


Myeloproliferative neoplasms (MPNs) are frequently driven by mutations within the C-terminal domain (C-domain) of calreticulin (CRT). CRTDel52 and CRTIns5 are recurrent mutations. Oncogenic transformation requires both mutated CRT and the thrombopoietin receptor (Mpl), but the molecular mechanism of CRT-mediated constitutive activation of Mpl is unknown. We show that the acquired C-domain of CRTDel52 mediates both Mpl binding and disulfide-linked CRTDel52 dimerization. Cysteine mutations within the novel C-domain (C400A and C404A) and the conserved N-terminal domain (N-domain; C163A) of CRTDel52 are required to reduce disulfide-mediated dimers and multimers of CRTDel52. Based on these data and published structures of CRT oligomers, we identify an N-domain dimerization interface relevant to both WT CRT and CRTDel52. Elimination of disulfide bonds and ionic interactions at both N-domain and C-domain dimerization interfaces is required to abrogate the ability of CRTDel52 to mediate cell proliferation via Mpl. Thus, MPNs exploit a natural dimerization interface of CRT combined with C-domain gain of function to achieve cell transformation.
Introduction
Myeloproliferative neoplasms (MPNs) comprising polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF) are hematopoietic stem cell disorders characterized by the overproduction of myeloid lineage cells (reviewed in Campbell and Green, 2006). Somatic mutations resulting from deletions or insertions in exon 9 of the CALR gene were identified in the majority of patients with PV, ET, and MF who were negative for mutations in Janus kinase 2 (JAK2) and in the thrombopoietin receptor/myeloproliferative leukemia protein (Mpl; Klampfl et al., 2013; Nangalia et al., 2013). A majority of CALR-mutated patients have one of two gene variants: type 1 with a 52-bp deletion (Del52) or type 2 with a 5-bp insertion (Ins5; Klampfl et al., 2013; Nangalia et al., 2013). The mutations change the sequence of the acidic C-terminus of CRT to a basic sequence and cause loss of the ER retention KDEL sequence (Klampfl et al., 2013; Nangalia et al., 2013).
Calreticulin (CRT) is an ER chaperone that functions in the folding and assembly of glycoproteins (Michalak et al., 2009; Raghavan et al., 2013). CRT has specificity for monoglucosylated N-linked glycans, which are transiently present during glycoprotein maturation in the ER. MPN-linked CRT mutants induce specific amplification of the megakaryocyte lineage of cells and increase platelet production (Araki et al., 2016; Balligand et al., 2020; Chachoua et al., 2016; Marty et al., 2016; Shide et al., 2017). CRT mutants mediate constitutive activation of Mpl and downstream signaling pathways (Araki et al., 2019; Araki et al., 2016; Chachoua et al., 2016; Elf et al., 2018; Elf et al., 2016; Kollmann et al., 2017; Marty et al., 2016; Shide et al., 2017).
Previous studies demonstrated a critical role of glycan-binding site residues of CRT in the recognition of sugars linked to N117 of Mpl (Chachoua et al., 2016; Elf et al., 2018; Masubuchi et al., 2020; Pecquet et al., 2019). The interaction is thought to initiate in the ER, where CRTDel52 acts as a chaperone for Mpl, and the complexes are cotrafficked to the cell surface with partially mature Mpl via the secretory pathway (Masubuchi et al., 2020; Pecquet et al., 2019). Secreted CRT does not mediate paracrine activation of Mpl (Han et al., 2016). This is possibly explained by the absence of immature monoglucosylated N117-linked glycan on cell surface Mpl. Since both WT and mutant CRT are, in principle, capable of glycan-mediated interactions with Mpl, glycan-binding alone cannot account for mutant CRT-mediated Mpl activation. The novel basic amino acids in the C-termini of mutant CRTs were shown to be critical for inducing cell proliferation and developing the ET phenotype (Araki et al., 2019; Elf et al., 2016; Shide et al., 2017). Studies of truncated mutants revealed that residues 376–383 from the C-terminus of CRTDel52 are required to activate Mpl-mediated signaling (Elf et al., 2018). However, the molecular contacts between Mpl and the C-terminal tail of CRT mutants remain undefined. Here, we demonstrate a direct role of the C-terminal tail of CRTDel52 in Mpl binding and in conferring Mpl specificity to CRTDel52.
Ligand-induced dimerization of receptor molecules is an established paradigm for signal transduction mediated by cytokine receptors (Baker et al., 2007; Moraga et al., 2014), including the erythropoietin receptor (Mohan et al., 2019; Syed et al., 1998) and Mpl (Matthews et al., 2011). While homooligomeric forms of MPN mutant CRT have been described and implicated in Mpl activation and cytokine-independent cell growth (Araki et al., 2019; Charonis et al., 2017), the nature of the productive oligomers of CRT mutants that may trigger Mpl dimerization and activation has not been established. Here, based on the occurrence of novel cysteine residues in the CRT mutant C-termini (Klampfl et al., 2013; Nangalia et al., 2013), we investigated the relevance of disulfide bond–mediated interactions in CRT multimerization in primary patient platelets and human cell lines expressing recombinant mutant CRT. In addition, we tested the relevance of noncovalent interactions relevant to CRT multimerization.
Results
Mutant CRTs form disulfide-stabilized multimers in MPN patient platelets
An antibody [anti-CRT(Cmut)] was raised against the C-terminal 22 residues of mutant CRT, within the novel mutant-specific C-termini. To assess mutant CRT multimerization in primary cells, which has not previously been undertaken, platelets were purified from the blood of healthy donors or MPN patients with known clinical characteristics, as summarized in Table 1. Anti-CRT(Cmut) specifically detected a 50-kD band in platelet lysates from patients with CRT mutants but not from patients with JAK2 mutants or healthy control individuals (Fig. 1 A; and Fig. S1, A and B, top panel). As expected, based on size, mutant CRT in samples 8744 and 4995-2, which correspond to CRTDel4 and CRTIns5 mutations, migrate more slowly than the CRTDel52 samples in the same blots (Fig. 1 A and Fig. S1 A, top panel). The levels of mutant CRT expression were somewhat variable between patients (Fig. S1, A and B).
Details of MPN patient samples used in this study.
| Patient ID | Age/sex | Mutation | Platelets (K/µl) | Disease |
|---|---|---|---|---|
| 5502-1 | 44/F | CRT 52-bp del | 622 | ET |
| 5502-5 | 44/F | CRT 52-bp del | 441 | ET |
| 8744 | 75/M | CRT 4-bp del | 559 | ET |
| 1526 | 75/F | CRT 52-bp del | 270 | ET |
| 6105 | 31/F | CRT 52-bp del | 341 | ET |
| 2648-2 | 70/M | CRT 52-bp del | 389 | ET |
| 2648-3 | 70/M | CRT 52-bp del | 399 | ET |
| 6102 | 34/M | CRT 52-bp del | 312 | PMF |
| 4995-2 | 45/F | CRT 5-bp ins | 300 | ET |
| 3829 | 48/M | CRT 52-bp del | 738 | ET |
| 7245 | 30/F | CRT 52-bp del | 433 | ET |
| 8742 | 56/F | CRT 52-bp del | 178 | PMF |
| 8251-2 | 72/F | CRT 5-bp ins | 558 | PMF |
| 1718 | 81/F | CRT 52-bp del | 261 | PMF |
| 2791-1 | 62/M | CRT 52-bp del | 91 | Post-ET MF |
| 1521 | 78/M | CRT 52-bp del | 273 | MF |
| 2028-1 | 59/M | CRT 52-bp del | 400 | Post-ET MF |
| 1244 | 71/M | JAK2V617F | 98 | PMF |
| 9813 | 84/M | JAK2V617F | 420 | PMF |
| 2161 | 48/M | JAK2V617F | 422 | PV |
| 4493 | 69/F | JAK2V617F | 172 | Post-PV MF |
| 2791-4 | 64/M | CRT 52-bp del | 68 | Post-ET MF |
| 7313-1 | 61/M | CRT 5-bp ins | 139 | MF |
For some patients, multiple blood collections on different dates were used for this study. These are indicated by hyphens and the internal blood collection number following the patient ID number. del, deletion; F, female; ins, insertion; M, male.

Mutant CRTs form disulfide-stabilized multimers in MPN patient platelets and are detectable in MPN patient serum. (A–C and E–G) Lysates from MPN patients or healthy donor platelets were probed by SDS-PAGE under reducing or nonreducing conditions (using 4–20% gradient gels) or by native-PAGE, as indicated, followed by immunoblotting with indicated antibodies. In E, boxes indicate hemoglobin (Hb) contamination. (D) CRT secretion in healthy donor or MPN patient serum was examined by IP and immunoblot analyses with anti-CRT (Cmut) (top panel) or anti-CRT (Thermo Fisher Scientific) antibodies (bottom panel). No antibody lanes are controls in the absence of anti-CRT antibodies to assess nonspecific precipitation. In all panels, HC indicates healthy control samples, and CRT mutant patient samples are indicated as Del52 or Del4 (characterized clinically as a 4-bp deletion in exon 9 resulting in a frameshift at K374 based on next-generation sequencing). The predicted size of Del4 is 427 aa compared with 411 aa for Del52. See Fig. S1 for additional replicate blots of platelet lysates. Ab, antibody; MW, molecular weight.
Mutant CRTs form disulfide-stabilized multimers in MPN patient platelets and are detectable in MPN patient serum. (A–C and E–G) Lysates from MPN patients or healthy donor platelets were probed by SDS-PAGE under reducing or nonreducing conditions (using 4–20% gradient gels) or by native-PAGE, as indicated, followed by immunoblotting with indicated antibodies. In E, boxes indicate hemoglobin (Hb) contamination. (D) CRT secretion in healthy donor or MPN patient serum was examined by IP and immunoblot analyses with anti-CRT (Cmut) (top panel) or anti-CRT (Thermo Fisher Scientific) antibodies (bottom panel). No antibody lanes are controls in the absence of anti-CRT antibodies to assess nonspecific precipitation. In all panels, HC indicates healthy control samples, and CRT mutant patient samples are indicated as Del52 or Del4 (characterized clinically as a 4-bp deletion in exon 9 resulting in a frameshift at K374 based on next-generation sequencing). The predicted size of Del4 is 427 aa compared with 411 aa for Del52. See Fig. S1 for additional replicate blots of platelet lysates. Ab, antibody; MW, molecular weight.

Mutant CRT is multimeric via disulfide bonds in MPN patient platelets. (A) SDS-PAGE (8% gels) under reducing conditions and immunoblots of platelet lysates from healthy donors and MPN patient platelets were probed with the indicated antibodies. The same lysates were loaded onto two gels and probed with anti-CRT(Cmut) (top panel) or anti-CRT(N) (middle panel) antibodies. The latter was reprobed with anti-vinculin (lower panel) antibody. Anti-CRT(Cmut) does not detect WT CRT in healthy control (HC) lysates. Lysate 4995 is a CRTIns5 mutant, which based on size (Fig. 2 A), should migrate more slowly than CRTDel52 (all other samples in the blot). (B) Lysates from MPN patients or healthy donor platelets were separated by reducing or nonreducing SDS-PAGE (4–20% gradient gels), and the same lysates were loaded onto two gels probed with anti-CRT(Cmut) (top panel) or anti-CRT(N) (middle panel) antibodies. The former was reprobed with anti-GAPDH antibody. Monomer and multimer CRT bands are indicated. Bands indicated as dimers or multimers are overrepresented under nonreducing conditions in the CRT mutant platelet lysates, whereas monomer bands are depleted under the same conditions for 8251-2 and 2648-3, which appear to have a high mutational load. The lower expression of mutant CRT in 2028-1 patient lysate precluded its detection under nonreducing conditions at greater than background levels. Anti-CRT(Cmut) does not detect WT CRT in HC lysates; hence, no bands are visualized in lanes labeled HC in immunoblots with anti-CRT(Cmut). Nonspecific bands that are detected in the reducing blots are marked as such. (C and D) Native immunoblots (8% gels) of platelet lysates (25 µg) from MPN patient platelets or same-day healthy donor platelets or RBC lysates were probed with anti-CRT(Cmut) (left panels) and anti-CRT(N) (right panels) antibodies. Oligomeric forms of mutant CRT are readily detectable at the top of the gel, particularly with samples such as 2648-2, which showed high expression of mutant CRT. Boxes indicate hemoglobin (Hb) contamination of platelets, verified using RBC lysates. CRT HC indicates HC samples; CRT mutant patient samples are indicated as Del52 or Ins5; JAK2 indicates JAK2 mutant patient samples; and RBC indicates RBC lysates. MW, molecular weight.
