. Three independent experiments. Data were analyzed by one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (F) Validation of an shRNA clone to knockdown rat VPS34. Rat cortical neurons were infected with lentiviruses carrying either VPS34-shRNA (TRCN0000025373; Millipore Sigma) or control-shRNA (pLKO.1 scrambled nontarget shRNA SHC002, Millipore Sigma) at an MOI of 2. At 6 days after infection, cell extracts were collected and analyzed by western blot. GAPDH was used as a loading control. (G) The levels of VPS34 protein were quantified in neurons infected with VPS34-shRNA and normalized to VPS34 levels in control-shRNA–infected neurons. ctrl-shRNA: 100%; VPS34-shRNA: 30.5 ± 8.50%. N = 3 independent experiments. Statistical significance was determined using unpaired two-tailed Student’s t test, **P < 0.01. Error bars are SEM. (H) DIV12 neurons were co-transfected with eGFP (filler) and either ctrl-shRNA or VPS34-shRNA. At DIV16, neurons were either treated with cLTP or left untreated and fixed 50 min after cLTP. The maximum width for each spine was quantified and the average size of the dendritic spines in the first 30 μm of secondary dendrites was calculated. ctrl-shRNA: 0.351 ± 0.007, N = 30 neurons; ctrl-shRNA with cLTP: 0.436 ± 0.010, N = 30 neurons; VPS34-shRNA: 0.347 ± 0.008, N = 30 neurons; VPS34-shRNA with cLTP: 0.360 ± 0.007, N = 30 neurons. Three independent experiments. Data were analyzed by one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. DIV, days in vitro. Source data are available for this figure: SourceData F2.
During LTP, dendritic spines undergo activity-dependent structural changes, which involve actin cytoskeleton reorganization, membrane trafficking, and membrane remodeling. LTP is associated with a structural increase in the dendritic spine head area (Nakahata and Yasuda, 2018; Bosch et al., 2014; Meyer et al., 2014). Thus, we tested whether dynamic PI(3)P synthesis is necessary for changes in dendritic spine width that accompany LTP. We employed three different approaches to decrease PI(3)P levels. Cultured hippocampal neurons were treated with either DMSO or 1 µM VPS34-IN1 for 30 min, followed by cLTP/mock stimulation, and then the width of dendritic spines within the first 30 µm of secondary dendrites were quantified. In DMSO-treated cells, cLTP resulted in a significant increase in dendritic spine width, whereas treatment with the VPS34 inhibitor completely blocked these structural changes in spines (Fig. 2, C and D). Similarly, treatment with 1 µM SAR405 also prevented the cLTP-dependent increase in dendritic spine width (Fig. 2 E).
As an alternative means to block PI(3)P synthesis, we employed shRNA to knockdown VPS34 in rat neurons (Fig. 2, F and G). As expected, cells expressing a scrambled (control) shRNA exhibited a significant increase in spine head area 50 min after cLTP induction. However, this structural plasticity was lost in VPS34 knockdown neurons (Fig. 2 H). Together, these data indicate that PI(3)P synthesis by VPS34 is critical for the structural changes that underlie the enduring enhancement of synaptic function following cLTP.
We next sought to determine the temporal window during which PI(3)P synthesis is required for cLTP-induced structural plasticity. We tested 2 conditions: pretreatment with VPS34-IN1 for 30 min prior to cLTP induction and initiation of VPS34-IN1 treatment 5 min after removal of the cLTP stimulus. Inhibition of VPS34 prior to cLTP induction effectively blocked the cLTP-dependent structural changes in dendritic spines. In contrast, when VPS34-IN1 was applied after cLTP induction, structural plasticity of spines was preserved (Fig. S2, A and B). These results suggest that early PI(3)P synthesis is critical for the cLTP-dependent structural remodeling of spines.

Sustained inhibition of PI(3)P synthesis is necessary to block the cLTP-dependent structural plasticity of spines. (A) Diagram of experiment. DIV16 hippocampal neurons were transfected with eGFP as a filler to visualize dendritic morphology. In the long treatment condition, 24 h after transfection, cells were either treated with DMSO or 1 μM VPS34-IN1 for 30 min, followed by a 5-min cLTP stimulus in the presence of the compounds where indicated. Neurons were then further incubated in the presence of DMSO or VPS34-IN1 for an additional 50 min before fixation. In the short treatment condition, treatment with DMSO or 1 μM VPS34-IN1 was initiated 5 min after the cLTP stimulus. (B) The maximum width for each spine was quantified, and the average size of the dendritic spines in the first 30 μm of secondary dendrites was calculated. DMSO (short): 0.360 ± 0.007, N = 30 neurons; DMSO (short) with cLTP: 0.417 ± 0.012, N = 30 neurons; DMSO (long): 0.353 ± 0.006, N = 30 neurons; DMSO (long) with cLTP: 0.453 ± 0.013, N = 30 neurons; VPS34-IN1 (short): 0.345 ± 0.008, N = 30 neurons; VPS34-IN1 (short) with cLTP: 0.425 ± 0.011, N = 30 neurons; VPS34-IN1 (long): 0.355 ± 0.009, N = 30 neurons; VPS34-IN1 (long) with cLTP: 0.351 ± 0.008, N = 30 neurons. Three independent experiments. Data were analyzed by one-way ANOVA with Tukey’s post hoc test, ***P < 0.005, ****P < 0.001. Error bars are SEM. DIV, days in vitro.
Sustained inhibition of PI(3)P synthesis is necessary to block the cLTP-dependent structural plasticity of spines. (A) Diagram of experiment. DIV16 hippocampal neurons were transfected with eGFP as a filler to visualize dendritic morphology. In the long treatment condition, 24 h after transfection, cells were either treated with DMSO or 1 μM VPS34-IN1 for 30 min, followed by a 5-min cLTP stimulus in the presence of the compounds where indicated. Neurons were then further incubated in the presence of DMSO or VPS34-IN1 for an additional 50 min before fixation. In the short treatment condition, treatment with DMSO or 1 μM VPS34-IN1 was initiated 5 min after the cLTP stimulus. (B) The maximum width for each spine was quantified, and the average size of the dendritic spines in the first 30 μm of secondary dendrites was calculated. DMSO (short): 0.360 ± 0.007, N = 30 neurons; DMSO (short) with cLTP: 0.417 ± 0.012, N = 30 neurons; DMSO (long): 0.353 ± 0.006, N = 30 neurons; DMSO (long) with cLTP: 0.453 ± 0.013, N = 30 neurons; VPS34-IN1 (short): 0.345 ± 0.008, N = 30 neurons; VPS34-IN1 (short) with cLTP: 0.425 ± 0.011, N = 30 neurons; VPS34-IN1 (long): 0.355 ± 0.009, N = 30 neurons; VPS34-IN1 (long) with cLTP: 0.351 ± 0.008, N = 30 neurons. Three independent experiments. Data were analyzed by one-way ANOVA with Tukey’s post hoc test, ***P < 0.005, ****P < 0.001. Error bars are SEM. DIV, days in vitro.
Increased PI(3)P synthesis drives the recruitment of SNX17 and SNX27 to endosomal compartments upon cLTP
SNX17 and SNX27 define two major endomembrane recycling pathways in non-neuronal cells (Steinberg et al., 2013; McNally et al., 2017). Both SNX17 and SNX27 belong to the PX-FERM subfamily of sorting nexins and are recruited to early endosomal membranes through their PX domains, which bind PI(3)P (Fig. S3 A) (Lunn et al., 2007; Gillooly et al., 2000; Ponting, 1996; Knauth et al., 2005). In addition to their PX domains, both proteins contain FERM domains. While the SNX17 FERM domain binds to cargoes containing NPxY/NxxY motifs (Ghai et al., 2013; Wang et al., 2018), the SNX27 FERM domain interacts with SNX1/2 on endosomal membranes (Yong et al., 2021). Unique to the PX-FERM subfamily, SNX27 also contains a PDZ domain, which is involved in cargo recognition (Fig. S3 A) (Steinberg et al., 2013). Notably, both SNX27 and SNX17 have been implicated in LTP. SNX27 is critical for LTP via its role in recycling AMPA receptors (Wang et al., 2013; Hussain et al., 2014; McMillan et al., 2021; Huo et al., 2020), while SNX17 is critical for LTP via its role in the recycling of β1-integrin (Rivero-Ríos et al., 2023).

PI(3)P and its lipid kinase VPS34 colocalize with SNX17 and SNX27 in dendrites. (A) Domain architecture of the SNX-FERM proteins used in this study. (B) Rat hippocampal neurons were transfected at DIV16 with dsRed-EEA1-FYVE and either GFP-SNX17 or GFP-SNX27 and fixed 24 h later. Scale bar, 5 µm. (C) The colocalization between dsRed-EEA1-FYVE– and either GFP-SNX17– or GFP-SNX27–positive puncta was analyzed using Mander’s colocalization coefficient (×100). GFP-SNX17: 79.55 ± 1.617%, N = 30 neurons; GFP-SNX27: 83.00 ± 1.573%, N = 30 neurons. (D) Rat hippocampal neurons were co-transfected at DIV16 with mScarlet-SNX27, mNeonGreen-SNX17, and HA-VPS34. 24 h later, cells were fixed, permeabilized, and immunostained with an anti-HA antibody. Scale bar, 2.5 µm. (E) The percentage of mNeonGreen-SNX17 overlapping with mScarlet-SNX27 was analyzed using Mander’s colocalization coefficient (×100). 81.22 ± 3.386%, N = 15 neurons. Three independent experiments. Error bar is SEM. (F) The percentage of mScarlet-SNX27 overlapping with mNeonGreen-SNX17 was analyzed using Mander’s colocalization coefficient (×100). 82.49 ± 2.480%, N = 15 neurons. Three independent experiments. Error bar is SEM. (G) The percentage of endosomes containing both mNeonGreen-SNX17 and mScarlet-SNX27 that colocalize with HA-VPS34 was analyzed using Mander’s colocalization coefficient (×100). 72.83 ± 3.323%, N = 15 neurons. Three independent experiments. Error bar is SEM. DIV, days in vitro.
PI(3)P and its lipid kinase VPS34 colocalize with SNX17 and SNX27 in dendrites. (A) Domain architecture of the SNX-FERM proteins used in this study. (B) Rat hippocampal neurons were transfected at DIV16 with dsRed-EEA1-FYVE and either GFP-SNX17 or GFP-SNX27 and fixed 24 h later. Scale bar, 5 µm. (C) The colocalization between dsRed-EEA1-FYVE– and either GFP-SNX17– or GFP-SNX27–positive puncta was analyzed using Mander’s colocalization coefficient (×100). GFP-SNX17: 79.55 ± 1.617%, N = 30 neurons; GFP-SNX27: 83.00 ± 1.573%, N = 30 neurons. (D) Rat hippocampal neurons were co-transfected at DIV16 with mScarlet-SNX27, mNeonGreen-SNX17, and HA-VPS34. 24 h later, cells were fixed, permeabilized, and immunostained with an anti-HA antibody. Scale bar, 2.5 µm. (E) The percentage of mNeonGreen-SNX17 overlapping with mScarlet-SNX27 was analyzed using Mander’s colocalization coefficient (×100). 81.22 ± 3.386%, N = 15 neurons. Three independent experiments. Error bar is SEM. (F) The percentage of mScarlet-SNX27 overlapping with mNeonGreen-SNX17 was analyzed using Mander’s colocalization coefficient (×100). 82.49 ± 2.480%, N = 15 neurons. Three independent experiments. Error bar is SEM. (G) The percentage of endosomes containing both mNeonGreen-SNX17 and mScarlet-SNX27 that colocalize with HA-VPS34 was analyzed using Mander’s colocalization coefficient (×100). 72.83 ± 3.323%, N = 15 neurons. Three independent experiments. Error bar is SEM. DIV, days in vitro.
Given that PI(3)P is a shared binding partner of both pathways, we hypothesized that PI(3)P may regulate both SNX17- and SNX27-dependent recycling during cLTP.
As a first approach, we investigated whether SNX17 and SNX27 localize to PI(3)P-positive endosomes in dendrites. As there is no suitable antibody available for immunolocalization of endogenous rat SNX27, we generated GFP-tagged constructs for SNX17 and SNX27. Note that the addition of an N-terminal tag to these proteins does not interfere with their ability to bind cargo or endosomal membranes (McNally et al., 2017; McMillan et al., 2021). Co-transfection of neurons with dsRed-EEA1-FYVE and either GFP-SNX17 or GFP-SNX27 for 24 h revealed that ∼80% of the SNX17- or SNX27-positive puncta overlap with PI(3)P (Fig. S3, B and C).
As an additional means to test the link between PI(3)P synthesis and the recruitment of SNX17 and SNX27 to endosomes, we analyzed the colocalization of these sorting nexins with VPS34. Neurons were co-transfected with HA-VPS34, mNeonGreen-SNX17, and mScarlet-SNX27 and imaged to assess triple colocalization. Given that SNX17 and SNX27 exhibit a high degree of overlap, with ∼80% colocalization (Fig. S3, D–F), we generated a binary mask of compartments that are positive for both SNX17 and SNX27 to define endosomes actively engaged in recycling and quantified their colocalization with HA-VPS34. Approximately 70% of these SNX17/SNX27-positive endosomes also contained VPS34 (Fig. S3, D and G), supporting a strong spatial association between PI(3)P synthesis and the recruitment of endocytic recycling machinery.
To investigate whether the increase in PI(3)P-positive compartments following cLTP results in enhanced endosomal recruitment of SNX17 and SNX27, we transiently transfected neurons with dsRed-EEA1-FYVE and GFP-SNX17 and treated them with or without cLTP. We then analyzed the numbers of PI(3)P and GFP-SNX17 puncta at 10 and 30 min after cLTP. We observed a significant increase in PI(3)P puncta over time (Fig. 3, A and B), which corresponded with a similar increase in GFP-SNX17 puncta (Fig. 3, A and C). Notably, SNX17 showed strong colocalization with PI(3)P under basal conditions and at 10 and 30 min after cLTP (Fig. 3, A and D). In parallel, we co-transfected neurons with dsRed-EEA1-FYVE and GFP-SNX27 and quantified puncta numbers 10 and 30 min after cLTP stimulation. The numbers of GFP-SNX27–positive puncta also increased upon cLTP and showed strong overlap with PI(3)P at each time point tested (Fig. 3, E–H).

Increased PI(3)P synthesis upon cLTP regulates the formation of SNX17- and SNX27-positive puncta. (A) Representative confocal images of DIV17 hippocampal neurons that were transfected at DIV16 with GFP-SNX17 and dsRed-EEA1-FYVE. 24 h later, neurons were either fixed in the absence of cLTP (baseline) or treated with cLTP for 5 min and fixed 10 or 30 min after the cLTP stimulus. Scale bar, 5 µm. (B) The number of dsRed-EEA1-FYVE–positive puncta in the first 30 μm of secondary dendrites was quantified at the indicated time points. Baseline: 0.241 ± 0.010, N = 30 neurons; cLTP 10 min: 0.327 ± 0.013, N = 30 neurons; cLTP 30 min: 0.361 ± 0.010, N = 30 neurons. Three independent experiments. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (C) The number of GFP-SNX17 puncta in the first 30 μm of secondary dendrites was quantified at the indicated time points. Baseline: 0.227 ± 0.009, N = 30 neurons; cLTP 10 min: 0.317 ± 0.013, N = 30 neurons; cLTP 30 min: 0.349 ± 0.010, N = 30 neurons. Three independent experiments. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (D) The colocalization between dsRed-EEA1-FYVE– and GFP-SNX17–positive puncta was analyzed using Mander’s colocalization coefficient (×100). Baseline: 84.25 ± 2.019%, N = 30 neurons; cLTP 10 min: 84.94 ± 2.041%, N = 30 neurons; cLTP 30 min: 82.87 ± 1.751%, N = 30 neurons. Three independent experiments. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test. Error bars are SEM. (E) Representative confocal images of DIV17 hippocampal neurons that were transfected at DIV16 with GFP-SNX27 and dsRed-EEA1-FYVE. 24 h later, neurons were either fixed in the absence of cLTP (baseline) or treated with cLTP for 5 min and fixed 10 or 30 min after the cLTP stimulus. Scale bar, 5 µm. (F) The number of dsRed-EEA1-FYVE–positive puncta in the first 30 μm of secondary dendrites was quantified at the indicated time points. Baseline: 0.268 ± 0.010, N = 30 neurons; cLTP 10 min: 0.353 ± 0.013, N = 30 neurons; cLTP 30 min: 0.389 ± 0.015, N = 30 neurons. Three independent experiments. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (G) The number of GFP-SNX27 puncta in the first 30 μm of secondary dendrites was quantified at the indicated time points. Baseline: 0.258 ± 0.010, N = 30 neurons; cLTP 10 min: 0.340 ± 0.013, N = 30 neurons; cLTP 30 min: 0.378 ± 0.014, N = 30 neurons. Three independent experiments. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (H) The colocalization between dsRed-EEA1-FYVE– and GFP-SNX27–positive puncta was analyzed using Mander’s colocalization coefficient (×100). Baseline: 84.60 ± 1.668%, N = 30 neurons; cLTP 10 min: 86.17 ± 1.483%, N = 30 neurons; cLTP 30 min: 85.68 ± 2.027%, N = 30 neurons. Three independent experiments. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test. Error bars are SEM. DIV, days in vitro.
