


The fission yeast, Schizosaccharomyces pombe, is an excellent eukaryote model organism for studying essential biological processes. Its genome contains ∼1,200 genes essential for cell viability, most of which are evolutionarily conserved. To study these essential genes, resources enabling conditional perturbation of target genes are required. Here, we constructed comprehensive arrayed libraries of plasmids and strains to knock down essential genes in S. pombe using dCas9-mediated CRISPRi. These libraries cover ∼98% of all essential genes in fission yeast. We estimate that in ∼60% of these strains, transcription of a target gene was repressed so efficiently that cell proliferation was significantly inhibited. To demonstrate the usefulness of these libraries, we performed metabolic analyses with knockdown strains and revealed flexible interaction among metabolic pathways. Libraries established in this study enable comprehensive functional analyses of essential genes in S. pombe and will facilitate the understanding of essential biological processes in eukaryotes.

Introduction
Fission yeast forward and reverse genetics are very powerful for studying genes involved in essential physiological processes, including the cell cycle, chromosomal segregation, and centromere functions (Fantes and Hoffman, 2016; Hoffman et al., 2015). Genes involved in essential processes are supposedly necessary for cell viability, and their deletion is lethal. The S. pombe genome contains 1,221 essential genes (PomBase, https://www.pombase.org/, accessed on August 24th, 2022, see Materials and methods) (Harris et al., 2022; Rutherford et al., 2024), deletion of which in haploid spores derived from heterozygous diploid deletion strains completely inhibits germination or cell proliferation under optimal growth conditions, i.e., nutrient-rich YES complete medium at 25°C and 32°C (Kim et al., 2010). The essentiality of such genes is well conserved among eukaryotes, consistent with the fact that molecular mechanisms underlying those essential processes are well conserved. For example, ∼80% (953/1221) of them are also essential in distantly related budding yeast, Saccharomyces cerevisiae (Kim et al., 2010).
Biological resources, including whole-genome sequences and comprehensive genetic deletion libraries, have facilitated genome-wide studies on the genetic functions of this organism (Kim et al., 2010; Wood et al., 2002). However, functional characterization of genes required for cell viability remains challenging because of lethality caused by the loss of their functions. To circumvent this issue, conditionally lethal mutations, such as temperature-sensitive mutations, must be employed. However, using conventional random mutagenesis, such conditional mutations can be isolated only by chance. According to the genome database of fission yeast (PomBase, https://www.pombase.org/, accessed on August 24th, 2022) (Harris et al., 2022; Rutherford et al., 2024), temperature- and cold-sensitive alleles are available for only 14% of the 1,221 essential genes of fission yeast. Systematic gene perturbation methods, including promoter replacement, 3′UTR disruption, and auxin-inducible protein degradation (Bähler et al., 1998; Ishikawa and Saitoh, 2023; Kanke et al., 2011; Roguev et al., 2008; Zhang et al., 2022), permit knockdown of an additional 13% of essential genes (Harris et al., 2022; Rutherford et al., 2024). Thus, in total, ∼300 essential genes have conditional knockdown alleles to date, whereas the remaining ∼900 essential genes are still difficult to access in genetic analyses.
In addition to the conventional methods described above, several CRISPR-based gene perturbation methods have been implemented recently in S. pombe (Chen et al., 2023; Ishikawa and Saitoh, 2023; Ishikawa et al., 2021; Jing et al., 2018; Zhao and Boeke, 2020). We introduced the gene perturbation method, CRISPR interference (CRISPRi), to S. pombe using catalytically inactive Cas9 (dCas9) (Ishikawa et al., 2021). Since this method has higher throughput than other techniques, it is a promising approach for constructing a large number of conditional knockdown strains in this organism (Ishikawa and Saitoh, 2023). CRISPRi can inhibit transcription of an arbitrary gene through specific binding to the target region by a ribonucleoprotein complex formed with dCas9 protein and single-guide RNA (sgRNA) (Bikard et al., 2013; Qi et al., 2013). CRISPRi for S. pombe can be switched on/off by changing the concentration of thiamine in the medium (Ishikawa et al., 2021). With this method, functions of essential genes can be downregulated conditionally, i.e., only in the absence of thiamine. A key to determining the efficiency of transcriptional repression by CRISPRi is the sgRNA targeting sequence, which binds in a complementary fashion to the targeted gene locus. Our observations using model genes, ura4+ and ade6+, suggested that sgRNAs binding to the non-template strand of the transcription start site (TSS) or the template strand ∼90 bp downstream of the TSS, effectively repress target transcription (Ishikawa et al., 2021). This discovery dramatically reduced the labor required to find effective targeting sequences for CRISPRi. In fact, this design protocol allowed us to construct knockdown strains for uncharacterized essential genes, and functional analyses using this approach proved feasible (Ishikawa et al., 2023).
CRISPRi strain libraries have been constructed either as pooled libraries or as arrayed libraries (Gilbert et al., 2014; Momen-Roknabadi et al., 2020; Smith et al., 2017b; Smith et al., 2016; Sun et al., 2023). A pooled library is a mixture of knockdown strains targeting different genes. Although pooled libraries have been successfully utilized for genetic screening, this form of library has two technical limitations (Gilbert et al., 2014; Momen-Roknabadi et al., 2020; Smith et al., 2016). First, its phenotypic evaluation is limited to fitness or abundance of each strain in the mixture, measured by deep sequencing. Second, direct access to individual knockdown strains is difficult or impossible. On the other hand, arrayed libraries, which are constituted with knockdown strains stocked separately, can potentially be applied to any phenotypic analysis, including fitness phenotypes, morphological phenotypes, e.g., cellular and organelle shape, population phenotypes, e.g., mating-type switch, and biochemical phenotypes such as metabolomic phenotypes. Moreover, in arrayed libraries, individual knockdown strains are directly accessible, and this facilitates broad applications for detailed individual analyses (Smith et al., 2017b). Thus, arrayed libraries offer greater flexibly in applications than pooled libraries. Currently, complete arrayed libraries of CRISPRi strains covering all annotated genes have not been established in any model organism (Sun et al., 2023), although partial arrayed libraries established for many bacterial species have been applied in large-scale analyses of fitness, cell morphology, and metabolomics (Anglada-Girotto et al., 2022; de Wet et al., 2020; Donati et al., 2021; Peters et al., 2016; Sun et al., 2023). In eukaryotes, only Saccharomyces cerevisiae has a partial arrayed library covering most essential genes and genes required for respiration (Smith et al., 2017b). As strains of this S. cerevisiae library were constructed with chromosomally integrated CRSIPRi modules, crossing library strains with a strain of interest and selecting strains of purpose are required for systematic analyses in other genetic backgrounds.
In this study, we constructed comprehensive arrayed libraries of plasmids and strains to perturb essential genes in S. pombe using a dCas9-mediated CRISPRi. To our knowledge, these are the first plasmid-based arrayed libraries for comprehensive knockdown of essential genes in eukaryotes, enabling the introduction of CRISPRi modules for each target gene to strains with various genetic backgrounds by simple transformation. In ∼60% of the strains in the library, transcription of a targeted gene was repressed so efficiently that cell proliferation was significantly inhibited, indicating that the library is applicable for genome-wide high-throughput studies. Additionally, the arrayed format of the library also allowed us to conduct complicated phenotypic analyses on cellular morphologies and metabolomes of arbitrarily selected knockdown strains, proving its feasibility for various applications. Metabolomic analyses in the present study provided biological insights into NAD biosynthesis and the interaction between glycolysis and the pentose-phosphate pathway. Thus, libraries constructed in this study open new avenues for comprehensive analyses of essential genes in S. pombe.
Results
sgRNA design for comprehensive library construction
To conduct comprehensive CRISPRi for essential genes, we optimized and automated the targeting sequence design of sgRNAs. Our previous study showed that the reverse targeting (RV) sequence that is complementary to the non-template strand nearest the TSS and the forward targeting (FW) sequence, which is complementary to the template strand, nearest a site ∼90 bp downstream from the TSS is suitable for dCas9-mediated CRISPRi in fission yeast. Notably, RV sequences tend to provide better transcriptional repression than FW sequences in a limited number of examples of CRISPRi for model target genes, ade6+, ura4+, his2+, and his7+ (Ishikawa et al., 2021). To test whether this tendency is generally applicable to other genes, greater numbers of essential genes were subjected to CRISPRi with RV and FW sequences, and their transcriptional repression efficiencies were examined (Fig. S1). As the genes examined in this study are essential for cell viability, their transcriptional repression is predicted to cause growth retardation and/or cell death. FW and RV targeting sequences for 23 genes were designed, and their abilities to inhibit colony formation in CRISPRi were examined. RV sequences inhibited colony formation in 11 of 23 tested genes, whereas FW sequences inhibited colony formation in only 2 of them (aro2 and fol3). These observations strongly suggest that RV targeting sequences are more likely to repress transcription of targeted genes than FW sequences; thus, RV sequences should be the first choice in attempting CRISPRi of arbitrary genes. Therefore, we decided to utilize RV sequences to conduct comprehensive CRISPRi for essential genes in S. pombe.

CRISPRi for a subset of essential genes using reverse and forward targeting sequences. Serial dilution (10-fold) of knockdown strains for the indicated target genes was spotted on EMM2 plates after induction of CRISPRi. Asterisks indicate inhibited colony formation. RV, reverse targeting sequence; FW, forward targeting sequence.
CRISPRi for a subset of essential genes using reverse and forward targeting sequences. Serial dilution (10-fold) of knockdown strains for the indicated target genes was spotted on EMM2 plates after induction of CRISPRi. Asterisks indicate inhibited colony formation. RV, reverse targeting sequence; FW, forward targeting sequence.
Automation of sgRNA design is necessary to facilitate the construction of CRISPRi-libraries. For this purpose, we devised a Python script to choose targeting sequences (Datas S1, S2, and S3). Briefly, the script reads names and sequences around the TSS (±300 bp of the TSS) of ∼1,200 genes listed in a text file named “query.csv” and searches the sequences for candidate targeting sequences using CRISPRdirect (https://crispr.dbcls.jp/) (Fig. 1 A). Among these candidates, RV sequences located from TSS-20 (20 bp upstream of the TSS) to TSS+20 (20 bp downstream of the TSS) are listed. As it was reported that the presence of homopolymers (>4 nucleotides, UUUU, GGGG, or AAAA) reduces CRISPRi efficiency in another model system (Gilbert et al., 2014), candidate sequences containing these homopolymer(s) were excluded from the list. The RV sequence nearest the TSS among the listed candidates was then employed for library construction. In case all candidate sequences were eliminated due to homopolymer sequences, the RV sequence nearest the TSS was selected from candidates positioned from TSS-14 to TSS+14, regardless of the presence of homopolymers. When RV sequences were not found within TSS±14, the RV sequence that did not contain homopolymer sequences and was nearest to the TSS among the candidates was employed. If necessary, FW sequences situated from TSS+60 to TSS+150 were listed, and the sequence nearest the TSS+90 was selected after the exclusion of those containing homopolymer(s). Positions of the TSS were obtained from the Eukaryotic Promoter Database (https://epd.expasy.org/epd) (Meylan et al., 2020). Positions of TSSs and distances between the TSS and the targeting sequence are available in Table S1.

