


Integrin endocytosis is essential for many fundamental cellular processes. Whether and how the internalization impacts cellular mechanics remains elusive. Whereas previous studies reported the contribution of the integrin activator, talin, in force development, the involvement of inhibitors is less documented. We identified ICAP-1 as an integrin inhibitor involved in mechanotransduction by co-working with NME2 to control clathrin-mediated endocytosis of integrins at the edge of focal adhesions (FA). Loss of ICAP-1 enables β3-integrin-mediated force generation independently of β1 integrin. β3-integrin-mediated forces were associated with a decrease in β3 integrin dynamics stemming from their reduced diffusion within adhesion sites and slow turnover of FA. The decrease in β3 integrin dynamics correlated with a defect in integrin endocytosis. ICAP-1 acts as an adaptor for clathrin-dependent endocytosis of integrins. ICAP-1 controls integrin endocytosis by interacting with NME2, a key regulator of dynamin-dependent clathrin-coated pits fission. Control of clathrin-mediated integrin endocytosis by an inhibitor is an unprecedented mechanism to tune forces at FA.
Introduction
Adhesive receptors, most notably integrins, help cells to perceive their microenvironment by sensing chemical, physical, and mechanical cues of extracellular matrix (ECM) through adhesive machineries called focal adhesions (FA), which act in concert with the actomyosin-based contractility system (Albiges-Rizo et al., 2009; Engler et al., 2006). Actomyosin-mediated contractility is a highly conserved mechanism for generating mechanical stress and governing cell shape, cell migration, cell differentiation, and morphogenesis (Murrell et al., 2015). Cellular contractility usually proceeds with the engagement of α5β1 and αvβ3 integrins in the context of cells exploring fibronectin (Fn)-based microenvironments (Schiller et al., 2013). These two integrins cooperate for the fine tuning of FA lifetime and adhesion strength in response to mechanical forces (Kuo et al., 2011; Schiller et al., 2013, 2011; Zamir et al., 2000; Rossier et al., 2012; Roca-Cusachs et al., 2009; Milloud et al., 2017; De Mets et al., 2019). Reinforcement of FA and cell contractility is likely coupled to the inhibition of FA disassembly, a processes involving FA targeting by microtubules, enhanced integrin endocytosis, calpain-mediated cleavage of talin, and loss of tension upon Rho kinase inhibition (Wehrle-Haller, 2012). α5β1 and αvβ3 integrins are functionally distinct, yet it remains unclear how these two Fn-binding integrin receptors are coordinately regulated to orchestrate assembly or disassembly of adhesion sites, to adjust adhesion lifetime, to adapt adhesion strength and force to the cellular environment.
The regulation of integrin function can be achieved at several levels, including ECM ligand engagement and recruitment of intracellular adaptors including integrin activators such as talin and integrin inhibitors like ICAP-1. These intracellular adaptors control integrin clustering and their activation switch, which is crucial for modulating integrin ligand-binding affinity and for serving as nucleation hubs for the assembly of larger signaling and structural scaffolds linked to actomyosin fibers (Legate and Fässler, 2009). Whereas talin is known to be important for building actin-bound mechanosensitive adhesive complexes (Klapholz and Brown, 2017; Shattil et al., 2010), the contribution of integrin inhibitors such as ICAP-1 in force transmission is still elusive.
ICAP-1 does not localize in FA, but it can accumulate at small adhesion sites or tips of filopodia (Jacquemet et al., 2019; Fournier et al., 2002, 2005; Millon-Frémillon et al., 2008). Thus, ICAP-1 interacts with partners described to be within and outside FA (Fournier et al., 2002; Millon-Frémillon et al., 2008). ICAP-1 interacts with the membrane-distal NPXY motif in the cytoplasmic tail of β1 integrin through a phosphotyrosine binding (PTB) domain (Zhang and Hemler, 1999; Degani et al., 2002). Further, it binds to the nucleoside diphosphate kinase NDPK/NME2/NM23-H2 (Fournier et al., 2002). Of note, ICAP-1, NME2, and β1 integrin are colocalized at the cell leading edge during the early stage of cell spreading on Fn, thereby suggesting a transitory function of this complex in adhesion site dynamics (Fournier et al., 2002). However, the biological relevance of ICAP-1/NME2 interaction in cell adhesion remains unknown. We have shown that ICAP-1 is a determinant for controlling FA dynamics since it impedes FA assembly by limiting both talin and kindlin interaction with β1 integrin (Millon-Frémillon et al., 2013; Millon-Frémillon et al., 2008). Furthermore, the physical interaction of ICAP-1 with the β1 integrin tail leading to the modulation of β1 integrin affinity state is required for down regulation of FA assembly. We have shown that ICAP-1 is involved in cell mechanical properties and cell differentiation in a β1 integrin-dependent manner (Brunner et al., 2011; Faurobert et al., 2013; Bouvard et al., 2007; Renz et al., 2015; Millon-Frémillon et al., 2008). We have also proposed ICAP-1 as a key modulator of cellular mechanoresponse in a β1-integrin–independent manner (Bouin et al., 2017). To delve deeper into the ICAP-1 functional properties, we interrogated β1-integrin–dependent and –independent contributions of ICAP-1 to the cell mechanical response. Using cellular models genetically engineered to lack only β1 integrin or ICAP-1 or both β1 integrin and ICAP-1 in conjunction with quantitative traction force microscopy, integrin dynamics, and proximity ligation assays, we provide converging evidence that points to a control by ICAP-1 of clathrin-mediated β3 integrin endocytosis and crucial tuning of cell mechanoresponses.
Results
The loss of ICAP-1 enables β3-integrin-mediated force generation independent of β1 integrin
To understand to what extent ICAP-1 regulates forces, we designed a quantitative traction force microscopy analysis of osteoblasts deleted for β1 integrin and/or ICAP-1. Deletion of β1 integrin or ICAP-1 in the respective cell lines was confirmed by Western blot (Fig. S1, A–C). Based on qPCR experiments and Western blot analysis (Fig. S1, D and E), we checked that the combined deletion of β1 integrin and ICAP-1 did not affect the total expression of β3 integrin. Cells were seeded on Fn-coated polyacrylamide hydrogels (PA hydro gels) with a Young’s modulus (E) of 5 kPa as previously described (Bouin et al., 2017). First, we observed that osteoblasts lacking β1 integrin (β1 integrin–/–-icap-1+/+) were defective in force generation (Fig. 1, A and B) similarly to what was reported for other cell lines (Danen et al., 2002; Schiller et al., 2013; Milloud et al., 2017). The β1 integrin-deficient cell line developed only 50% of the total forces generated by β1 integrin+/+- icap-1+/+ cells (Fig. 1 B). Secondly and unexpectedly, we found that additional loss of ICAP-1 in β1 integrin-deficient osteoblasts restored the traction force potential (Fig. 1, A and B). This result was confirmed by the rescue of myosin light-chain phosphorylation (pp-MLC) as judged by immunofluorescence staining (Fig. 1, C–E; and Fig. S1 F) and Western blot analysis (Fig. S1 G) using phospho site-specific antibodies against myosin light chain. In addition, while β1 integrin–/– cells displayed limited spreading as evidenced by the projected area of actin cytoskeleton, normal cell spreading was rescued in the double-mutant (β1 integrin–/–-icap-1–/–) cells that typically assembled thick actin stress fibers highly decorated with ppMLC (Fig. 1, C–E; and Fig. S1 F). Of note, the loss of ICAP-1 increased slightly force developed by osteoblasts although the difference with control cells was non-significant (Fig. 1, A and B). Contractility is an important aspect of cell migration, thus set out to explore the role of ICAP-1 in the adaptation of cell speed as a function of substrate stiffness. Cells were seeded on Fn-coated PA hydrogels of increasing rigidity and imaged for 3 h to monitor migration velocity (Fig. S2 A). β1 integrin+/+- icap-1+/+ and β1-deficient cells showed a stiffness-dependent increase in cell velocity. However, additional loss of ICAP-1 uncoupled the correlation between cell speed and substrate rigidity. As the first steps of Fn fibrillogenesis are known to depend on actomyosin contractility (Wu et al., 1995), we investigated whether the traction force observed in β1 integrin–/–-icap-1–/– cells was correlated with Fn fibrillogenesis. As previously described (Danen et al., 2002), the lack of β1 integrin drastically impaired the formation of a stretched meshwork of Fn fibrils (Fig. 1, F and G). However, additional loss of ICAP-1, which was associated with increased traction forces (Fig. 1 A), rescued the formation and organization of thick and numerous Fn fibrils connecting to large β3 integrin adhesions as compared to the short and less dense Fn fibrils observed in the case of β1 integrin deletion alone (Fig. 1, F and G).
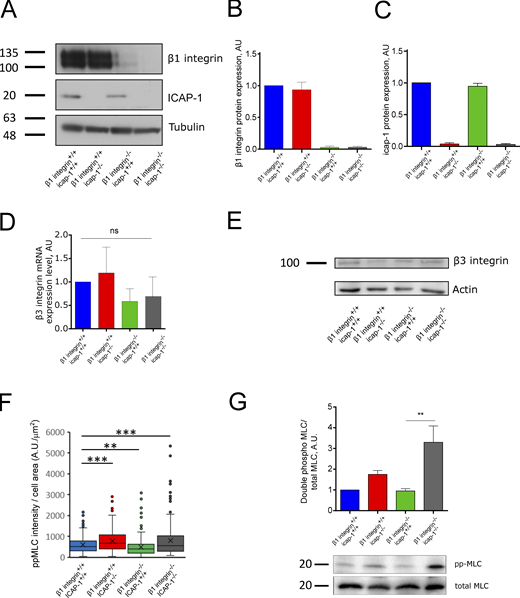
β3 integrin expression level is unchanged upon loss of β1 integrin. (A–C) Western blot of total cell lysate (A) and quantification (B and C) confirmed the deletion of β1 integrin or ICAP-1 in osteoblast cells. Tubulin is used as loading control. (E) The deletion of β1 integrin and ICAP-1 do not affect the expression of β3 integrin. Actin is used as loading control. (D) The expression of β3 integrin mRNA is not changed upon deletion of β1 integrin or ICAP-1. Luminescence signal is normalized to the β1 integrin+/+- icap-1+/+. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (F) Intensity of ppMLC staining decorating the stress fibers, normalized to cell area. Error bars represent SD. N ≥ 211 cells. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (G) The level of the double phosphorylation (T18/S19) of the MLC was assessed and quantified via Western blot against the total level of MLC of cell lysates of cells spread for 4 h on fibronectin-covered glass. Error bars represent SD. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. Source data are available for this figure: SourceData FS1.

