


PU.1 (encoded by Spi1), an ETS-family transcription factor with many hematopoietic roles, is highly expressed in the earliest intrathymic T cell progenitors but must be down-regulated during T lineage commitment. The transcription factors Runx1 and GATA3 have been implicated in this Spi1 repression, but the basis of the timing was unknown. We show that increasing Runx1 and/or GATA3 down-regulates Spi1 expression in pro–T cells, while deletion of these factors after Spi1 down-regulation reactivates its expression. Leveraging the stage specificities of repression and transcription factor binding revealed an unconventional but functional site in Spi1 intron 2. Acute Cas9-mediated deletion or disruption of the Runx and GATA motifs in this element reactivates silenced Spi1 expression in a pro–T cell line, substantially more than disruption of other candidate elements, and counteracts the repression of Spi1 in primary pro–T cells during commitment. Thus, Runx1 and GATA3 work stage specifically through an intronic silencing element in mouse Spi1 to control strength and maintenance of Spi1 repression during T lineage commitment.

Introduction
PU.1, encoded by Spi1, is an E twenty-six (ETS)–family transcription factor with multiple roles in the development and function of myeloid and lymphoid hematopoietic lineages (Carotta et al., 2010; Rothenberg et al., 2019; Singh et al., 1999). Expression levels of Spi1 are highest in macrophages, granulocytes, and dendritic cells and moderate in lymphoid progenitors, reducing expression in B cells and turning off entirely in natural killer and T cells (Yoshida et al., 2019). Through pioneering-like activity and differential cofactor recruitment, PU.1 can control specification of several blood cell types (Heinz et al., 2010; Heinz et al., 2013; Hosokawa et al., 2018b; Ostuni et al., 2013; Rothenberg et al., 2019). Dysregulation of Spi1 expression and translocation of the Spi1 gene can induce malignant transformation in multiple hematopoietic lineages (Rosenbauer et al., 2004; Seki et al., 2017; Vangala et al., 2003). Thus, Spi1 expression must be tightly regulated in a lineage-specific manner. Spi1-deficient hematopoietic stem cells fail to contribute to the T cell lineage in bone marrow (BM) chimera mice (Dakic et al., 2005; Scott et al., 1994), but Spi1 expression must be shut off during T lineage commitment. The mechanism of this repression has remained obscure until now.
T cells develop from multipotent precursors in the thymus, where microenvironmental Notch signaling educates progenitors to become T cells (Hosokawa and Rothenberg, 2021; Radtke et al., 2013; Romero-Wolf et al., 2020). The earliest T progenitors in the thymus lack expression of the mature T cell markers CD4 and CD8 (double negative [DN]). The expression profiles of Kit, CD44, and CD25 in DN thymocytes distinguish developmental stages (Hosokawa et al., 2021; Hosokawa and Rothenberg, 2021; Rothenberg et al., 2008; Yang et al., 2010; Yui and Rothenberg, 2014). The earliest T cell precursors in the thymus, Kithi DN1 cells (here “DN1,” Kithi+CD44+CD25−, or ETP), and their DN2a (Kithi+CD44+CD25+) descendants, still have access to non–T cell fates and proliferate to expand the intrathymic progenitor pool (Lu et al., 2005). They lose multipotency in transition from DN2a to DN2b (Kitint+CD44+CD25+), becoming committed to the T lineage (Fig. 1 A). Next, TCRβ gene rearrangement at the DN3 (KitloCD44loCD25+) stage triggers pre-TCR–dependent progression to DN4 (KitloCD44loCD25−) and double-positive (CD4+CD8+) stages, leading to full TCR expression (Hosokawa and Rothenberg, 2018; Yui and Rothenberg, 2014).

Expression profiles of GATA3, PU.1, and Bcl11b around the T lineage commitment checkpoint. (A) Schematic of early T cell development is shown with expression kinetics of the transcription factors Spi1 (PU.1), Runx1, Gata3, and Bcl11b. (B) Intracellular staining for GATA3 and PU.1 was performed using DN thymocytes from Bcl11b-YFP reporter mice, gated as in Fig. S1 A. Representative profiles of isotype control (left), GATA3 (middle), and PU.1 (right) with Bcl11b-YFP in DN1, DN2a, and DN2b are shown with the percentages of cells in each quadrant. Results are representative of three independent experiments. (C) Intracellular staining profiles of GATA3 (left) and PU.1 (right) in DN1, DN2a-YFP−, DN2a-YFP+, and DN2b are shown. The gray-filled histogram shows isotype control staining. GATA3, DN2a YFP+ and DN2b curves overlap; PU.1, DN1 and DN2a YFP− curves overlap. Results are representative of three independent experiments.
Expression profiles of GATA3, PU.1, and Bcl11b around the T lineage commitment checkpoint. (A) Schematic of early T cell development is shown with expression kinetics of the transcription factors Spi1 (PU.1), Runx1, Gata3, and Bcl11b. (B) Intracellular staining for GATA3 and PU.1 was performed using DN thymocytes from Bcl11b-YFP reporter mice, gated as in Fig. S1 A. Representative profiles of isotype control (left), GATA3 (middle), and PU.1 (right) with Bcl11b-YFP in DN1, DN2a, and DN2b are shown with the percentages of cells in each quadrant. Results are representative of three independent experiments. (C) Intracellular staining profiles of GATA3 (left) and PU.1 (right) in DN1, DN2a-YFP−, DN2a-YFP+, and DN2b are shown. The gray-filled histogram shows isotype control staining. GATA3, DN2a YFP+ and DN2b curves overlap; PU.1, DN1 and DN2a YFP− curves overlap. Results are representative of three independent experiments.
The chromatin landscape and transcriptome profiles change dynamically at the transition from precommitment, DN1 and DN2a, pro–T cells to T lineage–committed, DN2b and DN3, pro–T cells (Hosokawa et al., 2021; Hosokawa and Rothenberg, 2021; Hu et al., 2018). Two transcription factors, PU.1 and Bcl11b, change expression reciprocally in this process. While Bcl11b turns on as cells undergo commitment (Kueh et al., 2016; Zhou et al., 2019), Spi1 is shut off during T lineage commitment, eliminating access to myeloid programs (Hosokawa et al., 2021; Hosokawa and Rothenberg, 2018; Yui and Rothenberg, 2014; Fig. 1 A). Conditional deletion of the Spi1 gene before commitment not only causes faster developmental progression but also reduces cell yield (Champhekar et al., 2015; Hosokawa et al., 2018b). Thus, PU.1 supports the expansion of the uncommitted DN1 and DN2a T progenitor pool (Hosokawa and Rothenberg, 2021). However, inefficient repression or abnormal activation of Spi1 after T lineage commitment can yield malignancy, directly or via its target genes (Boehm et al., 1991; Homminga et al., 2011; Hosokawa and Rothenberg, 2021; Rosenbauer et al., 2006; Seki et al., 2017; Yui and Rothenberg, 2014).
Regulatory mechanisms controlling Spi1 expression kinetics during early T cell development are not fully understood. Multiple reports have suggested roles for Runx1 and GATA3 as mediators of Spi1 repression in mouse pro–T cells. When Runx1 was deleted conditionally by Mx1-Cre activation in mice, early T cell development was significantly blocked at precommitment stages with increased Spi1 expression (Growney et al., 2005; Huang et al., 2008), and Runx1 deletion in pro–T cells developing in vitro also caused Spi1 up-regulation (Hosokawa et al., 2018b). GATA3 overexpression severely repressed Spi1 in fetal pro–T cells (Taghon et al., 2007), while shRNA knockdown of Gata3 caused up-regulation of Spi1 in fetal thymus- or liver-derived pro–T cells (Scripture-Adams et al., 2014). However, Runx1 and Gata3 perturbation in progenitor cells also strongly reduces pro–T cell yields, and their expression increases little during commitment. Thus, how Runx1 and GATA3 cause this stage-specific Spi1 repression has remained uncertain.
Multiple cis-regulatory elements around the Spi1 locus could play roles. The promoter, a major upstream regulatory element (URE), and conserved noncoding element 4 (CE4) and CE5 have all been suspected to play roles in cell type–specific regulation of Spi1 in myeloid, B, and T cells (Hoogenkamp et al., 2007; Li et al., 2001; Okuno et al., 2005; Rosenbauer et al., 2006; Rosenbauer et al., 2004; Zarnegar et al., 2010). The URE, a highly conserved noncoding genomic region 14 kb upstream of the transcriptional start site of Spi1 (−14 kb; Li et al., 2001; Okuno et al., 2005), showed enhancer activity in myeloid and B cells (Rosenbauer et al., 2006; Rosenbauer et al., 2004), while its T lineage role appeared bivalent or repressive (Hoogenkamp et al., 2007; Huang et al., 2008; Montecino-Rodriguez et al., 2018; Zarnegar et al., 2010). URE KO mice showed a partial defect in early T cell development, characterized by increased Spi1 expression in DN1 as well as DN3 stages of adult mice (Rosenbauer et al., 2006). Thus, the bifunctional URE region can damp Spi1 expression in pro–T cells, but whether it explains the switch to repression is less clear. CE4 and CE5, upstream (−9.2 and −10.3 kb) conserved noncoding regions, were identified as T cell–specific silencer and myeloid-specific enhancer elements using reporter assays with cell lines (Zarnegar et al., 2010). However, CE4 element function has not been demonstrated in its native context.
Here, we describe a new regulatory element through which GATA3 and Runx1 appear to work stage specifically to silence Spi1 in T lineage commitment. Using pro–T cell differentiation cultures with acute, stage-specific perturbations, we verify roles and structural requirements of GATA3 and Runx1 for Spi1 repression. Highly stage-specific binding of Runx1 and GATA3 in pro–T cells during commitment identifies a Spi1 intronic site that is functionally important for the magnitude and maintenance of Spi1 repression in a pro–T cell line and primary pro–T cells, with greater impact than CE4 or other candidate repression elements. Taken together, this evidence identifies a direct mechanism through which Runx1 and GATA3 mediate the stage-specific repression of Spi1.
Results
Runx1 and GATA3 protein expression relative to Spi1 expression around T lineage commitment
Gata3 turns on soon after precursors enter the thymus, reaching a plateau in late DN2a stage (Yoshida et al., 2019; Zhou et al., 2019; Fig. 1 A). Pro–T cells express Runx1 more highly than other hematopoietic cell types (Yoshida et al., 2019), and both Runx1 mRNA and protein are highest across commitment (Shin et al., 2021; Zhou et al., 2019). In DN pro–T cells, shortly before Spi1 expression decreases, expression of Gata3 and Runx1 mRNA increases slightly (Yoshida et al., 2019; Zhang et al., 2012).
To determine whether GATA3 protein levels could be rate limiting for PU.1 silencing in early pro–T cells, we conducted intracellular staining for GATA3 and PU.1. We used thymocytes from Bcl11b-YFP reporter mice (Kueh et al., 2016) to monitor Bcl11b up-regulation, marking T lineage commitment in late DN2a stage (Fig. 1 A), and compared DN1 (Kithi+CD25−), DN2a (Kithi+CD25+), and DN2b (Kitint+CD25+) stages among the DN, Lin−CD44+Kit+ thymocytes (Fig. S1 A). The results (Fig. 1, B and C) confirmed that GATA3 levels were very low in the DN1 stage, slightly up-regulated in the DN2a Bcl11b− (YFP−) stage, and further increased in the DN2a Bcl11b+ (YFP+) and DN2b stages. PU.1 was highly expressed in the DN1 and DN2a Bcl11b− stages but then shifted lower in DN2a cells as they began to express Bcl11b (Fig. 1, B and C) and had clearly dropped by DN2b stage (cf. Ungerbäck et al., 2018; Yui et al., 2010). Given the high protein stability of PU.1 (Kueh et al., 2013), the slight reduction seen in Bcl11b+ DN2a cells is consistent with the onset of repression. In separate work (Shin et al., 2021), we found that Runx1 protein also detectably increased from the DN1 to DN2a/b stages. Thus, GATA3 and Runx1 protein levels beyond a certain threshold could influence the timing of PU.1 repression.
