


Autophagy is a conserved eukaryotic lysosomal degradation pathway that responds to environmental and cellular cues. Autophagy is essential for the meiotic exit and sporulation in budding yeast, but the underlying molecular mechanisms remain unknown. Here, we show that autophagy is maintained during meiosis and stimulated in anaphase I and II. Cells with higher levels of autophagy complete meiosis faster, and genetically enhanced autophagy increases meiotic kinetics and sporulation efficiency. Strikingly, our data reveal that the conserved phosphatase Cdc14 regulates meiosis-specific autophagy. Cdc14 is activated in anaphase I and II, accompanying its subcellular relocation from the nucleolus to the cytoplasm, where it dephosphorylates Atg13 to stimulate Atg1 kinase activity and thus autophagy. Together, our findings reveal a meiosis-tailored mechanism that spatiotemporally controls meiotic autophagy activity to ensure meiosis progression, exit, and sporulation.
Introduction
Meiosis depends on a carefully orchestrated sequence of intracellular processes that produce viable gametes after a diploid cell undergoes two rounds of nuclear division, wherein targeted proteolysis provides a rapid and irreversible mechanism for promoting transitions between meiotic stages (Neiman, 2011; King et al., 1996). Autophagy is a conserved lysosomal degradation pathway discovered initially as a survival mechanism to cellular stress, while emerging evidence indicates that it is also involved in selective regulation of cell development and differentiation (Politi et al., 2014; Rojansky et al., 2016; Sato and Sato, 2011; Wang et al., 2020b; Maria Fimia et al., 2007). In budding yeast, an important model system for research on meiosis, autophagy is required for meiotic entry (Piekarska et al., 2010; Sarkar et al., 2014; Wen et al., 2016; Enyenihi and Saunders, 2003; Straub et al., 1997) and exit (Wang et al., 2020b; Wang et al., 2020a). Specifically, loss of autophagy during meiotic cell divisions results in delayed onset of meiosis II (metaphase II and anaphase II), failed exit from meiosis, and cell death (Wang et al., 2020b). In the absence of autophagy, the cell overduplicates the spindle pole body (SPB, the yeast centrosome), the nucleation center for the spindle, and the membrane of the prospore (the precursor of the daughter cell), resulting in abnormal chromosome segregation and prospore membrane formation (Wang et al., 2020b). However, the underlying mechanisms remain elusive.
At anaphase I, autophagy coordinates with proteasome to rapidly destroy the RNA-binding protein Rim4 (Wang et al., 2020b; Carpenter et al., 2018), which forms amyloid-like aggregates to sequester a subset of mRNAs of mid-late meiotic genes, e.g., the B-type cyclin CLB3 (Berchowitz et al., 2015). Thus, autophagy activity at anaphase I is crucial for releasing Rim4-suppressed translation (Wang et al., 2020b). The essential role of autophagy in Rim4 clearance (anaphase I) and meiosis exit (anaphase II) implies that autophagy during meiotic divisions, minimally at anaphase I and II, should be secured. What, then, is the mechanism? As a major degradation pathway, autophagy must be tightly regulated to avoid excess or insufficient autophagic degradation. Consistent with this, dysregulation of autophagy is associated with various diseases and disorders (Mizushima and Komatsu, 2011; Meijer and Codogno, 2009; Guo et al., 2013). Among various mechanisms that regulate autophagy in response to extra- and intracellular stimuli is posttranslational modification. In particular, alteration of protein phosphorylation state can rapidly switch autophagy initiation on and off (Funakoshi et al., 1997; Scott et al., 2000; Kamada et al., 2000; Memisoglu et al., 2019; Mao et al., 2013; Pengo et al., 2017; Davis et al., 2016; Licheva et al., 2021) as well as influence cargo selection (Aoki et al., 2011; Pfaffenwimmer et al., 2014; Gatica et al., 2018; Stolz et al., 2014). To date, several protein kinases have been identified and mechanistically characterized in regulating autophagy, including mTOR complex 1 (mTORC1), a master suppressor of autophagy initiation (Kim and Guan, 2015). In sharp contrast, our knowledge of the role of protein phosphatases in autophagy regulation remains much more limited (Klionsky, 2005) and is missing during meiotic divisions.
Intriguingly, in mammalian mitosis, autophagy is regulated by the mitotic master kinase CDK1 (Cdc28 in yeast), which replaces the role of mTORC1 in phosphorylating autophagy initiation factors such as ULK1 (Atg1), ATG13 (Atg13), and ATG14 (Atg14) (Odle et al., 2020). Whether CDK1 activates or suppresses mitotic autophagy during mitosis remains controversial (Yamasaki et al., 2020; Li et al., 2020; Odle et al., 2021). Similarly, we do not yet know whether CDK1 modulates autophagy during meiosis, a specialized cell division program that differs from mitosis. Nonetheless, TORC1 activity is suppressed during meiosis in budding yeast to allow meiotic entry and progression (Yamamoto, 2004; van Werven and Amon, 2011; Harigaya and Yamamoto, 2007), while Cdc28 (CDK1) drives meiosis progression. The main phosphatase that counteracts Cdc28 is Cdc14. Therefore, Cdc28 and Cdc14, activated in metaphase and anaphase, respectively, could shape autophagy activity in coordination with meiotic progression. Previously, it was reported that inactivation of Cdc14 in haploid vegetative (mitotic) yeast cells is associated with a reduction in autophagy after TORC1 inactivation (Kondo et al., 2018); however, it is unclear whether Cdc14 regulates autophagy during meiosis.
Here, we found that meiosis benefits from a unique profile of autophagy flux that is distinct from autophagy during quiescence or starvation. During anaphase I and II, Cdc14 relocates from the nucleolus into the cytoplasm, where it dephosphorylates Atg13 to activate Atg1 and thus autophagy. In addition, we identified two motifs of Atg13 required for Cdc14–Atg13 binding and six serine residues of Atg13 that are critical targets of Cdc14 phosphatase activity, providing molecular insights into the Cdc14–Atg13 interaction. Together, our findings reveal a meiosis-specific mechanism of Cdc14-mediated upregulation of autophagy that modulates meiotic progression and sporulation.
Results
Meiosis features a unique autophagy profile
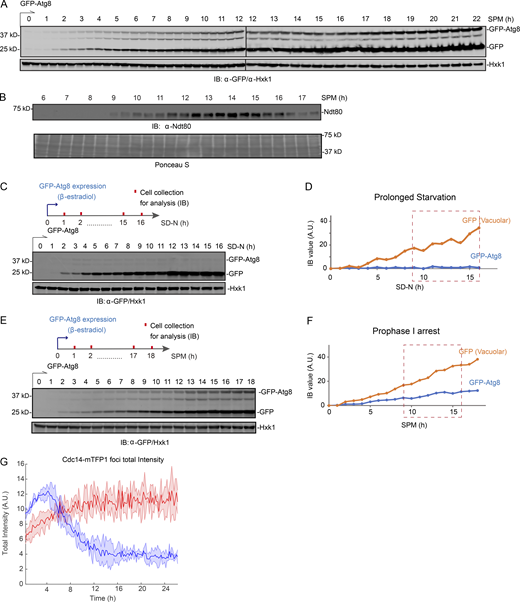
To investigate how autophagy is regulated during meiosis, we monitored autophagy flux from meiotic entry to exit and early sporulation. We used a GFP-Atg8 processing assay that quantifies the delivery of GFP-Atg8 on the inner autophagosomal membrane to the vacuole (Fig. 1 A) by immunoblotting (IB: α-GFP; Figs. S1 A and 1 B, top panel). Atg8 is degraded in the vacuole lumen, leaving behind free GFP, which is more resistant to hydrolysis and thereby is an indicator of autophagy (Klionsky et al., 2016). After meiosis was triggered, we found that autophagy flux (free GFP level) remained relatively stable at early time points, but fluctuated during the meiotic divisions, marked by expression of NDT80, a meiotic transcription factor required for nuclear divisions, and determined by immunofluorescence (IF) of Tub1 (Fig. 1 B, ∼10 to ∼16 h). This fluctuation was specifically linked to meiotic divisions and was not observed after prolonged nitrogen starvation (Fig. S1, C and D) or in ndt80Δ meiotic cells that were forced to arrest before meiotic divisions in prophase I (Fig. S1, E and F).

Meiosis features a unique temporal pattern of autophagy. (A) Schematic of GFP-Atg8 induction and cell collection for IB and IF staining (α-Tubulin) during meiosis and sporulation. Z4EV is a β-estradiol–activated transcription factor, and ZD is a Z4EV-driven promoter. (B) Top, graph of cellular GFP-Atg8/GFP (with a representative IB image) and Ndt80 protein levels (determined by IB, Fig. S1, A and B) during meiosis and sporulation as in A. Bottom and middle, representative images and graph of meiosis stages determined by IF of Tub1 (α-Tubulin) and DAPI staining (n > 100 cells; two independent experiments). Dashed box (red), time window of meiotic divisions. Scale bar, 5 µm. (C–G) Time-lapse FM analysis. (C) Representative time-lapse (FM) image of a cell undergoing meiosis. Cell expresses Whi5-mKOκ, mNG-Atg8, Cdc14-mTFP1, and Tup1-mNeptune2.5. Symbols mark Atg8 puncta (white arrowhead), nucleus (yellow circle), and cell (white circle). Scale bar, 5 µm. (D and E) Total mNG-Atg8 (D) and the vacuolar mNG-Atg8 (E) signal (line) with the standard deviations (shade) from time-lapse FM of starved (SD-N) mitotic cells (grey, n = 549 cells), quiescent cells (green, n = 327 cells), meiotic cells (blue, n = 323 cells), and meiotic ndt80Δ cells (red, n = 231 cells). (F and G) Cdc14-mTFP1 foci intensity (F) and mNG-Atg8 fluorescence intensity (G) in WT (blue) and ndt80Δ (red) meiotic cells. Cells were in silico aligned to the onset of premeiotic DNA replication (Whi5 nuclear export). A.U., arbitrary unit.
Meiosis features a unique temporal pattern of autophagy. (A) Schematic of GFP-Atg8 induction and cell collection for IB and IF staining (α-Tubulin) during meiosis and sporulation. Z4EV is a β-estradiol–activated transcription factor, and ZD is a Z4EV-driven promoter. (B) Top, graph of cellular GFP-Atg8/GFP (with a representative IB image) and Ndt80 protein levels (determined by IB, Fig. S1, A and B) during meiosis and sporulation as in A. Bottom and middle, representative images and graph of meiosis stages determined by IF of Tub1 (α-Tubulin) and DAPI staining (n > 100 cells; two independent experiments). Dashed box (red), time window of meiotic divisions. Scale bar, 5 µm. (C–G) Time-lapse FM analysis. (C) Representative time-lapse (FM) image of a cell undergoing meiosis. Cell expresses Whi5-mKOκ, mNG-Atg8, Cdc14-mTFP1, and Tup1-mNeptune2.5. Symbols mark Atg8 puncta (white arrowhead), nucleus (yellow circle), and cell (white circle). Scale bar, 5 µm. (D and E) Total mNG-Atg8 (D) and the vacuolar mNG-Atg8 (E) signal (line) with the standard deviations (shade) from time-lapse FM of starved (SD-N) mitotic cells (grey, n = 549 cells), quiescent cells (green, n = 327 cells), meiotic cells (blue, n = 323 cells), and meiotic ndt80Δ cells (red, n = 231 cells). (F and G) Cdc14-mTFP1 foci intensity (F) and mNG-Atg8 fluorescence intensity (G) in WT (blue) and ndt80Δ (red) meiotic cells. Cells were in silico aligned to the onset of premeiotic DNA replication (Whi5 nuclear export). A.U., arbitrary unit.
Autophagy in meiotic cells, starved cells, and meiotic ndt80Δ cells. (A) IB of cell lysates with indicated antibodies showing GFP-Atg8 processing. Whole cell extracts derived from cells during nonsynchronized meiosis were treated as diagrammed in Fig. 1 A and analyzed by IB with indicated antibodies. GFP-Atg8 expression was induced upon SPM incubation (t = 0 h). (B) Top: IB (α-Ndt80) showing Ndt80 level in the same samples prepared as in A. Bottom: Proteins transferred to the blotting membrane were stained with Ponceau S before IB. (C and D) IB of whole cell extracts derived from cells under starvation (SD-N) with indicated antibodies. (C) Top: Schematic of GFP-Atg8 induction and cell collection. Bottom: IB images. (D) Quantification of IB intensity for GFP and GFP-Atg8 (normalized by Hxk1 IB intensity) over time course. The window of time matching meiotic divisions in Fig. 1 B (9–16 h) is marked by red box (dashed). GFP-Atg8 expression was induced by 1 µM β-estradiol upon SD-N starvation (t = 0 h). (E and F) IB of whole cell extracts derived from ndt80Δ cells under meiosis/sporulation condition (SPM) with indicated antibodies. (E) Top: Schematic of GFP-Atg8 induction and cell collection. Bottom: IB images. (F) Quantification of IB (α-GFP) intensity for GFP and GFP-Atg8 (normalized by Hxk1 IB intensity) over time course. The window of time matching meiotic divisions in Fig. 1 B (9–16 h) is marked by red box (dashed). GFP-Atg8 expression was induced by 1 µM β-estradiol upon SPM incubation (t = 0 h). (G) Time-lapse FM analysis of Cdc14-mTFP1 foci intensity during quiescence (red) and meiosis (blue). Shown are Cdc14-mTFP1 foci signal (line) with the standard deviations (shade). A.U., arbitrary unit. Source data are available for this figure: SourceData FS1.

Autophagy in meiotic cells, starved cells, and meiotic ndt80Δ cells. (A) IB of cell lysates with indicated antibodies showing GFP-Atg8 processing. Whole cell extracts derived from cells during nonsynchronized meiosis were treated as diagrammed in Fig. 1 A and analyzed by IB with indicated antibodies. GFP-Atg8 expression was induced upon SPM incubation (t = 0 h). (B) Top: IB (α-Ndt80) showing Ndt80 level in the same samples prepared as in A. Bottom: Proteins transferred to the blotting membrane were stained with Ponceau S before IB. (C and D) IB of whole cell extracts derived from cells under starvation (SD-N) with indicated antibodies. (C) Top: Schematic of GFP-Atg8 induction and cell collection. Bottom: IB images. (D) Quantification of IB intensity for GFP and GFP-Atg8 (normalized by Hxk1 IB intensity) over time course. The window of time matching meiotic divisions in Fig. 1 B (9–16 h) is marked by red box (dashed). GFP-Atg8 expression was induced by 1 µM β-estradiol upon SD-N starvation (t = 0 h). (E and F) IB of whole cell extracts derived from ndt80Δ cells under meiosis/sporulation condition (SPM) with indicated antibodies. (E) Top: Schematic of GFP-Atg8 induction and cell collection. Bottom: IB images. (F) Quantification of IB (α-GFP) intensity for GFP and GFP-Atg8 (normalized by Hxk1 IB intensity) over time course. The window of time matching meiotic divisions in Fig. 1 B (9–16 h) is marked by red box (dashed). GFP-Atg8 expression was induced by 1 µM β-estradiol upon SPM incubation (t = 0 h). (G) Time-lapse FM analysis of Cdc14-mTFP1 foci intensity during quiescence (red) and meiosis (blue). Shown are Cdc14-mTFP1 foci signal (line) with the standard deviations (shade). A.U., arbitrary unit. Source data are available for this figure: SourceData FS1.
