


The myosin super-relaxed (SRX) state is central to striated muscle metabolic and functional regulation. In skeletal muscle, SRX myosin are predominantly colocalized with myosin-binding protein C (MyBP-C) in the sarcomere C-zone. To define how cardiac MyBP-C (cMyBP-C) and its specific domains contribute to stabilizing the SRX state in cardiac muscle, we took advantage of transgenic cMyBP-C null mice and those expressing cMyBP-C with a 271-residue N-terminal truncation. Utilizing super-resolution microscopy, we determined the lifetime and subsarcomeric location of individual fluorescent-ATP turnover events within isolated cardiac myofibrils. The proportion of SRX myosin demonstrated a gradient along the half-thick filament, highest in the P- and C-zones (72 ± 9% and 71 ± 6%, respectively) and lower in the D-zone (45 ± 10%), which lies farther from the sarcomere center and lacks cMyBP-C, suggesting a possible role for cMyBP-C in stabilizing the SRX. However, myofibrils from cMyBP-C null mice demonstrated an ∼40% SRX reduction, not only within the now cMyBP-C-free C-zone (49 ± 9% SRX), but also within the D-zone (22 ± 5% SRX). These data suggest that the influence of cMyBP-C on the SRX state is not limited to the C-zone but extends along the thick filament. Interestingly, myofibrils with N-terminal truncated cMyBP-C had an SRX content and spatial gradient similar to the cMyBP-C null, indicating that the N terminus of cMyBP-C is necessary for cMyBP-C’s role in enhancing the SRX gradient along the entire thick filament. Given that SRX myosin exist as a gradient along the thick filament that is highest in the C-zone, even in the absence of cMyBP-C or its N-terminus, an inherent bias must exist in the structure of the thick filament to stabilize the SRX state.
Introduction
Striated muscle (i.e., skeletal and cardiac) activation requires calcium binding to the thin filament regulatory complex (i.e., troponin and tropomyosin) which shifts the position of the complex to allow force-generating myosin motors access to actin-binding sites along the thin filament (Fig. 1 A; McKillop and Geeves, 1993; Lehman et al., 1995). Recently, a myosin thick filament–based regulatory mechanism has been proposed, wherein myosin heads can adopt an energetically economical super-relaxed (SRX) state that is also effectively incapable of binding to the thin filament (Stewart et al., 2010; Fig. 1 A). The SRX state is biochemically distinct with a very low ATP hydrolysis rate of ∼0.005 s−1 or about 1 ATP molecule every 200 s. This rate is fivefold lower than that of the disordered-relaxed (DRX) myosin, which can bind to the thin filament and generate force once muscle is calcium-activated (Stewart et al., 2010). The SRX biochemical state may be inherent to each of myosin’s two heads (Anderson et al., 2018; Rohde et al., 2018; Chu et al., 2021; Walklate et al., 2022) but is further stabilized when the two heads are folded back and interact with each other and the myosin tail (i.e., interacting heads motif or IHM; Yang et al., 2020; Fig. 1 A). This structural motif has been observed in high-resolution, 3-D electron microscopic reconstructions of native cardiac thick filaments (Zoghbi et al., 2008; Al Khayat et al., 2013). Interestingly, a population of cardiac myosin remains sequestered in the mechanically inactive SRX state during calcium activation of the sarcomere (Hooijman et al., 2011), possibly serving as a “reserve pool,” from which myosin can be recruited in response to physiological demands. Importantly, cardiac myosin mutations associated with hypertrophic cardiomyopathy (HCM) have been linked to a reduced proportion of myosin in the SRX state (McNamara et al., 2017; Toepfer et al., 2019). If so, this may explain the hypercontractility of hearts in HCM patients and support the therapeutic basis of Mavacamten, an FDA-approved small molecule that stabilizes the SRX state as a means of restoring the normal proportion of SRX myosin and thus normal contractility (Green et al., 2016; Anderson et al., 2018).

Structure and regulation of myosin, cMyBP-C, and the sarcomere. (A) Myosin’s biochemical states. (B) Cartoon of the sarcomere, showing thick and thin filaments, delineation of C-, D-, P-, and bare zones, as well as myosin and cMyBP-C (yellow). (C) Domain structure of cMyBP-C, truncated variant (ΔC0C1f), and null (t/t).
Structure and regulation of myosin, cMyBP-C, and the sarcomere. (A) Myosin’s biochemical states. (B) Cartoon of the sarcomere, showing thick and thin filaments, delineation of C-, D-, P-, and bare zones, as well as myosin and cMyBP-C (yellow). (C) Domain structure of cMyBP-C, truncated variant (ΔC0C1f), and null (t/t).