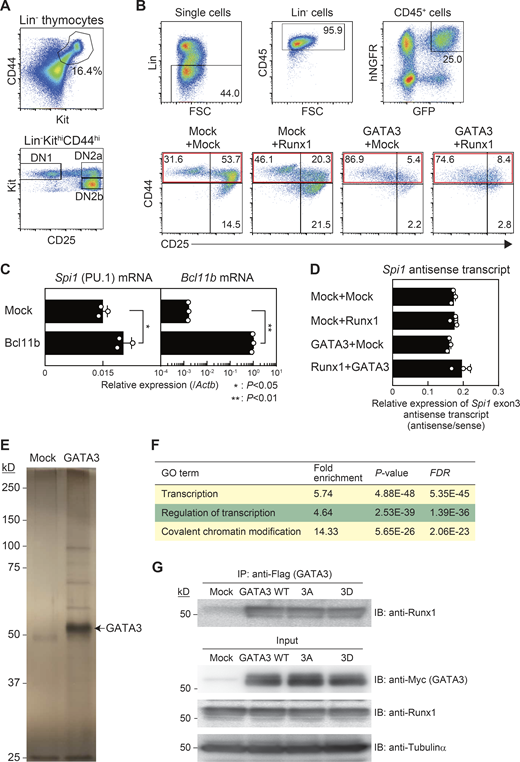
Effects of Runx1, GATA3, or Bcl11b introduction on Spi1 expression in precommitment pro–T stages.(A) Flow cytometric analysis of thymocytes from Bcl11b-YFP reporter mice was performed. Representative Kit/CD44 profile in Lin− cells (top) and CD25/Kit profile in Lin−KithiCD44hi cells (bottom) are shown. Results are representative of three independent experiments. (B) Flow cytometric analysis of retrovirus-infected Lin−CD45+CD44+GFP+hNGFR+ precommitment cells was performed at 3 d after introduction using protocol A. Gating strategy for Lin−CD45+GFP+hNGFR+ cells is shown (top). Representative profiles of CD44/CD25 in Lin−CD45+GFP+hNGFR+ cells are shown (bottom). Gates to isolate CD44+ cells for sorting are labeled with red rectangles. Results are representative of three independent experiments. (C) Retrovirus encoding Bcl11b was infected into precommitment cells, and then Lin−CD45+CD44+GFP+hNGFR+ cells were sorted at 3 d after introduction from protocol A. Expression levels of Spi1 and Bcl11b were analyzed by RT-qPCR. The relative expression (/Actb) is shown with SD. *, P < 0.05; **, P < 0.01 by two-sided Student’s t test. Data are based on three biological replicates. (D) Retrovirus-infected Lin−CD45+CD44+GFP+hNGFR+ precommitment cells were sorted at 3 d after introduction (protocol A). Strand-specific cDNAs around the exon 3 region of the Spi1 locus were synthesized as described previously (Ebralidze et al., 2008). Ratio of antisense transcripts against sense transcripts (antisense/sense) of Spi1 exon3 is shown with SD. Data are based on three biological replicates. (E) Myc- and Flag-tagged (double epitope–tagged) GATA3 was retrovirally transduced into a DN3-like cell line, Scid.adh.2c2. Total extracts from Myc-Flag-GATA3–expressing Scid.adh.2c2 cells were subjected to two-step affinity purification followed by SDS-PAGE and silver staining. All of the visible bands were subjected to mass spectrometry analysis. Data are representative of two independent experiments. (F) Gene Ontology (GO) annotation of proteins identified from their GATA3-interacting peptides was performed using the DAVID analysis tool (http://david.ncifcrf.gov/). Top three GO terms for GATA3-interacting molecules (Table S1) are shown. (G) Total extracts from Scid.adh.2c2 cells transduced with Myc-Flag-GATA3 WT, -3A, or -3D were subjected to immunoprecipitation (IP) with anti-Flag mAb followed by immunoblotting (IB) with anti-Runx1 antibodies. To measure protein levels in the inputs, nuclear or cytoplasmic lysates were subjected to IB with anti-Myc (GATA3), anti-Runx1, or anti-tubulinα antibodies, respectively (input). Data are representative of three independent experiments.
Effects of Runx1, GATA3, or Bcl11b introduction on Spi1 expression in precommitment pro–T stages.(A) Flow cytometric analysis of thymocytes from Bcl11b-YFP reporter mice was performed. Representative Kit/CD44 profile in Lin− cells (top) and CD25/Kit profile in Lin−KithiCD44hi cells (bottom) are shown. Results are representative of three independent experiments. (B) Flow cytometric analysis of retrovirus-infected Lin−CD45+CD44+GFP+hNGFR+ precommitment cells was performed at 3 d after introduction using protocol A. Gating strategy for Lin−CD45+GFP+hNGFR+ cells is shown (top). Representative profiles of CD44/CD25 in Lin−CD45+GFP+hNGFR+ cells are shown (bottom). Gates to isolate CD44+ cells for sorting are labeled with red rectangles. Results are representative of three independent experiments. (C) Retrovirus encoding Bcl11b was infected into precommitment cells, and then Lin−CD45+CD44+GFP+hNGFR+ cells were sorted at 3 d after introduction from protocol A. Expression levels of Spi1 and Bcl11b were analyzed by RT-qPCR. The relative expression (/Actb) is shown with SD. *, P < 0.05; **, P < 0.01 by two-sided Student’s t test. Data are based on three biological replicates. (D) Retrovirus-infected Lin−CD45+CD44+GFP+hNGFR+ precommitment cells were sorted at 3 d after introduction (protocol A). Strand-specific cDNAs around the exon 3 region of the Spi1 locus were synthesized as described previously (Ebralidze et al., 2008). Ratio of antisense transcripts against sense transcripts (antisense/sense) of Spi1 exon3 is shown with SD. Data are based on three biological replicates. (E) Myc- and Flag-tagged (double epitope–tagged) GATA3 was retrovirally transduced into a DN3-like cell line, Scid.adh.2c2. Total extracts from Myc-Flag-GATA3–expressing Scid.adh.2c2 cells were subjected to two-step affinity purification followed by SDS-PAGE and silver staining. All of the visible bands were subjected to mass spectrometry analysis. Data are representative of two independent experiments. (F) Gene Ontology (GO) annotation of proteins identified from their GATA3-interacting peptides was performed using the DAVID analysis tool (http://david.ncifcrf.gov/). Top three GO terms for GATA3-interacting molecules (Table S1) are shown. (G) Total extracts from Scid.adh.2c2 cells transduced with Myc-Flag-GATA3 WT, -3A, or -3D were subjected to immunoprecipitation (IP) with anti-Flag mAb followed by immunoblotting (IB) with anti-Runx1 antibodies. To measure protein levels in the inputs, nuclear or cytoplasmic lysates were subjected to IB with anti-Myc (GATA3), anti-Runx1, or anti-tubulinα antibodies, respectively (input). Data are representative of three independent experiments.
Transduction of Runx1 and Gata3 represses Spi1 expression in precommitment pro–T cells
To verify whether Runx1 and/or GATA3 levels control Spi1 expression in pro–T cells, we tested whether their overexpression in DN1 and DN2a pro–T cells could accelerate the natural silencing of Spi1. Runx and GATA3 perturbations are demonstrably toxic in vitro, leaving some uncertainty as to whether gene-expression changes seen in previous studies reflected direct gene regulation or population selection (Guo et al., 2008; Huang et al., 2008; Scripture-Adams et al., 2014; Taghon et al., 2007; Telfer et al., 2004). We used BM progenitor cells from Bcl2 transgenic (Tg) mice to maximize viable recovery, culturing them on OP9 Delta-like 1 (DLL1; a Notch ligand) stroma to initiate T cell development in vitro (see Materials and methods). On day 3 of culture, when most of the cells were still in precommitment stages (DN1 and DN2a), they were infected with retroviral vectors expressing Runx1 and/or Gata3 with GFP or human nerve growth factor receptor (hNGFR) markers, respectively. We sorted Lin−CD45+CD44+GFP+hNGFR+ precommitment cells for analysis at 3 d postinfection (dpi; day 6 of culture overall; Fig. 2, A and B; and Fig. S1 B; protocol A). Introduction of Gata3 into these precommitment cells, which still had high Spi1 expression, profoundly repressed Spi1, while Runx1 transduction had a moderate but consistent effect. The combination of Runx1 and Gata3 introduction further intensified repression of Spi1 (Fig. 2 C). In contrast, while Spi1 down-regulation coincided most closely with the timing of Bcl11b-YFP activation during development, overexpression of Bcl11b did not repress Spi1 expression (Fig. S1 C). This supports previous indications that Bcl11b does not itself regulate Spi1 in mouse pro–T cells (Hosokawa et al., 2018a; Longabaugh et al., 2017).

Introduction of Runx1 and Gata3 represses Spi1 expression in precommitment stages. (A) An experimental scheme for introduction of Runx1 and/or Gata3 in precommitment pro–T cells is shown (protocol A). (B) BM progenitors obtained from Bcl2 Tg mice were co-cultured on OP9-DLL1. Flow cytometric analysis of T progenitors was performed on day 3, before retrovirus infection. A representative CD44/CD25 profile in Lin−CD45+ cells is shown (left). Schematic of the pro–T cell development trajectory is shown (right). Results are representative of three independent experiments. (C) Retrovirus-infected Lin−CD45+CD44+GFP+hNGFR+ precommitment cells were sorted at 3 d after introduction. Expression levels of Spi1 (left), Runx1 (middle), and Gata3 (right) were analyzed by RT-qPCR. The relative expression (/Actb) is shown with SD. **, P < 0.01 by two-sided Student’s t test. Data are representative of two independent experiments and based on three biological replicates. (D) Precommitment cells were introduced with GATA3 WT, phospho-deficient (3A), phospho-mimic (3D; Hosokawa et al., 2016; Hosokawa et al., 2013b), methylation-deficient (R261K), or methylation-mimic (R261F) GATA3 mutants (Hosokawa et al., 2015; Nakayama et al., 2017) as protocol A. Expression levels of Spi1 (left) and Gata3 (right) were analyzed by RT-qPCR on 3 d after introduction. The relative expression (/Actb) is shown with SD. **, P < 0.01 by two-sided Student’s t test. Data are representative of two independent experiments and based on four biological replicates. (E) Reads per kilobase per million reads (RPKM) values for Spi1 in WT or DN form of Runx1 introduced DN3-like cell lines are shown with SD (Hosokawa et al., 2018b).
Introduction of Runx1 and Gata3 represses Spi1 expression in precommitment stages. (A) An experimental scheme for introduction of Runx1 and/or Gata3 in precommitment pro–T cells is shown (protocol A). (B) BM progenitors obtained from Bcl2 Tg mice were co-cultured on OP9-DLL1. Flow cytometric analysis of T progenitors was performed on day 3, before retrovirus infection. A representative CD44/CD25 profile in Lin−CD45+ cells is shown (left). Schematic of the pro–T cell development trajectory is shown (right). Results are representative of three independent experiments. (C) Retrovirus-infected Lin−CD45+CD44+GFP+hNGFR+ precommitment cells were sorted at 3 d after introduction. Expression levels of Spi1 (left), Runx1 (middle), and Gata3 (right) were analyzed by RT-qPCR. The relative expression (/Actb) is shown with SD. **, P < 0.01 by two-sided Student’s t test. Data are representative of two independent experiments and based on three biological replicates. (D) Precommitment cells were introduced with GATA3 WT, phospho-deficient (3A), phospho-mimic (3D; Hosokawa et al., 2016; Hosokawa et al., 2013b), methylation-deficient (R261K), or methylation-mimic (R261F) GATA3 mutants (Hosokawa et al., 2015; Nakayama et al., 2017) as protocol A. Expression levels of Spi1 (left) and Gata3 (right) were analyzed by RT-qPCR on 3 d after introduction. The relative expression (/Actb) is shown with SD. **, P < 0.01 by two-sided Student’s t test. Data are representative of two independent experiments and based on four biological replicates. (E) Reads per kilobase per million reads (RPKM) values for Spi1 in WT or DN form of Runx1 introduced DN3-like cell lines are shown with SD (Hosokawa et al., 2018b).
Antisense transcripts from intron 3 of the Spi1 locus reportedly act as negative modulators of Spi1 expression (Ebralidze et al., 2008). Runx1 and GATA3 might reduce Spi1 expression by stimulating expression of these antisense transcripts. However, precommitment cells with and without overexpression of Runx1 and/or Gata3 had comparable ratios of sense and antisense transcripts of exon3 of the Spi1 gene (Fig. S1 D). Therefore, Spi1 repression by Runx1 and GATA3 does not appear to be mediated by up-regulation of antisense transcripts of Spi1 locus.