β3 integrin expression level is unchanged upon loss of β1 integrin. (A–C) Western blot of total cell lysate (A) and quantification (B and C) confirmed the deletion of β1 integrin or ICAP-1 in osteoblast cells. Tubulin is used as loading control. (E) The deletion of β1 integrin and ICAP-1 do not affect the expression of β3 integrin. Actin is used as loading control. (D) The expression of β3 integrin mRNA is not changed upon deletion of β1 integrin or ICAP-1. Luminescence signal is normalized to the β1 integrin+/+- icap-1+/+. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (F) Intensity of ppMLC staining decorating the stress fibers, normalized to cell area. Error bars represent SD. N ≥ 211 cells. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (G) The level of the double phosphorylation (T18/S19) of the MLC was assessed and quantified via Western blot against the total level of MLC of cell lysates of cells spread for 4 h on fibronectin-covered glass. Error bars represent SD. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. Source data are available for this figure: SourceData FS1.

Osteoblasts are able to exert traction force on fibronectin-coated substrate in the absence of β1 integrin and ICAP-1. (A and B) Representative traction forces maps (A) and quantification (B) of the total force applied on fibronectin-coated polyacrylamide gel with a defined rigidity of 5 kPa. β1 integrin KO cells exert less force than the other osteoblasts mutants. The additional deletion of ICAP-1 led to generation of traction forces revealing a novel pathway independent of β1 integrins to generate traction forces on fibronectin. Scale bar, 10 μm. Error bars represent SD. N ≥ 60 cells. ns, adjusted P value >0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (C) Immunofluorescence staining of the ppMyosin (ppMLC antibody, red) and F-actin (phalloidin, green) in the four osteoblasts cell lines showed that deletion of ICAP-1 alone does not change the organization of acto-myosin cytoskeleton but increases slightly the intensity and the thickness of the stress fibers (see quantification of the ppMLC area in E). Deletion of β1 integrin leads to a decrease of the cell area (D) and disorganization and decrease of thickness and number of the ppMLC decorated stress fibers. Scale bar, 20 µm. (D) Quantified cell spreading area from staining of fluorescent F-actin. Error bars represent SD. N ≥ 429 cells. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (E) Quantified ppMLC staining, normalized to cell area. Error bars represent SD. N ≥ 211 cells. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (F) Osteoblasts cells were spread in serum-free medium on uncoated glass for 24 h. Immunofluorescence staining of the extracellular fibronectin (cellular fibronectin antibody, red), F-actin (phalloidin, blue) and β3 integrins (Luc.A5 antibody, green) in the four osteoblasts cell lines were analyzed by fluorescent confocal microscopy. The β1+/+ cell lines orchestrate FN fibrillogenesis events and β1 integrin−/−/icap-1+/+ cells demonstrated very poor organization of synthetized fibronectin. β1 integrin−/−/icap-1−/− on the other side showed significant amount of FN fibrillogenesis. Scale bar, 20 µm. (G) FN fibrils length of thresholded images of deposited and organized FN fibrillogenesis were processed and quantified. 20 images per condition were analyzed. Quantifications reveal that the additional loss of ICAP-1 in β1 KO cell line increases the fibril length compared to the β1 KO cell line, expressing ICAP-1. Error bars represent SD. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001.
Osteoblasts are able to exert traction force on fibronectin-coated substrate in the absence of β1 integrin and ICAP-1. (A and B) Representative traction forces maps (A) and quantification (B) of the total force applied on fibronectin-coated polyacrylamide gel with a defined rigidity of 5 kPa. β1 integrin KO cells exert less force than the other osteoblasts mutants. The additional deletion of ICAP-1 led to generation of traction forces revealing a novel pathway independent of β1 integrins to generate traction forces on fibronectin. Scale bar, 10 μm. Error bars represent SD. N ≥ 60 cells. ns, adjusted P value >0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (C) Immunofluorescence staining of the ppMyosin (ppMLC antibody, red) and F-actin (phalloidin, green) in the four osteoblasts cell lines showed that deletion of ICAP-1 alone does not change the organization of acto-myosin cytoskeleton but increases slightly the intensity and the thickness of the stress fibers (see quantification of the ppMLC area in E). Deletion of β1 integrin leads to a decrease of the cell area (D) and disorganization and decrease of thickness and number of the ppMLC decorated stress fibers. Scale bar, 20 µm. (D) Quantified cell spreading area from staining of fluorescent F-actin. Error bars represent SD. N ≥ 429 cells. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (E) Quantified ppMLC staining, normalized to cell area. Error bars represent SD. N ≥ 211 cells. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (F) Osteoblasts cells were spread in serum-free medium on uncoated glass for 24 h. Immunofluorescence staining of the extracellular fibronectin (cellular fibronectin antibody, red), F-actin (phalloidin, blue) and β3 integrins (Luc.A5 antibody, green) in the four osteoblasts cell lines were analyzed by fluorescent confocal microscopy. The β1+/+ cell lines orchestrate FN fibrillogenesis events and β1 integrin−/−/icap-1+/+ cells demonstrated very poor organization of synthetized fibronectin. β1 integrin−/−/icap-1−/− on the other side showed significant amount of FN fibrillogenesis. Scale bar, 20 µm. (G) FN fibrils length of thresholded images of deposited and organized FN fibrillogenesis were processed and quantified. 20 images per condition were analyzed. Quantifications reveal that the additional loss of ICAP-1 in β1 KO cell line increases the fibril length compared to the β1 KO cell line, expressing ICAP-1. Error bars represent SD. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001.

ICAP-1 is required for cells to adapt cell velocity as function of substrate stiffness independently on the presence of β1 integrin and is involved in β3 integrin FA translocation to the cell center. (A) Osteoblast cells were spread on FN-coated PAA gels of different rigidities (3, 15, and 60 kPa). Cell migration was monitored for 5 h using time-lapse microscopy. The cell velocity was determined by individually tracking 200–300 cells in three independent experiments. ICAP-1 deficient cells displayed a constant migration velocity whatever the substrate rigidity as compared to the β1 integrin+/+- icap-1+/+ cells, independently of the presence of β1 integrin highlighting the crucial role of ICAP-1 in rigidity sensing. Error bars represent SD. N ≥ 199 cells. *, P < 0.05; **, P < 0.005; ***, P < 0.0005. (B) Note that the additional deletion of ICAP-1 in cells depleted in β1 integrin induces an increase of β3 integrin FA translocation to the cell center. Quantification of the percentage of FA whose distance to the nearest cell contour point is >12.5% of the average diameter of the cell. Error bars represent SD. N ≥ 208 cells. ****, P value ≤ 0.0001.

ICAP-1 is required for cells to adapt cell velocity as function of substrate stiffness independently on the presence of β1 integrin and is involved in β3 integrin FA translocation to the cell center. (A) Osteoblast cells were spread on FN-coated PAA gels of different rigidities (3, 15, and 60 kPa). Cell migration was monitored for 5 h using time-lapse microscopy. The cell velocity was determined by individually tracking 200–300 cells in three independent experiments. ICAP-1 deficient cells displayed a constant migration velocity whatever the substrate rigidity as compared to the β1 integrin+/+- icap-1+/+ cells, independently of the presence of β1 integrin highlighting the crucial role of ICAP-1 in rigidity sensing. Error bars represent SD. N ≥ 199 cells. *, P < 0.05; **, P < 0.005; ***, P < 0.0005. (B) Note that the additional deletion of ICAP-1 in cells depleted in β1 integrin induces an increase of β3 integrin FA translocation to the cell center. Quantification of the percentage of FA whose distance to the nearest cell contour point is >12.5% of the average diameter of the cell. Error bars represent SD. N ≥ 208 cells. ****, P value ≤ 0.0001.
These results indicate that ICAP-1 plays a role in cellular mechanotransduction and that traction strength on a compliant Fn substrate does not strictly correlate with the presence of β1 integrin and is likely related to another integrin receptor regulated by ICAP-1. As Fn-coated surface mediates RGD binding to α5β1 and αvβ3 integrins (Leiss et al., 2008) which can exert both specific and redundant functions (Ballestrem et al., 2001; Danen et al., 2002), we investigated whether the increase in traction forces in β1 integrin−/−-icap-1−/− cells was dependent on β3 integrin engagement. Silencing of β3 integrin by RNA interference (Fig. 2 F) not only led to a significant decrease in cell spreading and ppMLC staining (Fig. 2, A and B) but also abolished traction forces generated in β1 integrin−/−-icap-1−/− cells when compared to β1 integrin+/+- icap-1+/+ osteoblasts (Fig. 2, C and D). These data position β3 integrin as a major driver for the spreading and tensile phenotypes observed on β1 integrin−/−-icap-1−/− cells. In line with this scheme, the contractile phenotype of β1 integrin−/−-icap-1−/− cells was correlated with an increase in β3 integrin expression at the cell surface as judged by FACS analysis (Fig.2 E) while the total amount of β3 integrin remained unchanged (Fig. S1, D and E). Altogether, our results suggest a new regulatory role for ICAP-1 in actomyosin contractility and force generation through β3 integrin.