As the SRX state may represent a key mechanism for both cardiac energetic and contractile regulation, defining the physiological and structural determinants of the SRX state is critically important. We recently described a super-resolution imaging approach in rat soleus myofibrils to visualize single, fluorescently labeled ATP being hydrolyzed by myosin within individual sarcomeres (Nelson et al., 2020; Fig. 1 B). Interestingly, SRX myosin was not uniformly distributed along the thick filament, but was predominantly found in the C-zone where myosin-binding protein C (MyBP-C) only exists. The flanking P- and D-zones are devoid of MyBP-C and demonstrated much lower frequencies of SRX myosin (Nelson et al., 2020). These data and a more recent study using a similar spatially resolved myofibrillar approach (Pilagov et al., 2023) support earlier, more globally measured ATP turnover studies that implicated MyBP-C in stabilizing the SRX state (McNamara et al., 2016; Toepfer et al., 2019).
MyBP-C is an ∼140-kD protein, encoded by three orthologous and muscle fiber-type specific genes (fast-type, slow-type, and cardiac). The cardiac isoform, cardiac MyBP-C (cMyBP-C), is composed of 11 Ig-like and Fn3-like domains, denoted as C0 through C10 (Fig. 1 C). cMyBP-C is anchored to the thick filament backbone through its C-terminal domains (C8–C10; Lee et al., 2015; Gilbert et al., 1996), whereas the N-terminal domains (C0–C2) are critical to cMyBP-C’s capacity to modulate cardiac contractility through interactions with the myosin head region and/or actin (van Dijk et al., 2014; Fig. 1 C). Mutations in cMyBP-C are the leading cause of HCM (Walsh et al., 2017) and are also associated with reductions in the SRX state (McNamara et al., 2017), further emphasizing a potential role for cMyBP-C in the regulation of the SRX state.
To define cMyBP-C’s role in stabilizing the SRX state and the involvement of its N terminus, we took advantage of genetically modified mouse cardiac muscle that was either devoid of cMyBP-C (McConnell et al., 1999) or that expressed cMyBP-C lacking the first 29 kD of the N terminus (Lynch et al., 2021). Here, we applied the super-resolution imaging of single fluorescently labeled ATP hydrolysis events in myofibrils from these mouse models to define the spatial distribution of SRX myosin in the presence, absence, and N-terminal truncation of cMyBP-C.
Materials and Methods
Mouse models
All experiments using animals detailed in this work were approved by the Institutional Animal Care and Use Committees at the University of Vermont and the University of Cincinnati and followed the policies of the Guide for the Use and Care of Laboratory Animals published by the National Institutes of Health. cMyBP-C null (t/t) mice were generated by insertion of a 2-kb neomycin resistance gene into exon 30 of MYBPC3 (McConnell et al., 1999). While truncated cMyBP-C (MyBP-CΔC0C1f) were generated by cardiomyocyte-specific replacement of endogenous cMyBP-C with an equivalent cDNA lacking the first 813 nucleotides, as previously described in Lynch et al. (2021). For wildtype (WT) controls, frozen hearts from FVB mice were ordered from Jackson Labs.
Preparation of mouse cardiac myofibrils
Mouse cardiac ventricular myofibrils were prepared from flash-frozen mouse hearts following a protocol modified from that described in Creed and Tong (2021). Briefly, the apex of the frozen heart was removed using a razorblade and rinsed twice to remove blood in 1 ml of K60 buffer (60 mM KCl, 2 mM MgCl2, and 20 mM MOPS, pH 7.4) with 30 mM BDM, 1 mM EGTA, and protease inhibitor cocktail (P8340; Sigma-Aldrich). The tissue was then placed in 1 ml K60 buffer and homogenized using a “Tissue Tearor” homogenizer (Biospec Products) operated for two rounds at 21,000 rpm for 30 s each. Tissue was collected by centrifugation at 1,000 × g for 10 min at 4°C, and then permeabilized in K60 buffer with 1% v/v Triton X-100, 1 mM EGTA, and a protease inhibitor cocktail for 30 min. Myofibrils were collected by centrifugation at 1,000 × g for 5 min at 4°C and resuspended in 1 ml of K60 buffer, followed by three more washes with K60 buffer with 1 mg/ml BSA.
