


Neuronal extracellular vesicles (EVs) are locally released from presynaptic terminals, carrying cargoes critical for intercellular signaling and disease. EVs are derived from endosomes, but it is unknown how these cargoes are directed to the EV pathway rather than for conventional endolysosomal degradation. Here, we find that endocytic machinery plays an unexpected role in maintaining a release-competent pool of EV cargoes at synapses. Endocytic mutants, including nervous wreck (nwk), shibire/dynamin, and AP-2, unexpectedly exhibit local presynaptic depletion specifically of EV cargoes. Accordingly, nwk mutants phenocopy synaptic plasticity defects associated with loss of the EV cargo synaptotagmin-4 (Syt4) and suppress lethality upon overexpression of the EV cargo amyloid precursor protein (APP). These EV defects are genetically separable from canonical endocytic functions in synaptic vesicle recycling and synaptic growth. Endocytic machinery opposes the endosomal retromer complex to regulate EV cargo levels and acts upstream of synaptic cargo removal by retrograde axonal transport. Our data suggest a novel molecular mechanism that locally promotes cargo loading into synaptic EVs.
Introduction
Neurons depend on complex and highly interconnected endosomal membrane trafficking pathways to sort physiologically and pathologically relevant cargoes (Winckler et al., 2018; Yarwood et al., 2020). One function of endosomal trafficking in neurons is to sort cargoes for release in extracellular vesicles (EVs; Blanchette and Rodal, 2020). EVs are small membrane-bound compartments that transport protein, lipid, and nucleic acid cargoes from EV-releasing cells to target cells (van Niel et al., 2018). In the nervous system, EV-mediated cargo transport regulates intercellular communication and contributes to neurodegenerative disease pathology (Budnik et al., 2016; Holm et al., 2018; Song et al., 2020). However, our understanding of how EV cargo traffic is spatially and temporally regulated within the polarized and complex morphology of neurons remains limited (Blanchette and Rodal, 2020).
Exosomes are a type of EV generated when endosomal multivesicular bodies (MVBs) undergo fusion with the plasma membrane to release their intralumenal vesicles. Alternatively, MVBs can be trafficked to the lysosome for cargo degradation (van Niel et al., 2018). It remains unclear how and where MVBs are generated in neurons, and how they are directed to an EV versus lysosomal fate. We previously described a trafficking pathway that controls the levels of EV cargoes in donor neuron synaptic terminals, regulated by endosome-plasma membrane recycling machinery including the retromer complex and Rab11 (Walsh et al., 2021). This suggests the existence of a specialized presynaptic “EV-permissive” recycling compartment, where cargo is protected from degradation en route to MVB formation and released from donor cells. However, the mechanisms by which this compartment is loaded are unknown.
The Drosophila larval neuromuscular junction (NMJ) is a powerful in vivo model for addressing the gaps in our understanding of synaptic EV biology (Ashley et al., 2018; Koles et al., 2012; Korkut et al., 2009; Korkut et al., 2013; Lauwers et al., 2018; Walsh et al., 2021). EVs are released from the presynaptic terminals of larval motor neurons and accumulate postsynaptically within the folds of the muscle membrane subsynaptic reticulum (SSR) or are taken up by muscles and glia (Fuentes-Medel et al., 2009; Koles et al., 2012). EVs released at the Drosophila NMJ likely represent exosomes, as presynaptic terminals contain MVBs positive for EV cargoes and release EVs similar in size to exosomes (Koles et al., 2012; Korkut et al., 2009; Walsh et al., 2021). Several endogenously and exogenously expressed transmembrane proteins have been characterized as neuronal EV cargoes at the Drosophila NMJ, including synaptotagmin-4 (Syt4), which drives functional and structural plasticity, as well as the Alzheimer’s disease-associated human amyloid precursor protein (hAPP; Korkut et al., 2013; Walsh et al., 2021). Thus, the Drosophila NMJ provides a unique opportunity to interrogate the mechanisms and functional significance of traffic of EV cargoes in an intact nervous system. Here, using this system, we have identified a novel and unexpected role for canonical endocytic machinery in locally maintaining EV cargoes in EV-permissive compartments at synaptic terminals, thereby promoting their release in synaptically derived EVs. Our work suggests that endocytic machinery regulates endosomal sorting in neurons to protect EV cargoes from retrograde transport-mediated depletion at synaptic terminals.
Results
EV cargoes are locally depleted at nwk mutant presynaptic terminals
EVs at the Drosophila larval NMJ are derived from neuronal membranes, and thus contain neuronal glycoproteins that can be detected using α-HRP antibodies (Fuentes-Medel et al., 2009; Snow et al., 1987; Walsh et al., 2021). During our studies of the endocytic FCH-BIN amphiphysin RVS (F-BAR) and SH3 domain-containing protein Nervous Wreck (Nwk), we found that nwk null mutants exhibited a strong reduction in the intensity of postsynaptic α-HRP puncta (within 3 µm of the boundary of motor neurons innervating muscles 6 and 7; Fig. 1 A). This suggests that in addition to its previously defined roles in synaptic morphogenesis, growth factor receptor trafficking, endocytosis, and synaptic transmission (Coyle et al., 2004; Del Signore et al., 2021; O’Connor-Giles et al., 2008; Rodal et al., 2008; Ukken et al., 2016), Nwk may also regulate EV cargo traffic at synapses.
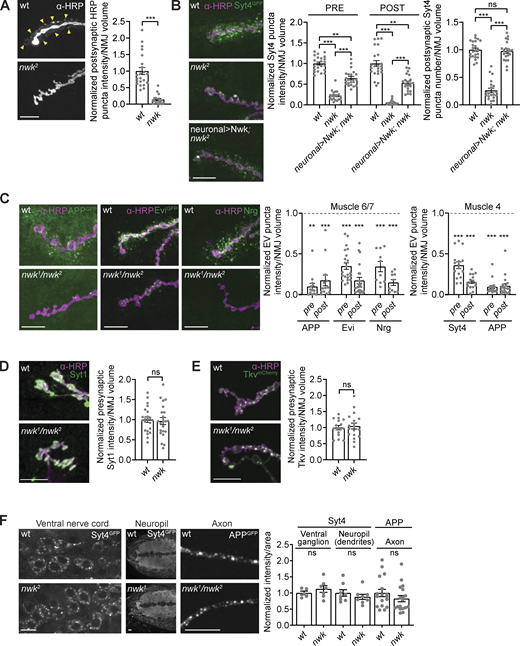
EV cargoes are locally depleted at nwk mutant synapses. (A)nwk mutant synapses lack α-HRP–labeled postsynaptic neuronal membrane puncta (indicated by arrowheads). Left: Representative images of muscle 6/7 NMJs. Right: Quantification of postsynaptic α-HRP puncta intensity. (B)nwk mutants cause neuron-autonomous pre- and postsynaptic depletion of the endogenously tagged EV cargo Syt4-GFP. Nwk was expressed using C380-GAL4, a neuron-specific driver. Left: Representative images of muscle 6/7 NMJs. Right: Quantification of Syt4-GFP puncta intensity and number. (C) EV cargoes APP-GFP, Evi-GFP, Nrg, and Syt4-GFP are reduced at nwk mutant synapses. Left: Representative images of muscle 6/7 NMJs. Middle: Quantification of EV cargo puncta intensity at muscle 6/7. Right: Quantification of EV cargo puncta intensity at muscle 4. Dashed line indicates mean EV cargo levels at control NMJs. (D and E) Levels of the non-EV cargoes Syt1 and Tkv-mCherry are unaffected at nwk mutant synapses. Left: Representative images of muscle 6/7 NMJs (Syt1) and muscle 4 NMJs (Tkv-mCherry). Right: Quantification of Syt1 or Tkv-mCherry puncta intensity. (F) EV cargo levels remain unchanged in cell bodies, neuropil, and axons in nwk mutants. Left: Representative images of Syt4-GFP in ventral ganglion cell bodies and neuropil, and APP-GFP in axons. Right: Quantification of Syt4-GFP or APP-GFP intensity. Data is represented as mean ± SEM; n is depicted by individual gray dots on the graphs and represents NMJs (A–E), or axons or brains (F). NMJ intensity measurements were normalized to presynaptic volume; all measurements were further normalized to the mean of their respective controls. All scale bars are 10 µm. Associated with Fig. S1. See Table S1 and Table S3 for detailed genotypes, sample sizes, and statistical analyses. wt, wild-type.
EV cargoes are locally depleted at nwk mutant synapses. (A)nwk mutant synapses lack α-HRP–labeled postsynaptic neuronal membrane puncta (indicated by arrowheads). Left: Representative images of muscle 6/7 NMJs. Right: Quantification of postsynaptic α-HRP puncta intensity. (B)nwk mutants cause neuron-autonomous pre- and postsynaptic depletion of the endogenously tagged EV cargo Syt4-GFP. Nwk was expressed using C380-GAL4, a neuron-specific driver. Left: Representative images of muscle 6/7 NMJs. Right: Quantification of Syt4-GFP puncta intensity and number. (C) EV cargoes APP-GFP, Evi-GFP, Nrg, and Syt4-GFP are reduced at nwk mutant synapses. Left: Representative images of muscle 6/7 NMJs. Middle: Quantification of EV cargo puncta intensity at muscle 6/7. Right: Quantification of EV cargo puncta intensity at muscle 4. Dashed line indicates mean EV cargo levels at control NMJs. (D and E) Levels of the non-EV cargoes Syt1 and Tkv-mCherry are unaffected at nwk mutant synapses. Left: Representative images of muscle 6/7 NMJs (Syt1) and muscle 4 NMJs (Tkv-mCherry). Right: Quantification of Syt1 or Tkv-mCherry puncta intensity. (F) EV cargo levels remain unchanged in cell bodies, neuropil, and axons in nwk mutants. Left: Representative images of Syt4-GFP in ventral ganglion cell bodies and neuropil, and APP-GFP in axons. Right: Quantification of Syt4-GFP or APP-GFP intensity. Data is represented as mean ± SEM; n is depicted by individual gray dots on the graphs and represents NMJs (A–E), or axons or brains (F). NMJ intensity measurements were normalized to presynaptic volume; all measurements were further normalized to the mean of their respective controls. All scale bars are 10 µm. Associated with Fig. S1. See Table S1 and Table S3 for detailed genotypes, sample sizes, and statistical analyses. wt, wild-type.