Increased PI(3)P synthesis upon cLTP regulates the formation of SNX17- and SNX27-positive puncta. (A) Representative confocal images of DIV17 hippocampal neurons that were transfected at DIV16 with GFP-SNX17 and dsRed-EEA1-FYVE. 24 h later, neurons were either fixed in the absence of cLTP (baseline) or treated with cLTP for 5 min and fixed 10 or 30 min after the cLTP stimulus. Scale bar, 5 µm. (B) The number of dsRed-EEA1-FYVE–positive puncta in the first 30 μm of secondary dendrites was quantified at the indicated time points. Baseline: 0.241 ± 0.010, N = 30 neurons; cLTP 10 min: 0.327 ± 0.013, N = 30 neurons; cLTP 30 min: 0.361 ± 0.010, N = 30 neurons. Three independent experiments. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (C) The number of GFP-SNX17 puncta in the first 30 μm of secondary dendrites was quantified at the indicated time points. Baseline: 0.227 ± 0.009, N = 30 neurons; cLTP 10 min: 0.317 ± 0.013, N = 30 neurons; cLTP 30 min: 0.349 ± 0.010, N = 30 neurons. Three independent experiments. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (D) The colocalization between dsRed-EEA1-FYVE– and GFP-SNX17–positive puncta was analyzed using Mander’s colocalization coefficient (×100). Baseline: 84.25 ± 2.019%, N = 30 neurons; cLTP 10 min: 84.94 ± 2.041%, N = 30 neurons; cLTP 30 min: 82.87 ± 1.751%, N = 30 neurons. Three independent experiments. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test. Error bars are SEM. (E) Representative confocal images of DIV17 hippocampal neurons that were transfected at DIV16 with GFP-SNX27 and dsRed-EEA1-FYVE. 24 h later, neurons were either fixed in the absence of cLTP (baseline) or treated with cLTP for 5 min and fixed 10 or 30 min after the cLTP stimulus. Scale bar, 5 µm. (F) The number of dsRed-EEA1-FYVE–positive puncta in the first 30 μm of secondary dendrites was quantified at the indicated time points. Baseline: 0.268 ± 0.010, N = 30 neurons; cLTP 10 min: 0.353 ± 0.013, N = 30 neurons; cLTP 30 min: 0.389 ± 0.015, N = 30 neurons. Three independent experiments. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (G) The number of GFP-SNX27 puncta in the first 30 μm of secondary dendrites was quantified at the indicated time points. Baseline: 0.258 ± 0.010, N = 30 neurons; cLTP 10 min: 0.340 ± 0.013, N = 30 neurons; cLTP 30 min: 0.378 ± 0.014, N = 30 neurons. Three independent experiments. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (H) The colocalization between dsRed-EEA1-FYVE– and GFP-SNX27–positive puncta was analyzed using Mander’s colocalization coefficient (×100). Baseline: 84.60 ± 1.668%, N = 30 neurons; cLTP 10 min: 86.17 ± 1.483%, N = 30 neurons; cLTP 30 min: 85.68 ± 2.027%, N = 30 neurons. Three independent experiments. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test. Error bars are SEM. DIV, days in vitro.
Since VPS34 inhibition leads to a loss of PI(3)P puncta in dendrites (Fig. 1, A and B), we tested whether PI(3)P produced by VPS34 is required for the cLTP-dependent recruitment of SNX17 and SNX27. We transiently transfected neurons with mNeonGreen-SNX17 and mScarlet-SNX27 and analyzed puncta numbers at 10 and 30 min following cLTP in the presence or absence of VPS34-IN1. VPS34-IN1 treatment significantly reduced the numbers of SNX17- and SNX27-positive puncta and also blocked the cLTP-dependent increase in puncta numbers (Fig. S4, A–C). mNeonGreen-SNX17 and mScarlet-SNX27 showed ∼80% colocalization under baseline conditions and at each time point tested (Fig. S4, D and E), which suggests that they are coordinately recruited to PI(3)P-positive endosomes. Together, these results suggest that PI(3)P generation by VPS34 is necessary for the increase in SNX17- and SNX27-positive puncta following cLTP.

Decreased PI(3)P levels block the cLTP-dependent increase in SNX17- and SNX27-positive puncta. (A) Representative confocal images of DIV17 hippocampal neurons that were transfected at DIV16 with mNeonGreen-SNX17 and mScarlet-SNX27. 24 h later, neurons were treated with either DMSO or 1 μM VPS34-IN1 for 30 min. Neurons were either fixed in the absence of cLTP (baseline) or treated with cLTP for 5 min and fixed 10 or 30 min after the cLTP stimulus. DMSO or VPS34-IN1 were maintained during the course of the experiment. Scale bar, 5 µm. (B) The number of mNeonGreen-SNX17 puncta in the first 30 μm of secondary dendrites was quantified at the indicated time points. DMSO baseline: 0.238 ± 0.013, N = 30 neurons; DMSO cLTP 10 min: 0.362 ± 0.013, N = 30 neurons; DMSO cLTP 30 min: 0.379 ± 0.014, N = 30 neurons; VPS34-IN1 baseline: 0.090 ± 0.007, N = 30 neurons; VPS34-IN1 cLTP 10 min: 0.114 ± 0.010, N = 30 neurons; VPS34-IN1 cLTP 30 min: 0.119 ± 0.010, N = 30 neurons. Three independent experiments. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (C) The number of mScarlet-SNX27 puncta in the first 30 μm of secondary dendrites was quantified at the indicated time points. DMSO baseline: 0.243 ± 0.012, N = 30 neurons; DMSO cLTP 10 min: 0.367 ± 0.013, N = 30 neurons; DMSO cLTP 30 min: 0.389 ± 0.014, N = 30 neurons; VPS34-IN1 baseline: 0.101 ± 0.007, N = 30 neurons; VPS34-IN1 cLTP 10 min: 0.124 ± 0.011, N = 30 neurons; VPS34-IN1 cLTP 30 min: 0.124 ± 0.010, N = 30 neurons. Three independent experiments. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (D) DIV16 hippocampal neurons were transfected with mNeonGreen-SNX17 and mScarlet-SNX27. 24 h later, neurons were either fixed in the absence of cLTP (baseline) or treated with cLTP for 5 min and fixed 0, 10, 30 or 60 min after the cLTP stimulus. The percentage of SNX17 that overlaps with SNX27 at the different time points was determined using Mander’s colocalization coefficient (×100). Baseline: 80.94 ± 2.058%, N = 26 neurons; 0 min after cLTP: 80.42 ± 1.678%, N = 28 neurons; 10 min after cLTP: 83.64 ± 1.582%, N = 30 neurons; 30 min after cLTP: 85.79 ± 1.474%, N = 30 neurons; 60 min after cLTP: 79.57 ± 1.512%, N = 24 neurons. Three independent experiments. Data were analyzed by one-way ANOVA with Tukey’s post hoc test. Error bars are SEM. (E) Same as D, but the percentage of SNX27 that overlaps with SNX17 at the different time points was determined using Mander’s colocalization coefficient (×100). Baseline: 90.52 ± 2.066%, N = 26 neurons; 0 min after cLTP: 89.63 ± 1.605%, N = 28 neurons; 10 min after cLTP: 91.05 ± 1.689%, N = 30 neurons; 30 min after cLTP: 90.43 ± 1.525%, N = 30 neurons; 60 min after cLTP: 88.47 ± 2.162%, N = 24 neurons. Three independent experiments. Data were analyzed by one-way ANOVA with Tukey’s post hoc test. Error bars are SEM. DIV, days in vitro.
Decreased PI(3)P levels block the cLTP-dependent increase in SNX17- and SNX27-positive puncta. (A) Representative confocal images of DIV17 hippocampal neurons that were transfected at DIV16 with mNeonGreen-SNX17 and mScarlet-SNX27. 24 h later, neurons were treated with either DMSO or 1 μM VPS34-IN1 for 30 min. Neurons were either fixed in the absence of cLTP (baseline) or treated with cLTP for 5 min and fixed 10 or 30 min after the cLTP stimulus. DMSO or VPS34-IN1 were maintained during the course of the experiment. Scale bar, 5 µm. (B) The number of mNeonGreen-SNX17 puncta in the first 30 μm of secondary dendrites was quantified at the indicated time points. DMSO baseline: 0.238 ± 0.013, N = 30 neurons; DMSO cLTP 10 min: 0.362 ± 0.013, N = 30 neurons; DMSO cLTP 30 min: 0.379 ± 0.014, N = 30 neurons; VPS34-IN1 baseline: 0.090 ± 0.007, N = 30 neurons; VPS34-IN1 cLTP 10 min: 0.114 ± 0.010, N = 30 neurons; VPS34-IN1 cLTP 30 min: 0.119 ± 0.010, N = 30 neurons. Three independent experiments. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (C) The number of mScarlet-SNX27 puncta in the first 30 μm of secondary dendrites was quantified at the indicated time points. DMSO baseline: 0.243 ± 0.012, N = 30 neurons; DMSO cLTP 10 min: 0.367 ± 0.013, N = 30 neurons; DMSO cLTP 30 min: 0.389 ± 0.014, N = 30 neurons; VPS34-IN1 baseline: 0.101 ± 0.007, N = 30 neurons; VPS34-IN1 cLTP 10 min: 0.124 ± 0.011, N = 30 neurons; VPS34-IN1 cLTP 30 min: 0.124 ± 0.010, N = 30 neurons. Three independent experiments. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (D) DIV16 hippocampal neurons were transfected with mNeonGreen-SNX17 and mScarlet-SNX27. 24 h later, neurons were either fixed in the absence of cLTP (baseline) or treated with cLTP for 5 min and fixed 0, 10, 30 or 60 min after the cLTP stimulus. The percentage of SNX17 that overlaps with SNX27 at the different time points was determined using Mander’s colocalization coefficient (×100). Baseline: 80.94 ± 2.058%, N = 26 neurons; 0 min after cLTP: 80.42 ± 1.678%, N = 28 neurons; 10 min after cLTP: 83.64 ± 1.582%, N = 30 neurons; 30 min after cLTP: 85.79 ± 1.474%, N = 30 neurons; 60 min after cLTP: 79.57 ± 1.512%, N = 24 neurons. Three independent experiments. Data were analyzed by one-way ANOVA with Tukey’s post hoc test. Error bars are SEM. (E) Same as D, but the percentage of SNX27 that overlaps with SNX17 at the different time points was determined using Mander’s colocalization coefficient (×100). Baseline: 90.52 ± 2.066%, N = 26 neurons; 0 min after cLTP: 89.63 ± 1.605%, N = 28 neurons; 10 min after cLTP: 91.05 ± 1.689%, N = 30 neurons; 30 min after cLTP: 90.43 ± 1.525%, N = 30 neurons; 60 min after cLTP: 88.47 ± 2.162%, N = 24 neurons. Three independent experiments. Data were analyzed by one-way ANOVA with Tukey’s post hoc test. Error bars are SEM. DIV, days in vitro.
Increased PI(3)P synthesis drives the recruitment of SNX17 and SNX27 to dendritic spines upon cLTP
We previously discovered that SNX17 is recruited to dendritic spines upon cLTP and correlates with dendritic spine enlargement (Rivero-Ríos et al., 2023). The strong colocalization between SNX17 and SNX27 after cLTP (Fig. S4, D and E) suggests that SNX27 may also be recruited to dendritic spines upon cLTP. To test this possibility, we performed live-cell imaging of mScarlet-SNX27 in neurons co-expressing soluble eGFP (Fig. S5 A). In the cLTP-treated cells, there was a marked increase in dendritic spine width 30 min after the stimulus (Fig. S5 B). Consistent with our findings for SNX17 (Rivero-Ríos et al., 2023), we observed increased recruitment of SNX27 to synapses following cLTP. The intensity of mScarlet-SNX27 at dendritic spines significantly increased 5 min after the stimulus, continued to rise at 10 min, and remained significantly elevated at all subsequent time points up to 30 min after cLTP (Fig. S5 C). This increased recruitment of SNX27 to spines was also accompanied by an increase in the number of mScarlet-SNX27–positive puncta following cLTP (Fig. S5 D). Similar to SNX17 (Rivero-Ríos et al., 2023), the expression of CaMKII-T286D was sufficient to promote the formation of mScarlet-SNX27–positive puncta and their subsequent recruitment to dendritic spines (Fig. S5, E and F).

SNX27 is recruited to dendritic spines upon cLTP, which is dependent on the CaMKII pathway. (A) Representative confocal images from DIV17 hippocampal neurons expressing mScarlet-SNX27 and eGFP (filler) under conditions of cLTP or HBS control (mock). Images of live neurons were captured before cLTP (baseline); during cLTP; and at 0, 5, 10, 15, 20, 25, and 30 min after the cLTP stimulus. Representative images of baseline, t = 10, and t = 30 are shown. Intensity presented in the fire LUT color scheme. Scale bar, 2.5 µm. (B) The endpoint of the experiment (30 min) was used to measure the cLTP-dependent increase in dendritic spine width. To calculate the increase in spine size, the final spine width was normalized to the baseline width for each spine. Mock: 1.024 ± 0.025, N = 98 spines across 15 cells; cLTP: 1.498 ± 0.056, N = 108 spines across 15 cells. Statistical significance was determined using unpaired two-tailed Student’s t test, ****P < 0.001. Error bars are SEM. (C) The mean intensity of mScarlet-SNX27 was measured in the spines that remained in the same plane at each time point following cLTP (or mock) and normalized to the baseline for each individual spine. Mock: N = 98 spines across 15 cells (baseline: 1,000, cLTP: 0.989 ± 0.023, 0: 1.037 ± 0.023, 5: 1.037 ± 0.025, 10: 0.991 ± 0.028, 15: 1.032 ± 0.027, 20: 1.022 ± 0.031, 25: 1.015 ± 0.029, 30: 1.017 ± 0.028); cLTP: N = 108 spines across 15 cells (baseline: 1,000, cLTP: 1.117 ± 0.034, 0: 1.252 ± 0.057, 5: 1.347 ± 0.070, 10: 1.506 ± 0.073, 15: 1.523 ± 0.065, 20: 1.566 ± 0.066, 25: 1.651 ± 0.073, 30: 1.673 ± 0.064). Two-way ANOVA with Sidak’s multiple comparison test was used to determine statistical significance, **P < 0.01, ****P < 0.001. Error bars are SEM. (D) The number of puncta containing mScarlet-SNX27 in 30 µm of dendritic spines at the indicated time points was quantified for the cLTP and mock conditions and normalized to the baseline for each dendrite. Mock: N = 15 dendrites (baseline: 1,000, 10: 1.020 ± 0.025, 30: 1.079 ± 0.028); cLTP: N = 15 dendrites (baseline: 1,000, 10: 1.312 ± 0.049, 30: 1.544 ± 0.074). Three independent experiments. Two-way ANOVA with Sidak’s multiple comparison test was used to determine statistical significance, ****P <0.001. Error bars are SEM. (E) DIV16 hippocampal neurons were co-transfected with mScarlet-SNX27 and either an empty pRK5 vector or pRK5-HA-CaMKII-T286D. 24 h later, neurons were fixed, permeabilized, and incubated with an anti-HA antibody. The mean intensity of mScarlet-SNX27 was measured in the dendritic spines present in the first 30 μm of secondary dendrites and normalized to mScarlet-SNX27 mean intensity in the dendritic shaft. Ctrl: 0.775 ± 0.035, N = 30 neurons; CaMKII-T286D: 0.909 ± 0.041, N = 30 neurons. Statistical significance was determined using unpaired two-tailed Student’s t test, *P < 0.05. Error bars are SEM. (F) Same as E, but the number of puncta containing mScarlet-SNX27 in 30 µm of dendritic spines was quantified. Ctrl: 0.257 ± 0.016, N = 30 neurons; CaMKII-T286D: 0.365 ± 0.018, N = 30 neurons. Statistical significance was determined using unpaired two-tailed Student’s t test, ****P < 0.001. Error bars are SEM. DIV, days in vitro.