Structural requirements for GATA3 and Runx1 repression of Spi1
To test whether GATA3 might repress Spi1 directly or whether it worked by inducing other repressors, we tested GATA3 mutants that can differentially affect function, localization, and organization of GATA3 complexes in type 2 T helper and type 2 innate lymphoid cells (Furusawa et al., 2013; Hosokawa et al., 2015; Hosokawa et al., 2016; Hosokawa et al., 2013a; Hosokawa et al., 2013b; Yamagata et al., 2000). Phosphorylation of GATA3 at S308, T315, and S316 inhibits its repressive action by dissociating histone deacetylase 2 (HDAC2) from the GATA3–nucleosome remodeling and deacetylase (NuRD) complex (Hosokawa et al., 2016; Hosokawa et al., 2013b) and can be mimicked by a triple phospho-mimetic mutant (GATA3 3D). Transcriptional activity of GATA3, in contrast, associates with methylation of R261 (Hosokawa et al., 2015). Here, whereas phospho-deficient (GATA3 3A), methylation-mimicking (GATA3 R261F) and methylation-deficient (GATA3 R261K) mutants (Hosokawa et al., 2015; Hosokawa et al., 2016) all repressed Spi1 similarly to GATA3 WT in precommitment, primary pro–T cells, the GATA3 3D mutant caused only minimal Spi1 repression (Fig. 2 D). This suggests that unphosphorylated GATA3 itself is directly involved in the repression mechanism.
Intact Runx1 was also needed to maintain repression, at least in the Scid.adh.2c2 line, a postcommitment pro–T cell that normally does not express Spi1 (Dionne et al., 2005; Hosokawa et al., 2018b). A strongly DNA-binding, truncated form of Runx (Runx1DN) lacking transactivation and repression domains competitively blocks both repression (Telfer et al., 2004) and activation (Hosokawa et al., 2018b) by endogenous Runx factors. Overexpression of full-length Runx1 in Scid.adh.2c2 cells did not alter the fully silent state of Spi1. However, introduction of the Runx1DN mutant caused endogenous Spi1 to become activated, disrupting the established silent state (Fig. 2 E). Thus, the ability of Runx1 to recruit other protein partners via non-DNA-binding domains is crucial for maintenance of Spi1 repression.
To identify the functional components of GATA3 complexes that control PU.1 repression in pro–T stages, we transduced Scid.adh.2c2 cells with Myc- and Flag-tagged GATA3 and recovered GATA3 complexes by two-step affinity purification followed by SDS-PAGE and silver staining (Fig. S1 E). Analysis by liquid chromatography and tandem mass spectrometry identified more than 150 molecules annotated as being involved in transcription or chromatin remodeling (Fig. S1 F and Table S1, sheet 1). While multiple subunits for the NuRD and BRG1-assocated factor (BAF) complexes were repeatedly detected, Runx1 had one of the highest signals in our mass spectrometry analyses, consistently in four separate samples (Table S1, sheet 2). The association between GATA3 and Runx1 was validated by coimmunoprecipitation with immunoblotting. Notably, even the GATA3 3D mutant (Fig. 2 D) was fully able to interact with Runx1, similarly to GATA3 WT and 3A mutant (Fig. S1 G), indicating that altered GATA3 activity itself caused by the 3D mutation accounted for its loss of repressive activity for Spi1.
Stage-specific deletion of Runx1 and Gata3 derepresses Spi1 in committed pro–T cells
To confirm the role of natural levels of Runx1 and GATA3 expression in Spi1 repression, we performed acute, stage-specific Runx1 and Gata3 disruption in primary cells after T lineage commitment using bicistronic retroviral vectors carrying sgRunx1 or sgGATA3 with CFP or hNGFR markers, respectively. These constructs induced acute, specific losses of the targeted proteins as detected by immunoblotting when transduced into Cas9-expressing Scid.adh.2c2 cells (Fig. S2 A). To test their impact in primary cells, BM progenitor cells from Cas9;Bcl2 Tg mice were co-cultured on OP9-DLL1 for 10 d, until nearly all cells had undergone commitment, and then they were transduced with the single guide RNA (sgRNA) against Runx1 and/or Gata3, using control sgRNA transductions for comparison (Fig. 3, A and B; protocol B). 4-dpi, doubly transduced, postcommitment Lin−CD45+CD44loCD25+CFP+hNGFR+ cells were sorted (Fig. S2 B), followed by RT quantitative PCR (RT-qPCR) analysis for Spi1 mRNA (Fig. 3 C). Expression of Spi1, which is normally low in these committed cells, was substantially up-regulated by Runx1 or Gata3 KO, and combined loss of Runx1 and Gata3 up-regulated Spi1 even more (Fig. 3 C). Earlier Runx1 and Gata3 deletion, around the time of T lineage commitment, had a similar effect. The deletion was initiated at day 4 (before T lineage commitment) and harvested on day 8 overall (Fig. 3 D; protocol C), in transition from DN2a to DN2b/DN3 (Fig. S2 C). Again, mRNA levels of Spi1 were clearly higher in Runx1 and/or Gata3 KO cells than in controls. This up-regulation was specific for Spi1, because Bcl11b, a positive target of GATA3 and Runx1 (Kueh et al., 2016), showed the opposite pattern (Fig. 3 E). Thus, normal endogenous Runx1 and GATA3 are important for both establishing and maintaining repression of Spi1 in the T cell lineage commitment transition.

Effects of Runx1 or GATA3 perturbations on Spi1 expression in postcommitment pro–T stages. (A) To test Cas9-mediated disruption, Scid.adh.2c2 cells already expressing Cas9 from a GFP+ vector were transduced with sgRunx1 or control in an hNGFR+ vector or with sgGata3 or control in a CFP+ vector. 3 d after sgRNA transduction, lysates from the retrovirus-infected Cas9-GFP+CFP+ or Cas9-GFP+hNGFR+ Scid.adh.2c2 cells were subjected to immunoblotting (IB) for Runx1 (top) and GATA3 (bottom), respectively. Two independent experiments were performed with similar results. (B) Flow cytometric analyses of retrovirus-infected Lin−CD45+CFP+hNGFR+ postcommitment primary cells were performed at 4 d after transduction using protocol B. Gating strategies for Lin−CD45+GFP+hNGFR+ cells are shown (top). Representative profiles of CD44/CD25 in Lin−CD45+GFP+hNGFR+ cells are shown (bottom). Gates to define CD44lo cells for sorting are labeled with red rectangles. Results are representative of three independent experiments. (C) Lin−CD45+CFP+hNGFR+ cells that had been transduced with sgRNAs before commitment were subjected to flow cytometric analysis at 4 d after transduction using protocol C. Gating strategies for Lin−CD45+GFP+hNGFR+ cells are shown (top). Representative profiles of CD44/CD25 in Lin−CD45+GFP+hNGFR+ cells are shown (bottom). CD25+ cells for sorting were labeled with red rectangles. The percentages of CD25+CD44lo cells are indicated with SD. **, P < 0.01 by two-sided Student’s t test for the indicated sample pairs. Data are representative of two independent experiments and based on three biological replicates in an experiment.
Effects of Runx1 or GATA3 perturbations on Spi1 expression in postcommitment pro–T stages. (A) To test Cas9-mediated disruption, Scid.adh.2c2 cells already expressing Cas9 from a GFP+ vector were transduced with sgRunx1 or control in an hNGFR+ vector or with sgGata3 or control in a CFP+ vector. 3 d after sgRNA transduction, lysates from the retrovirus-infected Cas9-GFP+CFP+ or Cas9-GFP+hNGFR+ Scid.adh.2c2 cells were subjected to immunoblotting (IB) for Runx1 (top) and GATA3 (bottom), respectively. Two independent experiments were performed with similar results. (B) Flow cytometric analyses of retrovirus-infected Lin−CD45+CFP+hNGFR+ postcommitment primary cells were performed at 4 d after transduction using protocol B. Gating strategies for Lin−CD45+GFP+hNGFR+ cells are shown (top). Representative profiles of CD44/CD25 in Lin−CD45+GFP+hNGFR+ cells are shown (bottom). Gates to define CD44lo cells for sorting are labeled with red rectangles. Results are representative of three independent experiments. (C) Lin−CD45+CFP+hNGFR+ cells that had been transduced with sgRNAs before commitment were subjected to flow cytometric analysis at 4 d after transduction using protocol C. Gating strategies for Lin−CD45+GFP+hNGFR+ cells are shown (top). Representative profiles of CD44/CD25 in Lin−CD45+GFP+hNGFR+ cells are shown (bottom). CD25+ cells for sorting were labeled with red rectangles. The percentages of CD25+CD44lo cells are indicated with SD. **, P < 0.01 by two-sided Student’s t test for the indicated sample pairs. Data are representative of two independent experiments and based on three biological replicates in an experiment.

Deletion of Runx1 and Gata3 induces derepression of Spi1 expression in T lineage–committed cells. (A) BM progenitors obtained from Cas9;Bcl2 Tg mice were co-cultured on OP9-DLL1 for 10 d. Postcommitment cells were retrovirally introduced sgRNAs against Runx1 and/or Gata3 and then analyzed 4 dpi (protocol B). (B) Flow cytometric analysis of BM-derived pro–T cells was performed on day 10, before sgRNA introduction. A representative CD44/CD25 profile in Lin−CD45+ cells is shown. The result is representative of three independent experiments. (C) sgRNA-introduced Lin−CD45+CD25+CD44loCFP+hNGFR+ postcommitment cells were sorted and subjected to RT-qPCR analysis at 4 d after sgRNA introduction. The relative expression (/Actb) of Spi1 is shown with SD. **, P < 0.01 by two-sided Student’s t test. Data are representative of two independent experiments and based on three biological replicates. (D) BM progenitors were co-cultured on OP9-DLL1 for 4 d. Precommitment cells were retrovirally introduced sgRNAs against Runx1 and/or Gata3 and then analyzed 4 dpi (protocol C). (E) sgRNA-introduced Lin−CD45+CFP+hNGFR+ cells (D) were sorted and subjected to RT-qPCR analysis. The relative expression (/Actb) of Spi1 and Bcl11b is shown with SD. **, P < 0.01 by two-sided Student’s t test. Data are representative of two independent experiments and based on three biological replicates.
Deletion of Runx1 and Gata3 induces derepression of Spi1 expression in T lineage–committed cells. (A) BM progenitors obtained from Cas9;Bcl2 Tg mice were co-cultured on OP9-DLL1 for 10 d. Postcommitment cells were retrovirally introduced sgRNAs against Runx1 and/or Gata3 and then analyzed 4 dpi (protocol B). (B) Flow cytometric analysis of BM-derived pro–T cells was performed on day 10, before sgRNA introduction. A representative CD44/CD25 profile in Lin−CD45+ cells is shown. The result is representative of three independent experiments. (C) sgRNA-introduced Lin−CD45+CD25+CD44loCFP+hNGFR+ postcommitment cells were sorted and subjected to RT-qPCR analysis at 4 d after sgRNA introduction. The relative expression (/Actb) of Spi1 is shown with SD. **, P < 0.01 by two-sided Student’s t test. Data are representative of two independent experiments and based on three biological replicates. (D) BM progenitors were co-cultured on OP9-DLL1 for 4 d. Precommitment cells were retrovirally introduced sgRNAs against Runx1 and/or Gata3 and then analyzed 4 dpi (protocol C). (E) sgRNA-introduced Lin−CD45+CFP+hNGFR+ cells (D) were sorted and subjected to RT-qPCR analysis. The relative expression (/Actb) of Spi1 and Bcl11b is shown with SD. **, P < 0.01 by two-sided Student’s t test. Data are representative of two independent experiments and based on three biological replicates.
DN2b/3 stage-specific binding of Runx1 and GATA3 at the intronic region of the Spi1 locus
Multiple cis-regulatory elements have been described around the Spi1 locus, especially evolutionarily conserved open chromatin regions, that reportedly control levels of Spi1 expression in various hematopoietic cell types, as discussed in the Introduction. In an unbiased search for genomic sites that could mediate active imposition of repression during commitment, we examined Runx1 and GATA3 binding around Spi1 by chromatin immunoprecipitation (ChIP) and deep sequencing (ChIP-seq). We compared their occupancy patterns before (DN1) and after (DN2b/3) T lineage commitment (Hosokawa et al., 2018b; Fig. 4 A) and compared these with sites changing chromatin accessibility (Yoshida et al., 2019) and looping interactions (Hu et al., 2018) around Spi1 in pre- and postcommitment pro–T cells (Fig. S3, A and B). Despite the strong impact of endogenous GATA3 on Spi1, conventional ChIP-seq conditions using formaldehyde (FA) crosslinking showed little, if any, GATA3 binding near this locus (Fig. S3 A). However, using a di(N-succinimidyl) glutarate (DSG) cross-linker that stabilizes Runx1-containing complexes (Hosokawa et al., 2020; Hosokawa et al., 2018b), GATA3 recruitment was clear (Fig. 4 A; see Materials and methods). Runx1 and GATA3 were found at URE, CE4 (Huang et al., 2008; Zarnegar et al., 2010), and the transcriptional start site of the neighboring Slc39a13 gene. However, none of these peaks appeared after commitment (Fig. 4 A, blue rectangles). A site at −17.8 kb previously reported to mediate repression by GATA1 in erythroid cells (Chou et al., 2009) showed slight binding, but only in DN1 cells, not DN2b/3 cells.

