


During meiosis, cohesin and meiosis-specific proteins organize chromatin into an axis-loop architecture, coordinating homologous synapsis, recombination, and ordered chromosome segregation. However, how the meiotic chromosome axis is assembled and differentiated with meiotic progression remains elusive. Here, we explore the dynamic recruitment of two long arms of the bivalent proteins, LAB-1 and LAB-2, in Caenorhabditis elegans. LAB proteins directly interact with the axis core HORMA complexes and weak interactions contribute to their recruitment. LAB proteins phase separate in vitro, and this capacity is promoted by HORMA complexes. During early prophase, synapsis oppositely regulates the axis enrichment of LAB proteins. After the pachytene exit, LAB proteins switch from a reciprocal localization pattern to a colocalization pattern, and the normal dynamic pattern of LAB proteins is altered in meiotic mutants. We propose that LAB recruitment senses axis differentiation, and phase separation of meiotic structures helps subdomain establishment and accurate segregation of the chromosomes.
Introduction
Meiosis produces haploid gametes from diploid germ cells through two consecutive rounds of chromosome segregation after a single round of DNA replication. Accurate meiotic chromosome segregation depends on the establishment and ordered dissolution of physical connections between sister chromatids and homologous chromosomes. During meiotic prophase, cohesin complexes maintain interactions between sister chromatids and recruit meiosis-specific proteins to form the chromosome axis, which organizes the chromatin into an axis-loop architecture and allows homologous chromosomes to undergo pairing, synapsis, recombination, and crossing over. Along with inter-sister cohesion, inter-homologous crossovers allow the proper alignment of homologous chromosomes with their sister kinetochores mono-oriented during meiosis I. Chromosome arm cohesion, but not centromeric cohesion, is removed by separase during anaphase I to allow homolog segregation. During meiosis II, sister kinetochores are bioriented, and centromeric cohesin cleavage results in the segregation of sister chromatids (Watanabe, 2012). Meiotic chromosome subdomain differentiation is thus essential for their ordered segregation.
Chromosome axis formation allows programmed DNA double-strand break formation and proper recognition of homologs and provides structural frames for the assembly of the synaptonemal complex (SC), which in turn control inter-homologous crossover formation (Zhang et al., 2021). The meiotic chromosome axis thus functions as a central structure to coordinate a series of critical meiotic events to ensure accurate chromosome segregation. Although meiotic chromosome axes have conserved roles, the identified subunits of the core structure are highly divergent across species (Gao and Colaiácovo, 2018; Zhang et al., 2021). For example, in mammals, SYCP2 and SYCP3 are core components of the axis, with HORMA proteins (HORMAD1 and HORMAD2) associating with them (Kouznetsova et al., 2005; Wojtasz et al., 2009; Yang et al., 2006). In Caenorhabditis elegans, the complexes formed by HORMA domain proteins (HTP-1/2/3 and HIM-3) play essential roles in axis formation (Couteau and Zetka, 2005; Goodyer et al., 2008; Martinez-Perez and Villeneuve, 2005; Severson et al., 2009; Zetka et al., 1999). However, how chromosome axis composition and properties are altered in response to various meiotic events remains not fully understood.
In holocentric C. elegans, ordered meiotic chromosome segregation is also achieved by sequentially removing sister chromatid cohesion on chromosome subdomains. During meiotic prophase, off-centered crossover formation divides chromosomes into long and short arms. The SC asymmetrically disassembles upon pachytene exit and is maintained on bivalent short arms until early diakinesis. Crossover-coupled axis differentiation results in a long-arm association of HTP-1/2 and LAB-1, which was suggested to function as shugoshin to protect inter-sister chromatid cohesion during meiosis I (de Carvalho et al., 2008; Martinez-Perez et al., 2008). On the other hand, bivalent short arms are enriched with the chromosomal passenger complex (CPC), whose activation triggers local cohesion removal during meiosis I (Kaitna et al., 2002; Rogers et al., 2002; Romano et al., 2003). However, how meiotic structures are coordinated to establish distinct chromosome subdomains remains elusive.
Multivalent weak interaction-mediated phase separation underlies the formation of biomolecular condensates that compartmentalize different groups of proteins or nucleic acid molecules and control various biological processes (Banani et al., 2017; Zhang et al., 2023). Phase-separated structures are actively regulated in cells and may have distinct states, including a highly dynamic liquid-like state, a less dynamic but reversible hydrogel state, and an irreversible amyloid state (Zhang et al., 2020a). A few meiotic structures have been suggested to be formed through phase separation, including the SC in C. elegans (Rog et al., 2017; Zhang et al., 2018, 2020b), the DSB formation machinery in budding yeast (Claeys Bouuaert et al., 2021), and RNA-containing pairing droplets in fission yeast (Ding et al., 2019). Whether phase separation may be involved in the formation of other structures on meiotic chromosomes remains to be investigated.
Here, we characterized the expression and localization patterns of two axis-associated proteins, LAB-1 and LAB-2 (ORF F56C9.11), during meiotic progression in C. elegans. Their dynamic localization patterns during a normal meiotic process and altered patterns in meiotic mutants suggest that LAB protein recruitment senses axis differentiation. Moreover, both LAB proteins phase separate in vitro, and phase separation may underlie the establishment of distinct chromosome subdomains required for accurate chromosome segregation.
Results
Identification of a novel axial component LAB-2
To better understand axis composition in C. elegans, we performed immunoprecipitation (IP) and mass spectrometry analysis to identify the binding proteins of HTP-3, the core component of the HORMA complex required for axis assembly (Goodyer et al., 2008; Severson et al., 2009). HTP-3::GFP IP recovered all the known HORMA proteins (Fig. 1 A). No SYP proteins were recovered from HTP-3 IPs, suggesting a lack of stable interactions between SC central region proteins and HORMA proteins, consistent with our previous work that axial proteins were not identified in SYP IPs (Zhang et al., 2020b). In addition to LAB-1, we also identified an uncharacterized protein, which we named LAB-2 (ORF F56C9.11), binding to the HORMA protein complex (Fig. 1 A). Similar to SC components, LAB proteins are only conserved among Caenorhabditis species (Fig. S1 A) and are likely co-evolved with the SC. Moreover, structure analysis suggested that both LAB-1 and LAB-2 contain unstructured regions at their C-termini (Fig. S1, B and C).

Identification and expression analysis of axis-associated proteins. (A) Mass spectrometry (MS) identification of HTP-3::GFP binding partners in vivo. Identified HTP-3-specific binding proteins are shown as red dots. Gray dots are proteins also present in the control IPs. Data represent the mean value of three independent replicates. (B) Expression patterns of GFP-tagged axial components HTP-3, HTP-1, LAB-2, and LAB-2 in the germline. Bar, 30 μm. (C) Localization of LAB-1::GFP and LAB-2::GFP (green) on diakinesis bivalents. Chromatin was stained with DAPI (magenta). Bar, 5 μm. The cartoon depicts the long-arm localization of LAB proteins. L, long arm; S, short arm. (D) LAB-2::GFP (green) localization in diakinesis nuclei of htp-3 mutants. Chromatin was stained with DAPI (magenta). Bar, 10 μm. (E) MS identification of LAB-2::GFP-binding proteins. LAB-2-specific binding proteins are shown as red dots, and the identified known axial components are labeled. Non-specific binding proteins are shown in gray. Data represent the mean value of three independent replicates. (F) Quantification of chromosome association of meiotic chromosome axis components at the indicated stages. Measurement was performed as in Fig. S2, and the numbers of nuclei analyzed are indicated on the top. Each data point represents a segment value or the mean value of multiple segments from a nucleus, and bars represent the mean ± SD. Statistical analysis was performed by the two-tailed unpaired t test.
Identification and expression analysis of axis-associated proteins. (A) Mass spectrometry (MS) identification of HTP-3::GFP binding partners in vivo. Identified HTP-3-specific binding proteins are shown as red dots. Gray dots are proteins also present in the control IPs. Data represent the mean value of three independent replicates. (B) Expression patterns of GFP-tagged axial components HTP-3, HTP-1, LAB-2, and LAB-2 in the germline. Bar, 30 μm. (C) Localization of LAB-1::GFP and LAB-2::GFP (green) on diakinesis bivalents. Chromatin was stained with DAPI (magenta). Bar, 5 μm. The cartoon depicts the long-arm localization of LAB proteins. L, long arm; S, short arm. (D) LAB-2::GFP (green) localization in diakinesis nuclei of htp-3 mutants. Chromatin was stained with DAPI (magenta). Bar, 10 μm. (E) MS identification of LAB-2::GFP-binding proteins. LAB-2-specific binding proteins are shown as red dots, and the identified known axial components are labeled. Non-specific binding proteins are shown in gray. Data represent the mean value of three independent replicates. (F) Quantification of chromosome association of meiotic chromosome axis components at the indicated stages. Measurement was performed as in Fig. S2, and the numbers of nuclei analyzed are indicated on the top. Each data point represents a segment value or the mean value of multiple segments from a nucleus, and bars represent the mean ± SD. Statistical analysis was performed by the two-tailed unpaired t test.

Analysis of LAB proteins conservation and structural features. (A) A heatmap showing the sequence similarity of the known SC components and LAB proteins in C. elegans and other Caenorhabditis species. (B) Secondary structural feature analysis of LAB proteins in Caenorhabditis species. Analysis was performed as in Zhang et al. (2020b). (C) 3D structures of LAB proteins predicted by AlphaFold.
Analysis of LAB proteins conservation and structural features. (A) A heatmap showing the sequence similarity of the known SC components and LAB proteins in C. elegans and other Caenorhabditis species. (B) Secondary structural feature analysis of LAB proteins in Caenorhabditis species. Analysis was performed as in Zhang et al. (2020b). (C) 3D structures of LAB proteins predicted by AlphaFold.
To examine the expression and localization of LAB-2, the green fluorescent protein (GFP) coding sequence was inserted before the stop codon of endogenous lab-2 by CRISPR/Cas9. The expression of LAB-2::GFP was germline-specific and its chromosome association increased as the germ cells progressed to late meiotic prophase (Fig. 1 B). Similar to the localization pattern of LAB-1, LAB-2 also localized to the long arms of diakinesis bivalents (Fig. 1 C).