Mutant CRT is multimeric via disulfide bonds in MPN patient platelets. (A) SDS-PAGE (8% gels) under reducing conditions and immunoblots of platelet lysates from healthy donors and MPN patient platelets were probed with the indicated antibodies. The same lysates were loaded onto two gels and probed with anti-CRT(Cmut) (top panel) or anti-CRT(N) (middle panel) antibodies. The latter was reprobed with anti-vinculin (lower panel) antibody. Anti-CRT(Cmut) does not detect WT CRT in healthy control (HC) lysates. Lysate 4995 is a CRTIns5 mutant, which based on size (Fig. 2 A), should migrate more slowly than CRTDel52 (all other samples in the blot). (B) Lysates from MPN patients or healthy donor platelets were separated by reducing or nonreducing SDS-PAGE (4–20% gradient gels), and the same lysates were loaded onto two gels probed with anti-CRT(Cmut) (top panel) or anti-CRT(N) (middle panel) antibodies. The former was reprobed with anti-GAPDH antibody. Monomer and multimer CRT bands are indicated. Bands indicated as dimers or multimers are overrepresented under nonreducing conditions in the CRT mutant platelet lysates, whereas monomer bands are depleted under the same conditions for 8251-2 and 2648-3, which appear to have a high mutational load. The lower expression of mutant CRT in 2028-1 patient lysate precluded its detection under nonreducing conditions at greater than background levels. Anti-CRT(Cmut) does not detect WT CRT in HC lysates; hence, no bands are visualized in lanes labeled HC in immunoblots with anti-CRT(Cmut). Nonspecific bands that are detected in the reducing blots are marked as such. (C and D) Native immunoblots (8% gels) of platelet lysates (25 µg) from MPN patient platelets or same-day healthy donor platelets or RBC lysates were probed with anti-CRT(Cmut) (left panels) and anti-CRT(N) (right panels) antibodies. Oligomeric forms of mutant CRT are readily detectable at the top of the gel, particularly with samples such as 2648-2, which showed high expression of mutant CRT. Boxes indicate hemoglobin (Hb) contamination of platelets, verified using RBC lysates. CRT HC indicates HC samples; CRT mutant patient samples are indicated as Del52 or Ins5; JAK2 indicates JAK2 mutant patient samples; and RBC indicates RBC lysates. MW, molecular weight.
In contrast to anti-CRT(Cmut), anti-CRT(N), a commercial antibody from Cell Signaling Technology (CST) directed against the N-terminus of CRT, detected CRT in all samples (Fig. 1 B; and Fig. S1, A and B, middle panels). As discussed below, in transfected human embryonic kidney (HEK) 293T cells, anti-CRT(N) can distinguish CRTDel52 from CRTWT based on the smaller size of CRTDel52. However, two distinct CRT bands were not readily detectable with anti-CRT(N) in lysates from MPN patient platelets (Fig. 1 B; and Fig. S1, A and B, middle panels), except for the most highly expressed samples, such as 2648-2 (Fig. S1 A), indicating that the detected protein corresponds to CRTWT. The C-terminal frameshift in all MPN mutant CRT results in a loss of the ER retention KDEL motif, causing their secretion from cells (Arshad and Cresswell, 2018; Garbati et al., 2016; Han et al., 2016; Liu et al., 2020), and indeed, the mutants but not WT CRT are detectable in patient serum by coimmunoprecipitation (coIP) analyses (Fig. 1 D). Enhanced secretion is expected to render all MPN CRT mutants more difficult to detect than the WT CRT using generic anti-CRT antibodies in platelet lysates.
Analyses of the platelet lysates on nonreducing gels indicated that the mutant CRT species are DTT sensitive, which was readily apparent in immunoblots with anti-CRT(Cmut) for most samples, and with both anti-CRT(Cmut) and anti-CRT(N) for the high-expressing samples 2648-3 and 8251-2 (Fig. 1, A and B; and Fig. S1 B; reducing gels [+DTT] compared with nonreducing gels [−DTT]). Notably, a band consistent with a dimer of CRT (∼100 kD) was visualized in the nonreducing gels with anti-CRT(Cmut) in several mutant CRT samples, in addition to several heterogeneous higher-molecular-weight species (Fig. 1 A and Fig. S1 B, top panel).
We also conducted immunoblots of the platelet lysates following native-PAGE. The major specific bands detected (not observed in healthy controls) had low mobility, migrated close to the stacking gel, and were detectable with both anti-CRT(Cmut) and anti-CRT(N) in the CRT mutant samples (Fig. 1, E and F; and Fig. S1, C and D). Some platelet preparations show a band above the 64 kD marker that likely represents a contamination of hemoglobin, based on the appearance of the same background in RBC preparations (Fig. 1, E and F, boxed; and Fig. S1 D, which shows the RBC lysate). This band was eliminated when additional steps were taken to minimize RBC contamination in a new set of patient platelet preparations, where only the low-mobility species were observed with anti-CRT(Cmut) (Fig. 1 G, right panel). Thus, CRT mutants form heterogeneous disulfide-bonded species in patient platelets, and the predominant species observed in native blots displays low mobility.
The C-terminal domain (C-domain) of CRTDel52 confers specificity for Mpl and forms disulfide-linked dimers
The novel C-domains of type I and type II MPN mutant CRT contain two or three cysteine residues (Fig. 2 A). To further study the C-domain–mediated multimerization and function, the C-domains of CRTWT, CRTDel52, and different truncation mutants of CRTDel52 (Fig. 2 A) were expressed as histidine and GB1 (his-GB1)–tagged proteins in HEK293T cells (Fig. 2 B). All constructs encoded the signal sequences of cyclooxygenase 2 (Cox2) to allow insertion into the ER lumen. Since the CRTDel52 mutation was the most frequent in our patient group (Table 1), our studies were focused on this mutant. The C-domain of CRTDel52 was expressed at higher levels than the C-domain of CRTWT (Fig. 2 B, lane 1 vs. 2). Truncations of CRTDel52 C-domain progressively reduced expression levels (Fig. 2 B, lanes 1–6). Using coIP assays, binding interactions were observed between Mpl and the isolated C-domains of all the CRTDel52 constructs, except the poorly expressed CRTDel52Δ36, whereas binding between Mpl and the C-domain of CRTWT was not observed (Fig. 2 B, lanes 7–12). The binding of C-domains to Mpl is specific, as protein G on the beads did not bind the C-domain constructs in the absence of anti-Mpl (Fig. 2 B, lysate + beads lanes).

The C-domain of CRTDel52 confers specificity for Mpl and forms disulfide-linked dimers. (A) Structure of CRTWT (Protein Data Bank accession no. 6ENY, subunit G; Blees et al., 2017) with a globular domain (yellow, residues 18–203 from N-domain and 301–335 from C-domain), a P-domain (blue, residues 206–300), and an α-helical (residues 336–386) C-domain (red). The C-terminus of the C-domain (pink, residues 387–417) is not resolved in the structure. Sequences of CRTDel52 C-domain truncation constructs are shown. The lower panel shows indicated C-domain sequences (line 1), secondary structure predictions performed using I-TASSER (Roy et al., 2010; lines 2 and 3; line 3 is the confidence score [conf. score] of the prediction), and the secondary structure based on 6eny or the CRTDel52 model discussed in this study (line 4). The sequence of the mutant-specific C-tail used to produce the anti-CRT(Cmut) antibody is underlined, and cysteine residues are in large font. (B and D) IP with anti-Mpl antibody or control IP (no antibody marked as lysate + beads) of lysates from HEK293T cells (B) or Ba/F3-Mpl cells (D) expressing Mpl along with his-GB1–tagged C-domain constructs (B and D) or full-length CRTDel52 (D), as indicated, and subsequent blots with the indicated antibodies. Data are representative of three independent experiments (from two separate transductions for D). (C and F) lysates from HEK293T cells expressing indicated CRTDel52 C-domain constructs were separated by SDS-PAGE under reducing (12.5% gels) or nonreducing (4–20% gradient gels) conditions and immunoblotted with indicated antibodies. Data are representative of three (C) or one (F) independent experiments (see also panel B for relative expression levels of WT CRT vs. CRTDel52 C-domain expression). (E) Proliferation of Ba/F3-Mpl cells expressing His and GB1-tagged C-domains or full-length untagged CRTDel52 or lacking CRTDel52 (Vec) as indicated in the absence of mouse IL-3. Data are averaged from two separate transductions of Ba/F3-Mpl cells with the relevant viruses in a total of four analyses. Ab, antibody; MW, molecular weight.
The C-domain of CRTDel52 confers specificity for Mpl and forms disulfide-linked dimers. (A) Structure of CRTWT (Protein Data Bank accession no. 6ENY, subunit G; Blees et al., 2017) with a globular domain (yellow, residues 18–203 from N-domain and 301–335 from C-domain), a P-domain (blue, residues 206–300), and an α-helical (residues 336–386) C-domain (red). The C-terminus of the C-domain (pink, residues 387–417) is not resolved in the structure. Sequences of CRTDel52 C-domain truncation constructs are shown. The lower panel shows indicated C-domain sequences (line 1), secondary structure predictions performed using I-TASSER (Roy et al., 2010; lines 2 and 3; line 3 is the confidence score [conf. score] of the prediction), and the secondary structure based on 6eny or the CRTDel52 model discussed in this study (line 4). The sequence of the mutant-specific C-tail used to produce the anti-CRT(Cmut) antibody is underlined, and cysteine residues are in large font. (B and D) IP with anti-Mpl antibody or control IP (no antibody marked as lysate + beads) of lysates from HEK293T cells (B) or Ba/F3-Mpl cells (D) expressing Mpl along with his-GB1–tagged C-domain constructs (B and D) or full-length CRTDel52 (D), as indicated, and subsequent blots with the indicated antibodies. Data are representative of three independent experiments (from two separate transductions for D). (C and F) lysates from HEK293T cells expressing indicated CRTDel52 C-domain constructs were separated by SDS-PAGE under reducing (12.5% gels) or nonreducing (4–20% gradient gels) conditions and immunoblotted with indicated antibodies. Data are representative of three (C) or one (F) independent experiments (see also panel B for relative expression levels of WT CRT vs. CRTDel52 C-domain expression). (E) Proliferation of Ba/F3-Mpl cells expressing His and GB1-tagged C-domains or full-length untagged CRTDel52 or lacking CRTDel52 (Vec) as indicated in the absence of mouse IL-3. Data are averaged from two separate transductions of Ba/F3-Mpl cells with the relevant viruses in a total of four analyses. Ab, antibody; MW, molecular weight.
The findings of Fig. 2 B parallel the previous findings of preferential interactions between the full-length versions of MPN mutant CRT and Mpl (Araki et al., 2019; Araki et al., 2016; Chachoua et al., 2016; Elf et al., 2018; Elf et al., 2016; Masubuchi et al., 2020; Pecquet et al., 2019). Correspondingly, in an anti-Mpl coIP with HEK293T cells transfected with plasmids encoding Mpl and full-length mutant CRT constructs, binding was detectable between CRT mutants and Mpl using anti-CRT(Cmut) (Fig. S2 A, top and middle panels for Mpl and anti-CRT(Cmut) blots). On the other hand, CRTWT signals probed with anti-CRT(Thermo), a commercial polyclonal antibody directed against the whole protein (Thermo Fisher Scientific), were essentially undetectable in the steady state following anti-Mpl coIPs (Fig. S2 A, lower panel). WT but not mutant protein overexpression was detectable with anti-CRT(Thermo) (Fig. S2 A, compare lanes 1–3 with lane 4). As noted above, anti-CRT(Cmut) allows for more sensitive detection of the mutants. It is also possible that anti-CRT(Thermo) epitopes reside within the C-terminus. Preferential binding between mutant CRT and Mpl was also detectable in patient platelets (Fig. S2, B and C).

