


ER takes trip on microtubule tips
The ER membrane hitches a ride on the tips of microtubules, as revealed by Susana Montenegro Gouveia.

STIM1 (green) hooks the ER to microtubule plus ends (red).
AKHMANOVA/ELSEVIER
Gouveia, Ilya Grigoriev, and Anna Akhmanova (Erasmus Medical Center, Rotterdam, Netherlands) are interested in the growing tips of microtubules and their protein binding partners. Many of these partners also associate with one particularly common tip-binding protein, called EB1. Gouveia thus used EB1 as bait to identify new plus end–binding proteins.
One of the most abundant proteins she isolated was an ER transmembrane protein called STIM1, which is well-known for its role in calcium signaling. When ER calcium levels drop, ER-bound STIM1 cuddles up to the plasma membrane. It then helps open channels in the plasma membrane that let in calcium, which then refills the ER stores.
The finding that STIM1 fastens to microtubules might help explain how the ER membrane gets close enough to the plasma membrane for STIM1 to open the channels: the membrane rides on growing microtubules. Gouveia showed that high levels of STIM1 caused ER membrane tubules to extend, while STIM1 depletion inhibited their extension.
The loss of ER calcium, which forces STIM1 molecules into immobile aggregates, separated STIM1 from microtubule tips. Thus, as for other tip-binding proteins, STIM1 must be able to diffuse freely to maintain tip association. In this case, however, the diffusion occurs within the ER membrane rather than in the cytoplasm.
Whether STIM1-mediated pulling of the ER membrane is required for calcium signaling is not clear. At least in fibroblasts, the link between STIM1 and microtubules was not necessary for channel opening. But in larger cells such as neurons, in which the ER must stretch farther to reach the periphery, the STIM1 link might be more vital. 
Grigoriev, I., et al. 2008. Curr. Biol. doi: https://doi.org/10.1016/j.cub.2007.12.050.
When inversin's gone, what's left?
The asymmetry of internal organs is essential to their function—a symmetrical heart, for example, would not pump properly. Research presented by Svetlana Makova and Martina Brueckner (Yale University, New Haven, CT) revealed that left–right asymmetry might be set up by unequal cell migration during early embryogenesis.

A calcium boost (bottom) shifts inversin (red) to the cytoplasm and removes E-cadherin (green) from the membrane, kicking off migration.
MAKOVA
Left–right asymmetry is initiated just before gastrulation at a part of the embryo called the node. The beating of cilia on node cells causes fluid around the embryo to flow in a defined leftward direction. Cells to the left of the node show a coincident rise in intracellular calcium, but the relationship of this calcium increase to downstream asymmetric organ development was unknown.
Makova and colleagues reported that the calcium influx triggered a pathway that increased cell mobility—a behavior that's necessary for normal gastrulation.
The pathway started with the relocalization of a protein called inversin. The team was studying inversin because internal organs are switched left to right in mice that lack this protein. In cultured epithelial cells, calcium caused inversin to move from the plasma membrane and nucleus to the cytoplasm. The treated cells adopted the shape and behavior of migratory cells, including loss of a cell–cell junction protein called E-cadherin. By expressing a cytoplasmic form of inversin, Makova showed that its repositioning there caused the migratory behavior.
Inversin also relocated to the cytoplasm in cells on the left-hand side of the mouse embryonic node, indicating that asymmetric cell migration at gastrulation might create asymmetric organs. What remains a mystery, however, is why mice that lack inversin show a complete reversal of organ orientation rather than two right sides. 
Schlueter, J., and T. Brand. 2007. Cytogenet. Genome Res. 117:256–267.
Lung cells stiffen up against stress
Lung epithelial cells' first response to shear stress is to toughen up their keratin frame, according to a talk by Karen Ridge (Northwestern University, Chicago, IL). Such stress can occur in patients receiving mechanical ventilation.

The keratin frame (green) of a lung cell stiffens in response to shear stress.
