


Neuregulins (NRGs) are EGF-like ligands associated with cognitive disorders. Unprocessed proNRG3 is cleaved by BACE1 to generate the mature membrane-bound NRG3 ligand, but the subcellular site of proNRG3 cleavage, mechanisms underlying its transport into axons, and presynaptic accumulation remain unknown. Using an optogenetic proNRG3 cleavage reporter (LA143-NRG3), we investigate the spatial-temporal dynamics of NRG3 processing and sorting in neurons. In dark conditions, unprocessed LA143-NRG3 is retained in the trans-Golgi network but, upon photoactivation, is cleaved by BACE1 and released from the TGN. Mature NRG3 then emerges on the somatodendritic plasma membrane from where it is re-endocytosed and anterogradely transported on Rab4+ vesicles into axons via transcytosis. By contrast, the BACE1 substrate APP is sorted into axons on Rab11+ vesicles. Lastly, by a mechanism we denote “trans-synaptic retention,” NRG3 accumulates at presynaptic terminals by stable interaction with its receptor ErbB4 on postsynaptic GABAergic interneurons. We propose that trans-synaptic retention may account for polarized expression of other neuronal transmembrane ligands and receptors.
Introduction
Neuregulins (NRGs) are a family of signaling proteins encoded by four genes (NRG1–NRG4) that harbor an EGF-like (EGF-L) domain necessary to bind ErbB3/4 receptor tyrosine kinases. The NRG/ErbB signaling pathway has been implicated in numerous aspects of neural development, and genetic variants of NRG1 and ERBB4 have been associated with risk for psychiatric disorders, especially schizophrenia (Buonanno and Fischbach, 2001; Falls, 2003; Mei and Nave, 2014). Although NRG3 is the most widely expressed NRG throughout central nervous system (CNS) development (Longart et al., 2004; Rahman et al., 2019; Zhang et al., 1997), only recently have its processing, subcellular distribution, functions in CNS neurons, and association with disease begun to be investigated. Processes regulated by NRG3 include interneuron migration (Bartolini et al., 2017) and neurite outgrowth (Rahman-Enyart et al., 2020), neuronal and oligodendrocyte survival (Carteron et al., 2006), glutamatergic transmission (Wang et al., 2018), and synapse formation/maturation onto ErbB4+ GABAergic interneurons (Exposito-Alonso et al., 2020; Müller et al., 2018; Vullhorst et al., 2017). Furthermore, NRG3 single-nucleotide polymorphisms have been associated with risks for psychiatric disorders (Avramopoulos, 2018; Kao et al., 2010; Loos et al., 2014; Morar et al., 2011; Paterson et al., 2017; Zeledon et al., 2015; Zhou et al., 2020), and NRG3 knockout mice exhibit alterations in neuronal network activity and behaviors associated with schizophrenia (Hayes et al., 2016; Müller et al., 2018; Wang et al., 2018). It is therefore imperative to understand the cellular processes that regulate NRG3 processing and targeting to specific neuronal compartments.
NRGs are translated as unprocessed transmembrane (TM) pro-proteins (proNRG) that require proteolytic cleavage at a juxtamembrane site located between the EGF-L and TM domains to become signaling-competent (Falls, 2003; Fleck et al., 2012; Willem, 2016). We recently showed that, based on their TM topology and signaling mode, NRGs can be broadly categorized into two groups: (1) single-pass TM NRGs (sp-NRGs), such as NRG1 (types I, II, and IV) and NRG2, which shed their ectodomains into the extracellular space and thus signal in autocrine or paracrine mode, and (2) dual-pass TM NRGs (dp-NRGs), such as NRG1 (type III) and NRG3, which harbor an additional TM domain near the amino terminus (TMN) that enables them to remain membrane-anchored upon processing and thus signal in juxtacrine mode (Vullhorst et al., 2017; Wang et al., 2001). Traditionally, NRGs had been thought of as axonal/presynaptic factors regardless of isoform and TM topology. However, we recently reported that sp-NRGs accumulate on cell bodies and proximal dendrites where they are shed by ADAM10 in response to N-methyl-D-aspartate receptor activation (Vullhorst and Buonanno, 2019; Vullhorst et al., 2015), whereas pro-forms of dp-NRGs are processed by BACE1 and accumulate in axons and presynaptic processes (Fleck et al., 2013; Müller et al., 2018; Vullhorst et al., 2017). While the aforementioned studies demonstrated selective NRG3 accumulation in axons, the underlying molecular mechanisms and transport machinery remain elusive.
Membrane proteins are usually carried to their proper subcellular domains through transport vesicles. Due to the highly polarized nature of neurons, multiple coordinated activities are necessary for the sorting, packaging, trafficking, and kinetics of vesicular cargo to specific neuronal domains (reviewed by Bentley and Banker [2016], Bourke et al. [2018], Lasiecka and Winckler [2011], Nabb et al. [2020], and Roy [2020]). Sorting to the somatodendritic domain is often mediated by recognition of sorting signals in the cytosolic domain of transmembrane protein cargo by adaptor proteins that are components of protein coats, most notably the adaptor protein 1 complex (Farias et al., 2012; reviewed by Bonifacino [2014] and Guardia et al. [2018]). By contrast, no such signals have been identified for sorting of axonal proteins, and the mechanisms involved in this process remain elusive (reviewed by Wisco et al. [2003], Lasiecka and Winckler [2011], and Bentley and Banker [2016]). Proposed mechanisms include direct transport from the TGN to the axon (Gu et al., 2003; Sampo et al., 2003; Li et al., 2016; Fletcher-Jones et al., 2019), delivery to the somatodendritic plasma membrane followed by endocytosis and subsequent transport to axons (i.e., transcytosis; Ascano et al., 2009; Buggia-Prevot et al., 2014; Yamashita et al., 2017), and selective retention resulting from interactions of their TM and/or intracellular domains with accessory and cytoskeletal proteins (Ribeiro et al., 2018; reviewed by Lasiecka and Winckler [2011] and Bentley and Banker [2016]). In some instances, proteolytic processing in the ER or TGN may unmask sorting signals (Di Martino et al., 2019; Welch and Munro, 2019) or promote conformational changes that generate protein fragments for differential transport to somatodendritic or axonal compartments (reviewed by Bentley and Banker [2016]). Once proteins are packaged into transport vesicles, the vesicles themselves are delivered to their corresponding domains by interactions with distinct molecular motors (Gumy and Hoogenraad, 2018; Maday et al., 2014; Zahavi et al., 2021). Different populations of transport vesicles are distinguished by their association with small GTPases such as members of the Rab and Arl families (Arimura et al., 2009; Kiral et al., 2018; Niwa et al., 2008; Parker et al., 2018).
To investigate the mechanisms that mediate the polarized accumulation of NRG3 at axonal terminals, here we combined a variety of molecular and cellular approaches, including the development of an optogenetic NRG3 reporter based on the light-oxygen-voltage sensing (LOV2) domain (see Dagliyan et al. [2019] and Wu et al. [2011]). Our results show that axonal NRG3 trafficking involves BACE1 cleavage-dependent release from the TGN, followed by transcytosis of processed NRG3 from the somatodendritic to the axonal compartment in Rab4-positive (Rab4+) vesicles, and selective retention at presynaptic terminals via trans-synaptic interaction with ErbB4 receptors at postsynaptic glutamatergic synapses on GABAergic interneurons. These findings underscore the importance of “trans-synaptic retention” as a mechanism, distinct from intracellular retention, to maintain polarized distribution of TM proteins via juxtacrine interactions between pre- and postsynaptic proteins.
Results
NRG3 is widely expressed in distinct hippocampal neuronal subtypes
We investigated the expression of NRG3 mRNA in excitatory (glutamatergic) and inhibitory (GABAergic) neuronal subtypes of the adult hippocampus using RNAscope, a multiplex fluorescent in situ hybridization approach (Wang et al., 2012). As shown in Fig. S1, A and B, NRG3 transcripts, relative to NRG1 and NRG2 mRNAs (Longart et al., 2004), are expressed more broadly and at relatively higher levels during prenatal and postnatal brain neurodevelopment. NRG3 transcripts are abundantly expressed in granule cell neurons of the dentate gyrus, as well as principal neurons in the Cornu Ammonis (CA) pyramidal cell layers (CA1–CA3) of the hippocampus. NRG3 expression in excitatory and inhibitory neurons was analyzed by co-hybridizing sections with markers for the vesicular glutamate transporter (VGluT1) and glutamic acid decarboxylase (GAD), respectively. Higher-magnification images taken from the CA1 pyramidal layer show that NRG3 is highly expressed in practically all VGluT1-positive (VGluT1+) glutamatergic neurons, as well as GAD+ GABAergic interneurons (Fig. S1, C–F), consistent with previous studies (Grieco et al., 2020; Rahman et al., 2019). Given the extensive expression of NRG3 in the hippocampus, we used primary dissociated hippocampal cultures that retain NRG3 expression in glutamatergic VGluT1+ (Fig. S1, G and H) and GABAergic GAD+ (Fig. S1, I and J) neurons to investigate its subcellular trafficking in central neurons.

NRG3 mRNA expression in distinct hippocampal neuron subtypes in the adult brain and in dissociated culture. Expression of NRG3 transcripts in distinct neuronal subtypes was analyzed in adult mouse hippocampal coronal sections and cultured rat hippocampal neurons using multiplex fluorescent in situ hybridization (RNAscope). (A) Low magnification micrograph showing merged signals for probes corresponding to NRG3 (white), VGluT1 (red; glutamatergic neurons) and GAD (green; GABAergic interneurons). (B) Abundant NRG3 expression is detected in granule neurons of the dentate gyrus (DG) and in pyramidal cell layer neurons (CA1–CA3). (C–F) Higher magnification micrograph of CA1 neurons showing merged (C) and separate signals for NRG3 (D), VGluT1 (E), and GAD (F); examples of glutamatergic neurons co-expressing NRG3 and VGluT (filled arrowheads) or GABAergic co-expressing NRG3 and GAD (open arrowheads) are indicated. (G–J) Micrographs of cultured dissociated hippocampal neurons co-hybridized with probes for NRG3 and VGluT1 (G and H) or NRG3 and GAD (I and J) illustrate NRG3 expression in both cultured excitatory and inhibitory neuron types. Sections and cultures were additionally stained with DAPI to label nuclei. Scale bars: A and B, 200 µm; C–J, 20 µm.

NRG3 mRNA expression in distinct hippocampal neuron subtypes in the adult brain and in dissociated culture. Expression of NRG3 transcripts in distinct neuronal subtypes was analyzed in adult mouse hippocampal coronal sections and cultured rat hippocampal neurons using multiplex fluorescent in situ hybridization (RNAscope). (A) Low magnification micrograph showing merged signals for probes corresponding to NRG3 (white), VGluT1 (red; glutamatergic neurons) and GAD (green; GABAergic interneurons). (B) Abundant NRG3 expression is detected in granule neurons of the dentate gyrus (DG) and in pyramidal cell layer neurons (CA1–CA3). (C–F) Higher magnification micrograph of CA1 neurons showing merged (C) and separate signals for NRG3 (D), VGluT1 (E), and GAD (F); examples of glutamatergic neurons co-expressing NRG3 and VGluT (filled arrowheads) or GABAergic co-expressing NRG3 and GAD (open arrowheads) are indicated. (G–J) Micrographs of cultured dissociated hippocampal neurons co-hybridized with probes for NRG3 and VGluT1 (G and H) or NRG3 and GAD (I and J) illustrate NRG3 expression in both cultured excitatory and inhibitory neuron types. Sections and cultures were additionally stained with DAPI to label nuclei. Scale bars: A and B, 200 µm; C–J, 20 µm.
Unprocessed proNRG3 accumulates in the TGN and cleaved N-terminal fragment (NTF) traffics to neurites
With the goal of identifying the subcellular compartment(s) where proNRG3 processing occurs and the mode of trafficking to axons following processing, we generated a proNRG3 construct tagged at its amino terminus with GFP and at its carboxyl terminus with mCherry, denoted GFPN-proNRG3-mCherryC. This construct enabled us to independently monitor the fates of the NTF and the C-terminal fragment (CTF) following the cleavage of proNRG3 by BACE1 between the EGF-L and TMC domains (Fig. 1 A). Henceforth, we use the term NTF to denote a peptide comprising the 68 amino-acid N-terminal intracellular domain (ICDN), the first TM domain (TMN), and the extracellular EGF-L domain required for ErbB binding, and CTF to denote a peptide that encompasses the second TM domain (TMC) and the C-terminal intracellular domain (ICDC) of proNRG3. As shown in Fig. 1 B, cultured hippocampal neurons transfected with GFPN–proNRG3–mCherryC showed extensive overlap of GFP and mCherry fluorescence in the perinuclear region, suggesting that unprocessed proNRG3 accumulates in the ER and/or Golgi. By contrast, neurites (i.e., dendrites and axons) were strongly positive only for the NTF but not the CTF, as confirmed by line scan densitometry. To determine if NTF-positive neurites include bona fide axons, we co-expressed a V5 epitope-tagged variant of proNRG3 and an axon-specific mutant of syntaphilin fused to red fluorescent protein (SNPHΔMTB), denoted hereafter as SNPH (Kang et al., 2008). Immunofluorescence microscopy 24 h after transfection, using anti-V5 to label the NTF and a NRG3-specific antibody raised against the ICDC (Vullhorst et al., 2017) to detect the CTF, revealed robust accumulation of the NTF but not the CTF in axons (Fig. 1 C). Quantification of mean NTF/CTF fluorescence intensities in SNPH+ axons vs. SNPH− neurites (i.e., dendrites) indicates an approximately threefold higher preference of the NTF to accumulate at both neuronal processes relative to the CTF (Fig. 1 D). Of note, this imaging approach did not allow us to evaluate the extent to which residual CTF signals in neurites represent unprocessed proNRG3 or the processed CTF. Notwithstanding, these observations strongly suggest that the bulk of proNRG3 is cleaved prior to trafficking of its NTF, harboring the signaling-competent EGF-L domain, to neurites.