ppMyosin area and traction force are dependent on β3 integrin and ICAP-1 expression. (A) β1 integrin KO osteoblast cell lines were treated with β3 integrin siRNA or scramble siRNA for 48 h and then let spread on FN coated glass for 24 h. Staining of myosin phosphorylation (ppMLC antibody, red) and β3 integrin (LucA.5, green) was performed and analyzed by fluorescent confocal microscopy. Silencing the expression of β3 integrin leads to the complete abolishment of the ppMLC-decorated stress fibers in the β1−/−/icap-1−/− and shrinkage of the cell area. Scale bar, 20 µm. (B) Quantification of the ppMLC area normalized to the cellular surface area after treatment with β3 integrin siRNA (si β3, clear colors) or with scramble siRNA (si Scr, dark colors). Customized particle analysis script from ImageJ was used after application of Unsharpen mask and Despecle filters. The error bars represent SD. N ≥ 42 cells. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (C and D) Representative traction forces maps (TFM) from β1 integrin+/+-icap-1−/− and β1 integrin−/−-icap-1−/− cell lines treated with β3 integrin siRNA (siβ3, bottom panels) or with scramble siRNA (si Scr, upper panels) and quantification (D) of the total force applied (nN) on fibronectin-coated polyacrylamide gel with a defined rigidity of 5 kPa. The additional silencing of β3 integrins in the β1−/−/icap-1−/− cells decimates the traction forces, confirming that, in absence of β1 integrin and ICAP-1, generation of strong cellular contractility at adhesion sites is dependent on β3 integrins. Scale bar, 10 μm. Error bars represent SD. N = 80 cells. ****, P value ≤ 0.0001. (E) FACS analysis of median fluorescence intensity of β3 integrin on cell surface in β1 integrin KO osteoblast cell lines. Error bars represent SD. (F) Western blot showing the efficiency of β3 integrin silencing in β1 integrin+/+-icap-1−/− and β1 integrin−/−-icap-1−/− cell lines. Source data are available for this figure: SourceData F2.
ppMyosin area and traction force are dependent on β3 integrin and ICAP-1 expression. (A) β1 integrin KO osteoblast cell lines were treated with β3 integrin siRNA or scramble siRNA for 48 h and then let spread on FN coated glass for 24 h. Staining of myosin phosphorylation (ppMLC antibody, red) and β3 integrin (LucA.5, green) was performed and analyzed by fluorescent confocal microscopy. Silencing the expression of β3 integrin leads to the complete abolishment of the ppMLC-decorated stress fibers in the β1−/−/icap-1−/− and shrinkage of the cell area. Scale bar, 20 µm. (B) Quantification of the ppMLC area normalized to the cellular surface area after treatment with β3 integrin siRNA (si β3, clear colors) or with scramble siRNA (si Scr, dark colors). Customized particle analysis script from ImageJ was used after application of Unsharpen mask and Despecle filters. The error bars represent SD. N ≥ 42 cells. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (C and D) Representative traction forces maps (TFM) from β1 integrin+/+-icap-1−/− and β1 integrin−/−-icap-1−/− cell lines treated with β3 integrin siRNA (siβ3, bottom panels) or with scramble siRNA (si Scr, upper panels) and quantification (D) of the total force applied (nN) on fibronectin-coated polyacrylamide gel with a defined rigidity of 5 kPa. The additional silencing of β3 integrins in the β1−/−/icap-1−/− cells decimates the traction forces, confirming that, in absence of β1 integrin and ICAP-1, generation of strong cellular contractility at adhesion sites is dependent on β3 integrins. Scale bar, 10 μm. Error bars represent SD. N = 80 cells. ****, P value ≤ 0.0001. (E) FACS analysis of median fluorescence intensity of β3 integrin on cell surface in β1 integrin KO osteoblast cell lines. Error bars represent SD. (F) Western blot showing the efficiency of β3 integrin silencing in β1 integrin+/+-icap-1−/− and β1 integrin−/−-icap-1−/− cell lines. Source data are available for this figure: SourceData F2.
β3-integrin-dependent traction force is associated with redistribution and higher lifetime of enlarged β3-integrin-positive focal adhesions
Stabilization of αvβ3 integrin-Fn bonds through actomyosin-mediated tension is required for cells to adjust cell contractility to the substrate stiffness (Schiller et al., 2013; De Mets et al., 2019). Based on the increase in surface β3 integrin expression levels in β1 integrin−/−-icap-1−/− cells (Fig. 2 E), we investigated whether the contractile behavior mediated by β3 integrin upon the loss of β1 integrin and ICAP-1 might be related to a change in β3 integrin-containing FA organization. All four osteoblast cells lines were able to form β3 integrin FAs on an Fn-coated substratum as revealed by immunostaining of β3 integrin (Fig. 3 A). Nevertheless, the deletion of β1 integrin noticeably reduced cell spreading (Fig. 1 D) without affecting the adhesion area of FAs occupied by β3 integrin even though it total adhesion area was increased with respect to the cell area (Fig. 3, A–C). These results suggest that β3 integrin alone failed to support the typical spreading of osteoblasts bound to Fn. Remarkably, the additional loss of ICAP-1 in β1-integrin-deficient cells restored cell spreading and increased the mean size of β3 integrin FAs when compared to β1 integrin null cells (Fig. 3, A and C). We noticed an increase of β3 integrin FA translocation to the cell center (Fig. S2 B) which is compatible with the rescue of Fn fibril organization in β1 integrin−/−-icap-1−/− cells (Fig. 1, F and G). These results revealed a major role for ICAP-1 in controlling β3 integrin clustering and function in the absence of β1 integrin. Next, we analyzed whether the contractile behavior mediated by β3 integrin upon the loss of β1 integrin and ICAP-1 might be linked to a change in β3-integrin-containing FA dynamics. To this end, the turnover of FA in osteoblast cell lines transfected with β3 integrin-eGFP was monitored in real time (Fig. 3 D). The life time of β3 integrin-eGFP FAs increased in association with their translocation to the cell center in β1 integrin−/−-icap-1−/− cells (Fig. 3, D and E; and Fig. S2 B).

Size and dynamic of β3 integrin FAs are dependent on β1 integrins and ICAP-1. (A) Staining area of β3 integrins was carried out on osteoblast cells spread on fibronectin-coated coverglass for 4 h using LucA5 antibody. Scale bar, 20 µm. (B) Quantification of the adhesive area of β3 integrins FA, normalized to the cellular surface area. Error bars represent SD. N ≥ 208 cells. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (C) Quantification of the mean of β3 integrin FAs area. The deletion of ICAP-1 in β1 deficient cell line (grey box) drives massive leap of β3 integrin stained FAs size. Error bars represent SD. N ≥ 208 cells. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (D) Representative time series of the lifetime of eGFP-β3 integrin FAs of the four osteoblastic cell lines. Asterix points out typical eGFP-β3 integrin FAs in the cells with typical disassembly lifetime. Note the translocation of FA to the cell center in β1 integrin−/−/icap-1−/− cells. Scale bar, 2 µm. (E) TIRF lifetime analysis on the eGFP-β3 integrin FAs. The lifetime of the eGFP-β3 integrin containing FA is increased in β1 integrin−/−/icap-1−/− cells. Spinning disk videos of eGFP-β3 integrin were taken for the duration of 2 h. The adhesion lifetime was analyzed by Focal Adhesion Analysis Server (FAAS; Berginski and Gomez, 2013) and verified visually. Error bars represent SD. N ≥ 50 focal adhesions. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (F) FRAP analysis on the eGFP-β3 integrin FAs reveal that the β3 integrin mobility at the plasma membrane is halted in the absence of ICAP-1 and β1 integrin. At least six FAs per cell lines were bleached and their recovery was monitored for 5 min. Error bars represent SD. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001.
Size and dynamic of β3 integrin FAs are dependent on β1 integrins and ICAP-1. (A) Staining area of β3 integrins was carried out on osteoblast cells spread on fibronectin-coated coverglass for 4 h using LucA5 antibody. Scale bar, 20 µm. (B) Quantification of the adhesive area of β3 integrins FA, normalized to the cellular surface area. Error bars represent SD. N ≥ 208 cells. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (C) Quantification of the mean of β3 integrin FAs area. The deletion of ICAP-1 in β1 deficient cell line (grey box) drives massive leap of β3 integrin stained FAs size. Error bars represent SD. N ≥ 208 cells. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (D) Representative time series of the lifetime of eGFP-β3 integrin FAs of the four osteoblastic cell lines. Asterix points out typical eGFP-β3 integrin FAs in the cells with typical disassembly lifetime. Note the translocation of FA to the cell center in β1 integrin−/−/icap-1−/− cells. Scale bar, 2 µm. (E) TIRF lifetime analysis on the eGFP-β3 integrin FAs. The lifetime of the eGFP-β3 integrin containing FA is increased in β1 integrin−/−/icap-1−/− cells. Spinning disk videos of eGFP-β3 integrin were taken for the duration of 2 h. The adhesion lifetime was analyzed by Focal Adhesion Analysis Server (FAAS; Berginski and Gomez, 2013) and verified visually. Error bars represent SD. N ≥ 50 focal adhesions. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (F) FRAP analysis on the eGFP-β3 integrin FAs reveal that the β3 integrin mobility at the plasma membrane is halted in the absence of ICAP-1 and β1 integrin. At least six FAs per cell lines were bleached and their recovery was monitored for 5 min. Error bars represent SD. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001.
β3 integrin exchange rates were further analyzed by fluorescent recovery after photobleaching (FRAP) using total internal reflection fluorescent (TIRF) microscopy on osteoblasts lines transfected with β3 integrin-eGFP (Fig. 3 F). Neither single loss of β1 integrin nor that of ICAP-1 had any significantly effect on β3 integrin exchange rate in FAs. In contrast, a threefold slower β3 integrin-eGFP exchange rate typified the oversized β3 integrin FAs observed in double β1 integrin−/−-icap-1−/− cells (Fig. 3 F), as compared to β1 null cells. Finally, the slower β3 integrin exchange rate was associated with an increase of β3 integrin-eGFP FA life time (Fig. 3, D–F; and Videos 1, 2, 3, and 4). In the context of β1 null cells, these data hint that the loss of ICAP-1 strongly affects the clustering and dynamics of β3 integrin in FAs. These findings reveal that the control of β3 integrin clustering and dynamics by ICAP-1 might modulate traction force generation.
β1 integrin+/+-icap-1+/+cells expressing β3 integrin-GFP were spread in 2-well LabTeks and left to spread for 4 h. Spinning disk videos were taken for the length of 2 h with 1 min frequency. Playback speed, 1 frame/s.
β1 integrin+/+-icap-1+/+cells expressing β3 integrin-GFP were spread in 2-well LabTeks and left to spread for 4 h. Spinning disk videos were taken for the length of 2 h with 1 min frequency. Playback speed, 1 frame/s.
β1 integrin+/+-icap-1−/−cells expressing β3 integrin-GFP were spread in 2-well LabTeks and left to spread for 4 h. Spinning disk videos were taken for the length of 2 h with 1 min frequency. Playback speed, 1 frame/s.
β1 integrin+/+-icap-1−/−cells expressing β3 integrin-GFP were spread in 2-well LabTeks and left to spread for 4 h. Spinning disk videos were taken for the length of 2 h with 1 min frequency. Playback speed, 1 frame/s.
β1 integrin−/−-icap-1+/+cells expressing β3 integrin-GFP were spread in 2-well LabTeks and left to spread for 4 h. Spinning disk videos were taken for the length of 2 h with 1 min frequency. Playback speed, 1 frame/s.
β1 integrin−/−-icap-1+/+cells expressing β3 integrin-GFP were spread in 2-well LabTeks and left to spread for 4 h. Spinning disk videos were taken for the length of 2 h with 1 min frequency. Playback speed, 1 frame/s.
β1 integrin−/−-icap-1−/−cells expressing β3 integrin-GFP were spread in 2-well LabTeks and left to spread for 4 h. Spinning disk videos were taken for the length of 2 h with 1 min frequency. Playback speed, 1 frame/s.
β1 integrin−/−-icap-1−/−cells expressing β3 integrin-GFP were spread in 2-well LabTeks and left to spread for 4 h. Spinning disk videos were taken for the length of 2 h with 1 min frequency. Playback speed, 1 frame/s.
β3-integrin-dependent contractility is associated with a defect of clathrin-mediated β3 integrin endocytosis
As endocytic membrane traffic regulates the availability of cell-surface receptors and associated signaling (Ceresa and Schmid, 2000; Scita and Di Fiore, 2010), we hypothesized that the concomitant increase in β3 integrin surface expression (Fig. 2 E) and the decrease in β3 integrin dynamics in β1/ICAP-1 double-mutant cells (Fig. 3, D–F) might stem from a defect in β3 integrin endocytosis. To test this hypothesis, we monitored the uptake of anti-integrin β3 antibodies by cells plated on an Fn substrate using confocal microscopy. Uptake was measured using β3 integrin-specific antibody (LucA.5) coupled with pH-rodo, which becomes fluorescent in the acidic environment of endocytic vesicles (Fig. 4 A). Quantification of intracellular vesicles positive for internalized β3 integrin (LucA5-positive) revealed a significant decrease in uptake (23%) in the absence ICAP-1 as compared to β1 integrin+/+- icap-1+/+ cells. An even more significant decrease (40%) was measured in osteoblasts deficient for both β1 integrin and ICAP-1 when compared to cells depleted in β1 integrin only (Fig. 4 A). Past studies have demonstrated that surface β3 integrins are constitutively internalized by clathrin-mediated endocytosis that depends on the AP-2 clathrin adaptor complex (Arjonen et al., 2012; Yu et al., 2015; Ezratty et al., 2009). Moreover, the large GTPase dynamins are known to be required for the scission of newly formed clathrin-coated vesicles (CCV) from the plasma membrane clathrin-coated pits (CCP). We applied proximity ligation assay (PLA), which allows the detection of close proximity, not only between ICAP-1 and β3 integrin (Fig. 4 B) but also between ICAP-1 and CCP components such as the α-adaptin of the clathrin adaptor complex, AP-2 and dynamin-2 (Fig. 4 C), supporting a role for ICAP-1 in β3 integrin endocytosis. Indeed, the decrease of β3 integrin endocytosis in the case of ICAP-1 deletion was associated with an increase of CCP containing β1 and β3 integrins (Fig. 4 D), specifying the contribution of ICAP-1 in the formation of CCVs.