RIDGE/AAAS
Mechanical ventilation, although a life-saving technique, exposes the lungs to unnatural forces that can induce rips in lung alveoli and cause hemorrhaging. If the patient survives, these problems may lead to severe scarring and thus reduced lung function. Mechanical ventilators cannot be simply turned down, as this would not provide sufficient oxygenation for the patient. Ridge, Robert Goldman, and colleagues are investigating the physiology and cell biology behind ventilator-associated lung trauma to begin steps toward alternative ways to reduce injuries.
According to Ridge, normal lung epithelial cells contained a meshwork of keratin fibers that was densely packed around the nucleus and became increasingly lacy toward the cell periphery. Following shear stress, the meshwork became a uniform density, built up by virtually all the cell's soluble keratin. The reinforcement of the peripheral keratin ramped up rapidly and remained in place up to 48 hours after the shear stress was removed.
The strengthened keratin mesh prevented cell ripping and detachment from the basement membrane, presumably due to its strain-induced stiffening. But cells that are continually exposed to stress eventually disassemble their keratin frame and undergo apoptosis, said Ridge. If the keratin disassembly is the trigger for cell death, the development of therapies that maintain keratin buildup might help reduce lung injuries resulting from ventilator use.
Sivaramakrishnan, S., et al. 2008. Proc. Natl. Acad. Sci. USA. 105:889–894.
Nuclear actin filaments found
Roshni Basu and Fred Chang (Columbia University, New York, NY) gave the audience a glimpse of elusive nuclear actin filaments. The results suggest that too many nuclear filaments are something to be avoided.
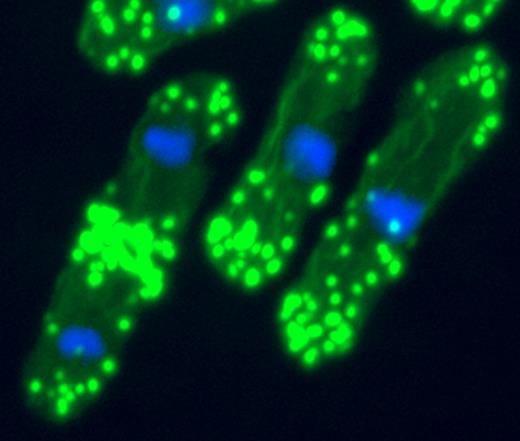
In dip1p mutants, actin filaments (green) appear in the nucleus (blue), unlike normal yeast cells (shown).
CHANG
Researchers in the contentious field of nuclear actin are starting to agree on one thing: actin in one form or another lurks in the nucleus. Actin oligomers seem to be needed for transcriptional elongation. Longer filaments may also exist, but definitive sightings are generally lacking.
Even Basu's new sightings were fortuitous. Basu was studying the formation of actin filaments by cytoplasmic nucleators called formins. She identified mutant yeast cells that lacked both a formin and its inhibitor, dip1p, and noticed that the mutant nuclei were strewn with a haze of filamentous actin. In some cells, thicker filaments resembling bars could be seen.
Nuclear filaments do not seem to be made by formins, as they were most abundant in cells lacking a formin. It's possible that nuclear actin filaments instead build up because of the potential for additional free actin monomers in these mutant cells.
The mutant cells had oddly shaped nuclei that jutted with dynamic protrusions. The group proposed that proper nuclear shape and stability require the inhibition of nuclear actin filaments, possibly by dip1p. Perhaps dip1p blocks an as-yet unidentified nuclear actin nucleator. Alternatively, it might export actin monomers from the nucleus, as has been noted in animal cells.
Martin, S.G., and F. Chang. 2006. Curr. Biol. 16:1161–1170.
Ruffles have little to do with stretching
As cells reach out to move forward, their membranes sometimes ruffle. It was thought that this ruffling is caused by the reaching arms falling short and folding back on themselves. But Michael Skalski (University of Guelph, Ontario, Canada) revealed that reaching and ruffling are not inextricably linked.

