


Cellular functions, such as division and migration, require cells to undergo robust shape changes. Through their contractility machinery, cells also sense, respond, and adapt to their physical surroundings. In the cytoplasm, the contractility machinery organizes into higher order assemblies termed contractility kits (CKs). Using Dictyostelium discoideum, we previously identified Discoidin I (DscI), a classic secreted lectin, as a CK component through its physical interactions with the actin crosslinker Cortexillin I (CortI) and the scaffolding protein IQGAP2. Here, we find that DscI ensures robust cytokinesis through regulating intracellular components of the contractile machinery. Specifically, DscI is necessary for normal cytokinesis, cortical tension, membrane–cortex connections, and cortical distribution and mechanoresponsiveness of CortI. The dscI deletion mutants also have complex genetic epistatic relationships with CK components, acting as a genetic suppressor of cortI and iqgap1, but as an enhancer of iqgap2. This work underscores the fact that proteins like DiscI contribute in diverse ways to the activities necessary for optimal cell function.
Introduction
Many biological processes require cells to sense and adapt to their physical surroundings. This adaptability allows cells to perform robust cell shape changes. Processes ranging from cytokinesis and cell migration to more complex events such as development require integration of internal and external mechanical cues to exert these physical changes (Blanchoin et al., 2014; Sinha et al., 2017; Umbarger, 1956). To integrate these internal and external cues, cells leverage their contractility machinery, which includes actin filaments, Myosin II (MyoII) motors, crosslinkers, and other scaffolding proteins. These proteins compose a mechanoresponsive system that senses and then responds to chemical and mechanical signals by accumulating locally to drive and ensure high fidelity cell shape changes (Kee et al., 2012; Luo et al., 2012; Schiffhauer et al., 2017; Srivastava et al., 2016; West-Foyle and Robinson, 2012).
Within the contractile machinery, MyoII and Cortexillin I (CortI) serve as key sensors and actuators of applied mechanical stress. These proteins provide the cells with the ability to quickly respond and adapt to external mechanical stimuli. These same mechanoresponsive proteins also accumulate at the cleavage furrow cortex of a dividing cell to promote contractility. Altogether, this mechanoresponsiveness of the contractility machinery provides a quality control function that ensures that the cell completes cytokinesis with high fidelity despite mechanical disturbances (Kee et al., 2012; Liu and Robinson, 2018; Murthy and Wadsworth, 2005; Nguyen et al., 2022; Yumura et al., 2008). Along with other actin crosslinkers, this system collectively bears the mechanical load at the cell cortex, and loss of these proteins leads to altered physical properties of cortex, resulting in changes in cell behaviors (Ren et al., 2009). Therefore, this machinery allows cells to exert robust control and modulation on multiple processes that involve cell shape modifications.
An interdependency of the cytoskeletal machinery exists within the contractile machinery. For example, mechanoreponsive proteins like MyoII and CortI cooperatively accumulate at sites of mechanical perturbations (Kee et al., 2012; Kothari et al., 2019a; Ren et al., 2009; Schiffhauer et al., 2017). In contrast, mechanisms exist to prevent overaccumulation and hyperactivity of this system. Two CortI-binding proteins, IQGAP1 and IQGAP2, provide a higher level of regulation on this mechanoresponsive system. IQGAP1 inhibits the mechanoresponsive accumulation of MyoII and CortI, while IQGAP2 relieves this repression. In the absence of IQGAP2, MyoII and CortI fail to accumulate in response to externally applied perturbations due to inhibition by IQGAP1 (Kee et al., 2012; Kothari et al., 2019b; Srivastava et al., 2016). These mechanical feedback systems spatially and temporally tune the level of MyoII and CortI accumulation and contractility.
Previously, we found that several key proteins of the mechanosensory system preassemble in the cytoplasm, forming mechanoresponsive (contains IQGAP2) and non-mechanoresponsive (contains IQGAP1) complexes that we term contractility kits (CKs; Kothari et al., 2019b). While the contractility network involves a much larger cohort of proteins, the proteins in the CKs encompass a smaller subset that allows for delivery of key cytoskeletal proteins such as CortI and MyoII to the cortex (Fig. 1 A). A likely benefit of being “pre-assembled” in the cytoplasm is that it may allow the CKs to respond more rapidly to mechanical disturbances and/or stimuli. IQGAP2 and IQGAP1 are key regulators of these CKs and aid in controlling the level of the proteins that accumulate at the cortex, i.e., they likely help establish the setpoint and responsiveness of the system. The CK concept, hence, accounts for how so many proteins can be directed rapidly and synchronously in response to discrete cues, such as from mechanical stresses. In addition to the CKs, we found that the CK network includes several other proteins, including Discoidin I (DscI; Kothari et al., 2019b). In Kothari et al. (2019b), using liquid chromatography–mass spectrometry, DscI was detected as a biochemical interactor of IQGAP2 in both cytosolic and cytoskeletal fractions of FLAG–GFP–IQGAP2 in iqg2-null cells. Furthermore, using fluorescence cross-correlation spectroscopy (FCCS), a method to measure in vivo interactions, the authors detected interactions of DscI with both CortI and IQGAP2 within the cytoplasm. Interestingly, DscI also was recovered as a genetic suppressor of cort1 null mutants through a suppressor screen where Dsc overexpression rescue the developmental phenotypes of cortI null cells (Nguyen and Robinson, 2020; Robinson and Spudich, 2000). These results indicate a possible role of Dsc proteins in the mechanobiome.

DscI is required for normal growth, cytokinesis, and cortical integrity. (A) The CKs pre-assemble in the cytoplasm and accumulate quickly to the cortex upon mechanical stimuli. (B and C)dsc1 null mutant clones generated in the KAx3 background were verified using Western analysis. Western blot showed deletion of DscI bands, quantified and normalized to WT level in C, in all dsc1 null mutant clones. Dynacortin (Dyn) is provided as a loading control. (D) Representative growth curves show that dsc1 null mutants have mild growth defects in suspension culture. Cell densities, as indicated, are plotted as a function of time. Growth curves for control cell lines are shown in black and those for dsc1 null mutant clones are shown in blue. Number of samples = 3 per growth curve; bars represent the SEM. Some SEM bars are smaller than the symbols. (E) Growth rates quantified during exponential growth and normalized to WT reveal significant reduction in suspension culture growth in different dsc1 mutant clones. Growth rate of each cell line was normalized to that of WT control. Number of samples analyzed, n = 6 from two independent experiments. (F) Quantification of the number of nuclei per cell when cells were 3 d in suspension culture. All but one of dsc1 mutants became more multinucleated, especially within 3–5 nuclei/cell. n = 4 per cell line, with 100–200 cells per cell line quantified per replicate. P values were calculated using the ANOVA followed by Fisher’s LSD test based on the single nuclei/cell fraction subset (**, P < 0.001). (G) Images of DIC channel and nuclear staining by Hoescht reveal a slight increase in cell size as well as number of nuclei in each cell in several dsc1 clones. DIC and Hoescht images for WT are reproduced in Fig. 4 C. (H) Graph shows fraction of WT and dsc1 cells having a WT-like U-shaped cleavage furrow, an aberrant V-shaped furrow, or an intermediate furrow during cytokinesis. An intermediate furrow is one that shows both a U-shaped and V-shaped furrow along different stages of cytokinesis. n = 28 for WT and n = 20 for dsc1. P value was calculated using the Comparison of Proportions test. (I) Images of different stages of cytokinesis of a WT cell and a dsc1 cell having a V-shaped furrow. Images were collected from cells at 70% confluency, and t = 0 is defined as the time when each video begins. Full video of WT and dsc1 dividing cells are provided in the Supplemental materials (Videos 1 and 2). (J) The diagram shows the manipulation of cells by MPA for cortical tension and mechanoresponsiveness analysis. For cortical tension measurements, the pressure is increased until the length (Lp) of the region of the cell pulled into the micropipette equals the radius of the pipette (Rp). At this equilibrium pressure (ΔPc), the radius of the cell at can be quantified, and the effective cortical tension Teff is calculated from the equation shown in the panel. (K)dsc1 mutant cells have a higher degree of deformation when aspirated at the same negative pressure compared to WT cells. Red arrows show the front of the cell inside the micropipette. Scale bar, 10 μm and applies to all panels. (L) The effective cortical tension values of WT, dsc1, and dsc1-complemented cells were quantified as detailed in the Materials and methods. Gray background indicates non-transformed cell lines. P values were calculated using Kruskal-Wallis followed by Wilcoxon test. Number of cells measured, n = 7–10 from three independent experiments. (M) When compressed using agarose overlay, which introduces mechanical stress to the cortex, dsc1 mutant cells expressing only GFP displayed a bleb-like morphology. This morphology was observed in both DIC and GFP channels. This phenotype was rescued to WT levels in dsc1:: GFP-DscIA cells. (N)dsc1 mutants displayed a significant reduction in solidity, a measure of smoothness of the cell periphery, when subjected to mechanical stress, as compared to WT control and rescue control (Kruskal-Wallis, P < 0.0001; Wilcoxon, P < 0.0001). Number of cells analyzed, n = 17–28 from two independent experiments. Solidity measurement was analyzed using ImageJ and calculated as the area of a particle divided by its convex hull area. Source data are available for this figure: SourceData F1.
DscI is required for normal growth, cytokinesis, and cortical integrity. (A) The CKs pre-assemble in the cytoplasm and accumulate quickly to the cortex upon mechanical stimuli. (B and C)dsc1 null mutant clones generated in the KAx3 background were verified using Western analysis. Western blot showed deletion of DscI bands, quantified and normalized to WT level in C, in all dsc1 null mutant clones. Dynacortin (Dyn) is provided as a loading control. (D) Representative growth curves show that dsc1 null mutants have mild growth defects in suspension culture. Cell densities, as indicated, are plotted as a function of time. Growth curves for control cell lines are shown in black and those for dsc1 null mutant clones are shown in blue. Number of samples = 3 per growth curve; bars represent the SEM. Some SEM bars are smaller than the symbols. (E) Growth rates quantified during exponential growth and normalized to WT reveal significant reduction in suspension culture growth in different dsc1 mutant clones. Growth rate of each cell line was normalized to that of WT control. Number of samples analyzed, n = 6 from two independent experiments. (F) Quantification of the number of nuclei per cell when cells were 3 d in suspension culture. All but one of dsc1 mutants became more multinucleated, especially within 3–5 nuclei/cell. n = 4 per cell line, with 100–200 cells per cell line quantified per replicate. P values were calculated using the ANOVA followed by Fisher’s LSD test based on the single nuclei/cell fraction subset (**, P < 0.001). (G) Images of DIC channel and nuclear staining by Hoescht reveal a slight increase in cell size as well as number of nuclei in each cell in several dsc1 clones. DIC and Hoescht images for WT are reproduced in Fig. 4 C. (H) Graph shows fraction of WT and dsc1 cells having a WT-like U-shaped cleavage furrow, an aberrant V-shaped furrow, or an intermediate furrow during cytokinesis. An intermediate furrow is one that shows both a U-shaped and V-shaped furrow along different stages of cytokinesis. n = 28 for WT and n = 20 for dsc1. P value was calculated using the Comparison of Proportions test. (I) Images of different stages of cytokinesis of a WT cell and a dsc1 cell having a V-shaped furrow. Images were collected from cells at 70% confluency, and t = 0 is defined as the time when each video begins. Full video of WT and dsc1 dividing cells are provided in the Supplemental materials (Videos 1 and 2). (J) The diagram shows the manipulation of cells by MPA for cortical tension and mechanoresponsiveness analysis. For cortical tension measurements, the pressure is increased until the length (Lp) of the region of the cell pulled into the micropipette equals the radius of the pipette (Rp). At this equilibrium pressure (ΔPc), the radius of the cell at can be quantified, and the effective cortical tension Teff is calculated from the equation shown in the panel. (K)dsc1 mutant cells have a higher degree of deformation when aspirated at the same negative pressure compared to WT cells. Red arrows show the front of the cell inside the micropipette. Scale bar, 10 μm and applies to all panels. (L) The effective cortical tension values of WT, dsc1, and dsc1-complemented cells were quantified as detailed in the Materials and methods. Gray background indicates non-transformed cell lines. P values were calculated using Kruskal-Wallis followed by Wilcoxon test. Number of cells measured, n = 7–10 from three independent experiments. (M) When compressed using agarose overlay, which introduces mechanical stress to the cortex, dsc1 mutant cells expressing only GFP displayed a bleb-like morphology. This morphology was observed in both DIC and GFP channels. This phenotype was rescued to WT levels in dsc1:: GFP-DscIA cells. (N)dsc1 mutants displayed a significant reduction in solidity, a measure of smoothness of the cell periphery, when subjected to mechanical stress, as compared to WT control and rescue control (Kruskal-Wallis, P < 0.0001; Wilcoxon, P < 0.0001). Number of cells analyzed, n = 17–28 from two independent experiments. Solidity measurement was analyzed using ImageJ and calculated as the area of a particle divided by its convex hull area. Source data are available for this figure: SourceData F1.
DscI belongs to the discoidin protein family, consisting of DscI and DscII, which are N-acetylgalactosamine-binding lectins found in Dictyostelium discoideum and expressed as Dictyostelium enters the developmental phase (Alexander et al., 1983; Frazier et al., 1975; Rosen et al., 1973). The DscI family is composed of three highly homologous dsc1 genes: dsc1A, C, and D. DscI was first suggested to play a role in mediating cell–cell and cell–substrate interactions, due to its lectin properties (Berger and Armant, 1982; Springer and Barondes, 1980; Springer et al., 1984). Structurally, DscI consists of two domains: an N-terminal discoidin domain and a C-terminal lectin domain. The N-terminal discoidin domain contains a motif that has diverse functions in mediating interactions with various molecules in both eukaryotes and prokaryotes. The C-terminal domain is an H-type lectin domain that facilitates hemaglutination activity (Baumgartner et al., 1998; Kiedzierska et al., 2007). Dsc was traditionally thought to be mainly involved in cell surface signaling and glycoconjugate binding and is required for aggregation and development since univalent anti-DscI blocked aggregation in WT cells (Springer and Barondes, 1980). Several other studies have indicated that DscI functions in the ECM and was detected in the ECM through a proteomics analysis (Huber and O'Day, 2015). While most studies have focused on DscI’s extracellular functions, some studies have alluded to its possible intracellular roles. In one study, using immunofluorescence, DscI was observed to reside in the cytoplasm of vegetative Dictyostelium cells (Alexander et al., 1992). In another, DscI was located intracellularly inside vesicles of developmental cells (Barondes et al., 1985).
In this study, we find that DscI has important roles intracellularly, specifically within the CK machinery. We found that dsc1 null single mutants have a mild cytokinesis defect, reduced cortical tension, and impaired CortI mechanoresponsiveness (the ability to accumulate in response to mechanical stress). Deletion of dsc1 also acts as genetic suppressor of cortI and iqgap1 null mutants, but as a genetic enhancer of iqgap2 null mutants. Furthermore, transcript and protein expression of DscI are tightly controlled by CortI and IQGAP1. In addition, DscI localizes to the cortex and helps with CortI cortical association. This association is crucial for the integrity of the cortex and the membrane since the deletion of dscI results in atypical outpouching of the cell cortex and plasma membrane. We also find that absence of DscI weakens the interactions of CortI with IQGAP2 and MyoII as assessed by FCCS. These observations support the notion that these proteins function in a complex, and DscI helps stabilize the assembly of CortI into this complex. In short, DscI is necessary for full function of the CK system. Overall, this work re-emphasizes the point that many proteins contribute in multiple diverse ways to the function of the cell, and it is increasingly apparent that proteins may not be described as providing a single function for the cell.
Results
DscI is crucial for normal growth and cytokinesis
To begin deciphering the roles of DscI, we first studied the cytokinetic defects of cells lacking DscI by generating knockout (KO) mutants using CRISPR/Cas9 (Sekine et al., 2018). A gRNA sequence complementary to a gene coding sequence close to the 5′ end that is identical in all three dsc1 paralogs was designed to direct gene specificity. KO mutants were confirmed using Western (Fig. 1 B and Fig. S1 A) and sequence analysis (Fig. S1 B). Western quantification showed complete absence of DscI protein in various clones (Fig. 1 C and Fig. S1 C) in two separate D. discoideum backgrounds. Sequence analysis of the clones generated in the Ax2 background showed that most mutant clones obtain mutations at the targeted dsc1A gene (Fig. S1 B). Sequencing analysis cannot be performed in the KAx3 background due to the duplication of the dsc1 genes on chromosome 2. In the mutants analyzed, only the gene sequence of dsc1A was altered, while that of dsc1C and dsc1D remained intact (not shown). This observation indicates that mutating dsc1A gene is sufficient to obliterate DscI protein expression in vegetative cells. Quantitative RT-PCR (qRT-PCR) experiments (Fig. 6 C) also confirm that dsc1 transcripts were absent in these cells. These results suggest that dsc1C and dsc1D are not expressed in vegetative cells. Therefore, from this point on, we refer to DscIA as DscI as it is likely the only functional paralog in vegetative cells.
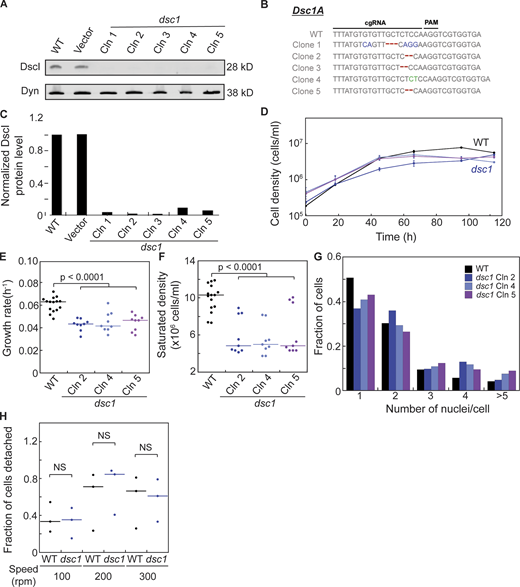
Cytokinetic defects in dsc1 null mutants generated in the Ax2 background. (A)dsc1 null mutant clones generated in the Ax2 background were verified using Western analysis. Western blot showed absence of DscI protein in all dsc1 null mutant clones. Dynacortin (Dyn) is provided as a loading control. (B) Sequencing analysis confirmed mutations on the dsc1A coding genes of dsc1 mutant clones. Sequences of WT and five separate clones are presented. Nucleotide changes are shown in blue, deletions in red, and insertions in green. (C) Quantification of DscI protein level of the Western blot provided in A. Average integrated intensity of each DscI band was background-subtracted and normalized to their corresponding Dyn control band. Each value was then normalized to WT. (D) Representative growth curves showed that dsc1 null mutants in Ax2 background also display mild growth defects in suspension culture. Cell densities, as indicated, were plotted as a function of time. Growth curve for WT (vector control) cell line is shown in black and those for dsc1 null mutant clones are shown in shades of blue and purple. dsc1 mutant clone 2,4 and 5 were used for this experiment. n = 3 per growth curve, bars represent SEM. (E) Growth rates quantified during exponential growth showed mild yet significant defects in growth in different dsc1 mutant clones. Number of cells analyzed, n = 9–15 from three independent experiments. (F)dsc1 mutants generally stopped growing at a significantly lower saturated density as compared to WT control. For E and F, P values were calculated using ANOVA followed by Fisher’s LSD test. Number of cells analyzed, n = 9–15 from two independent experiments. (G) Quantification of the number of nuclei per cell when cells were 3 d in suspension culture. All dsc1 mutant clones analyzed became more multinucleated compared to WT control. n = 2 per cell line with 100–200 cells per cell line quantified per replicate. (H) Graph shows quantification of fraction of cells detaching when WT and dsc1 cells at equal densities were rotated at different speeds on 6-well non-culture-treated plates. Amount of cells detaching was normalized to the seeding cell number, and the ratio of cell detached over total cell number was determined. P values were calculated using ANOVA followed by Fisher’s LSD test. Number of samples analyzed, n = 3. Source data are available for this figure: SourceData FS1.