α-HRP antibodies are likely to detect many neuronal proteins undergoing a variety of trafficking itineraries (Snow et al., 1987), only a small fraction of which result in EV sorting. Therefore, we next tested the role of nwk in the traffic of specific established EV cargoes. First, using endogenously tagged Syt4-GFP (Walsh et al., 2021), we found that loss of nwk led to reduced mean postsynaptic Syt4-GFP levels, together with a decreased number of postsynaptic Syt4-GFP puncta (Fig. 1 B), similar to the α-HRP signal. Using this single, defined cargo, we also detected a striking reduction presynaptically (Fig. 1 B), which was surprising since endocytic mutants would be expected to accumulate cargoes at the plasma membrane. These phenotypes were rescued by neuronal re-expression of Nwk using the binary GAL4/UAS (upstream activating sequence) system (Fig. 1 B). Taken together, these results suggest that Nwk cell autonomously regulates Syt4 levels in EV donor neurons, resulting in reduced release of Syt4 in EVs. Loss of nwk similarly caused a dramatic decrease in pre- and postsynaptic levels of several other known EV cargoes, including neuronal GAL4-driven hAPP (hAPP-EGFP [enhanced GFP]; Walsh et al., 2021), the neuronal GAL4-driven long isoform of evenness interrupted (Evi) tagged with EGFP (Evi-EGFP; Korkut et al., 2009), and the endogenous neuronal isoform of neuroglian (Nrg; Walsh et al., 2021; Fig. 1 C and Fig. S1, A–C). This phenotype was not specific to muscle 6/7, as we observed a similar effect at the NMJ on muscle 4 (Fig. 1 C; and Fig. S1, D and E). Therefore, the effects of nwk mutants are consistent for multiple neurons and EV cargoes. These observations highlight a new role for Nwk in regulating the levels of EV cargo proteins in donor synapses, and thus in the recipient postsynaptic cell.
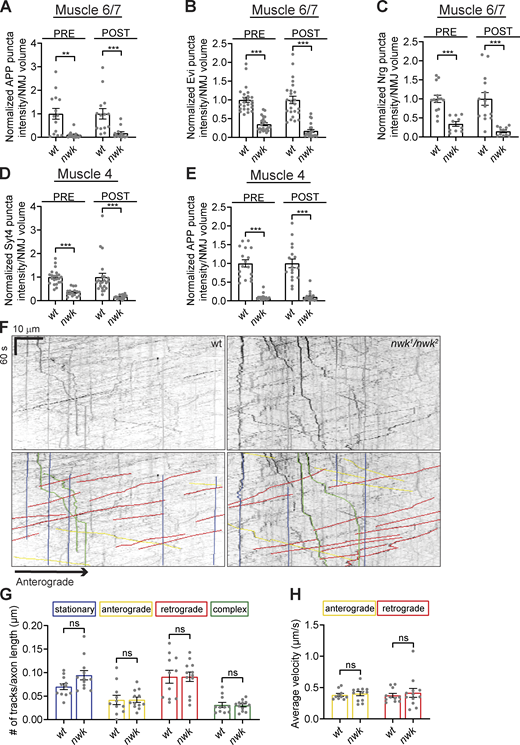
EV cargo levels are reduced at nwk mutant synaptic terminals but show no change in axonal trafficking. (A–E)Fig. 1 C datasets shown with corresponding controls. (A) Quantification of APP-GFP pre- and postsynaptic puncta intensity at muscle 6/7 NMJs. (B) Quantification of Evi-GFP pre- and postsynaptic puncta intensity at muscle 6/7 NMJs. (C) Quantification of Nrg pre and postsynaptic puncta intensity at muscle 6/7 NMJs. (D) Quantification of Syt4-GFP pre- and postsynaptic puncta intensity at muscle 4 NMJs. (E) Quantification of APP-GFP pre- and postsynaptic puncta intensity at muscle 4 NMJs. (F) Top: Representative kymographs of APP-GFP puncta dynamics in control and nwk mutant live axons. Bottom: Overlayed colored lines indicate stationary (blue), anterograde (yellow), retrograde (red), and complex (green) APP-GFP tracks. (G) Quantification of number of stationary (blue), anterograde (yellow), retrograde (red), and complex (green) APP-GFP puncta tracks in control and nwk mutant axons. (H) Quantification of average velocity of anterograde (yellow) and retrograde (red) APP-GFP puncta movement in control and nwk mutant axons. Data is represented as mean ± SEM; n is depicted by individual gray dots on the graphs and represents NMJs (A–E) and animals (G and H). NMJ intensity measurements were normalized to presynaptic volume (A–E). Number of tracks were normalized to axon length (G). Measurements in A–E were further normalized to the mean of their respective controls. Associated with Fig. 1. See Table S1 and Table S3 for detailed genotypes, sample sizes, and statistical analyses. wt, wild-type.
EV cargo levels are reduced at nwk mutant synaptic terminals but show no change in axonal trafficking. (A–E)Fig. 1 C datasets shown with corresponding controls. (A) Quantification of APP-GFP pre- and postsynaptic puncta intensity at muscle 6/7 NMJs. (B) Quantification of Evi-GFP pre- and postsynaptic puncta intensity at muscle 6/7 NMJs. (C) Quantification of Nrg pre and postsynaptic puncta intensity at muscle 6/7 NMJs. (D) Quantification of Syt4-GFP pre- and postsynaptic puncta intensity at muscle 4 NMJs. (E) Quantification of APP-GFP pre- and postsynaptic puncta intensity at muscle 4 NMJs. (F) Top: Representative kymographs of APP-GFP puncta dynamics in control and nwk mutant live axons. Bottom: Overlayed colored lines indicate stationary (blue), anterograde (yellow), retrograde (red), and complex (green) APP-GFP tracks. (G) Quantification of number of stationary (blue), anterograde (yellow), retrograde (red), and complex (green) APP-GFP puncta tracks in control and nwk mutant axons. (H) Quantification of average velocity of anterograde (yellow) and retrograde (red) APP-GFP puncta movement in control and nwk mutant axons. Data is represented as mean ± SEM; n is depicted by individual gray dots on the graphs and represents NMJs (A–E) and animals (G and H). NMJ intensity measurements were normalized to presynaptic volume (A–E). Number of tracks were normalized to axon length (G). Measurements in A–E were further normalized to the mean of their respective controls. Associated with Fig. 1. See Table S1 and Table S3 for detailed genotypes, sample sizes, and statistical analyses. wt, wild-type.
To test if this effect was specific to EV cargoes, we measured the presynaptic levels of two transmembrane proteins that are not normally trafficked into EVs: the calcium sensor synaptotagmin-1 (Syt1), which is associated with synaptic vesicles (Littleton et al., 1993), and the bone morphogenetic protein (BMP) signaling receptor thickveins (Tkv), which localizes to the plasma membrane and endosomes (Deshpande et al., 2016; Smith et al., 2012). We found that presynaptic levels of both endogenous Syt1 and neuronally GAL4-driven UAS-Tkv-mCherry were similar between nwk mutants and controls (Fig. 1, D and E), suggesting that reduction of Syt4, hAPP, Evi, and Nrg in nwk mutants is specific to the EV trafficking itinerary.
We next asked whether Nwk functions to maintain EV cargo levels specifically at synaptic terminals or throughout the neuron. We measured the mean intensity of Syt4-GFP in the cell bodies and dendrites within the larval ventral nerve cord, and the mean intensity of hAPP-EGFP (as the Syt4-GFP signal was too faint for this analysis) in the region of the motor neuron axon closest to muscle 4 synaptic terminals. We found that nwk mutants had similar levels of Syt4-GFP in cell bodies and dendrites compared to controls, and similar levels of hAPP-EGFP in their axons compared to controls (Fig. 1 F). We next used live imaging to ask whether nwk mutants exhibit defects in the velocity or directionality of EV cargo transport in axons. We found no change in the dynamics of hAPP-EGFP puncta in nwk mutant axons compared to controls (Fig. S1, F–H), suggesting that changes to axonal transport are unlikely to account for the depletion of EV cargoes we observed at nwk mutant synaptic terminals. Together, these results suggest that Nwk specifically and locally regulates the levels of EV cargo proteins at synapses.
nwk mutants phenocopy loss of EV cargo function
We next asked if depletion of EV cargoes at nwk mutant synapses causes a loss of EV cargo function. At the NMJ, traffic of Syt4 into EVs is thought to regulate multiple forms of activity-dependent synaptic growth and plasticity (Barber et al., 2009; Harris et al., 2018; Korkut et al., 2013; Piccioli and Littleton, 2014; Yoshihara et al., 2005). In this system, high-frequency stimulation causes an increase in the frequency of miniature excitatory junction potentials (mEJPs). This functional plasticity is termed high-frequency stimulation-induced miniature release (HFMR) and depends on presynaptically derived Syt4 (Korkut et al., 2013). To directly test whether nwk mutants exhibit a loss of Syt4 function and, therefore, phenocopy syt4 mutant defects, we measured the responses of nwk mutant synapses to high-frequency stimulation, and found that HFMR was strongly reduced in nwk mutants (Fig. 2, A–C), similar to syt4 mutants (Yoshihara et al., 2005). This phenotype could be rescued by neuronal but not muscle GAL4-driven expression of Nwk, indicating that HFMR specifically requires presynaptic Nwk (Fig. 2 C).