A spreading cell uses SNAP23 (green) to form both lamellipodia and ruffles, but these two protrusions are otherwise independent.
SKALSKI
Skalski and Mark Coppolino investigate how cell motility is driven by membrane trafficking proteins called SNAREs that hook together two membrane domains. Membrane trafficking, specifically exocytosis, is necessary for the formation of lamellipodia—the arms that stretch out and pull the cell forward. But which of the many SNARE family members might be responsible for hooking exocytic vesicles to the plasma membrane was unknown.
Because lamellipodia form by creating new contacts with the extracellular matrix (ECM), Skalski chose to look first at three SNAREs that form a complex upon cell adhesion to the ECM. Inhibition of one of these, SNAP23, prevented both lamellipodia formation and membrane ruffling. But inhibition of the other two left membrane ruffling intact and prevented only lamellipodia formation and cell spreading.
Skalski reported that he has now found a SNARE member that is needed to form ruffles but not lamellipodia. This SNARE was not on the ECM induction shortlist but does form a complex with SNAP23, possibly explaining why ruffles were prevented by SNAP23 inhibition.
With their own dedicated SNARE complex, membrane ruffles can no longer be dismissed as mere folds of failed lamellipodia. Other groups have suggested that these underappreciated protrusions might be involved in pinocytosis—a theory that now warrants closer analysis.
Skalski, M., and M.G. Coppolino. 2005. Biochem. Biophys. Res. Commun. 335:1199–1210.
McNiven, M.A. 2006. Trends Cell Biol. 16:487–492.
Dead cells squeezed out the top
A dying epithelial cell is squeezed out by its neighbors. Jody Rosenblatt (University of Utah, Salt Lake City, UT) described some microtubule gymnastics that send the cell out in the right direction, away from where it might cause damage.
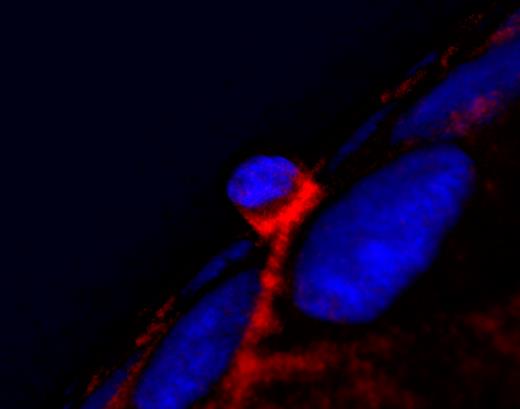
Apical extrusion of a dying cell (center) requires contraction of basal actin (red) in the surrounding cells.
ROSENBLATT
Epithelia act as barriers, so when an epithelial cell dies, it has to be removed carefully to avoid compromising that barrier. Rosenblatt previously showed that the barrier is maintained by an unusual cell extrusion process. Upon sensing an as-yet unidentified apoptotic signal, the dying cell's neighbors contract, which draws them closer together and squeezes out the deadbeat in the middle.
Rosenblatt is now studying directionality in this extrusion process. Using zebrafish and cell culture monolayers as models, she found that a dying epithelial cell is usually squeezed out the apical side. The neighbors contracted around their base, pushing the dying cell upwards. Active myosin, which contracts the actin network, was seen along the basolateral surface of the contracting cells.
The actomyosin network is normally at the top of cells, just below tight junctions. But Rosenblatt showed that, before contraction, the contractile cytoskeleton moved basally. The relocation was done by microtubules, which reoriented their plus ends toward the basal surface. What leads to the microtubule rearrangements is not clear. Work from Ron Vale's lab at UCSF has shown that plus ends localize a myosin-activating protein called RhoGEF2. Rosenblatt found that this same RhoGEF was needed for contraction.
Apical extrusion sends cells into the lumen of the epithelium, where the corpses can be easily disposed. But cells could be forced to extrude in the basal direction using drugs that freeze microtubules into their original position, at the top of the cell. Basal extrusion was also seen occasionally in normal epithelia. Rosenblatt is now interested in whether cancer cells that harbor mutations that block cell death are still able to extrude. If these cells escape basally into the underlying tissue, they might initiate metastasis.