Mutant CRT preferentially interacts with Mpl in HEK293T cells and MPN patient platelets. (A) Lysates from HEK293T cells expressing full-length untagged WT or mutant CRTs (Ins5 or Del52) and Mpl or control cells expressing Mpl alone (Vec) were immunoprecipitated with anti-Mpl antibody, and subsequent immunoblotting was undertaken with the indicated antibodies. Data shown are representative of two independent experiments. (B and C) Lysates from indicated healthy donor (HC) or MPN patient platelets (Del52) were immunoprecipitated with anti-Mpl antibody (B) or anti-CRT(Cmut) antibody (C) and subsequently analyzed by immunoblotting with the indicated antibodies. CRT mutant patients are indicated as Del52. In all panels, nonspecific interactions in the absence of primary antibody are shown by the lysate + beads lanes. Ab, antibody; MW, molecular weight.
Mutant CRT preferentially interacts with Mpl in HEK293T cells and MPN patient platelets. (A) Lysates from HEK293T cells expressing full-length untagged WT or mutant CRTs (Ins5 or Del52) and Mpl or control cells expressing Mpl alone (Vec) were immunoprecipitated with anti-Mpl antibody, and subsequent immunoblotting was undertaken with the indicated antibodies. Data shown are representative of two independent experiments. (B and C) Lysates from indicated healthy donor (HC) or MPN patient platelets (Del52) were immunoprecipitated with anti-Mpl antibody (B) or anti-CRT(Cmut) antibody (C) and subsequently analyzed by immunoblotting with the indicated antibodies. CRT mutant patients are indicated as Del52. In all panels, nonspecific interactions in the absence of primary antibody are shown by the lysate + beads lanes. Ab, antibody; MW, molecular weight.
In lysates of transfected HEK293T cells, a band consistent with the size of a C-domain dimer was detected under nonreducing conditions for the CRTDel52-C but not its cysteine mutant CRTDel52-C(C44A/C48A) (Fig. 2 C, right panel, lanes 2 and 3). Thus, the mutant C-domain, when expressed on its own, is capable of forming disulfide-linked dimers. Binding interactions between Mpl and the CRTDel52 full-length and C-domain constructs were also observed in Ba/F3 cells. The efficiency of Mpl binding to the C-domain constructs appears to be at least as high as that for full-length CRTDel52 (Fig. 2 D; lane 2 compared with lane 4), even though expression of the C-domain constructs is not directly detectable in Ba/F3 lysates. MPN mutant CRT constructs are known to induce proliferation of Ba/F3 cells in an Mpl-dependent and a cytokine-independent manner (Chachoua et al., 2016; Pecquet et al., 2019), as also shown in Fig. 2 E. Consistent with previous findings (Elf et al., 2016), the CRTDel52 C-domain alone is insufficient to mediate proliferation (Fig. 2 E). Thus, while the CRTDel52 C-domains confer Mpl binding specificity and dimerize, additional interactions mediated by CRTDel52 are required for functional interactions with Mpl. The relative expression level of the CRTDel52 C-domain is higher than the WT C-domain both in the presence and in the absence of Mpl (Fig. 2, B and F, respectively), consistent with the higher predicted helical content of the CRTDel52 C-domain (Fig. 2 A).
Truncations of C-terminal cysteines of full-length CRTDel52 alter but do not abrogate disulfide-linked interactions
To ask if C-domain cysteines were sufficient for full-length CRTDel52 multimerization, we generated the successive truncations of the C-terminal sequences (Fig. 2 A) within full-length CRTDel52 and expressed those constructs as N-terminal his-GB1–tagged proteins. CRTDel52Δ12 and CRTDel52Δ19 were expressed at low levels, and their protein loads had to be increased to achieve similar protein expression as the other constructs (Fig. 3 A; note the varying intensities of the endogenous CRT bands). In nonreducing SDS-PAGE gels (Fig. 3 B), bands consistent with the size of dimers/multimers were observed for CRTDel52, CRTDel52Δ12, CRTDel52Δ19, and CRTDel52Δ28, even though all the truncation constructs of CRTDel52 lacked the two C-terminal cysteines. However, the specific band indicated as dimers for CRTDel52 was absent in CRTDel52Δ12, CRTDel52Δ19, and CRTDel52Δ28, and instead, slower mobility bands were observed (Fig. 3 B, lane 3 compared with lanes 4–6). These findings suggest that the presence of two C-terminal cysteines in CRTDel52 contribute to the induction of distinct disulfide-linked species. Additionally, the proportion of monomer bands to total CRT bands progressively increases with increased truncation size (Fig. 3 C). In native gels, monomers are undetectable for CRTDel52Δ12 and CRTDel52Δ19, begin to appear with the CRTDel52Δ28 truncation, and are the predominant species for CRTDel52Δ36 (Fig. 3 D). Together the findings of Fig. 3 suggest that novel C-domain cysteines and additional cysteines contribute to the formation of CRTDel52 disulfide-linked dimers/multimers. Further, the CRTDel52Δ28 truncation is needed to partially destabilize CRTDel52 multimers, and the CRTDel52Δ36 truncation is needed to fully destabilize CRTDel52 multimers, suggesting that both covalent and noncovalent interactions mediated by the C-domains contribute to multimer formation.

Truncations of C-terminal cysteines of full-length CRTDel52 alter but do not abrogate disulfide-linked interactions. (A, B, and D) Lysates from HEK293T cells expressing N-terminal his-GB1–tagged full-length or C-terminally truncated CRTDel52 constructs were separated by SDS-PAGE under reducing (10% gels; A) or nonreducing (10% gels; B) conditions or by native-PAGE (4–20% gradient gels; D) and immunoblotted with the anti-CRT(N) antibody. Different amounts of lysates were loaded to achieve similar protein expression of different truncated constructs: CRTWT, 0.5 µg lysates; CRTDel52, 5 µg lysates; CRTDel52Δ12, 18 µg lysates; CRTDel52Δ19, 18 µg lysates; CRTDel52Δ28, 3 µg lysates; CRTDel52Δ36, 1.8 µg lysates; or a plasmid lacking CRT (Vec), 10 µg lysates. The endogenous CRT band serves as the lysate loading controls. Species consistent with the size of endogenous CRT, his-GB1-CRT monomers, dimers, multimers, and loading wells are indicated. (C) Quantification of CRT monomer/monomer + multimer (total) bands from B averaged over six independent blots from five independent transfections. Data show mean ± SD, with statistical significance assessed via ordinary one-way ANOVA. MW, molecular weight.
Truncations of C-terminal cysteines of full-length CRTDel52 alter but do not abrogate disulfide-linked interactions. (A, B, and D) Lysates from HEK293T cells expressing N-terminal his-GB1–tagged full-length or C-terminally truncated CRTDel52 constructs were separated by SDS-PAGE under reducing (10% gels; A) or nonreducing (10% gels; B) conditions or by native-PAGE (4–20% gradient gels; D) and immunoblotted with the anti-CRT(N) antibody. Different amounts of lysates were loaded to achieve similar protein expression of different truncated constructs: CRTWT, 0.5 µg lysates; CRTDel52, 5 µg lysates; CRTDel52Δ12, 18 µg lysates; CRTDel52Δ19, 18 µg lysates; CRTDel52Δ28, 3 µg lysates; CRTDel52Δ36, 1.8 µg lysates; or a plasmid lacking CRT (Vec), 10 µg lysates. The endogenous CRT band serves as the lysate loading controls. Species consistent with the size of endogenous CRT, his-GB1-CRT monomers, dimers, multimers, and loading wells are indicated. (C) Quantification of CRT monomer/monomer + multimer (total) bands from B averaged over six independent blots from five independent transfections. Data show mean ± SD, with statistical significance assessed via ordinary one-way ANOVA. MW, molecular weight.
Disulfide-linked CRTDel52 dimer and multimer formation is C-domain and N-terminal domain (N-domain) dependent
To further elucidate the mode of CRTDel52 multimerization, we generated various cysteine mutants of CRTDel52 as untagged constructs and examined their multimerization in transfected HEK293T cells. Anti-CRT(Cmut) did not detect WT CRT expressed in HEK293T cells and was specific for the mutants (Fig. 4 A). As noted above, CRTDel52 formed dimers (∼100-kD band) and higher-order species, which were detected in immunoblots under nonreducing conditions using anti-CRT(Cmut) (Fig. 4 B, lanes 3–4). The double mutant CRTDel52-2CA [CRTDel52(C400A/C404A)] that lacks the novel cysteines in the mutant C-terminus formed fewer higher-order multimer structures and more lower-order structures compared with CRTDel52 (Fig. 4 B, lanes 5 and 6 compared with lanes 3 and 4; quantification in Fig. 4 C) when protein loads were similar (Fig. 4 A, lanes 5 and 6). Notably, the bands indicated as dimers were more intense but migrated more slowly for CRTDel52-2CA compared with the corresponding CRTDel52 construct (Fig. 4 B, lanes 3 and 4 compared with 5 and 6), suggesting that CRTDel52 dimers are more compact due to the presence of the two C-terminal cysteines, C400 and C404.

Disulfide-linked CRTDel52 dimer and multimer formation is C-domain and N-domain dependent. (A, B, D, and E) Lysates from HEK293T cells overexpressing untagged full-length CRTWT or expressing CRTDel52, CRTDel52-2CA [CRTDel52(C400A/C404A)], CRTDel52-3CA [CRTDel52(C163A/C400A/C404A)], CRTDel52-CA [CRTDel52(C163A)], or control transfected cells (Vec) were separated by SDS-PAGE under reducing (8% gels; A and D) or nonreducing (4–20% gradient gels; B and E) conditions and immunoblotted with indicated antibodies. Full-gel panels and two protein loads are shown for each condition. Data are representative of two to three independent experiments. Species consistent with the size of CRT monomers, dimers, and multimers are indicated. In panel D where CRTWT and CRTDel52 constructs are resolved, the migration position of each band is indicated as endogenous CRT and CRTDel52. (C) Quantifications of blots from B to calculate monomer/monomer + multimer (total) mutant CRT following nonreducing SDS-PAGE are shown, averaged over three independent experiments (two protein loads each). Consistent ratios are quantified at two different protein loads of each construct (labeled as 1 or 2). Data show mean ± SD, with statistical significance assessed via ordinary one-way ANOVA. (F) Cell lysates from indicated transfected cells were separated by native-PAGE (8% gels) and immunoblotted with indicated antibody. Bands corresponding to CRTDel52 monomers, multimers, and loading wells are indicated. MW, molecular weight.
Disulfide-linked CRTDel52 dimer and multimer formation is C-domain and N-domain dependent. (A, B, D, and E) Lysates from HEK293T cells overexpressing untagged full-length CRTWT or expressing CRTDel52, CRTDel52-2CA [CRTDel52(C400A/C404A)], CRTDel52-3CA [CRTDel52(C163A/C400A/C404A)], CRTDel52-CA [CRTDel52(C163A)], or control transfected cells (Vec) were separated by SDS-PAGE under reducing (8% gels; A and D) or nonreducing (4–20% gradient gels; B and E) conditions and immunoblotted with indicated antibodies. Full-gel panels and two protein loads are shown for each condition. Data are representative of two to three independent experiments. Species consistent with the size of CRT monomers, dimers, and multimers are indicated. In panel D where CRTWT and CRTDel52 constructs are resolved, the migration position of each band is indicated as endogenous CRT and CRTDel52. (C) Quantifications of blots from B to calculate monomer/monomer + multimer (total) mutant CRT following nonreducing SDS-PAGE are shown, averaged over three independent experiments (two protein loads each). Consistent ratios are quantified at two different protein loads of each construct (labeled as 1 or 2). Data show mean ± SD, with statistical significance assessed via ordinary one-way ANOVA. (F) Cell lysates from indicated transfected cells were separated by native-PAGE (8% gels) and immunoblotted with indicated antibody. Bands corresponding to CRTDel52 monomers, multimers, and loading wells are indicated. MW, molecular weight.
Additionally, MPN-linked CRT mutants have one free cysteine within the globular domain C163, which is also present in the WT CRT. Ala-substitution of C163 in CRTDel52-CA [CRTDel52(C163A)] mutant resulted in a dimer and multimer pattern similar to CRTDel52 based on the mobility of the dimer (Fig. 4 B, lanes 9 and 10), but the larger multimeric species was lower order compared with CRTDel52. However, for the triple cysteine mutant CRTDel52-3CA [CRTDel52(C163A/C400A/C404A)], monomer species were predominant (Fig. 4, B, lanes 7 and 8; and Fig. 4 C). These results indicate that all three mutations are needed to significantly inhibit the formation of disulfide-linked dimers and multimers of CRTDel52. In nonreducing blots for the triple mutant CRTDel52-3CA, in addition to the intense band corresponding to monomers, there are faint bands corresponding to disulfide-linked dimers and oligomers. The only cysteines in CRTDel52-3CA are C105 and C137, which form a disulfide-bridge within the N-domain (Chouquet et al., 2011; Kozlov et al., 2010). We suggest that the residual oligomerization of CRTDel52-3CA may be caused by formation of intermolecular disulfide bonds via C105 and C137 that undergo aberrant thiol–disulfide exchange.