In sporulation medium (SPM), some cells will enter quiescence instead of meiosis (Argüello-Miranda et al., 2018). In addition, meiosis kinetics differs at the level of single cells (Nachman et al., 2007). Given that, we performed time-lapse fluorescence microscopy (FM) in a strain endogenously expressing mNG-Atg8 to track autophagy in single cells undergoing meiosis, quiescence, or nitrogen starvation during proliferation. In addition, we tracked Tup1-mNeptune2.5, a marker for nuclear divisions (Keleher et al., 1992); Whi5-mKOκ, a CDK1(Cdc28) activity marker that is sequestered in the cytoplasm during meiotic divisions but remains nuclear during premeiotic G1 and meiotic exit (Argüello-Miranda et al., 2018); and Cdc14-mTFP1, whose release from the nucleolus marks anaphase I and II (Fig. 1 C and Video 1). We found that meiotic cells rapidly upregulated the level of Atg8 (Fig. 1 D) and thereby the vacuolar delivery of Atg8 (Fig. 1 E), indicating that autophagy is active through meiosis. In contrast, quiescent cells marked by constant nucleolar Cdc14 (Fig. S1 G and Video 1) and starved mitotic cells downregulated average Atg8 levels (Fig. 1 D) and decreased the vacuolar delivery of Atg8 after an initial response (Fig. 1 E).
Autophagy and meiotic progression at the single-cell level. A representative cell undergoing meiosis expressing the Cdc28-activity marker Whi5-mKOκ, the autophagy reporter mNG-Atg8, the phosphatase Cdc14-mTFP1, and nuclear marker Tup1-mNeptune2.5. The experimental sampling rate was 6 min, 3 frames/s.
Autophagy and meiotic progression at the single-cell level. A representative cell undergoing meiosis expressing the Cdc28-activity marker Whi5-mKOκ, the autophagy reporter mNG-Atg8, the phosphatase Cdc14-mTFP1, and nuclear marker Tup1-mNeptune2.5. The experimental sampling rate was 6 min, 3 frames/s.
To test whether meiotic nuclear divisions were required for the autophagy profile observed in meiotic cells, we analyzed WT and ndt80Δ strains, which presented similar upregulation of Atg8 levels during the first 5 h in SPM (before meiotic divisions; Fig. 1 D); however, only WT cells continued to upregulate Atg8 until 12 h, with tight single-cell variation (Fig. 1D). In silico synchronization of WT cells to the onset of premeiotic DNA replication (t = 0 h) showed that meiotic divisions occur between ∼3 and ∼8 h (Fig. 1 F). During this time window, Atg8 levels oscillate with tight variation in WT cells, but not in arrested ndt80Δ cells (Fig. 1 G). Thus, we concluded that autophagy was specifically regulated during meiotic divisions.
Autophagy influences meiosis kinetics and sporulation
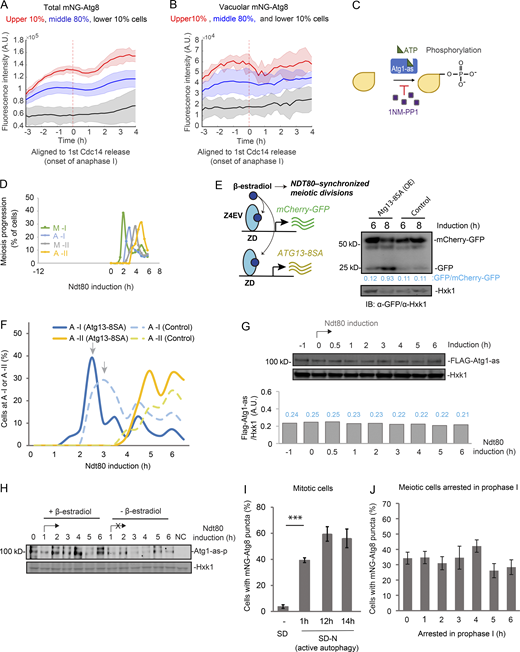
During meiosis, the level of Atg8 and its vacuolar delivery, especially the latter, exhibit intriguing variation in single cells (Fig. 2 A). Remarkably, the fast cells to complete meiosis also had higher levels of Atg8 and autophagy flux during meiotic divisions (Fig. S2, A and B). Moreover, the faster onset of meiosis I and II both correlate with higher level of Atg8, but not Whi5 (Fig. 2 B). Previously, we demonstrated that inhibition of autophagy by 1NM-PP1 during meiotic divisions delays meiosis II onset and prevents meiosis exit (Wang et al., 2020b; Fig. S2 C, inhibition strategy). Here, we induced expression of a gain-of-function mutant of ATG13 (Atg13-8SA; Kamada et al., 2010) in GAL-NDT80–synchronized meiosis (described in Fig. 2 C; herein called synchronized meiosis; Carlile and Amon, 2008; Benjamin et al., 2003; Wang et al., 2020b), to upregulate autophagy immediately before meiotic divisions (Fig. S2, D and E). Remarkably, autophagy upregulation during meiotic divisions led to a faster cellular accumulation of Clb3 (Fig. 2 D), a meiosis II–specific cyclin (Carlile and Amon, 2008); faster meiosis progression as determined by IF analysis of Tub1 and DAPI staining (Figs. 2 E, S2 F, and 2 F); and significantly increased sporulation efficiency (Fig. 2 G). Together, our data demonstrate that active autophagy during meiotic divisions positively influences meiosis kinetics and sporulation.

Autophagy level influences meiosis kinetics and sporulation. (A) Heatmap analysis of total (top) and vacuolar (bottom) mNG-Atg8 levels of individual meiotic cells that completed meiosis, aligned to the onset of premeiotic DNA replication. (B) Left: Correlation plots of Atg8 (mNG-Atg8) level with the onset time of meiosis I (first Cdc14 release; top left) and meiosis II (second Cdc14 release; bottom left). Right: Correlation plots of Whi5 nuclear level with the onset time of meiosis I (top right) and meiosis II (bottom right). Cells transferred to SPM (0 h). (C) Schematic of simultaneously induced expression of Atg13-8SA (pZD: Atg13-8SA, for autophagy enhanced) and NDT80 (pGAL: NDT80, for synchronized metaphase I entry) by 1 µM β-estradiol. For GAL-NDT80−synchronized meiosis, 1 µM β-estradiol was added to meiotic cells after 12 h in SPM to synchronize the entry of meiotic divisions, depicted as t = 0 h of NDT80 induction. (D–G) Effects of Atg13-8SA on synchronized meiotic kinetics and sporulation. Control, strain without pZD: Atg13-8SA. (D and E) IB of Clb3-FLAG/Hxk1 in control and Atg13-8SA cells (D); IF of Tub1 showing percentage of control (dashed lines) and Atg13-8SA (solid lines) cells reaching metaphase I (M-I, green) and metaphase II (M-II, grey; E) at indicated time points. n ≥ 100. Arrows mark M-II peaks, Atg13-8SA shortens the time to reach M-II peak. (F) Percentage of tri-/tetranucleated cells (DAPI staining) after 6 h of NDT80 induction. n ≥ 300. Statistically significant differences: **, P ≤ 0.01; error bars represent standard deviations of three replicate experiments. (G) Sporulation efficiency (%) in control or Atg13-8SA cells after 48 h in SPM (n ≥ 300 cells; t test; ***, P ≤ 0.001). 1NM-PP1 treatment (5 µM, autophagy inhibition) at entry of metaphase I (NDT80 expression) abolished sporulation. A.U., arbitrary unit. Source data are available for this figure: SourceData F2.
Autophagy level influences meiosis kinetics and sporulation. (A) Heatmap analysis of total (top) and vacuolar (bottom) mNG-Atg8 levels of individual meiotic cells that completed meiosis, aligned to the onset of premeiotic DNA replication. (B) Left: Correlation plots of Atg8 (mNG-Atg8) level with the onset time of meiosis I (first Cdc14 release; top left) and meiosis II (second Cdc14 release; bottom left). Right: Correlation plots of Whi5 nuclear level with the onset time of meiosis I (top right) and meiosis II (bottom right). Cells transferred to SPM (0 h). (C) Schematic of simultaneously induced expression of Atg13-8SA (pZD: Atg13-8SA, for autophagy enhanced) and NDT80 (pGAL: NDT80, for synchronized metaphase I entry) by 1 µM β-estradiol. For GAL-NDT80−synchronized meiosis, 1 µM β-estradiol was added to meiotic cells after 12 h in SPM to synchronize the entry of meiotic divisions, depicted as t = 0 h of NDT80 induction. (D–G) Effects of Atg13-8SA on synchronized meiotic kinetics and sporulation. Control, strain without pZD: Atg13-8SA. (D and E) IB of Clb3-FLAG/Hxk1 in control and Atg13-8SA cells (D); IF of Tub1 showing percentage of control (dashed lines) and Atg13-8SA (solid lines) cells reaching metaphase I (M-I, green) and metaphase II (M-II, grey; E) at indicated time points. n ≥ 100. Arrows mark M-II peaks, Atg13-8SA shortens the time to reach M-II peak. (F) Percentage of tri-/tetranucleated cells (DAPI staining) after 6 h of NDT80 induction. n ≥ 300. Statistically significant differences: **, P ≤ 0.01; error bars represent standard deviations of three replicate experiments. (G) Sporulation efficiency (%) in control or Atg13-8SA cells after 48 h in SPM (n ≥ 300 cells; t test; ***, P ≤ 0.001). 1NM-PP1 treatment (5 µM, autophagy inhibition) at entry of metaphase I (NDT80 expression) abolished sporulation. A.U., arbitrary unit. Source data are available for this figure: SourceData F2.
Meiotic autophagy is coupled to meiosis progression. (A and B) Shown are total mNG-Atg8 (A) and vacuolar mNG-Atg8 signal (line; B) with the standard deviations (shade) aligned to first Cdc14 release (anaphase I, marked by red dashed line). Cells from experiments in Fig. 1 C were divided into three groups based on how fast they went through meiosis (pink, 10% of the total, the fastest; gray, 10% of the total, the slowest; blue, 80% of the total, the middle; total, n = 303 cells; Kolmogorov–Smirnov test, P = 0.05). (C) Schematic of the chemical-genetic strategy for inhibiting the kinase activity of Atg1-as. Atg1 was genetically mutated to its Shokat allele, Atg1-M102G, with the bulky gatekeeper residue in the ATP-binding pocket of Atg1 replaced to create a functional ATP-analog–sensitive allele of Atg1 (Atg1-as). 1NM-PP1, a membrane-permeable ATP analog, can specifically inhibit Atg1-as kinase activity in a live cell. (D) Graph of Gal-NDT80–synchronized meiosis showing the percentage of cells at metaphase I (M-I), anaphase I (A-I), metaphase II (M-II), and anaphase II (A-II) at indicated time points. Cells derived every 0.5 h after NDT80 induction (t = 0 h) were fixed and subjected to IF staining (IF: α-tubulin/DAPI) as described in Materials and methods. (E) IB of meiotic cell lysates with indicated antibodies, showing the effect of induced Atg13-8SA (OE) on mCherry-GFP cleavage at indicated time relative to NDT80 induction. Left: Schematic of simultaneously induced expression of Atg13-8SA, mCherry-GFP, and NDT80 by 1 µM β-estradiol at 12 h in SPM (t = 0 h, induction). Atg13-8SA stimulates mCherry-GFP cleavage. (F) IF of Tub1 showing percentage of control (dashed lines) and Atg13-8SA (solid lines) cells reaching anaphase I (A-I, blue) and anaphase II (A-II, yellow) at indicated time points. Arrows mark A-I peaks; Atg13-8SA shortens the time to reach A-I peak; pattern of tubulin was quantified every 0.5 h after NDT80 induction (t = 0 h; n ≥ 100 cells at each time point). (G) Whole cell extracts derived from Gal-NDT80–synchronized meiotic cells were analyzed by IB with α-FLAG and α-Hxk1 antibodies. Graph at the bottom shows the quantification of FLAG-Atg1-as IB intensity with value (blue) in arbitrary units normalized by Hxk1 IB intensity. FLAG-Atg1-as level during meiotic divisions is relatively stable. (H) Atg1-as cells during synchronized meiosis (+ β-estradiol) or prophase I arrest (– β-estradiol) were collected at indicated time points. After Atg1 kinase assay, as diagrammed in Fig. 3 A, the whole-cell lysates were subjected to IB with indicated antibodies. NC, no supplemented ATP. (I and J) Percentage of cells with mNG-Atg8 puncta under vegetative growth (SD; I), nitrogen starvation (SD-N; J) or prophase I arrest (12 h in SPM as t = 0 h; I). n ≥ 300 cells at each time point from three replicate experiments; t test; ***, P ≤ 0.001. A.U., arbitrary unit. Source data are available for this figure: SourceData FS2.

Meiotic autophagy is coupled to meiosis progression. (A and B) Shown are total mNG-Atg8 (A) and vacuolar mNG-Atg8 signal (line; B) with the standard deviations (shade) aligned to first Cdc14 release (anaphase I, marked by red dashed line). Cells from experiments in Fig. 1 C were divided into three groups based on how fast they went through meiosis (pink, 10% of the total, the fastest; gray, 10% of the total, the slowest; blue, 80% of the total, the middle; total, n = 303 cells; Kolmogorov–Smirnov test, P = 0.05). (C) Schematic of the chemical-genetic strategy for inhibiting the kinase activity of Atg1-as. Atg1 was genetically mutated to its Shokat allele, Atg1-M102G, with the bulky gatekeeper residue in the ATP-binding pocket of Atg1 replaced to create a functional ATP-analog–sensitive allele of Atg1 (Atg1-as). 1NM-PP1, a membrane-permeable ATP analog, can specifically inhibit Atg1-as kinase activity in a live cell. (D) Graph of Gal-NDT80–synchronized meiosis showing the percentage of cells at metaphase I (M-I), anaphase I (A-I), metaphase II (M-II), and anaphase II (A-II) at indicated time points. Cells derived every 0.5 h after NDT80 induction (t = 0 h) were fixed and subjected to IF staining (IF: α-tubulin/DAPI) as described in Materials and methods. (E) IB of meiotic cell lysates with indicated antibodies, showing the effect of induced Atg13-8SA (OE) on mCherry-GFP cleavage at indicated time relative to NDT80 induction. Left: Schematic of simultaneously induced expression of Atg13-8SA, mCherry-GFP, and NDT80 by 1 µM β-estradiol at 12 h in SPM (t = 0 h, induction). Atg13-8SA stimulates mCherry-GFP cleavage. (F) IF of Tub1 showing percentage of control (dashed lines) and Atg13-8SA (solid lines) cells reaching anaphase I (A-I, blue) and anaphase II (A-II, yellow) at indicated time points. Arrows mark A-I peaks; Atg13-8SA shortens the time to reach A-I peak; pattern of tubulin was quantified every 0.5 h after NDT80 induction (t = 0 h; n ≥ 100 cells at each time point). (G) Whole cell extracts derived from Gal-NDT80–synchronized meiotic cells were analyzed by IB with α-FLAG and α-Hxk1 antibodies. Graph at the bottom shows the quantification of FLAG-Atg1-as IB intensity with value (blue) in arbitrary units normalized by Hxk1 IB intensity. FLAG-Atg1-as level during meiotic divisions is relatively stable. (H) Atg1-as cells during synchronized meiosis (+ β-estradiol) or prophase I arrest (– β-estradiol) were collected at indicated time points. After Atg1 kinase assay, as diagrammed in Fig. 3 A, the whole-cell lysates were subjected to IB with indicated antibodies. NC, no supplemented ATP. (I and J) Percentage of cells with mNG-Atg8 puncta under vegetative growth (SD; I), nitrogen starvation (SD-N; J) or prophase I arrest (12 h in SPM as t = 0 h; I). n ≥ 300 cells at each time point from three replicate experiments; t test; ***, P ≤ 0.001. A.U., arbitrary unit. Source data are available for this figure: SourceData FS2.
