


Multivesicular bodies (MVBs) contain intralumenal vesicles that are delivered to lysosomes for degradation or released extracellularly for intercellular signaling. Here, we identified Caenorhabditis elegans filamin FLN-2 as a novel regulator of MVB biogenesis. FLN-2 co-localizes with V-ATPase subunits on MVBs, and the loss of FLN-2 affects MVB biogenesis, reducing the number of MVBs in C. elegans hypodermis. FLN-2 associates with actin filaments and is required for F-actin organization. Like fln-2(lf) mutation, inactivation of the V0 or V1 sector of V-ATPase or inhibition of actin polymerization impairs MVB biogenesis. Super-resolution imaging shows that FLN-2 docks V-ATPase-decorated MVBs onto actin filaments. FLN-2 interacts via its calponin-homology domains with F-actin and the V1-E subunit, VHA-8. Our data suggest that FLN-2 mediates the docking of MVBs on the actin cytoskeleton, which is required for MVB biogenesis.
Introduction
Multivesicular bodies (MVBs) contain intralumenal vesicles (ILVs) formed by a membrane invagination process on endosomes. Cargoes are sorted into the internal vesicles of nascent MVBs, which mature or detach from endosomes to become free MVBs (Hanson and Cashikar, 2012; Scott et al., 2014). MVBs deliver the cargoes to lysosomes for degradation or release them from the cell as exosomes after fusion with the plasma membrane. Together, the degradative and secretory MVB pathways regulate a wide array of functions (Hanson and Cashikar, 2012; Mathieu et al., 2019; van Niel et al., 2018).
Endosomal sorting complexes required for transport (ESCRTs) play key roles in MVB biogenesis in both degradative and secretory MVB pathways. In the canonical ESCRT pathway, ubiquitinated cargo and PtdIns3P recruit ESCRT-0 to endosomal membranes for cargo concentration and engagement of downstream ESCRTs (Gruenberg and Stenmark, 2004; Hanson and Cashikar, 2012). ESCRT-I, -II, and -III sequentially mediate formation and scission of ILVs (Hanson and Cashikar, 2012). Multiple other factors regulate cargo sorting and/or ILV formation in coordination with or independently of ESCRT components (Hanson and Cashikar, 2012; Hessvik and Llorente, 2018). Lipids such as ceramide, phosphatidic acid, and sphingosine-1-phosphate (S1P) are involved in exosomal MVB formation (Hessvik and Llorente, 2018; Kajimoto et al., 2013; Trajkovic et al., 2008). Vacuolar-type ATPase (V-ATPase) acidifies the lumen of various endomembrane compartments (Futai et al., 2019). In cell-free assays, inactivation of V-ATPase affects ILV formation and inhibits the generation of endosomal carrier vesicles from early endosomes (Clague et al., 1994; Falguieres et al., 2008). Thus, MVB biogenesis may require endosomal acidification by V-ATPase, although the mechanism is unclear. The membrane-bound V0 sector of V-ATPase has also been implicated in regulated exocytosis via a role in membrane fusion (Di Giovanni et al., 2010; Poea-Guyon et al., 2013). In Caenorhabditis elegans, the V0 “a” subunit VHA-5 promotes apical secretion of Hedgehog-related proteins through MVBs, probably by mediating membrane fusion independent of V1 (Liegeois et al., 2006).
The actin cytoskeleton has roles in both biogenesis and regulated exocytosis of MVBs. In MVB biogenesis, cloud-like F-actin patches are nucleated and assembled on early endosomes, which may help to segregate multivesicular membranes from the tubular recycling region on early endosomes (Morel et al., 2009; Muriel et al., 2016). Moreover, F-actin formation induced by S1P signaling promotes cargo sorting into exosomal ILVs, thus contributing to the biogenesis of secretory MVBs (Kajimoto et al., 2018). In regulated exocytosis, cortical actin networks act as docking sites to both trap and mobilize secretory MVBs from and to the fusion site (Li et al., 2018; Sinha et al., 2016). It is unclear if actin filaments provide docking sites for MVB biogenesis.
Filamins are F-actin crosslinkers that organize cortical actin filaments into dynamic 3D structures and link them to cellular membranes (Modarres and Mofradt, 2014; Zhou et al., 2021). Filamins also act as scaffolds for various signaling molecules implicated in cell motility, transcription, and mechanical sensing (Lamsoul et al., 2020; Zhou et al., 2010). Filamin gene mutations are associated with diseases including malformation of nervous system, heart, and skeleton (Bandaru et al., 2021; Rosa et al., 2019; Zhou et al., 2021). Here, we identified C. elegans filamin FLN-2 as a novel regulator of MVB biogenesis. FLN-2 associates with VHA-5–positive MVBs, and the loss of its function affects MVB biogenesis. Our data suggest that FLN-2 is a bridging molecule that docks MVBs on the actin cytoskeleton to facilitate MVB biogenesis.
Results and discussion
VHA-5–positive MVBs are impaired in fln-2 mutants
The V0 “a” subunit VHA-5 localizes to the apical membrane stacks and on the limiting membrane of MVBs in C. elegans hypodermis (Liegeois et al., 2006). Accordingly, VHA-5::RFP exhibits a punctate staining pattern at the apical region of the main hypodermal syncytium hyp7 (Fig. 1 A; Liegeois et al., 2006). VHA-5 vesicles are not labeled by SCAV-3 (lysosome membrane protein) or lysotracker red, consistent with an endosomal identity (Fig. S1, A and B″; Li et al., 2016). In a forward genetic screen, we isolated three recessive mutations (qx416, qx439, and qx463), which diminished the VHA-5::RFP signal in hyp7 (Figs. 1, B–D; and S1, C–H, and K). All three mutations affect fln-2, which encodes a divergent C. elegans homolog of human filamin A, an F-actin cross-linker (Fig. S1 C). FLN-2 expression restores VHA-5::RFP fluorescence in qx416 mutants (Fig. S1, I–K). FLN-2 contains 3 N-terminal actin-binding calponin-homology (CH) domains and 22 immunoglobulin-like (IgG) repeats (Fig. S1 C). The qx416, qx439, and qx463 mutations are all predicted to truncate the FLN-2 protein, removing 16 out of 22 Ig repeats (Fig. S1 C); thus, they are probably loss-of-function mutations. The qx416 allele was used in later experiments.

FLN-2 is required for MVB formation. (A–D) Confocal fluorescence images of the epidermis in adult day 2 WT (A–A″) and fln-2 (B–C″) co-expressing VHA-5::RFP and VHA-8::GFP. (D) Mean fluorescence intensity; n = 17 worms per strain. (E–H) TEM images of epidermal MVBs in WT (E), fln-2 (F), cup-5 (G), and cup-5;fln-2 (H) at adult day 2. Blue arrows, light MVBs; red arrows, dark MVBs. (I–L) MVB number/µm2 epidermal surface (I and L), ILV number (J), and light MVB diameter (K) in the indicated strains. Around 4–6 cross-sections were scored in each of the 5 worms per group in (I and L) and n ≥ 30 light MVBs were scored per group in J and K. (M–N″) DIC (M) and confocal fluorescence images of epidermis in WT adults expressing FLN-2::GFP (M′) or FLN-2::GFP and VHA-5::RFP (N–N″). Yellow arrowheads, strip-like structures; white arrowheads, vesicular-tubular structures. D and I–L show mean ± SD. **, P < 0.01; ***, P < 0.001; N.S, not significant, by unpaired two-tailed Student’s t test. Scale bars, 5 μm (A–C″ and M–N″), 500 nm (E–H).
FLN-2 is required for MVB formation. (A–D) Confocal fluorescence images of the epidermis in adult day 2 WT (A–A″) and fln-2 (B–C″) co-expressing VHA-5::RFP and VHA-8::GFP. (D) Mean fluorescence intensity; n = 17 worms per strain. (E–H) TEM images of epidermal MVBs in WT (E), fln-2 (F), cup-5 (G), and cup-5;fln-2 (H) at adult day 2. Blue arrows, light MVBs; red arrows, dark MVBs. (I–L) MVB number/µm2 epidermal surface (I and L), ILV number (J), and light MVB diameter (K) in the indicated strains. Around 4–6 cross-sections were scored in each of the 5 worms per group in (I and L) and n ≥ 30 light MVBs were scored per group in J and K. (M–N″) DIC (M) and confocal fluorescence images of epidermis in WT adults expressing FLN-2::GFP (M′) or FLN-2::GFP and VHA-5::RFP (N–N″). Yellow arrowheads, strip-like structures; white arrowheads, vesicular-tubular structures. D and I–L show mean ± SD. **, P < 0.01; ***, P < 0.001; N.S, not significant, by unpaired two-tailed Student’s t test. Scale bars, 5 μm (A–C″ and M–N″), 500 nm (E–H).

