


The nuclear pore complex (NPC) physically interacts with chromatin and regulates gene expression. The Saccharomyces cerevisiae inner ring nucleoporin Nup170 has been implicated in chromatin organization and the maintenance of gene silencing in subtelomeric regions. To gain insight into how Nup170 regulates this process, we used protein–protein interactions, genetic interactions, and transcriptome correlation analyses to identify the Ctf18-RFC complex, an alternative proliferating cell nuclear antigen (PCNA) loader, as a facilitator of the gene regulatory functions of Nup170. The Ctf18-RFC complex is recruited to a subpopulation of NPCs that lack the nuclear basket proteins Mlp1 and Mlp2. In the absence of Nup170, PCNA levels on DNA are reduced, resulting in the loss of silencing of subtelomeric genes. Increasing PCNA levels on DNA by removing Elg1, which is required for PCNA unloading, rescues subtelomeric silencing defects in nup170Δ. The NPC, therefore, mediates subtelomeric gene silencing by regulating PCNA levels on DNA.
Introduction
Nuclear pore complexes (NPCs) are large proteinaceous assemblies embedded in the nuclear envelope (NE) that serve as the only conduits for nucleocytoplasmic transport (Aitchison and Rout, 2012). The yeast NPC, an over-500-protein, 50-megadalton complex, is made up of an assembly of ∼30 different proteins, termed nucleoporins (Alber et al., 2007; Kim et al., 2018), that are each present in multiple copies per NPC. These nucleoporins assemble to form higher-order modular structures called spokes, and eight coaxially arranged spokes form a symmetrical, cylindrical channel. Each spoke is composed of different subcomplexes arranged to form the outer, inner, and membrane rings, cytoplasmic filaments, and a nuclear basket (Alber et al., 2007; Kim et al., 2018). The central channel of the NPC is composed of nucleoporins rich in disordered Phe-Gly (FG) repeat motifs that line the central channel and confer selectivity to transport of cargos between the nucleus and cytoplasm (Aitchison and Rout, 2012).
NPCs also function as positional beacons or scaffolds in the NE with various roles in chromatin organization and gene regulation. Thus, they influence heterochromatin states, nucleosome organization, chromatin boundaries, and telomere localization. These influences, in turn, lead to the regulation of transcriptional activation, transcriptional repression, transcriptional memory, and subtelomeric silencing (Ptak et al., 2014; Van de Vosse et al., 2013; Dilworth et al., 2001; Kadota et al., 2020; Light et al., 2010; Galy et al., 2000).
During S phase, newly synthesized DNA is packaged into nucleosomes by replication-coupled nucleosome assembly, which prevents DNA damage and preserves the integrity of the genome (Serra-Cardona and Zhang, 2018). Replication forks act as organizing centers for a host of chromatin modifiers that are required to reassemble chromatin after replication. Impairment of replication fork progression during DNA replication by endogenous or exogenous factors can result in DNA damage and trigger DNA damage response pathways. Some of the stalled forks become spatially segregated to the nuclear periphery, and the NPC has been implicated in the resolution of these stalled forks and in restarting DNA replication (Lamm et al., 2021; Whalen and Freudenreich, 2020). However, the precise role that the NPCs play in this process remains poorly understood.
The nucleoporin Nup170 is a major component of the NPC’s inner ring in Saccharomyces cerevisiae. There, it is the outermost layer of the inner ring, adjacent to and forming interactions with the pore membrane (Fig. 1). Despite its core position, it is in principle accessible to the cyto- and nucleoplasm and proteins therein (Akey et al., 2022; Hakhverdyan et al., 2021). From there, it interacts with chromatin and it is implicated in the maintenance of heterochromatin and genome stability (Kerscher et al., 2001; Van de Vosse et al., 2013). Nup170 is required for telomere tethering to the NE, and disruption of Nup170 leads to altered chromatin structure and the upregulation of many genes, the majority of which are located in the otherwise silenced subtelomeric regions of chromosomes (Van de Vosse et al., 2013). Nup155, the mammalian ortholog of Nup170, also associates with and modulates chromatin functions, suggesting a potentially conserved role (Breuer and Ohkura, 2015; Kehat et al., 2011). Although disruption of Nup170 has profound effects on various aspects of chromatin activities, the mechanistic underpinnings of its contributions to chromatin regulation remain unclear.

Nup170 is accessible from the nucleoplasm . In situ cryo-electron tomography structure of S. cerevisiae NPC fit with eightfold radially symmetric atomic models of nucleoporins (left, EMD-24258; PDB: 7N9F) viewed from the nuclear side. Closer examination of inner ring (inset right) shows accessibility to the nuclear side of the NPC. Figure generated by UCSF Chimera (Pettersen et al., 2004).
Nup170 is accessible from the nucleoplasm . In situ cryo-electron tomography structure of S. cerevisiae NPC fit with eightfold radially symmetric atomic models of nucleoporins (left, EMD-24258; PDB: 7N9F) viewed from the nuclear side. Closer examination of inner ring (inset right) shows accessibility to the nuclear side of the NPC. Figure generated by UCSF Chimera (Pettersen et al., 2004).
To gain insight into the roles of Nup170 in chromatin organization and gene regulation, we employed molecular systems biology approaches to identify the alternative proliferating cell nuclear antigen (PCNA) loader Ctf18-RFC complex as a mediator of Nup170’s chromatin modification functions. Here, we report that the Ctf18-RFC complex physically and functionally interacts with Nup170, and PCNA levels on chromatin are reduced in cells lacking Nup170. We show loss of subtelomeric silencing in nup170Δ cells is largely rescued by increasing PCNA levels on DNA by removal of the PCNA unloader Elg1. Our results reveal a surprising new role for NPCs in the maintenance of PCNA levels on DNA and the regulation of chromatin functions.
Results
Systems biology approaches identify the Ctf18-RFC complex as a mediator of Nup170’s gene regulatory functions
To understand the underlying mechanism(s) of the gene regulatory functions of Nup170, we defined a protein–protein interaction map of transient and indirect vicinal interactors of the NPC that are centered around Nup170. A chimeric Nup170-GFP fusion protein was affinity-purified from whole-cell extracts. We have shown that when isolating NPCs (and other complexes), adjusting the buffer conditions can dramatically influence our ability to isolate highly purified intact NPCs, individual complexes within the NPC, or NPCs with vicinal components attached (Hakhverdyan et al., 2015). Thus, to enable the identification of vicinal proteins that may interact with the NPC in a more transient manner, we used less stringent isolation conditions and identified co-enriching proteins by mass spectrometry (MS; Fig. S1 A). This analysis revealed 557 non-nucleoporin potential interacting proteins of Nup170, and therefore represent putative candidate facilitators of Nup170’s role in regulating chromatin (Fig. 2 A and Table S1). These included previously identified Nup170 interacting chromatin structure remodeling (RSC) complex proteins Arp9, Rsc2, Rsc4, and Rsc7, and with less stringent search criteria, silencing factor sirtuin 4 (Sir4), Sir3, Rsc1, Rsc6, Rsc9, and Rsc59, consistent with previous results (Van de Vosse et al., 2013). To identify proteins with putative functions related to Nup170’s role in gene expression, we sequentially parsed this list through the following requirements: (1) functional relevance revealed by a genetic interaction with Nup170 and (2) among these genetically interacting genes, the requirement that its deletion mutant phenocopy nup170Δ as measured by gene expression profiles. Importantly, nup170Δ is viable as it has high structural redundancy with its surrounding nucleoporins, and thus nuclear transport proceeds virtually unimpaired in this strain (Aitchison et al., 1995; Akey et al., 2022; Hakhverdyan et al., 2021; Kim et al., 2018; Rajoo et al., 2018). There are 415 reported genetic interactors of Nup170 (Table S2) in BioGRID (https://thebiogrid.org/; Stark et al., 2006), and 60 of these genetic interactors were also identified as non-nucleoporin vicinal interactors of Nup170 (Fig. S1 B), suggesting some of these proteins may function with, or redundantly to, Nup170. We assessed the correlation of gene expression profiles using a compendium of transcriptomic profiles of ∼1,500 individual deletion strains (Kemmeren et al., 2014) called the “deleteome.” This transcriptome correlation analysis identified 40 individual gene deletions (Fig. 2 A) whose expression profiles were significantly correlated with that from the nup170Δ strain. Among genetic and physically interacting proteins, the deletions with significant transcriptome correlation scores were chromosome transmission fidelity 18 (Ctf18; Mayer et al., 2001) and ubiquitin-specific protease 3 (Ubp3; Baker et al., 1992). We prioritized Ctf18 for further study based on its nuclear localization and direct and defined roles in chromatin organization, telomere maintenance, and telomere recruitment to the nuclear periphery (Gellon et al., 2011; Hiraga et al., 2006; Kubota et al., 2011; Liu et al., 2020; Ogiwara et al., 2007a; Stokes et al., 2020). Ubp3 has also been implicated in subtelomeric gene silencing (Moazed and Johnson, 1996), and it remains for future study.
