


Toll-like receptors 7 (TLR7) and 8 (TLR8) each sense single-stranded RNA (ssRNA), but their activation results in different immune activation profiles. Attempts to selectively target either TLR7 or TLR8 have been hindered by their high degree of homology. However, recent studies revealed that TLR7 and TLR8 bind different ligands resulting from the processing of ssRNA by endolysosomal RNases. We demonstrate that by introducing precise 2′ sugar-modified bases into oligoribonucleotides (ORNs) containing known TLR7 and TLR8 binding motifs, we could prevent RNase-mediated degradation into the monomeric uridine required for TLR8 activation while preserving TLR7 activation. Furthermore, a novel, optimized protocol for CRISPR-Cas9 knockout in primary human plasmacytoid dendritic cells showed that TLR7 activation is dependent on RNase processing of ORNs and revealed a previously undescribed role for RNase 6 in degrading ORNs into TLR ligands. Finally, 2′ sugar-modified ORNs demonstrated robust innate immune activation in mice. Altogether, we identified a strategy for creating tunable TLR7-selective agonists.
Introduction
Innate immune activation is crucial for the generation of potent antigen-specific adaptive immunity. Receptors for innate agonists such as Toll-like receptors (TLRs) play critical roles in the process by eliciting the release of a broad range of cytokines that drive or sustain inflammatory responses (Akira et al., 2006; Medzhitov, 2007). TLR7 and TLR8 are two highly homologous TLR family members that reside in endosomes and detect viral single-stranded RNA (ssRNA) to initiate antiviral immune responses (Diebold et al., 2004; Heil et al., 2004; Lund et al., 2004). TLR7/8 agonists used as vaccine adjuvants have been shown to induce superior CD8 T cell responses in preclinical models (Kranz et al., 2016; Lynn et al., 2020; Wille-Reece et al., 2006). Due to their robust immunostimulatory potential, TLR7/8 agonists are currently evaluated as cancer immunotherapy and vaccine adjuvants in the clinic (Bhagchandani et al., 2021). Despite their similarities, TLR7 and TLR8 induce distinct cytokine profiles due in part to their distinct cell type expression patterns (Heil et al., 2004; Hornung et al., 2002). In humans, TLR7 is primarily expressed in plasmacytoid dendritic cells (pDCs). Its activation results predominantly in signaling through transcription factor IRF7, leading to the potent induction of IFN⍺ (Kawai et al., 2004). In contrast, TLR8 is more broadly expressed in human myeloid cells including monocytes, macrophages, and conventional DCs (cDCs), and its activation preferentially results in NF-κB translocation that drives the release of proinflammatory cytokines such as TNF⍺ and IL-6, which are associated with systemic adverse events and tolerability issues (Aluri et al., 2021; Bender et al., 2020; Guiducci et al., 2013). Therefore, modulating the selectivity of an agonist to preferentially stimulate TLR7 or TLR8 would enable fine-tuning of proinflammatory cytokine and chemokine profiles and potentially improve their therapeutic benefit.
While both TLR7 and TLR8 detect ssRNA, recent work has identified differences in their binding specificities, exposing potential opportunities for designing ssRNA agonists with differential activation of TLR7 or TLR8. Importantly, studies showed that both TLR7 and TLR8 bind ssRNA degradation products. Crystal structures of TLR7 and TLR8 revealed that each receptor contains two binding sites for ssRNA degradation products, termed site 1 and site 2 (Shibata et al., 2016; Tanji et al., 2015; Zhang et al., 2016). For TLR7, site 1 binds guanosine (G) and site 2 binds a trinucleotide consisting of uridine (U) flanked by two nucleotides (NUN), with a preference for pyrimidines in the N positions. In contrast, TLR8 site 1 binds uridine and site 2 binds UN dinucleotides with a preference for UG, UA, or UU. This illustrates the challenge in generating TLR7-selective RNA ligands; site 2 of TLR7 requires an ssRNA containing uridine, but TLR8 will be activated by the degradation of any uridine-containing ssRNA into monomeric uridine and UN dimers (Forsbach et al., 2008). Recent reports have identified two endosomal RNases, RNase 2 and RNase T2, as key players in this degradation step for TLR8 (Greulich et al., 2019; Ostendorf et al., 2020). Furthermore, RNase T2 expression was demonstrated to be required for the activation of TLR7 by RNA ligands in murine macrophages and pDCs (Liu et al., 2021). Nucleotide modifications can prevent RNase degradation (Eckstein, 2014; Kawasaki et al., 1993; Ostendorf et al., 2020) as well as modify TLR7 and 8 activation (Freund et al., 2019). Therefore, we hypothesized that nucleotide modifications could be used to modulate the activity of endosomal RNases in differentially processing TLR7 and TLR8 ligands from ssRNAs.
In this study, we used rational design to create oligoribonucleotides (ORNs) that have diminished TLR8 recognition with preserved TLR7-stimulating activity. This was achieved by strategically placing 2′ modifications on select nucleotides of sequences containing TLR7- and TLR8-binding motifs, which slowed and potentially abrogated the ability of endosomal RNases to process the ORNs into monomeric uridine capable of binding to and activating TLR8. In addition, by first establishing and then implementing a protocol for generating knockouts (KOs) in primary human pDCs, we found that RNase T2 and RNase 6 were necessary for the activity of the TLR7-selective ORNs. Finally, we demonstrated that these ORNs stimulated IFN⍺ release and DC maturation in vivo. Together, our findings indicate that site-specific modifications can modulate the cleavage kinetics of ssRNAs into ligands that limit TLR8 activation and preferentially trigger TLR7, thereby enabling a strategy for the generation of TLR7-, or potentially TLR8-, selective agonists for potential use as single agents or in combination with mRNA-based and other therapeutics.
Results
2′ chemical modification strategies for modulating cleavage of ORNs by RNases
RNases act by catalyzing a nucleophilic attack of the 2′-OH at the 3′ phosphate within a phosphodiester (PD) bond of an RNA, leading to release of a 3′ nucleotide with a 5′-OH and the formation of a 2′,3′ cyclophosphate at the 5′ nucleotide (Fig. 1 A). Chemical modifications can stabilize the RNA backbone by slowing or preventing this reaction (Fig. 1, B and C). Indeed, substitutions of the 2′OH with a 2′O-methyl (2′OMe) effectively blocks RNA cleavage by RNase T2 and RNase 2, while 2′fluoro (2′F) modifications are generally described as providing resistance to RNase degradation (Kawasaki et al., 1993; Ostendorf et al., 2020). Additionally, substitution of one oxygen with a sulfur at the 3′ phosphate creates a phosphorothioate (PS) linkage, which stabilizes RNA relative to PD backbones (Eckstein, 2014).

2′ RNA modifications for modulating cleavage of ORNs by RNases. (A) RNase cleavage of unmodified RNA leads to release of a 3′ nucleotide with a 5′-OH and a 5′ nucleotide with a 2′,3′-cyclophosphate intermediate. (B) RNA modifications limit RNase-mediated cleavage 3′ to the modified nucleotide. (C) RNA modifications tested herein. (D) Approach for the incorporation of site-specific modifications in ORNs to direct generation of known TLR7 binding site two ligands without further cleavage into TLR8 ligands. Bold red font indicates modified nucleotides.
2′ RNA modifications for modulating cleavage of ORNs by RNases. (A) RNase cleavage of unmodified RNA leads to release of a 3′ nucleotide with a 5′-OH and a 5′ nucleotide with a 2′,3′-cyclophosphate intermediate. (B) RNA modifications limit RNase-mediated cleavage 3′ to the modified nucleotide. (C) RNA modifications tested herein. (D) Approach for the incorporation of site-specific modifications in ORNs to direct generation of known TLR7 binding site two ligands without further cleavage into TLR8 ligands. Bold red font indicates modified nucleotides.
To design ORNs that reduce TLR8 activity while maintaining TLR7 agonism, we engineered “foundation sequences” containing both TLR7 and TLR8 binding motifs with site-specific modifications to slow or stop RNase-mediated degradation into TLR8 binding site motifs (Fig. 1, B–D). As described below, we leveraged three sequences from published studies (Forsbach et al., 2008; Zhang et al., 2018). These foundation sequences carry one or more N1U2N3 motifs for TLR7 binding site 2, where N represents a cytidine or uridine. To prevent degradation into the TLR8 ligand monomeric uridine, we modified N1 with either a 2′OMe or 2′F and modified N3 only if followed by another uridine. We avoided modifying U2 under the hypothesis that its modification may inhibit TLR7 binding as the 2′-OH of U2 is deeply embedded in TLR7 binding site 2 (Zhang et al., 2016). We also designed ORNs with PS backbone substitutions for further stabilization. This resulted in the creation of 20 total ORNs divided among three discrete sequence-specific suites, which are listed in detail in Table 1.