The β3 integrin dependent contractility is associated with the defect of β3 integrin endocytosis. (A) The β3 integrin uptake was measured using β3 integrin specific antibody (LucA.5), coupled with pH-Rhodo. The volume of the endocytotic vesicles per cell volume was measured at 37 and 4°C. Note the decrease of β3 integrin endocytosis in cell lines deleted in ICAP-1. The quantification was performed after 45 min incubation. Error bars represent SD. N ≥ 127 cells. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (B) Representative images of PLA performed with antibodies against ICAP-1 and β3 integrin. Red dots denote regions of signal amplification indicating the close proximity between ICAP-1 and β3 integrin. PLA performed on ICAP-1 deficient cell line is used as control. Nuclei are stained in blue with DAPI. (C) Representative images of proximity ligation assays performed with antibodies against ICAP-1 and AP2 or Dynamin 2. Red dots denote regions of signal amplification indicating that ICAP-1 belongs to clathrin endocytosis machinery. PLA performed on ICAP-1 deficient cell line is used as control. Nuclei are stained in blue with DAPI. Scale bar, 20 µm. (D) PLA performed with antibodies against AP2 and β1 integrin or β3 integrin and quantification of the number of PLA spots per cell shows that deficiency in ICAP-1 leads to increase of the association of β1 and β3 integrins with AP2. N ≥ 25 cells/condition from three independent experiments. Scale bars, 20 µm. Error bars represent SD. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (E) Immunofluorescence staining of the phospho-myosin (ppMLC antibody, red) and β3 integrin (LucA.5 antibody, green). Representative micrographs of the four osteoblast cell lines treated with clathrin siRNA (si Clat, right) or scramble siRNA (si Scr, left). Inhibition of clathrin expression in β1 integrin−/−/icap-1+/+ cell line rescues cell spreading through β3 integrin-mediated FA and development of acto-myosin cytoskeleton (see also graphs F–H). Scale bar, 20 µm. (F) Quantification of cell spreading area of the four osteoblast cell lines treated with clathrin siRNA (si Clathrin, light colors) or scramble siRNA (si Scr, dark colors). Error bars represent SD. N ≥ 27 cells. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (G) Quantification of area of β3 integrin containing FAs normalized to the cellular surface area of the four osteoblast cell lines treated with clathrin siRNA (si Clathrin, light colors) or scramble siRNA (si Scr, dark colors). Error bars represent SD. N ≥ 27 cells. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (H) Quantification of phospho-myosin staining area normalized to the cellular surface area of the four osteoblast cell lines treated with clathrin siRNA (si Clathrin, light colors) or scramble siRNA (si Scr, dark colors). Error bars represent SD. N ≥ 27 cells. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001.
The β3 integrin dependent contractility is associated with the defect of β3 integrin endocytosis. (A) The β3 integrin uptake was measured using β3 integrin specific antibody (LucA.5), coupled with pH-Rhodo. The volume of the endocytotic vesicles per cell volume was measured at 37 and 4°C. Note the decrease of β3 integrin endocytosis in cell lines deleted in ICAP-1. The quantification was performed after 45 min incubation. Error bars represent SD. N ≥ 127 cells. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (B) Representative images of PLA performed with antibodies against ICAP-1 and β3 integrin. Red dots denote regions of signal amplification indicating the close proximity between ICAP-1 and β3 integrin. PLA performed on ICAP-1 deficient cell line is used as control. Nuclei are stained in blue with DAPI. (C) Representative images of proximity ligation assays performed with antibodies against ICAP-1 and AP2 or Dynamin 2. Red dots denote regions of signal amplification indicating that ICAP-1 belongs to clathrin endocytosis machinery. PLA performed on ICAP-1 deficient cell line is used as control. Nuclei are stained in blue with DAPI. Scale bar, 20 µm. (D) PLA performed with antibodies against AP2 and β1 integrin or β3 integrin and quantification of the number of PLA spots per cell shows that deficiency in ICAP-1 leads to increase of the association of β1 and β3 integrins with AP2. N ≥ 25 cells/condition from three independent experiments. Scale bars, 20 µm. Error bars represent SD. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (E) Immunofluorescence staining of the phospho-myosin (ppMLC antibody, red) and β3 integrin (LucA.5 antibody, green). Representative micrographs of the four osteoblast cell lines treated with clathrin siRNA (si Clat, right) or scramble siRNA (si Scr, left). Inhibition of clathrin expression in β1 integrin−/−/icap-1+/+ cell line rescues cell spreading through β3 integrin-mediated FA and development of acto-myosin cytoskeleton (see also graphs F–H). Scale bar, 20 µm. (F) Quantification of cell spreading area of the four osteoblast cell lines treated with clathrin siRNA (si Clathrin, light colors) or scramble siRNA (si Scr, dark colors). Error bars represent SD. N ≥ 27 cells. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (G) Quantification of area of β3 integrin containing FAs normalized to the cellular surface area of the four osteoblast cell lines treated with clathrin siRNA (si Clathrin, light colors) or scramble siRNA (si Scr, dark colors). Error bars represent SD. N ≥ 27 cells. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (H) Quantification of phospho-myosin staining area normalized to the cellular surface area of the four osteoblast cell lines treated with clathrin siRNA (si Clathrin, light colors) or scramble siRNA (si Scr, dark colors). Error bars represent SD. N ≥ 27 cells. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001.
Given the link between ICAP-1 and clathrin-mediated endocytosis of β3 integrin, we questioned whether the loss of clathrin might impact cell spreading, like the loss of ICAP-1. As expected, whereas β1 integrin-deficient cells displayed a decrease in their spreading capacity compared to β1 integrin+/+- icap-1+/+ cells, additional knockdown of the clathrin heavy chain (Fig. S3) restored their ability to spread (Fig. 4, E and F), in conjunction with the formation of larger β3 integrin-positive FAs (Fig. 4, E and G) and ppMLC-enriched actin stress fibers (Fig. 4, E and H), thus phenocopying the effects of the ICAP-1 loss in those cells. Importantly, there was no additive effect of clathrin deletion over ICAP-1 loss (β1 integrin−/−-icap-1−/− cells and β1 integrin+/+-icap-1−/− cells) with respect to cell spreading, β3 integrin-positive FA size and ppMLC staining (Fig. 4, E–H). These data demonstrate that the loss of clathrin and resulting impairment in clathrin-mediated endocytosis impacts β3 integrin signaling. Accordingly, the increase in β3 integrin clustering concurs with the increase in cell spreading and reorganization of the actomyosin cytoskeleton. In addition, the non-sensitivity of cells devoid of ICAP-1 to alteration of the clathrin-based endocytic machinery and the close proximity of ICAP-1 with AP2 and dynamin support a potential role of ICAP-1 in clathrin-mediated integrin endocytosis.
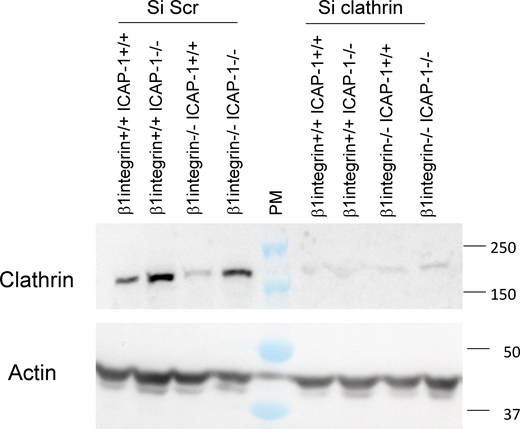
Efficiency of clathrin silencing in osteoblast cell lines. Clathrin and actin are visualized by Western blot after treatment with siRNA against clathrin as compared to Scramble conditions. Source data are available for this figure: SourceData FS3.

Efficiency of clathrin silencing in osteoblast cell lines. Clathrin and actin are visualized by Western blot after treatment with siRNA against clathrin as compared to Scramble conditions. Source data are available for this figure: SourceData FS3.
ICAP-1 controls β3 integrin endocytosis through NME-dependent scission of endocytic clathrin-coated pits
Next, we investigated the mechanism by which ICAP-1 might affect β3 integrin endocytosis. We have previously reported an interaction between ICAP-1 and nucleoside diphosphate kinase NME2 (Fournier et al., 2002), known to catalyze the synthesis of nucleoside triphosphates including GTP from nucleoside diphosphates and ATP (Boissan et al., 2018). Genetic and functional studies have demonstrated the ability of the related NME1 and NME2 NDPKs to fuel dynamin with GTP to support clathrin-dependent endocytosis (Boissan et al., 2014; Krishnan et al., 2001; Dammai et al., 2003; Nallamothu et al., 2008). Based on these findings, we investigated whether NME might impact β3 integrin dynamics. As NME2 can work in a hexameric complex with NME1 to be recruited to CCPs through their physical interaction with dynamin (Boissan et al., 2014), both NME1 and NME2 were knocked down in osteoblasts (Fig. 5 A). Deletion of NME1/2 in β1-integrin-depleted osteoblasts decreased β3 integrin–GFP turnover as evidenced by FRAP experiments (Fig. 5 B), thus recapitulating the deletion of ICAP-1. We further addressed whether ICAP-1 might impact the localization and function of NME2 before membrane scission. First, PLA showed that NME2 and α-adaptin as well as dynamin (Fig. 5, C and D) exist in close proximity to each other in agreement with the known functional link between NME and CCP (Boissan et al., 2014). Secondly, PLA confirmed the close proximity between NME2 and ICAP-1 in β1 integrin+/+- icap-1+/+ osteoblasts in contrast to ICAP-1–deficient cells (Fig. 5, C and D) confirming the proximity of these two proteins as previously observed based on classical immunofluorescence (Fournier et al., 2002). More importantly, we noticed a decrease of PLA signal between AP-2 and NME2 and between dynamin and NME2 upon ICAP-1 loss (Fig. 5, C and D), suggesting that ICAP-1 acts as a linker to connect NME2 and CCP components. Similarly, we found that the proximity between NME2 and β1 or β3 integrin was disrupted in the absence of ICAP-1 (Fig. 5, E and F), indicating the requirement for ICAP-1 in keeping β integrin and NME2 in close vicinity. Interestingly, ICAP-1 loss had a negligible effect on transferrin receptor/NME2 PLA signal, confirming the specificity of ICAP-1’s function in maintaining NME2/integrin proximity (Fig. 5, E and F). In addition, we detected specific interactions between NME1/2 and ICAP-1, β1, and β3 occurring at α-adaptin–marked CCPs in β1 integrin+/+- icap-1+/+ osteoblasts (Fig. S4 A).

