


Spatiotemporally dynamic microtubule acetylation underlies diverse physiological and pathological events. Despite its ubiquity, the molecular mechanisms that regulate the sole microtubule acetylating agent, α-tubulin-N-acetyltransferase-1 (α-TAT1), remain obscure. Here, we report that dynamic intracellular localization of α-TAT1 along with its catalytic activity determines efficiency of microtubule acetylation. Specifically, we newly identified a conserved signal motif in the intrinsically disordered C-terminus of α-TAT1, consisting of three competing regulatory elements—nuclear export, nuclear import, and cytosolic retention. Their balance is tuned via phosphorylation by CDK1, PKA, and CK2, and dephosphorylation by PP2A. While the unphosphorylated form binds to importins and resides both in cytosol and nucleus, the phosphorylated form binds to specific 14-3-3 adapters and accumulates in the cytosol for maximal substrate access. Unlike other molecules with a similar phospho-regulated signal motif, α-TAT1 uniquely uses the nucleus as a hideout. This allosteric spatial regulation of α-TAT1 function may help uncover a spatiotemporal code of microtubule acetylation in normal and aberrant cell behavior.
Introduction
Acetylation of lysine-40 of α-tubulin is an evolutionarily conserved posttranslational modification in Eukarya (L’Hernault and Rosenbaum, 1985; Diggins and Dove, 1987; Janke and Montagnac, 2017), which is involved in diverse physiological and pathological states (Schatten et al., 1988; Boggs et al., 2015; Esteves et al., 2019). Acetylation is mainly observed in polymerized microtubules (L’Hernault and Rosenbaum, 1983; Maruta et al., 1986; Piperno et al., 1987) and may provide structural flexibility to stabilize microtubules against bending forces (Sudo and Baas, 2010; Portran et al., 2017; Xu et al., 2017; Eshun-Wilson et al., 2019). In cultured cells, microtubule acetylation mediates focal adhesion dynamics, adaptation to extracellular matrix rigidity, as well as regulation of tissue stiffness (Joo and Yamada, 2014; Bance et al., 2019; Swiatlowska et al., 2020; Seetharaman et al., 2021; Coleman et al., 2021). Additionally, acetylated microtubules regulate touch sensation in Mus musculus, Caenorhabditis elegans, and Drosophila melanogaster, suggesting a role in mechano-response (Zhang et al., 2002; Shida et al., 2010; Morley et al., 2016; Yan et al., 2018). Microtubule acetylation has been implicated in axonal transport in neurons (Lin et al., 2017; Fourcade et al., 2017; Morelli et al., 2018), migration in cancer cells (Boggs et al., 2015; Lee et al., 2018; Castro-Castro et al., 2012; Oh et al., 2017), autophagy (Esteves et al., 2019; Geeraert et al., 2010; McLendon et al., 2014), podosome stabilization in osteoclasts (Destaing et al., 2005), and viral infections (Elliott and O’Hare, 1998; Husain and Harrod, 2011; Sabo et al., 2013; Zan et al., 2017). α-tubulin-N-acetyltransferase-1 (α-TAT1) is the only known acetyltransferase for α-tubulin (Kalebic et al., 2013b; Kim et al., 2013) in mammals. α-TAT1 predominantly catalyzes α-tubulin in stable polymerized microtubules (Shida et al., 2010; Szyk et al., 2014) and may have additional effects on microtubules independent of its catalytic activity (Kalebic et al., 2013a).
Although microtubule acetylation is spatiotemporally regulated downstream of many molecular signaling pathways, little is known about how these pathways converge on α-TAT1 to achieve such dynamic patterns. In the present study, we used computational sequence analyses and live cell microscopy to identify a conserved motif in the intrinsically disordered C-terminus of α-TAT1, consisting of a nuclear export signal (NES) and a nuclear localization signal (NLS), that mediates its spatial distribution. We show that cytosolic localization of α-TAT1 is critical for microtubule acetylation and that nuclear localization of α-TAT1 is inhibited by serine-threonine kinases, cyclin-dependent kinase 1 (CDK1), PKA, and casein kinase 2 (CK2), and promoted by phosphatase 2A (PPP2CA). We also identified specific 14-3-3 isoforms as binding partners of α-TAT1. Our findings establish a novel regulatory role of the intrinsically disordered C-terminus in controlling α-TAT1 function by its intracellular localization downstream of kinase and phosphatase activities.
Results
α-TAT1 localization mediates microtubule acetylation
α-TAT1 has an N-terminal catalytic domain that shows homology to other acetyltransferases, while its C-terminus was not resolved in crystal structures (Friedmann et al., 2012; Fig. 1 a). Based on its amino acid sequence (Fig. S1 a), α-TAT1 C-terminus was predicted to be intrinsically disordered by both IUPred2A (Mészáros et al., 2018) and PrDOS (Ishida and Kinoshita, 2007) prediction servers (Fig. 1 a and Fig. S1 b). To explore if the intracellular localization of α-TAT1 is regulated, we used the α-TAT1 amino acid sequence (Fig. S1 a) with the prediction program NetNES (la Cour et al., 2004) and identified a putative NES in α-TAT1 C-terminus (Fig. 1 a and Fig. S1 c). Additionally, PSORT-II (Horton et al., 2007) subcellular localization program predicted a putative class 4 NLS (Kosugi et al., 2009) in its C-terminus (Fig. 1 a). The putative NES and NLS are conserved in all human α-TAT1 isoforms as well as in mammalian α-TAT1 (Fig. S1, d and e), but not in C. elegans (Fig. S1 f). To examine any intracellular distribution pattern of α-TAT1, we expressed mVenus-α-TAT1 in HeLa cells and observed nuclear exclusion in most, but not all, cells (Fig. 1 b). Based on visual categorization, ratiometric, and colocalization correlation analyses (see Materials and methods; Fig.S2 a), we determined that nearly 80% of cells showed cytosolic distribution and 20% showed lack of nuclear exclusion (diffused pattern or nuclear enrichment) of mVenus-α-TAT1 (Fig. 1, c–e). While we used a region of interest (ROI)–based ratiometric approach (see Materials and methods), which may have subjective variation based on the position of the ROI, the nuc/cyto ratio from ROIs showed a strong correlation with nuc/cyto ratios from whole cell ratiometric approach (Fig. S2 b). In addition, unlike the ratiometric approach, both the categorical and colocalization analyses take the whole cell signal into account, and the consistent pattern of distribution across all three approaches demonstrates the robustness of our observation. The nuc/cyto ratio did not show a strong correlation with mVenus-α-TAT1 expression levels (Fig. S2 c), suggesting that the intracellular localization was not an artifact of exogenous expression of mVenus-α-TAT1. Both the nuc/cyto ratio values and Pearson’s R colocalization values broadly showed a normal distribution (Fig. S2 d). Immunostaining of mouse embryonic fibroblasts (MEFs) with an anti–α-TAT1 antibody showed a predominantly cytosolic signal (Fig. 1 f and Fig. S2 e, left panels), suggesting that endogenous α-TAT1 also showed nuclear exclusion. MEFs from α-TAT1 knock-out (KO) mice (Aguilar et al., 2014) did not show any signal (Fig. S2 e, right panels), demonstrating specificity of the antibody. Time-lapse microscopy showed spontaneous changes in fluorescence intensity of mVenus-α-TAT1 in cell nuclei and cytosol (Fig. S2, f and g), suggesting that α-TAT1 localization is dynamic.

Intracellular distribution of α-TAT1 mediates its function. (a–i) (a) Cartoon showing α-TAT1 with predicted NES and NLS in intrinsically disordered C-terminus, adapted from PDB accession no. 4GS4; (b) intracellular distribution of mVenus-α-TAT1; red dotted lines outline nuclei as identified by H2B-mCherry (cytosolic and diffused) or DAPI (nucleus enriched) in lower panel; (c) categorical analysis (318 cells; Nuc: Nucleus enriched, Diff: Diffused, Cyto: Cytosolic); (d) ratiometric analysis (184 cells) and (e) colocalization analysis (180 cells) of mVenus-α-TAT1 localization in HeLa cells; (f) immunostaining with anti–α-TAT1 antibody showing intracellular localization of endogenous α-TAT1 in MEF cells; red dotted lines outline nuclei as identified by DAPI; (g) cartoon showing α-TAT1 mutants used in (h) immunofluorescence assays showing levels of acetylated α-tubulin and total α-tubulin, transfected cells are indicated with red arrowheads; (i) ratio of acetylated α-tubulin to total tubulin intensities with exogenous expression of α-TAT1 and its mutants, normalized against that of non-transfected cells (WT: 50, catalytic domain: 44, NLS-catalytic domain: 48 cells). Scale bar = 10 µm. ***, P < 0.001 or as shown, Student’s t test.
Intracellular distribution of α-TAT1 mediates its function. (a–i) (a) Cartoon showing α-TAT1 with predicted NES and NLS in intrinsically disordered C-terminus, adapted from PDB accession no. 4GS4; (b) intracellular distribution of mVenus-α-TAT1; red dotted lines outline nuclei as identified by H2B-mCherry (cytosolic and diffused) or DAPI (nucleus enriched) in lower panel; (c) categorical analysis (318 cells; Nuc: Nucleus enriched, Diff: Diffused, Cyto: Cytosolic); (d) ratiometric analysis (184 cells) and (e) colocalization analysis (180 cells) of mVenus-α-TAT1 localization in HeLa cells; (f) immunostaining with anti–α-TAT1 antibody showing intracellular localization of endogenous α-TAT1 in MEF cells; red dotted lines outline nuclei as identified by DAPI; (g) cartoon showing α-TAT1 mutants used in (h) immunofluorescence assays showing levels of acetylated α-tubulin and total α-tubulin, transfected cells are indicated with red arrowheads; (i) ratio of acetylated α-tubulin to total tubulin intensities with exogenous expression of α-TAT1 and its mutants, normalized against that of non-transfected cells (WT: 50, catalytic domain: 44, NLS-catalytic domain: 48 cells). Scale bar = 10 µm. ***, P < 0.001 or as shown, Student’s t test.

The intrinsically disordered C-terminus of αTAT1 has a putative NES and NLS. (a–f) (a) Amino acid sequence of αTAT1 isoform 5 used as the query; the putative NES and NLS are shown in bold; (b) Anchor2, IUPred2, and PrDOS prediction suggesting that αTAT1 C-terminus is disordered; (c) NetNES prediction for αTAT1 showing putative NES; (d) sequence alignment of NES in human αTAT1 isoforms (upper panel) and in mammalian αTAT1; (e) sequence alignment of NLS in human αTAT1 isoforms (upper panel) and in mammalian αTAT1; (f) sequence alignment of human and C. elegans αTAT1; putative 14-3-3 binding residues in the C. elegans αTAT1 are indicated in red boxes; NES and NLS sequences in the human isoform are underlined.
The intrinsically disordered C-terminus of αTAT1 has a putative NES and NLS. (a–f) (a) Amino acid sequence of αTAT1 isoform 5 used as the query; the putative NES and NLS are shown in bold; (b) Anchor2, IUPred2, and PrDOS prediction suggesting that αTAT1 C-terminus is disordered; (c) NetNES prediction for αTAT1 showing putative NES; (d) sequence alignment of NES in human αTAT1 isoforms (upper panel) and in mammalian αTAT1; (e) sequence alignment of NLS in human αTAT1 isoforms (upper panel) and in mammalian αTAT1; (f) sequence alignment of human and C. elegans αTAT1; putative 14-3-3 binding residues in the C. elegans αTAT1 are indicated in red boxes; NES and NLS sequences in the human isoform are underlined.

αTAT1 undergoes dynamic nuclear transport. (a–g) (a) Strategies to quantify intracellular localization of mVenus-α-TAT1; (b) ratiometric analysis with ROI or whole cell values for the same 24 cells (left panel), and their Pearson's correlation (right panel), ***, P < 0.001; (c) Nuc/Cyto ratio of mVenus-αTAT1 versus expression levels in transiently transfected HeLa cells (n = 184 cells); (d) normal probability plot of Nuc/Cyto (upper panel, n = 184 cells) and Pearson’s R value (lower panel, n = 180 cells); (e) immunostaining with anti–α-TAT1 antibody in MEF cells from WT and α-TAT1 KO mice; nuclei (lower panel) are indicated with red arrowheads; (f) spontaneous temporal changes in mVenus-α-TAT1 distribution; (g) temporal changes in nuclear fluorescence intensity of mVenus-α-TAT1 in HeLa cells; each line indicates normalized nuclear intensity of mVenus- α-TAT1 in a single cell; n = 20 cells. Scale bar = 10 µm.
αTAT1 undergoes dynamic nuclear transport. (a–g) (a) Strategies to quantify intracellular localization of mVenus-α-TAT1; (b) ratiometric analysis with ROI or whole cell values for the same 24 cells (left panel), and their Pearson's correlation (right panel), ***, P < 0.001; (c) Nuc/Cyto ratio of mVenus-αTAT1 versus expression levels in transiently transfected HeLa cells (n = 184 cells); (d) normal probability plot of Nuc/Cyto (upper panel, n = 184 cells) and Pearson’s R value (lower panel, n = 180 cells); (e) immunostaining with anti–α-TAT1 antibody in MEF cells from WT and α-TAT1 KO mice; nuclei (lower panel) are indicated with red arrowheads; (f) spontaneous temporal changes in mVenus-α-TAT1 distribution; (g) temporal changes in nuclear fluorescence intensity of mVenus-α-TAT1 in HeLa cells; each line indicates normalized nuclear intensity of mVenus- α-TAT1 in a single cell; n = 20 cells. Scale bar = 10 µm.