SNX27 is recruited to dendritic spines upon cLTP, which is dependent on the CaMKII pathway. (A) Representative confocal images from DIV17 hippocampal neurons expressing mScarlet-SNX27 and eGFP (filler) under conditions of cLTP or HBS control (mock). Images of live neurons were captured before cLTP (baseline); during cLTP; and at 0, 5, 10, 15, 20, 25, and 30 min after the cLTP stimulus. Representative images of baseline, t = 10, and t = 30 are shown. Intensity presented in the fire LUT color scheme. Scale bar, 2.5 µm. (B) The endpoint of the experiment (30 min) was used to measure the cLTP-dependent increase in dendritic spine width. To calculate the increase in spine size, the final spine width was normalized to the baseline width for each spine. Mock: 1.024 ± 0.025, N = 98 spines across 15 cells; cLTP: 1.498 ± 0.056, N = 108 spines across 15 cells. Statistical significance was determined using unpaired two-tailed Student’s t test, ****P < 0.001. Error bars are SEM. (C) The mean intensity of mScarlet-SNX27 was measured in the spines that remained in the same plane at each time point following cLTP (or mock) and normalized to the baseline for each individual spine. Mock: N = 98 spines across 15 cells (baseline: 1,000, cLTP: 0.989 ± 0.023, 0: 1.037 ± 0.023, 5: 1.037 ± 0.025, 10: 0.991 ± 0.028, 15: 1.032 ± 0.027, 20: 1.022 ± 0.031, 25: 1.015 ± 0.029, 30: 1.017 ± 0.028); cLTP: N = 108 spines across 15 cells (baseline: 1,000, cLTP: 1.117 ± 0.034, 0: 1.252 ± 0.057, 5: 1.347 ± 0.070, 10: 1.506 ± 0.073, 15: 1.523 ± 0.065, 20: 1.566 ± 0.066, 25: 1.651 ± 0.073, 30: 1.673 ± 0.064). Two-way ANOVA with Sidak’s multiple comparison test was used to determine statistical significance, **P < 0.01, ****P < 0.001. Error bars are SEM. (D) The number of puncta containing mScarlet-SNX27 in 30 µm of dendritic spines at the indicated time points was quantified for the cLTP and mock conditions and normalized to the baseline for each dendrite. Mock: N = 15 dendrites (baseline: 1,000, 10: 1.020 ± 0.025, 30: 1.079 ± 0.028); cLTP: N = 15 dendrites (baseline: 1,000, 10: 1.312 ± 0.049, 30: 1.544 ± 0.074). Three independent experiments. Two-way ANOVA with Sidak’s multiple comparison test was used to determine statistical significance, ****P <0.001. Error bars are SEM. (E) DIV16 hippocampal neurons were co-transfected with mScarlet-SNX27 and either an empty pRK5 vector or pRK5-HA-CaMKII-T286D. 24 h later, neurons were fixed, permeabilized, and incubated with an anti-HA antibody. The mean intensity of mScarlet-SNX27 was measured in the dendritic spines present in the first 30 μm of secondary dendrites and normalized to mScarlet-SNX27 mean intensity in the dendritic shaft. Ctrl: 0.775 ± 0.035, N = 30 neurons; CaMKII-T286D: 0.909 ± 0.041, N = 30 neurons. Statistical significance was determined using unpaired two-tailed Student’s t test, *P < 0.05. Error bars are SEM. (F) Same as E, but the number of puncta containing mScarlet-SNX27 in 30 µm of dendritic spines was quantified. Ctrl: 0.257 ± 0.016, N = 30 neurons; CaMKII-T286D: 0.365 ± 0.018, N = 30 neurons. Statistical significance was determined using unpaired two-tailed Student’s t test, ****P < 0.001. Error bars are SEM. DIV, days in vitro.
Given that PI(3)P is required for the endosomal localization of both SNX17 and SNX27, we tested whether PI(3)P also drives their recruitment to dendritic spines. Since 30-min VPS34 inhibition led to a near-complete loss of mNeonGreen-SNX17– and mScarlet-SNX27–positive puncta (Fig. S4, A–C), we applied a shorter, 10-min VPS34-IN1 treatment immediately following cLTP induction (Fig. 4 A). Unlike the prolonged inhibition, this brief treatment did not significantly affect the number of SNX17- or SNX27-positive puncta or their fluorescence intensity within dendritic spines compared with DMSO-treated cells (Fig. S6, A–D). Furthermore, this short treatment did not block the cLTP-dependent increase in SNX17 or SNX27 puncta (Fig. S6, E and F). However, in the cells that were treated with VPS34-IN1 for 10 min, the intensities of SNX17 and SNX27 in dendritic spines remained at baseline levels (Fig. 4, B–E), suggesting that although existing PI(3)P is sufficient to maintain endosomal pools of SNX17 and SNX27, de novo PI(3)P synthesis following cLTP is required to drive their accumulation in spines. Note that initiating VPS34 inhibition 5 min after cLTP does not block structural plasticity (Fig. S2, A and B), and we detect an increase in PI(3)P-positive puncta immediately after the cLTP stimulus (Fig. 1 D). Together, these findings suggest that early PI(3)P production (during or shortly after cLTP) is sufficient to support spine remodeling. It is possible that initial recruitment of SNX17 and SNX27 occurs within this early window, allowing spine plasticity to proceed despite subsequent VPS34 inhibition. Alternatively, the brief post-cLTP inhibition may limit or delay further accumulation of SNX17 and SNX27 at spines without disrupting the threshold needed for structural changes.

Brief inhibition of PI(3)P synthesis upon cLTP blocks the recruitment of SNX17 and SNX27 to dendritic spines. (A) Diagram of experiment. DIV16 hippocampal neurons were transfected with mNeonGreen-SNX17 or mScarlet-SNX27 along with a filler to visualize dendritic morphology. 24 h later, cells were treated with or without cLTP stimulation. cLTP-treated cells were washed and incubated in HBS containing either DMSO or 1 μM VPS34-IN1 for 10 min. Cells were fixed, and the intensity of SNX17 or SNX27 at dendritic spines and in the dendritic shaft was quantified. (B) Example confocal images of DIV17 neurons expressing mNeonGreen-SNX17 and mCherry (filler) under the indicated conditions. mNeonGreen-SNX17 intensity is presented in the fire LUT color scheme to facilitate the visualization of dendritic spines. Scale bar, 2.5 µm. (C) Example confocal images of DIV17 neurons expressing mScarlet-SNX27 and eGFP (filler) under the indicated conditions. mScarlet-SNX27 intensity is presented in the fire LUT color scheme to facilitate the visualization of dendritic spines. Scale bar, 2.5 µm. (D) The mean intensity of mNeonGreen-SNX17 was measured in the dendritic spines present in the first 30 μm of secondary dendrites and normalized to mNeonGreen-SNX17 mean intensity in the dendritic shaft. Baseline: 0.657 ± 0.026, N = 30 neurons; cLTP+DMSO: 0.823 ± 0.036, N = 30 neurons; cLTP+VPS34-IN1: 0.673 ± 0.026, N = 30 neurons. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ***P < 0.005. Error bars are SEM. (E) The mean intensity of mScarlet-SNX27 was measured in the dendritic spines present in the first 30 μm of secondary dendrites and normalized to mScarlet-SNX27 mean intensity in the dendritic shaft. Baseline: 0.759 ± 0.035, N = 30 neurons; cLTP+DMSO: 0.976 ± 0.060, N = 30 neurons; cLTP+VPS34-IN1: 0.729 ± 0.049, N = 30 neurons. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, **P < 0.01. Error bars are SEM. DIV, days in vitro.
Brief inhibition of PI(3)P synthesis upon cLTP blocks the recruitment of SNX17 and SNX27 to dendritic spines. (A) Diagram of experiment. DIV16 hippocampal neurons were transfected with mNeonGreen-SNX17 or mScarlet-SNX27 along with a filler to visualize dendritic morphology. 24 h later, cells were treated with or without cLTP stimulation. cLTP-treated cells were washed and incubated in HBS containing either DMSO or 1 μM VPS34-IN1 for 10 min. Cells were fixed, and the intensity of SNX17 or SNX27 at dendritic spines and in the dendritic shaft was quantified. (B) Example confocal images of DIV17 neurons expressing mNeonGreen-SNX17 and mCherry (filler) under the indicated conditions. mNeonGreen-SNX17 intensity is presented in the fire LUT color scheme to facilitate the visualization of dendritic spines. Scale bar, 2.5 µm. (C) Example confocal images of DIV17 neurons expressing mScarlet-SNX27 and eGFP (filler) under the indicated conditions. mScarlet-SNX27 intensity is presented in the fire LUT color scheme to facilitate the visualization of dendritic spines. Scale bar, 2.5 µm. (D) The mean intensity of mNeonGreen-SNX17 was measured in the dendritic spines present in the first 30 μm of secondary dendrites and normalized to mNeonGreen-SNX17 mean intensity in the dendritic shaft. Baseline: 0.657 ± 0.026, N = 30 neurons; cLTP+DMSO: 0.823 ± 0.036, N = 30 neurons; cLTP+VPS34-IN1: 0.673 ± 0.026, N = 30 neurons. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ***P < 0.005. Error bars are SEM. (E) The mean intensity of mScarlet-SNX27 was measured in the dendritic spines present in the first 30 μm of secondary dendrites and normalized to mScarlet-SNX27 mean intensity in the dendritic shaft. Baseline: 0.759 ± 0.035, N = 30 neurons; cLTP+DMSO: 0.976 ± 0.060, N = 30 neurons; cLTP+VPS34-IN1: 0.729 ± 0.049, N = 30 neurons. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, **P < 0.01. Error bars are SEM. DIV, days in vitro.

Brief 10-min inhibition of VPS34 does not affect the formation of SNX17- and SNX27-positive puncta or their recruitment to dendritic spines. (A) DIV16 hippocampal neurons were co-transfected with mNeonGreen-SNX17 and mCherry (filler). 24 h later, cells were incubated in HBS containing either DMSO or 1 μM VPS34-IN1 for 10 min. Cells were fixed, and the number of mNeonGreen-SNX17–positive puncta in 30 µm of dendritic spines was quantified. DMSO: 0.291 ± 0.014, N = 30 neurons; VPS34-IN1: 0.281 ± 0.014, N = 30 neurons. Unpaired two-tailed Student’s t test determined that there are no significant differences between the DMSO and VPS34-IN1 conditions. Error bars are SEM. (B) Same as B, but the mean intensity of mNeonGreen-SNX17 was measured in the dendritic spines present in the first 30 μm of secondary dendrites and normalized to mNeonGreen-SNX17 mean intensity in the dendritic shaft. DMSO: 0.691 ± 0.035, N = 30 neurons; VPS34-IN1: 0.683 ± 0.037, N = 30 neurons. Unpaired two-tailed Student’s t test determined that there are no significant differences between the DMSO and VPS34-IN1 conditions. Error bars are SEM. (C) DIV16 hippocampal neurons were co-transfected with mScarlet-SNX27 and eGFP (filler). 24 h later, cells were incubated in HBS containing either DMSO or 1 μM VPS34-IN1 for 10 min. Cells were fixed, and the number of mScarlet-SNX27-positive puncta in 30 µm of dendritic spines was quantified. DMSO: 0.291 ± 0.014, N = 30 neurons; VPS34-IN1: 0.279 ± 0.014, N = 30 neurons. Unpaired two-tailed Student’s t test determined that there are no significant differences between the DMSO and VPS34-IN1 conditions. Error bars are SEM. (D) Same as C, but the mean intensity of mScarlet-SNX27 was measured in the dendritic spines present in the first 30 μm of secondary dendrites and normalized to mScarlet-SNX27 mean intensity in the dendritic shaft. DMSO: 0.750 ± 0.036, N = 30 neurons; VPS34-IN1: 0.781 ± 0.038, N = 30 neurons. Unpaired two-tailed Student’s t test determined that there are no significant differences between the DMSO and VPS34-IN1 conditions. Error bars are SEM. (E) DIV16 hippocampal neurons were transfected with mNeonGreen-SNX17 and mCherry (filler). 24 h later, cells were treated with or without cLTP stimulation. cLTP-treated cells were washed, and incubated in HBS containing either DMSO or 1 μM VPS34-IN1 for 10 min. Cells were fixed, and the number of mNeonGreen-SNX17–positive puncta in 30 µm of dendritic spines was quantified. Baseline: 0.253 ± 0.013, N = 30 neurons; cLTP+DMSO: 0.343 ± 0.020, N = 30 neurons; cLTP+VPS34-IN1: 0.319 ± 0.024, N = 30 neurons. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, *P < 0.05, **P < 0.01. Error bars are SEM. (F) DIV16 hippocampal neurons were transfected with mScarlet-SNX27 and eGFP (filler). 24 h later, cells were treated with or without cLTP stimulation. cLTP-treated cells were washed and incubated in HBS containing either DMSO or 1 μM VPS34-IN1 for 10 min. Cells were fixed, and the number of mScarlet-SNX27–positive puncta in 30 µm of dendritic spines was quantified. Baseline: 0.246 ± 0.013, N = 30 neurons; cLTP+DMSO: 0.373 ± 0.020, N = 30 neurons; cLTP+VPS34-IN1: 0.361 ± 0.021, N = 30 neurons. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. DIV, days in vitro.
Brief 10-min inhibition of VPS34 does not affect the formation of SNX17- and SNX27-positive puncta or their recruitment to dendritic spines. (A) DIV16 hippocampal neurons were co-transfected with mNeonGreen-SNX17 and mCherry (filler). 24 h later, cells were incubated in HBS containing either DMSO or 1 μM VPS34-IN1 for 10 min. Cells were fixed, and the number of mNeonGreen-SNX17–positive puncta in 30 µm of dendritic spines was quantified. DMSO: 0.291 ± 0.014, N = 30 neurons; VPS34-IN1: 0.281 ± 0.014, N = 30 neurons. Unpaired two-tailed Student’s t test determined that there are no significant differences between the DMSO and VPS34-IN1 conditions. Error bars are SEM. (B) Same as B, but the mean intensity of mNeonGreen-SNX17 was measured in the dendritic spines present in the first 30 μm of secondary dendrites and normalized to mNeonGreen-SNX17 mean intensity in the dendritic shaft. DMSO: 0.691 ± 0.035, N = 30 neurons; VPS34-IN1: 0.683 ± 0.037, N = 30 neurons. Unpaired two-tailed Student’s t test determined that there are no significant differences between the DMSO and VPS34-IN1 conditions. Error bars are SEM. (C) DIV16 hippocampal neurons were co-transfected with mScarlet-SNX27 and eGFP (filler). 24 h later, cells were incubated in HBS containing either DMSO or 1 μM VPS34-IN1 for 10 min. Cells were fixed, and the number of mScarlet-SNX27-positive puncta in 30 µm of dendritic spines was quantified. DMSO: 0.291 ± 0.014, N = 30 neurons; VPS34-IN1: 0.279 ± 0.014, N = 30 neurons. Unpaired two-tailed Student’s t test determined that there are no significant differences between the DMSO and VPS34-IN1 conditions. Error bars are SEM. (D) Same as C, but the mean intensity of mScarlet-SNX27 was measured in the dendritic spines present in the first 30 μm of secondary dendrites and normalized to mScarlet-SNX27 mean intensity in the dendritic shaft. DMSO: 0.750 ± 0.036, N = 30 neurons; VPS34-IN1: 0.781 ± 0.038, N = 30 neurons. Unpaired two-tailed Student’s t test determined that there are no significant differences between the DMSO and VPS34-IN1 conditions. Error bars are SEM. (E) DIV16 hippocampal neurons were transfected with mNeonGreen-SNX17 and mCherry (filler). 24 h later, cells were treated with or without cLTP stimulation. cLTP-treated cells were washed, and incubated in HBS containing either DMSO or 1 μM VPS34-IN1 for 10 min. Cells were fixed, and the number of mNeonGreen-SNX17–positive puncta in 30 µm of dendritic spines was quantified. Baseline: 0.253 ± 0.013, N = 30 neurons; cLTP+DMSO: 0.343 ± 0.020, N = 30 neurons; cLTP+VPS34-IN1: 0.319 ± 0.024, N = 30 neurons. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, *P < 0.05, **P < 0.01. Error bars are SEM. (F) DIV16 hippocampal neurons were transfected with mScarlet-SNX27 and eGFP (filler). 24 h later, cells were treated with or without cLTP stimulation. cLTP-treated cells were washed and incubated in HBS containing either DMSO or 1 μM VPS34-IN1 for 10 min. Cells were fixed, and the number of mScarlet-SNX27–positive puncta in 30 µm of dendritic spines was quantified. Baseline: 0.246 ± 0.013, N = 30 neurons; cLTP+DMSO: 0.373 ± 0.020, N = 30 neurons; cLTP+VPS34-IN1: 0.361 ± 0.021, N = 30 neurons. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. DIV, days in vitro.