In the htp-3 mutant background, where HORMA proteins fail to assemble onto chromosomes (Kim et al., 2014), LAB-2::GFP was diffusely distributed in germ cells during the entire meiotic prophase (Fig. 2 D and data not shown), suggesting a requirement of HORMA complexes for its chromosome recruitment. Moreover, LAB-2::GFP IP and mass spectrometry analysis also identified HORMA domain proteins, but no other known chromosome-associated proteins, as the binding partners of LAB-2 (Fig. 1 E), further suggesting that HORMA complexes are responsible for LAB-2 chromosome association. Similarly, chromosome association of LAB-1 also requires HORMA complexes (Ferrandiz et al., 2018; Tzur et al., 2012). These findings suggest that LAB-1 and LAB-2 are axis-associated proteins mediated by HORMA complexes in vivo.
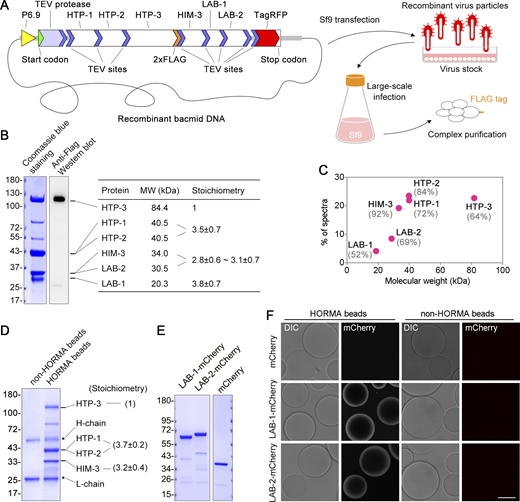
LAB proteins interact directly with the HORMA complex. (A) A cartoon depicts the expression and purification of the HORMA complexes with the SmartBac system, which uses recombinant baculoviruses to express large multiprotein complexes in Sf9 insect cells. All components were translated from a single transcript and separated by tobacco etch virus (TEV)-mediated cleavage. Two copies of FLAG tag were incorporated at the C-terminus of HTP-3 and the complexes were purified with anti-FLAG agarose beads. (B) PAGE analysis of purified HORMA complexes with LAB-1 and LAB-2 coexpressed. The molecular weight (MW) of each component was indicated. Relative protein stoichiometry was calculated based on band densitometric values from four replicates and normalized with their molecular weights. (C) MS spectra abundances of the HORMA complex proteins. MS coverage was indicated. (D) PAGE analysis of agarose bead-bounded HORMA complexes. HORMA complexes without LAB proteins were expressed in Sf9 cells as depicted in A. Relative protein stoichiometry was measured from three replicates and indicated in the parentheses. (E) PAGE analysis of mCherry and mCherry-tagged LAB proteins purified from E. coli. (F) Recruitment of LAB-1-mCherry and LAB-2-mCherry to HORMA-associated agarose beads. 5 μM mCherry or mCherry-tagged proteins were used. Bar, 50 μm. Source data are available for this figure: SourceData F2.
LAB proteins interact directly with the HORMA complex. (A) A cartoon depicts the expression and purification of the HORMA complexes with the SmartBac system, which uses recombinant baculoviruses to express large multiprotein complexes in Sf9 insect cells. All components were translated from a single transcript and separated by tobacco etch virus (TEV)-mediated cleavage. Two copies of FLAG tag were incorporated at the C-terminus of HTP-3 and the complexes were purified with anti-FLAG agarose beads. (B) PAGE analysis of purified HORMA complexes with LAB-1 and LAB-2 coexpressed. The molecular weight (MW) of each component was indicated. Relative protein stoichiometry was calculated based on band densitometric values from four replicates and normalized with their molecular weights. (C) MS spectra abundances of the HORMA complex proteins. MS coverage was indicated. (D) PAGE analysis of agarose bead-bounded HORMA complexes. HORMA complexes without LAB proteins were expressed in Sf9 cells as depicted in A. Relative protein stoichiometry was measured from three replicates and indicated in the parentheses. (E) PAGE analysis of mCherry and mCherry-tagged LAB proteins purified from E. coli. (F) Recruitment of LAB-1-mCherry and LAB-2-mCherry to HORMA-associated agarose beads. 5 μM mCherry or mCherry-tagged proteins were used. Bar, 50 μm. Source data are available for this figure: SourceData F2.
Distinct chromosome association patterns of the axial components during meiotic prophase
We noticed that the chromosome association of LAB-2 was highly enriched during the late meiotic prophase. This expression pattern was similar to LAB-1 but distinct from HORMA proteins (Fig. 1 B). To precisely assess the chromosome association of meiotic axis proteins, the max fluorescence intensity for each axis segment and the corresponding background intensity were measured at different meiotic stages, and the ratio of the two values was referred to as relative axis loading (Fig. S2). During early pachytene, the axis loading of LAB proteins was significantly lower than HORMA proteins HTP-3 and HTP-1. When cells progressed to late pachytene, LAB protein axis loading became similar to HORMA proteins. While at diakinesis, the axis loading of LAB proteins was significantly higher than HORMA proteins (Fig. 1 F). These observations suggest that the composition or stoichiometry of axial components undergoes a consistent change during meiotic progression.

Schematic diagram of the fluorescence measurement of chromosome-associated proteins. Raw images were captured with a 60× objective lens and axis segments of interest (exampled by yellow arrows) were saved as text images by ImageJ. Further calculation was performed in batch with an in-house R script to find the max intensity value of a 3 × 3 pixels region (corresponding to 0.4 × 0.4 μm) and the background intensity of each segment. Bar, 1 μm.
Schematic diagram of the fluorescence measurement of chromosome-associated proteins. Raw images were captured with a 60× objective lens and axis segments of interest (exampled by yellow arrows) were saved as text images by ImageJ. Further calculation was performed in batch with an in-house R script to find the max intensity value of a 3 × 3 pixels region (corresponding to 0.4 × 0.4 μm) and the background intensity of each segment. Bar, 1 μm.
LAB proteins directly interact with HORMA proteins
To get insight into the mechanisms of the chromosome recruitment of LAB proteins, we used the SmartBac system to express and purify HORMA complexes with or without LAB proteins from insect cells (Fig. 2 A; Zhai et al., 2019). The complexes were affinity purified through the 2xFlag tag fused to HTP-3 C-terminus. HTP-3 migrated at a 120 kD position in SDS-PAGE gel, which is higher than its expected size (Fig. 2 B), but similar to the endogenous protein examined in our previous study (Gao et al., 2016). When LAB proteins were coexpressed, both LAB-1 and LAB-2 were copurified (Fig. 2, B and C), suggesting direct interactions of LAB proteins with the HORMA complexes. Protein stoichiometry estimation suggested that the binding of LAB-1 to the HORMA domain protein complexes appears stoichiometric. However, given the known stoichiometry of the HTP-3:HIM-3 complex (1:2–4; Kim et al., 2014; Woglar et al., 2020), which is also consistent with our analysis (Fig. 2 D), the binding of LAB-2 to the HORMA complexes is likely substoichiometric (Fig. 2 B). Furthermore, we also purified mCherry and mCherry-tagged LAB proteins from Escherichia coli (Fig. 2 D). By incubating these purified proteins with agarose beads coupled with insect-expressed HORMA complexes (Fig. 2 E), we observed a clear mCherry fluorescence signal at the surface of the agarose beads for both LAB proteins under microscopy examination. In contrast, such a signal was not observed for HORMA beads incubated with mCherry or non-HORMA beads incubated with mCherry-tagged LAB proteins (Fig. 2 F). This observation further suggests direct interactions between HORMA complexes and LAB proteins.
To further look into the interactions between HORMA complexes and LAB proteins, binary interactions between HORMA proteins and LAB proteins were examined in 293T cells. LAB-1 showed variable interactions with HORMA proteins and exhibited the strongest interactions with HTP-1/2 (Fig. S3 A), consistent with the previous finding that HTP-1/2 is responsible for LAB-1 chromosome recruitment (Ferrandiz et al., 2018). In comparison, LAB-2 had only weak interactions with HORMA proteins and LAB-1, which were much weaker than the interactions between HORMA proteins (HIM-3 and HTP-1; Fig. S3 B). Unlike the recruitment of LAB-1, the chromosome recruitment of LAB-2 was not highly dependent on HTP-1/2, and htp-2 RNAi in htp-1 mutant background did not cause a depletion of LAB-2 from the chromosomes (Fig. S3 C). Moreover, chromosome recruitment of LAB-2 remained in him-3 and lab-1 single mutants (Fig. S3 D). These analyses suggest that a HORMA complex may provide multiple binding sites for LAB recruitment.
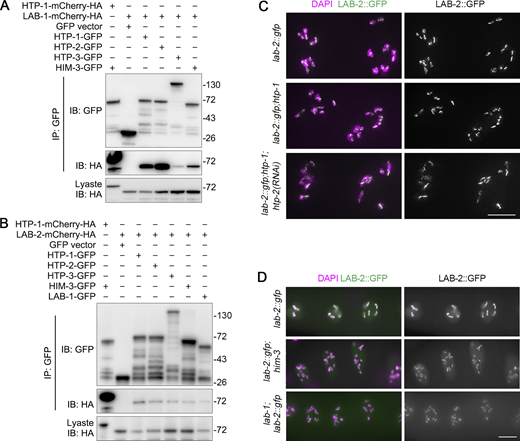
HORMA complexes may provide multi-binding sites for LAB proteins. (A and B) Analysis of interactions of LAB-1 (A) or LAB-2 (B) with HORMA proteins. GFP-tagged HORMA proteins were binary co-expressed with LAB-1-mCherry-HA or LAB-2-mCherry-HA in 293T cells, and their interactions were analyzed by immunoprecipitation and Western blotting. The interaction between HTP-1 and HIM-3 was used as a positive control. (C) Chromosome association of LAB-2::GFP (green) in diakinesis nuclei of the indicated genotypes. Chromatin was stained with DAPI (magenta). Bar, 5 μm. (D) Chromosome association of LAB-2::GFP (green) in diakinesis nuclei of the indicated genotypes. Chromatin was stained with DAPI (magenta). Bar, 10 μm. Source data are available for this figure: SourceData FS3.