List of ORNs
| Suite | ORN ID | Backbone | Modification | Sequence |
|---|---|---|---|---|
| pUC | pUC | PD | -- | U-C-U-C-U-C-U-C-U-C-U-C-U-C-U-C-U-C-U-C |
| pUC-O | PD | OMe | U-mC-U-mC-U-mC-U-mC-U-mC-U-mC-U-mC-U-mC-U-mC-U-C | |
| pUC-F | PD | F | U-fC-U-fC-U-fC-U-fC-U-fC-U-fC-U-fC-U-fC-U-fC-U-C | |
| pUC-U2′O | PD | OMe | mU-C-mU-C-mU-C-mU-C-mU-C-mU-C-mU-C-mU-C-mU-C-mU-C-U | |
| pUC-U2′F | PD | F | fU-C-fU-C-fU-C-fU-C-fU-C-fU-C-fU-C-fU-C-fU-C-fU-C | |
| pUC-PS | PS | -- | U*C*U*C*U*C*U*C*U*C*U*C*U*C*U*C*U*C*U*C | |
| pU2C | pUUC | PD | -- | C-C-U-U-C-U-U-C-U-U-C-U-U-C-U-U-C-U-U-C |
| pUUC-F | PD | F | C-C-fU-U-C-fU-U-C-fU-U-C-fU-U-C-fU-U-C-fU-U-C | |
| pUUC-O | PD | OMe | C-C-mU-U-C-mU-U-C-mU-U-C-mU-U-C-mU-U-C-mU-U-C | |
| pUUC-PS | PS | -- | C*C*U*U*C*U*U*C*U*U*C*U*U*C*U*U*C*U*U*C | |
| U4.G | U4.G-PS | PS | -- | C*C*G*A*G*C*C*G*C*U*U*U*U*C*C*C |
| U4.G-PS-OO | PS | OMe-OMe | C*C*G*A*G*C*C*G*C*mU-U*mU-U*C*C*C | |
| U4.G-PS-FF | PS | F-F | C*C*G*A*G*C*C*G*C*fU-U*fU-U*C*C*C | |
| U4.G-PS-OF | PS | OMe-F | C*C*G*A*G*C*C*G*C*mU-U*fU-U*C*C*C | |
| U4.G-PS-FO | PS | F-OMe | C*C*G*A*G*C*C*G*C*fU-U*mU-U*C*C*C | |
| U4.G-PD | PD | -- | C-C-G-A-G-C-C-G-C-U-U-U-U-C-C-C | |
| U4.G-PD-OO | PD | OMe-OMe | C-C-G-A-G-C-C-G-C-mU-U-mU-U-C-C-C | |
| U4.G-PD-FF | PD | F-F | C-C-G-A-G-C-C-G-C-fU-U-fU-U-C-C-C | |
| U4.G-PD-OF | PD | OMe-F | C-C-G-A-G-C-C-G-C-mU-U-fU-U-C-C-C | |
| U4.G-PD-FO | PD | F-OMe | C-C-G-A-G-C-C-G-C-fU-U-mU-U-C-C-C | |
| Control | pUUG | PD | -- | G-G-U-U-G-U-U-G-U-U-G-U-U-G-U-U-G-U-U-G |
ORNs are listed by foundation sequence. Foundation sequence pUC (5′-UCUCUCUCUCUCUCUCUCUC-3′) and foundation sequence pUUC (5′-CCUUCUUCUUCUUCUUCUUC-3′) were both previously shown to have TLR7 activity when codelivered with guanosine (Zhang et al., 2018). For pUC, we engineered 2′ modifications on the cytidines to maintain an unmodified U2 in any resulting CUC products, or on the uridines to assess site-specificity. For pUUC, we included 2′ modifications on U1 in the UUC motif to inhibit the generation of monomeric uridine. In addition, we engineered both sequences with a PS backbone. For foundation sequence U4.G (5′-CCGAGCCGCUUUUCCC-3′; Forsbach et al., 2008), we introduced 2′ modifications on U1 and U3. We also created 2′ modified versions with a PS backbone throughout. Within the sequences, “-” indicates the standard PD backbone, and “*” indicates the PS backbone. Bold font indicates the modified nucleotides, where “f_” is a 2′fluoro (“F”) and “m_” is a 2′O-methyl (“OMe”) modification.
2′-RNA modifications protect ORNs from RNase-mediated degradation and result in reduced TLR8 activation
We first sought to identify the RNases responsible for the generation of TLR8 ligands by determining the expression of endosomal RNases in TLR8-expressing myeloid cells. Using publicly available single-cell (sc)RNA-seq and conventional microarray datasets, we confirmed that RNase T2 (RNASET2) and RNase 2 (RNASE2) were expressed in primary human monocytes, as previously reported (Ostendorf et al., 2020; Fig. 2 A). In addition, RNase 6 (RNASE6) was strongly expressed by primary human and murine monocytes, indicating it may also play a role in RNA processing for TLR8. Interestingly, the commonly used human monocytic cell line, THP-1, showed notable differences in RNase gene expression relative to primary monocytes. Specifically, THP-1 cells expressed higher RNASE2 and lower RNASE6 transcript levels, underscoring the need to study the biology of these RNases in primary cells.
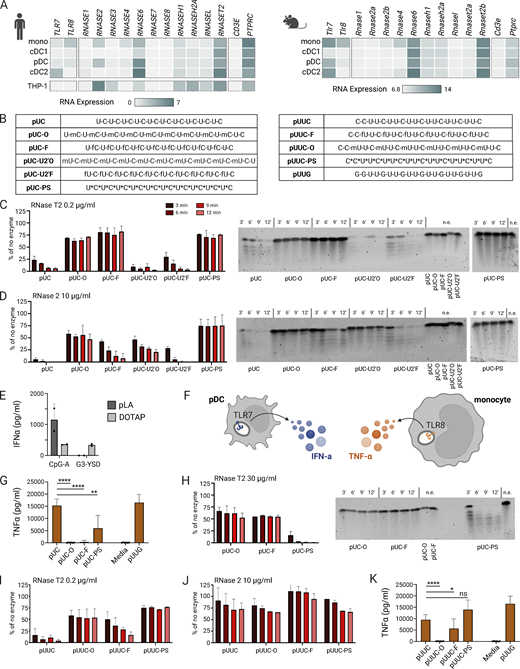
2′ RNA modifications protect from RNase T2- and RNase 2-mediated cleavage and limit TLR8 activity. (A) Upper left: RNA expression of endosomal RNases in human dendritic cells and monocytes from single-cell RNA-seq data (NCBI GEO accession no. GSE94820; Villani et al., 2017). Lower left: RNA expression in the THP-1 human monocytic cell line from bulk RNA-seq data. Right: RNA expression of endosomal RNases in mouse dendritic cells and monocytes from microarray data (NCBI GEO accession no. GSE35458; Haniffa et al., 2012). Colors represent average expression level. (B) Table of ORNs referenced in this figure. (C and D) Quantification of in vitro cleavage assay gels shown as a percentage relative to the no enzyme (n.e.) negative control (n = 2 in two experiments for all). ORNs were incubated with recombinant RNase T2 or RNase 2 at the indicated concentrations for time points shown, then run on a 15% TBE-Urea gel. For each reaction with enzyme, 25% of each reaction was run while 12.5% of the reaction was run for the n.e. controls. Representative in vitro cleavage assay gels are shown (right). (E) IFN⍺ in cell culture supernatants 16 h after pLA or DOTAP delivery of known innate immune receptor agonists to hPBMCs. The TLR9 agonist CpG-A (ODN2216) served as a readout for endosomal delivery, and the cGAS ligand G3-YSD (Herzner et al., 2016) served as a readout for cytosolic delivery. Both agonists were used at a final concentration of 1 µg/ml. (F) Expected cytokine outcome after TLR7 or TLR8 activation. (G) TNF⍺ measured by ELISA in cell culture supernatants of hPBMCs ∼16 h after treatment with 0.6 µg/ml of pUC ORNs delivered with pLA (n = 6 donors in two experiments). (H–J) Quantification of in vitro cleavage assay gels as in C and D. (K) TNF⍺ as in G after treatment with pUUC ORNs delivered with pLA (six donors in two experiments). All bar graphs show mean ± SD. Statistics were determined by one-way ANOVA with Benjamini, Krieger, and Yekutieli correction for multiple comparisons; *P ≤ 0.05, **P ≤ 0.01, ****P ≤ 0.0001. Source data are available for this figure: SourceData F2.