SNX17 and SNX27 define two distinct pathways for structural plasticity and act by promoting the recycling of different sets of cell surface proteins
The presence of SNX17 and SNX27 in the same compartments and their similar recruitment to dendritic spines upon cLTP raise the question of how these pathways work alongside each other.
To investigate how these pathways function relative to one another in neurons, we used an shRNA-mediated approach to knock down either rat SNX17 or SNX27. The knockdown efficiency was validated by lentiviral infection in rat cortical neurons (Fig. S7, A–C), as was the specificity of each shRNA construct to its corresponding target (Fig. S7, A–C).
Validation of shRNA constructs. (A) Validation of shRNA clones to knockdown SNX17 and SNX27 in rat cortical neurons. Neurons were infected with lentiviruses carrying either control-shRNA (pLKO.1 scrambled nontarget shRNA SHC002, Millipore Sigma), SNX17-shRNA (TRCN0000190340, Millipore Sigma) or SNX27-shRNA (TRCN0000253473, Millipore Sigma) at an MOI of 2. At 6 days after infection, cell extracts were collected and analyzed by western blot. GAPDH was used as a loading control. (B) SNX17 protein levels were quantified in neurons infected with lentiviruses carrying SNX17- or SNX27-shRNAs and normalized to the protein levels in control-shRNA–infected neurons. ctrl-shRNA: 100%; SNX17-shRNA: 32.2 ± 3.52%, SNX27-shRNA: 100.3 ± 5.85%. N = 3 independent experiments. Statistical significance was determined using ANOVA with Tukey’s post hoc comparisons, ****P < 0.001. Error bars are SEM. (C) The protein levels of SNX27 were quantified in neurons infected with lentiviruses carrying SNX17- or SNX27-shRNAs and normalized to the protein levels in control-shRNA–infected neurons. ctrl-shRNA: 100%; SNX17-shRNA: 102.3 ± 9.21%, SNX27-shRNA: 37.96 ± 0.29%. N = 3 independent experiments. Statistical significance was determined using ANOVA with Tukey’s post hoc comparisons, ***P < 0.005. Error bars are SEM. (D) Validation of an shRNA clone to knockdown rat VPS26B. Rat cortical neurons were infected with lentiviruses carrying either control-shRNA (pLKO.1 scrambled nontarget shRNA SHC002; Millipore Sigma) or VPS26B-shRNA (TRCN0000306336; Millipore Sigma) at an MOI of 2. At 6 days after infection, cell extracts were collected and analyzed by western blot. GAPDH was used as a loading control. (E) HEK293 cells stably expressing the tet repressor (TR-HEK293) were either transfected with control-shRNA or SNX27-shRNA in the absence or presence of eGFP, GFP-SNX27, or shRNA-resistant GFP-SNX27 (SNX27-R), as indicated. Five days after infection, cells were treated with 1 μg/ml of doxycycline to promote the expression of the eGFP-tagged constructs. 24 h later, extracts were collected and analyzed by western blot. Source data are available for this figure: SourceData FS7.
Validation of shRNA constructs. (A) Validation of shRNA clones to knockdown SNX17 and SNX27 in rat cortical neurons. Neurons were infected with lentiviruses carrying either control-shRNA (pLKO.1 scrambled nontarget shRNA SHC002, Millipore Sigma), SNX17-shRNA (TRCN0000190340, Millipore Sigma) or SNX27-shRNA (TRCN0000253473, Millipore Sigma) at an MOI of 2. At 6 days after infection, cell extracts were collected and analyzed by western blot. GAPDH was used as a loading control. (B) SNX17 protein levels were quantified in neurons infected with lentiviruses carrying SNX17- or SNX27-shRNAs and normalized to the protein levels in control-shRNA–infected neurons. ctrl-shRNA: 100%; SNX17-shRNA: 32.2 ± 3.52%, SNX27-shRNA: 100.3 ± 5.85%. N = 3 independent experiments. Statistical significance was determined using ANOVA with Tukey’s post hoc comparisons, ****P < 0.001. Error bars are SEM. (C) The protein levels of SNX27 were quantified in neurons infected with lentiviruses carrying SNX17- or SNX27-shRNAs and normalized to the protein levels in control-shRNA–infected neurons. ctrl-shRNA: 100%; SNX17-shRNA: 102.3 ± 9.21%, SNX27-shRNA: 37.96 ± 0.29%. N = 3 independent experiments. Statistical significance was determined using ANOVA with Tukey’s post hoc comparisons, ***P < 0.005. Error bars are SEM. (D) Validation of an shRNA clone to knockdown rat VPS26B. Rat cortical neurons were infected with lentiviruses carrying either control-shRNA (pLKO.1 scrambled nontarget shRNA SHC002; Millipore Sigma) or VPS26B-shRNA (TRCN0000306336; Millipore Sigma) at an MOI of 2. At 6 days after infection, cell extracts were collected and analyzed by western blot. GAPDH was used as a loading control. (E) HEK293 cells stably expressing the tet repressor (TR-HEK293) were either transfected with control-shRNA or SNX27-shRNA in the absence or presence of eGFP, GFP-SNX27, or shRNA-resistant GFP-SNX27 (SNX27-R), as indicated. Five days after infection, cells were treated with 1 μg/ml of doxycycline to promote the expression of the eGFP-tagged constructs. 24 h later, extracts were collected and analyzed by western blot. Source data are available for this figure: SourceData FS7.
Knockdown of either SNX17 or SNX27 in cultured hippocampal neurons prevented the cLTP-dependent increase in dendritic spine width (Fig. 5, A and B), which supports previous findings that these pathways are involved in LTP regulation (Loo et al., 2014; Rivero-Ríos et al., 2023). SNX17-dependent recycling requires the Retriever complex (McNally et al., 2017), while SNX27 relies on the Retromer complex (Steinberg et al., 2013; Temkin et al., 2017). Therefore, we utilized shRNA clones to knock down the Retriever subunit VPS26C, which we had previously validated (Rivero-Ríos et al., 2023), and the Retromer subunit VPS26B (Fig. S7 D), the primary VPS26 paralog in neurons (Simoes et al., 2021; Kim et al., 2008; Bugarcic et al., 2011; Qureshi et al., 2022). We found that knockdown of either VPS26C or VPS26B also blocked the cLTP-dependent increase in dendritic spine width (Fig. 5 C), which further supports a key role for both endocytic recycling pathways in regulating LTP.

PI(3)P synthesis is required for SNX17- and SNX27-dependent structural plasticity of dendritic spines. (A) Representative confocal images of dendritic spines in DIV16 hippocampal neurons co-transfected at DIV12 with eGFP (filler) and either ctrl-, SNX17-, or SNX27-shRNAs. Neurons were either treated with cLTP or left untreated and fixed 50 min after cLTP. Scale bar, 5 µm. (B) The maximum width for each spine was quantified, and the average size of the dendritic spines in the first 30 μm of secondary dendrites was calculated. ctrl-shRNA: 0.374 ± 0.007, N = 30 neurons; ctrl-shRNA with cLTP: 0.475 ± 0.012, N = 30 neurons; SNX17-shRNA: 0.360 ± 0.008, N = 30 neurons; SNX17-shRNA with cLTP: 0.351 ± 0.008, N = 30 neurons; SNX27-shRNA: 0.354 ± 0.008, N = 30 neurons; SNX27-shRNA with cLTP: 0.358 ± 0.005, N = 30 neurons. Three independent experiments. Data were analyzed by one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (C) DIV16 hippocampal neurons co-transfected at DIV12 with eGFP (filler) and either ctrl-, VPS26C-, or VPS26B-shRNAs. Neurons were either treated with cLTP or left untreated and fixed 50 min after cLTP. Dendritic spine width was calculated as in B. ctrl-shRNA: 0.376 ± 0.006, N = 30 neurons; ctrl-shRNA with cLTP: 0.472 ± 0.012, N = 30 neurons; VPS26C-shRNA: 0.371 ± 0.007, N = 30 neurons; VPS26C-shRNA with cLTP: 0.378 ± 0.007, N = 30 neurons; VPS26B-shRNA: 0.384 ± 0.009, N = 30 neurons; VPS26B-shRNA with cLTP: 0.381 ± 0.008, N = 30 neurons. Three independent experiments. Data were analyzed by one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (D) Rat hippocampal neurons were co-transfected at DIV12 with SNX17-shRNA and a shRNA-resistant SNX17 construct (SNX17-R). At DIV16, neurons were treated with either DMSO or 1 μM VPS34-IN1 for 30 min, followed by a 5-min cLTP stimulus in the presence of the compounds where indicated. Neurons were further incubated in the presence of DMSO or VPS34-IN1 for 50 min before fixation. The width of dendritic spines in the first 30 µm of secondary dendrites was quantified. DMSO: 0.359 ± 0.009, N = 30 neurons; DMSO with cLTP: 0.430 ± 0.010, N = 30 neurons; VPS34-IN1: 0.364 ± 0.009, N = 30 neurons; VPS34-IN1 with cLTP: 0.369 ± 0.007, N = 30 neurons. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (E) Rat hippocampal neurons were co-transfected at DIV12 with SNX27-shRNA and a shRNA-resistant SNX27 construct (SNX27-R). At DIV16, neurons were treated with either DMSO or 1 μM VPS34-IN1 for 30 min, followed by a 5-min cLTP stimulus in the presence of the compounds where indicated. Neurons were further incubated in the presence of DMSO or VPS34-IN1 for 50 min before fixation. The width of dendritic spines in the first 30 µm of secondary dendrites was quantified. DMSO: 0.340 ± 0.006, N = 30 neurons; DMSO with cLTP: 0.420 ± 0.009, N = 30 neurons; VPS34-IN1: 0.347 ± 0.011, N = 30 neurons; VPS34-IN1 with cLTP: 0.338 ± 0.007, N = 30 neurons. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. DIV, days in vitro.
PI(3)P synthesis is required for SNX17- and SNX27-dependent structural plasticity of dendritic spines. (A) Representative confocal images of dendritic spines in DIV16 hippocampal neurons co-transfected at DIV12 with eGFP (filler) and either ctrl-, SNX17-, or SNX27-shRNAs. Neurons were either treated with cLTP or left untreated and fixed 50 min after cLTP. Scale bar, 5 µm. (B) The maximum width for each spine was quantified, and the average size of the dendritic spines in the first 30 μm of secondary dendrites was calculated. ctrl-shRNA: 0.374 ± 0.007, N = 30 neurons; ctrl-shRNA with cLTP: 0.475 ± 0.012, N = 30 neurons; SNX17-shRNA: 0.360 ± 0.008, N = 30 neurons; SNX17-shRNA with cLTP: 0.351 ± 0.008, N = 30 neurons; SNX27-shRNA: 0.354 ± 0.008, N = 30 neurons; SNX27-shRNA with cLTP: 0.358 ± 0.005, N = 30 neurons. Three independent experiments. Data were analyzed by one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (C) DIV16 hippocampal neurons co-transfected at DIV12 with eGFP (filler) and either ctrl-, VPS26C-, or VPS26B-shRNAs. Neurons were either treated with cLTP or left untreated and fixed 50 min after cLTP. Dendritic spine width was calculated as in B. ctrl-shRNA: 0.376 ± 0.006, N = 30 neurons; ctrl-shRNA with cLTP: 0.472 ± 0.012, N = 30 neurons; VPS26C-shRNA: 0.371 ± 0.007, N = 30 neurons; VPS26C-shRNA with cLTP: 0.378 ± 0.007, N = 30 neurons; VPS26B-shRNA: 0.384 ± 0.009, N = 30 neurons; VPS26B-shRNA with cLTP: 0.381 ± 0.008, N = 30 neurons. Three independent experiments. Data were analyzed by one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (D) Rat hippocampal neurons were co-transfected at DIV12 with SNX17-shRNA and a shRNA-resistant SNX17 construct (SNX17-R). At DIV16, neurons were treated with either DMSO or 1 μM VPS34-IN1 for 30 min, followed by a 5-min cLTP stimulus in the presence of the compounds where indicated. Neurons were further incubated in the presence of DMSO or VPS34-IN1 for 50 min before fixation. The width of dendritic spines in the first 30 µm of secondary dendrites was quantified. DMSO: 0.359 ± 0.009, N = 30 neurons; DMSO with cLTP: 0.430 ± 0.010, N = 30 neurons; VPS34-IN1: 0.364 ± 0.009, N = 30 neurons; VPS34-IN1 with cLTP: 0.369 ± 0.007, N = 30 neurons. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (E) Rat hippocampal neurons were co-transfected at DIV12 with SNX27-shRNA and a shRNA-resistant SNX27 construct (SNX27-R). At DIV16, neurons were treated with either DMSO or 1 μM VPS34-IN1 for 30 min, followed by a 5-min cLTP stimulus in the presence of the compounds where indicated. Neurons were further incubated in the presence of DMSO or VPS34-IN1 for 50 min before fixation. The width of dendritic spines in the first 30 µm of secondary dendrites was quantified. DMSO: 0.340 ± 0.006, N = 30 neurons; DMSO with cLTP: 0.420 ± 0.009, N = 30 neurons; VPS34-IN1: 0.347 ± 0.011, N = 30 neurons; VPS34-IN1 with cLTP: 0.338 ± 0.007, N = 30 neurons. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. DIV, days in vitro.
To determine whether the role of SNX17 and SNX27 in structural plasticity depends on PI(3)P, we performed rescue experiments using shRNA-resistant versions of both proteins. The shRNA-resistant GFP-SNX17 (SNX17-R) had been previously validated (Rivero-Ríos et al., 2023). For SNX27, we generated an shRNA-resistant GFP-SNX27 (SNX27-R) by introducing six silent mutations within the shRNA target sequence and validated its resistance to knockdown in HEK293 cells (Fig. S7 E). Cultured hippocampal neurons were co-transfected with SNX17-shRNA and SNX17-R, and 24 h later, treated with either DMSO or VPS34-IN1 for 30 min prior to cLTP induction. In DMSO-treated neurons, SNX17-R fully rescued the defects in dendritic spine enlargement caused by SNX17 knockdown. In contrast, in VPS34-IN1–treated neurons, reintroduction of SNX17-R failed to rescue these defects (Fig. 5 D). Similarly, SNX27-R restored cLTP-induced structural plasticity only in DMSO-treated cells, but not when VPS34 was inhibited (Fig. 5 E).
The requirement of both SNX17 and SNX27 for spine structural plasticity raises the question of whether these proteins act cooperatively or in parallel pathways. To address this, we conducted rescue experiments using the shRNA-resistant constructs. Importantly, transient transfection of shRNA-resistant GFP-SNX17 in cultured neurons rescued the defects in dendritic spine enlargement following LTP caused by SNX17 knockdown (Fig. 6 A), but not by SNX27 knockdown (Fig. 6 B). Conversely, shRNA-resistant GFP-SNX27 rescued the defects in cLTP-induced spine enlargement resulting from SNX27 knockdown (Fig. 6 B), but not those from SNX17 knockdown (Fig. 6 A). These findings indicate that both the SNX17–Retriever and SNX27–Retromer pathways are required for the cLTP-related structural changes at spines and that each pathway is unable to compensate for the loss of the other.