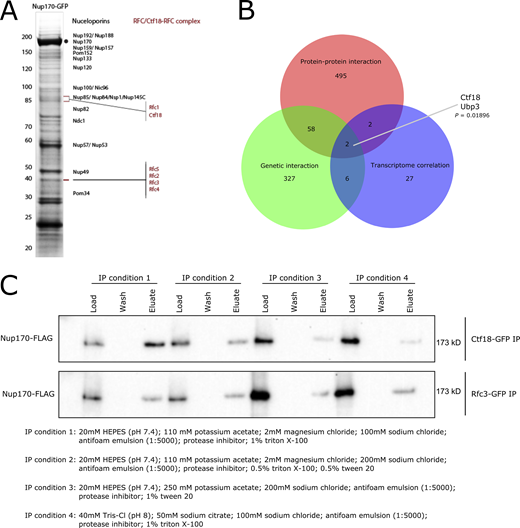
Identification of the Ctf18-RFC complex as a mediator of Nup170’s gene regulatory functions. (A) Nup170-GFP was affinity-purified using a GFP nanobody and copurifying proteins were resolved by SDS-PAGE and visualized by staining with Coomassie blue. The numbers on the left correspond to molecular weight markers (in kD). The gel bands were cut, processed by in-gel digestion, and analyzed by MS. Proteins identified by MS are listed in Table S1. (B) Venn diagram showing overlap between candidates identified by protein–protein interaction, genetic interaction, and transcriptome correlation analyses. The P value is derived from a hypergeometric test. (C) Ctf18-GFP and Rfc3-GFP fusion proteins were affinity-purified from the cell lysate containing Nup170-3FLAG under various extraction conditions. Eluates were analyzed by immunoblotting using anti-FLAG antibody to detect co-immunoprecipitating Nup170. Source data are available for this figure: SourceData FS1.
Identification of the Ctf18-RFC complex as a mediator of Nup170’s gene regulatory functions. (A) Nup170-GFP was affinity-purified using a GFP nanobody and copurifying proteins were resolved by SDS-PAGE and visualized by staining with Coomassie blue. The numbers on the left correspond to molecular weight markers (in kD). The gel bands were cut, processed by in-gel digestion, and analyzed by MS. Proteins identified by MS are listed in Table S1. (B) Venn diagram showing overlap between candidates identified by protein–protein interaction, genetic interaction, and transcriptome correlation analyses. The P value is derived from a hypergeometric test. (C) Ctf18-GFP and Rfc3-GFP fusion proteins were affinity-purified from the cell lysate containing Nup170-3FLAG under various extraction conditions. Eluates were analyzed by immunoblotting using anti-FLAG antibody to detect co-immunoprecipitating Nup170. Source data are available for this figure: SourceData FS1.

Systems biology approaches identify the Ctf18-RFC complex as a mediator of Nup170’s gene regulatory functions. (A) Schematic of our approach to identify and prioritize candidates that mediate gene regulatory roles of the nucleoporin Nup170. The LC-MS analysis of affinity-purified Nup170-GFP identified 557 putative, transient, vicinal non-nucleoporin interactors, 60 of which have known genetic interactions with NUP170. Among genetically and physically interacting candidates, transcriptomic profiles of ctf18∆ and ubp3∆ strains were significantly similar to the nup170∆ strain in a compendium of deletion strain transcriptomes. (B) Heatmap showing gene expression profiles of the nup170∆ strain and the three (ctf18∆, ctf8∆, and dcc1∆) viable deletion strains for Ctf18-RFC complex constituents. The P values are from hypergeometric tests. (C) Cytoscape network illustrating the interaction between Nup170 and the Ctf18-RFC complex.
Systems biology approaches identify the Ctf18-RFC complex as a mediator of Nup170’s gene regulatory functions. (A) Schematic of our approach to identify and prioritize candidates that mediate gene regulatory roles of the nucleoporin Nup170. The LC-MS analysis of affinity-purified Nup170-GFP identified 557 putative, transient, vicinal non-nucleoporin interactors, 60 of which have known genetic interactions with NUP170. Among genetically and physically interacting candidates, transcriptomic profiles of ctf18∆ and ubp3∆ strains were significantly similar to the nup170∆ strain in a compendium of deletion strain transcriptomes. (B) Heatmap showing gene expression profiles of the nup170∆ strain and the three (ctf18∆, ctf8∆, and dcc1∆) viable deletion strains for Ctf18-RFC complex constituents. The P values are from hypergeometric tests. (C) Cytoscape network illustrating the interaction between Nup170 and the Ctf18-RFC complex.
Ctf18 is a subunit of the highly conserved heptameric alternative DNA sliding clamp loader replication factor C (RFC) complex (Mayer et al., 2001). The canonical RFC complex consists of five essential proteins of the AAA + ATPase family containing one large (Rfc1) and four small (Rfc2–5) subunits (Bowman et al., 2004; Yao and O’Donnell, 2012). In the alternative clamp loader Ctf18-RFC complex, Rfc1 is replaced by Ctf18, and its C-terminus is bound to two additional proteins, Ctf8 and defective in sister chromatid cohesion 1 (Dcc1; Grabarczyk et al., 2018). Consistent with a functional relationship between the Ctf18-RFC complex and Nup170, each of the four small subunits—Rfc2, Rfc3, Rfc4, and Rfc5—were present in our Nup170 vicinal interactome dataset (Fig. 2 C and Table S1). Moreover, expression profiles for deletion strains of the two non-essential components DCC1 and CTF8 were significantly correlated with that of nup170Δ and ctf18Δ (Fig. 2, A–C), while Rfc2–5 are essential proteins, and therefore transcriptome profiles cannot be obtained for their deletions.
To confirm the physical interaction between Nup170 and the Ctf18-RFC complex, we performed reciprocal affinity capture of Ctf18-GFP and Rfc3-GFP under different extraction conditions and probed the eluates for Nup170-3FLAG by immunoblot. Consistent with our affinity purification MS data, we found that Nup170-3FLAG co-purified with both Ctf18-GFP and Rfc3-GFP (Fig. S1 C), confirming the likely vicinal association between the Ctf18-RFC complex and Nup170.
The Ctf18-RFC complex is recruited to a subset of NPCs lacking the nuclear basket nucleoporins Mlp1 and Mlp2
Accumulating evidence suggests that structurally heterogeneous NPCs exist within a single cell, which may in turn reflect specific NPC functions (Akey et al., 2022; Fernandez-Martinez and Rout, 2021). Indeed, recent studies by Akey et al., (2022) suggest that at least three major structural forms of NPCs exist in a single NE, which may aid in functional specialization of the nuclear periphery. The three isoforms of the NPC contain either a single or double outer ring on their nucleoplasmic side or lack the nuclear basket. Separately, Nup170 has been shown to be a constituent of a distinct “Snup” complex of nucleoporins that interacts with Sir4 to recruit subtelomeres to the nuclear periphery (Lapetina et al., 2017). The Snup complex is physically distinct from mature NPCs and lacks the flexible-connector nucleoporins of the inner ring complex (Nup53 and Nup59) and also the nuclear basket nucleoporins (Nup60, Nup1, and Mlp1). Given this background, we asked two questions. (1) Is the Ctf18-RFC complex recruited to all NPCs? And if not, (2) can we define the composition of the NPC isoform(s) with which it interacts? To answer the first question, we used a split GFP system (Cabantous and Waldo, 2006) where two proteins of interest are separately tagged with two non-fluorescent parts of GFP (GFP1-10 and GFP11; Fig. 3 A). This system has been used to study in vivo, dynamic, and sub-stoichiometric protein–protein interactions (Hu and Kerppola, 2003; Hu et al., 2002). If the two proteins physically interact, the two GFP fragments are brought into close proximity, assemble, and reconstitute fluorescence (Fig. 3 A; Hu et al., 2002; Smoyer et al., 2016). Using Nup170 as bait, we tested each component of the Ctf18-RFC complex and observed several GFP puncta per cell that localized to the NE and overlapped with a portion of NPCs labeled with Nup188-mCherry (Fig. 3, B and C). These results indicate a close physical interaction between Nup170 and the Ctf18-RFC complex in vivo and that the Ctf18-RFC complex is recruited to only a subset of NPCs (Fig. 3, B and C). These findings are likely specific for a subset of the total Ctf18-RFC complex because despite the high abundance of some members, e.g., Rfc3 is present at ∼5,000 copies per cell (Ho et al., 2018), only a few GFP foci were observed. As a control, we did not observe GFP puncta when Rfc3 was tested in combination with the FG-containing Nup49 (Fig. 3, B and C; Wente et al., 1992), suggesting that portions of the central core of the NPC are inaccessible to the Ctf18-RFC complex.