Atg1 kinase activity is upregulated in anaphase I and II
How is autophagy coordinated during meiotic divisions? To answer this question, we investigated whether autophagy initiation, indicated by Atg1 kinase activity (Mizushima, 2010; Kamada et al., 2010), increases at a specific meiotic stage. To biochemically evaluate Atg1 kinase activity through synchronized meiotic divisions, we adapted a functional analog–sensitive allele of Atg1, Atg1-M102G (Atg1-as; Kamber et al., 2015); the mutant protein can accommodate a bulky ATP γS analog (6-PhEt-ATP-γ-S) as a substrate to phosphorylate Atg1 substrates in cell lysates (Fig. 3 A; Allen et al., 2007). Although Atg1 phosphorylates many substrates to enable autophagy initiation, e.g., Atg13 and Atg9 (Papinski et al., 2014; Kamber et al., 2015; Rao et al., 2016), the best-studied reaction is Atg1 autophosphorylation, which further activates Atg1 to initiate autophagy (Yeh et al., 2010; Yeh et al., 2011). Based on Atg1-as autophosphorylation, we found that meiotic divisions activated Atg1 to a level comparable to nitrogen starvation (Fig. 3 B), in which Atg1 activation and autophagy upregulation are well established.

Atg1 kinase activity is upregulated in anaphase I and II. (A) Schematic of the chemical-genetic strategy for monitoring Atg1-as (analog-sensitive Atg1, Atg1-M102G) kinase activity in vitro. Atg1-as thiophosphorylates its substrates with a bulky ATPγS analog (N6-PhEt-ATP-γ-S). Thiophosphorylated substrates of Atg1-as can then be alkylated with para-nitrobenzyl mesylate (PNBM) and detected by IB using anti-thiophosphate ester (α-thioP) antibodies. (B) Atg1-as cells under synchronized meiosis (SPM, t = 1 h after NDT80 induction), mitosis (SD, log phase), and starvation (SD-N, t = 20 min) were collected and treated as in A. Following Atg1 kinase assay, the whole-cell lysates were subjected to IB with indicated antibodies. Hxk1, hexokinase isoenzyme 1 (loading control); Atg1-as-p, Atg1-as autophosphorylation. *, unidentified proteins. NC, no supplemented ATP in starved cell lysates. (C) IB of Atg1-as kinase activity in Atg1-as or Atg1 cells, as in B, during synchronized meiosis. (D) Meiotic kinetics of Atg1-as cells in C determined by IF of Tub1, showing upregulated Atg1-as activity at onset of A-I (2 h), A-II (4 h), and post–A-II (6 h). Source data are available for this figure: SourceData F3.
Atg1 kinase activity is upregulated in anaphase I and II. (A) Schematic of the chemical-genetic strategy for monitoring Atg1-as (analog-sensitive Atg1, Atg1-M102G) kinase activity in vitro. Atg1-as thiophosphorylates its substrates with a bulky ATPγS analog (N6-PhEt-ATP-γ-S). Thiophosphorylated substrates of Atg1-as can then be alkylated with para-nitrobenzyl mesylate (PNBM) and detected by IB using anti-thiophosphate ester (α-thioP) antibodies. (B) Atg1-as cells under synchronized meiosis (SPM, t = 1 h after NDT80 induction), mitosis (SD, log phase), and starvation (SD-N, t = 20 min) were collected and treated as in A. Following Atg1 kinase assay, the whole-cell lysates were subjected to IB with indicated antibodies. Hxk1, hexokinase isoenzyme 1 (loading control); Atg1-as-p, Atg1-as autophosphorylation. *, unidentified proteins. NC, no supplemented ATP in starved cell lysates. (C) IB of Atg1-as kinase activity in Atg1-as or Atg1 cells, as in B, during synchronized meiosis. (D) Meiotic kinetics of Atg1-as cells in C determined by IF of Tub1, showing upregulated Atg1-as activity at onset of A-I (2 h), A-II (4 h), and post–A-II (6 h). Source data are available for this figure: SourceData F3.
Strikingly, although Atg1 protein levels remained stable (Fig. S2 G), Atg1 kinase activity (autophosphorylation) periodically increased during synchronized meiosis, peaking ∼2, ∼4, and ∼6 h after NDT80 induction (Fig. 3 C), determined as anaphase I onset, anaphase II, and post–anaphase II, respectively (Fig. 3 D). As a negative control, Atg1 autophosphorylation in Atg1 WT cells was not detected (Fig. 3 C). This periodic upregulation of Atg1 kinase activity disappeared in cells arrested in prophase I due to lack of NDT80 expression (Fig. S2 H). Thus, we conclude that Atg1 is active during meiotic cell divisions and is stimulated at anaphase I and II.
Autophagy initiation is upregulated in anaphase I and II
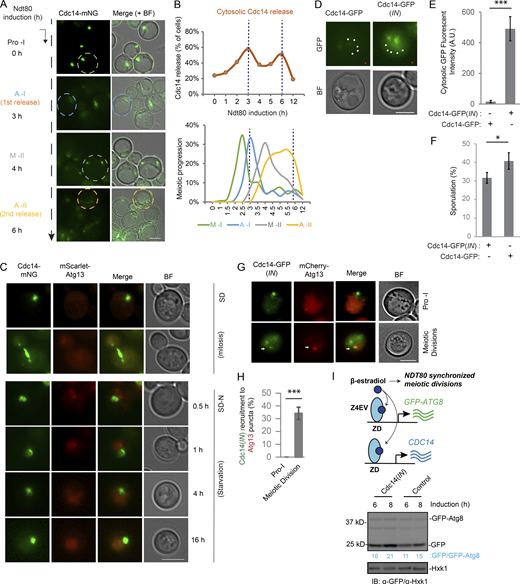
Upon autophagy initiation, the pre-autophagosomal structure (PAS) coordinately recruits (and activates) Atg1 and several other autophagy-related (ATG) proteins (e.g., Atg13 and Atg9), thereby forming cytosolic puncta marked by these ATG proteins (Suzuki et al., 2001; Mizushima, 2010; Stjepanovic et al., 2014; Yamamoto et al., 2016; Kawamata et al., 2008). Therefore, we performed FM to examine Atg1-mNG puncta formation during synchronized meiosis and under other growth conditions as comparisons (Fig. 4 A). Consistent with the pattern of Atg1 autophosphorylation (Fig. 3, C and D), the proportion of cells with Atg1-mNG puncta increased at anaphase I and II onset in an Ndt80-dependent manner, to levels comparable to (or higher than) those observed in starved mitotic cells (Fig. 4, B and C). Thus, Atg1 was periodically stimulated and recruited to PAS at anaphase I and II.

Autophagy initiation is upregulated in anaphase I and II. (A and B) FM analysis of Atg1-mNG cells under synchronized meiosis, prophase I arrest, mitotic log phase, starvation (1 h, SD-N), and extended starvation (12/14 h, SD-N). Shown are representative FM images (A) and percentage of cells with Atg1-mNG puncta (B; n ≥ 300 cells at each time point from three replicate experiments; t test). (C) Meiotic kinetics of experiment A determined by IF of Tub1, showing upregulated Atg1-mNG puncta formation at onset of A-I (2 h), A-II (4 h), and post–A-II (6 h). (D–F) FM analysis of mNG-Atg8 puncta and Tub1-mCherry in live cells during synchronized meiosis. Meiotic stages were identified by the specific Tub1-mCherry distribution pattern depicted on top. (D) Representative images from composite of five planes (Z-step size, 0.5 µm). Scale bar, 5 µm. (E and F) Quantitation of C (n ≥ 300 cells at each meiotic stage from three replicate experiments, t test; **, P ≤ 0.01; ***, P ≤ 0.001). Red line in F indicates the median.
Autophagy initiation is upregulated in anaphase I and II. (A and B) FM analysis of Atg1-mNG cells under synchronized meiosis, prophase I arrest, mitotic log phase, starvation (1 h, SD-N), and extended starvation (12/14 h, SD-N). Shown are representative FM images (A) and percentage of cells with Atg1-mNG puncta (B; n ≥ 300 cells at each time point from three replicate experiments; t test). (C) Meiotic kinetics of experiment A determined by IF of Tub1, showing upregulated Atg1-mNG puncta formation at onset of A-I (2 h), A-II (4 h), and post–A-II (6 h). (D–F) FM analysis of mNG-Atg8 puncta and Tub1-mCherry in live cells during synchronized meiosis. Meiotic stages were identified by the specific Tub1-mCherry distribution pattern depicted on top. (D) Representative images from composite of five planes (Z-step size, 0.5 µm). Scale bar, 5 µm. (E and F) Quantitation of C (n ≥ 300 cells at each meiotic stage from three replicate experiments, t test; **, P ≤ 0.01; ***, P ≤ 0.001). Red line in F indicates the median.
Using FM, we next monitored cytosolic puncta formation of mNG-Atg8, which marks PAS and the following autophagosome maturation (Klionsky et al., 2016); the meiotic stage of each live cell was determined based on the characteristic morphology of spindle microtubules labeled with Tub1-mCherry (Fig. 4 D). As shown in Fig. 4 E, the percentage of cells with mNG-Atg8 puncta was significantly higher in anaphase I and II (57 ± 1.5% and 74 ± 7.4%, respectively) than in metaphase I and II (39 ± 5.6% and 29 ± 3.8%, respectively). Furthermore, the number of mNG-Atg8 puncta per cell was significantly higher in anaphase I and II (Fig. 4 F). Notably, only 4 ± 1.4% of log-phase mitotic cells (SD medium) form Atg8 puncta, while starvation (SD-N, medium lacking nitrogen) significantly increases the percentage (Fig. S2 I, ∼40% 1 h after starvation). On the other hand, mNG-Atg8 puncta formation in cells arrested in prophase I due to lack of NDT80 expression was active but lacked oscillation (Fig. S2 J, ∼35%). Collectively, these results indicate that PAS and autophagosome biogenesis remain active during meiotic divisions and are upregulated at anaphase I and II.
Cdc14 stimulates Atg1 activity in anaphase to assist autophagy initiation
During meiosis in budding yeast, Cdc14 is periodically activated and released from nucleolus at anaphase I and II (Fig. S3, A and B; D’Amours and Amon, 2004, Marston et al., 2003; Visintin et al., 2003; Yoshida and Toh-e, 2002). The tight spatiotemporal correlation between Atg1 activity and Cdc14 behavior raised the possibility that Cdc14 activates Atg1 and stimulates autophagy initiation at PAS. Even though Cdc14 phosphatase is only transiently in contact with its substrates (Rudolph, 2007; Wang et al., 2004), we found, using FM, that 12 ± 1.5% of analyzed cells contain Atg13 (mScarlet-Atg13) puncta (PAS) with Cdc14 (Cdc14-mNG) partially colocalized or attached (Fig. 5 A, bottom, super resolution FM; and Fig. 5 B). Consistently, a deep-learning based approach that plots the intensity of the Cdc14 signal over the distance from Atg13 puncta (Fig. 5 C, cartoon) found that ∼10% of Atg13 puncta showed Cdc14 enrichment (n = 19,060 puncta analyzed after NDT80 induction; Fig. 5 C). In line with this observation, Cdc14-mNG was also recruited to another PAS marker, Atg1 (Atg1-mScarlet), with a similar efficiency, in a manner that depended on meiotic divisions driven by NDT80 expression (Fig. 5, D and E, 12 ± 1.1%). As a control, we observed no recruitment of Cdc14 to PAS (Atg13 puncta) during mitotic proliferation (log-phase) or nitrogen starvation (Fig. S3 C), suggesting that this phenomenon might be meiosis specific. Thus, cytosolic Cdc14 is positioned to activate Atg1 at PAS during meiotic divisions.
Cdc14 stimulates autophagy during meiosis. (A and B) Cdc14-mNG relocation from nucleolus to cytosol at anaphase I and II. (A) Representative FM images of synchronized meiotic cells at indicated time points with Cdc14-mNG localized in nucleolus and cytosol; symbols mark representative cell at prophase I (Pro-I, green circle), anaphase I (A-I, blue circle), metaphase II (M-II, gray circle), and anaphase II (A-II, yellow circle). Scale bar, 5 μm. (B) Top, quantitative analysis of A showing percentage of cells with Cdc14-mNG cytosolic release during synchronized meiosis (n ≥ 300). Bottom: Graph of meiotic progression determined by IF of Tub1. Dashed lines mark the first and second Cdc14 release peaks at anaphase I and II, respectively. (C) Representative FM images showing Cdc14-mNG and mScarlet-Atg13 fluorescence signal in cells under vegetative growth condition (SD, log-phase cells) or nitrogen starvation condition (SD-N). Scale bar, 5 µM. n ≥ 300 cells. Cdc14-mNG was not recruited to mScarlet-Atg13 puncta (SD-N). (D and E) FM analysis of cytosolic Cdc14-GFP and Cdc14-GFP (IN) at anaphase II. (D) Representative FM images. Scale bar, 5 µm. (E) Quantitation of GFP fluorescence signal. The fluorescence intensity measured at the white dots was normalized to the background signal (red dot) and shown as average (n = 10 cells, 5 white dots/cell, t test; ***, P ≤ 0.001). (F) Percentage of synchronized meiotic cells showing sporulation after 48 h in SPM with indicated conditions (n ≥ 300 cells; t test; *, P ≤ 0.05). Cd14-GFP(IN) was induced by 1 µM β-estradiol at 12 h in SPM. (G and H) FM analysis of Cdc14 GFP (IN) recruitment to mCherry-Atg13 cytosolic puncta at prophase I and during synchronized meiosis divisions. (G) Representative FM images. Scale bar, 5 µm. (H) Quantitation (n ≥ 300 cells; t test; ***, P ≤ 0.001). (I) Top: Schematic of simultaneously induced expression ofCDC14, GFP-ATG8, and NDT80 at arrested prophase I due to lack of NDT80 expression. Bottom: IB analysis of whole cell lysates with indicated antibodies, showing increased GFP-Atg8 processing (free GFP accumulation) in response to Cdc14(IN). The ratios of GFP/GFP-Atg8 (IB intensity) are listed (blue). A.U., arbitrary unit. Source data are available for this figure: SourceData FS3.

