Leukemia-driving mutations are thought to arise in hematopoietic stem cells (HSC), yet the natural history of their spread is poorly understood. We genetically induced mutations within endogenous murine HSC and traced them in unmanipulated animals. In contrast to mutations associated with clonal hematopoiesis (such as Tet2 deletion), the leukemogenic KrasG12D mutation dramatically accelerated HSC contribution to all hematopoietic lineages. The acceleration was mediated by KrasG12D-expressing multipotent progenitors (MPP) that lacked self-renewal but showed increased proliferation and aberrant transcriptome. The deletion of osteopontin, a secreted negative regulator of stem/progenitor cells, delayed the early expansion of mutant progenitors. KrasG12D-carrying cells showed increased CXCR4-driven motility in the bone marrow, and the blockade of CXCR4 reduced the expansion of MPP in vivo. Finally, therapeutic blockade of KRASG12D spared mutant HSC but reduced the expansion of mutant MPP and their mature progeny. Thus, transforming mutations facilitate their own spread from stem cells by reprogramming MPP, creating a preleukemic state via a two-component stem/progenitor circuit.
Introduction
The blood comprises diverse cell lineages including platelets, erythrocytes, short-lived myeloid cells including monocytes and granulocytes, and long-lived lymphocytes. This entire spectrum is continuously generated in the bone marrow (BM) of mammals from adult hematopoietic stem cells (HSC), the unique multipotent cell type capable of self-renewal (Eaves, 2015). The rare “top-level” HSC give rise to short-term HSC (ST-HSC) and multipotent progenitors (MPP), the non-renewing multipotent cell types that progressively differentiate into lineage- and cell type–committed progenitors and mature cells (Laurenti and Göttgens, 2018). Although originally based on transplantation studies, this HSC-driven hierarchical model of hematopoiesis has been recently confirmed to reflect native hematopoiesis in unmanipulated animals (Pucella et al., 2020) and humans (Sankaran et al., 2022). In particular, genetic labeling and tracing of endogenous murine HSC revealed their continuous multilineage contribution to hematopoiesis in the steady state (Busch et al., 2015; Chapple et al., 2018; Sawai et al., 2016; Säwen et al., 2018). Importantly, this process can be dynamically modulated by environmental stimuli and pathological conditions (Collins et al., 2021): for example, the multilineage contribution of HSC is accelerated by interferon response (Sawai et al., 2016), whereas bacterial infections spare HSC but accelerate the differentiation of MPP (Fanti et al., 2023).
The well-balanced hierarchy of hematopoiesis can be disrupted by genetic lesions that arise in HSC and/or their progeny. Thus, age-related clonal hematopoiesis (ARCH) involves the expansion of specific cell clones within the hematopoietic system. ARCH is common in elderly humans who are otherwise healthy, although it is thought to increase the risk of leukemia and cardiovascular disease (Ahmad et al., 2023; Warren and Link, 2020; Weeks and Ebert, 2023). Cell clones expanded in ARCH often carry inactivating mutations in epigenetic regulators such as methylcytosine dioxygenase TET2 or DNA methyltransferase DNMT3A. These mutations are thought to arise in HSC and increase their competitive fitness (Yamashita et al., 2020), especially in the inflammatory environment (Avagyan and Zon, 2023; King et al., 2020). In contrast, another set of mutations is found almost exclusively in neoplastic conditions including myelodysplastic syndrome, leukemia, and lymphoma (Kishtagari et al., 2020). These mutations often block differentiation (e.g., those in key hematopoietic transcription factors such as RUNX1) or enhance signaling cascades leading to cell proliferation, such as activating mutation of the FLT3 receptor or of RAS proteins (KRAS or NRAS). Indeed, stepwise accumulation of these two classes of mutations in HSC is thought to cause eventual emergence of fully transformed leukemic clones (Miles et al., 2020; Morita et al., 2020; Schwede et al., 2023, Preprint).
The distinct pathogenic consequences of ARCH-associated (e.g., Tet2 loss) and leukemia-associated (e.g., RAS activation) mutations can be recapitulated in mouse models. For example, the deletion of Tet2 causes myeloproliferative disease with a long latency (Li et al., 2011; Moran-Crusio et al., 2011; Quivoron et al., 2011), which can be accelerated by microbial stimulation (Meisel et al., 2018). In contrast, the activation of RAS signaling in hematopoietic cells, e.g., by inducing the activating KRASG12D mutation, causes a variety of leukemia types (Braun et al., 2004; Chan et al., 2004; Dail et al., 2010; Sabnis et al., 2009; Tarnawsky et al., 2017). In these models, mutations are simultaneously present in the entire hematopoietic system, circumventing the natural process as it occurs in human patients. To understand the early effects of ARCH- and leukemia-associated mutations on normal hematopoiesis, it is imperative to model their stepwise initiation and progression. To this end, the mutations need to be introduced specifically into endogenous HSC, and their spread throughout the hematopoietic system should be characterized by lineage tracing.
Here, we used Cre recombination to induce prototypical leukemia-associated (KRASG12D) or ARCH-associated (TET2 loss) mutations specifically in HSC and trace the mutant HSC population in their native hosts. In contrast to minimal effects of ARCH mutations, KrasG12D dramatically accelerated HSC contribution in the steady state, with the mutation rapidly spreading throughout the hematopoietic system prior to leukemia development. Unexpectedly, this accelerated contribution commenced at the MPP stage and was facilitated by a profound reprogramming of progenitor physiology, including their increased CXCR4-driven motility. The results emphasize the ability of leukemia-associated mutations to reprogram the normal hematopoietic differentiation, converting HSC into their “reservoirs” and progenitors into “super-spreaders.”
Results
KrasG12D mutation in HSC accelerates their contribution to hematopoiesis
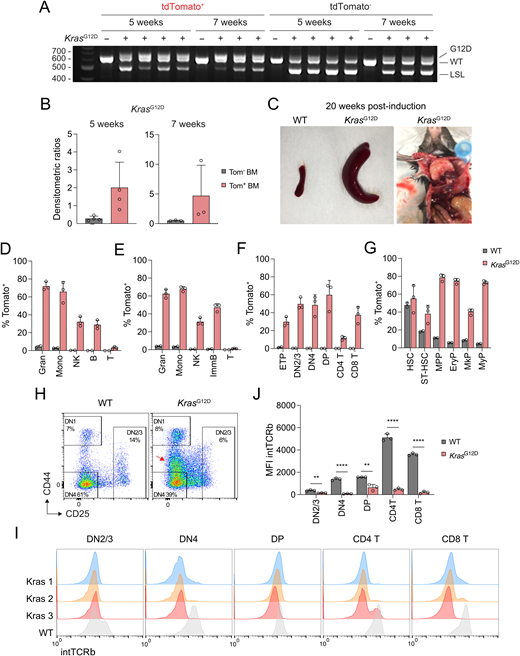
We have previously generated Pdzk1ip1-CreER transgenic mice expressing tamoxifen-inducible Cre recombinase (CreER) specifically in HSC. When crossed with a reporter allele expressing red fluorescent protein tdTomato (Tom) in a Cre-inducible manner (R26Tom), the resulting reporter mice allow specific labeling of HSC with Tom and tracing their contribution to progenitors and mature cells (Sawai et al., 2016). We crossed Pdzk1ip1-CreER R26Tom/Tom reporter mice with KrasG12D Lox-Stop-Lox (LSL) strain (Jackson et al., 2001), in which Cre recombination activates the expression of the constitutively active KRASG12D protein. Importantly, KRASG12D is expressed from the endogenous Kras allele and thus is under its normal transcriptional regulation (Tuveson et al., 2004) (Fig. 1 A). In the resulting Pdzk1ip1-CreER R26Tom/TomKrasG12D reporter mice, tamoxifen-induced Cre recombination is predicted to activate both KrasG12D and Tom in HSC, allowing the tracing of mutation-carrying HSC (Fig. 1 A). To confirm that, Pdzk1ip1-CreER R26Tom/TomKrasG12D mice were administered a single dose of tamoxifen, and 3 wk later used to sort lineage marker–negative (Lin−) Sca-1+ c-Kit+ (LSK) population, which contains HSC (CD150+ CD48−), ST-HSC (CD150− CD48−), and MPP (CD150− CD48+). Tom+ but not Tom− LSK cells showed extensive recombination of the KrasG12D allele, validating Tom expression as a marker of mutation-carrying cells at this time point (Fig. 1 B). The recombined KrasG12D allele became detectable in total BM Tom− cells at 5 wk (in one out of four mice) and 7 wk (in all mice) (Fig. S1 A), likely reflecting a strong selection for rare Tom− Kras-mutant cells over time. However, KrasG12D recombination was much more extensive in Tom+ versus Tom− cells at these time points (Fig. S1 B), supporting the correlation between Kras mutation and Tom expression within the first several weeks after induction. As expected, tamoxifen-treated Pdzk1ip1-CreER R26Tom/TomKrasG12D mice became moribund with a median survival of 18 wk (Fig. 1 C), showing features of acute T-lymphoblastic leukemia (T-ALL) including splenomegaly and massively enlarged thymus (Fig. S1 C). We then tested the effect of KrasG12D induction in HSC on hematopoiesis within 4–12 wk after induction (Fig. 1 D). We found that the accrual of Tom+ cells was dramatically accelerated in the peripheral blood (PB) of KrasG12D compared with wild-type (WT) reporter mice, with significant increases in most lineages by 4 wk after induction, and in all lineages including T cells by 8 wk (Fig. 1 E).