2′ RNA modifications protect from RNase T2- and RNase 2-mediated cleavage and limit TLR8 activity. (A) Upper left: RNA expression of endosomal RNases in human dendritic cells and monocytes from single-cell RNA-seq data (NCBI GEO accession no. GSE94820; Villani et al., 2017). Lower left: RNA expression in the THP-1 human monocytic cell line from bulk RNA-seq data. Right: RNA expression of endosomal RNases in mouse dendritic cells and monocytes from microarray data (NCBI GEO accession no. GSE35458; Haniffa et al., 2012). Colors represent average expression level. (B) Table of ORNs referenced in this figure. (C and D) Quantification of in vitro cleavage assay gels shown as a percentage relative to the no enzyme (n.e.) negative control (n = 2 in two experiments for all). ORNs were incubated with recombinant RNase T2 or RNase 2 at the indicated concentrations for time points shown, then run on a 15% TBE-Urea gel. For each reaction with enzyme, 25% of each reaction was run while 12.5% of the reaction was run for the n.e. controls. Representative in vitro cleavage assay gels are shown (right). (E) IFN⍺ in cell culture supernatants 16 h after pLA or DOTAP delivery of known innate immune receptor agonists to hPBMCs. The TLR9 agonist CpG-A (ODN2216) served as a readout for endosomal delivery, and the cGAS ligand G3-YSD (Herzner et al., 2016) served as a readout for cytosolic delivery. Both agonists were used at a final concentration of 1 µg/ml. (F) Expected cytokine outcome after TLR7 or TLR8 activation. (G) TNF⍺ measured by ELISA in cell culture supernatants of hPBMCs ∼16 h after treatment with 0.6 µg/ml of pUC ORNs delivered with pLA (n = 6 donors in two experiments). (H–J) Quantification of in vitro cleavage assay gels as in C and D. (K) TNF⍺ as in G after treatment with pUUC ORNs delivered with pLA (six donors in two experiments). All bar graphs show mean ± SD. Statistics were determined by one-way ANOVA with Benjamini, Krieger, and Yekutieli correction for multiple comparisons; *P ≤ 0.05, **P ≤ 0.01, ****P ≤ 0.0001. Source data are available for this figure: SourceData F2.
To determine the cleavage kinetics of each ORN variant (Fig. 2 B), we performed in vitro assays with recombinant RNase T2 or RNase 2. We first assessed RNase-mediated processing with the simplest foundation sequence, pUC. pUC ORNs with 2′-modified cytidines (pUC-O and pUC-F) showed robust protection against RNase T2 cleavage compared with the unmodified pUC (Fig. 2 C). This observation is consistent with previous work demonstrating that RNase T2 preferentially cleaves before uridine (Greulich et al., 2019; Ostendorf et al., 2020). We also evaluated pUC ORNs with 2′ modified uridines (pUC-U2′O and pUC-U2′F). These ORNs were not protected from RNase T2 cleavage, which demonstrates that 2′ modifications suppress cleavage in a site-specific manner. In contrast, pUC ORNs with 2′ modified cytidines or uridines both showed partial protection from RNase 2 cleavage, in line with literature showing a capacity for RNase 2 to cleave after either pyrimidine (Ostendorf et al., 2020; Fig. 2 D). Interestingly, pUC ORNs with 2′F modifications appeared less protected from RNase 2 cleavage than those with 2′OMe modifications, suggesting differential tolerance of RNase 2 to each modification.
To assess whether the reduction in RNase cleavage of these novel, chemically modified ORNs impacted TLR8 activation relative to their foundational counterparts, we transfected each ORN into human peripheral blood mononuclear cells (hPBMCs) isolated from healthy donors using poly-L-arginine (pLA). pLA is a polymeric carrier that exclusively delivers nucleic acid cargoes to endosomes for endosomal TLR activation. Notably, pLA does not release the ligands to cytosolic receptors (Coch et al., 2013). This is in contrast to the commonly used transfection reagent 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), which can deliver cargoes such as the cGAS agonist G3-YSD into the cytosol (Fig. 2 E). As a readout of TLR8 activation, we measured TNF⍺ production in response to the ORNs, which in hPBMCs is primarily driven by TLR8-expressing monocytes (Gorden et al., 2005; Hornung et al., 2002; Fig. 2 F).
We then measured the TNF⍺ response from hPBMCs transfected with each of the pUC ORNs. A dose-response was produced using pUUG to determine the optimal concentration of ORNs to deliver to cells (Fig. S1, A and B). A concentration of 0.6 µg/ml achieved robust levels of IFN⍺ and TNF⍺ while higher doses resulted in loss of signal due to toxicity. Delivery of unmodified pUC resulted in TNF⍺ induction comparable with the well-characterized TLR7- and TLR8-activating RNA pUUG, demonstrating that this ORN activates TLR8 (Fig. 2 G). Interestingly, both pUC-O and pUC-F dramatically reduced TNF⍺ induction despite the apparent susceptibility of pUC-F to RNase 2-mediated partial degradation (Fig. 2 D). This result suggests that RNase 2 plays a less significant role in the degradation of these ORNs in primary monocytes, further evidenced by (1) low RNase 2 expression in primary monocytes and (2) lower efficiency of RNase 2 compared with RNase T2 in processing pUC. Additionally, the expected product from RNase 2 processing pUC would be C-U, which is a suboptimal TLR8 ligand.

Dose exploration of ORNs delivered to hPBMCs with pLA. Related to Fig. 2. (A and B) IFN⍺ (A) and TNF⍺ (B) in cell culture supernatants measured by ELISA. hPBMCs were stimulated overnight (16 h) with pUUG at various doses, and delivered with pLA (n = 3 in one experiment). All bar graphs show mean ± SD.
Dose exploration of ORNs delivered to hPBMCs with pLA. Related to Fig. 2. (A and B) IFN⍺ (A) and TNF⍺ (B) in cell culture supernatants measured by ELISA. hPBMCs were stimulated overnight (16 h) with pUUG at various doses, and delivered with pLA (n = 3 in one experiment). All bar graphs show mean ± SD.
Surprisingly, while pUC-PS demonstrated strong protection from both RNase T2 and RNase 2 cleavage (Fig. 2, C and D), it retained TLR8 activation capacity (Fig. 2 G). We hypothesized that PS backbones may be less effective than 2′ modifications at abrogating cleavage. To test this, we assayed pUC-O, pUC-F, and pUC-PS using a higher RNase T2 concentration (Fig. 2 H). Indeed, the 2′ modified ORNs remained resistant to cleavage while pUC-PS was efficiently degraded.
Next, we confirmed these results with another sequence, pUUC. We found that 2′OMe and 2′F modifications of U1 in the UUC motifs also increased resistance to RNase T2, although to a lesser extent than with 2′F modification (Fig. 2 I). As with pUC, the protection was less clear with RNase 2 and the PS backbone showed limited impact on TLR8 activation (Fig. 2, J and K). This pattern supports our observation that the PS backbone offers protection from RNase cleavage at low concentrations. However, the cytokine assay suggests that the PS modification is not as protective as 2′ modifications in a cellular context where the kinetics and RNase concentrations likely vary. Taken together, these data demonstrate that ORNs engineered with site-specific 2′ modifications abrogate the ability of RNase T2 (and to a lesser, but significant, extent RNase 2) to produce the RNA degradation products required for TLR8 stimulation.
2′ modifications inhibit RNase 6-mediated degradation of ORNs
The robust expression of RNase 6 in human monocytes supported a previously uncharacterized role for RNase 6 in degrading ssRNAs into TLR8 ligands. Therefore, we asked whether 2′ modifications would confer resistance to degradation by RNase 6 in a recombinant cleavage assay. Again, starting with the simplest sequence, we found that pUC-O and pUC-F inhibited or delayed cleavage by RNase 6 compared with unmodified pUC (Fig. 3, A–C). pUC ORNs with 2′ modifications on the uridines rather than cytidines (pUC-U2′F, pUC-U2′O) exhibited greater resistance to degradation by RNase 6, which was consistent with recent reports that RNase 6 preferentially cleaves after uridine (Prats-Ejarque et al., 2016). In contrast to the results observed for RNase T2 and RNase 2, 2′ modified pUUC ORNs were degraded by RNase 6 in vitro. The degradation kinetics appeared delayed compared with unmodified pUUC, especially for pUUC-O, but not inhibited (Fig. 3, D and E). These results may be explained by differences in cut-site preference between the enzymes and the prevalence of accessible RNase 6 cut-sites even in the modified pUUC ORNs. Taken together, these results support a model in which our engineered site-specific modifications slow the generation of TLR8 ligands by all three endosomal RNases.
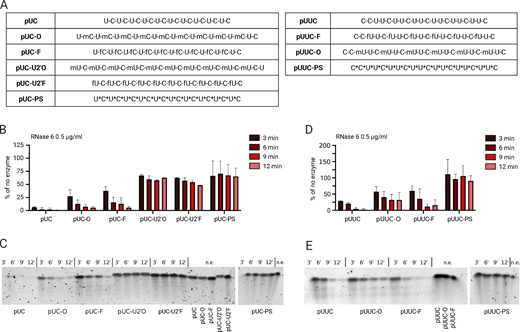
2′ RNA modifications protect ORNs from RNase 6 cleavage. (A) Table of ORNs referenced in this figure. (B and D) Quantification of in vitro cleavage assay gels shown as a percentage relative to the no enzyme (n.e.) negative control (n = 2 in two experiments for all). pUC (B) and pUUC (D) ORNs were incubated with recombinant RNase 6 at the indicated concentration for the indicated time points, then run on a 15% TBE-Urea gel. (C and E) In vitro cleavage assay gels for pUC (C) and pUUC (E) ORNs are shown. All bar graphs show mean ± SD. Source data are available for this figure: SourceData F3.