Cytokinetic defects in dsc1 null mutants generated in the Ax2 background. (A)dsc1 null mutant clones generated in the Ax2 background were verified using Western analysis. Western blot showed absence of DscI protein in all dsc1 null mutant clones. Dynacortin (Dyn) is provided as a loading control. (B) Sequencing analysis confirmed mutations on the dsc1A coding genes of dsc1 mutant clones. Sequences of WT and five separate clones are presented. Nucleotide changes are shown in blue, deletions in red, and insertions in green. (C) Quantification of DscI protein level of the Western blot provided in A. Average integrated intensity of each DscI band was background-subtracted and normalized to their corresponding Dyn control band. Each value was then normalized to WT. (D) Representative growth curves showed that dsc1 null mutants in Ax2 background also display mild growth defects in suspension culture. Cell densities, as indicated, were plotted as a function of time. Growth curve for WT (vector control) cell line is shown in black and those for dsc1 null mutant clones are shown in shades of blue and purple. dsc1 mutant clone 2,4 and 5 were used for this experiment. n = 3 per growth curve, bars represent SEM. (E) Growth rates quantified during exponential growth showed mild yet significant defects in growth in different dsc1 mutant clones. Number of cells analyzed, n = 9–15 from three independent experiments. (F)dsc1 mutants generally stopped growing at a significantly lower saturated density as compared to WT control. For E and F, P values were calculated using ANOVA followed by Fisher’s LSD test. Number of cells analyzed, n = 9–15 from two independent experiments. (G) Quantification of the number of nuclei per cell when cells were 3 d in suspension culture. All dsc1 mutant clones analyzed became more multinucleated compared to WT control. n = 2 per cell line with 100–200 cells per cell line quantified per replicate. (H) Graph shows quantification of fraction of cells detaching when WT and dsc1 cells at equal densities were rotated at different speeds on 6-well non-culture-treated plates. Amount of cells detaching was normalized to the seeding cell number, and the ratio of cell detached over total cell number was determined. P values were calculated using ANOVA followed by Fisher’s LSD test. Number of samples analyzed, n = 3. Source data are available for this figure: SourceData FS1.
Cytokinesis and cell growth are required for cells to multiply and require extensive cell shape changes, and cell growth may be impaired due to defects in cytokinesis. We used suspension growth, a highly restrictive growth condition for impaired cytokinesis (Adachi, 2001; Uyeda et al., 2000), to assess whether DscI is essential for cell growth and for cytokinesis. We found that dsc1 mutants grow slightly more slowly (Fig. 1 D and Fig. S1 D), yielding a mild, yet significant, growth rate reduction (Fig. 1 E, and Fig. S1 E) as compared to WT controls. These mutants also saturate at a lower cell density than WT parental cells (Fig. S1 F).
Another prominent defect of cytokinesis is the presence of multinucleated cells, especially when grown in suspension culture (Park et al., 2018; Rivero et al., 2002). When cells cannot complete cytokinesis successfully, these uncompleted events lead to the accumulation of multiple nuclei within a cell. Within 3 d in suspension, with the exception of one clone, cells lacking DscI accumulated more nuclei within the cytoplasm, largely in the range of 3–5 nuclei/cell (Fig. 1, F and G; and Fig. S1 G). As a result, dsc1 null cells generally appear bigger than WT cells due to the increase in multinucleation (Fig. 1 G). Defects in both growth and multinucleation were observed across multiple mutant clones in two different strain backgrounds. Among more than seven mutants tested, only one mutant did not display increased multinucleation. This difference is likely due to clonal variation as these mutants were subcloned from the parental cell line during the process of mutant generation. dsc1 mutant clone 4 was randomly selected for subsequent experiments. Importantly, when the dsc1 mutant was complemented with DscIA, both growth and multinucleation defects were rescued, confirming that these defects are attributed to the absence of DscIA (Fig. S2, A–D).
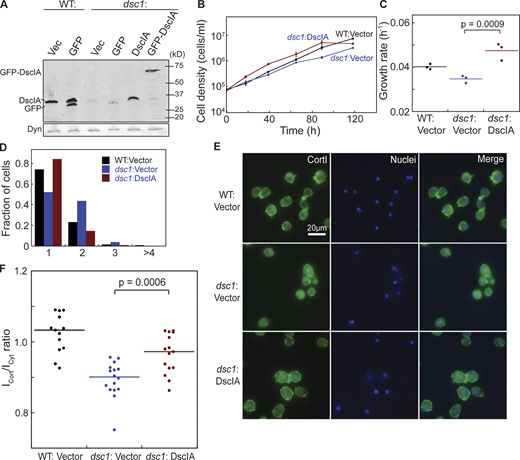
Exogenous expression of DscIA rescues cytokinetic defects of dsc1 null mutants. (A) Western analysis verified expression of exogenous DscIA and GFP-DscIA in WT and dsc1 null background (KAx3 background). (B) Representative growth curves in suspension culture revealed that exogenous expression of DscIA rescues growth defect of dsc1 null mutants. Cell densities, as indicated, were plotted as a function of time. Growth curve for WT expressing empty plasmid is shown in black; that for dsc1 null expressing empty plasmid is shown in blue with round symbols; and that for dsc1 null expressing DscIA is shown in blue with square symbols. n = 3 per growth curve, bars represent SEM. (C) Growth rates quantified during exponential growth showed an increase in growth in dsc1 null complemented cells compared to null cells. P values were calculated using ANOVA followed by Fisher’s LSD test. (D) Quantification of the number of nuclei per cell when cells were 3 d in suspension culture. (E and F) Representative confocal immunofluorescence images of WT and dsc1 mutants stained with anti-CortI (green) and Hoescht for nuclei (blue) show that expression of DscIA in dsc1 null cells restored the level of CortI at the cortex, quantified in panel F (number of cells analyzed, n = 13–16), back to WT level. P values were derived for ANOVA followed by Fisher’s LSD test. Source data are available for this figure: SourceData FS2.