Imaging and antibody labeling conditions
Fluorescence imaging was performed as described (Nelson et al., 2020). To identify the sarcomere center (M-line) where myomesin resides, prior to imaging, each myofibril preparation was incubated with anti-myomesin antibody (cat #EPR17322-9; Abcam) at a 1:5,000 final dilution for 15 min on ice, followed by Alexa Fluor 647 goat-anti-mouse IgG (cat #A21237; Life Technologies) at a final concentration of 1:5,000. After 5 min on ice, labeled myofibrils were flowed into flowcells that were made using plasma-cleaned glass. The flowcell was then incubated at room temperature for 20 min to allow myofibrils to adhere to the coverglass surface. Finally, 100 µl of relaxing buffer (120 mM KOAc, 5 mM K-phosphate, 4 mM EGTA, 4 mM MgCl2, 50 mM MOPS, 4 mM ATP, and 10 nM BODIPY-ATP, pH 6.8; cat #A12410; Thermo Fisher Scientific) was flowed into the flowcell, and samples were imaged using a custom-built dual camera super-resolution microscope, described previously (Nelson et al., 2020), for 50 min at 10 FPS. From each myofibril preparation, three to four recordings were made, with each flowcell being used for only a single imaging session.
Fluorescent ATP binding event analysis
The lifetime and position of each BODIPY-ATP binding event were measured separately to ensure accuracy. BODIPY-ATP binding events (green channel) were manually documented in image stacks using ImageJ (Schindelin et al., 2012). First, image stacks were integrated into 1FPS using the “grouped Z-project” function. Image stacks were then converted into a stack of kymographs using the “Reslice” function, and the events were documented using rectangular selections and the “overlay” function. Event lifetimes were determined manually from kymographs (Fig. 2 A) to account for visually apparent blinking behavior, counting only events with visually apparent beginnings and endings (i.e., not cut off by the beginning or end of the recording). Super-resolution position information was determined using the ImageJ plugin “D.O.M.” (Katrukha, 2020). Finally, BODIPY-ATP binding lifetimes (from kymograph analysis) were connected to subpixel localizations in the sarcomere, as the kymograph X, Y, and slice coordinates can be directly related to Timestamp, X, and Y coordinates, respectively, in the DOM output files.

ATP binding events and overall lifetime distributions. (A) Kymograph showing multiple fluorescent nucleotide binding events (white streaks). (B) Overall lifetime distributions for WT, t/t, and ΔC0C1f transgenic. Inset shows the proportions of SRX and DRX events. Error bars denote the standard error of the fit. Both t/t and ΔC0C1f lifetime distributions are significantly different than WT (P = 0.00000002 and P = 0.0002 by KS test, respectively). * denotes P < 0.05. WT events are combined from 19 technical replicates taken from 6 biological replicates. t/t events are combined from 16 technical replicates taken from 5 biological replicates. ΔC0C1f events are combined from 10 technical replicates taken from 3 biological replicates.
ATP binding events and overall lifetime distributions. (A) Kymograph showing multiple fluorescent nucleotide binding events (white streaks). (B) Overall lifetime distributions for WT, t/t, and ΔC0C1f transgenic. Inset shows the proportions of SRX and DRX events. Error bars denote the standard error of the fit. Both t/t and ΔC0C1f lifetime distributions are significantly different than WT (P = 0.00000002 and P = 0.0002 by KS test, respectively). * denotes P < 0.05. WT events are combined from 19 technical replicates taken from 6 biological replicates. t/t events are combined from 16 technical replicates taken from 5 biological replicates. ΔC0C1f events are combined from 10 technical replicates taken from 3 biological replicates.
P1 is the proportion of relaxed events, t1 is the time constant for the relaxed DRX population, and t2 is the time constant of the SRX population. Lifetime distributions were fit by direct optimization of the log-likelihood estimates as implemented in the “fitdistr” function included in the “MASS” library for the statistical programming language R (Venables and Ripley, 2002) using the “L-BFGS-B” method (Byrd et al., 1995). For sliding window analysis (Fig. 3 D), nucleotide binding events were subsetted into multiple overlapping 160-nm-wide windows, centered on each point, and each containing between 37 and 180 events per window. Within each window, the fraction of SRX events was determined by fitting the double exponential model described above. For convergence reasons when fitting sliding window and zone-specific lifetime distributions, t1 and t2 were constrained to the values obtained from the overall lifetime fits.
Binding event lifetime distributions were compared using a Kolmogorov–Smirnov test, while sarcomere lengths were compared using a Student’s t test.
To localize M-line positions, myomesin-labeled (red channel) images were integrated into blocks of 1,000 frames using the “Grouped Z-project” function in ImageJ. Each M-line was tracked using the ImageJ plugin “FilamentJ” (Smith et al., 2010) using default settings, except for “Deform Iterations,” which was set to 500. Finally, for each BODIPY-ATP binding event, the subsarcomeric location was determined as the distance to the nearest M-line at the corresponding time point, after correction for channel registration (described below).