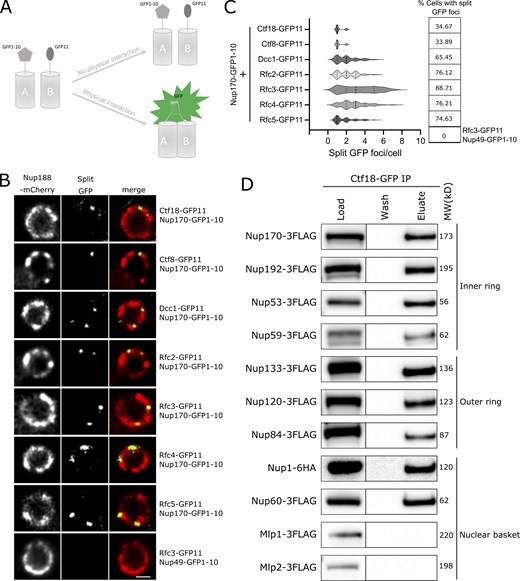
Ctf18-RFC is recruited to a subset of NPCs lacking the nuclear basket nucleoporins Mlp1 and Mlp2. (A) Schematic showing the principle of the split-GFP assay. GFP1-10 and GFP11 are fused to proteins A and B, respectively, and when A and B interact, GFP1-10 and GFP11 associate to form a full-length GFP that can fluoresce. (B) Images of the nuclei of cells co-expressing Nup170–GFP1-10 and GFP11 tagged components of the Ctf18-RFC complex. The bottom panel image shows a cell co-expressing Nup49–GFP1-10 and Rfc3-GFP11. In these cells, the NPCs are marked by Nup188-mCherry. Single plane images from the acquired z-stacks are shown. Scale bar = 1 µm. (C) Violin plots showing the number of split GFP foci per cell for strains expressing Nup170–GFP1-10 and GFP11 tagged components of the Ctf18-RFC complex. The table on the right shows percentages of cells containing split GFP foci for the corresponding strains in the violin plots. The split GFP foci were not detected in cells co-expressing Nup49–GFP1-10 and Rfc3-GFP11 (bottom row of the table). (D) Ctf18-GFP fusion protein was affinity-purified from cell lysates containing 3FLAG-tagged nucleoporins or Nup1-6HA. Eluates were analyzed by immunoblotting using anti-FLAG or anti-HA antibodies to detect indicated nucleoporins. MW, molecular weight. Source data are available for this figure: SourceData F3.
Ctf18-RFC is recruited to a subset of NPCs lacking the nuclear basket nucleoporins Mlp1 and Mlp2. (A) Schematic showing the principle of the split-GFP assay. GFP1-10 and GFP11 are fused to proteins A and B, respectively, and when A and B interact, GFP1-10 and GFP11 associate to form a full-length GFP that can fluoresce. (B) Images of the nuclei of cells co-expressing Nup170–GFP1-10 and GFP11 tagged components of the Ctf18-RFC complex. The bottom panel image shows a cell co-expressing Nup49–GFP1-10 and Rfc3-GFP11. In these cells, the NPCs are marked by Nup188-mCherry. Single plane images from the acquired z-stacks are shown. Scale bar = 1 µm. (C) Violin plots showing the number of split GFP foci per cell for strains expressing Nup170–GFP1-10 and GFP11 tagged components of the Ctf18-RFC complex. The table on the right shows percentages of cells containing split GFP foci for the corresponding strains in the violin plots. The split GFP foci were not detected in cells co-expressing Nup49–GFP1-10 and Rfc3-GFP11 (bottom row of the table). (D) Ctf18-GFP fusion protein was affinity-purified from cell lysates containing 3FLAG-tagged nucleoporins or Nup1-6HA. Eluates were analyzed by immunoblotting using anti-FLAG or anti-HA antibodies to detect indicated nucleoporins. MW, molecular weight. Source data are available for this figure: SourceData F3.
To assay for the NPC isoform(s) associating with the Ctf18-RFC complex, we affinity-purified Ctf18 and probed the eluate for a panel of nucleoporins. In addition to Nup170, we tested for members of the inner ring complex (Nup192, Nup53, and Nup59), the outer ring complex (Nup133, Nup120, and Nup84), and the nuclear basket (Nup1, Nup60, Mlp1, and Mlp2), but not the cytoplasmic nucleoporins. All tested inner ring, outer ring, and nucleoplasmic FG nucleoporins copurified with Ctf18-GFP (Fig. 3 D). However, we did not detect a physical association between Ctf18-GFP and the nuclear basket proteins Mlp1 and Mlp2. To validate the absence of Mlp1 and Mlp2 on Ctf18-RFC interacting NPCs in vivo, we endogenously tagged Mlp1, Mlp2, or Nup188 with mCherry in cells expressing Nup170–GFP1-10 and Rfc3-GFP11 and determined the percentage of split GFP puncta that overlapped with mCherry signal. We found a high percentage (median: 71.43, mean: 60.94) of split GFP spots overlapped with Nup188-mCherry signal (defined by the centroid of the GFP signal being on or within the signal produced by mCherry; Fig. S2), whereas percentages of split GFP spots overlapping with Mlp1-mCherry (median: 13.39, mean: 27.87) or Mlp2-mCherry (median: 0, mean: 22.80) were remarkably small (Fig. S2). Taken together, these results suggest that Ctf18-RFC preferentially interacts with a subset of NPCs that lack Mlp1 and 2, which, based on the co-purification of Nup53, Nup59, and Nup1, are distinct from the Snup complex (Lapetina et al., 2017).

Ctf18-RFC interacting NPCs lack Mlp1 and Mlp2. (A) Images of cells co-expressing Nup170–GFP1-10 and Rfc3-GFP11 in strains where Mlp1, Mlp2, or Nup188 are endogenously tagged with mCherry. Scale bar = 2 µm. (B) Mean of percentages of splitGFP foci overlapping with mCherry signal. (C) Median of percentages of splitGFP foci overlapping with mCherry signal. In B and C, the error bars represent 95% confidence intervals. Number of cells analyzed: Mlp1, 237; Mlp2, 273; and Nup188, 265.
Ctf18-RFC interacting NPCs lack Mlp1 and Mlp2. (A) Images of cells co-expressing Nup170–GFP1-10 and Rfc3-GFP11 in strains where Mlp1, Mlp2, or Nup188 are endogenously tagged with mCherry. Scale bar = 2 µm. (B) Mean of percentages of splitGFP foci overlapping with mCherry signal. (C) Median of percentages of splitGFP foci overlapping with mCherry signal. In B and C, the error bars represent 95% confidence intervals. Number of cells analyzed: Mlp1, 237; Mlp2, 273; and Nup188, 265.
The interaction between Ctf18 and NPCs peaks during S phase
Chromatin immunoprecipitation (ChIP) analyses have shown that the Ctf18-RFC complex localizes to replication forks and is bound to chromatin throughout S phase (Crabbé et al., 2010; Lengronne et al., 2006; Liu et al., 2020). To test if the observed Ctf18-RFC interaction with NPCs differed through the cell cycle, we analyzed the localization of Ctf18-GFP during various stages of the cell cycle in a strain in which NPCs were marked by endogenously tagged Nup188-mCherry. Ctf18-GFP appeared mainly diffused in the nucleoplasm during G1 but concentrated into a few foci during S phase. After completion of S phase, Ctf18-GFP was redistributed and appeared diffuse in the nucleus during G2/M phase (Fig. 4, A and B). We quantified the localization of Ctf18-GFP foci relative to Nup188-mCherry in individual cells and found that ∼63% of total foci overlapped with the Nup188-mCherry signal (Fig. 4 C). To further evaluate cell cycle–dependent interactions between Ctf18 and the NPC, we affinity-purified Ctf18-GFP from cells harvested at different timepoints after release from G1 arrest and probed eluates for Nup170-3FLAG by immunoblotting. Consistent with our microscopy data, we found that the recovery of Nup170 with Ctf18 was highest during S phase (Fig. 4 D and Fig. S3).
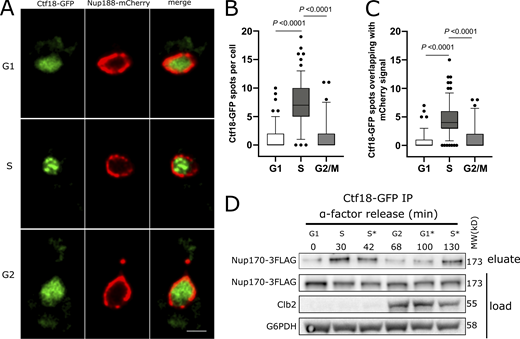
Ctf18 interaction with the NPC is peaks during Sphase. (A) Cells expressing genomically encoded Nup188-mCherry and Ctf18-GFP were arrested in G1 phase by α-factor and images were acquired at regular intervals for 110 min after release from arrest. Representative images are shown for different cell cycle stages. Scale bar = 2 µm. (B) Box plot showing the number of Ctf18-GFP spots per cell at the G1 (n = 140), S (n = 155), and G2/M (n = 69) cell cycle stages. (C) Box plots showing the number of Ctf18-GFP spots overlapping with NPCs at those stages. In B and C, the P values are from Wilcoxon rank sum tests. The bars represent the 5th and 95th percentiles, and the dots are outliers. (D) Cells producing Ctf18-GFP and Nup170-3FLAG were arrested in G1 and samples were collected at indicated time points after release. The Ctf18-GFP fusion protein was affinity-purified from cell lysates and the eluate was analyzed by immunoblotting to detect Nup170-3FLAG. The cell cycle stages are indicated based on G1 arrest and Clb2 (indicative of G2 stage) levels in cell lysates. Cell cycle stages with an asterisk (*) are based on time after release from G1 arrest or after G2 stage. Anti-FLAG, Clb2, and G6PHD antibodies were used for immunoblotting. MW, molecular weight. Source data are available for this figure: SourceData F4.