Designing targeting sequences. (A) An algorithm to design targeting sequences for CRISPRi. Rev sequences targeting sites closest to the TSS were selected, while avoiding homopolymers, poly-N (>4 nucleotides, UUUU, GGGG, and AAAA), if possible (See the Results for details). Targeting sequences designed by this algorithm are categorized into three classes, RV-1, RV-2, and RV-3. RV-1, RV sequences located ±20 of the TSS without homopolymers; RV-2, RV sequences situated ±14 of the TSS with homopolymers; RV-3, RV sequences distantly located (less than −20 or greater than +20) from the TSS without homopolymers. (B) Coverage of the knockdown-library. Coverages are shown by the gene ontology category “biological processes.” “const. & chara.” denotes genes for which knockdown strain were constructed and for which proliferation phenotypes (colony formation ability and MGR) were characterized. “const” indicates genes for which knockdown strains were constructed, but not phenotypically characterized. “not const.” identifies genes for which knockdown strains were not constructed.
Designing targeting sequences. (A) An algorithm to design targeting sequences for CRISPRi. Rev sequences targeting sites closest to the TSS were selected, while avoiding homopolymers, poly-N (>4 nucleotides, UUUU, GGGG, and AAAA), if possible (See the Results for details). Targeting sequences designed by this algorithm are categorized into three classes, RV-1, RV-2, and RV-3. RV-1, RV sequences located ±20 of the TSS without homopolymers; RV-2, RV sequences situated ±14 of the TSS with homopolymers; RV-3, RV sequences distantly located (less than −20 or greater than +20) from the TSS without homopolymers. (B) Coverage of the knockdown-library. Coverages are shown by the gene ontology category “biological processes.” “const. & chara.” denotes genes for which knockdown strain were constructed and for which proliferation phenotypes (colony formation ability and MGR) were characterized. “const” indicates genes for which knockdown strains were constructed, but not phenotypically characterized. “not const.” identifies genes for which knockdown strains were not constructed.
Double-stranded oligo DNAs with the selected targeting sequences, which are listed in Table S2, were inserted into the sgRNA gene encoded on a CRISPRi vector, pSPdCas9, which contains an expression cassette for a dCas9 gene placed under a thiamine-controllable promoter (Ishikawa et al., 2021). Wild-type S. pombe cells were then transformed individually with the resulting plasmids. In this study, we refer to these collections of plasmids and strains as the plasmid library and the knockdown library, respectively. These libraries cover ∼98% (1194/1221) of all essential genes in S. pombe (Table S3) and 92–100% of essential genes in each gene ontology biological process, which were defined by the Quick Little Tool (QuiLT) (Harris et al., 2022) (Fig. 1 B). Knockdown strains and plasmids of genes for which the TSS was not determined (27 genes) were not constructed.
Characterization of the knockdown-library on cell proliferation
To characterize the efficiency of CRISPRi, a subset of the knockdown library, which comprises 508 strains covering 41.6% (508/1221) of all essential genes, was tested for colony formation ability and proliferation rate after induction of CRISPRi as follows (Fig. 2 A). This subset covers 11 of the 14 gene ontology processes of essential genes in S. pombe, i.e., cell catabolism, cellular component biogenesis, cell cycle, chromatin organization, DNA metabolism, lipid metabolism, membrane organization, protein folding, signaling, small molecule metabolism, and unknown (Fig. 1 B). To measure colony formation ability, transformed cells were cultivated in liquid synthetic minimal EMM2 medium without thiamine, in which the dCas9 protein for CRISPRi is actively expressed. The resulting cell cultures were spotted on a solid EMM2 medium lacking thiamine. After 72 h of incubation at 33°C, colony sizes and densities were compared with those of a nonsense control strain (n.s.) transformed with the pSPdCas9 vector carrying nonsense sgRNA, which does not match any region of the S. pombe genome. Representative results of the spot test are shown in Fig. 2 B and comprehensive results are in Figs. S2, S3, and S4. 56.3% (286/508) of the strains in the subset showed clear inhibition of colony formation (Figs. S2, S3, and S4). Among them, 70.6% (202/286) did not form visible colonies. For quantitative evaluation of cell proliferation, maximum growth rates (MGR, divisions/hour) of transformants in liquid media were measured by continuous monitoring the OD600 of individual cell cultures in 96-well plates containing EMM2 liquid medium lacking thiamine at 33°C (Ishikawa et al., 2023). Representative results of the MGR measurement are shown in Fig. 2 C and comprehensive results are in Fig. 3 A and Table S3. The MGR of the nonsense control strain was 0.213 ± 0.008 (mean ± SD, 24 biological replicates), whereas 55.3% (281/508) of the subset showed reduced MGR (<0.197, lower than the mean MGR of the nonsense control by twofold of the standard deviation). Among them, 33.1% (93/281) of these strains stopped cell proliferation completely (MGR < 0) (Fig. 3 A). Several strains showed MGRs lower than 0, and this indicates that their OD600 decreased during measurement. A decrease of the OD600 might occur due to cell lysis or shortening. Collectively, colony formation and/or cell proliferation were greatly impaired in 61.2% (311/508) of the subset strains, 82.3% (256/311) of which were affected in both colony formation and MGR (Fig. 3 B). This indicates that targeting sequences designed by our method effectively repressed transcription of targeted genes; thus, the algorithm for designing the CRISPRi targeting sequence proposed in the previous study (Ishikawa et al., 2021) is generally applicable to most S. pombe genes.

Procedure to construct and evaluate the knockdown library. (A) Procedure to construct and evaluate the library. Plasmids carrying the dCas9 gene and sgRNA genes were modified by cloning targeting sequences specific to essential genes. A parent strain of the fission yeast was independently transformed with each plasmid that contains a specific targeting sequence. Knockdown strains that target different essential genes were stored separately in a 96-well format without inducing CRISPRi due to inhibition with thiamine. To evaluate the influence of genetic perturbation in knockdown strains, CRISPRi was induced by removing thiamine, followed by characterization of proliferation through colony formation and time-course measurement of OD600. (B) Examples of colony formation assays for 48 knockdown strains are shown. The left panel indicates target genes subjected to transcriptional repression by CRISPRi. Light blue indicates genes for which colony formation was inhibited after knockdown. The right panel displays photos showing colony formation of each knockdown strain. Cells after CRISPRi induction were grown on EMM2 plate at 33°C for 72 h. n.s., nonsense control strain that carries a plasmid with a nonsense sgRNA gene. (C) Examples of MGR measurements for 48 knockdown strains are shown. The left panel indicates target genes subjected to CRISPRi. Light green indicates genes for which MGR was significantly reduced after gene knockdown. The center panel indicates a growth curve drawn by measuring the time course of OD600. The right panel indicates MGRs calculated from time course measurements of OD600. Knockdown strains after CRISPRi induction were grown in EMM2 liquid medium with OD600 measurements every 30 min. A dark green line or plot denotes data of the nonsense control strain (n.s.). A light green line or plot shows data of strains with reduced MGR (affected). A gray line and plot show data of strains with unaffected MGR (unaffected).
Procedure to construct and evaluate the knockdown library. (A) Procedure to construct and evaluate the library. Plasmids carrying the dCas9 gene and sgRNA genes were modified by cloning targeting sequences specific to essential genes. A parent strain of the fission yeast was independently transformed with each plasmid that contains a specific targeting sequence. Knockdown strains that target different essential genes were stored separately in a 96-well format without inducing CRISPRi due to inhibition with thiamine. To evaluate the influence of genetic perturbation in knockdown strains, CRISPRi was induced by removing thiamine, followed by characterization of proliferation through colony formation and time-course measurement of OD600. (B) Examples of colony formation assays for 48 knockdown strains are shown. The left panel indicates target genes subjected to transcriptional repression by CRISPRi. Light blue indicates genes for which colony formation was inhibited after knockdown. The right panel displays photos showing colony formation of each knockdown strain. Cells after CRISPRi induction were grown on EMM2 plate at 33°C for 72 h. n.s., nonsense control strain that carries a plasmid with a nonsense sgRNA gene. (C) Examples of MGR measurements for 48 knockdown strains are shown. The left panel indicates target genes subjected to CRISPRi. Light green indicates genes for which MGR was significantly reduced after gene knockdown. The center panel indicates a growth curve drawn by measuring the time course of OD600. The right panel indicates MGRs calculated from time course measurements of OD600. Knockdown strains after CRISPRi induction were grown in EMM2 liquid medium with OD600 measurements every 30 min. A dark green line or plot denotes data of the nonsense control strain (n.s.). A light green line or plot shows data of strains with reduced MGR (affected). A gray line and plot show data of strains with unaffected MGR (unaffected).

Evaluation of the knockdown library by colony formation. Left panels are lists of target genes. Light blue indicates genes for which colony formation was inhibited by CRISPRi of the target gene. Orange/n.s. indicates the nonsense control strain with a nonsense sgRNA. The right panels indicate the results of colony formation after induction of CRISPRi. Spots of knockdown strains are arranged in the same way as the left panels. After induction of CRISPRi for indicated genes, strains were spotted on EMM2 plates and incubated at 33°C for 72 h. Fig. S2 B is identical to Fig. 2 B.
Evaluation of the knockdown library by colony formation. Left panels are lists of target genes. Light blue indicates genes for which colony formation was inhibited by CRISPRi of the target gene. Orange/n.s. indicates the nonsense control strain with a nonsense sgRNA. The right panels indicate the results of colony formation after induction of CRISPRi. Spots of knockdown strains are arranged in the same way as the left panels. After induction of CRISPRi for indicated genes, strains were spotted on EMM2 plates and incubated at 33°C for 72 h. Fig. S2 B is identical to Fig. 2 B.