To better characterize the accelerated differentiation of Kras-mutant HSC, we analyzed mice at 2- to 7-wk endpoints. At 2 wk after induction, mature immune cells in the spleen (Fig. 1 F) and BM (Fig. 1 G) showed the expected absence of labeling in both KrasG12D and WT reporter mice. However, the labeling of all cell types in KrasG12D mice became significantly increased at 3 wk and reached high levels by 5 wk, showing an ∼20-fold increase in myeloid cells, ∼60–80-fold increase in natural killer (NK) cells, and ∼120–160-fold increase in B cells compared with WT (Fig. 1, F and G). The resulting labeling efficiencies of 60–70% at 7 wk (Fig. S1, D and E) are typically attained in WT mice by 8–9 mo after labeling (Sawai et al., 2016). As described previously (Sawai et al., 2016), WT reporters showed minimal labeling of the early thymic progenitors (ETP) or the downstream thymocyte subsets at 3–5 wk (Fig. 1 H) and up to 7 wk (Fig. S1 F). In contrast, all KrasG12D reporter mice showed detectable (∼5%) ETP labeling at 3 wk and massive (>50%) labeling at 5 wk, along with comparably high labeling in CD4−CD8− double-negative (DN2/3 and DN4) and CD4+CD8+ double-positive (DP) thymocytes. At 7 wk, the labeling of DP thymocytes reached or exceeded that of HSC and also spread into mature single-positive CD4+ and CD8+ thymocytes (Fig. S1, F and G). This massive expansion likely is a consequence of enhanced HSC contribution to thymopoiesis, although the expansion of a very rare labeled thymic progenitor cannot be ruled out. The activation of Kras was shown to circumvent the β-selection checkpoint, which ensures that only those thymocytes that productively rearranged their TCRβ chain progress from CD25+ DN3 to CD25− DN4 and DP stages (Kortum et al., 2013). Accordingly, the DN3 population in the thymi of induced KrasG12D reporter mice was reduced in favor of a CD25int population (Fig. S1 H); moreover, the acquisition of intracellular TCRβ at the DN4 and subsequent stages was abolished in these mice (Fig. S1, I and J). These data suggest a failure of β-selection in KrasG12D-expressing thymocytes, likely contributing to the subsequent development of T-ALL. Overall, the acquisition of activating Kras mutation by HSC causes a dramatic acceleration of their contribution to all hematopoietic lineages, leading to the rapid spread of the mutation prior to leukemogenesis.
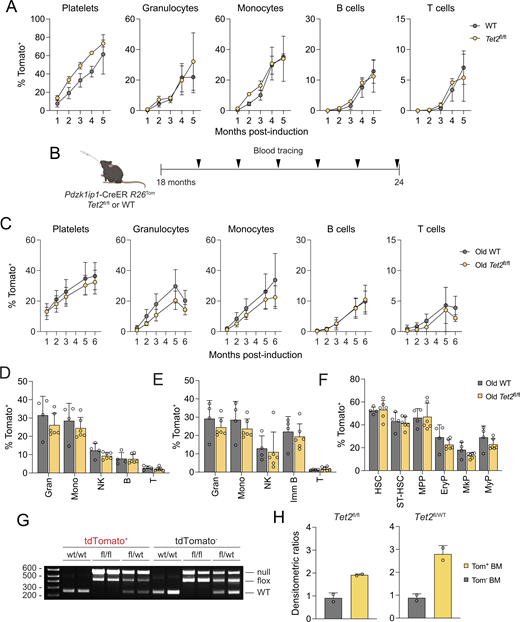
To test whether similar acceleration is caused by mutations associated with ARCH, we generated Pdzk1ip1-CreER R26Tom/Tom reporter animals that also carried LoxP-flanked (floxed) conditional null allele of Tet2 (Tet2fl/fl) (Moran-Crusio et al., 2011). No difference in the accrual of Tom+ cells was observed in Tet2fl/fl reporter mice versus controls (Fig. S2 A). Similarly, no accelerated accrual of Tom+ cells was observed in aged (18-mo-old) Tet2fl/fl reporter mice (Fig. S2 B), which showed the same global reduction of multilineage HSC contribution as WT reporters (Jang et al., 2023) (Fig. S2, C–F). The recombined Tet2fl allele was predominant in the Tom+ fraction of total BM cells in both homozygous Tet2fl/fl and heterozygous Tet2fl/wt reporter mice at 6 mo after induction (Fig. S2, G and H). However, the recombined allele was also clearly detectable in the Tom− fraction, and the difference between Tom+ and Tom− fractions was not as strong as for KrasG12D, possibly due to the extended timeframe of tracing.
Because the injection of microbial mimic Pam3CSK4 facilitates myeloproliferation in mice with global Tet2 deletion (Meisel et al., 2018), Tet2fl/fl or WT reporter mice were administered tamoxifen together with 3 daily doses of Pam3CSK4 (Fig. S3 A). Pam3CSK4-treated Tet2fl/fl reporter mice showed no consistent increase in Tom+ cells in the PB over 6 mo compared with untreated Tet2fl/fl mice, likely due to the high labeling variability in the latter (Fig. S3 B). However, compared with Pam3CSK4-treated WT reporter mice, Pam3CSK4-treated Tet2fl/fl mice showed a significant increase of Tom+ cells in platelets (at 1–6 mo) and in myeloid cells (at 4–6 mo). This increased labeling was confirmed in myeloid cells at the 6-mo endpoint (Fig. S3 C) and was also evident in MPP and myeloid progenitors (MyP) (Fig. S3, D and E). As noted above, the tracing of Tet2-deficient cells may underestimate their expansion in the Tom− fraction. Even with this caveat, the data suggest that ARCH-associated mutations such as Tet2 deletion have a smaller effect on HSC contribution than Kras activation, but can modestly enhance it following inflammatory stimulation.
KrasG12D mutation in HSC generates hypercompetitive multilineage progenitors
We sought to map the stage at which the expansion of Kras-mutant cells is commenced following Kras activation in HSC. At 2 wk, the fractions of Tom+ stem/progenitor cells were comparable between KrasG12D and WT reporter mice (Fig. 2 A). At 3 wk, KrasG12D reporter mice showed increased fractions of Tom+ cells among erythroid progenitors (EryP) and MyP. Tom+ cells were also increased among Flt3− MPP (MPP3) and Flt3+ MPP (MPP4 [Pietras et al., 2015]), albeit not reaching significance in the former. At 5 wk, Tom+ cells among MPP3, MPP4, and all downstream progenitors were significantly increased, approaching the level of HSC labeling (Fig. 2 A) and exceeding it by 7 wk (Fig. S1 G). Similarly, labeled MPP (hereafter representing combined MPP3 and MPP4), EryP, and MyP were significantly increased in absolute numbers at 3 and 5 wk in KrasG12D reporter mice (Fig. 2 B). In contrast, Tom+ HSC and ST-HSC were not increased at any point and even showed a minor decrease at 3 wk, suggesting that the expansion of Kras-mutant cells commences at the MPP stage.