Loss of FLN-2 disrupts VHA-5 – positive vesicles. (A–B″) Confocal fluorescence images of the apical epidermis in WT adults co-expressing VHA-5::RFP and SCAV-3::GFP (A–A″) or expressing VHA-5::GFP and stained by lysotracker red (B–B″). (C) Schematic diagram showing the fln-2 gene structure and the FLN-2A domain structure. Black boxes indicate exons and lines designate introns. The red, green, and blue arrows indicate the mutation sites identified in qx416, qx439, and qx463, respectively. (D–K) Confocal fluorescence images of VHA-5::RFP in the hypodermis of WT (D), qx439 (E and F), qx463 (G and H), qx416 (I) and qx416 (J) expressing FLN-2 at adult day 2. The average fluorescence intensity of VHA-5 in the indicated strains is quantified in K. 15 animals were scored in each strain. Data are shown as mean ± SD. Unpaired two-tailed Student’s t test was performed to compare mutant datasets with WT or datasets that are linked by lines. ***, P < 0.001. (L–M′) DIC and confocal fluorescence images of WT adults expressing FLN-2::GFP in the head and tail regions. (N–O″) Confocal fluorescence images of the epidermis (N–N″) or the excretory canal (O–O″) in WT adults co-expressing FLN-2::RFP and VAB-10A::GFP (N–N″) or VHA-5::RFP (O–O″). FLN-2 overlaps with VAB-10A on the stripe-like fibrous organelles (N–N″). In the excretory canal cell, FLN-2 is aligned at the apical side of VHA-5 as indicated by arrowheads (N–N″). Scale bars, 5 μm.
Loss of FLN-2 disrupts VHA-5 – positive vesicles. (A–B″) Confocal fluorescence images of the apical epidermis in WT adults co-expressing VHA-5::RFP and SCAV-3::GFP (A–A″) or expressing VHA-5::GFP and stained by lysotracker red (B–B″). (C) Schematic diagram showing the fln-2 gene structure and the FLN-2A domain structure. Black boxes indicate exons and lines designate introns. The red, green, and blue arrows indicate the mutation sites identified in qx416, qx439, and qx463, respectively. (D–K) Confocal fluorescence images of VHA-5::RFP in the hypodermis of WT (D), qx439 (E and F), qx463 (G and H), qx416 (I) and qx416 (J) expressing FLN-2 at adult day 2. The average fluorescence intensity of VHA-5 in the indicated strains is quantified in K. 15 animals were scored in each strain. Data are shown as mean ± SD. Unpaired two-tailed Student’s t test was performed to compare mutant datasets with WT or datasets that are linked by lines. ***, P < 0.001. (L–M′) DIC and confocal fluorescence images of WT adults expressing FLN-2::GFP in the head and tail regions. (N–O″) Confocal fluorescence images of the epidermis (N–N″) or the excretory canal (O–O″) in WT adults co-expressing FLN-2::RFP and VAB-10A::GFP (N–N″) or VHA-5::RFP (O–O″). FLN-2 overlaps with VAB-10A on the stripe-like fibrous organelles (N–N″). In the excretory canal cell, FLN-2 is aligned at the apical side of VHA-5 as indicated by arrowheads (N–N″). Scale bars, 5 μm.
Most fln-2(lf) mutant worms contain very faint VHA-5::RFP signals, while 7–15% of mutants contain aggregated VHA-5::RFP puncta (Figs. 1, B–C″; and S1, E–H). Mean VHA-5::RFP fluorescence intensity is significantly reduced in fln-2 mutants (Figs. 1 D and S1 K). The V1-E subunit VHA-8::GFP and the V1-A subunit VHA-13::GFP co-localize with VHA-5::RFP on apical vesicles in hyp7, and GFP fluorescence is reduced in fln-2 mutants (Figs. 1, A–D and S2, A–D). Although the fluorescent signal is reduced, the protein level of VHA-5::RFP or VHA-8::GFP is unaffected in fln-2 mutants (Fig. S2, E–G). Thus, loss of FLN-2 affects formation or maintenance of VHA-5–positive vesicles or their association with V-ATPase.
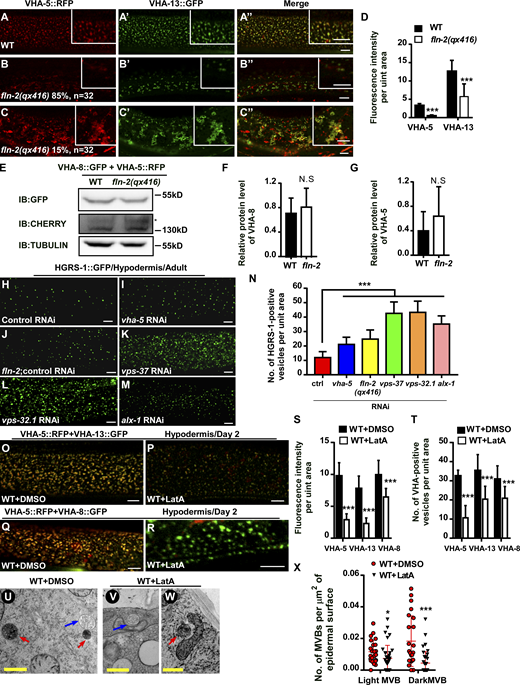
FLN-2 and actin cytoskeleton are required for MVB formation. (A–D) Confocal fluorescence images of the epidermis in WT (A–A″) and fln-2 (B–C″) co-expressing VHA-5::RFP and VHA-13::GFP at adult day 2. Average fluorescence intensity of VHA-5 and VHA-13 is quantified in D. At least 17 animals were scored in each strain. (E–G) The protein levels of VHA-5::RFP and VHA-8::GFP are unaltered in fln-2. Three independent immunoblot experiments were performed; E shows a representative result. (H–N) Confocal fluorescence images of the epidermis in WT animals expressing HGRS-1::GFP with the indicated RNAi treatments at adult day 2. The number of HGRS-1–positive vesicles per unit area (238 μm2) was quantified and shown in N. At least 14 animals were scored in each strain. (O–R) Merged confocal fluorescence images of the epidermis in WT day 2 adults co-expressing VHA-5::RFP and VHA-13::GFP (O and P) or VHA-8::GFP (Q and R) treated with either DMSO or LatA. (S and T) Mean fluorescence intensity and number of VHA-5–, VHA-13–, or VHA-8–positive vesicles per unit area (134 μm2) under the indicated treatments. 15 animals were scored in each treatment condition. (U–X) Representative TEM images of MVBs in the epidermis of WT day 2 adults treated with DMSO (U) or LatA (V and W). The number of MVBs per µm2 of epidermis in the indicated treatments is shown in X. 23 and 32 cross sections were quantified under DMSO and LatA treatment, respectively. In D, F, G, N, S, T, and X, data are shown as mean ± SD. Unpaired two-tailed Student’s t test was performed to compare mutant datasets with WT or control treatment or datasets that are linked by lines. *, P < 0.05; ***, P < 0.001. Scale bars, 5 μm in A–C″, H–M, and O–R; 500 nm in U–W. Source data are available for this figure: SourceData FS2.
FLN-2 and actin cytoskeleton are required for MVB formation. (A–D) Confocal fluorescence images of the epidermis in WT (A–A″) and fln-2 (B–C″) co-expressing VHA-5::RFP and VHA-13::GFP at adult day 2. Average fluorescence intensity of VHA-5 and VHA-13 is quantified in D. At least 17 animals were scored in each strain. (E–G) The protein levels of VHA-5::RFP and VHA-8::GFP are unaltered in fln-2. Three independent immunoblot experiments were performed; E shows a representative result. (H–N) Confocal fluorescence images of the epidermis in WT animals expressing HGRS-1::GFP with the indicated RNAi treatments at adult day 2. The number of HGRS-1–positive vesicles per unit area (238 μm2) was quantified and shown in N. At least 14 animals were scored in each strain. (O–R) Merged confocal fluorescence images of the epidermis in WT day 2 adults co-expressing VHA-5::RFP and VHA-13::GFP (O and P) or VHA-8::GFP (Q and R) treated with either DMSO or LatA. (S and T) Mean fluorescence intensity and number of VHA-5–, VHA-13–, or VHA-8–positive vesicles per unit area (134 μm2) under the indicated treatments. 15 animals were scored in each treatment condition. (U–X) Representative TEM images of MVBs in the epidermis of WT day 2 adults treated with DMSO (U) or LatA (V and W). The number of MVBs per µm2 of epidermis in the indicated treatments is shown in X. 23 and 32 cross sections were quantified under DMSO and LatA treatment, respectively. In D, F, G, N, S, T, and X, data are shown as mean ± SD. Unpaired two-tailed Student’s t test was performed to compare mutant datasets with WT or control treatment or datasets that are linked by lines. *, P < 0.05; ***, P < 0.001. Scale bars, 5 μm in A–C″, H–M, and O–R; 500 nm in U–W. Source data are available for this figure: SourceData FS2.