Ctf18 interaction with the NPC is peaks during Sphase. (A) Cells expressing genomically encoded Nup188-mCherry and Ctf18-GFP were arrested in G1 phase by α-factor and images were acquired at regular intervals for 110 min after release from arrest. Representative images are shown for different cell cycle stages. Scale bar = 2 µm. (B) Box plot showing the number of Ctf18-GFP spots per cell at the G1 (n = 140), S (n = 155), and G2/M (n = 69) cell cycle stages. (C) Box plots showing the number of Ctf18-GFP spots overlapping with NPCs at those stages. In B and C, the P values are from Wilcoxon rank sum tests. The bars represent the 5th and 95th percentiles, and the dots are outliers. (D) Cells producing Ctf18-GFP and Nup170-3FLAG were arrested in G1 and samples were collected at indicated time points after release. The Ctf18-GFP fusion protein was affinity-purified from cell lysates and the eluate was analyzed by immunoblotting to detect Nup170-3FLAG. The cell cycle stages are indicated based on G1 arrest and Clb2 (indicative of G2 stage) levels in cell lysates. Cell cycle stages with an asterisk (*) are based on time after release from G1 arrest or after G2 stage. Anti-FLAG, Clb2, and G6PHD antibodies were used for immunoblotting. MW, molecular weight. Source data are available for this figure: SourceData F4.
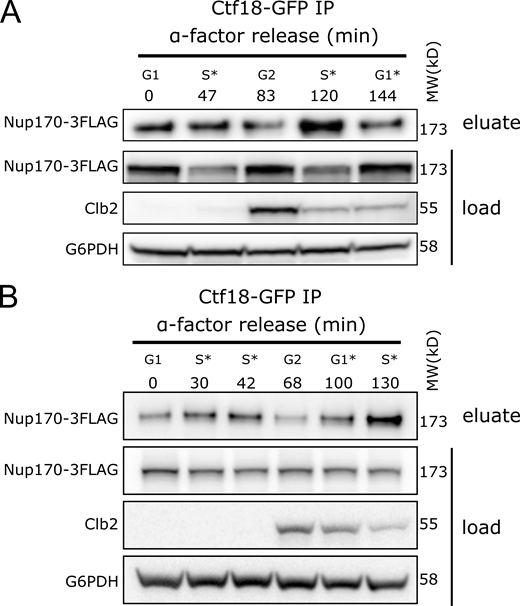
Ctf18 interaction with the NPC peaks during Sphase. (A and B) Cells producing Ctf18-GFP and Nup170-3FLAG were arrested in G1 and samples were collected at indicated time points after release. The Ctf18-GFP fusion protein was affinity-purified from cell lysates and the eluate was analyzed by immunoblotting to detect Nup170-3FLAG. The cell cycle stages are indicated based on G1 arrest and Clb2 (indicative of G2 stage) levels in cell lysates. Anti-FLAG, Clb2, and G6PDH antibodies were used for immunoblotting. A and B are biological replicates, and B is the technical replicate of the Fig. 4 D. MW, molecular weight. Source data are available for this figure: SourceData FS3.
Ctf18 interaction with the NPC peaks during Sphase. (A and B) Cells producing Ctf18-GFP and Nup170-3FLAG were arrested in G1 and samples were collected at indicated time points after release. The Ctf18-GFP fusion protein was affinity-purified from cell lysates and the eluate was analyzed by immunoblotting to detect Nup170-3FLAG. The cell cycle stages are indicated based on G1 arrest and Clb2 (indicative of G2 stage) levels in cell lysates. Anti-FLAG, Clb2, and G6PDH antibodies were used for immunoblotting. A and B are biological replicates, and B is the technical replicate of the Fig. 4 D. MW, molecular weight. Source data are available for this figure: SourceData FS3.
Nup170 functions in silencing of subtelomeric genes and mating type loci and DNA repair, but does not affect sister chromatid cohesion
Nup170 plays a role in telomere positioning to the nuclear periphery and controls subtelomeric silencing (Van de Vosse et al., 2013). Similar to Nup170, the Ctf18-RFC complex is required for telomere positioning to the nuclear periphery and helps facilitate silencing of a telomeric ADE2 reporter gene (Hiraga et al., 2006; Suter et al., 2004). Indeed, the expression profiles of the NUP170 deletion strain (nup170Δ) and deletion strains of the three non-essential components of Ctf18-RFC complex—ctf18Δ, ctf8Δ, and dcc1Δ—were all highly enriched for upregulated subtelomeric genes (hypergeometric test, false discovery rate [FDR]–adjusted P values <0.05; Fig. 5 A). These results further confirm the roles of Nup170 and the Ctf18-RFC complex in the regulation of subtelomeric gene silencing. The Ctf18-RFC complex has been also implicated in maintaining silencing at mating type loci (Suter et al., 2004). Similarly, our own prior microarray analysis showed dramatic derepression of MFA1 and MFA2 genes, which are normally silenced in Mat α cells (8.9- and 3.6-fold increase in nup170Δ cells compared with WT, respectively; Van de Vosse et al., 2013). Similar results were observed in deleteome studies (2.5- and 1.5-fold, respectively; Kemmeren et al., 2014). These results suggest that like subtelomeric genes, silencing at mating type loci also involves both Nup170 and Ctf18.

Nup170 functions in subtelomeric silencing and replication checkpoint but does not affect SCC. (A) Enrichment of differentially expressed genes in the nup170∆, ctf18∆, dcc1∆, and ctf8∆ strains from the deleteome compendium according to gene distance from chromosome ends. X axis: distance from chromosome ends in 25-kb bins. Y axis: negative log10 of the enrichment test’s FDR-adjusted P value. Dashed lines represent significance cutoff. (B) Log-phase cultures of the indicated strains were equalized in cellular density, serially diluted 10-fold, and spotted onto plates containing YPD with or without 100 mM HU. Plates were scanned after 2 d at 30°C. (C) SCC was assessed in three biological replicates for the indicated strains containing a lacO tandem repeat inserted at the TRP1 locus on chromosome IV and LacI-GFP. The number of GFP spots in each cell was scored and percentages of cells with two GFP spots are plotted.
Nup170 functions in subtelomeric silencing and replication checkpoint but does not affect SCC. (A) Enrichment of differentially expressed genes in the nup170∆, ctf18∆, dcc1∆, and ctf8∆ strains from the deleteome compendium according to gene distance from chromosome ends. X axis: distance from chromosome ends in 25-kb bins. Y axis: negative log10 of the enrichment test’s FDR-adjusted P value. Dashed lines represent significance cutoff. (B) Log-phase cultures of the indicated strains were equalized in cellular density, serially diluted 10-fold, and spotted onto plates containing YPD with or without 100 mM HU. Plates were scanned after 2 d at 30°C. (C) SCC was assessed in three biological replicates for the indicated strains containing a lacO tandem repeat inserted at the TRP1 locus on chromosome IV and LacI-GFP. The number of GFP spots in each cell was scored and percentages of cells with two GFP spots are plotted.
The Ctf18-RFC complex has been implicated in the replication checkpoint response pathway (Crabbé et al., 2010; Naiki et al., 2001). To test if Nup170 functions like the Ctf18-RFC complex in response to DNA damage, we evaluated the sensitivity of cells lacking Nup170 or the components of the Ctf18-RFC complex to hydroxyurea (HU), a ribonucleotide reductase inhibitor that arrests cells in S phase and induces replication stress (Alvino et al., 2007). We found that similar to the ctf18Δ, ctf8Δ, and dcc1Δ strains, cells lacking Nup170 were more sensitive to HU compared with WT (Fig. 5 B).
One of the main functions of the Ctf18-RFC complex is to establish sister chromatid cohesion (SCC) after DNA replication as defective SCC can lead to genomic instability (Hanna et al., 2001; Mayer et al., 2001). We investigated whether the nup170Δ strain also exhibited an SCC defect and whether that could explain the defect in subtelomeric silencing and the strain’s sensitivity to HU. To visualize SCC, we used a strain containing lacO-repeats integrated at 12.5 kb from the centromere of chromosome IV that binds a lac repressor-GFP fusion protein (Sanchez et al., 1999). To assess SCC, cells were arrested in G1 phase using α-factor followed by release into nocodazole-containing growth medium to arrest again in G2/M phase. As expected, ctf18Δ showed SCC defects (Fig. 5 C; Hanna et al., 2001; Mayer et al., 2001); however, SCC defects were not observed in nup170Δ cells. These results suggest that defects in chromatin silencing and the replication checkpoint associated with functional Nup170 and Ctf18 are distinct from major defects associated with Ctf18’s role in SCC.