2′ RNA modifications protect ORNs from RNase 6 cleavage. (A) Table of ORNs referenced in this figure. (B and D) Quantification of in vitro cleavage assay gels shown as a percentage relative to the no enzyme (n.e.) negative control (n = 2 in two experiments for all). pUC (B) and pUUC (D) ORNs were incubated with recombinant RNase 6 at the indicated concentration for the indicated time points, then run on a 15% TBE-Urea gel. (C and E) In vitro cleavage assay gels for pUC (C) and pUUC (E) ORNs are shown. All bar graphs show mean ± SD. Source data are available for this figure: SourceData F3.
ORN 2′ modifications simultaneously reduce TLR8 activity while allowing for the generation of TLR7 ligands
While the 2′ modifications slow or abrogate the production of TLR8-specific ligands by RNases, the sequences were engineered to still permit the generation of TLR7 binding site 2 ligands (Fig. 1 D). To determine the TLR7 activity of each ORN in hPBMCs, we harnessed the known biology that IFN⍺, in response to ssRNA agonists, is primarily driven by TLR7-expressing pDCs (Fig. 2 F; and Fig. 4, A and B). Although pUC and pUUC can produce the ligands for binding site 2, their degradation cannot generate the guanosine needed for TLR7 binding site 1 as their sequences are composed entirely of Us and Cs (Fig. 4 C). Therefore, to prime TLR7 site 1, it was necessary to codeliver each ORN with exogenous 2′,3′-cGMP, representing the high-affinity RNase-degradation product of guanosine-containing RNA (Zhang et al., 2018). When delivered with exogenous 2′,3′-cGMP using pLA, the 2′ modified pUC and pUUC ORNs induced varying levels of IFN⍺, indicating TLR7 activity (Fig. 4 D). We next delivered each ORN to hPBMCs depleted of pDCs, the predominant TLR7-expressing cell, and observed the IFN⍺ levels return to baseline, further confirming the dependence of IFN⍺ induction on TLR7-expressing pDCs. In contrast, pDC depletion had minimal impact on the levels of TNF⍺, confirming that it is most likely produced by distinct TLR8-expressing myeloid cells in PBMCs (Fig. 4 E). Taken together, these data demonstrate that site-specific 2′ modifications can allow for TLR selectivity by slowing or inhibiting degradation of ORNs into TLR8 ligands while retaining the ORNs’ ability to activate TLR7.

2′ RNA modifications reduce TLR8 activity while allowing for generation of TLR7 ligands. (A) Gating strategy to identify pDCs in hPBMCs. (B) Confirmation of pDC depletion from hPBMCs (n = 12 donors in four experiments). (C) Table of ORNs referenced in this figure. (D) IFN⍺ in supernatants 16 h after pLA delivery of ORNs to hPBMCs with or without pDC depletion (n = 3 donors in one experiment). 0.6 µg/ml ORNs were delivered with 5 µg/ml 2′3′-cGMP. pUUG is used as a positive control. (E) TNF⍺ in supernatants 16 h after pLA delivery of 0.6 µg/ml ORNs to hPBMCs with or without pDC depletion (n = 6 donors in two experiments).
2′ RNA modifications reduce TLR8 activity while allowing for generation of TLR7 ligands. (A) Gating strategy to identify pDCs in hPBMCs. (B) Confirmation of pDC depletion from hPBMCs (n = 12 donors in four experiments). (C) Table of ORNs referenced in this figure. (D) IFN⍺ in supernatants 16 h after pLA delivery of ORNs to hPBMCs with or without pDC depletion (n = 3 donors in one experiment). 0.6 µg/ml ORNs were delivered with 5 µg/ml 2′3′-cGMP. pUUG is used as a positive control. (E) TNF⍺ in supernatants 16 h after pLA delivery of 0.6 µg/ml ORNs to hPBMCs with or without pDC depletion (n = 6 donors in two experiments).
CRISPR-Cas9 KO in human pDCs demonstrates a requirement for RNase T2 and RNase 6 for TLR7 activation
We next sought to investigate whether the ORNs activated TLR7 in human pDCs in an RNase-dependent manner. Human and mouse pDCs highly expressed RNase T2 (RNASET2, Rnaset2b) and RNase 6 (RNASE6, Rnase6) but not RNase 2 (RNASE2, Rnase2a/Rnase2b; Fig. 2 A), suggesting that RNase 6 and RNase T2 play a bigger role in TLR7 activation in pDCs. To determine the relative contributions of RNase T2 and RNase 6 for ORN-directed activation of TLR7 in human cells, we developed a novel protocol to disrupt genes in primary human pDCs in vitro using CRISPR-Cas9 gene editing. We screened 80 conditions to deliver Cas9 ribonucleoprotein (RNP) complexes into hPBMCs by nucleofection (Fig. S2 A) and assessed KO efficiency by quantifying the level of β2M depletion 3 days after nucleofection with validated β2M-specific Cas9-RNPs (Freund et al., 2020; Fig. S2, B and C). The top nine conditions were selected based on the maintenance of cell viability and β2M depletion, and each was subsequently validated using enriched pDCs to assess KO efficiency, viability, and responsiveness to TLR7 stimulation (Fig. S2, D–F). Using the highest performing nucleofection condition (buffer P3, program CA-137), we generated pDCs depleted of TLR7, RNASET2, RNASE6, and RNASET2/RNASE6, each with comparable recovery (Fig. 5, A and B). The CRISPR-modified pDCs were treated overnight with TLR9 agonist CpG-A as a control or pUUC-F, the most potent, and TLR7-selective ORN when delivered with pLA in the presence of exogenous 2′,3′-cGMP. No perturbation impacted CpG-A/TLR9-dependent IFN⍺ secretion, indicating the cells retained the ability to be stimulated (Fig. 5 C). In contrast, TLR7-depleted pDCs produced minimal IFN⍺ when treated with pUUC-F, which phenocopies the data from the pDC depletion assay (Fig. 4 D) and confirms the TLR7 dependency of IFN⍺ induction by pUUC-F. RNASET2-depleted pDCs treated with pUUC-F exhibited reduced but detectable IFN⍺ compared with pDCs that received non-targeting control (NTC) single guide RNA (sgRNA). In contrast, pUUC-F-treated RNASE6-depleted pDCs produced higher IFN⍺ levels compared with NTC pDCs. We hypothesize that this difference was due to RNase T2 cleavage of pUUC-F to a series of UUC trinucleotides which were not degraded further in the absence of RNase 6 (Fig. 5 D). These UUC degradation products have been shown to be the preferred ligands of TLR7 site 2 in structural studies (Zhang et al., 2018). Strikingly, the residual IFN⍺ signal in the RNASET2-depleted pDCs was almost completely ablated in the RNASET2/RNASE6-depleted pDCs, indicating that the processing of pUUC-F is dependent on both RNase T2 and RNase 6 for optimal ligand production and TLR7 activation in human pDCs. Since the guanosine for TLR7 binding site 1 was exogenously delivered, our results indicate that TLR7 binding site 2 also senses degradation products.

Optimization of a CRISPR-Cas9 KO protocol in primary human pDCs. Related to Fig. 5. (A) Workflow to screen for optimal nucleofection conditions with the Lonza Primary Cell Optimization Kit. hPBMCs were cultured for 3 days in media containing 10 ng/ml IL-3. (B) Gating strategy to assess the viability and KO efficiency of pDCs among hPBMCs after 3 days of culture. (C) Recovery of pDCs relative to media control (left) and β2 M KO efficiency in pDCs (right) measured on day 3 (n = 1). In the β2 M KO efficiency plot, the frequency of KO is not shown for conditions with <10 gated pDCs (dark gray fill). (D) Recovery (left) and β2M KO efficiency (right) in a validation screen of the top nucleofection conditions performed as in C (n = 3 in two experiments). (E) Normalized IFN⍺ levels in enriched pDC cultures. Cells were nucleofected with non-targeting control sgRNA (N) or TLR7 sgRNA (T), cultured for 3 days, then stimulated overnight with the TLR7 agonist Imiquimod (n = 2 in one experiment). (F) Purity of pDCs after immunomagnetic negative selection from elutriated leukopaks (one representative donor of four).
Optimization of a CRISPR-Cas9 KO protocol in primary human pDCs. Related to Fig. 5. (A) Workflow to screen for optimal nucleofection conditions with the Lonza Primary Cell Optimization Kit. hPBMCs were cultured for 3 days in media containing 10 ng/ml IL-3. (B) Gating strategy to assess the viability and KO efficiency of pDCs among hPBMCs after 3 days of culture. (C) Recovery of pDCs relative to media control (left) and β2 M KO efficiency in pDCs (right) measured on day 3 (n = 1). In the β2 M KO efficiency plot, the frequency of KO is not shown for conditions with <10 gated pDCs (dark gray fill). (D) Recovery (left) and β2M KO efficiency (right) in a validation screen of the top nucleofection conditions performed as in C (n = 3 in two experiments). (E) Normalized IFN⍺ levels in enriched pDC cultures. Cells were nucleofected with non-targeting control sgRNA (N) or TLR7 sgRNA (T), cultured for 3 days, then stimulated overnight with the TLR7 agonist Imiquimod (n = 2 in one experiment). (F) Purity of pDCs after immunomagnetic negative selection from elutriated leukopaks (one representative donor of four).