By transmission electron microscopy (TEM), VHA-5 was previously observed on the limiting membranes of both electron-lucent and electron-dark MVBs, which are called light and dark MVBs, respectively (Liegeois et al., 2006). We examined hypodermal MVBs in fln-2 mutants by TEM. The density (number per unit area) of light MVBs is significantly reduced in fln-2 hypodermis, while dark MVBs almost disappear (Fig. 1, E, F, I, and L). The diameter of light MVBs in fln-2(qx416) worms is unchanged, but the number of ILVs in light MVBs is lower (Fig. 1, J and K). CUP-5 is a lysosomal Ca2+ channel homologous to human TRPML1. Loss of CUP-5 affects lysosomal acidity and degradation activity (Hersh et al., 2002; Miao et al., 2020; Sun et al., 2011; Sun et al., 2020; Treusch et al., 2004). In cup-5(bp510) mutants, dark MVBs accumulate in the hypodermis while the density of light MVBs is reduced slightly (Fig. 1, G and L). In cup-5;fln-2 double mutants, there are fewer light and dark MVBs per unit of epidermal surface, as in fln-2 single mutants (Fig. 1, H and L). Thus, MVB biogenesis is impaired in fln-2 mutants.
FLN-2 and V-ATPase act in the same pathway to regulate MVB biogenesis
We constructed FLN-2::GFP and FLN-2::RFP reporter strains by CRISPR/Cas9. FLN-2 is expressed in the hypodermis including the main body syncytium hyp7 and the smaller head and tail hypodermal cells (Figs. 1, M and M′; and S1, L–M′). In hyp7, FLN-2::GFP shows both vesicular and parallel stripe-like patterns (Fig. 1 M′). FLN-2::GFP co-localizes with VHA-5::RFP on the vesicular structures (Fig. 1, N and N′). The parallel stripe-like pattern of FLN-2 overlaps with VAB-10A, a component of the epidermal attachment structures called fibrous organelles (FOs), which suggests that FLN-2 also localizes to the FOs (Fig. S1, N–N″; Zhang and Labouesse, 2010). FLN-2 is also expressed in the kidney-like excretory canal cell, which is important for osmoregulation (Fig. S1 O; Liegeois et al., 2006; Sundaram and Buechner, 2016). In the excretory canal, FLN-2::GFP is aligned at the apical side of VHA-5::RFP (Fig. S1, O–O″).
The V1 subunits VHA-8 and VHA-13 overlap with VHA-5 on the apical vesicles in hyp7 (Figs. 1, A–A″; and 2, A–A″ and D–D″). RNAi of vha-8 or vha-13 disrupts VHA-5::RFP vesicles and reduces the intensity of VHA-13::GFP (by vha-8 RNAi) or VHA-8::GFP (by vha-13 RNAi; Fig. 2, B–F). In vha-5 RNAi worms, VHA-8::GFP and VHA-13::GFP lose the bright vesicular localization pattern and become diffuse in the cytosol (Fig. 2, G–J). By TEM, the density of both light and dark MVBs is reduced in vha-5 RNAi and vha-8 RNAi worms as in fln-2(qx416) mutants, and the MVB density is not further reduced in double mutants defective in both fln-2 and vha-5 or vha-8 (Fig. 2, K–R). These data suggest that V-ATPase is required for MVB biogenesis and it acts in the same pathway with FLN-2.

V-ATPase is important for MVB formation. (A–F) Confocal fluorescence images of epidermis in control RNAi (A–A″ and D–D″), vha-8 RNAi (B–B″), or vha-13 RNAi worms (E–E″) co-expressing VHA-5::RFP and VHA-13::GFP (A–B″) or VHA-8::GFP (D–E″) at adult day 1. (C and F) Average fluorescence; n = 16 worms per group. (G–J) Confocal images of epidermis in control RNAi and vha-5 RNAi day 1 adults expressing VHA-8::GFP (G and H) or VHA-13::GFP (I and J). (K–R) TEM images and quantification of MVBs in day 2 adult epidermis. Blue arrows, light MVBs; red arrows, dark MVBs. For each strain, four to six cross-sections were scored in each of three to five worms. (C, F, and R) show mean ± SD. **, P < 0.01; ***, P < 0.001; N.S, not significant, by unpaired two-tailed Student’s t test. Scale bars, 5 μm (A–B″, D–E″, and G–J), 500 nm (K–Q).
V-ATPase is important for MVB formation. (A–F) Confocal fluorescence images of epidermis in control RNAi (A–A″ and D–D″), vha-8 RNAi (B–B″), or vha-13 RNAi worms (E–E″) co-expressing VHA-5::RFP and VHA-13::GFP (A–B″) or VHA-8::GFP (D–E″) at adult day 1. (C and F) Average fluorescence; n = 16 worms per group. (G–J) Confocal images of epidermis in control RNAi and vha-5 RNAi day 1 adults expressing VHA-8::GFP (G and H) or VHA-13::GFP (I and J). (K–R) TEM images and quantification of MVBs in day 2 adult epidermis. Blue arrows, light MVBs; red arrows, dark MVBs. For each strain, four to six cross-sections were scored in each of three to five worms. (C, F, and R) show mean ± SD. **, P < 0.01; ***, P < 0.001; N.S, not significant, by unpaired two-tailed Student’s t test. Scale bars, 5 μm (A–B″, D–E″, and G–J), 500 nm (K–Q).
FLN-2 and ESCRT complex act at different steps in MVB biogenesis
ESCRT complex is crucial for MVB biogenesis. The ESCRT-0 component HGRS-1/Hrs is recruited to the endosomal surface to initiate MVB formation (Gruenberg and Stenmark, 2004; Hanson and Cashikar, 2012). We found that HGRS-1 RNAi affects VHA-5::RFP-positive vesicles and makes the FLN-2::GFP signal more diffuse (Fig. 3, B–B″, G, and H). VHA-5::RFP vesicles are similarly affected in worms defective in vps-37 (encoding the ESCRT-I component VPS-37), vps-32.1 (encoding the ESCRT-III component VPS-32.1), or alx-1 (encoding the ESCRT associated protein ALX-1; Fig. 3, C, D, E, and H). In vps-37, vps-32.1, and alx-1 RNAi worms, the FLN-2::GFP signal is much lower (Fig. 3, C′–E″, and I). By TEM, the density of light MVBs is reduced in hypodermis of hgrs-1 RNAi worms, while dark MVBs almost disappear (Fig. 3, J–M). The diameter of light MVBs increases but the ILV number decreases (Fig. 3, N and O). Thus, ESCRT complex is important for the formation of VHA-5–positive MVBs.

ESCRT complex is required for MVB biogenesis. (A–E″) Confocal fluorescence images of epidermis in WT day 2 adults co-expressing VHA-5::RFP and FLN-2::GFP with the indicated RNAi treatments. (F and G) Line scan analyses of VHA-5::RFP and FLN-2::GFP along the dotted lines in (A″) and (B″). (H and I) Mean fluorescence intensity of VHA-5 (H) and FLN-2 (I) in the indicated RNAi treatments. n ≥ 15 worms per group. (J–L) TEM images of MVBs in the epidermis of control RNAi (J and K) and hgrs-1 RNAi (L) day 2 adults. Blue arrows, light MVBs; red arrows; dark MVBs. (M–O) MVB number/µm2 epidermal surface (M), ILV number (N) and light MVB diameter (O) in control RNAi and hgrs-1 RNAi worms. 4–5 cross sections were scored in each of 5 worms per group (M). n ≥ 59 light MVBs were scored per group (N and O). (P–R) Merged fluorescence images of epidermis in WT adults co-expressing HGRS-1::GFP and VHA-5::RFP (P) or FLN-2::RFP (Q). (R and S) Line scan analyses along the white lines in (P and Q). H, I, and M–O show mean ± SD. ***, P < 0.001, by unpaired two-tailed Student’s t test. Scale bars, 5 μm (A–E″, P, and Q), 500 nm (J–L).