PCNA levels decline at stalled replication forks in nup170Δ cells
The Ctf18-RFC complex is involved in both loading and unloading PCNA onto DNA (Bermudez et al., 2003; Bylund and Burgers, 2005); however, its PCNA unloading activity is not well understood, and it is unclear how this occurs in vivo. Previous studies in ctf18Δ cells showed that PCNA levels decline at replication forks in cells synchronously progressing through S phase or arrested in HU (Lengronne et al., 2006; Liu et al., 2020), suggesting that Ctf18-RFC functions as the net loader. We asked whether, similar to the Ctf18-RFC complex, Nup170 was also required for maintaining PCNA levels at replication forks. To analyze PCNA binding to DNA, we synchronized WT and nup170Δ cells in G1 phase using α-factor followed by release into HU-containing growth medium to further arrest cells in S phase. We performed PCNA ChIP sequencing (ChIP-seq) to assess whether Nup170 played a role in PCNA binding. While the genome-wide binding pattern of PCNA was the same in the presence or absence of Nup170, showing high confidence binding to autonomously replicating sequence (ARS) sites in both nup170Δ and WT cells (Fig. 6 and Fig. S4; Jaccard similarity coefficient = 0.78), the recovery of chromatin was ∼22% less from nup170Δ relative to WT cells. To further evaluate PCNA binding to chromatin, we quantified PCNA binding by ChIP quantitative PCR (qPCR) for four ARS sites, which confirmed significant decreases in PCNA binding to ARS sites in nup170Δ compared with WT cells (Fig. 6).

PCNA levels on chromatin decline in nup170Δ cells. Cells were synchronized in G1 and released into HU-containing YPD medium to arrest them in S phase, and ChIP was performed using an anti-PCNA antibody. Normalized PCNA binding profiles along chromosomes III, VI, and IX obtained by ChIP-seq analysis visualized with the Integrative Genomics Viewer. For visualization, sequencing reads from nup170Δ cells were downsampled by 22% and plotted to reflect the relative recovery of chromatin compared with WT cells. Inset shows PCNA enrichment at the indicated autonomously replicating sequence (ARS) sites analyzed by qPCR. Means ± SD from four experiments are shown; *P < 0.05, **P < 0.01. The P is from a two-tailed Student’s t test.
PCNA levels on chromatin decline in nup170Δ cells. Cells were synchronized in G1 and released into HU-containing YPD medium to arrest them in S phase, and ChIP was performed using an anti-PCNA antibody. Normalized PCNA binding profiles along chromosomes III, VI, and IX obtained by ChIP-seq analysis visualized with the Integrative Genomics Viewer. For visualization, sequencing reads from nup170Δ cells were downsampled by 22% and plotted to reflect the relative recovery of chromatin compared with WT cells. Inset shows PCNA enrichment at the indicated autonomously replicating sequence (ARS) sites analyzed by qPCR. Means ± SD from four experiments are shown; *P < 0.05, **P < 0.01. The P is from a two-tailed Student’s t test.

PCNA binding to all chromosomes in nup170Δ and WT cells. Cells were synchronized in G1 and released into HU-containing YPD medium to arrest them in S phase, and ChIP was performed using an anti-PCNA antibody. PCNA binding profiles along chromosomes obtained by ChIP-seq analysis are visualized with the Integrative Genomics Viewer.
PCNA binding to all chromosomes in nup170Δ and WT cells. Cells were synchronized in G1 and released into HU-containing YPD medium to arrest them in S phase, and ChIP was performed using an anti-PCNA antibody. PCNA binding profiles along chromosomes obtained by ChIP-seq analysis are visualized with the Integrative Genomics Viewer.
Deleting ELG1 in nup170Δ rescues subtelomeric silencing
We reasoned that if the subtelomeric silencing defect in nup170Δ cells stems from the reduction of PCNA levels on DNA, then increasing the PCNA levels on DNA would reverse this defect. Enhanced level of genomic instability 1 (Elg1)-RFC complex is an alternative RFC complex for unloading PCNA from chromatin, and its removal elevates PCNA levels on chromatin (Bellaoui et al., 2003; Kubota et al., 2013). Consistent with our hypothesis, analysis of the deleteome compendium data revealed that loss of Elg1 results in the decreased expression of genes significantly enriched in subtelomeric regions (Fig. 7 A; hypergeometric test, FDR-adjusted P values <0.05), essentially mirroring the increased expression observed for subtelomeric genes when Nup170 is absent (Fig. 7 B; Pearson’s correlation coefficient = −0.18, P = 0.002). These data are consistent with a previous report showing that an ELG1 deletion results in increased silencing of an ADE2 marker gene inserted into a subtelomeric region of chromosome X (Smolikov et al., 2004).

Increasing PCNA levels on DNA in nup170∆ cells rescues subtelomeric silencing defects. (A) Enrichment of differentially expressed genes in the nup170∆ and elg1∆ strains according to gene distance from chromosome ends. X axis: distance from telomere in 25-kb bins. Y axis: negative log10 of the enrichment test’s FDR-adjusted P value. Dashed lines represent significance cutoff. (B) Heatmap showing expression profiles of subtelomeric genes in the nup170∆ and elg1∆ strains. (C) Gene expression levels of four subtelomeric genes were measured in the WT, elg1∆, nup170∆, and nup170∆ elg1∆ strains by RT-qPCR, and RQ values are plotted. The RQmin/RQmax values are presented as error bars. Data from at least three biological replicates are presented. (D) PCNA enrichment at the indicated ARS sites was analyzed by qPCR. Means ± SD from three experiments are shown; *P < 0.05, **P < 0.01.
Increasing PCNA levels on DNA in nup170∆ cells rescues subtelomeric silencing defects. (A) Enrichment of differentially expressed genes in the nup170∆ and elg1∆ strains according to gene distance from chromosome ends. X axis: distance from telomere in 25-kb bins. Y axis: negative log10 of the enrichment test’s FDR-adjusted P value. Dashed lines represent significance cutoff. (B) Heatmap showing expression profiles of subtelomeric genes in the nup170∆ and elg1∆ strains. (C) Gene expression levels of four subtelomeric genes were measured in the WT, elg1∆, nup170∆, and nup170∆ elg1∆ strains by RT-qPCR, and RQ values are plotted. The RQmin/RQmax values are presented as error bars. Data from at least three biological replicates are presented. (D) PCNA enrichment at the indicated ARS sites was analyzed by qPCR. Means ± SD from three experiments are shown; *P < 0.05, **P < 0.01.
To test if increasing PCNA levels on DNA in nup170Δ enhanced silencing, we measured the expression of a subset of subtelomeric genes by RT-qPCR in nup170Δ and nup170Δelg1Δ strains. COS4, COS10, GIT1, and TOG1 were selected based on their subtelomeric locations in the genome and previous observations that their silencing depends on Nup170 (Van de Vosse et al., 2013; Kemmeren et al., 2014). Deletion of ELG1 in the nup170Δ strain reversed the effect of diminished subtelomeric silencing, with three of the four genes tested showing a significantly reduced expression in nup170Δelg1Δ cells compared with nup170Δ (Fig. 7 C). Furthermore, ChIP qPCR data confirmed that PCNA levels on DNA were elevated in the nup170Δelg1Δ strain compared with nup170Δ (Fig. 7 D).
PCNA binds to chromatin assembly factor 1 (CAF-1), a three-protein complex comprising Rlf2, Cac2, and Msi1, which recruits histones to accomplish DNA replication- or DNA repair-coupled nucleosome assembly and facilitates heterochromatin establishment and gene silencing (Gaillard et al., 1996; Young et al., 2020; Moggs et al., 2000; Essers et al., 2005; Gaillard et al., 1996; Young et al., 2020). We asked if the loss of subtelomeric silencing in nup170Δ cells resulting from a decrease in PCNA levels on chromatin was due to their effects on CAF-1 complex. To test this, we first compared subtelomeric gene expression profiles between nup170Δ and strains lacking CAF-1 complex components. Our results showed a significant correlation between nup170Δ and the CAF-1 mutants (Fig. S5 A), suggesting that, at least in part, the effects of reduced PCNA on the loss of subtelomeric silencing are due to their effects on the CAF-1 complex. However, our correlation analysis revealed that the extent of similarity in subtelomeric gene expression profiles between nup170Δ and CAF-1 complex mutants was lower than that between nup170Δ and Ctf18-RFC complex mutants (Fig. S5 A). To test this further, next we constructed double mutants lacking Nup170 and CAF-1 proteins and performed RT-qPCR for the selected subtelomeric genes. Our data revealed that the double mutants had a synergistic loss of subtelomeric gene silencing (Fig. S5 B). We also tested double mutants lacking Elg1 and individual components of CAF-1 and found that in all cases, the addition of elg1Δ alleviated silencing defects associated with CAF-1 complex mutants (Fig. S5 C).

Functional relationship between Nup170, Ctf18-RFC complex, CAF-1 complex, and Elg1. (A) Heatmap showing similarities in subtelomeric genes expression profiles between nup170Δ and mutants lacking components of CAF-1 and Ctf18-RFC complex. Correlation scores (R) are listed below the heatmap. (B and C) Gene expression levels of four subtelomeric genes were measured by RT-qPCR in strains lacking components of CAF-1 complex individually or when combined either with nup170Δ (in A) or elg1Δ (in B). RQ values from three biological replicates are presented as heatmaps. (D) Log-phase cultures of the indicated strains were equalized in cellular density, serially diluted 10-fold, and spotted onto plates containing YPD with or without 100 mM HU. Plates were scanned after 2 d at 30°C.