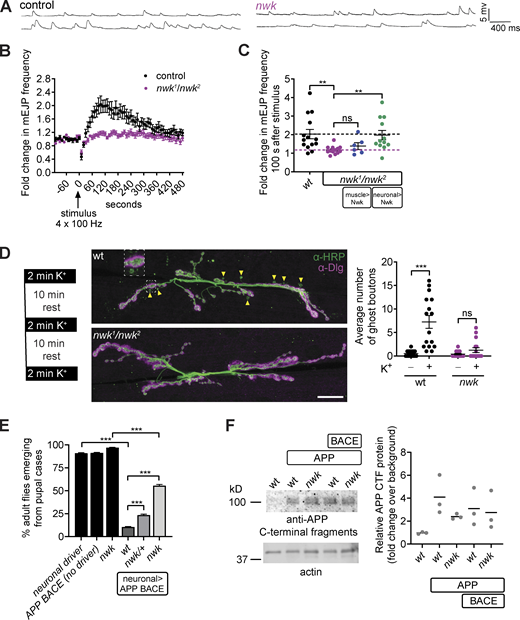
nwk mutants exhibit loss of EV cargo function. (A–C) Syt4-dependent functional plasticity (HFMR) is abolished in nwk mutants. (A) Representative traces of mEJPs before (top trace) and after (bottom trace) high-frequency stimulation (4 × 100 Hz). (B) Timecourse of mEJP frequency after stimulation. (C)nwk mutant HFMR phenotype is cell autonomous to neurons. Scatter plot of mEJP frequency 100 s after high-frequency stimulation. Black dashed line represents wild-type (wt) mean value, and the pink dashed line represents nwk mean value. C57-GAL4 and Vglut-GAL4 are muscle and neuron-specific drivers, respectively. (D) Syt4-dependent structural plasticity (ghost bouton budding) is abolished in nwk mutants. Left: Paradigm for spaced stimulation with high K+/Ca2+. Middle: Representative maximum intensity projections of confocal stacks at muscle 6/7 labeled with α-HRP (presynaptic) and α-Dlg (postsynaptic) antibodies. Arrowheads point to ghost boutons, containing HRP but not Dlg, enlarged in inset. Scale bar is 20 µm. Right: Quantification of ghost boutons with and without stimulation. (E) APP BACE-induced eclosion defect is suppressed in nwk mutants. Graph shows the percentage of adult flies emerging from pupal cases. APP and BACE were expressed with C155-GAL4, a neuron-specific driver. (F) GAL4C155-driven APP levels are comparable in wt and nwk heads: Immunoblot of Drosophila head extracts with α-APP C-terminal fragment and α-actin antibodies. Quantification shows fold change of APP relative to actin levels. Data is represented as mean ± SEM (B, C, and D) or mean ± standard error of the proportion (E); n represents NMJs (B–D), pupal cases (E), or biological replicates (F), and in C, D, and F, n is depicted by individual gray dots on the graphs. See Table S1 and Table S3 for detailed genotypes, sample sizes, and statistical analyses. Source data are available for this figure: SourcedataF2.
nwk mutants exhibit loss of EV cargo function. (A–C) Syt4-dependent functional plasticity (HFMR) is abolished in nwk mutants. (A) Representative traces of mEJPs before (top trace) and after (bottom trace) high-frequency stimulation (4 × 100 Hz). (B) Timecourse of mEJP frequency after stimulation. (C)nwk mutant HFMR phenotype is cell autonomous to neurons. Scatter plot of mEJP frequency 100 s after high-frequency stimulation. Black dashed line represents wild-type (wt) mean value, and the pink dashed line represents nwk mean value. C57-GAL4 and Vglut-GAL4 are muscle and neuron-specific drivers, respectively. (D) Syt4-dependent structural plasticity (ghost bouton budding) is abolished in nwk mutants. Left: Paradigm for spaced stimulation with high K+/Ca2+. Middle: Representative maximum intensity projections of confocal stacks at muscle 6/7 labeled with α-HRP (presynaptic) and α-Dlg (postsynaptic) antibodies. Arrowheads point to ghost boutons, containing HRP but not Dlg, enlarged in inset. Scale bar is 20 µm. Right: Quantification of ghost boutons with and without stimulation. (E) APP BACE-induced eclosion defect is suppressed in nwk mutants. Graph shows the percentage of adult flies emerging from pupal cases. APP and BACE were expressed with C155-GAL4, a neuron-specific driver. (F) GAL4C155-driven APP levels are comparable in wt and nwk heads: Immunoblot of Drosophila head extracts with α-APP C-terminal fragment and α-actin antibodies. Quantification shows fold change of APP relative to actin levels. Data is represented as mean ± SEM (B, C, and D) or mean ± standard error of the proportion (E); n represents NMJs (B–D), pupal cases (E), or biological replicates (F), and in C, D, and F, n is depicted by individual gray dots on the graphs. See Table S1 and Table S3 for detailed genotypes, sample sizes, and statistical analyses. Source data are available for this figure: SourcedataF2.
We next tested whether Nwk is required for Syt4-dependent structural plasticity. Spaced potassium stimulation of wild-type Drosophila synapses leads to the Syt4-dependent formation of nascent synapses, termed ghost boutons, which contain presynaptic markers but have not yet assembled postsynaptic components such as Discs Large (Dlg; Ataman et al., 2008; Korkut et al., 2013; Piccioli and Littleton, 2014). In contrast to the significant increase in ghost bouton formation following spaced stimulation in control animals, nwk mutants did not show an increase (Fig. 2 D). Together, these results indicate that nwk mutants exhibit a loss of Syt4 function in activity-dependent synaptic growth and plasticity. Since nwk mutants have normal Syt4 levels in cell bodies and neuropil, these results further suggest that Syt4 specifically requires presynaptic localization for its functions.
As loss of nwk led to a decrease in hAPP at NMJs (Fig. 1 C and Fig. S1, A and E), we tested if loss of nwk correlates with a reduction in hAPP-induced toxicity. GAL4-mediated neuronal overexpression of hAPP and the amyloidogenic protease beta secretase (BACE) leads to defects including decreased eclosion (Chakraborty et al., 2011; Greeve et al., 2004; Mhatre et al., 2014; Fig. 2 E). Remarkably, this eclosion defect was significantly suppressed in both nwk heterozygous and nwk homozygous mutant backgrounds, suggesting that loss of even one copy of nwk can reduce the toxicity of hAPP and BACE in the adult nervous system (Fig. 2 E). The suppression was not due to broad changes in hAPP levels in adult fly head protein extracts, again suggesting a local and specific effect (Fig. 2 F). Together these results show that the depletion of EV cargoes Syt4 and hAPP at nwk mutant synaptic terminals correlates with a loss of function for these EV cargos in the nervous system.
Endocytic machinery regulates EV cargo traffic at synapses
Nwk is a conserved membrane-remodeling protein that physically interacts with components of the canonical clathrin-associated endocytic machinery (Almeida-Souza et al., 2018; Del Signore et al., 2021; O’Connor-Giles et al., 2008; Rodal et al., 2008; Xiao et al., 2018; Xiao and Schmid, 2020). At Drosophila synapses, this machinery controls synaptic vesicle recycling, synaptic transmission, and growth factor receptor trafficking (Deshpande and Rodal, 2015; Kaempf and Maritzen, 2017). We hypothesized that like Nwk, other endocytic proteins may also regulate EV cargo traffic at synapses. We therefore investigated EV cargo traffic in mutants of endocytic proteins of diverse functions including regulation of membrane lipid composition, membrane remodeling, and the cytoskeleton. Loss of the lipid phosphatase synaptojanin (Synj) or presynaptic knockdown of the endocytic adapter Dap160/intersectin led to a depletion of Syt4-GFP both pre- and postsynaptically (Fig. 3 A; and Fig. S2, A and B). Further, neuronal expression of a dominant negative dynamin mutant (ShiK44A) or presynaptic knockdown of the membrane-deforming protein Endophilin A (EndoA) caused reduction of postsynaptic α-HRP puncta (Fig. 3 B; and Fig. S2, D and E). By contrast, both pre- and postsynaptic Syt4-GFP levels were slightly increased in null mutants of the BAR-SH3 sorting nexin SH3PX1/Snx9, despite its physical interaction with Nwk (Ukken et al., 2016; Fig. 3 A and Fig. S2 C). These results indicate that maintenance of EV cargo levels at synapses is a function shared by some but not all Nwk-interacting endocytic machinery.
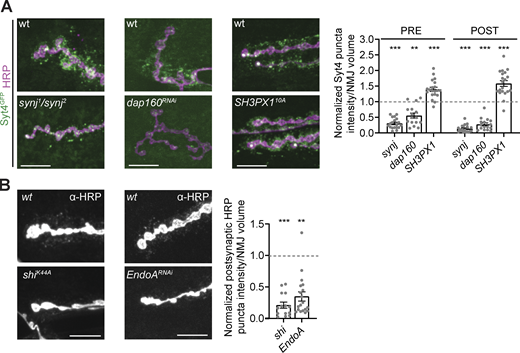
A subset of endocytic machinery is required for EV cargo trafficking. Pre- and postsynaptic EV cargo levels are reduced in multiple endocytic mutants. (A) Left: Representative images of muscle 6/7 NMJs. Right: Quantification of Syt4-GFP puncta intensity. (B) Left: Representative images of α-HRP debris on muscle 6/7 NMJs. Right: Quantification of α-HRP debris intensity. Data is represented as mean ± SEM; n is depicted by individual gray dots on the graphs and represents NMJs. NMJ intensity measurements were normalized to presynaptic volume; all measurements were further normalized to the mean of their respective controls, which are indicated with a dashed line. All scale bars are 10 µm. Associated with Fig. S2. See Table S1 and Table S3 for detailed genotypes, sample sizes, and statistical analyses. wt, wild-type.
A subset of endocytic machinery is required for EV cargo trafficking. Pre- and postsynaptic EV cargo levels are reduced in multiple endocytic mutants. (A) Left: Representative images of muscle 6/7 NMJs. Right: Quantification of Syt4-GFP puncta intensity. (B) Left: Representative images of α-HRP debris on muscle 6/7 NMJs. Right: Quantification of α-HRP debris intensity. Data is represented as mean ± SEM; n is depicted by individual gray dots on the graphs and represents NMJs. NMJ intensity measurements were normalized to presynaptic volume; all measurements were further normalized to the mean of their respective controls, which are indicated with a dashed line. All scale bars are 10 µm. Associated with Fig. S2. See Table S1 and Table S3 for detailed genotypes, sample sizes, and statistical analyses. wt, wild-type.