NRG3 NTF is targeted to axons following BACE1-mediated processing. (A) Diagram illustrating the domain organization of NGFP-proNRG3-mCherryC used to analyze the subcellular distribution of proNRG3 (top) and processed NTF/CTF (bottom) following BACE1 cleavage. The NTF (green outline) encompasses a short cytoplasmic N-terminal sequence, the first transmembrane domain (TMN), the extracellular EGF-L domain that extends down to the BACE1 cleavage site (red arrowhead) in the juxtamembrane region upstream of TMC, as well as GFP at the N-terminus. The CTF (red outline) encompasses sequences downstream of the BACE1 cleavage site and includes mCherry (mCh) at the C-terminus. A V5-epitope–tagged variant lacking GFP/mCherry moieties (proNRG3/V5; epitope tag [yellow] located in the NTF upstream of the EGF-L domain) was used in some experiments. Domains are not drawn to scale. (B) Left: Representative image of a neuron transfected with NGFP-proNRG3-mCherryC showing overlapping GFP/mCh fluorescence in the soma (ROI1; see also corresponding line graph on right), but only GFP fluorescence in neurites (ROI2 and corresponding line graph), suggesting that processing occurs prior to NTF transport into neurites. (C) Representative overview image, magnified region of interest (ROI), and densitometric line scan of an axon from a neuron co-transfected with proNRG3/V5 and the axonal marker SNPH (pseudocolored magenta). Axons show NRG3 puncta positive for the V5-tagged NTF, but not for the CTF labeled with a C-terminal antibody. (D) Integrated fluorescence intensities for the NTF and CTF in SNPH− dendrites and SNPH+ axons. Data represent the mean ± SEM from 18 neurites (three independent experiments). (E) Representative images showing extensively overlapping NRG3 NTF and CTF signals in the TGN (identified with anti-TGN38) in a NGFP-proNRG3-mCherryC-transfected neuron treated with either vehicle or the BACE1 inhibitor BACE-IV (1 µM) for 24 h. (F) Quantitative analysis of experiment shown in E. Data are plotted as Mander’s overlap coefficients and represent the mean ± SEM from four independent experiments (n = 14 for all groups). (G and H) Subcellular distribution of BACE1 and NRG3 in a neuron co-expressing mCherry-tagged BACE1 and NRG3/V5 reveals overlap in the TGN (G) but not in neurites (H; arrowheads in magnified ROI). (I) Schematic illustration of the subcellular distribution of proNRG3, NRG3 NTF and NRG3 CTF. ****, P < 0.0001 (unpaired t test). Scale bars: B, C, and H, 20 μm; B (ROI1), E, and G, 10 μm; B (ROI2), C, and H (inset), 5 μm.
NRG3 NTF is targeted to axons following BACE1-mediated processing. (A) Diagram illustrating the domain organization of NGFP-proNRG3-mCherryC used to analyze the subcellular distribution of proNRG3 (top) and processed NTF/CTF (bottom) following BACE1 cleavage. The NTF (green outline) encompasses a short cytoplasmic N-terminal sequence, the first transmembrane domain (TMN), the extracellular EGF-L domain that extends down to the BACE1 cleavage site (red arrowhead) in the juxtamembrane region upstream of TMC, as well as GFP at the N-terminus. The CTF (red outline) encompasses sequences downstream of the BACE1 cleavage site and includes mCherry (mCh) at the C-terminus. A V5-epitope–tagged variant lacking GFP/mCherry moieties (proNRG3/V5; epitope tag [yellow] located in the NTF upstream of the EGF-L domain) was used in some experiments. Domains are not drawn to scale. (B) Left: Representative image of a neuron transfected with NGFP-proNRG3-mCherryC showing overlapping GFP/mCh fluorescence in the soma (ROI1; see also corresponding line graph on right), but only GFP fluorescence in neurites (ROI2 and corresponding line graph), suggesting that processing occurs prior to NTF transport into neurites. (C) Representative overview image, magnified region of interest (ROI), and densitometric line scan of an axon from a neuron co-transfected with proNRG3/V5 and the axonal marker SNPH (pseudocolored magenta). Axons show NRG3 puncta positive for the V5-tagged NTF, but not for the CTF labeled with a C-terminal antibody. (D) Integrated fluorescence intensities for the NTF and CTF in SNPH− dendrites and SNPH+ axons. Data represent the mean ± SEM from 18 neurites (three independent experiments). (E) Representative images showing extensively overlapping NRG3 NTF and CTF signals in the TGN (identified with anti-TGN38) in a NGFP-proNRG3-mCherryC-transfected neuron treated with either vehicle or the BACE1 inhibitor BACE-IV (1 µM) for 24 h. (F) Quantitative analysis of experiment shown in E. Data are plotted as Mander’s overlap coefficients and represent the mean ± SEM from four independent experiments (n = 14 for all groups). (G and H) Subcellular distribution of BACE1 and NRG3 in a neuron co-expressing mCherry-tagged BACE1 and NRG3/V5 reveals overlap in the TGN (G) but not in neurites (H; arrowheads in magnified ROI). (I) Schematic illustration of the subcellular distribution of proNRG3, NRG3 NTF and NRG3 CTF. ****, P < 0.0001 (unpaired t test). Scale bars: B, C, and H, 20 μm; B (ROI1), E, and G, 10 μm; B (ROI2), C, and H (inset), 5 μm.
ProNRG3 is processed by BACE1 in the TGN
BACE1 activity has been reported in multiple subcellular compartments including endosomes, synaptic vesicles, and the TGN (Choy et al., 2012; Das et al., 2013; Sannerud et al., 2011; Zhang and Song, 2013). To explore where BACE1 cleaves proNRG3, hippocampal neurons transfected with NGFP-proNRG3-mCherryC were treated for 24 h with 1 µM BACE-IV, an antagonist with marked preference for BACE1 over BACE2, and labeled with anti-TGN38 to identify the TGN. As shown in Fig. 1 E, BACE1 inhibition markedly increased NTF/CTF fluorescence intensities in the TGN, as well as their colocalization with TGN38 (Fig. 1 F; Mander’s overlap coefficient for control: CTF; 0.24 ± 0.02 and NTF; 0.27 ± 0.03 vs. BACE-IV: CTF; 0.68 ± 0.05 and NTF; 0.73 ± 0.05), suggesting that cleavage is required for TGN exit. Furthermore, co-transfection with proNRG3/V5 (a proNRG3 construct tagged with V5 that lacks other fluorescent markers; see Methods) and mCherry-tagged BACE1 showed the highest degree of proNRG3/BACE1 colocalization in the TGN (Fig. 1 G), and to a much lesser extent at individual puncta in neurites (Fig. 1 H). Taken together, these findings suggest that the principal subcellular compartment for proNRG3 processing by BACE1 is the TGN, prior to NTF sorting into neurites (Fig. 1 I).
Temporal analysis of proNRG3 processing by BACE1 using photoactivatable LOV2
To follow proNRG3 trafficking after BACE1 cleavage in the TGN with subcellular resolution and in real-time, we generated fluorescent light activatable NGFP-proNRG3-mCherryC cleavage reporters (LA-NRG3) by inserting the Avena sativa phototropin 1 LOV2/Jα domain (see Dagliyan et al. [2019] and Wu et al. [2011]) adjacent to the BACE1 site. The goal was to “cage” proNRG3 in a cleavage-resistant conformation under dark conditions (see Zimmerman et al. [2016]), which in response to brief pulses of 458 nm (blue) light, would be uncaged to make proNRG3 accessible to BACE1 processing (see schema in Fig. 2 A). Importantly, like cultured neurons, HEK293 cells expressing either WT proNRG3 and treated with the BACE1 inhibitor BACE-IV (1 µM) or a cleavage-resistant version of NRG3 (crNRG3) accumulate unprocessed proNRG3 in the TGN (Fig. S2, A–C). Conversely, BACE1 over-expression increases NTF signals at the cell membrane (Fig. S2, D and E). Therefore, we concluded that HEK293 cells were suitable to assess photoactivation of BACE1-mediated cleavage of LA-NRG3 variants in the TGN.

Light-dependent processing of LA 143 -NRG3 and its trafficking to somato-dendritic compartments and axons. (A) Illustration of light-induced conformational changes in LA-NRG3 that enable BACE1 processing of proNRG3. (B) Representative postfixation images of neurons transfected with LA143-NRG3 before (dark, top) and 30 min after photoactivation (light, bottom); overview grayscale images on the left show the NRG3 NTF. GFP (NTF) and mCherry (CTF) fluorescence overlap extensively in the TGN (labeled with anti-TGN38) under dark conditions whereas photoactivation reduces both GFP and mCherry signals in the TGN and promotes the dispersal of the NTF throughout the neuron including in neurites (arrowheads). (C) Quantitative analysis of results shown in B. Bars represent integrated pixel densities for the NTF and CTF before (dark) and after (light) photoactivation in the soma (left) and neurites (right). Data represent the mean ± SEM of three independent experiments (n = 10–11 cells). (D) Time-lapse images illustrating the appearance of punctate NTF (arrowheads) but not CTF signals in a neurite 5 min after photoactivation. m, min. (E) Representative images (top) and densitometric line scans (bottom) of SNPH+ axons from neurons transfected with LA143-NRG3, illustrating the lack of both NTF and CTF before photoactivation (dark) and appearance of NTF but not CTF 3 h after photoactivation (light). Note that for this experiment, LA143-NRG3 was tagged with mCherry at the N-terminus; rabbit polyclonal anti-NRG3 against the ICDC was used after fixation to label the untagged CTF. Signals for NTF, CTF, and SNPH are pseudocolored respectively in green, red, and magenta to maintain consistency with other panels. (F and G) Quantification of pre- and postphotoactivation NTF/CTF fluorescence, showing increased NTF but not CTF signals in both axons (F) and dendrites (G) after photoactivation. Data are plotted as integrated pixel densities and represent the mean ± SEM from three independent experiments (n = 8–12 neurites). (H) Fluorescence intensities of NTF/CTF signals in the soma/TGN are unchanged after photoactivation in neurons expressing cleavage-resistant LA-crNRG3. Data represent the mean ± SEM from three independent experiments (n = 5 neurons). ****, P < 0.0001; ***, P < 0.001; ns, P > 0.05 (one-way ANOVA with Tukey’s post-hoc test). Scale bars: B (overview), 20 μm; B (ROIs), 10 μm; D and E, 5 µm.
Light-dependent processing of LA 143 -NRG3 and its trafficking to somato-dendritic compartments and axons. (A) Illustration of light-induced conformational changes in LA-NRG3 that enable BACE1 processing of proNRG3. (B) Representative postfixation images of neurons transfected with LA143-NRG3 before (dark, top) and 30 min after photoactivation (light, bottom); overview grayscale images on the left show the NRG3 NTF. GFP (NTF) and mCherry (CTF) fluorescence overlap extensively in the TGN (labeled with anti-TGN38) under dark conditions whereas photoactivation reduces both GFP and mCherry signals in the TGN and promotes the dispersal of the NTF throughout the neuron including in neurites (arrowheads). (C) Quantitative analysis of results shown in B. Bars represent integrated pixel densities for the NTF and CTF before (dark) and after (light) photoactivation in the soma (left) and neurites (right). Data represent the mean ± SEM of three independent experiments (n = 10–11 cells). (D) Time-lapse images illustrating the appearance of punctate NTF (arrowheads) but not CTF signals in a neurite 5 min after photoactivation. m, min. (E) Representative images (top) and densitometric line scans (bottom) of SNPH+ axons from neurons transfected with LA143-NRG3, illustrating the lack of both NTF and CTF before photoactivation (dark) and appearance of NTF but not CTF 3 h after photoactivation (light). Note that for this experiment, LA143-NRG3 was tagged with mCherry at the N-terminus; rabbit polyclonal anti-NRG3 against the ICDC was used after fixation to label the untagged CTF. Signals for NTF, CTF, and SNPH are pseudocolored respectively in green, red, and magenta to maintain consistency with other panels. (F and G) Quantification of pre- and postphotoactivation NTF/CTF fluorescence, showing increased NTF but not CTF signals in both axons (F) and dendrites (G) after photoactivation. Data are plotted as integrated pixel densities and represent the mean ± SEM from three independent experiments (n = 8–12 neurites). (H) Fluorescence intensities of NTF/CTF signals in the soma/TGN are unchanged after photoactivation in neurons expressing cleavage-resistant LA-crNRG3. Data represent the mean ± SEM from three independent experiments (n = 5 neurons). ****, P < 0.0001; ***, P < 0.001; ns, P > 0.05 (one-way ANOVA with Tukey’s post-hoc test). Scale bars: B (overview), 20 μm; B (ROIs), 10 μm; D and E, 5 µm.
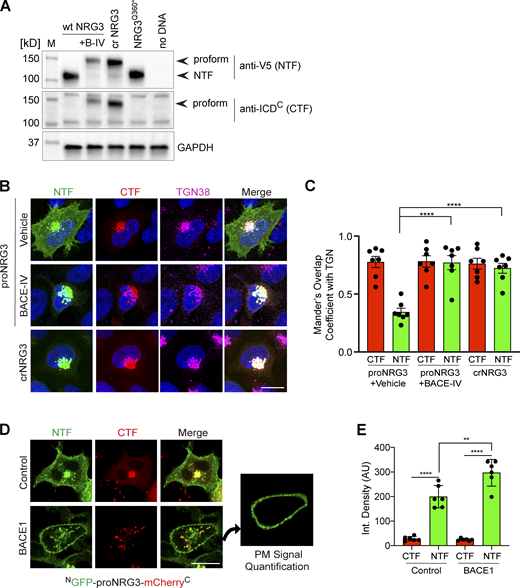
BACE1 cleaves proNRG3 in the TGN of transfected HEK293 cells. (A) Western blot analysis of cells co-transfected with BACE1-mCherry and either proNRG3/V5, crNRG3/V5 or truncated NRG3(Q360*)/V5 (resembling the NTF following BACE cleavage of proNRG3) using anti-V5 for the NTF and anti-NRG3 ICDC antibody for the CTF. Note that BACE1 inhibition with BACE-IV (B-IV; 1 µM for 24 h) blocks generation of the NRG3 NTF in cells expressing proNRG3, and that the NRG3 NTF is absent from cells expressing crNRG3/V5. (B) Cells transfected with proNRG3/V5 and treated with vehicle (DMSO, top) show widespread V5 immunoreactivity indicative of processed NRG3 NTF throughout the cell body in addition to the TGN (labeled with anti-TGN38) while the CTF is detected exclusively in the TGN, indicative of unprocessed proNRG3. By contrast, cells transfected with proNRG3/V5 and treated with 1 µM BACE-IV for 24 h (middle), as well as cells transfected with crNRG3/V5 (bottom), show extensive overlap of NTF and CTF signals in the TGN and low NTF signals elsewhere. (C) Quantitative analysis of results shown in B. Data plotted as Mander’s coefficients for CTF/NTF with TGN38, representing the mean ± SEM from three independent experiments (n = 7 cells). (D) Representative images of HEK293 cells co-transfected with NGFP-proNRG3-mCherryC and an empty vector (top) or a vector expressing BACE1 (bottom). The panel additionally illustrates how PM signals were quantified via isolation of the peripheral signal (right). Of note, single frames were used for this analysis to minimize the contribution of intracellular compartments. (E) Quantitative analysis of results shown in D. Increased PM signals for the NTF (but not the CTF) in cells co-transfected with BACE1 suggest augmented proNRG3 processing. Data are plotted as integrated pixel density and represent the mean ± SEM from three independent experiments (n = 6 cells). **, P < 0.01; ****, P < 0.0001 (C, nonparametric t test; E, one-way ANOVA). Scale bars: 10 μm. Source data are available for this figure: SourceData FS2.