Identification of the DN2b/3-specific Runx1 and GATA3 binding site at the Spi1 locus. (A) BM progenitors obtained from B6 mice were co-cultured on OP9-DLL1 for 5 or 14 d. Lin−CD45+CD25−Kithi+CD44+ DN1 cells and Lin−CD45+CD25+KitloCD44lo DN2b/3 cells were sorted on days 5 and 14, respectively, and subjected to ChIP-seq analysis for Runx1 (Hosokawa et al., 2018b) and GATA3. Representative ChIP-seq tracks in DN1 and DN2b/3 cells around the Spi1 locus are shown with the conservation track. Previously reported cis-regulatory elements and a DN2b/3-specific Runx1 and GATA3 binding site are labeled with blue and magenta rectangles, respectively. Data are representative of two independent experiments. Chr2, chromosome 2. (B) Representative ATAC-sequencing tracks in DN1, DN2a, DN2b and DN3 cells around the Spi1 locus are shown (Yoshida et al., 2019). (C) The short interspersed nuclear element (SINE) and LINE element tracks around the Spi1 locus in mouse (mm10) are shown with the conservation track. A position for the DN2b/3-specific Runx1 and GATA3 binding site is labeled with a magenta rectangle.
Identification of the DN2b/3-specific Runx1 and GATA3 binding site at the Spi1 locus. (A) BM progenitors obtained from B6 mice were co-cultured on OP9-DLL1 for 5 or 14 d. Lin−CD45+CD25−Kithi+CD44+ DN1 cells and Lin−CD45+CD25+KitloCD44lo DN2b/3 cells were sorted on days 5 and 14, respectively, and subjected to ChIP-seq analysis for Runx1 (Hosokawa et al., 2018b) and GATA3. Representative ChIP-seq tracks in DN1 and DN2b/3 cells around the Spi1 locus are shown with the conservation track. Previously reported cis-regulatory elements and a DN2b/3-specific Runx1 and GATA3 binding site are labeled with blue and magenta rectangles, respectively. Data are representative of two independent experiments. Chr2, chromosome 2. (B) Representative ATAC-sequencing tracks in DN1, DN2a, DN2b and DN3 cells around the Spi1 locus are shown (Yoshida et al., 2019). (C) The short interspersed nuclear element (SINE) and LINE element tracks around the Spi1 locus in mouse (mm10) are shown with the conservation track. A position for the DN2b/3-specific Runx1 and GATA3 binding site is labeled with a magenta rectangle.

GATA3 and Runx1 modestly bind to the intronic region of the Spi1 locus in DN2b/3 and Scid.adh.2c2 cells. (A) Sensitivity of GATA3 DNA-binding pattern to ChIP-seq cross-linking conditions. FA+DSG-cross-linked DN1 and DN2b/3 cells and FA cross-linked DN2b/3 cells were subjected to ChIP-seq analysis for GATA3. ChIP-seq tracks around the Spi1 locus are shown with representative ATAC-sequencing tracks in long-term hematopoietic stem cells (LT-HSC), short-term (ST-) HSC, DN1, DN2a, DN2b, DN3, and double-positive (DP) cells (Yoshida et al., 2019) and the conservation track. The Spi1 locus is highlighted in light blue. (B) Changes in higher-order chromatin looping around the Spi1 locus before and after pro–T cell commitment, from published Hi-C results of (Hu et al., 2018). Map positions are in mm9 coordinates (top) and therefore show some offset from mm10 coordinates in other figure panels. Gene models are at bottom (RefSeq genes). Tracks for A/B compartment flip and change in domain score are empty due to lack of changes in this genomic region. Active transcription units are indicated by “RNASeq DN2” track, and strongly open chromatin is shown by DNase sequencing from DN2 cells in the study. Arc plots pool data from hematopoietic stem cells to DN2 cells as “precommitment” due to the constancy of their patterns, and data from DN3 to double positive as “postcommitment”. Arcs shown are those that connect any promoters to any DNase-sensitive region. Data display was downloaded from the Washington University St. Louis epigenome browser (http://epigenomegateway.wustl.edu/browser/?genome=mm9&session=bxT0F5m0YY). New arcs appearing during commitment that involve the Spi1 locus are indicated by arrows, and their anchor points in the Spi1 locus or the Spi1-Slc39a13 intergenic region (site of the URE) are indicated by blue stars. Dark blue star indicates the intron 2 anchor point. (C) DN1, DN2b/3, and Scid.adh.2c2 cells were subjected to ChIP-seq analysis for Runx1 and GATA3 (GSE103953 and GSE93755; Hosokawa et al., 2018b). ChIP-seq tracks around the Spi1 locus are shown. Previously reported cis-regulatory elements and a DN2b/3-specific Runx1 and GATA3 binding site are labeled with blue and magenta rectangles, respectively.
GATA3 and Runx1 modestly bind to the intronic region of the Spi1 locus in DN2b/3 and Scid.adh.2c2 cells. (A) Sensitivity of GATA3 DNA-binding pattern to ChIP-seq cross-linking conditions. FA+DSG-cross-linked DN1 and DN2b/3 cells and FA cross-linked DN2b/3 cells were subjected to ChIP-seq analysis for GATA3. ChIP-seq tracks around the Spi1 locus are shown with representative ATAC-sequencing tracks in long-term hematopoietic stem cells (LT-HSC), short-term (ST-) HSC, DN1, DN2a, DN2b, DN3, and double-positive (DP) cells (Yoshida et al., 2019) and the conservation track. The Spi1 locus is highlighted in light blue. (B) Changes in higher-order chromatin looping around the Spi1 locus before and after pro–T cell commitment, from published Hi-C results of (Hu et al., 2018). Map positions are in mm9 coordinates (top) and therefore show some offset from mm10 coordinates in other figure panels. Gene models are at bottom (RefSeq genes). Tracks for A/B compartment flip and change in domain score are empty due to lack of changes in this genomic region. Active transcription units are indicated by “RNASeq DN2” track, and strongly open chromatin is shown by DNase sequencing from DN2 cells in the study. Arc plots pool data from hematopoietic stem cells to DN2 cells as “precommitment” due to the constancy of their patterns, and data from DN3 to double positive as “postcommitment”. Arcs shown are those that connect any promoters to any DNase-sensitive region. Data display was downloaded from the Washington University St. Louis epigenome browser (http://epigenomegateway.wustl.edu/browser/?genome=mm9&session=bxT0F5m0YY). New arcs appearing during commitment that involve the Spi1 locus are indicated by arrows, and their anchor points in the Spi1 locus or the Spi1-Slc39a13 intergenic region (site of the URE) are indicated by blue stars. Dark blue star indicates the intron 2 anchor point. (C) DN1, DN2b/3, and Scid.adh.2c2 cells were subjected to ChIP-seq analysis for Runx1 and GATA3 (GSE103953 and GSE93755; Hosokawa et al., 2018b). ChIP-seq tracks around the Spi1 locus are shown. Previously reported cis-regulatory elements and a DN2b/3-specific Runx1 and GATA3 binding site are labeled with blue and magenta rectangles, respectively.
Notably, however, we saw a small but reproducible peak of Runx1 and GATA3 association at +3.7 kb within intron 2 of Spi1, and these signals were highly DN2b/3-stage specific (Fig. 4 A, magenta rectangle). Runx1 and GATA3 were also detected here in Scid.adh.2c2 cells (Fig. S3 C). Despite the modest signals, this site was the only one in the gene-to-gene interval containing Spi1 (Slc39a13-Mybpc3) that showed DN2b/DN3-specific occupancy by Runx1 or GATA3. The sequence of this region included multiple Runx motifs and a GATA motif (see below). Runx1, at least, appeared to bind to this site independently of GATA3, as shown by acute knockouts and ChIP-qPCR assays (Fig. S4 A).

Potential silencer activity of the DN2b/3-specific looping site around the Spi1 locus. (A) Test for interdependence of GATA3 and Runx1 binding to candidate Spi1 regulatory elements. sgRNA-introduced Lin−CD45+hNGFR+CFP+ postcommitment cells were purified using protocol B, and then the binding of Runx1 and GATA3 at the previously reported cis-regulatory elements and the DN2b/3-specific Runx1 and GATA3 binding site around the Spi1 locus were determined by ChIP assay with qPCR analysis. The mean values (percent input) are shown with SD. Data are based on two independent experiments. UTR, untranslated region. (B) Representative ChIP-seq tracks in DN1 and DN2b/3 cells and ATAC-sequencing tracks in DN1, DN2a, DN2b, and DN3 cells are shown with the conservation track over an extended region around the Spi1 locus. Runx1 and GATA3 binding sites around the DN2b/3-specific looping site (+145, +167, and +211 kb) are labeled with magenta rectangles. Data are representative of two independent experiments. (C) Efficient disruption of candidate distal regulatory elements. Cas9-introduced Scid.adh.2c2 cells were infected with sgRNAs against two sides of the targeted genomic regions (+145-, +167-, and +211-kb sites in B). GFP+CFP+hNGFR+ cells were subjected to single-cell sorting and then expanded for 2 wk, as in Fig. 5 A. Genomic DNA from each clone was isolated and subjected to qPCR analysis to confirm deletion of the targeted genomic regions. The relative intensity (/Gapdh promoter) is shown with SD. Data are based on five independent clones from each sgRNA transduction. (D) Relative expression levels of Spi1, against Actb, are shown with SD. Circles indicate independent clones. **, P < 0.01 by two-sided Student’s t test. Data are based on five independent clones.
Potential silencer activity of the DN2b/3-specific looping site around the Spi1 locus. (A) Test for interdependence of GATA3 and Runx1 binding to candidate Spi1 regulatory elements. sgRNA-introduced Lin−CD45+hNGFR+CFP+ postcommitment cells were purified using protocol B, and then the binding of Runx1 and GATA3 at the previously reported cis-regulatory elements and the DN2b/3-specific Runx1 and GATA3 binding site around the Spi1 locus were determined by ChIP assay with qPCR analysis. The mean values (percent input) are shown with SD. Data are based on two independent experiments. UTR, untranslated region. (B) Representative ChIP-seq tracks in DN1 and DN2b/3 cells and ATAC-sequencing tracks in DN1, DN2a, DN2b, and DN3 cells are shown with the conservation track over an extended region around the Spi1 locus. Runx1 and GATA3 binding sites around the DN2b/3-specific looping site (+145, +167, and +211 kb) are labeled with magenta rectangles. Data are representative of two independent experiments. (C) Efficient disruption of candidate distal regulatory elements. Cas9-introduced Scid.adh.2c2 cells were infected with sgRNAs against two sides of the targeted genomic regions (+145-, +167-, and +211-kb sites in B). GFP+CFP+hNGFR+ cells were subjected to single-cell sorting and then expanded for 2 wk, as in Fig. 5 A. Genomic DNA from each clone was isolated and subjected to qPCR analysis to confirm deletion of the targeted genomic regions. The relative intensity (/Gapdh promoter) is shown with SD. Data are based on five independent clones from each sgRNA transduction. (D) Relative expression levels of Spi1, against Actb, are shown with SD. Circles indicate independent clones. **, P < 0.01 by two-sided Student’s t test. Data are based on five independent clones.
Interestingly, this site would not have been identified by assay of transposase-accessible chromatin (ATAC) accessibility or conservation. The URE and CE4 regions are ATAC accessible in DN1 stage and gradually close during pro–T progression, and a neighboring intron 2 site (at +5.0 kb) appears constitutively open. But the +3.7-kb site of the Spi1 locus appears “closed” both before and after commitment (Yoshida et al., 2019; Fig. 4 B, magenta rectangle). This region also falls within a long interspersed nuclear element (LINE element; Fig. 4 C, magenta rectangle). Still, this site was close to the intron 2 anchor for a chromatin looping interaction that appears newly during commitment (Fig. S3 B, dark blue star).