ESCRT complex is required for MVB biogenesis. (A–E″) Confocal fluorescence images of epidermis in WT day 2 adults co-expressing VHA-5::RFP and FLN-2::GFP with the indicated RNAi treatments. (F and G) Line scan analyses of VHA-5::RFP and FLN-2::GFP along the dotted lines in (A″) and (B″). (H and I) Mean fluorescence intensity of VHA-5 (H) and FLN-2 (I) in the indicated RNAi treatments. n ≥ 15 worms per group. (J–L) TEM images of MVBs in the epidermis of control RNAi (J and K) and hgrs-1 RNAi (L) day 2 adults. Blue arrows, light MVBs; red arrows; dark MVBs. (M–O) MVB number/µm2 epidermal surface (M), ILV number (N) and light MVB diameter (O) in control RNAi and hgrs-1 RNAi worms. 4–5 cross sections were scored in each of 5 worms per group (M). n ≥ 59 light MVBs were scored per group (N and O). (P–R) Merged fluorescence images of epidermis in WT adults co-expressing HGRS-1::GFP and VHA-5::RFP (P) or FLN-2::RFP (Q). (R and S) Line scan analyses along the white lines in (P and Q). H, I, and M–O show mean ± SD. ***, P < 0.001, by unpaired two-tailed Student’s t test. Scale bars, 5 μm (A–E″, P, and Q), 500 nm (J–L).
HGRS-1::GFP labels abundant puncta in hyp7 (Fig. 3, P and Q). The inactivation of VPS-37, VPS-32.1, or ALX-1, which act downstream of HGRS-1 in the ESCRT pathway, increases the numbers of HGRS-1 vesicles (Fig. S2, H and K–N). Similarly, more HGRS-1 puncta are observed in vha-5 RNAi worms or fln-2 mutants than in controls (Fig. S2, I, J, and N). HGRS-1 puncta do not overlap with FLN-2– or VHA-5–positive vesicles in hyp7, which suggests that HGRS-1 and FLN-2 or VHA-5 are enriched on different vesicular compartments or subregions (Fig. 3, P–S). Together, these data suggest that FLN-2 and ESCRT complex act at different steps to regulate formation and/or maintenance of MVBs.
Disrupting the actin cytoskeleton affects MVB biogenesis
Filamins are well-characterized F-actin cross-linkers that organize the actin cytoskeleton in diverse processes (Lamsoul et al., 2020; Zhou et al., 2021). FLN-2::RFP associates with actin filaments visualized by the actin probe ABD::GFP (Fig. 4, A and B; Bosher et al., 2003; Liegeois et al., 2007). In fln-2(qx416), ABD::GFP is diffuse and disorganized, and its overall fluorescence intensity is lower (Fig. 4, C–F). This suggests that FLN-2 is important for actin filament organization. VHA-5::RFP vesicles stay close to the actin filaments (Fig. 4, G and I). Latrunculin A (LatA) treatment, which inhibits actin polymerization, causes reduced ABD::GFP and VHA-5::RFP signals (Fig. 4, H–J). The fluorescence intensity of VHA-8::GFP and VHA-13::GFP, and the numbers of VHA-5, -8, or -13–positive vesicles, are also reduced by LatA (Fig. S2, O–T). By TEM, the density of hypodermal light and dark MVBs is reduced after LatA treatment (Fig. S2, U–X). Thus, the actin cytoskeleton is important for the formation and/or maintenance of MVBs.

FLN-2 docks VHA-5 vesicles on the actin cytoskeleton. (A and B) Confocal fluorescence images (A) and line scan analysis (B) of the epidermis in WT adults co-expressing FLN-2::RFP and the actin probe ABD::GFP. (C–F) Confocal images of the epidermis in WT (C) and fln-2 (D) day 2 adults expressing ABD::GFP. (E) Line scan analyses. (F) Mean fluorescence intensity of ABD::GFP; n ≥ 17 worms per group. (G–J) Merged images of the epidermis in WT day 2 adults co-expressing VHA-5::RFP and ABD::GFP treated with DMSO (G) or LatA (H). (I) Line scan analyses. (J) Mean fluorescence intensity of VHA-5::RFP and ABD::GFP; n = 16 worms per group. (K–S) SIM fluorescence images of epidermis WT adults co-expressing VHA-5::RFP, ABD::GFP, and FLN-2::BFP (K–R). Boxed area in K is magnified in L–R. (S) Three different co-localization patterns of FLN-2, VHA-5, and actin as indicated in R. Yellow, red, and white arrows indicate VHA-5-, FLN-2-, and F-actin–positive structures, respectively. (T and U) Quantification of FLN-2-actin attachment (T) and position of VHA-5 vesicles relative to FLN-2 and actin (U). 10 (T) and 20 (U) different regions were scored in 8 and 15 worms, respectively. F, J, T, and U show mean ± SD. **, P < 0.01; ***, P < 0.001, by unpaired two-tailed Student’s t test. Scale bars, 5 μm (A–A″, C, D, G, and H), 1 μm (K–R).
FLN-2 docks VHA-5 vesicles on the actin cytoskeleton. (A and B) Confocal fluorescence images (A) and line scan analysis (B) of the epidermis in WT adults co-expressing FLN-2::RFP and the actin probe ABD::GFP. (C–F) Confocal images of the epidermis in WT (C) and fln-2 (D) day 2 adults expressing ABD::GFP. (E) Line scan analyses. (F) Mean fluorescence intensity of ABD::GFP; n ≥ 17 worms per group. (G–J) Merged images of the epidermis in WT day 2 adults co-expressing VHA-5::RFP and ABD::GFP treated with DMSO (G) or LatA (H). (I) Line scan analyses. (J) Mean fluorescence intensity of VHA-5::RFP and ABD::GFP; n = 16 worms per group. (K–S) SIM fluorescence images of epidermis WT adults co-expressing VHA-5::RFP, ABD::GFP, and FLN-2::BFP (K–R). Boxed area in K is magnified in L–R. (S) Three different co-localization patterns of FLN-2, VHA-5, and actin as indicated in R. Yellow, red, and white arrows indicate VHA-5-, FLN-2-, and F-actin–positive structures, respectively. (T and U) Quantification of FLN-2-actin attachment (T) and position of VHA-5 vesicles relative to FLN-2 and actin (U). 10 (T) and 20 (U) different regions were scored in 8 and 15 worms, respectively. F, J, T, and U show mean ± SD. **, P < 0.01; ***, P < 0.001, by unpaired two-tailed Student’s t test. Scale bars, 5 μm (A–A″, C, D, G, and H), 1 μm (K–R).
FLN-2 docks VHA-5 vesicles on actin filaments
We examined the subcellular localization of VHA-5, FLN-2, and actin filaments by super-resolution microscopy (Fig. 4 K). We co-expressed VHA-5:RFP, FLN-2::BFP, and ABD::GFP in the hypodermis and found that all VHA-5::RFP puncta are positive for FLN-2::BFP, while FLN-2::BFP-positive but VHA-5::RFP-negative structures are observed (Fig. 4, L, M, P, and U). FLN-2 is closely associated with actin filaments. Over 95% of FLN-2::BFP-positive structures are attached to actin filaments or overlap with ABD::GFP, while almost all actin filaments are attached to FLN-2 (Fig. 4, M–O, and T). Unlike FLN-2::BFP, most VHA-5::RFP vesicles stay adjacent to but do not directly associate with actin filaments (Fig. 4, L, N, Q, and U). We analyzed the relationship of VHA-5 vesicles with FLN-2 and actin. VHA-5 vesicles, which stay close to actin filaments, appear to attach to actin via FLN-2 (Fig. 4, R and S, vesicles #2 and #3). Some VHA-5 vesicles (e.g., #1 in Fig. 4, R and S) appear to dock directly on an actin filament. In this scenario, FLN-2::BFP is present on both the VHA-5::RFP-labeled vesicular structure and the ABD::GFP-labeled filamentous structure (Fig. 4, R and S). Out of >500 VHA-5–positive vesicles scored, 100% were labeled by FLN-2::BFP and 94% were attached to actin filaments by FLN-2. These data suggest that FLN-2 acts as a bridge between VHA-5–positive vesicles and the actin cytoskeleton, and the latter may be a platform for MVB assembly or maturation. In line with this, RNAi of vha-5, vha-8, or vha-13, which affect MVB biogenesis, does not disrupt F-actin organization or FLN-2 association with actin filaments (Fig. S3, A–F).