ICAP-1 is required for NME recruitment in clathrin-coated pits and for keeping the proximity between integrin and NME. (A) Western blot analysis showing the efficiency of SiNME1/2 in osteoblast cells. (B) TIRF/FRAP analysis shows that deletion of NME1/2 complex by siRNAs (siNME1/2, light colors) or scramble siRNA (si Scr, dark colors) impedes the turnover of eGFP-β3 integrins at the plasma membrane in β1 integrin−/−/icap-1+/+ cell line. Six FAs per cell were bleached for each experiment. eGFP-β3 integrin recovery was monitored for 5 min. 19 cells ≤ N ≤ 107 cells. Error bars represent SD. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (C and D) Representative images of PLAs (C) and PLA assay quantification (D) of the number of PLA spots per cell performed with antibodies against NME and ICAP-1 or AP2 or Dynamin 2. Red dots denote regions of signal amplification consistent with NME/ICAP-1 interaction, NME/AP2 interaction, and NME/dynamin 2 interaction in β1 integrin+/+- icap-1+/+ osteoblast cells (left). PLA performed on ICAP-1 deficient cell line is used as control. The deletion of ICAP-1 induces a decrease of red dots in all three cases indicating the crucial role of ICAP-1 for keeping NME in clathrin endocytosis machinery (right). Nuclei are stained in blue with DAPI. Scale bar, 20 µm. Error bars represent SD. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (E and F) Representative images (E) of PLAs with quantification (F) performed with antibodies against NME and β1 integrin or β3 integrin or transferrin receptor. Red dots denote regions of signal amplification consistent with NME/integrin proximity in β1 integrin+/+- icap-1+/+ osteoblast cells (left). The deletion of ICAP-1 induces a decrease of the number of red dots indicating the crucial role of ICAP-1 for keeping NME and integrin vicinity (right). PLA performed on ICAP-1 deficient cell line is used as control. Nuclei are stained in blue with DAPI. Scale bar, 20 µm. Error bars represent SD. N ≥ 25 cells/condition from three independent experiments. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. Source data are available for this figure: SourceData F5.
ICAP-1 is required for NME recruitment in clathrin-coated pits and for keeping the proximity between integrin and NME. (A) Western blot analysis showing the efficiency of SiNME1/2 in osteoblast cells. (B) TIRF/FRAP analysis shows that deletion of NME1/2 complex by siRNAs (siNME1/2, light colors) or scramble siRNA (si Scr, dark colors) impedes the turnover of eGFP-β3 integrins at the plasma membrane in β1 integrin−/−/icap-1+/+ cell line. Six FAs per cell were bleached for each experiment. eGFP-β3 integrin recovery was monitored for 5 min. 19 cells ≤ N ≤ 107 cells. Error bars represent SD. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (C and D) Representative images of PLAs (C) and PLA assay quantification (D) of the number of PLA spots per cell performed with antibodies against NME and ICAP-1 or AP2 or Dynamin 2. Red dots denote regions of signal amplification consistent with NME/ICAP-1 interaction, NME/AP2 interaction, and NME/dynamin 2 interaction in β1 integrin+/+- icap-1+/+ osteoblast cells (left). PLA performed on ICAP-1 deficient cell line is used as control. The deletion of ICAP-1 induces a decrease of red dots in all three cases indicating the crucial role of ICAP-1 for keeping NME in clathrin endocytosis machinery (right). Nuclei are stained in blue with DAPI. Scale bar, 20 µm. Error bars represent SD. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. (E and F) Representative images (E) of PLAs with quantification (F) performed with antibodies against NME and β1 integrin or β3 integrin or transferrin receptor. Red dots denote regions of signal amplification consistent with NME/integrin proximity in β1 integrin+/+- icap-1+/+ osteoblast cells (left). The deletion of ICAP-1 induces a decrease of the number of red dots indicating the crucial role of ICAP-1 for keeping NME and integrin vicinity (right). PLA performed on ICAP-1 deficient cell line is used as control. Nuclei are stained in blue with DAPI. Scale bar, 20 µm. Error bars represent SD. N ≥ 25 cells/condition from three independent experiments. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001. Source data are available for this figure: SourceData F5.

NME/β3 integrin interaction occurs at α-adaptin–marked CCPs and does not need β1 integrin in osteoblasts. (A) PLA signal using NME antibody in combination with ICAP-1 antibody (4d1d6) or ICAP-1 antibody (9b10) or β3 integrin antibody or β1 integrin antibody in WT osteoblasts. Immunolabeling of α-adaptin was performed after PLA. Insets are higher magnification of boxed regions. Insets show PLA signals (NME/ICAP-1 or NME/β1 integrin or NME/β3 integrin) colocalizing with α-adaptin-positive CCPs (arrowheads). PLA signal indicates a close proximity, possibly in situ interaction of NME with ICAP-1, β1, and β3 integrins at CCPs. Scale bar, 10 μm. Inset scale bar, 5 μm. (B and C) Representative images of PLAs (B) and PLA assay quantification (C) of the number of PLA spots per cell performed with antibodies against NME and β3 integrin. Red dots denote regions of signal amplification consistent with NME/β3 integrin proximity in osteoblast cells deficient or not in β1 integrin. PLA performed on ICAP-1 and β1 integrin deficient cell line is used as control. Scale bar, 20 μm. Error bars represent SD. N = 15 cells/condition from three independent experiments. ns, adjusted P value > 0.05.

NME/β3 integrin interaction occurs at α-adaptin–marked CCPs and does not need β1 integrin in osteoblasts. (A) PLA signal using NME antibody in combination with ICAP-1 antibody (4d1d6) or ICAP-1 antibody (9b10) or β3 integrin antibody or β1 integrin antibody in WT osteoblasts. Immunolabeling of α-adaptin was performed after PLA. Insets are higher magnification of boxed regions. Insets show PLA signals (NME/ICAP-1 or NME/β1 integrin or NME/β3 integrin) colocalizing with α-adaptin-positive CCPs (arrowheads). PLA signal indicates a close proximity, possibly in situ interaction of NME with ICAP-1, β1, and β3 integrins at CCPs. Scale bar, 10 μm. Inset scale bar, 5 μm. (B and C) Representative images of PLAs (B) and PLA assay quantification (C) of the number of PLA spots per cell performed with antibodies against NME and β3 integrin. Red dots denote regions of signal amplification consistent with NME/β3 integrin proximity in osteoblast cells deficient or not in β1 integrin. PLA performed on ICAP-1 and β1 integrin deficient cell line is used as control. Scale bar, 20 μm. Error bars represent SD. N = 15 cells/condition from three independent experiments. ns, adjusted P value > 0.05.
The functional link between integrin and NME was further supported by the distribution of NME/integrin complexes in the vicinity of vinculin-stained FAs (Fig. 6, A and B), showing a role for NME/integrin association close to adhesion sites. Indeed, for each proximity interaction, the fraction of hits was significantly higher (N = 96 for β1 integrin, P < 0.0001; N = 52 for β3 integrin, P = 0.0014; Welch t tests) than when using randomized images. This finding indicates that hits are unlikely due to stochastic distribution of duolink spots in the cell and thus suggests that a correlation exists between duolink spots and spatial distribution of FAs. Whereas the presence of ICAP-1 was necessary to maintain NME close to both β1 and β3 integrins (Fig. 5, E and F), β1 integrin was not required for NME/β3 integrin (Fig. 6 C) or NME/ICAP-1 proximity (Fig. S4, B and C). Indeed, the proximity between β3 integrin and NME was retained even in cells lacking β1 integrin (Fig. 6, C and D). As NME2 is crucial for GTP-loading on dynamin (Boissan et al., 2014), and dynamin function precedes the transient recruitment of the clathrin uncoating protein auxilin immediately after CCP scission (Massol et al., 2006), we analyzed whether inhibition of NME1/2 could affect the burst of auxilin recruitment, a hallmark of CCP scission (Massol et al., 2006). Consistent with this hypothesis, recruitment of auxilin to CCPs was strongly impaired when NME2 and NME1 were deleted (Fig. S5, A and B). In addition, like NME1/2 loss, the loss of ICAP-1 in osteoblasts also led to a decrease of the auxilin burst at the rim of FA (Fig. 6, E and F). Altogether, these data indicate that ICAP-1 is required for NME function at CCPs upstream of dynamin and auxilin-dependent steps to allow optimal formation of clathrin-coated vesicles. The consequences of the inhibition of clathrin-dependent endocytosis on force generation were assessed by TFM after clathrin and NME1/2 knock-down in β1 integrin−/− cells and β1 integrin−/−-icap-1−/− cells. While deletion of clathrin and NME mimicked ICAP-1 deletion in β1 integrin−/− cells cultured on a stiff substrate by rescuing stress fibers and P-myosin (Fig. 4, F–H) and by decreasing β3 integrin turnover (Fig. 5 B), TFM experiments performed on soft gels (5 kPa) did not reveal any significant change in force generation after blocking clathrin-mediated endocytosis in cells devoid of β1 integrin. These results reveal an additional activity of ICAP-1 on β3 integrin in soft environments that is not supported by NME and clathrin.