CRISPR-Cas9 KO in human pDCs demonstrates requirement for RNase T2 and RNase 6 for TLR7 activation. (A) Workflow for generating KOs in primary human pDCs in vitro using CRISPR-Cas9 gene editing (see also Fig. S2). (B) Recovery of pDCs 3 days after nucleofection. The percentage of live cells recovered on day 3 is shown relative to the number of cells nucleofected on day 0 (n = 4 in four experiments, indicated by symbols). (C) IFN⍺ measured by ELISA in cell culture supernatants after overnight incubation of pDC KOs with CpG-A as a control or pUUC-F + 2′3′ cGMP using pLA (n = 3–4 in four experiments). All bar graphs show mean ± SD. Statistics determined by two-way ANOVA with multiple comparisons test (two-way linear step-up procedure of Benjamini, Krieger, and Yekutieli), *P < 0.05, **P < 0.01, ns = not significant. (D) Expected cleavage products in CRISPR-Cas9 KO pDCs treated with pUUC-F. Red triangles listed above and below the ORN sequence indicate predicted cleavage sites (filled red triangle on top for RNase T2, empty red triangle below for RNase 6). Arrows indicate relative amounts of the predicted degradation product in each KO compared to the non-targeting control. Red down arrows indicate reduced levels, while green up arrows predict increased production of the cleavage product. Preferred TLR7 trinucleotide “UUC” is indicated in blue font.
CRISPR-Cas9 KO in human pDCs demonstrates requirement for RNase T2 and RNase 6 for TLR7 activation. (A) Workflow for generating KOs in primary human pDCs in vitro using CRISPR-Cas9 gene editing (see also Fig. S2). (B) Recovery of pDCs 3 days after nucleofection. The percentage of live cells recovered on day 3 is shown relative to the number of cells nucleofected on day 0 (n = 4 in four experiments, indicated by symbols). (C) IFN⍺ measured by ELISA in cell culture supernatants after overnight incubation of pDC KOs with CpG-A as a control or pUUC-F + 2′3′ cGMP using pLA (n = 3–4 in four experiments). All bar graphs show mean ± SD. Statistics determined by two-way ANOVA with multiple comparisons test (two-way linear step-up procedure of Benjamini, Krieger, and Yekutieli), *P < 0.05, **P < 0.01, ns = not significant. (D) Expected cleavage products in CRISPR-Cas9 KO pDCs treated with pUUC-F. Red triangles listed above and below the ORN sequence indicate predicted cleavage sites (filled red triangle on top for RNase T2, empty red triangle below for RNase 6). Arrows indicate relative amounts of the predicted degradation product in each KO compared to the non-targeting control. Red down arrows indicate reduced levels, while green up arrows predict increased production of the cleavage product. Preferred TLR7 trinucleotide “UUC” is indicated in blue font.
Guanosine-containing ORNs bypass the requirement for exogenous delivery of binding site 1 ligands for optimal TLR7 activation
We then determined whether site-specific modifications of an ORN sequence containing guanosines and U-containing trinucleotides, the ligands for site 1 and site 2 of TLR7, could still limit TLR8 activation while maintaining TLR7 activity. The foundation sequence U4.G-PS robustly induced both IFN⍺ and TNF⍺, in agreement with published data (Forsbach et al., 2008; Fig. 6, A and B). As observed with pUC and pUUC, site-specific 2′ modifications on U4.G-PS resulted in a dramatic decrease in TLR8 activation. While the addition of 2′ modifications on U4.G-PS also resulted in a loss of IFN⍺ production for three of the ORNs, U4.G-PS-FF retained robust IFN⍺ production relative to its unmodified counterpart. Strikingly, U4.G-PS-FF stimulated IFN⍺ levels comparable with pUUG, a known and potent activator of both TLR7 and TLR8. To test whether the PS backbone was necessary for achieving TLR7 selectivity with this sequence, we also delivered a suite of U4.G ORNs formulated with a PD backbone. Similar to the PS backbone suite, we observed that the addition of 2′OMe modifications at specified sites was able to reduce TLR8 activity, but also limited TLR7 activity. In contrast, only the PS backbone paired with the “-FF” modifications resulted in a loss of TNF⍺ while maintaining strong IFN⍺ production.

Guanosine-containing ORNs bypass the requirement for exogenously delivered binding site 1 ligand for optimal TLR7 activation. (A) Table of ORNs referenced in this figure. (B) IFN⍺ and TNF⍺ in cell culture supernatants measured by ELISA. hPBMCs were stimulated overnight (16 h) with 0.6 µg/ml of ORNs delivered with pLA or R848 (n = 3 in one experiment). (C–E) Quantification of in vitro cleavage assay gels shown as a percentage relative to the no enzyme (n.e.) negative control (n = 2 in two experiments for all). ORNs were incubated with recombinant RNase T2, RNase 6, or RNase 2 at the indicated concentrations for time points shown and then run on a 15% TBE-Urea gel. 25% of each reaction with enzyme was run while 12.5% of the reaction was run for the n.e. controls.
Guanosine-containing ORNs bypass the requirement for exogenously delivered binding site 1 ligand for optimal TLR7 activation. (A) Table of ORNs referenced in this figure. (B) IFN⍺ and TNF⍺ in cell culture supernatants measured by ELISA. hPBMCs were stimulated overnight (16 h) with 0.6 µg/ml of ORNs delivered with pLA or R848 (n = 3 in one experiment). (C–E) Quantification of in vitro cleavage assay gels shown as a percentage relative to the no enzyme (n.e.) negative control (n = 2 in two experiments for all). ORNs were incubated with recombinant RNase T2, RNase 6, or RNase 2 at the indicated concentrations for time points shown and then run on a 15% TBE-Urea gel. 25% of each reaction with enzyme was run while 12.5% of the reaction was run for the n.e. controls.
To confirm the hypothesized mechanism of action that the 2′ modifications were impacting RNase cleavage efficiency, we performed in vitro cleavage assays with the U4.G-PS ORN suite. As expected with a full PS backbone that provides broad resistance to RNase cleavage (Eckstein, 2014), higher concentrations of the recombinant RNases were required for observable degradation of the U4.G-PS ORNs (Fig. 6, C–E). Nevertheless, the 2′ modifications on U1 and U3 within the UUUU motif increased resistance to RNase T2 (Fig. 6 C), in line with Fig. 2 H. The 2′ modified U4.G-PS ORNs showed less additional resistance to RNase 2 or RNase 6 (Fig. 6, D and E), perhaps due to the prevalence of consensus cut-sites for both enzymes in the sequence of U4.G flanking the UUUU motif. Taken together, the data indicate that site-specific 2′ modifications on ORNs designed to produce binding site 1 and 2 ligands can increase selectivity for TLR7.
Systemic delivery of ORNs induces innate immune activation in vivo
Having shown the ability of our engineered ORNs to enhance TLR7 selectivity in hPBMCs using pLA (Fig. 2 and Fig. 6), we next sought to assess the immunostimulatory activity of our top TLR7-selective ORNs in vitro and in vivo when codelivered with a messenger RNA (mRNA) for potential use as a tunable adjuvant in a clinical context. We first delivered U4.G-PS-FF and pUUC-F to hPBMCs with DOTAP, which is more widely used than pLA in preclinical studies in vivo and delivers cargo to both the endosomes and the cytosol (Simberg et al., 2004; Fig. 2 E). To mimic the effects of a codelivered mRNA-based therapeutic, each ORN was cotransfected with 5-methoxyuridine (5moU)-modified mRNA encoding enhanced green fluorescent protein (eGFP). The substitution of unmodified uridines with 5moU prevents innate immune activation by the mRNA (Karikó et al., 2005). However, since the mRNA contains unmodified guanosines, we reasoned that its degradation could serve as a source of the monomeric guanosine (2′,3′-cGMP) required for binding to TLR7 site 1. Indeed, codelivery of U4.G-PS-FF or pUUC-F with 5moU-modified mRNA using DOTAP both induced an IFN⍺ response comparable with the pUUG control, but with a weak TNF⍺ response, supporting their TLR7 selectivity (Fig. 7, A and B). Taken together, the data demonstrate that U4.G-PS-FF and pUUC-F consistently induce robust levels of IFN⍺ and demonstrate TLR7 selectivity when delivered with either pLA or DOTAP.

Systemic delivery of ORNs induces innate activation in vivo. (A) IFN⍺ and TNF⍺ measured by ELISA in cell culture supernatants of hPBMCs ∼16 h (h) after treatment with 0.6 µg/ml of select ORNs delivered with DOTAP + 5 µg/ml of 5-moU-modified mRNA (three donors in three experiments). Red font indicates top-performing TLR7-selective ORNs. (B) Table of ORNs referenced in this figure. (C) Experimental timeline. Mice were injected i.v. with DOTAP complexed with top-performing ORNs pUUC-F (10 µg) and U4.G-PS-F (20 µg). 5-moU-modified mRNA, alone (20 µg) was used as a control, or as a source of guanosine when paired with pUUC-F (10 µg pUUC-F + 10 µg mRNA). The TLR9 agonist CpG-A (10 µg) was used as a positive control for innate stimulation. (D and E) Quantification of IFN⍺ (D) and other cytokines (E) in sera 6 h after injection (n = 3–5 mice per group). Heatmap color represents the group average for each cytokine on a log2 scale. (F) Representative gating strategy for splenic DC subsets in mice. (G) Top: Representative histograms of CD86 expression in splenic DC subsets 24 h after injection. Numbers indicate the frequency of CD86+ for each DC subset. Bottom: Summary graphs. All bar graphs show mean ± SD.