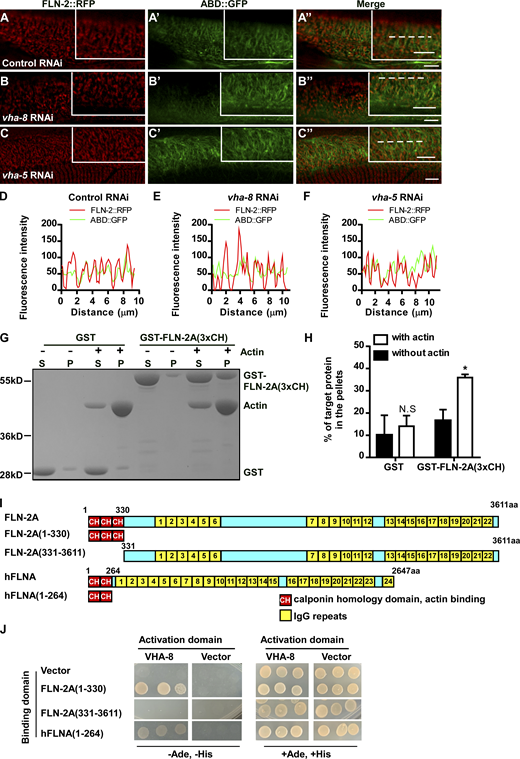
Inactivation of vha-5 and vha-8 does not affect the association of FLN-2 with actin. (A–F) Confocal fluorescence images of the epidermis in WT co-expressing FLN-2::RFP and ABD::GFP with the indicated RNAi treatments. (D–F) Line scan analyses of FLN-2::RFP and ABD::GFP along the dotted lines (A″, B″, and C″) in the indicated RNAi treatments. Scale bars, 5 μm. (G and H) In vitro F-actin co-sedimentation assay showing that GST-tagged FLN-2A(3xCH), but not GST, co-precipitates with F-actin. Three independent experiments were performed. Quantification is shown in H. Data are presented as mean ± SD. Unpaired two-tailed Student’s t test was performed to compare the two datasets (with actin versus without actin). *, P < 0.05; N.S not significant. (I) Schematic diagram showing domains of FLN-2A and human filamin A (hFLNA) and the truncations that were used in yeast-2 hybrid analyses. (J) Interactions between FLN-2A or hFLNA and VHA-8 were examined by Y2H analyses. FLN-2A(1-330, 3xCH) and hFLNA(1-264, 2xCH), but not FLN-2A(331-3611, Δ3xCH), interacted with VHA-8. Source data are available for this figure: SourceData FS3.
Inactivation of vha-5 and vha-8 does not affect the association of FLN-2 with actin. (A–F) Confocal fluorescence images of the epidermis in WT co-expressing FLN-2::RFP and ABD::GFP with the indicated RNAi treatments. (D–F) Line scan analyses of FLN-2::RFP and ABD::GFP along the dotted lines (A″, B″, and C″) in the indicated RNAi treatments. Scale bars, 5 μm. (G and H) In vitro F-actin co-sedimentation assay showing that GST-tagged FLN-2A(3xCH), but not GST, co-precipitates with F-actin. Three independent experiments were performed. Quantification is shown in H. Data are presented as mean ± SD. Unpaired two-tailed Student’s t test was performed to compare the two datasets (with actin versus without actin). *, P < 0.05; N.S not significant. (I) Schematic diagram showing domains of FLN-2A and human filamin A (hFLNA) and the truncations that were used in yeast-2 hybrid analyses. (J) Interactions between FLN-2A or hFLNA and VHA-8 were examined by Y2H analyses. FLN-2A(1-330, 3xCH) and hFLNA(1-264, 2xCH), but not FLN-2A(331-3611, Δ3xCH), interacted with VHA-8. Source data are available for this figure: SourceData FS3.
FLN-2 interacts via its calponin-homology domains with F-actin and VHA-8
Via alternative splicing, fln-2 encodes 22 different protein isoforms: 8 of them contain both actin-binding CH domains and multiple IgG repeats, while the rest contain IgG repeats only (Fig. 5 A). We fused the three CH domains of the FLN-2A isoform with GST and performed in vitro F-actin co-sedimentation assays. More GST-FLN-2A(3xCH) is pelleted when F-actin is included (Fig. S3, G and H), consistent with the role of FLN-2 in F-actin organization (Zhou et al., 2010). We examined whether FLN-2 interacts directly with MVB components. By yeast 2-hybrid (Y2H) analysis, FLN-2A(3xCH) interacts with VHA-8 but not VHA-5 or VHA-13 (Fig. 5 B). FLN-2A(331-3611), which lacks the three CH domains, does not interact with VHA-8, while the CH domains of human filamin A interact with VHA-8 (Fig. S3, I and J). The FLN-2D isoform contains 16 IgG repeats but lacks the N-terminal CH domains and does not interact with VHA-8, VHA-5, or VHA-13 by Y2H (Fig. 5, A and C). VHA-8-HIS is pulled down by GST-FLN-2A(3xCH) but not by GST, while VHA-13-HIS fails to interact with either GST-FLN-2A(3xCH) or GST (Fig. 5 D). Together, these data suggest that FLN-2 binds both F-actin and VHA-8 through its CH domains.
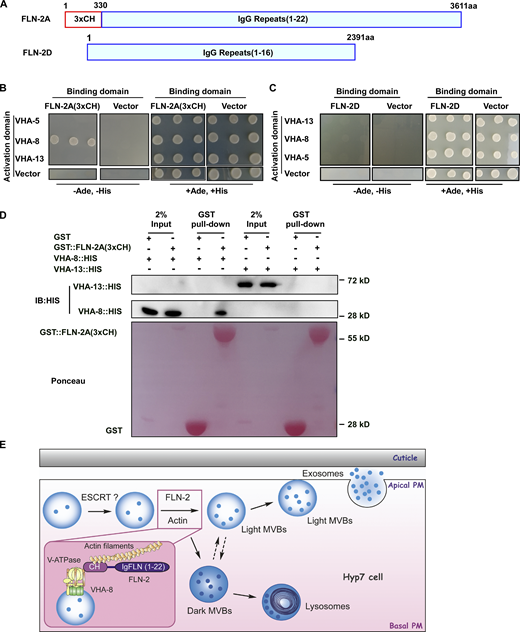
FLN-2 interacts with VHA-8 and actin.(A) Domains of FLN-2A and FLN-2D. (B–D) Y2H (B and C) and GST (D) pulldown analyses of interactions between FLN-2A(3xCH) or FLN-2D and VHA-5/8/13 subunits. FLN-2A(3xCH) interacts with VHA-8 but not VHA-13 or VHA-5. n ≥ 3 independent GST pull-down experiments; D is a representative result. IB, immunoblot. (E) Model of FLN-2 and actin in MVB biogenesis. FLN-2 facilitates MVB biogenesis by acting as a cross-linking protein to organize actin filaments and as a bridging molecule to dock MVBs on the actin cytoskeleton. ESCRT components may function upstream of FLN-2 to regulate MVB formation. Source data are available for this figure: SourceData F5.
FLN-2 interacts with VHA-8 and actin.(A) Domains of FLN-2A and FLN-2D. (B–D) Y2H (B and C) and GST (D) pulldown analyses of interactions between FLN-2A(3xCH) or FLN-2D and VHA-5/8/13 subunits. FLN-2A(3xCH) interacts with VHA-8 but not VHA-13 or VHA-5. n ≥ 3 independent GST pull-down experiments; D is a representative result. IB, immunoblot. (E) Model of FLN-2 and actin in MVB biogenesis. FLN-2 facilitates MVB biogenesis by acting as a cross-linking protein to organize actin filaments and as a bridging molecule to dock MVBs on the actin cytoskeleton. ESCRT components may function upstream of FLN-2 to regulate MVB formation. Source data are available for this figure: SourceData F5.