Functional relationship between Nup170, Ctf18-RFC complex, CAF-1 complex, and Elg1. (A) Heatmap showing similarities in subtelomeric genes expression profiles between nup170Δ and mutants lacking components of CAF-1 and Ctf18-RFC complex. Correlation scores (R) are listed below the heatmap. (B and C) Gene expression levels of four subtelomeric genes were measured by RT-qPCR in strains lacking components of CAF-1 complex individually or when combined either with nup170Δ (in A) or elg1Δ (in B). RQ values from three biological replicates are presented as heatmaps. (D) Log-phase cultures of the indicated strains were equalized in cellular density, serially diluted 10-fold, and spotted onto plates containing YPD with or without 100 mM HU. Plates were scanned after 2 d at 30°C.
Taken together, these results indicate that the subtelomeric silencing defect observed in nup170Δ cells is due to reduced PCNA levels and that increasing those levels largely restores silencing. We also investigated whether increasing PCNA levels in nup170Δ cells rescued the replication checkpoint defect; it did not, because nup170Δelg1Δ cells remained susceptible to growth on HU (Fig. S5 D).
Discussion
Many studies in yeast and metazoans have established roles for the NPC in chromatin organization, gene regulation, and DNA repair (reviewed in Lamm et al., 2021; Pascual-Garcia and Capelson, 2021; Ptak et al., 2014; Sumner and Brickner, 2022). However, in many cases, the physical and functional relationship between NPCs and chromatin-modifying factors that regulate these processes remain poorly understood. In yeast, nucleoporin Nup170 has been shown to play a role in nucleosome organization and genome stability, and the loss of Nup170 leads to altered subtelomeric chromatin structure and loss of subtelomeric silencing (Kerscher et al., 2001; Van de Vosse et al., 2013). We also previously showed that Nup170 physically and functionally interacts with normally silenced subtelomeric chromatin and the silencing factor Sir4, establishing the NPC as an important regulator of chromatin silencing (Van de Vosse et al., 2013). However, the mechanistic underpinnings of this complex relationship were unclear. To investigate this further, we identified genetic and physical interactors of Nup170 and identified mutants that phenocopy the transcriptome profile associated with loss of Nup170. This approach identified a physical and functional relationship between NPCs and the PCNA loader Ctf18-RFC complex and uncovered a novel role for the NPC in mediating PCNA levels on DNA and controlling heterochromatin.
PCNA is a DNA replication processivity factor that encircles DNA and is tethered to replicative DNA polymerases δ and ε (Boehm et al., 2016). In addition to its major role at replication forks during DNA replication, PCNA also serves as a loading platform for proteins involved in establishing SCC, in remodeling chromatin, and in DNA repair (Mailand et al., 2013). The dynamics of PCNA on DNA are important to its function, progressing with leading and lagging strands, but also coordinating nucleosome assembly with DNA replication in concert with its loading and unloading on DNA. PCNA is loaded onto DNA by the Rfc1-RFC and Ctf18-RFC complexes, although Ctf18-RFC is not essential and loads PCNA onto DNA apparently less efficiently than its Rfc1-RFC counterpart (Bermudez et al., 2003). A third complex, Elg1-RFC, unloads PCNA from DNA, which is also important for histone deposition, DNA repair, and telomere silencing (Smolikov et al., 2004; Kubota et al., 2013; Ogiwara et al., 2007b; Gali et al., 2018). Recently, it was suggested that upon replication fork stalling, Pol ε decouples from the Cdc45-MCM-GINS complex, which surrounds the leading strand of DNA and recruits DNA Pol ε (Tanaka and Araki, 2013), and recruits additional Ctf18-RFC complexes (Fujisawa et al., 2017). Recruitment of additional Ctf18-RFC may trigger new rounds of PCNA loading that can further recruit factors required for DNA repair and restart DNA synthesis. Thus, an intricate balance of PCNA levels on DNA that is tailored for its various activities is necessary for it to perform its functions. Disrupting the NPC (in particular Nup170) alters this balance by reducing PCNA levels on chromatin and causes defects in chromatin silencing and DNA repair, but not sister chromatid cohesion.
Ctf18-RFC’s interaction with NPCs peaks during S phase of the cell cycle (Fig. 4, A–D), a phase that is also prone to DNA damage due to actively replicating DNA. In the absence of Nup170, we detected reduced levels of PCNA on stalled replication forks, and this was associated with sensitivity to replication stress, suggesting that Nup170 facilitates PCNA levels on DNA upon induction of DNA damage, thus enabling DNA repair. Intriguingly, Ctf18-RFC associates with NPCs even in the absence of Nup170 (data not shown), suggesting a critical role for Nup170 in regulating the function of Ctf18-RFC but not its recruitment to the NPCs. In the absence of Nup170, inefficient loading of PCNA by the Ctf18-RFC complex and/or unresolved replication forks leading to fork collapse are expected to affect genomic integrity (Alexander and Orr-Weaver, 2016).
In support of our model that reduced PCNA levels on chromatin also lead to defects in chromatin silencing, we showed that mutations in Elg1, which reversed the loss of PCNA on DNA observed in nup170Δ cells, also reversed the subtelomeric silencing defects resulting from loss of Nup170. Taken together, our data point toward a mechanism in which the NPC plays a role in establishing heterochromatin by regulating the Ctf18-RFC complex activities through the regulation of PCNA levels on DNA (Fig. 8).

Cartoon illustrating the role of the NPC and Nup170 in DNA damage repair and heterochromatin formation.
Cartoon illustrating the role of the NPC and Nup170 in DNA damage repair and heterochromatin formation.
PCNA also physically tethers CAF-1 to sites of replication and repair. CAF-1 is a histone chaperone and serves to deposit histone H3 and H4 onto newly synthesized DNA. Mutations in CAF-1 result in a dramatic reduction in DNA-bound histone H3 levels, which leads to reduced levels of Sir2 and Sir4 at silent loci (Tamburini et al., 2006), and mutations that disrupt PCNA–CAF-1 association lead to defects in heterochromatin formation and subtelomeric silencing (Kaufman et al., 1997; Zhang et al., 2000; Krawitz et al., 2002). We wondered if PCNA establishes silencing entirely through its interaction with CAF-1 complex, and therefore, by facilitating nucleosome assembly and establishment of heterochromatin, then mutants lacking CAF-1 components should resemble the phenotypes of nup170Δ. Indeed, our comparative transcriptome analysis showed significant correlation in expression of subtelomeric genes from nup170Δ cells and strains lacking CAF-1 components. However, the levels of similarity in subtelomeric gene expression profiles between nup170Δ and rlf2Δ, cac2Δ, and msi1Δ were less than those between nup170Δ and strains lacking members of Ctf18-RFC complex. This suggests that the CAF-1 complex only partly contributes to Nup170’s subtelomeric silencing defects. This is further supported by the finding that double mutants lacking Nup170 and individual components of the CAF-1 complex had augmented loss of subtelomeric gene silencing. The finding that even in the absence of CAF-1 complex, elevated levels of PCNA are sufficient to establish subtelomeric silencing is intriguing and suggests that PCNA may recruit other factors, independent of CAF-1, that can circumvent silencing defects associated with defective CAF-1.
Structural analyses of NPCs from several model organisms suggest significant similarities in the core scaffold regions; however, there is considerable variability in the peripheral regions of NPCs (Akey et al., 2022; Fernandez-Martinez and Rout, 2021). Architectural diversity in NPCs across organisms associated with differences in the stoichiometry of the structural modules or by gain or loss of components reflects adaptations during evolution suited for each organism. Structural heterogeneity within individual cells can be also envisioned to enable the variety of functions NPCs perform. For example, the nuclear basket proteins Mlp1 and Mlp2 are excluded from NPCs juxtaposed to the nucleolus (Galy et al., 2004); core components of the spindle pole body coimmunoprecipitated with Mlp2, but not Mlp1 (Niepel et al., 2005); and the Snup complex has been shown to contain most nucleoporins, but lacks components of the nuclear basket (Nup60, Nup1, and Mlp1) and inner ring (Nup53 and Nup59) and interacts with Sir4 (Lapetina et al., 2017). Moreover, at least three structural variants of NPCs appear to exist within a single cell (Akey et al., 2022). These NPC isoforms contain either a single or double outer ring on the nucleoplasmic side, with an additional subpopulation of NPCs lacking a nuclear basket. Functionally, the nuclear basket nucleoporins Nup2 and Mlp1 have been shown to play a role in gene activation and transcriptional memory (Brickner et al., 2019; Tan-Wong et al., 2009; Dilworth et al., 2005), whereas Nup170 is involved in the maintenance of subtelomeric silencing and heterochromatin formation (Van de Vosse et al., 2013). Our finding that Ctf18 is recruited to a small subpopulation of NPCs that lack the nuclear basket proteins Mlp1 and Mlp2 suggests that the absence of these nuclear basket nucleoporins may render these NPCs more accessible to chromatin. These findings shed light on the functional specialization of NPCs possessing or lacking a nuclear basket. Our data are consistent with a model in which Nup170’s functional relationship with chromatin occurs via specialized NPCs lacking a nuclear basket.