The analysis of Tom+ cells among IL-7R+ lymphoid progenitors (LyP) in KrasG12D reporter mice revealed a widespread upregulation of IL-7R among all Flt3+ progenitors. These included Sca-1− c-Kit− and c-Kitlo/int progenitors harboring the so-called common LyP, as well as Sca-1− c-Kit+ MyP and Sca-1+ c-Kit+ MPP4 (Fig. 2, C–E). Importantly, Tom+ cells were dramatically expanded among IL-7R+ cells in all these populations at 3 wk after induction in KrasG12D reporter mice (Fig. 2 F). These data are consistent with the induction of IL-7R expression by the Ras signaling pathway (Li et al., 2010) and reveal widespread lymphoid priming of mutant progenitors starting from the MPP4 stage.
We measured the proliferation rate of stem/progenitor populations by injecting mice with 1 mg 5-ethynyl-2′-deoxyuridine (EdU) and detecting its incorporation 2 h later by flow cytometry (Akinduro et al., 2018). We compared the fractions of EdU+ cells in Tom+ and Tom− cell fractions in KrasG12D reporter mice 5 wk after tamoxifen treatment, as well as in total cells in WT reporter mice (given the low fraction of Tom+ cells at that stage) (Fig. 3 A). As expected, HSC and ST-HSC showed low but detectable EdU incorporation, which was progressively increased in MPP and erythromyeloid progenitors in WT mice (Fig. 3 B). Notably, only Tom+ MPP showed a significantly increased EdU incorporation in KrasG12D reporter mice when compared both to Tom− counterparts from the same mice and to the same population from WT mice (Fig. 3 B). This significant increase was observed in both MPP3 and MPP4 analyzed separately (Fig. 3 C). Moreover, a similar increase in EdU incorporation in MPP (but not in HSC) was observed at 3 wk, the earliest time point of Tom+ cell expansion in KrasG12D reporter mice (Fig. 3 D). We repeated these experiments at 3 wk after induction using two injections of 1 mg EdU over the 48-h period and stained cells for EdU incorporation along with DNA content. Again, HSC and ST-HSC showed minimal differences between Tom+ and Tom− cell fractions in KrasG12D reporter mice or total cells in WT reporter mice (Fig. 3 E). In contrast, MPP showed a significant increase in EdU+ cells and a reciprocal decrease in EdU− 2n cells, i.e., cells that did not enter the S-phase within 48 h (Fig. 3, F and G). These data suggest that the induction of KrasG12D in HSC does not affect their proliferation rate, but increases that of the downstream MPP population.
To test whether this increased proliferation rate reflected the acquisition of self-renewal capacity, we sorted Tom+ HSC or MPP from KrasG12D reporter mice 5 wk after tamoxifen treatment and transferred them into lethally irradiated CD45.1 congenic recipients along with supporting total CD45.1 BM cells. Whereas the transfer of 50 HSC/recipient resulted in durable multilineage reconstitution by CD45.2+ donor cells, the transfer of 1,000 MPP/recipient yielded a small fraction of donor-derived lymphocytes at 1 mo, and no detectable reconstitution thereafter (Fig. 3 H). Thus, Kras activation in HSC causes a rapid expansion of mutant MPP, which show increased proliferation but are incapable of self-renewal.
KrasG12D-expressing MPP have a distinct transcriptome
To characterize the KrasG12D-carrying HSC and their immediate progeny, we analyzed them by cellular indexing of transcriptomes and epitopes (CITE-seq). CITE-seq enables combined single-cell RNA sequencing of multiple samples by labeling them using oligonucleotide-conjugated antibody “hashtags” (Stoeckius et al., 2018). Pdzk1ip1-CreER R26Tom/TomKrasG12D or WT reporter mice were treated with tamoxifen and analyzed at 3 wk, the earliest time point for the expansion of Kras-mutant progenitors. Lin− BM cells were isolated, hashtagged, and used to sort the LSK population. For KrasG12D reporter mice, Tom+ LSK cells were sorted; because WT reporter mice have very few Tom+ MPP at this time point (Fig. 2 A), total LSK cells were sorted (Fig. 4 A). Hashtagged LSK cells from two individual mice per genotype were pooled and subjected to sequencing. Because the standard Uniform Manifold Approximation and Projection could not fully resolve the closely related HSC and MPP (Fig. S4 A), we applied K-nearest neighbor-based Network graph drawing Layout (KNetL) (Feng et al., 2022; Khodadadi-Jamayran and Tsirigos, 2020, Preprint) to yield nine distinct clusters (Fig. S4 B). One of these clusters (cluster 3) represented contaminating mature cells including MHC class II–expressing dendritic cells (Fig. S4 C and Table S1) and was removed, whereas two closely related clusters (clusters 2 and 6) were merged, yielding seven final clusters (Fig. 4 B). Based on the expression of characteristic marker genes (Fig. 4 C and Table S1), the clusters were identified as HSC (cluster 1), three clusters of MPP (clusters 2&6, 4, and 5), an additional cluster of MPP with a strong proliferation signature (prolif. MPP, cluster 9), MyP (cluster 7), and megakaryocyte/erythroid progenitors (MEP, cluster 8).
The majority of cells in the resulting dataset (10,594 cells) were derived from one KrasG12D mouse (3,472 cells) and two WT mice (2,829 and 3,461 cells) (Fig. 4 D). The contributions of individual KrasG12D and WT mice to the HSC cluster 1 were comparable to the entire dataset (Fig. 4 D), suggesting a similarity of KrasG12D and WT HSC transcriptomes. However, MPP clusters 2&6 and 4 were comprised predominantly WT and Kras-mutant cells, respectively (Fig. 4 D). Accordingly, pairwise comparison between WT and Kras cells within the HSC cluster showed minimal differences, with only 6 differentially expressed genes (DEG) showing a greater than twofold difference with statistical significance (adjusted P value <0.05) (Fig. 4 E and Table S2). These included the upregulation of Ras targets Junb and Egr1 and downregulation of the differentiation marker Cd52 (Fig. S4 D and Table S2). In contrast, MPP clusters showed 758 significant DEG (Fig. 4 E and Table S2), which included downregulation of many HSC-related genes (e.g., Meis, Tal1, Pbx1, Mpl, Pdzk1ip1) in mutant MPP (Fig. S4 D). Conversely, mutant MPP showed the upregulation of lymphoid development–associated genes (sterile Ighv transcripts, Rag2, Ccr9, Ebf1, Dntt) and downregulation of Dach1, a negative marker of LyP (Amann-Zalcenstein et al., 2020) (Fig. S4, D and E; and Table S2). Many genes associated with proliferation (e.g., Ccna2, Cdca8, Cenpw, Mcm10, Cdk1) were also upregulated in mutant MPP (Fig. S4, D and F; and Table S2). The extent of differential expression was lower in proliferative MPP (400 DEG), although the same patterns were observed (e.g., downregulation of Pbx1, Mpl, and Dach1, and upregulation of Ighv and Rag2) (Fig. 4 E and Table S2). On the other hand, much less differential expression was observed in MEP (99 DEG) and MyP (18 DEG) (Fig. 4 E and Table S2). Collectively, these data support the notion that Kras mutation has a minimal effect on the transcriptome of HSC, but strongly affects the transcriptome of mutant MPP, which show enhanced differentiation, lymphoid priming, and proliferation.
Osteopontin facilitates the early stages of Kras-mutant progenitor expansion
The rapid takeover of the MPP population by Kras-mutant cells raised the possibility that they outcompete normal cells by mechanisms beyond increased proliferation. We noted that the Kras-specific MPP cluster showed the increased expression of Tnf (encoding the inflammatory cytokine TNF-α) and Spp1 (encoding osteopontin) (Fig. S4 D and Fig. 5 A). Osteopontin is a secreted glycoprotein that is produced by stromal cells and restricts the size of the normal HSC and progenitor pool (Grassinger et al., 2009; Stier et al., 2005). To test its potential role in the expansion of mutant MPP, we crossed Pdzk1ip1-CreER R26Tom WT or KrasG12D mice onto the osteopontin-null (Spp1−/−) background.