Parallel immunoblots with anti-CRT(N) allowed visualization of CRTWT, which under reducing and nonreducing conditions migrated more slowly than all CRTDel52 constructs, consistent with its larger size (Fig. 4, D and E). The endogenous CRTWT was detected in all transfections. Bands corresponding to dimers and oligomers detected under nonreducing conditions resembled those detected with anti-CRT(Cmut) (Fig. 4 E). Slower migration of CRTDel52-2CA dimer bands than CRTDel52 (Fig. 4 E, lanes 3 and 4 compared with 5 and 6), and the pattern and relative mobilities of CRTDel52-CA compared with CRTDel52 were again rather noticeable (Fig. 4 E, lanes 3 and 4 compared with 9 and 10). Finally, disulfide-linked oligomers were practically absent for the triple mutant CRTDel52-3CA. Together, these findings implicate C-terminal cysteines (C400 and C404) and N-domain C163 in disulfide-mediated dimerization and multimerization of CRTDel52 mutant.
In native-PAGE gels, however, all four CRTDel52 constructs formed higher-order species (Fig. 4 F). Nonetheless, signals corresponding to monomeric species were detectable only in the CRTDel52-2CA and CRTDel52-3CA lysates (Fig. 4 F). We concluded from these analyses that both covalent and noncovalent interactions contribute to CRTDel52 multimerization. Together with the truncation mutant data (Fig. 3), these findings indicate that disulfide-dependent interactions contribute to dimer and multimer stability, but that loss of S-S bonds is not sufficient to fully block multimer formation.
Reducing and nonreducing SDS-PAGE analyses indicated that disulfide-linked dimers and multimers of CRTDel52 were present not only in CRTDel52 transfected cells but also in cell media (Fig. S3 A). Finally, oligomers of CRTIns5 constructs were observed, similarly to CRTDel52, although the overall pattern was more complex than that of CRTDel52 because of the presence of an additional C-terminal cysteine in CRTIns5 mutant (Fig. 2 A; and Fig. S3, B and C). Further studies were focused on CRTDel52 mutant.

Secreted forms of mutant CRT multimerize, and CRTIns forms disulfide-linked multimers similar to CRTDel52. (A–C) HEK293T cells were transiently transfected with plasmids encoding full-length his-GB1–tagged CRTDel52 (A) or untagged CRTWT, CRTIns5, CRTIns5-2CA [CRTIns5(C419A/C423A)], CRTIns5-3CA [CRTIns5(C163A/C419A/C423A)], or plasmid lacking CRT (Vec; B and C). Cell media from untransfected (HEK) or CRTDel52-transfected cells were purified on a nickel column and the eluates analyzed by immunoblot following SDS-PAGE under reducing or nonreducing conditions. (B and C) Lysates from indicated cells were separated by SDS-PAGE under reducing (8% gels) or nonreducing (4–20% gradient gels) conditions and immunoblotted with indicated antibodies. Data are representative of two (A) and three (B and C) sets of analyses. In blots following nonreducing SDS-PAGE, species consistent with the size of CRT monomers, dimers, and endogenous CRT are indicated. MW, molecular weight.
Secreted forms of mutant CRT multimerize, and CRTIns forms disulfide-linked multimers similar to CRTDel52. (A–C) HEK293T cells were transiently transfected with plasmids encoding full-length his-GB1–tagged CRTDel52 (A) or untagged CRTWT, CRTIns5, CRTIns5-2CA [CRTIns5(C419A/C423A)], CRTIns5-3CA [CRTIns5(C163A/C419A/C423A)], or plasmid lacking CRT (Vec; B and C). Cell media from untransfected (HEK) or CRTDel52-transfected cells were purified on a nickel column and the eluates analyzed by immunoblot following SDS-PAGE under reducing or nonreducing conditions. (B and C) Lysates from indicated cells were separated by SDS-PAGE under reducing (8% gels) or nonreducing (4–20% gradient gels) conditions and immunoblotted with indicated antibodies. Data are representative of two (A) and three (B and C) sets of analyses. In blots following nonreducing SDS-PAGE, species consistent with the size of CRT monomers, dimers, and endogenous CRT are indicated. MW, molecular weight.
To explore the possible formation of disulfide crosslinks between CRT mutants and Mpl, which contains 15 cysteines and thus at least one unpaired cysteine in its extracellular domain, we analyzed lysates from cells expressing CRTDel52 alone, Mpl alone, or both. Under nonreducing conditions, we did not observe new disulfide-linked species in cell lysates containing Mpl and CRTDel52 vs. CRTDel52 alone (Fig. S4 A, top panel) or in lysates containing Mpl alone vs. Mpl and CRTDel52 (Fig. S4 B, top panel). This result indicates that CRTDel52 does not form disulfide crosslinks with Mpl during association into a signaling complex. However, in the native gel, fast-mobility CRTDel52 bands were upshifted to slow-mobility species in cell lysates coexpressing Mpl and CRTDel52 (Fig. S4 A, lower panel), resembling native gels of platelet lysates (Fig. 1, E and G; and Fig. S1, C and D), which may also contain Mpl. Thus, Mpl binding induces a distinct shift in CRTDel52 migration in native gels, suggesting the formation of high-molecular-weight complexes.

Influences of Mpl and CRTDel52-D165K and CRTDel52-D166K mutations upon CRTDel52 multimerization. (A–C) HEK293T cells were transiently transfected with plasmids encoding CRTDel52 alone or in combination with His-FLAG Mpl or His-FLAG Mpl alone (A and B) or untagged full-length CRTDel52, CRTDel52-D165K, CRTDel52-D166K, or CRTDel52-3CA (CRTDel52(C163A/C400A/C404A)) or a control vector (Vec; C). Cell lysates from indicated cells were separated by SDS-PAGE under reducing (8% gels) or nonreducing (4–20% gradient gels) conditions or by native-PAGE (8% gels) and immunoblotted with anti-FLAG or anti-CRT(Cmut) antibodies. In blots following SDS-PAGE under nonreducing and native conditions, species consistent with the size of mutant CRT monomers, dimers, and multimers are indicated. Location of loading wells is also indicated. Data are representative of two sets of analyses. MW, molecular weight.
Influences of Mpl and CRTDel52-D165K and CRTDel52-D166K mutations upon CRTDel52 multimerization. (A–C) HEK293T cells were transiently transfected with plasmids encoding CRTDel52 alone or in combination with His-FLAG Mpl or His-FLAG Mpl alone (A and B) or untagged full-length CRTDel52, CRTDel52-D165K, CRTDel52-D166K, or CRTDel52-3CA (CRTDel52(C163A/C400A/C404A)) or a control vector (Vec; C). Cell lysates from indicated cells were separated by SDS-PAGE under reducing (8% gels) or nonreducing (4–20% gradient gels) conditions or by native-PAGE (8% gels) and immunoblotted with anti-FLAG or anti-CRT(Cmut) antibodies. In blots following SDS-PAGE under nonreducing and native conditions, species consistent with the size of mutant CRT monomers, dimers, and multimers are indicated. Location of loading wells is also indicated. Data are representative of two sets of analyses. MW, molecular weight.
A working model for a CRTDel52 dimer, including ionic interactions mediated by the N-domain
To bring all the studied CRT mutations into a structural context, we generated a molecular model for monomeric and dimeric CRTDel52 based on available crystal structures of the human CRT, as described in the Molecular modeling section of Materials and methods. It is known that CRTWT can form oligomers following heat shock, exposure to low calcium, exposure to low pH conditions, truncation of the acidic C-domain, or following specific mutations, including H170A (Jeffery et al., 2011; Jørgensen et al., 2003; Rizvi et al., 2004). Based on the hypothesis that CRTWT and its cancer-linked mutants can exploit similar dimerization interfaces, we selected the most stable dimers found in crystals of human CRT D71K mutants (Protein Data Bank accession no. 5LK5; Moreau et al., 2016) that could account for our experimental data (Figs. 1–4). One such dimer is stabilized via intermolecular ionic interactions between N-domain loop residues 160–167 (Fig. 5 A, “N-N” dimer). Another dimer is formed through the tight packing of antiparallel α-helices from C-domains (Fig. 5 B, “C-C” dimer). We selected the N-N dimer as a prime target for further analysis, as its dimerization interface contained C163, which, based on our data (Fig. 4), is involved in disulfide-mediated CRTDel52 dimerization. In the crystal structure of the N-N dimer, two C163 are not in direct contact, but could move closer to each other and form a disulfide bond following minor loop rearrangements in SDS or other destabilizing conditions. In addition, the C-termini of molecules in the N-N dimer are much closer to each other than in the C-C dimer and can be further connected through two intermolecular disulfides between C400 and C404 of CRTDel52 (Fig. 5 C). Examination of the dimerization interface formed by loop residues 160–167 revealed four intermolecular ionic pairs between opposing subunits that can stabilize the N-N dimer: two D165-K142 and two D165-R162 interactions. Additionally, this interface contains H170 that forms intramolecular hydrogen bonds with D166, which stabilize the loop conformation. We previously showed that the H170A mutant of murine CRT (H153A in mature protein numbering) formed dimers linked by disulfide bonds (Jeffery et al., 2011). In the present work, we found that H170A mutations of human CRTWT also induced disulfide-mediated oligomerization (Fig. 6, A and B).

Proposed structural models for CRTDel52 dimers and tetramers. (A and B) Two major dimerization modes were observed in the crystal structure of the 10-mer complex of CRT D71K mutant (Protein Data Bank accession no. 5LK5; Moreau et al., 2016): dimerization via N-domain loops rich on charged residues that form intermolecular ionic bridges (N-N dimer between subunits E-G and J-X; A) and tight packing of antiparallel α-helices (C-C dimer between subunits G-J and E-X; B). Contacting residues at C-domain helix–helix and N-domain loop–loop interfaces are shown by sticks colored blue for C atoms. Red arrows indicate N344, a glycosylation site. (C) A molecular model of a proposed CRTDel52 dimer. Subunit 1 is colored light green and subunit 2, light blue. Each subunit contains a globular N-domain and a C-domain composed of long and short α-helices connected by a 14-residue loop. The P-domain is omitted. Ca2+ ions bound to the high-affinity site are shown by cyan spheres. Residues from the carbohydrate recognition site are shown by purple sticks (for C atoms). Cysteines participating in the formation of predicted intermolecular disulfide bonds (two C400-C404) and two C163 residues are shown by orange spheres. Residues forming H-bonds and ion pairs (K142, R162, D165, D166, and H170) are shown by sticks colored green (for C atoms) for subunit E and blue for subunit G. Inset highlights interactions at the contact interface. (D) A homology model of CRTDel52 tetramer lacking P-domains. Mutated residues (367–406) from the novel C-tail of CRTDel52 are shown by sticks. Cysteine residues from each subunit predicted to form intermolecular disulfide bonds are shown by orange spheres. Residues (Y109, D135, D317, and W319) located in glycan-binding pockets are shown by red spheres.
Proposed structural models for CRTDel52 dimers and tetramers. (A and B) Two major dimerization modes were observed in the crystal structure of the 10-mer complex of CRT D71K mutant (Protein Data Bank accession no. 5LK5; Moreau et al., 2016): dimerization via N-domain loops rich on charged residues that form intermolecular ionic bridges (N-N dimer between subunits E-G and J-X; A) and tight packing of antiparallel α-helices (C-C dimer between subunits G-J and E-X; B). Contacting residues at C-domain helix–helix and N-domain loop–loop interfaces are shown by sticks colored blue for C atoms. Red arrows indicate N344, a glycosylation site. (C) A molecular model of a proposed CRTDel52 dimer. Subunit 1 is colored light green and subunit 2, light blue. Each subunit contains a globular N-domain and a C-domain composed of long and short α-helices connected by a 14-residue loop. The P-domain is omitted. Ca2+ ions bound to the high-affinity site are shown by cyan spheres. Residues from the carbohydrate recognition site are shown by purple sticks (for C atoms). Cysteines participating in the formation of predicted intermolecular disulfide bonds (two C400-C404) and two C163 residues are shown by orange spheres. Residues forming H-bonds and ion pairs (K142, R162, D165, D166, and H170) are shown by sticks colored green (for C atoms) for subunit E and blue for subunit G. Inset highlights interactions at the contact interface. (D) A homology model of CRTDel52 tetramer lacking P-domains. Mutated residues (367–406) from the novel C-tail of CRTDel52 are shown by sticks. Cysteine residues from each subunit predicted to form intermolecular disulfide bonds are shown by orange spheres. Residues (Y109, D135, D317, and W319) located in glycan-binding pockets are shown by red spheres.