HORMA complexes may provide multi-binding sites for LAB proteins. (A and B) Analysis of interactions of LAB-1 (A) or LAB-2 (B) with HORMA proteins. GFP-tagged HORMA proteins were binary co-expressed with LAB-1-mCherry-HA or LAB-2-mCherry-HA in 293T cells, and their interactions were analyzed by immunoprecipitation and Western blotting. The interaction between HTP-1 and HIM-3 was used as a positive control. (C) Chromosome association of LAB-2::GFP (green) in diakinesis nuclei of the indicated genotypes. Chromatin was stained with DAPI (magenta). Bar, 5 μm. (D) Chromosome association of LAB-2::GFP (green) in diakinesis nuclei of the indicated genotypes. Chromatin was stained with DAPI (magenta). Bar, 10 μm. Source data are available for this figure: SourceData FS3.
Weak interactions contribute to the axis recruitment of LAB proteins
To test if weak interactions might contribute to the axis recruitment of LAB proteins in vivo, we analyzed their sensitivity to 1,6-hexanediol, an aliphatic alcohol that disrupts weak hydrophobic interactions (Kroschwald et al., 2017; Rog et al., 2017). 10% 1,6-hexanediol treatment had only a minor impact on the axis loading of HTP-1::GFP, consistent with its stable interaction within the HORMA complexes. However, the same treatment significantly reduced the axis loading of LAB-1::GFP and LAB-2::GFP (Fig. 3, A and B), suggesting that weak interactions contribute to their axis recruitment. However, while the axis association of LAB proteins was completely disrupted at pachytene by 1,6-hexanediol exposure, partial retention of axis association was observed at late meiotic prophase (Fig. 3 A). We hypothesize that this axis-retained pool of LAB proteins may have more stable interactions with the HORMA complexes, and multitype or multilayer interactions may contribute to their axis loading.

Localization of LAB proteins can be disrupted by 1,6-hexanediol. (A) Localization of GFP-tagged HTP-1, LAB-1, and LAB-2 (green) in pachytene and diakinesis nuclei with or without 10% 1,6-hexanediol treatment. Chromatin was stained with DAPI (magenta). Bar, 20 μm. (B) Quantification of axis loading of HTP-1, LAB-1, and LAB-2 at late pachytene (LP) and diakinesis stages with or without 1,6-hexanediol treatment. Each data point represents a segment value or the mean value of multiple segments from the same nucleus, and bars represent the mean ± SD. The numbers of nuclei analyzed are indicated. Statistical analysis was performed by the two-tailed unpaired t test. (C) Chromosome reassociation of LAB-2::GFP (green) in diakinesis nuclei after 1,6-hexanediol washout. Chromatin was stained with DAPI (magenta). Bar, 20 μm. (D) Quantification of axis loading of LAB-2::GFP in diakinesis nuclei after 1,6-hexanediol washout. Each data point represents a segment value or the mean value of multiple segments from the same nucleus, and bars represent the mean ± SD. The numbers of nuclei analyzed are indicated. Statistical analysis was performed by the two-tailed unpaired t test.
Localization of LAB proteins can be disrupted by 1,6-hexanediol. (A) Localization of GFP-tagged HTP-1, LAB-1, and LAB-2 (green) in pachytene and diakinesis nuclei with or without 10% 1,6-hexanediol treatment. Chromatin was stained with DAPI (magenta). Bar, 20 μm. (B) Quantification of axis loading of HTP-1, LAB-1, and LAB-2 at late pachytene (LP) and diakinesis stages with or without 1,6-hexanediol treatment. Each data point represents a segment value or the mean value of multiple segments from the same nucleus, and bars represent the mean ± SD. The numbers of nuclei analyzed are indicated. Statistical analysis was performed by the two-tailed unpaired t test. (C) Chromosome reassociation of LAB-2::GFP (green) in diakinesis nuclei after 1,6-hexanediol washout. Chromatin was stained with DAPI (magenta). Bar, 20 μm. (D) Quantification of axis loading of LAB-2::GFP in diakinesis nuclei after 1,6-hexanediol washout. Each data point represents a segment value or the mean value of multiple segments from the same nucleus, and bars represent the mean ± SD. The numbers of nuclei analyzed are indicated. Statistical analysis was performed by the two-tailed unpaired t test.
Moreover, the disruption of LAB-2::GFP localization by 1,6-hexanediol was reversible, and 1,6-hexanediol washout allowed its reassociation onto bivalent long arms (Fig. 3, C and D), suggesting a weak interaction-mediated self-assembly property of LAB-2 in vivo.
LAB proteins exhibit phase separation properties in vitro
Weak interactions drive phase separation of biomolecules in cells to control various biological processes and many phase-separated structures can be dissolved by 1,6-hexanediol treatment (Kroschwald et al., 2017; Rog et al., 2017; Strom et al., 2017). We proposed that LAB proteins may undergo scaffolded phase separation on the meiotic axis, contributing to their enrichment on bivalent arms during the late meiotic prophase. We set a phase separation assay to examine the phase separation capacity of fluorescent protein-tagged LAB proteins purified from E. coli (Fig. S4 A and Fig. 4 A). While even at a high concentration of 30 μM, no droplet formation was observed for mCherry protein (Fig. S4 A). However, both LAB-1 and LAB-2 formed droplets at 3 μM or higher concentrations under fluorescence and white light (DIC) observations (Fig. 4 A). In addition to mCherry-tagged LABs, LAB proteins fused with GFP tag also form droplets in the phase separation assay (Fig. S4, B and C), further confirming their phase separation capacities.
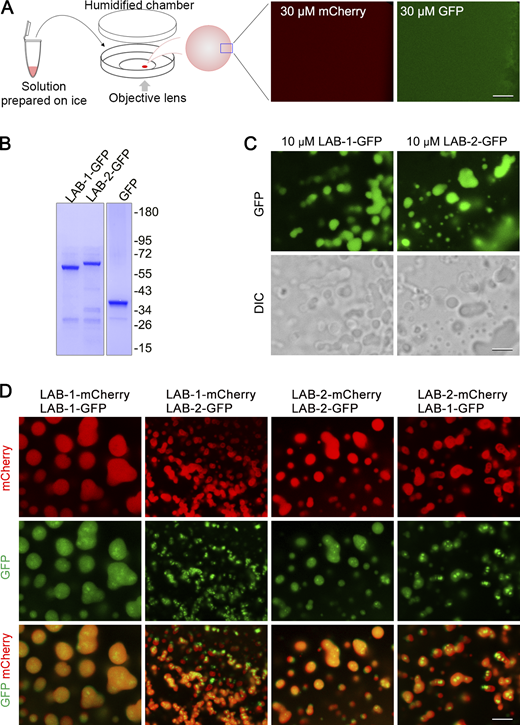
Phase separation analysis of fluorescent protein-tagged LAB proteins. (A) Left, a cartoon depicts the setup of phase separation observation under a confocal microscope. Solutions of purified proteins were prepared on ice, and their phase separation properties were examined under a confocal microscope at 20°C. Right, mCherry or GFP protein does not phase separate at 30 μM. Bar, 5 μm. (B) SDS-PAGE and Coomassie blue staining of GFP and GFP-tagged LAB proteins purified from E. coli. (C) Phase separation analysis of GFP and GFP-tagged LAB proteins at the indicated concentrations. Bar, 5 μm. (D) Phase separation analysis of mixed LAB proteins fused with different fluorescent proteins. Bar, 5 μm. Source data are available for this figure: SourceData FS4.
Phase separation analysis of fluorescent protein-tagged LAB proteins. (A) Left, a cartoon depicts the setup of phase separation observation under a confocal microscope. Solutions of purified proteins were prepared on ice, and their phase separation properties were examined under a confocal microscope at 20°C. Right, mCherry or GFP protein does not phase separate at 30 μM. Bar, 5 μm. (B) SDS-PAGE and Coomassie blue staining of GFP and GFP-tagged LAB proteins purified from E. coli. (C) Phase separation analysis of GFP and GFP-tagged LAB proteins at the indicated concentrations. Bar, 5 μm. (D) Phase separation analysis of mixed LAB proteins fused with different fluorescent proteins. Bar, 5 μm. Source data are available for this figure: SourceData FS4.

LAB proteins phase separate in vitro. (A) Droplet formation of mCherry-tagged LAB-1 and LAB-2 at the indicated concentrations. The differential interference contrast (DIC) images of the droplets were shown for the 10 μM condition. Bar, 5 μm. (B) FRAP analysis of droplets formed by mCherry-tagged LAB proteins. The time points (second) after photobleaching are labeled. PB, pre-bleaching image. Bar, 5 μm. (C) FRAP quantification for LAB-1 and LAB-2 droplets. The line and shade represent the mean ± SD of the measurements from the indicated number of experiments. (D and E) Fusion of droplets formed by mCherry-tagged LAB-1 (D) or LAB-2 (E). Duration of time was indicated (second). Droplets undergoing fussing were circled with dotted lines. Bar, 5 μm. (F) PAGE analysis of HORMA complex (without LABs) expressed and purified from Sf9 cells. (G and H) HORMA complexes promote phase separation of mCherry-tagged LAB-1 (G) and LAB-2 (H). The elution from the same purification procedure performed on Sf9 cells transfected with the empty vector was used as the control. The molar concentration of HORMA complexes was represented by the concentration of HTP-3. Bar, 5 μm. Source data are available for this figure: SourceData F4.