The functional specialization of NPCs is an intriguing way by which cells confer regulatory mechanisms of gene expression through spatial and temporal compartmentation at the nuclear periphery. Our results reveal how one subset of NPCs, facilitated by Nup170’s interactions with the Ctf18-RFC complex, impacts PCNA levels on chromatin and thereby regulates heterochromatin formation and gene expression. Further comprehensive structural and functional analyses will be necessary to identify and characterize the NPC isoforms. There are likely many additional gene regulatory mechanisms similarly influenced by Nup170 and other nucleoporins given the substantial network of chromatin modifying and/or regulatory proteins that we have uncovered as physically and functionally linked to Nup170. Further understanding of these mechanisms will provide new avenues and opportunities to understand the NPC’s involvement in a variety of diseases and developmental disorders, as well as strategies employed by infectious agents that target NPC components.
Materials and methods
S. cerevisiae strains and growth conditions
Unless otherwise specified, all S. cerevisiae strains used in this study are derived from the S288C background and are listed in Table S3. Cells were grown in a YPD medium (1% yeast extract, 2% peptone, and 2% glucose) or synthetic complete medium containing 2% glucose. To arrest cells in G1, cell cultures were grown to an OD600 of 0.6 and then α-factor (T6901; Sigma-Aldrich) was added (5 g/ml and 100 ng/ml for BAR1 and bar1∆ cells, respectively) and grown for 2.5 h. Cells were washed twice in YPD and then resuspended in YPD, and samples were harvested at various time points. All experiments were performed at 30°C. Yeast transformations were performed using the lithium acetate/polyethylene glycol method (Gietz and Woods, 2002). Strains harboring genomic insertions and deletions were made using a plasmid/PCR-based one-step genomic integration method, and correct integration was verified by PCR using gene-specific primers. The following plasmids were used to construct strains: pJR200 for 3 × FLAG tagging (Ranish et al., 2004); pYM14 for 6HA tagging (Janke et al., 2004); pHIPZ-Pex14-mKate2 for zeocin-based deletion (Chen et al., 2018); and pSJ1256 for GFP1-10 tagging (Smoyer et al., 2016). For GFP11 tagging, the natR cassette was amplified from pYM42 (Janke et al., 2004) using a forward primer (5′-GGTGGAGGTTCTGGAGGAGGTAGTAGAGATCATATGGTTTTGCATGAATATGTTAATGCTGCTGGTATTACTTAAGGATCCCCGGGTTAATT-3′), containing GFP11 and a linker sequence, and a reverse primer (5′-AGCTCGATTACAACAGGTGT-3′). The resulting 1,393 base pair PCR fragment was used as a template for gene-specific tagging.
Transcriptome correlation analysis
Using the deleteome compendium of 1,484 yeast deletion transcriptomic profiles (Kemmeren et al., 2014), we identified gene deletion strains with profiles similar to the nup170Δ strain. This was done by first listing the genes in the signature of the nup170Δ strain (genes with expression values that differed significantly from WT strains) and then performing correlation tests to compare their log2 fold-change values against those from each deletion strain in the deleteome. For strains showing a significant positive correlation with the nup170Δ strain, we then performed a reciprocal correlation test where the signature of the deletion strain was correlated against the corresponding genes in the nup170Δ profile. Deletion strains showing significant correlations in both cases (FDR-corrected P values <0.05) were considered similar to the nup170Δ strain. To help eliminate potential false positives and focus on high-confidence results, we selected strains whose correlation with the nup170Δ signature had an FDR-corrected P value in the lowest 5% of all such values. We found that 40 deletion strains passed this criterion, suggesting that the deleted proteins in these strains may have functional overlap with NUP170 because their deletion resulted in transcriptomic shifts similar to those in the nup170Δ strain. All analyses on deleteome strain gene expression data, including all correlation tests, were performed using the R software package.
Affinity purification
To perform affinity capture of C-terminally GFP-tagged proteins, cells were grown in YPD to a final OD600 of 1–1.5 and harvested by centrifugation. The cell pellets were washed twice with 20 mM Hepes-KOH, pH 7.4, followed by a wash with 20 mM Hepes-KOH, pH 7.4, 1.2% polyvinylpyrrolidone, and 1:100 protease inhibitor cocktail. The cell pellet was pushed through a syringe directly into liquid nitrogen to flash-freeze and make “yeast noodles.” Yeast noodles were cryomilled into fine powder using a 50-ml stainless steel jar and a ball mill (PM100; Retsch Planetary Ball Mill). To prevent sample heating, the milling jar was placed inside a custom-made Teflon jar insulator during the entire milling process. Each sample was subjected to three cycles of milling (6 min each at 450 rpm with reverse rotation every 45 s, with immersion in liquid nitrogen between each cycle) and the resulting yeast powder was stored at −80°C. Unless specified otherwise, yeast protein lysates were prepared by resuspending 200 mg of cell powder in 450 μl of extraction buffer containing 20 mM Hepes Buffer (pH 7.4), 110 mM CH3CO2K, 2 mM MgCl2, 100 mM NaCl, antifoam-B emulsion (1:5,000), 1:100 protease inhibitor cocktail, and 1% Triton X-100. When scaling up, the same ratio of cell powder to extraction buffer was maintained. Cell powder was homogenously dispersed by sonication (4°C, 1 A, 60 s at 10-s intervals) using a QSonica Q700 equipped with a 4-tip microprobe. The resuspension was clarified by centrifugation at 16,000 g for 10 min at 4°C. The clarified protein lysate was mixed with 1.5 mg magnetic beads (Dynabead M-270 Epoxy, Thermo Fisher Scientific) conjugated with in-house GFP nanobody (Fridy et al., 2014), and the mixture was incubated with gentle agitation for 20 min at 4°C. The beads were washed four times with the extraction buffer. The captured protein complexes were eluted from the beads by adding 35 μl 1.5 × lithium dodecyl sulfate sample buffer (NuPAGE) and incubated at room temperature for 10 min. The load samples from the cleared cell lysates were prepared by TCA precipitation.
Proteomics sample preparation and liquid chromatography/mass spectrometry (LC/MS) analysis
In-gel digestion
Protein complexes from the eluate were resolved by SDS-PAGE and stained with Coomassie blue (Imperial Protein Stain, ThermoFisher Scientific). Protein bands of interest between 250 and 20 kD were excised, and the gel slices were transferred into 3 × washed 0.5-ml microcentrifuge tubes with slits on the bottom. These shredder tubes were then placed into 1.5-ml collector tubes, and the assembly was spun in a benchtop centrifuge at 20,000 g until all gel slices were shredded and collected into the collector tubes. Gel pieces were destained by incubating in 100 μl of 100 mM ammonium bicarbonate/acetonitrile (1:1 vol/vol) for 30 min followed by 5-min incubation in 500 μl of neat acetonitrile, and the supernatant solution was discarded. The destained gel pieces were submerged in 10 mM DTT/100 mM ammonium bicarbonate solution for 30 min at 56°C on a thermomixer, followed by washing with neat acetonitrile as before to remove the reducing solution. Immediately, freshly prepared 55 mM iodoacetamide/100 mM ammonium bicarbonate solution was added to the gel pieces to carry out alkylation in the dark for 40 min followed by a wash in neat acetonitrile. Gel pieces were dried in a speed-vac for 20 min and rehydrated in 50 μl of sequencing-grade trypsin (Promega, 1.3 ng/μl solution in 10 mM ammonium bicarbonate/10% acetonitrile). After the trypsin solution was completely absorbed, the gel pieces were completely covered in a minimal required volume of 10 mM ammonium bicarbonate/10% acetonitrile, and tryptic digestion was carried out overnight at 37°C in an air-circulating incubator. The next day, a 1:2 (vol/vol) of 5% formic acid in acetonitrile was added to each tube and incubated for 15 min at 37°C. Supernatants containing extracted peptides were then transferred into fresh tubes, and gel pieces were resuspended into 100 μl of 1:1 acetonitrile/5% formic acid for the final extraction, incubated on a shaker for 10 min at 37°C, and spun down quickly, and the second supernatants were carefully transferred into the same tubes containing the first extract. The digests were dried completely in a speed-vac for 10 min, resuspended in 0.1% trifluoroacetic acid, and desalted using C18 Zip tips (Millipore), following the manufacturer’s protocol. The desalted peptide digests were dried in a speed vac and analyzed by LC/MS.
LC/MS analysis and protein search
LC/MS analysis was performed at the Proteomics Core (Fred Hutch). Peptides were analyzed by LC/electrospray ionization MS/MS with a Thermo Fisher Scientific Easy-nLC II (Thermo Fisher Scientific) nano high-performance LC system coupled to a hybrid Orbitrap Elite ETD (Thermo Fisher Scientific) mass spectrometer. In-line, desalting was accomplished using a reversed-phase trap column (100 μm × 20 mm) packed with Magic C18AQ (5-μm 200 Å resin; Michrom Bioresources) followed by peptide separations on a reversed-phase column (75 μm × 250 mm) packed with Magic C18AQ (5-μm 100 Å resin; Michrom Bioresources) directly mounted on the electrospray ion source. A 60-min gradient from 7 to 35% acetonitrile in 0.1% formic acid at a flow rate of 400 nl/min was used for chromatographic separations. The heated capillary temperature was set to 300°C and a spray voltage of 2,750 V was applied to the electrospray tip. The Orbitrap Elite instrument was operated in the data-dependent mode, switching automatically between MS survey scans in the Orbitrap (automatic gain control target value 1,000,000, resolution 120,000, and injection time 250 ms) with MS/MS spectra acquisition in the dual linear ion trap. The 10 most intense ions from the Fourier-transform full scan were selected for fragmentation in the dual linear ion trap by collision-induced dissociation with a normalized collision energy of 35%. Selected ions were dynamically excluded for 10 s with a list size of 500 and exclusion mass by mass width ± 10 ppm. Raw LC/MS data were searched with MaxQuant (v.1.6.1.0; Cox and Mann, 2008) against a custom protein database composed of 6,729 total yeast protein sequences. MaxQuant search parameters were as follows: peptide spectral match FDR = 0.01; protein identification FDR = 0.01; fixed peptide modifications = oxidation (M) and acetylation (protein N-terminal); MS/MS tolerance = 0.5 Da. Less stringent analysis used the Trans Proteomic Pipeline search engine (Deutsch et al., 2010) FDR = 0.1. The common coeluting S. cerevisiae contaminants as listed on the CRAPOME (Mellacheruvu et al., 2013) were excluded from the list and proteins that were identified using FDR = 0.01 and with four or more unique peptides were included in downstream analyses. Nuclear proteins were identified based on their cellular component annotation on PantherDB (Mi et al., 2021).