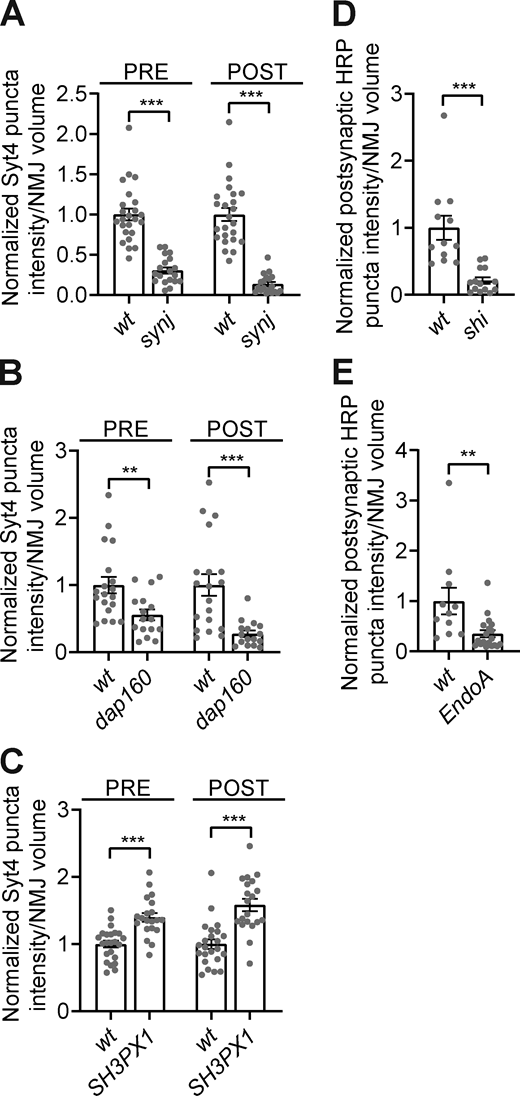
A subset of endocytic machinery is required for synaptic EV traffic.Fig. 3, A and B datasets shown with corresponding controls. (A) Quantification of pre- postsynaptic Syt4-GFP puncta intensity in control (wt) and synj mutants. (B) Quantification of pre- and postsynaptic Sy4-GFP puncta intensity in control and upon neuronal knockdown of dap160. (C) Quantification of pre- and postsynaptic Syt4-GFP puncta intensity in control and SH3PX1 mutants. (D) Quantification of postsynaptic HRP puncta intensity in control and upon neuronal expression of ShiK44A. (E) Quantification of postsynaptic HRP puncta intensity in control and upon neuronal knockdown of EndoA. Data is represented as mean ± SEM; n is depicted by individual gray dots on the graphs and represents NMJs. NMJ intensity measurements were normalized to presynaptic volume; all measurements were further normalized to the mean of their respective controls. Associated with Fig. 3. See Table S1 and Table S3 for detailed genotypes, sample sizes, and statistical analyses.
A subset of endocytic machinery is required for synaptic EV traffic.Fig. 3, A and B datasets shown with corresponding controls. (A) Quantification of pre- postsynaptic Syt4-GFP puncta intensity in control (wt) and synj mutants. (B) Quantification of pre- and postsynaptic Sy4-GFP puncta intensity in control and upon neuronal knockdown of dap160. (C) Quantification of pre- and postsynaptic Syt4-GFP puncta intensity in control and SH3PX1 mutants. (D) Quantification of postsynaptic HRP puncta intensity in control and upon neuronal expression of ShiK44A. (E) Quantification of postsynaptic HRP puncta intensity in control and upon neuronal knockdown of EndoA. Data is represented as mean ± SEM; n is depicted by individual gray dots on the graphs and represents NMJs. NMJ intensity measurements were normalized to presynaptic volume; all measurements were further normalized to the mean of their respective controls. Associated with Fig. 3. See Table S1 and Table S3 for detailed genotypes, sample sizes, and statistical analyses.
The canonical function of endocytic proteins is to control clathrin-mediated endocytosis, which is a primary mechanism for synaptic vesicle recycling at the Drosophila larval NMJ (Heerssen et al., 2008; Kasprowicz et al., 2008) and is required for synaptic morphogenesis via endocytic traffic of growth factor receptors (Choudhury et al., 2016; Dwivedi et al., 2021). We asked if clathrin and its heterotetrameric AP-2 adaptor complex were similarly involved in synaptic EV cargo traffic. We found that mutants lacking the AP-2σ subunit exhibited a significant decrease in pre- and postsynaptic levels of Syt4-GFP (Figs. 4 A and S3 A). Surprisingly, we found mutants lacking either the AP-2α or the AP-2µ subunit exhibited only a mild decrease in presynaptic levels of Syt4-GFP, and no significant change postsynaptically (Figs. 4 A and S3 B). This suggests that the levels of EV cargo at synapses specifically depend on the AP-2σ subunit, or that there is redundancy between the AP-2α and AP-2μ subunits, as has been previously suggested (Gu et al., 2013). To decipher between these possibilities, we measured Syt4-GFP levels in AP-2α; AP-2μ double mutants and found a striking decrease in pre- and postsynaptic levels of Syt4-GFP, similar to nwk mutants (Figs. 4 A and S3 B). Thus, EV cargo levels are dependent on the AP-2 clathrin adaptor complex with functional redundancies among the AP-2α and AP-2μ subunits.
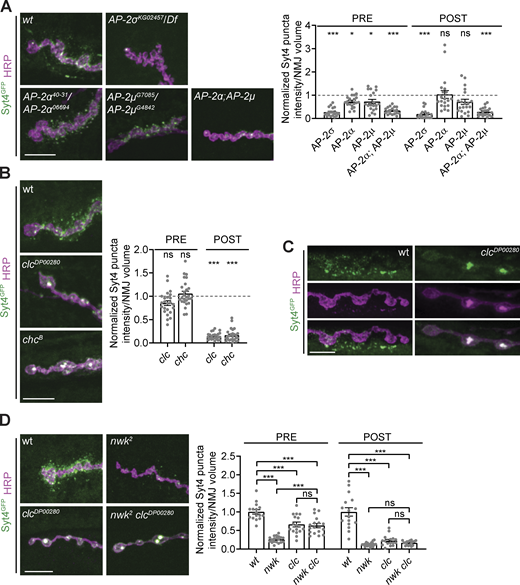
EV cargo trafficking requires clathrin-associated endocytic machinery. (A) Pre- and postsynaptic EV cargo levels are reduced in clathrin adaptor AP-2 mutants. Left: Representative images of muscle 6/7 NMJs. Right: Quantification of Syt4-GFP puncta intensity. (B) EV cargoes show altered localization in clathrin mutants and are not released from neurons. Left: Representative images of muscle 6/7 NMJs. Right: Quantification of Syt4-GFP puncta intensity. (C) Airyscan microscopy of clc mutant muscle 6/7 NMJs show that EV cargo accumulations extend into the interior of the bouton. (D) EV cargo accumulation in clathrin mutants is epistatic to its depletion in nwk mutants. Left: Representative images of muscle 6/7 NMJs. Right: Quantification of Syt4-GFP puncta intensity. Data is represented as mean ± SEM; n is depicted by individual gray dots on the graphs and represents NMJs. NMJ intensity measurements were normalized to presynaptic volume; all measurements were further normalized to the mean of their respective controls, which is indicated with a dashed line. All scale bars are 10 µm. Associated with Fig. S3. See Table. S1 and Table. S3 for detailed genotypes, sample sizes, and statistical analyses. wt, wild-type.
EV cargo trafficking requires clathrin-associated endocytic machinery. (A) Pre- and postsynaptic EV cargo levels are reduced in clathrin adaptor AP-2 mutants. Left: Representative images of muscle 6/7 NMJs. Right: Quantification of Syt4-GFP puncta intensity. (B) EV cargoes show altered localization in clathrin mutants and are not released from neurons. Left: Representative images of muscle 6/7 NMJs. Right: Quantification of Syt4-GFP puncta intensity. (C) Airyscan microscopy of clc mutant muscle 6/7 NMJs show that EV cargo accumulations extend into the interior of the bouton. (D) EV cargo accumulation in clathrin mutants is epistatic to its depletion in nwk mutants. Left: Representative images of muscle 6/7 NMJs. Right: Quantification of Syt4-GFP puncta intensity. Data is represented as mean ± SEM; n is depicted by individual gray dots on the graphs and represents NMJs. NMJ intensity measurements were normalized to presynaptic volume; all measurements were further normalized to the mean of their respective controls, which is indicated with a dashed line. All scale bars are 10 µm. Associated with Fig. S3. See Table. S1 and Table. S3 for detailed genotypes, sample sizes, and statistical analyses. wt, wild-type.
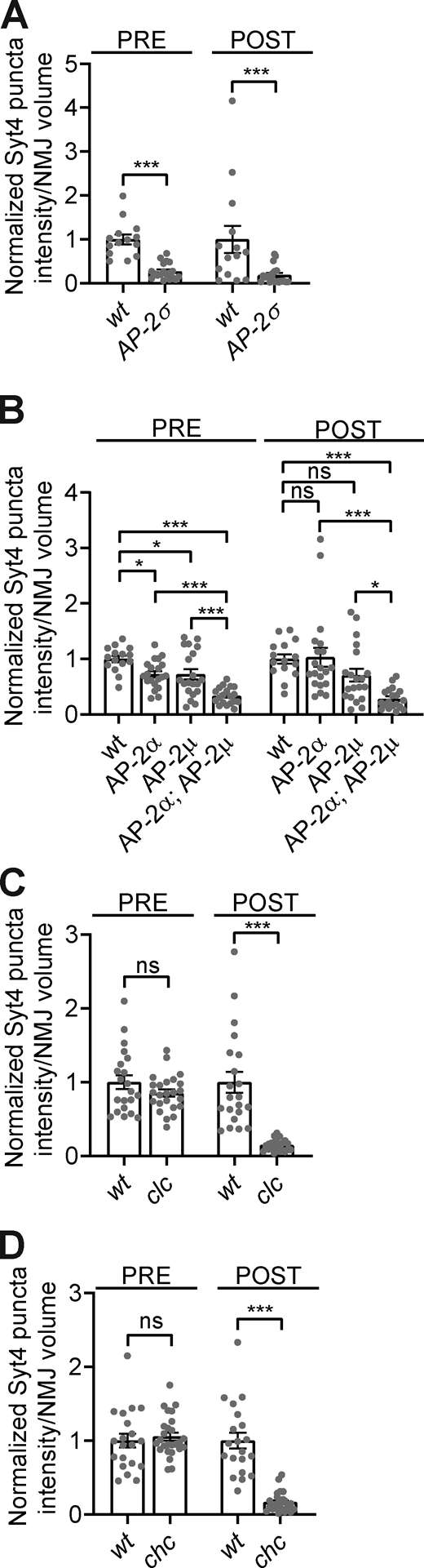
EV traffic is dependent on clathrin-associated endocytic machinery.Fig. 4, A and B datasets shown with corresponding controls. (A) Quantification of Syt4-GFP pre- and postsynaptic puncta intensity in control (wt) and AP-2σ mutants. (B) Quantification of Syt4-GFP pre- and postsynaptic puncta intensity in control, AP-2α, AP-2μ, and AP-2α;AP-2μ mutants. (C) Quantification of Syt4-GFP pre- and postsynaptic puncta intensity in control and clc mutants. (D) Quantification of Syt4-GFP pre- and postsynaptic puncta intensity in control and chc mutants. Data is represented as mean ± SEM; n is depicted by individual gray dots on the graphs and represents NMJs. NMJ intensity measurements were normalized to presynaptic volume; all measurements were further normalized to the mean of their respective controls. Associated with Fig. 4. See Table S1 and Table S3 for detailed genotypes, sample sizes, and statistical analyses.