LAB proteins phase separate in vitro. (A) Droplet formation of mCherry-tagged LAB-1 and LAB-2 at the indicated concentrations. The differential interference contrast (DIC) images of the droplets were shown for the 10 μM condition. Bar, 5 μm. (B) FRAP analysis of droplets formed by mCherry-tagged LAB proteins. The time points (second) after photobleaching are labeled. PB, pre-bleaching image. Bar, 5 μm. (C) FRAP quantification for LAB-1 and LAB-2 droplets. The line and shade represent the mean ± SD of the measurements from the indicated number of experiments. (D and E) Fusion of droplets formed by mCherry-tagged LAB-1 (D) or LAB-2 (E). Duration of time was indicated (second). Droplets undergoing fussing were circled with dotted lines. Bar, 5 μm. (F) PAGE analysis of HORMA complex (without LABs) expressed and purified from Sf9 cells. (G and H) HORMA complexes promote phase separation of mCherry-tagged LAB-1 (G) and LAB-2 (H). The elution from the same purification procedure performed on Sf9 cells transfected with the empty vector was used as the control. The molar concentration of HORMA complexes was represented by the concentration of HTP-3. Bar, 5 μm. Source data are available for this figure: SourceData F4.
Importantly, the droplets formed by LAB proteins have dynamic properties as indicated by fluorescence recovery after photobleaching (FRAP; Fig. 4, B and C) and droplet fusion (Fig. 4, D and E). In the FRAP analysis, we observed a more rapid fluorescence recovery for LAB-1-mCherry than LAB-2-mCherry (Fig. 4 C), which may suggest more dynamic movements of the molecules in LAB-1 droplets. Fluorescence in LAB-2-mCherry droplets failed to achieve a full recovery after photobleaching (Fig. 4 C), likely a result of gelation after droplet formation. However, during droplet formation and growth, droplet fusion events were frequently observed for both LAB proteins (Fig. 4, D and E), exhibiting a typical phase separation characteristic.
Interestingly, while unmixed fluorescent protein-tagged LABs form homogeneous droplets in phase separation assays, the mixture of GFP- and mCherry-tagged LAB proteins caused the formation of heterogeneous droplets, with GFP-tagged proteins preferentially aggregating inside the mCherry-tagged protein droplets (Fig. S4 D). This phenomenon may be caused by the weak dimerization property of the GFP tag, which increases the interacting valency of the fused proteins. Furthermore, the heterogeneity of the phase-separated droplets was more pronounced when different LABs were mixed, suggesting different phase-separation properties of LAB-1 and LAB-2.
HORMA complexes promote phase separation of LAB proteins in vitro
Unlike SC central region proteins, which form polycomplexes with dynamic properties at pachytene when the core axis protein HTP-3 is lacking (Rog et al., 2017), LAB-2::GFP did not form aggregates in htp-3 mutants, suggesting that the diffused LAB-2 might not reach a concentration that allowed its phase separation. We hypothesized that the HORMA complexes on the chromosome axis might locally enrich LAB proteins, thereby promoting LAB phase separation. To test this, four-component HORMA complexes were purified from Sf9 cells (Fig. 4 F). Adding 0.15 μM HORMA complexes (assessed by HTP-3 concentration, corresponding to ∼1 μM HTP-1/2 and HIM-3 as assessed in Fig. 2) induced the phase separation of LAB proteins at 2.5 μM. On the contrary, the addition of the empty elution buffer (not shown) or the control elution from the same purification procedure did not induce the phase separation of LAB proteins at the same concentration (Fig. 4, G and H).
LAB-1 and LAB-2 promote proper chromosome remodeling during late meiotic prophase
Previous studies showed that LAB-1 protects meiotic inter-sister chromatid cohesion and thus promotes accurate meiotic chromosome segregation (de Carvalho et al., 2008; Tzur et al., 2012). The lab-1 mutation causes elevated embryonic lethality (Emb) and high incidence of males (Him), phenotypes indicative of meiotic chromosome segregation errors. To understand if LAB-1 and LAB-2 may have similar functions during meiosis, we generated a lab-2 null mutant using CRISPR/Cas9 (Fig. S5, A and B) and compared its phenotypes with that of the wild-type and lab-1 mutants at 20°C and 25°C culture conditions, respectively (Fig. 5 A–D).
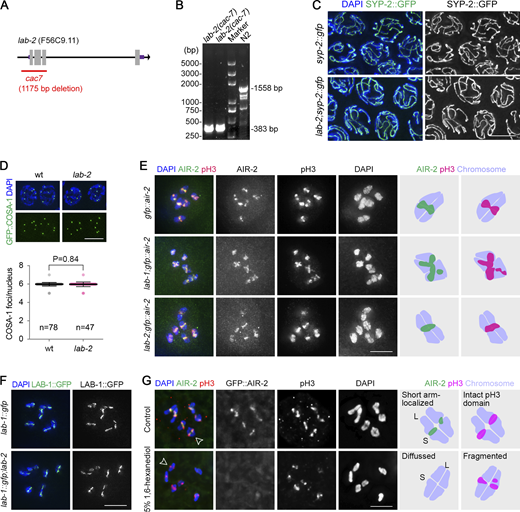
Cytological analysis synapsis, crossover formation, and AIR-2/phospho-H3 localization in lab-2 mutants and AIR-2/phospho-H3 localization upon 1,6-hexanediol treatment. (A) Schematic of a lab-2 deletion mutant created by CRISPR. An 1175-bp deletion removed a part of the lab-2 promoter (506 bp) and the first three exons. (B) The genotyping result of lab-2 mutants by PCR. Primers used were 5′-AACGGTTCTGGATCATCGTC-3′ and 5′-AAAGATTGGTCGCTTCGAGA-3′. (C) SC formation (indicated by SYP-2::GFP, green) is normal during pachytene in lab-2 mutants. Gonads dissected from the indicated genotypes were fixed and stained with DAPI (blue). Bar, 5 μm. (D) Quantification of GFP::COSA-1 focus number in late pachytene nuclei of wild type and lab-2 mutants. Bars represent the mean ± SD, and the numbers of nuclei analyzed are indicated. Statistical analysis was performed by the two-tailed unpaired t test. Scale bar, 5 μm. (E) Immunostaining of Ser10-phosphorylated histone H3 (pH3, red) in −1 oocytes of the transgenic worm expressing GFP::AIR-2 (green). Chromatin was stained with DAPI (blue). Bar, 5 μm. The cartoons on the right depict the representative localization patterns of AIR-2 and pH3 on bivalents of the indicated genotypes. (F) LAB-1::GFP (green) localization on diakinesis bivalents of the indicated genotypes. Chromatin was stained with DAPI (blue). Bar, 5 μm. (G) Immunostaining of pH3 (red) in −1 oocytes of the transgenic worm expressing GFP::AIR-2 (green). Dissected gonads were treated with or without 5% 1,6-hexanediol 10 min before fixation. Chromatin was stained with DAPI (blue). The cartoons on the right depict the localization patterns of AIR-2 and pH3 on bivalents indicated by the open arrowheads. Bar, 5 μm. Source data are available for this figure: SourceData FS5.
Cytological analysis synapsis, crossover formation, and AIR-2/phospho-H3 localization in lab-2 mutants and AIR-2/phospho-H3 localization upon 1,6-hexanediol treatment. (A) Schematic of a lab-2 deletion mutant created by CRISPR. An 1175-bp deletion removed a part of the lab-2 promoter (506 bp) and the first three exons. (B) The genotyping result of lab-2 mutants by PCR. Primers used were 5′-AACGGTTCTGGATCATCGTC-3′ and 5′-AAAGATTGGTCGCTTCGAGA-3′. (C) SC formation (indicated by SYP-2::GFP, green) is normal during pachytene in lab-2 mutants. Gonads dissected from the indicated genotypes were fixed and stained with DAPI (blue). Bar, 5 μm. (D) Quantification of GFP::COSA-1 focus number in late pachytene nuclei of wild type and lab-2 mutants. Bars represent the mean ± SD, and the numbers of nuclei analyzed are indicated. Statistical analysis was performed by the two-tailed unpaired t test. Scale bar, 5 μm. (E) Immunostaining of Ser10-phosphorylated histone H3 (pH3, red) in −1 oocytes of the transgenic worm expressing GFP::AIR-2 (green). Chromatin was stained with DAPI (blue). Bar, 5 μm. The cartoons on the right depict the representative localization patterns of AIR-2 and pH3 on bivalents of the indicated genotypes. (F) LAB-1::GFP (green) localization on diakinesis bivalents of the indicated genotypes. Chromatin was stained with DAPI (blue). Bar, 5 μm. (G) Immunostaining of pH3 (red) in −1 oocytes of the transgenic worm expressing GFP::AIR-2 (green). Dissected gonads were treated with or without 5% 1,6-hexanediol 10 min before fixation. Chromatin was stained with DAPI (blue). The cartoons on the right depict the localization patterns of AIR-2 and pH3 on bivalents indicated by the open arrowheads. Bar, 5 μm. Source data are available for this figure: SourceData FS5.

Phenotypes of lab mutants. (A) Brood sizes (fertilized eggs) of the indicated genotypes at 20°C and 25°C. The numbers of P0 worms analyzed are indicated at the top. Bars represent the mean ± SD. Statistical analysis was performed by the two-tailed unpaired t test. (B) Adult brood sizes of the indicated genotypes at 20°C and 25°C. The numbers of P0 worms analyzed are indicated at the top. Bars represent the mean ± SD. Statistical analysis was performed by the two-tailed unpaired t test. (C) Percentage of viable embryos of the indicated genotypes at 20°C and 25°C. The number of embryos scored is indicated. Asterisks indicate statistical difference compared with wild type (**** P < 0.0001, by Fisher’s exact test). (D) Percentage of the male progeny of the indicated genotypes at 20°C and 25°C. The numbers of adults scored are indicated. Asterisks indicate statistical difference compared with wild type (**** P < 0.0001, by Fisher’s exact test). (E) SYP-2::GFP (green) distribution on bivalent arms at the indicated stages. Chromatin was stained with DAPI (blue). Bar, 5 μm. The cartoons depict the different distribution patterns of SYP-2. L, bivalent long arm; S, bivalent short arm. Nucleus numbers exhibiting different SYP-2::GFP distribution patterns are indicated in parentheses (data of two biological replicates).