Cdc14 stimulates autophagy during meiosis. (A and B) Cdc14-mNG relocation from nucleolus to cytosol at anaphase I and II. (A) Representative FM images of synchronized meiotic cells at indicated time points with Cdc14-mNG localized in nucleolus and cytosol; symbols mark representative cell at prophase I (Pro-I, green circle), anaphase I (A-I, blue circle), metaphase II (M-II, gray circle), and anaphase II (A-II, yellow circle). Scale bar, 5 μm. (B) Top, quantitative analysis of A showing percentage of cells with Cdc14-mNG cytosolic release during synchronized meiosis (n ≥ 300). Bottom: Graph of meiotic progression determined by IF of Tub1. Dashed lines mark the first and second Cdc14 release peaks at anaphase I and II, respectively. (C) Representative FM images showing Cdc14-mNG and mScarlet-Atg13 fluorescence signal in cells under vegetative growth condition (SD, log-phase cells) or nitrogen starvation condition (SD-N). Scale bar, 5 µM. n ≥ 300 cells. Cdc14-mNG was not recruited to mScarlet-Atg13 puncta (SD-N). (D and E) FM analysis of cytosolic Cdc14-GFP and Cdc14-GFP (IN) at anaphase II. (D) Representative FM images. Scale bar, 5 µm. (E) Quantitation of GFP fluorescence signal. The fluorescence intensity measured at the white dots was normalized to the background signal (red dot) and shown as average (n = 10 cells, 5 white dots/cell, t test; ***, P ≤ 0.001). (F) Percentage of synchronized meiotic cells showing sporulation after 48 h in SPM with indicated conditions (n ≥ 300 cells; t test; *, P ≤ 0.05). Cd14-GFP(IN) was induced by 1 µM β-estradiol at 12 h in SPM. (G and H) FM analysis of Cdc14 GFP (IN) recruitment to mCherry-Atg13 cytosolic puncta at prophase I and during synchronized meiosis divisions. (G) Representative FM images. Scale bar, 5 µm. (H) Quantitation (n ≥ 300 cells; t test; ***, P ≤ 0.001). (I) Top: Schematic of simultaneously induced expression ofCDC14, GFP-ATG8, and NDT80 at arrested prophase I due to lack of NDT80 expression. Bottom: IB analysis of whole cell lysates with indicated antibodies, showing increased GFP-Atg8 processing (free GFP accumulation) in response to Cdc14(IN). The ratios of GFP/GFP-Atg8 (IB intensity) are listed (blue). A.U., arbitrary unit. Source data are available for this figure: SourceData FS3.

Cdc14 stimulates atg1 activity in anaphase to assist autophagy initiation. (A and B) FM analysis of synchronized meiotic cells expressing Cdc14-mNG and mScarlet-Atg13. (A) Representative images; white arrow, mScarlet-Atg13 puncta that recruits Cdc14-mNG; bottom panel, high-resolution spinning-disk confocal FM and amplified area (white box); scale bars, 5 µm; zoom in (3×) scale bar, 2 µm. (B) Percentage of cells with Cdc14-mNG recruited to mScarlet-Atg13 puncta; only cells that carry mScarlet-Atg13 puncta were analyzed (n ≥ 300 cells, three replicate experiments, t test). (C) Top: Schematic depiction of the algorithmic quantification of Cdc14-mNG pixel intensity around Atg13 foci. Bottom: Cluster analysis showing the fluorescence intensity of Cdc14mNG/Atg13-mScarlet (line) with the standard deviations (shade) as a function of distance from the center of the Atg13 foci. Whereas four clusters can be identified in the experimental strain (Cdc14-mNG), with two of them (blue and green) showing significant enrichment of Cdc14 around the Atg3 foci, all clusters in the control (no Cdc14-mNG) showed no such dependence. In both experimental and control strains all clusters showing mScarlet-Atg13 intensity enrichment as predicted. A total of 19,000 foci from confocal FM images were analyzed by deep learning methods. (D and E) FM analysis of cells expressing Cdc14-mNG and Atg1-mScarlet during prophase I (before NDT80 induction), meiotic divisions (after NDT80 induction), and prolonged prophase I (no NDT80 induction). (D) Representative FM images; white arrows mark Cdc14-mNG recruitment to Atg1-mScarlet puncta; scale bar, 5 µm. (E) Percentage of cells with Cdc14-mNG recruited to Atg1-mScarlet puncta; only cells that carry Atg1-mScarlet puncta were analyzed (n ≥ 300 cells, three replicate experiments, t test). (F) Top: Schematic of induced CDC14(IN) expression (pZD: CDC14) during synchronized meiosis. Bottom: Atg1-as activity in response to Cdc14(IN) expression during synchronized meiosis examined as in Fig. 3 C. (G) Left: Schematic of simultaneously induced expression of CDC14, mCherry-GFP, and NDT80 (synchronized meiosis). Right: IB analysis with indicated antibodies, showing increase of mCherry-GFP cleavage due to Cdc14(IN). A.U., arbitrary unit. Source data are available for this figure: SourceData F5.
Cdc14 stimulates atg1 activity in anaphase to assist autophagy initiation. (A and B) FM analysis of synchronized meiotic cells expressing Cdc14-mNG and mScarlet-Atg13. (A) Representative images; white arrow, mScarlet-Atg13 puncta that recruits Cdc14-mNG; bottom panel, high-resolution spinning-disk confocal FM and amplified area (white box); scale bars, 5 µm; zoom in (3×) scale bar, 2 µm. (B) Percentage of cells with Cdc14-mNG recruited to mScarlet-Atg13 puncta; only cells that carry mScarlet-Atg13 puncta were analyzed (n ≥ 300 cells, three replicate experiments, t test). (C) Top: Schematic depiction of the algorithmic quantification of Cdc14-mNG pixel intensity around Atg13 foci. Bottom: Cluster analysis showing the fluorescence intensity of Cdc14mNG/Atg13-mScarlet (line) with the standard deviations (shade) as a function of distance from the center of the Atg13 foci. Whereas four clusters can be identified in the experimental strain (Cdc14-mNG), with two of them (blue and green) showing significant enrichment of Cdc14 around the Atg3 foci, all clusters in the control (no Cdc14-mNG) showed no such dependence. In both experimental and control strains all clusters showing mScarlet-Atg13 intensity enrichment as predicted. A total of 19,000 foci from confocal FM images were analyzed by deep learning methods. (D and E) FM analysis of cells expressing Cdc14-mNG and Atg1-mScarlet during prophase I (before NDT80 induction), meiotic divisions (after NDT80 induction), and prolonged prophase I (no NDT80 induction). (D) Representative FM images; white arrows mark Cdc14-mNG recruitment to Atg1-mScarlet puncta; scale bar, 5 µm. (E) Percentage of cells with Cdc14-mNG recruited to Atg1-mScarlet puncta; only cells that carry Atg1-mScarlet puncta were analyzed (n ≥ 300 cells, three replicate experiments, t test). (F) Top: Schematic of induced CDC14(IN) expression (pZD: CDC14) during synchronized meiosis. Bottom: Atg1-as activity in response to Cdc14(IN) expression during synchronized meiosis examined as in Fig. 3 C. (G) Left: Schematic of simultaneously induced expression of CDC14, mCherry-GFP, and NDT80 (synchronized meiosis). Right: IB analysis with indicated antibodies, showing increase of mCherry-GFP cleavage due to Cdc14(IN). A.U., arbitrary unit. Source data are available for this figure: SourceData F5.
Next, we induced expression of extra Cdc14 (pZD: CDC14) immediately before synchronized entry into metaphase I, herein referred to as Cdc14(IN), to increase cytosolic Cdc14 levels during meiotic divisions (Fig. S3, D and E). This strategy ensures WT sporulation efficiency while avoiding the potential adverse effects of extreme Cdc14 overexpression during early meiosis (Fig. S3 F). Remarkably, Cdc14(IN) enhanced the recruitment of Cdc14 to Atg13 puncta (Fig. S3, G and H, 34 ± 4.8%; as opposed to 12% ± 1.5% in Fig. 5 B), stimulated Atg1 activity (Fig. 5 F), and increased vacuolar processing of GFP-Atg8 (Fig. S3 I) and mCherry-GFP (Fig. 5 G), a bulk autophagy reporter. These findings suggest that during meiotic divisions, cytosolic Cdc14 can modulate autophagy by activating Atg1.
Cdc14 dephosphorylates Atg13 to activate Atg1
To establish whether Cdc14 directly activates Atg1, we examined the effect of recombinant Cdc14 on Atg1 kinase activity in vitro. First, we confirmed that recombinant Cdc14 is active using a colorimetric phosphatase assay (Fig. S4, A and B). 6His-3FLAG-Cdc14 exhibited concentration- and time-dependent phosphatase activity (Fig. S4 C) that was inhibited by phosphatase inhibitors (PIs; Fig. S4 D). Cdc14m (6His-3FLAG-Cdc14-C283S) did not exhibit detectable phosphatase activity even at 80 μg/ml (1.12 µM; Fig. S4 E). Next, we immunoisolated FLAG-Atg1 complex from yeast cell lysates (Fig. S4 F) and treated it with recombinant Cdc14. Each cell contains ∼8,550 molecules of Cdc14 (∼0.4 µM; Ghaemmaghami et al., 2003), which remains cytosolic for ∼20 min during meiotic divisions. In a dose-dependent manner ([Cdc14] ranging from 0.3 to 0.7 µM), Atg1 autophosphorylation (Atg1 activity) increased in response to 20 min of Cdc14 phosphatase activity (Fig. 6 A). In addition, PIs fully abolished the effect of Cdc14 on Atg1 autophosphorylation (Fig. 6 A). This observation suggests that Cdc14 can dephosphorylate Atg1/Atg1-associated proteins to activate Atg1.
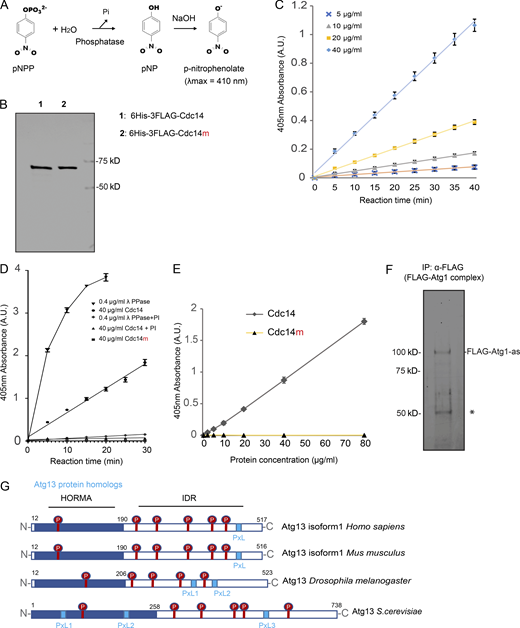
Recombinant Cdc14 carries phosphatase activity in vitro. (A) Schematic of the colorimetric phosphatase activity measured spectrophotometrically at 410 nm by determining the amount of pNP produced after the hydrolysis of Pi from pNPP. The addition of NaOH after a specified assay time (e.g., 10 min) serves to stop the phosphatase reaction while simultaneously converting the product p-nitrophenol into the yellow-colored p-nitrophenolate (λmax = 410 nm). (B) The indicated proteins were prepared from E. coli, as detailed in Materials and methods, for recombinant protein expression and purification and analyzed by SDS-PAGE followed by Coomassie Blue staining. (C) Phosphatase activity of recombinant 6His-3FLAG-Cdc14 protein with indicated concentrations was assayed as in A. Shown are the average and standard deviation, from three replication experiments, of p-nitrophenolate absorbance at 405 nm, plot to incubation time. (D) Phosphatase activity of 40 µg/ml recombinant 6His-3FLAG-Cdc14, 40 µg/ml 6His-3FLAG-Cdc14m, or 0.4 µg/ml λ phosphatase assayed in the presence or absence of PI as in A, shown as C. Note that PI almost abolished phosphatase activity in 6His-3FLAG-Cdc14 and λ phosphatase. (E) Phosphatase activity of recombinant 6His-3FLAG-Cdc14 and 6His-3FLAG-Cdc14m (enzymatically dead Cdc14 mutant, Cdc14-C283S) protein was assayed for 30 min as in A. Shown are the average and standard deviation, from three replication experiments, of p-nitrophenolate absorbance at 405 nm, plot to protein concentration. Note that 6His-3FLAG-Cdc14m yielded no detectable absorbance over the background. (F) IP (α-FLAG) of Atg1 (FLAG-Atg1-as) complex from cell lysates derived from the FLAG-Atg1-as cells arrested at prophase I. After washing, the Atg1 (FLAG-Atg1-as) complex was eluted from the resin with FLAG peptide. Eluted proteins were resolved by SDS-PAGE and visualized by Sypro Ruby staining. IB confirmed Flag-Atg1-as protein identity. *, IgG. (G) Protein domain organization of Atg13 from various species, with the location of protein domains annotated by residue number. Highlighted are the Serine-Proline (SP, in red) and Proline-x-Leucine (PxL, light blue box) motif. HORMA, Hop1p, Rev7p, and MAD2. Note that IDR harbors at least one PxL motif and the majority of SP sites. A.U., arbitrary unit. Source data are available for this figure: SourceData FS4.