First, we tested the contribution of normal HSC in Pdzk1ip1-CreER R26TomSpp1−/− or control Spp1+/+ mice without the KrasG12D allele. Given the relatively slow labeling of normal progenitors in WT reporter mice, we analyzed Tom+ cells 12 wk after tamoxifen administration. The initial labeling of HSC was comparable between Spp1−/− and Spp1+/+ mice as determined by BM biopsy (Fig. S5 A). At the endpoint, we observed a comparable labeling of HSC and a minor <1.5-fold reduction in the labeling of downstream populations in Spp1−/− reporter mice. The reduction reached statistical significance in B cells and their progenitors, and in some additional progenitor populations (megakaryocyte progenitors [MkP], MyP, DN2/3, and DP thymocytes) but not in MPP or in mature myeloid cells (Fig. S5, B–E). We then analyzed tamoxifen-induced KrasG12D reporter mice that were Spp1+/+ or Spp1−/−, which also showed comparable HSC labeling at the start of tracing (Fig. S5 F). Subsequent tracing revealed the expected acceleration of label accrual in KrasG12D mice that was comparable between Spp1+/+ and Spp1−/− reporters at the 7-wk endpoint (Fig. S5, G–J). Thus, osteopontin has a minor effect on the steady-state differentiation of normal HSC and is dispensable for the long-term expansion of Kras-mutant cells.
We then tested an earlier role of osteopontin by examining the KrasG12D reporter mice at 3 wk after labeling, when the expansion of Kras-mutant cells first becomes apparent. We observed a highly significant reduction of Tom+ cells in all mature cells, in all stages of B-cell development in the BM, and in the thymic ETP of Spp1−/− reporter mice (Fig. 5, B–D). We also observed a modest less than twofold reduction in the fraction of Tom+ HSC, potentially suggesting some effect of osteopontin on the maintenance of mutant HSC (Fig. 5 E). Irrespectively, the reduction in the labeling of all downstream progenitors was much more pronounced, suggesting its key role in the reduced labeling of mature cells (Fig. 5 E). Of note, these experiments could not establish the relevant source of osteopontin, which may be derived from mutant MPP, from stromal cells, or both; nor did they identify the specific stage of differentiation affected by osteopontin. Nevertheless, these results suggest a role of osteopontin in the initial expansion of Kras-mutant progenitors, possibly by favoring them over normal progenitors.
KrasG12D-expressing stem/progenitor cells show enhanced motility
We sought to define additional mechanisms whereby mutation-carrying progenitors outcompete their normal counterparts. Stem and progenitor cells in their BM niche display CXCR4-dependent motility that facilitates their retention in the BM and the interaction with stroma (Beck et al., 2014; Upadhaya et al., 2020). To analyze the behavior of Kras-mutant cells in their BM niche, we examined Pdzk1ip1-CreER R26TomKrasG12D or WT mice 2–3 wk after tamoxifen treatment by two-photon laser scanning microscopy (Fig. 6 A). We thinned the bone wall of tibiae in anesthetized mice and imaged the underlying BM continuously for 60 min with a frame rate of 45–55 s. HSC and their progeny were defined as Tom+ and distinguished from autofluorescent macrophages (Upadhaya et al., 2020) (Fig. 6 B, Video 1, and Video 2). At 2–3 wk after tamoxifen treatment, Tom+ cells comprise a mixture of HSC, various progenitors, and some Lin+ cells; thus, HSC and MPP cannot be distinguished by this method. Notably, the composition of Tom+ cells is comparable between KrasG12D and WT reporter mice at 2 wk (Fig. 2, A and B). We found that labeled cells in KrasG12D reporter mice showed higher motility as defined by track reconstruction in the xy plane (Fig. 6, B and C), track and displacement velocities (Fig. 6 D), and mean squared displacement (MSD [Fooksman et al., 2010]) over time (Fig. 6 E). Critically, all these differences were significant both at 3 and at 2 wk.
To model the increased motility of Kras-mutant progenitor cells in vitro, we tested Lin− BM cells from KrasG12D or WT reporter mice in a transwell migration assay toward the CXCR4 ligand CXCL12 (Fig. 6 F). Cells were additionally enriched for MPP by depletion with antibodies against CD11c, CD115, CD16/32, and CD41. Tom+ cells showed lower migration than Tom− cells in WT mice, likely because they comprise HSC and early progenitors that are less mobile in vitro than the more differentiated Tom− cells. Importantly, Tom+ cells from KrasG12D mice showed enhanced migration toward CXCL12, whereas the migration of Tom− cells was similar to WT (Fig. 6 G). Accordingly, we found that the expression of CXCR4 was significantly increased on Tom+ MPP but not on HSC from KrasG12D mice (Fig. 6, H and I). Collectively, these results suggest that Kras-mutant cells show increased motility in their BM niche in vivo; moreover, MPP-enriched cells show increased CXCR4-dependent migration in vitro, corresponding to the increased CXCR4 expression on MPP.
CXCR4-dependent motility of Kras-mutant progenitors contributes to their expansion in vivo
To prove that the increased in vivo motility of Kras-mutant progenitors is driven by CXCR4, we used LY2510924, a potent selective peptide antagonist of CXCR4 (Peng et al., 2015). We imaged Tom+ stem/progenitor cells in live tamoxifen-induced KrasG12D or WT reporter mice for 60 min as above, administered LY2510924 i.v., and continued the imaging for an additional 60 min. After LY2510924 administration to WT reporter mice, Tom+ cells rounded up and ceased to move (Video 3 and Fig. 7 A). The same change of morphology and reduction of motility were observed in Tom+ cells from KrasG12D mice, although not to the level of WT mice (Video 4 and Fig. 7 A). The reduced motility of Tom+ cells after LY2510924 administration in both KrasG12D and WT reporter mice was evident from shortened tracks (Fig. 7 B), reduced track and displacement velocities (Fig. 6 C), and reduced MSD of Tom+ cells (Fig. 7 D). Thus, LY2510924 blocks CXCR4 and reduces the motility of Kras-mutant cells in the BM.
To test whether CXCR4 blockade affects the rapid expansion of Kras-mutant cells from MPP in vivo, KrasG12D or WT reporter mice at 2 wk after tamoxifen treatment were treated with LY2510924 or with vehicle (PBS) for 1 wk and analyzed at the 3-wk endpoint (Fig. 7 E). As expected, HSC labeling was similar between KrasG12D and WT mice and was unaffected by the treatment (Fig. 7 F). However, the highly elevated labeling of MPP and downstream progenitors in KrasG12D reporters was significantly reduced by LY2510924 (Fig. 7 F). Furthermore, the inhibitor significantly reduced the labeling of mature cells in the spleen of treated KrasG12D reporter mice (Fig. 7 G). A similar trend (albeit with higher variability) was observed in the BM (Fig. 7 H). Thus, CXCR4 mediates the increased motility of Kras-mutant progenitors in the BM and contributes to their rapid expansion in vivo.