Requirement of ICAP-1 for NME function in CCP upstream of dynamin and auxillin steps to allow optimal clathrin-coated vesicle budding in the vicinity of focal adhesion. (A) Highly inclined illumination fluorescence microscopy analysis was performed at the basal face of adherent cell on FN coated coverslide. Left panel shows cells immunostained for vinculin (green) and β1 integrin/NME duolink signal (PLA event, red), and the right panel shows cells stained for vinculin (green) and β3 integrin/NME duolink signal (PLA event, red). Scale bar, 5 µm. (B) Boxplot representation of the fraction of the hits estimated for β1 integrin/NME-vinculin and for β3 integrin/NME-vinculin and for randomized images (rand.). (C and D) Representative images of PLA with quantification performed with antibodies against NME and β3 integrin. Red dots denote regions of signal amplification revealing ICAP-1/β3 integrin proximity in β1 integrin deficient cells. PLA performed on ICAP-1 deficient cell line is used as control. Nuclei are stained in blue with DAPI. N ≥ 25 cells/condition from three independent experiments. Scale bar, 20 µm. (E and F) Spinning disk highly magnified video micrographs (every 4 s) of an FA, (vinculin - green) and auxillin bursts in red (indicated by yellow arrow) in β1 integrin+/+-ICAP-1+/+ cells. The quantification in F shows that ICAP-1 deletion slows down the auxillin bursts/FA by 50%. At least 35 FAs and 3 squares in at least 4 cells per condition were imaged in 3 independent experiments. Error bars represent SD. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001.
Requirement of ICAP-1 for NME function in CCP upstream of dynamin and auxillin steps to allow optimal clathrin-coated vesicle budding in the vicinity of focal adhesion. (A) Highly inclined illumination fluorescence microscopy analysis was performed at the basal face of adherent cell on FN coated coverslide. Left panel shows cells immunostained for vinculin (green) and β1 integrin/NME duolink signal (PLA event, red), and the right panel shows cells stained for vinculin (green) and β3 integrin/NME duolink signal (PLA event, red). Scale bar, 5 µm. (B) Boxplot representation of the fraction of the hits estimated for β1 integrin/NME-vinculin and for β3 integrin/NME-vinculin and for randomized images (rand.). (C and D) Representative images of PLA with quantification performed with antibodies against NME and β3 integrin. Red dots denote regions of signal amplification revealing ICAP-1/β3 integrin proximity in β1 integrin deficient cells. PLA performed on ICAP-1 deficient cell line is used as control. Nuclei are stained in blue with DAPI. N ≥ 25 cells/condition from three independent experiments. Scale bar, 20 µm. (E and F) Spinning disk highly magnified video micrographs (every 4 s) of an FA, (vinculin - green) and auxillin bursts in red (indicated by yellow arrow) in β1 integrin+/+-ICAP-1+/+ cells. The quantification in F shows that ICAP-1 deletion slows down the auxillin bursts/FA by 50%. At least 35 FAs and 3 squares in at least 4 cells per condition were imaged in 3 independent experiments. Error bars represent SD. ns, adjusted P value > 0.05; *, P value ≤ 0.05; **, P value ≤ 0.01; ***, P value ≤ 0.001; ****, P value ≤ 0.0001.

Requirement of NME in CCP upstream of dynamin and auxillin steps to allow optimal clathrin-coated vesicle budding. (A) Still image and kymographs of simultaneous two-color TIRF-M time series of mock- or siRNA-treated BSC-1 cells overexpressing mRFP-LCa and GFP-auxilin (1 image/s). In mock-treated cells, vertical arrowhead points to a burst of GFP-auxilin coincident with mRFP-LCa signal disappearing. In siRNA-treated cells, arrowhead points to mRFP-LCa signal disappearing without associating GFP-auxilin. (B) Percentage of disappearing mRFP-LCa CCPs ending with a burst of GFP-auxilin in mock- and siRNA-treated BSC-1 cells. At least 300 events were analyzed for each condition.

Requirement of NME in CCP upstream of dynamin and auxillin steps to allow optimal clathrin-coated vesicle budding. (A) Still image and kymographs of simultaneous two-color TIRF-M time series of mock- or siRNA-treated BSC-1 cells overexpressing mRFP-LCa and GFP-auxilin (1 image/s). In mock-treated cells, vertical arrowhead points to a burst of GFP-auxilin coincident with mRFP-LCa signal disappearing. In siRNA-treated cells, arrowhead points to mRFP-LCa signal disappearing without associating GFP-auxilin. (B) Percentage of disappearing mRFP-LCa CCPs ending with a burst of GFP-auxilin in mock- and siRNA-treated BSC-1 cells. At least 300 events were analyzed for each condition.
In summary, ICAP-1 regulates integrin endocytosis by controlling NME association to integrin-containing CCPs and is required for NME-dependent scission of CCPs. More importantly, ICAP-1 might regulate cellular force by controlling integrin endocytosis through NME2-control of dynamin function. Nevertheless, our results suggest that unlike clathrin and NME1/2, ICAP-1 plays an additional role to rescue force in β1 integrin deleted cells cultured on a soft substratum.
Discussion
Integrin trafficking contributes to both cell movement and adhesion receptor signaling (Caswell et al., 2009; Moreno-Layseca et al., 2019; Lock et al., 2019). Although essential for many cellular processes, the sequence of molecular events during clathrin-mediated integrin endocytosis remains elusive. Our present results implicate a critical contribution of the integrin inhibitor, ICAP-1, in cellular mechanotransduction by working in concert with NME2 NDPK to control clathrin-mediated endocytosis of integrins at the edge of FAs. This new mechanism illustrates the importance of integrin inhibitors in mechanosensing and mechanoresponsiveness (Bouin et al., 2017; Lerche et al., 2020). Mechanistically, ICAP-1 acts as an adaptor protein in integrin endocytosis. ICAP-1 mediates cargo selection by positioning NME close to the integrin receptor, together with AP2 and dynamin in order to fuel dynamin at CCP allowing execution of vesicle fission to ensure integrin turnover (Fig. 7). It has been previously described that the cell mechanical state regulates clathrin coat dynamics at the plasma membrane (Ferguson et al., 2017; Baschieri et al., 2018). Our study provides further insights into the importance of controlling integrin turnover to limit the maturation of FAs and tune cell contractility. In addition, our results add another piece of evidence to the conclusion that both β1 and β3 integrins can control actomyosin contractility (Roca-Cusachs et al., 2009; Schiller et al., 2013). While our results clearly confirm the efficiency of β1 integrin deletion to abolish traction forces, they also suggest the inefficiency of β3 integrin to take over in the presence of ICAP-1. Thus, suppression of ICAP-1 appears to be necessary to unlock a potential inhibition applied to β3 integrin to permit its translocation to the cell center, generate cell contractile response and ensure Fn fibrillogenesis. Despite the main function of β1 integrin in traction force and fibrillogenesis, β3 integrin has already been reported to reinforce force (Roca-Cusachs et al., 2009) and to stimulate the initial steps of αvβ3-mediated Fn fibril formation (Danen et al., 2002). Moreover, the ability of αvβ3 integrin to drive Fn fibrillogenesis is supported by previous data demonstrating the role of integrin-αvβ3 to trigger Fn assembly and CAF invasion (Attieh et al., 2017). Our results demonstrate that ICAP-1 and NME share the task of β3 integrin endocytosis control to regulate integrin cell surface and cell tensional homeostasis.