Potential silencer activity of the DN2b/3-specific Runx1 and GATA3 binding site in the Spi1 locus
To test whether the DN2b/3-specific Runx1 and GATA3 binding site at +3.7 kb of the Spi1 locus could actually mediate Runx1- and GATA3-dependent silencer activity, we designed sgRNAs to disrupt the DN2b/3-specific Runx1 and GATA3 occupancy region and tested their impact first in the Scid.adh.2c2 pro–T cell line. Scid.adh.2c2 cells expressing Cas9-GFP were retrovirally transduced in parallel with pairs of sgRNAs flanking each of four different Runx1 and GATA3 binding regions around the Spi1 locus. Besides the +3.7-kb region, we targeted the URE, CE4, and the neighboring Slc39a13 promoter (Slc39a13 pro.). Cas9+ doubly transduced (GFP+ CFP+ hNGFR+) cells were cloned by single-cell sorting (Fig. 5 A), and we identified clones with complete loss of the targeted genomic regions by genomic qPCR analysis (Fig. 5 B). Deletion of 567 bp of the +3.7-kb site (sgInt.2) induced markedly up-regulated Spi1 expression in Scid.adh.2c2 cells (Fig. 5 C). In contrast, expression levels of Spi1 were comparable among cells transduced with sgControl (sgRNA against luciferase), sgSlc39a13 promoter, and sgURE, whereas disruption of CE4 (sgCE4) led to only a slight increase in Spi1 expression (Fig. 5 C). Therefore, in this DN3-like cell line, the DN2b/3-specific Runx1 and GATA3 binding site at +3.7 kb of the Spi1 locus was required to maintain repression of Spi1.

Activation of Spi1 is induced by deletion of the DN2b/3-specific Runx1 and GATA3 binding site in a DN3-like cell line. (A) A DN3-like cell line, Scid.adh.2c2, was used for CRISPR-Cas9–mediated deletion of the specific genomic regions around Spi1 locus. First, Scid.adh.2c2 cells were introduced Cas9-GFP, and then they were infected second retrovirus infection for sgRNAs against two sides of the targeted genomic regions. GFP+CFP+hNGFR+ cells were subjected to single-cell sorting and expanded for 2 wk. (B) Genomic DNA from each clone was isolated and subjected to qPCR analysis to confirm deletion of the targeted genomic regions. The relative intensity (/Gapdh promoter) is shown with SD. Data are based on three independent clones. (C) Relative expression levels of Spi1 against Actb are shown with SD. Circles indicate independent clones. **, P < 0.01; *, P < 0.05 by two-sided Student’s t test. Data are based on more than five independent clones.
Activation of Spi1 is induced by deletion of the DN2b/3-specific Runx1 and GATA3 binding site in a DN3-like cell line. (A) A DN3-like cell line, Scid.adh.2c2, was used for CRISPR-Cas9–mediated deletion of the specific genomic regions around Spi1 locus. First, Scid.adh.2c2 cells were introduced Cas9-GFP, and then they were infected second retrovirus infection for sgRNAs against two sides of the targeted genomic regions. GFP+CFP+hNGFR+ cells were subjected to single-cell sorting and expanded for 2 wk. (B) Genomic DNA from each clone was isolated and subjected to qPCR analysis to confirm deletion of the targeted genomic regions. The relative intensity (/Gapdh promoter) is shown with SD. Data are based on three independent clones. (C) Relative expression levels of Spi1 against Actb are shown with SD. Circles indicate independent clones. **, P < 0.01; *, P < 0.05 by two-sided Student’s t test. Data are based on more than five independent clones.
Because GATA3 binding to the +3.7-kb element seemed weak, we examined the wider Spi1 locus neighborhood for any additional sites where GATA3 binding increased from DN1 to DN2b/3. Of special interest were potential downstream anchor sites for the commitment-associated looping noted above (Fig. S3 B). Indeed, elements at +145 kb and +211 kb showed higher GATA3 binding signals in DN2b/3 stages than DN1 (Fig. S4 B), and another strong GATA3 peak near the potential loop anchor was seen at +167 kb. All three showed Runx1 and GATA3 co-occupancy in postcommitment cells with various degrees of stage specificity. In direct tests, disruption of the +145-kb genomic region induced weak but reproducible derepression of Spi1 in Scid.adh.2c2 clones (Fig. S4, C and D), similar to CE4 disruption, whereas disruption of the +167-kb and +211-kb elements had no effect. Therefore, Runx1 and GATA3 may be involved in establishment of stage-specific repressive chromatin looping architectures around the Spi1 locus, including the +3.7-kb, +145-kb, and CE4 elements.
Roles of Runx and GATA motifs in the DN2b/3-specific Runx1 and GATA3 binding site in PU.1 repression in primary pro–T cells
In primary pro–T cells, clones could not be grown out and selected for deletion of the complete element, but a Cas9-mediated point mutation strategy could be highly effective to test the role of the +3.7-kb element. There are three Runx motifs and one GATA motif around the center of the +3.7-kb element (Figs. 6 A and S5 A). Thus, we induced direct mutations in these Runx and GATA motifs at the +3.7-kb element in primary Cas9;Bcl2 Tg pro–T cells. Cells were cultured and transduced according to protocol B using sgRNAs targeting each motif (Fig. 6 A, red arrowheads) and analyzing the postcommitment, multiply transduced cells at day 14 overall (Fig. S5 B). Disruption of individual motifs had significant but minor effects on Spi1 expression. In contrast, transduction of the mixture of sgRNAs against all four motifs induced prominent derepression of Spi1 (Fig. 6 B), albeit not to full precommitment expression levels. PU.1 protein levels in individual cells were up-regulated throughout the population when all four motifs were disrupted (Fig. 6 C), while the cells introduced with the sgRNA mixture had individually undergone different combinations of point mutations or small deletions (Fig. S5 C).

Stage-specific derepression of Spi1 is induced by mutations on the Runx and GATA motifs in the Spi1 +3.7-kb element. (A) The sequence of the DN2b/3-specific Runx1 and GATA3 binding site at intron 2 of the Spi1 gene (chromosome 2; 91,100,260–91,100,519) is indicated. Runx motifs and a GATA motif are labeled with green and blue boxes, respectively. Red arrowheads indicate targeted sites for CRISPR-Cas9–mediated direct mutations. (B) sgRNA-introduced Lin−CD45+CD25+CD44loCFP+hNGFR+ postcommitment cells were sorted and subjected to RT-qPCR analysis at 4 d after sgRNA introduction (protocol B). The relative expression (/Actb) of Spi1 is shown with SD. **, P < 0.01; *, P < 0.05 by two-sided Student’s t test. Data are representative of two independent experiments and based on more than five biological replicates. (C) sgControl- or sg4 motifs mix–introduced Lin−CD45+CD25+CD44loCFP+hNGFR+ postcommitment cells were subjected to intracellular staining for PU.1, at 4 d after sgRNA introduction (protocol B). Representative profiles for PU.1 are shown with isotype control staining (gray). Results are representative of four independent experiments. (D) BM progenitors obtained from Cas9;Bcl2 Tg mice were co-cultured on OP9-DLL1 for 2 d. Precommitment cells were retrovirally introduced with sgRNAs and then analyzed 6 (DN2a/2b stages) or 12 (DN2b/3 stages) dpi (protocol D). For cell surface phenotypes, see Fig. S5 D. (E) sgRNA-introduced Lin−CD45+CD44+CFP+hNGFR+ DN2a/2b cells (left) and Lin−CD45+CD25+CD44loCFP+hNGFR+ DN2b/3 cells (right) were sorted on 6 or 12 d after sgRNA introduction, respectively, and subjected to RT-qPCR analysis. The relative expression (/Actb) of Spi1 is shown with SD. **, P < 0.01 by two-sided Student’s t test. Data are representative of two independent experiments and based on three biological replicates.
Stage-specific derepression of Spi1 is induced by mutations on the Runx and GATA motifs in the Spi1 +3.7-kb element. (A) The sequence of the DN2b/3-specific Runx1 and GATA3 binding site at intron 2 of the Spi1 gene (chromosome 2; 91,100,260–91,100,519) is indicated. Runx motifs and a GATA motif are labeled with green and blue boxes, respectively. Red arrowheads indicate targeted sites for CRISPR-Cas9–mediated direct mutations. (B) sgRNA-introduced Lin−CD45+CD25+CD44loCFP+hNGFR+ postcommitment cells were sorted and subjected to RT-qPCR analysis at 4 d after sgRNA introduction (protocol B). The relative expression (/Actb) of Spi1 is shown with SD. **, P < 0.01; *, P < 0.05 by two-sided Student’s t test. Data are representative of two independent experiments and based on more than five biological replicates. (C) sgControl- or sg4 motifs mix–introduced Lin−CD45+CD25+CD44loCFP+hNGFR+ postcommitment cells were subjected to intracellular staining for PU.1, at 4 d after sgRNA introduction (protocol B). Representative profiles for PU.1 are shown with isotype control staining (gray). Results are representative of four independent experiments. (D) BM progenitors obtained from Cas9;Bcl2 Tg mice were co-cultured on OP9-DLL1 for 2 d. Precommitment cells were retrovirally introduced with sgRNAs and then analyzed 6 (DN2a/2b stages) or 12 (DN2b/3 stages) dpi (protocol D). For cell surface phenotypes, see Fig. S5 D. (E) sgRNA-introduced Lin−CD45+CD44+CFP+hNGFR+ DN2a/2b cells (left) and Lin−CD45+CD25+CD44loCFP+hNGFR+ DN2b/3 cells (right) were sorted on 6 or 12 d after sgRNA introduction, respectively, and subjected to RT-qPCR analysis. The relative expression (/Actb) of Spi1 is shown with SD. **, P < 0.01 by two-sided Student’s t test. Data are representative of two independent experiments and based on three biological replicates.

Effects of mutations of the Runx and GATA motifs in the Spi1 +3.7-kb element in primary pro–T cells. (A) Runx1 (top) and GATA3 (bottom) motif sequence logos from JASPAR reference motif are shown. (B) Flow cytometric analyses of sgRNA-introduced Lin−CD45+CFP+hNGFR+ cells were performed as protocol B. Representative CD44/CD25 profiles are shown, with magenta rectangles indicating sorting gates. Results are representative of three independent experiments. (C) Mutation types induced in the +3.7-kb element by Cas9-mediated disruption using the sg4 motifs mix. Genomic DNA samples from the sgControl (sgCont.) or sg4 motifs mix–introduced Lin−CD45+CFP+hNGFR+ cells (B) were subjected to PCR analysis using FW and RV primers (indicated with arrows). The gel of PCR amplicons shows that different cells in the population experienced different extents of deletions. Each band was TA-cloned and sequenced and all were found to be altered from WT. Summary of mutations found in cells transduced with the sg4 motifs mix is shown. (D) Flow cytometric analyses of sgRNA-introduced Lin−CD45+CFP+hNGFR+ cells were performed using protocol D. Representative CD44/CD25 profiles 6 (left) or 12 (right) d after sgRNA infection are shown. CD25+ (6 d) and CD44lo (12 d) cells for sorting were labeled with red rectangles. Results are representative of three independent experiments. (E) sgRNA-introduced Lin−CD45+CD44+CFP+hNGFR+ DN2a/2b cells and Lin−CD45+CD25+CD44loCFP+hNGFR+ DN2b/3 cells were subjected to RT-qPCR analysis as protocol D. The relative expression levels (/Actb) of Spi1 are shown with SD. **, P < 0.01 by two-sided Student’s t test. Data are representative of two independent experiments and based on three biological replicates within an experiment. (F) Characterization of the GATA3 ChIP-seq peaks detected under different conditions of cross-linking. Percentages of GATA3 ChIP peaks from FA or DSG+FA (see Materials and methods) cross-linked DN2b/3 cells on promoters, ATAC-open regions, and repeat sequence elements and P values for enrichment of GATA motif are shown.