α-TAT1 preferentially acetylates polymerized microtubules, which are typically cytosolic. Based on this, we hypothesized that spatial regulation of α-TAT1 may control its function. Exogenous expression of mVenus-α-TAT1 or its catalytic domain (residues 1–236; Shida et al., 2010; Friedmann et al., 2012; Fig. 1 g) was sufficient to significantly increase α-tubulin acetylation in HeLa cells compared to non-transfected cells (Fig. 1, h and i). To test whether nuclear localization may sufficiently sequester α-TAT1 from microtubules, we tethered the NLS from cMyc to α-TAT1 catalytic domain and thus localized it to the nucleus (Fig. 1, g and h top panel). Exogenous expression of NLS-mVenus-α-TAT1(1-236) did not increase α-tubulin acetylation levels compared to non-transfected cells (Fig. 1, h and i), suggesting that nuclear sequestration of α-TAT1 inhibits its ability to acetylate microtubules.
α-TAT1 undergoes exportin 1 (Exp1) dependent nuclear export
Since α-TAT1 was predominantly cytosolic, we speculated that it is actively exported out of the nucleus and/or that α-TAT1 binds to a protein that keeps the complex out of the nucleus. We began testing the first possibility by assessing involvement of the nuclear export machinery. Exp1 mediates nuclear export of many proteins (Kutay and Güttinger, 2005; Hutten and Kehlenbach, 2007). GFP-α-TAT1, but not GFP, coimmunoprecipitated with endogenous Exp1 (Fig. 2 a), indicating an interaction between these two proteins. Treatment with 100 nM Leptomycin-B (LMB), an inhibitor of Exp1 mediated nuclear export (Sun et al., 2013), significantly decreased nuclear exclusion of mVenus-α-TAT1 compared with vehicle (Fig. 2, b–e). LMB treatment induced significant reduction in α-tubulin acetylation levels in HeLa cells within 4 h compared to vehicle (Fig. 2, f and g). Inhibition of nuclear export was initiated within an hour of LMB treatment (Fig. 2, h and i), although some cells were refractory to the treatment (Fig. 2 i). Decreased LMB concentrations (1 and 10 nM) had a comparable impact as 100 nM dosage (Fig. 2, j and k). Furthermore, immunostaining with anti–α-TAT1 antibody showed an increase in nuclear localization of α-TAT1 in MEFs treated with 100 nM LMB compared with those treated with vehicle (Fig. 2 l). Taken together, our data suggest that α-TAT1 undergoes nuclear export in an Exp1-dependent manner and that this export facilitates α-TAT1 function. The residual exclusion of α-TAT1 could be due to Exp1-independent nuclear export pathways or association with other cytosolic proteins, which was further investigated below.
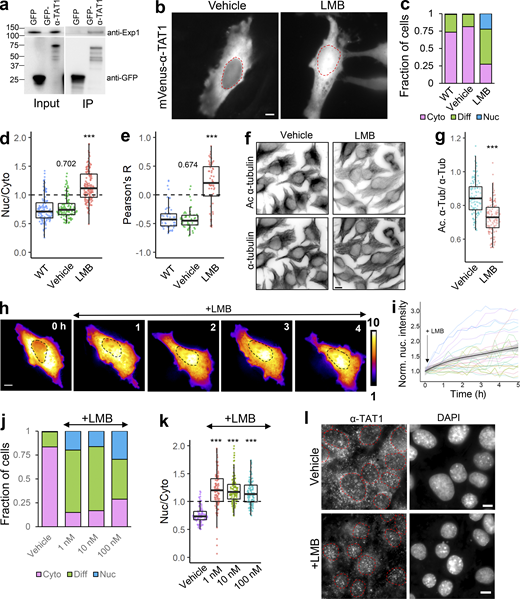
α-TAT1 undergoes Exp1 mediated nuclear export. (a–l) (a) Coimmunoprecipitation of endogenous Exp1 with GFP-α-TAT1 (molecular weights in kD are indicated); (b) intracellular distribution of mVenus-α-TAT1 with vehicle (EtOH) and 100 nM LMB treatment; nuclei are indicated in red dotted lines; (c) categorical analysis (WT: 263, vehicle: 300, LMB: 255 cells); (d) ratiometric analysis (WT: 125, vehicle: 116, LMB: 163 cells) and (e) colocalization analysis (WT: 55, vehicle: 54, LMB: 52 cells) of mVenus-α-TAT1 localization with vehicle and LMB treatment; (f) immunofluorescence images showing acetylated and total α-tubulin in HeLa cells with vehicle or LMB treatment; (g) ratio of acetylated to total α-tubulin with vehicle or LMB treatment (vehicle:120, LMB: 130 cells); (h) changes in intracellular localization of mVenus-α-TAT1 on LMB treatment; (i) changes in nuclear intensity of mVenus-α-TAT1 on LMB treatment (black line shows the mean and gray line indicate the 95% confidence interval; n = 26); (j) categorical analysis (vehicle: 274, LMB 1 nM: 329, LMB 10 nM: 291, LMB 100 nM: 295 cells) and (k) ratiometric analysis of mVenus-α-TAT1 localization on vehicle (103) or LMB treatment (1 nM: 121, 10 nM: 122 and 100 nM: 103 cells); (l) immunostaining with anti–α-TAT1 antibody showing intracellular localization of endogenous α-TAT1 in MEF cells on treatment with vehicle (EtOH) or 100 nM LMB; red dotted lines outline nuclei as identified by DAPI. Scale bar = 10 µm. ***, P < 0.001 or as shown, Student’s t test. For categorical analyses, Nuc: Nucleus enriched, Diff: Diffused, Cyto: Cytosolic. Source data are available for this figure: SourceData F2.
α-TAT1 undergoes Exp1 mediated nuclear export. (a–l) (a) Coimmunoprecipitation of endogenous Exp1 with GFP-α-TAT1 (molecular weights in kD are indicated); (b) intracellular distribution of mVenus-α-TAT1 with vehicle (EtOH) and 100 nM LMB treatment; nuclei are indicated in red dotted lines; (c) categorical analysis (WT: 263, vehicle: 300, LMB: 255 cells); (d) ratiometric analysis (WT: 125, vehicle: 116, LMB: 163 cells) and (e) colocalization analysis (WT: 55, vehicle: 54, LMB: 52 cells) of mVenus-α-TAT1 localization with vehicle and LMB treatment; (f) immunofluorescence images showing acetylated and total α-tubulin in HeLa cells with vehicle or LMB treatment; (g) ratio of acetylated to total α-tubulin with vehicle or LMB treatment (vehicle:120, LMB: 130 cells); (h) changes in intracellular localization of mVenus-α-TAT1 on LMB treatment; (i) changes in nuclear intensity of mVenus-α-TAT1 on LMB treatment (black line shows the mean and gray line indicate the 95% confidence interval; n = 26); (j) categorical analysis (vehicle: 274, LMB 1 nM: 329, LMB 10 nM: 291, LMB 100 nM: 295 cells) and (k) ratiometric analysis of mVenus-α-TAT1 localization on vehicle (103) or LMB treatment (1 nM: 121, 10 nM: 122 and 100 nM: 103 cells); (l) immunostaining with anti–α-TAT1 antibody showing intracellular localization of endogenous α-TAT1 in MEF cells on treatment with vehicle (EtOH) or 100 nM LMB; red dotted lines outline nuclei as identified by DAPI. Scale bar = 10 µm. ***, P < 0.001 or as shown, Student’s t test. For categorical analyses, Nuc: Nucleus enriched, Diff: Diffused, Cyto: Cytosolic. Source data are available for this figure: SourceData F2.
Nuclear export of α-TAT1 is mediated by a C-terminal NES
Our data demonstrate that α-TAT1 function is linked to Exp1 mediated nuclear export. To examine whether nuclear export of α-TAT1 is regulated by its catalytic activity, we expressed a catalytic dead mutant, mVenus-α-TAT1(D157N) (Friedmann et al., 2012), or the catalytic domain, mVenus- α-TAT1(1-236), in HeLa cells. mVenus-α-TAT1(D157N) did not display any loss of nuclear exclusion, whereas mVenus-α-TAT1(1-236) displayed a complete loss of nuclear exclusion (Fig. 3, a–e). The C-terminus, mVenus-α-TAT1(236-323), displayed a distribution pattern comparable to that of WT (Fig. 3, a–e). In addition, LMB treatment significantly reduced the nuclear exclusion of mVenus-α-TAT1 C-terminus (Fig. 3, f and g). Exclusion of α-TAT1 C-terminus (size ≈ 38 kD) further indicates that nuclear exclusion of mVenus-α-TAT1 (size ≈ 63 kD) is not simply due to size exclusion of passive diffusion into nuclei and demonstrates that nuclear exclusion of α-TAT1 is a transferable property mediated by its C-terminus. Since the catalytic dead mutant showed reduced nuclear localization, while the catalytic domain showed increased nuclear enrichment, we further examined any role of catalytic activity in nuclear localization by using a dead catalytic domain. mVenus-α-TAT1(1–236) and mVenus-α-TAT1(1–236, D157N) showed virtually identical distribution patterns (Fig. 3, a, b, h, and i), suggesting that α-TAT1 catalytic activity does not mediate its localization.

Intracellular distribution of α-TAT1 is mediated by its C-terminal NES. (a–i) (a) Cartoon showing α-TAT1 mutant design; (b) images showing intracellular distribution of indicated mVenus-α-TAT1 mutants; red dotted lines outline nuclei; (c) categorical analysis (WT: 257, Cat. Dead: 289, Cat. Dom: 271, C-term: 290, ΔNES: 243, and NES/A: 321 cells); (d) ratiometric analysis (WT: 103, Cat. Dead: 153, Cat. Dom: 145, C-term: 138, ΔNES: 124 and NES/A: 153 cells) and (e) colocalization analysis (WT: 68, Cat. Dead: 85, Cat. Dom: 65, C-term: 60, ΔNES: 62 and NES/A: 48 cells); (f) ratiometric analysis (vehicle: 93, LMB: 107 cells) and (g) colocalization analysis (vehicle: 98, LMB: 53) of intracellular distribution of mVenus-α-TAT1 C-term with vehicle or 100 nM LMB; (h) ratiometric analysis (Cat. Domain: 63, Dead Cat. Domain: 61 cells) and (i) colocalization analysis (Cat. Domain: 42, Dead Cat. Domain: 42 cells) of intracellular distribution of mVenus-α-TAT1 mutants. Scale bar = 10 µm. ***, P < 0.001 or as shown, Student’s t test. For categorical analyses, Nuc: Nucleus enriched, Diff: Diffused, Cyto: Cytosolic.
Intracellular distribution of α-TAT1 is mediated by its C-terminal NES. (a–i) (a) Cartoon showing α-TAT1 mutant design; (b) images showing intracellular distribution of indicated mVenus-α-TAT1 mutants; red dotted lines outline nuclei; (c) categorical analysis (WT: 257, Cat. Dead: 289, Cat. Dom: 271, C-term: 290, ΔNES: 243, and NES/A: 321 cells); (d) ratiometric analysis (WT: 103, Cat. Dead: 153, Cat. Dom: 145, C-term: 138, ΔNES: 124 and NES/A: 153 cells) and (e) colocalization analysis (WT: 68, Cat. Dead: 85, Cat. Dom: 65, C-term: 60, ΔNES: 62 and NES/A: 48 cells); (f) ratiometric analysis (vehicle: 93, LMB: 107 cells) and (g) colocalization analysis (vehicle: 98, LMB: 53) of intracellular distribution of mVenus-α-TAT1 C-term with vehicle or 100 nM LMB; (h) ratiometric analysis (Cat. Domain: 63, Dead Cat. Domain: 61 cells) and (i) colocalization analysis (Cat. Domain: 42, Dead Cat. Domain: 42 cells) of intracellular distribution of mVenus-α-TAT1 mutants. Scale bar = 10 µm. ***, P < 0.001 or as shown, Student’s t test. For categorical analyses, Nuc: Nucleus enriched, Diff: Diffused, Cyto: Cytosolic.
Exp1 typically binds with short stretches of hydrophobic, often leucine rich, NES (la Cour et al., 2004; Kutay and Güttinger, 2005) that are often found in disordered regions of the cargo proteins. As previously mentioned, NetNES suggested the presence of a conserved NES between V286 and L297, while a Hidden Markov Model predicted that the NES encompassed the residues between L282 and L297 in α-TAT1 C-terminus (Fig. S1 c). Interestingly, this region is also predicted to be a site of protein–protein interactions by ANCHOR2 prediction software (Fig. S1 b, left panel). Truncation of this putative NES, α-TAT1-ΔNES, abrogated its nuclear exclusion (Fig. 3, a–e). Alanine substitution of the hydrophobic residues in the NES predicted by NetNES Hidden Markov Method algorithm (Fig. S1 c) significantly reduced nuclear exclusion of α-TAT1 (Fig. 3, a–e), suggesting that these residues contribute to nuclear export of α-TAT1. Taken together, our data suggest that α-TAT1 has an NES in its C-terminus.