Kras-targeted therapy abolishes the expansion of mutant cells but spares their HSC reservoir
Our data suggest that HSC serve as a reservoir of the KrasG12D mutation, which rapidly spreads to multiple lineages via hypercompetitive progenitors. To test whether the chemical inhibition of mutant KRAS may impair the maintenance or spread of the mutation, we first used a combination therapy with mirdametinib (PD0325901) and pictilisib (GDC-0941). These small molecules inhibit RAS-activated MEK/ERK and PI(3)K signaling pathways, respectively, and have shown efficiency against KrasG12D-driven leukemia (Dail et al., 2014). 2 wk after tamoxifen induction, Pdzk1ip1-CreER R26Tom/Tom WT or KrasG12D mice were treated daily with PD0325901 and GDC-0941 for 4 days followed by GDC-0941 alone for 3 days, and the fraction of Kras-mutant Tom+ cells was examined in the spleen and BM (Fig. 8 A). In this experiment, the fraction of Tom+ HSC showed a significant ∼1.3-fold expansion in untreated KrasG12D versus WT reporter mice, and this expansion was abolished by the treatment (Fig. 8 B). However, Tom+ MPP and erythromyeloid progenitors showed the expected >10-fold expansion in KrasG12D reporters, and this expansion was reduced ∼2-fold and ∼4-fold, respectively (Fig. 8 B). Accordingly, the expansion of Tom+ cells within mature splenic myeloid and lymphoid lineages of treated KrasG12D reporter mice was reduced three- to fourfold, reaching significance in granulocytes, monocytes, and T cells (Fig. 8 C). The minor reduction of mutant HSC is unlikely to contribute to the effect, because (1) HSC contribution to differentiation is extremely low at this early time point (Upadhaya et al., 2018); and (2) only MPP and downstream progenitors underwent a major expansion commensurate with that of mature cells (Fig. 8, B and C).
To directly target the driver mutation, we used a recently developed small molecule inhibitor of the mutant KRASG12D protein, MRTX1133. Unlike PD0325901/GDC-0941, MRTX1133 targets the mutation itself rather than its downstream pathways, exhibits a >1,000-fold selectivity for KRASG12D-expressing versus KRASWT cells, and does not have any off-target effects at therapeutic concentrations used herein (Wang et al., 2022; Wei et al., 2024). Pdzk1ip1-CreER R26Tom/TomKrasG12D mice 3 wk after tamoxifen induction were treated with MRTX1133 twice a day for 1 wk and euthanized (Fig. 8 D). Consistent with the high specificity of MRTX1133 for KRASG12D, the treatment did not affect the BM cellularity (Fig. 8 E) or the fractions of total (i.e., combined Tom+ and Tom−) stem/progenitor cells within the BM (Fig. 8 F) in KrasG12D reporter mice. Furthermore, MRTX1133-treated KrasG12D reporter mice showed no reduction of Tom+ Kras-mutant cells in HSC or ST-HSC (Fig. 8 G). In contrast, Tom+ cells were significantly reduced greater than threefold among MPP and downstream progenitors (Fig. 8 G) and among all mature lineages including T cells (Fig. 8 H). Accordingly, Tom+ cells were virtually eliminated in the thymus of KrasG12D reporter mice (Fig. 8 I). Thus, Kras inhibitors selectively impair the expansion of mutant multipotent and lineage-committed progenitors, although an effect on HSC cannot be completely ruled out.
Finally, we tested the durability of the inhibitor effect by treating the KrasG12D or WT reporter mice between 3 and 4 wk after induction with MRTX1133, and allowing them to recover for 3 wk (Fig. 9 A). We chose MRTX1133 over PD0325901/GDC-0941 given its above-mentioned specificity for KRASG12D, as well as its exclusive effect on mutant MPP but not on HSC (Fig. 8 G). PB tracing showed that MRTX1133 had no effect on the accrual of Tom+ cells in WT reporter mice immediately after the treatment (4 wk) or following the recovery (7 wk) (Fig. 9, B and C). In contrast, MRTX1133 treatment of KrasG12D reporter mice reduced the expanded fraction of Tom+ cells ~2-fold in granulocytes, ~4-fold in monocytes, and ~3-fold in lymphocytes at 4 wk (Fig. 9 B). These data are consistent with the endpoint analysis of MRTX1133-treated KrasG12D reporter mice at 4 wk (Fig. 8 H) and confirm the specificity of MRTX1133 for Kras-mutant cells. Following the 3-wk recovery, the reduction of Tom+ cells in MRTX1133-treated KrasG12D reporter mice was no longer significant in granulocytes and diminished to <1.3-fold in monocytes (Fig. 9 C). Accordingly, Tom+ cells were no longer reduced among myeloid cells in the spleen of MRTX1133-treated KrasG12D reporter mice at the postrecovery endpoint (Fig. 9 D). The fraction of Tom+ cells was still reduced ~2-fold in lymphocytes, likely due to the longer lifespans of the latter. Within the stem/progenitor compartment, Tom+ cells were unaffected in HSC, and reduced ∼1–5-fold among ST-HSC and MPP and ∼1.2-fold among committed progenitors (Fig. 9 E), in contrast to the much stronger reduction immediately after the treatment (Fig. 8 G). Similarly, the massive expansion of Tom+ thymocytes was no longer reduced in MRTX1133-treated mice (Fig. 9 F). The caveats of this experiment include the reduced concordance of KrasG12D induction and Tom expression at 7 wk (Fig. S1 A), and a trend toward higher labeling of HSC in the KrasG12D versus WT reporters (Fig. 8 G and Fig. 9 E), which may mask some effects of MRTX1133 on HSC. Nevertheless, collectively these data suggest that (1) therapeutic inhibition of Kras signaling reduces the early expansion of Kras-mutant cells; (2) mutant MPP and downstream progenitors are diminished by the therapy but rebound after its cessation; and (3) mutant HSC are less affected by the therapy than MPP, thus providing a durable reservoir of the mutation.
Discussion
We employed HSC-specific inducible Cre recombination to trace the natural history of leukemia-associated mutations as they progress from HSC to mature lineages in unmanipulated animals. This system revealed that the transforming KrasG12D mutation dramatically accelerated multilineage HSC contribution, equilibrating the labeling of mutant HSC and their progeny within 5–7 wk compared with 1–2 years required for normal HSC (Jang et al., 2023). This was in contrast to the deletion of Tet2, which did not affect the net contribution of the HSC population to steady-state hematopoiesis. Importantly, our tracing may have been affected by the incomplete correlation of Tet2 deletion with reporter expression, and/or missed the expansion of individual clones (Schiroli et al., 2024). On the other hand, our results are consistent with the persistence of TET2 mutations in healthy humans with ARCH for many years, and with very slow latency or absence of leukemogenesis in Tet2-deficient mice, respectively (Ko et al., 2011; Li et al., 2011; Moran-Crusio et al., 2011; Quivoron et al., 2011). Notably, a combination of HSC-specific Tet2 deletion with inflammatory stimulation resulted in a significant increase in HSC contribution to MyP and mature cells, in agreement with previous reports (Abegunde et al., 2018; Cai et al., 2018; Meisel et al., 2018) and supporting the emerging role of inflammation in ARCH and myeloid malignancy (Avagyan and Zon, 2023; Balandrán et al., 2023; Jakobsen et al., 2024; King et al., 2020). Thus, transforming mutations such as activated Kras appear distinct from ARCH-associated mutations in their major effect on HSC contribution in the steady state.
Unexpectedly, Kras-mutant HSC showed only limited transcriptomic abnormalities and no increased proliferation, unlike the hyperproliferative HSC reported in mice with a constitutive KrasG12D activation (Sabnis et al., 2009). However, HSC in the latter study were defined as Flt3− LSK cells; this definition would include MPP3, which are shown herein to undergo expansion and hyperproliferation following KrasG12D activation. Indeed, the frequency of functional HSC in this system was reduced, while their contribution to hematopoiesis was increased (Sabnis et al., 2009), consistent with our data. In contrast to HSC, mutant MPP (including both MPP3 and MPP4) showed rapid expansion, increased proliferation, and a distinct transcriptional signature characterized by increased differentiation and lymphoid priming. The latter was associated with a particularly rapid contribution of mutant HSC to lymphopoiesis, compared with the slow and stochastic contribution of normal HSC (Upadhaya et al., 2018). This is consistent with the activation of multiple signaling pathways including JAK/STAT, ERK, and mTOR by KRASG12D in hematopoietic stem/progenitor cells, resulting in hyperproliferation in vitro (Van Meter et al., 2007). An important question raised by these findings is why KRAS activation has a relatively minor effect on HSC but a dramatic effect on MPP. One likely explanation is the glycolysis-driven relative metabolic quiescence of HSC (Kasbekar et al., 2023; Snoeck, 2017), which may “resist” the enhanced signaling induced by KRASG12D. However, this may not be the sole explanation, because even the more active downstream progenitors were affected less than MPP in terms of their proliferation or transcriptome. One potentially relevant mechanism is Flt3 signaling, which is induced at the HSC-to-MPP transition and is subsequently downregulated in committed progenitors. While Kras activation may be insufficient to fully substitute for the absent Flt3 signaling in HSC, it may synergize with the physiological Flt3 signaling in MPP to enhance their proliferation and lineage priming, as shown in immature lymphocytes (Li et al., 2010). Indeed, both normal and aberrantly activated Flt3 signaling activate Kras (Köthe et al., 2013), and Ras pathway activation accounts for the resistance of myeloid leukemia to FLT3 inhibitors (McMahon et al., 2019). Collectively, these data emphasize the distinction between stem cells as the cells of origin of the mutation, and their progeny as targets of the mutation’s activity.