BACE1 cleaves proNRG3 in the TGN of transfected HEK293 cells. (A) Western blot analysis of cells co-transfected with BACE1-mCherry and either proNRG3/V5, crNRG3/V5 or truncated NRG3(Q360*)/V5 (resembling the NTF following BACE cleavage of proNRG3) using anti-V5 for the NTF and anti-NRG3 ICDC antibody for the CTF. Note that BACE1 inhibition with BACE-IV (B-IV; 1 µM for 24 h) blocks generation of the NRG3 NTF in cells expressing proNRG3, and that the NRG3 NTF is absent from cells expressing crNRG3/V5. (B) Cells transfected with proNRG3/V5 and treated with vehicle (DMSO, top) show widespread V5 immunoreactivity indicative of processed NRG3 NTF throughout the cell body in addition to the TGN (labeled with anti-TGN38) while the CTF is detected exclusively in the TGN, indicative of unprocessed proNRG3. By contrast, cells transfected with proNRG3/V5 and treated with 1 µM BACE-IV for 24 h (middle), as well as cells transfected with crNRG3/V5 (bottom), show extensive overlap of NTF and CTF signals in the TGN and low NTF signals elsewhere. (C) Quantitative analysis of results shown in B. Data plotted as Mander’s coefficients for CTF/NTF with TGN38, representing the mean ± SEM from three independent experiments (n = 7 cells). (D) Representative images of HEK293 cells co-transfected with NGFP-proNRG3-mCherryC and an empty vector (top) or a vector expressing BACE1 (bottom). The panel additionally illustrates how PM signals were quantified via isolation of the peripheral signal (right). Of note, single frames were used for this analysis to minimize the contribution of intracellular compartments. (E) Quantitative analysis of results shown in D. Increased PM signals for the NTF (but not the CTF) in cells co-transfected with BACE1 suggest augmented proNRG3 processing. Data are plotted as integrated pixel density and represent the mean ± SEM from three independent experiments (n = 6 cells). **, P < 0.01; ****, P < 0.0001 (C, nonparametric t test; E, one-way ANOVA). Scale bars: 10 μm. Source data are available for this figure: SourceData FS2.
We began by systematically testing in HEK293 cells 16 LA-NRG3 reporters that differed in the location where the LOV2/Jα domain was inserted upstream of the BACE1 cleavage site and the length of the LOV2 Jα peptide (Fig. S3 A). NTF fluorescence signal intensities were measured at the plasma membrane (PM) and in the TGN before photoactivation (see Fig. S3, B–D). While most reporter variants did not significantly affect subcellular distribution, a reporter harboring the LOV2 domain between proNRG3 residues I354 and P358 and a slightly shortened Jα helix (LA143-NRG3∆ILSDP) exhibited pronounced retention in the TGN under dark conditions and was released to the PM following blue-light illumination (Figs. S3, B–D); hereafter we denote LA143-NRG3∆ILSDP as LA143-NRG3 for simplicity. Importantly, using live-cell imaging, we observed that pretreatment of transfected HEK293 cells with BACE-IV prevented the accumulation of LA143-NRG3 at the PM following photoactivation. However, following BACE-IV washout, GFP fluorescence accumulated at the PM, thus confirming that release of LA143-NRG3 from the TGN after photoactivation is dependent upon BACE1-mediated NRG3 processing (Fig. S4 A) Interestingly, unlike the NTF, CTF fluorescence signals did not increase at the PM upon photoactivation and BACE-IV washout, suggesting distinct sorting modes for the NRG3 NTF and CTF upon BACE1-mediated processing (see also Fig. 1). We additionally confirmed LA143-NRG3 processing by immunoblotting, which revealed release of the GFP-NTF upon light activation (Fig. S4, B and C).

Generation and validation of a photoactivatable proNRG3 cleavage reporter (LA-NRG3). (A) Schematic overview of proNRG3-LOV2/Jα fusion proteins tested to derive a configuration with photoactivatable BACE1 processing properties. Fusion constructs varied with regards to the LOV2/Jα insertion site and number of deleted residues in proNRG3 (carboxyl-terminal to L362 [no deletion], P358 [ΔTDHL], S353 [ΔILSDP], or P349 [ΔKTDS]), as well as the extent of carboxyl-terminal deletions of the LOV2 Jα helix, ranging from 0 in LA144 to 8 in LA136 (superscript numbers indicate the overall length of the LOV2/Jα moiety). All fusion protein are based on NGFP-proNRG3-mCherryC (shown above; BACE1 site indicated). LOV2/Jα–encoding residues are shown in blue. Superscript numbers denote amino acid positions relative to their native proteins (proNRG3 and A. sativa phototropin 1, respectively). (B) Effect of LOV2/Jα insertion on NRG3 accumulation at the PM in transfected HEK293 cells. GFP fluorescence intensities at TGN and PM were measured under dark conditions and used to derive TGN over PM ratios illustrating the extent of retention in the TGN. The LA143-NRG3∆ILSDP variant (bold) showed the highest ratio indicative of maximum retention. As controls, TGN retention of proNRG3 lacking the LOV2/Jα domain after 24 h of BACE1 inhibition by BACE-IV (1 µM) is included. Data are plotted as signal intensity ratios and represent the mean ± SEM from three independent experiments (n = 7 cells). (C) Representative images showing subcellular NRG3 NTF/CTF distribution in HEK293 cells transfected with LA144-NRG3362, LA143-NRG3362, LA143-NRG3∆TDHL, and LA143-NRG3∆ILSDP under dark conditions. Note the very low NTF signal for LA143-NRG3∆ILSDP in the PM. (D) Quantitative analysis of experiment shown in C. TGN retention values are plotted as ratios of integrated green or red fluorescence pixel intensities in the TGN over the PM and support the notion that LA143-NRG3∆ILSDP accumulates in the TGN as an unprocessed protein. Data represent the mean ± SEM from three independent experiments (n = 8 cells). Scale bar: 10 μm. *, P < 0.05; **, P < 0.01; ****, P < 0.0001 (one-way ANOVA).

Generation and validation of a photoactivatable proNRG3 cleavage reporter (LA-NRG3). (A) Schematic overview of proNRG3-LOV2/Jα fusion proteins tested to derive a configuration with photoactivatable BACE1 processing properties. Fusion constructs varied with regards to the LOV2/Jα insertion site and number of deleted residues in proNRG3 (carboxyl-terminal to L362 [no deletion], P358 [ΔTDHL], S353 [ΔILSDP], or P349 [ΔKTDS]), as well as the extent of carboxyl-terminal deletions of the LOV2 Jα helix, ranging from 0 in LA144 to 8 in LA136 (superscript numbers indicate the overall length of the LOV2/Jα moiety). All fusion protein are based on NGFP-proNRG3-mCherryC (shown above; BACE1 site indicated). LOV2/Jα–encoding residues are shown in blue. Superscript numbers denote amino acid positions relative to their native proteins (proNRG3 and A. sativa phototropin 1, respectively). (B) Effect of LOV2/Jα insertion on NRG3 accumulation at the PM in transfected HEK293 cells. GFP fluorescence intensities at TGN and PM were measured under dark conditions and used to derive TGN over PM ratios illustrating the extent of retention in the TGN. The LA143-NRG3∆ILSDP variant (bold) showed the highest ratio indicative of maximum retention. As controls, TGN retention of proNRG3 lacking the LOV2/Jα domain after 24 h of BACE1 inhibition by BACE-IV (1 µM) is included. Data are plotted as signal intensity ratios and represent the mean ± SEM from three independent experiments (n = 7 cells). (C) Representative images showing subcellular NRG3 NTF/CTF distribution in HEK293 cells transfected with LA144-NRG3362, LA143-NRG3362, LA143-NRG3∆TDHL, and LA143-NRG3∆ILSDP under dark conditions. Note the very low NTF signal for LA143-NRG3∆ILSDP in the PM. (D) Quantitative analysis of experiment shown in C. TGN retention values are plotted as ratios of integrated green or red fluorescence pixel intensities in the TGN over the PM and support the notion that LA143-NRG3∆ILSDP accumulates in the TGN as an unprocessed protein. Data represent the mean ± SEM from three independent experiments (n = 8 cells). Scale bar: 10 μm. *, P < 0.05; **, P < 0.01; ****, P < 0.0001 (one-way ANOVA).
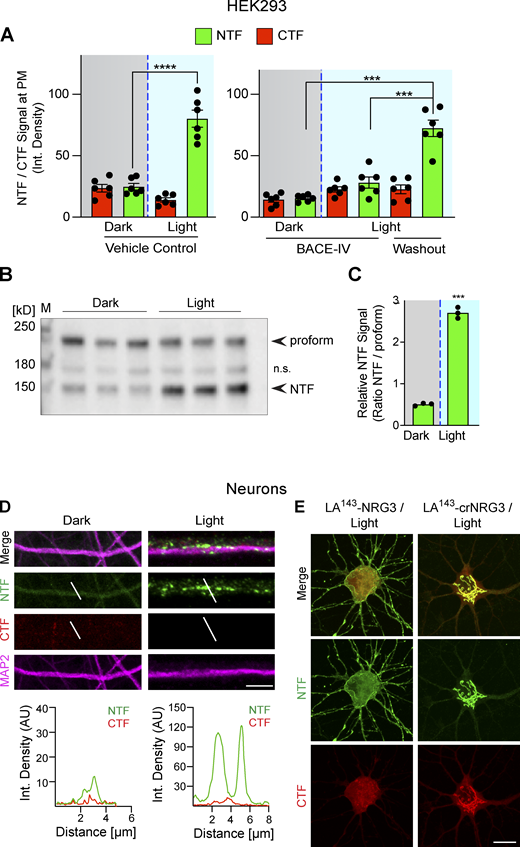
Photoactivation triggers LA143-NRG3 processing and accumulation of its NTF in dendrites. (A) BACE1-dependent translocation of the NRG3 NTF from the TGN to the PM following photoactivation. Data are plotted as integrated NTF/CTF pixel densities at the PM before (gray background) and after (blue background) photoactivation in HEK293 cells pre-treated for 24 h with vehicle (left) or 1 μM BACE-IV (right). Note the increase in PM signal following drug washout. Data represent the mean ± SEM of n = 6–8 cells from three independent experiments. (B) Blue-light illumination increases NTF signal levels in HEK293 cells, thus demonstrating that LA143-NRG3 is processed upon photoactivation. Western blot results are from three independent experiments. (C) Densitometric analysis of results shown in B; data are plotted as ratios of NTF over unprocessed LA143-NRG3 signals. (D) Neurons were transfected with LA143-NRG3 and imaged without (Dark) or following (Light) photoactivation. Representative post-fixation images (top) and corresponding line scan densitometry (bottom) illustrate the lack of NTF/CTF signals under dark conditions, and the appearance of NTF but not CTF signals upon photoactivation in MAP2+ dendrites. (E) Neurons were transfected with WT or noncleavable LA143-NRG3 (LA143-crNRG3) and photoactivated. Representative images illustrate how for WT LA143-NRG3, the NTF distributes broadly throughout the neuron cell body and neurites while the CTF remained in the cell body. By contrast, LA143-crNRG3 remained confined to the cell body (presumably in the TGN). Data represent the mean ± SEM from three independent experiments. Scale bars: D, 10 μm; E, 8 μm. ****, P < 0.0001; ***, P < 0.001 (A, one-way ANOVA; C, non-parametric t test). Source data are available for this figure: SourceData FS4.

Photoactivation triggers LA143-NRG3 processing and accumulation of its NTF in dendrites. (A) BACE1-dependent translocation of the NRG3 NTF from the TGN to the PM following photoactivation. Data are plotted as integrated NTF/CTF pixel densities at the PM before (gray background) and after (blue background) photoactivation in HEK293 cells pre-treated for 24 h with vehicle (left) or 1 μM BACE-IV (right). Note the increase in PM signal following drug washout. Data represent the mean ± SEM of n = 6–8 cells from three independent experiments. (B) Blue-light illumination increases NTF signal levels in HEK293 cells, thus demonstrating that LA143-NRG3 is processed upon photoactivation. Western blot results are from three independent experiments. (C) Densitometric analysis of results shown in B; data are plotted as ratios of NTF over unprocessed LA143-NRG3 signals. (D) Neurons were transfected with LA143-NRG3 and imaged without (Dark) or following (Light) photoactivation. Representative post-fixation images (top) and corresponding line scan densitometry (bottom) illustrate the lack of NTF/CTF signals under dark conditions, and the appearance of NTF but not CTF signals upon photoactivation in MAP2+ dendrites. (E) Neurons were transfected with WT or noncleavable LA143-NRG3 (LA143-crNRG3) and photoactivated. Representative images illustrate how for WT LA143-NRG3, the NTF distributes broadly throughout the neuron cell body and neurites while the CTF remained in the cell body. By contrast, LA143-crNRG3 remained confined to the cell body (presumably in the TGN). Data represent the mean ± SEM from three independent experiments. Scale bars: D, 10 μm; E, 8 μm. ****, P < 0.0001; ***, P < 0.001 (A, one-way ANOVA; C, non-parametric t test). Source data are available for this figure: SourceData FS4.
Light-activated processing and trafficking of proNRG3 in neurons
Consistent with our observations in HEK293 cells, transfected hippocampal neurons kept under dark conditions accumulate LA143-NRG3 in the TGN (Fig. 2 B). However, 30 min after photoactivation, LA143-NRG3 signals in the TGN were markedly reduced, as evidenced by the simultaneous reduction of both GFP (NTF) and mCherry (CTF) fluorescence. Quantitative analyses confirmed reduced NTF and CTF signals in the TGN upon photoactivation and a parallel increase of NTF, but not CTF, signals in neurites (Fig. 2 C). Furthermore, time-lapse imaging revealed the trafficking and accumulation of punctate NTF, but not CTF, signals in neurites within 5 min following photoactivation (Fig. 2 D). To determine if these neurites included axons, neurons were co-transfected with LA143-NRG3 and SNPH, photoactivated, fixed, and labeled (Fig. 2 E). Quantitative analysis of fluorescence signals confirmed the accumulation of the NTF, but not the CTF, in axonal processes following photoactivation (integrated pixel densities for CTF: 11.3 ± 1.4 [dark] vs. 14.9 ± 2.8 [light]; NTF: 13.3 ± 2.4 [dark] vs. 64.7 ± 9.9 [light]; Fig. 2 F). In addition to axons, we found that the NTF, but not the CTF, was also targeted to MAP2+ dendrites upon photoactivation (integrated pixel densities for CTF: 14 ± 1.0 [dark] vs. 11.4 ± 1.1 [light]; NTF: 12.2 ± 1.5 [dark] vs. 62.3 ± 4.7 [light]; Figs. 2 G and S4 D). Importantly, to exclude the possibility that photoactivation exerts non-specific effects on NRG3 processing and trafficking, we used a BACE1 cleavage-resistant version of LA143-NRG3 (LA143-crNRG3) that harbors a mutation in the juxtamembrane cleavage site (see also Fig. S2). Unlike cleavable LA143-NRG3, this variant remained confined to the TGN after photoactivation, confirming that photoactivation specifically enabled BACE1 cleavage (compare Fig. 2, C and H; see also Fig. S4 E). Taken together, our results from HEK293 cells and primary neurons show that proNRG3 processing by BACE1 takes place in the TGN and that, in the absence of cleavage, the NRG3 NTF fails to be released from the TGN. Furthermore, our data indicate that upon processing, the NRG3 NTF initially distributes broadly throughout the neuron.
The NRG3 NTF is trafficked to axons via transcytosis
We were intrigued by the manner in which the NRG3 NTF quickly and broadly spreads throughout the neuron upon BACE1 processing and release from the TGN, particularly in light of the highly restricted presynaptic NRG3 accumulation at steady-state (Müller et al., 2018; Vullhorst et al., 2017). In HEK293 cells, we regularly observed strong fluorescence signals at invaginating PM sites, suggesting that the NTF is trafficked to, and then endocytosed from, the cell surface (Fig. 3 A). To further explore this possibility, we used an approach originally adopted to image surface trafficking of AMPA-type glutamate receptors (Sekine-Aizawa and Huganir, 2004). The method consisted of inserting a high-affinity α-bungarotoxin (BTX) binding sequence (BBS) directly upstream of the extracellular EGF-L domain of GFP–proNRG3 (GFP-proNRG3BBS), which allowed us to measure the binding of cell-impermeant Alexa 555–labeled BTX to surface GFP-proNRG3BBS in transfected HEK293 cells (Fig. 3 B). We confirmed that, like WT proNRG3, GFP-proNRG3BBS is processed by BACE1 in transfected HEK293 cells (Fig. 3 C). Indeed, following a 30-min incubation with BTX, we observed a high degree of BTX/GFP overlap in vesicles (Fig. 3 D). This co-localization was the result of direct BTX binding to GFP-NRG3BBS, not passive uptake by pinocytosis, as evidenced by the extremely low BTX signals and lack of colocalization in cells transfected with control GFP-proNRG3 lacking the BBS. Furthermore, co-transfection of HEK293 cells with proNRG3 and the early endosomal marker Rab5 (Fig. 3 E) revealed extensive co-localization, thereby confirming that recently endocytosed NRG3 NTF traffics into early endosomal compartments.