SNX17 and SNX27 define two distinct pathways for structural plasticity by recycling specific cargoes. (A) Rat hippocampal neurons were co-transfected at DIV12 with either ctrl- or SNX17-shRNA and the indicated constructs. At DIV16, neurons were either treated with cLTP or left untreated and fixed 50 min after cLTP. The width of dendritic spines in the first 30 µm of secondary dendrites were quantified. ctrl-shRNA + eGFP: 0.393 ± 0.011, N = 30 neurons; ctrl-shRNA + eGFP with cLTP: 0.515 ± 0.012, N = 30 neurons; SNX17-shRNA + eGFP: 0.370 ± 0.006, N = 30 neurons; SNX17-shRNA + eGFP with cLTP: 0.334 ± 0.014, N = 30 neurons; SNX17-shRNA + SNX17-R: 0.373 ± 0.008, N = 30 neurons; SNX17-shRNA + SNX17-R with cLTP: 0.509 ± 0.012, N = 30 neurons; SNX17-shRNA + SNX27-R: 0.367 ± 0.006, N = 30 neurons; SNX17-shRNA + SNX27-R with cLTP: 0.356 ± 0.006, N = 30 neurons. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (B) Rat hippocampal neurons were co-transfected at DIV12 with either ctrl- or SNX27-shRNA and the indicated constructs. At DIV16, neurons were either treated with cLTP or left untreated and fixed 50 min after cLTP. The width of dendritic spines in the first 30 µm of secondary dendrites were quantified. ctrl-shRNA + eGFP: 0.409 ± 0.008, N = 30 neurons; ctrl-shRNA + eGFP with cLTP: 0.503 ± 0.013, N = 30 neurons; SNX27-shRNA + eGFP: 0.402 ± 0.008, N = 30 neurons; SNX27-shRNA + eGFP with cLTP: 0.398 ± 0.010, N = 30 neurons; SNX27-shRNA + SNX27-R: 0.397 ± 0.009, N = 30 neurons; SNX27-shRNA + SNX27-R with cLTP: 0.478 ± 0.010, N = 30 neurons; SNX27-shRNA + SNX17-R: 0.388 ± 0.009, N = 30 neurons; SNX27-shRNA + SNX17-R with cLTP: 0.380 ± 0.006, N = 30 neurons. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (C) DIV11 rat cortical neurons were infected with lentiviruses carrying ctrl-, SNX17-, or SNX27-shRNAs, and the surface levels of β1-integrin and GluA1 were determined at DIV17 using surface biotinylation assays. Knockdown of SNX17 and SNX27 was validated by western blot of the lysate, and GAPDH was used as a loading control. (D) The levels of surface β1-integrin protein were quantified and expressed as a percentage of ctrl-shRNA. ctrl-shRNA: 100%, SNX17-shRNA: 48.350 ± 4.901%, SNX27-shRNA: 129.400 ± 6.202%. N = 4 independent experiments. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (E) The levels of surface GluA1 protein were quantified and expressed as a percentage of ctrl-shRNA. ctrl-shRNA: 100%, SNX17-shRNA: 113.200 ± 5.011%, SNX27-shRNA: 51.820 ± 0.696%. N = 3 independent experiments. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. DIV, days in vitro. Source data are available for this figure: SourceData F6.
SNX17 and SNX27 define two distinct pathways for structural plasticity by recycling specific cargoes. (A) Rat hippocampal neurons were co-transfected at DIV12 with either ctrl- or SNX17-shRNA and the indicated constructs. At DIV16, neurons were either treated with cLTP or left untreated and fixed 50 min after cLTP. The width of dendritic spines in the first 30 µm of secondary dendrites were quantified. ctrl-shRNA + eGFP: 0.393 ± 0.011, N = 30 neurons; ctrl-shRNA + eGFP with cLTP: 0.515 ± 0.012, N = 30 neurons; SNX17-shRNA + eGFP: 0.370 ± 0.006, N = 30 neurons; SNX17-shRNA + eGFP with cLTP: 0.334 ± 0.014, N = 30 neurons; SNX17-shRNA + SNX17-R: 0.373 ± 0.008, N = 30 neurons; SNX17-shRNA + SNX17-R with cLTP: 0.509 ± 0.012, N = 30 neurons; SNX17-shRNA + SNX27-R: 0.367 ± 0.006, N = 30 neurons; SNX17-shRNA + SNX27-R with cLTP: 0.356 ± 0.006, N = 30 neurons. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (B) Rat hippocampal neurons were co-transfected at DIV12 with either ctrl- or SNX27-shRNA and the indicated constructs. At DIV16, neurons were either treated with cLTP or left untreated and fixed 50 min after cLTP. The width of dendritic spines in the first 30 µm of secondary dendrites were quantified. ctrl-shRNA + eGFP: 0.409 ± 0.008, N = 30 neurons; ctrl-shRNA + eGFP with cLTP: 0.503 ± 0.013, N = 30 neurons; SNX27-shRNA + eGFP: 0.402 ± 0.008, N = 30 neurons; SNX27-shRNA + eGFP with cLTP: 0.398 ± 0.010, N = 30 neurons; SNX27-shRNA + SNX27-R: 0.397 ± 0.009, N = 30 neurons; SNX27-shRNA + SNX27-R with cLTP: 0.478 ± 0.010, N = 30 neurons; SNX27-shRNA + SNX17-R: 0.388 ± 0.009, N = 30 neurons; SNX27-shRNA + SNX17-R with cLTP: 0.380 ± 0.006, N = 30 neurons. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (C) DIV11 rat cortical neurons were infected with lentiviruses carrying ctrl-, SNX17-, or SNX27-shRNAs, and the surface levels of β1-integrin and GluA1 were determined at DIV17 using surface biotinylation assays. Knockdown of SNX17 and SNX27 was validated by western blot of the lysate, and GAPDH was used as a loading control. (D) The levels of surface β1-integrin protein were quantified and expressed as a percentage of ctrl-shRNA. ctrl-shRNA: 100%, SNX17-shRNA: 48.350 ± 4.901%, SNX27-shRNA: 129.400 ± 6.202%. N = 4 independent experiments. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. (E) The levels of surface GluA1 protein were quantified and expressed as a percentage of ctrl-shRNA. ctrl-shRNA: 100%, SNX17-shRNA: 113.200 ± 5.011%, SNX27-shRNA: 51.820 ± 0.696%. N = 3 independent experiments. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test, ****P < 0.001. Error bars are SEM. DIV, days in vitro. Source data are available for this figure: SourceData F6.
We reasoned that the lack of compensatory effect between SNX17 and SNX27 might be attributed to the exclusive recycling of certain cell surface proteins by each pathway. To test this, we chose two cell surface proteins known to play roles in LTP: the adhesion molecule β1-integrin and the AMPA receptor subunit GluA1. To determine whether these cargoes are specific to one recycling pathway, we performed surface biotinylation assays. Neurons were transduced with lentiviruses carrying shRNAs for SNX17, SNX27, or a scrambled control, and the surface levels of β1-integrin and GluA1 were assessed. Knockdown of SNX17, but not SNX27, resulted in reduced surface β1-integrin levels (Fig. 6, C and D), whereas surface GluA1 levels were decreased only by SNX27 knockdown (Fig. 6, C and E). In contrast, knockdown of WASHC5, encoding the WASH complex subunit strumpellin, which functions in both recycling pathways, led to reduced surface levels of both β1-integrin and GluA1 (Fig. S8, A–C).
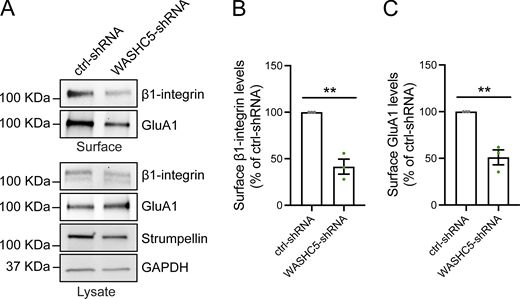
The WASH complex regulates the cell surface levels of SNX17- and SNX27-dependent cargoes. (A) DIV11 rat cortical neurons were infected with lentiviruses carrying ctrl- or WASHC5-shRNAs, and the surface levels of β1-integrin and GluA1 were determined at DIV17 using surface biotinylation assays. The total levels of β1-integrin, GluA1, and strumpellin were validated by western blot of the lysate, and GAPDH was used as a loading control. (B) The levels of surface β1-integrin protein were quantified and expressed as a percentage of ctrl-shRNA. ctrl-shRNA: 100%, VPS34-shRNA: 41.50 ± 8.006%. N = 3 independent experiments. Statistical significance was determined using unpaired two-tailed Student’s t test, **P < 0.01. Error bars are SEM. (C) The levels of surface GluA1 protein were quantified and expressed as a percentage of ctrl-shRNA. ctrl-shRNA: 100%, VPS34-shRNA: 51.02 ± 8.005%. N = 3 independent experiments. Statistical significance was determined using unpaired two-tailed Student’s t test, **P < 0.01. Error bars are SEM. DIV, days in vitro. Source data are available for this figure: SourceData FS8.
The WASH complex regulates the cell surface levels of SNX17- and SNX27-dependent cargoes. (A) DIV11 rat cortical neurons were infected with lentiviruses carrying ctrl- or WASHC5-shRNAs, and the surface levels of β1-integrin and GluA1 were determined at DIV17 using surface biotinylation assays. The total levels of β1-integrin, GluA1, and strumpellin were validated by western blot of the lysate, and GAPDH was used as a loading control. (B) The levels of surface β1-integrin protein were quantified and expressed as a percentage of ctrl-shRNA. ctrl-shRNA: 100%, VPS34-shRNA: 41.50 ± 8.006%. N = 3 independent experiments. Statistical significance was determined using unpaired two-tailed Student’s t test, **P < 0.01. Error bars are SEM. (C) The levels of surface GluA1 protein were quantified and expressed as a percentage of ctrl-shRNA. ctrl-shRNA: 100%, VPS34-shRNA: 51.02 ± 8.005%. N = 3 independent experiments. Statistical significance was determined using unpaired two-tailed Student’s t test, **P < 0.01. Error bars are SEM. DIV, days in vitro. Source data are available for this figure: SourceData FS8.
PI(3)P is required for the recycling of SNX17- and SNX27-dependent cargoes both under basal conditions and upon cLTP
The requirement for PI(3)P synthesis for the endosomal localization of SNX17 and SNX27 in dendrites, as well as for their subsequent recruitment to spines upon cLTP, suggests that PI(3)P may regulate the recycling of cell surface receptors important for cLTP. As an initial approach to test this, we examined the colocalization of β1-integrin and GluA1 with VPS34. Neurons were transfected with HA-VPS34, and 24 h later, live labeled for 15 min with antibodies targeting the extracellular epitopes of β1-integrin and GluA1. Neurons were then washed and incubated at 37°C to allow internalization of the antibody-bound cargoes, followed by an acid wash to remove any remaining surface-bound antibodies. β1-integrin and GluA1 showed ∼60% overlap (Fig. 7, A–C), which indicates that they traffic through shared endosomal compartments. To determine whether these compartments are associated with PI(3)P synthesis, we generated a mask of the overlap between β1-integrin and GluA1 and assessed its colocalization with HA-VPS34. Approximately 80% of these shared compartments also contained VPS34 (Fig. 7, A and D), which suggests that PI(3)P synthesis occurs in endosomes engaged in recycling SNX17 and SNX27 cargoes.
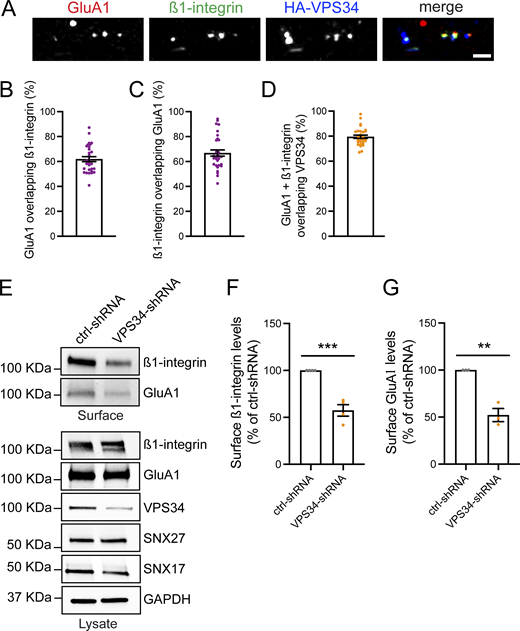
PI(3)P regulates the cell surface levels of SNX17- and SNX27-dependent cargoes. (A) Representative confocal images of DIV16 rat hippocampal neurons transfected at DIV15 with HA-VPS34. 24 h later, neurons were incubated live with antibodies against surface β1-integrin and surface GluA1 for 15 min at 37°C. Cells were then washed and further incubated at 37°C for 10 min. Antibodies remaining on the neuronal surface were stripped with an acid wash (0.5 M NaCl + 0.2 N acetic acid, 1 min, 4°C), followed by fixation, blocking, and incubation with secondary antibodies. Neurons were then permeabilized, blocked, and stained with an anti-HA antibody. Scale bar, 2.5 µm. (B) The percentage of GluA1 overlapping with β1-integrin was analyzed using Mander’s colocalization coefficient (×100). 61.90 ± 1.999%, N = 30 neurons. Three independent experiments. Error bar is SEM. (C) The percentage of β1-integrin overlapping with GluA1 was analyzed using Mander’s colocalization coefficient (×100). 66.80 ± 2.514%, N = 30 neurons. Three independent experiments. Error bar is SEM. (D) The percentage of endosomes containing both β1-integrin and GluA1 that colocalize with HA-VPS34 was analyzed using Mander’s colocalization coefficient (×100). 79.48 ± 1.283%, N = 30 neurons. Three independent experiments. Error bar is SEM. (E) DIV11 rat cortical neurons were infected with lentiviruses carrying ctrl- or VPS34-shRNAs, and the surface levels of β1-integrin and GluA1 were determined at DIV17 using surface biotinylation assays. The total levels of β1-integrin, GluA1, VPS34, SNX17, and SNX27 were validated by western blot of the lysate, and GAPDH was used as a loading control. (F) The levels of surface β1-integrin protein were quantified and expressed as a percentage of ctrl-shRNA. ctrl-shRNA: 100%, VPS34-shRNA: 57.40 ± 6.130%. N = 4 independent experiments. Statistical significance was determined using unpaired two-tailed Student’s t test, ***P < 0.005. Error bars are SEM. (G) The levels of surface GluA1 protein were quantified and expressed as a percentage of ctrl-shRNA. ctrl-shRNA: 100%, VPS34-shRNA: 52.180 ± 7.024%. N = 3 independent experiments. Statistical significance was determined using unpaired two-tailed Student’s t test, **P < 0.01. Error bars are SEM. DIV, days in vitro. Source data are available for this figure: SourceData F7.
PI(3)P regulates the cell surface levels of SNX17- and SNX27-dependent cargoes. (A) Representative confocal images of DIV16 rat hippocampal neurons transfected at DIV15 with HA-VPS34. 24 h later, neurons were incubated live with antibodies against surface β1-integrin and surface GluA1 for 15 min at 37°C. Cells were then washed and further incubated at 37°C for 10 min. Antibodies remaining on the neuronal surface were stripped with an acid wash (0.5 M NaCl + 0.2 N acetic acid, 1 min, 4°C), followed by fixation, blocking, and incubation with secondary antibodies. Neurons were then permeabilized, blocked, and stained with an anti-HA antibody. Scale bar, 2.5 µm. (B) The percentage of GluA1 overlapping with β1-integrin was analyzed using Mander’s colocalization coefficient (×100). 61.90 ± 1.999%, N = 30 neurons. Three independent experiments. Error bar is SEM. (C) The percentage of β1-integrin overlapping with GluA1 was analyzed using Mander’s colocalization coefficient (×100). 66.80 ± 2.514%, N = 30 neurons. Three independent experiments. Error bar is SEM. (D) The percentage of endosomes containing both β1-integrin and GluA1 that colocalize with HA-VPS34 was analyzed using Mander’s colocalization coefficient (×100). 79.48 ± 1.283%, N = 30 neurons. Three independent experiments. Error bar is SEM. (E) DIV11 rat cortical neurons were infected with lentiviruses carrying ctrl- or VPS34-shRNAs, and the surface levels of β1-integrin and GluA1 were determined at DIV17 using surface biotinylation assays. The total levels of β1-integrin, GluA1, VPS34, SNX17, and SNX27 were validated by western blot of the lysate, and GAPDH was used as a loading control. (F) The levels of surface β1-integrin protein were quantified and expressed as a percentage of ctrl-shRNA. ctrl-shRNA: 100%, VPS34-shRNA: 57.40 ± 6.130%. N = 4 independent experiments. Statistical significance was determined using unpaired two-tailed Student’s t test, ***P < 0.005. Error bars are SEM. (G) The levels of surface GluA1 protein were quantified and expressed as a percentage of ctrl-shRNA. ctrl-shRNA: 100%, VPS34-shRNA: 52.180 ± 7.024%. N = 3 independent experiments. Statistical significance was determined using unpaired two-tailed Student’s t test, **P < 0.01. Error bars are SEM. DIV, days in vitro. Source data are available for this figure: SourceData F7.