Effects of mutations of the Runx and GATA motifs in the Spi1 +3.7-kb element in primary pro–T cells. (A) Runx1 (top) and GATA3 (bottom) motif sequence logos from JASPAR reference motif are shown. (B) Flow cytometric analyses of sgRNA-introduced Lin−CD45+CFP+hNGFR+ cells were performed as protocol B. Representative CD44/CD25 profiles are shown, with magenta rectangles indicating sorting gates. Results are representative of three independent experiments. (C) Mutation types induced in the +3.7-kb element by Cas9-mediated disruption using the sg4 motifs mix. Genomic DNA samples from the sgControl (sgCont.) or sg4 motifs mix–introduced Lin−CD45+CFP+hNGFR+ cells (B) were subjected to PCR analysis using FW and RV primers (indicated with arrows). The gel of PCR amplicons shows that different cells in the population experienced different extents of deletions. Each band was TA-cloned and sequenced and all were found to be altered from WT. Summary of mutations found in cells transduced with the sg4 motifs mix is shown. (D) Flow cytometric analyses of sgRNA-introduced Lin−CD45+CFP+hNGFR+ cells were performed using protocol D. Representative CD44/CD25 profiles 6 (left) or 12 (right) d after sgRNA infection are shown. CD25+ (6 d) and CD44lo (12 d) cells for sorting were labeled with red rectangles. Results are representative of three independent experiments. (E) sgRNA-introduced Lin−CD45+CD44+CFP+hNGFR+ DN2a/2b cells and Lin−CD45+CD25+CD44loCFP+hNGFR+ DN2b/3 cells were subjected to RT-qPCR analysis as protocol D. The relative expression levels (/Actb) of Spi1 are shown with SD. **, P < 0.01 by two-sided Student’s t test. Data are representative of two independent experiments and based on three biological replicates within an experiment. (F) Characterization of the GATA3 ChIP-seq peaks detected under different conditions of cross-linking. Percentages of GATA3 ChIP peaks from FA or DSG+FA (see Materials and methods) cross-linked DN2b/3 cells on promoters, ATAC-open regions, and repeat sequence elements and P values for enrichment of GATA motif are shown.
Next, we tested whether the activity mediated by the +3.7-kb element is generally damping or stage specific. We disrupted all four Runx and GATA motifs early in the culture and then compared effects on Spi1 levels expressed in precommitment stages (6 d) and later in postcommitment stages (12 d; Fig. 6 D; protocol D). Disruption with the four sgRNAs did not affect progression of DN stages, defined by CD44 and CD25 expression, and Spi1 showed some developmental down-regulation in commitment in both experimental samples and controls (Fig. S5, D and E). However, the high expression of Spi1 at 6 dpi (mostly DN2a/2b) was very similar in control and experimental samples, whereas by postcommitment stages at 12 dpi (DN2b/3, day 14 of overall culture), the four sgRNAs substantially alleviated Spi1 repression compared with controls (Fig. 6 E). Thus, the intronic site of postcommitment stage-specific Runx1 and GATA3 binding was needed to strengthen or maintain repression of Spi1 expression, stage specifically, after commitment.
Finally, to determine whether the +3.7-kb site was required to mediate repressive action of Runx1 or GATA3 on Spi1, we disrupted the four motifs in precommitment pro–T cells and then transduced the cells with Runx1 or Gata3 (Fig. 7 A; protocol E). Runx1- and GATA3-mediated repression of Spi1 expression was significantly weakened in the cells transduced with the mixed sg4 motifs compared with controls (Fig. 7 B). Although introduction of Gata3 was still able to cause some repression of Spi1, the attenuation of this effect via a single intronic element was notable considering not only the intact +145-kb and CE4 elements but also the multiple potent gene network impacts of Runx1 and GATA3 on other regulators in the cells (Scripture-Adams et al., 2014; Shin et al., 2021; Fig. 7 B). Taken together, postcommitment stage-specific binding of Runx1 and GATA3 to the +3.7-kb element of the Spi1 locus may play a substantial role in timing of Spi1 repression during early T cell development.

Runx1 and GATA3 repress Spi1 expression partly via the +3.7-kb element of the Spi1 locus. (A) BM progenitors obtained from Cas9;Bcl2 Tg mice were co-cultured on OP9-DLL1. Precommitment cells were retrovirally introduced with sg4 motifs mix on day 2, then with Runx1 or Gata3 on day 3. Retrovirus-transduced cells were analyzed 3 d after the last infection (protocol E). (B) Retrovirus-transduced Lin− CD45+ CD44+ CFP+ hNGFR+ GFP+ cells were sorted on 3 d after introduction and subjected to RT-qPCR analysis as protocol E. The relative expression (/Actb) of Spi1 is shown with SD. **, P < 0.01; *, P < 0.05 by two-sided Student’s t test. Data are representative of two independent experiments and based on three biological replicates.
Runx1 and GATA3 repress Spi1 expression partly via the +3.7-kb element of the Spi1 locus. (A) BM progenitors obtained from Cas9;Bcl2 Tg mice were co-cultured on OP9-DLL1. Precommitment cells were retrovirally introduced with sg4 motifs mix on day 2, then with Runx1 or Gata3 on day 3. Retrovirus-transduced cells were analyzed 3 d after the last infection (protocol E). (B) Retrovirus-transduced Lin− CD45+ CD44+ CFP+ hNGFR+ GFP+ cells were sorted on 3 d after introduction and subjected to RT-qPCR analysis as protocol E. The relative expression (/Actb) of Spi1 is shown with SD. **, P < 0.01; *, P < 0.05 by two-sided Student’s t test. Data are representative of two independent experiments and based on three biological replicates.
Discussion
PU.1 has an indispensable role for the earliest T lineage precursors in the thymus (Champhekar et al., 2015; Dakic et al., 2005), but its expression must be shut off during T lineage commitment, at the transition from DN2a to DN2b, and both the mechanism and coordination of this timing have needed explanation. Despite evidence that Runx1 and GATA3 play roles in the repression of Spi1 in pro–T cells, possibly via the URE and other cis-regulatory elements around the Spi1 locus (Huang et al., 2008; Rosenbauer et al., 2006; Scripture-Adams et al., 2014; Zarnegar et al., 2010), both Runx1 and GATA3 are expressed for many cell divisions before Spi1 is silenced. Similarly, the chromatin accessibility and transcription factor binding dynamics at these previously reported genomic elements have failed to explain how Spi1 expression is repressed specifically during pro–T cell lineage commitment. This problem motivated our search for new Runx1–GATA3–Spi1 interactions that could mediate this stage-specific functional change. Here, we exploited CRISPR-Cas9–mediated acute, stage-specific deletions of Runx1, Gata3, and/or potential cis-regulatory elements in an in vitro T cell development culture system. We confirmed that precommitment overexpression of Runx1 and GATA3 markedly accelerated Spi1 repression, whereas stage-specific deletion of Runx1 and Gata3 derepressed or significantly weakened repression of Spi1. Using stage-specific Runx1 and GATA3 binding as a search criterion, we identified a novel element in the second intron of Spi1, not described before, which was shown to mediate stage-specific silencer activity dependent on the integrity of Runx1 and GATA3 binding sites. While GATA3 and Runx1 binding to the previously described CE4 element (Zarnegar et al., 2010) and a far downstream element at +145 kb may also contribute, their effects were weaker in a cell line model. Disruption of the +3.7-kb element alone was sufficient to give Spi1 at least 50% protection from the repressive effects of overexpressed GATA3 or Runx1. Thus, the repression of Spi1 by Runx1 and GATA3 is controlled at least in part via their stage-specific binding to this Spi1 intronic site.
The striking feature of Runx1 and GATA3 action in repression of Spi1 has been the stage-specific functional response occurring with seemingly little change in levels of the two candidate repressive factors. However, both Runx factors and GATA3 have been noted for the shifting of their genomic binding site choices from one stage to another in pro–T cells (Hosokawa et al., 2021; Hosokawa et al., 2018b; Shin et al., 2021; Zhang et al., 2012). Runx1 protein expression levels increase little between precommitment DN2a and postcommitment DN2b stages (Shin et al., 2021), and recent data show that Runx3 actually collaborates with Runx1 to repress Spi1 during commitment, with their combined activities virtually constant before and after Spi1 repression begins (Shin et al., 2021). However, both factors show the same stage-specific increased binding to the +3.7-kb site during commitment (Shin et al., 2021). GATA3 protein levels do increase slightly during commitment, in a way well coordinated with the change in expression of PU.1, but seemingly by less than threefold. Thus, despite little absolute change in total Runx and GATA3, their repression of Spi1 correlates with their occupancy of the +3.7-kb site, possibly connected with a new chromatin loop anchored in intron 2. Recruitment could depend on additional partners not yet defined.
Deletion of Runx1 and Gata3 before T lineage commitment had stronger effects on progression of pro–T cell development and Spi1 repression than deletion of the intronic stage-specific Runx1 and GATA3 target site. This could reflect additional repressive inputs from more distal regulatory sites, such as the +145-kb site near the distal end of a new commitment-associated loop, three to five genes away from the Spi1 locus (Fig. S3 B; and Fig. S4, B–D). Repressive activity of GATA3 could also be unmasked if it undergoes signal-dependent dephosphorylation during commitment (Fig. 2 D). Alternatively, as Runx1 and GATA3 are critical regulators of multiple other genes, their levels could also influence Spi1 expression via indirect mechanisms (Fig. 7 B). In principle, they could repress activators of Spi1 such as Cebpa (Laiosa et al., 2006; Wang et al., 2006; Yeamans et al., 2007) and Lmo2 (Cleveland et al., 2013). However, Cebpa is expressed at very low levels in T lineage progenitors, and Lmo2 is turned off before the DN2a stage, well before Spi1 levels start going down (Hosokawa and Rothenberg, 2021; Yui and Rothenberg, 2014; Zhou et al., 2019). Thus, these indirect mechanisms may contribute to, but not fully explain, the stage specificity.
Mutation of the potential repression element enabled Spi1 to begin expression in a nonexpressing pro–T cell line, under conditions where deletions of other elements suspected to mediate repression had much less effect. Most notably, Cas9 targeting of the three Runx and one GATA motifs in the +3.7-kb element also enabled primary pro–T cells to sustain readily measurable Spi1 expression in a developmental stage where controls had virtually extinguished it. Thus, the integrity of this element was important for full developmental stage-specific repression. However, it was atypical of elements normally sought through genomic methods. The binding of Runx1 and GATA3, though reproducible under the conditions used, was very weak. This could reflect the ATAC-inaccessible chromatin state at this site both before and after commitment. While the potential silencer region was not conserved among mammals, this region was perfectly overlapped by a LINE element. LINE elements are known to acquire roles in species-specific gene expression via activation and repression of proximal genes (Robbez-Masson and Rowe, 2015). Furthermore, a recent report on 3D structure of the human SPI1 locus in monocytes suggest interactions of the promoter with the intron 2 region of SPI1 as well as with the URE (Schuetzmann et al., 2018). Hence, stage-specific repression of human SPI1 could be regulated in part via similar or independent human-specific elements, even though the SPI1 locus overall has multiple conserved cis-regulatory elements.
In summary, we identified a potential stage-specific silencer region for Spi1, which appears to be controlled by Runx1 and GATA3, in early T cell development. Acute deletion of Runx1 and Gata3 by CRISPR-Cas9 revealed stage-specific roles of Runx1 and GATA3 in Spi1 repression in pro–T cell stages. A postcommitment stage-specific Runx1 and GATA3 occupancy site at the Spi1 locus was elucidated by comparative ChIP-seq analysis using precommitment and postcommitment pro–T cells, in the same T lineage pathway but with a few days difference on the trajectory. Acute deletion evidence then supported a strong role for this site in physiological Spi1 repression. Thus, the developmentally based strategy used in this study could be useful to identify other functionally important cis-regulatory elements, whether or not they were in open chromatin or phylogenetically conserved.
Materials and methods
Mice
C57BL/6 (referred to as B6), B6.Cg-Tg(BCL2)25Wehi/J (Bcl2-Tg; Strasser et al., 1991), and B6.Gt(ROSA)26Sortm1.1(CAG-cas9*,-EGFP)Fezh/J (Cas9; Platt et al., 2014) mice were purchased from The Jackson Laboratory. Bcl11b-YFP (backcrossed to C57BL/6 mice 10 times) mice were described previously (Ng et al., 2018). All animals were bred and maintained in the California Institute of Technology Laboratory Animal Facility under specific pathogen–free conditions, and the protocol supporting animal breeding for this work was reviewed and approved by the Institute Animal Care and Use Committee of the California Institute of Technology.