Recruitment of NME by ICAP-1 to regulate cellular force by promoting inactive integrin internalization. Scheme representing the sequence of molecular events during clathrin-mediated integrin endocytosis occurring at the edge of FA. ICAP-1 acts as an adaptor protein in integrin endocytic process. ICAP-1 mediates cargo selection by positioning NME close to integrin (1), AP2, and dynamin (2) in order to fuel dynamin at the clathrin-coated pits (3) allowing execution of subsequent membrane deformation and vesicle fission (4) to ensure the turnover of integrins.
Recruitment of NME by ICAP-1 to regulate cellular force by promoting inactive integrin internalization. Scheme representing the sequence of molecular events during clathrin-mediated integrin endocytosis occurring at the edge of FA. ICAP-1 acts as an adaptor protein in integrin endocytic process. ICAP-1 mediates cargo selection by positioning NME close to integrin (1), AP2, and dynamin (2) in order to fuel dynamin at the clathrin-coated pits (3) allowing execution of subsequent membrane deformation and vesicle fission (4) to ensure the turnover of integrins.
Clathrin-dependent trafficking of integrins is essential for FA disassembly, a process dictated by the intracellular domains of both α and β integrin subunits which control the nature of the ligand and the activation status of integrin (Margadant et al., 2011; De Franceschi et al., 2016; Ezratty et al., 2009; Arjonen et al., 2012). By targeting the cytoplasmic domain of β integrin subunit, ICAP-1 directly participates to FA disassembly (Bouvard et al., 2003). However, as supported by 2-hybrid approaches and in vitro interaction assays (Fournier et al., 2002), ICAP-1 must also interact with NME2 in order to drive integrin internalization. Different approaches including proteomic studies have highlighted that NME form a complex with both β1 and β3 integrin complexes on the one hand (Kuo et al., 2011; Schiller et al., 2011), and with AP2 (Marino et al., 2013) on the other hand, indicating a functional contribution of NME in dynamin-mediated scission of CCPs (Boissan et al., 2014). By uncovering the impact of integrin internalization on force generation through ICAP-1/NME complex formation, we provide an additional mechanistic facet of the otherwise multifunctional protein ICAP-1, which is known to regulate FA disassembly, cell contractility, Fn fibrillogenesis, and cell migration (Millon-Frémillon et al., 2008; Faurobert et al., 2013; Bouin et al., 2017; Lisowska et al., 2018; Brunner et al., 2011). The versatility of ICAP-1 needs to be considered to explain the lack of rescue of force generation upon clathrin or NME1/2 deletion in β1 null cells on soft substrates. While β1 null cells do not form large β3 FAs and do not produce traction force when plated on a soft substrate (TFM conditions), ICAP-1 deletion releases the interaction site for kindlin which favors β3 integrin clustering, a step required for efficient linkage of integrin to the actin cytoskeleton and the development of traction force (Ye et al., 2013). Our assumption is that integrin internalization by the ICAP-1/NME1 complex in CCPs is spatially controlled and requires adhesion sites with minimal clustering of integrins to be effective. According to our experiments (Figs. 4, F–H and 5 B), clathrin and NME deletion mimics ICAP-1 deletion in stiff conditions since substrate stiffness is enough to induce integrin clustering (Fig. 4, F–H). Integrin clustering might be important to correctly position the CCP machinery. This hypothesis is supported by the distribution of NME/integrin complexes (Fig. 6, A and B) and bursts of auxilin (Fig. 6, E and F) in the vicinity of vinculin-stained FA. This result emphasizes a role for the ICAP-1/NME/integrin trio close to adhesion sites in a stiffness-dependent manner. Of note, ICAP-1 knock-out revealed a phenotype in bone which might support an important role of ICAP-1 in stiff conditions (Bouvard et al., 2007).
So far it was unknown whether individual integrin classes like α5β1 and αvβ3 interacting with the same ligand like Fn are internalized using common or specific routes. Because the deletion of ICAP-1 led to an increase of both AP2/β1 and AP2/β3 complexes, the partnership between ICAP-1 and NME2 likely contributes to both β1 and β3 internalizations. Concomitantly, the control of CCP scission by ICAP-1/NME complex directly regulates cell mechanics which has previously been shown to result from the cooperation between β1 and β3 integrin (Schiller et al., 2013). However, we cannot exclude a compensatory effect between β1 and β3 integrins to generate force since the proximity between NME with ICAP-1 and β3 integrin is not conditioned by the presence of β1 integrin.
Importantly, many other endocytic accessory proteins or co-adaptors associate with integrins and CCPs (Yap and Winckler, 2015). Dab2 and Numb are PTB domain-containing proteins that accumulate at or near FA shortly before their disassembly (Chao and Kunz, 2009; Nishimura and Kaibuchi, 2007; Ezratty et al., 2009; Teckchandani et al., 2009). The physiological relevance of having many adaptors or co-adaptors like ICAP-1, Numb, or Dab2 in the same cell might respond either to integrin specificity or to physical properties of the microenvironment. As an illustration, Dab2 is not found at β3 integrin-mediated FA sites formed on RGD-coated glass. In addition, it has been shown that the development of actomyosin contractility inhibits Dab2 binding to β3 integrin. However the loss of cell–matrix force development is a key determinant for Dab2 binding to β3 integrin clusters and the ongoing endocytosis of β3 integrin under soft conditions such as on mobile RGD membranes (Yu et al., 2015). This suggests a specificity of some adaptors depending on the microenvironment stiffness. So far, no direct interaction between ICAP-1 and β3 integrin has been demonstrated, possibly due to the lack of proper post-translational modifications in classical pull-down assays. Moreover, the nature of this interaction might be complexified by the requirement of a third partner to bridge β3 integrin and ICAP-1. However, PLA experiments show proximity not only between β3 integrin and ICAP-1 but also between β3 integrin and NME, even in the absence of β1 integrin. ICAP-1 might take over for Dab2 in stiffer microenvironment as ICAP-1 and Dab2 share the same distal NPXY motif to interact with integrin. Regarding β1 integrin endocytosis, Dab2 and clathrin are not found to activate the β1 integrin clusters observed in cells upon adhesion on mobile RGD membranes (Yu et al., 2015). This suggests that the integrin turnover might be regulated differentially with respect to the physical properties of the microenvironment, possibly involving ICAP-1 and Dab2 in stiff and soft microenvironments, respectively. Whether specific mechanosensitive pathways and distinct PTB domain integrin adapters are required to control integrin internalization or endocytic frustration (Lock et al., 2019; Baschieri et al., 2018), depending on the physical properties of the microenvironment will be the next avenue to explore.
As migratory and adhesive behavior is mediated by β1 and β3 integrin cooperation (Schiller et al., 2013), the regulation of their respective intracellular trafficking in a coordinated manner might be likely essential for rapidly and efficiently adapting the responsiveness of migratory cells to extracellular guidance cues. Whether ICAP-1 coordinates commonly β1 and β3 integrin endocytic process to adapt integrin dynamics and traction force generation in a context-dependent manner is a pending question.
Materials and methods
Antibodies and chemicals
Human plasma Fn was purchased from Sigma-Aldrich. Anti β3 integrin antibody was purchased from Emfret (Clone LucA.5, #M030-0), the double phosphorylated (T18/S19) myosin light-chain antibody and clathrin antibody were obtained from Cell Signaling, #3674 and #4796, respectively, and the unmodified myosin light-chain antibody and tubulin were purchased from Sigma-Aldrich, #M4401 and #T4026, respectively. The polyclonal anti-β3 integrin was kindly provided by M.H. Ginsberg (University of California San Diego, San Diego, CA). The Fn antibody was purchased from Millipore (#AB2033). The transferrin antibody was purchased from Abcam (#ab82411). The HRP-conjugated antibodies were obtained from Jackson ImmunoResearch—F(ab')₂ Anti-Rabbit HRP (#711-036-152) or anti-mouse IgG, light-chain HRP (#115-035-174).The fluorescent secondary antibodies conjugated with AlexaFluor 488 (#A-11063), AlexaFluor 546 (#A-11003), or AlexaFluor 633 (#A-21053) were obtained from Thermo Fisher Scientific. Mouse monoclonal anti-NME2 was purchased from Kamiya Biomedical Company. Rabbit polyclonal pan-NME antibodies (recognizing both NME1 and NME2 isoforms) were prepared by affinity purification using purified human recombinant NME1 and NME2 proteins coupled to NHS-activated HiTrap columns. Mouse monoclonal anti-ICAP-1α antibodies (4D1D6 and 9B10) were prepared using recombinant His-tagged ICAP-1α protein as antigen (Fournier et al., 2002). Phalloidin coupled with Atto 647 was also purchased from Thermo Fisher Scientific (#A22287).
Cell culture
Immortalized osteoblasts from icap-1−/−; β1 integrin flox/flox mice were generated as described previously (Bouvard et al., 2007). These cells were infected or not by adenoCre viruses from gene transfer vector core (University of Iowa) in order to obtain β1 integrin-null cells. The icap-1 null cells were incubated with retroviral particles to obtain rescued cells expressing ICAP-1 WT. The cells were selected with 1 mg/ml puromycin to produce cell populations with heterogeneous ICAP-1 expression levels. Cells were maintained in culture in DMEM (#31966-021; Life Technologies) supplemented with 10% FBS (#S1810-500; Dominique Dutscher), 100 U/ml penicillin, and 100 µg/ml streptomycin (#P06-07100; PAN Biotech) at 37°C in a 5% CO2-humidified chamber. For all experiments, cells were washed with PBS (#L0615-500; Dominique Dutcher), detached using trypsin (#L0615-500; Dominique Dutcher), and treated with 1 mg/ml trypsin inhibitor (#T6522; Sigma-Aldrich). Cells were then plated in DMEM containing 10% FBS for 4 h and then the appropriate analysis was carried out. Where needed, a serum-free medium OptiMEM was used (#51985-026; Life Technologies) as substitute. The four osteoblast clones—icap-1+/+; icap-1−/−; β1 integrinfloxed/floxed; β1 integrinfloxed/floxed; and icap-1−/−—were infected using lentiviral infection system from Invitrogen with pLenti–murine β3 integrin-GFP vector.
Western blotting analysis
Cells were plated on 50% confluence and left to spread overnight. The next day, the dishes were washed twice with ice cold PBS and lysed in cold RIPA buffer, supplemented with 1× cOmplete protease inhibitors, 5 mM NaF, and 2 mM Na-orthovanadate. After protein quantification through Pierce BCA Protein Assay (#23227; Thermo Fisher Scientific), the samplers were mixed with Laemmli sample buffer (0.4% SDS, 20% glycerol, 120 mM Tris-Cl [pH 6.8], and 0.02% [w/v] bromophenol blue) and loaded on electrophoretic PAA gels. Following the standard wet blotting protocol, the nitrocellulose membranes (#10600003; Amersham) were probed with the appropriate primary antibodies, diluted in 5% BSA in Tween-TBS (TTBS), and incubated overnight. The membrane was subsequently incubated with the appropriate secondary antibodies, also diluted in 5% BSA in TTBS for 1 h and then developed using Clarity ECL kit (#170-5061; Biorad) and recorded with ChemiDoc Imaging System and analyzed with ImageLab software.
Traction force microscopy
The poly-acrylamide hydrogels with defined rigidity of 5 kPa and containing fluorescent microbeads (#F8783; Life Technologies) were cast in 2-well LabTeks (#154461; Thermo Fisher Scientific), coated with BindSilane (#GE17-1330-01; Sigma-Aldrich) and covered with coverslip, coated with Sigmacote (#SL2; Sigma-Aldrich). After the polymerization of the polyacrylamide the wells were flooded with water and the coverslips were detached gently. For the functionalization a protocol from (Przybyla et al., 2016) was used. Briefly, solution of tetramethacrylate, N6, and Irgacure was deposed on the gels and baked under UV light (312 nm) for 5 min. Then, Fn (5 μg/ml) was deposed on the gels and incubated at 4°C overnight. Cells were allowed to adhere and spread 4 h in DMEM containing 10% FBS and then placed in 4% FBS. Just before the acquisition, the membrane was stained with red fluorescent membrane marker PKH26 (# PKH26GL; Sigma-Aldrich). Images were taken using spinning disk microscope iMIC EMCCD Andromeda (FEI) equipped with heating chamber, CO2 installation, using 40× magnification oil objective (Plan-Apochromat 40×/1.4 Oil-DIC M27 WD 0.13 mm). A fluorescent image of the beads with the cell spread on and fluorescent image of the cell membrane was obtained. Then, the culture medium was replaced with pure solution of trypsin and after verification that cells were completely detached the second image of the fluorescent beads were taken. Isolated cells were randomly chosen for each experimental condition. Force calculations were performed as previously described (Tseng et al., 2011). Briefly, the displacement fields describing the deformation of the PA substrate are determined from the analysis of fluorescent beads images before and after removal of the adhering cells with trypsin treatment. The displacement field is obtained by a two-step method consisting of particle image velocimetry followed by individual bead tracking (Butler et al., 2002; Sabass et al., 2008). A special procedure is used to evaluate displacements in the area of the adhesive pattern where gel deformation is expected to be largest. Depending on the pattern shape, traction forces may be strongly localized leading to large displacements in very small areas. In this case, failure to correctly track a few beads in such areas would significantly alter the calculated force magnitude. Therefore, the pattern area is divided into smaller windows that are allowed to overlap, before applying the cross-correlation and tracking analysis. Reducing the size of the windows makes it possible to retrieve larger displacements with cross-correlation and, using overlapped windows, we can avoid missing beads close to the windows boundaries. All image processing and analysis were performed using Matlab (Gao and Kilfoil, 2009). To calculate cell-induced traction stress from displacement data, we have used the Fourier-transform traction cytometry (FTTC) method (Sabass et al., 2008). We kept the regularization parameter at small values (λ < ∼10−9) in order to maintain the best spatial resolution, which is estimated to be about 50 nm in our case.