Consistent with this idea, surface biotinylation assays demonstrated that decreased PI(3)P levels result in diminished surface expression of both SNX17 and SNX27 cargoes. Specifically, VPS34-shRNA caused approximately a 42.6% reduction in surface β1-integrin levels (Fig. 7, E and F) and a 47.8% reduction in surface GluA1 levels (Fig. 7, E and G).
We next sought to determine whether the surface levels of β1-integrin and GluA1 are regulated by synaptic activity in a PI(3)P-dependent manner. To assess the trafficking of these proteins in response to cLTP, we used surface labeling with antibodies targeting the extracellular epitopes of β1-integrin or GluA1. Neurons were treated with or without cLTP, live labeled for 15 min, and then fixed and immunostained for MAP2. cLTP induction resulted in a significant increase in the surface levels of both β1-integrin (Fig. 8, A and B) and GluA1 (Fig. 8, C and D). As expected, SNX17 knockdown blocked the cLTP-dependent increase in surface β1-integrin levels, while SNX27 knockdown had no effect (Fig. 8, A and B). Conversely, SNX27 knockdown, but not SNX17 knockdown, prevented the cLTP-dependent increase in surface GluA1 (Fig. 8, C and D). Knockdown of VPS34 blocked the cLTP-dependent increase in the surface levels of both β1-integrin (Fig. 9, A and B) and GluA1 (Fig. 9, C and D). Taken together, these studies demonstrate that activity-dependent PI(3)P synthesis coordinates SNX17- and SNX27-dependent cargo recycling, which is crucial for the structural changes in dendritic spines necessary for enduring forms of synaptic plasticity.

SNX17 and SNX27 promote the recycling of different cargoes upon cLTP. (A) Representative confocal images of surface β1-integrin levels of DIV17 hippocampal neurons that were infected at DIV11 with lentiviruses carrying either ctrl-, SNX17-, or SNX27-shRNAs. Neurons were treated in the presence or absence of cLTP and live labeled with an anti-surface β1-integrin antibody for 15 min, followed by fixation and immunostaining for MAP2. Scale bar, 5 µm. (B) The mean intensity of β1-integrin in the first 50 µm of secondary dendrites was quantified, and values were normalized to crtl-shRNA. ctrl-shRNA: 1,000 ± 0.025, N = 40 neurons; ctrl-shRNA cLTP: 1.168 ± 0.034, N = 40 neurons; SNX17-shRNA: 0.720 ± 0.034, N = 40 neurons; SNX17-shRNA cLTP: 0.725 ± 0.046, N = 40 neurons; SNX27-shRNA: 1.003 ± 0.036, N = 40 neurons; SNX27-shRNA cLTP: 1.191 ± 0.028, N = 40 neurons. Three independent experiments. Data were analyzed by one-way ANOVA with Tukey’s post hoc test, **P < 0.01. Error bars are SEM. (C) Representative confocal images of surface GluA1 levels of DIV17 hippocampal neurons that were infected at DIV11 with lentiviruses carrying either ctrl-, SNX17-, or SNX27-shRNAs. Neurons were treated in the presence or absence of cLTP and live labeled with an anti-surface GluA1 antibody for 15 min, followed by fixation and immunostaining for MAP2. Scale bar, 5 µm. (D) The mean intensity of surface GluA1 in the first 50 µm of secondary dendrites was quantified, and values were normalized to crtl-shRNA. ctrl-shRNA: 1,000 ± 0.025, N = 40 neurons; ctrl-shRNA cLTP: 1.135 ± 0.033, N = 40 neurons; SNX17-shRNA: 0.946 ± 0.031, N = 40 neurons; SNX17-shRNA cLTP: 1.097 ± 0.035, N = 40 neurons; SNX27-shRNA: 0.757 ± 0.033, N = 40 neurons; SNX27-shRNA cLTP: 0.706 ± 0.025, N = 40 neurons. Three independent experiments. Data were analyzed by one-way ANOVA with Tukey’s post hoc test, *P < 0.05, **P < 0.01. Error bars are SEM. DIV, days in vitro.
SNX17 and SNX27 promote the recycling of different cargoes upon cLTP. (A) Representative confocal images of surface β1-integrin levels of DIV17 hippocampal neurons that were infected at DIV11 with lentiviruses carrying either ctrl-, SNX17-, or SNX27-shRNAs. Neurons were treated in the presence or absence of cLTP and live labeled with an anti-surface β1-integrin antibody for 15 min, followed by fixation and immunostaining for MAP2. Scale bar, 5 µm. (B) The mean intensity of β1-integrin in the first 50 µm of secondary dendrites was quantified, and values were normalized to crtl-shRNA. ctrl-shRNA: 1,000 ± 0.025, N = 40 neurons; ctrl-shRNA cLTP: 1.168 ± 0.034, N = 40 neurons; SNX17-shRNA: 0.720 ± 0.034, N = 40 neurons; SNX17-shRNA cLTP: 0.725 ± 0.046, N = 40 neurons; SNX27-shRNA: 1.003 ± 0.036, N = 40 neurons; SNX27-shRNA cLTP: 1.191 ± 0.028, N = 40 neurons. Three independent experiments. Data were analyzed by one-way ANOVA with Tukey’s post hoc test, **P < 0.01. Error bars are SEM. (C) Representative confocal images of surface GluA1 levels of DIV17 hippocampal neurons that were infected at DIV11 with lentiviruses carrying either ctrl-, SNX17-, or SNX27-shRNAs. Neurons were treated in the presence or absence of cLTP and live labeled with an anti-surface GluA1 antibody for 15 min, followed by fixation and immunostaining for MAP2. Scale bar, 5 µm. (D) The mean intensity of surface GluA1 in the first 50 µm of secondary dendrites was quantified, and values were normalized to crtl-shRNA. ctrl-shRNA: 1,000 ± 0.025, N = 40 neurons; ctrl-shRNA cLTP: 1.135 ± 0.033, N = 40 neurons; SNX17-shRNA: 0.946 ± 0.031, N = 40 neurons; SNX17-shRNA cLTP: 1.097 ± 0.035, N = 40 neurons; SNX27-shRNA: 0.757 ± 0.033, N = 40 neurons; SNX27-shRNA cLTP: 0.706 ± 0.025, N = 40 neurons. Three independent experiments. Data were analyzed by one-way ANOVA with Tukey’s post hoc test, *P < 0.05, **P < 0.01. Error bars are SEM. DIV, days in vitro.

PI(3)P synthesis is necessary for the recycling of SNX17 and SNX27 cargoes upon cLTP. (A) Representative confocal images of surface β1-integrin levels of DIV17 hippocampal neurons that were infected at DIV11 with lentiviruses carrying either ctrl- or SNX27-shRNAs. Neurons were treated in the presence or absence of cLTP and live labeled with an anti-surface β1-integrin antibody for 15 min, followed by fixation and immunostaining for MAP2. Scale bar, 5 µm. (B) The mean intensity of β1-integrin in the first 50 µm of secondary dendrites was quantified, and values were normalized to crtl-shRNA. ctrl-shRNA: 1,000 ± 0.031, N = 40 neurons; ctrl-shRNA cLTP: 1.179 ± 0.038, N = 40 neurons; VPS34-shRNA: 0.770 ± 0.029, N = 40 neurons; VPS34-shRNA cLTP: 0.779 ± 0.031, N = 40 neurons. Three independent experiments. Data were analyzed by one-way ANOVA with Tukey’s post hoc test, ***P < 0.005, ****P < 0.001. Error bars are SEM. (C) Representative confocal images of surface GluA1 levels of DIV17 hippocampal neurons that were infected at DIV11 with lentiviruses carrying either ctrl- or VPS34-shRNAs. Neurons were treated in the presence or absence of cLTP and live labeled with an anti-surface GluA1 antibody for 15 min, followed by fixation and immunostaining for MAP2. Scale bar, 5 µm. (D) The mean intensity of surface GluA1 in the first 50 µm of secondary dendrites was quantified, and values were normalized to crtl-shRNA. ctrl-shRNA: 1,000 ± 0.033, N = 40 neurons; ctrl-shRNA cLTP: 1.171 ± 0.052, N = 40 neurons; VPS34-shRNA: 0.653 ± 0.027, N = 40 neurons; VPS34-shRNA cLTP: 0.688 ± 0.031, N = 40 neurons. Three independent experiments. Data were analyzed by one-way ANOVA with Tukey’s post hoc test, **P < 0.01, ****P < 0.001. Error bars are SEM. DIV, days in vitro.
PI(3)P synthesis is necessary for the recycling of SNX17 and SNX27 cargoes upon cLTP. (A) Representative confocal images of surface β1-integrin levels of DIV17 hippocampal neurons that were infected at DIV11 with lentiviruses carrying either ctrl- or SNX27-shRNAs. Neurons were treated in the presence or absence of cLTP and live labeled with an anti-surface β1-integrin antibody for 15 min, followed by fixation and immunostaining for MAP2. Scale bar, 5 µm. (B) The mean intensity of β1-integrin in the first 50 µm of secondary dendrites was quantified, and values were normalized to crtl-shRNA. ctrl-shRNA: 1,000 ± 0.031, N = 40 neurons; ctrl-shRNA cLTP: 1.179 ± 0.038, N = 40 neurons; VPS34-shRNA: 0.770 ± 0.029, N = 40 neurons; VPS34-shRNA cLTP: 0.779 ± 0.031, N = 40 neurons. Three independent experiments. Data were analyzed by one-way ANOVA with Tukey’s post hoc test, ***P < 0.005, ****P < 0.001. Error bars are SEM. (C) Representative confocal images of surface GluA1 levels of DIV17 hippocampal neurons that were infected at DIV11 with lentiviruses carrying either ctrl- or VPS34-shRNAs. Neurons were treated in the presence or absence of cLTP and live labeled with an anti-surface GluA1 antibody for 15 min, followed by fixation and immunostaining for MAP2. Scale bar, 5 µm. (D) The mean intensity of surface GluA1 in the first 50 µm of secondary dendrites was quantified, and values were normalized to crtl-shRNA. ctrl-shRNA: 1,000 ± 0.033, N = 40 neurons; ctrl-shRNA cLTP: 1.171 ± 0.052, N = 40 neurons; VPS34-shRNA: 0.653 ± 0.027, N = 40 neurons; VPS34-shRNA cLTP: 0.688 ± 0.031, N = 40 neurons. Three independent experiments. Data were analyzed by one-way ANOVA with Tukey’s post hoc test, **P < 0.01, ****P < 0.001. Error bars are SEM. DIV, days in vitro.
Discussion
The studies reported here reveal that the signaling lipid PI(3)P plays a key role in driving coordinate activation of the parallel SNX17- and SNX27-dependent endocytic recycling pathways in neurons. We found that synaptic activity promotes PI(3)P generation by VPS34, a process dependent on CaMKII signaling. PI(3)P mediates the recruitment of the SNX17–Retriever and SNX27–Retromer pathways to endosomes. Furthermore, activity-dependent PI(3)P synthesis during LTP induction promotes the subsequent recruitment of SNX17–Retriever/SNX27–Retromer to dendritic spines, thus promoting the recycling of SNX17 and SNX27 cargoes to the plasma membrane. SNX17 and SNX27 define two parallel endocytic recycling pathways that recycle different sets of cell surface proteins necessary for LTP. Specifically, the SNX17 pathway regulates the recycling of β1-integrin, while the SNX27 pathway is responsible for GluA1 recycling. Inhibition of PI(3)P synthesis blocks the activity-dependent recycling of these cargoes and prevents the cLTP-dependent structural changes in dendritic spines (Fig. 10).

Model of PI(3)P-mediated regulation of endocytic recycling upon cLTP. Increased PI(3)P synthesis upon cLTP promotes endocytic recycling through the SNX17 and SNX27 pathways and is necessary for the cLTP-dependent structural changes in dendritic spines. Glycine-mediated cLTP (1) activates the CaMKII pathway (2) and promotes the recruitment of the lipid kinase VPS34 to endosomes, leading to an increase in PI(3)P-positive endosomes (3). This rise in PI(3)P levels facilitates the recruitment of the SNX17–Retriever and SNX27–Retromer pathways to dendritic spines (4), leading to increased recycling of cell surface proteins (5). SNX17 and SNX27 mediate the recycling of distinct sets of cell surface proteins, including β1-integrin and the AMPA receptor subunit GluA1, respectively, that are necessary for the cLTP response (6). Created with https://BioRender.com.
Model of PI(3)P-mediated regulation of endocytic recycling upon cLTP. Increased PI(3)P synthesis upon cLTP promotes endocytic recycling through the SNX17 and SNX27 pathways and is necessary for the cLTP-dependent structural changes in dendritic spines. Glycine-mediated cLTP (1) activates the CaMKII pathway (2) and promotes the recruitment of the lipid kinase VPS34 to endosomes, leading to an increase in PI(3)P-positive endosomes (3). This rise in PI(3)P levels facilitates the recruitment of the SNX17–Retriever and SNX27–Retromer pathways to dendritic spines (4), leading to increased recycling of cell surface proteins (5). SNX17 and SNX27 mediate the recycling of distinct sets of cell surface proteins, including β1-integrin and the AMPA receptor subunit GluA1, respectively, that are necessary for the cLTP response (6). Created with https://BioRender.com.
The recycling of AMPA-type glutamate receptors from endosomes to the plasma membrane is a well-established hallmark of LTP (Park, 2018). However, in cultured neurons, stimuli that induce LTP lead to an overall increase in the recycling of cargo and membrane components (Park et al., 2004). A recent quantitative analysis of the neuronal surface proteome demonstrated that multiple types of proteins undergo rapid externalization in response to cLTP (van Oostrum et al., 2020). Although the specific trafficking pathways activated by cLTP were not directly examined, our findings support a model in which LTP stimulation engages at least two major recycling pathways: SNX17–Retriever and SNX27–Retromer. Whether additional endomembrane recycling pathways are involved in LTP remains to be determined.
LTP engages both the SNX17 and SNX27 recycling pathways through a shared upstream regulator—the signaling lipid PI(3)P—which increases in response to synaptic NMDAR activation and downstream CaMKII signaling. Our data indicate that cLTP promotes the recruitment of VPS34 to endosomal compartments, where it drives activity-dependent PI(3)P synthesis. However, the mechanism underlying VPS34 activation and recruitment remains unclear. Given the rapid appearance of PI(3)P puncta upon cLTP, it is unlikely that increased VPS34 activity is due to new protein synthesis. Indeed, we did not detect changes in total VPS34 expression following cLTP. Notably, the VPS34 complex is regulated by PTMs, including phosphorylation (Ohashi et al., 2019). Since LTP triggers multiple signaling pathways (Li et al., 2016), it is possible that LTP-mediated PTMs may promote VPS34 recruitment to endosomes. While we did not observe changes in VPS34 phosphorylation after cLTP, its recruitment to endosomes may instead be regulated indirectly, potentially via PTMs of VPS34-interacting partners or upstream regulators of endosomal trafficking.
PI(3)P interacts with multiple proteins that contain PX or FYVE domains (Kutateladze, 2007). Therefore, it is possible that additional PI(3)P effectors may contribute to the roles of this lipid in synaptic function. For instance, another PI(3)P-binding sorting nexin, SNX16 (Chandra et al., 2019), controls endosomal maturation and distribution in neurons (Wang et al., 2019), although its role in synaptic plasticity remains unknown.