EV traffic is dependent on clathrin-associated endocytic machinery.Fig. 4, A and B datasets shown with corresponding controls. (A) Quantification of Syt4-GFP pre- and postsynaptic puncta intensity in control (wt) and AP-2σ mutants. (B) Quantification of Syt4-GFP pre- and postsynaptic puncta intensity in control, AP-2α, AP-2μ, and AP-2α;AP-2μ mutants. (C) Quantification of Syt4-GFP pre- and postsynaptic puncta intensity in control and clc mutants. (D) Quantification of Syt4-GFP pre- and postsynaptic puncta intensity in control and chc mutants. Data is represented as mean ± SEM; n is depicted by individual gray dots on the graphs and represents NMJs. NMJ intensity measurements were normalized to presynaptic volume; all measurements were further normalized to the mean of their respective controls. Associated with Fig. 4. See Table S1 and Table S3 for detailed genotypes, sample sizes, and statistical analyses.
The importance of the AP-2 clathrin adaptor complex in EV cargo regulation prompted us to test directly whether EV cargo levels at synapses are dependent on clathrin itself. We examined Syt4-GFP in hypomorphic mutants of clathrin light chain (clc) or clathrin heavy chain (chc). We found no change in presynaptic levels of Syt4-GFP in clc and chc mutants compared to controls, but its localization was drastically shifted into large α-HRP–positive presynaptic accumulations (Fig. 4, B and C; and Fig. S3, C and D). This change in Sy4-GFP presynaptic localization in clc and chc mutants was associated with a striking decrease in postsynaptic levels of Syt4-GFP compared to controls (Fig. 4 B; and Fig. S3, C and D), suggesting that Syt4-GFP is not released in EVs from clathrin mutant synapses. The inactivation of clathrin is known to lead to the formation of large cisternal membrane compartments derived from bulk membrane uptake in synapses (Heerssen et al., 2008; Kasprowicz et al., 2008), which may correspond to the Syt4-GFP and HRP-positive accumulations that we see in clc and chc synapses. As nwk mutants exhibit a significant depletion of EV cargoes both pre- and postsynaptically (Fig. 1, B and C), we wondered whether these presynaptic accumulations of Syt4-GFP in clc and chc mutants would be sensitive or resistant to loss of nwk. We found that simultaneous loss of clc and nwk phenocopied loss of clc alone with regards to levels and localization of Syt4-GFP pre- and postsynaptically (Fig. 4 D). This suggests that the Syt4-GFP accumulations in clc mutant synapses are not accessible to the mechanism by which EV cargoes are depleted at nwk mutant synapses. Although the grossly disrupted membrane structures found at clathrin mutant synaptic terminals make it difficult to interpret the specific role of clathrin itself in EV cargo traffic, our finding that loss of canonical clathrin adaptors recapitulates the phenotypes of other endocytic mutants suggests that a clathrin-associated mechanism regulates maintenance of EV cargoes at synapses.
Nwk opposes the function of Vps35/retromer in EV cargo trafficking
We next explored the mechanisms by which Nwk-associated membrane-remodeling machinery controls the distribution of EV cargoes between release in EVs versus synaptic depletion. One possibility is that this machinery acts at the plasma membrane in a noncanonical role to regulate fusion of EV-containing MVBs (similar to functions of dynamin at chromaffin granule fusion pores (Anantharam et al., 2011; Wong et al., 2015) and that in its absence these MVBs are targeted for degradation. Alternatively, endocytic machinery could serve a canonical function in endocytic loading of cargoes into an EV-permissive precursor compartment that populates MVBs destined for EV release. Mutation of the retromer complex component Vps35 causes accumulation of EV cargoes in these precursor compartments and a concomitant increased release of EVs (Walsh et al., 2021). If the Nwk-dependent endocytic machinery acts to promote EV fusion with the plasma membrane, we would expect to see recovery of presynaptic cargo levels in Vps35;nwk double mutants relative to nwk single mutants, but reduced postsynaptic recovery, as MVBs would not be released (Fig. 5 A, “release”). On the other hand, if Nwk plays a more canonical role in loading MVB precursors prior to their release, we would expect to see recovery of both pre- and postsynaptic EV cargo levels in Vps35;nwk mutants relative to nwk single mutants (Fig. 5 A, “sorting”). We found that loss of Vps35 in the nwk mutant background significantly increased Syt4-GFP levels both pre- and postsynaptically, and also significantly increased the number of postsynaptic Syt4-GFP puncta (Fig. 5 B) compared to the nwk mutant alone, suggesting that Nwk is involved in loading EV precursors and maintaining presynaptic cargo levels, rather than in fusion of MVBs and release of EVs from the neuron.
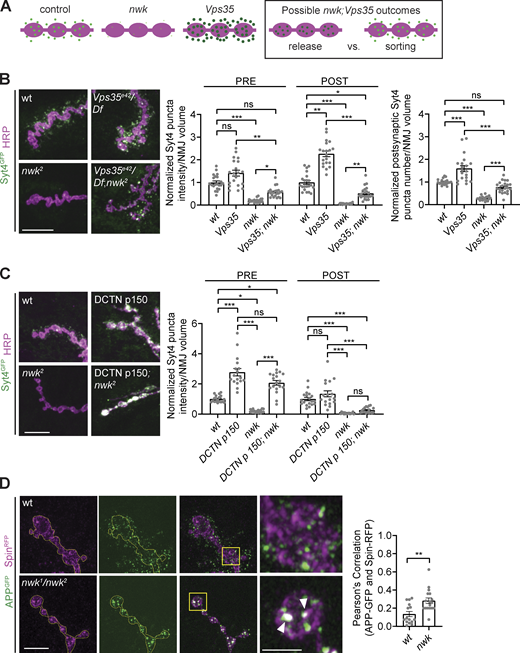
Nwk opposes retromer in loading EV precursors and acts upstream of dynactin-mediated retrograde transport. (A) Cartoon depicting potential EV phenotype outcomes for nwk; Vps35 experiment, testing if Nwk regulates MVB–plasma membrane fusion or EV cargo sorting. (B) EV cargoes are increased both pre- and postsynaptically in nwk; Vps35 double mutants relative to nwk mutants. Left: Representative images of muscle 6/7 NMJs. Right: Quantification of Syt4-GFP puncta intensity and number. (C) EV cargoes are retained at the synapse upon p150-DCTN overexpression, but these compartments are not competent for EV release. Left: Representative images of muscle 6/7 NMJs. Right: Quantification of Syt4-GFP puncta intensity. (D) EV cargo APP-GFP exhibits enhanced colocalization with the late endosome/lysosome marker Spin in nwk mutants. Left: Airyscan microscopy of muscle 6/7 NMJs. Outlines of presynaptic region are shown in yellow. Middle: Zoom of boutons of yellow boxed regions of NMJ. Arrowhead points to colocalized spots of APP-GFP and Spin-RFP. Right: Pearson’s correlation of APP-GFP and Spin-RFP signal within the presynaptic region only. Data is represented as mean ± SEM; n is depicted by individual gray dots on the graphs and represents NMJs. NMJ intensity measurements were normalized to presynaptic volume; all measurements were further normalized to the mean of wild-type (wt) controls. Scale bars are 10 µm (B and C), 5 µm (D, three rows of panels on left), and 2 µm (D, enlarged insets on right). Associated with Fig. S4. See Table S1 and Table S3 for detailed genotypes, sample sizes, and statistical analyses.
Nwk opposes retromer in loading EV precursors and acts upstream of dynactin-mediated retrograde transport. (A) Cartoon depicting potential EV phenotype outcomes for nwk; Vps35 experiment, testing if Nwk regulates MVB–plasma membrane fusion or EV cargo sorting. (B) EV cargoes are increased both pre- and postsynaptically in nwk; Vps35 double mutants relative to nwk mutants. Left: Representative images of muscle 6/7 NMJs. Right: Quantification of Syt4-GFP puncta intensity and number. (C) EV cargoes are retained at the synapse upon p150-DCTN overexpression, but these compartments are not competent for EV release. Left: Representative images of muscle 6/7 NMJs. Right: Quantification of Syt4-GFP puncta intensity. (D) EV cargo APP-GFP exhibits enhanced colocalization with the late endosome/lysosome marker Spin in nwk mutants. Left: Airyscan microscopy of muscle 6/7 NMJs. Outlines of presynaptic region are shown in yellow. Middle: Zoom of boutons of yellow boxed regions of NMJ. Arrowhead points to colocalized spots of APP-GFP and Spin-RFP. Right: Pearson’s correlation of APP-GFP and Spin-RFP signal within the presynaptic region only. Data is represented as mean ± SEM; n is depicted by individual gray dots on the graphs and represents NMJs. NMJ intensity measurements were normalized to presynaptic volume; all measurements were further normalized to the mean of wild-type (wt) controls. Scale bars are 10 µm (B and C), 5 µm (D, three rows of panels on left), and 2 µm (D, enlarged insets on right). Associated with Fig. S4. See Table S1 and Table S3 for detailed genotypes, sample sizes, and statistical analyses.