Spatial offsets between red and green imaging channels were calculated by imaging multicolor-fluorescent beads (Tetraspeck, Thermo Fisher Scientific) and analysis using the Particle Image Velocimetry plugin for ImageJ (Tseng et al., 2012), which calculates relative offsets between the two images by maximizing cross-correlation between corresponding regions in the two images.
Online supplemental material
Results
Single fluorescent ATP turnover events
When myofibrils from WT mouse hearts were imaged under relaxing conditions (no calcium, 4 mM ATP) with the addition of 10 nM BODIPY-ATP, discrete single, fluorescent-nucleotide binding events were observed within myofibrils (Fig. 3 A). We interpret these events as the specific binding of a fluorescent ATP molecule, followed by hydrolysis and product release (fluorescent ADP). As in other studies, sarcomeric ATP binding is attributed to myosin as it is the predominant ATPase in the sarcomere (Stewart et al., 2010). Individual event lifetimes were determined by kymograph analysis (Fig. 2 A) and are a direct measure of the ATPase cycle time, given that the product release steps of the cycle are rate-limiting. Similar to our previous studies using the identical approach in slow skeletal muscle (Nelson et al., 2020), fluorescent ATP binding lifetime distributions were best fit as the sum of two exponential processes (Fig. 2 B), with time constants of 25 ± 4 and 161 ± 11 s (N = 972 events from 19 myofibrils from six mice). These time constants are similar to those reported in the literature for various striated muscles and thus indicative of the DRX and SRX states, respectively (Hooijman et al., 2011). However, cardiac myofibrils demonstrated a greater overall fraction (57 ± 5%) of SRX events, as in other reports for mouse cardiac muscles (Toepfer et al., 2020), but approximately twofold greater than that of skeletal muscles (Stewart et al., 2010; Nelson et al., 2020; Pilagov et al., 2023).
Nucleotide binding event locations
Utilizing super-resolution imaging methodologies, we determined the subsarcomeric location of each event (Fig. 3 B). The well-defined sarcomeric geometry allowed us to assign each event to a sarcomere zone based on the distance to the nearest M-line, i.e., the sarcomere center. In mammalian striated muscle, the myosin-containing A-band is defined by the thick filament length, extending 800 nm on either side of the M-line. Sarcomere lengths, as measured from M-line to M-line averaged 1.75 ± 0.07 μm (N = 177 sarcomeres), and a combined spatial resolution of 21 nm allowed us to parse each half of the A-band into four zones (Lee et al., 2015), specifically, the C-zone (155–500 nm from M-line), comprised of both myosin and cMyBP-C, while the flanking P-zone (80–155 nm from M-line) and D-zones (>500 nm from M-line) are both devoid of cMyBP-C (Fig. 1 B). Finally, the region nearest (<80 nm) to the M-line is referred to as the “bare zone,” as this region does not contain myosin heads or MyBP-C. A plot of each event’s lifetime versus its distance from the M-line (Fig. 3 B) shows no clear trend in lifetimes, but suggests elevated event frequencies near the M-line and thick filament tip (Fig. 3 C, left). Sliding window analysis coupled with the fitting of the double exponential model shows that the proportion of SRX events is highest near the P-zone/C-zone boundary, with a spatial gradient decreasing toward more distal portions of the C- and D-zone (Fig. 3 D, WT). When binding events within WT myofibrils were binned according to their corresponding zones and their distributions fit with the double exponential model (Fig. 3 E and Fig. S1, WT), we found that the P- and C-zone demonstrated nearly identical proportions of SRX events (74 ± 8% [N = 20] and 71 ± 6% [N = 177], respectively; P = 0.43), significantly higher than the D-zone with 45 ± 10% SRX (N = 168; P = 0.02), verifying a spatial differential in SRX location. Of the events that could be unambiguously assigned a subsarcomeric location, 85% localized to the myosin-containing P-, C-, or D-zone. Curiously, 13% of spatially resolved events (53/418 events) shared ATPase rates similar to the myosin DRX and SRX states but occurred in the bare zone. Given that this region is devoid of myosin heads, these events may arise from some other M-line localized ATPase, of which there are many (e.g., creatine kinase; Lange et al., 2020). As these bare zone events may arise from a non-myosin ATPase, they were omitted from further analysis.