Rosenblatt, J., et al. 2001. Curr. Biol. 11:1847–1857.
Centromeres fight physical forces
Heterochromatin may keep a stressed nucleus in shape, according to a model presented by Megan King and Günter Blobel (Rockefeller University, New York, NY).

Heterochromatin might counter microtubule forces that keep the nucleus (blue) in the center of a yeast cell.
NURSE
The shape of the nucleus is defined by the shape of its envelope—a favorite subject of the Blobel lab. King discussed her work on a novel inner nuclear envelope protein called Ima1, which was originally discovered in a proteomics screen. Having struggled to identify Ima1 function in mammalian cells, King turned to the simpler fission yeast system.
In yeast, Ima1 formed distinct foci on the envelope in association with proteins that hook the nucleus to the yeast version of the centrosome. Movies revealed that the Ima1 foci oscillated similarly to these attachment points, where microtubules meet the envelope. The oscillations reflect the constant recentering of the nucleus by microtubules, which push back against it from both ends of the cell.
When King knocked out Ima1, the mutant yeast nuclei became misshapen and distended. She imagined that Ima1 was part of a structural fortification that prevents the poking microtubules from deforming the envelope. “The nucleus is relatively huge. It probably takes a lot of force to move it,” she said. “Something within the envelope must be strong enough to withstand that, to keep the membrane intact.”
To identify that strong something, King considered a candidate Ima1-binding partner—DNA. She showed that Ima1, which has a DNA-binding Zn-finger domain, was enriched at heterochromatic centromeres, which huddle up against the yeast nuclear envelope during interphase. She proposed that Ima1 links this dense chromatin to spots on the envelope where microtubules bind and push. “Chromatin might be the physical anchor that deals with the force from microtubules,” she suggested.
In mammalian cells, Ima1 formed many more nuclear foci, possibly reflecting a greater number of attachment points between the nucleus and the cytoskeleton, including actin and intermediate filaments as well as microtubules.
Schirmer, E.C., et al. 2003. Science. 301:1380–1382.
Cancer's construction site
Cancers build their own unique extracellular matrix (ECM) highway that promotes directional cell migration, according to a presentation by Ning Yang and Andreas Friedl (University of Wisconsin, Madison, WI). The foreman in charge of construction is a matrix receptor protein commonly up-regulated in tumors.

Tumors construct extracellular matrix fibers into a parallel arrangement (bottom) that allows cancer cells to make a getaway.
YANG
The receptor is syndecan-1, a transmembrane proteoglycan that interacts with numerous components of the ECM and promotes growth factor signaling. Yang and colleagues previously found that syndecan-1 can promote cancer growth and is strongly induced in stromal fibroblasts of invasive breast carcinomas.
This growth promotion might be caused by a difference in ECM composition, the team imagined. To test this theory, they grew normal and syndecan-1-expressing breast fibroblasts under 3D culture conditions that allowed ECM formation. The syndecan-1–expressing fibroblasts built an entirely different ECM structure. Whereas normal cells built a crisscrossing meshwork of ECM, the syndecan-1 expressers laid down their ECM fibers in parallel.
The syndecan-1–induced ECM did not prompt carcinoma cell proliferation, as the authors had originally hypothesized. But it did promote their attachment—possibly because it contained more fibronectin, which helps attach the cells to the matrix. The parallel fiber arrangement also encouraged the migration of breast carcinoma cells in the longitudinal direction of the fibers.
Parallel ECM fiber production by primary tumor-associated fibroblasts had been observed before, but their cause was unknown. It now remains to be determined how high levels of syndecan-1 direct the construction of these cancer cell escape routes.
Amatangelo, M., et al. 2005. Am. J. Pathol. 167:475–488.
Provenzano, P.P. 2006. BMC Med. 4:38.