Exogenous expression of DscIA rescues cytokinetic defects of dsc1 null mutants. (A) Western analysis verified expression of exogenous DscIA and GFP-DscIA in WT and dsc1 null background (KAx3 background). (B) Representative growth curves in suspension culture revealed that exogenous expression of DscIA rescues growth defect of dsc1 null mutants. Cell densities, as indicated, were plotted as a function of time. Growth curve for WT expressing empty plasmid is shown in black; that for dsc1 null expressing empty plasmid is shown in blue with round symbols; and that for dsc1 null expressing DscIA is shown in blue with square symbols. n = 3 per growth curve, bars represent SEM. (C) Growth rates quantified during exponential growth showed an increase in growth in dsc1 null complemented cells compared to null cells. P values were calculated using ANOVA followed by Fisher’s LSD test. (D) Quantification of the number of nuclei per cell when cells were 3 d in suspension culture. (E and F) Representative confocal immunofluorescence images of WT and dsc1 mutants stained with anti-CortI (green) and Hoescht for nuclei (blue) show that expression of DscIA in dsc1 null cells restored the level of CortI at the cortex, quantified in panel F (number of cells analyzed, n = 13–16), back to WT level. P values were derived for ANOVA followed by Fisher’s LSD test. Source data are available for this figure: SourceData FS2.
A majority of dsc1 cells displayed aberrant morphology during different stages of cytokinesis when grown on surfaces. Normally, WT Dictystostelium cells undergo a characteristic sequence of steps that include cleavage furrow formation, ingression, and abcission during cytokinesis. A WT cleavage furrow displays a characteristic U shape (Fig. 1, H and I; Videos 1 and 2) that gradually ingresses to form a thin bridge, which is then severed. In dsc1 cells, only 30% of the cells maintain WT-like furrow morphology. 30% of dsc1 cells have abnormally shaped furrows, which is more V-shaped and less visible throughout cytokinesis (Fig. 1, H and I; Videos 1 and 2). The rest of dsc1 cells display a mix of U-shaped and V-shaped morphologies, described as an “intermediate furrrow morphology.” Since DscI is implicated in cell-substrate adhesion, this abnormal V-shaped morphology may be due to defects in adhesion as dsc1 cells divide. This adhesion defect, however, was not observed across the whole population of cells, which is primarily composed of interphase cells (Fig. S1 G). Hence, it is likely that in vegetative cells, dsc1 is important for adhesion primarily during cytokinesis. Altogether, these observations reveal an important role of DscI in ensuring cytokinesis fidelity.
Video of a dividing WT (KAx3) cell. Each frame was collected every 2 s with a 40× (NA 1.30) oil objective and imaged by DIC using an Olympus IX71 microscope. Playback speed is 10 frames per second.
Video of a dividing WT (KAx3) cell. Each frame was collected every 2 s with a 40× (NA 1.30) oil objective and imaged by DIC using an Olympus IX71 microscope. Playback speed is 10 frames per second.
Video of a dividing dsc1 null cell. Each frame was collected every 2 s with a 40× (NA 1.30) oil objective and imaged by DIC using an Olympus IX71 microscope. Playback speed is 10 frames per second.
Video of a dividing dsc1 null cell. Each frame was collected every 2 s with a 40× (NA 1.30) oil objective and imaged by DIC using an Olympus IX71 microscope. Playback speed is 10 frames per second.
A crucial role of the contractile machinery is to provide the cortical mechanical properties, which are largely defined by the actin cytoskeleton (Efremov et al., 2015; Fritzsche et al., 2016; Luo and Robinson, 2011; Ren et al., 2009). Since DscI interacts with several members of the contractile machinery, to test if DscI impacts cortical mechanics, we used micropipette aspiration (MPA) to measure the degree of deformability in dsc1 null cells (Fig. 1 J). The dsc1 null cells were more deformable when aspiration pressure was applied to the cortex (Fig. 1 K). In the example shown (Fig. 1 K), the WT cell required 0.20 nN/μm2 pressure to reach the critical pressure at which the length of the cell inside the pipette (Lp) is equal to the pipette radius (Rc). In contrast, the dsc1 mutant cell only required 0.05 nN/μm2 of pressure to deform to the same extent. The average effective cortical tension, a measure of deformability, of dsc1 cells is 0.66 nN/μm, which is significantly lower than that of WT cells (1.0 nN/μm; Fig. 1 L). Furthermore, when these cells are mechanically challenged using agarose overlay, dsc1 null cells displayed aberrant membrane morphology, characterized by significant cortical outpouching, i.e., bleb-like structures (Fig. 1 M). The degree of bleb-like formation is quantified as changes in solidity of the cell membrane (Fig. 1 N). The bleb-like structures on the mechanically challenged membrane usually indicate accumulated tension at the cortex due to increase MyoII activity and/or weakened linkages between the cortex and membrane (Charras et al., 2006; Chikina et al., 2019; Ghosh et al., 2021; Luo et al., 2012). These defects in cortical functions were rescued in dsc1 null-complemented cells (Fig. 1, L–N), indicating that they are specifically attributed to the absence of DscI. Altogether, these results indicate another function of DscI is to ensure cortical mechanical integrity.
Balanced expression of dscI directs cortI cortical distribution
DscI has cytoplasmic and membrane-associated distributions in developing cells (Alexander et al., 1992; Barondes et al., 1985; Fukuzawa and Ochiai, 1993). Given the cortical phenotypes, including reduced cortical tension, we examined the subcellular distribution of DscI in vegetative WT cells. Using immunofluorescence imaging, we found that the majority of DscI stays in the cytoplasm while a portion of the protein enriches at the cell cortex (Fig. 2 A). This observation is consistent with the fact that many DscI interactors also localize to the cortex, notably IQGAP2 and CortI (Kothari et al., 2019b). However, unlike CortI, which localizes to the cleavage furrow cortex, immunofluorescence imaging in fixed and live cells showed that DscIA does not concentrate at the cleavage furrow as vegetative cells undergo cytokinesis (Fig. 2, B and C; Video 3; and Fig. S4, A and B; Kee et al., 2012; Liu and Robinson, 2018; Murthy and Wadsworth, 2005; Yumura et al., 2008).

A portion of DscI localizes to the cortex, and Dsc is needed for CortI cortical distribution. (A) Representative images of immunofluorescence imaging of DscI (green) using anti-DscI antibody and nuclei (blue) labeled with Hoescht in WT and dsc1 cells. Images were collected using confocal microscopy. DscI signal was present in the cytoplasm and slightly enriched in the cortex of WT cells. This signal was absent in dsc1 null cells, confirming the antibody’s specificity for DscI. (B) Representative images of immunofluorescence imaging of DscI (green) using anti-DscI antibody and nuclei (blue) labeled with Hoescht in dividing WT cells. (C) Time series of a dsc1 null-complemented cell completing cytokinesis. DscIA was fused with GFP and images were collected with a GFP channel on an Olympus IX71 microscope, equipped with a 40× (NA 1.30) oil objective at room temperature using Zen software and analyzed with ImageJ. t = 0 is defined as the time when the video began. The video is provided in the Supplemental materials (Video 3). (D and F) Representative confocal immunofluorescence images of WT, dsc1 null, DscIA-overexpressed, vector control (Vec) and dscI null-complemented cells stained with anti-CortI (green) and Hoescht to label nuclei (blue). Images were collected using a Zeiss AxioObserver with 780-Quasar confocal module microscope, equipped with a C-Apochromat 40× (NA 1.2) water objective at room temperature using Zen software and analyzed with ImageJ. Vector encoding mCherry was used as the vector (Vec) control. (E) Western blot verifying overexpression of DscIA in WT background cells is shown. Dynacortin (Dyn) is provided as a loading control. (G) CortI cortical localization for D and F was quantified as the ratio of mean signal intensity at the cortex (Icort) to the mean signal intensity of the cytoplasm (Icyt). P values were calculated using the ANOVA followed by Fisher’s LSD test. Number of cells analyzed, n = 15–25 from two independent experiments. (H) Summary diagram shows that a fraction of intracellular DscI localizes to the cortex, and DscI helps with cortical distribution of CortI. Source data are available for this figure: SourceData F2.
A portion of DscI localizes to the cortex, and Dsc is needed for CortI cortical distribution. (A) Representative images of immunofluorescence imaging of DscI (green) using anti-DscI antibody and nuclei (blue) labeled with Hoescht in WT and dsc1 cells. Images were collected using confocal microscopy. DscI signal was present in the cytoplasm and slightly enriched in the cortex of WT cells. This signal was absent in dsc1 null cells, confirming the antibody’s specificity for DscI. (B) Representative images of immunofluorescence imaging of DscI (green) using anti-DscI antibody and nuclei (blue) labeled with Hoescht in dividing WT cells. (C) Time series of a dsc1 null-complemented cell completing cytokinesis. DscIA was fused with GFP and images were collected with a GFP channel on an Olympus IX71 microscope, equipped with a 40× (NA 1.30) oil objective at room temperature using Zen software and analyzed with ImageJ. t = 0 is defined as the time when the video began. The video is provided in the Supplemental materials (Video 3). (D and F) Representative confocal immunofluorescence images of WT, dsc1 null, DscIA-overexpressed, vector control (Vec) and dscI null-complemented cells stained with anti-CortI (green) and Hoescht to label nuclei (blue). Images were collected using a Zeiss AxioObserver with 780-Quasar confocal module microscope, equipped with a C-Apochromat 40× (NA 1.2) water objective at room temperature using Zen software and analyzed with ImageJ. Vector encoding mCherry was used as the vector (Vec) control. (E) Western blot verifying overexpression of DscIA in WT background cells is shown. Dynacortin (Dyn) is provided as a loading control. (G) CortI cortical localization for D and F was quantified as the ratio of mean signal intensity at the cortex (Icort) to the mean signal intensity of the cytoplasm (Icyt). P values were calculated using the ANOVA followed by Fisher’s LSD test. Number of cells analyzed, n = 15–25 from two independent experiments. (H) Summary diagram shows that a fraction of intracellular DscI localizes to the cortex, and DscI helps with cortical distribution of CortI. Source data are available for this figure: SourceData F2.
Video of a dividing dsc1:GFP-DscIA cell. Each frame was collected every 5 s with a 40× (NA 1.30) oil objective and imaged by DIC using an Olympus IX71 microscope. Playback speed is 3 frames per second.
Video of a dividing dsc1:GFP-DscIA cell. Each frame was collected every 5 s with a 40× (NA 1.30) oil objective and imaged by DIC using an Olympus IX71 microscope. Playback speed is 3 frames per second.
Previous studies established that CortI, in particular, serves as a cortical actin crosslinker and actin-membrane tether, and cortI null mutants have similar aberrant cell behaviors and cortical mechanics as observed for dsc1 (Kee et al., 2012; Simson et al., 1998; Stock et al., 1999; Weber et al., 1999). DscI may then help CortI carry out its cortical functions. Indeed, we observed that in dsc1 nulls, the cortical localization of CortI is severely disrupted (Fig. 2, D and G; and Fig. S2, E and F). Since DscI also interacts with other cytoskeletal proteins such as IQGAP2 (Kothari et al., 2019b), we investigated whether localization of IQGAP2 and MyoII is impacted in dsc1 null cells. However, the cortical localization of these proteins was unchanged in dsc1 null cells (Fig. S3, A–D). We then asked if overexpression of DscIA would affect the distribution of CortI. We found that when DscI is overexpressed, CortI cortical distribution is also significantly disrupted (Fig. 2, E–G).

The absence of DscI in the cell has no effect on the cortical localization of MyoII and IQGAP2. (A) Representative confocal immunofluorescence images of WT and dsc1 mutants stained with anti-myoII heavy chain My6 antibody (green) and Hoescht for nuclei (blue) revealed that the amount of MyoII at the cortex is unaffected in the dscI mutants. (B) Cortical MyoII localization was quantified as the ratio of mean signal intensity at the cortex (Icort) to the mean signal intensity of the cytoplasm (Icyt). No significant difference in this ratio between WT (n = 11) and dsc1 (n = 17) cells was present. (C) Representative DIC and GFP images of WT cells expressing GFP-alone control, iqgap2 expressing GFP-IQGAP2, and iqgap2; dsc1 expressing GFP-IQGAP2 are provided. A thin layer of IQGAP2 is present underneath the membrane in iqgap2::GFP-IQGAP2 and in iqgap2; dsc1 ::GFP-IQGAP2, but not in the GFP-alone cells. (D) Quantification of cortical IQGAP2 showed that there was no significance difference in the amount of IQGAP2 at the cortex between cells lacking DscI expression and positive control. All P values were derived for ANOVA followed by Fisher’s LSD test. Number of cells analyzed, n = 12–13.

The absence of DscI in the cell has no effect on the cortical localization of MyoII and IQGAP2. (A) Representative confocal immunofluorescence images of WT and dsc1 mutants stained with anti-myoII heavy chain My6 antibody (green) and Hoescht for nuclei (blue) revealed that the amount of MyoII at the cortex is unaffected in the dscI mutants. (B) Cortical MyoII localization was quantified as the ratio of mean signal intensity at the cortex (Icort) to the mean signal intensity of the cytoplasm (Icyt). No significant difference in this ratio between WT (n = 11) and dsc1 (n = 17) cells was present. (C) Representative DIC and GFP images of WT cells expressing GFP-alone control, iqgap2 expressing GFP-IQGAP2, and iqgap2; dsc1 expressing GFP-IQGAP2 are provided. A thin layer of IQGAP2 is present underneath the membrane in iqgap2::GFP-IQGAP2 and in iqgap2; dsc1 ::GFP-IQGAP2, but not in the GFP-alone cells. (D) Quantification of cortical IQGAP2 showed that there was no significance difference in the amount of IQGAP2 at the cortex between cells lacking DscI expression and positive control. All P values were derived for ANOVA followed by Fisher’s LSD test. Number of cells analyzed, n = 12–13.
These collective observations indicate that DscI levels reside near an optimal sweetspot, not too little or not too much, in WT cells to ensure a normal CortI accumulation at the cortex (Fig. 2 H).
CortI requires dscI to mechanorespond and accumulate at the cleavage furrow cortex
Many proteins within the CK network such as MyoII, CortI, and IQGAP2 can mechanorespond, which is defined as the ability of the protein to accumulate locally in response to applied mechanical stress (Effler et al., 2006; Kee et al., 2012; Ren et al., 2009). Since DscI helps maintain cortical integrity and is partially localized at the cortex, this raised the question whether DscI itself can mechanorespond. Therefore, we used MPA to aspirate dsc1 mutant cells expressing GFP-DscIA and examined if GFP-DscIA accumulates at the site of aspiration. No GFP-DscIA was detected at the cortex inside the pipette (Fig. 3 A). The quantified ratio of cortical GFP signal intensity inside the pipette to the opposite cortex (Ip/Io) is not different than that of the GFP control and is significantly lower than that of GFP-MyoII, a positive control (Fig. 3 D).