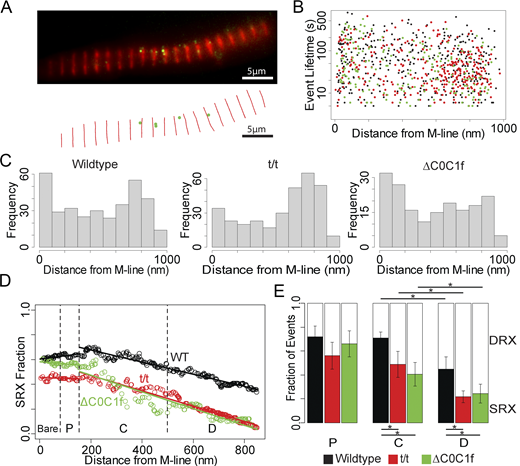
Event localizations and distributions. (A) Top: Combined fluorescence image showing anti-myomesin (red) and BODIPY-ATP (green), combined from 100 s of acquisition. Bottom: X-Y plot of resulting localization data for the same interval. (B) Scatterplot showing distance from M-line and lifetime for each localized event. WT (black), ΔC0C1f (green), and t/t (red). (C) Histograms showing event frequency as a function of distance from the M-line. (D) Fitting the double-exponential model within a sliding window shows a gradient in SRX abundance for all three mouse models. Linear regressions were fitted through data in the bare and P-zones and separately through the C- and D-zones for all three models. Fit parameters are reported in Table S1. (E) Fractions of SRX/DRX events for events when binned by zone with lifetime distribution plots for these results in Fig. S1. * denotes P < 0.05 by KS test. Error bars denote the standard error of the fit. The C-zones of both t/t and ΔC0C1f were significantly different than WT (P = 0.02 and P = 0.01, respectively), as were the D-zones (P = 0.0007 and P = 0.04). D-zone event distributions were significantly different from the C-zone for WT (P = 0.04), t/t (P = 0.04), and ΔC0C1f (P = 0.048). WT events are combined from 13 technical replicates taken from 4 biological replicates. t/t events are combined from 11 technical replicates taken from 4 biological replicates. ΔC0C1f events are combined from 7 technical replicates taken from 3 biological replicates.
Event localizations and distributions. (A) Top: Combined fluorescence image showing anti-myomesin (red) and BODIPY-ATP (green), combined from 100 s of acquisition. Bottom: X-Y plot of resulting localization data for the same interval. (B) Scatterplot showing distance from M-line and lifetime for each localized event. WT (black), ΔC0C1f (green), and t/t (red). (C) Histograms showing event frequency as a function of distance from the M-line. (D) Fitting the double-exponential model within a sliding window shows a gradient in SRX abundance for all three mouse models. Linear regressions were fitted through data in the bare and P-zones and separately through the C- and D-zones for all three models. Fit parameters are reported in Table S1. (E) Fractions of SRX/DRX events for events when binned by zone with lifetime distribution plots for these results in Fig. S1. * denotes P < 0.05 by KS test. Error bars denote the standard error of the fit. The C-zones of both t/t and ΔC0C1f were significantly different than WT (P = 0.02 and P = 0.01, respectively), as were the D-zones (P = 0.0007 and P = 0.04). D-zone event distributions were significantly different from the C-zone for WT (P = 0.04), t/t (P = 0.04), and ΔC0C1f (P = 0.048). WT events are combined from 13 technical replicates taken from 4 biological replicates. t/t events are combined from 11 technical replicates taken from 4 biological replicates. ΔC0C1f events are combined from 7 technical replicates taken from 3 biological replicates.

Zone-specific lifetime distributions and fits. Fluorescent nucleotide binding events were parsed into P-, C-, and D-zones according to distance from the M-line (see text and Fig. 3 E). Lifetime distributions were then fit with a double exponential model, as described in Materials and methods.
Zone-specific lifetime distributions and fits. Fluorescent nucleotide binding events were parsed into P-, C-, and D-zones according to distance from the M-line (see text and Fig. 3 E). Lifetime distributions were then fit with a double exponential model, as described in Materials and methods.
Deletion of cMyBP-C
Given prior reports that SRX myosin may be stabilized by MyBP-C (McNamara et al., 2017; Toepfer et al., 2019; Nelson et al., 2020), we took advantage of an established cMyBP-C transgenic mouse line that is devoid of cMyBP-C (cMyBP-Ct/t; McConnell et al., 1999) to determine if eliminating cMyBP-C alters the overall proportion of SRX and whether any positional bias for the remaining SRX still exists within the sarcomere. The cMyBP-Ct/t hearts were hypertrophied but exhibited no apparent sarcomeric abnormalities, consistent with prior reports of preserved sarcomere ultrastructure in this model (McConnell et al., 1999). Although myocytes from cMyBP-Ct/t mice have been reported to exhibit highly variable sarcomere lengths (Toepfer et al., 2019), the myofibrils used in our analysis demonstrated sarcomere lengths of 1.67 ± 0.07 μm (N = 199 sarcomeres), not significantly different than WT (P = 0.20).