Seeking the source of heterogeneity
In some developmental systems, a population of genetically identical cells receiving apparently identical extracellular cues gives rise to heterogeneous cell types. Kevin Janes and colleagues (Harvard Medical School, Boston, MA) are now attempting to understand the origin of heterogeneity using a new technique called stochastic sampling.
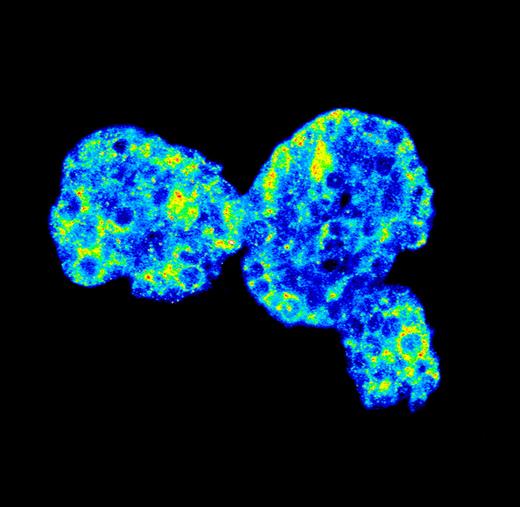
Across a population of “identical” mammary epithelial cells, FOXO target genes are highly expressed (yellow/red) in some cells and barely expressed (blue) in others.
JANES
The technique uses quantitative mRNA expression profiling to compare random small groups of ∼10 cells from an apparently homogeneous population. To pick out true transcriptional differences from noise, the researchers included a strict control. They made an mRNA mix from over 100 cells from the test population and then isolated samples of the mix equivalent to 10 cells' worth. Following PCR amplification, only those genes that had comparable expression levels in all of the ten-cell-sized samples were eligible for further analysis. Approximately 8,000 genes made it through.
The team used identical-looking, growth-arrested mammary epithelial cells as their test population. These cells are thought to express target genes of FOXO transcription factors at heterogeneous levels. Using statistical analysis of repeated samplings of ∼10 cells, the researchers deduced that FOXO targets were indeed expressed to high levels in some cells and low levels in others. And when one FOXO target was expressed strongly, so were many other FOXO targets.
Including the FOXO targets, a total of 25–30% of genes exhibited transcriptional heterogeneity, showing that the “identical” cells actually varied considerably.
The source of heterogeneity in any developmental system might be resolvable with this technique, suggests Janes. Using a fluorescent reporter of FOXO localization, for example, he can test whether all cells with nuclear FOXO also express the target genes or whether heterogeneity begins here.
Translation at focal adhesions
Nearby translation builds up focal adhesions, according to Amber Wells and Rob Singer (Albert Einstein College of Medicine, Bronx, NY).

The tagging of β-actin mRNA (green) reveals its translation sites (red) in living cells.
RODRIGUEZ
The Singer lab has shown that focal adhesions, where cells attach to their matrix, attract mRNAs. The loitering transcripts encode several adhesion components, including vinculin, talin, and β-actin. Ribosomes also linger nearby, prompting the group to wonder whether translation occurs at adhesions.
A recently developed technique to visualize translation in living cells now allows them to address this question. In this system, a tetracysteine tag on the target mRNA is translated first. The tag is then visualized using fluorescent dyes. By tagging β-actin, Wells revealed that this mRNA is translated in the periphery of migrating fibroblasts, where focal adhesions form.
The local newly made β-actin, Wells suggested, might build up actin stress fibers, which feed into adhesion sites. When she displaced its translation by removing the transcript's localization sequence, smaller, possibly immature adhesion sites predominated.
The group is still unraveling the effects of the local translation on the adhesions. The β-actin transcript is accompanied by an RNA-binding protein called ZBP, which represses translation until it is released from the mRNA. Expression of a ZBP mutant that is not released slowed cell spreading and created more adhesions that were oddly shaped.