α-TAT1 has a phospho-inhibited NLS in its C-terminus
Our observations thus far with predominant nuclear exclusion of mVenus-α-TAT1 indicate that the putative NLS (P316AQRRRT322) is either non-functional or basally inhibited. NLS-mediated nuclear import is often phospho-regulated (Nardozzi et al., 2010). To identify putative phospho-sites in α-TAT1, we ran its amino-acid sequence through the NetPhos prediction server, which identified over 30 putative phospho-sites in α-TAT1 (Fig. S3 a). Nine of these residues have been reported to be phosphorylated in phospho-proteomic studies (Fig. S3 b). Since α-TAT1(1-284) did not display nuclear exclusion (Fig. 3, b–e), we reasoned that the phospho-sites which inhibit nuclear localization of α-TAT1 might be located between F285 and R323. NetPhos prediction server identified S294, T303, S315, and T322 as potential phospho-sites in this region (Fig. S3 a, red box), wherein only S315 and T322 have been reported to be phosphorylated (Fig. S3 b) and they also flank the putative NLS (Fig. 4 a). Importin-α binds with NLS enriched in basic residues through a charge-based interaction (Kosugi et al., 2009). Phosphorylation of amino acids adjacent to such an NLS may inhibit Importin-α binding by disrupting the charge balance in the NLS region (Harreman et al., 2004). Alanine substitution of T322, but not of S315, significantly increased nuclear localization of mVenus-α-TAT1 (Fig. 4, a–e). Alanine substitution of both S315 and T322, α-TAT1(ST/A), showed more nuclear localization than T322 alone (Fig. 4, a–e), suggesting that T322 phosphorylation inhibits nuclear localization of α-TAT1, while S315 may play a cooperative role. Substitution of S315 with acidic residues (S315D) appeared to boost nuclear exclusion of α-TAT1, whereas substitution of T322 with acidic residues (T322E) or both (ST/DE) displayed increased diffused pattern, but not increased nuclear accumulation (Fig. S3, c and d). This may be because these acidic residues, unlike phosphate moieties, do not sufficiently counter the basic residues in the NLS or that phospho-T322, and to a lesser extent, phospho-S315 phosphorylation may be involved in protein–protein interactions, such as with 14-3-3 adaptor proteins, which inhibit nuclear localization of α-TAT1. To confirm whether T322 is indeed phosphorylated, we performed a Western blot with an antibody against the phospho-peptide RRXS*/T* (* indicating phosphorylated residue) since T322 within the NLS (PAQRRRT) followed this motif. We observed anti-RRXS*/T* signal with GFP-α-TAT1 but not with GFP-α-TAT1(T322A; Fig. 4 f), suggesting that T322 is indeed phosphorylated.
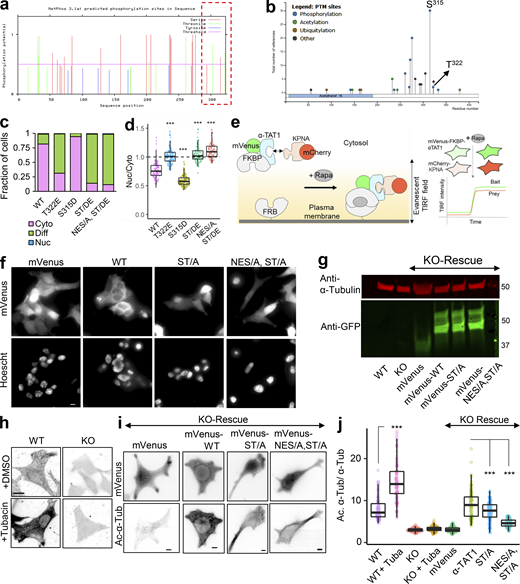
αTAT1 localization is mediated by its T322 residue. (a–j) (a) NetPhos predicted phosphosites (F285-R323 are highlighted in red box); (b) posttranslational modifications in α-TAT1 curated by PhosphoSitePlus; (c) categorical and (d) ratiometric analyses of indicated α-TAT1 mutants; (e) cartoon of live cell protein–protein interaction assay, Rapa: Rapamycin; (f) images showing intracellular localization of mVenus and mVenus-tagged α-TAT1 mutants; (g) Western blot showing expression levels of mVenus and mVenus-tagged α-TAT1 mutants in KO cells rescued with indicated constructs (molecular weights in kD are indicated); (h) acetylated α-tubulin levels in WT and KO cells, with or without Tubacin treatment; (i) mVenus and acetylated α-tubulin levels in KO-rescue cells; (j) ratio of acetylated to total α-tubulin in MEFs as indicated. Scale bar: 10 µm. ***, P < 0.001, Student’s t test. For categorical analyses, Nuc: Nucleus enriched, Diff: Diffused, Cyto: Cytosolic. Source data are available for this figure: SourceData FS3.
αTAT1 localization is mediated by its T322 residue. (a–j) (a) NetPhos predicted phosphosites (F285-R323 are highlighted in red box); (b) posttranslational modifications in α-TAT1 curated by PhosphoSitePlus; (c) categorical and (d) ratiometric analyses of indicated α-TAT1 mutants; (e) cartoon of live cell protein–protein interaction assay, Rapa: Rapamycin; (f) images showing intracellular localization of mVenus and mVenus-tagged α-TAT1 mutants; (g) Western blot showing expression levels of mVenus and mVenus-tagged α-TAT1 mutants in KO cells rescued with indicated constructs (molecular weights in kD are indicated); (h) acetylated α-tubulin levels in WT and KO cells, with or without Tubacin treatment; (i) mVenus and acetylated α-tubulin levels in KO-rescue cells; (j) ratio of acetylated to total α-tubulin in MEFs as indicated. Scale bar: 10 µm. ***, P < 0.001, Student’s t test. For categorical analyses, Nuc: Nucleus enriched, Diff: Diffused, Cyto: Cytosolic. Source data are available for this figure: SourceData FS3.
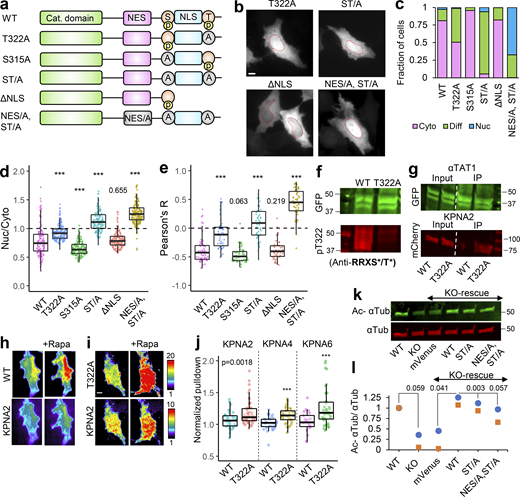
α-TAT1 has a C-terminal phospho-inhibited NLS. (a–l) (a) Cartoon showing α-TAT1 mutant design (S: S315 residue, T: T322 residue, A: Alanine, p: phosphate moiety); (b) intracellular distribution of mVenus-α-TAT1 mutants as indicated; nuclei are outlined in red dotted lines; (c) categorical analysis (WT: 212, T322A: 246, S315A: 197, ST/A: 190, ΔNLS: 163, NES/A, ST/A: 186 cells); (d) ratiometric analysis (WT: 135, T322A: 170, S315A: 146, ST/A: 137, ΔNLS: 162, NES/A, ST/A: 195 cells) and (e) colocalization analysis (WT: 63, T322A: 54, S315A: 53, ST/A: 51, ΔNLS: 51, NES/A, ST/A: 55 cells) of intracellular localization of mVenus-α-TAT1 mutants; (f) Western blot showing staining with anti-GFP and anti-RRXS*/T* antibodies in HEK cells with exogenous expression of GFP-α-TAT1 and GFP-α-TAT1(T322A); (g) coimmunoprecipitation assay of mCherry-KPNA2 with GFP- α-TAT1 and GFP-α-TAT1(T322A); (h) images showing changes in TIRF intensity in Chemically induced dimerization based protein–protein interaction assay for mCherry-KPNA2 with mVenus-FKBP-α-TAT1 and (i) mVenus-FKBP-α-TAT1(T322A), Rapa: Rapamycin; (j) normalized corecruitment levels of indicated baits of mCherry-KPNA2 (WT: 36, T322A: 51), mCherry-KPNA4 (WT: 32, T322A: 33); and mCherry-KPNA6 (WT: 21, T322A: 40); (k) Western blot showing acetylated α-tubulin and α-tubulin levels in WT and α-TAT1-KO MEFS, and KOs rescued with mVenus or indicated mVenus- α-TAT1 mutants; (l) relative acetylation of α-Tubulin in WT and α-TAT1-KO MEFs, and KOs rescued with mVenus or indicated mVenus-α-TAT1 mutants; values from two Western blots (indicated by different colours) were normalized to WT MEF in each. Scale bar = 10 µm, ***, P < 0.001 or as shown, Student’s t test. For categorical analyses, Nuc: Nucleus enriched, Diff: Diffused, Cyto: Cytosolic. For f, g, and k, molecular weights in kD are indicated. Source data are available for this figure: SourceData F4.
α-TAT1 has a C-terminal phospho-inhibited NLS. (a–l) (a) Cartoon showing α-TAT1 mutant design (S: S315 residue, T: T322 residue, A: Alanine, p: phosphate moiety); (b) intracellular distribution of mVenus-α-TAT1 mutants as indicated; nuclei are outlined in red dotted lines; (c) categorical analysis (WT: 212, T322A: 246, S315A: 197, ST/A: 190, ΔNLS: 163, NES/A, ST/A: 186 cells); (d) ratiometric analysis (WT: 135, T322A: 170, S315A: 146, ST/A: 137, ΔNLS: 162, NES/A, ST/A: 195 cells) and (e) colocalization analysis (WT: 63, T322A: 54, S315A: 53, ST/A: 51, ΔNLS: 51, NES/A, ST/A: 55 cells) of intracellular localization of mVenus-α-TAT1 mutants; (f) Western blot showing staining with anti-GFP and anti-RRXS*/T* antibodies in HEK cells with exogenous expression of GFP-α-TAT1 and GFP-α-TAT1(T322A); (g) coimmunoprecipitation assay of mCherry-KPNA2 with GFP- α-TAT1 and GFP-α-TAT1(T322A); (h) images showing changes in TIRF intensity in Chemically induced dimerization based protein–protein interaction assay for mCherry-KPNA2 with mVenus-FKBP-α-TAT1 and (i) mVenus-FKBP-α-TAT1(T322A), Rapa: Rapamycin; (j) normalized corecruitment levels of indicated baits of mCherry-KPNA2 (WT: 36, T322A: 51), mCherry-KPNA4 (WT: 32, T322A: 33); and mCherry-KPNA6 (WT: 21, T322A: 40); (k) Western blot showing acetylated α-tubulin and α-tubulin levels in WT and α-TAT1-KO MEFS, and KOs rescued with mVenus or indicated mVenus- α-TAT1 mutants; (l) relative acetylation of α-Tubulin in WT and α-TAT1-KO MEFs, and KOs rescued with mVenus or indicated mVenus-α-TAT1 mutants; values from two Western blots (indicated by different colours) were normalized to WT MEF in each. Scale bar = 10 µm, ***, P < 0.001 or as shown, Student’s t test. For categorical analyses, Nuc: Nucleus enriched, Diff: Diffused, Cyto: Cytosolic. For f, g, and k, molecular weights in kD are indicated. Source data are available for this figure: SourceData F4.
One possible explanation for increased nuclear localization of T322A mutant is that phospho-T322 mediates nuclear export of α-TAT1. Truncation of the putative NLS including T322, α-TAT1-ΔNLS, did not increase nuclear localization (Fig. 4, a–e), indicating that T322 did not mediate nuclear export of α-TAT1. Furthermore, alanine-substituted NES along with S315A and T322A, α-TAT1(NES/A, ST/A), not only abrogated nuclear exclusion but considerably increased nuclear accumulation of α-TAT1 (Fig. 4, a–e), suggesting additive effects of NES inhibition on the nuclear localization. The α-TAT1(NES/A, ST/DE) mutant also showed diffused pattern but not nuclear accumulation (Fig. S3, c and d). Taken together, our observations suggest that phospho-T322 inhibits NLS, and that the NES and NLS act independently of one another.
T332 phosphorylation inhibits association of α-TAT1 NLS to Importin-α
NLS mediates nuclear import by binding to Importin-α molecules (also called karyopherin-α), which in turn heterodimerize with Importin-β and undergo nuclear translocation (Goldfarb et al., 2004). Human Importin-α is broadly classified into three subfamilies, exemplified by KPNA2 (Importin-α1), KPNA4 (Importin-α3), and KPNA6 (Importin-α7; Oka and Yoneda, 2018). Class 4 NLS has been reported to bind to all three subfamilies (Kosugi et al., 2009). To examine whether the putative α-TAT1 NLS binds to Importin-α, we performed a coimmunoprecipitation assay with mCherry-KPNA2 and GFP-α-TAT1 or GFP-α-TAT1(T322A). Consistent with our observations, we observed increased pull-down of mCherry-KPNA2 by GFP-α-TAT1(T322A) compared with WT (Fig. 4 g). To further examine if the NLS interacted with other karyopherin-α, we performed a chemical-induced dimerization-based protein–protein interaction assay in HeLa cells (see Materials and methods, Fig. S3 e). Consistent with our model of phospho-inhibited NLS, we observed increased binding of mCherry-KPNA2, mCherry-KPNA4, and mCherry-KPNA6 by mVenus-FKBP-α-TAT1(T322A) compared with mVenus-FKBP-α-TAT1 (Fig. 4, h and i).