The NRG3 NTF is endocytosed from the PM. (A) Airyscan high-resolution time-lapse images of GFP-proNRG3 transfected HEK293 cells showing endocytosis of the NTF over the course of 30 s (red arrowhead; see also Video 1). (B) Schematic illustration of the experimental approach used to monitor NRG3 NTF endocytosis. Top: GFP-tagged proNRG3 and BACE1 processed NRG3 NTF harboring a BBS upstream of the EGF-L domain (denoted GFP-proNRG3BBS and GFP-NRG3BBS NTF, respectively). Bottom: Schematic illustration showing surface uptake and internalization of fluorescently labeled BTX (BTX-A555) by the GFP-NRG3BBS NTF. Note that in experiments including GFP-tagged SNPH (see H and I), a proNRG3BBS variant without GFP but with a V5 tag upstream of the EGF-L domain was used instead. However, for consistency across panels, NRG3 is always shown in green, with other markers shown in red (BTX, Rab5) or magenta (SNPH). (C) Western blot of transfected HEK293 cells showing BACE1 dependent GFP-proNRG3BBS processing. Note how BACE1 inhibition by BACE-IV reduces NTF signals and causes the accumulation of the unprocessed proform detected with antibodies against GFP (top) and the NRG3 ICDC (bottom). (D) Still frames from live-cell imaging of HEK293 cells expressing GFP-proNRG3 (top) or GFP-proNRG3BBS (bottom). Cells were surface-labeled with BTX–Alexa 555 for 30 min prior to imaging. Note the extensive overlap between BTX and GFP signals for GFP-proNRG3BBS, but not for the negative control GFP-proNRG3, in the micrographs and the corresponding densitometric line scans. The summary graph shows GFP and BTX–Alexa 555 colocalization (right). Data are plotted as Mander’s overlap coefficient and represent the mean ± SEM from three independent experiments (n = 8 cells). (E) Representative image and line scan densitometry illustrating extensive NRG3/Rab5 colocalization in a HEK293 cell co-transfected with V5-tagged proNRG3 and GFP-tagged Rab5 (arrowheads). Single-channel images shown in grayscale. (F) Representative image of a neuron transfected with GFP-proNRG3BBS and labeled for 30 min with BTX–Alexa 555. The magnified area in the lower panel shows a primary neurite, with single-channel images shown in grayscale. Note the extensive overlap between GFP and BTX–Alexa 555 indicative of recent NRG3 NTF endocytosis. Similar results were obtained in three independent experiments. (G) Representative overview image and magnified area of a GFP-proNRG3BBS–transfected and BTX–Alexa 555-treated neuron (5 min), additionally labeled with anti-MAP2 to demonstrate NRG3 endocytosis in dendrites. Location of the densitometric line scan shown on the right is indicated in the merged magnified image. (H) Similarly, neurons were transfected with V5-tagged NRG3BBS and GFP-SNPH, incubated for 5 or 30 min with BTX–Alexa 555, fixed, and labeled with anti-V5. Note that very little BTX signal was detected in SNPH+ axons after 5 min whereas double NTF+/BTX+ puncta were abundant after 30 min. (I) Quantitative co-localization analysis of experiments shown in G and H confirms extensive overlap of BTX and NRG3 NTF signals in MAP2+ dendrites at 5 min, and in SNPH+ axons at 30 min but not 5 min, indicating that transcytosis of the NRG3 NTF occurs chiefly in the somatodendritic compartment. Data plotted as Mander’s overlap coefficients, representing the mean ± SEM from three independent experiments (n = 16 ROIs). ****, P < 0.0001 (unpaired t test). Scale bars: A, E, H, and F and G (insets), 5 μm; D, 10 μm; F and G, 20 µm. Source data are available for this figure: SourceData F3.
The NRG3 NTF is endocytosed from the PM. (A) Airyscan high-resolution time-lapse images of GFP-proNRG3 transfected HEK293 cells showing endocytosis of the NTF over the course of 30 s (red arrowhead; see also Video 1). (B) Schematic illustration of the experimental approach used to monitor NRG3 NTF endocytosis. Top: GFP-tagged proNRG3 and BACE1 processed NRG3 NTF harboring a BBS upstream of the EGF-L domain (denoted GFP-proNRG3BBS and GFP-NRG3BBS NTF, respectively). Bottom: Schematic illustration showing surface uptake and internalization of fluorescently labeled BTX (BTX-A555) by the GFP-NRG3BBS NTF. Note that in experiments including GFP-tagged SNPH (see H and I), a proNRG3BBS variant without GFP but with a V5 tag upstream of the EGF-L domain was used instead. However, for consistency across panels, NRG3 is always shown in green, with other markers shown in red (BTX, Rab5) or magenta (SNPH). (C) Western blot of transfected HEK293 cells showing BACE1 dependent GFP-proNRG3BBS processing. Note how BACE1 inhibition by BACE-IV reduces NTF signals and causes the accumulation of the unprocessed proform detected with antibodies against GFP (top) and the NRG3 ICDC (bottom). (D) Still frames from live-cell imaging of HEK293 cells expressing GFP-proNRG3 (top) or GFP-proNRG3BBS (bottom). Cells were surface-labeled with BTX–Alexa 555 for 30 min prior to imaging. Note the extensive overlap between BTX and GFP signals for GFP-proNRG3BBS, but not for the negative control GFP-proNRG3, in the micrographs and the corresponding densitometric line scans. The summary graph shows GFP and BTX–Alexa 555 colocalization (right). Data are plotted as Mander’s overlap coefficient and represent the mean ± SEM from three independent experiments (n = 8 cells). (E) Representative image and line scan densitometry illustrating extensive NRG3/Rab5 colocalization in a HEK293 cell co-transfected with V5-tagged proNRG3 and GFP-tagged Rab5 (arrowheads). Single-channel images shown in grayscale. (F) Representative image of a neuron transfected with GFP-proNRG3BBS and labeled for 30 min with BTX–Alexa 555. The magnified area in the lower panel shows a primary neurite, with single-channel images shown in grayscale. Note the extensive overlap between GFP and BTX–Alexa 555 indicative of recent NRG3 NTF endocytosis. Similar results were obtained in three independent experiments. (G) Representative overview image and magnified area of a GFP-proNRG3BBS–transfected and BTX–Alexa 555-treated neuron (5 min), additionally labeled with anti-MAP2 to demonstrate NRG3 endocytosis in dendrites. Location of the densitometric line scan shown on the right is indicated in the merged magnified image. (H) Similarly, neurons were transfected with V5-tagged NRG3BBS and GFP-SNPH, incubated for 5 or 30 min with BTX–Alexa 555, fixed, and labeled with anti-V5. Note that very little BTX signal was detected in SNPH+ axons after 5 min whereas double NTF+/BTX+ puncta were abundant after 30 min. (I) Quantitative co-localization analysis of experiments shown in G and H confirms extensive overlap of BTX and NRG3 NTF signals in MAP2+ dendrites at 5 min, and in SNPH+ axons at 30 min but not 5 min, indicating that transcytosis of the NRG3 NTF occurs chiefly in the somatodendritic compartment. Data plotted as Mander’s overlap coefficients, representing the mean ± SEM from three independent experiments (n = 16 ROIs). ****, P < 0.0001 (unpaired t test). Scale bars: A, E, H, and F and G (insets), 5 μm; D, 10 μm; F and G, 20 µm. Source data are available for this figure: SourceData F3.
In neurons, proteins are transported to axons either directly via TGN-derived vesicles or indirectly via transcytosis, a process by which proteins are initially delivered to the somatodendritic PM and then endocytosed and anterogradely transported to the axon (Ascano et al., 2009; Barford et al., 2017; Buggia-Prevot et al., 2014; Yamashita et al., 2017). Using neurons transfected with GFP-proNRG3BBS and treated with Alexa 555–labeled BTX, we found abundant double BTX+/GFP+ puncta throughout the cell body and in neurites (Fig. 3 F). To estimate the extent to which endocytosis of GFP-NRG3BBS occurs in dendrites, neurons were fixed and stained for MAP2 (Fig. 3, G and I). Likewise, neurons co-transfected with SNPH (see above) were used to assess NRG3 endocytosis in axons (Fig. 3, H and I). Extensive BTX/NTF overlap was observed in dendrites as early as 5 min following BTX incubation (Mander’s overlap: 0.8 ± 0.02). By contrast, BTX/NTF colocalization in axons was marginal after 5 min (0.09 ± 0.01) but increased dramatically after 30 min (0.6 ± 0.04; Fig. 3 I). These data suggest that the NTF is initially endocytosed from the somatodendritic PM, consistent with the idea that axonal targeting of NRG3 is mediated by transcytosis.
Next, we further explored NRG3 NTF endocytosis in neurons by analyzing its co-localization with the early endosome marker Rab5. As shown in Fig. 4, A–C, NRG3+/Rab5+ vesicles were mostly observed in the neuronal soma and MAP2+ dendrites (Mander’s overlap coefficient: 0.53 ± 0.041), and to a much lesser extent in axons (0.13 ± 0.02), supporting the notion that NRG3 endocytosis occurs primarily in the somatodendritic compartment. Furthermore, inhibition of endocytosis with the small-molecule inhibitors Pitstop 2 (blocks clathrin-dependent endocytosis; von Kleist et al., 2011) or Dynole (blocks dynamin-dependent endocytosis; Hill et al., 2009) reduced NRG3/Rab5 colocalization (Fig. 4 D; 0.6 ± 0.04 in vehicle vs. 0.5 ± 0.03 in Dynole and 0.3 ± 0.03 in Pitstop 2) in soma and dendrites. Interestingly, Pitstop 2, and to some extent, Dynole, reduced axonal NRG3 accumulation 24 h after transfection, at a time when robust signals were observed in control neurons treated with vehicle (Fig. 4 E). Taken together, these results strongly suggest that, following processing in the TGN, the bulk of the NRG3 NTF is first targeted to the somatodendritic PM from where it is endocytosed into Rab5+ early endosomes prior to anterograde transport into axons.

The NRG3 NTF is trafficked via early and recycling endosomes. (A) Representative image of a neuron transfected with V5-tagged NRG3 and GFP-tagged Rab5 and additionally labeled for MAP2 to identify dendrites. Magnified area and line scan densitometry illustrate extensive NTF/Rab5 overlap. (B) By contrast, many NTF+ puncta in live-imaged axons lacked corresponding strong punctate signals for Rab5 (white arrowheads), while conversely strongly Rab5+ puncta generally lacked corresponding NTF signals (blue arrowheads). (C) Quantitative NRG3/Rab5 co-localization analysis in MAP2+ dendrites and SNPH+ axons. Data plotted as Mander’s overlap coefficients, representing the mean ± SEM from three independent experiments (n = 16 ROIs). (D) Reduced NTF/Rab5 co-localization in neurons co-transfected with NRG3-mCherry and GFP-Rab5 and treated for 6 h with endocytosis inhibitors Dynole (DYN; 10 µM) or Pitstop 2 (PTS; 20 µM). (E) Likewise, axonal NTF accumulation is similarly reduced in DYN/PTS-treated neurons transfected with NRG3/V5 and SNPH. Data in D are plotted as Mander’s overlap coefficients and in E as integrated density, and each represents the mean ± SEM from three independent experiments (n = 14–16 ROIs). ****, P < 0.0001; **, P < 0.01 (one-way ANOVA with Tukey’s post-hoc test). Scale bars: A, 20 µm; A (ROI) and B, 5 µm.
The NRG3 NTF is trafficked via early and recycling endosomes. (A) Representative image of a neuron transfected with V5-tagged NRG3 and GFP-tagged Rab5 and additionally labeled for MAP2 to identify dendrites. Magnified area and line scan densitometry illustrate extensive NTF/Rab5 overlap. (B) By contrast, many NTF+ puncta in live-imaged axons lacked corresponding strong punctate signals for Rab5 (white arrowheads), while conversely strongly Rab5+ puncta generally lacked corresponding NTF signals (blue arrowheads). (C) Quantitative NRG3/Rab5 co-localization analysis in MAP2+ dendrites and SNPH+ axons. Data plotted as Mander’s overlap coefficients, representing the mean ± SEM from three independent experiments (n = 16 ROIs). (D) Reduced NTF/Rab5 co-localization in neurons co-transfected with NRG3-mCherry and GFP-Rab5 and treated for 6 h with endocytosis inhibitors Dynole (DYN; 10 µM) or Pitstop 2 (PTS; 20 µM). (E) Likewise, axonal NTF accumulation is similarly reduced in DYN/PTS-treated neurons transfected with NRG3/V5 and SNPH. Data in D are plotted as Mander’s overlap coefficients and in E as integrated density, and each represents the mean ± SEM from three independent experiments (n = 14–16 ROIs). ****, P < 0.0001; **, P < 0.01 (one-way ANOVA with Tukey’s post-hoc test). Scale bars: A, 20 µm; A (ROI) and B, 5 µm.
Axonal transport of the NRG3 NTF is mediated by Rab4+ vesicles
Based on prior neuronal transcytosis studies (Bel et al., 2009; de Hoop et al., 1995; Lee et al., 2012), we hypothesized that following endocytosis into Rab5+ early endosomes the NRG3 NTF is sorted into Rab4+ or Rab11+ vesicles, as these transport vesicles are known to mediate anterograde trafficking of axonal cargo. As shown in Fig. 5, A–C, axonal processes from neurons co-transfected with proNRG3 N-terminally tagged with mCherry and GFP-tagged Rab4 or Rab11 revealed that the NTF preferentially localizes to Rab4+ vesicles (Mander’s overlap coefficients for NRG3/Rab4: 0.59 ± 0.04 vs. NRG3/Rab11: 0.20 ± 0.02; Fig. 5 F). We found that axonal Rab4+ vesicles harboring NRG3 cargo were transported anterogradely (Video 2), consistent with studies reporting the importance of Rab4 anterograde transport for regulating synapse organization (Dey et al., 2017). In stark contrast, neurons expressing amyloid precursor protein (APP), another well-known transmembrane BACE1 substrate (Cai et al., 2001), showed preferential colocalization with Rab11 rather than Rab4 (Fig. 5, D–F; Mander’s overlap coefficient for APP/Rab11: 0.49 ± 0.04 vs. APP/Rab4: 0.21 ± 0.02), consistent with prior studies (Buggia-Prevot et al., 2014; Das et al., 2016).