Here, we show that docking on the actin cytoskeleton is required for MVB biogenesis, and we identified C. elegans filamin FLN-2 as a key regulator in this process. Our data suggest that FLN-2 is both a cross-linking protein that organizes actin filaments and a bridging molecule that docks MVBs on the actin cytoskeleton, and thus facilitates the formation and/or maintenance of MVBs (Fig. 5 E). Similar to filamins in other species, FLN-2 may solidify and promote the orthogonal crosslinking of actin filaments, resulting in actin gel networks that support MVB biogenesis (Stossel et al., 2001). FLN-2 may also act as a scaffold to recruit factors involved in MVB assembly or ILV formation. Our data suggest that actin and V-ATPase are required for MVB biogenesis. The actin cytoskeleton may anchor VHA-5–positive MVBs for assembly and/or maturation. Actin may also provide a scaffold to organize factors critical for MVB formation, as in regulated exocytosis (Li et al., 2018). Previous work identified mutations in VHA-5 that affect V0-specific function, leading to defects in apical secretion but not in the biogenesis of MVBs (Liegeois et al., 2006). We found that inactivation of VHA-5, VHA-8, or VHA-13 affected MVB formation or maintenance, which suggests that both V0 and V1 functions are important in MVB biogenesis. In addition to mediating MVB-actin tethering via VHA-8–FLN-2 interaction, V-ATPase subunits may recruit factors essential for MVB biogenesis, as in endocytic transport and membrane fusion processes, or they may promote ILV formation by maintaining lumenal acidity (Clague et al., 1994; Di Giovanni et al., 2010; Falguieres et al., 2008; Hurtado-Lorenzo et al., 2006). However, as VHA-5 vesicles were not readily stained by lysotracker red, further work is needed to determine whether lumenal acidity contributes to MVB biogenesis.
Our work reveals a novel role of the versatile filamin proteins in MVB biogenesis, which may provide additional insights into the pathogenesis of filamin-related diseases. FLN-2–VHA-8/V1-E interaction should be further explored, especially in filamin- and V-ATPase–related diseases. More work is also needed to address how the docking of MVBs on the actin cytoskeleton by FLN-2 promotes MVB biogenesis and how this is connected to ESCRT function in cargo sorting and ILV formation.
Materials and methods
C. elegans strains
C. elegans strains were cultured and maintained using standard protocols. N2 Bristol was used as the wild-type strain except for polymorphism mapping, in which the Hawaiian CB4856 strain was used. Fluorescent protein knock-in at the endogenous loci was accomplished by CRISPR/Cas9-mediated genome editing. Insertion of single-copy arrays at the specific genomic region on chromosome IV was achieved by CRISPR/Cas9. The following C. elegans strains were used in this work: LG I, cas602 (VAB-10A::GFP, Cas9 knock-in); LG II, mcSi52 (Pvha-5VHA-5::mRFP, single-copy insertion); LG III, cup-5(bp510); LG IV, qxSi41 [Pvha-8VAB-10B (ABD,1-290)::GFP, single-copy insertion], qxSi47 (Pvha-8VHA-8::GFP, single-copy insertion), qx801 (VHA-5::GFP::MTn, Cas9 knock-in); LG V, qxIs430 (Pscav-3SCAV-3::GFP); and LG X, fln-2(qx416, qx439, qx463), qx504 (FLN-2::GFP, Cas9 knock-in), qx531 (FLN-2::tagRFP-T, Cas9 knock-in), qx673 (FLN-2::ceBFP, Cas9 knock-in). Transgenic reporters: Phgrs-1HGRS-1::GFP, yqEx199 (Pvha-13VHA-13::GFP).
We obtained mcSi52 from Dr. Michel Labouesse (Institut de Biologie Paris, Paris, France), cup-5(bp510) from Dr. Hong Zhang (Institute of Biophysics, Chinese Academy of Sciences, Beijing, China), HGRS-1::GFP reporter from R. Legouis (Institute for Integrative Biology of the Cell, Gif-sur-Yvette, France), and cas602 from Dr. Guangshuo Ou (Tsinghua University, Beijing, China).
Isolation, mapping, and cloning of fln-2
The qx416, qx439, and qx463 mutations were isolated from a forward genetic screen for animals showing abnormal VHA-5::RFP distribution at adult stages. qx416 was mapped to linkage group X (LG X) between genetic map positions 0.41 and 1.732 by single nucleotide polymorphism mapping. A transgene containing fosmid WRM0620dC04 restored the VHA-5::RFP pattern in qx416 mutants. WRM0620dC04 contained two genes, fln-2 and c23f12.9. Sequencing of qx416 identified a C-to-T mutation in fln-2, which causes a premature stop codon at Gln1316. The qx439 and qx463 mutations are allelic to qx416. Sequencing analyses identified C-to-T mutations in qx439 and qx463 that cause premature stop codons at Arg1199 and Arg1433, respectively. The qx416, qx439, and qx463 mutations were backcrossed with N2 worms four times before further analysis.
Knock-in tags and single-copy arrays were inserted by CRISPR/Cas9. Tag insertion at the endogenous loci by Cas9-mediated homologous recombination was performed as previously described (Paix et al., 2014). Briefly, single guide RNA (sgRNA) for a specific genomic targeting site was cloned into pDD162 vector (pDD162-Peft-3::Cas9-Pu6::sgRNA), which expresses the Cas9 protein and sgRNA. Repair templates containing desired editing sequences and homologous arms with the targeted region were used to achieve site-directed editing. Here, dpy-10 was used as a selection marker for co-conversion events in the F1 progeny as reported previously. To insert GFP or tagRFP-T at the FLN-2 locus, a single guide RNA (sgRNA1: 5′-ATGATTGCGTCCTTTATGTTTGG-3′) and linear homologous repair templates encompassing 30–35 nt of homology arms were used. To insert ceBFP at the C-terminus of FLN-2, two sgRNAs (sgRNA1: 5′-ATGATTGCGTCCTTTATGTTTGG-3′; and sgRNA2: 5′-TATGGGCTTGGAACACCCATTGG-3′) and a repair template plasmid with ceBFP containing ∼1 kb homology arms were used to increase the editing efficiency. Following injection, dpy-10 was used as a selection marker for co-conversion events in the F1 progeny, as reported previously. Dpy or roller F1 worms were picked and screened for tag insertion by PCR. Positive candidates were confirmed by sequencing. Single-copy insertion of VHA-8 and F-actin reporter was performed as described before (Takayanagi-Kiya et al., 2016). In brief, a Pvha-8VHA-8::GFP::UNC-54 3′UTR or Pvha-8VAB-10B(ABD)::GFP::UNC-54 3′UTR cassette was cloned into a donor vector containing LG-IV–targeted homology arms and a hygromycin resistance gene as the selection marker. Injected worms were screened on hygromycin plates for array insertion. Pmyo-2GFP was co-injected to exclude the candidates with extrachromosomal arrays, which were also hygromycin-resistant. The positive recombinants were sequenced to confirm correct insertion. All strains obtained were outcrossed with the N2 wild-type strain four times before further analysis.
RNAi experiments
RNAi bacterial clones from Ahringer RNAi library (Source BioScience) were used for the RNAi experiments, and L4440 empty vector strain was used as control. Feeding RNAi was performed using standard procedures. For most RNAi experiments, 30–100 synchronized L1 larvae were cultured on RNAi plates and examined for corresponding phenotypes at the stage indicated in the figure legends. For RNAi that caused severe lethality or larval arrest (i.e., vha-8 or vha-13 RNAi), synchronized L1 larvae were cultured on OP50 plates for about 17 h and worms were washed and transferred to corresponding RNAi plates. Phenotypes were examined at the stage indicated in the figure legends.
Microscopy and imaging analysis
Differential interference contrast (DIC) and fluorescence images were taken by an inverted confocal microscope (LSM 880; Carl Zeiss) with 488 (emission filter BP 503–530) and 543 (emission filter BP 560–615) lasers. A 63× objective (NA 1.30; Plan-Neofluar) was used with Immersol 518F oil (Carl Zeiss). Images were processed and viewed using ZEN software (Carl Zeiss). Super-resolution imaging of MVB, FLN-2, and F-actin was performed on a Delta Vision OMX V3 imaging system (Applied Precision). Laser lines at 405, 488, and 561 nm were used for excitation, and images were captured with a 100× objective (NA 1.4; Olympus UplanSApo) and charge-coupled device (CCD) cameras (Evolve 512×512; Photometrics) in the presence of immersion oil using conventional mode. Images were processed by deconvolution methods using softWoRx 5.0 software (Applied Precision) with the following settings: Wiener filter enhancement 0.9; Wiener filter smoothing 0.8. Representative images shown are prepared in ImageJ software.
All images were taken at 20°C. To quantify the fluorescence intensity of VHA-5::RFP, VHA-8::GFP, VHA-13::GFP, ABD::GFP, and FLN-2::GFP, images of strains for comparison were captured with a 63× objective using the same exposure setting. Mean fluorescence intensity was determined by ImageJ software (National Institute of Health). At least 15 animals in each strain were scored. For line scan analyses, fluorescence intensity values along the dotted or solid lines of a given image were extracted with ImageJ software and plotted using Graphpad Prism 7.