Nwk acts upstream of dynactin-mediated retrograde transport
Our data suggest that endocytic machinery protects EV cargoes from degradation by loading them into an EV-permissive precursor compartment. We hypothesized that in nwk mutants, these EV cargoes may be depleted from the synapse via retrograde traffic to the cell body for lysosomal degradation. To test this, we overexpressed a dominant negative dynactin (DCTN) truncated p150/Glued subunit to inhibit dynein/dynactin-mediated retrograde transport (Allen et al., 1999). In otherwise wild-type synapses expressing DCTN-p150Δ, we found that presynaptic Syt4-GFP levels more than doubled (Fig. 5 C), indicating that EV cargoes do undergo retrograde transport and build up at the synapse in the absence of dynein-mediated transport. Interestingly, there was no concomitant increase in postsynaptic Syt4-EGFP levels (Fig. 5 C), indicating that the cargoes destined for retrograde transport are located in a non-EV permissive compartment. We then used this system to test whether in nwk mutants EV cargoes are depleted from the synapse due to retrograde transport. We overexpressed DCTN-p150Δ in the nwk mutant background and found the same striking increase in presynaptic Syt4-GFP levels as in wild-type synapses (Fig. 5 C). This result both provides further evidence that delivery of EV cargoes to synapses is not compromised in nwk mutants and indicates that they are largely being depleted from synapses by retrograde transport. Further, postsynaptic Syt4-GFP levels remained low relative to wild-type controls (Fig. 5 C), indicating that in nwk mutants synaptic EV cargoes that are destined for retrograde transport are in a non-EV permissive compartment.
To explore the nature of this compartment, we examined presynaptic colocalization of EV cargoes with the late endosome/lysosome marker Spinster (Spin; Sweeney and Davis, 2002). Expression of Spin-RFP did not modify the nwk mutant EV phenotype at synaptic terminals, as we still observed a pre- and postsynaptic depletion of hAPP-EGFP (Fig. S4 A). Notably, the residual hAPP-EGFP puncta in nwk mutant presynaptic terminals exhibited increased colocalization with Spin-RFP compared to controls (Fig. 5 D), with no change in colocalization with the presynaptic membrane-marking α-HRP signal as a negative control (Fig. S4 B), indicating that EV cargoes are shifted to degradative compartments in nwk mutants. Finally, we observed a mild decrease in Spin-RFP intensity at nwk mutant NMJs (Fig. S4 C), indicating that degradative compartments are not expanded in these mutant synapses, and further suggesting that sorting defects occur upstream of endolysosomal maturation. Overall, our data suggest a working model in which synaptic depletion of EV cargoes in nwk mutants results from a shift of cargoes toward a degradative compartment that cannot produce EVs and is sent to the cell body via retrograde transport (Fig. 6).
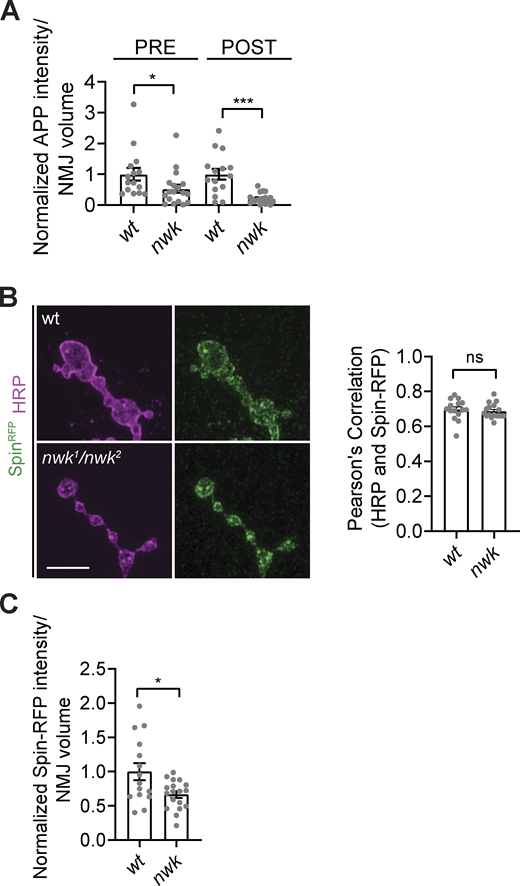
EV cargo levels are reduced at nwk mutant synapses, and remaining EV cargo puncta specifically colocalize with degradative compartments. (A) Quantification of APP-GFP pre- and postsynaptic puncta intensity in control (wt) and nwk mutants. (B) Presynaptic α-HRP and Spin-RFP colocalize to the same degree at control and nwk mutant NMJs. (Left) Representative Airyscan images of muscle 6/7 NMJs with α-HRP and Spin-RFP. (Right) Pearson’s correlation of presynaptic α-HRP and Spin-RFP. (C) Quantification of presynaptic Spin-RFP puncta intensity in control and nwk mutants. Data is represented as mean ± SEM; n is depicted by individual gray dots on the graphs and represents NMJs. NMJ intensity measurements were normalized to presynaptic volume; all measurements were further normalized to the mean of their respective controls. Scale bar is 5 µm. Associated with Fig. 5. See Table S1 and Table S3 for detailed genotypes, sample sizes, and statistical analyses.
EV cargo levels are reduced at nwk mutant synapses, and remaining EV cargo puncta specifically colocalize with degradative compartments. (A) Quantification of APP-GFP pre- and postsynaptic puncta intensity in control (wt) and nwk mutants. (B) Presynaptic α-HRP and Spin-RFP colocalize to the same degree at control and nwk mutant NMJs. (Left) Representative Airyscan images of muscle 6/7 NMJs with α-HRP and Spin-RFP. (Right) Pearson’s correlation of presynaptic α-HRP and Spin-RFP. (C) Quantification of presynaptic Spin-RFP puncta intensity in control and nwk mutants. Data is represented as mean ± SEM; n is depicted by individual gray dots on the graphs and represents NMJs. NMJ intensity measurements were normalized to presynaptic volume; all measurements were further normalized to the mean of their respective controls. Scale bar is 5 µm. Associated with Fig. 5. See Table S1 and Table S3 for detailed genotypes, sample sizes, and statistical analyses.

Endocytic machinery protects EV cargoes from local synaptic depletion. At wild-type synapses, proper traffic and function of EV cargoes depend on a robust flux through the recycling pathway (left, orange arrows). However, endocytic mutant synapses exhibit a depletion of EV cargoes, likely due to reduced recycling flux, resulting in aberrant sorting for degradation upstream of dynactin/dynein mediated retrograde transport (right). EE is early endosome, RE is recycling endosome.
Endocytic machinery protects EV cargoes from local synaptic depletion. At wild-type synapses, proper traffic and function of EV cargoes depend on a robust flux through the recycling pathway (left, orange arrows). However, endocytic mutant synapses exhibit a depletion of EV cargoes, likely due to reduced recycling flux, resulting in aberrant sorting for degradation upstream of dynactin/dynein mediated retrograde transport (right). EE is early endosome, RE is recycling endosome.
Discussion
Here we report a novel function of canonical endocytic machinery in regulating trafficking of EV cargoes at synapses in vivo. We find that endocytic mutants exhibit a local depletion of EV cargoes at synaptic terminals, correlating with a loss of Syt4-dependent synaptic plasticity at the larval NMJ and a reduction in APP-dependent toxicity in the adult nervous system. Mechanistically, the endocytic machinery opposes the retromer complex in sorting cargoes into EV-precursor compartments and acts upstream of dynactin-mediated retrograde transport. Together, our data support a working model in which the endocytic machinery protects EV cargoes from local depletion at synaptic terminals, consequently preserving cargo function and promoting its release in EVs. Our results suggest a new interpretation for previously reported phenotypes of endocytic mutants and uncover a new pathway for investigating and therapeutically intervening in EV traffic.
EV traffic is genetically separable from canonical functions of the endocytic machinery
Our discovery that EV cargoes are depleted from endocytic mutant synaptic terminals was surprising, as the expected phenotype for loss of endocytic machinery is accumulation of EV cargoes at the plasma membrane. Indeed, the phenotypes we observed for endocytic mutants (reduction in cargo levels) and retromer (increase in cargo levels; Walsh et al., 2021) at synapses are opposite to what has been found for EV-relevant cargoes in other cell types. For example, in several Caenorhabditis elegans cell types, Wntless/Evi accumulates near the plasma membrane in dpy-23/AP-2 mutants and is depleted in vps-35 mutants (Pan et al., 2008; Yang et al., 2008). Similarly, EV cargoes are trapped at the plasma membrane upon inhibition of endocytosis in PC12 cells and sympathetic neuronal cell bodies (Escudero et al., 2014), or in HeLa cells (Janvier and Bonifacino, 2005; Lau and Chou, 2008). Our results underscore the unique properties of trafficking in distal neuronal processes such as synapses, due to their extreme morphology and limited lysosomal capacity, and highlight the importance of using in vivo models to study neuronal EVs (Blanchette and Rodal, 2020; Ferguson, 2018).
Our new findings also add to the diverse functions of the endocytic machinery in neurons, including synaptic vesicle recycling, release site clearance, synaptic transmission, signaling receptor trafficking, and synaptic growth (Chanaday et al., 2019; Deshpande and Rodal, 2015). If EV cargo trafficking defects in endocytic mutants were indirectly caused by impairment of these canonical roles, then we would expect the severity of EV phenotypes to consistently scale with defects in these other functions. However, we find that mutants with more severe defects in synaptic vesicle recycling (e.g., AP-2α; Gonzalez-Gaitan and Jackle, 1997) exhibit very mild EV phenotypes, while mutants with mild synaptic vesicle cycling phenotypes (e.g., nwk and dap160; Del Signore et al., 2021; Koh et al., 2004; Marie et al., 2004) have severe EV defects. In a similar vein, AP-2α, AP-2μ, and dap160 mutants exhibit severe synaptic growth phenotypes (Choudhury et al., 2016; Dwivedi et al., 2021; Koh et al., 2004; Marie et al., 2004), likely due to the failure to downregulate BMP receptor signaling (Deshpande and Rodal, 2015). Among these, only loss of dap160 has a severe EV defect. By contrast, nwk mutants have a relatively mild synaptic growth phenotype (Coyle et al., 2004) and a severe EV phenotype. Vps35/retromer mutants also exhibit synaptic vesicle cycling defects (Inoshita et al., 2017) and BMP pathway-dependent synaptic overgrowth (Korolchuk et al., 2007), but conversely accumulate excess EV cargoes rather than depleting them (Walsh et al., 2021). Thus, synaptic vesicle endocytosis, synaptic growth signaling, and EV trafficking can be genetically uncoupled.