Evaluation of the knockdown library by colony formation. Left panels are lists of target genes. Light blue indicates genes for which colony formation was inhibited by CRISPRi of the target gene. Orange/n.s. indicates the nonsense control strain with a nonsense sgRNA. Right panels indicate the results of colony formation after induction of CRISPRi. Spots of knockdown strains are arranged in the same way as the left panels. After induction of CRISPRi for indicated genes, strains were spotted on EMM2 plates and incubated at 33°C for 72 h.
Evaluation of the knockdown library by colony formation. Left panels are lists of target genes. Light blue indicates genes for which colony formation was inhibited by CRISPRi of the target gene. Orange/n.s. indicates the nonsense control strain with a nonsense sgRNA. Right panels indicate the results of colony formation after induction of CRISPRi. Spots of knockdown strains are arranged in the same way as the left panels. After induction of CRISPRi for indicated genes, strains were spotted on EMM2 plates and incubated at 33°C for 72 h.

Evaluation of the knockdown library by colony formation. Left panels are lists of target genes. Light blue indicates genes for which colony formation was inhibited by CRISPRi of the target gene. Orange/n.s. indicates the nonsense control strain with a nonsense sgRNA. Right panels indicate the results of colony formation after induction of CRISPRi. Spots of knockdown strains are arranged in the same way as the left panels. After induction of CRISPRi for indicated genes, strains were spotted on EMM2 plates and incubated at 33°C for 72 h.
Evaluation of the knockdown library by colony formation. Left panels are lists of target genes. Light blue indicates genes for which colony formation was inhibited by CRISPRi of the target gene. Orange/n.s. indicates the nonsense control strain with a nonsense sgRNA. Right panels indicate the results of colony formation after induction of CRISPRi. Spots of knockdown strains are arranged in the same way as the left panels. After induction of CRISPRi for indicated genes, strains were spotted on EMM2 plates and incubated at 33°C for 72 h.

Characterization of the knockdown library. (A) MGR of each knockdown strain is plotted in order of the highest value. (B) Venn diagram of phenotypic classification of knockdown strains. CF, knockdown strains in which colony formation was inhibited after CRISPRi of a target gene. MGR, knockdown strains in which MGR was significantly decreased after CRISPRi. (C) Histogram showing the number of sgRNAs and the distance from the TSS. The distance from the TSS indicates the length in base pairs (bp) between the TSS and the center of the 20-nt targeting sequence on the genome. (D) Comparison of the ability to inhibit cell proliferation between two groups of sgRNAs. Statistical significance was tested with the chi-square test and its P value is indicated. A percentage (number of sgRNAs) is indicated in each box. −4 to 25 bp, sgRNAs for which the targeting sequence was located −4 to 25 bp from the TSS; others, other sgRNAs; n, number of sgRNAs. (E) Histogram showing the number of sgRNAs and GC content. GC contents were calculated with 20-nt targeting sequences of sgRNAs. (F) Comparison of the ability to inhibit cell proliferation between two groups of sgRNAs whose GC contents were 10–30% and 35–65%. Statistical significance was tested with the chi-square test and its P value is indicated. A percentage (number of sgRNAs) is indicated in each box. n, number of sgRNAs. (G) Comparison of the ability to inhibit cell proliferation between three classes of sgRNA, RV-1, RV-2, and RV-3. Statistical significance was tested with the chi-square test and its P value is indicated. A percentage (number of sgRNAs) is indicated in each box. RV-1, RV sequences located ±20 of the TSS without homopolymers; RV-2, RV sequences situated ±14 of the TSS with homopolymers; RV-3, RV sequences distantly located (less than −20 or greater than +20) from the TSS without homopolymers; n, number of sgRNAs. (H) The ability of sgRNA targeting genes belonging to the gene ontology category “biological process” that was defined using QuiLT. Statistical significance was tested with the chi-square test between the indicated gene ontology group and others. P values are shown. n, number of sgRNAs in each group. (I) Percentage of mRNA levels of target genes after CRISPRi. P values were calculated using the unpaired, two-tailed Welch’s t test. Filled circles indicate actual data points. Black lines indicate medians. effective, sgRNAs that inhibited colony formation and/or MGR after CRISPRi; ineffective, sgRNAs that inhibited neither colony formation nor MGR. n, number of sgRNA.
Characterization of the knockdown library. (A) MGR of each knockdown strain is plotted in order of the highest value. (B) Venn diagram of phenotypic classification of knockdown strains. CF, knockdown strains in which colony formation was inhibited after CRISPRi of a target gene. MGR, knockdown strains in which MGR was significantly decreased after CRISPRi. (C) Histogram showing the number of sgRNAs and the distance from the TSS. The distance from the TSS indicates the length in base pairs (bp) between the TSS and the center of the 20-nt targeting sequence on the genome. (D) Comparison of the ability to inhibit cell proliferation between two groups of sgRNAs. Statistical significance was tested with the chi-square test and its P value is indicated. A percentage (number of sgRNAs) is indicated in each box. −4 to 25 bp, sgRNAs for which the targeting sequence was located −4 to 25 bp from the TSS; others, other sgRNAs; n, number of sgRNAs. (E) Histogram showing the number of sgRNAs and GC content. GC contents were calculated with 20-nt targeting sequences of sgRNAs. (F) Comparison of the ability to inhibit cell proliferation between two groups of sgRNAs whose GC contents were 10–30% and 35–65%. Statistical significance was tested with the chi-square test and its P value is indicated. A percentage (number of sgRNAs) is indicated in each box. n, number of sgRNAs. (G) Comparison of the ability to inhibit cell proliferation between three classes of sgRNA, RV-1, RV-2, and RV-3. Statistical significance was tested with the chi-square test and its P value is indicated. A percentage (number of sgRNAs) is indicated in each box. RV-1, RV sequences located ±20 of the TSS without homopolymers; RV-2, RV sequences situated ±14 of the TSS with homopolymers; RV-3, RV sequences distantly located (less than −20 or greater than +20) from the TSS without homopolymers; n, number of sgRNAs. (H) The ability of sgRNA targeting genes belonging to the gene ontology category “biological process” that was defined using QuiLT. Statistical significance was tested with the chi-square test between the indicated gene ontology group and others. P values are shown. n, number of sgRNAs in each group. (I) Percentage of mRNA levels of target genes after CRISPRi. P values were calculated using the unpaired, two-tailed Welch’s t test. Filled circles indicate actual data points. Black lines indicate medians. effective, sgRNAs that inhibited colony formation and/or MGR after CRISPRi; ineffective, sgRNAs that inhibited neither colony formation nor MGR. n, number of sgRNA.
To investigate whether an sgRNA’s ability to inhibit cell proliferation correlates with a certain feature of targeting sequences, targeting sequences were classified into two groups: “effective” and “ineffective” sequences. “Effective” sequences reduced MGR and/or colony formation (311 sequences), whereas “ineffective” sequences did not (197 sequences). Between these two groups, distance from the TSS and GC content of targeting sequences were compared (Fig. 3, C–F). A histogram of distance from the TSS indicates that targeting sequences located −4 to 25 bp from TSS are more enriched in the “effective” group than in the “ineffective” group (Fig. 3 C). Consistently, 67.8% of 357 sequences located in this region inhibited cell proliferation (and were classified into the “effective” group), whereas the percentage of the effective group decreased to 45.7% among the 151 sequences located outside of this region (Fig. 4 D). Similarly, targeting sequences with no <35% GC content are more enriched in the “effective” groups than in the “ineffective” group (Fig. 3 E). 66.8% of 392 sequences with 35–65% GC content inhibited cell proliferation (“efficient” group), while a smaller proportion (42.2%) of 116 sequences with lower GC content was classified in the “effective” group (Fig. 3 F). Collectively, these results suggest that target sequences that effectively inhibit cell proliferation tend to have higher (>35%) GC content and/or become located in the proximity of the TSS (−4 to 25 bp from TSS).

Cellular morphological analysis of fas2-KD and mis6-KD strains. (A) Fluorescence microscopy of knockdown strains. Cells of indicated strains before (0 h, + thiamine) and after (24, 48 h, −thiamine) induction of CRISPRi were fixed and stained with DAPI, followed by fluorescence microscopy. A scale bar indicates 10 μm. Arrows indicate cells with unequally segregated nuclei. An asterisk indicates a cell with lagging chromosomes. Brightness and contrast are adjusted so that each photo shows a similar signal intensity. n.s., nonsense control strain with nonsense sgRNA; fas2-KD, fas2 knockdown strain; mis6-KD, mis6 knockdown strain. (B) Fluorescence microscopy of temperature-sensitive strains. Cells of indicated strains cultured at the permissive temperature (26°C) or restrictive temperature (36°C) were fixed and stained with DAPI, followed by fluorescence microscopy. A scale bar indicates 10 μm. Arrows indicate cells with unequal nuclei. An asterisk indicates a cell with lagging chromosomes. Brightness and contrast are adjusted so that each photo shows similar signal intensity. wt, wild-type strain; fas2ts, temperature-sensitive mutant of fas2 gene; mis6ts, temperature-sensitive mutant of mis6 gene. Parentheses indicate mutant alleles. (C) Morphological classification utilized in quantification in panels D and E. normal, normal cells; lagging, cells with lagging chromosomes; unequal, cells with unequal nuclei. (D) Frequency of cells showing the indicated morphological phenotypes in knockdown strains. Mitotic and postmitotic cells were examined and classified into categories shown in panel C, under fluorescence microscopy after fixation and DAPI staining. Data are shown as sums of two independent experiments. time, time (h) after induction of CRISPRi; n, number of cells observed for quantification. (E) Frequency of cells showing the indicated morphological phenotypes in temperature-sensitive strains. Mitotic and post-mitotic cells were examined and classified into categories shown in panel C, under fluorescence microscopy after fixation and DAPI staining. Data are shown as sums of two independent experiments. temp., culture temperature; n, number of cells observed for quantification. (F) Serial dilution (10-fold) spot test. Cell suspensions of indicated strains were spotted on EMM2 medium after induction of CRISPRi for 48 h and then incubated at 33°C for 72 h. n.s., nonsense control strain with nonsense sgRNA; fas2-KD, fas2 knockdown strain; mis6-KD, mis6 knockdown strain.