Recombinant Cdc14 carries phosphatase activity in vitro. (A) Schematic of the colorimetric phosphatase activity measured spectrophotometrically at 410 nm by determining the amount of pNP produced after the hydrolysis of Pi from pNPP. The addition of NaOH after a specified assay time (e.g., 10 min) serves to stop the phosphatase reaction while simultaneously converting the product p-nitrophenol into the yellow-colored p-nitrophenolate (λmax = 410 nm). (B) The indicated proteins were prepared from E. coli, as detailed in Materials and methods, for recombinant protein expression and purification and analyzed by SDS-PAGE followed by Coomassie Blue staining. (C) Phosphatase activity of recombinant 6His-3FLAG-Cdc14 protein with indicated concentrations was assayed as in A. Shown are the average and standard deviation, from three replication experiments, of p-nitrophenolate absorbance at 405 nm, plot to incubation time. (D) Phosphatase activity of 40 µg/ml recombinant 6His-3FLAG-Cdc14, 40 µg/ml 6His-3FLAG-Cdc14m, or 0.4 µg/ml λ phosphatase assayed in the presence or absence of PI as in A, shown as C. Note that PI almost abolished phosphatase activity in 6His-3FLAG-Cdc14 and λ phosphatase. (E) Phosphatase activity of recombinant 6His-3FLAG-Cdc14 and 6His-3FLAG-Cdc14m (enzymatically dead Cdc14 mutant, Cdc14-C283S) protein was assayed for 30 min as in A. Shown are the average and standard deviation, from three replication experiments, of p-nitrophenolate absorbance at 405 nm, plot to protein concentration. Note that 6His-3FLAG-Cdc14m yielded no detectable absorbance over the background. (F) IP (α-FLAG) of Atg1 (FLAG-Atg1-as) complex from cell lysates derived from the FLAG-Atg1-as cells arrested at prophase I. After washing, the Atg1 (FLAG-Atg1-as) complex was eluted from the resin with FLAG peptide. Eluted proteins were resolved by SDS-PAGE and visualized by Sypro Ruby staining. IB confirmed Flag-Atg1-as protein identity. *, IgG. (G) Protein domain organization of Atg13 from various species, with the location of protein domains annotated by residue number. Highlighted are the Serine-Proline (SP, in red) and Proline-x-Leucine (PxL, light blue box) motif. HORMA, Hop1p, Rev7p, and MAD2. Note that IDR harbors at least one PxL motif and the majority of SP sites. A.U., arbitrary unit. Source data are available for this figure: SourceData FS4.

Cdc14 dephosphorylates Atg13 to activate Atg1. (A) FLAG-Atg1-as protein (complex, as in Fig. S4 F) was affinity-purified (IP: α-FLAG) from extracts of meiotic cells arrested at prophase I and then treated as indicated ([Cdc14] 0.3–0.7 µM), followed by Atg1-as kinase assay as diagrammed (Fig. 3 A). Shown are IB of FLAG-Atg1-as complex after kinase assay with indicated antibodies. (B) Top: Schematic of 3HA-Atg13 isolation (IP: α-HA) and binding to Cdc14/Cdc14m. Extracts of 3HA-Atg13 cells were from vegetative growth. Bottom: IB showing bead-immobilized 3HA-Atg13 pull-down recombinant FLAG-Cdc14m. (C) IB of 3HA-Atg13-p dephosphorylation (band shift on gel) in cell lysates after Cdc14-GFP(IN) (top two panels) or Cdc14m-GFP(IN) (bottom two panels) was induced during synchronized meiotic divisions. Signals of 3HA-Atg13-p/3HA-Atg13 in cell lysates were enriched by IP (α-HA), followed by IB (α-HA). (D and E) Top: Schematic of experimental design. Bead-immobilized 3HA-Atg13-p from IP (α-HA) of cell lysates derived from vegetative growth condition was treated by recombinant Cdc14 to enable dephosphorylation, or as indicated, followed by incubation with Flag-Atg1-as cell lysates to pull down Flag-Atg1-as (D) or with GFP-Atg1-as cell lysates to stimulate Atg1 autophosphorylation (E). Samples were resolved by SDS-PAGE followed by IB with indicated antibodies. NC in D, no FLAG-Atg1-as cell lysate; NC in E, no supplemented ATP. *, Unidentified protein. #, 3HA-Atg13-p (lanes 1–5) and unidentified proteins (lanes 6–8) that migrate together. Source data are available for this figure: SourceData F6.
Cdc14 dephosphorylates Atg13 to activate Atg1. (A) FLAG-Atg1-as protein (complex, as in Fig. S4 F) was affinity-purified (IP: α-FLAG) from extracts of meiotic cells arrested at prophase I and then treated as indicated ([Cdc14] 0.3–0.7 µM), followed by Atg1-as kinase assay as diagrammed (Fig. 3 A). Shown are IB of FLAG-Atg1-as complex after kinase assay with indicated antibodies. (B) Top: Schematic of 3HA-Atg13 isolation (IP: α-HA) and binding to Cdc14/Cdc14m. Extracts of 3HA-Atg13 cells were from vegetative growth. Bottom: IB showing bead-immobilized 3HA-Atg13 pull-down recombinant FLAG-Cdc14m. (C) IB of 3HA-Atg13-p dephosphorylation (band shift on gel) in cell lysates after Cdc14-GFP(IN) (top two panels) or Cdc14m-GFP(IN) (bottom two panels) was induced during synchronized meiotic divisions. Signals of 3HA-Atg13-p/3HA-Atg13 in cell lysates were enriched by IP (α-HA), followed by IB (α-HA). (D and E) Top: Schematic of experimental design. Bead-immobilized 3HA-Atg13-p from IP (α-HA) of cell lysates derived from vegetative growth condition was treated by recombinant Cdc14 to enable dephosphorylation, or as indicated, followed by incubation with Flag-Atg1-as cell lysates to pull down Flag-Atg1-as (D) or with GFP-Atg1-as cell lysates to stimulate Atg1 autophosphorylation (E). Samples were resolved by SDS-PAGE followed by IB with indicated antibodies. NC in D, no FLAG-Atg1-as cell lysate; NC in E, no supplemented ATP. *, Unidentified protein. #, 3HA-Atg13-p (lanes 1–5) and unidentified proteins (lanes 6–8) that migrate together. Source data are available for this figure: SourceData F6.
In the Atg1 kinase complex, dephosphorylation of Atg13 generally enhances the Atg13–Atg1 interaction and activates Atg1 at PAS (Kamada et al., 2000; Memisoglu et al., 2019; Yeasmin et al., 2016). From the amino acid sequence of Saccharomyces cerevisiae Atg13, we observed several Cdc14 preferred dephosphorylation sites (i.e., SP sites; Gray et al., 2003; Bremmer et al., 2012) and putative Cdc14 docking sites (PxL sites; Fig. S4 G). Therefore, we investigated whether Cdc14 binds to and dephosphorylates Atg13. The Cdc14/substrate interaction is labile in coimmunoprecipitation (co-IP) assays (Flint et al., 1997; Bradshaw and Dennis, 2009). To circumvent this limitation, we used the catalytically dead mutant Cdc14m, which stabilizes Cdc14 binding to its bona fide substrates (Bloom et al., 2011; Chen et al., 2013). We found that bead-immobilized 3HA-Atg13 pull-down supplemented recombinant Cdc14m, but not Cdc14 (Fig. 6 B), indicating that Atg13 could be a substrate of Cdc14 during meiosis. Consistent with this idea, induction of Cdc14(IN) at metaphase I entry stimulated hypophosphorylation of Atg13 during meiotic divisions (Fig. 6 C), whereas Cdc14m(IN) had no effect.
To directly probe whether Atg13 is a substrate of Cdc14 phosphatase, we reconstituted Cdc14-mediated Atg13 dephosphorylation in vitro. As expected, treating immunoprecipitated hyperphosphorylated Atg13-p (3HA-Atg13-p) with recombinant Cdc14 accelerated the gel migration of the band corresponding to Atg13-p on the gel to a similar degree as λ phosphatase treatment (Fig. 6 D, lanes 1, 2, and 5, red asterisk). By contrast, neither Cdc14m nor Cdc14 with PIs detectably dephosphorylated 3HA-Atg13-p (Fig. 6 D, lanes 3 and 4).
Next, we investigated whether Atg13 dephosphorylation by Cdc14 activates Atg13, and hence Atg1. Atg13 after Cdc14 treatment exhibited enhanced binding to Atg1 (FLAG-Atg1-as; Fig. 6 D, lanes 1 and 2) and activated Atg1 (GFP-Atg1-as) in cell lysates (Fig. 6 E, GFP-Atg1-as autophosphorylation, lane 1 and 3) in a manner dependent on Cdc14 phosphatase activity (Fig. 6 E, lanes 2, 4, and 5). Together, these results demonstrate that Cdc14 dephosphorylates Atg13, enhancing the Atg13/Atg1 interaction and Atg1 kinase activity.
Two PxL motifs in Atg13 are required for Cdc14 docking
Atg13 contains three PxL motifs that are potentially accessible to Cdc14 docking (Figs. 7 A and S5 A; Kataria et al., 2018). Mutating a bona fide Cdc14 docking PxL motif into AxG will disrupt substrate–Cdc14 interaction (Kataria et al., 2018). Therefore, we immunoprecipitated 3HA-tagged WT and PxL mutants (PxL1m, PxL2m, and PxL3m) of Atg13-p from cell lysates and probed to bind supplemented recombinant Cdc14m. Only WT and PxL2m, but not PxL1m or PxL3m of Atg13-p, pulled down Cdc14m, suggesting that the PxL1 and PxL3 sites are required for Cdc14 binding (Fig. 7 B).

Two PxL motifs in Atg13 are required for Cdc14 docking. (A) Top: Schematic of Atg13 (S. cerevisiae) with six Cdc14 preferred target sites (SP1−6: S129-P, S348-P, S454-P, S535-P, S541-P, and S646-P, in red) and three predicted docking motifs (PxL1−3: P76-PL, P207-IL, and P476-DL, in blue). HORMA, Hop1p, Rev7p, and MAD2. Bottom: Sequence alignment of the PxL3 (P476-DL) region among Atg13 homologs from indicated species. Symbols mark identical (yellow shade) and similar residues (*). (B) IB of recombinant FLAG-tagged Cdc14/Cdc14m binding to bead-immobilized 3HA-Atg13 variant proteins that were purified by IP (α-HA) from vegetative cell lysates. Top: Schematic of experimental design. (C and D) FM analysis of GFP-tagged Cdc14(IN) recruitment to the puncta of mScarlet-tagged Atg13 variants, during synchronized meiotic divisions. (C) Representative FM images. Scale bar, 5 µm. White arrows mark recruitment of Cdc14-GFP to mScarlet-Atg13 variants. (D) Quantitation (n ≥ 300 cells, t test; ***, P ≤ 0.001). (E) Sporulation was triggered in nonsynchronized meiotic cells expressing indicated ATG13 variants, which were carried by pRS303 vector in atg13Δ strain. Shown is the percentage of cells sporulated after 48 h in SPM (n ≥ 300 cells; t test; ***, P ≤ 0.001). Source data are available for this figure: SourceData F7.
Two PxL motifs in Atg13 are required for Cdc14 docking. (A) Top: Schematic of Atg13 (S. cerevisiae) with six Cdc14 preferred target sites (SP1−6: S129-P, S348-P, S454-P, S535-P, S541-P, and S646-P, in red) and three predicted docking motifs (PxL1−3: P76-PL, P207-IL, and P476-DL, in blue). HORMA, Hop1p, Rev7p, and MAD2. Bottom: Sequence alignment of the PxL3 (P476-DL) region among Atg13 homologs from indicated species. Symbols mark identical (yellow shade) and similar residues (*). (B) IB of recombinant FLAG-tagged Cdc14/Cdc14m binding to bead-immobilized 3HA-Atg13 variant proteins that were purified by IP (α-HA) from vegetative cell lysates. Top: Schematic of experimental design. (C and D) FM analysis of GFP-tagged Cdc14(IN) recruitment to the puncta of mScarlet-tagged Atg13 variants, during synchronized meiotic divisions. (C) Representative FM images. Scale bar, 5 µm. White arrows mark recruitment of Cdc14-GFP to mScarlet-Atg13 variants. (D) Quantitation (n ≥ 300 cells, t test; ***, P ≤ 0.001). (E) Sporulation was triggered in nonsynchronized meiotic cells expressing indicated ATG13 variants, which were carried by pRS303 vector in atg13Δ strain. Shown is the percentage of cells sporulated after 48 h in SPM (n ≥ 300 cells; t test; ***, P ≤ 0.001). Source data are available for this figure: SourceData F7.
Surprisingly, all three PxL mutants decreased mScarlet-Atg13 puncta formation during meiotic divisions (Fig. S5 B), indicating a reduction in autophagy initiation derived from reduced Atg1 activation. Rapamycin stimulates Atg1 by inhibiting TORC1 (Kamada et al., 2000). In line with down-regulated TORC1 activity during meiosis, rapamycin has a moderate effect on meiotic Atg13 puncta formation (Fig. S5 B); however, Cdc14(IN) in combination with rapamycin largely restores Atg13 puncta formation in PxLm cells (Fig. S5 B), suggesting that PxLm-induced defects in autophagy initiation are due to a weakened Cdc14–Atg13 interaction and thereby reduced Atg1 activation. Next, we examined the recruitment of Cdc14 to Atg13 puncta in rapamycin-treated meiotic cells expressing mScarlet-Atg13 and Cdc14-GFP (IN). Consistent with our pull-down results (Fig. 7 B), PxL1m and PxL3m reduced the recruitment of cytosolic Cdc14-GFP to mScarlet-Atg13 puncta during meiotic divisions, whereas PxL2m had a less pronounced but still significant effect (Fig. 7, C and D: WT, 26 ± 0.8%; PxL1m, 2 ± 0.6%; PxL2m, 17 ± 1.8%; and PxL3m, 4 ± 0.8%). Collectively, our data demonstrate that the PxL1 and PxL3 sites in Atg13 are required for Cdc14–Atg13 interaction and autophagy initiation; the role of PxL2 in Cdc14–autophagy connection is less critical. Consistent with this observation, Pxl1m and Pxl3m, but not PxL2m, significantly decreased autophagy levels during meiosis (Fig. S5 C) and decreased sporulation efficiency by ∼60% relative to WT, and PxL123m (triple mutations) failed to sporulate altogether (Fig. 7 E).
