Lysosomes are degradation and signaling organelles central to metabolic homeostasis. It remains unclear whether and how harmful metabolites compromise lysosome function in the etiopathology of metabolic disorders. Combining Caenorhabditiselegans and mouse models, we demonstrate that homocysteine, an intermediate in methionine-cysteine metabolism and the cause of the life-threatening disease homocystinuria, disrupts lysosomal functions. In C. elegans, mutations in cystathionine β-synthase cause strong buildup of homocysteine and developmental arrest. We reveal that homocysteine binds to and homocysteinylates V-ATPase, causing its inhibition and consequently impairment of lysosomal degradative capacity. This leads to enormous enlargement of lysosomes with extensive cargo accumulation and lysosomal membrane damage in severe cases. Cbs-deficient mice similarly accumulate homocysteine, displaying abnormal or damaged lysosomes reminiscent of lysosomal storage diseases in multiple tissues. These findings not only uncover how a metabolite can damage lysosomes but also establish lysosomal impairment as a critical contributing factor to homocystinuria and homocysteine-related diseases.
Introduction
Lysosomes are single-membrane–limited organelles that play a central role in cellular metabolism. Lysosomes are characterized by their acidic lumen (pH 4.5–5.0), which is generated and maintained by the multi-subunit lysosomal V-ATPase and co-regulated by several ion transporters localized on lysosomal membranes (Mindell, 2012; Vasanthakumar and Rubinstein, 2020). In the acidic lumen, >60 different lysosomal hydrolases are activated, which act coordinately to digest extra- and intracellular materials generated by endocytosis, phagocytosis, and autophagy. The degradation products are exported to the cytoplasm by lysosomal membrane transporters and reused as building blocks to sustain cellular homeostasis. Thus, lysosomes are the degradation and recycling centers of the cell. On the other hand, lysosomes serve as major signaling hubs for metabolic regulation in the cell. A variety of protein kinases, including mTOR, AMPK, PKC, and GSK3, reside on lysosomal surfaces to sense and transduce nutritional, energy, and growth factor cues; together, they coordinate anabolism with catabolism under normal and stress conditions (Goul et al., 2023; Li et al., 2016b; Su et al., 2024, Preprint). Dysfunction of lysosomes, caused either by deficiencies in lysosomal hydrolases, transporters, and structural proteins or by mutations of non-lysosomal proteins required for appropriate transport of lysosomal enzymes, leads to lysosomal storage diseases (LSDs) that manifest as accumulation of undigested materials (Ballabio and Bonifacino, 2020; Parenti et al., 2015; Platt et al., 2012; Yang and Wang, 2021). Moreover, lysosome dysfunction plays a role in metabolic disorders, such as obesity and diabetes, which perturb mTOR-dependent signaling and hence cell homeostasis (Ballabio and Bonifacino, 2020; Goul et al., 2023; Yang and Wang, 2021).
While lysosomes act as the major metabolic organelles, it is largely unknown how their functions and homeostasis might be affected by abnormally accumulated intermediates resulting from defective metabolism of major nutrients, including AAs, sugars, and fatty acids. As the building blocks of proteins, AAs are of particular importance to lysosomal homeostasis and function. Cytosolic and lysosomal AAs are sensed by AA receptors/transporters, and the signals are further transduced to mTORC1 located on the lysosomal surface (Ballabio and Bonifacino, 2020; Goul et al., 2023). A sufficient AA supply enables activation of mTOR, which promotes protein synthesis but inhibits lysosome biogenesis and autophagy. In contrast, AA deprivation leads to mTOR inactivation, which in turn promotes TFEB-dependent lysosomal biogenesis and autophagy (Ballabio and Bonifacino, 2020; Goul et al., 2023; Yang and Wang, 2021). Moreover, the export of AAs out of lysosomes during lysosomal degradation plays an essential role in lysosome reformation, defects of which reduce the abundance of lysosomes (Gan et al., 2019; Jouandin et al., 2022; Liu et al., 2012). In mammals, AAs also undergo oxidation, serving as an alternative energy supply. In addition, AAs are constantly converted from one to another by transamination or other mechanisms (Bröer, 2023; Chandel, 2021). These metabolic processes generate a wide variety of metabolic intermediates, which, if aberrantly accumulated, lead to aminoacidopathies or organic acidemias/acidurias (Wajner and Goodman, 2011). Such metabolic disorders often manifest as severe developmental retardation and intellectual disability (Wajner and Goodman, 2011; Zhou et al., 2019). To date, it is unknown whether and how AA metabolic intermediates cause lysosome damage and dysfunction. Furthermore, it is not understood how lysosome dysfunction contributes to life-threatening organic acidemias/acidurias.