Systemic delivery of ORNs induces innate activation in vivo. (A) IFN⍺ and TNF⍺ measured by ELISA in cell culture supernatants of hPBMCs ∼16 h (h) after treatment with 0.6 µg/ml of select ORNs delivered with DOTAP + 5 µg/ml of 5-moU-modified mRNA (three donors in three experiments). Red font indicates top-performing TLR7-selective ORNs. (B) Table of ORNs referenced in this figure. (C) Experimental timeline. Mice were injected i.v. with DOTAP complexed with top-performing ORNs pUUC-F (10 µg) and U4.G-PS-F (20 µg). 5-moU-modified mRNA, alone (20 µg) was used as a control, or as a source of guanosine when paired with pUUC-F (10 µg pUUC-F + 10 µg mRNA). The TLR9 agonist CpG-A (10 µg) was used as a positive control for innate stimulation. (D and E) Quantification of IFN⍺ (D) and other cytokines (E) in sera 6 h after injection (n = 3–5 mice per group). Heatmap color represents the group average for each cytokine on a log2 scale. (F) Representative gating strategy for splenic DC subsets in mice. (G) Top: Representative histograms of CD86 expression in splenic DC subsets 24 h after injection. Numbers indicate the frequency of CD86+ for each DC subset. Bottom: Summary graphs. All bar graphs show mean ± SD.
Next, we determined whether U4.G-PS-FF and pUUC-F were capable of inducing TLR7-mediated innate immune activation in vivo using mouse models. Of note, mouse TLR stimulation differs from that of humans due to altered TLR8 binding preferences (Gorden et al., 2006; Martinez et al., 2010) and a wider distribution of TLR7 expression (Fig. 2 A). Thus, this system allows only a comparative assessment of TLR7 activation. We injected mice with ORN/mRNA-DOTAP complexes and collected sera 6 h after injection to measure systemic cytokines (Fig. 7 C). After 24 h, spleens were harvested and processed to measure DC activation. The TLR9 agonist CpG-A was used as a positive control for DC activation and IFN⍺ induction. In agreement with our human in vitro studies (Fig. 6 B and Fig. 7 A), U4.G-PS-FF induced a robust IFN⍺ response (Fig. 7 D). As expected, delivery of pUUC-F alone resulted in minimal induction of IFN⍺, likely due to the absence of localized guanosine which is needed to bind TLR7 site 1 for its activation. On the other hand, codelivery of this ORN with a 5moU-modified mRNA encoding eGFP as a source of guanosine dramatically boosted the IFN⍺ response to levels similar to that elicited by CpG-A. Importantly, injection of 5moU-modified mRNA-DOTAP alone did not induce IFN⍺, confirming that the 5moU-modified bases prevent innate immune activation. To take a broader look at innate immune activation, we analyzed the induction of additional serum cytokines by Luminex (Fig. 7 E). Codelivery of U4.G-PS-FF or pUUC-F with 5moU-modified mRNA in DOTAP-induced cytokine levels similar to those observed with CpG-A. In particular, these ORNs stimulated the production of IFN-inducible protein IP-10 as well as chemokines involved in myeloid cell recruitment such as CCL2, CCL4, and CCL5. This could be explained by the broader expression of TLR7 in mouse myeloid cells compared with humans (Fig. 2 A). Interestingly, we again observed low-level activation of cytokines, including IFN⍺, when pUUC-F was delivered without mRNA. One possibility is that a limited amount of guanosine nucleosides, potentially from degraded RNA released by dying cells, are taken up by DCs in vivo which would provide a source of TLR7 binding site 1 ligand to achieve activation. Nevertheless, there was a robust increase in innate stimulation when pUUC-F was codelivered with the 5moU-modified mRNA.
Lastly, we found that our modified ORNs could induce splenic DC maturation in vivo (Fig. 7, F and G; see Table S1 for antibody list). Both U4.G-PS-FF and pUUC-F induced robust upregulation of the costimulatory marker CD86 in cDC1s, cDC2s, and pDCs. This is consistent with a broader TLR7 expression in mice that includes pDCs and cDCs. Again, when pUUC-F was codelivered with 5moU-modified mRNA, we observed a considerable increase in CD86 expression across all DC subsets analyzed, indicating that the splenic DCs were activated by both of the systemically delivered ORNs. Notably, eGFP expression among splenic myeloid cells was maintained whether the eGFP mRNA was delivered with an ORN or alone (Fig. S3). Together, these results not only confirm that the engineered ORNs require a source of guanosine for robust TLR7 activation, but they also indicate that these ORNs can be delivered systemically to stimulate innate immunity in vivo.

Codelivery of ORNs and eGFP-encoding mRNA does not inhibit mRNA expression. Related to Fig. 7. eGFP expression among splenic myeloid populations measured by flow cytometry 24 h after i.v. injection of DOTAP complexes with eGFP mRNA (10 µg) ± selected ORNs (10 µg) (n = 3–5 mice per group).
Codelivery of ORNs and eGFP-encoding mRNA does not inhibit mRNA expression. Related to Fig. 7. eGFP expression among splenic myeloid populations measured by flow cytometry 24 h after i.v. injection of DOTAP complexes with eGFP mRNA (10 µg) ± selected ORNs (10 µg) (n = 3–5 mice per group).
Discussion
In this study, we engineered ORNs to selectively stimulate TLR7 while avoiding TLR8 activation. TLR7 selectivity was achieved with strategic positioning of ribose 2′ modifications to limit site-specific degradation of ORNs by endosomal RNases, thereby preventing the unwanted release of monomeric uridine required for activation of TLR8. Together, this provides a basis for the discovery of novel, TLR7-selective agonists that stimulate robust IFN⍺ production without eliciting the release of proinflammatory molecules such as TNF⍺.
The data presented here uncovers several new aspects of endosomal RNases in pDCs and other innate immune cells. We showed that similar to TLR8 and murine TLR7, human TLR7 requires RNase processing of the engineered ORNs to generate degradation products that bind site 1 and site 2 for activation (Greulich et al., 2019; Liu et al., 2021; Ostendorf et al., 2020). These findings parallel recent reports that demonstrate a requirement for DNase II-mediated digestion of CpG DNA for optimal activation of TLR9 (Chan et al., 2015). The role of these RNases is somewhat paradoxical: some degradation is required to generate the NUN trinucleotide for TLR7 site 2, but complete degradation would eliminate this trinucleotide and thus limit TLR7 activation. While our ORNs were designed to avoid degradation into TLR8 ligands, additional optimization would likely increase the stability of the NUN trinucleotide. In line with this, we observed that RNase 6 depletion in pDCs improved TLR7 activation by pUUC-F, suggesting that approaches to selectively inhibit RNases could further enhance TLR7 targeting and activation strength of the ORNs described here and elsewhere. In fact, an important point to note is that our PS ORNs were not stereochemically pure. The PS stereochemistry of antisense oligonucleotides has been shown to impact their efficacy at least in part due to varying resistance to RNases (Iwamoto et al., 2017). Therefore, it is possible that the PS ORNs could be further optimized by assessing which stereochemical permutation is most resistant to the relevant RNases. Our data also uncover differences between RNases including cell-type dependent expression patterns, cut-site preferences, and ability to cleave modified ORNs. Specifically, we highlight the previously uncharacterized role of RNase 6 in producing degradation products to stimulate TLR7 in immune cells. Further studies are warranted to identify additional RNA modifications and their effects on the relevant RNases and TLRs. For example, a recent study used site-specific 2′OMe modifications to drive TLR8 selectivity of ORNs (Nicolai et al., 2022), while another used 2′OMe-modified antisense oligonucleotides (ASOs) to modulate TLR7 and TLR8 activity (Alharbi et al., 2020). Approaches that can identify the exact sequence of the degradation products from the in vitro cleavage assays from both studies would also extend the ability to formulate rational designs for TLR selective activation. Finally, future efforts will aim to explore alternative delivery methods and ways to provide a localized TLR7 site 1 ligand.
In addition to engineering TLR7-selective ORNs, we developed a protocol for CRISPR-Cas9 KOs in primary human pDCs. pDCs are a major source of IFN⍺ in vivo and therefore hold great interest as a target for vaccination, antiviral treatments, and cancer immunotherapies. However, primary human pDCs are challenging to study due to their low frequency (<1%) in human blood, sensitivity to manipulation, and relatively short culture timelines, all of which create challenges for genetic manipulation. While immortalized human pDC cell lines are available, they have aberrations that make them poorly suited to study their functional regulation; for example, Cal-1 cells do not produce IFN⍺ in response to TLR7 stimulation (Maeda et al., 2005). Our protocol enables targeted KOs in this important cell type and paves the way for future studies into the functional regulation of human pDCs.