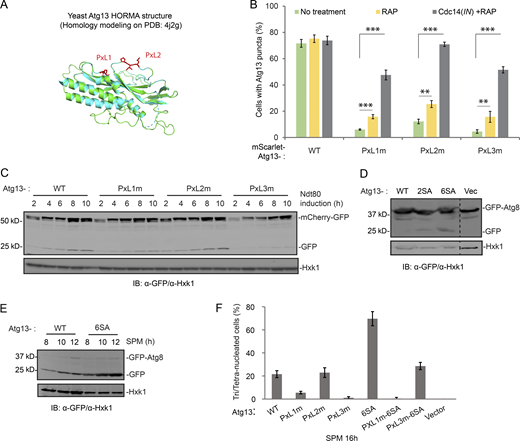
Two PxL motifs in Atg13 are critical for the function of Atg13 in autophagy. (A) A PyMOL cartoon representation of the homology model of Atg13 (S. cerevisiae) residues 7–267 (green) made by SWISS-MODEL homology modeling (Waterhouse et al., 2018) superimposed on the cartoon of the crystal structure of Lachancea thermotolerans Atg13 HORMA domain (cyan; Protein Data Bank accession no. 4J2G). PxL1 and PxL2 sites mutated in this study are indicated as sticks in red. (B) The effects of rapamycin (RAP) or Cdc14(IN) plus rapamycin on cytosolic puncta formation of mScarlet-tagged Atg13 (variants) during synchronized meiosis. Shown are the percentage of cells with Atg13 (variants) puncta counted at t = 6 h after NDT80 induction (n ≥ 300, t test), by FM analysis. If applied, rapamycin was added to SPM 2 h before NDT80 induction; Cdc14(IN) (PZEV-CDC14-GFP) was induced simultaneously with NDT80. Colors of bars indicate the experimental conditions: no treatment (green); rapamycin (yellow); and rapamycin plus Cdc14(IN) (gray). (C) IB of cell extracts from synchronized meiotic cells expressing ATG13 (variants) with indicated antibodies. The mCherry-GFP (pZD: mCherry-GFP) and NDT80 were simultaneously induced by 1 µM β-estradiol. ATG13 variants carried by pRS303 or empty vector were introduced into atg13Δ cells. Note that mCherry-GFP processing was reduced by PxL1m (A76-PG) and by PxL3m (A476-DG), but not by PxL2m (A207-IG). (D) IB of extracts from cells expressing indicated ATG13 variants during log-phase (SD). ATG13 variants carried by pRS303 or empty vector were introduced into atg13Δ cells, in which GFP-Atg8 (pZD: GFP-ATG8) was induced by β-estradiol for 4 h before collection of cells. 2SA, Atg13-S129A-S454A; 6SA, Atg13-S129A-S348 A-S454A-S535 A-S541A-S646A. Note that the GFP-Atg8 processing levels in 2SA and 6SA cells increased, indicated by increased free GFP generation normalized by GFP-Atg8. (E) IB of cell extracts from nonsynchronized meiotic ATG13 and Atg13-6SA cells with indicated antibodies. ATG13 or Atg13-6SA carried by pRS303 was introduced into atg13Δ cells, which harbor inducible GFP-Atg8 (pZD: GFP-ATG8). After initiating sporulation for 6 h in SPM, GFP-Atg8 expression was induced; next, cells were harvested at indicated time points. Note that free GFP generation increased in Atg13-6SA cells. (F) Percentage of cells tri-/tetranucleated (three or four DAPI dots) at 16 h in SPM (nonsynchronized meiosis). From the experiment of Fig. 8 B. Cells collected at indicated time points were fixed and subjected to DAPI staining as described in Materials and methods. Note that 6SA restored the rate of tri-/tetranucleation in PxL3m cells to WT level. Source data are available for this figure: SourceData FS5.

Two PxL motifs in Atg13 are critical for the function of Atg13 in autophagy. (A) A PyMOL cartoon representation of the homology model of Atg13 (S. cerevisiae) residues 7–267 (green) made by SWISS-MODEL homology modeling (Waterhouse et al., 2018) superimposed on the cartoon of the crystal structure of Lachancea thermotolerans Atg13 HORMA domain (cyan; Protein Data Bank accession no. 4J2G). PxL1 and PxL2 sites mutated in this study are indicated as sticks in red. (B) The effects of rapamycin (RAP) or Cdc14(IN) plus rapamycin on cytosolic puncta formation of mScarlet-tagged Atg13 (variants) during synchronized meiosis. Shown are the percentage of cells with Atg13 (variants) puncta counted at t = 6 h after NDT80 induction (n ≥ 300, t test), by FM analysis. If applied, rapamycin was added to SPM 2 h before NDT80 induction; Cdc14(IN) (PZEV-CDC14-GFP) was induced simultaneously with NDT80. Colors of bars indicate the experimental conditions: no treatment (green); rapamycin (yellow); and rapamycin plus Cdc14(IN) (gray). (C) IB of cell extracts from synchronized meiotic cells expressing ATG13 (variants) with indicated antibodies. The mCherry-GFP (pZD: mCherry-GFP) and NDT80 were simultaneously induced by 1 µM β-estradiol. ATG13 variants carried by pRS303 or empty vector were introduced into atg13Δ cells. Note that mCherry-GFP processing was reduced by PxL1m (A76-PG) and by PxL3m (A476-DG), but not by PxL2m (A207-IG). (D) IB of extracts from cells expressing indicated ATG13 variants during log-phase (SD). ATG13 variants carried by pRS303 or empty vector were introduced into atg13Δ cells, in which GFP-Atg8 (pZD: GFP-ATG8) was induced by β-estradiol for 4 h before collection of cells. 2SA, Atg13-S129A-S454A; 6SA, Atg13-S129A-S348 A-S454A-S535 A-S541A-S646A. Note that the GFP-Atg8 processing levels in 2SA and 6SA cells increased, indicated by increased free GFP generation normalized by GFP-Atg8. (E) IB of cell extracts from nonsynchronized meiotic ATG13 and Atg13-6SA cells with indicated antibodies. ATG13 or Atg13-6SA carried by pRS303 was introduced into atg13Δ cells, which harbor inducible GFP-Atg8 (pZD: GFP-ATG8). After initiating sporulation for 6 h in SPM, GFP-Atg8 expression was induced; next, cells were harvested at indicated time points. Note that free GFP generation increased in Atg13-6SA cells. (F) Percentage of cells tri-/tetranucleated (three or four DAPI dots) at 16 h in SPM (nonsynchronized meiosis). From the experiment of Fig. 8 B. Cells collected at indicated time points were fixed and subjected to DAPI staining as described in Materials and methods. Note that 6SA restored the rate of tri-/tetranucleation in PxL3m cells to WT level. Source data are available for this figure: SourceData FS5.
Genomic replacement of ATG13 by the phospho-defective mutants Atg13-2SA or Atg13-6SA (with two [2SA] or six [6SA] serine mutated to alanine at the six Cdc14 preferred SP sites) is sufficient to stimulate autophagy in cells under nutrient-rich conditions (SD), with 6SA having a more significant effect (Fig. S5 D). Furthermore, 6SA boosted autophagy during meiosis (Fig. S5 E). Because ATG13-6SA mimics the product of Cdc14-mediated Atg13 dephosphorylation, we asked whether it could rescue the defect in PxL1m or PxL3m mutants that failed to bind Cdc14 and potentially remained hyperphosphorylated. Indeed, autophagic degradation of mCherry-GFP (Fig. 8 A), meiosis kinetics (Figs. 8 B and S5 F), and sporulation (Fig. 8 C) were all restored to the WT level in PxL3m-6SA cells, demonstrating that PxL3 is a bona fide Cdc14-docking site on Atg13 that enables Atg13 dephosphorylation. Interestingly, 6SA did not rescue PxL1m. Thus, Cdc14 docking at the PxL1 site of Atg13 might play a nonenzymatic structural role; alternatively, it might mediate dephosphorylation of other ATG proteins (e.g., Atg1) by Cdc14 at the PAS. Notably, PxL3, which resides in an intrinsically disordered region (IDR), is a conserved PxL site of Atg13 and likely has the best access to the six SP sites due to their physical proximity and flexibility (Fig. 7 A).

The PxL3 motif is required for Cdc14-mediated Atg13 dephosphorylation. (A) IB of extracts from synchronized meiotic cells with indicated antibodies. Cell with ATG13 variants simultaneously expresses mCherry-GFP (pZD: mCherry-GFP) and NDT80, induced by 1 µM β-estradiol. 6SA restores mCherry-GFP processing in PxL3m to the WT level. (B) Meiotic kinetics in nonsynchronized meiotic cells expressing ATG13 variants, as indicated, by IF of Tub1. Symbols mark M-II peak position in WT (red dashed line), delayed M-II (red arrow), faster M-II (black arrow), and M-II similar to WT (gray arrow). (C) Sporulation was triggered in nonsynchronized meiotic cells expressing indicated ATG13 variants, which were carried by pRS303 vector in atg13Δ strain. Shown are the percentage of cells sporulated after 48 h in SPM (n ≥ 300 cells; t test; **, P ≤ 0.01; ***, P ≤ 0.001; N.S., not significant). (D) Signal transduction model of Cdc14 stimulating autophagy spatiotemporally. At anaphase I and II, active Cdc14 relocates from nucleolus to cytosol and dephosphorylates Atg13 at PAS to activate atg1 and hence autophagy. Source data are available for this figure: SourceData F8.
The PxL3 motif is required for Cdc14-mediated Atg13 dephosphorylation. (A) IB of extracts from synchronized meiotic cells with indicated antibodies. Cell with ATG13 variants simultaneously expresses mCherry-GFP (pZD: mCherry-GFP) and NDT80, induced by 1 µM β-estradiol. 6SA restores mCherry-GFP processing in PxL3m to the WT level. (B) Meiotic kinetics in nonsynchronized meiotic cells expressing ATG13 variants, as indicated, by IF of Tub1. Symbols mark M-II peak position in WT (red dashed line), delayed M-II (red arrow), faster M-II (black arrow), and M-II similar to WT (gray arrow). (C) Sporulation was triggered in nonsynchronized meiotic cells expressing indicated ATG13 variants, which were carried by pRS303 vector in atg13Δ strain. Shown are the percentage of cells sporulated after 48 h in SPM (n ≥ 300 cells; t test; **, P ≤ 0.01; ***, P ≤ 0.001; N.S., not significant). (D) Signal transduction model of Cdc14 stimulating autophagy spatiotemporally. At anaphase I and II, active Cdc14 relocates from nucleolus to cytosol and dephosphorylates Atg13 at PAS to activate atg1 and hence autophagy. Source data are available for this figure: SourceData F8.
Discussion
This study found that in anaphase I and II, Cdc14 stimulates autophagy initiation by dephosphorylating Atg13 to activate Atg1. Thus, Cdc14 links the developmentally regulated progress of meiotic divisions to autophagy, which promotes meiotic progression and sporulation.
Autophagy level during meiosis
Meiosis involves dynamic synthesis and destruction of intracellular structures and requires tightly controlled autophagy. Remarkably, cells with higher levels of autophagic flux during meiotic divisions complete meiosis faster (Figs. 2 B and S2, A and B), indicating the importance of autophagy upregulation during meiosis. Thus, Cdc14 likely represents a strategy to boost autophagy above a critical level as cells progress through meiotic divisions. Consistent with this, enhanced autophagy due to induced expression of Atg13-8SA or Atg13-6SA during meiotic divisions robustly improved meiosis kinetic and increased sporulation efficiency (8SA: Fig. 2, D–F; and Figs. S2 F, S6 A, 8 B, and S5 F).
Thus far, we did not observe any meiotic defects associated with high autophagy levels. However, high autophagy levels, thereby fast meiotic progression, and increased sporulation might reduce the ability of meiotic cells to react to the fluctuating environment in the natural world, e.g., the need to exit meiosis and return to vegetative growth when a nutrient restriction disappears. It is also unclear whether autophagy levels affect the spore viability/quality. To understand how autophagy levels, with their associated benefits and costs, influence specific cellular processes during meiosis and sporulation, the current quantitative approaches for autophagy measurement need to be improved; e.g., GFP-Atg8 processing relies on constant vacuolar protease activity that might fluctuate under certain circumstances, while Atg1 activation and PAS formation mostly reflect autophagy initiation.
Autophagy upregulation at anaphase I and II
At anaphase I, we previously identified Rim4, a RNA-binding protein that essentially regulates meiotic translation, as a meiosis-specific autophagy substrate (Wang et al., 2020b). Timely translation of Rim4-sequestered transcripts relies on rapid removal of Rim4, which conceptually favors upregulation of autophagy at anaphase I. Except for Rim4, little is known about autophagy substrates during meiotic divisions. However, a recent report (Otto et al., 2021) illustrated an upregulation of ER-phagy (autophagy mediated ER degradation) that features timed expression of the autophagy receptor ATG40 at anaphase II, suggesting that autophagy at anaphase II could facilitate subcellular structure remodeling and organelle inheritance. In line with this speculation is one striking phenotype we previously observed from meiotic autophagy inhibition, i.e., overduplication of SPB after anaphase II. The members of the Cdc14 family of phosphatases regulate the duplication cycle of SPBs in yeast and centrosomes in humans (Wu et al., 2008; Avena et al., 2014; Elserafy et al., 2014). Thus, Cdc14 might coordinate autophagy upregulation with SPB dynamics at meiotic anaphases, with a mechanism yet to be revealed.
Molecular mechanism of autophagy regulation during meiosis
Our data demonstrate that Cdc14 controls Atg13 phosphorylation to stimulate autophagy initiation. Given that CDK1 regulates mitotic autophagy by phosphorylating Atg13 and other ATG proteins (Odle et al., 2020; Li et al., 2020), a role of CDK1/Cdc28 to counteract Cdc14 in regulation of meiotic autophagy (e.g., through Atg13 phosphorylation) is an appealing hypothesis. Consistently, all six mutated phosphorylation sites in our gain-of-function mutant, Atg13-6SA, carry the minimal consensus of Cdc28 targets, i.e., SP sites (Ubersax et al., 2003). Nonetheless, among the six SP sites, Ser646 was a TORC1 target both in vitro and in vivo (Hu et al., 2019; Kamada et al., 2010); Ser535 and Ser541 are targeted by Atg1 (Rao et al., 2016; Hu et al., 2019). Therefore, the phosphorylation-based mechanism for meiotic autophagy regulation might include factors other than Cdc28 and Cdc14, e.g., like Ime2 (a meiosis-specific kinase), Atg1, or the TORC1 kinase. Although TORC1 inhibition is a prerequisite for yeast meiosis entry, the low remaining levels of TORC1 activity might be sufficient to regulate Atg13. Moreover, PP2A and PP2C phosphatases stimulate autophagy under certain stress conditions, e.g., starvation and genotoxin treatment, respectively (Yeasmin et al., 2016; Memisoglu et al., 2019). Thus, the potential contributions of several kinases (e.g., Ime2, Cdc28, and TORC1) and phosphatases (e.g., PP2A and PP2C) in modulating autophagy during meiosis need to be studied. We speculate that phosphorylation/dephosphorylation of ATG proteins orchestrated by their kinases and phosphatases fine-tune and connect meiotic autophagy to meiosis progression and sporulation.
Besides posttranslational modification, autophagy can be regulated transcriptionally and at the level of protein translation (He and Klionsky, 2009; Acevo-Rodríguez et al., 2020; Di Malta et al., 2019). Our time-lapse cell imaging analysis showed that Atg8 levels increased at meiosis entry and during meiotic divisions (Fig. 1, D and G), consistent with a previous ribosome profiling study of yeast meiosis (Brar et al., 2012). The second Atg8 upregulation requires expression of NDT80 (Fig. 1 G), the master transcription factor for mid-late meiotic gene expression. Studies in the future will elucidate how the levels of Atg8 and other ATG proteins are regulated during meiosis.