DscI facilitates the mechanoresponsiveness of CortI. (A) GFP images show localization of GFP-MyoII, GFP-alone, and GFP-DscIA as cells are aspirated with a micropipette (from left to right). GFP-MyoII serves as a positive control. (B) GFP images show localization of GFP-CortI in cells with and without DscI as these cells are aspirated with a micropipette. (C) GFP images show localization of GFP-MyoII in cells with and without DscI as these cells are aspirated with a micropipette. GFP-alone serves as a negative control. For A–C, the genetic background of each cell line is indicated in the upper edge of each image, and GFP-alone serves as a negative control. Micropipette suction pressure was applied to the right side of each cell. (D) The degree of mechanoresponsiveness of GFP-tagged proteins is quantified as the GFP intensity ratio (Ip/Io) of the cortex inside the pipette (Ip) to the cortex on the opposite side of the cell (Io). P values were derived from Kruskal Wallis followed by the Wilcoxon test. In A–D, “labeled” indicates the protein labeled by GFP, and “background” indicates the genetic background (WT or specific single or double null) expressing the labeled protein. Number of cells analyzed, n = 7–10 from four independent experiments.
DscI facilitates the mechanoresponsiveness of CortI. (A) GFP images show localization of GFP-MyoII, GFP-alone, and GFP-DscIA as cells are aspirated with a micropipette (from left to right). GFP-MyoII serves as a positive control. (B) GFP images show localization of GFP-CortI in cells with and without DscI as these cells are aspirated with a micropipette. (C) GFP images show localization of GFP-MyoII in cells with and without DscI as these cells are aspirated with a micropipette. GFP-alone serves as a negative control. For A–C, the genetic background of each cell line is indicated in the upper edge of each image, and GFP-alone serves as a negative control. Micropipette suction pressure was applied to the right side of each cell. (D) The degree of mechanoresponsiveness of GFP-tagged proteins is quantified as the GFP intensity ratio (Ip/Io) of the cortex inside the pipette (Ip) to the cortex on the opposite side of the cell (Io). P values were derived from Kruskal Wallis followed by the Wilcoxon test. In A–D, “labeled” indicates the protein labeled by GFP, and “background” indicates the genetic background (WT or specific single or double null) expressing the labeled protein. Number of cells analyzed, n = 7–10 from four independent experiments.
We then tested if DscIA is required for other mechanoresponsive proteins to accumulate at sites of mechanical stress. We used MPA to apply stress to GFP-CortI expressing cortI single and cortI; dsc1 double mutants. GFP-CortI failed to accumulate at the cortex inside the pipette when dsc1 was deleted (Fig. 3 B). This reduction in CortI’s mechanoresponsiveness is reflected in the significant difference in the Ip/Io values between GFP-CortI signal in cortI and cortI; dsc1 background (Fig. 3 D). In fact, the level of GFP-CortI accumulation at the aspirated cortex in the double mutant is similar to that of the GFP-alone control. Furthermore, CortI accumulation at the cleavage furrow cortex is also significantly reduced when DscIA is absent (Fig. S4, D and E). In contrast, dsc1 deletion does not inhibit GFP-MyoII’s mechanoresponsiveness or ability to localize to the cleavage furrow cortex (Fig. 3, C and D; and Fig. S4, C and E). Interestingly, overexpression of DscI does not inhibit CortI’s mechanoresponsiveness, although it does significantly reduce cortical CortI (Fig. S4, F and G). This observation suggests that once a certain threshold of CortI’s ability to associate with the cortex is reached, CortI can mechanorespond.

DscI facilitates CortI’s cleavage furrow accumulation. (A) DIC and GFP images show localization of GFP-MyoII, GFP alone, and GFP-DscIA as cells form cleavage furrows during cytokinesis (from left to right). GFP-MyoII serves as a positive control. (B) The ratio of the background-corrected mean GFP signal intensity of both sides of the furrow (If) to that of both of the poles (Ip) was measured. Number of cells analyzed, n = 4–6 from three independent experiments. (C and D) GFP images show accumulation of GFP-CortI and GFP-MyoII at the cleavage furrow in cells with and without DscI. (E) Quantification of If/Ip ratio for GFP-CortI and GFP-MyoII signals at the cleavage furrows as cells of different genetic backgrounds divide. If/Ip ratio for vector expression in dsc1 cells (shaded) is reproduced from B for direct comparison. All P values were derived by ANOVA followed by Fisher’s LSD test. (F) GFP images show localization of GFP-CortI in cortI null–complemented cells with DscIA overexpression as these cells were aspirated by micropipette. Number of cells analyzed, n = 4–9 from two independent experiments. Vec, vector. (G) The degree of mechanoresponsiveness of GFP-tagged proteins was quantified as the GFP intensity ratio (Ip/Io) of the cortex inside the pipette (Ip) to the cortex on the opposite side of the cell (Io). P values were derived from ANOVA followed by Fisher’s LSD test. Number of cells analyzed, n = 8–9 from two independent experiments.

DscI facilitates CortI’s cleavage furrow accumulation. (A) DIC and GFP images show localization of GFP-MyoII, GFP alone, and GFP-DscIA as cells form cleavage furrows during cytokinesis (from left to right). GFP-MyoII serves as a positive control. (B) The ratio of the background-corrected mean GFP signal intensity of both sides of the furrow (If) to that of both of the poles (Ip) was measured. Number of cells analyzed, n = 4–6 from three independent experiments. (C and D) GFP images show accumulation of GFP-CortI and GFP-MyoII at the cleavage furrow in cells with and without DscI. (E) Quantification of If/Ip ratio for GFP-CortI and GFP-MyoII signals at the cleavage furrows as cells of different genetic backgrounds divide. If/Ip ratio for vector expression in dsc1 cells (shaded) is reproduced from B for direct comparison. All P values were derived by ANOVA followed by Fisher’s LSD test. (F) GFP images show localization of GFP-CortI in cortI null–complemented cells with DscIA overexpression as these cells were aspirated by micropipette. Number of cells analyzed, n = 4–9 from two independent experiments. Vec, vector. (G) The degree of mechanoresponsiveness of GFP-tagged proteins was quantified as the GFP intensity ratio (Ip/Io) of the cortex inside the pipette (Ip) to the cortex on the opposite side of the cell (Io). P values were derived from ANOVA followed by Fisher’s LSD test. Number of cells analyzed, n = 8–9 from two independent experiments.
Altogether, DscIA does not accumulate locally in response to mechanical stress, but its presence is crucial for CortI’s, but not MyoII’s, mechanoresponsiveness.
DscI is antagonized by non-mechanoresponsive CK components
Previous genetic dissection demonstrated that IQGAP proteins, including IQGAP1 and IQGAP2, modulate the activity and mechanoresponsivenes of MyoII and CortI (Kee et al., 2012; Srivastava and Robinson, 2015). IQGAP1 is a negative regulator as it inhibits the mechanoresponsive accumulation of CortI and MyoII. IQGAP2, in contrast, relieves this repression, allowing the proteins to accumulate in response to mechanical stress (Kee et al., 2012; Kothari et al., 2019a; Ren et al., 2014; Srivastava and Robinson, 2015). These IQGAP proteins pre-assemble into antagonistic CKs with MyoII and CortI in the cytoplasm and actively fine-tune the level of the contractile system at the cortex (Kothari et al., 2019b). Therefore, we asked if DscI functions in the same regulatory pathway, and potentially the same CK complexes. To address this question, we used CRISPR/Cas9 to generate double mutants that lack dsc1 and cortI, iqgap1, or iqgap2 and examined their genetic interactions using suspension growth. We first examined the genetic relationship between dsc1 and cortI. We grew WT, dsc1 and cortI single-mutant, and cortI; dsc1 double-mutant cells in suspension and analyzed their growth rates. Strikingly, deleting dsc1 in cortI null cells significantly improved their growth, compared to cortI single mutants (Fig. 4 A). The average growth rate of cortI; dsc1 cells (k = 0.052 h−1) was significantly faster than the cortI cells (k = 0.027 h−1) and similar to the growth rate measured for the dsc1 cells (k = 0.053 h−1; Fig. 4 B). Consistent with the improved growth rate, cortI; dsc1 double-mutant cells were also less multinucleated when compared to cortI single-mutant cells (Fig. 4 C). Compared to the WT, cortI single-mutant cells are severely multinucleated with the majority of cells enlarged, having more than five nuclei. However, the cortI; dsc1 double-mutant cells had lower multinucleation and normal cell size (Fig. 4, C and D). These observations indicate an antagonistic relationship between DscI and CortI.

DscI is antagonized by CortI and IQGAP1, which serves as a negative regulator of CK mechanoresponsiveness. (A) Representative growth curves in suspension culture of WT (blue), dsc1 (red), and cortI (green) single mutants and cortI; dsc1 (brown) double mutant. Growth curve of cortI; dsc1 shows faster growth, as compared to the cortI single mutant. n = 3 per growth curve; bars represent SEM. (B) The quantified growth rates for the exponential growth phase for each cell line shown in A are presented. The average growth rate of cortI; dsc1 (0.051 h−1) was significantly higher than that of cortI (0.026 h−1) and was lower than dsc1 single mutants (0.059 h−1). P values were calculated using ANOVA followed by Fisher’s LSD test. Number of samples analyzed, n = 6 from two independent experiments. (C) Representative images of DIC and nuclei show relative cell size and number of nuclei/cell between different cell lines. cortI single mutant displayed an enlarged cell size due to a higher degree of multinucleation. Cells were less multinucleated in the cortI; dsc1 double mutant strain. DIC and Hoescht images for WT are reproduced from Fig. 1 G. (D) Quantification of the number of nuclei/cell for WT, dsc1, cortI, and cortI;dsc1 after 2 d in suspension. The cortI mutant displayed a high number of multinucleated cells, especially in the 3–10 nuclei/cell range. Note that among these cell lines, only cortI accumulated >10 nuclei. The cortI; dsc1 double mutant was less multinucleated as compared to the cortI single mutant, with most cells having 1–2 nuclei/cell, similar to dsc1 single mutant cells. The data provided represent two replicates, with 90–115 cells per cell line for each replicate. P value was calculated using the Comparison of Proportions test. (**, P < 0.001; n.s, not significant). (E) Representative growth curves in suspension culture of WT (blue), dsc1 (red), and iqgap1 (orange) single mutant cells and the iqgap1; dsc1 double (black) mutant cells. Growth in suspension of iqgap1; dsc1 was faster than that of iqgap1 and slower than that of dsc1 single mutant cells. n = 6 per growth curve, bars represent SEM. (F) Growth rates of the exponential growth phase for each cell line. The average growth rate of iqgap1; dsc1 (0.041 h−1) was higher than that of iqgap1 (0.026 hr−1) and lower than dsc1 (0.048 h−1). P values were derived using ANOVA followed by Fisher’s LSD test. Number of samples analyzed, n = 6 from two independent experiments. (G) Summary diagram shows the genetic relationship between DscI, CortI, and IQGAP1. While IQGAP1 is a known inhibitor of CortI, it also antagonizes DscI. On the other hand, DscI is also antagonized by CortI, whose roles are important for balanced cellular mechanics and cytokinesis regulation.
DscI is antagonized by CortI and IQGAP1, which serves as a negative regulator of CK mechanoresponsiveness. (A) Representative growth curves in suspension culture of WT (blue), dsc1 (red), and cortI (green) single mutants and cortI; dsc1 (brown) double mutant. Growth curve of cortI; dsc1 shows faster growth, as compared to the cortI single mutant. n = 3 per growth curve; bars represent SEM. (B) The quantified growth rates for the exponential growth phase for each cell line shown in A are presented. The average growth rate of cortI; dsc1 (0.051 h−1) was significantly higher than that of cortI (0.026 h−1) and was lower than dsc1 single mutants (0.059 h−1). P values were calculated using ANOVA followed by Fisher’s LSD test. Number of samples analyzed, n = 6 from two independent experiments. (C) Representative images of DIC and nuclei show relative cell size and number of nuclei/cell between different cell lines. cortI single mutant displayed an enlarged cell size due to a higher degree of multinucleation. Cells were less multinucleated in the cortI; dsc1 double mutant strain. DIC and Hoescht images for WT are reproduced from Fig. 1 G. (D) Quantification of the number of nuclei/cell for WT, dsc1, cortI, and cortI;dsc1 after 2 d in suspension. The cortI mutant displayed a high number of multinucleated cells, especially in the 3–10 nuclei/cell range. Note that among these cell lines, only cortI accumulated >10 nuclei. The cortI; dsc1 double mutant was less multinucleated as compared to the cortI single mutant, with most cells having 1–2 nuclei/cell, similar to dsc1 single mutant cells. The data provided represent two replicates, with 90–115 cells per cell line for each replicate. P value was calculated using the Comparison of Proportions test. (**, P < 0.001; n.s, not significant). (E) Representative growth curves in suspension culture of WT (blue), dsc1 (red), and iqgap1 (orange) single mutant cells and the iqgap1; dsc1 double (black) mutant cells. Growth in suspension of iqgap1; dsc1 was faster than that of iqgap1 and slower than that of dsc1 single mutant cells. n = 6 per growth curve, bars represent SEM. (F) Growth rates of the exponential growth phase for each cell line. The average growth rate of iqgap1; dsc1 (0.041 h−1) was higher than that of iqgap1 (0.026 hr−1) and lower than dsc1 (0.048 h−1). P values were derived using ANOVA followed by Fisher’s LSD test. Number of samples analyzed, n = 6 from two independent experiments. (G) Summary diagram shows the genetic relationship between DscI, CortI, and IQGAP1. While IQGAP1 is a known inhibitor of CortI, it also antagonizes DscI. On the other hand, DscI is also antagonized by CortI, whose roles are important for balanced cellular mechanics and cytokinesis regulation.
We next tested how DscI interacts with IQGAP1. We knocked dsc1A out of the iqgap1 null cells and found that DscI and IQGAP1 also work antagonistically. Suspension growth curves reveal that iqgap1; dsc1 cells (k = 0.04 h−1) grow faster than iqgap1 single-mutant cells (k = 0.027 h−1), although these double-mutant cells still grow slightly slower than dsc1 single-mutant cells (k = 0.049 h−1; Fig. 4, E and F). Interestingly, these interactions are very similar to those observed between DscI and CortI. The magnitudes of changes in growth and multinucleation are comparable between the two double mutants (cortI; dsc1 and iqgap1; dsc1) and were rescuable back to the single-mutant rates (Fig. S5, A and B). Overall, DscI activity is inhibited by IQGAP1 and CortI.
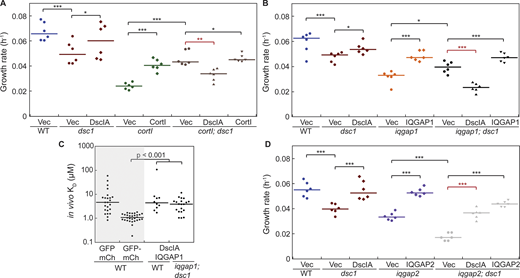
DscIA has antagonistic genetic interactions with CortI and IQGAP1 and synergistic genetic interactions with IQGAP2. (A) Quantification of growth rates during exponential growth in suspension of WT (blue), dsc1 (red) and cortI (green) single mutants, and double mutant (brown) expressing empty plasmids together with their corresponding complemented backgrounds. Expression of DscIA and CortI had opposite effects on growth of cortI; dsc1 double mutant cells. Exogenous expression of DscIA in the absence of CortI was deleterious to growth. Exogenous expression of DscIA rescued the growth defect of dsc1 null, while CortI partially rescued the growth defect of cortI null. Number of cell samples, n = 6 from two independent experiments. (B) Quantification of growth rates during exponential growth in suspension of WT (blue), dsc1 (red) and iqgap1 (orange) single mutants, and double mutant (black) expressing empty plasmids together with their corresponding complemented backgrounds. Exogenous expression of DscIA in the absence of IQGAP1, similar to that of CortI, was also deleterious to growth of iqgap1; dsc1 cells. Exogenous expression of IQGAP1 rescued growth of the double mutant cells. Number of cell samples, n = 6 from two independent experiments. (C) FCCS measurement of in vivo KD values demonstrated that DscIA and IQGAP1 do not detectably interact in the cytoplasm in either the WT or in the iqgap1; dsc1 complemented backgrounds. Positive and negative controls (shaded) in WT background are reproduced from Fig. 7 C. Compared to the negative control, the P values of the interactions in the WT background was 0.64 and in the iqgap1; dsc1-complemented background was 0.16. P values were derived from an ANOVA followed by Fisher’s LSD test. Number of cells analyzed, n = 10–30 from two independent experiments. (D) Quantification of growth rates during exponential growth in suspension of WT (blue), dsc1 (red) and iqgap2 (purple) single mutants, and double mutant (gray) expressing empty plasmids together with their corresponding complemented backgrounds. Exogenous expression of DscIA and IQGAP2 rescued the growth of iqgap2; dsc1 double mutant cells, returning growth to the single mutant levels. All growth rate values in these experiments are detailed in Table S2. All P values were derived by ANOVA followed by Fisher’s LSD test (*, P < 0.05; **, P < 0.001; ***, P < 0.0001). Number of cell samples, n = 6 from two independent experiments.