Distinct axonal sorting of BACE1 substrates NRG3 and APP. (A) Representative grayscale images of a neuron transfected with mCherry-NRG3 and GFP-Rab4. Arrowheads point to examples of overlapping NRG3 (left) and Rab4 (right) axonal puncta. (B–E) Representative images (top) and corresponding densitometric line scans (bottom), illustrating the extent of co-localization of the NRG3 NTF with Rab4 (B corresponds to ROI shown in A) and Rab11 (C), and of APP (mCherry) with Rab4 (D) and Rab11 (E). Images were taken 16 h after transfection. (F) Quantitative analysis of NRG3 and APP colocalization with Rab4+/Rab11+ trafficking vesicles in axons that illustrate preferential NRG3 trafficking in Rab4+ vesicles vs. APP trafficking in Rab11+ vesicles. Data are plotted as Mander’s overlap coefficients and represent the mean ± SEM of three independent experiments (n = 14 ROIs). (G) The role of Rab4 and Rab11 GTPase activity in vesicular trafficking of NRG3 and APP was explored in neurons transfected with mCherry-tagged NRG3 or APP and either WT or DN variants of GFP Rab4 and GFP Rab11. Integrated NRG3 NTF pixel density was reduced in axons expressing DN-Rab4 relative to axons expressing WT Rab4. Data represent the mean ± SEM from three independent experiments (n = 10 ROIs). (H) mCherry-tagged BACE1 co-localizes preferentially with GFP-tagged Rab11 in neurons imaged 12–16 h following transfection. Data are plotted as Mander’s overlap coefficients and represent the mean ± SEM of three independent experiments (n = 10 ROIs). (I) DN-Rab11, but not DN-Rab4, significantly reduces axonal trafficking of BACE1. Data are plotted as integrated pixel densities and represent the mean ± SEM of three independent experiments (n = 10 ROIs). (J) Schematic illustration of preferential anterograde trafficking of the NRG3 NTF on Rab4+ transport vesicles and of APP on Rab11+ transport vesicles. ****, P < 0.0001; ***, P < 0.001; **, P < 0.01; ns, P > 0.05 (unpaired t test). Scale bars: A, 20 μm; B, C, D, and E, 5 μm.
Distinct axonal sorting of BACE1 substrates NRG3 and APP. (A) Representative grayscale images of a neuron transfected with mCherry-NRG3 and GFP-Rab4. Arrowheads point to examples of overlapping NRG3 (left) and Rab4 (right) axonal puncta. (B–E) Representative images (top) and corresponding densitometric line scans (bottom), illustrating the extent of co-localization of the NRG3 NTF with Rab4 (B corresponds to ROI shown in A) and Rab11 (C), and of APP (mCherry) with Rab4 (D) and Rab11 (E). Images were taken 16 h after transfection. (F) Quantitative analysis of NRG3 and APP colocalization with Rab4+/Rab11+ trafficking vesicles in axons that illustrate preferential NRG3 trafficking in Rab4+ vesicles vs. APP trafficking in Rab11+ vesicles. Data are plotted as Mander’s overlap coefficients and represent the mean ± SEM of three independent experiments (n = 14 ROIs). (G) The role of Rab4 and Rab11 GTPase activity in vesicular trafficking of NRG3 and APP was explored in neurons transfected with mCherry-tagged NRG3 or APP and either WT or DN variants of GFP Rab4 and GFP Rab11. Integrated NRG3 NTF pixel density was reduced in axons expressing DN-Rab4 relative to axons expressing WT Rab4. Data represent the mean ± SEM from three independent experiments (n = 10 ROIs). (H) mCherry-tagged BACE1 co-localizes preferentially with GFP-tagged Rab11 in neurons imaged 12–16 h following transfection. Data are plotted as Mander’s overlap coefficients and represent the mean ± SEM of three independent experiments (n = 10 ROIs). (I) DN-Rab11, but not DN-Rab4, significantly reduces axonal trafficking of BACE1. Data are plotted as integrated pixel densities and represent the mean ± SEM of three independent experiments (n = 10 ROIs). (J) Schematic illustration of preferential anterograde trafficking of the NRG3 NTF on Rab4+ transport vesicles and of APP on Rab11+ transport vesicles. ****, P < 0.0001; ***, P < 0.001; **, P < 0.01; ns, P > 0.05 (unpaired t test). Scale bars: A, 20 μm; B, C, D, and E, 5 μm.
The GTPase activity of Rab proteins is important to regulate vesicular transport (Ascano et al., 2009; Kiral et al., 2018; Mignogna and D’Adamo, 2018). Therefore, we tested if Rab4 and Rab11 activities are required for axonal transport of NRG3 and APP. Neurons transfected with the dominant-negative Rab4 mutant S22N (DN-Rab4) indeed showed reduced axonal NRG3 NTF accumulation in Rab4 vesicles, whereas the dominant-negative Rab11 mutant S25N (DN-Rab11) was without effect (Fig. 5 G). Conversely, DN-Rab11, but not DN-Rab4, decreased axonal APP signals, confirming the selective and differential role of Rab GTPases in axonal transport of NRG3 and APP. Interestingly, whereas BACE1 processing of NRG3 predominantly occurs in the TGN (see above), BACE1 cleavage of APP has been reported to occur in axons (Buggia-Prevot et al., 2014). Therefore, we investigated the distribution of axonal BACE1 in Rab4 and Rab11 vesicles and found that, like APP, BACE1 was mostly detected in Rab11+ but not in Rab4+ vesicles (Fig. 5 H). Furthermore, BACE1 axonal transport was significantly attenuated by DN-Rab11, but not by DN-Rab4 (Fig. 5 I). Taken together, these findings reveal that the two major BACE1 substrates NRG3 and APP are differentially processed and transported into axons (Fig. 5 J).
NRG3 ICDN and TMN domains are necessary for transcytosis and axonal transport
Based on the available literature, we were unable to identify sequences in NRG3 conforming to canonical signals that promote preferential sorting via endosomal Rab5+ vesicles or axonal targeting via Rab4+ vesicles. We reasoned that if NRG3 harbors presumptive motifs that regulate its transport in Rab5+ and Rab4+ vesicles, they are likely to reside in the ICDN and/or TMN, but not the TMC or ICDC, because of the separation of the CTF from the NTF following cleavage by BACE1 in the TGN. We therefore generated a series of NRG3 mutants with deleted, swapped-out, or minimal ICDN, TMN, TMC, or ICDC domains, shown schematically in Fig. 6 A. Initially, we confirmed that protein is expressed from these constructs by immunofluorescence microscopy in transfected HEK293 cells (Fig. S5, A and B). Then, we analyzed co-localization of these mutant NRG3 proteins with Rab5+ endosomal vesicles in MAP2+ dendrites of transfected hippocampal neurons (Fig. 6 B).

Distinct requirements of the ICD N and TM N domains for NRG3 association with Rab5+ early endosomal and Rab4+ transport vesicles. (A) Schematic representation of NRG3 variants and mutants used for deletion experiments. All constructs harbor a V5 epitope tag upstream of the EGF-L domain used for detection. Note that hFB-NRG3 is based on a naturally occurring variant encoding a single-pass TM protein that results from the use of an alternative 5′ exon lacking ICDN and TMN sequences (Carteron et al., 2006). The truncated NRG3Q360* mutant essentially encompasses the NTF following BACE1 cleavage. N and C termini are indicated. (B) Representative images (above) and corresponding line scan densitometry (below) of MAP2+ dendrites showing Rab5 co-localization with NRG3 mutants harboring the TMN domain (NRG3Q360*, ICDN-TMN, TMN-ICDC) but not with variants/mutants lacking the TMN (hFB-NRG3, ICDN-TMC, TMC-ICDC). (C) Summary analysis of experiments shown in B. Data are plotted as Mander’s overlap coefficients and represent the mean ± SEM of three independent experiments (n = 7 ROIs). (D and E) mCherry was C-terminally fused to either the NRG3 TMN (D) or TMC (E). Representative images and line scan densitometry show co-localization of TMN-mCherry (mCh) but not TMC-mCherry in MAP2+ dendrites (blue in merged images). (F) Summary analysis of data shown in D and E; data plotted as described in C, representing the mean ± SEM of three independent experiments (n = 16 ROIs). (G) Representative images and line scan densitometry illustrating co-localization of NRG3 mutants harboring the TMN and ICDN, but not of NRG3 mutants lacking the ICDN, with axonal Rab4+ vesicles. (H) Summary analysis of data shown in G. Data are plotted as integrated density values and represent as mean ± SEM of three independent experiments. ****, P < 0.001 (C and H: one-way ANOVA with Tukey’s post-hoc test; F: unpaired t test). Scale bars: 5 μm.
Distinct requirements of the ICD N and TM N domains for NRG3 association with Rab5+ early endosomal and Rab4+ transport vesicles. (A) Schematic representation of NRG3 variants and mutants used for deletion experiments. All constructs harbor a V5 epitope tag upstream of the EGF-L domain used for detection. Note that hFB-NRG3 is based on a naturally occurring variant encoding a single-pass TM protein that results from the use of an alternative 5′ exon lacking ICDN and TMN sequences (Carteron et al., 2006). The truncated NRG3Q360* mutant essentially encompasses the NTF following BACE1 cleavage. N and C termini are indicated. (B) Representative images (above) and corresponding line scan densitometry (below) of MAP2+ dendrites showing Rab5 co-localization with NRG3 mutants harboring the TMN domain (NRG3Q360*, ICDN-TMN, TMN-ICDC) but not with variants/mutants lacking the TMN (hFB-NRG3, ICDN-TMC, TMC-ICDC). (C) Summary analysis of experiments shown in B. Data are plotted as Mander’s overlap coefficients and represent the mean ± SEM of three independent experiments (n = 7 ROIs). (D and E) mCherry was C-terminally fused to either the NRG3 TMN (D) or TMC (E). Representative images and line scan densitometry show co-localization of TMN-mCherry (mCh) but not TMC-mCherry in MAP2+ dendrites (blue in merged images). (F) Summary analysis of data shown in D and E; data plotted as described in C, representing the mean ± SEM of three independent experiments (n = 16 ROIs). (G) Representative images and line scan densitometry illustrating co-localization of NRG3 mutants harboring the TMN and ICDN, but not of NRG3 mutants lacking the ICDN, with axonal Rab4+ vesicles. (H) Summary analysis of data shown in G. Data are plotted as integrated density values and represent as mean ± SEM of three independent experiments. ****, P < 0.001 (C and H: one-way ANOVA with Tukey’s post-hoc test; F: unpaired t test). Scale bars: 5 μm.

Validation of expression of mutant NRG3 constructs and of goat polyclonal anti-NRG3 antibody used in this study. (A) Representative images showing expression of V5-tagged NRG3Q360*, hFB-NRG3, ICDN-TMN, ICDN-TMC, ICDC-TMC, and ICDC-TMN in transfected HEK293 cells. Cells were fixed and stained with anti-V5; DAPI nuclear stain was also included. (B) Still frames of live-cell imaging experiments in HEK293 cells co-transfected with GFP-NRG3 and either TMN-mCherry or TMC-mCherry. Note that TMN-mCherry signals overlap extensively with GFP-NRG3 including in the cell periphery (white arrowheads) whereas TMC-mCherry distribution is mostly non-overlapping with GFP-NRG3 (white/blue arrowheads). (C and D) Goat anti-NRG3 antibody labeling specificity was verified in sagittal sections from adult WT (C) and NRG3 knockout (D) mouse brains. Note the absence of mossy fiber staining in CA3 stratum lucidum (sl) in the KO section. Scale bars: A and B, 10 µm; C and D, 100 µm.

Validation of expression of mutant NRG3 constructs and of goat polyclonal anti-NRG3 antibody used in this study. (A) Representative images showing expression of V5-tagged NRG3Q360*, hFB-NRG3, ICDN-TMN, ICDN-TMC, ICDC-TMC, and ICDC-TMN in transfected HEK293 cells. Cells were fixed and stained with anti-V5; DAPI nuclear stain was also included. (B) Still frames of live-cell imaging experiments in HEK293 cells co-transfected with GFP-NRG3 and either TMN-mCherry or TMC-mCherry. Note that TMN-mCherry signals overlap extensively with GFP-NRG3 including in the cell periphery (white arrowheads) whereas TMC-mCherry distribution is mostly non-overlapping with GFP-NRG3 (white/blue arrowheads). (C and D) Goat anti-NRG3 antibody labeling specificity was verified in sagittal sections from adult WT (C) and NRG3 knockout (D) mouse brains. Note the absence of mossy fiber staining in CA3 stratum lucidum (sl) in the KO section. Scale bars: A and B, 10 µm; C and D, 100 µm.
As anticipated, the truncated NRG3 variant Q360*, which essentially encompasses the entire BACE1-processed NTF, extensively colocalized with Rab5 (Fig. 6 B; left). By contrast, the naturally expressed hFB-NRG3 splice variant that encodes a sp-NRG3 lacking the ICDN and TMN domains (Carteron et al., 2006) failed to accumulate in neurites or to colocalize with Rab5, suggesting that sequences in the ICDN and/or TMN domains are necessary for NRG3 endocytosis. Since proNRG3 and hFB-NRG3 variants share most sequences, with the exception of ICDN and TMN domains, we expressed a minimal construct encompassing only these two domains. Interestingly, as shown in Fig. 6 B, the 91 amino acids encompassing these two NRG3 domains were sufficient to target expression in neurites and promote extensive colocalization with Rab5+ vesicles. Conversely, minimal NRG3 constructs encompassing either the ICDN alone or CTF failed to colocalize with Rab5 (Fig. 6 B). Mander’s overlap coefficients with Rab5 were similar for WT NRG3, NRG3Q360*, ICDN-TMN, and TMN-ICDC (0.44 ± 0.03, 0.37 ± 0.05, 0.42 ± 0.03, and 0.42 ± 0.03, respectively), whereas co-localization was significantly reduced compared with WT NRG3 for hFB-NRG3, ICDN-TMC, and ICDC-TMC constructs lacking the TMN domain (0.14 ± 0.02, 0.14 ± 0.01, and 0.10 ± 0.02; Fig. 6 C). Taken together, these findings indicated that the ICDN and/or TMN domains are necessary for NRG3 NTF accumulation in Rab5+ endosomes. To specifically test the role of the TMN, we generated minimal mutants encompassing the TMN or TMC domains carboxyl-terminally fused to mCherry; their expression was initially confirmed in HEK293 cells (Fig. S5 B). We found that, despite similar length and hydrophobicity of both TM domains, only TMN-mCherry colocalized with Rab5+ vesicles whereas TMC-mCherry did not (Fig. 6, D and E). In fact, the extent of overlap with Rab5 was similar to that of NRG3Q360* (Mander’s overlap coefficient for TMN: 0.42 ± 0.03 vs. NRG3: 0.44 ± 0.03; Fig. 6, C and F). These results suggest that either specific TMN residues or its membrane topology support endosomal sorting of the NRG3 NTF. While this finding was surprising, previous studies have demonstrated that in proteins lacking cytosolic sorting signals, TM domains can alternatively mediate endocytosis (Banfield, 2011; Gonzalez Montoro et al., 2017; Mercanti et al., 2010; Singh and Mittal, 2016; see also Discussion).
Next, we used the aforementioned NRG3 deletion and chimeric constructs to identify domains necessary for co-localization with Rab4+ vesicles in axons. Interestingly, variants harboring both the ICDN and TMN accumulated on axonal Rab4+ vesicles (NRGQ360* and ICDN-TMN), whereas variants lacking the ICDN domain did not (TMN-ICDC and TMN-mCherry; Fig. 6, G and H). These results indicate that, whereas the TMN is sufficient to drive NRG3 endocytosis into Rab5+ vesicles, both the ICDN and TMN are necessary for subsequent sorting into Rab4+ vesicles and, by extension, for transport into axons.
Retention of the NRG3 NTF at axonal puncta requires trans-synaptic interactions with ErbB4 at glutamatergic synapses onto GABAergic interneurons
We previously hypothesized that based on in vitro studies using dissociated hippocampal neurons transfected with proNRG3, accumulation of its NTF on axons results from its juxtacrine interaction with ErbB4 receptors expressed on the soma and dendrites of inhibitory GABAergic interneurons (Vullhorst et al., 2017). Consistent with this notion, using adult rat hippocampal sections (Fig. 7 A) and cultured neurons (Fig. 7 B) labeled with ErbB4 and KO-validated NRG3 antibodies (see Fig. S5, C and D), we observed NRG3 puncta that extensively colocalized with the presynaptic glutamatergic marker vGluT1 and that overlapped with postsynaptic ErbB4 puncta on the soma and dendrites of GABAergic interneurons. To stringently test the idea that persistent presynaptic accumulation of the NRG3 NTF requires juxtacrine interactions with ErbB4, we generated an adeno-associated virus (AAV) harboring a potent shRNA (ErbB4_592) to knock down ErbB4 expression in cultured GABAergic interneurons (Fig. 7, C and D). Because in these experiments we targeted ErbB4, we identified GABAergic interneurons by the presence of NRG2 puncta, which we previously showed selectively accumulate on the soma of inhibitory interneurons but not on excitatory glutamatergic neurons (Vullhorst et al., 2017). Consistent with our hypothesis, we found strong attenuation of NRG3 signals associated with NRG2+ GABAergic interneurons transduced with ErbB4 shRNA, but not with nontargeting control (NTC) shRNA (Fig. 7, E and F). Moreover, NRG3 signal intensities in NRG2+ neurons expressing ErbB4 shRNA were similar to control neurons lacking NRG2, presumably glutamatergic neurons, and ErbB4 knockdown was without effect on these neurons. Lastly, we tested whether interference with anterograde NRG3 transport by inhibition of endogenous Rab4 function similarly reduces presynaptic NRG3 accumulation on ErbB4+ neurons (see Fig. 5). Toward this goal, cultures were transduced at DIV7 (days in vitro) with AAVs expressing WT or DN Rab4, and fixed and labeled for NRG3 and ErbB4 on DIV15. As shown in Fig. 7, G and H, presynaptic NRG3 signal levels on ErbB4+ neurons were indeed strongly reduced in DN-Rab4 cultures compared with WT-Rab4 control cultures (integrated pixel densities: 209 ± 16 [DN-Rab4] vs. 378 ± 27 [WT-Rab4]). Taken together with our prior observations (Vullhorst et al., 2017; see Discussion), these findings firmly establish that the polarized neuronal accumulation of NRG3 puncta on axonal processes is achieved by Rab4-dependent anterograde transport and juxtacrine binding to postsynaptic ErbB4 receptors. These studies, therefore, identify a novel trans-synaptic mechanism that results in the polarized expression of surface TM protein at axonal processes that could be shared by numerous other neuronal proteins.