IFN⍺ and IFNβ are the best-characterized members of the type I IFN family. They play a key role in antiviral immunity and are induced in most cell types upon pathogen sensing by pattern recognition receptors. They signal through the heterodimeric receptor IFNAR1/2, resulting in the transcriptional upregulation of numerous IFN-stimulated genes that have direct antiviral activity. Type I IFNs also have the capacity to promote innate and adaptive immune responses, including enhancement of crosspresentation by DCs and promotion of CD8+ T cell cytotoxicity and survival (Le Bon et al., 2006; Kolumam et al., 2005; McNab et al., 2015; Müller et al., 1994; Oh et al., 2011; Schoggins et al., 2011). Type I IFNs can also be induced upon sensing of damaged or dying cells and have been shown to be critical in cancer immunosurveillance, and have also been implicated in the mechanism of multiple anticancer therapies (Deng et al., 2014; Diamond et al., 2011; Dunn et al., 2005; Parker et al., 2016; Schiavoni et al., 2011; Sistigu et al., 2014). Furthermore, recent studies have demonstrated their role in cancer vaccines (Audsley et al., 2021; Baharom et al., 2022; Kranz et al., 2016). Conversely, type I IFNs can also have anti-inflammatory functions (Ivashkiv and Donlin, 2014). A striking example is IFNβ treatment driving T and B cells with regulatory function in patients with multiple sclerosis (Zafranskaya et al., 2007). The cytokine subtype, i.e., the IFN⍺ isoform(s) or IFNβ, as well as the level and duration of induction, may contribute to the different outcomes (Ng et al., 2016). These considerations have fueled the development of strategies targeting the IFN⍺ pathway more specifically. The ORNs presented here preferentially target TLR7, which is predominantly expressed by pDCs, resulting in acute induction of high amounts of IFN⍺, but limited IFNβ. This approach also offers the opportunity to harness other pDC functions in the context of a vaccine. Indeed, in addition to being specialized in producing high levels of IFN⍺, pDCs can contribute to CD8 T cell priming by transferring antigens to cDC1 through exosomes (Fu et al., 2020) and even stimulate CD4 T cells by direct antigen presentation (Abbas et al., 2020).
Additionally, TLR7-selective ORNs are well-suited to counter the delivery limitations of current small-molecule TLR7 agonists. An ssRNA platform allows for easy coformulation with antigen-encoding mRNAs via lipid or nanoparticle encapsulation, which can be delivered intravenously to the cells of interest such as pDCs as an alternative to systemic and nonspecific delivery of small molecule agonists (Hou et al., 2021). Recently, lipid delivery systems have seen rapid advances and widespread adoption due to the success of modified mRNA-lipid nanoparticle vaccines against SARS-CoV-2 (Hogan and Pardi, 2022). This pairing allows independent titration of antigen and adjuvant to create a highly tunable system. Additionally, our in vivo data showed tolerability at a dose that induced strong systemic IFN⍺ and DC maturation in mice. It is important to note that there are differences between humans and mice in response to RNA stimuli, so further studies are needed to better predict the safety, tolerability, and therapeutic index of these ORNs in humans.
In summary, this study demonstrates proof of concept for rationally designing synthetic ORNs that selectively target one of two highly homologous endosomal TLRs for the goal of stimulating a specific cytokine repertoire. The strategy employs existing and newly uncovered knowledge of innate immune receptors and relevant RNases for purposeful placement of site-specific modifications within an ORN sequence. We propose that this general strategy can be extended and further optimized to design additional ORNs, for example, to selectively activate TLR8 for research and therapeutic aims.
Materials and methods
Reagents
RNA production
ORNs were synthesized and RNase-free HPLC-purified by Integrated DNA Technologies (IDT).
Biological resources
Human specimens
Buffy coats and leukopaks were collected from voluntary healthy human donors participating in the Genentech blood donor program after written, informed consent. This program is approved by the WIRB-Copernicus Group Institutional Review Board.
Mice
6–8-wk old female C57BL/6 (B6) mice were purchased from Charles River Labs. Animals were maintained under specific pathogen-free conditions. All animal experiments were performed under protocols approved by the Genentech Institutional Animal Care and Use Committee.
Statistical analyses
Statistical details such as the number of replicates and statistical tests used are described in figure legends.
Method details
PBMC isolation
hPBMCs were obtained from buffy coats from healthy adult donors. Buffy coat samples were first diluted with an equal volume of 1× phosphate-buffered saline (PBS). Next, PBMCs were isolated via density gradient centrifugation with Lymphoprep density gradient media (STEMCELL Technologies) and SepMate-50 tubes (STEMCELL Technologies). Samples were centrifuged at 1,000 g for 15 min. After centrifugation, the upper layers were collected by pouring into a new tube, diluted with PBS, and centrifuged (400 g, 5 min). To lyse red blood cells, cell pellets were resuspended with 10 ml ammonium-chloride-potassium (ACK) lysis buffer and incubated for 10 min at room temperature. After lysis, cells were washed with PBS and passed through two 70-µm cell strainers. Viable cell counts were determined with a Vi-CELL cell counter (Beckman Coulter). PBMCs were used fresh, not frozen. Cells were plated into 96-well round-bottom plates at a concentration of 4 × 105 cells per well in a final volume of 200 µl R10 media (RPMI media containing 10% heat-inactivated fetal bovine serum, 1% L-glutamine, and 1× pen/strep). For CRISPR-Cas9 KO experiments, cells were acquired from fraction 5 of elutriated leukopaks on the day of collection.
pDC depletion of hPBMCs
PBMCs were isolated as described above. pDCs were depleted using the CD303 Microbead Kit, human (Miltenyi Biotec) according to the manufacturer’s instructions. pDC depletion was confirmed by flow cytometry.
PBMC stimulation
To generate p-L-arginine-RNA complexes, RNA and p-L-arginine (>70,000 kD; Sigma #P3892) were mixed in a 1.9:1 mass ratio in PBS (15 µl per reaction). Where indicated, 2′,3′-cGMP (eMolecules) was included in the complex, with p-L-arginine mass increased accordingly. After mixing by pipetting up and down, the reaction was incubated for 10 min at room temperature, pipetted up and down again, and then immediately added to cells plated in 185 µl R10 media for a final volume of 200 µl per well. Complexes were left with the cells overnight (∼16–18 h).
To generate DOTAP-RNA complexes, master mixes containing DOTAP (Roche) and Opti-MEM (Thermo Fisher Scientific) or RNA and Opti-MEM were made and incubated for 5 min at room temperature. The two master mixes were then combined at a final RNA:DOTAP mass ratio of 1:5. The mixtures were then incubated for 20 min at room temperature before being added to the cells. Cells were incubated with DOTAP complexes for 2 h at 37°C, then centrifuged (400 g, 5 min), washed 1× with PBS, and resuspended in R10 media. Where indicated, CleanCap 5moU mRNA encoding eGFP (TriLink Biotechnologies) was complexed together with the ORNs.
In vivo delivery of ORNs
ORNs were complexed with DOTAP (Roche) at a mass ratio of 1:5 RNA:DOTAP and prepared in PBS for a final injection volume of 300 µl per mouse. Complexes were allowed to form for 20–30 min at room temperature, then delivered intravenously via tail vein injection. Where indicated, ORNs were codelivered with CleanCap 5moU eGFP mRNA (TriLink Biotechnologies). The VacciGrade mouse-preferred TLR9 agonist CpG-A ODN1585 (InvivoGen) was used as a positive control for innate activation and cytokine induction.
Cytokine detection
Human
Cell culture supernatants were collected 15–18 h after stimulation and stored at −80°C. Cytokines in cell culture supernatants were detected with the BD OptEIA human TNF ELISA Set (BD Biosciences) and the Human IFN Alpha Multi-Subtype ELISA Kit (PBL Assay Science). For Fig. 7 A, TNF⍺ was measured by Luminex using R&D kits.
Mouse
Peripheral blood was collected by retro-orbital bleed 6 h after injection. Blood was allowed to clot by incubating for at least 5 min at room temperature and then centrifuged to separate the serum (2,650 g, 5 min, room temperature). Sera were stored at −80°C until cytokine assays were performed. IFN⍺ and IFNβ were detected with the VeriKine-HS Mouse Interferon Alpha All Subtype ELISA Kit (PBL Assay Science) and VeriKine-HS Mouse Interferon Beta Serum ELISA kit (PBL Assay Science), respectively. Additionally, serum cytokines were assayed by Luminex with the ProcartaPlex Cytokine and Chemokine 26-Plex Mouse Panel 1 (Invitrogen).
Analysis of RNase gene expression
Human
Human scRNA-seq data was downloaded as log-transformed and filtered TPM counts from the Broad Single Cell Portal study “Atlas of human blood dendritic cells and monocytes” (https://singlecell.broadinstitute.org/single_cell/study/SCP43/atlas-of-human-blood-dendritic-cells-and-monocytes#study-download; Villani et al., 2017). Expression was averaged across single cells for each cell type as defined by the study metadata. Bulk RNA-seq expression data from the human monocytic cell line THP-1 was drawn from a published Genentech dataset (Klijn et al., 2015). Gene expression was calculated as normalized reads per kilobase per million (nRPKM), where normalization refers to an extra step for DESeq2 size factor adjustment.