Cellular morphological analysis of fas2-KD and mis6-KD strains. (A) Fluorescence microscopy of knockdown strains. Cells of indicated strains before (0 h, + thiamine) and after (24, 48 h, −thiamine) induction of CRISPRi were fixed and stained with DAPI, followed by fluorescence microscopy. A scale bar indicates 10 μm. Arrows indicate cells with unequally segregated nuclei. An asterisk indicates a cell with lagging chromosomes. Brightness and contrast are adjusted so that each photo shows a similar signal intensity. n.s., nonsense control strain with nonsense sgRNA; fas2-KD, fas2 knockdown strain; mis6-KD, mis6 knockdown strain. (B) Fluorescence microscopy of temperature-sensitive strains. Cells of indicated strains cultured at the permissive temperature (26°C) or restrictive temperature (36°C) were fixed and stained with DAPI, followed by fluorescence microscopy. A scale bar indicates 10 μm. Arrows indicate cells with unequal nuclei. An asterisk indicates a cell with lagging chromosomes. Brightness and contrast are adjusted so that each photo shows similar signal intensity. wt, wild-type strain; fas2ts, temperature-sensitive mutant of fas2 gene; mis6ts, temperature-sensitive mutant of mis6 gene. Parentheses indicate mutant alleles. (C) Morphological classification utilized in quantification in panels D and E. normal, normal cells; lagging, cells with lagging chromosomes; unequal, cells with unequal nuclei. (D) Frequency of cells showing the indicated morphological phenotypes in knockdown strains. Mitotic and postmitotic cells were examined and classified into categories shown in panel C, under fluorescence microscopy after fixation and DAPI staining. Data are shown as sums of two independent experiments. time, time (h) after induction of CRISPRi; n, number of cells observed for quantification. (E) Frequency of cells showing the indicated morphological phenotypes in temperature-sensitive strains. Mitotic and post-mitotic cells were examined and classified into categories shown in panel C, under fluorescence microscopy after fixation and DAPI staining. Data are shown as sums of two independent experiments. temp., culture temperature; n, number of cells observed for quantification. (F) Serial dilution (10-fold) spot test. Cell suspensions of indicated strains were spotted on EMM2 medium after induction of CRISPRi for 48 h and then incubated at 33°C for 72 h. n.s., nonsense control strain with nonsense sgRNA; fas2-KD, fas2 knockdown strain; mis6-KD, mis6 knockdown strain.
In our automatic sgRNA design protocol, RV targeting sequences are categorized into three types of sequences, RV-1, RV-2, and RV-3, in order of priority (Fig. 1 A). RV-1 sequences are located ±20 bp from the TSS and do not contain homopolymers, RV-2 sequences contain homopolymers and are situated ±14 bp from the TSS, and RV-3 sequences are located distantly (less than −20 or >20 bp) from the TSS and do not contain homopolymers. 66.9% of 362 RV-1 target sequences inhibited cell proliferation (Fig. 3 G). This proportion is significantly higher than that of RV-2 (48.2% of 46 sequences), indicating that the presence of homopolymers reduces CRISPRi efficiency in fission yeast, as reported in human tissue culture (Gilbert et al., 2014). The proportion of “effective” sequences in RV-3 (43.9% of 82 sequences) was as low as that in RV-2 compared with that in RV-1, consistent with the above-mentioned result that targeting sequences in the region −4 to 25 bp from TSS are enriched in the “effective” group.
Gene ontology category “biological process” of target genes, which was classified with QuiLT, may reveal a correlation between gene function and susceptibleness to CRISPRi (Fig. 3 H). Among the genes examined, CRISPRi of genes related to cell cycle were more prone to inhibit cell proliferation. The group of cell-cycle related genes contains 24 genes encoding subunits of proteasome (rpt1,2,5; rpn2,3,6,7,8,9,11,12; pre1,3,4,5,8,10; pup1,2,3; mts4, scl1, pts1, and pam1). Interestingly, all sgRNAs targeting these 24 genes inhibited cell proliferation (Fig. S2 C and Table S3). Thus, proteasome is susceptible to CRISPRi, and a high occupancy (24/53) of proteasome genes in the cell-cycle group may explain why CRISPRi-targeting genes in this group were more prone to inhibit cell proliferation. In contrast, genes classified in the gene ontology category, “lipid metabolism and unknown” tended to be more robust toward CRISPRi than other genes (Fig. 3 H).
Transcriptional repression in knockdown strains
As shown above, in ∼60% of the essential genes examined, CRISPRi repressed transcription so efficiently as to retard cell proliferation and/or colony formation significantly. For the remaining genes, CRISPRi may have failed to repress transcription. Alternatively, for these genes, the minimal levels of mRNAs to support cell proliferation may be so low that cells could proliferate even with robust transcriptional repression of the target gene. To examine these possibilities, mRNA levels were quantified in 48 knockdown strains randomly selected from those harboring the “effective” and “ineffective” sgRNAs. These knockdown strains were cultivated in an EMM2 medium lacking thiamine for 26 h, during which CRISPRi for each target gene was induced, and then total RNA samples were prepared. mRNA levels of these genes quantified by RT-qPCR are shown in Fig. 3 I. The “effective” group showed lower median mRNA levels (39.9% of a nonsense sgRNA control) than did the “ineffective” group (74.3%). More than 80% (83.3%, 20/24) of the “effective” group members reduced mRNA levels to <60% of the nonsense control, whereas 75% (18/24) of the “ineffective” group members showed transcription levels >60%. This is consistent with our previous observation that cell growth was not impaired when mRNA levels of some essential genes were not <60% of the control (Ishikawa et al., 2023). These results indicate that the inefficiency of transcriptional repression is a primary reason for the unaffected phenotype in the latter group. To achieve more efficient transcriptional repression in this group, optional CRISPRi methods reported previously may be useful (Ishikawa et al., 2023). Simultaneous application of two sgRNAs for a single target gene can enhance CRISPRi efficiency (Ishikawa et al., 2023). While this method includes trial-and-error steps to select optimal sgRNAs empirically, this is not suitable for high-throughput applications. For some target genes, it was difficult to design the desired targeting sequence because of the limitations of the PAM (5′-NGG). Utilization of a dCas9 variant, dSpG, that has a short PAM (5′-NG) may help to design better sgRNAs (Ishikawa et al., 2023). It should be noted that some TSSs are possibly misannotated in the database and that such misannotation would result in the failure of transcriptional repression by CRISPRi. Accumulation of precise TSS data will improve sgRNA design for CRISPRi.
An arrayed knockdown library enables complicated phenotypical analyses
While many CRISPRi libraries have been constructed as pooled libraries to date, isolating individual strains from a library in this form is difficult, and they are not suitable for analyses of complicated phenotypes of cell morphology and/or metabolomes of individual knockdown strains. In our arrayed knockdown library, each strain is stored separately; therefore, we can select knockdown strains arbitrarily and analyze their morphologies and metabolomes individually. As a proof of principle, we conducted these phenotypic analyses with selected strains as described below.
First, we examined whether gene knockdown by CRISPRi causes expected phenotypes. We analyzed morphological phenotypes of knockdown strains of two essential genes, fas2+ and mis6+. The fas2+/lsd1+ and mis6+ genes encode the α subunit of a fatty acid synthase, conserved in fungi, and an inner centromere protein, conserved in eukaryotes, respectively (Saitoh et al., 1996, 1997). While temperature-sensitive hypomorphic mutations in both mis6 and fas2 cause unequal separation of daughter nuclei during mitosis, defects causing the mitotic phenotype are different. Sister chromatids are segregated unequally in mis6 mutant cells, whereas nuclear components other than chromosomes are supposed to be separated unequally in fas2 mutant cells (Saitoh et al., 1996, 1997). To test whether this phenotype is reproduced by knockdown strains in our library, we observed the cellular morphology of the fas2- and mis6-knockdown (KD) strains with DAPI staining (Fig. 4). Before induction of CRISPRi, they look indistinguishable from the nonsense control. Expectedly, fas2-KD cells started showing the unequal nuclear division phenotype 24 h after CRISPRi induction (Fig. 4 A). This phenotype was identical to that observed in fas2/lsd1 temperature-sensitive mutant cells (Saitoh et al., 1996) (Fig. 4 B). Similarly, 48 h after induction of CRISPRi, the mis6-KD strain produced unequally divided nuclei and lagging chromosomes, which were observed in the mis6 temperature-sensitive mutant cells (Saitoh et al., 1997) (Fig. 4 B). The frequency of morphological anomalies observed in knockdown strains of the library was lower than but comparable with that of temperature-sensitive mutants (Fig. 4, C–E). The frequency of cells showing mitotic phenotypes observed in the mis6-KD strain was not so high as that of fas2-KD cells (Fig. 4 D). Consistent with this result, CRISPRi for fas2 inhibited colony formation nearly completely in a spot test, whereas mis6-KD impaired formation to a lesser extent (Fig. 4 F). In the fas2-KD strain, the proportion of cells with unequal nuclei was reduced from 24 to 48 h (Fig. 4 D). This suggests that cells less susceptible to fas2-knockdown were selected during this period.
Second, we investigated metabolic influences caused by the perturbation of essential metabolic genes using our knockdown strains. We performed a metabolomic analysis in knockdown strains of essential genes, qns1+ and fba1+, which serve important functions in nicotinamide adenine dinucleotide (NAD) production and glycolysis, respectively. While perturbation of genes in metabolic pathways is expected to cause accumulation of metabolic intermediates and reduction of final products in general, this expectation has not been experimentally proven in most metabolic pathways in S. pombe. Here, we explored the influence of gene perturbation for those two essential genes by comparing the nonsense control and our knockdown strains. As expected from their different metabolic functions, the qns1-KD and fba1-KD strains formed distinct metabolic profiles after induction of CRISPRi, which were demonstrated by principal component analysis of their metabolites (Fig. S5 A). A list of measured water-soluble metabolites and their quantities in each biological replicate are available in Table S4.