Phenotypes of lab mutants. (A) Brood sizes (fertilized eggs) of the indicated genotypes at 20°C and 25°C. The numbers of P0 worms analyzed are indicated at the top. Bars represent the mean ± SD. Statistical analysis was performed by the two-tailed unpaired t test. (B) Adult brood sizes of the indicated genotypes at 20°C and 25°C. The numbers of P0 worms analyzed are indicated at the top. Bars represent the mean ± SD. Statistical analysis was performed by the two-tailed unpaired t test. (C) Percentage of viable embryos of the indicated genotypes at 20°C and 25°C. The number of embryos scored is indicated. Asterisks indicate statistical difference compared with wild type (**** P < 0.0001, by Fisher’s exact test). (D) Percentage of the male progeny of the indicated genotypes at 20°C and 25°C. The numbers of adults scored are indicated. Asterisks indicate statistical difference compared with wild type (**** P < 0.0001, by Fisher’s exact test). (E) SYP-2::GFP (green) distribution on bivalent arms at the indicated stages. Chromatin was stained with DAPI (blue). Bar, 5 μm. The cartoons depict the different distribution patterns of SYP-2. L, bivalent long arm; S, bivalent short arm. Nucleus numbers exhibiting different SYP-2::GFP distribution patterns are indicated in parentheses (data of two biological replicates).
The production of fertilized eggs was reduced in both lab-1 and lab-2 single mutants compared with the wild type at both temperatures, and a more severe reduction was observed in lab-2 and lab-1;lab-2 mutants compared with lab-1 single mutants at 25°C (Fig. 5 A). Similarly, adult brood sizes were significantly reduced in all lab mutants compared with the wild type at both temperatures, and lab-1;lab-2 double mutants showed more severe reduction compared with the single mutants at 25°C (Fig. 5 B). Unlike lab-1 mutants, lab-2 single mutants showed only mild Emb and Him phenotypes at the elevated temperature (Fig. 5, C and D), suggesting their distinct molecular functions, although they were both required for producing the normal adult brood sizes.
Cytological analyses showed that LAB-2 was not required for synapsis and crossover formation (Fig. S5, C and D), suggesting the reduced brood sizes of lab-2 might be associated with meiotic defects during later stages in meiosis. Interestingly, abnormal SC redistribution on bivalent arms during late meiotic prophase was observed in both lab-1 and lab-2 mutants. However, the phenotypes are not identical. Specifically, in lab-1 mutants, the SC central region failed to enrich on bivalent short arms upon pachytene exit and localized to both arms at diakinesis (Fig. 5 E; de Carvalho et al., 2008). However, in lab-2 mutants, the SC central region is still restricted to the short arms upon pachytene exit but is then redistributed to both arms during diakinesis (Fig. 5 E), suggesting a remodeling defect in the mutants.
Proper meiotic chromosome remodeling results in bivalent short-arm localization of CPC, which promotes local cohesion removal and homolog segregation during meiosis I (Kaitna et al., 2002; Rogers et al., 2002; Romano et al., 2003). As a substrate of CPC, histone H3 is also phosphorylated, and this phosphorylated histone marker (pH3) is enriched only on the short arms (Hsu et al., 2000). While lab-1 mutation caused AIR-2 and pH3 to localize to both bivalent arms, lab-2 mutation did not cause obvious mislocalization of AIR-2 and pH3 (Fig. S5 E), suggesting that LAB-2 is only required for some aspects of the remodeling process, consistent with the mild phenotypes observed in lab-2 compared with that of lab-1 mutants. Furthermore, the bivalent long-arm localization of LAB-1 during late meiotic prophase was not affected in lab-2 mutants (Fig. S5 F).
LAB-2 shows unique dynamic patterns during the meiotic prophase
Colocalization analysis of LAB-2::GFP with SC central region protein SYP-5 showed that they fully colocalized during early prophase but surprisingly showed reciprocal localization during late prophase, and the transition took place during early- to mid-diplotene state (Fig. 6, A and B). This dynamic localization pattern is distinct from the HORMA proteins. LAB-1 has been shown to exhibit a reciprocal localization pattern with the SC at pachytene exit (de Carvalho et al., 2008), and the colocalization of LAB-2 with the SC suggests LAB proteins might not colocalize at pachytene exit. To confirm this, we crossed lab-1::gfp into a lab-2::mScarlet strain. Indeed, LAB-1::GFP and LAB-2::mScarlet showed a reciprocal localization at pachytene exit, although they became fully colocalized at diakinesis (Fig. 6 C). Thus, the altered localization of LAB-2 during the meiotic prophase represents a unique dynamic pattern that is different from the SC central region proteins and other known axial components in C. elegans. The different dynamic patterns of LAB proteins may also explain the differences in chromosome remodeling defects observed in their mutants. A multistep axis differentiation likely occurs during meiotic progression, causing the dynamic localization of LAB proteins (detailed in Discussion).

LAB proteins have distinct dynamic patterns during the meiotic prophase. (A) Immunostaining of SYP-5 (red) in gonads dissected from lab-2::gfp worms. Yellow arrowheads indicate examples where SYP-5 colocalizes with LAB-2::GFP (green), and open arrowheads indicate chromosome domains where SYP-5 and LAB-2::GFP show reciprocal localization. Chromatin was stained with DAPI (blue). Bar, 5 μm. (B) Schematic of LAB-2 and SC localization during meiotic progression. L, long arm; S, short arm; CO, crossover. (C) Localization of LAB-1::GFP (green) and LAB-2::mScarlet (red) on chromosomes at late pachytene and diakinesis stages. Chromatin was stained with DAPI (blue). Bar, 5 μm. The cartoons on the right depict their relative localization. (D) LAB-1::GFP (green) localization on chromosomes during mid-late pachytene in the indicated genotypes. Chromatin was stained with DAPI (blue). Arrowheads indicate chromosomes enriched for LAB-1::GFP. Boxed regions are enlarged in the right panels. Bar, 5 μm. The cartoon on the right depicts the enrichment of LAB-1 on the unsynapsed chromosomes. (E) LAB-2::GFP (green) localization on chromosomes during mid-late pachytene in the indicated genotypes. Chromatin was stained with DAPI (blue). Arrowheads indicate chromosomes lacking LAB-2::GFP. Boxed regions are enlarged in the right panels. Bar, 5 μm. The cartoon on the right depicts the absence of LAB-2 on the unsynapsed chromosomes.
LAB proteins have distinct dynamic patterns during the meiotic prophase. (A) Immunostaining of SYP-5 (red) in gonads dissected from lab-2::gfp worms. Yellow arrowheads indicate examples where SYP-5 colocalizes with LAB-2::GFP (green), and open arrowheads indicate chromosome domains where SYP-5 and LAB-2::GFP show reciprocal localization. Chromatin was stained with DAPI (blue). Bar, 5 μm. (B) Schematic of LAB-2 and SC localization during meiotic progression. L, long arm; S, short arm; CO, crossover. (C) Localization of LAB-1::GFP (green) and LAB-2::mScarlet (red) on chromosomes at late pachytene and diakinesis stages. Chromatin was stained with DAPI (blue). Bar, 5 μm. The cartoons on the right depict their relative localization. (D) LAB-1::GFP (green) localization on chromosomes during mid-late pachytene in the indicated genotypes. Chromatin was stained with DAPI (blue). Arrowheads indicate chromosomes enriched for LAB-1::GFP. Boxed regions are enlarged in the right panels. Bar, 5 μm. The cartoon on the right depicts the enrichment of LAB-1 on the unsynapsed chromosomes. (E) LAB-2::GFP (green) localization on chromosomes during mid-late pachytene in the indicated genotypes. Chromatin was stained with DAPI (blue). Arrowheads indicate chromosomes lacking LAB-2::GFP. Boxed regions are enlarged in the right panels. Bar, 5 μm. The cartoon on the right depicts the absence of LAB-2 on the unsynapsed chromosomes.
LAB proteins have different axis-binding preferences during early prophase
The reciprocal localization of LAB proteins at late pachytene/pachytene exit suggests they may have different axis-binding preferences regarding synapsis during early prophase. To test this, lab-1::gfp and lab-2::gfp were crossed into him-8 mutants, in which X chromosome pairing is abolished without affecting autosomal paring or synapsis (Phillips et al., 2005). Interestingly, LAB-1:GFP in him-8 mutants was highly enriched on two chromosomes in each nucleus during mid-late pachytene, likely corresponding to the separated X chromosomes (Fig. 6 D). However, in lab-2::gfp;him-8 worms, there was a clear lack of LAB-2::GFP signal on chromosomes with condensed DAPI morphology before the diplotene stage (Fig. 6 E). These observations suggest that LAB-1 and LAB-2 have different preferences for axial binding in pachytene nuclei with partial synapsis. The disparate localization of LABs to chromosomes with different synapsis statuses in pachytene mirrors their distinct localizations to the long and short arms upon pachytene exit, suggesting that localization of LAB proteins differentially senses axis differentiation in these stages at whole and subdomain chromosome levels.
LAB proteins show distinct localization patterns on bivalent arms in remodeling defective mutants
Our previous work found that the lack of SC central region protein SYP-5 does not significantly affect pachytene crossover designation but causes the premature disassembly of the SC at pachytene exit and defective chromosome remodeling at late prophase (Zhang et al., 2020b). Using syp-5 mutants allowed us to investigate how LAB proteins behave under such a remodeling defective condition. While LAB-1::GFP retained a long-arm localization in syp-5 mutants, the localization of LAB-2::GFP was altered, localizing to both bivalent arms at diakinesis (Fig. 7, A and B). The separated localization of LAB proteins in syp-5 mutants may also be explained by multisteps of axis differentiation post crossover designation. LAB-1 likely senses an earlier step of axis differentiation that is normally established in syp-5 mutants, while LAB-2 senses a later step of axis differentiation that fails to be established in the mutants.