N-domain dimer interface residues influence CRT multimerization. (A, C, and E) Lysates from HEK293T cells expressing indicated untagged full-length CRTWT or CRTDel52 constructs or control transfected cells (Vec) were separated by native-PAGE (8% gels; top left panels) or SDS-PAGE under reducing (8% gels; bottom left panels) or nonreducing (4–20% gradient gels; right panels) conditions and immunoblotted with the indicated antibodies. (B, D, and F) Left panels: WT or mutant CRT monomer bands from native-PAGE immunoblots in panels A, C, and E were normalized relative to the corresponding total CRT signal from the reducing SDS-PAGE immunoblots. Data show mean ± SD; the normalized signals were log-transformed, and the statistical significance was assessed via one-way repeated measures ANOVA (D and F) or one-sample t test (B). Right panels: Quantification of CRT monomer/monomer + multimer (total) bands from the nonreducing SDS-PAGE immunoblots. Data show mean ± SD, and the statistical significance was assessed via one-way repeated measures ANOVA (D and F) or one-sample t test (B). Data were averaged over five independent blots from four to five independent transfections (B and D) or two independent blots from two independent transfections (F), each with two to three protein loads. MW, molecular weight.
N-domain dimer interface residues influence CRT multimerization. (A, C, and E) Lysates from HEK293T cells expressing indicated untagged full-length CRTWT or CRTDel52 constructs or control transfected cells (Vec) were separated by native-PAGE (8% gels; top left panels) or SDS-PAGE under reducing (8% gels; bottom left panels) or nonreducing (4–20% gradient gels; right panels) conditions and immunoblotted with the indicated antibodies. (B, D, and F) Left panels: WT or mutant CRT monomer bands from native-PAGE immunoblots in panels A, C, and E were normalized relative to the corresponding total CRT signal from the reducing SDS-PAGE immunoblots. Data show mean ± SD; the normalized signals were log-transformed, and the statistical significance was assessed via one-way repeated measures ANOVA (D and F) or one-sample t test (B). Right panels: Quantification of CRT monomer/monomer + multimer (total) bands from the nonreducing SDS-PAGE immunoblots. Data show mean ± SD, and the statistical significance was assessed via one-way repeated measures ANOVA (D and F) or one-sample t test (B). Data were averaged over five independent blots from four to five independent transfections (B and D) or two independent blots from two independent transfections (F), each with two to three protein loads. MW, molecular weight.
In addition, in CRTDel52 mutants, but not in the WT CRT, two symmetrical “N-C” dimerization interfaces could be formed between the cross-talking N-domain of one molecule and the C-domain of another molecule (Fig. 5 C). A slight decrease of the α-helix kink at A352 would move the C-terminal part of the α-helix (residues 366–383) closer to the N-domain glycan recognition site. As a result, ionic and hydrophobic interactions may be formed between positively charged and nonpolar residues from the C-domain of one molecule (residues M377, R376, M380, and R381) and negatively charged and aromatic residues from the N-domain glycan-binding site of the second molecule (residues F74, Y109, D125, E127, Y128, and D317). These interactions may explain the significant role of residues 376–383 for multimerization of CRTDel52 constructs, as their truncation in CRTDel52Δ36 eliminated oligomer formation (Fig. 3). Thus, the N-N dimerization mode was selected for the working model of the CRTDel52 dimer, as it is compatible with our current experimental data. Furthermore, CRTDel52 tetramers and larger oligomers can be easily formed by combining both types of dimerization modes: through C-domain helix–helix interface and N-domain loop–loop interface (Fig. 5 D).
Based on the proposed model of CRTDel52 dimer (Fig. 5 C), we tested the structural role of D165-K142 and D165-R162 salt bridges at the globular domain dimer interface. The D165K mutations alone (CRTDel52-D165K) did not significantly influence the oligomerization potential of CRTDel52 (Fig. S4 C). However, the combination of D165K and CRTDel52-3CA mutations induced more monomeric species in the CRTDel52-3CA/D165K mutant compared with CRTDel52-3CA in native blots but not in nonreducing blots (Figs. 6, C and D), indicating the impact of combined disruptions of both dimer interfaces. This was not observed with the D166K mutation, for which enhanced levels of disulfide-linked species were induced (with both CRTDel52-3CA/D166K and the double mutant CRTDel52-3CA/D165KD166K [CRTDel52-3CA-2DK] compared with CRTDel52-3CA; Fig. 6, C and D). Since the only remaining cysteines in all CRTDel52-3CA constructs are C105 and C137, which form an intramolecular disulfide in WT CRT (Chouquet et al., 2011; Kozlov et al., 2010), these findings suggest that repulsion among several positively charged residues in the D166K mutant, following disruption of its interaction with H170, induces rearrangements that bring the C105-C137 disulfide from one subunit of the N-N dimer closer (from 18 Å to 5 Å) to that from the other subunit to enhance intermolecular disulfides via thiol–disulfide exchange. Correspondingly, the H170A mutation on the CRTDel52-3CA background also induces disulfide-linked dimers and multimers (Fig. 6, E and F) that are expected to correspond to enhanced intermolecular disulfides mediated by C105-C137 rearrangements. Overall, the findings of Fig. 6 support the relevance of the dimer model of Fig. 5 C to Del52 multimerization and suggest that MPN mutations exploit a natural dimerization interface of CRTWT known to be induced by ER stress conditions.
Large C-domain truncations or combined N-domain and C-domain dimer interface mutations are required to abrogate CRTDel52-mediated cell proliferation
Ba/F3-Mpl cells were transduced with viruses encoding the series of untagged C-domain–truncated constructs of CRTDel52 and its point mutants to compare their proliferation-inducing activities. We observed reduced abilities of CRTDel52Δ12 and CRTDel52Δ19 to mediate Ba/F3 cell proliferation (although statistically nonsignificant), while CRTDel52Δ28 and CRTDel52Δ36 were unable to promote cell growth, similar to CRTWT (Fig. 7 A; all constructs shown in Fig. 7 are untagged). These results deviate from those of Elf et al. (2018), where the transforming capacity of CRT mutant was abolished only after the most severe truncation of its C-terminus in CRTDel52Δ36. We could not unambiguously detect expression of the truncated untagged CRT constructs over interfering background bands in two independent sets of retroviral infection of Ba/F3-Mpl cells (data not shown). Thus, we reassessed expression and functional activities of N-terminal histidine and GB1-tagged versions in Ba/F3-Mpl cells following plasmid nucleofections (Fig. S5). Proliferation mediated by tagged versions of CRTDel52Δ28 and CRTDel52Δ36 was again impaired under conditions where expression of both those constructs were detectable at higher levels than of CRTDel52 (Fig. S5 A). Thus, while removal of the novel C-terminal cysteines has a small effect on CRTDel52-mediated proliferation, a larger truncation is needed to completely abrogate CRTDel52-mediated proliferation (Fig. 7 A and Fig S5 A). The basis for differences in results with CRTDel52Δ28 between our studies and those of Elf et al. remain unclear but could relate to protein expression levels achieved, although we were unable to measure functional activities of CRTDel52Δ28 with two different expression systems.

Large C-domain truncations or combined N-domain and C-domain dimer interface mutations are required to abrogate CRTDel52-mediated cell proliferation.(A–D) Cytokine-independent proliferation of Ba/F3-Mpl cells expressing full-length untagged CRTWT, CRTDel52, indicated CRTDel52 mutants, or control cells (Vec). Data are averaged from three separate viral transductions of Ba/F3-Mpl cells and a total of 5 independent proliferation experiments (A), eight separate viral transductions of Ba/F3-Mpl cells and a total of 10–13 independent experiments (B), two to three separate viral transductions of Ba/F3-Mpl cells and a total of 3–6 experiments (C), or two separate retroviral transductions of Ba/F3-Mpl cells and a total of 5 independent experiments (D). Mean ± SEM is shown, with statistical significance assessed via ordinary one-way ANOVA from the indicated days of proliferation assay. Statistically significant means are indicated as *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001. (E–G) Lysates from Ba/F3-Mpl cells expressing indicated constructs or control cells expressing Mpl alone (Vec) were directly loaded for immunoblotting analyses (labeled as lysates) or immunoprecipitated with anti-CRT(Cmut) antibody (for CRTDel52 and its variants) or with anti-CRT(Thermo) antibody (for CRTWT), and subsequent immunoblotting was undertaken with the indicated antibodies. Results are representative of three (E and F) or four (G) independent experiments. Nonspecific interactions in the absence of primary antibody are shown by the lysate + beads lanes. Ab, antibody; MW, molecular weight.
Large C-domain truncations or combined N-domain and C-domain dimer interface mutations are required to abrogate CRTDel52-mediated cell proliferation.(A–D) Cytokine-independent proliferation of Ba/F3-Mpl cells expressing full-length untagged CRTWT, CRTDel52, indicated CRTDel52 mutants, or control cells (Vec). Data are averaged from three separate viral transductions of Ba/F3-Mpl cells and a total of 5 independent proliferation experiments (A), eight separate viral transductions of Ba/F3-Mpl cells and a total of 10–13 independent experiments (B), two to three separate viral transductions of Ba/F3-Mpl cells and a total of 3–6 experiments (C), or two separate retroviral transductions of Ba/F3-Mpl cells and a total of 5 independent experiments (D). Mean ± SEM is shown, with statistical significance assessed via ordinary one-way ANOVA from the indicated days of proliferation assay. Statistically significant means are indicated as *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001. (E–G) Lysates from Ba/F3-Mpl cells expressing indicated constructs or control cells expressing Mpl alone (Vec) were directly loaded for immunoblotting analyses (labeled as lysates) or immunoprecipitated with anti-CRT(Cmut) antibody (for CRTDel52 and its variants) or with anti-CRT(Thermo) antibody (for CRTWT), and subsequent immunoblotting was undertaken with the indicated antibodies. Results are representative of three (E and F) or four (G) independent experiments. Nonspecific interactions in the absence of primary antibody are shown by the lysate + beads lanes. Ab, antibody; MW, molecular weight.

Assessments of cytokine-independent proliferation induced by truncated his-GB1–tagged CRTDel52 mutants and secretory efficiencies of selected untagged CRTDel52 mutants. (A) Ba/F3-Mpl cells were electroporated with pcDNA-CoxG plasmids encoding N-terminal his-GB1–tagged full-length CRTWT, CRTDel52, CRTDel52Δ12, CRTDel52Δ19, CRTDel52Δ28, CRTDel52Δ36, or a control vector (Vec) and subsequently selected with Zeocin at 0.2 mg/ml. Left panel: Lysates from Ba/F3-Mpl cells expressing the indicated constructs were immunoblotted with anti-CRT(N) antibody (single analysis). Right panel: Cells were subsequently cultured in the absence of mouse IL-3, and proliferation was measured based on cell counting on the indicated days. Data are averaged from two independent proliferation experiments undertaken following a single electroporation of Ba/F3-Mpl cells. (B) IP of proteins from media and cell lysates of retrovirally transduced Ba/F3 cells expressing the indicated CRTDel52 constructs (those used for the analyses of Fig. 7). Media/cell ratios of CRTDel52 recovery were quantified and averaged across multiple experiments. Each data point in the graph represents an independent experiment. (C) Cell lysates from B were digested with PNGase F or EndoH as indicated before immunoblotting analyses. Data are representative of one to two independent experiments. MW, molecular weight.