In addition to PI(3)P binding, other mechanisms may contribute to the cLTP-dependent recruitment of SNX17 and SNX27 to endosomes and, subsequently, to dendritic spines. In non-neuronal cells, the ability of several SNX proteins, including SNX27, to bind membranes is regulated by phosphorylation (Lenoir et al., 2018; Mao et al., 2021). Although further work is necessary to determine whether SNX17 and SNX27 are phosphorylated in neurons, it is possible that LTP-mediated PTMs may contribute to their recruitment. Moreover, other proteins that interact with or regulate these sorting nexins could be modulated by LTP and potentially contribute to their recruitment. One intriguing possibility is the lipid kinase PIKfyve, which converts PI(3)P to PI(3,5)P2 (Zolov et al., 2012; Rivero-Ríos and Weisman, 2022). In non-neuronal cells, VPS34 and PIKfyve coordinate a phosphoinositide cascade to regulate SNX17-dependent recycling (Giridharan et al., 2022). While PIKfyve has been implicated in some forms of synaptic plasticity, including homeostatic downscaling and long-term depression (McCartney et al., 2014), its role in LTP has yet to be explored. Another candidate is the ESCPE complex component SNX1/2, which has been shown to interact with SNX27 to promote neuronal growth and brain development in zebrafish (Yong et al., 2021).
Further proteomic and functional studies will be necessary to fully understand the roles of SNX17 and SNX27 in neurons. Notably, proteomic analysis of the SNX27 interactome identified >200 potential cargoes in rat neurons, including proteins implicated in excitotoxicity, epilepsy, intellectual disabilities, and working memory deficits (McMillan et al., 2021). Conducting a similar proteomic analysis of the SNX17 interactome in neurons will be essential to identify other SNX17-dependent cargoes that participate in LTP.
Understanding the functional interplay between SNX17 and SNX27 cargoes will provide critical insights into the regulation of synaptic plasticity. Although both pathways originate from PI(3)P-containing endosomes, they may direct distinct sets of cargoes to spatially segregated domains within dendritic spines. Such compartmentalization could enable fine-tuned control of local membrane composition and receptor availability in response to synaptic activity.
Moreover, the identification of novel cargoes for both SNX17 and SNX27 may reveal new mechanisms by which these sorting nexins regulate synaptic function and plasticity. Despite their similar roles in LTP, the SNX17 and SNX27 pathways may have distinct roles in presynaptic function or other forms of synaptic plasticity. Notably, the fact that dysfunction in these pathways is associated with different neurological disorders (Saitoh, 2022) suggests that they play at least partially distinct roles in the brain.
Finally, alterations in endosomal recycling are associated with neurodegenerative diseases, including Alzheimer’s disease (Small et al., 2005; Small et al., 2017) and Parkinson’s disease (Vilariño-Güell et al., 2011; Zimprich et al., 2011; Zavodszky et al., 2014; McGough et al., 2014). Importantly, reduced levels of PI(3)P have been observed in brain tissue from AD patients and in AD-related mouse models (Morel et al., 2013). These decreased PI(3)P levels disrupt the trafficking of the amyloid precursor protein (Morel et al., 2013), which may in part explain the role of PI(3)P in AD pathology. However, the role for PI(3)P in LTP described here may also contribute to its involvement in AD. Several AD-related mouse models exhibit alterations in LTP (Chapman et al., 1999; Nalbantoglu et al., 1997; Palop et al., 2007; Lanté et al., 2015; Barrow et al., 2000; Parent et al., 1999; Schneider et al., 2001; Auffret et al., 2009; Trinchese et al., 2004; Chang et al., 2006; Song et al., 2014; Richards et al., 2003; Oddo et al., 2003; Kimura and Ohno, 2009), which suggest that altered PI(3)P levels may affect synaptic plasticity and cognitive function in AD. Future studies using AD-relevant cellular and animal models will be necessary to address this possibility. Of note, however, upregulating the SNX27–Retromer pathway has demonstrated therapeutic potential for AD (Li et al., 2020). Pharmacological approaches that target PI(3)P synthesis and turnover, either alone or in combination with strategies to enhance the SNX17 or SNX27 pathways, may open new avenues for treating AD and related neurodegenerative diseases.
Materials and methods
DNA constructs and site-directed mutagenesis
For knockdown experiments, we used the following shRNA lentiviral plasmids: SNX17 (pLKO.1-puro with shRNA target sequence 5′-CCAGATGACTTGATCGGATAT-3′, TRCN0000190340; Millipore Sigma), SNX27 (pLKO.005-puro with shRNA target sequence 5′-AGCTGGAGAACCAGGTAATAG-3′, TRCN0000253473; Millipore Sigma), VPS26B (pLKO.005-puro with shRNA target sequence 5′-TGCAGCCTAGGTAGGGATAAG-3′, TRCN0000306336; Millipore Sigma), VPS26C (pGIPZ vector with shRNA target sequence 5′-TAATCTTGATGTCCAGAGT-3′, V3LMM_455807; Horizon Discovery), VPS34 (pLKO.1-puro with shRNA target sequence 5′-CGTCAAGATCAGCTTATTCTT-3′, TRCN0000025373; Millipore Sigma), Strumpellin (pLKO.005-puro with shRNA target sequence 5′-CATCGCTCCTTTGAGTATATA-3′, TRCN0000264509; Millipore Sigma), and scrambled control (MISSION pLKO.1 scrambled nontarget shRNA SHC002; Millipore Sigma).
pmCherry-C1 (cat. no. 632524) and pAcGFP-N1 (cat. no. 632469) were purchased from Clontech. dsRed-EEA1-FYVE was a generous gift from Dr. Daniel D. Billadeau (Mayo Clinic, Rochester, MN, USA) and has been previously described by Singla et al. (2019). pRK5-HA-CaMKII-T286D was a generous gift from Dr. Gentry Patrick (University of California at San Diego, San Diego, CA, USA). HA-VPS34 was a gift from Do-Hyung Kim (University of Minnesota, St. Paul, MN, USA) (Addgene plasmid 86749), and the 6xHis-SNAP-PI(3)P probe was a gift from Hannes Maib (University of Sheffield, Sheffield, UK) and David Murray (University of Dundee, Dundee, UK) (plasmid 211508; Addgene).
The generation of pTO-GFP-SNX17 and the shRNA-resistant version of pTO-GFP-SNX17 was previously described (Rivero-Ríos et al., 2023).
To generate mNeonGreen-SNX17, the region containing the GS linker and SNX27 was PCR amplified from pTO-GFP-SNX17 using primers 5′-CAAGGAATTCGGGGGCGGTTCAGGGGGT-3′ and 5′-TATCGATAAGCTTGATATCGACCGGTTTACAGATCCTCATCTCCAATGCC-3′, which add restriction sites for EcoRI and AgeI at 5 and -3′, respectively. mNeonGreen was PCR amplified from a modified pAAV vector, pAAV-eno-mNeonGreen (Rivero-Ríos et al., 2023), using the oligos 5′-CCGCCACTACCACCGGGTACGGTACCGCCACCATGGTG-3′ and 5′-AACCGCCCCCGAATTCCTTGTACAGCTCGTCC-3′ to delete the stop codon and add an EcoRI site at the 3′ end. Note that the mNeonGreen fragment contains a KpnI site at 5′. Both PCR products were digested with the corresponding restriction enzymes and purified. pAAV-eno-mNeonGreen was digested with KpnI and EcoRI to release the mNeonGreen fragment, and the purified vector was used for the ligation reaction. This generated the pAAV-eno-mNeonGreen-SNX17.
To generate GFP-SNX27, SNX27 was amplified from rat whole brain cDNA with oligos 5′-CGCTCGCTTACTCGCAAGA-3′ and 5′-GCTAAGAGGCTGAGGGCAAA-3′, and a DNA fragment with a 5′ KpnI site containing GFP and an 18-base pair GS linker was ordered from Twist Bioscience. To add regions of overlap, SNX27 was amplified with primers 5′-CAGGGGGTGGAAGCGGTGGTGCGGACGAGGACGGGGAAGGG-3′ and 5′-GTCGAGGCTGATCAGCGGGTTTAAACGCTAGGTGGCCAC-3′. The pCDNA4:TO vector (Rivero-Ríos et al., 2023) was digested with ApaI and KpnI, and NEBuilder HiFi DNA Assembly Cloning Kit (New England Biolabs) was used to assemble the digested vector with the GFP and SNX27 constructs. This generated the vector pTO-GFP-SNX27.
pTO-GFP-SNX27 was used to generate an shRNA-resistant version of GFP-SNX27 by introducing six silent mutations into the target sequence of the SNX27 shRNA clone using two rounds of mutagenesis with the Q5-site directed mutagenesis kit (New England Biolabs). Specifically, the original sequence 5′-AGCTGGAGAACCAGGTAATAG-3′ was mutated to 5′-AACTTGAAAATCAAGTGATAG-3′. A first round of mutagenesis with oligos 5′-CAGGTAATAGCGTTTGAATGGGATGAGATGC-3′ and 5′-GTTTTCAAGTTGCCCTTCTTCAGTGCAGG-3′ generated the sequence 5′-AACTTGAAAACCAGGTAATAG-3′, and a second round of mutagenesis with oligos 5′-CAAGTGATAGCGTTTGAATGGGATGAGATGC-3′ and 5′-ATTTTCAAGTTGCCCTTCTTCAGTGCAGG-3′ resulted in the final sequence. This generated the pCDNA4:TO-GFP-SNX27-R vector (GFP-SNX27-R).
To generate mScarlet-SNX27, the region containing the GS linker and SNX27 was PCR amplified from pTO-GFP-SNX27 using primers 5′-CAAGGAATTCGGGGGCGGTTCAGGGGGT-3′ and 5′-TATCGATAAGCTTGATATCGACCGGTCTAGGTGGCCACATCC-3′ to add EcoRI and AgeI restriction sites. A DNA fragment containing the mScarlet2 sequence was ordered from Twist Bioscience and PCR amplified with primers 5′-CCGCCACTACCACCGGGTACCGCTAGCGCCACCATGGTG-3′ and 5′-AACCGCCCCCGAATTCCTTGTACAGCTCGTCC-3′ to add NheI and EcoRI restriction sites. Both PCR products were digested with the corresponding restriction enzymes and purified. pAAV-eno-mNeonGreen was digested with KpnI and EcoRI to release the mNeonGreen fragment, and the purified vector was used for the ligation reaction. This generated the pAAV-eno-mScarlet-SNX27.
DNA was prepared from bacterial cultures grown at 37°C using a Midiprep kit (PureYield Plasmid Midiprep System, Promega) according to the manufacturer’s instructions. The identity of the constructs was verified by sequencing the entire coding region.
Primary neuron culture and transfection
All experimental protocols were approved by the University of Michigan Committee on the Use and Care of Animals. Primary hippocampal neuron cultures were prepared as previously described (Henry et al., 2017). Briefly, timed pregnant Sprague–Dawley rats were obtained from Charles River Laboratories. Hippocampi were dissected from postnatal day 1–2 rat pups of either sex, enzymatically digested using a papain-containing solution, and plated at a density of 60,000 cells in poly-D-lysine–coated 35-mm glass-bottom Petri dishes (MatTek). Cultures were maintained at 37°C and 5% CO2 in growth medium (Neurobasal A [Invitrogen] supplemented with B27 [Invitrogen] and Glutamax [Invitrogen]).
For surface biotinylation and western blot experiments, cortical tissue was dissected from postnatal day 1–2 rat pups of either sex, enzymatically digested, and triturated. Neurons were plated at a density of 6,000,000 cells on 35-mm dishes and maintained at 37°C and 5% CO2 in growth medium.
Neurons were transfected at the indicated days in vitro using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Neurons were used 4 days after shRNA transfection or 1 day after transfection of other constructs.
Chemically induced LTP
Under baseline conditions, neurons were incubated in HEPES-buffered saline (HBS) containing 119 mM NaCl, 5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 30 mM Glucose, and 10 mM HEPES, pH 7.4. cLTP in hippocampal neurons was achieved via brief (5 min) exposure to a Mg2+-free HBS solution supplemented with: 0.4 mM Glycine (Thermo Fisher Scientific), 0.02 mM Bicuculline (Abcam), and 0.003 mM Strychnine (Tocris). After glycine stimulation, neurons were washed with warm HBS and imaged or fixed at the indicated time points.
To evaluate the effect of compounds on the cLTP-dependent increase in VPS34-positive compartments, the following reagents were used: DMSO, 10 μM BAPTA-AM (Calbiochem), 100 μM D-APV (Sigma-Aldrich), 10 μM nifedipine (EMD Millipore), 10 μM AIP (Sigma-Aldrich), 2 μM KT5720 (Millipore Sigma), and 10 μM U0126 (LC Laboratories). Neurons were pretreated for 30 min before cLTP, and the compounds were maintained during the whole experiment.
To evaluate the effect of decreased PI(3)P synthesis on cLTP, the following compounds were used: DMSO, 1 μM VPS34-IN1 (EMD Millipore), and 1 μM SAR405 (MedChemExpress).
Fixation, surface labeling, and immunocytochemistry
Transfected cells were fixed with 4% paraformaldehyde/4% sucrose in PBS with 1 mM MgCl2 and 0.1 mM CaCl2 (PBS-MC) for 15 min and stored at 4°C before image acquisition.
To label surface β1-integrin or surface GluA1, neurons were incubated live with 10 µg/ml of β1-integrin antibody clone HM β1-1 (Armenian hamster mAb, 102202; BioLegend) or 1 µg/ml of GluR1-NT antibody clone RH95 (mouse mAb, MAB2263; Millipore Sigma), respectively, for 15 min at 37°C. Neurons were then fixed with 4% paraformaldehyde and 4% sucrose for 20 min, blocked with 2% BSA in PBS-MC for 1 h, and incubated with fluorescent secondary antibody. Secondary antibodies included goat anti-Armenian hamster Alexa Fluor 488 (1:1,000, ab173003; Abcam) for β1-integrin and goat anti-mouse Alexa Fluor 488 (1:1,000, A-11029; Invitrogen) for GluA1. To stain for MAP2, neurons were permeabilized with 0.1% Triton X-100 for 10 min and blocked with 2% BSA before incubation with MAP2 rabbit mAb (1:200, ab183830; Abcam) overnight and, later, donkey anti-rabbit Alexa Fluor 594 antibody (1:1,000, A-21207; Invitrogen) for 1 h.
For internalized β1-integrin and GluA1 labeling, after live labeling (15 min), cells were washed twice with PBS-MC and incubated at 37°C for 10 min to allow internalization of surface-bound antibodies. Remaining surface-bound antibodies were stripped using 0.5 M NaCl/0.2 N acetic acid for 1 min on ice. After washing, cells were fixed, blocked, and incubated with secondary antibodies as described above. For HA-VPS34 staining, neurons were then permeabilized with 0.1% Triton X-100 for 10 min, blocked with 2% BSA, and incubated overnight with HA rabbit mAb (1:500, 3724; Cell Signaling), followed by incubation with donkey anti-rabbit Alexa Fluor 405 antibody (1:1,000, A-48258; Invitrogen) for 1 h.
Purification and labeling of the PI(3)P recombinant biosensor
The purification of the PI(3)P probe was performed as previously described (Maib et al., 2024). The 6xHis-SNAP-PI(3)P vector was transformed into Escherichia coli strain BL21 (DE3), and transformed bacteria were grown in LB medium at 37°C to OD600 = 0.8. Protein expression was induced by adding 0.5 mM IPTG (Invitrogen) and incubating at 18°C overnight. Bacterial pellets were resuspended in 750 μl standard buffer (20 mM HEPES, 250 mM NaCl, and 0.5 mM TCEP). The lysate was transferred to a 2-ml tube, and 250 μl of 0.1-mm zirconia beads were added for cell lysis using a MiniBeadBeater-8 (six cycles of 30-s agitation followed by cooling on ice). After lysis, the samples were cleared by centrifugation, and the soluble fraction was incubated with 50 μl of pre-equilibrated Dynabeads His-tag (Invitrogen) for 1 h with gentle rotation at 4°C. Beads were washed three times with standard buffer, and bound proteins were eluted with 100 μl standard buffer containing 250 mM imidazole. Imidazole was removed by overnight dialysis against standard buffer at 4°C, and the sample was concentrated using a 10-kDa MWCO Amicon spin filter (Millipore Sigma). The biosensor was then labeled with SNAP-Surface Alexa Fluor 488 (NEB) at a 2:1 M excess for 3 h on ice, aliquoted, and stored at −80°C.