Cdc14 as a regulator of meiotic autophagy
Although belonging to the tyrosine-protein phosphatase family, Cdc14 is a dual phosphatase with a structurally defined binding preference for a PxL motif (Kataria et al., 2018) and prefers dephosphorylating serine-proline (SP) motifs (Bremmer et al., 2012; Eissler et al., 2014). This pattern discriminates Cdc14 from the PP2A and PP2C phosphatases and might explain why Cdc14 is recruited to PAS, a structurally undefined meshwork of the Atg1 protein complex, PI3K complex, Atg2–Atg18 complex, and Atg9 vesicles (Suzuki et al., 2001; Kawamata et al., 2008). In addition to Atg13, several PAS-localized ATG proteins (e.g., Atg1 and Atg18) also have PxL sites in their LCD regions flanked by several SP sites. Hence, Cdc14 at PAS might involve binding and dephosphorylating multiple PAS components to regulate autophagy.
Kondo et al. (2018) previously reported that inactivating a temperature-sensitive cdc14-1 allele in haploid mitotic yeast cells led to reduced ATG13/ATG8 upregulation and a hyperphosphorylated Atg13 profile when TORC1 is inactivated due to starvation. In contrast, overexpression of Cdc14 induces autophagy in nutrient-rich conditions. Thus, Cdc14 might dephosphorylate Atg13, or nonexclusively influence ATG protein levels, to upregulate autophagy in starved mitotic cells. For the former possibility, the caveats are that Atg13-8SA did not improve autophagy in Cdc14-1 cells, whereas deleting PP2A, a phosphatase implicated in stimulating starvation-trigged autophagy, in a Cdc14-1 background unexpectedly rescued autophagy (Kondo et al., 2018). In addition, we did not observe Cdc14 recruited to Atg13 puncta in starved mitotic cells (Fig. S3 C). Nevertheless, there are many ways to reconcile these findings. For example, Cdc14 might dephosphorylate Atg13 in a way unrelated to PAS sites in starved mitotic cells. Alternatively, mitotic Cdc14 might mainly regulate starvation-triggered autophagy by facilitating induction of ATG8 and ATG13 expression. These speculations remind us how little we know about the phosphatase network (e.g., Cdc14 and PP2A) and its autophagy connections during meiosis and mitosis, and the complexity of related molecular mechanisms.
Materials and methods
S. cerevisiae strain construction
S. cerevisiae strains are derivatives of W303 (ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 can1-100; Table S1). Unless otherwise indicated, the analog sensitive version of atg1(Atg1-as) was created by gatekeeper residue change (M102G) as described earlier (Blethrow et al., 2004; Kamber et al., 2015), and introduced into the background of listed strains to allow conditional autophagy inhibition by 1NM-PP1.
Deletion strains were constructed in a parent background by PCR-mediated knockout with one of the following drug resistance or prototrophic marker cassettes: pFA6a-kanMX6/pFA6a-NAT (Longtine et al., 1998), pKlURA/pCgHIS (Goldstein and McCusker, 1999). C- or N-terminal tagging at the endogenous genomic locus was introduced by PCR-mediated epitope tagging as previously described (Denic and Weissman, 2007). Specifically, strains with GAL-NDT80 GAL4-ER were constructed by replacing the endogenous NDT80 promoter with the inducible GAL1,10 promoter as described (Carlile and Amon, 2008). Strains with ndt80Δ were constructed with pFA6a-NAT marker cassette replacement of NDT80 coding sequence. Strains with β-estradiol–inducible GFP-ATG8, CDC14-GFP, mCherry-GFP, or Atg13-8SA transgene were made by homologous recombination of a Z4EV expression cassette with a Z4EV-driven (ZD) promoter (McIsaac et al., 2013) upstream of the GFP-ATG8, Cdc14-GFP mCherry-GFP, or ATG13-8SA open reading frame at the LEU2 locus. TUB1-mCherry, CDC14-mTFP, CDC14-mNeonGreen, ATG1-as-mNeonGreen, ATG1-as-mScarlet, GFP-ATG1-as, FLAG-ATG1-as, and CLB3-3FLAG were generated at their endogenous genome locus by transformation with PCR tagging cassettes of indicated C-terminal tags using the PEG/lithium acetate yeast transformation kit (Millipore-Sigma, YEAST1). Plasmids with 2XGFP-ATG8 in pRS303, mNeonGreen-ATG8 in pRS305, CDC14-GFP in pRS305, CDC14m-GFP in pRS305, mCherry-ATG13 in pRS303, 3HA-ATG13 in pRS303, mScarlet-ATG13 in pRS303, and mScarlet-ATG8 in pRS303, driven by their endogenous promoters, were integrated into the genomic locus of HIS3/LEU2 (Wang et al., 2020b).
The OAM835 strain was constructed for microfluidics experiments, which resulted from mating haploids obtained by tetrad dissection or by transformation with PCR tagging cassettes or plasmids using the PEG/lithium acetate protocol. This CDC14-mTFP1/WHI5-mKOk/TUP1-mNeptune2.5/mNeonGreen-ATG8 strain had WT kinetics for proliferation and sporulation.
Strains harboring ATG13-6SA, ATG13-2SA, or ATG13-PxL mutants were made by introducing mutated pRS303-harbored ATG13 or HA (or mScarlet)-ATG13 into HIS locus of parent strains, in which endogenous ATG13 coding sequence was deleted by PCR-mediated knockout with URA/NAT marker cassette.
Media
The following media were used in this study: YPD (2% peptone, 1% yeast extract, and 2% glucose), YPA (2% peptone, 1% yeast extract, and 2% potassium acetate), SD (0.67% yeast nitrogen base, 2% glucose, and auxotrophic amino acids and vitamins), and standard SPM (0.6% potassium acetate, pH 8.5). The SD-dropout medium was made with dropout stock powder lacking histidine, tryptophan, leucine, uracil, and/or methionine. The nitrogen-starvation experiments in yeast cells were performed in a synthetic minimal medium lacking nitrogen, SD-N (0.17% yeast nitrogen base without amino acids, 2% glucose).
Plasmid construction
Plasmids used in this study are listed in Table S2. pRS303-ATG13 was constructed by subcloning PCR-amplified regions of genomic ATG13 with its promoter and terminator into NotI/SalI restriction sites of pRS303. 3HA, mScarlet, or mCherry fragments were fused to the N-terminus of ATG13 by Gibson assembly cloning. Mutagenesis of all six Serine sites and three PxL sites, as shown in Table S2, were introduced by overlap extension (OE) PCR. pRS303-mScarlet-ATG8 was constructed by replacing the 2xGFP region in pRS303-2xGFP-ATG8 with mScarlet via Gibson assembly cloning.
Vectors for bacterial expression of 6His-3FLAG-tagged Cdc14 were created by subcloning PCR amplified regions of the genomic CDC14 encoding region into the NdeI/XhoI restriction sites of pET28a. In addition, C283S mutation was introduced into pET28-CDC14 by OE PCR.
Plasmids for C-terminal fluorophore tagging of WHI5-mKOk, TUP1-mNeptune2.5, and CDC14-mTFP1 were confirmed by sequencing and restriction analysis and were originally reported in Argüello-Miranda et al. (2018). pRS304 ATG8p-mNeonGreen-ATG8-ATG8ter was created by replacing the PacI-2xyeGFP-BamHI sequence in pRS304-ATG8p-2xyeGFP-ATG8-ATG8ter with a PacI-mNeonGreen-BamHI sequence amplified from pYLB10 (Argüello-Miranda et al., 2018).
Protein expression and purification
Recombinant protein purification
pET-based protein expression in BL21 (DE3) Escherichia coli cells was induced by IPTG as described previously (Wang et al., 2010). 6His-3Flag-Cdc14 and 6His-3Flag-Cdc14m were expressed at 16°C overnight. Cells were collected by centrifugation, resuspended in bacteria lysate buffer (BLB; 20 mM Tris-HCl, pH 8.0, 500 mM NaCl, 20 mM imidazole, 10% glycerol, 5 mM 2-ME, and 1 mM PMSF), and lysed using a High-Pressure Cell Press Homogenizer (Avestin Emulsiflex-C5). The lysate was supplemented with 0.1 mg/ml DNase I, 1 mM MgCl2, and 0.1% Triton X-100 and cleared by centrifugation at 10,000 g for 10 min at 4°C. The supernatant was applied to an NTA-Ni column (30230; Qiagen) and washed three times with BLB. The 6xHis-proteins were eluted with 20 mM Tris-HCl, pH 8.0, 500 mM NaCl, 300 mM imidazole, 10% glycerol, and 5 mM 2-ME. The eluted proteins were separated by Superdex 200 Increase 10/300 GL column (GE Healthcare) equilibrated in size-exclusive buffer (SEC; 50 mM Tris/HCl, pH 7.5, 500 mM NaCl, 10% glycerol, and 5 mM 2-ME). Finally, purified proteins were concentrated in SEC and frozen in liquid nitrogen.
IP of proteins expressed in yeast
Cells grown in SPM or YPD medium were pelleted at 3,000 g for 5 min at 4°C and washed with ice-cold distilled water containing 1 mM PMSF. The pellets were resuspended in ice-cold yeast lysis buffer (YLB; 50 mM Hepes-KOH, pH 6.8, 150 mM KOAc, 2 mM MgCl2, 1 mM CaCl2, 0.2 M sorbitol, 10 mM PMSF, and 2× protease inhibitor cocktails [Roche, 11873580001]), dropped into liquid nitrogen, ground using a Retsch ball mill (PM100 or Retsch MM400), and stored at −80°C for immunoprecipitation (IP), as well as for atg1 kinase assay and IB.
To isolate the FLAG-Atg1-as complex, frozen cell lysate powder (∼500 OD600 units) was thawed in 2 ml of 1× IP buffer (50 mM Hepes-KOH, pH 6.8, 150 mM K2OAc, 2 mM MgOAc, 1 mM CaCl2, 15% glycerol, 1% NP-40, 1× protease inhibitor cocktail, and 1× PIs) and cleared twice by centrifugation at 1,000 g for 5 min at 4°C. 10 μl of Protein G Dynabeads (Invitrogen) that were loaded with mouse anti-FLAG M2 antibody (F3165; Sigma-Aldrich) was then added to the cell extract and incubated for 3 h at 4°C with constant agitation. The beads were collected and washed five times with IP buffer containing 1% NP-40 and 1× PIs, and bound proteins were eluted with 25 μl of 1 mg/ml 3× FLAG peptide (F4799; Sigma-Aldrich) in IP buffer containing 1% NP-40 and 1× protease inhibitors at 4°C. Eluates were aliquoted, frozen in liquid nitrogen, and stored at −80°C. The purified FLAG-Atg1 complex was resolved by SDS-PAGE and analyzed by Sypro Ruby staining (S12000; Thermo Fisher Scientific).
To purify 3HA-Atg13 and its variants, 0.4 g frozen cell lysates from cells grown in SD medium (Fig. 4, C, E, and F) or SPM (Fig. 4 D) was thawed on ice and supplemented with 0.8 ml IP buffer. Extracts were cleared twice by centrifugation at 1,000 g for 5 min at 4°C. Supernatants were then incubated with 5 μl protein G magnetic beads with bound anti-HA antibodies for 3 h at 4°C with constant agitation. After washing five times in IP buffer, resins were resuspended in 100 μl of IP buffer and used for (a) phosphatase treatment (Fig. 4, E and F); (b) Cdc14 pull-down (Fig. 4 C); and (c) IB (Fig. 4 D).
Sporulation, meiotic division synchronization, and vegetative growth
A single colony of yeast strains from the YPD plate was picked, spread on YPG (3% glycerol) plates, and grown at 30°C for 2 d. Colonies grown out were spread to YPD plates and grown until cells formed a lawn (∼24 h). Cells on plates were collected and suspended in YPA liquid medium (OD600 = 0.3) and grown for 14–16 h at 30°C. Cells were then pelleted, washed twice with water, and resuspended in SPM to final OD600 = 3. For meiotic division synchronization, after incubation in SPM for 12 h, strains containing GAL-NDT80 GAL4-ER were released from prophase I arrest by addition of 1 μM β-estradiol to induce NDT80 expression. To assess sporulation efficiency, cells with matured spores were counted after 48 h in SPM at room temperature. At least 300 cells of each strain were counted by bright-field microscopy (Olympus BX40, 40× objective). Two-sided t test was used to calculate the significance P value. The F test was used to test the null hypothesis that the variance of two populations are equal.
For single-cell time-lapse imaging, the OAM835 strain was inoculated to 0.03 OD600 and grown for 23 h or until the OD600 reached 2.5. 70 μl of the culture was sonicated (4–6 s at 3 W) and transferred to a Y04C CellASIC microfluidic device (http://www.cellasic.com/) set to 25°C. Two pulses of cells were loaded (8 psi for 5 s) and then exposed to a meiosis-inducing medium (0.6% potassium acetate, delivered at 0.6 psi flow rate, pH 8.0 adjusted with 0.125 M Na2CO3 immediately before the experiment) for 24 h.
For vegetative growth experiments, cells were cultured in YPD or SD medium to log phase (OD = 0.6∼1.0). To induce expression of pZD promoter-controlled genes (CDC14 variants, Atg13-8SA, GFP-Atg8, and mCherry-GFP), 1 μM β-estradiol (10 mM stock in ethanol; E2758; Sigma-Aldrich) was added. To inhibit Atg1-as, 5 μM of 1-NM-PP1 was added (10 mM stock in DMSO; B1299; APExBIO) as needed. For extended long-time starvation, cells grown to log phase in SD medium were washed and transferred to SD-N medium for 16–18 h.
IB analysis
Three OD600 yeast cells were collected by centrifugation, resuspended in 100 μl of 2× SDS sample buffer (SSB), and boiled at 95°C for 5 min (2× SSB: 62.5 mM Tris-HCl, pH 6.8, 2% SDS, 0.05% BPB, 10% glycerol, 5% 2-Mercaptoethanol, 1× Protease Inhibitor Cocktail [11873580001; Roche], and 1 mM PMSF). Proteins were separated by SDS-PAGE (90 min at 150 V) using 4–20% Criterion TGX Stain-Free gel (5678095; Bio-Rad) and electroblotted onto nitrocellulose membranes (1620115; Bio-Rad) using a Trans-blot SD semidry transfer cell (1703940; Bio-Rad). After blocking with 5% skim milk in TBS, 0.1% Tween 20, cells were incubated with respective primary antibodies followed by HRP- or StarBright B700–conjugated secondary antibodies. For detection and quantitative analysis, the IB images were captured by ChemiDoc MP imaging system (12003154; Bio-Rad), and analyzed using Image Lab (v6.0.1) software (Bio-Rad).
We used the following antibodies: recombinant monoclonal anti-thiophosphate ester rabbit IgG [51-8] (ab92570; Abcam), monoclonal anti-GFP mouse IgG (11814460001; 1:5,000; Roche), polyclonal anti-hexokinase 1 rabbit IgG (169073; 1:10,000; United States Biological), polyclonal anti-Ndt80 rabbit IgG (1:10,000), monoclonal anti-HA mouse IgG1 (21683; 1:3,000; Thermo Fisher Scientific), monoclonal anti-FLAG M2 mouse IgG (F3165; 1:5,000; Millipore-Sigma), polyclonal anti-FLAG rabbit IgG (SAB4301135; 1:5,000; Millipore-Sigma), StarBright B700–labeled goat anti-mouse IgG secondary antibody (12004158; 1:5,000; Bio-Rad), HRP-conjugated goat anti-mouse IgG (STAR207P; Bio-Rad), and StarBright B700–labeled goat anti-rabbit IgG (12004161; 1:5,000; Bio-Rad).