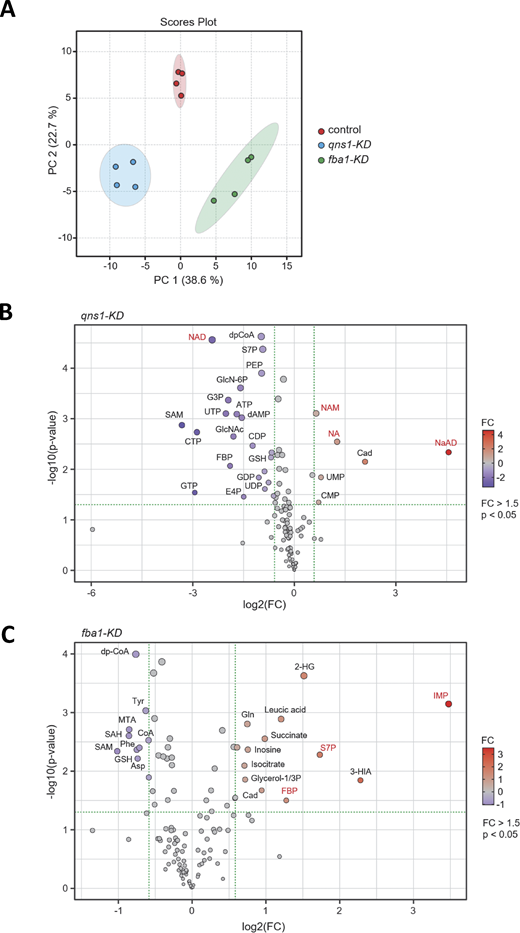
Statistical analysis of metabolomic data in knockdown strains. (A) PC (principal component) analysis of metabolites in control and knockdown strains. Ellipses of clusters show 95% confidence regions for each sample group. Control, nonsense control strain with nonsense sgRNA; qns1-KD, qns1+ knockdown strain; fba1-KD, fba1+ knockdown strain. See also Table S4. (B and C) Volcano plots showing P values versus fold change (FC) from metabolomic data of qns1-KD (B) and fba1-KD (C) strains. Metabolites marked in red are shown in Fig. 5. P values were calculated with unpaired, two-tailed Welch’s t tests. qns1-KD, qns1+ knockdown strain; fba1-KD, fba1+ knockdown strain; n, number of biological replicates NaAD, nicotinic acid adenine dinucleotide; NAM, nicotinamide; NA, nicotinic acid; NAD, nicotinamide adenine dinucleotide; Cad, cadaverine; dpCoA, dephospho-CoA; S7P, sedoheptulose-7-phosphate; PEP, phosphoenolpyruvate; G3P, glyceraldehyde-3-phosphate; FBP, fructose-1,6-bisphosphate; E4P, erythrose-4-phosphate; GlcN-6P, glucosamine-6-phosphate; MTA, 5-methylthioadenosine; 2-HG, 2-hydroxyglutarate; 3-HIA, 3-hydroxyisovaleric acid; IMP, inosine monophosphate.
Statistical analysis of metabolomic data in knockdown strains. (A) PC (principal component) analysis of metabolites in control and knockdown strains. Ellipses of clusters show 95% confidence regions for each sample group. Control, nonsense control strain with nonsense sgRNA; qns1-KD, qns1+ knockdown strain; fba1-KD, fba1+ knockdown strain. See also Table S4. (B and C) Volcano plots showing P values versus fold change (FC) from metabolomic data of qns1-KD (B) and fba1-KD (C) strains. Metabolites marked in red are shown in Fig. 5. P values were calculated with unpaired, two-tailed Welch’s t tests. qns1-KD, qns1+ knockdown strain; fba1-KD, fba1+ knockdown strain; n, number of biological replicates NaAD, nicotinic acid adenine dinucleotide; NAM, nicotinamide; NA, nicotinic acid; NAD, nicotinamide adenine dinucleotide; Cad, cadaverine; dpCoA, dephospho-CoA; S7P, sedoheptulose-7-phosphate; PEP, phosphoenolpyruvate; G3P, glyceraldehyde-3-phosphate; FBP, fructose-1,6-bisphosphate; E4P, erythrose-4-phosphate; GlcN-6P, glucosamine-6-phosphate; MTA, 5-methylthioadenosine; 2-HG, 2-hydroxyglutarate; 3-HIA, 3-hydroxyisovaleric acid; IMP, inosine monophosphate.
The qns1+ gene was predicted to encode glutamine-dependent NAD synthetase, which catalyzes the reaction producing NAD from nicotinic acid adenine dinucleotide (NaAD) (Harris et al., 2022; Katsyuba et al., 2018) (Fig. 5 A). Many eukaryotes, including humans, have a de novo NAD biosynthesis pathway (kynurenine pathway) starting from tryptophan (Castro-Portuguez and Sutphin, 2020). In S. pombe, which does not have the kynurenine pathway, ingested nicotinic acid (NA) was proposed to be anabolized through the NAD synthesis pathway, which converges with the kynurenine pathway at nicotinic acid mononucleotide (NAMN) in humans (Li and Bao, 2007) (Fig. 5 A). The enzyme encoded by the qns1+ gene, glutamine-dependent NAD synthetase, was predicted to catalyze the last reaction in these pathways, converting NaAD to NAD.

Manipulation of metabolic pathways using CRISPRi. (A) NAD biosynthetic pathway. NA, nicotinic acid; NAMN, nicotinic acid mononucleotide; NaAD, nicotinic acid adenine dinucleotide: NAD; nicotinamide adenine dinucleotide; NAM, nicotinamide. Qns1 is an enzyme that putatively catalyzes the conversion of NaAD to NAD. Red, metabolites increased with knockdown of the qns1+ gene; blue, metabolites that decreased with qns1+-knockdown; black, metabolites that were not quantified. (B) Quantification of metabolites. Cells after induction of CRISPRi for the qns1+ gene were subjected to quantification of indicated metabolites with LC-MS/MS. Data show relative metabolite levels with mean ± SD from four biological replicates. P values were calculated using the unpaired, two-tailed Welch’s t test. control, nonsense control strain with nonsense sgRNA; qns1-KD, qns1+ knockdown strain; n, number of biological replicates. (C) A part of glycolysis and associated pathways. Fba1 is fructose-bisphosphate aldolase, which catalyzes the splitting of FBP to DHAP and G3P. Glc, glucose; G6P, glucose-6-phosphate; F6P, fructose-6-phosphate; FBP, fructose-1,6-bisphosphate; G3P, glyceraldehyde-3-phosphate; DHAP, dihydroxyacetone phosphate; R5P, ribulose/ribose-5-phosphate; S7P, sedoheptulose-7-phosphate; PRPP, phosphoribosylpyrophosphate; IMP, inosine monophosphate. Red, metabolites that increased with knockdown of the fba1+ gene. Green, metabolites unaffected by fba1+-knockdown (Quantification of these metabolites, except for that of G3P, is shown in Fig. S6). Black, metabolites that were not quantified. Thick gray arrows indicate a putative rerouting pathway upon fba1 knockdown. (D) Quantification of metabolites. Cells after induction of CRISPRi for the fba1+ gene were subjected to quantification of indicated metabolites with LC-MS/MS. Data show relative metabolite levels with mean ± SD from four biological replicates. P values were calculated using the unpaired, two-tailed Welch’s t test (N.S.; P > 0.05). N.S., not significant; control, nonsense control strain with nonsense sgRNA; fba1-KD, fba1+ knockdown strain; n, number of biological replicates.
Manipulation of metabolic pathways using CRISPRi. (A) NAD biosynthetic pathway. NA, nicotinic acid; NAMN, nicotinic acid mononucleotide; NaAD, nicotinic acid adenine dinucleotide: NAD; nicotinamide adenine dinucleotide; NAM, nicotinamide. Qns1 is an enzyme that putatively catalyzes the conversion of NaAD to NAD. Red, metabolites increased with knockdown of the qns1+ gene; blue, metabolites that decreased with qns1+-knockdown; black, metabolites that were not quantified. (B) Quantification of metabolites. Cells after induction of CRISPRi for the qns1+ gene were subjected to quantification of indicated metabolites with LC-MS/MS. Data show relative metabolite levels with mean ± SD from four biological replicates. P values were calculated using the unpaired, two-tailed Welch’s t test. control, nonsense control strain with nonsense sgRNA; qns1-KD, qns1+ knockdown strain; n, number of biological replicates. (C) A part of glycolysis and associated pathways. Fba1 is fructose-bisphosphate aldolase, which catalyzes the splitting of FBP to DHAP and G3P. Glc, glucose; G6P, glucose-6-phosphate; F6P, fructose-6-phosphate; FBP, fructose-1,6-bisphosphate; G3P, glyceraldehyde-3-phosphate; DHAP, dihydroxyacetone phosphate; R5P, ribulose/ribose-5-phosphate; S7P, sedoheptulose-7-phosphate; PRPP, phosphoribosylpyrophosphate; IMP, inosine monophosphate. Red, metabolites that increased with knockdown of the fba1+ gene. Green, metabolites unaffected by fba1+-knockdown (Quantification of these metabolites, except for that of G3P, is shown in Fig. S6). Black, metabolites that were not quantified. Thick gray arrows indicate a putative rerouting pathway upon fba1 knockdown. (D) Quantification of metabolites. Cells after induction of CRISPRi for the fba1+ gene were subjected to quantification of indicated metabolites with LC-MS/MS. Data show relative metabolite levels with mean ± SD from four biological replicates. P values were calculated using the unpaired, two-tailed Welch’s t test (N.S.; P > 0.05). N.S., not significant; control, nonsense control strain with nonsense sgRNA; fba1-KD, fba1+ knockdown strain; n, number of biological replicates.
Given that Qns1 converts NaAD to NAD, amounts of these metabolites were expected to be altered in qns1-KD cells. Therefore, these metabolites were quantified in the qns1-KD strain. After induction of CRISPRi, qns1-KD and nonsense control cells were broken down in methanol, and their water-soluble metabolites were quantified by liquid chromatography with tandem mass spectrometry (LC-MS/MS) (Fig. 5 B). In qns1-KD cells, the amount of NAD was reduced to about one-fifth of that in control cells, whereas the amount of NaAD increased by 23.5-fold in a specific manner (Fig. S5 B), consistent with the predicted function of Qns1. NAD is consumed by enzymes and converted to nicotinamide (NAM), which is utilized to regenerate NAD through the Preiss-Handler pathway after conversion to NA (Castro-Portuguez and Sutphin, 2020; Preiss and Handler, 1958a, 1958b). NAM and NA also accumulated after the knockdown of qns1+ (Fig. 5 B). Apart from the NAD synthesis pathway, high-energy phosphates (GTP, CTP UTP, and ATP) were reduced in the qns1-KD strain (Fig. S5 B). This is consistent with the reduction of NAD that is required for glycolysis and the TCA (tricarboxylic acid) cycle, which supports ATP production.