Immunoblotting
Proteins were separated on 4–12% gradient SDS-PAGE gels and transferred to nitrocellulose membranes. Membranes were blocked in blocking buffer (Tris-buffered saline with 0.1% Tween, with 4% milk powder) and the following antibodies were used for immunodetection: mouse monoclonal anti-FLAG-HRP (A8592; Sigma-Aldrich), mouse monoclonal anti-HA (H3663; Sigma-Aldrich), rabbit polyclonal anti-Clb2 (sc-9071; Santa Cruz), and rabbit polyclonal anti-G-6-PDH (A9521; Sigma-Aldrich). When required, an appropriate HRP-conjugated secondary antibody was used.
ChIP, ChIP qPCR, and ChIP-seq
ChIP was performed as previously described (Smith et al., 2007; Wan et al., 2009) with the following modifications. Briefly, cells were arrested in G1, washed twice with YPD, and resuspended in YPD containing 200 mM HU followed by incubation for 40 min at 30°C. The cells were crosslinked with 1% formaldehyde and the crosslinking reaction was then quenched by incubation with 125 mM glycine. Cells were then disrupted by a BeadBeater using glass beads, and then chromatin was sheared to an average size of ∼400 bp using a Covaris S2 sonicator and the cell lysate was clarified by centrifugation at 16,000 g. The DNA concentration in each sample was measured using a Qubit Flex Fluorometer (Thermo Fisher Scientific). Anti-PCNA antibody (GTX64144; GeneTex) was conjugated with Dynabeads Protein G (10004D; Thermo Fisher Scientific) per the manufacturer’s instructions. The sheared chromatin (2 µg DNA in 400 μl) was incubated with 1.5 g of the conjugated beads overnight on a rotating platform at 4°C. The eluate and input samples were reverse crosslinked, treated with Proteinase K, and the DNA was prepared using the ChIP DNA Clean & Concentrator kit (cat# D5205; Zymo research). The purified DNA samples were used for ChIP qPCR and ChIP-seq analyses. Real-time qPCR was performed using QuantStudio 5 Real-Time PCR System (Thermo Fisher Scientific) and PowerUp SYBR green master mix. Primers for ARS605, ARS606, ARS607, and ARS609 were previously described (Liu et al., 2020). For ChIP-seq, the library was prepared using NEBNext Ultra II DNA Library Prep Kit (Cat# E7645S; Illumina), and sequencing was performed using an Illumina HiSeq 4000. Paired-end reads were aligned to the yeast genome (Ensembl version 103) using STAR (Dobin et al., 2013), and alignment files were filtered with SAMtools (Li et al., 2009) using the 1804 flag and excluding reads with MAPQ scores below 30 as well as orphan reads and read pairs mapping to different chromosomes. Duplicate reads were marked and removed using Picard (https://github.com/broadinstitute/picard) and SAMtools. Alignment files for immunoprecipitated and non-immunoprecipitated control samples containing read pairs passing these criteria were then input to MACS2 (Zhang et al., 2008) for peak calling and to generate normalized, bedGraph-formatted read pileup profiles for visualization in the Integrative Genomics Viewer (Robinson et al., 2011). To reflect the differences in PCNA-based chromatin recovery, normalized sequencing data were downsampled accordingly relative to WT cells before plotting.
RNA isolation and RT-qPCR
Cells were harvested from exponentially growing cultures and the RNA was isolated using MasterPure Yeast RNA Purification Kit. The cDNA was prepared using SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific). The real-time qPCR was performed as described above using gene-specific primer pairs listed in Table S4. RFC1 was used as a reference gene and the data were analyzed using Thermo Fisher Scientific’s Relative Quantification (RQ) tool.
SCC assay
LacI-GFP/lac operator repeat strains (Mayer et al., 2001; Sanchez et al., 1999) were grown to log phase in YPD, cells were collected and washed with synthetic complete media lacking histidine. After washing, cells were resuspended in synthetic complete media containing 15 mM 3-amino-1,2,4-triazole (Sigma-Aldrich) and 5 µg/ml α-factor (T6901; Sigma-Aldrich), and grown for 2.5 h. After G1 arrest, cells were resuspended in YPD containing 15 µg/ml nocodazole (M1404; Sigma-Aldrich) to arrest them in G2/M. Cells were fixed in 4% paraformaldehyde, and the number of GFP spots in each cell was scored.
Fluorescence microscopy and quantification
Images were acquired on a DeltaVision Elite high-resolution microscope (GE Healthcare) equipped with a 100 × 1.4 NA objective lens (Olympus). Fluorescence excitation was driven by an Insight SSI solid-state light engine (Cytiva) and fluorescence emission was collected by a CoolSnap HQ2 CCD camera (Photometrics). Acquired images were deconvolved using Huygens Professional Software (Scientific Volume Imaging BV) using theoretically determined point spread functions. For the cell cycle colocalization experiment, cells coexpressing Ctf18-GFP and Nup188-mCherry were grown in YPD and arrested in G1 by adding α-factor. After washing the cells, a slide was prepared as described previously (Mast et al., 2016) and images were acquired at regular intervals for 110 min. Object-based colocalization analysis was performed using Imaris (Bitplane). The fluorescence signal from Nup188-mCherry was processed with the “Surface” function, and the Ctf18-GFP fluorescence signal was processed with the “Spots” function. The “Find Spots Close to Surface” function was used to identify Ctf18-GFP spots proximal to Nup188-mCherry. The “Spots Close to Surface” function finds spots that are closer to the surface than the user-specified threshold (0 μm). The distance is calculated as the minimal distance from the center of the spot to any point from the surface.
Statistical analysis
Statistical tests performed and the number of replicates are specified in the figure legends. When testing for differences between data sets, Shapiro–Wilk tests were used to determine whether both data sets were normally distributed. If so, t tests were used for the comparison; otherwise, Wilcoxon rank sum tests were used.
Online supplemental material
Fig. S1 contains SDS-PAGE gel image used for LC-MS; Venn diagram showing overlap between LC-MS hits, genetic interaction, and transcriptome analysis data; and immunoblots confirming the physical interaction of Nup170 with Ctf18 and Rfc3. Fig. S2 contains microscopy images and quantitation showing Ctf18-RFC interacting NPCs lack Mlp1 and Mlp2. Fig. S3 contains additional immunoblots showing that the Ctf18 interaction with the NPC peaks during S phase. Fig. S4 shows PCNA binding to all chromosomes in nup170Δ and WT. Fig. S5 contains various analyses showing functional relationships between Nup170, Ctf18-RFC complex, CAF-1 complex, and Elg1. Table S1 contains a list of Nup170 interacting candidates identified by MaxQuant. Table S2 contains 415 reported genetic interactors of Nup170 in BioGRID. Tables S3 and S4 contain lists of S. cerevisiae strains and RT-qPCR primers, respectively.
Data availability
The ChIP-seq data generated in this study have been deposited to the SRA database https://www.ncbi.nlm.nih.gov/sra with the accession number PRJNA917682. The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD041496.
Acknowledgments
We thank Philip Hieter from the University of British Columbia, Vancouver, BC, Canada, for providing Y819 strain and Jeff Ranish, Institute for Systems Biology, Seattle, WA, USA, for providing pJR200 plasmid. We thank Phil Gafken and Lisa Jones of Proteomics & Metabolomics Shared Resource of Fred Hutch for help in MS analysis. We thank Paul Olivier and other members of the Aitchison laboratory for thoughtful discussions.
This work was supported by grants P41GM109824 and R01 GM112108 from the National Institutes of Health to J.D. Aitchison and M.P. Rout.
Author contributions: S.K. Choudhry: conceptualization, methodology, investigation, data curation, project administration, writing of original draft, reviewing, and editing; M L. Neal: investigation, methodology, formal analysis, reviewing, and editing; S. Li and T. Van Eeuwen: investigation; A.T. Navare and F.M. Mast: investigation, reviewing, and editing; R.W. Wozniak: investigation, reviewing, and editing; M.P. Rout: funding acquisition, supervision, reviewing, and editing; J.D. Aitchison: conceptualization, methodology, supervision, project administration, funding acquisition, writing of original draft, reviewing, and editing.