To test our model of spatial regulation of α-TAT1 function, we utilized WT and KO MEFs (Aguilar et al., 2014). KO MEFs showed significantly lower levels of acetylated microtubules than WT MEFs (Fig. 4, k and l; and Fig. S3, h and j). We used lentiviral transduction to stably express mVenus, mVenus-α-TAT1, mVenus-α-TAT1(ST/A), and mVenus-α-TAT1(NES/A, ST/A) in KO cells, each of which localized as expected (Fig. S3 f). These cells were further sorted using flow cytometry to isolate cell populations with similar levels of mVenus fluorescence to ensure comparable expression levels of the introduced proteins (Fig. S3 g). Pharmacological inhibition of histone deacetylase-6 by Tubacin, a known deacetylase for α-Tubulin, increased microtubule acetylation in WT MEFs but not KO MEFs (Fig. S3, h and j), further demonstrating that α-TAT1 is the primary microtubule acetyltransferase in mice. Rescue with mVenus-α-TAT1, but not mVenus, restored acetylation in KO MEFs (Fig. 4, k and l; and Fig. S3, i and j). Moreover, KO MEFs expressing mVenus-α-TAT1(ST/A) or mVenus-α-TAT1(NES/A, ST/A) showed reduced acetylation compared with mVenus-α-TAT1 (Fig. 4, k and l; and Fig. S3, i and j), validating our model.
Serine/threonine kinase activities promote cytosolic localization of α-TAT1 and microtubule (MT) acetylation
Since our data suggest that the α-TAT1 NLS is phospho-inhibited, we hypothesized that α-TAT1 localization may be mediated by serine-threonine kinases. Treatment with 100 nM Staurosporin, a pan-kinase inhibitor, significantly increased nuclear localization of α-TAT1 (Fig. S4, a and b), suggesting that phosphorylation negatively regulated nuclear localization of α-TAT1. To narrow down the specific kinases that may regulate α-TAT1 localization, we performed a preliminary kinase-inhibitor screening assay where we treated HeLa cells expressing mVenus-α-TAT1 with several serine-threonine kinase inhibitors (Fig. S4, a and b). Based on this screen, we identified RO-3306 (inhibitor for CDK1 and CDK2 [Vassilev et al., 2006; Jorda et al., 2018]), PD0332991 (also called Pablociclib, inhibitor for CDK4 and CDK6 [Fry et al., 2004; Toogood et al., 2005]), Silmitasertib (also called CX-4945, an inhibitor for CK2 (Ferguson et al., 2011), and mCFP-PKI (peptide-based inhibitor for PKA [Kim et al., 2020]) as potential inhibitors of nuclear exclusion of mVenus-α-TAT1 (Fig. S4, and b). In addition, LJI308 (inhibitor for Ribosomal S9 kinase) appeared to promote nuclear exclusion of mVenus-α-TAT1 (Fig. S4, a and b). The preliminary screening assays were not performed in parallel, and to validate our initial observations, we repeated the kinase inhibition assay with these inhibitors in parallel. Unlike DMSO (vehicle), treatment with Staurosporin, RO-3306, PD0332991, and Silmitasertib significantly increased nuclear localization of mVenus-α-TAT1 (Fig. 5, a and b; and Fig. S4 c). In these experiments, we did not observe an increase in nuclear exclusion of mVenus-α-TAT1 in cells treated with LJI308 (Fig. 5 b and Fig. S4 c). Compared to coexpression with mCFP, mCFP-PKI considerably increased nuclear localization of mVenus-α-TAT1 (Fig. 5 c and Fig. S4 d). These data demonstrate that nuclear localization of α-TAT1 is inhibited by CDKs, CK2, and PKA. To examine the specificity of kinases, we further treated HeLa cells with exogenous expression of mVenus-α-TAT1 with alternate pharmacological inhibitors BMS-265246 (inhibitor for CDK1 and CDK2), AT7519 (inhibitor for CDK1, CDK2, CDK4, CDK6, and CDK9), Ellagic acid (inhibitor for CK2), and TTP22 (inhibitor for CK2), all of which increased nuclear localization of mVenus-α-TAT1 (Fig. 5 d and Fig. S4 e). Treatment of cells expressing GFP-α-TAT1 with RO-3306 showed decreased phosphorylation of T322 compared with untreated ones (Fig. S4 f). Additionally, unlike exogenous expression of WT CDK1 and CK2α, dominant negative Cdk1(D146N) or dominant negative CK2α(K68A) increased nuclear localization of mVenus-α-TAT1 (Fig. 5 e and Fig. S4, g–i).
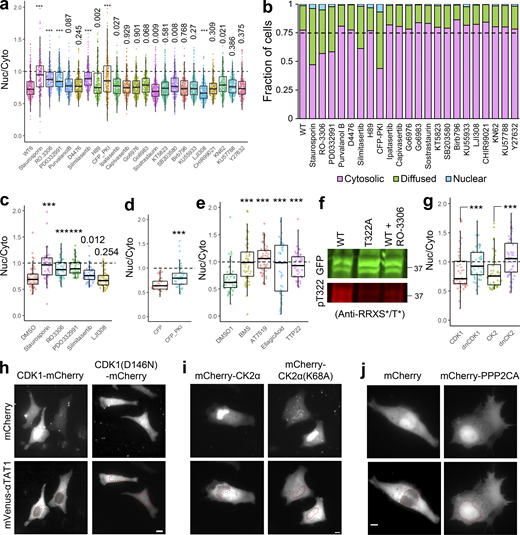
αTAT1 localization is mediated by Serine-Threonine kinases. (a–j) (a) Ratiometric and (b) categorical analysis of mVenus-α-TAT1 localization on kinase inhibitor screening assay; (c) ratiometric analysis of vehicle (DMSO) and kinase inhibitors; (d) over-expression of CFP and CFP-PKI; (e) vehicle (DMSO) and kinase inhibitors; (f) Western blot showing staining with anti-GFP and anti-RRXS*/T* antibodies in HEK cells with exogenous expression of GFP-α-TAT1 and GFP-α-TAT1(T322A) and GFP-α-TAT1 treated overnight with 10 μM RO-3306 (molecular weights in kD are indicated); (g) ratiometric analyses of over-expression of mVenus-α-TAT1 localization in cells coexpressing mCherry-tagged CDK1, dnCDK1 (D146N), CK2, and dnCK2 (K68A); images showing mVenus-α-TAT1 localization in cells coexpressing (h) CDK1-mCherry, CDK1(D146N)-mCherry, (i) mCherry-CK2 and mCherry-CK2(K68A), and (j) mCherry and mCherry-PPP2CA. Scale bar: 10 µm. ***, P < 0.001 or as indicated, Student’s t test. For categorical analyses, Nuc: Nucleus enriched, Diff: Diffused, Cyto: Cytosolic. Source data are available for this figure: SourceData FS4.
αTAT1 localization is mediated by Serine-Threonine kinases. (a–j) (a) Ratiometric and (b) categorical analysis of mVenus-α-TAT1 localization on kinase inhibitor screening assay; (c) ratiometric analysis of vehicle (DMSO) and kinase inhibitors; (d) over-expression of CFP and CFP-PKI; (e) vehicle (DMSO) and kinase inhibitors; (f) Western blot showing staining with anti-GFP and anti-RRXS*/T* antibodies in HEK cells with exogenous expression of GFP-α-TAT1 and GFP-α-TAT1(T322A) and GFP-α-TAT1 treated overnight with 10 μM RO-3306 (molecular weights in kD are indicated); (g) ratiometric analyses of over-expression of mVenus-α-TAT1 localization in cells coexpressing mCherry-tagged CDK1, dnCDK1 (D146N), CK2, and dnCK2 (K68A); images showing mVenus-α-TAT1 localization in cells coexpressing (h) CDK1-mCherry, CDK1(D146N)-mCherry, (i) mCherry-CK2 and mCherry-CK2(K68A), and (j) mCherry and mCherry-PPP2CA. Scale bar: 10 µm. ***, P < 0.001 or as indicated, Student’s t test. For categorical analyses, Nuc: Nucleus enriched, Diff: Diffused, Cyto: Cytosolic. Source data are available for this figure: SourceData FS4.

CDK1 and CK2 mediate α-TAT1 localization and MT acetylation. (a–n) (a) Intracellular distribution of mVenus-α-TAT1 in cells treated with DMSO (vehicle) or kinase inhibitors as indicated; nuclei are outlined in red dotted lines; (b) colocalization analyses of intracellular localization of mVenus-α-TAT1 in cells treated with DMSO (52 cells), Staurosporine (46 cells), RO-3306 (47 cells), PD0332991 (47 cells), Silmitasertib (47 cells), and LJI308 (46 cells); (c) coexpression of CFP (41 cells) or CFP-PKI (44 cells) (d) treated with DMSO (44 cells), BMS265246 (43 cells), AT7519 (45 cells), Ellagic acid (52 cells), or TTP22 (42 cells) and (e) coexpression of CDK1-mCherry (100 cells), CDK1(D146N)-mCherry (121 cells), mCherry-CK2α (112 cells), and mCherry-CK2α(K68A) (141 cells); (f) immunofluorescence images showing acetylated α-tubulin and α-tubulin; (g) ratio of acetylated to total α-tubulin in HeLa cells treated with vehicle (105 cells), RO-3306 (101 cells), BMS265246 (103 cells), and AT7519 (104 cells); (h) immunofluorescence images showing acetylated α-tubulin and α-tubulin; (i) ratio of acetylated to total α-tubulin in HeLa cells treated with vehicle (114 cells), Ellagic acid (120 cells), Silmitasertib (105 cells), and TTP22 (108 cells); (j) intracellular distribution of mVenus-α-TAT1 in HeLa cells treated with vehicle (DMSO) or PP2A agonist iHAP1; nuclei are outlined in red dotted lines; (k) colocalization analyses of intracellular localization of mVenus-α-TAT1 in cells treated with DMSO (62 cells) or iHAP1 (70 cells); (l) coexpression of mCherry (68 cells) or mCherry-PPP2CA (74 cells); (m) immunofluorescence images showing acetylated α-tubulin and α-tubulin; (n) ratio of acetylated to total α-tubulin in HeLa cells treated with vehicle (98 cells) or iHAP1 (69 cells). Scale bar = 10 µm. ***, P < 0.001 or as shown, Student’s t test.
CDK1 and CK2 mediate α-TAT1 localization and MT acetylation. (a–n) (a) Intracellular distribution of mVenus-α-TAT1 in cells treated with DMSO (vehicle) or kinase inhibitors as indicated; nuclei are outlined in red dotted lines; (b) colocalization analyses of intracellular localization of mVenus-α-TAT1 in cells treated with DMSO (52 cells), Staurosporine (46 cells), RO-3306 (47 cells), PD0332991 (47 cells), Silmitasertib (47 cells), and LJI308 (46 cells); (c) coexpression of CFP (41 cells) or CFP-PKI (44 cells) (d) treated with DMSO (44 cells), BMS265246 (43 cells), AT7519 (45 cells), Ellagic acid (52 cells), or TTP22 (42 cells) and (e) coexpression of CDK1-mCherry (100 cells), CDK1(D146N)-mCherry (121 cells), mCherry-CK2α (112 cells), and mCherry-CK2α(K68A) (141 cells); (f) immunofluorescence images showing acetylated α-tubulin and α-tubulin; (g) ratio of acetylated to total α-tubulin in HeLa cells treated with vehicle (105 cells), RO-3306 (101 cells), BMS265246 (103 cells), and AT7519 (104 cells); (h) immunofluorescence images showing acetylated α-tubulin and α-tubulin; (i) ratio of acetylated to total α-tubulin in HeLa cells treated with vehicle (114 cells), Ellagic acid (120 cells), Silmitasertib (105 cells), and TTP22 (108 cells); (j) intracellular distribution of mVenus-α-TAT1 in HeLa cells treated with vehicle (DMSO) or PP2A agonist iHAP1; nuclei are outlined in red dotted lines; (k) colocalization analyses of intracellular localization of mVenus-α-TAT1 in cells treated with DMSO (62 cells) or iHAP1 (70 cells); (l) coexpression of mCherry (68 cells) or mCherry-PPP2CA (74 cells); (m) immunofluorescence images showing acetylated α-tubulin and α-tubulin; (n) ratio of acetylated to total α-tubulin in HeLa cells treated with vehicle (98 cells) or iHAP1 (69 cells). Scale bar = 10 µm. ***, P < 0.001 or as shown, Student’s t test.
Since exogenous expression of nuclear-localized catalytic domain of α-TAT1 had failed to induce MT acetylation (Fig. 1, g and h), we reasoned that pharmacological inhibition of CDKs and CK2 may inhibit its function and reduce MT acetylation. HeLa cells treated with CDK1 inhibitors (RO-3306, BMS-265246, or AT7519) or CK2 inhibitors (Ellagic acid, Silmitasertib, or TTP22) showed significantly reduced MT acetylation levels compared with DMSO (vehicle) treatment (Fig. 5, f–i). The basal level of acetylation as measured by Ac. α-Tub/α-Tub in these experiments appears to be lower than those in Fig. 2 g. This is probably because of differences in experimental conditions including different dye-conjugated to secondary antibodies, as well as different fluorescence microscopy parameters (exposure time, excitation light strength etc.). Of note, experimental conditions were kept identical for the data grouped together. Taken together, these data demonstrate that inhibition of CDK1 (and potentially other CDKs) and CK2 increase nuclear localization of mVenus-α-TAT1 and inhibit MT acetylation.