cMyBP-Ct/t myofibrils demonstrated a nearly 40% reduction in the overall SRX content compared with WT (35 ± 5% versus 57 ± 5% SRX, respectively, P < 0.001; N = 882 events from 16 myofibrils from five mice; Fig. 2 B). This reduction in SRX was distributed across both the C- and D-zone (N = 117 and 216, respectively), such that the C-zone continued to exhibit approximately twofold higher proportion of SRX myosin than the D-zone (49 ± 9% versus 22 ± 5%, respectively; P = 0.02, Fig. 3 E), preserving the gradient that was present in WT myofibrils across these zones (Fig. 3 D). The SRX fraction in the P-zone in cMyBP-Ct/t sarcomeres (N = 24) was not different from those of the WT P-zone (P = 0.57).
N-terminal cMyBP-C truncation
The overall reduction in SRX abundance observed in the cMyBP-Ct/t myofibrils (Fig. 2 B) suggests that the presence of cMyBP-C contributes to SRX stabilization. If so, then are cMyBP-C N-terminal domains critical to this stabilization? We again took advantage of an existing cMyBP-C transgenic mouse line that expresses an N-terminal truncated cMyBP-C, lacking the C0 and C1 domains and the first 17 amino acids of the M-domain (cMyBP-CΔC0-C1f; Lynch et al., 2021; Fig. 1 C). As previously characterized, hearts express the full complement of cMyBP-C, of which 85% is the N-terminal truncation (Lynch et al., 2021). As with the cMyBP-Ct/t null myofibrils, the cMyBP-CΔC0-C1f sarcomere lengths were no different than WT (1.75 ± 0.06 versus 1.75 ± 0.07 μm, respectively; P = 0.38). Similar to the cMyBP-Ct/t myofibrils, the cMyBP-CΔC0-C1f myofibrils demonstrated a significant 45% reduction in the overall SRX content compared with WT (31 ± 7% versus 57 ± 5%, respectively; P = 0.02; N = 469 events from 10 myofibrils from three mice; Fig. 2 B). This reduction in SRX occurred uniformly in both the C- and D-zone (N = 101 and 80, respectively), such that the C-zone continued to exhibit approximately twofold higher proportion of SRX myosin compared with the D-zone (40 ± 10% versus 24 ± 8%, respectively; P = 0.20; Fig. 3 E), demonstrating a nearly identical spatial gradient in SRX abundance compared with the cMyBP-Ct/t myofibrils (Fig. 3 D). Once again, the SRX fraction of P-zone events (Fig. 3 E) was not significantly different than in the WT (N = 25; P = 0.93).
Discussion
The SRX state appears to be common to all mammalian striated muscle myosin and might serve a significant role in modulating muscle contractility (Brunello et al., 2020). However, different muscle types show substantial differences in the overall proportion of SRX, evidenced by the twofold difference between the present data in mouse cardiac myofibrils compared with our previously published data (Nelson et al., 2020) in slow-twitch, rat soleus (57 versus 28% SRX, respectively), although other studies have reported SRX content as high as 48% in human skeletal muscles (Wilson et al., 2020). The higher SRX content in cardiac tissue may represent a unique aspect of cardiac myosin. Unlike skeletal muscle, cardiac SRX myosin remain sequestered upon calcium activation (Hooijman et al., 2011), potentially acting as a “reserve pool” of myosin that can be recruited for additional contractile capability in response to physiological demand.
In both the slow-twitch, rat soleus (Nelson et al., 2020), and mouse cardiac myofibrils studied here, SRX myosin are highly abundant in the C-zone where MyBP-C resides. Where they differ is in the abundance of SRX in their P- and D-zone, which are devoid of MyBP-C. Specifically, in the cardiac myofibril P- and D-zone, a significant SRX proportion (72 and 45%, respectively) are present compared with their minimal presence (13 and 9%, respectively) in the soleus. With the potential that myosin may exist in a dynamic equilibrium between SRX and DRX states (Nag et al., 2017), these data suggest this equilibrium might be shifted more toward the SRX state in mouse cardiac myosin than in skeletal muscle, perhaps enhanced within the context of the cardiac thick filament (Gollapudi et al., 2021). Additionally, this dynamic equilibrium appears to be further shifted by the presence of cMyBP-C, which in some manner stabilizes the SRX (Fig. 2 B).