Mouse
Mouse microarray data was downloaded from NCBI GEO accession no. GSE35458 (Haniffa et al., 2012). Raw data was quantile normalized and log2 transformed with the limma package in R.
In vitro cleavage assays
Cleavage assays were performed as described by Ostendorf et al. (2020) with the following modifications: 0.5 µg of each RNA was incubated with recombinant RNase T2 (Origene) in a 15 µl reaction. A four-point time course was performed where reactions were incubated for 3, 6, 9, or 12 min. Reactions were stopped with 100 mM Tris-HCL (pH 7.5). Upon addition of 2× TBE-Urea sample buffer (Thermo Fisher Scientific), the reactions were incubated for 3 min at 70°C. Samples were then loaded into 15% Criterion TBE-Urea polyacrylamide gels (Bio-Rad) and run for 1.5 h at 120 V. Gels were stained with SYBR Gold Nucleic Acid Gel Stain (Invitrogen) for ORN with PD backbones or SYBR Green II Nucleic Acid Gel Stain (Invitrogen) for ORN with PS backbones, both diluted 1:10,000 in 1× TBE for 15 min at room temperature. Then, gels were washed three times in deionized water for 15 min per wash. Images were acquired on a ChemiDoc MP Imaging System (Bio-Rad). Quantification of gels was performed using Bio-Rad Image Lab software. The intensity of the full-length band was quantified and each experimental condition was normalized to the no enzyme negative control to achieve a relative intensity measurement.
Screening conditions for CRISPR-Cas9 KO in primary human pDCs
hPBMCs were isolated as described above. After isolation, cells were washed 2× with at least 5 ml of 1× PBS, then resuspended in primary cell nucleofection buffers from the Primary Cell Optimization 4D-Nucleofector X Kit (Lonza) at a concentration of 3 × 106 PBMCs in 20 µl buffer for each condition. TruCut V2 Cas9 (5 mg/ml; Thermo Fisher Scientific) was combined with sgRNA (100 µM β2M sg2; Freund et al., 2020) at a 1:1 vol ratio and incubated for at least 10 min at room temperature to allow ribonucleoprotein (RNP) complexes to form. For each nucleofection condition, 20 µl of cells was combined with 1 µl RNP complexes and 1 µl of Alt-R Cas9 electroporation enhancer (IDT) and then pipetted into one well of a nucleocuvette strip (Lonza). Cells were nucleofected with a 4D-Nucleofector X (Lonza) with preselected programs suggested in the optimization kit. Immediately after nucleofection, prewarmed R10 media containing 10 ng/ml recombinant human IL-3 (R&D Systems) for pDC survival was added to the cells. PBMCs were plated in 96-well round-bottom plates (1 × 106 cells per well) and cultured in an incubator (37°C, 5% CO2) for 3 d. On the third day, cells were stained for flow cytometry to assess pDC recovery and efficiency of β2M KO.
CRISPR-Cas9 KO of RNASET2, RNASE6, and TLR7 in primary human pDCs
Primary human pDCs were isolated from fraction 5 of elutriated leukopaks by immunomagnetic negative selection using the EasySep Human Plasmacytoid DC Isolation Kit (STEMCELL Technologies) according to the manufacturer’s instructions. After initial negative selection, cells were counted and then underwent a second round of negative selection. Purified pDCs were washed twice in 1× PBS, counted, centrifuged, and resuspended in 100 µl P3 (Lonza). sgRNA and Cas9 RNPs were prepared with 2 µl of each sgRNA (100 µM stocks) and a 1:1 vol ratio with TruCut V2 Cas9 (5 mg/ml; Thermo Fisher Scientific). sgRNA sequences were sourced from IDT with the sequences listed in Table S2. Less than 3 million pDCs were added to each RNP condition in 20 µl P3 buffer, then transferred to a nucleofection cassette, and treated with the program CA-137. Immediately after nucleofection, complete media (RPMI with 10% FBS, pen/strep, β-mercaptoethanol, non-essential amino acids, sodium pyruvate, HEPES) with 10 ng/ml recombinant human IL-3 (R&D Systems) was added to pDCs. Cells were transferred to a 96-well round-bottom plate and cultured in an incubator (37°C, 5% CO2) for 3 days. On the third day, cells were counted, plated at 125,000 cells per well, and treated overnight with the human-preferred TLR9 agonist CpG-A ODN2216 (5 µg/ml, no carrier; InvivoGen), pUUC-F (0.6 µg/ml, delivered with pLA), or media control.
Preparation of a single-cell suspension from murine spleen
Mice were sacrificed 24 h after injection. Spleens were collected and placed in 5 ml of Hanks-Based Cell Dissociation Buffer (Gibco) with 12.5 µg/ml Liberase (Roche) and 50 µg/ml DNase I (Invitrogen). Spleens were mechanically disrupted with a syringe plunger, and then incubated for 15 min at room temperature. Cell suspensions were passed through a 100-μm filter. 5 ml MACS buffer (PBS with 0.5% BSA and 2 mM EDTA) was used to rinse the filter and added to cell suspensions. Cells were centrifuged (400 g, 5 min, room temperature) and then resuspended in 2 ml ACK lysis buffer and incubated for 8 min at room temperature. 13 ml MACS buffer was added to stop the lysis and then cells were centrifuged (400 g, 5 min, room temperature). Cell pellets were resuspended in PBS and an aliquot was taken to count with a Vi-CELL BLU (Beckman-Coulter).
Staining cells for flow cytometry analysis
Antibodies for flow cytometry were purchased from BioLegend or BD Biosciences. A list of all antibodies used for flow cytometry can be found in Table S1. Cells were acquired on a five-laser FACSymphony (BD Biosciences). Unstained cells and single-stained UltraComp eBeads (Invitrogen) were used for compensation controls. Data were analyzed with FlowJo Software (Tree Star, Inc).
Human
Cells were centrifuged (550 g, 4 min, room temperature), washed with 1× PBS, then resuspended in Human TruStain FcX (BioLegend) diluted 1:200 in PBS, and incubated for 10 min on ice.
Mouse
From each sample, 10 million splenocytes were transferred to a 96-well V-bottom for staining. Cells were centrifuged (550 g, 2 min, room temperature), then resuspended in 2.5 µg/ml TruStain FcX (BioLegend), and incubated for 10 min on ice. Cells were washed with PBS, then resuspended in the antibody cocktail and incubated for 20 min on ice. Cells were washed 1.5× with FACS buffer (PBS with 0.5% BSA and 0.05% sodium azide), centrifuged, and resuspended in FACS buffer for acquisition. Prior to acquisition, DAPI (BD Biosciences) was added at a final concentration of 0.1 µg/ml for the detection of dead cells.
Online supplemental material
Fig. S1 shows the dose exploration of ORNs delivered to hPBMCs with pLA. Fig. S2 shows the optimization of the CRISPR-Cas9 KO protocol for primary human pDCs. Fig. S3 shows eGFP expression in murine splenic myeloid populations upon codelivery of ORNs and eGFP-encoding mRNA in vivo. Table S1 shows detailed information about all flow cytometry antibodies used in this work. Table S2 shows sgRNA sequences used for CRISPR-Cas9 KO experiments.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Acknowledgments
We thank the blood donors for their participation and the Genentech FACS laboratory and Luminex core for technical advice and support. We thank S. Bauernfried for the critical reading of the manuscript. We thank our colleagues in gCell and gCellGenomics for making the THP-1 cell line expression data available. Figures were prepared with BioRender.
This work was supported by Genentech, Inc., a member of the Roche group. All authors are current or past employees of Genentech/Roche and may hold Roche stock or stock options. A.-J. Tong, R. Leylek, A.-M. Herzner, S. Wichner, E.C. Freund, and L. Delamarre are inventors on a pending patent related to this work.
Author contributions: Conceptualization: A.-J. Tong, R. Leylek, A.-M. Herzner, and E.C. Freund; Formal Analysis: A.-J. Tong, R. Leylek, A.-M. Herzner, D. Rigas, and E.C. Freund; Investigation: A.-J. Tong, R. Leylek, A.-M. Herzner, D. Rigas, S. Tahtinen, and E.C. Freund; Methodology: A.-J. Tong, R. Leylek, A.-M. Herzner, S. Wichner, and E.C. Freund; Project Administration: C. Blanchette, C.C. Kemball, B. Haley, E.C. Freund, and L. Delamarre; Resources: C. Blanchette, C.C. Kemball, B. Haley, and L. Delamarre; Supervision: C.C. Kemball, I. Mellman, B. Haley, E.C. Freund, and L. Delamarre; Validation: A.-J. Tong, R. Leylek, S. Wichner, and E.C. Freund; Visualization: A.-J. Tong, R. Leylek, and E.C. Freund; Writing—Original Draft: A.-J. Tong, R. Leylek, A.-M. Herzner, E.C. Freund, and L. Delamarre; Writing—Review & Editing: all authors.