Phosphatase 2A (PP2A) promotes nuclear localization of α-TAT1 and inhibits MT acetylation
PP2A is a ubiquitous serine/threonine phosphatase that has broad substrate specificity (Mumby, 2007). Based on our observation that inhibition of kinases increased nuclear localization of α-TAT1, we sought to examine whether activation of serine/threonine phosphatase may do the same. Treatment of cells with iHAP1, an agonist for PP2A (Morita et al., 2020), significantly decreased cytoplasmic localization of mVenus-α-TAT1 (Fig. 5, j and k). Since iHAP1 may directly inhibit microtubule polymerization (Vit et al., 2022), we further examined the role of PP2A in α-TAT1 localization by overexpression of mCherry-PPP2CA (PP2A catalytic subunit α). mCherry-PPP2CA, but not of mCherry alone, significantly decreased nuclear exclusion of mVenus-α-TAT1 (Fig. 5 l and Fig. S4 j). Treatment of HeLa cells with iHAP1 also inhibited MT acetylation (Fig. 5, m and n), further demonstrating a role of PP2A in regulating α-TAT1 function.
14-3-3 proteins inhibit nuclear localization of α-TAT1
14-3-3 protein binding has been reported to negatively regulate nuclear import by inhibiting binding of importins to NLS (Sekimoto et al., 2004; Mancini et al., 2009; Huang et al., 2018). 14-3-3 binding to proteins is mediated by phosphorylated serine and threonine residues (Yaffe et al., 1997). Part of the putative NLS sequence “PAQRRRTR” bears similarity to 14-3-3 binding motif RXX(pS/pT)XP (Yaffe et al., 1997), and 14-3-3-Pred (Madeira et al., 2015), a 14-3-3 interaction prediction server, identified T322 as a potential 14-3-3 binding site. In a previous mass spectrometry analysis of α-TAT1 in HEK cells, we identified 14-3-3-β and 14-3-3-ζ as potential interactors (Seetharaman et al., 2021). To examine whether α-TAT1 bound to 14-3-3 proteins, we coexpressed GFP-α-TAT1 with HA-tagged 14-3-3-β or 14-3-3-ζ in HEK-293T cells and performed a coimmunoprecipitation assay. GFP-α-TAT1, but not GFP alone, coprecipitated with HA-14-3-3-β and HA-14-3-3-ζ (Fig. 6 a). Exogenous expression of a 14-3-3 inhibitor peptide R18 (Petosa et al., 1998; Masters and Fu, 2001) or Difopein (a dimerized form of R18; Masters and Fu, 2001) significantly increased nuclear localization of mVenus-α-TAT1 in HeLa cells (Fig. 6, b–d). To identify which 14-3-3 isoforms interacted with α-TAT1, we performed the chemical-induced dimerization-based protein–protein interaction assay in HeLa cells (see Materials and methods, Fig. S3 e). Based on this assay, we found that mVenus-FKBP-α-TAT1 interacted with all seven isoforms of 14-3-3 proteins (Fig. 6, e, g, and i and Fig. S5 a). To further examine whether any 14-3-3 isoform specifically interacted with T322 and S315 in α-TAT1, we performed the protein–protein interaction assay with mVenus-FKBP-α-TAT1(T322A) and mVenus-FKBP-α-TAT1(ST/A), both of which showed significantly decreased interaction with 14-3-3 isoforms β, γ, ε, and ζ, but not η, θ, or σ (Fig. 6, f, h, and i and Fig. S5 a). The mVenus-FKBP-α-TAT1 mutants showed comparable plasma-membrane recruitment levels (Fig. S5 b). The kinetics of corecruitment of mCherry-14-3-3ζ closely followed that of mVenus-FKBP-α-TAT1 (Fig. S5 c), suggesting that increase in plasma membrane localization of 14-3-3 proteins was due to their association with α-TAT1.
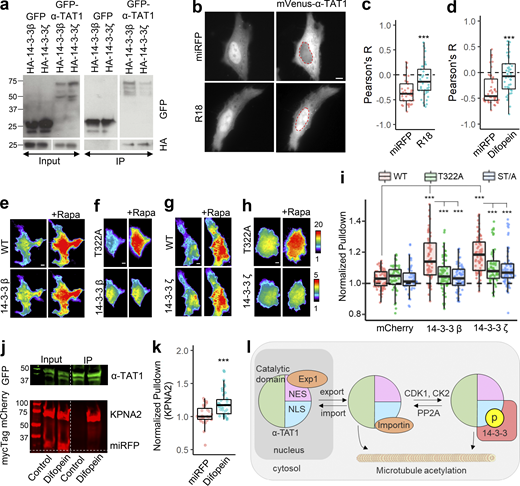
14-3-3 proteins interact with α-TAT1 through T322. (a–k) Coimmunoprecipitation of GFP-α-TAT1 with HA-14-3-3β and HA-14-3-3ζ proteins; (b) intracellular distribution of mVenus-α-TAT1 in cells coexpressing miRFP703 or miRFP703-14-3-3i; nuclei are outlined in red dotted lines; (c) colocalization analysis of mVenus-α-TAT1 in cells coexpressing miRFP703 (45 cells) or miRFP703-R18 (45 cells); (d) colocalization analysis of mVenus-α-TAT1 in cells coexpressing myc-miRFP703 (55 cells) or myc-miRFP703-Difopein (52 cells); (e) images showing changes in TIRF intensity in CID based protein–protein interaction assay for mCherry-14-3-3β with mVenus-FKBP-α-TAT1 and (f) mVenus-FKBP-α-TAT1(T322A), (g) mCherry-14-3-3ζ with mVenus-FKBP-α-TAT1 or with (h) mVenus-FKBP-α-TAT1(T322A), Rapa: Rapamycin; (i) normalized corecruitment levels by indicated baits of mCherry (α-TAT1: 79, T322A: 45, ST/A: 49), mCherry-14-3-3ζ (α-TAT1: 70, T322A: 58, ST/A: 79; complete panel: Fig. S5 a); (j) coimmunoprecipitation of KPNA2 with GFP-α-TAT1 coexpressed with myc-miRFP703 or myc-miRFP703-Difopein in HEK cells (molecular weights in kD are indicated); (k) normalized corecruitment levels of mCherry-KPNA2 by mVenus-FKBP-α-TAT1 in cells coexpressing myc-miRFP703(27) or myc-miRFP703-Difopein(39) (complete panel: Fig. S5 d); (l) proposed model of spatial regulation of α-TAT1 function (p: phosphate moiety). Scale bar = 10 µm. ***, P < 0.001 or as shown, Student’s t test. Source data are available for this figure: SourceData F6.
14-3-3 proteins interact with α-TAT1 through T322. (a–k) Coimmunoprecipitation of GFP-α-TAT1 with HA-14-3-3β and HA-14-3-3ζ proteins; (b) intracellular distribution of mVenus-α-TAT1 in cells coexpressing miRFP703 or miRFP703-14-3-3i; nuclei are outlined in red dotted lines; (c) colocalization analysis of mVenus-α-TAT1 in cells coexpressing miRFP703 (45 cells) or miRFP703-R18 (45 cells); (d) colocalization analysis of mVenus-α-TAT1 in cells coexpressing myc-miRFP703 (55 cells) or myc-miRFP703-Difopein (52 cells); (e) images showing changes in TIRF intensity in CID based protein–protein interaction assay for mCherry-14-3-3β with mVenus-FKBP-α-TAT1 and (f) mVenus-FKBP-α-TAT1(T322A), (g) mCherry-14-3-3ζ with mVenus-FKBP-α-TAT1 or with (h) mVenus-FKBP-α-TAT1(T322A), Rapa: Rapamycin; (i) normalized corecruitment levels by indicated baits of mCherry (α-TAT1: 79, T322A: 45, ST/A: 49), mCherry-14-3-3ζ (α-TAT1: 70, T322A: 58, ST/A: 79; complete panel: Fig. S5 a); (j) coimmunoprecipitation of KPNA2 with GFP-α-TAT1 coexpressed with myc-miRFP703 or myc-miRFP703-Difopein in HEK cells (molecular weights in kD are indicated); (k) normalized corecruitment levels of mCherry-KPNA2 by mVenus-FKBP-α-TAT1 in cells coexpressing myc-miRFP703(27) or myc-miRFP703-Difopein(39) (complete panel: Fig. S5 d); (l) proposed model of spatial regulation of α-TAT1 function (p: phosphate moiety). Scale bar = 10 µm. ***, P < 0.001 or as shown, Student’s t test. Source data are available for this figure: SourceData F6.

14-3-3 binding inhibits αTAT1 association with Importin-α. (a–d) (a) Normalized corecruitment levels by indicated baits of mCherry (α-TAT1: 79, T322A: 45, ST/A: 49), mCherry-14-3-3β (α-TAT1: 64, T322A: 63, ST/A: 66), mCherry-14-3-3γ (α-TAT1: 50, T322A: 57, ST/A: 64), mCherry-14-3-3ε (α-TAT1: 65, T322A: 52, ST/A: 58), mCherry-14-3-3ζ (α-TAT1: 70, T322A: 58, ST/A: 79), mCherry-14-3-3η (α-TAT1: 48, T322A: 50, ST/A: 58), mCherry-14-3-3θ (α-TAT1: 53, T322A: 68, ST/A: 53), mCherry-14-3-3σ (α-TAT1: 53, T322A: 56, ST/A: 73); (b) normalized recruitment levels of baits (α-TAT1: 90, T322A: 97, ST/A: 105); (c) kinetics of mVenus-FKBP-α-TAT1 and mCherry-14-3-3-ζ translocation to plasma membrane; n = 24 cells, means ± 95% confidence interval are shown; (d) normalized corecruitment levels in cells coexpressing myc-miRFP or myc-miRFP-Difopein by mVenus-FKBP-α-TAT1 of indicated preys mCherry-KPNA2 (miRFP: 27, Difopein:39), mCherry-KPNA4 (miRFP: 25, Difopein:37), mCherry-KPNA6 (miRFP: 26, Difopein:38). ***, P < 0.001 or as shown, Student’s t test.
14-3-3 binding inhibits αTAT1 association with Importin-α. (a–d) (a) Normalized corecruitment levels by indicated baits of mCherry (α-TAT1: 79, T322A: 45, ST/A: 49), mCherry-14-3-3β (α-TAT1: 64, T322A: 63, ST/A: 66), mCherry-14-3-3γ (α-TAT1: 50, T322A: 57, ST/A: 64), mCherry-14-3-3ε (α-TAT1: 65, T322A: 52, ST/A: 58), mCherry-14-3-3ζ (α-TAT1: 70, T322A: 58, ST/A: 79), mCherry-14-3-3η (α-TAT1: 48, T322A: 50, ST/A: 58), mCherry-14-3-3θ (α-TAT1: 53, T322A: 68, ST/A: 53), mCherry-14-3-3σ (α-TAT1: 53, T322A: 56, ST/A: 73); (b) normalized recruitment levels of baits (α-TAT1: 90, T322A: 97, ST/A: 105); (c) kinetics of mVenus-FKBP-α-TAT1 and mCherry-14-3-3-ζ translocation to plasma membrane; n = 24 cells, means ± 95% confidence interval are shown; (d) normalized corecruitment levels in cells coexpressing myc-miRFP or myc-miRFP-Difopein by mVenus-FKBP-α-TAT1 of indicated preys mCherry-KPNA2 (miRFP: 27, Difopein:39), mCherry-KPNA4 (miRFP: 25, Difopein:37), mCherry-KPNA6 (miRFP: 26, Difopein:38). ***, P < 0.001 or as shown, Student’s t test.
14-3-3 binding may regulate protein–protein interactions by altering protein structures or blocking protein interaction sites. To examine whether 14-3-3 binding affects α-TAT1 association with karyopherin-α, we performed a coimmunoprecipitation assay of GFP-α-TAT1 and mCherry-KPNA2 with coexpression of myc-miRFP703 or myc-miRFP703-Difopein. We observed increased pulldown of mCherry-KPNA2 by GFP-α-TAT1 in the presence of myc-miRFP703-P2A-Difopein compared with myc-miRFP703 (Fig. 6 j). Using the chemical-induced dimerization live-cell pulldown assay, we observed increased pulldown of KPNA2 and KPNA6, but not of KPNA4, by mVenus-FKBP-α-TAT1 in cells coexpressing myc-miRFP703-Difopein compared with those expressing myc-miRFP703 (Fig. 6 k and Fig. S5 d). These data suggest that α-TAT1 interacts with specific 14-3-3 isoforms through T322 and that 14-3-3 proteins mediate nuclear exclusion of α-TAT1. Based on these observations, we propose a model of spatial regulation of α-TAT1 wherein a balance of phospho-regulated nuclear transport and 14-3-3 association mediates α-TAT1 localization and MT acetylation (Fig. 6 l).