Assessments of cytokine-independent proliferation induced by truncated his-GB1–tagged CRTDel52 mutants and secretory efficiencies of selected untagged CRTDel52 mutants. (A) Ba/F3-Mpl cells were electroporated with pcDNA-CoxG plasmids encoding N-terminal his-GB1–tagged full-length CRTWT, CRTDel52, CRTDel52Δ12, CRTDel52Δ19, CRTDel52Δ28, CRTDel52Δ36, or a control vector (Vec) and subsequently selected with Zeocin at 0.2 mg/ml. Left panel: Lysates from Ba/F3-Mpl cells expressing the indicated constructs were immunoblotted with anti-CRT(N) antibody (single analysis). Right panel: Cells were subsequently cultured in the absence of mouse IL-3, and proliferation was measured based on cell counting on the indicated days. Data are averaged from two independent proliferation experiments undertaken following a single electroporation of Ba/F3-Mpl cells. (B) IP of proteins from media and cell lysates of retrovirally transduced Ba/F3 cells expressing the indicated CRTDel52 constructs (those used for the analyses of Fig. 7). Media/cell ratios of CRTDel52 recovery were quantified and averaged across multiple experiments. Each data point in the graph represents an independent experiment. (C) Cell lysates from B were digested with PNGase F or EndoH as indicated before immunoblotting analyses. Data are representative of one to two independent experiments. MW, molecular weight.
Both CRTDel52-2CA and CRTDel52-3CA showed nonsignificant reductions in the ability to induce cytokine-independent proliferation of Ba/F3-Mpl cells compared with CRTDel52 (Fig. 7 B), despite the higher expression of the mutant (Fig. 7 E, top panel, lysate lanes). Notably, we also observed decreased binding of CRTDel52-2CA and CRTDel52-3CA to Mpl relative to CRTDel52 (Fig. 7 E, lanes marked as IP). The N-domain CRTDel52-D165K and CRTDel52-D166K single mutants showed small reductions in the ability to mediate Ba/F3-Mpl cell proliferation compared with CRTDel52 (Fig. 7 C). Notably, the combination of D165K, D166K, or both with the CRTDel52-3CA mutation resulted in marked abolishment of cell proliferation (Fig. 7 C). The impaired abilities of CRTDel52-3CA/D165K and CRTDel52-3CA/D166K to mediate cytokine-independent cell proliferation correlated with almost complete impairment in Mpl binding (Fig. 7 F, IP lanes). Parallel results were obtained with CRTDel52-3CA/H170A (Fig. 7, D and G). Among all mutants, the expression level of the combined mutant CRTDel52-3CA/2DK (3CA + D165K + D166K) was rather low. Thus, the impaired cell proliferation induced by this mutant could be partially caused by its low expression. In contrast, the expression of CRTDel52-3CA, CRTDel52-3CA/D165K, CRTDel52-3CA/D166K, and CRTDel52-3CA/H170A mutants was higher than that of CRTDel52 (Fig. 7, F and G, top panels), indicating an enhanced stability of these mutants. Therefore, loss of stability could not explain their functional loss.
Since the secretory efficiencies of mutants could affect function, we also assessed levels of secretion by comparing the media/cell ratios of different CRTDel52 mutants. We found that secretion efficiencies were reduced for several single mutants, although only significantly for CRTDel52-H170A (Fig. S5 B). These mutants, however, were largely functional for mediating cell proliferation (Fig. 7, C and D). Furthermore, secretion efficiencies of the functionally defective CRTDel52-3CA/D165K, CRTDel52-3CA/D166K, CRTDel52-3CA/2DK, and CRTDel52-3CA/H170A mutants (Fig. 7) were not impaired relative to CRTDel52 (Fig. S5 B). Thus, the functional impairments of these mutants did not relate to low secretory efficiencies.
Examination of Fig. 7, G and F; Fig. S5 B; and Fig. S5 C revealed the presence of additional slow-migrating bands, particularly for CRTDel52-D166K and CRTDel52-2DK, and to a lower extent, for CRTDel52-H170A mutants in SDS-PAGE immunoblots. The slower migrating bands were sensitive to both EndoH and PNGase F digestions (Fig. S5 C), suggesting that they corresponded to glycosylated forms of CRTDel52. The only predicted N-linked glycosylation site on CRTDel52 is N344, which is buried between interacting C-helices of the C-C dimer (Fig. 5 B; two N344 are indicated by arrows). We suggest that D166K and H170A mutations destabilize the conformation of the loop 160–167 within the N-N dimer. This in turn could influence the tetramer structure and destabilize the packing of C-helices, thus partly exposing N344 for glycosylation. These findings support the relevance of the multimer model of Fig. 5 D to CRTDel52 oligomerization.
Stable dimers are observable with purified CRTDel52
To further examine the formation of disulfide-linked dimers of CRTDel52 as a building block for its multimerization, we analyzed SDS-PAGE gels of purified CRTDel52 by Coomassie staining and mass spectroscopy. A FLAG-His–tagged version of CRTDel52 was expressed and purified from HEK293T cells using anti-FLAG beads. Protein analysis by Coomassie staining revealed the presence of bands consistent with the size of CRTDel52 monomers and dimers, while higher-order oligomers were not clearly visible, probably due to their lower abundance. The identity of CRTDel52 monomers and dimers was verified by mass spectrometric analyses. Absence of the dimer bands in reducing gels confirmed that CRTDel52 dimers are stabilized by disulfide crosslinking (Fig. 8 A). These data demonstrate that CRTDel52 indeed exists in monomeric and disulfide-linked dimeric forms. The observed dimers are likely functionally relevant, as their destabilization by mutations in both N- and C-domain dimerization interfaces abrogates the ability of CRTDel52 to induce cytokine-independent activation of the Mpl-mediated signaling pathway (Fig. 7).
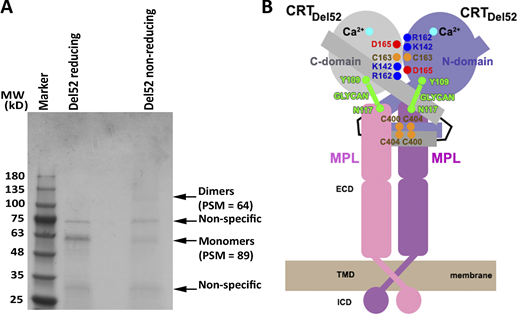
Observation of disulfide-linked CRTDel52 dimers and a cartoon showing a proposed heterotetrameric complex Mpl2-(CRTDel52)2.(A) A purified FLAG-His–tagged CRTDel52 was analyzed by Coomassie staining via reducing or nonreducing SDS-PAGE gels. Indicated monomer and dimer bands were excised and subjected to mass spectrometric analyses. Peptide-spectrum match (PSM) values for FLAG-His–tagged CRTDel52-derived peptides are indicated (see also Data S1). The gel is representative of two purifications, and MS analyses were conducted on one of the purified samples. (B) The proposed mechanism of oncogenic activation of Mpl involves formation of a heterotetramer from the preformed Mpl dimer and the CRTDel52 dimer stabilized by disulfides and ionic interactions at C-domain and N-domain dimerization interfaces. The C-domain (residues 357–411) of CRTDel52 contributes to the specificity of Mpl binding in addition to glycan-binding site residues of the CRT N-domain that mediate Mpl recruitment. ECD, extracellular domain; ICD, intracellular domain; MW, molecular weight; TMD, transmembrane domain.
Observation of disulfide-linked CRTDel52 dimers and a cartoon showing a proposed heterotetrameric complex Mpl2-(CRTDel52)2.(A) A purified FLAG-His–tagged CRTDel52 was analyzed by Coomassie staining via reducing or nonreducing SDS-PAGE gels. Indicated monomer and dimer bands were excised and subjected to mass spectrometric analyses. Peptide-spectrum match (PSM) values for FLAG-His–tagged CRTDel52-derived peptides are indicated (see also Data S1). The gel is representative of two purifications, and MS analyses were conducted on one of the purified samples. (B) The proposed mechanism of oncogenic activation of Mpl involves formation of a heterotetramer from the preformed Mpl dimer and the CRTDel52 dimer stabilized by disulfides and ionic interactions at C-domain and N-domain dimerization interfaces. The C-domain (residues 357–411) of CRTDel52 contributes to the specificity of Mpl binding in addition to glycan-binding site residues of the CRT N-domain that mediate Mpl recruitment. ECD, extracellular domain; ICD, intracellular domain; MW, molecular weight; TMD, transmembrane domain.
Discussion
In this work, we observed that the novel C-domain in MPN mutant CRT induces CRT dimerization and the formation of higher-order oligomers in pathologically relevant MPN patient–derived platelets (Fig. 1) and in transfected HEK293T cells (Figs. 2, 3, 4 and 6). Reduction of oligomeric bands under disulfide-reducing conditions indicates that the intermolecular disulfides stabilize multimers both in transfected cells and in MPN patient–derived platelets. However, the N-domain–mediated ionic interactions also contribute to the MPN CRT multimerization.
The data presented here are consistent with the model of dimers of CRTDel52 with two dimerization interfaces, the first involving interactions between the distal parts of C-terminal tails and the second involving association of globular domains via loop residues 160DIRCKDDEFTH170 (Fig. 5 A). The novel C-domains of the MPN-linked CRT mutant CRTDel52 contains two cysteine residues, C400 and C404 (Fig. 2 A), whose mutations to alanines abrogate the formation of dimers of isolated C-domains (Fig. 2 C). In addition, C163 residue from the globular N-domain may contribute to the formation of intermolecular disulfides in the CRTDel52-2CA mutant, but ionic interactions rather than C163 within the globular domain likely play a deterministic role in formation of the productive Mpl-activating dimer of CRTDel52.
Based on available crystal structures of CRT oligomers, we propose a model of CRTDel52 dimer that is consistent with our experimental data, wherein dimer stabilizing interactions occur through N-domains (four ionic bridges involving D165) and through C-tails crosslinked by two C400–C404 disulfides (Fig. 5 A, and related discussion). The combination of D165K with the triple cysteine mutant in CRTDel52-3CA significantly increases the fraction of the monomeric form of the protein. This is not observed with CRTDel52-3CA/D166K, which appears to induce structurally modified dimers (Fig. 6 B), which, nonetheless, are not compatible with Mpl binding and activation.
Previous findings have indicated preferential binding of full-length CRT mutants to Mpl in transfected cells (Araki et al., 2019; Araki et al., 2016; Chachoua et al., 2016; Elf et al., 2018; Elf et al., 2016; Masubuchi et al., 2020; Pecquet et al., 2019). Here, we observed the binding of Mpl not only to the full-length CRTDel52 (Fig. S2 A) but also to its isolated C-domain (Fig. 2, B and D). This is direct evidence that the novel C-terminus of CRTDel52 confers the Mpl binding specificity. Cysteine residues within the mutant C-domains are not absolutely required for Mpl binding either to the full-length CRTDel52 or to its C-domain (Figs. 2 B and 7 E). However, mutations C400A/C404A in CRTDel52 reduced its ability to bind Mpl (Fig. 7, E–G). Additional disruption of intermolecular ionic interactions involving the globular N-domain that are predicted to stabilize CRTDel52 dimers (Fig. 5 A) further decreases CRTDel52 binding to Mpl (Fig. 7, F and G).
The formation of soluble Mpl2-(CRTDel52)2 heterotetramers (∼200 kD) was previously suggested by size exclusion chromatography (Pecquet et al., 2019). This could be achieved by binding of a (CRTDel52)2 dimer (Fig. 8 A) to two interacting Mpl molecules, thus stabilizing the structure of the activated Mpl dimer that initiates persistent cytokine-independent JAK2/STAT5 signaling pathways in cells (Fig. 8 B). In this type of model, two interaction interfaces are predicted to be key determinants of mutant CRT-Mpl binding: (1) C-domain–dependent interactions conferring the Mpl targeting specificity to CRT mutants (consistent with Fig. 2) and (2) interactions between monoglucosylated N117 of Mpl and carbohydrate-binding site of the mutant CRT (consistent with published data; Chachoua et al., 2016; Pecquet et al., 2019). The loss of the majority or a subset of contacts with the first site is consistent with the impaired abilities of CRTDel52Δ28 and CRTDel52Δ36 to mediate cell proliferation (Fig. 7 A; Elf et al., 2016). Additionally, proliferation impairment observed with several dimer interface mutants of CRTDel52 (Fig. 7, C and D) is accompanied by altered dimer conformation observed with those mutants (Fig. 6). While multimers (higher order than dimers) can be formed by CRTDel52 (Fig. 5 D) and may bind Mpl, we suggest that a signaling dimer (Fig. 8 A) with a unique conformation (Fig. 5 C) comprises an essential activating unit among all possible disulfide-driven and multimeric structures that could be formed.