EV trafficking functions of the endocytic machinery are also unlikely to arise from indirect effects on neurotransmitter release. For example, loss of EndoA leads to decreased EV cargo levels, but EndoA mutants have no defects in synaptic transmission (Verstreken et al., 2002). Interestingly, nwk and SH3PX1 physically interact and share a role in synaptic transmission (Cao et al., 2013; Coyle et al., 2004; Ukken et al., 2016). However, this shared role is not linked to their EV functions, as we found that SH3PX1 mutants exhibit a significant increase in EV cargo levels, opposite to nwk mutants. SH3PX1 could directly inhibit the EV cargo sorting activity of Nwk, or else it may act independently on recycling endosomes to restrict EV traffic, as suggested by studies of its functions in other cell types (Bendris and Schmid, 2017; Knaevelsrud et al., 2013; Zhang et al., 2019). Overall, our results indicate that the role of endocytic machinery in regulating EV cargo traffic is genetically separable from its roles in synaptic transmission, synaptic vesicle endocytosis, and synaptic growth, and instead reflects an independent and novel function of these conserved molecules at synaptic terminals.
Endocytic machinery sorts cargoes into EV-precursor compartments
What then is the mechanism by which endocytic machinery regulates EV cargo levels? Our data indicate that Nwk functions in an opposing pathway to the retromer complex to load cargoes into EV-permissive compartments. One possibility is that this occurs via the canonical role of the clathrin/AP-2 endocytic machinery at the plasma membrane (Kaksonen and Roux, 2018). Our data differentiating the phenotypes of clathrin and AP-2 mutants provide some insights into the specific mechanisms involved: We find that clathrin is strictly required to form functional EV precursors, and in its absence, cargoes likely accumulate in large cisternae. By contrast, loss of AP-2 adaptors and other endocytic machinery leads to cargo depletion, perhaps due to a slowed but still partially functional endocytic pathway (Chen and Schmid, 2020; Dickman et al., 2005). Indeed, efficient endocytic internalization could be required for effective EV loading by increasing the flux of cargo through the endosome–plasma membrane recycling pathway that populates these EV precursor compartments (Walsh et al., 2021; Fig. 6, orange arrows). In support of this hypothesis, loss of nwk phenocopies mutation of the recycling endosome regulator Rab11, including presynaptic depletion of EV cargoes in opposition to retromer, and loss of Syt4 physiological function (Ashley et al., 2018; Koles et al., 2012; Korkut et al., 2013; Walsh et al., 2021). Finally, our observation that AP-2α and AP-2μ exhibit partial redundancy indicates that hemicomplexes of AP-2 may be sufficient to sort EV cargo (Gu et al., 2013), or that other adaptors such as AP180 may also function in this pathway (Bao et al., 2005).
Alternatively, endocytic machinery may be acting in a noncanonical role on intracellular compartments to sort EV cargoes. Endocytic proteins are required to reform synaptic vesicles from endosomal intermediates, particularly during elevated activity (Heerssen et al., 2008; Kasprowicz et al., 2008; Kasprowicz et al., 2014; Kittelmann et al., 2013; Kononenko et al., 2014; Watanabe et al., 2014). Clathrin localizes directly to endosomes, where it regulates endosomal cargo sorting and intralumenal vesicle formation (Raiborg et al., 2001; Raiborg et al., 2006; Sachse et al., 2002; Shi et al., 2009; Wenzel et al., 2018). Further, the mammalian Nwk homolog FCHSD2, in addition to its role at the internalization step of endocytosis (Almeida-Souza et al., 2018; Xiao et al., 2018), suppresses recycling and lysosomal degradation of signaling receptors (Xiao and Schmid, 2020). Retromer functions directly on endosomes and genetically opposes the functions of Nwk, suggesting that Nwk may also act on endosomes. The extreme density of endocytic machinery at synaptic membranes makes it difficult to detect a small endosomally localized fraction, although Nwk does exhibit partial colocalization with Rab11 (Rodal et al., 2008). Future studies will be necessary to discriminate between whether clathrin-mediated endocytic machinery indirectly loads EV-precursor endosomes via canonical endocytosis, or if it instead acts on them directly.
EV cargo sorting for release occurs upstream of retrograde transport
Our data provide new insights into the overall trafficking itineraries of neuronal EV cargoes. The following observations suggest that EV precursor compartments are locally loaded at synapses by the endocytic machinery: (1) We observed no defect in retrograde or anterograde axonal traffic that could account for reduced synaptic levels. (2) Endocytic machinery is abundant and active at synaptic terminals, and endocytic mutants lead to local loss of EV cargoes at synapses. (3) Dynactin mutants cause synaptic accumulation of EV cargo-containing compartments, indicating that these compartments are generated at synapses.
Our data also indicate that once cargoes are destined for retrograde transport, they are no longer EV permissive, and therefore dynein-mediated transport does not regulate release (as has been hypothesized in other systems; Heisler et al., 2018). Indeed, trapping these compartments at nwk mutant synapses is sufficient to restore presynaptic levels of EV cargoes but does not allow the release of these cargoes from the neuron. Thus, Nwk-mediated cargo sorting occurs upstream of retrograde transport, and the missorted cargoes in the nwk mutant are no longer EV-competent and are destined for synaptic removal, likely for degradation in the cell body. In support of this, we find enhanced colocalization between residual EV cargo puncta and the late endosome/lysosome marker Spin at nwk mutant synaptic terminals. Degradative lysosomes are enriched in cell bodies relative to axons and synaptic terminals (Ferguson, 2018; Lie et al., 2021), and there is also evidence of local protein degradation along axons or at axon terminals (Farfel-Becker et al., 2019; Jin et al., 2018). Our data suggest for the first time that synaptic EV cargoes are degraded after transport rather than on-site, although we cannot exclude the possibility of separate pools of cargoes: Some that are locally degraded, and some that are targeted for retrograde transport. Future in vivo investigations focused on the role of retrograde trafficking machinery and their adaptors may yield insights into additional points by which neurons control degradation of EV cargoes at distinct subcellular locations (Heisler et al., 2018; van Niel et al., 2018).
Implications for the functions of neuronal EVs and for EV-directed disease therapies
Although there has been an explosion of research identifying the contents of EVs and the diverse functions of their cargoes, the field has been lacking approaches to specifically manipulate their biogenesis and release. Our findings provide new tools to ask where EV cargoes act in the neuron, to locally manipulate EV cargo levels at synapses, and to define the functions of the EV trafficking route. We found that loss of nwk leads to a local decrease in EV cargo levels presynaptically (and as a result, postsynaptically), with a corresponding loss of EV cargo function, indicating that levels of EV cargoes at synaptic terminals, but not elsewhere in the neuron, correlate with EV cargo function. Importantly, to date, endogenous EV cargo function at the Drosophila NMJ has not been tested in trafficking mutants that are demonstrated to specifically disrupt EV release without impacting levels in the donor cell (Ashley et al., 2018; Korkut et al., 2009; Korkut et al., 2013; Lauwers et al., 2018). Our results suggest that in addition to the trans-synaptic signaling functions that have been previously proposed, EV trafficking could play an equally important role for cargoes whose primary function is in the donor neuron, as a proteostatic mechanism to maintain levels at specific subcellular locations such as synapses.
Given that local depletion of EV cargoes at nwk mutant synapses correlates with a loss of their physiological functions, EV cargo misregulation may also play unrecognized roles in the previously reported phenotypes of endocytic mutants. These may therefore need to be reinterpreted in the context of local synaptic loss of a multitude of EV cargoes (Holm et al., 2018). For example, Shits mutants have been used in numerous Drosophila circuit-mapping studies to block synaptic transmission at the restrictive temperature (Kitamoto, 2001); however, these mutants also show defects at the permissive temperature (Dickman et al., 2006), which could inadvertently cause chronic loss of EV cargoes important for that circuit. Further, neurological phenotypes of mice mutant for endocytic machinery could be consistent with EV signaling defects (Holm et al., 2018; Malakooti et al., 2020; Milosevic et al., 2011).
Finally, our results suggest new interpretations of neurological disease mechanisms. Defects in EV and endosomal trafficking are linked to neurodegenerative diseases such as Alzheimer’s disease (Becot et al., 2020; Song et al., 2020; Winckler et al., 2018). Aged neurons exhibit increased APP endocytosis, leading to endosomal dysfunction and synapse loss (Burrinha et al., 2021). Our data predict that this may also promote APP loading into the EV pathway. This is consistent with previous reports suggesting that upregulation of endocytic machinery exacerbates Alzheimer’s disease phenotypes (Keating et al., 2006; Ren et al., 2008; Yu et al., 2018), and that loss of endocytic machinery can suppress these phenotypes (Zhu et al., 2013). Downregulation of APP, either genetically or by enhancing lysosomal biogenesis, can suppress generation of amyloid beta and amyloid plaques in Alzheimer’s disease (Hung et al., 2021; Xiao et al., 2015). We report a new and synapse-specific mechanism for targeting APP: Loss of nwk severely reduces synaptic levels of APP and ameliorates its toxicity in the nervous system while causing only mild defects in the synaptic vesicle cycle and synaptic growth. Our data suggest that therapeutically targeting EV-sensitive components of endocytic machinery could be a strategy to reduce pathological synaptic EV cargoes in neurological disease.
Materials and Methods
Drosophila culture
Flies were cultured using standard media and techniques. Flies used for experiments were maintained at 25°C, except for RNAi experiments (Dap160 and EndoA) that were maintained at 29°C, and the chcB and clc nwk experiments that were maintained at 20°C. For detailed information on fly stocks used, see Table S1, and for detailed genotype information for each figure panel, see Table S3.
Immunohistochemistry
Wandering third instar larvae from density-controlled crosses were dissected in HL3.1 (Feng et al., 2004) and fixed in HL3.1 with 4% paraformaldehyde for 10 min. For detailed information on antibodies used in this study, see Table S2. Washes and antibody dilutions were conducted using PBS containing 0.2% Triton X-100 (0.2% PBX). Primary antibody incubations were conducted overnight at 4°C, and secondary antibody incubations for 1–2 h at room temperature. α-HRP incubations were conducted either overnight at 4°C or for 1–2 h at room temperature. Prior to imaging, fillets were mounted on slides with Vectashield (Vector Labs). α-GFP nanobodies (Nanotag Biotechnologies) were used to amplify Syt4-GFP signal only for Fig. 4 C.