Discussion
One of the bottlenecks in elucidating the role of microtubule acetylation in biological phenomena is the knowledge gap of how upstream molecular signaling pathways control α-TAT1 function to modulate MT acetylation. Autoacetylation of lysine residues is proposed to promote α-TAT1 catalytic activity (Kalebic et al., 2013a). Similarly, TAK1-dependent phosphorylation of α-TAT1 Serine-237 has been reported to stimulate its catalytic property (Shah et al., 2018). In neurons, p27kip1 directly binds to α-TAT1 and stabilizes it against proteasomal degradation (Morelli et al., 2018), thus enhancing MT acetylation. Our study demonstrates that intracellular α-TAT1 localization is a dynamic process that is orchestrated on a delicate balance of nuclear export and import, and that is directly reflected in MT acetylation levels (Fig. 6 l). Based on decrease in MT acetylation in the presence of kinase inhibitors or KO-rescue with α-TAT1(NES/A, ST/A), we posit that the effects of spatial regulation are comparable with previously reported regulatory mechanisms (Morelli et al., 2018; Kalebic et al., 2013a; Shah et al., 2018), suggesting a significant role in tuning MT acetylation levels. To our knowledge, this is the first study to identify molecular mechanisms that spatially regulate α-TAT1 function. In addition, we demonstrated a hitherto unknown role of the inherently disordered α-TAT1 C-terminus and identified its interactions with specific 14-3-3 proteins and serine-threonine kinases, namely CDK1 and CK2. Our observation that spatial sequestration of α-TAT1 from cytosolic MTs modulates acetylation dynamics suggests a role of the nucleus as a reservoir or sequestration chamber to control protein access of substrates.
We have demonstrated active nuclear export of α-TAT1 by Exp1 through an NES rich in hydrophobic residues, which was critical for efficient microtubule acetylation. In addition, we have identified an NLS consistent with non-canonical class 4 NLS (Kosugi et al., 2009). Interestingly, position 7 of this NLS, which should not be an acidic residue, is occupied by Threonine-322. Since phosphorylation of threonine can significantly increase its net negative charges, it is ideally situated to function as an ON/OFF switch for the NLS. Although we have identified Threonine-322 to be the critical phospho-residue that inhibits nuclear import, Serine-315 appears to provide additional inhibition. The increased nuclear localization of ST/A mutant over T322A mutant raises the possibility that S315 and T322 may aggregate signals from different signaling pathways to fine-tune α-TAT1 localization. That both the NES and NLS are well conserved across all human α-TAT1 isoforms as well as mammalian α-TAT1 suggests a physiological role. However, on sequence alignment with C. elegans’ α-TAT1, we observed similarity in the catalytic domain, but not in the C-terminus. In addition, the C. elegans’ α-TAT1 did not appear to have either an NES or an NLS, although it did have three putative 14-3-3 binding sites (Fig. S1 f), which may mediate its spatial regulation.
Our data demonstrate that cytosolic localization of α-TAT1 is mediated by kinase and phosphatase action, possibly on Threonine-322 and Serine-315. T322 site (SPAQRRRT*R) matches the known substrate recognition site for PKA (RRXS*/T*), whereas S319 matches the consensus motif for CDK1 substrate (S*/T*P or S*/T*XXR/K). Neither S319 nor T322 match the consensus motif for CK2 substrate (S*/T*DXE). However, CK2 has been recently reported to bind to the C-terminus of α-TAT1, phosphorylate S236, and regulate its activity (You et al., 2022). Nevertheless, our data demonstrate a role of CDK1 and CK2 in regulating α-TAT1 localization. It is possible that these kinases indirectly facilitate phosphorylation of T322 through PKA or some other kinase. Specifically, our study shows a role of CDK1 and CK2 as well as PP2A in coordinating spatial distribution of α-TAT1 and downstream MT acetylation, which provides a possible mechanism for the changes in α-TAT1 localization and microtubule acetylation observed at different stages of the cell cycle (Nekooki-Machida et al., 2018). α-TAT1 KO MEFs are highly proliferative (Aguilar et al., 2014), and loss of α-TAT1 has been reported to cause mitotic errors (Chien et al., 2016). Since there is a loss of nuclear membrane integrity at the onset of mitosis, it is unclear whether the spatial regulation that we describe may play a role in mitosis. However, the C-terminus may still act as a signal aggregation hub to mediate both α-TAT1 activity and specific localization.
We demonstrated Threonine-322 to be a putative binding site for 14-3-3 β, γ, ε, and ζ proteins, which are also involved in cellular DNA damage response and cell cycle progression (Chen et al., 1999; Andersen et al., 2012; Pennington et al., 2018). 14-3-3s interact with phospho-serines or phospho-threonines in intrinsically disordered regions and may mediate nuclear transport of proteins by masking NES or NLS (Pennington et al., 2018). Furthermore, 14-3-3 proteins may significantly alter the structure of their binding partners to align along their rigid α-helical backbone to expose or hide critical binding sites (Yaffe, 2002). Of course, based on our data, we cannot determine whether phospho-inhibition of α-TAT1 association with Importin-α is due to disruptions in electrostatic interactions or due to masking of the NLS through 14-3-3 binding to T322 or a combination of these phenomena. Indeed, 14-3-3 association may also stabilize phosphorylated T322 by masking it from PP2A.
While there are pharmacological agents that promote increased MT acetylation through inhibition of deacetylases, currently there are no available pharmacological inhibitors of α-TAT1 itself, although there appears to be a considerable interest in the development of such inhibitors (Kwon et al., 2020). Identifying a small chemical target to the C-terminus signal motif to alter the subcellular localization, instead of the catalytic activity, may open up a novel approach for inhibiting MT acetylation. α-TAT1 does not acetylate histones in vitro (Shida et al., 2010). Additionally, since an acetylation-deficient α-tubulin mutant (K40A) appears to mimic most phenotypes of α-TAT1 deficiency and an acetylation mimic mutant (K40Q) rescues α-TAT1 deficiency (Shida et al., 2010; Morley et al., 2016), α-tubulin is thought to be the predominant substrate for α-TAT1. Nevertheless, it is possible that nuclear import of α-TAT1 facilitates interactions with presently unidentified substrates located in the nucleus or that the C-terminus of α-TAT1 facilitates other protein–protein interactions. In a similar vein, lysine residues in cyclins, CDK1, CK2, 14-3-3 proteins, and Exp1 are acetylated (Choudhary et al., 2009; Andersen et al., 2011; Deota et al., 2019), and it is tempting to speculate that these acetylation events may be directly or indirectly mediated by α-TAT1.
It is worthwhile to consider that a significant number of posttranslational modifications of α-TAT1 appear on its intrinsically disordered C-terminus (Fig. S3 b). Disordered regions may act as a signaling hub by interacting with multiple proteins, thus facilitating complex formation and acting as integrators of signaling pathways (Wright and Dyson, 2015). We demonstrated the presence of an NES, NLS, phosphorylation sites, and putative 14-3-3 binding sites within the α-TAT1 C-terminus. This signal motif is well-conserved across mammalian species as well as in all the human isoforms, suggesting a critical role of the α-TAT1 C-terminus in its function. Indeed, numerous cancer-associated mutations curated in COSMIC (Tate et al., 2019) and TCGA Research Network databases in the ATAT1 gene are located in the intrinsically disordered C-terminal region, suggesting a physiological role. More specifically, there are a considerable number of deletions, frame shifts, and missense mutations encompassing the NES and the NLS regions, which may be expected to affect the spatial distribution of α-TAT1. Whether these mutations underlie the pathogenesis in these cancers remains to be examined. Considering the role of microtubule acetylation in a wide array of cellular activities, it is possible that loss of spatial regulation of α-TAT1 may be relevant in other diseases as well.
Kinase-mediated regulation of nuclear export and nuclear import has previously been reported in transcription regulators (Oeckinghaus and Ghosh, 2009; Spencer et al., 2013; Regot et al., 2014; Deneke et al., 2016). In particular, regulation of Cdc25 localization by Checkpoint kinase 1–mediated phosphorylation of and subsequent recruitment of 14-3-3-β to an NLS-proximal phospho-site is virtually the same as our proposed model (Fig. 6 l) of α-TAT1 localization (Deneke et al., 2016; Uchida et al., 2004; Jiang et al., 2003), suggesting that such kinase-mediated balancing of nuclear export and import is a general strategy for protein localization. This is particularly intriguing in the context of the apparent role of α-TAT1 or MT acetylation in DNA damage response since Checkpoint kinase 1 association with 14-3-3 is triggered by DNA damage (Chen et al., 1999; Dunaway et al., 2005; Ryu and Kim, 2020). Localization of proteins is often regulated (Shimada et al., 2000; Volmat et al., 2001; Formstecher et al., 2001; Yang et al., 2017; Lasker et al., 2020; McKinsey et al., 2000; Mihaylova et al., 2011), and their aberrant regulation has been linked to diseases (Chen et al., 1995; Jiao et al., 2008; Hung and Link, 2011; Wang and Li, 2014). For example, class IIa histone deacetylases may be accumulated in the cytosol by 14-3-3 proteins, which inhibits acetylation of transcription factors (Mihaylova et al., 2011). Cytosolic retention of p42/44 MAP kinases by PEA-15 inhibits its effects on transcription and proliferation (Formstecher et al., 2001). Unlike these proteins, the nuclear localization of α-TAT1 limits the access of the enzyme to its substrates, namely polymerized microtubules, which exclusively reside in the cytosol.
In conclusion, we propose a model for spatial sequestration of α-TAT1 as one of the major regulatory mechanisms of microtubule acetylation (Fig. 6 l). The model consists of three key characteristics: presence of an NES that facilitates Exp1 mediated nuclear export, presence of an NLS to mediate nuclear import, and finally, modulation of this nuclear import by kinases. Further examination of the role of specific kinases on α-TAT1 localization may advance our understanding of its function in both cellular processes and pathologies, helping identify new therapeutic targets in the future.
Materials and methods
Cell culture and transfection
HeLa (ATCC) and HEK-293T (catalog # CRL-3216; ATCC) cells were cultured in DMEM basal media and passaged every third day of culture. For optimal growth, the media were supplemented with 10% (vol/vol) fetal bovine serum, L-glutamine, and penicillin/streptomycin. WT and α-TAT1 KO MEFs were a generous gift from Dr. Maxence Nachury (University of California, San Francisco, San Francisco, CA; Aguilar et al., 2014) and were cultured in DMEM basal media supplemented with 10% (vol/vol) fetal bovine serum, L-glutamine, penicillin/streptomycin, non-essential amino acids, and 0.05 mM β-mercaptoethanol. KO MEFs rescues with mVenus, mVenus-α-TAT1, mVenus-α-TAT1(ST/A), or mVenus-α-TAT1(NES/A, ST/A) were selected using the Sony SH800 cell sorter using manufacturer’s instructions to select cell populations with the same mVenus fluorescence thresholds to ensure similar expression levels of the proteins of interest. The cells were maintained under standard cell culture conditions (37 °C and 5% CO2) and were checked for mycoplasma contamination prior to use in experiments. In addition, the KO-rescue cells were cultured in medium containing 1 µg/ml of puromycin for selection; effective puromycin dosage was ascertained by testing on KO cells. FuGENE 6 reagent (Promega) was used for transient transfection of HeLa cells according to the manufacturer’s instructions. For immunoprecipitation assays and generation of lentiviral particles, HEK cells were transfected using polyethyleneimine (PEI).
DNA plasmids
H2B-mCFP in Clontech pmCFP-C1 vector was a generous gift from Dr. Sergi Regot (Johns Hopkins University, Baltimore, MD) and was subcloned into pmCherry-C1 vector (Clontech) to generate H2B-mCherry plasmid construct. EGFP-α-TAT1 (isoform 5) in Clontech EGFP-C1 vector was a generous gift from Dr. Antonina Roll-Mecak (National Institutes of Health, Bethesda, MD). The α-TAT1 construct was subcloned into the pTriEx-4 vector (Novagen) using PCR and restriction digestion with mVenus at the N terminus and α-TAT1 at the C terminus. mCherry-PKI construct was previously generated in our lab through PCR (Kim et al., 2020) and was subcloned into the pmCFP vector (Clontech) to generate the mCFP-PKI plasmid construct. GFP-αTAT1 (isoform 1) in GFP-C1 Clontech vector was a gift from Dr. Philippe Chavrier (Institut Curie, Paris, France) and Dr. Guillaume Montagnac (Sorbonne University, Paris, France). HA-14-3-3-β and HA-14-3-3-ζ plasmids in pcDNA3 vector backbone were a gift from Dr. Michael Yaffe (Massachusetts Institute of Technology, Cambridge, MA). 14-3-3-γ, 14-3-3-ε, 14-3-3-η, 14-3-3-θ, and 14-3-3-σ plasmids in pBiEx1 vector backbone were a gift from Dr. Douglas Robinson (Johns Hopkins University, Baltimore, MD). All 14-3-3 isoforms were subcloned into pTriEx-4 vector with an N-terminal mCherry tag. pRS3 (CK2α, CK2β) was a gift from David Litchfield (University of Western Ontario, London, Canada; plasmid # 27092; Addgene). pUHD-Cdk1 WT HA was a gift from Greg Enders (University of Pennsylvania, Philadelphia, PA; plasmid # 27652; Addgene). pBABE-zeo-PPP2CA was a gift from William Hahn (Dana Farber Cancer Institute, Boston, MA; plasmid # 10689; Addgene). CK2-α, CDK1, and PPP2CA were subcloned into pTriEx-4 vector. pCMVTNT-T7-KPNA2 (plasmid # 26678; Addgene), pCMVTNT-T7-KPNA4 (plasmid # 26680; Addgene), and pCMVTNT-T7-KPNA6 (plasmid # 26682; Addgene) were gifts from Bryce Paschal (University of Virginia, Charlottesville, VA). KPNA2, KPNA4, and KPNA6 were subcloned into pTriEX4 plasmid vector. As indicated in the results and figure legends, tags of compatible fluorescent proteins including Cerulean, mVenus, mCherry, and miRFP703 were appended to facilitate detection of the proteins of interest. Unless specified otherwise, the termini of tagging were positioned as in the orders they were written. Lentiviral plasmids were generated based on a modified Puro-Cre vector (plasmid # 17408; Addgene, mCMV promoter, and no Cre encoding region). Truncations of α-TAT1 were generated by PCR. Point mutations of α-TAT1 were generated using overlapping PCR. 14-3-3 inhibitor peptides R18, and Difopein was generated by PCR and appended to the C-terminus of miRFP703-P2A in a pTriEX4 vector so that the fluorescent protein could be used to detect expression but may not cause any steric hindrance of the inhibitor peptide. The open reading frames of all DNA plasmids were verified by Sanger sequencing.