The progeny of Kras-mutant HSC completely took over the MPP compartment within 1 wk, showing a dramatic competitive advantage over their normal counterparts. This is likely facilitated by the observed increase in their proliferation rate; however, the proliferation was accompanied by the correspondingly increased differentiation into downstream lineages, but not by the acquisition of self-renewal capacity. Therefore, additional mechanisms such as the observed upregulation of Tnf and Spp1 may confer an advantage specifically to mutation-carrying MPP. TNF-α (encoded by Tnf) and other TNF family members may modulate stroma-derived factors such as IL-7 (Feng et al., 2023); however, given its complex role in HSC biology (Yamashita and Passegué, 2019), its role in Kras-mutant MPP would have to be explored in the future. Here, we found that the global deletion of Spp1 (encoding osteopontin) delayed the initial expansion of KrasG12D MPP, albeit it did not prevent it in the longer term. This was in contrast to the minor impact of osteopontin loss on the HSC-driven hematopoietic flux in the steady state. Osteopontin is a complex glycoprotein that is secreted by stromal cells (particularly osteoblasts) and processed by thrombin, with the resulting fragments binding to CD44 and integrins on stem/progenitor cells and restricting their growth (Cao et al., 2019; Grassinger et al., 2009; Stier et al., 2005). It is possible that Kras activation may facilitate the resistance of mutant MPP to stroma-derived osteopontin. In addition, given that osteopontin was ectopically expressed in Kras-mutant MPP, this expression may facilitate their expansion by inhibiting the proliferation of normal MPP. Of note, thrombin-cleaved osteopontin facilitates lymphopoiesis (Guidi et al., 2017; Kanayama et al., 2017), in agreement with the observed lymphoid priming of mutant MPP. This scenario is also consistent with the elevated levels of osteopontin in myeloid leukemia patients (Liersch et al., 2012) and its cell-intrinsic role in the mouse model of myeloid leukemia (Zhou et al., 2022) and in other Kras-driven tumors such as lung adenocarcinoma (Giopanou et al., 2020; Wang et al., 2017). Even if indirect, our data suggest that mutation-carrying progenitors may utilize a secreted mediator such as osteopontin to outcompete their normal counterparts.
Another mechanism associated with the competitive advantage of Kras-mutant progenitors was their increased motility driven by the chemokine receptor CXCR4. Whereas activated Kras is known to increase the motility of tumor cells (Okudela et al., 2004; Pollock et al., 2005), our data demonstrate the same phenomenon in Kras-mutant progenitor cells prior to overt transformation. Most progenitor cells in the BM are motile in a CXCR4-dependent manner, including HSC (Upadhaya et al., 2020) and B-cell progenitors (Beck et al., 2014). In the latter case, CXCR4-driven motility allowed progenitors to resist the high shear force of the blood flow and hence facilitated their retention in the BM. We found that KrasG12D-expressing cells manifested increased motility in their native BM niche before and during their expansion, even though this analysis did not capture specific cell types. This motility was CXCR4-dependent, corresponding to the elevated expression of CXCR4 on mutant MPP. Most importantly, in vivo pharmacological blockade of CXCR4 reduced the motility and delayed the expansion of KrasG12D MPP and their contribution to mature lineages. Indeed, motility facilitates a continuous interaction with multiple stromal cells (Upadhaya et al., 2020) and hence the reception of signals derived from them, such as CXCL12 (Greenbaum et al., 2013) and membrane-bound Kit ligand (Ding et al., 2012). In addition, hyperactive CXCR4 may alter the BM stromal compartment (Zehentmeier et al., 2022), which may further impair the function of normal stem/progenitor cells. Because CXCR4-dependent motility and maintenance appear linked, these results suggest that mutation-carrying progenitors may outcompete normal cells by “outrunning” them in their BM niche. However, a motility-independent role of CXCR4 in the maintenance and expansion of Kras-mutant progenitors is entirely possible. CXCR4 represents a promising therapeutic target in leukemia (Cancilla et al., 2020) and in particular is important for the propagation of T-ALL in their BM niche (Hawkins et al., 2016; Passaro et al., 2015; Pitt et al., 2015). Our data suggest that it may also be important for the initial spread of mutant cells in the preleukemic state.
Collectively, our system reveals a striking effect of a common transforming mutation on the normal HSC-driven hematopoiesis long before the onset of leukemia. Surprisingly, this mutation did not affect HSC behavior but reprogrammed MPP into hypercompetitive cells that hijack normal hematopoiesis. In this “two-component” model, HSC serve as a source and long-term reservoir of the mutation, whereas the reprogrammed progenitors rapidly spread the mutation into all lineages. This model is consistent with the reconstructions of single-cell trajectories in human leukemia, which suggest that mutations arise in stem cells and are selected at the level of progenitors (Beneyto-Calabuig et al., 2023; Nam et al., 2022; Van Egeren et al., 2021). Moreover, it underscores the ability of current mutation-targeting therapies to expunge mutant progenitors and their progeny, but not the upstream reservoir of the mutation (Pollyea and Jordan, 2017; Schepers et al., 2015). The latter likely serves as a basis for subsequent disease relapses and should represent the key target of next-generation therapies for hematological malignancies.
Materials and methods
The antibodies, animal strains, reagents, software, and genotyping primers are listed in Table S3.
Animals
All animal studies were performed according to the investigator’s protocol approved by the Institutional Animal Care and Use Committee of New York University Grossman School of Medicine (NYUGSoM). Pdzk1ip1-CreER transgenic mouse strain was crossed with the Cre-inducible R26Tom reporter strain on C57BL/6 genetic background as described previously (Sawai et al., 2016; Upadhaya et al., 2018). All R26Tom mice were homozygous for the reporter allele. Mice of either sex were used, as no major differences in the efficiency or specificity of labeling were observed between them. Pdzk1ip1-CreER R26Tom/Tom reporter animals were crossed with Tet2flox mice ([Moran-Crusio et al., 2011] maintained by the Aifantis Lab at NYUGSoM). Pdzk1ip1-CreER R26Tom/Tom mice were also crossed with LSL-KrasG12D mice (Jackson et al., 2001) on pure C57BL/6 background (strain 008179; Jackson Laboratories) to generate reporter mice heterozygous for the KrasG12D allele. In addition, Pdzk1ip1-CreER R26Tom/+KrasG12D mice were crossed with Spp1−/− mice (Liaw et al., 1998) on pure C57BL/6 background (strain 004936; Jackson Laboratories).
Animal procedures
To induce Cre recombination, mice were administered with the different concentrations of tamoxifen (Sigma-Aldrich) in sunflower oil by gavage: 0.5 mg (KrasG12D and control reporter mice) or 1 mg (Tet2fl/fl and control reporter mice). PB sampling and BM biopsy were conducted as described previously (Sawai et al., 2016). Briefly, blood (∼100 µl) was collected by submandibular vein puncture with a sterile disposable lancet. BM biopsy was performed on isoflurane-anesthetized mice by inserting a 28.5G insulin syringe needle into the joint surface of the femur through the patellar tendon and then into the bone cavity. Up to 20 µl of the BM suspension (∼106 cells) was harvested. At the endpoint, animals were sacrificed and single-cell suspensions were prepared from the BM (femora and tibiae), spleens, and thymi, counted, and analyzed by flow cytometry.