In contrast to qns1-KD, in which amounts of metabolites changed as expected, CRISPRi of fba1+ altered the metabolome in a somewhat unexpected manner. Knockdown of the fba1+ gene influenced amounts of metabolites not only in glycolysis but also in associated pathways, the pentose phosphate pathway and purine nucleotide biosynthesis (Fig. S5 C; and Fig. 5, C and D), as detailed below. Fba1 is a fructose-bisphosphate aldolase that catalyzes the reversible split of fructose-1,6-bisphosphate (FBP) to dihydroxyacetone phosphate (DHAP) and glyceraldehyde 3-phosphate (G3P) in glycolysis (Fig. 5 C) (Pirovich et al., 2021). Fission yeast has a Class II aldolase that exhibits no sequence similarity to human Class I aldolases, whereas Class I and II aldolases catalyze the same reaction and share structural features (Mutoh and Hayashi, 1994; Pirovich et al., 2021). As expected, fba1-KD caused an accumulation of its substrate, FBP, by 2.4-fold. However, contrary to expectations, fba1-KD did not reduce the level of the product, G3P (Fig. 5 D). Interestingly, the fba1-KD strain accumulated metabolites in the pentose phosphate pathway (sedoheptulose-7-phosphate, S7P) and in the purine nucleotide biosynthesis pathway (inosine monophosphate, IMP). In comparison with the nonsense control, amounts of S7P and IMP increased by 3.3- and 11.1-fold, respectively (Fig. 5 D). Amounts of other metabolites, glucose-6-phosphate (G6P), fructose-6-phosphate (F6P), ribulose/ribose-5-phosphate (R5P), erythrose-4-phosphate (E4P), phosphoribosyl pyrophosphate (PRPP), and pyruvate were not significantly altered after knockdown of the fba1+ gene (Fig. S6). Accumulation of S7P indicated that fba1-KD may reroute the glycolysis pathway to produce G3P without fructose-bisphosphate aldolase (Fba1) activity. In fba1-KD cells, accumulated FBP may be converted to S7P, and consequently to G3P, through fructose 1,6-bisphosphatase, which produces F6P from FBP, and through the pentose phosphate pathway (Fig. 5 C). Alternatively, in this condition, glucose metabolism may be channeled into the pentose phosphate pathway rather than FBP production in glycolysis. Unexpected alterations in metabolite levels in fba1-KD presumably reflect complex interrelationships among metabolic processes. Collectively, the arrayed form of the knockdown library allowed us to explore complicated phenotypes of cellular morphology and the metabolome, which provided biological insights into NAD biosynthesis and the flexible pathway interaction between glycolysis and the pentose phosphate pathway. These results indicated that our library has the potential for expansion for comprehensive functional analyses of such complicated phenotypes.

Quantification of metabolites, amounts of which were not altered by knockdown of the fba1 + gene. After induction of CRISPRi for the fba1+ gene, cells were subjected to quantification of indicated metabolites using LC-MS/MS. Data show relative metabolite levels with mean ± standard deviation from four biological replicates. P values were calculated using unpaired, two-tailed Welch’s t tests (N.S.; P > 0.05). N.S., not significant; control, nonsense control strain with nonsense sgRNA; fba1-KD, fba1-knockdown strain; n, number of biological replicates; G6P, glucose-6-phosphate; F6P, fructose-6-phosphate; R5P, ribulose/ribose-5-phosphate; E4P, erythrose-4-phosphate; PRPP, phosphoribosylpyrophosphate.
Quantification of metabolites, amounts of which were not altered by knockdown of the fba1 + gene. After induction of CRISPRi for the fba1+ gene, cells were subjected to quantification of indicated metabolites using LC-MS/MS. Data show relative metabolite levels with mean ± standard deviation from four biological replicates. P values were calculated using unpaired, two-tailed Welch’s t tests (N.S.; P > 0.05). N.S., not significant; control, nonsense control strain with nonsense sgRNA; fba1-KD, fba1-knockdown strain; n, number of biological replicates; G6P, glucose-6-phosphate; F6P, fructose-6-phosphate; R5P, ribulose/ribose-5-phosphate; E4P, erythrose-4-phosphate; PRPP, phosphoribosylpyrophosphate.
Discussion
Traditionally, physiological roles of essential genes are analyzed by perturbing their expression/function with conditionally regulatable methods, including utilization of temperature-sensitive alleles, auxin-inducible protein degradation, promoter replacement, and untranslated region disruption (Bähler et al., 1998; Ishikawa and Saitoh, 2023; Kanke et al., 2011; Roguev et al., 2008; Zhang et al., 2022). Though these methods have greatly improved our understanding of essential genes, they cover only a limited number of essential genes in S. pombe. In this report, we constructed comprehensive arrayed libraries of knockdown strains and knockdown plasmids that cover ∼98% of essential genes of S. pombe. About 60% of tested knockdown strains showed inhibited colony formation and/or reduced MGR after induction of CRISPRi (Fig. 3 B), indicating that these libraries are suitable for genome-wide comprehensive studies of essential genes. The arrayed form of the knockdown library allowed us to explore complicated phenotypes of cellular morphologies and metabolomes, demonstrating its utility for various applications. Moreover, the plasmid library facilitates the construction of new arrayed knockdown libraries of S. pombe strains with other genetic backgrounds; therefore, it enables comprehensive studies of genetic interactions that have been difficult to perform by conventional techniques, as discussed below. Thus, the comprehensive plasmid library and knockdown library provided in this study expand our access to genetics of essential genes in this organism.
To date, the majority of the CRISPRi libraries have been constructed as pooled libraries, which are composed of sgRNAs covering large numbers of target genes (Gilbert et al., 2014; Momen-Roknabadi et al., 2020; Smith et al., 2016), whereas the arrayed library maintains each knockdown strain and/or sgRNA separately (Smith et al., 2017b). Pooled libraries are utilized for screens by evaluating fitness or populational abundance of knockdown strains, with deep sequencing in human tissue culture and budding yeast (Gilbert et al., 2014; McGlincy et al., 2021; Momen-Roknabadi et al., 2020). This approach has been successfully applied to genetic screens. In human tissue culture, it identified essential genes, tumor suppressors, and regulators of differentiation (Gilbert et al., 2014), while it revealed genes supporting metabolic pathways in budding yeast (Momen-Roknabadi et al., 2020). Since this library format is evaluated by deep sequencing, phenotype-based screens with pooled libraries require technical breakthroughs as a method developed for a molecular phenotype in an abundance of target mRNAs or proteins (Muller et al., 2020). Thus, currently, pooled libraries are not applicable to systematic analysis of phenotypes that cannot be evaluated with deep sequencing. In contrast, arrayed libraries, like the libraries constructed in this study, can potentially be applied to any phenotypic analyses previously established for individual genetic analyses. Therefore, the libraries established here will provide insights that cannot be obtained with other methods, including traditional genetics and pooled knockdown libraries.
Another characteristic of our libraries is that they are built upon plasmid-based CRISPRi, which is useful for systematic genetic interaction analyses. Conventionally, such analyses have been conducted by crossing knockout strains with a strain of interest, followed by selection with genetic markers (Roguev et al., 2008). However, this method is not applicable to sterile strains, e.g., mating-, meiosis-, and sporulation-defective mutants. Our plasmid-based approach to genetic knockdown can potentially be utilized to test genetic interaction even in sterile mutants by simple transformation, expanding the flexibility of targets in genetic interaction analyses. Additionally, the experimental procedure to select expected strains after transformation is simpler than that after crossing since transformation procedures circumvent the shuffling of chromosomes through meiosis. Thus, plasmid-based gene perturbation resources are superior in flexibility and simplicity to chromosomally modified resources, facilitating their applications in comprehensive functional analyses. Therefore, the comprehensive plasmid library provided in this study is valuable for systematic genetic interaction analyses in S. pombe.
Notably, with some genes, essentiality depends on genetic background, and exploring genetic interactions that suppress lethality upon loss of function of essential genes advanced our understanding of cellular evolution and genotype–phenotype relationships (Li et al., 2019; Liu et al., 2015; Takeda et al., 2019). Our resources may be utilized to promote these studies in S. pombe. While we estimate that ∼60% of knockdown strains in our library impaired cell proliferation after CRISPRi induction, the remaining 40% may also show clear phenotypes in combination with other genetic backgrounds. Our resources are also useful for seeking genetic backgrounds that exacerbate phenotypes of knockdown strains.
CRISPRi is a gene perturbation method with high specificity to target genes as we previously summarized (Ishikawa and Saitoh, 2023). It was reported that the frequency of off-target effects of CRISPRi is much lower than that of RNAi (Evers et al., 2016; Smith et al., 2017a; Stojic et al., 2018). In the present study, we employed targeting sequences that are unique 20-nt sequences in the genome and are situated next to the PAM. Although it is technically possible that the dCas9–sgRNA complex could bind to unexpected sites in the genome through partial sequence similarity, it seems unlikely that such undesired binding of the complex would repress the expression of off-target genes. Mismatches in the 20-nt targeting sequences significantly reduce the efficiency of transcriptional repression by CRISPRi (Bikard et al., 2013; Gilbert et al., 2014). Moreover, our previous and present studies indicate that unless the targeting sequence could bind to the narrow region of a promoter, i.e., −4 to 25 bp form the TSS in the reverse direction, Fig. 3 C, transcription of the off-target genes would not be repressed effectively (Ishikawa et al., 2021, 2023).
Pilot metabolomic analyses with individual strains suggested that the knockdown libraries presented here may reveal complex interrelationships among metabolic pathways. While perturbation of genes involved in metabolic pathways is expected to cause accumulation of metabolic intermediates and reduction of final products in general, this expectation has not been experimentally proven in most metabolic pathways in S. pombe. In the present study, the accumulation of NaAD, a substrate of the reaction catalyzed by Qns1, and the reduction of NAD, the product of the reaction, were empirically confirmed in qns1-KD cells. The drastic increase of NaAD and decrease of NAD in qns1-KD cells clearly indicated that NaAD is consumed solely for NAD production by Qns1 in vivo (Fig. 5). Unexpectedly, knockdown of the fba1+ gene did not result in reduction of G3P, which is the product of the reaction catalyzed by Fba1 (Fig. 5, C and D), suggesting the presence of regulatory mechanisms rewiring the network of glucose carbon metabolic pathways to circumvent the requirement of Fba1 for G3P production when Fba1 activity is diminished. Accumulation of S7P in the fba1-KD strain (Fig. 5 D) suggested this rewiring may occur through the pentose phosphate pathway. Consistently, it has been reported that ribose derived from uridine can be utilized as an energy source through the non-oxidative pentose phosphate pathway, from which its carbon flows into glycolysis and the TCA cycle in mammalian cells (Nwosu et al., 2023; Skinner et al., 2023). Thus, present and previous studies support a flexible connection between glycolysis and the pentose phosphate pathway.