DscIA has antagonistic genetic interactions with CortI and IQGAP1 and synergistic genetic interactions with IQGAP2. (A) Quantification of growth rates during exponential growth in suspension of WT (blue), dsc1 (red) and cortI (green) single mutants, and double mutant (brown) expressing empty plasmids together with their corresponding complemented backgrounds. Expression of DscIA and CortI had opposite effects on growth of cortI; dsc1 double mutant cells. Exogenous expression of DscIA in the absence of CortI was deleterious to growth. Exogenous expression of DscIA rescued the growth defect of dsc1 null, while CortI partially rescued the growth defect of cortI null. Number of cell samples, n = 6 from two independent experiments. (B) Quantification of growth rates during exponential growth in suspension of WT (blue), dsc1 (red) and iqgap1 (orange) single mutants, and double mutant (black) expressing empty plasmids together with their corresponding complemented backgrounds. Exogenous expression of DscIA in the absence of IQGAP1, similar to that of CortI, was also deleterious to growth of iqgap1; dsc1 cells. Exogenous expression of IQGAP1 rescued growth of the double mutant cells. Number of cell samples, n = 6 from two independent experiments. (C) FCCS measurement of in vivo KD values demonstrated that DscIA and IQGAP1 do not detectably interact in the cytoplasm in either the WT or in the iqgap1; dsc1 complemented backgrounds. Positive and negative controls (shaded) in WT background are reproduced from Fig. 7 C. Compared to the negative control, the P values of the interactions in the WT background was 0.64 and in the iqgap1; dsc1-complemented background was 0.16. P values were derived from an ANOVA followed by Fisher’s LSD test. Number of cells analyzed, n = 10–30 from two independent experiments. (D) Quantification of growth rates during exponential growth in suspension of WT (blue), dsc1 (red) and iqgap2 (purple) single mutants, and double mutant (gray) expressing empty plasmids together with their corresponding complemented backgrounds. Exogenous expression of DscIA and IQGAP2 rescued the growth of iqgap2; dsc1 double mutant cells, returning growth to the single mutant levels. All growth rate values in these experiments are detailed in Table S2. All P values were derived by ANOVA followed by Fisher’s LSD test (*, P < 0.05; **, P < 0.001; ***, P < 0.0001). Number of cell samples, n = 6 from two independent experiments.
Given the genetic interactions between DscI and IQGAP1 along with genetic and biochemical interactions between CortI and DscI, we next tested if DscI and IQGAP1 interact in the cytoplasm. We used FCCS in live interphase cells. This technique allows us to quantitatively characterize protein–protein interactions in a defined confocal volume within the cytoplasm (Bacia et al., 2006; Kothari et al., 2019b). The interaction of separately expressed GFP and mCherry acts as a negative control with a median apparent KD of 5.0 μM while that of fusion GFP-mCherry acts as a positive control with a median KD of 1.1 μM. The apparent KD measured by FCCS between DscI and IQGAP1 in both WT and complemented-null backgrounds are similar to each other and the negative control, suggesting that IQGAP1 and DscI do not appreciably biochemically interact in vivo (Fig. S5 C). Both IQGAPs are a conserved family of proteins characterized by a GAP-like domain that can bind Rac family small GTPases. Structurally, mammalian IQGAP proteins have an N-terminal calponin homology (CH) motif, which functions as an F-actin binding domain (Brill et al., 1996; Hart et al., 1996). In addition, the mammalian IQGAPs have multiple isoleucine-glutamine (IQ) (calponin-binding) domains and a tryptophan-tryptophan (WW) protein-interaction motif. However, in Dictyostelium, neither IQGAP1 nor IQGAP2 contain the CH, WW, or IQ domains (Adachi et al., 1997). Both IQGAP1 and IQGAP2 are present in the cytoplasm and accumulate at the cortex, and during cytokinesis, they enrich at the cleavage furrow cortex (Faix et al., 2001; Mondal et al., 2010). In terms of sequence, they are 52% identical and 70% similar in amino acid sequence, accounting for why DscI interacts with IQGAP2, but not IQGAP1.
Together, these observations indicate that DscI regulates the localization and function of CortI, but DscI itself is antagonistically regulated by components of the CKs, including CortI and IQGAP1 (Fig. 4 G). Furthermore, the FCCS results indicate that this regulation does not require IQGAP1 and DscI to interact biochemically directly or indirectly.
IQGAP2 acts synergistically with dscI
DscI was initially implicated in the CKs by the discovery of it serving as a molecular interactor of IQGAP2, the alleviator of IQGAP1’s inhibition of CortI’s and MyoII’s mechanoresponsiveness (Kee et al., 2012; Kothari et al., 2019a; Kothari et al., 2019b; Ren et al., 2014). Therefore, we tested if and how DscI and IQGAP2 interact. We generated iqgap2; dsc1 double-mutant cells and analyzed their phenotype. Using the suspension growth assay, we found that the iqgap2; dsc1 double-mutant cells have the most severe growth defects, having the lowest growth rates (k = 0.018 h−1) among the cell lines tested (Fig. 5, A and B). Further, the double mutants were fully rescuable to single-mutant phenotypes (Fig. S5 D). Furthermore, these cells have a high rate of multinucleation. Differential interference contrast (DIC)– and nuclei-stained images comparing iqgap2; dsc1 to WT and iqgap2 single-mutant cells reveal that iqgap2; dsc1 cells are larger and more multinucleated (Fig. 5 C). While nearly 40% of dsc1 and 50% of iqgap2 single mutants were multinucleated, more than 70% of iqgap2; dsc1 cells were multinucleated (Fig. 5 D). We observed many examples of iqgap2; dsc1 cells with more than 40 nuclei (Fig. 5 C). This extreme level of multinucleation was not observed in any other single or double mutants analyzed in this study. In addition, movies of iqgap2; dsc1 cells undergoing cytokinesis show that these cells display cleavage furrow morphology characteristic of traction-mediated cytofission (Fig. 5 E, and Videos 4 and 5). The cleavage furrow formed in this process is more elongated and highly similar to furrows formed by myoII null cells as they undergo traction-mediated cytofission (Neujahr et al., 1997). Traction-mediated cytofission is less efficient than mitosis-coupled cytokinesis and explains why iqgap2; dsc1 cells are oversized and multinucleated, as observed in Fig. 5 C. We then tested if the double mutants impact cortical tension. The iqgap2; dsc1 double-mutant cells had a significant reduction in cortical tension as compared to wild type (28% of WT) and the single mutants (Fig. 5 F). Note that the measured effective cortical tension of iqgap2 cells is very similar to that of published data (Kee et al., 2012). Overall, the iqgap2; dsc1 double mutant has more severe cytokinetic and mechanical phenotypes.

Deletion of dsc1 acts as an enhancer of iqgap2, which encodes a positive regulator of CK mechanoresponsiveness. (A) Representative growth curves from suspension culture of WT (blue), dsc1 (red), and iqgap2 (purple) single mutants and the iqgap2; dsc1 (gray) double mutant. Among these, iqgap2; dsc1 had the poorest growth, followed by iqgap2 and dsc1 single mutants and then WT. n = 6 per growth curve, bars represent SEM. (B) Growth rates quantified from the exponential growth phase had significant differences between these cell lines. P values were derived for ANOVA followed by Fisher’s LSD test. Number of samples analyzed, n = 6 from two independent experiments. (C) Representative images of DIC and nuclei stained by Hoescht of WT, iqgap2 and iqgap2; dsc1 mutant cell lines. Both iqgap2 and iqgap2; dsc1 had increased cell size and number of nuclei per cell as compared to WT. Scale bar, 40 μm and applies to all panels. (D) Quantification of the number of nuclei/cell revealed that iqgap2; dsc1 cells had the greatest amount of multinucleation. Note that this double mutant strain accumulated more than 20 nuclei in cells, a level not observed in any of the single mutants. The data shown represent two replicates, with 133–182 cells per cell line for each replicate. P value was calculated using the Comparison of Proportions test (*, P < 0.05; **, P < 0.001; ***, P < 0.0001). (E) Sequence of images show stages of cytokinesis of iqgap2 and iqgap2; dsc1 mutant cells. Images were collected on cells in the exponential growth phase. t = 0 is defined as when the videos start. Full videos are provided in the Supplemental materials (Videos 4, and 5). (F) The dot plot graph shows comparison of the effective cortical tension between WT, dsc1 null, iqgap2 null, and iqgap2; dsc1 double mutant cells. Effective cortical tension values for WT and dsc1 cells (shaded) are reproduced from Fig. 1 J to allow for side-by-side comparison. P values were calculated using Kruskal-Wallis followed by the Wilcoxon test. Number of cells analyzed, n = 4–10 from two independent experiments. (G) Summary diagram shows the genetic relationship between DscI and IQGAP2. This interaction indicates that both IQGAP2 and DscI are required for normal cytokinesis and that they work in parallel and/or collaboratively to ensure cytokinesis fidelity.
Deletion of dsc1 acts as an enhancer of iqgap2, which encodes a positive regulator of CK mechanoresponsiveness. (A) Representative growth curves from suspension culture of WT (blue), dsc1 (red), and iqgap2 (purple) single mutants and the iqgap2; dsc1 (gray) double mutant. Among these, iqgap2; dsc1 had the poorest growth, followed by iqgap2 and dsc1 single mutants and then WT. n = 6 per growth curve, bars represent SEM. (B) Growth rates quantified from the exponential growth phase had significant differences between these cell lines. P values were derived for ANOVA followed by Fisher’s LSD test. Number of samples analyzed, n = 6 from two independent experiments. (C) Representative images of DIC and nuclei stained by Hoescht of WT, iqgap2 and iqgap2; dsc1 mutant cell lines. Both iqgap2 and iqgap2; dsc1 had increased cell size and number of nuclei per cell as compared to WT. Scale bar, 40 μm and applies to all panels. (D) Quantification of the number of nuclei/cell revealed that iqgap2; dsc1 cells had the greatest amount of multinucleation. Note that this double mutant strain accumulated more than 20 nuclei in cells, a level not observed in any of the single mutants. The data shown represent two replicates, with 133–182 cells per cell line for each replicate. P value was calculated using the Comparison of Proportions test (*, P < 0.05; **, P < 0.001; ***, P < 0.0001). (E) Sequence of images show stages of cytokinesis of iqgap2 and iqgap2; dsc1 mutant cells. Images were collected on cells in the exponential growth phase. t = 0 is defined as when the videos start. Full videos are provided in the Supplemental materials (Videos 4, and 5). (F) The dot plot graph shows comparison of the effective cortical tension between WT, dsc1 null, iqgap2 null, and iqgap2; dsc1 double mutant cells. Effective cortical tension values for WT and dsc1 cells (shaded) are reproduced from Fig. 1 J to allow for side-by-side comparison. P values were calculated using Kruskal-Wallis followed by the Wilcoxon test. Number of cells analyzed, n = 4–10 from two independent experiments. (G) Summary diagram shows the genetic relationship between DscI and IQGAP2. This interaction indicates that both IQGAP2 and DscI are required for normal cytokinesis and that they work in parallel and/or collaboratively to ensure cytokinesis fidelity.
Video of a dividing iqgap2 null cell. Each frame was collected every 2 s with a 40× (NA 1.30) oil objective and imaged by DIC using an Olympus IX71 microscope. Playback speed is 10 frames per second.
Video of a dividing iqgap2 null cell. Each frame was collected every 2 s with a 40× (NA 1.30) oil objective and imaged by DIC using an Olympus IX71 microscope. Playback speed is 10 frames per second.
Video of a dividing iqgap2; dsc1 null cell. Each frame was collected every 2 s with a 40× (NA 1.30) oil objective and imaged by DIC using an Olympus IX71 microscope. Playback speed is 10 frames per second.
Video of a dividing iqgap2; dsc1 null cell. Each frame was collected every 2 s with a 40× (NA 1.30) oil objective and imaged by DIC using an Olympus IX71 microscope. Playback speed is 10 frames per second.
Overall, IQGAP2 and DscI have synergistic genetic interactions where deletion of one enhances the deletion of the other. This interaction indicates that both IQGAP2 and DscI are required for normal cytokinesis and that they work in parallel and/or collaboratively to ensure cytokinesis fidelity. This finding is particularly interesting since previous reports established that IQGAP2 is required for CortI to properly accumulate at the cleavage furrrow cortex of dividing cells (Faix et al., 2001). Collectively, based on these genetic interactions, protein distributions, and the in vivo protein interactions measured in our prior FCCS studies (Kothari et al., 2019b), IQGAP2 and DscI collaborate and function together in the same complexes to ensure proper cortical localization of CortI (Fig. 5 G).
DscI expression is downregulated in the absence of the non-mechanoresponsive CK components
DscI’s genetic interactions establish that DscI is negatively regulated by CortI and IQGAP1. One possible mode of regulation is through modification of transcription and protein expression. Previous studies have established the relationship between protein expression and function for cytoskeletal proteins. For instance, mammalian α-actinin regulates non-muscle myoIIB expression (Barai et al., 2021). Imbalanced expression of different MyoII isoforms also leads to changes in cell mechanics, including tension and elasticity during cell morphodynamics (Weißenbruch et al., 2021). Since well-regulated expression of DscI is important for cell function, we tested whether DscI expression is affected by deletion of any of these CK interactors. We used Western analysis to assess protein levels of DscI in iqgap1, cortI, and iqgap2 single KO cells. Since protein level of DscI is density-dependent (Clarke et al., 1987; Jain et al., 1992; Wetterauer et al., 1995), Western analysis was performed on cells seeded at equally low and high densities. Lysates of cells seeded for 8 h were analyzed by Western blot with anti-DscI antibodies. At both densities, the levels of DscI protein are significantly lower in iqgap1 and cortI cells. In iqgap2 mutants, DscI levels were reduced at the lower density of cells (Fig. 6 A). Quantification reveals that iqgap1 and cortI cells express DscI at 30 and 17% of WT levels at high density. At low density, the values for iqgap1, cortI, and iqgap2 mutants are 30, 10, and 55% of WT control, respectively (Fig. 6 B). The low levels of DscI protein expression could be explained by either reduced transcription, translation, or decreased protein stability. To address these possibilities, dsc1 transcript levels in these mutants were assessed. RNA from cells at an equal high density was extracted and analyzed using qRT-PCR with primers targeting all paralogs of DscI. The dsc1 RNA levels align tightly with protein levels. Among the mutants tested, cortI cells had the lowest level of dsc1 transcript, followed by iqgap1 cells. The iqgap2 null cells had unchanged levels of dsc1 transcript compared to WT control (Fig. 6 C). These observations indicate that DscI protein expression is regulated, in part, at the level of transcription and/or mRNA stability. These results suggest that CortI and IQGAP1, components of the non-mechanoresponsive CKs, also negatively regulate DscI through mechanisms that alter mRNA and protein levels.