Quantification of the overlap of FLN-2, actin, and VHA-5–positive vesicles
Fluorescence images of adult worms co-expressing VHA-5::RFP, FLN-2::BFP, and ABD::GFP were captured on a Delta Vision OMX(3D-SIM) microscope with the conventional mode due to the weak fluorescence signal of VHA-5::RFP. 10 regions with distinguishable and countable FLN-2/F-actin structures (FLN-2 or F-actin structures were sometimes too dense or irregular in shape and thus uncountable) were selected and manually quantified for FLN-2 attachment with actin. 8 animals were quantified, and in total 215 FLN-2–positive structures and 184 F-actin structures were scored. To quantify the positioning of VHA-5–positive vesicles in relation to FLN-2 or actin, 20 regions from 15 animals were selected and manually quantified. For VHA-5 vesicles, three different co-localization patterns were observed: (1) not adjacent to actin; (2) docks on actin via FLN-2; (3) overlaps with FLN-2. A total number of 526 VHA-5 vesicles were scored and the percentages of each co-localization pattern were quantified.
Quantification of HGRS-1– and VHA-positive vesicles
After RNAi treatments, fluorescence images of day 2 adults expressing HGRS-1::GFP were captured using a 63× objective on a confocal microscope (LSM 880; Carl Zeiss) with the same exposure setting. The number of HGRS-1–positive vesicles within a unit area (238 μm2) was manually counted. At least 17 animals were quantified for each RNAi condition.
After LatA or DMSO treatment, fluorescence images of day 2 adults co-expressing VHA-5::RFP and VHA-13::GFP or VHA-8::GFP were captured using a 63× objective on a confocal microscope (LSM 880; Carl Zeiss) with the same exposure setting. The number of VHA-5–, VHA-8–, or VHA-13–positive vesicles within the unit area (134 μm2) was manually counted. At least 15 animals were scored in each treatment condition.
TEM analysis
Day 2 adult C. elegans were rapidly frozen using a high-pressure freezer (EM HPM100; Leica Biosystems). A freezing substitution was performed in anhydrous acetone containing 1% osmium tetroxide. The samples were kept sequentially at −90°C for 72 h, −60°C for 12 h, and −30°C for 10 h and were finally brought to 0°C in a freeze-substitution unit (EM AFS2; Leica Biosystems). The samples were then washed three times (15 min each time) in fresh anhydrous acetone and further fixed in anhydrous acetone containing 0.5% uranyl acetate overnight at 4°C. The samples were gradually infiltrated with Embed-812 resin in the following steps: resin/acetone 1:3 for 3 h, 1:1 for 5 h, 3:1 overnight, and 100% resin for 2, 12, 12 h. Samples were then embedded at 60°C for 48 h. The fixed samples were cut into 70 nm sections with a microtome EM UC7 (Leica Biosystems). Sections were observed with a transmission electron microscope (JEM-1400 JEOL Japan) operated at 80 kV, and digital images were captured with a 4K×2.7K pixels CCD camera (Gatan). To analyze MVBs in the epidermis, 4–9 cross sections per worm were examined in 3–5 animals for each genotype/treatment. The total surface area of the epidermis in each section was measured by ImageJ software. The numbers of MVBs or ILVs within light MVBs were manually counted. Diameters of MVBs were measured by ImageJ software.
LatA treatment
NGM plates were supplemented with 1 μM LatA (ab144290; Abcam) and then spotted with OP50 E. coli strain. 100–300 synchronized L1 larvae of each genotype were then cultured on the LatA-supplemented plates for about 72 h, after which the MVB phenotypes were examined. Worms cultured on DMSO-supplemented plates to a similar stage (2-day post L4) were used as control. DMSO or LatA-supplemented plates were kept in the dark during the whole process.
Immunoblotting
To examine protein levels of VHA-5::RFP and VHA-8::GFP, ∼200 adult day 2 worms of each genotype were lysed by 3 rounds of freeze–thaw in 30 μl 1× SDS loading buffer followed by boiling at 100°C for 10 min. The resulting whole worm lysate samples were resolved by SDS-PAGE and blotted with anti-GFP (mouse; 11814460001; 1:1,000; Roche), anti-CHERRY (mouse; KM8017; 1:1,000; Sungene Biotech) and anti-α-tubulin (mouse; T5168; 1:5,000; Sigma-Aldrich) antibodies. The intensity of the protein band was quantified by ImageJ software. The relative protein level of VHA-5 and VHA-8 was calculated by dividing the band intensity of VHA-5::RFP or VHA-8::GFP by TUBULIN (internal control). Three independent experiments were performed and quantified in each strain.
F-actin co-sedimentation assay
The in vitro F-actin co-sedimentation assay was performed as previously described (Nakamura et al., 2007). Briefly, 3 μM of purified GST-FLN-2(ABD) or GST proteins were mixed with or without 5 μM G-actin in F-actin polymerization buffer (20 mM Tris-HCl, pH 7.5, 0.5 mM ATP, 5 mM MgCl2, 120 mM NaCl, and 0.2 mM DTT) and incubated at 25°C for 1 h. The solutions were then centrifuged at 200,000 g (TLA-100; Beckman) for 30 min to pellet F-actin. Proteins in the supernatants or pellets were solubilized in SDS loading buffer and analyzed by SDS-PAGE. Protein bands were visualized by Coomassie Blue staining. For quantification, the band intensity of GST and GST-FLN-2A(3xCH) in the supernatant and pellet of each group was quantified by ImageJ. The percentage of the target protein (GST or GST-FLN-2A[3xCH]) in the pellet was calculated by dividing the protein amount in the pellet by the total protein amount (supernatant plus pellet). Three independent experiments were performed and quantified. Unpaired two-tailed Student’s t test was performed to compare the two datasets (with actin versus without actin).
Y2H assay
The Y2H assay was performed according to the manufacturer’s instructions (Clonetech). Briefly, cDNA of target genes was cloned into pGADT7 and pGBKT7 vectors which contain the activation domain (AD) and DNA binding domain (DB) of Gal4, respectively. Specific pairs of constructs expressing AD and DB fusion proteins were then transformed into the AH109 yeast strain, and transformants were selected on a synthetic complete (SC) medium lacking leucine and tryptophan (SC-Leu-Trp). Individual clones were streaked on SC-Ade-His-Leu-Trp plates to test for the activation of the reporter genes HIS3 and ADE2.
GST pull-down assay
To examine protein interaction between FLN-2(3xCH) and VHA-8 or VHA-13, equal micromolar amounts of purified GST or GST-FLN-2(3xCH) recombinant proteins were immobilized on glutathione–agarose beads and incubated with equal amounts of VHA-8-His or VHA-13-His in GST-binding buffer (25 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 0.1% NP-40) for 2 h. The beads were then washed with GST-binding buffer seven times, and the bound proteins were eluted with SDS loading buffer and analyzed by Western blot. VHA-8 or VHA-13 was detected by anti-His antibody (mouse, sc-8036, 1:1,000; Santa Cruz), and GST-fusion proteins were visualized by Ponceau S staining.