Diakinesis chromosome association of LAB proteins in meiotic mutants. (A and B) Localization of LAB-1::GFP (A) or LAB-2::GFP (B) on diakinesis bivalents in wild type or syp-5 mutant background. Chromatin was stained with DAPI (blue). The cartoons on the right depict the localization patterns of LAB-1 or LAB-2 on bivalents indicated by the open arrowheads. Bar, 5 μm. (C) Localization of LAB-2::GFP (green) and SYP-5 (red) on early diakinesis bivalents in the indicated genotypes. Chromatin was stained with DAPI (blue). Bar, 5 μm. (D and E) Chromosome association of LAB-1::GFP (D) or LAB-2::GFP (E) in diakinesis nuclei of the indicated genotypes and treatments (with or without 5% 1,6-hexanediol incubation for 10 min). Chromatin was stained with DAPI (blue). Bar, 10 μm. (F and G) Quantification of relative chromosome loading of LAB-1::GFP (F) or LAB-2::GFP (G) in diakinesis nuclei of the indicated genotypes and treatments. Bars represent the mean ± SD, and the numbers of nuclei analyzed are indicated on the top. Statistical analysis was performed by the two-tailed unpaired t test.
Diakinesis chromosome association of LAB proteins in meiotic mutants. (A and B) Localization of LAB-1::GFP (A) or LAB-2::GFP (B) on diakinesis bivalents in wild type or syp-5 mutant background. Chromatin was stained with DAPI (blue). The cartoons on the right depict the localization patterns of LAB-1 or LAB-2 on bivalents indicated by the open arrowheads. Bar, 5 μm. (C) Localization of LAB-2::GFP (green) and SYP-5 (red) on early diakinesis bivalents in the indicated genotypes. Chromatin was stained with DAPI (blue). Bar, 5 μm. (D and E) Chromosome association of LAB-1::GFP (D) or LAB-2::GFP (E) in diakinesis nuclei of the indicated genotypes and treatments (with or without 5% 1,6-hexanediol incubation for 10 min). Chromatin was stained with DAPI (blue). Bar, 10 μm. (F and G) Quantification of relative chromosome loading of LAB-1::GFP (F) or LAB-2::GFP (G) in diakinesis nuclei of the indicated genotypes and treatments. Bars represent the mean ± SD, and the numbers of nuclei analyzed are indicated on the top. Statistical analysis was performed by the two-tailed unpaired t test.
The lack of LAB-1 causes abnormal chromosome remodeling, and SC central region proteins localize to both bivalent arms. Immunostaining of SYP-5 in lab-1;lab-2::gfp worms showed that SYP-5 presents on both arms, while LAB-2::GFP remains localized only at bivalent long arms (Fig. 7 C). This observation suggests that LAB-2 localization is independent of LAB-1-mediated remodeling events.
Meiotic recombination alters the axis recruitment of LAB proteins during late prophase
In spo-11 mutants, programmed DNA double-strand break formation is lacking, and homologous chromosomes fail to undergo interhomolog recombination and crossover formation, generating univalent chromosomes at diakinesis (Dernburg et al., 1998). To understand how lack of meiotic recombination might affect the axis association of LAB proteins, we crossed lab-1::gfp and lab-2::gfp into the spo-11 mutants. Both LAB proteins localized to the univalents at diakinesis (Fig. 7, D and E). However, quantification analysis showed that spo-11 mutation oppositely affected their recruitments. While LAB-1::GFP showed increased chromosome recruitment in spo-11 mutants compared to the wild type (Fig. 7 F), LAB-2::GFP recruitment was reduced in spo-11 mutants (Fig. 7 G). Moreover, exposure with 5% 1,6-hexanediol also revealed enhanced LAB-1 association and reduced LAB-2 association on diakinesis chromosomes in the mutant background (Fig. 7, D–G). Thus, in the absence of meiotic recombination, the undifferentiated axis core may provide more stable interactions with LAB-1, enhancing further LAB-1 recruitment. However, although LAB-2 may have impaired direct interactions with the axis in spo-11 mutants, the axes are the only available recruitment sites, and LAB-2 is still abundantly recruited.
Weak interactions mediate CPC recruitment on bivalent short arms
Bivalent short-arm localization of CPC promotes cohesion removal and homolog segregation during meiosis I (Kaitna et al., 2002; Rogers et al., 2002; Romano et al., 2003). As a substrate of CPC, histone H3 is also phosphorylated, and this phosphorylated histone marker is enriched only on the short arms (Hsu et al., 2000). In syp-5 mutants, the CPC kinase AIR-2 diffuses in the nucleus, and the phosphorylated histone H3 distributes on both bivalent arms, showing delayed chromosome segregation during meiosis I (Liu et al., 2021; Zhang et al., 2020b), suggesting that spatially restricted CPC activity is essential for efficient and accurate meiotic chromosome segregation. Given that LAB proteins have phase separation properties that may contribute to their enrichment on bivalent long arms, we tested whether a similar mechanism might be involved in CPC recruitment. Consistently, chromosome recruitment of AIR-2::GFP was abolished by 1,6-hexanediol treatment, suggesting weak interactions are involved in the recruitment of AIR-2 on bivalent short arms. Moreover, the signal of phosphorylated histone H3 also showed disrupted organization upon 1,6-hexanediol treatment, although the overall signal intensity was not altered (Fig. S5 G).
Discussion
In conclusion, the dynamic localization of axis-associated LAB proteins allows the reveal of multi-states of axis differentiation during meiotic prophase. Weak interaction-mediated structure formation on bivalent arms may underlie the establishment of chromosome subdomains critical for accurate meiotic chromosome segregation.
HORMA proteins and cohesin complexes are the core components of the meiotic chromosome axis in C. elegans. The HORMA protein complex is assembled through a hierarchical interaction network in which HTP-3 C-terminus interacts with other HORMA domain proteins (Kim et al., 2014). However, the HORMA complexes may exist in various forms with different conformations, compositions, and posttranslational modifications (PMTs) during meiotic progression, accompanied by altered interaction stabilities with LAB proteins. Based on the localization patterns of LAB proteins, we propose that there are at least four states of HORMA complexes during meiotic prophase (Fig. 8 A). State 1 represents HORMA complexes within the unsynapsed chromosome axis during early prophase, and LAB proteins are not recruited. Upon synapsis, SC central region assembly may cause conformation changes of HORMA proteins, allowing axis recruitment of LAB-2 (State 2). During mid- to late-pachytene, off-centered crossovers are designated, a process that is coupled with the asymmetric distribution of CO regulators (e.g., ZHP-1/2), kinases (e.g., PLK-2), and PMTs (e.g., phospho-SYP-4 and phospho-HIM-3; Nadarajan et al., 2017; Sato-Carlton et al., 2020; Zhang et al., 2018). The axis is differentiated into a third state, which gradually causes the short-arm localization of SC central region proteins at the pachytene exit. However, LAB proteins are differentially localized at this point, with LAB-1 enriched on the desynapsed bivalent long arms and LAB-2 enriched on the SC-associated short arms. After the entry into diplotene, the axis undergoes a further differentiation, allowing the switch of LAB-2 from SC-associated short arms to bivalent long arms (State 4). In meiotic mutants (e.g., syp-5 and spo-11 mutants), the abnormal localization patterns of LAB proteins also suggest disrupted axis differentiation (Fig. 8 B).

Models for the dynamics and coordination of various meiotic structures on chromosome axis. (A) Formation and coordination of various meiotic structures during meiotic prophase in the wild type. The localization patterns of the SC and LAB proteins suggest the HORMA complexes may have at least four states at the meiotic prophase. Black arrows indicate promoting regulation, and blue arrows indicate redistribution directions. (B) Summarized localization patterns of the indicated proteins or structures on early and late diakinesis bivalents in syp-5, lab-1, lab-2, and spo-11 mutants.
Models for the dynamics and coordination of various meiotic structures on chromosome axis. (A) Formation and coordination of various meiotic structures during meiotic prophase in the wild type. The localization patterns of the SC and LAB proteins suggest the HORMA complexes may have at least four states at the meiotic prophase. Black arrows indicate promoting regulation, and blue arrows indicate redistribution directions. (B) Summarized localization patterns of the indicated proteins or structures on early and late diakinesis bivalents in syp-5, lab-1, lab-2, and spo-11 mutants.
The distinct localization patterns of LAB proteins during meiotic progression suggest that they likely have distinct binding sites within the HORMA complexes, and changes in the complexes during axis differentiation can have different impacts on LAB interactions. Due to the weak interaction property, a minor change in the binding affinity of LAB proteins with the axis following axis differentiation can quickly cause the specific enrichment of LAB proteins on chromosome subdomains. This may also explain the enrichment of LAB proteins on specific chromosomes in synapsis-defective mutants during early prophase. Furthermore, the phase separation property of LAB proteins allows their unlimited axis recruitment, explaining their high enrichments on bivalent arms compared with HORMA proteins during late prophase. The cohesin protectors GSP-1/GSP-2 phosphatases (PP1 homologs in C. elegans) may be recruited to the bivalent long arms through LAB-1 condensates. Consistently, only weak interactions were detected between LAB-1 and GSP-1/2 (Tzur et al., 2012).
Axis-associated LABs were not completely removed by 1,6-hexanediol exposure during late meiotic prophase, suggesting the retained pool of LABs may have more stable interactions with the HORMA complexes. Given that 1,6-hexanediol treatments significantly disrupted the majority of LABs, there may be two layers of LAB recruitments, with one pool of LABs stably interacting with the HORMA complexes and another pool recruited by multivalent weak interactions between LAB proteins. Stable interactions may be regulated by several factors, including changes in complex composition, HORMA protein conformations, and PMTs. It is worth noting that the meiotic chromosome axis is dynamically regulated in vivo, and our insect purification and in vitro analysis may mimic a single snapshot of the axis status of the meiotic chromosome in vivo.
Previous findings and our current work suggest structures on the meiotic chromosomes are highly coordinated (de Carvalho et al., 2008; Ferrandiz et al., 2018; Martinez-Perez et al., 2008). For example, LAB-2 axis association is SC dependent during early prophase, and LAB-1 and SC central region proteins show reciprocal localization at late meiotic prophase. A subset of the meiotic axis-associated structures may be formed by phase separation and are sensitive to 1,6-hexanediol disruption, as exemplified by the SC (Zhang et al., 2023). Although our data revealed direct interactions between the LABs and HORMA complexes, it is still possible that axis recruitment of LAB proteins may also depend on other axis-associated structures at specific meiotic stages, and 1,6-hexanediol treatment may indirectly affect the axis recruitment of LABs by disrupting the dependent structures.