Transcript and protein levels of DscI are dependent on CortI and IQGAP1 while the transcript level of MyoII is dependent on DscI. (A) Western analysis with DscI antibody showed reduction in DscIA protein level in cortI and iqgap1 null mutants. Each cell line was seeded at two different densities 10-fold apart 6 h before analysis. 70 µg of whole cell lysates were loaded on 10% acrylamide gel. Dynacortin (Dyn) is provided as a loading control. (B) Quantification of the DscI protein levels after normalization to WT revealed reduced protein expression, particularly in iqgap1 and cortI null cells. Number of independent experiments, n = 3. Levels were quantified using integrated density of signal normalized by area and background in ImageJ, normalized against WT, and plotted. (C) qRT-PCR analysis with dscIA primers showed reduction in dsc1A transcript levels in cortI and iqgap1 null mutants. Relative quantification of normalized RNA levels was performed using the ΔΔCt method (Rao et al., 2013). All P values were derived by ANOVA followed by Fisher’s LSD test. (*, P < 0.05; **, P < 0.001; ***, P < 0.0001). Number of independent experiments, n = 4–5. (D) Example Western blot with antibodies against MyoII, CortI, and DscI in WT and dscI null cells is shown. Each cell line was seeded at three different densities 10-fold apart 6 h before analysis. 30 µg of whole cell lysates were loaded on 10% acrylamide gel. Dynacortin is provided as a loading control. (E) qRT-PCR analysis with primers complementary to CortI, MyoII, IQGAP1 and IQGAP2 was performed to measure transcript levels of cytoskeletal proteins in dsc1 null mutants. Relative quantification of normalized RNA levels was performed using the ΔΔCt method. RNA level for each protein in dscI cells was normalized to that of the WT parental cells. Number of independent experiments, n = 3–4. P values displayed above each dataset were derived by ANOVA followed by Fisher’s LSD test. Source data are available for this figure: SourceData F6.
Transcript and protein levels of DscI are dependent on CortI and IQGAP1 while the transcript level of MyoII is dependent on DscI. (A) Western analysis with DscI antibody showed reduction in DscIA protein level in cortI and iqgap1 null mutants. Each cell line was seeded at two different densities 10-fold apart 6 h before analysis. 70 µg of whole cell lysates were loaded on 10% acrylamide gel. Dynacortin (Dyn) is provided as a loading control. (B) Quantification of the DscI protein levels after normalization to WT revealed reduced protein expression, particularly in iqgap1 and cortI null cells. Number of independent experiments, n = 3. Levels were quantified using integrated density of signal normalized by area and background in ImageJ, normalized against WT, and plotted. (C) qRT-PCR analysis with dscIA primers showed reduction in dsc1A transcript levels in cortI and iqgap1 null mutants. Relative quantification of normalized RNA levels was performed using the ΔΔCt method (Rao et al., 2013). All P values were derived by ANOVA followed by Fisher’s LSD test. (*, P < 0.05; **, P < 0.001; ***, P < 0.0001). Number of independent experiments, n = 4–5. (D) Example Western blot with antibodies against MyoII, CortI, and DscI in WT and dscI null cells is shown. Each cell line was seeded at three different densities 10-fold apart 6 h before analysis. 30 µg of whole cell lysates were loaded on 10% acrylamide gel. Dynacortin is provided as a loading control. (E) qRT-PCR analysis with primers complementary to CortI, MyoII, IQGAP1 and IQGAP2 was performed to measure transcript levels of cytoskeletal proteins in dsc1 null mutants. Relative quantification of normalized RNA levels was performed using the ΔΔCt method. RNA level for each protein in dscI cells was normalized to that of the WT parental cells. Number of independent experiments, n = 3–4. P values displayed above each dataset were derived by ANOVA followed by Fisher’s LSD test. Source data are available for this figure: SourceData F6.
Feedback control of the CKs helps ensure robust assembly and function (Kee et al., 2012; Kothari et al., 2019a; Ren et al., 2009). Because DscI’s expression depends on CortI and IQGAP1, we next tested if DscI presence is necessary for expression of other key cytoskeletal proteins in the CKs. Using Western analysis and qRT-PCR to quantify both protein and transcript levels of various key cytoskeletal proteins in dsc1 null cells, we found that myoII (mhcA) transcript levels are ∼60% of WT, while its protein level is unchanged (Fig. 6, D and E). Protein and/or transcript levels of CortI, IQGAP1, and IQGAP2 are not different in dsc1 cells compared to WT. This finding indicates that DscI is required for WT myoII transcript levels, but not protein levels.
Dsc stabilizes the mechanoresponsive CKs
So far, we established DscI is both a biochemical and genetic interactor of components of the mechanoresponsive CKs, IQGAP2, and CortI (Kothari et al., 2019b). It is, therefore, possible that DscI functions by being a component of the mechanoresponsive CKs. We used co-immunoprecipitation to test for interactions between DscIA and other components of the complex. In lysates where GFP-DscIA was pulled down using GFP-trap beads, both CortI and MyoII proteins were detected in the elution fraction while they were not observed in the GFP pull-down control (Fig. 7 A). Thus, CortI and MyoII co-immunoprecipitated with GFP-DscIA. Similarly, in cells where both mCh-IQGAP2 and GFP-DscIA are expressed, mCh-IQGAP2 co-immunoprecipitated with GFP-DscIA (Fig. 7 B). Furthermore, the interaction between DscIA and MyoII was also detected using FCCS in cells that express GFP-DscIA and mCh-MyoII in various genetic backgrounds (Fig. 7 C). The mean in vivo KDs of this interaction in WT, dscI null–, and myoII null–complemented backgrounds are 2.8, 1.9, and 2.9 μM, respectively. These KDs are all significantly lower than that measured for uncoupled mCh and GFP (negative control). Collectively, these results indicate that DscIA is a component of the mechanoreponsive CKs, which include CortI, MyoII, and IQGAP2.

DscI is a component of the mechanoresponsive CKs. (A and B) Western blot of co-immunoprecipitation studies of GFP-DscIA in cell lysates of indicated cell lines is shown. FT, flow-through; IQG2, IQGAP2. No antibody for IQGAP2 is available so cell lines expressing mCh-fused IQGAP2 and mouse anti-mCh antibody (Novus Biologicals, 25157SS) were used. In A, bands boxed in the same color (orange or blue) are from the same Western blot. (C) Results from FCCS analysis examining interactions between DscIA and MyoII in different cellular backgrounds are shown. The apparent, or in vivo, KD for unlinked mCherry and GFP (negative control) and fusion GFP-mCherry (positive control) was 5.0 and 1.1 µM, respectively. P values were calculated using the log-transformed data and the ANOVA followed by Fisher’s LSD test (*, P < 0.05; ***, P < 0.0001). Number of cells analyzed, n = 19–30 from two independent experiments. (D) FCCS analysis confirmed the positive interaction between CortI and IQGAP2 in cortI-complemented background (with an apparent KD = 1.2; Kothari et al., 2019b). Median in vivo KDs for negative (=3.9 and 3.8) and positive (=1.1 and 1.1) controls in cortI-complemented and cortI; dsc1 double mutant background were similar to that of the WT background (C). In the absence of dsc1, CortI and IQGAP2 interacted with an apparent KD of 2.0, which is significantly weaker than that measured in the presence of DscI. Number of cells analyzed, n = 18–28 from two independent experiments. (E) FCCS-detected interactions between CortI and MyoII in the WT background (with KD = 1.5). The affinity between CortI and MyoII was significantly weakened in dsc1 null mutants (with KD = 2.6). Positive and negative controls (shaded) in WT background are reproduced from C for direct comparison. Positive and negative controls were also measured in dsc1 background (in dsc1: KD for negative control = 5.2, KD for positive control = 1.3). All P values were calculated on the log-transformed data using an Kruskal-Wallis followed by the Wilcoxon test (*, P < 0.05; **, P < 0.001; ***, P < 0.0001). P values signifying the statistical differences in the interactions between the presence and absence of DscI are highlighted in red. Number of cells analyzed, n = 24–36 from two independent experiments. (F) FCCS data in this study show that DscI helps stabilize the associations between key proteins in the mechanoresponsive CKs. Combined with previous proteomic analysis and co-immunoprecipitation results, it is likely that these proteins, including DscI, function in the same complex, and the absence of one affects the stability of other interactions within the assembly. Source data are available for this figure: SourceData F7.
DscI is a component of the mechanoresponsive CKs. (A and B) Western blot of co-immunoprecipitation studies of GFP-DscIA in cell lysates of indicated cell lines is shown. FT, flow-through; IQG2, IQGAP2. No antibody for IQGAP2 is available so cell lines expressing mCh-fused IQGAP2 and mouse anti-mCh antibody (Novus Biologicals, 25157SS) were used. In A, bands boxed in the same color (orange or blue) are from the same Western blot. (C) Results from FCCS analysis examining interactions between DscIA and MyoII in different cellular backgrounds are shown. The apparent, or in vivo, KD for unlinked mCherry and GFP (negative control) and fusion GFP-mCherry (positive control) was 5.0 and 1.1 µM, respectively. P values were calculated using the log-transformed data and the ANOVA followed by Fisher’s LSD test (*, P < 0.05; ***, P < 0.0001). Number of cells analyzed, n = 19–30 from two independent experiments. (D) FCCS analysis confirmed the positive interaction between CortI and IQGAP2 in cortI-complemented background (with an apparent KD = 1.2; Kothari et al., 2019b). Median in vivo KDs for negative (=3.9 and 3.8) and positive (=1.1 and 1.1) controls in cortI-complemented and cortI; dsc1 double mutant background were similar to that of the WT background (C). In the absence of dsc1, CortI and IQGAP2 interacted with an apparent KD of 2.0, which is significantly weaker than that measured in the presence of DscI. Number of cells analyzed, n = 18–28 from two independent experiments. (E) FCCS-detected interactions between CortI and MyoII in the WT background (with KD = 1.5). The affinity between CortI and MyoII was significantly weakened in dsc1 null mutants (with KD = 2.6). Positive and negative controls (shaded) in WT background are reproduced from C for direct comparison. Positive and negative controls were also measured in dsc1 background (in dsc1: KD for negative control = 5.2, KD for positive control = 1.3). All P values were calculated on the log-transformed data using an Kruskal-Wallis followed by the Wilcoxon test (*, P < 0.05; **, P < 0.001; ***, P < 0.0001). P values signifying the statistical differences in the interactions between the presence and absence of DscI are highlighted in red. Number of cells analyzed, n = 24–36 from two independent experiments. (F) FCCS data in this study show that DscI helps stabilize the associations between key proteins in the mechanoresponsive CKs. Combined with previous proteomic analysis and co-immunoprecipitation results, it is likely that these proteins, including DscI, function in the same complex, and the absence of one affects the stability of other interactions within the assembly. Source data are available for this figure: SourceData F7.
If DscIA is a component of the mechanoresponsive CKs, we hypothesized that removing one component of the complex might weaken the interactions between other members of the complex. Therefore, we asked whether DscI helps stabilize interactions between other CK components. Using FCCS, we measured the association between CortI and IQGAP2 in complemented cortI single and cortI; dsc1 double mutants. The measured in vivo binding affinity (KD = 1.2 μM) for this association in the cortI null-complemented background matches our previous measurements (Kothari et al., 2019b). However, the interaction between IQGAP2 and CortI was weakened in cortI; dsc1 null cells where the apparent in vivo affinity is decreased, yielding a KD of 2.0 μM (Fig. 7 D). Thus, DscI helps stabilize the association between IQGAP2 and CortI. We next assessed if DscI is crucial for maintaining the interaction between CortI and MyoII. In the WT background, these two proteins interact in the cytoplasm with an apparent in vivo KD of 1.5 μM, consistent with previously reported data (Kothari et al., 2019b). In the dsc1 mutant, however, the binding affinity KD is significantly higher (KD = 2.6 μM; Fig. 7 E). This significant change in KD indicates that binding between MyoII and CortI is strengthened by DscI. Overall, DscI helps stabilize the associations between key proteins in the mechanoresponsive CKs. Importantly, these results align with the hypothesis that these proteins, including DscI, function in the same complex, and the absence of one affects the stability of other interactions within the assembly (Fig. 7 F).
Discussion
In this study, we found that DscI is crucial to ensure proper cortical mechanics and integrity, which in turn are necessary for complex cell shape changes like cytokinesis. While we find that the majority of DscI stays in the cytoplasm, a fraction of it localizes to the cortex. At the cortex, DscI helps recruit CortI, thereby maintaining normal cortical tension and membrane–cortex linkage. DscI is also crucial for CortI to exert its mechanoresponsiveness. In short, DscI functions as a quality control mechanism for CortI, acting to ensure its proper localization and function.
DscI also acts as a genetic interactor of integral proteins of the CKs. DscI has an antagonistic genetic relationship with CortI and IQGAP1, but a synergistic (or additive) genetic relationship with IQGAP2 (Fig. 8 A). A possible explanation for these genetic interactions is that DscIA functions in parallel with IQGAP2 to aid in localizing CortI to the cortex. Previous studies indicate IQGAP2 also facilitates proper distribution of CortI. In cells lacking IQGAP2, CortI fails to localize to the cleavage furrow, similar to what is observed in dsc1 null cells (Kee et al., 2012). On the other hand, without CortI to regulate it, DscI is deleterious to cells, as cortI; dscI double mutants grow better than cortI single mutants. In contrast, overexpression of DscI also impairs proper CortI localization. These observations suggest that even though DscI acts to support the CKs, its expression is controlled by CortI and IQGAP1. Owing to its multiple cellular roles and functions, it is perhaps not surprising that DscI’s expression is tightly controlled within the cell. Both deletion and overexpression of DscI lead to changes in cell mechanical properties and behaviors such as growth, cytokinesis, and cortical integrity. We also find that DscI is controlled at the level of protein and mRNA stability by CortI and IQGAP1, rendering another layer of regulation of this system to ensure its proper function (Fig. 8 B). Notably, DscI is not the first protein in the cytoskeletal system that is tightly regulated and whose loss or overexpression causes defects to the system. Dynacortin is another example. Dynacortin is a cortical actin-bundling protein whose overexpression leads to defects in growth, cytokinesis fidelity, and cortical mechanics while loss of dynacortin causes altered cell mechanics and cleavage furrow ingression kinetics (Girard et al., 2004; Robinson and Spudich, 2000; Zhang and Robinson, 2005). We are continuously finding that this concept of a sweet spot (not too little, not too much, just right) is a recurring theme in mechanobiology (Schiffhauer et al., 2019).