The nematode Caenorhabditiselegans, which normally lives on Escherichia coli strains in the laboratory, provides an excellent system to investigate the effects of accumulated metabolites on intracellular organelles, including lysosomes and mitochondria (Han et al., 2017; Yang and Wang, 2021; Zhou et al., 2022). Using unbiased genetic screening, we here reveal that mutations of C. elegans cystathionine (Cth) β-synthase (CBS) lead to a strong accumulation of homocysteine (Hcy), an intermediate in methionine (Met)-cysteine metabolism (Kruger, 2017; Maron and Loscalzo, 2009), which causes lysosomal dysfunction characterized by impaired degradative capacity, extensive cargo accumulation, marked lysosomal enlargement, and compromised lysosomal membrane integrity. We demonstrate that Hcy impairs lysosomes by binding to and homocysteinylating lysosomal V-ATPase, resulting in significantly reduced V-ATPase activity. We further reveal that Cbs knockout mice accumulate high levels of Hcy and exhibit abnormally enlarged or damaged lysosomes, reminiscent of LSDs, in multiple tissues. Together these findings demonstrate how lysosomal function can be impaired by a pathogenic intermediate of AA metabolism. In humans, CBS mutations lead to homocystinuria (HCU) with a strong accumulation of Hcy. The harmful buildup of Hcy causes severe complications with developmental delay, intellectual disability, skeletal defects, vascular problems, and eye problems (Maron and Loscalzo, 2009; Moll and Varga, 2015). There is no cure for HCU, and its underlying cellular mechanisms are not understood. Thus, our findings establish a direct mechanistic link between pathologically accumulated Hcy and lysosomal dysfunction, providing new insights into the cellular basis of life-threatening disorder HCU as well as other Hcy-related human diseases.
Results
Loss of cbs-1 causes lysosomal enlargement and developmental defects
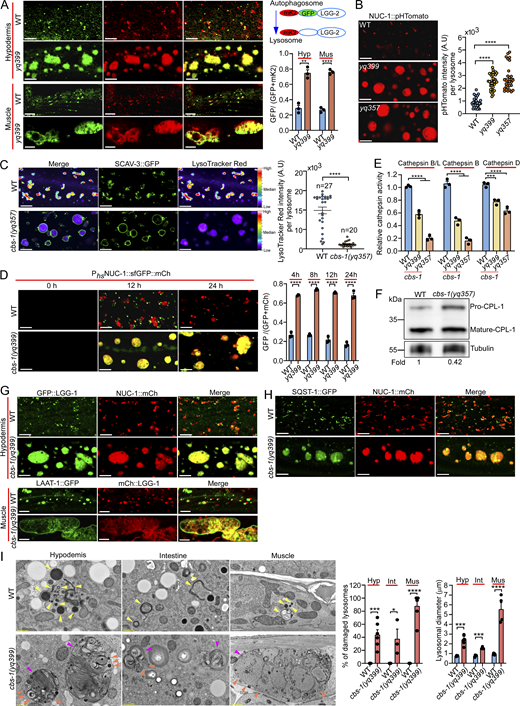
To explore how lysosome function is regulated in living animals, we sought to identify C. elegans mutants that are defective in lysosomal degradation of autophagic cargos. To this end, we performed ethyl methanesulfonate (EMS) mutagenesis of worms expressing GFP-tagged LGG-2 (GFP::LGG-2), the C. elegans homolog of mammalian LC3/ATG8 (Wu et al., 2015), and screened for mutants that exhibited an abnormal GFP::LGG-2 signal in adults fed on OP50 E. coli, which is the normal laboratory diet of C. elegans. A mutant, yq399, accumulated giant GFP::LGG-2-positive structures in the hypodermis and body wall muscles in a development-dependent manner, compared with the small GFP::LGG-2 puncta in WT (N2) animals (Fig. 1, A and B). The giant GFP::LGG-2 structures were also positive for mCherry (mCh)-fused NUC-1 (NUC-1::mCh), a lysosomal DNase (Li et al., 2016a; Miao et al., 2020) (Fig. 1 C), and were co-labeled with GFP-fused SCAV-3 (SCAV-3::GFP), a homolog of the mammalian lysosomal membrane protein LIMP2 (Fig. 1 D) (Li et al., 2016a). In contrast, the YFP::2xFYVE-positive endosomes in the hypodermis in yq399 mutants were not obviously affected compared with that in WT (Fig. S1 A). These results suggest that the yq399 mutation caused lysosomal abnormalities. Strikingly, yq399 mutants exhibited greatly shortened body length (Fig. 1 E), and their lifespan was strongly reduced compared with the WT (Fig. 1 F), suggesting a potential link between the abnormal lysosomes and the impaired animal development and survival.