Sequence alignment
Protein sequence alignment was performed using Clustal-W (Madeira et al., 2019; https://www.ebi.ac.uk/Tools/msa/clustalo/).
Nuclear transport and kinase inhibitors
LMB was purchased from LC Laboratories (catalog # L6100). SB203580 (catalog # S8307; Sigma-Aldrich), Doramapimod (catalog # S1574; BIRB 796, Selleck Chemicals), CHIR99021 (catalog # SML1046; Sigma-Aldrich), Sostrastaurin (catalog # S2791; Selleck Chemicals), RO-3306 (catalog # S7747; Selleck Chemicals), Ipatasertib (RG7440, catalog # S2808; Selleck Chemicals), Capivasertib (AZD5363, catalog # S8019; Selleck Chemicals), Silmitasertib (CX 4945, catalog # S2248; Selleck Chemicals), and KU-55933 (catalog # SML1109; Sigma-Aldrich) were generous gifts from Dr. Sergi Regot (Johns Hopkins University, Baltimore, MD). SB239063 (catalog # S0569; Sigma-Aldrich) was a generous gift from Dr. Jun Liu (Johns Hopkins University, Baltimore, MD). The rest of the kinase inhibitors were purchased as indicated: Staurosporine (catalog # 569397; Sigma-Aldrich), LJI308 (catalog # SML1788; Sigma-Aldrich), Y-27632 (catalog # Y-5301; LC Laboratories), Gö 6976 (catalog # 365250; Sigma-Aldrich), Gö 6983 (catalog # G1918; Sigma-Aldrich), H-89 (catalog # B1427; Sigma-Aldrich), D4476 (catalog # 1770; BioVision), KN-62 (catalog # S7422; Selleck Chemicals), KU-57788 (catalog # HY-11006; MedChemExpress), Purvalanol B (catalog # SYN-1070; AdipoGen Life Sciences), KT-5823 (catalog # 10010965; Cayman Chemicals), PD0332991 (catalog # PZ0199; Sigma-Aldrich), BMS-265246 (catalog # S2014; Selleck Chemicals), AT7519 (catalog # S1524; Selleck Chemicals), TTP22 (catalog # S6536; Selleck Chemicals), and Ellagic acid (catalog # S5516; Selleck Chemicals). All kinase inhibitors were applied for 4 h prior to imaging or fixation or immunofluorescence assays. iHAP1 was purchased from Selleck Chemicals (catalog # S9695) and applied overnight. For phospho-T322 Western blot assay, cells were incubated with RO-3306 CDK1 inhibitor overnight.
Immunofluorescence assays
For immunostaining of α-TAT1, cells were fixed using freshly prepared 4% paraformaldehyde at room temperature for 10 min, washed twice with PBS, blocked, and permeabilized in 10% goat serum in PBS with 0.1% Triton X-100 at room temperature for an hour and then incubated with polyclonal antibody against α-TAT1 (Rabbit, PA5114922; Thermo Fisher Scientific) in the above blocking buffer at room temperature for 1 h, washed three times in PBS, and incubated with secondary antibody (catalog # A-11008; Invitrogen AlexaFluor 488 Goat anti-Rabbit) and DAPI (catalog # D1306; Invitrogen) in the above blocking buffer for 1 h at room temperature. After that, they were washed three times in PBS, and the images were captured by microscopy. For immunostaining of acetylated and total α-tubulin, cells were fixed using ice-cold methanol for 10 min, washed thrice with cold PBS, blocked with 2% BSA in PBS for 1 h, and then incubated overnight at 4°C with monoclonal antibodies against α-tubulin (Rat, MAB1864; MilliporeSigma) and acetylated α-Tubulin (Mouse, T7451; MilliporeSigma). The next day, the samples were washed thrice with cold PBS and incubated with secondary antibodies (catalog # A-21096; Invitrogen AlexaFluor 680 Goat anti-Rat and catalog # A-11032; Invitrogen AlexaFluor 594 Goat anti-Mouse or catalog # A-11001; Invitrogen AlexaFluor 488 Goat anti-Mouse) for 1 h at room temperature, after which they were washed thrice with PBS, and images were captured by microscopy. For HeLa cells transiently transfected with mVenus-α-TAT1, mVenus-α-TAT1 catalytic domain, and NLS-mVenus-α-TAT1 catalytic domain, fixation and immunostaining were performed 24 h after transfection. For LMB treatment, HeLa cells were treated with 100 nM LMB or equal volume of vehicle (EtOH) and incubated for 4 h, followed by methanol fixation and immunostaining. For kinase inhibitors, HeLa cells were treated with RO-3306 (10 µM), BMS-265246 (100 nM), AT7519 (1 µM), Ellagic acid (1 µM), TTP22 (1 µM) or Silmitasertib (10 µM), or vehicle (DMSO) for 4 h followed by fixation and immunostaining. For iHAP1 treatment, HeLa cells were treated with iHAP1 (10 µM) or vehicle (DMSO) overnight followed by fixation and immunostaining. For Tubacin treatment, MEF cells were treated with 2 µM Tubacin or vehicle (DMSO) and incubated for 4 h. KO-rescue cells were treated with DMSO for 4 h.
Western blot assays
Cell lysates were prepared by scraping cells using lysis buffer (radioimmunoprecipitation assay buffer [# 9806S; Cell Signaling Technology] and mixed with protease/phosphatase inhibitor cocktail [# 5872S; Cell Signaling Technology]). Cell lysates were rotated on a wheel at 4°C for 15 min and centrifuged for 10 min at 15,000g and 4°C to pellet the cell debris, mixed with NuPAGE LDS Sample Buffer (# NP0007; Thermo Fisher Scientific) with protease and phosphatase inhibitors, and boiled 5 min at 95°C before loading onto polyacrylamide gels. Gels were transferred to polyvinylidene difluoride (PVDF) membranes, which were blocked with TBST (0.1% Tween) and 5% BSA and incubated overnight with the primary antibody and 1 h with LiCOR IR-dye conjugated secondary antibodies, after which bands were revealed using Odyssey imaging system. For quantification of relative α-tubulin acetylation in KO-rescue in Western blots (Fig. 4 l), we performed background subtraction and determined the integrated intensity of each band. Relative acetylation was determined by the ratio of acetylated-α-tubulin to total α-tubulin and normalized to the values for WT for each blot, shown in different colors. Antibodies used: anti-GFP (mouse, sc9996; Santa Cruz), anti-RRXS*/T* (rabbit, 9624S; Cell Signaling Technology), anti–α-tubulin (rat, MAB1864; MilliporeSigma), and anti-acetylated Lysine-40 α-Tubulin (mouse, T6793; MilliporeSigma). mVenus was detected using anti-GFP (rabbit, 50430-2-AP; Proteintech). Secondary IR-dye conjugated antibodies (680RD Goat anti-Rabbit, catalog # 926-68071; 800CW Donkey anti-Mouse, catalog # 926-32212; 680RD Donkey anti-Mouse, catalog # 926-68072; 680RD Goat anti-Rat, catalog # 926-68076; 800CW Goat anti-Rat, catalog # 926-32219; 800CW Goat anti-Rabbit, catalog # 926-32211) were purchased from LiCOR.
Immunoprecipitation assays
Immunoprecipitation of GFP, GFP-αTAT1, and GFP-αTAT1(T322A) were performed using GFP-Trap nanobody conjugated agarose beads (# 5872S; Chromotek) according to the manufacturer’s instructions. Briefly, HEK293T cells were transiently transfected with GFP or GFP-αTAT1 constructs and other plasmid constructs as indicated. Cell lysates were prepared by scraping cells using lysis buffer (radioimmunoprecipitation assay buffer [# 9806S; Cell Signaling Technology], mixed with protease/phosphatase inhibitor cocktail [# 5872S; Cell Signaling Technology]). Cell lysates were rotated on a wheel at 4°C for 15 min and centrifuged for 10 min at 15,000 g and 4°C to pellet the cell debris. A small volume of the supernatant was used as the soluble input. Soluble detergent extracts were diluted with wash buffer (10 mM Tris/Cl, pH 7.5, 150 mM NaCl, 0.5 mM EDTA, protease/phosphatase inhibitor cocktail) and incubated with GFP-Trap agarose beads for 1 h at 4°C. Samples were then centrifuged and washed thrice with wash buffer. The agarose beads and the soluble input were then mixed with Laemmli buffer (in assay with GFP-αTAT1 and HA-14-3-3) or NuPAGE LDS Sample Buffer (in assays with GFP-αTAT1 and mCherry-KPNA) with protease and phosphatase inhibitors and boiled 5 min at 95°C before loading onto polyacrylamide gels. Gels were transferred for Western blot onto PVDF membranes, which were blocked with TBST (0.1% Tween) and 5% milk or BSA and incubated overnight with the primary antibody and 1 h with secondary antibodies. For HRP-conjugated secondary antibody used in the coimmunoprecipitation assay of GFP-αTAT1 and HA-14-3-3 proteins, bands were revealed with ECL chemiluminescent substrate (Biorad). Two different Western blots were used to visualize GFP-Input and HA-14-3-3 proteins due to similar molecular weights. For LiCOR IR-dye conjugated secondary antibodies used in the coimmunoprecipitation assays of GFP-αTAT1 and mCherry-KPNA proteins, bands were revealed using Odyssey imaging system. Antibodies used: GFP-HRP (NB600-313, Novus Biologicals), anti-HA (rat, 11867423001; Merck), anti-Exp1 (mouse, 611832; BD Transduction Laboratories), anti-GFP (mouse, sc9996; Santa Cruz), anti-mCherry (rabbit, 43590S; Cell Signaling Technology), and anti-myc Tag (rabbit, 16286-1-AP; Proteintech). Secondary HRP-conjugated antibodies (Goat-anti-rat, catalog # 112-035-003 and goat anti-mouse, catalog # 115-035-003) were purchased from Jackson ImmunoResearch. Secondary IR-dye conjugated antibodies (680RD Goat anti-Rabbit, catalog # 926-68071; 800CW Donkey anti-Mouse, catalog # 926-32212; 680RD Donkey anti-Mouse, catalog # 926-68072; 680RD Goat anti-Rat, catalog # 926-68076; 800CW Goat anti-Rat, catalog # 926-32219; 800CW Goat anti-Rabbit, catalog # 926-32211) were purchased from LiCOR.
Chemically inducible corecruitment assay
mVenus-FKBP was fused to α-TAT1, while FRB is tethered to the inner leaflet of plasma membrane using the CAAX-region of K-Ras. Upon rapamycin addition, FKBP binds to FRB, which brings the bait (mVenus-FKBP-α-TAT1) and the prey capable of binding (mCherry-KPNA or mCherry-14-3-3 isoforms) to the plasma membrane. Recruitment of the bait and the prey to the plasma membrane was detected by total internal reflection fluorescence (TIRF) microscopy as an increased fluorescence signal (Fig. S3 e). For quantification, after background subtraction, corecruitment levels of prey were measured by increase in mCherry (prey) signal normalized to the intensity before rapamycin addition. Only cells showing at least 30% increase in mVenus (bait) intensity after rapamycin addition were considered.
Microscopy and image analyses
All epifluorescence imaging was performed with an Eclipse Ti microscope (Nikon) with a 100× objective (1.49 N.A., 1.0× zoom, and 4 × 4 binning) and Zyla 4.2 sCMOS camera (Andor), driven by NIS Elements software (Nikon). All TIRF imaging was performed with an Eclipse Ti microscope (Nikon) with a 100× objective (1.49 N.A., 1.0× zoom, and 4 × 4 binning) and ORCA-FusionBT sCMOS camera (Hamamatsu) and driven by NIS Elements software (Nikon). Time-lapse imaging for α-TAT1 localization (spontaneous or with LMB treatment) was performed at 15-min intervals. Time-lapse imaging for the chemically inducible corecruitment assay was performed at 2-min intervals. All live cell imaging was conducted at 37°C, 5% CO2, and 90% humidity with a stage top incubation system (Tokai Hit). Vitamin and phenol red-free media (US Biological) supplemented with 2% fetal bovine serum were used in imaging to reduce background and photobleaching. Inhibitors and vehicles were present in the imaging media during imaging. For visualization of nuclei with Hoechst staining, cells were incubated with 250 ng/ml Hoechst dye (catalog # H3570; Invitrogen) for at least 10 min before imaging, and imaging was performed with same concentration of Hoechst dye in the imaging medium. All image processing and analyses were performed using Metamorph (Molecular Devices) and FIJI software (National Institutes of Health).