Wells hypothesized that these stickier adhesions resulted from the translational blockade of other ZBP-binding mRNAs. Transcripts for many focal adhesion proteins contain putative ZBP1-binding sites; perhaps some are required for adhesion disassembly. Regulated local translation might thus allow each adhesion site to respond quickly and independently to local cues that either fortify it or tear it down.
Rodriguez, A.J., et al. 2006. J. Cell Biol. 175:67–76.
How microtubule motors deal with tau
Road bumps knock off some microtubule motors and set others in reverse, said Ram Dixit and Erika Holzbaur (University of Pennsylvania, Philadelphia, PA). Differences in tolerance for these obstacles might keep the cell's traffic flow where it belongs.

Dynein motors (green, arrowheads) reverse direction when they meet a tau cluster (red).
HOLZBAUR/AAAS
Most studies of microtubule-based motors in vitro rely on nice clean tracks for them to ride. But Dixit gave them obstacles to deal with in the form of the microtubule-associated protein tau. Tau helps neuronal microtubules grow and keeps them stable in the axon, but its continued presence, Dixit figured, might create problems for motor proteins.
The problems varied by motor, as Dixit showed using single-molecule tracking studies. Kinesin was bothered enough to fall right off microtubule tracks when it encountered a spot of tau. Once in a while, though, it paused rather than jumping off. Dynein, on the other hand, was less perturbed. Although it sometimes changed directions, dynein was more likely than kinesin to simply continue on past the tau obstruction. The group believes that other microtubule-bound impediments probably have similar effects on the motors.
The structural differences that account for the motors' unique reactions to tau await investigation. But Dixit pointed out that the larger, lankier dynein complex has been seen to move laterally across the microtubule surface—a feat that kinesin does not seem capable of.
In neurons, tau is abundant in axons but is essentially absent from the cell body and dendrites. This distribution might allow kinesin to escape the cell body easily with its axon-bound cargo—mitochondria and other organelles. Encounters with axonal tau might then cause the motor to fall off and deposit its cargo, while not interfering with dynein's path from axon to cell body.
Tau is abnormally expressed in the cell body early in the development of Alzheimer's disease. The new findings imply that this mispositioning might kill off neurons by depriving axons of precious cargo.
Dixit, R., et al. 2008. Science. doi: https://doi.org/10.1126/science.1152993.
Mitochondria team up for the big push
The merger of companies can make them more productive. According to a presentation by Kasturi Mitra and Jennifer Lippincott-Schwartz (National Institute of Health, Bethesda, MD), the merger of mitochondria might similarly boost productivity in ATP synthesis. The boost might be needed to meet the nucleotide demands of DNA replication.

Mitochondria (white) fuse into a giant network prior to S phase, which produces more ATP.
KASTURI
Mitochondria are not lonely organelles; they are constantly fusing together and popping apart. At cell division, mitochondria must be segregated into daughter cells, so Mitra was interested in whether these fusion and fission events occurred at particular points in the cell cycle.
Using a mammalian cell line, Mitra found that a cell's mitochondria fused together into one giant network before S phase and then disassembled upon S phase entry. Inhibiting the fusion prevented S phase via a pathway that depended on the p53 tumor suppressor.
The pathway from failed fusion to p53 activation is unknown. So too is the reason for mitochondrial fusion. The team has some clues, however. They showed that mitochondrial fusion increased ATP production, which might help meet the extra demand for this nucleotide during DNA synthesis. They now want to test whether an ATP boost alone drives G1 cells into S phase.
ATP production might not be the only reason for fusion. Photobleaching and microirradiation experiments showed that the inner matrix of the merged mitochondria was continuous throughout the network. The continuous matrix might provide mitochondria, which do not have their own DNA repair machinery, with the opportunity to complement damaged genomes by sharing good copies. As fusion occurs just before S phase, genome complementation might help to ensure that functional mitochondria are delivered to each daughter cell at the upcoming division.
Detmer, S.A., and D.C. Chan. 2007. Nat. Rev. Mol. Cell Biol. 8:870–879.