Cell culture of primary pro–T cells
For in vitro differentiation of pro–T cells, BM hematopoietic progenitors were used for input to OP9-DLL1 co-cultures (Schmitt and Zúñiga-Pflücker, 2002). In the conditions used, most cells underwent commitment around days 6–7 of culture. Vectors carrying genes for gain of function assays or CRISPR guide RNAs for knockout assays were introduced at different time points, depending on whether the cells were to be targeted before, after, or during commitment. Cells for gain-of-function experiments were harvested 3 d after transduction, and cells for loss-of-function experiments were harvested 4 d after transduction in most cases. The Bcl2 transgene (Tg) was present in the input cells for all these manipulations because it enhances viable recovery of cells under regulatory perturbations, allowing the RNA and protein expression to be measured in a more representative fraction of the responding cells, without altering early T cell development of controls (Yui et al., 2010).
BM was removed from the femurs and tibiae of 2–3-mo-old mice. Suspensions of BM cells were prepared and stained for lineage-specific, mature-cell markers (Lin) using the biotin-conjugated lineage antibodies CD11b (eBioscience; 13–0112-86), CD11c (13–0114-85), Gr-1 (13–5931-86), TER-119 (13–5921-85), NK1.1 (13–5941-85), CD19 (13–0193-85), and CD3ε (13–0031-082). They were then incubated with streptavidin-coated magnetic beads (Miltenyi Biotec) and passed through a magnetic column (Miltenyi Biotec) to remove the Lin+ cells. Then, the resulting enriched hematopoietic progenitors were cultured on OP9-DLL1 monolayers using OP9 medium (α-MEM, 20% FBS, 50 µM β-mercaptoethanol, and penicillin-streptomycin-glutamine) supplemented with 10 ng/ml IL-7 (Pepro Tech) and 10 ng/ml Flt3L (Pepro Tech). On day 7, cultured cells were disaggregated, filtered through 40-µm nylon mesh, and recultured on new OP9 monolayers with medium containing 5 ng/ml IL-7 and 5 ng/ml Flt3L. In cultures that were continued for longer times, cells were passaged onto fresh OP9-DLL1 monolayers at day 10 and maintained up to day 14 in 1 ng/ml each of IL-7 and Flt3L.
While the developmental trajectory of the cells in these cultures is extremely reproducible between experiments and experimenters, the exact degrees of progression of the cells along the pathway at a given time point often vary slightly from experiment to experiment. For precise measurements of fold change, to establish biological reproducibility with minimum confounding effects of timing on the expression of highly dynamic genes, biological replicate cell samples were prepared separately from three independent animals, purified separately, vector transduced separately, and cultured separately, but in parallel over the same absolute time course. Additional supporting information was obtained in independent experiments.
Cell culture of a pro–T cell-like line
Scid.adh.2c2 cells (Dionne et al., 2005) were cultured in RPMI1640 with 10% FBS (Sigma-Aldrich), sodium pyruvate (Gibco), nonessential amino acids (Gibco), penicillin-streptomycin-glutamine (Gibco), and 50 µM β-mercaptoethanol (Sigma-Aldrich).
Transduction of Runx1 and Gata3 into pro–T cells
Gain-of-function experiments were performed before commitment using protocol A (Fig. 2 A). BM progenitor cells from Bcl2-Tg animals were used to seed in vitro differentiation cultures as above. At day 3, the cells were transduced with retroviral vectors encoding reporters (GFP and hNGFR) and the indicated cDNAs (details of vector construction below) and then returned to OP9-DLL1 culture for harvest on day 6 overall. A modification of this protocol was used for gain of function of Runx1 and GATA3 coupled with disruption of the +3.7-kb site motifs as detailed in Fig. 7 A (protocol E). Conditions for Cas9-mediated disruption are described in the next section. The methods used to generate the virus supernatant and for infection were described previously (Hosokawa et al., 2018a). For RT-qPCR analysis, retrovirus-infected Lin−CD45+CD44+GFP+hNGFR+ (precommitment) cells were sorted on a BD FACSAria.
CRISPR-Cas9–mediated acute deletion of target genes in primary cells and a cell line
Acute deletions of Gata3 or Runx1 or their presumptive target sites in primary cells were performed using protocols B, C (Fig. 3, A and D), D, or E (Figs. 6 D and 7 A) to induce deletion in postcommitment, mid-commitment, or precommitment stages, respectively. To generate input cells, Cas9 mice were first bred to Bcl2-Tg mice to generate heterozygotes for both transgenes. BM cells from these Cas9;Bcl2-Tg animals were then used to seed in vitro differentiation cultures as above. At day 10 (for postcommitment effects, protocol B), day 4 (for mid-commitment effects, protocol C), or day 2 (for precommitment effects, protocol D), the cells were transduced with retroviral vectors encoding reporters (GFP, CFP and hNGFR) and the indicated cDNAs or guide RNAs (sgRNAs) as detailed below, and then returned to OP9-DLL1 culture for harvest 4 d later, at day 14 overall (protocol B), day 8 overall (protocol C), or day 6 overall (half of the cultures in protocol D). The other half of the cultures treated early, via protocol D, were continued to day 12 overall to monitor postcommitment effects of precommitment deletion. The methods used to generate the virus supernatant and for infection were as described previously (Hosokawa et al., 2018a). For RT-qPCR analysis, retrovirus-infected Lin− CD45+ CD44+ GFP+ hNGFR+ (precommitment) or Lin− CD45+ CD44lo CFP+ hNGFR+ (postcommitment) cells were sorted on a BD FACSAria.
A DN3-like cell line, Scid.adh.2c2, previously transduced to express Cas9-GFP, was infected with sgRNA-hNGFR and/or sgRNA-CFP vectors (Hosokawa et al., 2018b). 3 d after sgRNA transduction, sgRNA-introduced GFP+CFP+hNGFR+ Scid.adh.2c2 cells were sorted, followed by immunoblotting, or subjected to single-cell sorting. Cytoplasmic and nuclear extracts, used to the detection of tubulinα, and Runx1 and GATA3, respectively, were prepared using NE-PER nuclear and cytoplasmic extraction reagents (Pierce). Lysates were run on 10% polyacrylamide gel, followed by immunoblotting. The antibodies used for the immunoblot analysis were anti-tubulinα (Sigma-Aldrich; T6199), anti-Runx1 (Abcam), anti-Myc (MBL; My3), and anti-GATA3 (Santa Cruz Biotechnology; HG3-31) mAbs.
Cloning
cDNAs for GATA3 WT and mutants were inserted into a multicloning site of the pMxs-IRES-hNGFR, as previously reported (Hosokawa et al., 2015; Hosokawa et al., 2016).
GATA3 residues S308, T315 and S316, which are important for histone deacetylase 2 association in their nonphosphorylated state, are near the C-terminal zinc finger (Hosokawa et al., 2016; Hosokawa et al., 2013b). GATA3 R261, where arginine methylation increases transcriptional activation potency, is near the N-terminal zinc finger.
cDNA for full-length Runx1 from the distal promoter and Runx1DN (Runx1 d190) were inserted into a multicloning site of the MSCV-IRES-GFP (Telfer et al., 2004). For CRISPR targeting, the sgRNA-expression vector (E42-dTet; Hosokawa et al., 2018b) was used as described previously. For double-KO experiments, versions of E42-dTet with truncated NGFR reporter were made as well the original vector with a CFP reporter (here, mTurquoise2) so that doubly transduced cells could be sorted unambiguously. For site disruption experiments, vectors for sgRNA against Runx motifs were made with an hNGFR reporter, whereas vectors for sgRNA against the GATA motif were made with a CFP reporter. 19- or 20-mer sgRNAs were designed using the CHOPCHOP (https://chopchop.cbu.uib.no; Labun et al., 2016) or Benchling (https://benchling.com) web tool and inserted into the empty sgRNA-expression vector by PCR-based insertion. Three sgRNA-expression vectors were generated for one gene, and pooled retroviral plasmids were used to make retroviral supernatant. The following sgRNA sequences were used in this study: control (Luciferase), 5′-GGCATTTCGCAGCCTACCG-3′; GATA3 #1, 5′-GAGATCCGTGCAGCAGAGG-3′; GATA3 #2, 5′-GTTCCAGGGCGAGGCGGTG-3′; GATA3 #3, 5′-TGGCCGGCCGCCAGAGAAG-3′; Runx1 #1, 5′-GCTCGTGCTGGCATCTACG-3′; Runx1 #2, 5′-AGCCCCGGCAAGATGAGCG-3′; Runx1 #3, 5′-AGCGGCGACCGCAGCATGG-3′; Slc39a13 pro. #1, 5′-GCAGATGCAGCGCTCCGGG-3′; Slc39a13 pro. #2, 5′-AGGGTTACAGTCCTGGAGA-3′; URE #1, 5′-CAAATGGCCCGAGGTCCGA-3′; URE #2, 5′-TACGATCTAGACAGTGGGT-3′; CE4 #1, 5′-TAGAGCTGAGCCTAAGTTC-3′; CE4 #2, 5′-CTGAGGAAGACTTCCAGGT-3′; Spi1 intron2 #1, 5′-CCTGTAATCCTCTCTCGGG-3′; Spi1 intron2 #2, 5′-CACGTGGACCAGAGGTCAC-3′; +145 kb #1, 5′-GGTGTAAGAAGCAGAGTCTG-3′; +145 kb #2, 5′-TTTGTTCACACTGGAAACTG-3′; +167 kb #1, 5′-GCCTACAGCCAGTGGTCGAG-3′; +167 #2, 5′-GGTACGGGTCCTGACGCCGG-3′; +211 #1, 5′-AGAAAGATTCCAATTCAACA-3′; +211 #2, 5′-TCTACTAGGTCTTACCAACT-3′; Runx motif 1, 5′-GATGAACTCAGTGTGGTTT-3′; Runx motif 2, 5′-CTCGACCTGCCCTTAAATG-3′; Runx motif 3, 5′-AAACTAATCTTTTCTCTGT-3′; and GATA motif, 5′-CTCTGTGGGTGCTTATCAC-3′.
Two-step affinity purification of GATA3 complexes from Scid.adh.2c2 cells
Scid.adh.2c2 cells were infected with either mock control (pMxs-IRES-GFP) or Myc-Flag-GATA3–containing retrovirus. 3 dpi, Myc-Flag–tagged GATA3-infected GFP+ cells were solubilized with the following protease inhibitor–containing immunoprecipitation buffer: 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 10% glycerol, 0.1% Tween, 1 mM EDTA, 10 mM NaF, 1 mM DTT, and a protease inhibitor cocktail (Roche Applied Science) and lysed on ice for 30 min with gentle shaking and sonicated on a Misonix S-4000 sonicator (Qsonica) for three cycles, amplitude 20 for 30 s, followed by a 30-s rest. The insoluble materials were removed by centrifugation, and immunoprecipitation with anti-Flag M2 agarose (Sigma-Aldrich) was performed overnight at 4°C. Immune complexes were eluted from the agarose by 3xFlag peptide (Sigma-Aldrich), and the eluted GATA3 complexes were subjected to a second immunoprecipitation with anti-Myc gel (MBL). Immune complexes were eluted from the gel with Myc peptide (MBL) and separated by SDS-PAGE. The bands were excised from the gel and subjected to a mass spectrometric analysis to identify corresponding proteins. The gel pieces were washed twice with 100 mM bicarbonate in acetonitrile, and the proteins were digested with trypsin. After adding 0.1% formic acid to the supernatant, the peptides were analyzed by liquid chromatography-tandem mass spectrometry with an Advance ultra-high-performance liquid chromatograph (Bruker) and an Orbitrap Velos Pro mass spectrometer (Thermo Fisher Scientific). The resulting tandem mass spectrometry dataset was analyzed using the Mascot software program (Matrix Science). Mascot score is the probability that the observed match is a random event (Mascot score >100 means the absolute probability <1e-10).
Flow cytometry analysis
Thymocytes were isolated from 4–6-wk-old Bcl11b-YFP reporter mice. Lin+ cells were depleted by staining with biotinylated antibodies against CD8α (eBioscience; 13–0081-86), TCRγδ (13–5711-85), TCRβ (13–5961-85), TER-119, NK1.1, Dx5 (13–5971-82), CD11c, and CD11b, then incubating cells with streptavidin magnetic beads, followed by passing them through an LS magnetic column (Miltenyi Biotec) in accordance with the manufacturer’s instructions. Purified DN cells were stained with c-Kit APCe780 (eBioscience; 17–1171-82), CD25 PE (12–0251-28), CD44 e450 (48–0441-82), and streptavidin PerCPCy5.5 (45–4317-80) and then subjected to intracellular staining for GATA3 AF647 (BD PharMingen; 560068) or PU.1 AF647 (Cell Signaling; 2240) using the BrdU Flow Kit (BD PharMingen).