Cells that were rounded up or showed a high degree of blebbing were excluded from analysis to minimize artifacts from mitotic, apoptotic, or dead cells. For categorical analysis of mVenus-α-TAT1 localization, images were visually inspected and classified as displaying either cytosolic, diffused, or nuclear-enriched localization of mVenus fluorescence signal (Fig. S2 a). For ratiometric analysis, the ratio of the fluorescence intensity from region of interest (≈ 10 µm diameter) inside the nucleus (as identified by H2B-mCherry or Hoechst staining) to that in a perinuclear area was used to minimize any volumetric artifacts (Fig. S2 a). For all ratiometric analyses, background subtraction based on a cell-free area on each image was performed prior to calculation of the ratio. For colocalization analysis, Coloc2 function in FIJI was used to calculate the Pearson’s correlation coefficient (also called Pearson’s R) value between the mVenus-α-TAT1 image and the nuclear marker (H2B-Cherry or Hoechst). Pearson’s R provides a normalized measurement of the covariance between the pixel-by-pixel levels of α-TAT1 (mVenus-αTAT1) signal and the nuclear (H2B-mCherry or Hoechst) signal. Pearson’s R values are not dependent on signal levels (below saturation) or signal-to-noise ratio and are amenable to statistical analyses using Student’s t test, which we have used here (Dunn et al., 2011). To specifically measure the Pearson’s R value of individual cells, we drew an ROI around the whole cell and used that ROI for analysis. We report the Pearson’s R value above threshold so that only the αTAT1 and nuclear signal are considered for the measurement, and not the cell-free background areas. For all analyses, only cells showing a signal-to-noise ratio ≥2 in mVenus channel were used (∼12 bit to 16 bit range). For experiments using H2B-mCherry as nuclear marker, only cells expressing H2B-mCherry were considered for quantification. Images containing any saturated pixels in any channel (65535 value) within the cell area were excluded. Normal distribution of nuc/cyto ratio values as well as Pearson’s R-value were confirmed using normal probability plot (Fig. S1 d).
For immunofluorescence assays with exogenous expression of mVenus-α-TAT1 plasmids, transfected cells were identified by the presence of mVenus fluorescence signal. The ratio of acetylated α-tubulin over α-tubulin (Ac. α-tub/α-tub) for transfected cells was normalized against that for non-transfected cells averaged over 20 untransfected cells from the same dish. For LMB or kinase inhibitors, tubacin treatment or KO-rescue cells and the ratio of acetylated α-tubulin over α-tubulin (Ac. α-tub/α-tub) for individual cells are shown.
Statistical analyses
Microsoft Excel (Microsoft) and R (R Core Team, 2020) language with RStudio (RStudio Team, 2019) with tidyverse package (Wickham et al., 2019) were used for statistical analyses. The exact number of samples for each data set is specified in the respective figure legends. Data were pooled from at least three independent experiments performed in parallel. For the kinase inhibitor screening assay, data were pooled from at least three independent experiments for each inhibitor, but these experiments were not performed in parallel. To validate the preliminary observations from the screening assay, experiments were repeated with the inhibitors of interest with appropriate vehicle controls and performed in parallel. Sample sizes were chosen based on the commonly used range in the field without performing any statistical power analysis. Normal probability plot (Fig. S1 d) was utilized to test normal distribution of the Nuc/Cyto ratio or Pearson’s R. For immunocytochemistry assays and chemically inducible corecruitment assays, data were assumed to be normally distributed, but this was not formally tested. P values were obtained from two-tailed Student’s t test; equal variance was assumed but not formally tested. For Fig. 4 l, P values were obtained from paired one-tailed Student’s t test assuming equal variance; equal variance was assumed but not formally tested. P values >0.001 are shown in the figures.
Primer sequences
Primer sequences are listed in Table 1.
| Primer name | Sequence |
|---|---|
| BamHI-ATAT1(M1)-Fw | 5′-ATATGGATCCATGGAGTTCCCGTTCGATGTG-3′ |
| ATAT1(P284)-Stop-NotI-Re | 5′-ATATGCGGCCGCTTAGGGGCGGAGGGGACCTCGTTC-3′ |
| ATAT1(S315)-Stop-NotI-Re | 5′-ATATGCGGCCGCttaGCTGCCCCCAGGGTCAGCAGCCAACAGAAGGCGGGCGGTAGGGTG-3′ |
| ATAT1(NLS R/A)-Stop-NotI-Re | 5′-ATATGCGGCCGCTTATGCGGTAGCAGCAGCTTGAGCTGGGCTGCCCCCAGGGTCAGCAGC-3′ |
| ATAT1(NES VL/A)-Re | 5′-GCATGCGCGTGCGGAACGTGCTGCCTCCTGCTCTGGTGCAAAGGGGCGGAGGGGACCTCG-3′ |
| ATAT1(NES VL/A)-Fw | 5′-GCACCAGAGCAGGAGGCAGCACGTTCCGCACGCGCATGCCCCCCACACCCTACCGCC-3′ |
| ATAT1(S236)-Stop-NotI-Re | 5′-ATATGCGGCCGCTTAGGAGTATGGCTTGATGTCTCC-3′ |
| ATAT1(D157N)-Re | 5′-CAGCAGCTTCTGTGAGGGTCGGTTAATTGCCAGTTGGTGCGGTTC-3′ |
| ATAT1(D157N)-Fw | 5′-GAACCGCACCAACTGGCAATTAACCGACCCTCACAGAAGCTGCTG-3′ |
| ATAT1(S315D,T322E)-Re | 5′-TATGCGGCCGCTTACCTTTCGCGACGACGTTGAGCTGGATCGCCCCCAGGGTCAGCAGCC-3′ |
| ATAT1(S315A,T322A)-Re | 5′-TATGCGGCCGCTTACCTCGCGCGACGACGTTGAGCTGGCGCGCCCCCAGGGTCAGCAGCC-3′ |
| BamHI-GS-ATAT1(S236)-Fw | 5′-ATATGGATCCGGATCTTCTAGTGACCGAGAATTTCTG-3′ |
| BamHI-GS-ATAT1(L282)-Fw | 5′-ATATGGATCCGGATCTCTCCGCCCCTTTGTGCCAGAG-3′ |
| ATAT1(L/F -> A)-Re | 5′-TGCTGCCTCCTGCTCTGGTGCTGCGGGGCGTGCGGGACCTCGTTCTGGTGAGTTTC-3′ |
| ATAT1(T322A)-Stop-NotI-Re | 5′-ATATGCGGCCGCTTACCTGGCGCGACGACGTTGAGCTGGGCTG-3′ |
| ATAT1(T322E)-Stop-NotI-Re | 5′-ATATGCGGCCGCTTACCTTTCGCGACGACGTTGAGCTGGGCTG-3′ |
| ATAT1(S315A)-Stop-NotI-Re | 5′-TATGCGGCCGCTTACCTGGTGCGACGACGTTGAGCTGGGGCGCCCCCAGGGTCAGCAGCC-3′ |
| ATAT1(S315D)-Stop-NotI-Re | 5′-TATGCGGCCGCTTACCTGGTGCGACGACGTTGAGCTGGDTCGCCCCCAGGGTCAGCAGCC-3′ |
| ATAT1(V286, L291-2/A)-Fw | 5′-GCACCAGAGCAGGAGGCAGCACGTTCCTTGCGCCTCTGCCCCCCACAC-3′ |
| ATAT1(R321)-Stop-NotI-Re | 5′-ATATGCGGCCGCTTAGCGACGACGTTGAGCTGGGCTG-3′ |
| BamHI-GS-KPNA4-Fw | 5′-ATATGGATCCGGATCTGCGGACAACGAGAAACTGGACAAC-3′ |
| BamHI-GS-14-3-3-β-Fw | 5′-ATATGGATCCGGTTCAATGACAATGGATAAAAGTGAG-3′ |
| 14-3-3-β-Stop-NotI-Re | 5′-ATATGCGGCCGCTTAGTTCTCTCCCTCCCCAGCGTCTCCTTCGTCTCCCTGGTTTTC-3′ |
| BamHI-GS-14-3-3-γ-Fw | 5′-ATATGGATCCGGTTCAATGGTGGACCGCGAGCAACTG-3′ |
| 14-3-3-γ-Stop-NotI-Re | 5′-ATATGCGGCCGCTTAATTGTTGCCTTCGCCGCCATC-3′ |
| BamHI-GS-14-3-3-ε-Fw | 5′-ATATGGATCCGGTTCAATGGATGATCGGGAGGATCTG-3′ |
| BamHI-GS-14-3-3-η-Fw | 5′-ATATGGATCCGGTTCAATGGGGGACCGGGAGCAGCTG-3′ |
| 14-3-3-η-Stop-NotI-Re | 5′-ATATGCGGCCGCTCAGTTGCCTTCTCCTGCTTCTTC-3′ |
| BamHI-GS-14-3-3-θ-Fw | 5′-ATATGGATCCGGTTCAATGGAGAAGACTGAGCTGATC-3′ |
| 14-3-3-θ-Stop-NotI-Re | 5′-ATATGCGGCCGCTTAGTTTTCAGCCCCTTCTGCCGCATCACATTCTTCTCCTGCACTGTCTGATGTCCAAAG-3′ |
| Cdk1(D146N)-Re | 5′-CCAAAAGCTCTGGCAAGGCCAAAATtAGCCAGTTTAATTGTTCCTTTGTC-3′ |
| Cdk1(D146N)-Fw | 5′-GACAAAGGAACAATTAAACTGGCTaATTTTGGCCTTGCCAGAGCTTTTGG-3′ |
| BamHI-Kozak-Cdk1(M1)-Fw | 5′-ATATGGATCCGCCACCATGGAAGATTATACCAAAATAGAG-3′ |
| Cdk1-NotI-Re | 5′-ATATGCGGCCGCACATCTTCTTAATCTGATTGTC-3′ |
| BamHI-GS-CK2alpha(M1)-Fw | 5′-ATATGGATCCGGTTCCATGTCGGGACCCGTGCCAAGC-3′ |
| CK2alpha(Q391)-Stop-Re | 5′-ATATGCGGCCGCTTACTGCTGAGCGCCAGCGGCAGC-3′ |
| CK2alpha(K68A)-Re | 5′-CTTCTTTTTTACTGGCTTGAGAATCGCAACAACAACTTTTTCATTATTTG-3′ |
| CK2alpha(K68A)-Fw | 5′-CAAATAATGAAAAAGTTGTTGTTGCGATTCTCAAGCCAGTAAAAAAGAAG-3′ |
| BamHI-GS-PPP2CA(M1)-Fw | 5′-ATATGGATCCGGTTCAATGGACGAGAAGGTGTTCACCAAG-3′ |
| PPP2CA-Stop-NotI-Re | 5′-ATATGCGGCCGCTTACAGGAAGTAGTCTGGGGTACGACG-3′ |
| BamHI-R18-Fw | 5′-ATATGGATCCCCTCACTGCGTGCCCAGAGAC-3′ |
| DIFOPEIN1-Re | 5′-GTGAGGAGCTCCATCAGCACTGTCGAGTCCTGCTGCTCCGGGCAGGCACATGTTGGCCTC-3′ |
| DIFOPEIN2-Re1 | 5′-TGGCCTCCAGGTCCAGCCAGCTCAGGTCTGGGCACGCAGTGAGGAGCTCCATCAGCAC-3′ |
| DIFOPEIN3-EcoRI-Re | 5′-ATATGAATTCCAGTCCTGCTGCTCCAGAGGGCAGGCACATGTTGGCCTCCAGGTCCAGCC-3′ |
Online supplementary material
This manuscript has five supplementary figures. Fig. S1 shows the computational prediction of putative NES and NLS in α-TAT1 C-terminus. Fig. S2 shows the quantitative approaches to characterize dynamic intracellular localization of α-TAT1. Fig. S3 shows the significance of phospho-inhibited NLS in α-TAT1 localization and function. Fig. S4 specifies the kinases and phosphatases involved in regulation of the α-TAT1 NLS. Fig. S5 characterizes the role of 14-3-3 association with α-TAT1 and its impact on KPNA binding.
Data availability
All data and constructs used in this project are available upon reasonable request.
Acknowledgments
We thank Allen Kim for discussions that led to initiation of this project, Amy F. Peterson for help with the kinase inhibitor assays. We thank Francesc Garcia-Gonzalo, Dipika Gupta, Yuta Nihongaki, and Helen D. Wu for constructive discussions, and Robert DeRose for proofreading.
This project was supported by American Heart Association fellowship 20POST35220046 (A. Deb Roy), discretionary funds (T. Inoue), the La Ligue contre le cancer (S-CR17017), and Centre National de la Recherche Scientifique and Institut Pasteur. S. Seetharaman is funded by the ITN PolarNet Marie Curie grant and Fondation pour la Recherche Médicale and is enrolled at the Ecole Doctorale Frontières du Vivant—Programme Bettencourt.
The authors declare no competing financial interests.
Author contributions: A. Deb Roy initiated the project and designed and performed most of the experiments and data analyses. G.S. Pillai and E.G. Gross performed experiments and data analyses under the guidance of A. Deb Roy and T. Inoue. A. Deb Roy and S.Seetharaman performed the immunoprecipitation assays under the guidance of S. Etienne-Manneville and T. Inoue. A. Deb Roy and T. Inoue wrote the final version of the manuscript based on contributions from all the authors.