We mapped the yq399 mutation to the linkage group X and identified a g.2263 G>A mutation in the cbs-1 gene, which resulted in an E481K substitution in the encoded CBS-1 protein (Fig. S1 B). CBS-1 is a homolog of the mammalian pyridoxal 5′ phosphate–dependent CBS (Fig. S1, B and C). Using CRISPR/Cas9, we generated another mutant, cbs-1(yq357), which caused an R80Amber premature stop codon in CBS-1. cbs-1(yq357) mutants exhibited similar lysosomal enlargement and developmental defects (Fig. 1, A–D and F). A knock-in CBS-1::GFP, generated by CRISPR/Cas9, exhibited diffusive pattern in the cytoplasm in the hypodermis and along body wall muscle fibers (Fig. S1 D). Transgenic expression of GFP- or mCh-fused CBS-1 driven by the cbs-1 promoter (Pcbs-1cbs-1::gfp or Pcbs-1cbs-1::mCh), which appeared to be cytoplasmic as the knock-in CBS-1::GFP, fully rescued the defective lysosomes in both hypodermis and muscles as well as the shortened body length in cbs-1(yq399) mutants (Fig. 1, E, G, and H). Heterologous expression of human CBS::GFP driven by the semo-1 promoter (Psemo-1hCBS::gfp), which was mainly expressed in the hypodermis, also strongly rescued the shortened body length and defective lysosomes in cbs-1(yq399) mutants (Fig. 1, E and G). This suggests that C. elegans CBS-1 and mammalian CBS share conserved functions in regulating lysosome properties. Taken together, these findings suggest that loss cbs-1 function leads to lysosomal abnormality and defective development.
Loss of cbs-1 causes lysosomal dysfunction by disrupting lysosomal acidification
To investigate how lysosomal function is affected in the absence of cbs-1, we employed a reporter of LGG-2 tandemly fused with GFP and mKate2 (mK2::GFP::LGG-2), which differentially labels autophagic compartments and lysosomes based on the pH sensitivity of GFP and mKate2. In cbs-1(yq399) mutants, the GFP (green) and mKate2 (red) signals were mostly overlapped in the enlarged lysosomes, in contrast to the majority of red puncta representing autolysosomes/lysosomes in WT hypodermis and muscles (Fig. 2 A). Given that GFP signals are normally quenched in the acidic lysosomal lumen (Shaner et al., 2007), these results suggest that the yq399 mutation probably impairs lysosomal acidification. Using a heat shock promoter to drive the expression of NUC-1 fused with pHTomato (NUC-1::pHTomato), a pH-sensitive fluorescence protein, which has a pKa close to 7.8 and thus exhibits increased fluorescence when the pH is increased (Li and Tsien, 2012; Sun et al., 2020), we found that lysosomes in cbs-1(yq399) and cbs-1(yq357) animals were more strongly labeled by NUC-1::pHTomato than the WT (Fig. 2 B), further suggesting that lysosomal pH is elevated. Consistent with this, the staining of enlarged lysosomes (labeled with SCAV-3::GFP) by LysoTracker Red in cbs-1(yq357) mutants was much weaker than in WT animals (Fig. 2 C). In addition, we traced lysosomal maturation in cbs-1(yq399) mutants by expressing a transgenic array of NUC-1 fused tandemly with sfGFP and Cherry under the control of a heat shock promoter (Phsp-16nuc-1::sfgfp::Cherry) (Miao et al., 2020). In WT hypodermal cells, sfGFP signals were quenched in lysosomes 24 h following heat shock–induced expression of NUC-1::sfGFP::Cherry, and sfGFP signals seldom overlapped with Cherry signals (Fig. 2 D). In cbs-1(yq399) mutants, in contrast, sfGFP signals were not quenched but overlapped well with Cherry in the enlarged lysosomes (Fig. 2 D), suggesting that lysosomal acidification is impeded. In agreement with this, the activities of several cathepsins, including cathepsin B/L, cathepsin B (CTSB), and cathepsin D (CTSD), were significantly reduced in cbs-1(yq399) and cbs-1(yq357) mutants (Fig. 2 E). Western blotting confirmed that the maturation of cathepsin L (CPL-1) was strongly reduced in cbs-1(yq357) mutants (Fig. 2 F). Consequently, autophagic cargos, including p62/SQST-1 (SQST-1::GFP) and GABARAP/LGG-1 (GFP::LGG-1 or mCh::LGG-1), accumulated in the enlarged lysosomes marked with NUC-1::mCh or LAAT-1::GFP (Fig. 2, G and H).