NRG3 accumulates on axonal terminals by trans-synaptically interacting with postsynaptic ErbB4 receptors. (A) Representative immunofluorescence histochemistry of hippocampal area CA1 from a P30 rat brain section co-labeled for NRG3 and ErbB4, showing extensive overlap of punctate NRG3 and ErbB4 signals on soma and dendrites of presumptive stratum pyramidale GABAergic interneurons (arrowheads). (B) Representative Airyscan immunofluorescence micrograph of a cultured DIV24 hippocampal GABAergic interneuron that reveals extensive co-localization of punctate NRG3 signals with ErbB4 and the glutamatergic presynaptic marker protein vGluT1 (arrowheads). Boxed area on the left is magnified on the right and separated by channels. (C) Successful knockdown of ErbB4 protein in hippocampal cultures transduced with an AAV expressing shRNA 592. Cultures transduced with NTC shRNA are included as negative controls. GFP, co-expressed from the shRNA construct, serves as a transduction control. (D) Representative micrographs of GABAergic interneurons transduced with NTC (top) or ErbB4_592 shRNA (ErbB4-KD, bottom), illustrating dramatic reduction of ErbB4 immunoreactivity. (E) Representative micrographs illustrating strongly attenuated NRG3 signals in a GABAergic interneuron (labeled for NRG2) transduced with ErbB4_592 (bottom), as compared to a neuron transduced with NTC shRNA (top). (F) Summary analysis of results shown in E. Data are plotted as integrated pixel densities and represent the mean ± SEM of 22–24 neurons from three independent experiments. (G) Representative micrographs illustrating reduced presynaptic NRG3 signals on ErbB4+ GABAergic interneurons in cultures transduced with an AAV for DN Rab4, as compared to cultures transduced with WT Rab4. (H) Summary analysis of results shown in G. Data are plotted as integrated pixel densities and represent the mean ± SEM of 36 neurons from three experiments. ****, P < 0.0001; ns, P > 0.05 (F: Kruskal-Wallis test with Dunn’s multiple comparisons test; H: Mann-Whitney test). Scale bars: A, 50 µm; B, 10 µm; D, E, and G, 20 µm. Source data are available for this figure: SourceData F7.
NRG3 accumulates on axonal terminals by trans-synaptically interacting with postsynaptic ErbB4 receptors. (A) Representative immunofluorescence histochemistry of hippocampal area CA1 from a P30 rat brain section co-labeled for NRG3 and ErbB4, showing extensive overlap of punctate NRG3 and ErbB4 signals on soma and dendrites of presumptive stratum pyramidale GABAergic interneurons (arrowheads). (B) Representative Airyscan immunofluorescence micrograph of a cultured DIV24 hippocampal GABAergic interneuron that reveals extensive co-localization of punctate NRG3 signals with ErbB4 and the glutamatergic presynaptic marker protein vGluT1 (arrowheads). Boxed area on the left is magnified on the right and separated by channels. (C) Successful knockdown of ErbB4 protein in hippocampal cultures transduced with an AAV expressing shRNA 592. Cultures transduced with NTC shRNA are included as negative controls. GFP, co-expressed from the shRNA construct, serves as a transduction control. (D) Representative micrographs of GABAergic interneurons transduced with NTC (top) or ErbB4_592 shRNA (ErbB4-KD, bottom), illustrating dramatic reduction of ErbB4 immunoreactivity. (E) Representative micrographs illustrating strongly attenuated NRG3 signals in a GABAergic interneuron (labeled for NRG2) transduced with ErbB4_592 (bottom), as compared to a neuron transduced with NTC shRNA (top). (F) Summary analysis of results shown in E. Data are plotted as integrated pixel densities and represent the mean ± SEM of 22–24 neurons from three independent experiments. (G) Representative micrographs illustrating reduced presynaptic NRG3 signals on ErbB4+ GABAergic interneurons in cultures transduced with an AAV for DN Rab4, as compared to cultures transduced with WT Rab4. (H) Summary analysis of results shown in G. Data are plotted as integrated pixel densities and represent the mean ± SEM of 36 neurons from three experiments. ****, P < 0.0001; ns, P > 0.05 (F: Kruskal-Wallis test with Dunn’s multiple comparisons test; H: Mann-Whitney test). Scale bars: A, 50 µm; B, 10 µm; D, E, and G, 20 µm. Source data are available for this figure: SourceData F7.
Discussion
Here, we have developed an optogenetic cleavage reporter, used in combination with other molecular/cellular approaches, to gain mechanistic insights into the relationship of proNRG3 processing by BACE1 and subcellular trafficking of the processed NRG3 NTF in central neurons. Although visualization of post-Golgi transport of vesicular cargo in neurons is challenging due to their morphological complexity, this optogenetically regulated reporter provided unprecedented evidence that proNRG3 processing by BACE1 occurs in the TGN, processing is required for exit from the TGN, and the processed NRG3 NTF initially emerges on the somatodendritic plasma membrane from where it is delivered to axons via Rab5+ early endosomal and Rab4+ anterograde transport vesicles by transcytosis. Lastly, presynaptic NRG3 accumulation on the surface of axonal terminals results from its trans-synaptic interaction with postsynaptic ErbB4 receptors clustered at glutamatergic synapses onto GABAergic interneurons (Fig. 8).