If cMyBP-C directly stabilizes the SRX state, then eliminating cMyBP-C by genetic knock-down should dramatically reduce the overall proportion of SRX, but more specifically in the C-zone where cMyBP-C exists. In the cMyBP-Ct/t null mouse, we did in fact observe a marked 40% reduction in the overall proportion of SRX myosin. However, the reduction in the SRX state population was not limited to the C-zone but rather distributed uniformly along most of the thick filament length, as a similar gradient in the SRX fraction observed in the WT myofibrils remained in the t/t myofibril C- and D-zone (i.e., similar WT and t/t slopes, Fig. 3 D). Myosin regulatory light chain (RLC) phosphorylation can shift SRX myosin into the DRX state (Stewart et al., 2010; Nag et al., 2017). However, the t/t myofibrils had reduced RLC phosphorylation (Fig. S2), which by itself, should have caused an increase in SRX content (Stewart et al., 2010). Therefore, the observed decrease in SRX content in the t/t myofibrils emphasizes the role of cMyBP-C in SRX regulation, and more importantly, that this regulation is not limited to the C-zone, given the reduction of SRX in the D-zone as well. This result suggests that cMyBP-C must also act through some long-range, indirect mechanism to stabilize the SRX state in the D-zone that is devoid of cMyBP-C. However, the lack of a change in the fraction of SRX in the P-zone of t/t myofibrils suggests that this stabilization may not be propagated throughout the entire thick filament and that other structural cues independent of cMyBP-C may dictate the SRX state in this spatially short (75 nm) zone (see discussion below).
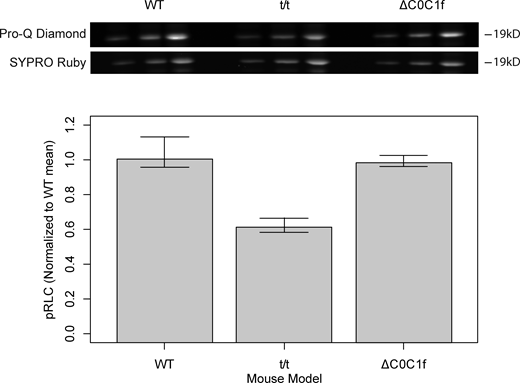
RLC phosphorylation. Top: SDS-PAGE gel was run on myofibril preparations from three mouse models (WT, t/t, and ΔC0C1f) under three loading conditions. The gel was then stained with Pro-Q Diamond (phosphoprotein stain), followed by SYPRO Ruby (total protein stain). Both stains were performed according to the manufacturer’s instructions (Molecular Probes/Invitrogen) and imaged in a Bio-Rad Chemidoc (Bio-Rad Products). Bottom: Fluorescence intensity ratios were determined using Gel Analysis Tools in ImageJ and normalized according to the mean intensity ratio for WT according to (Pro-Q Intensity:SYPRO Intensity)/(WT Pro-Q Intensity:WT SYPRO Intensity). Error bars denote SD. Values shown in the bar chart represent six technical replicates from two biological replicates for each condition. Source data are available for this figure: SourceData FS2.
RLC phosphorylation. Top: SDS-PAGE gel was run on myofibril preparations from three mouse models (WT, t/t, and ΔC0C1f) under three loading conditions. The gel was then stained with Pro-Q Diamond (phosphoprotein stain), followed by SYPRO Ruby (total protein stain). Both stains were performed according to the manufacturer’s instructions (Molecular Probes/Invitrogen) and imaged in a Bio-Rad Chemidoc (Bio-Rad Products). Bottom: Fluorescence intensity ratios were determined using Gel Analysis Tools in ImageJ and normalized according to the mean intensity ratio for WT according to (Pro-Q Intensity:SYPRO Intensity)/(WT Pro-Q Intensity:WT SYPRO Intensity). Error bars denote SD. Values shown in the bar chart represent six technical replicates from two biological replicates for each condition. Source data are available for this figure: SourceData FS2.
With the presence of cMyBP-C further stabilizing the SRX state, we asked whether this activity can be attributed to the N-terminal region, given its ability to modulate actomyosin contractile function both in in vitro motility (Razumova et al., 2006; Weith et al., 2012) and transgenic mouse studies (Lynch et al., 2021). Interestingly, myofibrils from the N-terminal truncated cMyBP-CΔC0-C1f mouse are almost indistinguishable from the cMyBP-Ct/t null, both in terms of the overall SRX proportion and SRX localization within the sarcomere (Fig. 3, D and E). The similarity between the cMyBP-CΔC0-C1f and cMyBP-Ct/t null data emphasizes the importance of cMyBP-C’s N terminus in cMyBP-C’s capacity to stabilize the SRX state along the length of the thick filament. Interestingly, in a previous study (Lynch et al., 2021), we reported no difference in SRX content in ventricular muscle from the cMyBP-CΔC0-C1f and WT mice, as measured by mantATP chase experiments in muscle bundles. The apparent discrepancy with the present results may reflect a methodological difference, whereby tension applied to the muscle bundles (Lynch et al., 2021) may trigger a shift in the equilibrium toward the myosin DRX state (discussed below).