To mimic microbial stimulation, Pdzk1ip1-CreER R26Tom/TomTet2fl/fl mice were injected i.p. with 0.1 mg of the TLR2 agonist Pam3CSK4 for 3 consecutive days starting from the day of tamoxifen treatment. To block CXCR4 in vivo, Pdzk1ip1-CreER R26Tom/Tom or Pdzk1ip1-CreER R26Tom/TomKrasG12D reporter mice were induced with tamoxifen, and 2 wk later were injected s.c. with LY2510924 (3 mg/kg) or PBS twice a day for 7 days, followed by euthanasia.
For small molecule treatments, Pdzk1ip1-CreER R26Tom/TomKrasG12D or WT mice were induced with tamoxifen, and 2 wk later were treated concomitantly with GDC-0941 (100 mg/kg) daily for 7 days and with PD0325901 (5 mg/kg) for the first 4 days via oral gavage, followed by euthanasia. The chemicals were dissolved in 0.5% carboxymethylcellulose sodium (CMC-Na). Control mice were treated in a similar way with the vehicle (CMC-Na) only. To directly inhibit the mutant KRASG12D protein, Pdzk1ip1-CreER R26Tom/TomKrasG12D or WT mice were induced with tamoxifen, and after 3 wk were administered with MRTX1133 (30 mg/kg) via i.p. injection twice a day for 7 days, and euthanized since the last injection or 3 wk later for the endpoint analysis. MRTX1133 was dissolved in 10% sulfobutylether-β-cyclodextrin (SBE-β-CD) in 50 mM citrate buffer, pH 5.0. Control mice were treated in a similar way with the vehicle (10% SBE-β-CD) only.
To test the reconstitution capacity of Kras-mutant progenitors, Pdzk1ip1-CreER R26Tom/TomKrasG12D mice were administered with tamoxifen and euthanized 5 wk later. Sorted Tom+ HSC (50 cells) or Tom+ MPP (1,000 cells) were mixed with 2 × 105 total BM cells from CD45.1 congenic mice and transferred into lethally irradiated CD45.1 congenic recipients. Two donor mice were induced, and donor cells from each mouse were transferred separately. PB was collected, stained with flow antibodies, and analyzed with the fractions of donor-derived Tom+ cells in the mature blood cell types up to 3 mo after transplantation.
Cell staining and flow cytometry
Cells from PB, BM, spleens, and thymi were suspended in 2% FBS-contained PBS after red blood cell lysis and wash. Cells were stained with indicated fluorochrome-conjugated antibodies (Table S3) for 20 or 60 min in 2% FBS-containing PBS. Samples were acquired on an Attune NxT flow cytometer (Thermo Fisher Scientific) and analyzed using FlowJo software (FlowJo, LLC). The definition of cell populations is described below.
For stem/progenitor staining, the lineage cocktail included antibodies of CD11b, CD11c, B220, TCRβ, Gr-1, NKp46, and Ter119, HSC (LSK CD150+ CD48−), ST-HSC (LSK CD150− CD48−), MPP (LSK CD150− CD48+), EryP (Lin− Sca-1− c-Kit+ CD150+ CD41−), MkP (Lin− Sca-1− c-Kit+ CD150+ CD41+), MyP (Lin− Sca-1− c-Kit+ CD150− CD41−), common myeloid progenitors (Lin− Sca-1− c-Kit+ CD16/32− CD34+), granulocyte–monocyte progenitors (Lin− Sca-1− c-Kit+ CD16/32+ CD34+), and MEP (Lin− Sca-1− c-Kit+ CD16/32− CD34−). The expression of IL-7Ra was measured in Sca-1− c-Kit− and c-Kitlo/int progenitors, Sca-1− c-Kit+ MyP, and Sca-1+ c-Kit+ MPP4. Subsets of B cells in the BM were identified as pre-pro B (IgD− IgM− B220+ CD43+ CD93+ CD19−), pro B (IgD− IgM− B220+ CD43+ CD93+ CD19+), pre B (IgD− IgM− B220+ CD43− CD93+ CD19+), and Imm B (IgD− IgM+ B220+ CD43− CD93+ CD19−). Thymocytes were determined as ETP (CD4− CD8− Lin− CD44+ CD25− c-Kit+), double-negative thymocytes 2/3 (DN2, CD4− CD8− Lin− CD25+), double-negative thymocytes 4 (DN4, CD4− CD8− CD44− CD25−), DP thymocytes (CD4+ CD8+ TCRβ−), CD4 T cells (CD4+ CD8− TCRβ+), and CD8 T cells (CD4− CD8+ TCRβ+). Immune cells in PB, BM, and spleen were defined as platelets (Plt, low forward and side scatter, Ter119− CD150+ CD41+), granulocytes (Neu, TCRβ− B220− NKp46− CD11b+ Ly6Cint Ly6G+), monocytes (Mono, TCRβ− B220− NKp46− CD11b+ Ly6Chi), NK cells (NK, TCRβ− B220− NKp46+), B cells (B, CD11b− Ly6C− Ly6G− NKp46− B220+), and T cells (T, CD11b− Ly6C− Ly6G− NKp46− TCRβ+).
EdU cell proliferation assay
Pdzk1ip1-CreER R26TomKrasG12D or WT mice were administered 0.5 mg of tamoxifen. 3 or 5 wk later, they were injected with 1 mg of EdU i.p. and euthanized 2 h later. EdU incorporation was measured by flow cytometry using Click-iT Plus EdU Alexa Fluor 488 Flow Cytometry Assay Kit. In some experiments, mice induced with tamoxifen 3 wk earlier were injected with 1 mg EdU on two consecutive days and euthanized 48 h after the first injection. Cells were stained for EdU incorporation as above, along with DNA content using FxCycle Violet Stain.
CITE-seq sample preparation
Pdzk1ip1-CreER R26Tom or Pdzk1ip1-CreER R26TomKrasG12D mice were treated with 0.5 mg of tamoxifen. After 3 wk after induction, mice were sacrificed and femora, tibiae, humerus, and pelvises were isolated. Bones were flushed with 2% FBS-contained PBS, and red blood cell lysis was performed. Lineage cells were depleted with a magnetic enrichment step (Streptavidin MicroBeads; Miltenyi) using a cocktail biotinylated antibodies against B220, TCRβ, NKp46, Ter119, CD11b, and Gr-1. After lineage depletion, the resulting cells were stained with TruStain FcX Fc Blocking Reagent (BioLegend) followed by incubation with PB-conjugated lineage antibodies, c-Kit, Sca-1, and live/dead cell dye, as well as TotalSeq B hashtags (BioLegend). For the KrasG12D group, ∼10,000 Tom+ LSK cells were sorted per mouse, and for the WT group, ∼10,000 total LSK cells were sorted per mouse. Cells from two individual mice per group (four hashtagged samples) or four different hashtag-connected samples were pooled, and the resulting cell mixture was prepared according to the 10x Genomics protocol and loaded onto the 10x Chromium Controller for gel beads-in-emulsion generation and barcoding. Libraries were prepared and sequenced using Illumina NovaSeq 6000 at the Genome Technology Core at NYUGSoM.
CITE-seq analysis
Reads from fastq files were processed using 10X Genomics’ Cellranger count pipeline (v.6.0.1). Demultiplexing of hashtags was performed using Seurat’s HTODemux method, with resulting singlets passed into iCellR for quality control (QC) and dimensionality reduction using KNetL with zoom setting set to 500 and sensitivity set to 500—resulting in nine clusters. QC settings included the following: mitochondrial reads for a given cell <5% of reads while retaining only those cells with between 200 and 6,000 genes, yielding 11,781 cells. One cluster was identified as a contaminant and removed, yielding 10,594 cells. For the pairwise comparison of selected populations, the iCellR (v.1.6.7) cluster-wise (cell type), condition-wise (WT, KRAS) comparisons were performed using the run.diff.exp method with “clustBase.condComp” argument. For each cell type and each gene independently, cells were grouped by condition with the Wilcoxon rank-sum test performed on the means of the two groups.