DscI acts as a modulator of the CK and is regulated by the non-mechanoresponsive CK components. (A) Schematic depiction of genetic interactions between DscI and key components of the CKs. DscI helps localize CortI to the cortex, which, in turn, is crucial for a balanced cellular mechanics (orange arrows). When components of the non-mechanoresponsive CKs are absent, DscI is deleterious to cytokinesis and is negatively regulated at the mRNA transcript and protein level (red blunt arrows). In contrast, DscI and IQGAP2 act in parallel genetic pathways to maintain normal cytokinesis (orange and blue arrows). (B) Mechanistic depiction of DscI intracellular activity and its regulation in the mechanobiome. DscI associates with the mechanoresponsive CK components and helps stabilize their assembly. It also acts to properly localize CortI to the cortex, ensuring WT cortical functions. DscI is negatively regulated by components of the non-mechanoresponsive CKs, which includes IQGAP1.
DscI acts as a modulator of the CK and is regulated by the non-mechanoresponsive CK components. (A) Schematic depiction of genetic interactions between DscI and key components of the CKs. DscI helps localize CortI to the cortex, which, in turn, is crucial for a balanced cellular mechanics (orange arrows). When components of the non-mechanoresponsive CKs are absent, DscI is deleterious to cytokinesis and is negatively regulated at the mRNA transcript and protein level (red blunt arrows). In contrast, DscI and IQGAP2 act in parallel genetic pathways to maintain normal cytokinesis (orange and blue arrows). (B) Mechanistic depiction of DscI intracellular activity and its regulation in the mechanobiome. DscI associates with the mechanoresponsive CK components and helps stabilize their assembly. It also acts to properly localize CortI to the cortex, ensuring WT cortical functions. DscI is negatively regulated by components of the non-mechanoresponsive CKs, which includes IQGAP1.
Furthermore, DscI helps stabilize the interactions between CortI, MyoII, and IQGAP2. These findings lead to a complex mechanistic explanation of the function of DscI in the contractility machinery, in which it acts as part of the mechanoresponsive CKs to fine-tune the accumulation, and hence govern the activity of the contractility machinery at the cortex. In this system, DscI acts as a component of the mechanoreponsive CKs, which function similarly to “packages” that are ready to be delivered to the cortex, where each component is then released. In these packages, DscI can be thought of as a chaperone that helps stabilize the complex and ensure proper delivery of the components, particularly CortI (Fig. 8 B).
Through our prior work, we already had indicators that suggested DscI contributed to the contractility system (Kothari et al., 2019b; Nguyen and Robinson, 2020; Robinson and Spudich, 2000). Interestingly, in our previous FCCS studies (Kothari et al., 2019b), DscI interacted with IQGAP2 only when CortI was present in the cell. In cortI null cells, DscI was not detected in the co-immunoprecipitated fraction, foreshadowing the role of CortI in regulating the expression of DscI. We now understand that in cortI null cells, DscI expression is reduced at the transcript and protein levels. We also found that DscI not only interacts with key constituents of the mechanoresponsive CKs, but also stabilizes the associations of these proteins with each other. Removing DscI from cells causes a significant decrease in the apparent binding affinities of CortI with MyoII and IQGAP2. Altogether, these results support the notion that DscI functions as part of the CKs and adds a layer of redundancy in regulating the contractility system. These observations are particularly interesting since for decades, DscI has been defined as a secreted/extracellular protein that facilitates membrane–substratum attachment and cell–cell adhesion in developing cells (Bastounis et al., 2016; Huber and O'Day, 2015; Springer et al., 1984). Our work here reveals that DscI also has important functions inside vegetative cells, where DscI remains largely intracellular (Alexander et al., 1992; Clarke et al., 1987). The differential roles of DscI inside and outside of a cell may be partially explained by its differential oligomeric state intracellularly versus extracellularly. DscI remains monomeric in the cytoplasm but assembles into a multimeric (presumably tri- or tetra-meric) state in the secreted multilamellar bodies (Fukuzawa and Ochiai, 1993; Mathieu et al., 2010).
The concept of functional synergy and/or cooperativity is becoming increasingly apparent in contractility systems. MyoII and CortI, the core elements of the mechanoresponsive machinery, both accumulate at the cleavage furrow cortex and exhibit cooperative accumulation at the point of mechanical perturbations. Deletion of one disrupts the stress-dependent accumulation of the other (Kee et al., 2012; Kothari et al., 2019a; Ren et al., 2009). Here, we see a similar story with DscI and IQGAP2. From the genetic interaction analysis, they have a synergistic genetic relationship, indicating that they act in parallel pathways. In this context, the term pathway reflects the genetic interactions. These interactions then help govern the CK complexes that are assembled in the cytoplasm. DscI and IQGAP2 also have common roles in ensuring proper accumulation of CortI at the cortex. In dsc1 null cells, significantly less CortI is found at the interphase cortex. Previously, cells lacking IQGAP2 also had impaired localization of CortI at the cleavage furrow cortex. This redundancy in mechanisms controlling CortI localization adds additional layers of spatial and temporal regulation of the mechanoaccumulation and contractility at the cortex.
Another concept in contractility regulation is that of feedback control, in which the system is structured as a contractility controller that also includes signaling inputs, for example, emanating from the mitotic spindle (Kee et al., 2012; Kothari et al., 2019a; Ren et al., 2009). Multiple modes of regulation through feedback control ensure robust assembly and function of the contractility machinery. While MyoII and CortI sit at the core of the contractility controller as the load-bearing and force-generating proteins, other regulators, such as IQGAP1, IQGAP2, and now DscI, are crucial for ensuring that this system does not over- or under-perform. IQGAP2 binds to CortI and MyoII and acts as positive regulator of this system, permitting the mechanoresponsiveness of these core proteins. To ensure that the system is not overly mechanoresponsive, IQGAP1 antagonizes IQGAP2, thereby controlling CortI and MyoII. As MyoII and CortI actively define the cortical mechanical properties and mechanoresponsiveness, their functions are then tightly regulated and fine-tuned by the feedback control of the IQGAPs (Dickinson et al., 2012; Kee et al., 2012; Pollard and O'Shaughnessy, 2019; Ren et al., 2014). Similarly, DscI fine-tunes CortI cortex levels and helps stabilize the assembly of the mechanoresponsive CKs, and in turn, the expression of DscI is regulated by non-mechanosponsive CK components. DscI also helps maintain the transcript levels of MyoII, but not protein level, consistent with the fact that many additional levels of regulation control the final protein amount. Nevertheless, this observation suggests that proteins within the cytoskeletal systems have layers of regulation both on transcript and protein levels, rendering its robustness in response to changes or challenges in the system. In an unpublished study, we find an additional major CK component, namely, the RNA-binding protein RNP1A, which binds mRNAs and helps set the levels of transcripts, encoding several cytoskeletal and CK-related proteins. Overall, the redundancy and feedback control in the cytoskeletal systems allow cells to ensure robust mechanics and mechanoresponsiveness, and high-fidelity shape change such as during cytokinesis.
The discovery and characterization of DscI, a traditional lectin, playing an intracellular function is intriguing, yet not necessarily surprising. In fact, DscI is not the first extracellular lectin protein discovered to have significant intracellular roles. Galectins are prime examples of this. These lectin proteins were first discovered based on their β-galactoside-binding activity and originally implicated in aiding in the formation of complex cell-surface glycans, which help mediate cell–cell recognition (Barondes et al., 1994; Johannes et al., 2018; Leffler et al., 2002). However, galectins were later found to reside inside the cell where they contribute to numerous intracellular functions, including having cytoskeletal regulatory roles. For example, Galectin-3 regulates the cytoskeleton through affecting Kras signaling (Reichert and Rotshenker, 2019), and Galectin-8 promotes cytoskeletal rearrangement through activation of Rho in trabecular meshwork cells (Diskin et al., 2012). Moreover, intracellular Galectin-9 sustains the rigidity of cortical actin cytoskeleton, thereby maintaining plasma membrane integrity (Querol Cano et al., 2019). Interestingly, both galectin and dsc proteins have roles as secreted proteins, but neither contain typical signal peptides required for classical secretion. Further, they are lectins that specifically bind to β-galactoside sugars (Cooper, 2002; Mathieu et al., 2010; Vasta et al., 2004; Vasta et al., 2017). The multiple levels of similarity from structures to functions between galectins and discoidins open up new avenues for follow-up studies in deciphering the roles of discoidins and their implications in intracellular function.
Understanding the functions of dsc is providing insights into a broader network that controls and drives the contractility system. Disruption of key proteins in this system often gives rise to aberrant cell behaviors and onsets of diseases (Conti et al., 2015; Nishida et al., 2017; Picariello et al., 2019; Schramek et al., 2014; Surcel et al., 2019). Since the contractility system is implicated in a much broader range of cellular behaviors, additional studies on the roles and functions of DscI in other cellular activities such as migration might lead to additional important findings. Finally, we, and the field, are increasingly finding that many sub-cellular systems are highly integrated, leading to the concept of the contractility machinery plugging into a grander mechanobiome that integrates with diverse cellular systems (Kothari et al., 2019a). Thus, elucidation of the molecular networks that drive this system will undoubtedly provide greater insights into normal cell behaviors as well as many diseases.
Materials and methods
Cell culture and maintenance
A complete list of strains generated and used in this study is provided in Table S1. Cells were grown at 22°C on 10-cm polystyrene plates in Hans’ enriched HL-5 media (1.4× HL-5 media with 8% final minimal media, penicillin, and streptomycin). WT-background strains were Ax2 and KAx3. Null mutants utilized were cortI, iqgap2, and iqgap1, and these strains were described in previous studies (Lee et al., 2010; Robinson and Spudich, 2000; Ruppel et al., 1994). Cells were maintained in exponential growth phase and split before reaching maximum confluency. Throughout our experimental designs, we made sure to seed control and mutant cells at the same density for each experiment since DscI expression is density-dependent (Clarke et al., 1987; Jain et al., 1992; Wetterauer et al., 1995).
Cells transformed with protein expression plasmids were grown in enriched 1.4× HL-5 media containing 15 μg/ml G418, 40 μg/ml hygromycin, or a combination of the drugs for double selection. For the agarose overlay assay, cells were maintained in MES buffer (50 mM MES, pH 6.8, 2 mM MgCl2, 0.2 mM CaCl2).
Reagents
Plasmids expressing the following proteins have been previously generated and described: GFP, mCherry, fusion GFP-mCherry, fusion GFP- and/or mCherry-tagged DscIA, CortI, IQGAP1, IQGAP2, and MyoII (Effler et al., 2006; Kee et al., 2012; Lee et al., 2010; West-Foyle et al., 2018; Zhou et al., 2010). For dsc1A plasmid generation, dsc1A coding sequence obtained from GFP-tagged dsc1A pDM181 plasmid was cloned into the pDM181 plasmid between the Sal I and Not I sites. gRNA sequence used for dsc1 gene CRISPR/Cas9 knockout is 5′-AGCATTTATGTGTGTTGCTCTCCA-3′. The transcript level of dsc1A was quantified using qRT-PCR with the forward primer 5′-ATGTCTACCCAAGGTTTAGT-3′ and the reverse primer 5′-AAACCACAACCAGCAACAATGTAT-3′. Both forward and reverse primers recognize regions that are identical in all three dsc1 paralogs (dsc1A, C, and D). The monoclonal mouse antibody 80-52-13 against DscI (cAMP binding domain) and the monoclonal mouse antibody 241-438-1 against cortI, both developed by Günther Gerisch (Max Planck Institute), were obtained from the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA; Stadler et al., 1984). DscI antibody was used at 1:1,000 dilution for Western analysis and at 1:100 dilution for immunocytochemistry. CortI antibody was used at 1:300 dilution for immunocytochemistry. The monoclonal mouse antibody My6 against MyoII heavy chain developed by James Spudich was used at 1:1,000 dilution for immunocytochemistry (Flicker et al., 1985).