On bivalent short arms, CPC and phosphorylated histones may also form phase-separated domains. The concentration of CPC within these domains can promote its local activity and significantly reduce its diffusion in the nucleoplasm and activity on other chromosome regions. These orchestrated controls of CPC activity ensure accurate meiotic chromosome segregation. Interestingly, recent work revealed that the inner centromere in mammalian mitotic cells is a biomolecular condensate scaffolded by CPC (Trivedi et al., 2019). Some conserved mechanisms may underlie the formation of chromosome subdomains to promote accurate chromosome segregation during different types of cell divisions. Observations in several mutants suggest that the SC maintenance on diakinesis bivalent arms may be required in guiding CPC recruitment. For example, the SC undergoes premature loss from the chromosomes in syp-5 mutants and the CPC failed to enrich on any bivalent arms (Zhang et al., 2020b). In lab-1 mutants, the SC is maintained on both bivalent arms, causing the enrichment of AIR-2 on both arms (de Carvalho et al., 2008). Similarly, the SC is maintained on diakinesis univalent chromosomes in spo-11 mutants, and AIR-2 then presents on all the chromosomes (Nabeshima et al., 2005). However, as revealed in the current work, although the SC is mislocalized to both bivalent arms in lab-2 mutants, AIR-2 and pH3 remain restricted to the short arms. Thus, the SC itself might not be enough to mediate the establishment of CPC-enriched domains and other factors are still required.
Materials and methods
C. elegans culture, genome editing, and RNAi
Bristol N2 was used as the wild-type strain in this study and all mutants were derived from the N2 background. Unless otherwise indicated, worms were maintained on NGM agar plates spread with E. coli OP50 according to the standard method (Stiernagle, 2006). CRISPR-Cas9 genomic editing was used to create lab-2 mutants and lab-1::gfp, lab-2::gfp, lab-2::mScarlet, and htp-1::gfp strains. Sets of plasmids expressing Cas9 (Peft-3::Cas9-SV40_NLS::tbb-2 3′UTR), guide RNAs, mCherry co-injection makers (pCFJ90 and pCFJ104), and plasmids containing the repair donor templates were coinjected into the gonads of young adults N2 hermaphrodites. The following sequences were used as the gRNA targets when generating the lines: 5′-CCATCTTTCAGCTCCCCGTAATT-3′ and 5′-AGTATTTGATAAGGCTATTGAGG-3′ for lab-2 null mutant creation, and used 5′-CCCGCTATGAATCAACTTCTTTC-3′ and 5′-CCAAATTGAGCTATTTTCTACTC-3′ for C-terminal tag insertion. The repair donor templates contained the desired sequences flanked by ∼1.5 kb of upstream and downstream homologous arms. F1 progeny expressing mCherry coinjection marker was singled and screened for successful genome edits via PCR and fluorescence microscope examination.
RNA interference of htp-2 was performed by feeding. To construct the RNAi clone, a segment of htp-2 exon was PCR-amplified with a pair of primers (forward primer: 5′-CTCGAGGGGGGGCCCGGTACCCCATCGAAGTGTTCTCTATGAAGTTC-3′; reverse primer: 5′-CTATAGGGCGAATTGGGTACCGTGTGTAAAAAGTGGAAGAATCGTCG-3′) and inserted into the pL4440 vector at the KpnI site through Gibson assembly. The constructed plasmids were transferred into HT115 bacteria and bacteria carrying the empty pL4440 vector were used as control. Quantitative real-time PCR analysis suggested both htp-1 and htp-2 were knocked down by the feeding RNAi clone.
C. elegans strains used in this study
The following strains were used in this study: AV630: meis8[pie-1p::gfp::cosa-1 + unc-119(+)] II; CA1230: htp-3(tm3655) I; ieSi6[htp-3p::htp-3::GFP + Cbr-unc-119(+)] II; unc-119(ed3) III; CV6: lab-1(tm1791) I/hT2[bli-4(e937) let-?(q782) qIs48] (I;III); MGC20: lab-2(cac7) III; MGC21: lab-2(cac8[lab-2::gfp]) III; MGC49: lab-2(cac8[lab-2::gfp]) III; him-8(e1489) IV; MGC51: lab-2(cac7) III; wgls227[syp-2::TY1::EGFP::3xFLAG(92C12) + unc-119(+)]; MGC53: syp-5(cac1) I; lab-2(cac8[lab-2::gfp]) III; MGC74: lab-2(cac8[lab-2::gfp]) III; spo-11(ok79) IV/nT1[unc-?(n754) let-?] (IV;V); MGC75: lab-2(cac8[lab-2::gfp]) III; htp-1(gk150) IV/nT1[unc-?(n754) let-? qIs50] (IV;V); MGC76: meis8[pie-1p::gfp::cosa-1 + unc-119(+)] II; lab-2(cac7) III; MGC79: lab-1(tm1791) I/hT2 [bli-4(e937) let-?(q782) qIs48] (I;III); lab-2(cac7) III; MGC115: htp-1(cac35[htp-1::gfp]) IV; MGC137: lab-1(tm1791) I/hT2 [bli-4(e937) let-?(q782) qIs48] (I;III); lab-2(cac8[lab-2::gfp]) III; MGC170: htp-3(tm3655) I/hT2 [bli-4(e937) let-?(q782) qIs48] (I,III); lab-2(cac8[lab-2::gfp]) III; MGC171: lab-2(cac8[lab-2::gfp]) III; him-3(me80) IV/nT1 [unc-?(n754) let-? qIs50] (IV;V); MGC256: lab-1(cac50[lab-1::gfp]) I; MGC267: lab-1(cac50[lab-1::gfp]) I; lab-2(cac49[lab-2::mScarlet]) III; MGC336: lab-1(cac50[lab-1::gfp]) I; him-8(e1489) IV; MGC557: lab-1(cac50[lab-1::gfp]) I; spo-11(ok79) IV/nT1[[unc-?(n754) let-?] (IV;V); MGC564: syp-5(cac1) I; lab-1(cac50[lab-1::gfp]) I; MGC578: lab-1(tm1791) I; wgls227[syp-2::TY1::EGFP::3xFLAG(92C12) + unc-119(+)]; MGC588: lab-2(cac-7) III; ojIs50[pie-1p::GFP::air-2 + unc-119(+)]; MGC597: lab-1(tm1791) I/hT2[bli-4(e937) let-?(q782) qIs48] (I;III); ojIs50[pie-1p::GFP::air-2 + unc-119(+)]; MGC588: lab-1(cac50[lab-1::gfp]) I; lab-2(cac-7) III; OP227: unc-119(ed3) III; wgIs227 [syp-2::TY1::EGFP::3xFLAG(92C12) + unc-119(+)]; WH371: unc-119(ed3) III; and ojIs50[pie-1p::GFP::air-2 + unc-119(+)].
Antibodies used in this study
The following antibodies were used at the indicated dilutions: rabbit anti-SYP-5 (1:1,000 for IF; Zhang et al., 2020b); rabbit anti–phospho-H3 (Ser10; 1:1,000 for IF; PA5-17869; Thermo Fisher Scientific); mouse anti-HA (1:2,000 for WB; SAB2702196; Sigma-Aldrich); mouse anti-FLAG (1:2,000 for WB; F3165; Sigma-Aldrich); chicken anti-GFP (1:2,000 for WB; ab13970; Abcam); HRP conjugated goat anti-mouse (1:5,000; #91196; Cell Signaling Technology); HRP conjugated rabbit anti-chicken (1:5,000; SA00001-6; Proteintech); and donkey anti-rabbit-Cy3 (1:200; 711-165-152; Jackson ImmunoResearch Laboratories).
Immunoprecipitation and mass spectrometry analysis
Immunoprecipitation of GFP-tagged HTP-3 and LAB-2 was performed with extracts of germ cell nuclei enriched by vortexing cut and differential centrifugation method (Gao et al., 2016). Nuclear extracts were incubated with GFP-Trap agarose beads (ChromoTek) for 3 h at 4°C. Immunoprecipitated proteins were eluted by boiling in 2% SDS in 20 mM Tris (pH 7.4) for 5 min. Eluted proteins were precipitated with the ProteoExtract Protein Precipitation Kit (Calbiochem) and digested with trypsin for MS analysis. To identify HTP-3::GFP or LAB-2::GFP binding proteins, we compared the IP data with at least three unrelated IPs that were performed identically. Proteins with an average spectral percentage >0.2% and detected in the IPs of at least two biological replicates were considered specific binding partners.
Immunofluorescence microscopy analysis of the C. elegans germline
Gonads were dissected from young adult worms (24 h post L4) in dissection buffer (25 mM Hepes at pH 7.4, 118 mM NaCl, 48 mM KCl, 2 mM EDTA, 5 mM EGTA, 0.1% Tween-20, and 10 mM NaN3) on coverslips and were transferred onto polylysine-coated slides by freeze crack on dry ice. Gonads were fixed first with −20°C methanol for 1 min and then 4% paraformaldehyde in PBS at room temperature for 30 min. After the block with 0.5% BSA (in PBST) for 1 h, gonads were sequentially incubated with the primary antibody (at 4°C overnight) and the secondary antibody (at room temperature for 2 h), each followed by three washes in PBST. Chromatin DNA was stained with 4′,6-diamidino-2-phenylindole, dihydrochloride (DAPI; Invitrogen), and the gonads were mounted with Vectashield mounting medium (Vector Laboratories). Fluorescence microscope images shown in Fig. 1 C, Fig. 5 E, Fig. 6, Fig. 7, A–C, Fig. S3 C, and Fig. S5 are maximum-intensity projections through 3D data stacks of whole nuclei that were captured at 200-nm intervals on a DeltaVision OMX microscope system with 60×/1.42 lens with SoftWoRx software (Applied Precision) in the conventional imaging mode. Images were deconvolved using a conservative algorithm with 10 iterations. Fluorescence microscope images shown in Fig. 2 F, Fig. 4, Fig. 7, D and E, and Fig. S4 were captured with a Leica DM6 B confocal microscope with 63×/1.40 lens with LAS X acquisition software (Leica Microsystems Inc.). All the other fluorescence microscopy images were wide-field images captured with a Nikon TS2-FL microscope with 60×/1.42 lens with NIS-Elements acquisition software (Nikon Instruments Inc.).