Deletion mutants of genes required for central carbon metabolism allowed improvement in an in silico model of metabolic pathways by revealing a novel reaction, though this approach had previously been intractable for essential genes (Nakahigashi et al., 2009). Recently, CRISPRi enabled systematic analyses of regulatory mechanisms of essential metabolic pathways by providing conditional knockdown strains of essential genes in E. coli (Donati et al., 2021). CRISPRi for essential and non-essential genes for small molecule metabolism was combined with proteomic and metabolomic analyses, revealing mechanisms buffering harmful effects caused by decreased levels of metabolic enzymes (Donati et al., 2021). Similarly, systematic gene knockdown and metabolomic analyses, which can be achieved using our knockdown libraries, may reveal unpredicted mechanisms regulating essential metabolic pathways to maintain metabolite homeostasis in eukaryotes. Systematic metabolomic analyses will facilitate not only understanding of metabolomics but also industrial applications to produce valuable metabolites. Our results show that some metabolites accumulate dramatically after CRISPRi (Fig. 5, NaAD, ∼24-fold; IMP, ∼11-fold). A list of such metabolomes in gene perturbation conditions would facilitate predictions of how metabolic engineering could maximize the production/accumulation of target metabolites. Conditional knockdown of essential genes is inevitable for manipulation of metabolic pathways because ∼18% of genes involved in small molecule metabolism are essential genes (Ishikawa and Saitoh, 2023). Our library covers 97% of the essential genes in this GO process (Fig. 1 B); therefore, this library can be used to establish efficient production of small molecules by manipulating essential metabolic processes.
Materials and methods
List of essential genes and annotation of the gene ontology category “biological process”
Essential genes subjected to CRISPRi in this study are listed as follows. First, we searched for genes that are annotated to the Fission Yeast Phenotype Ontology term “ inviable vegetative cell population” (FYPO:0002061) using the advanced search tool with parameters “include genes from these genotypes: single locus, haploid,” and “expression level: Null (deletion),” at a comparative knowledgebase for Schizosaccharomyces pombe, PomBase (Harris et al., 2013, 2022; Rutherford et al., 2024). This returned ∼1,400 genes, including genes in which deletion mutants are viable, depending on conditions. To remove these genes, we utilized the Quick Little Tool, QuiLT (Harris et al., 2022), which further classified the ∼1,400 genes into “deletion viability: inviable” and “deletion viability: variable.” By selecting the former group, a list of 1,221 essential genes, the deletion of which is invariably lethal, was obtained and subjected to CRISPRi (Table S3). PomBase on 24 August 2022 was used as a primary data source.
Gene ontology (GO) biological processes described in this study (Fig. 1 B and Table S3) reflect classification with QuiLT (Harris et al., 2022; The Gene Ontology Consortium, 2019). When a gene is annotated to multiple GO terms, QuiLT selected one according to a set priority: signaling (GO:0023052), gene expression (GO:0010467), chromatin organization (GO:0006325), protein folding (GO:0006457), cellular component biogenesis (GO:0044085), DNA metabolic process (GO:0006259), cell cycle (GO:0007049), cytoskeleton organization (GO:0007010), membrane organization (GO:0061024), organelle localization (GO:0051640), lipid metabolic process (GO:0006629), small molecule metabolic process (GO:0044281), generation of precursor metabolites and energy (GO:0006091), transport (GO:0006810), cellular catabolic process (GO:0044248), detoxification (GO:0098754), other (i.e., none of the above), and unknown (as described in https://www.pombase.org/documentation/quick-little-tool).
Media and fission yeast strains
Fission yeast cells were grown with Edinburgh Minimal Media 2 (EMM2), which contains 2% glucose, following standard culture conditions (Moreno et al., 1991). To inhibit CRISPRi induction, EMM2 was supplemented with 20 μM thiamine, and this medium is referred to as EMM2+T20. Knockdown strains carrying the plasmid for dCas9-mediated CRISPRi were prepared by the transformation of strain sp685 (h−leu1−) using the standard lithium acetate method (Moreno et al., 1991). Transformants were selected on EMM2+T20 plates utilizing a LEU2 marker coded on the plasmid. A wild-type strain (972, h−) and a temperature-sensitive mutant (sp1469, h−leu1−mis6-302) were from our laboratory collection. Another temperature-sensitive mutant (MY609, h−lsd1-H518) was provided by the National BioResource Project (https://yeast.nig.ac.jp/yeast/top.xhtml).
Automated design of efficient targeting sequences for dCas9-mediated CRISPRi
A Python script and example data for choosing targeting sequences are provided as electronic supplementary material (Datas S1, S2, and S3), and a flowchart of its algorithm is shown in Fig. 1 A. Briefly, using CRISPRdirect (https://crispr.dbcls.jp/), candidates for a targeting sequence were selected from the nucleotide sequence around the TSS (transcription start site) of a target gene, which included 300 bp upstream and downstream from the TSS, and the reverse targeting sequence closest to the TSS and the forward targeting sequence closest to 90 bp downstream of the TSS were chosen among candidates, which are unique 20-nt sequences in the genome situated next to the PAM. Parameters for CRISPRdirect search are database; “pombe,” PAM sequence; “NGG” and format; “txt.” Oligo DNA sequences inserted in CRISPRi plasmids are listed in Table S2. TSS positions of target genes, which were obtained from the EPD, and the distance between the TSS and the targeting sequence are available in Table S1. When a target gene has multiple TSSs, the one with the highest CAGE signal in the gene was employed.
Modification of the targeting sequence in the sgRNA
Conditional gene knockdown was conducted using a plasmid, pSPdCas9, which was devised for dCas9-mediated CRISPRi in S. pombe (Ishikawa et al., 2021). This plasmid contained sgRNA and dCas9 genes. Transcription of the dCas9 gene was controlled by the nmt1-41 promoter, which is inhibited by supplementing 15 μM thiamine (Forsburg, 1993). The targeting sequence of the sgRNA gene coded in the pSPdCas9 plasmid was modified as described below. Briefly, the sgRNA gene contains a cloning site of the targeting/spacer sequence, which was cleaved by a restriction enzyme, BbsI, and modified by ligating a short double-stranded (ds) oligo DNA with an arbitrary targeting sequence. Oligo dsDNAs containing targeting sequences were prepared by annealing two 20-μM, single-stranded oligo DNAs in annealing buffer (10 mM Tris-HCl (pH8.0), 50 mM NaCl, 1 mM EDTA) with a thermal cycler under following conditions: 95°C for 2 min, gradual cooling to 53°C at a rate of −2°C/min, 53°C for 10 min, gradual cooling to 47°C, 47°C for 10 min, and gradual cooling to 25°C. These oligo dsDNAs were independently ligated to the pSPdCas9 plasmid cleaved by BbsI. The intact plasmid, pSPdCas9, without the modification, has the sgRNA gene containing a BbsI cloning site in its targeting sequence region, and this plasmid is utilized as a negative control expressing the nonsense sgRNA.
High-throughput induction of dCas9 expression
Before induction of CRISPRi, knockdown strains were grown on EMM2+T20 plates for about 24 h at 33°C. To induce dCas9-mediated CRISPRi, cells were extensively washed to remove thiamine as follows. Aliquots of each yeast knockdown strain (2 mm in diameter) were resuspended in 500 μl of sterilized water in a 96-well filter plate (1-ml well, 0.45 μm Supor membrane, #8129; PALL). Then, water was removed by vacuum aspiration with a vacuum manifold (Wel-Vac 200, #4-379-1; Matsuura Seisakusho). This cell-washing procedure was repeated six times, and cells were resuspended in 500 μl of sterilized water. The cell suspension was diluted 10-fold with EMM2 liquid medium without thiamine in a 96-well DeepWell plate (#260251; Thermo Fisher Scientific) and this was subjected to incubation at 33°C for 6 h with shaking at 900 rpm. The resulting culture was diluted to 9.4 × 104 cells/ml in 500 μl of EMM2 medium and was then further incubated at 33°C for 20 h with shaking at 900 rpm.
Cell proliferation assays
Cell proliferation efficiency of knockdown strains was evaluated by two methods: colony formation ability and maximum growth rate (MGR) measurements. Colony formation ability was tested by spotting 100-fold diluted cell cultures on EMM2 plates after induction of CRISPRi, followed by incubation at 33°C for 72 h. To quantify growth rates, knockdown strains were subjected to time-course measurements at OD600 after CRISPRi induction. After induction, cell cultures of knockdown strains were diluted twofold with EMM2 in a 96-well plate, and this was incubated at 33°C for 24 h with shaking. The resulting cultures were further incubated after 10-fold dilution with EMM2 medium for 48 h at 33°C with monitoring of the OD600 every 30 min in a plate reader HiTS (Scinics Co.Ltd.). Using the results of the latter 48 h, the growth rate was calculated as the slope of the line best fit to every nine data points of log2 OD600 and time (h) using the least squares method. The maximum growth rate (divisions/hour; MGR) was the highest growth rate during 48 h. Here, the growth rate is defined as how many times cells divide per hour. For example, an MGR of 0.2 indicates that cells take 5 h (1/0.2) to divide once.
Cell morphological analysis
Cellular morphology after induction of CRISPRi for fas2+ and mis6+ genes was analyzed as follows. Briefly, log cultures of the nonsense control, fas2-KD, and mis6-KD strains were prepared with EMM2+T20 medium. To induce CRISPRi, cells of these cultures were extensively washed with sterilized water and grown in the absence of thiamine for 48 h. During this time, these cell cultures were diluted twice to prevent their reaching the stationary phase. For fluorescence microscopy, cells were fixed with 2.5% of glutaraldehyde and were observed in the presence of 25 μg/ml DAPI with an imaging system (EVOS FL AMF4300; Thermo Fisher Scientific, Inc.) including a built-in processing software and an imaging sensor, which was equipped with oil-immersion 100x objective lens (N.A. = 1.35). Morphological analyses of temperature-sensitive mutants were conducted as follows. Wild type (972), fas2ts (lsd1-H518) mutant, and mis6ts (mis6-302) mutant cells were grown in liquid YPD medium at 26°C until early log phase (∼2 × 106 cells/ml), and then the temperature was shifted to 36°C. After additional incubation for 6 h, cells were fixed and examined with fluorescence microscopy. Brightness and contrast were adjusted so that each photo shows a similar signal intensity.