Model of subcellular NRG3 trafficking in central neurons. Schematic diagram depicting the intricate interplay between NRG3 proteolytic processing, transcytosis, and ErbB4-dependent retention that result in the presynaptic accumulation of NRG3 at glutamatergic synapses onto ErbB4+ GABAergic interneurons. The process begins in the TGN where unprocessed proNRG3 is cleaved by BACE1 to separate its constituent NTF (green) and CTF (red) moieties (inset 1). Next, the NRG3 NTF trafficks to the somatodendritic cell surface from where it is re-internalized, likely via clathrin-dependent endocytosis based on data obtained with the inhibitor Pitstop (see Fig. 4), into Rab5+ endosomes (inset 2). It is then sorted into Rab4+ vesicles and anterogradely transported into axons, likely by moving on microtubule tracks via kinesin motors. Finally, the NRG3 NTF re-emerges on the axonal cell surface and is retained at presynaptic terminals by trans-synaptic interaction with ErbB4 receptors concentrated at postsynaptic densities of glutamatergic synapses (inset 3). Although trafficking of the CTF following BACE1 cleavage was not explored in this study, low CTF-only signals suggest quick turnover, possibly by lysosomal degradation (although other functions are conceivable). See main text for further discussion.
Model of subcellular NRG3 trafficking in central neurons. Schematic diagram depicting the intricate interplay between NRG3 proteolytic processing, transcytosis, and ErbB4-dependent retention that result in the presynaptic accumulation of NRG3 at glutamatergic synapses onto ErbB4+ GABAergic interneurons. The process begins in the TGN where unprocessed proNRG3 is cleaved by BACE1 to separate its constituent NTF (green) and CTF (red) moieties (inset 1). Next, the NRG3 NTF trafficks to the somatodendritic cell surface from where it is re-internalized, likely via clathrin-dependent endocytosis based on data obtained with the inhibitor Pitstop (see Fig. 4), into Rab5+ endosomes (inset 2). It is then sorted into Rab4+ vesicles and anterogradely transported into axons, likely by moving on microtubule tracks via kinesin motors. Finally, the NRG3 NTF re-emerges on the axonal cell surface and is retained at presynaptic terminals by trans-synaptic interaction with ErbB4 receptors concentrated at postsynaptic densities of glutamatergic synapses (inset 3). Although trafficking of the CTF following BACE1 cleavage was not explored in this study, low CTF-only signals suggest quick turnover, possibly by lysosomal degradation (although other functions are conceivable). See main text for further discussion.
Optogenetic control of proNRG3 processing in the TGN and post-Golgi trafficking in single cells
We previously showed that proNRG3, like CRD-NRG1, is a dual-pass TM protein that is processed by BACE1 (Vullhorst et al., 2017). Here we took advantage of the light-sensitive properties of the LOV2 domain of A. sativa phototropin 1 to generate LA143-NRG3, a fluorescent proNRG3 reporter whose processing is controlled by blue-light illumination. Using this light-activatable construct, in combination with pharmacological approaches, we provide direct evidence that BACE1 cleavage is required for exit of processed NRG3 from the Golgi, and that, in the absence of cleavage, dual-pass TM proNRG3 is retained in the TGN. It is noteworthy that proNRG3 processing by BACE1 occurs in the TGN but not in other compartments such as the ER, endosomes, plasma membrane, dendrites or axons, despite the presence of BACE1 in these subcellular domains (Choy et al., 2012; Das et al., 2016; Sannerud et al., 2011; Zhang and Song, 2013).
The mechanistic basis for retention of unprocessed proNRG3 in the TGN is currently not understood. Much of what is known about retention mechanisms originates from Golgi resident proteins like glycosylases, glycosyltransferases, and SNAREs that, like proNRG3 and CRD-NRG1, are single-pass TM proteins (see Ribeiro et al. [2018]). Some proteins destined for, or retained in, the Golgi harbor in their cytoplasmic tails specific amino acid motifs that serve as retention signals (Banfield, 2011). However, we were unable to identify any of these types of putative retention signals anywhere in the proNRG3 CTF using LocSigDB, an online prediction tool Negi et al., 2015). Alternatively, TM conformation, length, and biochemical properties can contribute to protein retention in the Golgi (Banfield, 2011; Sharpe et al., 2010; Singh and Mittal, 2016). Therefore, unprocessed NRG3 might harbor such a conformational Golgi retention signal that is disrupted upon BACE1 cleavage, resulting in the separation of its NTF and CTF domains.
Regardless of the exact mechanism of TGN retention, our optogenetic, molecular, and pharmacological data demonstrate that exit of the NRG3 NTF from the TGN and subsequent transport to the cell surface and axons is closely linked to BACE1 processing. By contrast, the fate of the NRG3 CTF following cleavage is likely short-lived, as CTF-only fluorescent signals were always faint. Our observation that it is the BACE1 processed NRG3 NTF (∼75–95 kD apparent molecular mass) that accumulates in both cultured neurons and in brain tissue (this work; Rahman et al., 2019; Vullhorst et al., 2017), and not unprocessed proNRG3 (∼130–140 kD apparent molecular mass) is fully supported by an independent study using NRG3 KO mice that reported a single specific ∼ 95 kD immunoreactive band on Western blots of adult WT mouse brains using an antibody raised against the NRG3 EGF-L domain (Müller et al., 2018). It is noteworthy that recent studies on presynaptic NRG3 functions at glutamatergic synapses onto ErbB4+ GABAergic interneurons implicated sequences located in its CTF rather than the NTF (Exposito-Alonso et al., 2020; Wang et al., 2018). Considering that in these studies presynaptic NRG3 was detected with antibodies against the NTF, and taken together with our findings that proNRG3 processing by BACE1 is a prerequisite for its exit from the TGN, that in Western blotting of brain and cultured neuron lysates the bulk of NRG3 protein migrates as a processed protein of apparent molecular mass of ∼75–95 kD as opposed to ∼130–140 kD for proNRG3 (Müller et al., 2018; Vullhorst et al., 2017; Wang et al., 2018; also see Fig. S2) and that presynaptic NRG3 clusters are comprised of the NTF but not the CTF (Vullhorst et al., 2017; this work), we believe the notion that presynaptic NRG3 functions as an unprocessed protein needs to be carefully re-evaluated. The functional importance of proteolytic processing to “unmask” the biological activities of dpNRGs is further supported by studies on CRD-NRG1 showing that genetic ablation of BACE1 phenocopies peripheral nerve myelination impairments observed in mice lacking CRD-NRG1 (Willem et al., 2006), and that transgenic re-expression of the CRD-NRG1 NTF is sufficient to rescue myelination in CRD-NRG1 mutant mice (Velanac et al., 2012). Whether the processed NRG3 CTF has functions of its own, as reported for NRG1 isoforms harboring the CTF-like long cytoplasmic tail (Bao et al., 2003; Navarro-Gonzalez et al., 2019), remains to be explored.
NRG3 NTF is trafficked into axons by transcytosis and requires ICD and TM sequences
Sorting signals or motifs that specifically target axonal-selective transport remain elusive (reviewed by Bentley and Banker [2016], Lasiecka and Winckler [2011], and Maday et al. [2014]). Instead, axonal polarity of some membrane proteins in absence of these signals relies on small GTPase-containing vesicles that preferentially deliver protein cargo and organelles into axons when complexed with distinct molecular motors (Arimura et al., 2009; Farias et al., 2017; Kiral et al., 2018; Niwa et al., 2008; Parker et al., 2018; Rosa-Ferreira and Munro, 2011). Our results show that, upon photoactivation, the bulk of processed LA143-NRG3 does not directly traffic to axons, but initially distributes broadly throughout the somatodendritic domain. Live-cell labeling experiments with fluorescent BTX in proNRG3BBS-expressing neurons furthermore reveal that somatodendritic NRG3 is transported to the cell surface from where it is endocytosed into Rab5+ early endosomes. When these observations are taken together with the finding that axonal NRG3 NTF co-localizes with Rab4+ vesicles and that axonal trafficking of NRG3 is impaired in neurons expressing dominant-negative Rab4, our data indicate that transcytosis is the primary route by which the NRG3 NTF is targeted to axons (Fig. 8). Thus, our findings add NRG3 to a growing list of neuronal proteins and growth factors, including APP, Trk receptors, cannabinoids receptors, transferrin, NgCAM, and Nrxn1α, that are targeted to axons via transcytosis (Ascano et al., 2009; Buggia-Prevot et al., 2014; Hemar et al., 1997; Leterrier et al., 2006; Ribeiro et al., 2018; Wisco et al., 2003; Yamashita et al., 2017).
We also evaluated NRG3 NTF domains required for sorting into distinct Rab+ vesicles. We found that association of NTF with Rab5+ early endosomes required the NRG3 TMN, whereas association with Rab4+ transport vesicles additionally required sequences located in the short ICDN domain (Fig. 6). The finding that TMN, but not TMC, could target mCherry to Rab5+ vesicles, and that a minimal ICDN-TM construct in which the TMN was replaced by the TMC failed to colocalize with Rab5, supports the notion that TM domains play prominent roles in sorting cargo to specific endosomal vesicle populations. Interestingly, the NRG3 TMN amino acid sequence (LCVVPLFIGFIGLGLSLMLLKWIVV) is 100% identical across more than 500 vertebrate species ranging from primates to bony fish and birds, underscoring the importance of TMN for faithful targeting of NRG3 NTF to axons. How the ICDN and TMN domains mediate association with distinct vesicle populations, and how the NRG3 NTF is sorted from Rab5+ endosomes to mostly Rab4+ vesicles, is presently not known but may involve other regulatory proteins. For example, Nrxn1α sorting from early endosomes to Rab11+ axonal transport vesicles is regulated by Sortilin-related CNS expressed 1 and Rip11 (Ribeiro et al., 2019). Interestingly, axonal Rab11+ vesicles have also been shown to co-transport APP and BACE1 (Buggia-Prevot et al., 2014; Das et al., 2016), an observation we confirmed in this work. Thus, despite both proNRG3 and APP being important BACE1 substrates that traffic to axons via transcytosis, the specific mechanisms underlying their axonal targeting differ with respect to the subcellular site of BACE1 processing and type of anterograde transport vesicle.
A potential limitation of this study, as is the case for numerous other cell biological experiments that require the transfection of cells with constructs expressing tagged proteins, is that over-expression of proteins can potentially affect their subcellular localization. To address this point, in prior studies and here we performed experiments using an expression vector that uses an attenuated, mutated CMV promoter to decrease expression levels, which we previously demonstrated minimizes potential over-expression artifacts (see Vullhorst et al. [2017]). Moreover, in this study we observed that localization did not differ when a variety of fluorescent proteins or sequences (i.e., V5, BTX binding site) were used to tag NRG3, strongly suggesting that the transfected protein was unlikely to be mis-localized.
The NRG3 NTF accumulates at presynaptic terminals by selective trans-synaptic retention
Polarized expression of channels in axons can result from the intracellular interactions of their accessory proteins with cytoskeletal elements that retain them at nodes of Ranvier, initial segments, or presynaptic sites (reviewed by Bentley and Banker [2016] and Maday et al. [2014]). The findings reported here, taken together with our prior study (Vullhorst et al., 2017), uncovered a distinct mechanism for axonal polarization that requires the interaction of surface TM proteins expressed on adjacent cells, which we denote as “trans-synaptic retention.” Several lines of evidence from both these studies support the idea that continued interactions between the NRG3 ligand (and CRD-NRG1) and its receptor ErbB4 are required to maintain polarized surface expression. First, we reported that a chimeric NRG3 construct, in which its EGF-L domain was replaced by the homologous domain of TGFα that binds ErbB1, but not ErbB4, fails to form presynaptic puncta that colocalize with ErbB4 on postsynaptic glutamatergic synapses. Second, we also showed that incubation of neurons with a soluble recombinant NRG1 EGF-L domain peptide disrupts juxtacrine NRG3/ErbB4 interactions and prevents NRG3 accumulation at axonal terminals. Third, we and others found that the developmental increase of ErbB4 puncta on GABAergic interneurons coincides with increased clusters of presynaptic NRG3 (Müller et al., 2018; Vullhorst et al., 2017). Lastly, NRG3 puncta fail to accumulate on GABAergic interneurons processes following ErbB4 knockdown, demonstrating that NRG3-ErbB4 trans-synaptic interactions are required for polarized expression of NRG3 (Fig. 7). Although it has long been appreciated that a multitude of adhesion proteins like NRLN/NRX, SynCAMs, and Ephrin-B/EphB2 interact trans-synaptically (McClelland et al., 2009; Ribeiro et al., 2018) and that sorting mechanisms can regulate the balance of their dendritic vs. axonal surface expression (Ribeiro et al., 2019), to our knowledge, a potential role of trans-synaptic retention as a mechanism that maintains their polarized expression has not been reported.
Importantly, the formation and maintenance of NRG3 clusters at presynaptic terminals are consistent with prior studies reporting that NRG3–ErbB4 interactions support glutamatergic synapse formation, maintenance, and function (Exposito-Alonso et al., 2020; Müller et al., 2018; Vullhorst et al., 2017; Wang et al., 2018). It is not clear at this time if a similar retention mechanism could occur at interaction sites between oligodendrocyte processes and axons, as NRG3 has been reported to promote oligodendrocyte survival (Carteron et al., 2006). Reports showing that NRG3 is a weak activator of ErbB4 tyrosine kinase activity (Hobbs et al., 2002; Müller et al., 2018; Rahman-Enyart et al., 2020) and that its effects on glutamatergic synapse formation do not require ErbB4 tyrosine kinase activity (Müller et al., 2018) are consistent with the idea that NRG3 may function to promote stable pre- and postsynaptic interactions at glutamatergic synapses during synapse formation and maturation. Considering the extensive similarities between NRG3 and CRD-NRG1 in terms of their TM topology, BACE1 processing, and axonal targeting (Vullhorst et al., 2017; this work), but also notable differences in the affinity of their EGF-L domains for ErbB4 (Jones et al., 1999), receptor activation (Hobbs et al., 2002; Müller et al., 2018), and additional processing of the CRD-NRG1 NTF (Fleck et al., 2013; Fleck et al., 2016), not observed for NRG3, it will be interesting to determine the extent to which these two dpNRGs engage in overlapping vs. distinct CNS processes.
In summary, our findings reveal the existence of multiple interlocking trafficking and retention mechanisms that together ensure proper delivery and retention of the mature processed NRG3 NTF in axons and axon terminals (Fig. 8). Taken together, our findings lend additional mechanistic support to the notion that dpNRGs and spNRGs differ in important ways, including their subcellular distribution (axons vs. somatodendritic ER-PM contact sites), sites of proteolytic processing (TGN vs. plasma membrane), identity of their cognate processing enzymes (BACE1 vs. ADAM10), and signaling modalities (cell-attached vs. paracrine/autocrine). These observations are likely to encourage further research into the distinct contributions of dpNRGs vs. spNRGs to neuronal processes known to be regulated by NRG3 and ErbB4 based on studies using mutant mice, such as neurotransmission, synaptic/critical period plasticity neuronal network activity (oscillations), and behaviors/cognitive functions in ErbB4 mutant mice (Bi et al., 2015; Hayes et al., 2016; Li et al., 2020; Shamir et al., 2012; Sun et al., 2016; Tan et al., 2018; Wang et al., 2018; Zeledon et al., 2015; Zhou et al., 2020), as well as their implications to NRG3 and ERBB4 polymorphisms associated with schizophrenia and its endophenotypes (Avramopoulos, 2018; Hayes et al., 2016; Kao et al., 2010; Li et al., 2020; Loos et al., 2014; Morar et al., 2011; Zeledon et al., 2015).
Materials and methods
Animals
Sprague–Dawley E19 rat embryos of either sex were used to prepare dissociated hippocampal neuron cultures. WT adult C57Bl6 mice (The Jackson Laboratory) were used for fluorescence in situ hybridization RNAscope technique (Wang et al., 2012) and as positive controls for the NRG3 antibody validation (see Fig. S5, C and D). Cryopreserved paraformaldehyde-fixed floating brain sections from NRG3 knockout mice were a kind gift from M. Pletnikov (Johns Hopkins University School of Medicine, Baltimore, MD; Hayes et al., 2016). WT adult Sprague–Dawley rats were used for the NRG3/ErbB4 histology shown in Fig. 7. All procedures were approved by the National Institute of Child Health and Human Development Animal Care and User Committee.
Drugs and primary antibodies
BACE-IV was from Calbiochem; Alexa 555-conjugated bungarotoxin was from Thermo Fisher Scientific; tubocurarine was from Tocris; Dynole-34-2 and Pitstop 2 were from Abcam. Goat polyclonal anti-NRG3 against the EGF-L domain was from Neuromics; rabbit polyclonal anti-NRG3 against the NRG3 C-terminus was described previously (Vullhorst et al., 2017). See also Table S1 for a complete list of primary antibodies used in this study. Secondary antibodies conjugated to fluorophores for immunofluorescence microscopy or to HRP for chemiluminescence-based Western blotting were from Thermo Fisher Scientific or Jackson Immunoresearch.
DNA constructs
NRG3 constructs
Full-length and Q360*-truncated variants of mouse NRG3 (GenBank accession number NM_008734.3) harboring the V5 epitope upstream of the EGF-L domain have been described previously (Vullhorst et al., 2017). A human NRG3 isoform harboring an alternative 5′ exon and lacking the sequence encoding the N-terminal transmembrane domain (hFB-NRG3; Carteron et al., 2006) was amplified by RT-PCR from human fetal brain total RNA (Cat-#738005-41; Lot-#: 1000808; Stratagene), cloned into the Gateway entry vector pENTR/D (Thermo Fisher Scientific), and tagged with a V5 epitope upstream of the EGF-L domain. Fluorescently tagged NRG3 constructs were generated by inserting synthetic DNA fragments harboring the desired GFP/mCherry sequences (“g-Blocks”; Integrated DNA Technologies) and included dual-fluorescence NGFP-proNRG3-mCherryC (see also Fig. 1 A), as well as single-fluorescence versions with GFP added to the N terminus or mCherry added to either the N or C terminus (NmCherry-NRG3 or NRG3-mCherryC).