Perhaps most surprising is that a gradient of SRX myosin along the thick filament length remains in cMyBP-Ct/t null (Fig. 3 D) so that a twofold higher proportion of SRX exists in the C-zone compared to the D-zone (Fig. 3 E). These data suggest that there must be an additional or alternative spatial or structural determinant that dictates the SRX gradient and effectively enriches SRX within the C-zone and apparently in the P-zone as well. One such structure is titin, which has super-repeats that differ in the P-, C-, and D-zone and underlies the patterning of cMyBP-C into 11 stripes (Tonino et al., 2019). Several domains of titin are exposed at the surface of the thick filament (Zoghbi et al., 2008), which may provide or modulate binding sites for myosin heads (Muhle-Goll et al., 2001), thus contributing to stabilization of the SRX state, independent of cMyBP-C. Additionally, the structure of the thick filament backbone changes along its length, particularly within the D-zone, as the thick filament tapers toward its tip (Sjostrom and Squire, 1977). In doing so, the landscape of myosin tails necessary for the folded SRX/IHM conformation may present more opportunities for heads to be stabilized into the SRX state within the P- and C-zone compared with the more distal thick filament D-zone.
What might be the functional consequence of such a SRX spatial gradient along the thick filament? Recently, a model of thick filament–dependent activation has been proposed (Linari et al., 2015), in which the SRX myosin constitutes the “off” state of the thick filament. Turning the thick filament “on” would then require that mechanical strain be sensed along the thick filament to trigger the release of myosin from the SRX state (Linari et al., 2015). Our data suggest that cardiac myosin motors are biased toward the DRX or on state within the D-zone, located at the ends of the myosin thick filament. These DRX myosin would be the most available and first to bind the thin filament and generate force upon muscle activation (Brunello et al., 2020). Once bound, these myosin heads would cause strain to be distributed along the entire thick filament length (Ma et al., 2018), facilitating the release of more centrally located SRX myosin motors, particularly those that are enriched by the presence of cMyBP-C in the C-zone. Therefore, with cMyBP-C having the capacity to shift the SRX/DRX equilibrium toward the SRX state, future therapeutic design may target the cMyBP-C as a means of altering cardiac contractility.
Acknowledgments
Henk L. Granzier served as editor.
The authors thank Mike Previs and Colleen Kelly (University of Vermont) for their contribution of biological samples, Guy Kennedy (Instrumentation and Model Facility, University of Vermont) for his technical expertises and imaging assistance, and James McNamara (Murdoch Children’s Research Institute) for insightful conversations.
This paper was funded by National Institutes of Health (NIH) grant HL150953 (to D.M. Warshaw) and was supported in part by a generous gift to D.M. Warshaw from Arnold and Mariel Goran. Dr. Sadayappan has received support from NIH grants R01 AR078001, R01 HL130356, R01 HL105826, R38 HL155775, and R01 HL143490; the American Heart Association 2019 Institutional Undergraduate Student (19UFEL34380251) and Transformation (19TPA34830084) awards; the PLN Foundation (PLN crazy idea); and the Leducq Foundation (Transatlantic Network 18CVD01, PLN-CURE).
Dr. Sadayappan provides consulting and collaborative research studies to the Leducq Foundation (CURE-PLAN), Red Saree Inc., Greater Cincinnati Tamil Sangam, Novo Nordisk, Pfizer, AavantioBio, AstraZeneca, MyoKardia, Merck, and Amgen, but such work is unrelated to the content of this article. The remaining authors declare no competing financial interest.
Author contributions: S. Nelson: Conceptualization, Methodology, Software, Formal analysis, Investigation, Resources, Data Curation, Writing—original draft, Writing – review and editing, and Visualization; S.B. Previs: Investigation, Resources, and Writing—review and editing; S. Sadayappan: Resources, Funding acquisition, and Writing—review and editing; C. Tong: Methodology, Funding acquisition, and Writing— review and editing; and D.M. Warshaw: Conceptualization, Writing—review and editing, Supervision, Project administration, and Funding acquisition.
References
This work is part of a special issue on Myofilament Function 2022.