Overall, the findings of this study reveal that disulfide-mediated homooligomerization is a fundamental feature of MPN CRT mutants. The proposed functionally relevant dimer likely exploits a natural dimerization interface between CRT N-domains containing multiple ionic interactions and may form disulfide bridges between novel C-tails and criss-cross noncovalent interactions between N- and C-domains of interacting CRT molecules (Fig. 5 C). Thus, cancer-linked mutations of CRT confer selective growth advantages to cells by inducing specificity for partially immature Mpl forms by facilitating their traffic in the dimeric state via the secretory pathway (Pecquet et al., 2019) and by stabilizing an activated dimeric conformation of the receptor via specific interactions with the signaling CRTDel52 dimer. Further mutagenesis studies are required to identify the corresponding contact residues between Mpl and CRTDel52.
Materials and methods
Healthy donor and patient samples
Blood was collected after written informed consent in accordance with University of Michigan institutional review board–approved protocols for a myeloproliferative diseases repository (HUM0006778) or the University of Michigan Platelet Pharmacology and Physiology Platelet core (HUM00107120).
DNA constructs
All primers used in this study are listed in Table S1. CRTIns5 (K385fs*47; Nangalia et al., 2013) was made using CRTWT (clone BC020493) as template by the QuikChange Site-Directed Mutagenesis Kit (Agilent). CRTDel52 (L367fs*46; Nangalia et al., 2013) was amplified from patient cDNA, and using ligation-independent cloning, transferred into the pGB1 vector (DelProposto et al., 2009).
Untagged CRTWT, CRTIns5, and CRTDel52 and their mutant constructs for mammalian expression
Full-length untagged CRTWT and CRTIns5 were cloned into the pMSCV (murine stem cell virus)–puro retroviral vector (Clontech) using XhoI and EcoRI sites. CRTDel52 in the pMSCV vector was generated by replacing an AccI–EcoRI fragment of pMSCV-CRTWT with an AccI–EcoRI fragment of pGB1-CRTDel52. Full-length CRTWT, CRTIns5, and CRTDel52 were subcloned into the pcDNA3.1/Zeo(−) (Invitrogen) from the pMSCV puromycin vectors via restriction digestion using XhoI and EcoRI sites.
Cysteine mutants of full-length CRTIns5 (C163A, C419A, and C423A) and CRTDel52 (C163A, C400A, and C404A) were generated using the corresponding untagged pcDNA and pMSCV plasmids with the QuikChange Site-Directed Mutagenesis Kit. D165K, D166K, and H170A mutations were also generated using the CRTWT or CRTDel52 and CRTDel52-3CA (CRTDel52(C163A/C400A/C404A)) untagged pcDNA and pMSCV plasmids using the QuikChange Site-Directed Mutagenesis Kit. C-terminal truncations of pMSCV-CRTDel52 (Δ12, Δ19, Δ28, and Δ36) plasmids were generated by introducing stop codons at indicated positions using the QuikChange Site-Directed Mutagenesis Kit. All residue numberings include the signal sequence.
His-GB1–tagged C-domain constructs for mammalian expression
The plasmid pcDNA3.1/Zeo(−) was used as the parent vector for construction of a ligation independent cloning (LIC)–compatible mammalian expression vector (pcDNA-COX2-G) for expression of his-GB1–tagged fusion proteins. The SspI restriction site in the plasmid was removed by site-directed mutagenesis and used as the parent vector for construction of the LIC region–containing plasmids. The SspI-minus pcDNA3.1 (−) vector was digested with XhoI and HindIII. The large DNA fragment from this digestion was gel purified. The LIC regions from pGB1 (DelProposto et al., 2009; encoding His6 and GB1 tag and TEV cleavage sites) were PCR amplified using the following oligonucleotides: 5′-GTCACTCGAGACCATGCACCATCATCATCATCATTCT-3′ and 5′GTCAAAGCTTGTCGACGGAGCTCGAATTCGGATC-3′. These oligonucleotides add an XhoI site followed by a modified Kozak sequence adjacent to the ATG translational start site of these regions and a HindIII site at the 3′ end of the LIC region. These fragments were digested and then ligated with the vector fragment. Positive clones were identified by PCR and confirmed by DNA sequencing. Using two rounds of PCR, pcDNA-COX2-G was constructed in which a secretion signal sequence was fused upstream of the polyhistidine sequence. The first round fused the signal sequence from Cox2 to the LIC region, and the second round introduced the XhoI restriction site with the modified Kozak sequence and ATG to the end of the signal sequence. The following oligonucleotides were used to add the signal sequence to the end of the fusion LIC region: 5′-GTGCGCAGTACTGGCTCTTTCTCACACTGCACACCATCATCATCATCAT-3′ and 5′-CCTCTCGAGACCATGCTGGCTCGTGCACTGCTTCTGTGCGCAGTACTGGCT-3′. The resulting fragments were digested with XhoI and HindIII, inserted into the vector, and sequenced.
Using LIC, CRTWT C-domain (residues MKDKQDEEQRLKEEEEDKKRKEEEEAEDKEDDEDKDEDEEDEEDKEEDEEEDVPGQAKDEL), and CRTDel52 C-domain (residues MKDKQDEEQRTRRMMRTKMRMRRMRRTRRKMRRKMSPARPRTSCREACLQGWTEA) were cloned into the pcDNA-COX2-G vector as N-terminal 6X his-GB1–tagged fusion proteins. Cysteine mutations (C44A and C48A) and truncations (Δ12, Δ19, Δ28, and Δ36) in the pcDNA-COX2-G-CRTDel52 C-domain plasmids were also generated using the QuikChange Site-Directed Mutagenesis Kit.
His-GB1–tagged full-length CRTWT, CRTDel52, and CRTDel52 truncation constructs for mammalian expression
The full-length CRTWT was also cloned into pcDNA-COX2-G vector using LIC. CRTDel52 in the pcDNA-COX2-G vector was generated by replacing the BsrGI-EcoRI fragment of pcDNA-COX2-G-CRTWT with the BsrGI-EcoRI fragment of pcDNA 3.1(−) CRTDel52. C-terminal truncations of his-GB1–tagged CRTDel52 pcDNA-COX2-G (Δ12, Δ19, Δ28, and Δ36) plasmids were generated by introducing a stop codon at indicated positions using the QuikChange Site-Directed Mutagenesis Kit.
His and FLAG-tagged full-length CRTDel52 for mammalian expression
The plasmid pcDNA3.1/Zeo(−) from Invitrogen was used as the parent vector for construction of LIC-compatible mammalian expression vector pcDNA-COX2-54 for expression of 6X His FLAG-tagged proteins. The SspI restriction site in pcDNA3.1/Zeo(−) was removed by site-directed mutagenesis, and this was used as the parent vector for construction of the LIC region–containing plasmids. The SspI-minus pcDNA3.1 (−) vector was digested with XhoI and HindIII. The large DNA fragment from this digestion was gel purified. The LIC regions from pMCSG54 (encoding His6 and FLAG tag and TEV cleavage sites; Midwest Center for Structural Genomics) were PCR amplified using the following oligonucleotides: 5′-GTCACTCGAGACCATGCACCATCATCATCATCATTCT-3′ and 5′-GTCAAAGCTTGTCGACGGAGCTCGAATTCGGATC-3′. These oligonucleotides add an XhoI site followed by a modified Kozak sequence adjacent to the ATG translational start site of these regions and a HindIII site at the 3′ end of the LIC region. These fragments were digested and then ligated with the vector fragment. Positive clones were identified by PCR and confirmed by DNA sequencing. Using two rounds of PCR, pcDNA-COX2-54 was constructed in which a secretion signal sequence was fused upstream of the polyhistidine sequence. The first round fused the signal sequence from Cox2 to the LIC region, and the second round introduced the XhoI restriction site with the modified Kozak sequence and ATG to the end of the signal sequence. The following oligonucleotides were used to add the signal sequence to the end of the fusion LIC region: 5′-GTGCGCAGTACTGGCTCTTTCTCACACTGCACACCATCATCATCATCAT-3′ and 5′-CCTCTCGAGACCATGCTGGCTCGTGCACTGCTTCTGTGCGCAGTACTGGCT-3′. The resulting fragments were digested with XhoI and HindIII, inserted into the vector, and sequenced. CRTDel52 was cloned into pcDNA-COX2-G54 vector using LIC as an N-terminal His6-FLAG fusion construct.
Mpl constructs
A plasmid encoding human Mpl (identical to reference sequence IMAGE:100016416 and accession no. BC153092) was procured from DNASU and used as the template for amplification of the DNA sequence encoding Mpl, which was then cloned into the pMSCV-neo vector (Clontech) using the EcoRI and XhoI sites for expression of untagged Mpl or into the pcDNA-COX2-54 vector via LIC for expression as an N-terminal His6-FLAG fusion.
Cell lines, transient HEK transfections, and BaF/3 infections
HEK293T cells (a kind gift from the laboratory of Dr. Kathleen Collins, University of Michigan, Ann Arbor, MI) were maintained in DMEM supplemented with 10% FCS and penicillin/streptomycin (100 U/ml, 100 mg/ml). Mouse pro–B cell line Ba/F3 (a kind gift from the laboratory of Dr. Moshe Talpaz) was maintained in RPMI 1640 medium supplemented with mouse IL-3 (BioLegend), 10% FCS, and penicillin/streptomycin (100 U/ml, 100 mg/ml).
HEK293T cells were transfected with pcDNA3.1 (−) encoding indicated constructs using polyethylenimine (Polysciences, Inc.) for 48 h. Retroviral supernatants encoding Mpl or CRT constructs were generated as previously described (Del Cid et al., 2010; Jeffery et al., 2011). Briefly, 5.5 µg pMSCV vector encoding Mpl or indicated CRT mutants were mixed with 0.5 µg VSV-G and 4 µg pCL-EcoDNA plasmids and incubated for 20 min at room temperature in a mixture containing Opti-MEM and X-tremeGENE (Sigma). Cell media were collected after 48 h and used for Ba/F3 cell infections. Ba/F3 cells were first transduced with a virus encoding untagged human Mpl to generate Ba/F3-Mpl cells and selected with 1 mg/ml geneticin (Thermo Fisher Scientific) for Mpl expression. Ba/F3-Mpl cells were further transduced with viruses encoding various indicated CRTs or a control virus and subsequently selected with 3 µg/ml puromycin (Thermo Fisher Scientific) for CRT expression. For some experiments, full-length tagged CRT constructs in the pcDNA vector were nucleofected into Ba/F3-Mpl cells using Cell Line Nucleofector Kit V (Lonza) and selected using 0.2 mg/ml Zeocin.
Antibodies
An anti-CRT mutant C-terminal antibody, anti-CRT(Cmut), was generated by GeneTel Laboratories using rabbits immunized with a 22-mer peptide (KMSPARPRTSCREACLQGWTEA) derived from the CRT mutant C-terminus and affinity purified.
Platelet isolation
Whole blood from healthy donors and MPN patients was collected in acid citrate dextrose anticoagulant tubes (BD Biosciences), and platelets were isolated as described previously (Greening et al., 2011), with some modifications. Platelet-rich plasma was separated by centrifugation of whole blood at 200 ×g for 15 min at room temperature with no brakes. Further, three fourths of the top layer containing platelet-rich plasma was extracted and treated with acid citrate dextrose (2.5% sodium citrate tribasic, 1.5% citric acid, 2.0% D-glucose) and apyrase (0.02 U/ml; Sigma) and centrifuged for 10 min at 2,000 ×g. The platelet pellet was resuspended in Tyrode’s buffer (10 mM Hepes, pH 7.4, 134 mM NaCl, 12 mM NaHCO3, 2.9 mM KCl, 0.34 mM Na2HPO4, 1 mM MgCl2, and 5 mM D-glucose) and rested at 37°C for 30 min before use. The rested platelets in Tyrode’s buffer were centrifuged at 2,000 ×g for 10 min and lysed in 500–800 µl of lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1% Triton X-100, 5 mM CaCl2, and protease inhibitor cocktail; Roche) using an end-over-end shaker for 30 min at 4°C followed by centrifugation at 16,800 ×g for 30 min.