PAA substrates
Polyacrylamide (PAA) substrates were prepared on 2-well LabTeks (Thermo Fisher Scientific) with 8% acrylamide/x% bis-acrylamide and 10 mM Hepes (pH 8.5) gel. After two sulfo-sanpah (Thermo Fisher Scientific) activation, gels were coated with 5 µg/ml fibronectin (1 µg/cm2) at 4°C overnight. We used three concentrations of bis-acrylamide in order to control the rigidity: 0.1% for 3 kPa, 0.2% for 15 kPa, and 0.4% for 60 kPa.
Random migration analysis
For migration assays, cells were plated on a 2-well LabTeks containing an Fn-coated PAA substrate (10 mm diameter, 100 nm thick) for 3 h in CO2-independent DMEM containing fibronectin-free 4% FCS. Cells are maintained at 37°C and imaged on an inverted microscope (Axiovert 200; Zeiss) equipped with a motorized stage, cooled CCD camera (CoolSnap HQ2; Roper Scientific), and a live-cell imaging 10× objective (EC Plan-Neofluar) for 5 h at a frequency of one image per 4 min. Cell velocity was obtained using the manual tracking plugin in ImageJ. 150–200 cells were analyzed from at least five different positions per experiment, and results were issued from three independent experiments.
Focal adhesion lifetime analysis
Cells stably expressing β3 integrin-GFP were spread in 2-well LabTeks and left to spread for 4 h. Spinning disk videos were taken for the length of 2 h with 1 min frequency. The lifetime analysis was performed with FA Analysis Server (Berginski and Gomez, 2013).
Immunofluorescence
Cells were plated at an approximate density of 6 × 104 per cm2 for 4 h, fixed with 4% PFA, permeabilized with 0.3% Triton X-100, and blocked with 10% goat-serum in PBS then with appropriate primary antibodies and after rinsing, with appropriate AlexaFluor-conjugated secondary antibody and AlexaFluor-conjugated phalloidin. Finally, the coverslips were mounted in Mowiol/DAPI solution. Images were acquired on an iMIC EMCCD Andromeda (FEI) spinning-disk microscope equipped with an objective Plan-Apochromat 40×/1.4 Oil-DIC M27, WD 0.13 mm.
siRNA-mediated downregulation of proteins in osteoblast cells
Cells were plated in 6-well plates at low density—6 × 103 cells per cm2 and left to spread overnight. The next day they were transfected with the appropriate siRNA using RNAiMAX system (Thermo Fisher Scientific). The medium was changed the next day, and a second hit with the same siRNA was performed. The transfected cells were used in 24 h after. All used siRNA were purchased from Dharmacon as follows: siRNA against β3 integrin – L-040746-01-0005; siRNA against clathrin – L-063954-00, siRNA against NME1 – L-040142-00, and siRNA against NME2 - L-040143-00; for all experiments we used a nontargeting siRNA as control – D-001810-10-20.
FRAP analysis
Cells stably expressing β3 integrin–GFP were spread in 2-well LabTeks and left to spread for 4 h. FRAP videos were taken using multimodal microscope for photo manipulations equipped with TIRF 63× objective (α-Plan-Apochromat 63×/1.46 Oil Korr M27, WD 0.1 mm). The analysis was carried out using build in FRAP analysis module in the FEI offline analysis.
Image analysis and statistical tests
For ppMLC staining or surface analysis, we measured the necessary signal using a thresholding method with manual correction when needed. More than 30 cells were measured in each condition that allowed us to do a non-parametric Kruskal–Wallis test (non-parametric) followed by Wilcoxon test with a Bonferroni correction when KW tests were significant (using GraphPad); experiments were carried out at least three times. For FA analysis, we measured the β3 integrin staining signal of at least 50 cells per experiment and three independent experiments using the particle analyzer of ImageJ software. Particles over 10 nm2 were analyzed. The total adhesive area per cell was analyzed by a Kruskal–Wallis test (non-parametric) followed by Wilcoxon test with a Bonferroni correction when KW tests were significant; the mean area of FA was analyzed by an ANOVA-2 analysis and Tukey’s HSD post-hoc tests (using GraphPad).
Fibronectin fibrillogenesis
100,000 cells were plated on LabTek glass slide 4 chambers and allowed to adhere for 24 h in serum-free medium made of OptiMEM. Then, cells were fixed with 4% PFA. Cells were stained with appropriate primary antibodies anti-Fn, anti-β3 integrin (LucA.5), and with appropriate AlexaFluor-conjugated secondary antibody and AlexaFluor-conjugated phalloidin. Then, the coverslips were mounted in Mowiol solution. The length of individual fibers was determined manually in ImageJ. To assess Fn coverage, the images were processed with fast Fourier transform bandpass filters to visualize all fibers, and the amount of fibronectin was measured by thresholding using Fiji (Schindelin et al., 2012).
Fluorescent integrin antibody uptake assays
LucA5 antibody was labeled using SiteClick Antibody Azido Modification Kit (Thermo Fisher Scientific) and Click-iT pHrodo iFL Red sDIBO Alkyne for Antibody Labeling (Thermo Fisher Scientific) following the manufacturer’s instructions. Cells were spread on Fn-coated LabTek slides in DMEM, supplemented with 10% FBS and 1% P/S. Labeled antibody (10 µg/ml) was added to the cells for 45 min at 37 or 4°C, and cells were fixed in 4% PFA and permeabilized. Actin staining was performed in order to detect cell contour. Images were acquired on an iMIC EMCCD Andromeda (FEI) spinning-disk microscope equipped with an objective Plan-Apochromat 40×/1.4 Oil-DIC M27, WD 0.13 mm, and analyzed using the 3D Object Counter in ImageJ. Statistical analysis was performed in R using one-way analysis of variance (ANOVA) and Tukey’s HSD post-hoc test.
In situ PLA
To monitor the subcellular localization of protein–protein interactions at single molecule resolution, an in situ PLA was performed as previously described (Söderberg et al., 2006). Cells grown on coverslips were fixed with cold methanol and then incubated with primary antibodies. Secondary antibodies tagged with short DNA oligonucleotides were added. Hybridization, ligation, amplification, and detection were realized according to the manufacturer’s protocol (Sigma-Aldrich). Briefly, secondary antibodies were incubated in preheated humidity chamber for 1 h at 37°C. Ligation was performed with a ligase-containing ligation solution for 30 min at 37°C. Finally, amplification step was performed with a polymerase-containing amplification solution for 1 h 40 min at 37°C. After the PLA reaction, coverslips were further incubated with AlexaFluor 488-conjugated IgGs and Cy5-conjugated IgGs to detect proteins corresponding to the primary antibodies used. PLA signal corresponds to the Cy3 fluorescence. Coverslips were analyzed on an inverted wide-field microscope (Leica DM4B, Camera Leica DFC 3000 G, Objectif X63 Leica Germany 506185).
Highly inclined illumination fluorescence microscopy (Imaging, 2010) was used to analyze the spatial distribution of β1/β3 integrin–NME proximity complexes revealed by the PLA technology in cells labeled for vinculin to assess the proximity of NME/integrin complexes with FA. Images have been analyzed by a homemade script in ImageJ (code available in Data S1) to detect duolink spots (using D. Sage watershed algorithm; Teaching by Doing, 2003) and FA, and assess their spatial proximity. The principle was to enlarge (by three pixels) regions corresponding to detected duolink spots following the method described in Montagnac et al. (2013). Then, the intensity of fluorescence in the second channel corresponding to the vinculin staining is measured to determine any colocalization. A “hit” is obtained in case of a positive result. The ratio of those hits to the total number of duolink spots is calculated for each cell. For each proximity complexes, the fraction of hits is significantly higher than when using randomized images. This indicates that the hits are unlikely to be due to stochastic distribution of duolink spots in the cell, and thus suggests that a correlation exists between duolink spots and FA spatial distribution.
Auxillin flashes measurement
The four osteoblast clones were electroporated using AMAXA nucleofector Kit V (protocol X-001) according to the manufacturer’s instructions in order to transfect them with two plasmids encoding, respectively, GFP-tagged Auxillin and the mCherry-tagged Paxillin. 100,000 cells per condition were plated on fluorodishes. In the following days, cells were imaged at 2 s intervals for 5 min using a spinning disk microscope (Andor) based on a CSU-W1 Yokogawa head mounted on the lateral port of an inverted IX-83 Olympus microscope equipped with a 60× 1.35 NA UPLSAPO objective lens and a laser combiner system, which included 491 and 561 nm 100 mW DPSS lasers (Andor). Images were acquired with a Zyla sCMOS camera (Andor). The system was steered by IQ3 software (Andor). Paxillin-positive FA were manually selected in Fiji and their area was measured. Auxillin flashes appearing at the border of FAs were manually counted. Subsequently, a square-shaped region of 2.5 × 2.5 µm2 was placed in the center of the cell in a region where FAs were absent in order to count Auxillin flashes outside of FAs. At least 35 FAs and 3 squares in at least 4 cells per condition were imaged in three independent experiments. Results are expressed as Auxillin flashes per second divided by the FA area or by the area of the square-shaped region of 2.5 × 2.5 µm2 and normalized to the value obtained in β1 integrin+/+- icap-1+/+ osteoblasts. Statistical analyses were performed in SigmaPlot using one-way ANOVA followed by All Pairwise Multiple Comparison Procedure (Holm-Sidak method).
Fluorescence-activated cell sorting
Cells were gently detached with trypsin and then treated with trypsin inhibitor (#T0256). Then, they were placed in round-bottom 96-well plate and blocked with 1% BSA in PBS for 30 min at 37°C. Then, cells were incubated with the appropriate antibodies and secondary antibodies as control diluted in PBS/1% BSA for 30 min on ice. After subsequent incubation with secondary antibodies cells were fixed in 4% PFA for 10 min and surface staining was detected with BD Accuri C6 flow cytometer and analyzed with the provided software.
Online supplemental material
Fig. S1 shows the characterization of osteoblast cells used in the study. Fig. S2 shows the role of ICAP-1 in cellular mechanotransduction. Fig. S3 shows the efficiency of clathrin silencing in osteoblast cell lines. Fig. S4 shows NME/β3 integrin complex at α-adaptin–marked CCPs and its independence with respect to β1 integrin. Fig. S5 shows the requirement of NME in CCP upstream of dynamin and auxillin steps to allow optimal clathrin-coated vesicle budding. Video 1 shows the lifetime of eGFP-β3 integrin FAs in β1 integrin+/+/icap-1+/+ cells. Video 2 shows the lifetime of eGFP-β3 integrin FAs in β1 integrin+/+/icap-1−/− cells. Video 3 shows the lifetime of eGFP-β3 integrin FAs in β1 integrin−/−/icap-1+/+ cells. Video 4 shows the lifetime of eGFP-β3 integrin FAs in β1 integrin−/−/icap-1−/− cells. Data S1 provides the homemade image script used in this paper.
Acknowledgments
We thank Isabelle Tardieux and Kate Miroshnikova for reviewing the manuscript. We thank Daniel Bouvard and Reinhard Fassler for providing osteoblast cells, Alexei Grichine, Jacques Mazzega, and Mylène Pezet for their technical assistance on the microcell Imaging platform from the Institute for Advanced Biosciences, and Agnieszka Kawska (http://www.IlluScientia.com) for artwork associated with Fig. 7. We thank Christiane Oddou for technical assistance. We are grateful to Dr. M.H. Ginsberg for providing polyclonal antibodies specific for β3 integrin.
A. Kyumurkov and M. Régent-Kloeckner are doctoral fellows funded from the French Ministère de l’Enseignement Supérieur et de la Recherche. M. Proponnet-Guerault is a recipient of a fellowship from the Ligue Nationale contre le Cancer (LNCC). This research was funded by the Agence Nationale de la Recherche grant (CODECIDE ANR-17-CE13-022, StrepB2brain, ANR-17-CE15-0026-01), by the Fondation pour la Recherche sur le Cancer and by the Fondation pour la Recherche Médicale grant (DEQ20170336702) to C. Albiges-Rizo and the Ligue Contre le Cancer Régionale Auvergne-Rhône-Alpes et Saône-et-Loire (17021) to A.-P. Bouin.
The authors declare no competing financial interests.
Author contributions: A. Kyumurkov and A.-P. Bouin did most of the experiments, analyzed the data, and prepared the figures. M. Proponnet-Guerault contributed in performing endocytosis experiments. M. Régent-Kloeckner contributed to performing quantitative analyses of phase-contrast live-cell imaging data. M. Balland and A. Nicolas contributed to generating TFM data. G. Montagnac, F. Baschieri, and P. Chavrier contributed to setting up Auxillin flash experiments. S. Manet contributed to FACS analysis and O. Destaing to TIRF and FRAP analyses. M. Boissan, C. Calmel, F. Waharte, and P. Chavrier contributed to performing PLA experiments. C. Albiges-Rizo, E. Planus, A.P. Bouin, G. Montagnac, P. Chavrier, O. Destaing, and M. Boissan contributed with material and critical discussions. C. Albiges-Rizo and E. Planus supervised the entire work. C. Albiges-Rizo and E. Planus initiated the topic and wrote the manuscript together with A.P. Bouin.