Plasmid construction
To generate FLN-2::GFP/tagRFP-T/ceBFP Cas9 knock-in strains, sgRNA1 and sgRNA2 were inserted into the pDD162 vector with primers PLLS229/230 and PLLS1333/1334 by PCR. Linear repair templates of GFP and tagRFP-T were amplified using primers PLLS231/232 and PLLS498/499. To generate ceBFP repair template plasmid, ∼1 kb of fln-2-5′ and fln-2-3′ homology arms were amplified using primers PLLS1327/1328 and PLLS1329/1330 and sequentially ligated to pPD49.26-ceBFP through Kpn I and Sac I sites. The above plasmids were further quick-changed using primers PLLS1337/1338 by PCR to introduce the synonymous substitution at the sgRNA site. To generate single-copy array insertion of Pvha-8VHA-8::GFP, vha-8 genomic DNA including the 1,155 bp promoter region was amplified using primers PLLS764/PTCH5 and ligated into pPD49.26-GFP through Bam HI-Kpn I sites. The whole Pvha-8VHA-8::GFP::UNC-54 3′UTR cassette was amplified using primers PJYH251/252 and ligated into pCFJ201-hygR CasSCI donor vector through Sph I-Spe I sites (Takayanagi-Kiya et al., 2016). To generate a single copy of Pvha-8VAB-10B(ABD,1-290aa)::GFP, the 1,155 bp promoter of vha-8 was firstly amplified using primers PLLS764/765 and ligated into pPD49.26-GFP through the Bam HI site, then a cDNA fragment of VAB-10B(ABD,1-290aa) was amplified using primers PLLS682/683 through Nhe I-Kpn I sites. As mentioned above, the whole Pvha-8VAB-10B(ABD,1-290aa)::GFP::UNC-54 3′UTR cassette was amplified using primers PJYH251/252 and ligated into pCFJ201-hygR CasSCI donor vector through Sph I-Spe I sites. To generate pGBKT7-FLN-2A(3xCH), a cDNA fragment of FLN-2A(1-330aa) was amplified using primers PLLS821/822 and ligated into pGBKT7 vector through Nde I-Bam HI sites. To generate pGBKT7-FLN-2D, cDNA of the FLN-2D isoform was amplified using primers PTCH144/145 and ligated into pGBKT7 vector through Nde I-Bam HI sites. To generate pGADT7-VHA-5/VHA-8/VHA-13, vha-5/vha-8/vha-13 cDNA was amplified using primers PTCH150/151, PTCH152/153, and PTCH156/157, respectively, and ligated into pGADT7 vector through Nde I-Bam HI sites. To generate pGBKT7-FLN-2A(Δ 3xCH), a cDNA fragment of FLN-2A(331-3611aa) was amplified using primers PLLS3613/3614 and ligated into pGBKT7 vector through Nde I-Bam HI sites.
To generate pGBKT7-hFLNA(2xCH), a cDNA fragment of hFLNA (1–264aa) was amplified using primers PLLS3615/3616 and ligated into pGBKT7 vector through Nde I-Bam HI sites. To generate GST::FLN-2A(3xCH), a cDNA fragment of FLN-2A(1–330aa) was amplified using primers PLLS1819/1822 and ligated into pGEX-6P1 vector through Bam HI-Not I sites. To generate VHA-8::HIS, vha-8 cDNA was amplified using primers PLLS1485/1457 and ligated into pET-21b through Nde I-Xho I sites. To generate VHA-13::HIS, vha-13 cDNA was amplified using primers PLLS1765/1766 and ligated into pET-21b through Nde I-Not I sites.
Primers used for plasmid construction
See Table 1 for the list of primers used for plasmid construction.
Primers used for plasmid construction
| Primers | Sequence (5′ to 3′) |
|---|---|
| PLLS229 | TGATTGCGTCCTTTATGTTGTTTTAGAGCTAGAAATAGC |
| PLLS230 | AACATAAAGGACGCAATCACAAGACATCTCGCAATAGGAG |
| PLLS1333 | ATGGGCTTGGAACACCCATGTTTTAGAGCTAGAAATAGC |
| PLLS1334 | ATGGGTGTTCCAAGCCCATCAAGACATCTCGCAATAGGAG |
| PLLS231 | GTTCCAAGCCCATATGGATCCGTTCCACCTCCG |
| PLLS232 | TGTAGTTTGGAATTGAGGTATCACTAATCAATACAATTTATTTGTATAGTTCATCCATGCCATG |
| PLLS498 | GTTCCAAGCCCATATGGATCCGTTCCACCTCCGACAAAGCACAAGGGCAGAAACCACATGGTGTCTAAGGGCGAAGAGCTG |
| PLLS499 | TGTAGTTTGGAATTGAGGTATCACTAATCAATACAAT TTACTTGTACAGCTCGTCCATGCCATT |
| PLLS1327 | TGGCTAGCGTCGACGGTACCGCTGTTTCTGCAGACACATCTG |
| PLLS1328 | AATAAGCTCTGACATGGTACCATGATTGCGTCCTTTATGTTTG |
| PLLS1329 | CACAAGCTTAATTAGGAGCTCATTGTATTGATTAGTGATACC |
| PLLS1330 | GACAGCGGCCGATGCGGAGCTCCCTCTCGCTGTTCACAAGTTCC |
| PLLS1337 | CGGACCGATCGGTGTTCCAAGCCCATATGGATCCGTT |
| PLLS1338 | GAACACCGATCGGTCCGAAATGGCCTGGTGGTGGCAG |
| PLLS764 | GCGGATCCAAAGTATTGTCCGCAAGGCACATC |
| PTCH5 | GCGGTACCGTCGAAGAAAGAACGGTTT |
| PJYH251 | CTCTAGAAAGTATAGGAACTTCGCATGCCATGATTACGCCAAGCTTGCATGC |
| PJYH252 | CAATAGAGGACCGCTACACACTAGTCGCGAGACGAAAGGGCCC |
| PLLS765 | GCGGATCCGTAAATTATTTTATTAGTAAAAAGG |
| PLLS682 | GCGCTAGCATGGTTGGAGAGAGGTATCGTAG |
| PLLS683 | GCGGTACCCTCCGGCTCGTGTGGAAGAGC |
| PLLS821 | GCCATATGATGGGCAAGCAGGGAGAAGAAGC |
| PLLS822 | GCGGATCCATTTATACAGAACAAAGTGCTGCAACAAG |
| PTCH144 | TACCAGATTACGCTCATATGATGCTGGCGGACCAACGTGTAC |
| PTCH145 | GCTCGAGCTCGATGGATCCTTAATGATTGCGTCCTTTATG |
| PTCH150 | TACCAGATTACGCTCATATGATGGGGTCGTTGTCGCGCTCG |
| PTCH151 | GCTCGAGCTCGATGGATCCTTAAGCTGCTTCAGCCTCCTGAAG |
| PTCH152 | TACCAGATTACGCTCATATGATGGGAATCAGCGACAACGAC |
| PTCH153 | GCTCGAGCTCGATGGATCCTTAGTCGAAGAAAGAACGGTTT |
| PTCH156 | TACCAGATTACGCTCATATGATGGCCGCAGAATCTTCG |
| PTCH157 | GCTCGAGCTCGATGGATCCTTAATCCTCGAGGTTTCTGAA |
| PLLS3613 | TGATCTCAGAGGAGGACCTGCATATGTCACTTGAGCAACAAGTAAAATA |
| PLLS3614 | TGCGGCCGCTGCAGGTCGACGGATCCTTAATGATTGCGTCCTTTATGTTTG |
| PLLS3615 | TGATCTCAGAGGAGGACCTGCATATGATGAGTAGCTCCCACTCTCGGGC |
| PLLS3616 | TGCGGCCGCTGCAGGTCGACGGATCCttaGAACTGGGACAGGTAGGTCATG |
| PLLS1819 | CTGTTCCAGGGGCCCCTGGGATCCATGGGCAAGCAGGGAGAAGAAG |
| PLLS1822 | GATCGTCAGTCAGTCACGATGCGGCCGCTTATACAGAACAAAGTGCTGCAACAAG |
| PLLS1485 | ATGCCATATGATGGGAATCAGCGACAACGACG |
| PLLS1457 | GCCTCGAGGTCGAAGAAAGAACGGTTTGG |
| PLLS1765 | GCATGCATATGATGGCCGCAGAATCTTCGTACG |
| PLLS1766 | GCGCGGCCGCATCCTCGAGGTTTCTGAAAGCG |
Statistical analysis
In the graphs, standard deviation (SD) was used as y-axis error bars. Data derived from different genetic backgrounds or drug treatments were compared by Student’s two-tailed unpaired t test. Data were considered statistically different at P < 0.05. P < 0.05 is indicated with single asterisks, P < 0.01 with double asterisks, and P < 0.001 with triple asterisks.
Online supplemental material
Acknowledgments
We thank Michel Labouesse (Institut de Biologie Paris, France) and Guangshuo Ou (Tsinghua University, China) for strains, Shuoguo Li and Center for Biological Imaging (CBI), Institute of Biophysics, for help with super-resolution imaging, and Isabel Hanson for editing services.
Some strains were from Caenorhabditis Genetics Center, which is funded by National Institutes of Health Office of Research Infrastructure Programs (P40OD010440). This work was supported by the National Key R&D Program of China (grant 2021YFA1300303) and the National Natural Science Foundation of China (grants 91754203, 32130028, and 31630018) to X. Wang.
The authors declare no competing financial interests.
Author contribution: X. Wang conceived and supervised the research. L. Shi performed most of the experiments. Y. Jian and M. Li performed EM analyses. T. Hao contributed experimental materials. C. Yang supervised EM experiments. X. Wang and L. Shi wrote the manuscript.