In vitro Cdc14 phosphatase activity assay
500 mM p-nitrophenyl phosphate (pNPP; NEB, P0757S) substrate solution was diluted to 20 mM in phosphatase reaction buffer (PRB; 20 mM Tris, pH 7.5, 4 mM MgCl2, 1 mM EGTA, 0.02% 2-ME, and 0.1 mg/ml BSA) to yield pNPP working stock. Recombinant 6His-3Flag-Cdc14/6His-3Flag-Cdc14m proteins at various concentrations (0.25–80 µg/ml prepared in PRB) were mixed with the pNPP working stock in 96-well plates to a final pNPP concentration of 10 mM. The reaction mix was incubated for 5–45 min at room temperature and stopped by 1 N (final concentration) NaOH. Absorbance at 405 nm was measured on a BioTek Gen5 microplate reader. 0.4 µg/ml λ phosphatase (P0753S; New England Biolabs) and 1× phosSTOP cocktail (4906845001; Millipore-Sigma) were added to control reactions.
In vitro dephosphorylation of 3HA-Atg13
20 μl of 3HA-Atg13, immobilized on protein G magnetic beads, was incubated with 50 µg/ml 6His-3FLAG-Cdc14, 6His-3FLAG-Cdc14m, or 10 µg/ml λ PPase in reaction buffer (50 mM Hepes-KOH, pH 6.8, 150 mM K2OAc, 350 mM NaCl, 2 mM MgOAc, 1 mM CaCl2, 15% glycerol, and 0.1% Triton X-100) in a 30°C water bath for 20 min. The reaction was stopped by adding 1× phosSTOP cocktail and used for Atg1 kinase assay (Fig. 6 E) or FLAG-Atg1 pulling down assay (Fig. 6 D).
Atg1 pull-down by 3HA-Atg13
Frozen cell lysates from FLAG-Atg1-as cells cultured in YPD to log phase (OD = 0.6∼1.0) were thawed, diluted with an equal volume of IP buffer (150 mM Hepes-KOH, pH 6.8, 150 mM K2OAc, 2 mM MgOAc, 1 mM CaCl2, 15% glycerol, 1% NP-40, 1× Roche protease inhibitor cocktail, and 1× phosSTOP cocktail) and cleared twice by centrifugation at 1,000 g for 5 min at 4°C. 20 μl magnetic beads coated with 3HA-Atg13 was incubated with 100 μl FLAG-Atg1-as cell extracts for 4 h at 4°C. Resins were collected and washed three times with ice-cold IP buffer. Proteins were eluted by boiling in SSB, separated by SDS-PAGE, and analyzed by IB using appropriate antibodies.
Cdc14/Cdc14m pull-down by 3HA-Atg13
20 μl of the bead-immobilized 3HA-Atg13 was incubated with 50 µg/ml 6His-3FLAG-Cdc14 or 6His-3FLAG-Cdc14m in 50 µl binding buffer (50 mM Hepes-KOH, pH 6.8, 150 mM K2OAc, 350 mM NaCl, 2 mM MgOAc, 1 mM CaCl2, 15% glycerol, 0.1% Triton X-100, 0.05% 2-ME, 1× Roche protease inhibitors, and 1× PhosSTOP cocktail) overnight at 4°C. The beads were washed three times with binding buffer, boiled in SSB to elute bead-bound proteins, and analyzed by SDS-PAGE and IB with appropriate antibodies.
Assay of Atg1-as kinase activity
In cell lysates
Cell lysates were mixed with 1× kinase buffer (150 mM K2OAc, 10 mM MgOAc, 0.5 mM EGTA, 5 mM NaCl, 20 mM Hepes-KOH, pH 7.3, and 5% glycerol) in equal volume (wt/vol) and thawed on ice. After two centrifugations at 1,000 g for 5 min at 4°C, supernatants were mixed with equal volumes of 2× kinase mix (kinase buffer, energy mix [90 mM creatine phosphate, 2.2 mM ATP, and 0.45 mg/ml creatine kinase], and 0.2 mM N6-phenylethyl-ATPγS [N6-PhEt-ATPγS, BLG-P026-05]) and incubated for 1.5 h at room temperature. Reactions were quenched with 20 mM EDTA and then alkylated with 2.5 mM paranitrobenzyl mesylate (Abcam) for 45 min at room temperature, boiled in SSB, and analyzed by IB. Thiophosphorylated substrates were identified by IB with a rabbit anti-thiophosphate ester primary antibody [51-8] (ab92570; Abcam) and StarBright B700–labeled goat anti-rabbit IgG secondary antibodies (Fig. 3, B, C, and F; and Fig. S2 H).
In cell lysates + 3HA-Atg13
After bead immobilization and phosphatase treatment as described above, 5 μl of 3HA-Atg13 on magnetic beads was incubated with 10 μl GFP-Atg1-as cell extracts in 1× kinase buffer for 15 min at room temperature with constant agitation, followed by atg1-as kinase assay (Fig. 6 E).
In purified Flag-Atg1-as complex
25 or 50 µg/ml recombinant Flag-Cdc14 was incubated with 2 μl Flag-Atg1-as complex, supplemented with 1× kinase buffer to 15 μl, for 20 min in a 30°C water bath, followed by Atg1-as kinase assay as described. 1× phosSTOP cocktail was added to inhibit Cdc14 phosphatase as control (Fig. 6 A).
FM
Tubulin IF analysis was described previously (Wang et al., 2020b). In brief, after NDT80 was induced, cells were fixed every 30 min by cold 3.7% formaldehyde in PBS. For nonsynchronized meiotic cells harboring ATG13 mutations, cells were fixed by cold 3.7% formaldehyde every 1 h from 7 h in SPM until 16 h. Fixed cells were washed with 0.1 M KPi (4.84 g/liter potassium phosphate dibasic and 9.83 g/liter potassium phosphate monobasic, pH 6.4) buffer and 1.2 M sorbitol citrate solution, and then digested by 1.4 mg/ml 20T zymolyase (120491; Amsbio) and glusulase (NEE154001EA; PerkinElmer). Cells were spread on glass slides coated with poly-L-lysine (P8920; Sigma-Aldrich), incubated with anti-tubulin Alexa Fluor 594 antibody (Dilution 1:200; Abcam) for 2 h, and mounted in ProLong Gold antifade reagent containing DAPI (P36935; Thermo Fisher Scientific). Spindle morphologies were then classified by the following morphological features. Metaphase I cells were defined as cells with a single DAPI mass spanned by thick, short, and bipolar meiotic spindle microtubules (∼2–3 μm in length). Anaphase I cells were defined as two parts of distinct (although not always separated) DAPI masses and a single long spindle that spans both DAPI masses. Metaphase II cells were defined as cells with two separate DAPI masses, with each spanned by a bipolar, thick, and short meiotic spindle. Anaphase II cells were defined as cells with four distinct (although not always separated) parts of DAPI masses with two long spindles (usually crossed). Images were acquired using a Zeiss Axio Imager Z2 microscope equipped with a Photometrics CoolSnap HQ2 CCD camera, a Zeiss Plan Apochromat 63×/0.9-NA oil-immersion objective, and filters for DAPI and Alexa Fluor 594. At least 100 cells were counted at each time point to determine the percentage of cells at each meiotic cell stage.
For live-cell imaging, 10 μl of cells growing in SD medium, SD-N medium, or SPM were spread on a slide and imaged immediately using a Zeiss Axio Imager Z2 microscope equipped with a Photometrics CoolSnap HQ2 CCD camera, a Zeiss Plan Apochromat 63×/0.9-NA oil-immersion objective, and filters for GFP/FITC and mCherry/Alexa Fluor 594. For better image resolution (Fig. 5 A) and reduced photobleaching, the strain harboring Cdc14-mNG/mScarlet-Atg13 in SPM was imaged using Nikon CSU-W1 SoRa spinning-disk confocal (resolution down to 120 nm) equipped with a Hamamatsu Orca-Fusion sCMOS camera, 100× oil-immersion objective, and filters for GFP/FITC and mCherry/TRITC. Strains harboring Cdc14-GFP(IN)/mScarlet-Atg13 mutants were imaged using a 3i spinning-disk confocal microscope, with a 63× oil-immersion objective with mTFP filter (473-488/10), mNeonGreen filter (515-542/27), mRuby filter (561-617/73), and GFP filter (413-525/80). Z-stack (5–10 planes, 0.5 µm/plane) images were acquired if needed. Images were processed using ImageJ except for the analysis of single-cell images as described above. Two-sided t test was used in the colocalization statistics.
Time-lapse microscopy, image processing and quantification of cellular features
Time-lapse images of microfluidics experiments were acquired using a motorized Zen software–operated Zeiss Observer Z1 microscope with temperature control, a Definite Focus 2.0 system, 40× Zeiss EC Plan-Neofluar 1.3-NA oil-immersion objective, and an AxioCam HR Rev 3 camera. At least three fields of five replicates were imaged. A set of custom dichroic mirrors and bandpass filters was used for fluorescence imaging, with exposure times of mTFP1 125 ms, mNeonGreen 175 ms, mKOκ 200 ms, mNeptune2.5 75 ms, and phase contrast 40 ms. LED light sources were kept at 25% intensity except for mNeptune2.5, at 50%.
ZEN pro software (Zeiss) was used to collect 2 × 2 binning noncompressed TIFF format images converted to double format before feature extraction. A 3 × 3 structured medfilt2 (Matlab function) filter removed shot noise. Cells were segmented with a previously described pipeline (Doncic et al., 2013; Wood and Doncic, 2019); background quantification was defined as the median intensity of the space not occupied by cells. A 2D Gaussian fit to the brightest pixel of nuclear proteins (Tup1 or Whi5) was used to calculate nuclear parameters (Doncic et al., 2013). Nucleus Cdc14 released was computed by a 2D gaussian filtered-based algorithm that labels the nucleus’s position using the brightest nuclear pixel associated with the Cdc14 signal. The time of Cdc14 release was defined by this algorithm as the time point at which the fluorescence intensity of the nuclear Cdc14 signal irreversibly decreases by 20%. mNeonGreen foci were detected with a two-step algorithm. First, the vacuole was segmented using a label-free intensity threshold–based algorithm directly from Gaussian-blurred phase–contrast images. Second, a dynamic intensity–based 2D Gaussian filter algorithm was used to quantify the number, size (in pixels), and other algorithmically defined Atg8 puncta properties. Foci were defined as vacuolar or nonvacuolar if they overlapped with the area algorithmically identified as the vacuole. The code for label-free vacuolar tracking and extraction and mNeonGreen-Atg8 foci quantification is available at: https://github.com/alejandrolvido/Vacuole_buster.
Deep learning–based intracellular foci enrichment analysis
For Cdc14-mNG fluorescent intensity enrichment analysis surrounding mScarlet-Atg13 foci, images were acquired using 3i Marianas CSUW-T1 Live-Cell Imaging Confocal microscope equipped with a Photometrics Prime 95B camera, a LaserStack (Model 3iL33), a Zeiss Plan Apochromat 63×/1.4-NA oil-immersion objective, and filters for mNG (C515-542/27)/mRuby (C561-617/73). The deep learning image segmentation algorithm Cellpose (Python) is used to produce detailed cell segmentation from confocal microscopy images. Cellpose segmentation was implemented using the command line option as described in https://cellpose.readthedocs.io/en/latest/command.html. The segmented images are used in turn to identify (Matlab) foci in a target channel and calculate the intensity of signals surrounding the foci in other channels. Atg13 foci were identified as the cytoplasm regions that were >3 standard deviations brighter than the average fluorescence intensity of cytoplasmic pixels. Concentric regions of 1 pixel around the Atg13 foci were defined algorithmically, and the fluorescence intensity of the mNeonGreen channel in cells bearing Cdc14-mNG or an isogenic control without Cdc14-mNG was quantified in each concentric region. The concentric regions expanded until they overlapped with the cellpose-defined pixel corresponding to the cell membrane. A k-means clustering algorithm was then used to classify foci according to the Cdc14-mNG pixel intensity in their surroundings. The output is plotted as signal intensity over distance from the foci perimeter pixels. The code for the Cellpose-based analysis is available at: https://github.com/alejandrolvido/Deep-learning-based-intracellular-foci-enrichment-analysis.
Online supplemental material
Fig. S1 shows autophagy in meiotic cells, starved cells, and meiotic ndt80Δ cells. Fig. S2 shows that meiotic autophagy is coupled to meiosis progression. Fig. S3 shows that Cdc14 stimulates autophagy during meiosis. Fig. S4 shows that recombinant Cdc14 carries phosphatase activity in vitro.Fig. S5 shows that two PxL motifs in Atg13 are critical for the function of Atg13 in autophagy. Table S1 lists strains used in this study. Table S2 lists plasmids used in this study. Table S3 lists primers used in this study. Video 1 shows autophagy and meiotic progression at the single-cell level.
Acknowledgments
We thank Beth Levine, the University of Texas Southwestern Medical Center for Autophagy Research and Benjamin Tu for imaging and experimental support. The authors would like to acknowledge the assistance of the University of Texas Southwestern Quantitative Light Microscopy Core, a Shared Resource of the Harold C. Simmons Cancer Center, supported in part by an NCI Cancer Center Support Grant, 1P30 CA142543-01, and an NIH Shared Instrumentation Award to Dr. K. Luby-Phelps (1S10OD028630-01). We thank Vladimir Denic (Harvard University, Boston, MA) and Soni Lacefield (Indiana University, Bloomington, IN) for reagents and protocols. We thank B. Levine, J. Seemann, J. Friedman, S. Lacefield, M. Henne, and members of the Wang lab for comments on the manuscript.
This work was supported by a grant from the National Institutes of Health to F. Wang (R01GM133899) and from the Welch Foundation to F. Wang (I-2019-20190330), as well as funding from Nancy Cain and Jeffrey A. Marcus Scholar in Medical Research, in Honor of Dr. Bill S. Vowell, to F. Wang, and the National Institute of General Medical Sciences of the National Institutes of Health (K99GM135487) to O. Argüello-Miranda.
The authors declare no competing financial interests.
Author contributions: Conceptualization, F. Wang and W. Feng; Methodology, F. Wang, W. Feng, and O. Argüello-Miranda; Investigation, F. Wang, W. Feng, O. Argüello-Miranda, and S. Qian; Writing—Original Draft, F. Wang; Writing—Review & Editing, F. Wang, O. Argüello-Miranda, and W. Feng; Funding Acquisition, F. Wang; Supervision, F. Wang.