For staining of sgRNA-introduced BM cells, surface antibodies against CD45 PECy7 (eBioscience; 25–0451-82), c-Kit APC, CD25 APC-e780 (47–0251-82), human-NGFR PE (12–9400-42), and a biotin-conjugated lineage cocktail (CD8α, CD11b, CD11c, Gr-1, TER-119, NK1.1, CD19, TCRβ, and TCRγδ) with streptavidin PerCPCy5.5 were used for staining.
Prior to cell surface staining, cells were treated with 2.4G2 cell supernatant. All of the cells were analyzed using flow cytometers, MacsQuant 10 (Miltenyi Biotec), LSRFortessa (BD PharMingen), or FACSAria (BD PharMingen) with FlowJo software (Tree Star).
ChIP library construction and characteristics
ChIP experiments and library preparation were performed as previously described (Hosokawa et al., 2018b; Ungerbäck et al., 2018). Briefly, 5–10 × 106 cells were fixed with 1 mg/ml DSG (Thermo Fisher Scientific) in PBS for 30 min at room temperature followed by an additional 10 min with addition of formaldehyde up to 1%. These conditions were previously found to give efficient detection of Runx1 binding with strong enrichment for Runx target motifs (Hosokawa et al., 2020; Hosokawa et al., 2018b). 5 μg anti-GATA3 mAbs (a mixture of 2.5 µg sc-268 [Santa Cruz Biotechnology] and 2.5 µg MAB26051 [R&D Systems]) or anti-Runx1 mAb (Abcam; ab23980) was prebound to Dynabeads anti-Mouse or anti-Rabbit Ig (Invitrogen) and then added to the diluted chromatin complexes. Precipitated chromatin fragments were cleaned up using Zymo ChIP DNA Clean & Concentrator and used as input for qPCR and/or ChIP-seq.
Note that GATA3 shows highly conditional genomic binding patterns at different stages within the T cell lineage, suggesting influence from factors other than its binding affinities for different genomic sequences (Wei et al., 2011; Zhang et al., 2012). For comparison in Fig. S3 A, a separate pair of DN2b/DN3 samples was also prepared for GATA3 ChIP-seq using fixation with 1% formaldehyde only for 10 min, as described previously (Ungerbäck et al., 2018). The patterns of GATA3 occupancy detected depended on the fixation used. GATA3 associations with many enhancer-like sites near suspected positive target genes were consistent, but GATA3 association with closed regions, promoters, and repeat sequence elements appeared systematically different between the two fixation conditions (compare with Fig. S3 A), with characteristics summarized in Fig. S5 F. Briefly, DSG+FA showed higher association with promoters and ATAC-open sites, whereas FA alone showed higher enrichment for strong GATA binding motifs but a substantially higher proportion of binding to repeat sequences. Thus, different fixation conditions could selectively stabilize interactions with different binding-modulatory chromatin contexts. Admixtures of functional sites with different “background” sequences are expected to alter relative enrichment, a key consideration for global statistical comparisons. However, in this study, the goal was maximal absolute recovery of candidates for functionally important sites within the Spi1 genomic neighborhood to maximize the chance for relevant repression sites to be detected.
Quantitative ChIP-PCR and ChIP-seq
For qPCR analysis, 7900HT Fast Real-time PCR System, QuantStudio3 (Applied Biosystems) or StepOnePlus systems (Applied Biosystems) were used with SYBR GreenER qPCR SuperMix (Invitrogen). The data are shown as the mean values (percent input). Primers are listed below. For deep-sequencing analysis, ChIP-seq libraries were constructed using NEBNext ChIP-Seq Library Preparation Kit (E6240, NEB) and sequenced on Illumina HiSeq2500 in single read mode with the read length of 50 nt. All analyses are based on results from two biologically separate replicates. ChIP-seq data were analyzed as described previously (Hosokawa et al., 2018b; Ungerbäck et al., 2018). BigWigs were generated from the aligned SAM or BED-file formats using Samtools (Li et al., 2009), Bedtools (Quinlan and Hall, 2010) and the UCSC genomeCoverageBed and bedGraphToBigWig and normalized to 1 million reads. For visualization of ChIP-seq tracks, bamToBed and genomeCoverageBed were used with the “-split” setting enabled. BigWig files were up-loaded to the UCSC genome browser (http://genome.ucsc.edu; Speir et al., 2016). Runx1 ChIP-seq data in DN1 and DN2b/3 cells and Runx1 and GATA3 ChIP-seq data in Scid.adh.2c2 cells used in this study were previously published (GEO accession numbers GSE103953 and GSE93755; Hosokawa et al., 2018b): Slc39a13 pro. forward (FW), 5′-CCTTCTGAGGATGCAGTTCC-3′; Slc39a13 pro. reverse (RV), 5′-CTGCTGACTCAGAGAGCCATAG-3′; URE FW, 5′-GGGCGCTTCCTGTTTTCT-3′; URE RV, 5′-CTGGGCAGGGTCAGAGTG-3′; CE4 FW, 5′-TTTGAGGTGCAGGAGACTGA-3′; CE4 RV, 5′-TGAATCTTACTGAACCCCGTCT-3′; intron 2 FW, 5′-CTCGACCTGCCCTTAAATGT-3′; and intron 2 RV, 5′-CCAGTGATAAGCACCCACAG-3′.
qPCR
Total RNA was isolated from samples of 2 × 105 cultured cells using a RNeasy Micro Kit (Qiagen). Reverse transcription was performed using the Superscript III kit (Invitrogen). Genomic DNA was isolated from 5 × 105 cultured cells using a NucleoSpin Tissue kit (Macherey-Nagel). For qPCR, the 7900HT Fast Real-time PCR System, QuantStudio3 (Applied Biosystems), or StepOnePlus system (Applied Biosystems) were used with SYBR GreenER qPCR SuperMix (Invitrogen). The data are shown as the relative expression normalized to Actb (for cDNA) or Gapdh promoter (genomic DNA) signals. The following primers were used in this study: PU.1 FW, 5′-TTCTGCACGGGGAGACAG-3′; PU.1 RV, 5′-GTCCACCCACCAGATGCT-3′; GATA3 FW, 5′-TATCCTCCGACCCACCAC-3′; GATA3 RV, 5′-AAGGGGCTGAGGTTCCAG-3′; Runx1 FW, 5′-CTCCGTGCTACCCACTCACT-3′; Runx1 RV, 5′-ATGACGGTGACCAGAGTGC-3′; Actb FW, TACAGCCCGGGGAGCAT-3′; Actb RV, 5′-ACACCCGCCACCAGTTC-3′; Bcl11b FW, 5′-TGGATGCCAGTGTGAGTTGT-3′; Bcl11b RV, 5′-GCTGCTTGCATGTTGTGC-3′; Slc39a13 pro. FW, 5′-CCTTCTGAGGATGCAGTTCC-3′; Slc39a13 pro. RV, 5′-CTGCTGACTCAGAGAGCCATAG-3′; URE FW, 5′-GGGCGCTTCCTGTTTTCT-3′; URE RV, CTGGGCAGGGTCAGAGTG-3′; CE4 FW, 5′-TTTGAGGTGCAGGAGACTGA-3′; 5′-CE4 RV, TGAATCTTACTGAACCCCGTCT-3′; intron 2 FW, 5′-CTCGACCTGCCCTTAAATGT-3′; intron 2 RV, 5′-CCAGTGATAAGCACCCACAG-3′; +145 kb FW, 5′-GGCACCTTTAACCCAGAGTTC-3′; +145 kb RV, 5′-AATGAATGACATCAGGCTACCA-3′; +167 kb FW, 5′-GAAGTTTGGGCCAGTCCAC-3′; +167 kb RV, 5′-TCTGGGCCAATGAACGTC-3′; +211 kb FW, 5′-CTGTAAACAGGAAACACTCAGCTC-3′; +211 kb RV, 5′-GTGGATGGCTTGCAGTAGG-3′; Ddb2 FW, 5′-TTGGCATCAAGGACAAACCT-3′; Ddb2 RV, 5′-AAACTGGTTGGTATTGAGATGGTT-3′; Pacsin3 FW, 5′-GAAGGCAGACAGCTCCATGT-3′; and Pacsin3 RV, 5′-GGGTCTGCTCATACTGGGTTT-3′.
Statistical analysis
The statistical significance of differences between datasets was determined by two-sided Student’s t test using Excel. Statistical details of experiments can be found in the figure legends. In all figures, error bars indicate SD.
Online supplemental material
Fig. S1 shows gating strategies for DN subsets in thymocytes and in vitro–differentiated pro–T cells, Spi1 expression levels in Bcl11b introduced pro–T cells, the ratio of sense and antisense Spi1 transcripts, proteomics data for GATA3-interacting molecules, and interactions between Runx1 and GATA3 mutants. Fig. S2 shows evidence for successful Runx1 and GATA3 disruption by Cas9 and gating strategies for DN subsets from in vitro–differentiated pro–T cells. Fig. S3 shows a comparison of GATA3 binding sites in different fixation conditions, a comparison of chromatin looping before and after T lineage commitment around the Spi1 locus, and a comparison of Runx1 and GATA3 binding patterns in phase 1, phase 2, and a DN3-like cell line. Fig. S4 shows a comparison of Runx1 and GATA3 binding patterns in GATA3- or Runx1-deficient pro–T cells around the Spi1 locus, Runx1 and GATA3 binding patterns around the postcommitment stage-specific distal chromatin looping sites, and Spi1 expression levels in +145, +167, or +211 kb element–disrupted DN3-like cell lines. Fig. S5 shows representative consensus motifs for Runx1 and GATA3, gating strategies for DN subsets in BM-derived pro–T cells with motif disruptions, validation of mutations in sg4 motifs mix-transduced pro–T cells, Spi1 expression levels in the cis-regulatory element disrupted cells, and a table comparing GATA3 occupancy site characteristics under different fixation conditions. Table S1 lists GATA3-interacting molecules in a pro–T cell-like cell line, Scid.adh.2c2.
Data availability
The GEO accession number for all new deep-sequencing data reported in this paper is GSE159960.
Acknowledgments
We thank D. Perez, J. Tijerina, P. Cannon, and R. Diamond for cell sorting and advice, I. Soto for mouse colony care, H. Amrhein and D. Trout for computational assistance, I. Antoshechkin for sequencing management, members of the Support Center for Medical Research and Education at Tokai University for their technical help, and the members of the Rothenberg group for valuable discussion and reagents.
E.V. Rothenberg was supported by the U.S. Public Health Service (grants R01 AI135200, R01AI083514, and R01HD076915). H. Hosokawa was supported by the Japan Society for the Promotion of Science KAKENHI grant JP19H03692, the Uehara Memorial Foundation, the Naito Foundation, the Takeda Science Foundation, the Yasuda Memorial Medical Foundation, the SENSHIN Medical Research Foundation, the Daiichi Sankyo Foundation of Life Science, the Tokyo Biochemical Research Foundation, a Princess Takamatsu Cancer Research Fund research grant, and the 2020 Tokai University School of Medicine Research Aid. T. Tanaka was supported by Japan Society for the Promotion of Science KAKENHI grant JP19H03708, the Uehara Memorial Foundation, the Naito Foundation, the Takeda Science Foundation, the Yasuda Memorial Medical Foundation, the SENSHIN Medical Research Foundation, the NOVARTIS Foundation (Japan) for the Promotion of Science, a Princess Takamatsu Cancer Research Fund research grant, the KOSE Cosmetology Research Foundation, and the Medical Institute of Bioregulation Kyushu University Cooperative Research Project Program. This work was also partly supported by the Tokai University General Research Organization Research and Study Project (H. Hosokawa), the L.A. Garfinkle Memorial Laboratory Fund and the Al Sherman Foundation, Provost and Division of Biology & Biological Engineering of Caltech special project funds, and the Albert Billings Ruddock Professorship (E.V. Rothenberg).
Author contributions: H. Hosokawa designed and supervised the study, performed experiments, analyzed data, and wrote the manuscript; M. Koizumi, K. Masuhara, and M. Romero-Wolf performed experiments and analyzed data; T. Tanaka performed experiments, analyzed data, and edited the manuscript; T. Nakayama provided unique biological reagents and helpful discussions and edited the manuscript; E.V. Rothenberg designed and supervised the study, analyzed data, and wrote the manuscript.