Image acquisition and analysis
Image acquisition
For analysis of EV and non-EV cargoes, NMJs (muscle 6/7 [segments A2 and A3] and muscle 4 [segments A2, A3, and A4]), brains (neuropil and cell bodies), and axons were imaged at room temperature. Z-stacks were acquired using a Nikon Ni-E upright microscope equipped with a Yokogawa CSU-W1 spinning-disk head, an Andor iXon 897U EMCCD camera, and Nikon Elements AR software. A 60× (NA 1.4) oil immersion objective was used to image NMJs, cell bodies, and fixed axons, and a 40× (NA 0.75) oil immersion objective for neuropils. For axonal trafficking, timelapse images of axon bundles proximal to the ventral nerve cord were taken with a 60× (NA 1.4) oil immersion objective. Nine Z slices were collected per frame (Step size 0.3 µm, with no acquisition delay between timepoints, resulting in a frame rate of 2.34–2.37 s/frame). Airyscan images in Fig. 4 C and Fig. 5 D were acquired at room temperature with Zen Blue software on a Zeiss LSM880 Fast Airyscan microscope in super resolution acquisition mode, using a 63× (NA 1.4) oil immersion objective. Image acquisition settings were identical for all images in each independent experiment.
Quantification of pre- and postsynaptic EV cargo levels at the NMJ
The 3D volumetric analyses of pre- and postsynaptic EV cargoes at NMJs were carried out using Volocity 6.0 software (Perkin Elmer). For each NMJ image, both type 1s and 1b boutons were retained for analysis while axons were cropped out. Presynaptic volume was determined through manual thresholding to the α-HRP signal (excluding objects smaller than 7 μm3 and closing holes), with a 3.3 μm dilation around this signal delineating the postsynaptic volume. Manual thresholding of the EV cargo signal was conducted to ensure measurements included only EV cargo signal above background muscle fluorescence. Postsynaptic objects smaller than 0.015 μm3 were excluded. EV cargo sum intensity measurements were calculated within these pre- and postsynaptic volumes and normalized to the presynaptic volume. Puncta number was quantified only within the postsynaptic volume.
Quantification of Syt4-GFP in cell bodies
This analysis was conducted in FIJI. Syt4-GFP intensity in even-skipped positive motor neurons was measured from a single middle slice through each cell body. Mean Syt4-GFP intensity was calculated by subtracting the mean intensity of an area of background outside of the cell body in the same slice from the mean intensity within the manually selected cell body region. Each data point corresponds to the average mean Syt4-GFP intensity across eight cell bodies per brain.
Quantification of Syt4-GFP levels in neuropil
This analysis was conducted in FIJI. Using single slices through the center of the ventral ganglion, a region within the neuropil was manually selected. Mean Syt4-GFP intensity within this region of neuropil was calculated by subtracting the mean intensity of an area of background outside of the ventral ganglion from the mean intensity within the selected neuropil region.
Quantification of APP-EGFP levels in fixed axons
APP-EGFP levels were measured using FIJI in the m4 axon region between the segmental axon bundle and synaptic boutons. Average intensity projections were manually cropped to exclude everything aside from the axon segment to be measured. Manual thresholding to the HRP signal was conducted to determine the region for measurement. To calculate mean APP-EGFP intensity in the axon, the mean intensity of three areas of background per image were averaged and subtracted from the mean APP-EGFP intensity within the HRP-thresholded region.
Quantification of live axonal trafficking of APP-GFP puncta
To quantify APP-GFP dynamics in live axons, maximum intensity projections of time course images were processed in FIJI to subtract background and adjust for XY drift using the StackReg plugin. Kymographs were generated from one to four axons per animal using the FIJI plugin KymographBuilder. Kymographs were blinded, and a number of tracks were manually counted. The minimum track length measured was 3 μm, with most tracks above 5 μm. Velocity was measured by calculating the slope of the identified tracks.
Quantification of APP-GFP and Spin-RFP colocalization
This analysis was done in FIJI. A 3D presynaptic mask was created by combining signals of the presynaptically enriched labels α-HRP and Spin-RFP. To generate masks, images were subjected to a Gaussian blur and thresholded using the intensity of the combined channels. Specific parameters were optimized empirically and kept consistent between control and mutant groups. Colocalization analysis was performed on 3D Airyscan images using a FIJI script that utilizes the Coloc2 plugin for ImageJ (https://imagej.net/Coloc_2). Background was subtracted using the rolling ball method to a radius of 50 pixels. NMJ intensities were measured in Volocity as described above.
Western blot
Heads (15 pooled per genotype) from Drosophila aged 3–10 d were homogenized in 50 µl 2× Laemmli buffer. 15 µl of extract was loaded in each lane and fractionated by SDS/PAGE and immunoblotted with α-APP-CTF (A8717; Sigma-Aldrich) and α-actin (JLA-20; DHSB). Blots were visualized using a Biorad Chemidoc system and quantified using FIJI with APP-CTF values normalized to the corresponding actin loading control.
Eclosion assay
Density-controlled crosses were grown at 25°C for 17 d, and the number of eclosed and uneclosed pupal cases were counted for each genotype. Eclosion rate was calculated as the number of pupal cases eclosed divided by the total number of pupal cases.
Ghost bouton budding
Ghost bouton budding experiments were carried out as previously described (Piccioli and Littleton, 2014). Wandering third instar larvae were dissected in HL3 saline solution (70 mM NaCl, 5 mM KCl, 0.2 CaCl2, 20 mM MgCl2, 10 mM NaHCO3, 5 mM trehalose, 115 mM sucrose, and 5 mM Hepes-NaOH, pH 7.2). Larval dissections were matched to a guide with consistent dissection pin locations varying by 5%. The pins were moved inward to 60% of the original size of each larva. Relaxed fillets were incubated three times for 2 min in 90 mM K solution (40 mM NaCl, 90 mM KCl, 1.5 CaCl2, 20 mM MgCl2, 10 mM NaHCO3, 5 mM trehalose, 5 mM sucrose, and 5 mM Hepes-NaOH, pH 7.2) spaced by 10 min in HL3 solution. After the third incubation, larvae were returned to HL3 solution and stretched to their original size. Ghost boutons were identified by the presence of a presynaptic bouton (HRP labeled) without the postsynaptic Dlg protein. Muscle 6/7 NMJs from segment 3 were included in analysis.
Electrophysiology
Wandering third instar larvae were dissected in HL3 saline (Stewart et al., 1994). Recordings were taken using an AxoClamp 2B amplifier (Axon Instruments). A recording electrode was filled with 3 M KCl and inserted into muscle 6 at abdominal segments A3 or A4. A stimulating electrode filled with saline was used to stimulate the severed segmental nerve using an isolated pulse stimulator (2100; A-M Systems). HFMR was induced by four trains of 100 Hz stimuli spaced 2 s apart in 0.3 mM extracellular Ca2+. mEJPs were recorded 2 min before and 10 min after HFMR induction. mEJP frequency at indicated time points was calculated in 10-s bins. Fold enhancement was calculated by normalizing to the baseline mEJP frequency recorded prior to HFMR induction. Analyses were performed using Clampfit 10.0 software (Molecular Devices) and Mini Analysis 6.0.3 (Synaptosoft, Inc.). Each n value represents a single muscle recording, with data generated from at least six individual larvae of each genotype arising from at least two independent crosses. Resting membrane potentials were between −50 and −75 mV and were not different between genotypes. Input resistances were between 5 and 10 MΩ and were not different between genotypes.
Statistical analyses
All graphing and statistical analyses were completed using GraphPad Prism. Datasets were first analyzed with the D’Agostino-Pearson normality test. Normally distributed datasets were analyzed with either an unpaired two-sided t-test (two groups) or a one-way ANOVA with Tukey’s multiple comparisons (more than two groups), while not normally distributed datasets were analyzed with either a two-sided Mann–Whitney test (two groups) or a Kruskal–Wallis test with Dunn’s multiple comparisons (more than two groups). Error bars report ± SEM. The eclosion experiment in Fig. 2 E was analyzed using a chi-squared test; error bars report standard error of the proportion. Detailed information about genotypes, sample sizes, and statistical analyses performed for each dataset can be found in Table S3. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Online supplemental material
Fig. S1 shows source data for EV cargo levels at nwk mutant synapses and axonal transport measurements. Fig. S2 shows source data for EV cargo levels at endocytic mutant synapses. Fig. S3 shows source data for EV cargo levels at clathrin and clathrin adaptor mutant synapses. Fig. S4 shows controls for EV cargo localization to degradative compartments at nwk mutant synapses. Table S1 lists Drosophila strains. Table S2 lists antibodies and reagents. Table S3 lists statistics by dataset.
Acknowledgments
We thank the Bloomington Drosophila Stock Center (Indiana University, Bloomington, IN, NIH P40OD018537) and the Developmental Studies Hybridoma Bank created by the National Institute of Child Health and Human Development of the National Institutes of Health. We thank Kate O’Connor-Giles (Brown University, Providence, RI) and Vivian Budnik (UMass Chan Medical School, Worcester MA) for fly lines and Sultana Bhuiyan, Steven Del Signore, Jack Cheng, Biljana Ermanoska, and Matthew Pescosolido for technical assistance and helpful discussions.
This work was supported by National Institute of Neurological Disorders and Stroke grants R01 NS103967 to A.A. Rodal, F32 NS110123 to C.R. Blanchette, F32 NS120909 to E.C. Dresselhaus, and T32 MH019929 to A.L. Scalera, and by Natural Sciences and Engineering Research Council of Canada to B.A. Stewart (RGPIN-06004).
The authors declare no competing financial interests.
Author contributions: C.R. Blanchette, A.L. Scalera, and A.A. Rodal designed the study and experiments. C.R. Blanchette, A.L. Scalera, K.P. Harris, Z. Zhao, E.C. Dresselhaus, K. Koles, A. Yeh, and J.K. Apiki conducted the experiments and performed analyses. C.R. Blanchette, A.L. Scalera, and A.A. Rodal wrote the manuscript. C.R. Blanchette, A.L. Scalera, K.P. Harris, Z. Zhao, E.C. Dresselhaus, K. Koles, A. Yeh, J.K. Apiki, B.A. Stewart, A.A. Rodal reviewed and/or edited the manuscript. B.A. Stewart and A.A. Rodal provided supervision.