Intravital imaging
Pdzk1ip1-CreER R26Tom/Tom or Pdzk1ip1-CreER R26Tom/TomKrasG12D mice were treated with 0.5 mg tamoxifen. 2 or 3 wk later, mice were examined by intravital two-photon laser scanning microscopy of the tibial BM as described previously (Upadhaya et al., 2020). Mice were anesthetized with isoflurane inhalation and secured in a supine position to an imaging plate heated to 37°C. An incision was made in the lower leg, and soft tissue was removed to expose the medial region of the tibia. The medial tibia was thinned to a thickness of ∼200 μm using a microdrill. A custom-built appliance with an opening was used to immobilize the leg and to expose the drilled bone to the microscope objective. A water-tight immersion well lined with vacuum grease was made on the appliance over the exposed bone and filled with warm lactated Ringer’s solution for the microscope objective. Mice were anesthetized with isoflurane inhalation during the entire intravital imaging session. To block CXCR4 in vivo, mice were imaged for 60 min as above, injected i.v. with 3 mg/kg of LY2510924 in PBS, and imaged for a further 60 min.
Two-photon images were acquired with an Olympus FV1000-MPE upright laser scanning microscope equipped with a 25× 1.05NA water immersion objective and Mai Tai DeepSee Ti:Sapphire pulsed laser (Spectra-Physics) that was tuned to 920 nm. The microscope was fitted with a custom-built incubated chamber to maintain a temperature of 37°C and block infiltrating room light. Movies were recorded for ∼60 min, and frames were acquired continuously without breaks. Each frame consisted of a 512 × 512 μm Z-stacked image that was 75–90 μm deep and acquired in 3-μm-thick slices. The amount of time elapsed between each consecutive frame was 45–55 s, depending on the depth of imaging.
Intravital imaging analysis
Image analysis was conducted using Imaris software 9.3. Cells were detected based on size and fluorescence and tracked three-dimensionally in autoregressive motion mode. Red tdTomato fluorescence was distinguished from background autofluorescence and channel bleed-through by creating ratio and subtracted channels. Drift correction was performed by three-dimensionally tracking sessile autofluorescent macrophages and correcting XYZ registrations over time. Track velocity was measured as the sum of the displacements between all consecutive time point pairs of a given cell track (e.g., t1 and t2, t2 and t3) divided by the total real-time duration of the track in minutes. Displacement velocity was measured as the distance between the first and last positions of a given cell track divided by the total real-time duration of the track in minutes. MSD is a composite measurement of all cell tracks in a given movie. For each time point of a movie converted to real-time minutes, the square of the distance between a cell’s initial position and the cell’s current position was averaged for all present cell tracks. For accuracy, only the linear portions of the MSD functions were analyzed.
Migration assay
Pdzk1ip1-CreER R26Tom or Pdzk1ip1-CreER R26TomKrasG12D mice were treated with 0.5 mg of tamoxifen. 3 wk later, mice were euthanized and lineage-negative cells were isolated by magnetic negative selection as described above for CITE-seq, but with additional antibodies to CD11c, CD115, CD16/32, and CD41 to deplete committed progenitors.
The migratory capacity was assayed using 6.5-mm transwells with 5.0 μm pore polycarbonate membrane (Corning). Cells were resuspended in RPMI medium supplemented with 0.5% BSA, penicillin/streptomycin, L-glutamine, and 50 μM 2-mercaptoethanol at 3 × 104 cells/100 μl and incubated for 2 h at 37°C. Medium containing CXCL12 (100 ng/ml in 600 μl) was added to the bottom wells; cells were added to the upper chamber and incubated for 4 h at 37°C. Cells migrated to the bottom wells were harvested, and the numbers of Tom+ or Tom− cells were determined by flow cytometry. Migration was measured as a percentage of migrated cells out of the total input for Tom+ or Tom− cells.
Detection of recombined Tet2fl/fl or KrasG12D allele
For Pdzk1ip1-CreER R26Tom/TomTet2fl/fl mice, Tom+ versus Tom− LSK cells were sorted at 3 wk after tamoxifen treatment, or Tom+ versus Tom− total BM cells were sorted at 6 mo after tamoxifen treatment. Genomic DNA was extracted by using Quick-DNA Miniprep Kit, and the recombination of Tet2fl/fl was tested by genomic PCR for the expected amplicon sizes: recombined (null) allele ∼550 bp, floxed allele ∼430 bp, and WT allele ∼250 bp. For Pdzk1ip1-CreER R26Tom/TomKrasG12D mice, Tom+ versus Tom− LSK cells were sorted at 3 wk after tamoxifen treatment, or Tom+ versus Tom− total BM cells were sorted at 5 or 7 wk after tamoxifen treatment. Genomic PCR was performed for the expected amplicon sizes: recombined (KrasG12D) allele ∼650 bp, LSL-cassette allele 500 bp, and WT allele 622 bp. Densitometric analysis of the gels was done in ImageJ2, by determining the density of a fixed area corresponding to each band, and subtracting the background density from the same lane.
Statistical analysis
Statistical significance was determined with the indicated tests using the Prism software (GraphPad).
Online supplemental material
Fig. S1 shows aberrant thymocyte development and T-cell lymphomas in mice with induced KrasG12D mutation in HSC. Fig. S2 shows the contribution of Tet2-deficient HSC to hematopoiesis in the steady state. Fig. S3 shows increased contribution of Tet2-deficient HSC to myelopoiesis after inflammatory stimulation. Fig. S4 shows additional analyses of the CITE-seq data. Fig. S5 shows additional characterization of HSC contribution in osteopontin-deficient mice. Video 1 shows intravital microscopy of Tom+ stem/progenitor cells in WT or KrasG12D reporter mice at 2 wk after tamoxifen induction. Video 2 shows intravital microscopy of Tom+ stem/progenitor cells in WT or KrasG12D reporter mice at 3 wk after tamoxifen induction. Video 3 shows intravital microscopy of Tom+ stem/progenitor cells in LY2510924-treated WT reporter mice at 3 wk after tamoxifen induction. Video 4 shows intravital microscopy of Tom+ stem/progenitor cells in LY2510924-treated KrasG12D reporter mice at 3 wk after tamoxifen induction. Table S1 lists DEG in CITE-seq cell clusters. Table S2 lists DEG between WT and Kras-mutant cells. Table S3 lists antibodies and reagents used in the study. Source data for Fig. 1 B, Fig. S1 A, and Fig. S2 G show uncropped gel images for the respective figures.
Data availability
Acknowledgments
We thank Makiko Hayashi and Thales Papagiannakopoulos for advice and help with MRTX1133 treatment. We acknowledge the use of NYU Genome Technology Center, which is partially supported by the Laura and Isaac Perlmutter Cancer Center.
This study was supported by the National Institutes of Health grant HL152637 (to B. Reizis, D.R. Fooksman), the Edward P. Evans Foundation (to B. Reizis), the Hirsch Foundation (to D.R. Fooksman), and SIRIC BRIO (to C.M. Sawai). Trainee support to R. Park was provided by the Medical Scientist Training Program’s National Institutes of Health training grant T32 GM149364.
Author contributions: G. Jang: conceptualization, data curation, formal analysis, investigation, methodology, project administration, validation, visualization, and writing—original draft. R. Park: formal analysis, investigation, visualization, and writing—original draft, review, and editing. E. Esteva: data curation, formal analysis, software, visualization, and writing—original draft. P.-F. Hsu: investigation. J. Feng: investigation. S. Upadhaya: writing—review and editing. C.M. Sawai: conceptualization, methodology, and writing—review and editing. I. Aifantis: resources and writing—review and editing. D.R. Fooksman: conceptualization, formal analysis, investigation, methodology, resources, software, supervision, validation, and writing—review and editing. B. Reizis: conceptualization, funding acquisition, project administration, supervision, and writing—original draft, review, and editing.
References
Author notes
Disclosures: The authors declare no competing interests exist.
J. Feng’s current affiliation is Department of Pharmacological Sciences, Icahn School of Medicine at Mount Sinai, New York, NY, USA.
S. Upadhaya’s current affiliation is Cancer Research Institute, New York, NY, USA.
Supplementary data
shows DEG in CITE-seq cell clusters.
shows antibodies and reagents.