For photoactivation experiments, 16 LA-NRG3 constructs were generated by inserting synthetic DNA fragments harboring the LOV2/Jα domain from A. sativa phototropin 1 between the EGF-L domain and the BACE1 cleavage site of NGFP-proNRG3-mCherryC (see also Figs. 2 A and S3 A). To achieve optimal photoactivation of BACE1-mediated proNRG3 cleavage, synthetic DNA fragments were generated that slightly differed in the position of the LOV2/Jα domain relative to proNRG3 and the length of the Jα helix itself. The proNRG3 insertion site was either L362, P358, S353, or P349; insertions at P358 and P349 were additionally accompanied by short deletions of respectively 4 (ΔTDHL) and 5 (ΔILSDP) residues located carboxyl-terminally to the insertion site (superscript numbers relative to full-length mouse proNRG3; GenBank accession number: NP_032760.1). Furthermore, for each of these insertions, four variants were made that harbored either the complete nine–amino acid Jα peptide (I539–P547; superscript numbers relative to phototropin 1) or carboxyl-terminally truncated versions (I539–L546; I539–A543; I539), resulting in LOV2 domains of different lengths (LA144, LA143, LA140, and LA136). The variant LA143-NRG3ΔILSDP exhibited maximal retention in the TGN under dark conditions and was henceforth named LA143-NRG3. For some experiments, LA143-NRG3 was modified to harbor only a single fluorescent tag.
For endocytosis experiments, a 13–amino acid BBS (Sekine-Aizawa and Huganir, 2004) was inserted as a synthetic DNA fragment upstream of the EGF-L domain in proNRG3 variants harboring either GFP or V5 at the N terminus; the resulting constructs were named, respectively, GFP-proNRG3BBS and V5-proNRG3BBS.
NRG3 constructs harboring ICDN/TMN, ICDN/TMC, TMC/ICDC, and TMN/ICDC domains were made as synthetic DNA fragments (see also Fig. 6 A). ICD/TM domains encompassed the following sequences: ICDN: M1-C68, TMN: V69-V91, TMC: V363-F385, ICDC: K386-K713; amino acid positions based on mouse NRG3 protein. For detection, the V5 epitope tag was added downstream (ICDN/TMN and ICDN/TMC) or upstream (TMC/ICDC and TMN/ICDC) of the respective NRG3 sequences. Minimal NRG3 constructs encompassing respectively the TMN and TMC domains were inserted upstream of mCherry. All mutant constructs included the potential N-terminal NRG3 signal peptide sequence N-MSEGAAGASPPGAASAAAAS-C.
All NRG3 constructs were assembled in entry vector pENTR/D and then transferred to pDESTDV3 (attenuated cytomegalovirus [CMV] promoter; Vullhorst et al., 2017) or pDESTD40 (WT CMV promoter) expression vectors using Gateway recombination (Thermo Fisher Scientific).
Other constructs
For identification of axons, we used GFP- and RFP-tagged versions of the syntaphilin variant SNPH, as previously described (Kang et al., 2008; Vullhorst et al., 2017). Human BACE1, C-terminally tagged with mCherry, and human APP, N-terminally tagged with mCherry, were derived from previously published constructs (Burgos et al., 2010; Prabhu et al., 2012).
The following constructs were obtained from Addgene; GFP-Rab4 (#49434); GFP-DN-Rab4 (#49475); GFP-Rab5 (#31733); dsRed-DN-Rab5 (#29688); GFP-Rab7 (#12605); dsRed-Rab11 (#12679); and dsRed-DN-Rab11 (#12680).
Potential target sequences for shRNA-mediated knockdown of ErbB4 were identified using an online design tool available from Thermo Fisher Scientific (web address: https://rnaidesigner.thermofisher.com/rnaiexpress/). Four high-scoring shRNA sequences (conserved in rat and mouse orthologs) were selected and cloned as double-stranded oligonucleotides between the EcoRI-BamHI sites of AAV vector pZacf (modified to co-express EGFP from the human synapsin promoter). shRNAs were first tested by Western blotting of whole-cell lysates from HEK293 cells co-transfected with the ErbB4 shRNA vectors and a vector expressing mouse ErbB4. This initial screen yielded a highly effective ErbB4 shRNA (ErbB4_592, target sequence: 5′-GGAAGATGCCATAAGTCTTGC-3′; numbers indicate the position of the first nucleotide relative to the target open reading frame). The vector was then packaged into AAV for transduction of cultured neurons (see below). The nontargeting control vector contained a sequence derived from the Photinus pyralis luciferase gene.
All constructs that were made or modified in-house were confirmed by sequencing.
HEK293 cell culture and transfections
HEK293 cells were propagated at 37°C/5% CO2 in DMEM growth medium supplemented with 10% FBS (Thermo Fisher Scientific). For imaging experiments, cells were plated on poly-D-lysine (PDL; MilliporeSigma) –coated coverglass in 24-well plates and transfected with 1.5 μg plasmid DNA per well using Xtreme HP transfection reagent (MilliporeSigma). For imaging experiments using LOV-based constructs, cells were imaged at 9–16 h after transfection, and for all other constructs cells were imaged 24 h after transfection. For NRG3 processing experiments (Figs. 3 C and S2 A), HEK293 cells were seeded in 12-well plates, transfected with 1 µg plasmid DNA per well using LipoD293 transfection reagent (SignaGen), and processed 24–48 h later for Western blotting. Rab transfection experiments were done by using 1.5 μg DNA of the respective plasmids per well of a 12-well plate using Xtreme HP transfection reagent (MilliporeSigma). Imaging was done after 16–20 h of transfection.
Hippocampal neuron cell culture and transfections
Dissociated hippocampal neurons were prepared from E19 Sprague–Dawley rat pups, plated on PDL-coated coverslips in 24-well plates, and maintained in Neurobasal/B27 medium (Thermo Fisher Scientific). Unless indicated otherwise, neurons were transfected at DIV10 with 1.5 μg plasmid DNA per well using Lipofectamine 2000 (Thermo Fisher Scientific), and equal mass ratios were used for experiments with two or three different plasmids. For LOV2-based constructs, cells were imaged 12–16 h after transfection. BACE1/NRG3 colocalization studies in the cell body were also done between 12 and 16 h after transfection. All other experiments were performed 24–48 h after transfection unless specified otherwise.
Immunofluorescence cytochemistry
Cells on coverslips were fixed in 4% PFA in PBS supplemented with 4% sucrose for 15 min at room temperature. After extensive washing with PBS, cells were blocked and permeabilized with 10% normal goat serum/0.1% Triton X-100 in PBS. For surface labeling, blocking solution without detergent was used. Cells were incubated with primary antibodies for 1–3 h at room temperature or overnight at +4°C with gentle rocking and for 1 h with donkey or goat secondary antibodies conjugated to Alexa or DyLight fluorophores (Thermo Fisher Scientific or Jackson Immunoresearch). DAPI stain was included to label nuclei (Thermo Fisher Scientific). Coverslips were again washed with PBS, dipped in water, and mounted on glass slides using Mowiol/DABCO mounting medium.
Fluorescent RNA in situ hybridization (RNAscope)
Adult mouse brains were fresh-frozen on powdered dry ice and stored at −80°C until use. Sections were cut at 12-µm thickness on a cryostat. Triple RNAscope fluorescent in situ hybridization of brain sections was performed, as reported previously (Erben and Buonanno, 2019), using probes for mouse NRG3, VGluT1, and GAD-1 (Cat-#s: 452171-O1 [channel 1], 416631-C2 [channel 2], 400951-C3 [channel 3]) following the manufacturer’s instructions (Advanced Cell Diagnostics). Cultured DIV10 hippocampal neurons, grown on PDL-coated coverslips and fixed with 4% PFA in PBS/4% sucrose for 15 min at room temperature, were similarly hybridized with the above mentioned RNAscope probes for either NRG3 and vGluT1 or NRG3 and GAD1.
Immunofluorescence histochemistry
Adult mouse or rat brains were fixed by transcardiac perfusion with 4% PFA in PBS (pH 7.4) and postfixed overnight in the same fixative. Then, 50-µm sections were cut on a vibratome. Floating sections were blocked in 10% normal donkey serum, 0.25% Triton X-100 in PBS for 1 h at room temperature and incubated with primary antibodies in 0.1 M phosphate buffer with 10% normal donkey serum and 0.25% Triton X-100 (dilution buffer) for 24 h at 4°C with gentle rocking. Slices were washed in PBS with 0.25% Triton X-100 for at least 40 min before incubation with fluorophore-conjugated donkey secondary antibodies (Jackson Immunoresearch) for 90 min at room temperature in dilution buffer. DAPI stain was included to label nuclei. After extensive washes in PBS with 0.25% Triton X-100, sections were mounted in Mowiol-DABCO on gelatin-coated glass slides.
Bungarotoxin uptake and endocytosis inhibition
24 h after transfection, neurons were pretreated with 150 μM tubocurarine chloride for >2 h to reduce non-specific binding and then labeled with 5 μM BTX conjugated to Alexa 555 (Thermo Fisher Scientific) for 1 h. Neurons were washed and subcellular BTX trafficking was monitored by live imaging for up to 90 min. In some experiments, dynamin- and clathrin-mediated endocytosis was inhibited by treating neurons with 1 μM dynole-34-2 or 5 μM Pitstop 2 for 3 h, respectively.
Preparation of AAVs for ErbB4 shRNA knockdown and WT/DN GFP-Rab4 expression
AAV-293 cells (Agilent), grown in DMEM medium with 10% FBS, were used for AAV production (serotype 1). Cells were seeded in 15-cm dishes and triple-transfected at 50–60% confluence with pHelper (Agilent), pAAV-RC1 (Cell Biolabs), and shRNA transfer vectors (see above). Medium was changed 2 h before transfection. For each dish, ∼15 μg DNA at equimolar ratios (6.6 μg pHelper, 4.4 μg pRC1, and 4.4 μg transfer vector) was first mixed with 60 μl polyethyleneimine (1 mg/ml) in 500 μl of DMEM medium (Thermo Fisher Scientific), incubated for 10 min, and then added to cells. Cultures were fed 48 h and harvested 72 h after transfection by scraping into gradient buffer (150 mM NaCl, 10 mM MgCl2, 10 mM Tris-Cl, pH 7.6). AAVs for ErbB4 shRNA knockdown were purified as follows: cells were lysed by multiple freeze–thaw cycles, aided by repeated passages through a 23-gauge needle. Lysates were then incubated with benzonase (MilliporeSigma) for 1 h at 37°C and cleared by centrifugation (15 min at 4,000 g, 4°C). Supernatants were layered on top of an iodixanol step gradient (15, 25, 40, and 58% [vol/vol] in gradient buffer; MilliporeSigma) and the remaining volume was filled with gradient buffer. Ultracentrifugation was done at 48,000 rpm using a type 70Ti fixed angle rotor (Beckman) for 2 h, 18°C. Viral particles were collected from the 40% iodixanol layer and stored at 4°C for immediate use or at −80°C for long-term storage. AAVs for WT and DN GFP-Rab4 were purified from conditioned culture supernatants using the AAV concentrator kit (Takara). All AAV preparations were titrated in cultured hippocampal neurons and used at dilutions that yielded widespread expression at moderate levels suitable for detection by Western blotting or low-to-moderate expression for immunocytochemistry and confocal microscopy.
Western blotting
Protein samples from cultured neurons were prepared by direct lysis in LDS sample buffer (Thermo Fisher Scientific). HEK293 protein samples were prepared by first lysing cells in buffer containing 50 mM Tris-Cl, pH 7.5, 150 mM NaCl, 1% Triton X-100, and protease inhibitors (Complete protease inhibitor cocktail; MilliporeSigma). Proteins were size-fractionated on 4–15% Mini-Protean TGX precast gels (Bio-Rad) and electroblotted onto nitrocellulose using semi-dry transfer (trans-blot turbo transfer system; Bio-Rad). Membranes were blocked with 3% BSA in Tris-buffered saline containing 0.1% Tween-20 (TBS/T) and incubated with primary antibody in blocking solution for several hours at room temperature or overnight at 4°C. After several washes with TBS/T, membranes were incubated with HRP-conjugated secondary antibodies (Jackson Immunoresearch) for 1 h at room temperature. Signals were detected by chemiluminescence (Super Signal West Pico PLUS; Thermo Fisher Scientific) using a ChemiDoc MP imager (Bio-Rad) and quantified using Image Lab software (Bio-Rad). For shRNA experiments, blots were reprobed with anti-GFP antibody N86/6 (NeuroMab) to estimate relative transduction efficiency between samples expressing ErbB4 shRNA 592 and the nontargeting control shRNA. Rhodamine-conjugated anti-tubulin and anti-GAPDH antibodies were used as loading controls (Bio-Rad). Densitometry analysis for LA143-NRG3 experiment was performed using ImageJ software.
Microscopy and image analyses
Confocal fluorescence microscopy of NRG3 mRNA in situ hybridization (RNAscope) and protein immunohistology was done on a LSM780 microscope (Zeiss). The low-magnification hippocampus overview micrograph shown in Fig. S1, A and B, was derived from tiled and stitched Z-projected image stacks taken with a 10× objective (numerical aperture [NA]: 0.8); the higher-magnification CA1 micrograph shown in Fig. S1, C–F, was obtained using a single-plane image taken with a 40× oil objective (NA: 1.3), as were the NRG3 immunohistochemistry micrographs shown in Figs. 7 A and S5, C and D.
Postfixation and live-cell imaging of HEK293 cells and cultured hippocampal neurons were done at 63× magnification. Live-cell imaging experiments were performed in growth medium at 37°C and 5% CO2 using an LSM780 confocal microscope (Zeiss), except for the endocytosis experiment shown in Fig. 3 A and Video 1 that was performed on an LSM880/Airyscan confocal microscope (Zeiss). Cells were imaged in culture medium without phenol red. Similarly, LA143-NRG3 photoactivation experiments were performed on an LSM 710 confocal microscope (Zeiss) with a 63× objective (NA: 1.4). Bungarotoxin uptake experiments were performed using an LSM780 confocal microscope with a 63× objective. LA143-NRG3 photoactivation experiments were done as described (Wu et al., 2011). In brief, during the light phase, cells were exposed to 458 nm laser pulses throughout image acquisition, while the laser was off during the dark phase. Laser intensity was set to 1–5% of maximal power. Pulse lengths were 1 s for HEK293 cells and 5 ms for cultured neurons; both conditions efficiently promoted photoactivation of LA-NRG3. Frames were acquired at 10-s intervals for HEK293 cells and at 30-s intervals for cultured neurons. Live-cell imaging of Rab4+ vesicles (Video 2) was done on a LSM780 microscope (Zeiss) with 63× objective (NA: 1.4). To demonstrate processing of LA143-NRG3 by blue light, HEK293 cells were transfected and light activation was performed 12–18 h after transfection in culture medium without phenol red. Light pulse lengths were 1 s at 20× magnification (NA: 0.75) using a TCS SP8 LSCM microscope (Leica). Cells were harvested and protein lysates were prepared for Western blotting after 30 min of light illumination.
Plasma membrane invagination showing NRG3 endocytosis. HEK293 cells were transfected with GFP-proNRG3 and imaged 16 h later in live-cell mode (60 s; 1 frame per second) using a high-resolution Airyscan/LSM880 microscope. The time-lapse movie shows invaginating plasma membrane (arrowhead) containing strong GFP signal indicative of NRG3-NTF endocytosis.
Plasma membrane invagination showing NRG3 endocytosis. HEK293 cells were transfected with GFP-proNRG3 and imaged 16 h later in live-cell mode (60 s; 1 frame per second) using a high-resolution Airyscan/LSM880 microscope. The time-lapse movie shows invaginating plasma membrane (arrowhead) containing strong GFP signal indicative of NRG3-NTF endocytosis.
Anterograde trafficking of Rab4+ vesicles in neurons. Cultured hippocampal neurons were transfected with GFP-Rab4 and imaged 24 h later. Live-cell imaging (60 s; 0.5 frames per second per second) shows anterograde trafficking of Rab4+ vesicles away from the cell body (arrowhead).
Anterograde trafficking of Rab4+ vesicles in neurons. Cultured hippocampal neurons were transfected with GFP-Rab4 and imaged 24 h later. Live-cell imaging (60 s; 0.5 frames per second per second) shows anterograde trafficking of Rab4+ vesicles away from the cell body (arrowhead).
For all quantitative analyses, images were acquired using laser intensities that avoided pixel saturation. For plasma membrane signal quantification in HEK293 cells, single-plane images were used; intracellular cell contents were masked and only the signal from the membrane was used for quantification (see Fig. S2 D). For all other analyses, image stacks were acquired and projected in the Z dimension using the maximum intensity method in the ZEN software (Zeiss). Images were adjusted for overall brightness and contrast using FIJI/Image J. For signal quantification within the TGN, the TGN38 signal was used to closely trace the organelle and exclude other cell compartments. MAP2+ and SNPH+ neurites were used for signal quantification in dendrites and axons, respectively. Data were plotted as integrated densities based on at least five neurites/cells from three independent experiments. For protein colocalization analyses, threshold was set at 50 and kept constant across analyses, and results were plotted as Mander’s overlap coefficients.
Statistical analyses
One-way ANOVA with Tukey’s post-hoc tests or Kruskal–Wallis with Dunn’s multiple comparison tests were used for independent multiple comparisons. P values for single comparisons were calculated using two-tailed unpaired t- or Mann–Whitney tests. Experiments that were not statistically analyzed were independently repeated a minimum of three times to ensure consistency and reproducibility.
Online supplemental material
Fig. S1 shows in situ hybridization fluorescent RNAscope experiments that analyze NRG3 mRNA expression in distinct hippocampal neuron subtypes in the adult brain and cultured hippocampal neurons. Fig. S2 shows experiments validating HEK293 cells as an appropriate model system to study BACE1-mediated proNRG3 cleavage in the TGN. Figs. S3 and S4 contain schematics and data related to the generation of the LA-NRG3 constructs and the validation of LA143-NRG3 in HEK293 cells and cultured neurons. Fig. S5 shows immunocytochemistry evidence of expression of various NRG3 constructs in HEK293 cells, as well as immunohistology of brain sections from WT and NRG3-KO mice to confirm the specificity of the anti-NRG3 antibody used in this study. Table S1 has a list of all antibodies used in this study. Video 1 shows NRG3 endocytosis at plasma membrane invaginations in HEK293 cells. Video 2 shows anterograde trafficking of Rab4+ vesicles in neurons.
Acknowledgments
We thank Vincent Schram and Carolyn Smith from the Porter Neuroscience Research Center imaging core for expert assistance with confocal microscopy, and Ricardo Murphy for preparation of neuronal primary cultures.
This work was supported by Eunice Kennedy Shriver National Institute of Child Health and Human Development with funding from the intramural research program grant NICHD ZIA-HD001607 (to J.S. Bonifacino) and NICHD ZIA-HD000711 (to A. Buonanno).
The authors declare no competing financial interests.
Author contributions: T. Ahmad, C.M. Guardia, D. Vullhorst, J.S. Bonifacino, and A. Buonanno conceived the study and designed experiments. The biochemical and cell biology experiments were performed by T. Ahmad (cell culture, live-imaging, generation and testing of light-sensitive LOV constructs, generation of BBS constructs, immunofluorescence cytochemistry, and imaging of most experiments), D. Vullhorst (generation of AAVs, immunofluorescence cytochemistry), C.M. Guardia (generation of Rab constructs), R. Chaudhuri (immunoblotting experiments) and I. Karavanova (neuronal cultures, in situ hybridization). T. Ahmad, D. Vullhorst, and N. Chaudhary collected, quantified, and performed statistical analyses of data. A. Buonanno, T. Ahmad, C.M. Guardia, and D. Vullhorst wrote the manuscript with input from J.S. Bonifacino. The study was supervised by A. Buonanno.