Immunoblotting
Cells were lysed in lysis buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1% Triton X-100, 5 mM CaCl2, and protease inhibitor cocktail; Roche) for 1 h at 4°C followed by centrifugation at 16,800 ×g for 30 min. For native, reducing, and nonreducing lysate blot experiments, transfected HEK293T cells were lysed in the presence of 10 mM N-ethylmaleimide (Sigma). Protein concentration was measured using bicinchoninic acid assay (Pierce). For native blots, lysates were added to the loading buffer without reducing agents and SDS. For nonreducing blots, lysates were added to the loading buffer with SDS but without any reducing agents. For reducing blots, lysates were added to the loading buffer with SDS and reducing agent (65 mM DTT). Antibodies used in the immunoblotting analyses were: for CRTWT, a rabbit anti-CRT(N) antibody (catalog no. 12238, 1:10,000 dilution; CST) or rabbit anti-CRT antibody (catalog no. PA3-900, 1:10,000 dilution; Thermo Fisher Scientific); for MPN CRT mutants, rabbit anti-CRT(Cmut) antibody; for Mpl, rabbit anti-c-Mpl (catalog no. 06944, 1:10,000 dilution; Millipore); for 6X His tag, mouse His-Tag antibody (catalog no. MA1-21315, 1:10,000 dilution; Thermo Fisher Scientific); for loading controls, rabbit anti-vinculin (catalog no. 13901, 1:10,000 dilution; CST), rabbit anti-GAPDH (catalog no. 5174, 1:10,000 dilution; CST), and mouse anti-FLAG (catalog no. F3165, 1:5,000 dilution; Sigma). Blots were incubated with a secondary antibody conjugated to horseradish peroxidase mouse anti-rabbit (catalog no. 211032171; Jackson ImmunoResearch) or goat anti-mouse (catalog no. 115035003; Jackson ImmunoResearch), and proteins were detected via chemiluminescence. In some experiments, 20 µg of cell lysates were digested with EndoH (P0702; New England Biolabs) or PNGase F (P0704; New England Biolabs) for 1 h at 37°C per the manufacturer’s protocol.
IP
CoIP was performed using transfected HEK293T cells or Ba/F3 Mpl CRT cells as described (Del Cid et al., 2010), with some modifications. The cells were first incubated with a membrane-permeable crosslinker dimethyl dithiobispropionimidate (4 mM) in PBS for 1 h on ice followed by incubation with 1 M Tris, pH 7.4, for 15 min to quench free crosslinker. Cells were lysed as described above. 500 µg cell lysate was incubated with custom anti-CRT(Cmut) (2 µg) or anti-CRT (PA3-900, 1:1,000; Thermo Fisher Scientific) or anti–c-Mpl (06944, 5 µg; Millipore) antibodies overnight at 4°C. In some experiments, 10 ml cell media from Ba/F3 Mpl CRT cells were cleared for cell debris and incubated with custom anti-CRT(Cmut) (2 µg) antibody overnight at 4°C. Similarly, healthy donor and MPN patient platelet lysates were lysed using the lysis buffer as described above but without crosslinking. Following clearance of the cell debris, 1 ml (0.5 mg) of the supernatants were incubated with either anti-CRT(Cmut) (2 µg) or anti-c-Mpl (06944, 5 µg; Millipore) or without antibodies and mixed gently at 4°C for 16 h, centrifuged to remove cell debris, and then incubated with protein G beads for 2 h; beads were washed with lysis buffer three times, and samples were eluted with Laemmli buffer. Samples were further denatured and separated by SDS-PAGE and immunoblotted as described above.
Protein purification and mass spectrometry (MS) analyses
HEK293T cells were transfected with full-length CRTDel52 in the pcDNA-COX2-G54 vector for 72 h using polyethylenimine in a 1% FBS–containing DMEM. Cells were lysed in the lysis buffer, as mentioned above, in the presence of 10 mM N-ethylmaleimide, and the cell lysates were incubated with anti-FLAG magnetic beads (M8823; Sigma) antibody for 3 h at 4°C. The beads were washed with wash buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 5 mM CaCl2, and protease inhibitor cocktail) six times and eluted in 150 ng/µl 3X FLAG peptide (F4799; Sigma) containing TBS (50 mM Tris, pH 7.4, and 150 mM NaCl). The sample was analyzed by SDS-PAGE under reducing and nonreducing conditions. Bands corresponding to monomers and dimers from the nonreducing gel were subjected to mass spectrometric analyses as described previously (Mohan et al., 2020). Briefly, the excised gels were resuspended in 50 µl of 0.1 M ammonium bicarbonate buffer (pH 8). 50 µl of 10 mM DTT were added, and samples were incubated at 45°C for 30 min to reduce cysteines. Samples were incubated with 65 mM 2-chloroacetamide in the dark for 30 min at room temperature for alkylation of cysteines and digested overnight with 1 µg sequencing grade, modified trypsin at 37°C with gentle agitation. The digestion was stopped by acidification, and peptides were desalted using SepPak C18 cartridges (manufacturer’s protocol; Waters). Samples were dried with a vacufuge and dissolved in 8 µl of 0.1% formic acid/2% acetonitrile solution. 2 µl of the peptide solution were resolved on a nano capillary reverse phase column (Acclaim PepMap C18, 2 μm, 50 cm; Thermo Fisher Scientific) using a 0.1% formic acid/2% acetonitrile (buffer A) and 0.1% formic acid/95% acetonitrile (buffer B) gradient at 300 nl/min over 180 min (2–22% buffer B for 110 min, 22–40% buffer B for 25 min, and 40–90% buffer B for 5 min followed by holding in 90% buffer B for 5 min and re-equilibration with buffer A for 25 min). Eluent was directly introduced into a Q Exactive HF mass spectrometer (Thermo Fisher Scientific) using an EasySpray source. MS1 scans were acquired at 60K resolution (automatic gain control target = 3 × 106; maximum ion injection time = 50 ms). Data-dependent collision-induced dissociation MS/MS spectra were acquired on the 20 most abundant ions following each MS1 scan (normalized collision energy ∼28%; automatic gain control target = 105; maximum ion injection time = 45 ms). Proteins were identified by searching the MS/MS data against Homo sapiens (UniProt; 20,353 reviewed entries; downloaded on June 29, 2019) and the CRTDel52 sequence using Proteome Discoverer version 2.4 (Thermo Fisher Scientific). Search parameters included MS1 mass tolerance of 10 ppm and fragment tolerance of 0.2 D; two missed cleavages were allowed; carbamidomethylation of cysteine was considered as fixed modification and oxidation of methionine; and deamidation of asparagine and glutamine was considered as potential modifications. The Target-Decoy PSM validator node of Proteome Discoverer was used to retain only high-confidence peptide hits (false discover rate ≤1%).
CRTDel52 was also purified from HEK cell media using His-GB1–tagged CRTDel52 pcDNA-COX2-G–transfected HEK cells. After 48–72 h, cell supernatants were centrifuged, diluted threefold in wash buffer (50 mM Tris, 150 mM NaCl, 1 mM CaCl2, pH 7.5), and incubated with Ni-NTA resin (QIAGEN) for 2 h at 4°C. The beads were washed with 5 mM imidazole in wash buffer (50 mM Tris, 150 mM NaCl, 10% glycerol, 1 mM CaCl2, pH 7.5). CRTDel52 was eluted from beads with 350 mM imidazole in wash buffer.
Cytokine-independent proliferation
Ba/F3-Mpl cells expressing CRT constructs were seeded at 106 cells/ml in the absence of mouse IL-3. Further, the proliferation rate of different cell lines was measured over 4–5 d by counting the live cells using a hemocytometer.
Molecular modeling
Modeling and verification of functionally relevant CRTDel52 dimers was performed in three steps: (1) modeling of a monomeric CRTDel52, (2) choice of dimerization mode from available crystal structures of CRT, and (3) verification of the functional relevance of the chosen CRTDel52 dimer by mutational and functional studies.
We first generated the monomer of CRTDel52 using available atomic structure of human CRT (Protein Data Bank accession no. 5LK5, subunit E, for residues 19–203 and 303–366; Moreau et al., 2016). The novel C-domain was modeled as an α-helix extension, using fragment 367–386 from the structure of human PLC editing module (Protein Data Bank accession no. 6ENY, subunit G; Blees et al., 2017), with subsequent residue substitutions in accordance with the new C-terminal sequence of CRTDel52 (Fig. 2 A). The distal C-terminal segment of CRTDel52 (399SCREACLQ406) was modeled as a two-turn α-helix based on secondary structure predictions by Iterative Threading Assembly Refinement (I-TASSER; Roy et al., 2010), while the connecting loop (residues 387–398) was modeled in an extended conformation. Residue substitutions and modeling of an 8-residue C-terminal α-helical segment together with the 14-residue connecting loop were performed using PyMOL molecular graphics system version 1.8.4.1 (Schrodinger, LLC).
Second, we examined crystal structures of CRT oligomers (Protein Data Bank accession nos. 5LK5, 3POS, and 3O0X) to identify dimerization modes that can account for the data described in Figs. 1, 2, 3, and 4, consistent with formation of intermolecular disulfides between N-domains (C163-C163) and between C-domains (two C400-C404 disulfides) of monomers. Relatively stable dimers observed in crystal structure 5LK5 were used to build the models of CRTDel52 dimers by molecular superposition. Two novel C-terminal α-helices of CRTDel52 were brought together to form two disulfide bonds (C400-C404) that were shown to stabilize CRTDel52 dimers (Figs. 2 and 4). To analyze the possible structures of higher-order oligomers of CRTDel52, we modeled tetramers of CRTDel52 (Fig. 5 D). The modeling was performed by superposition of CRTDel52 monomers with CRT trimer (subunits E, G, and J) from the crystal structure of 10-meric CRT complex for K71K mutant (Protein Data Bank accession no. 5LK5; Moreau et al., 2016). To add the fourth CRTDel52 subunit, we superposed it with an additional subunit X obtained by symmetry transformation of the crystal structure. In the resulting structure of a CRT tetramer, two subunit pairs are connected through C-terminal α-helices, while two other subunit pairs are connected through N-domain loops. The final verification of the proposed models by mutagenesis and other experimental studies is described in the Results.
Data sharing statement
MS data are uploaded as a supplementary file (Data S1). Other data will be made available on request.
Statistical analysis
All statistical analyses were performed in GraphPad Prism 8.0c as indicated in the figure legends.
Online supplemental material
Fig. S1 shows that mutant CRT is multimeric via disulfide bonds in MPN patient platelets. Fig. S2 shows that mutant CRT preferentially interacts with Mpl in HEK293T cells and MPN patient platelets. Fig. S3 shows that secreted forms of mutant CRT multimerize and CRTIns forms disulfide-linked multimers similar to CRTDel52. Fig. S4 shows the influences of Mpl and CRTDel52-D165K mutations upon CRTDel52 multimerization. Fig. S5 shows assessments of cytokine-independent proliferation induced by truncated his-GB1–tagged CRTDel52 mutants and secretory efficiencies of selected untagged CRTDel52 mutants. Table S1 lists the primers used in the study. Data S1 shows MS data on purified CRTDel52 monomers and dimers.
Acknowledgments
We are grateful to all the blood donors for their contribution to this work. We thank Venkatesha Basrur, Kevin Conlin, Alexey Nesvizhskii, and the Department of Pathology Proteomics Resource facility for the mass spectrometric analysis; Clay Brown and the Center for Structural Biology for plasmid constructions; and the University of Michigan DNA Sequencing Core for DNA sequencing. We are grateful to Harihar Mohan, Angela Danielski, Amanpreet Kaur, and Maha Hamed for their contributions to the project.
This work was funded by National Institutes of Health grant R01 AI123957 to M. Raghavan and the University of Michigan Fast Forward Protein Folding Diseases Initiative. I.D. Pogozheva was supported by the National Science Foundation Division of Biological Infrastructure (award 1855425).
The authors declare no competing financial interests.
Author contributions: A. Venkatesan helped design experiments, performed experiments, helped analyze data, and wrote sections of the manuscript. J. Geng helped design experiments, performed experiments related to Figs. 3 and 7, helped analyze data, and edited the manuscript. M. Kandarpa collected patient samples, purified platelets, and edited the manuscript. I.D. Pogozheva performed the molecular modeling, wrote some sections, and edited the manuscript. M. Talpaz directs the University of Michigan MPN repository and edited the manuscript. S.J. Wijeyesakere helped construct and sequence some CRT variants. A. Bhide helped construct and sequence some CRT variants described in this manuscript. M. Raghavan designed the study, analyzed data, and wrote sections of the manuscript.