Quantification (Fig. 4, D and E) was conducted by classifying mitotic and post-mitotic cells into three categories, normal, lagging (lagging chromosomes), and unequal (unequal nuclei) (Fig. 4 C). Cells containing daughter nuclei either with or without septa were considered postmitotic cells. Quantitative data are shown as the sum of two independent experiments.
Quantification of metabolites by LC-MS/MS
CRISPRi for the qns1+ and fba1+ genes was induced as described below. Briefly, yeast strains (qns1-KD, fba1-KD, and nonsense control) grown on EMM2+T20 were extensively washed with sterilized water, and 200 μl of the cell suspension were added to 2 ml of EMM2 medium, followed by incubation at 33°C for 6 h. The resulting cell culture was diluted to 9.4 × 104 cells/ml in 60 ml of EMM2 medium, and the cells were further cultured at 33°C for 20 h. About 2 × 108 induced cells were collected by centrifugation at 2 krpm for 2 min at room temperature. After discarding the supernatant, the cell pellet was washed with 1 ml of ice-cold sterilized water by resuspending and removing the supernatant after centrifugation. The resulting cell pellets were resuspended with 1 ml of ice-cold 100% methanol. These cell suspensions were stored at −80°C until quantification of metabolites by liquid chromatography with tandem mass spectrometry (LC-MS/MS), as detailed below.
A widely targeted metabolomic analysis was done as described below (Nishimura, 2020; Yamada et al., 2020). In brief, each frozen sample in a 1.5-ml plastic tube was homogenized in 200 μl of 50% methanol with glass beads using a microtube homogenizer (TAITEC Corp.) at 41.6 Hz for 2 min. Homogenates were mixed with 400 μl of methanol, 100 μl of H2O, and 200 μl of CHCl3 and vortexed for 20 min at RT. Samples were centrifuged at 20,000 × g for 15 min at 4°C. The supernatant was mixed with 350 μl of H2O, vortexed for 10 min at RT, and centrifuged at 20,000 × g for 15 min at 4°C. The aqueous phase was collected, dried in a vacuum concentrator, and redissolved in 2 mM ammonium bicarbonate (pH 8.0). Chromatographic separations were carried out using an Acquity UPLC H-Class System (Waters) under reverse-phase conditions with an ACQUITY UPLC HSS T3 column (100 × 2.1 mm, 1.8 μm particle size, Waters) and under HILIC conditions using an ACQUITY UPLC BEH Amide column (100 × 2.1 mm, 1.7 μm particle size; Waters). Ionized compounds were detected using a Xevo TQD triple quadrupole mass spectrometer coupled to an electrospray ionization source (Waters). Peak areas of target metabolites were analyzed using MassLynx 4.1 software (Waters). Metabolite signals were normalized to the total ion signals of the corresponding sample. P values were calculated using the unpaired, two-tailed Welch’s t test in Microsoft Excel. Further statistical analysis, including PCA (principal component analysis), was performed in MetaboAnalyst 5.0 (Pang et al., 2021). Data were normalized to the median per sample.
To characterize the nonsense control strain, we compared metabolomic profiles of the nonsense control (h−leu1− [pSPdCas9]) and a wild-type strain (strain 972, h−). Some portions of metabolite levels differed between these strains (Fig. S7 and Table S5). Therefore, metabolomic analyses demonstrated in this study are shown as comparisons between the nonsense control strain and knockdown strains to investigate metabolomic alteration caused by CRISPRi of target genes.

Statistical analysis of metabolomic data in control strains. (A) PC analysis of metabolites in wild-type (h−) and nonsense control (h−leu1− [pSPdCas9]) strains. Ellipses of clusters show the 95% confidence regions for each sample group. See also Table S5. control, nonsense control strain that carries a plasmid with a nonsense sgRNA gene. (B) Volcano plots showing P values versus fold change (FC) from metabolomic data of nonsense control strain (h−leu1− [pSPdCas9]). Results suggested sgRNA and dCas9 synthesis (as well as plasmid maintenance) may alter cellular metabolism. P values were calculated with unpaired two-tailed Welch’s t tests. IMP, inosine monophosphate; 2-HG, 2-hydroxyglutarate; FAD, flavine adenine dinucleotide; NAD, nicotinamide adenine dinucleotide; NADP, nicotinamide adenine dinucleotide phosphate; NaAD, nicotinic acid adenine dinucleotide; OPH, ophthalmic acid; GSH, glutathione; MTA, 5-methylthioadenosine; F6P, fructose-6-phosphate; SAH, S-adenosyl-homocysteine; Orn, ornithine. Control, nonsense control strain that carries a plasmid with a nonsense sgRNA gene.
Statistical analysis of metabolomic data in control strains. (A) PC analysis of metabolites in wild-type (h−) and nonsense control (h−leu1− [pSPdCas9]) strains. Ellipses of clusters show the 95% confidence regions for each sample group. See also Table S5. control, nonsense control strain that carries a plasmid with a nonsense sgRNA gene. (B) Volcano plots showing P values versus fold change (FC) from metabolomic data of nonsense control strain (h−leu1− [pSPdCas9]). Results suggested sgRNA and dCas9 synthesis (as well as plasmid maintenance) may alter cellular metabolism. P values were calculated with unpaired two-tailed Welch’s t tests. IMP, inosine monophosphate; 2-HG, 2-hydroxyglutarate; FAD, flavine adenine dinucleotide; NAD, nicotinamide adenine dinucleotide; NADP, nicotinamide adenine dinucleotide phosphate; NaAD, nicotinic acid adenine dinucleotide; OPH, ophthalmic acid; GSH, glutathione; MTA, 5-methylthioadenosine; F6P, fructose-6-phosphate; SAH, S-adenosyl-homocysteine; Orn, ornithine. Control, nonsense control strain that carries a plasmid with a nonsense sgRNA gene.
Quantification of mRNA
mRNA levels of target genes in randomly selected knockdown strains were quantified with reverse transcription-quantitative polymerase chain reaction (RT-qPCR). Yeast cells after induction with CRISPRi were prepared as described above (quantification of metabolites), though here the final culture volume was changed to 10 ml. The resulting yeast cells (0.9–1.5 × 108 cells in 10 ml of culture) were collected by centrifugation at 2 krpm for 2 min at room temperature. Cell pellets were washed with 1 ml of sterilized water and frozen in liquid nitrogen. Frozen pellets were stored at −80°C until total RNA preparation. Total RNA was purified using a nucleic acid purification device, Maxwell RSC (Promega Corp.), with the simplyRNA Tissue Kit (AS1340; Promega Corp.) according to manufacturer instructions with modifications, as follows. Frozen pellets were resuspended in 100 μl of homogenization solution with 1-thioglycerol (1-TG) included in the kit. To these cell suspensions, glass beads were added to the meniscus, followed by beating with a vortex mixer bearing a multiple sample head at maximum speed for 5 min at 4°C. After chilling lysate tubes on ice for 1 min, 250 μl of homogenization solution with 1-TG was added to each lysate. Insoluble components in lysates were removed by centrifugation at 14 krpm for 1 min at 4°C. 200 μl of supernatants were subjected to total RNA purification using the nucleic acid purification device with a standard protocol for the SimplyTissue/Cell kit. This procedure includes DNase treatment to remove genomic DNA. cDNA was prepared by using the ReverTra Ace qPCR RT kit according to manufacturer instructions (TOYOBO Co., Ltd.). cDNA was quantified by qPCR with FastStart Essential DNA Green Master Mix (Roche, Ltd.) and a real-time PCR instrument LightCycler (Roche, Ltd.) as previously reported (Ishikawa et al., 2023). Results are shown as values relative to the act1+ mRNA level. DNA sequences of primers utilized for qPCR are available in the Supplementary Information (Table S6).
Statistical analyses
Statistical significance of data presented in Fig. 3, D and F–H were evaluated by chi-square test. P values in Fig. 3 I; Fig. 5, B and D; and Fig. S6 were calculated using the unpaired, two-tailed Welch’s t test. Data distribution of these figures was assumed to be normal but this was not formally tested.
Reagent availability
Fission yeast strains and plasmids used in this study will be distributed from the National BioResource Project (https://yeast.nig.ac.jp/yeast/top.xhtml). Knockdown strains and their results in cell proliferation analyses are listed in Table S3.
Online Supplemental material
Fig. S1 shows CRISPRi for a subset of essential genes using reverse and forward targeting sequences. Figs. S2, S3, and S4 show the evaluation of the knockdown library by colony formation. Fig. S5 shows a statistical analysis of metabolomic data in knockdown strains. Fig. S6 shows quantification of metabolites, amounts of which were not altered by knockdown of the fba1+ gene. Fig. S7 shows a statistical analysis of metabolomic data in control strains. Table S1 shows TSS positions and distances between TSSs and target DNA. Table S2 shows oligo DNA with targeting sequences. Table S3 shows the strain list of the knockdown library. Table S4 shows metabolomic profiles for knockdown strains. Table S5 shows metabolomic profiles for the nonsense control strain. Table S6 shows qPCR primers. Data S1 is a Pdf file Python script. Data S2 is an Excel file example input data (query.csv) for the Python script. Data S3 is a text file example output data of the Python script.
Data availability
All data are incorporated into the article and its online supplementary material.
Acknowledgments
We thank our colleagues Dr. Yusuke Toyoda, Fumie Masuda, Yuri Okabe, Yoshito Fukushima, Taishi Kita, Tomoki Oomachi, Yuki Ootsubo, and Hidefumi Tokunou for their valuable discussions.
This study was partly supported by grant from the Kurume University Millennium Box Foundation for the Promotion of Science, a grant from the Ishibashi Foundation for the Promotion of Science, the Startup Award for Researchers with Life Events at Kurume University, the Kakihara Foundation for Science and Technology (3-9-14) to K. Ishikawa, the Fukuoka Bio-valley Project to K. Ishikawa and S. Saitoh, Grants-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of Science (17K07394 and 20K06648 to S. Saitoh), and by the MEXT-supported program for strategic research foundation at private universities from the Ministry of Education, Culture, Sports, Science and Technology, Japan. This work was carried out by the joint research program of the Institute for Molecular and Cellular Regulation, Gunma University.
Author contributions: K. Ishikawa: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Visualization, Writing - original draft, Writing - review & editing, S. Soejima: Investigation, T. Nishimura: Formal analysis, Investigation, Visualization, Writing - review & editing, S. Saitoh: Conceptualization, Funding acquisition, Methodology, Software, Supervision, Writing - original draft, Writing - review & editing.