RNA isolation and qRT-PCR
Cells were harvested at 5 × 106 cells/ml, and the cell count was determined. Cells were then spinned at 2,000 rpm for 3 min and lysed with 1 ml TRIzol reagent (15596026; Thermo Fisher Scientific) per 1 × 107 cells. 1.6 ml of chloroform was added per 8 ml of TRIzol used, and samples were incubated at room temperature for 3 min. After spinning for 15 min at 3,000 rcf, the top aqueous phase was collected, and RNA was precipitated using isopropyl alcohol. RNA pellets were washed once in 75% ethanol, air-dried, and stored in RNase-free water for qRT-PCR analysis.
qRT-PCR was performed using Verso 1-step RT-qPCR kit (AB4104A). All primer sequences for qRT-PCR analysis are included below. All experiments were conducted on a BIO-RAD CFX Opus 96 Real-Time PCR system. The program used was the following: Reverse Transcription—50°C for 15 min, 95°C for 15 min; PCR—95°C for 15 s, 60°C for 30 s, 72°C for 30 s; melt curve—95°C for 30 s, 60°C for 30 s, and then increased by 0.5°C per 10 s.
Relative quantification of normalized RNA levels was performed using the ΔΔCt method, in which the differences in Ct value between target genes and the control gene GAPDH for each cell line were calculated, and then normalized against WT control cell line.
qRT-PCR primers (used at 200 nM):
For IQGAP1: Forward: 5′-GAAGTCGGTGAACCAGAAGA-3′ and reverse: 5′-CACCTTTGGCAACGAGTGAA-3′.
For IQGAP2: Forward: 5′-GGTTGGGGTAGTGGTATG-3′ and reverse: 5′-CACCGACACACAAGTTAC-3′.
For Dsc1: Forward: 5′-ATGTCTACCCAAGGTTTAGT-3′ and reverse: 5′-AAACCACAACCAGCAACAATGTAT-3′.
For MyoII: Forward: 5′-GCCGATAAGAGTAAGAAGAC-3′ and reverse: 5′-GGATTGTTCAGCTTCAGATT-3′.
For CortI: Forward: 5′-GGCTCGTATGGAACTCAGAG-3′ and reverse: 5′-AAGAGACCAAGACCTTGAGC-3′.
For GAPDH: Forward: 5′-CCCAGAATGTAGAGTTTTAG-3′ and reverse: 5′-CTACAATGTATTCAGCACC-3′.
Plasmid transformation and exogenous protein expression
Cells were harvested during the exponential growth phase on plates and washed once with ice-cold E-Pore buffer. Cells are then suspended in cold E-Pore buffer at a concentration of 0.5–1.0 × 107 cells/ml. A mixture containing 1–4 µg of plasmid and 350 µl of suspended cells was transferred to a cold 4-mm electroporation cuvette. Electroporation was performed by one pulse using Genepulser-II electroporator (Bio-Rad) at 1.1 kV and 3.0 µF capacitance. Pulsed cells were transferred to a Petri dish containing 10 ml of cold Hans’ enriched HL-5 media. After 8–16 h, cells were placed in corresponding drug media for transformant selection. Colonies of transformants should appear around 6 d after electroporation. Protein expression was verified using fluorescence imaging and/or Western analysis.
CRISPR/Cas9 to generate KO mutants
A detailed protocol for using CRISPR/Cas9 system to target specific genes and generate KO mutants in D. discoideum is described in Sekine et al. (2018). gRNA guide sequences targeting dsc1 gene were designed using Cas-Designer. The gRNA sequence is detailed in the Reagents section. Customized gRNA oligos were obtained from Integrated DNA Technologies. This selected gRNA sequence is complementary to a 24 nt-long dsc1 coding region near the 3′ end that is identical in all three dsc1 paralogs (dsc1A, C, and D). gRNA sequence was then cloned into pTM1285 plasmid obtained from NBRP Nenkin.
Background cells were transformed with pTM1285 plasmid containing dsc1 gRNA using electroporation, using the transformation protocol described above. After 8–16 h of transformation, we replaced the media with Hans’ enriched HL-5 media containing 15 µg of G418 and cultured the transformed cells for 3 more days. After that, floating dead cells were washed off with Hans’ enriched HL-5 media. Remaining live cells were plated on SM-5 plates with cultured Klebsiella aerogenes and incubated at room temperature for 3–4 d until visible, isolated plaques formed. These individual plaques were separately transferred to 48-well plates containing Hans’ enriched HL-5 media in the absence of G418. After approximately 2–3 d when colonies grew, they were transferred to 10-cm Petri dishes. Since the cells should be expressing Cas9 and gRNA only transiently, it was important to test a subpopulation of these cells for G418 sensitivity to ensure the absence of pTM1285 plasmid. Retainment of the plasmid can potentially induce off-target gene modifications.
Suspension growth assay
Cells were seeded in 10 ml Hans’ enriched HL-5 media in 150-ml Erlenmeyer flasks at a starting cell concentration of 1.0 × 105 cells/ml. The flasks were rotated on a shaker at 180 rpm in a 22°C incubator. Cell concentration was measured and recorded roughly every 24 h using a hemocytometer. It is important to note that because of the time lag required for the winner cells to populate the cultures, we normally acquire and analyze data from a second cycle of suspension culture. As cells reached the later point of exponential growth phase, they were split down to a concentration of 0.5 − 1 × 105 cells/ml. Afterwards, the cell density continued to be measured and recorded roughly every 24 h (the exact time was recorded).
Multinucleation assay
Cells that have been in suspension at 180 rpm for 2–3 d were seeded at ∼70% confluency on glass coverslips in Hans’ enriched HL-5 media and allowed to adhere for 15–30 min. Next, media was removed, and cells were fixed with ice-cold fixative solution (2% PFA + 0.01% Triton X-100 in 1.5× HL-5 media) for 10 min. They were then washed quickly with 1× PBS, and the nuclei were stained with 10 µg/ml Hoechst for 10 min. Afterwards, cells were washed three times, 10 min each time, with 1× PBT (1× PBS + 0.01% Triton X-100). Before visualization, cells were mounted on coverslips using Invitrogen Prolong Diamond Antifade Mountan. Cells were imaged using a MolDev ImageXpress High-content Imager, equipped with a 10× dry objective (NA 0.25), and the number of nuclei per cell was recorded for 100–200 cells.
MPA, effective cortical tension, and mechanoresponsiveness quantification
For the mechanoresponsiveness measurement, cells at exponential growth phase were slowly aspirated to ∼0.80 nN/μm2 and held for at least 100 s. For quantification, the ratio of the background-corrected mean signal intensity of the cortex inside the pipette (Ip) to that of the opposite cortex outside the pipette (Io) was calculated.
Western blot quantification
Nitrocellulose membranes were imaged using 700- and 800-nm laser channels with the auto-exposure feature on Li-COR ODYSSEY CLx system. The blot images contain 16-bit data, allowing for a wide dynamic range in the signal. A rectangular shape was drawn around each protein band, and these bands were quantified using the integrated density measurement and normalized against the background signal. These values were then normalized against the corresponding Dynacortin band values. In some experiments, the normalized protein expression values were normalized against the respective control, typically a WT parental control, and plotted on a dotplot.
Agarose compression assay
Thin sheets of 2% agarose gel in MES buffer (recipe above) were prepared as described previously (Kee et al., 2012; Miao et al., 2017). Agarose sheets were cut into small pieces that fit inside an 8-well chamber. Cells were seeded at an equal density of 1 × 106 cells/ml in 8-well imaging chambers, which led to ∼70% confluency. Cells were then allowed to adhere for 30 min. Cell media was replaced with MES buffer. Afterwards, the agarose sheet was carefully placed inside the chamber on top of the cells. MES buffer was slowly aspirated from the chamber, allowing the agarose sheet to gradually press on the cells. Cells were imaged using an Olympus IX71 microscope, equipped with a 40× (NA 1.30) oil objective, at room temperature, immediately to avoid evaporation and any further complications caused by extended compression. Images were acquired with DIC and GFP channels using Metamorph software, and then analyzed using ImageJ.
Immunocytochemistry
Cells were seeded at an equal density of 1 × 106 cells/ml on a detachable 8-well chamber slide and allowed to adhere for 30 min. Media was removed and cells were fixed in −20°C methanol for 3 min. Residual methanol was quickly washed with 1× PBT (1× PBS + 0.05% Triton X-100), and cells were blocked in blocking buffer (3% BSA in 1× PBS) for 1 h at room temperature. Proteins of interest were detected using corresponding primary antibodies diluted in blocking buffer for 1–3 h at room temperature. Afterwards, cells were washed three times with 1× PBT (1× PBS + 0.05% Triton X-100), followed by room temperature incubation with appropriate secondary antibodies for 1 h. For nuclei detection, cells were washed once with 1× PBS and stained with 1 µg/ml Hoescht for 10 min. Cells were then washed three times, 10 min each time, with 1× PBT. Before visualization, cells were mounted on coverslips using Invitrogen Prolong Diamond Antifade Mountant.
FCCS
Co-immunoprecipitation of GFP-DscIA
Approximately 3 × 107 logarithmically growing cells were harvested, washed once in 1× PBS, and resuspended in 300 µl of freshly made lysis buffer (100 mM PIPES, pH 6.8, 2.5 mM EGTA, 1 mM MgCl2, 1 mM ATP, 0.1% Triton X-100, 100 mM NaCl, and protease inhibitor cocktail). Cell lysates were incubated on ice for 30 min, with gentle inversion every 10 min. Lysates were then centrifuged at 15,000 g for 5 min at 4°C. The supernatant was diluted with 200 µl dilution buffer (100 mM PIPES, pH 6.8, 2.5 mM EGTA, 1 mM MgCl2, 1 mM ATP, 100 mM NaCl, and protease inhibitor cocktail). 50 µl of the diluted lysates were saved as Input. Diluted lysates were then transferred to tubes containing 25 µl of GFP-trap agarose beads (Chromotek gta-10) and rotated at 4°C for 1 h. These beads were previously equilibrated according to the manufacturer’s instructions. Afterwards, beads were sedimented by centrifugation at 2,500 g for 5 min. 50 µl of the supernatant was saved as flow-through and the rest was discarded. Beads were washed two times in 500 µl wash buffer (dilution buffer + 0.01% Triton X-100). Beads were then resuspended in 70 µl 2× sample suffer plus 10% β-mercaptoethanol and boiled at 95°C for 5 min. Beads were sedimented at 2,500 g for 2 min, and samples were transferred to another tube. Approximately 30 µl of dilution buffer was added to final samples to bring the sample buffer to 1×. 50 µl 2× sample buffer plus 10% β-mercaptoethanol was added to each input and flow-through fraction and boiled at 95°C for 5 min. 20 µl of each sample was loaded on a Western gel for analysis.
Statistical analysis
Data sets that appeared to be normally distributed (but this was not formally tested) were statistically analyzed using ANOVA followed by Fisher’s least significant difference (LSD) test. Calculated P values that are lower than or equal to 0.05 were considered to be statistically significant. Data sets that were skewed and/or contained significant outliers were statistically analyzed using the Kruskal–Wallis test followed by the Mann-Whitney-Wilcoxon test. P values are either provided in each graph or denoted as *, P < 0.05; **, P < 0.001; ***, P < 0.0001. The above statistical tests were performed using the statistical test functions provided by Kaleidagraph. For data sets that considered differences in proportions (fractions), the Comparison of Proportions test was used (http://medcalc.org).
Online supplemental material
Supplemental materials include Figs. S1, S2, S3, S4, and S5, Tables S1 and S2, and Videos 1, 2, 3, 4, and 5. Fig. S1 shows cytokinetic defects in dsc1 null mutants generated in the Ax2 background. Fig. S2 shows exogenous expression of DscIA rescues cytokinetic defects of dsc1 null mutants. Fig. S3 shows the absence of DscI in the cell has no effect on the cortical localization of Myosin II and IQGAP2. Fig. S4 shows that DscI facilitates CortI’s cleavage furrow accumulation. Fig. S5 shows DscIA has antagonistic genetic interactions with CortI and IQGAP1 and synergistic genetic interactions with IQGAP2. Table S1 provides a summary of all D. discoideum cell strains used in this study. Table S2 provides growth rates of background and null-complemented cell lines used in the genetic interaction assays. Videos 1, 2, 3, 4, and 5 show dividing WT (KAx3), dsc1 null, dsc1:GFP-DscIA, iqgap2 null, and iqgap2; dsc1 null cells, respectively.
Data availability
Cell lines and reagents generated in this study will be made available to the scientific community upon request.
Acknowledgments
We thank the members of the Robinson lab for helpful discussions and feedback. We also thank the Johns Hopkins University Microscope Facility, particularly Barbara Smith and Hoku West-Foyle, for providing equipment and technical assistance for FCCS experiments.
This work was supported by National Institutes of Health grant R01 GM66817.
D.N. Robinson is exploring forming a biotech company. No other disclosures were reported.
Author contributions: L.T.S. Nguyen designed the studies, carried out experiments, conducted image and data analyses, performed statistical tests, and drafted the manuscript. D.N. Robinson helped design the study, analyzed data, and edited the manuscript. Both authors read and approved the final manuscript.