Cell staining with the PI(3)P recombinant biosensor
Immunofluorescence with the PI(3)P biosensor was done as described previously (Maib et al., 2024). Briefly, cultured hippocampal neurons were fixed with 4% paraformaldehyde/0.2% glutaraldehyde in PBS prewarmed to 37°C for 20 min, followed by two quick washes and incubation with 50 mM NH4Cl in PBS for 20 min. Neurons were then transferred onto ice and washed with ice-cold PIPES buffer (pH 7.2), followed by incubation with 500 nM labeled biosensor in PIPES containing 5% BSA and 0.5% saponin. Afterward, neurons were washed three times with ice-cold PIPES and postfixed with 2% PFA in PBS on ice for 10 min, then returned to room temperature for another 10 min. Finally, cells were washed with 50 mM NH4Cl in PBS at room temperature and stored in PBS at 4°C.
Image acquisition and analysis
After fixation or immunostaining, neurons were maintained in PBS-MC and imaged within a week. Images were acquired at room temperature with a Leica Stellaris 5 confocal microscope under an oil immersion 63× objective (z-series, 0.5-μm intervals). Single excitation for each wavelength separately was used for all acquisitions. Leica Stellaris 5 is equipped with a 405-nm diode laser, a 445-nm diode laser, and a white light laser tunable from 470 to 680 nm. The same laser settings and exposure times were used for the acquisition of individual experiments. Images were analyzed and processed using ImageJ.
The JACoP plugin of ImageJ was used to quantify the colocalization of SNX17 or SNX27 with PI(3)P. Manders’ overlap coefficient was obtained after thresholding the images, and the percentage of colocalization was obtained by multiplying the coefficient by 100. For triple colocalization experiments with VPS34, a mask representing the overlap between SNX17 and SNX27 (or between β1-integrin and GluA1) was first generated, and the colocalization of this mask with HA-VPS34 was calculated using JACoP.
Dendritic spine width was quantified manually by an observer blind to the experimental conditions. The maximum spine head width was determined using the line tool (in ImageJ), and the average width of the spines present in the first 30 μm after the first dendritic branchpoint was calculated.
The total numbers and the width of PI(3)P-, VPS34-, SNX17-, or SNX27-positive puncta in the first 30 μm of secondary dendrites were quantified using the Analyze Particles tool in ImageJ.
To measure SNX17 or SNX27 recruitment to dendritic spines, the mean intensity of mNeonGreen-SNX17 or mScarlet-SNX27 in the spines present in the first 30 μm of secondary dendrites was quantified and normalized to SNX17 or SNX27 mean intensity in the dendritic shaft.
To analyze the surface levels of β1-integrin or GluA1, the mean intensity in 50-μm segments of dendrites in MAP2-stained cells was measured.
To prepare representative images, the brightness and contrast were adjusted uniformly over all images of the respective assays using Photoshop 2021.
Live-cell imaging and analysis
To visualize the formation of PI(3)P-positive puncta, neurons were co-transfected with dsRed-EEA1-FYVE and eGFP. 24 h later, the media was replaced with fresh HBS for 10 min, followed by treatment with cLTP or with a mock cLTP stimulation with HBS for 5 min. After cLTP (or mock), neurons were washed with HBS and maintained in warm HBS media for 30 min.
For observation of SNX27 dynamics, neurons were transfected with mScarlet-SNX27 and eGFP 24 h before the experiment. Media was replaced with fresh HBS 10 min before imaging. Neurons were then treated with cLTP or with a mock cLTP stimulation with HBS for 5 min, followed by washing with warm HBS and addition of new HBS media.
Images were acquired with an AiryScan Zeiss LSM880 scanning confocal microscope with a 63× Plan-Apo oil immersion objective. The microscope is controlled with Zen software (Zeiss). The temperature was maintained at 37°C using a microscope incubation chamber. Z-stacks (0.5-μm intervals) were acquired before adding the cLTP stimulus (baseline), 2.5 min into the cLTP stimulus (during cLTP), directly after removing the stimulus (time 0), and every 5 min till 30 min after cLTP.
For each time point, the total numbers of dsRed-EEA1-FYVE- or mScarlet-SNX27-positive puncta in the first 30 μm of a secondary dendrite per neuron were quantified using ImageJ, and data were normalized to the baseline number of puncta for each neuron.
eGFP was used to identify and draw individual dendritic spines in the first 30 μm of one secondary dendrite per neuron. The mean intensity of mScarlet-SNX27 in individual spines that could be detected in the different time points was measured using ImageJ. The intensity at the different time points after cLTP was normalized to the baseline intensity for each spine.
Lentivirus-mediated shRNA knockdown
To test the knockdown efficiency for the SNX17, SNX27, VPS26B, VPS34, and strumpellin shRNA clones in primary cortical neurons, transduction-ready viral particles were produced by the University of Michigan Vector Core with a concentration of 107 transduction units per ml. Neurons were infected at an MOI of 2, without polybrene. After overnight incubation, the virus-containing medium was replaced with a saved conditioned medium. Experiments were performed 6 days after lentivirus transduction.
Generation of the TR-HEK293 cell line and validation of shRNA-resistant SNX27
To validate shRNA-resistant GFP-SNX27, we used a HEK293 cell line that stably expresses the tet repressor (TR-HEK293). The generation of this line was previously described (Rivero-Ríos et al., 2023). Briefly, HEK293 cells were cultured in 10-cm dishes to 70% confluence and transfected with 5 μg of the pCDNA6:TR vector (Invitrogen), which contains a blasticidin resistance cassette, using Lipofectamine 2000. Transfected cells were selected in the presence of 5 μg/ml blasticidin (Thermo Fisher Scientific) and cultured for 10 days. Cells were then sorted into a 96-well plate (1 cell/well) at the University of Michigan Flow Cytometry Core. Six clonal lines were expanded, and transient transfection with the pTO-eGFP vector was performed. Transfected cells were tested with 1 μg/ml doxycycline (Sigma-Aldrich) added for 12 h to allow for eGFP expression and a clone was chosen that had no detectable baseline eGFP.
TR-HEK293 was co-transfected with either control- or SNX27-shRNA together with pTO vectors expressing GFP, GFP-SNX27, or shRNA-resistant GFP-SNX27. This allows for the detection of the protein expression levels with an anti-GFP antibody. While shRNA expression occurs for 7 days, doxycycline addition for the last 24 h allows for the controlled expression of the GFP-fused target.
Surface biotinylations
Surface biotinylation assays in cultured cortical neurons were performed as described (Rivero-Ríos et al., 2023). Briefly, cells were washed with wash buffer (PBS containing 2.5 mM MgCl2 and 1 mM CaCl2) and incubated with 0.2 mg/ml NHS-SS-Biotin (Pierce) for 15 min. Neurons were then washed with wash buffer before being quenched in quenching buffer (50 mM Tris and 100 mM NaCl, pH 7) for 10 min. Neurons were lysed in 2% Triton X-100 containing protease inhibitors (Roche). Equal protein amounts were incubated with Dynabeads MyOne Streptavidin C1 (Thermo Fisher Scientific) for 1 h at 4°C before being washed and analyzed by western blot. All steps were carried out at 4°C to prevent the internalization of the cell surface proteins.
Immunoprecipitation of VPS34
Cells were collected in ice-cold RIPA buffer supplemented with phosphatase inhibitors (Sigma-Aldrich) and protease inhibitors (Roche). Lysates were cleared by centrifugation, and the supernatant was incubated with 5 μg VPS34 rabbit mAb (MA5-35160; Thermo Fisher Scientific) for 1 h at 4°C. For immunoprecipitation, 50 μl Dynabeads Protein A were used per sample. The beads were washed once with RIPA buffer and then incubated with the antibody-bound lysate for 1 h at 4°C. Beads were washed three times with RIPA buffer, and bound proteins were eluted by adding 40 μl 2× sample buffer containing β-mercaptoethanol, followed by heating at 75°C for 10 min.
Cell extracts and western blot
Cells were lysed in RIPA buffer (Pierce) containing protease inhibitors (Roche). Extracts were sonicated, boiled, and centrifuged at 10,000 g for 10 min. A BCA assay (Pierce) was used to determine protein concentrations, and equal amounts of protein were loaded into Mini-PROTEAN TGX Precast gels (Bio-Rad) and then transferred to nitrocellulose membranes. Membranes were blocked with Tris-HCl–buffered saline containing 5% BSA and 0.1% Tween for 1 h at room temperature and incubated with primary antibodies in blocking solution overnight at 4°C. Antibodies used for immunoblotting included SNX17 rabbit pAb (1:1,000, HPA043867; Atlas Antibodies), SNX27 rabbit pAb (1:1,000, Ab241128; Abcam), VPS34 rabbit mAb (1:1,000, MA5-35160; Thermo Fisher Scientific), VPS26B rabbit pAb (1:500, NBP1-92575; Novus Biologicals), SPG8/Strumpellin rabbit pAb (1:1000, A304-809A; Thermo Fisher Scientific), GFP rabbit mAb (1:1,000, Ab32146; Abcam), Phospho-(Ser/Thr) rabbit pAb (1:1000, 9631; Cell Signaling), β1-integrin goat pAb (1:1,000, AF2405; R&D Systems), GluA1 rabbit pAb (1:1,000, ABN241; Millipore Sigma), and GAPDH rabbit mAb (1:2,000, 2118; Cell Signaling). Following washes in TBS containing 0.1% Tween-20, the blots were incubated with HRP-conjugated secondary antibodies for 1 h and developed using chemiluminescence with an Amersham ECL western blotting detection reagent (Cytiva). Chemiluminescence signals were detected using a Bio-Rad ChemiDoc Imaging system. Immunoblots were analyzed using ImageLab software.
Hippocampal slice preparation
Transverse hippocampal slices were prepared from 6- to 8-wk-old wild-type mice. Mice were deeply anesthetized with isoflurane, decapitated, and the whole brain was rapidly removed and placed in an ice-cold modified artificial cerebrospinal fluid (ACSF) containing (in mM) 80 NaCl, 2.5 KCl, 1.25, NaH2PO4, 0.5 CaCl2, 3.5 MgCl2, 25 NaHCO3, 75 sucrose, 1.3 sodium ascorbate, and 3.0 sodium pyruvate, with pH at 7.4 and osmolality at 310–320 mOsm, bubbled with 95% O2 and 5% of CO2. Brain slices (300 μm) were cut sagittally by a vibratome (VT1200S; Leica). The slice was then incubated for about 1.5 h at 33°C in oxygenated (95% O2 and 5% CO2) cutting solution, which contains (in mM) 145 NaCl, 3 KCl, 1.5 CaCl2, 3.5 MgCl2, 10 D-glucose, and 10 NaHCO3 adjusted to pH 7.2–7.4 with NaOH and osmolality at 310–320 mOsm.
LTP recording
Acute hippocampal slices were incubated for at least 1.5 h in standard ACSF prior to recording. The ACSF contained (in mM) 124 NaCl, 1.2 NaH2PO4, 4.3 KCl, 25 NaHCO3, 10 glucose, 2 CaCl2, and 1 MgCl2. The pH was adjusted to 7.2–7.4 with NaOH, and osmolality was maintained at 310–320 mOsm, bubbled with 95% O2 and 5% of CO2. Following incubation, slices were transferred to a recording chamber continuously perfused with oxygenated ACSF and maintained at 25–26.5°C. Field excitatory postsynaptic potentials were recorded in the stratum radiatum of the CA1 region using ACSF-filled borosilicate glass pipettes (3–5 MΩ; Sutter Instrument). Presynaptic stimulation was delivered via a tungsten electrode placed in the CA3 stratum radiatum to activate Schaffer collateral axons using a constant-current stimulus isolation unit (WPI). Synaptic input–output curves were obtained by delivering stimulation pulses ranging from 50 to 250 μA in 50 μA increments. For LTP experiments, stimulation intensity was set to 50% of the maximum field excitatory postsynaptic potential response. After establishing a stable baseline for 10 min, LTP was induced using two high-frequency stimulation trains (100 Hz, 1 s each) separated by a 30-s interval. Data were acquired and analyzed using pClamp 10.0 software (Molecular Devices) via a MultiClamp 700B amplifier and Digidata 1550B digitizer.
Statistical analyses
All experiments were repeated at least three independent times. Microsoft Excel software was used for calculations, and the results were plotted and analyzed using GraphPad Prism software version 10. Data are expressed as means ± SEM. Statistical tests and the size of the samples are described in the respective figure legends. Data distribution was assumed to be normal, but this was not formally tested. Comparisons were made using unpaired two-tailed Student’s t test or one-way ANOVA with Tukey’s post hoc comparisons. Two-way ANOVA with Sidak’s multiple comparison test was used to analyze mScarlet-SNX27 recruitment to dendritic spines. P values are shown in asterisks where *: P value < 0.05, **: P value < 0.01, ***: P value < 0.005, and ****: P value < 0.001.
Online supplemental material
Fig. S1 shows that cLTP promotes the recruitment of VPS34 to endosomal compartment as well as the formation of endogenous PI(3)P in endosomes. Fig. S2 shows that sustained inhibition of PI(3)P synthesis is necessary to block the cLTP-dependent structural plasticity of spines. Fig. S3 shows VPS34 and its lipid product PI(3)P colocalize with SNX17 and SNX27 in dendrites. Fig. S4 shows that PI(3)P synthesis is necessary for the cLTP-dependent increase in SNX17- and SNX27-positive puncta. Fig. S5 shows that SNX27 is recruited to dendritic spines upon cLTP. Fig. S6 shows that brief 10-min inhibition of VPS34 does not affect the formation of SNX17- and SNX27-positive puncta or their recruitment to dendritic spines. Fig. S7 shows the validation of the SNX17-, SNX27-, and VPS26B-shRNA constructs, as well as the validation of the shRNA-resistant SNX27 construct. Fig. S8 shows that the WASH complex subunit strumpellin is necessary to recycle SNX17- and SNX27-dependent cargoes.
Data availability
All data are available from the corresponding authors upon request.
Acknowledgments
We thank Cindy Carruthers, Christian Althaus, and Takayuki Hayami in the Sutton lab for preparing neuronal cultures. We thank the members of the Weisman and Sutton labs for their insights and suggestions. We thank Carole Parent (University of Michigan, Ann Arbor, MI, USA) for insightful discussions and for providing access to her AiryScan Zeiss LSM880 scanning confocal microscope. We also thank Daniel D. Billadeau (Mayo Clinic, Rochester, MN, USA) for providing the dsRed-EEA1-FYVE construct, Gentry Patrick (University of California at San Diego, San Diego, CA, USA) for the pRK5-HA-CaMKII-T286D construct, Do-Hyung Kim (University of Minnesota, St. Paul, MN, USA) for the HA-VPS34 construct, and Hannes Maib (University of Sheffield, Sheffield, UK) and David Murray (University of Dundee, Dundee, UK) for the 6xHis-SNAP-PI(3)P probe.
This work was supported by the National Institutes of Health (NIH) under award numbers R01-NS129198 to Lois S. Weisman and Michael A. Sutton, R01-NS097498 to Michael A. Sutton, and R01NS118769 and R56DK139477 to Bo Duan. Pilar Rivero-Ríos was in part supported by an Alzheimer’s Association Research Fellowship, by a Michigan Life Sciences Postdoctoral Fellowship (University of Michigan), by an NIH/National Institute of Aging Michigan Alzheimer’s Disease Research Center grant P30AG072931, and by the University of Michigan Alzheimer’s Disease Center (Berger Endowment).
Author contributions: Pilar Rivero-Rios: conceptualization, formal analysis, funding acquisition, investigation, methodology, validation, visualization, and writing—original draft, review, and editing. Tunahan Uygun: formal analysis, investigation, visualization, and writing—review and editing. Garrett D. Chavis: investigation and writing—review and editing. Hankyu Lee: formal analysis, investigation, and writing—review and editing. Bo Duan: methodology, resources, and writing—review and editing. Michael A. Sutton: conceptualization, funding acquisition, methodology, project administration, resources, supervision, and writing—original draft, review, and editing. Lois S. Weisman: conceptualization, data curation, funding acquisition, project administration, resources, supervision, validation, visualization, and writing—original draft, review, and editing.