Axis loading quantification
For quantification of the axis loading, gonads dissected from young adult worms (24 h post L4) expressing GFP-tagged axial proteins were fixed and stained with DAPI. Fluorescent images were captured with a 60× objective equipped with a Nikon TS2-FL microscope and images from at least two biological replicates were collected for analysis. Axis segments of interest were converted to “Text image” with ImageJ (National Institutes of Health, Bethesda, MD, USA). Only isolated axis segments captured on the focal plane were selected for analysis, and an isolated segment was the only one present in the selected box with a minimal 15 pixels on any side. In diakinesis nuclei, only segments on bivalent long arms were selected. All possible isolated axis segments were measured to avoid bias. Further calculation was performed in batches with in-house R scripts. First, the max intensity value of a 3 × 3 pixels region (corresponds to 0.4 × 0.4 μm) on a measured segment was identified by scanning the text image. Then, pixel values of the text image were sorted in descending order and the mean value of the second half of the pixels was used as the corresponding background intensity. The ratio between the max intensity of each segment and the corresponding background intensity was defined as the relative axis loading.
HORMA complex expression and purification
HORMA complexes with or without LAB-1/LAB-2 were expressed and purified through the SmartBac system (Zhai et al., 2019). The coding sequences of the HORMA complexes were codon-optimized and synthesized (Sangon Biotech). TEV protease cleavage sites were inserted between subunits of the complex. A copy of the TEV protease coding sequence is present upstream of the inserted sequence, providing the protease required for polypeptide cleavage in the cells. For affinity purification, two copies of the FLAG tag were inserted at the C-terminal end of HTP-3. The sequence containing all the subunits was inserted into the 4V1R vector at the gap created by NcoI and EcoRI digestion. The generated plasmid was transformed into DH10Bac competent E. coli (Angyubio) for bacmid generation via recombination. The transformed E. coli were grown on LB agar plates containing 50 μg/ml kanamycin, 7 μg/ml gentamicin, 10 μg/ml tetracycline, 100 μg/ml Bluo-gal, and 40 μg/ml IPTG. Recombinant clones (white) were picked and cultured in the LB medium containing 50 μg/ml kanamycin, 7 μg/ml gentamicin, and 10 μg/ml tetracycline for further plasmid purification and verification.
For the production of the first generation of baculovirus (P1 virus), the bacmid DNA was extracted and transfected into 2 ml Sf9 cells (0.5 × 106 cells/ml) cultured in a six-well plate in SIM SF Expression Medium (Sino biological) at 27°C using Escort IV Transfection Reagent (L3287; Sigma-Aldrich). The expression of red fluorescent protein in Sf9 cells is indicative of Bacmid expression. 4 d after the transfection, baculovirus was further amplified by adding 1 ml of the transfected Sf9 cells into a 50-ml culture of SF9 cells (1.4 × 106 cells/ml) in a flask, which was cultured at 27°C with shaking at 120 rpm for another 3 d. The amplified virus (P2 virus) was cleared by centrifugation at 2,000 rpm for 10 min and the supernatant baculoviral stock was tittered.
For large-scale expression and purification, 200 ml of Sf9 cells (1–2 × 106 cells/ml) was infected with 2 ml P2 virus. 3 d after infection, cells were harvested by centrifugation at 4,000 rpm for 10 min at 4°C. After one wash with ice-cold PBS, cells were lysed with the lysis buffer (150 mM NaCl, 50 mM Tris-HCl, 1 mM β-sodium glycerophosphate, 1 mM EDTA, 1 mM Na3VO4, 0.8 mM EGTA, 2 mM NaF, and 2.5 mM sodium pyrophosphate, pH 7.4) supplemented with protease inhibitor mixture (Roche Diagnostics). Cell debris was removed by centrifugation at 11,000 g for 30 min at 4°C. Cleared lysate was incubated with anti-FLAG agarose beads (Abmart) for 3 h at 4°C. After three washes with the washing buffer, proteins were eluted with the elution buffer (50 mM Tris-HCl, 150 mM NaCl, 2 mg/ml FLAG peptides [GL Biochem Ltd.], pH 7.4). Eluted proteins were further concentrated with Amicon Ultra centrifugal filters (30 KD; Merck Millipore).
LAB protein purification
Codon-optimized coding sequences of LAB-1 and LAB-2 fused with a hexahistidine (6xHis) tag at the N-termini and a GFP or mCherry tag at the C-termini were cloned into pET-30a plasmid with the NdeI and HindIII digestion sites. BL-21 bacteria were transformed with the LAB protein-expressing plasmids or tag-only plasmids. Protein expression was induced with 0.5 mM IPTG at 16°C for 16 h. Induced proteins were extracted by sonication with Diagenode Bioruptor (Diagenode Inc.) in lysis buffer (50 mM Tris, 500 mM NaCl, 1% Triton X-100, and 10 mM imidazole, pH 7.4) supplied with 1mM PMSF and protease inhibitor mixture. Cell debris was removed at 11,000 g for 30 min at 4°C. 6xHis tagged proteins in the supernatants were captured with the HisPurTMNi-NTA Resin column (Thermo Fisher Scientific). After washing the column with wash buffer (50 mM Tris, 500 mM NaCl, and 30 mM Imidazole, pH 7.4), proteins were eluted with elution buffer (50 mM Tris, 500 mM NaCl, and 250 mM imidazole, pH 7.4). Eluted proteins were diluted with 50 mM Tris buffer to reduce NaCl concentration to 150 mM. Purified proteins were further concentrated with Amicon Ultra centrifugal filters (30 KD). Protein purification was analyzed by SDS-PAGE and protein concentration was measured.
Protein coexpression and interaction analysis in 293T cells
Coding sequences of axial proteins were cloned into mammalian expression plasmids with a cytomegalovirus promoter and fused with a GFP or mCherry-HA tag. Plasmids were cotransfected into 293T cells cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum at 37°C in a humidified atmosphere with 5% CO2. Polyethyleneimine reagent (Sigma-Aldrich) was used for plasmid transfection. Cells were lysed after 24 h in cell lysis buffer composed of 50 mM HEPES (pH 7.4), 1 mM EGTA, 3 mM MgCl2, 300 mM KCl, 10% glycerol, 1% Nonidet P-40, 1 mM dithiothreitol, and a protease inhibitor mixture. Interactions between cotransfected proteins were examined by IP and Western blot analysis. GFP-Trap magnetic beads (ChromoTek) were used to perform IPs.
1,6-hexanediol exposure
For 1,6-hexanediol exposure, gonads were dissected from young adults (24 h post-L4) in 20 µl PBS buffer containing 0.05% Tween-20 (PBST). Ten volumes (200 µl) of 11% or 5.5% 1,6-hexanediol in PBST were added to the dissection buffer to achieve a final concentration of 10% or 5% 1,6-hexanediol, respectively. The solution was mixed by gentle pipetting and incubated for 10 min. Gonads were picked and transferred to a new coverslip and were fixed and stained as performed previously (Gao et al., 2016). For the examination of axis reassociation after 1,6-hexanediol washout, gonads incubated in 10% 1,6-hexanediol for the desired time were picked with a 20 µl pipette tip and transferred into a 200 µl PBST droplet. After a quick mix, the gonads were left to rest for 10 min and were then fixed and stained.
In vitro phase separation assay
For phase separation examination, purified proteins were diluted with phase separation buffer (50 mM Tris-HCl, 150 mM NaCl, pH 7.4) to the desired concentrations on ice. A humidified chamber was prepared with a 35-mm capped glass-bottomed cell culture dish (MatTek Corporation). A drop of 2 μl protein solution was placed on the glass bottom of the dish and examined with a confocal microscope at room temperature (20°C). Phase separation preferentially occurred at the droplet’s edge and the phase separation process was recorded.
Statistical analysis
Statistical difference between two sets of data was determined in GraphPad Prism using a two-tailed unpaired Student’s t test and significance was assumed by P < 0.05. Data distribution was assumed to be normal, but this was not formally tested. Error bars represent the standard deviation of the data sets and the number of independent data points (n) are indicated on the plots.
Online supplemental material
Fig. S1 shows the analysis of LAB protein conservation and structural features. Fig. S2 shows a schematic diagram illustrating the fluorescence measurement of chromosome-associated proteins. Fig. S3 shows that HORMA complexes may provide multiple binding sites for LAB proteins. Fig. S4 shows the phase separation analysis of fluorescent protein-tagged LAB proteins. Fig. S5 shows the cytological analysis of synapsis, crossover formation, and AIR-2/phospho-H3 localization in lab-2 mutants and AIR-2/phospho-H3 localization upon 1,6-hexanediol treatment.
Data availability
Data are available in the article itself and its supplementary materials. The reagents and in-house R scripts used for data analysis are available from the corresponding author upon reasonable request.
Acknowledgments
We thank M.P. Colaiácovo (Harvard Medical School, Boston, MA, USA) for providing the lab-1 mutants.
This work was supported by grants from the National Natural Science Foundation of China (32022018, 31871360, 31900557, and 32370780) and the National Key R&D Program of China 2021YFA1101001. Some strains were provided by the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health Office of Research Infrastructure Programs (P40 OD010440).
Author contributions: Conceptualization, J. Gao; Methodology, J. Gao, R. Wang, and J. Li; Investigation, R. Wang, J. Li, Y. Tian, Y. Sun, Y. Zhang, M. Liu, R. Zhang, L. Zhao, Q. Li, X. Meng, and J. Gao; Writing, J. Gao, R. Wang, and J. Zhou; Funding Acquisition, J. Gao, R. Wang, and J. Zhou; Resources, J. Gao and J. Zhou; Supervision, J. Gao.