Using transmission electron microscopy (TEM) analysis, we further confirmed that cbs-1(yq399) mutants contained greatly enlarged lysosomes in multiple tissues, which accumulated a variety of undigested materials, including mitochondria (Fig. 2 I). TEM analysis of cbs-1(yq357) animals expressing SCAV-3::APEX::sfGFP (Li et al., 2024) confirmed that the enlarged lysosomes had the lysosomal membrane protein SCAV-3 (Fig. S2 C). Notably, a high proportion of these enlarged lysosomes were broken (Fig. 2 I). Together these results suggest that loss of cbs-1 impedes lysosomal acidification and consequently causes lysosomal dysfunction, abnormal storage of undegraded cargos, and breaking of lysosomal membranes.
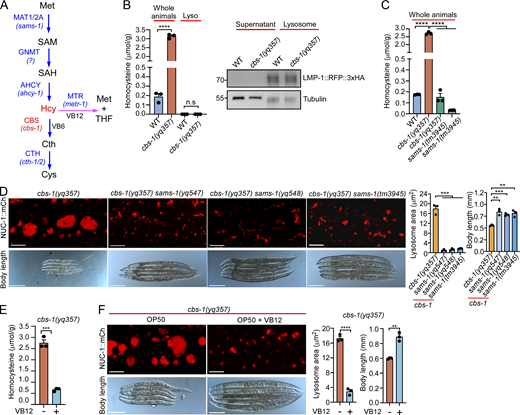
Hcy accumulation is causal to lysosomal dysfunction
Human CBS converts Hcy to Cth in the metabolism of Met (Fig. 3 A), and mutation of CBS leads to the Hcy-accumulating disease HCU (Kruger, 2017; Maron and Loscalzo, 2009; Moll and Varga, 2015). In C. elegans cbs-1(yq357) mutants, Hcy levels were greatly increased compared with WT (Fig. 3 B). However, further analysis revealed no obvious Hcy accumulation in lysosomes in cbs-1(yq357) animals (Fig. 3 B). To determine whether Hcy accumulation is responsible for the lysosomal defects in cbs-1 mutants, we generated double mutants of cbs-1(yq357) with the tm3945 deletion mutant of the sams-1 gene, which encodes the homolog of human MAT1A/MAT2A and catalyzes the conversion of Met to S-adenosylmethionine (SAM) in the Met-to-cysteine metabolism pathway (Fig. 3 A). Notably, no accumulation of Hcy was detected in sams-1(tm3945) single mutants and cbs-1(yq357) sams-1(tm3945) double mutants (Fig. 3 C). Consistent with this, the double mutants exhibited no marked enlargement of lysosomes, and their body length was significantly increased compared with cbs-1(yq357) single mutants (Fig. 3 D). In addition, we carried out an EMS mutagenesis screening for suppressors of the cbs-1(yq357) mutants and identified two additional sams-1 mutations, yq547 (M1R) and yq548 (G107E) (Fig. S2 A). Double mutants of sams-1(yq547) or sams-1(yq548) with cbs-1(yq357) similarly displayed no obvious lysosomal enlargement and had increased body lengths (Fig. 3 D and Fig. S2 B). Together, these results suggest the strongly elevated Hcy is probably responsible for lysosomal enlargement and dysfunction in cbs-1 mutants.
C. elegans is routinely fed on the B-type OP50 E. coli, which are deficient in vitamin B12 (VB12) (Revtovich et al., 2019; Zhou et al., 2022). To corroborate the above conclusion, we supplied VB12 to the OP50 E. coli to allow for Hcy metabolism through the VB12-dependent pathway (Fig. 3 A). VB12 significantly reduced Hcy accumulation and suppressed the lysosomal enlargement and shortened body length in cbs-1(yq357) animals (Fig. 3, E and F). Thus, in the absence of cbs-1, the strongly accumulated Hcy is causal to lysosomal dysfunction and defective development.
Dietary bacteria affect lysosomal health in the absence of cbs-1
It was reported that E. coli K12 strains probably have lower Met levels than the B-type strain OP50 (Neve et al., 2020). To investigate if the manifestation of lysosomal defects depends on host genes and dietary bacteria, we tested if feeding cbs-1(yq357) animals on HT115 E. coli, which is routinely used for RNAi, improves lysosomal health. As reported previously for the E. coli K12 strain MG1655 (Neve et al., 2020), HT115 E. coli contained lower Met levels than the OP50 strain (Fig. 4 A). HT115 feeding markedly reduced Hcy levels in cbs-1(yq357) animals compared with OP50 feeding (Fig. 4 B). Consistent with this, cbs-1(yq357) mutants fed on HT115 bacteria exhibited mostly normal lysosomes (marked with SCAV-3::GFP and NUC-1::mCh), and their body length and lifespan were significantly increased (Fig. 4, C and D). Supplementation of Met to HT115, however, restored the lysosomal enlargement in cbs-1(yq357) mutants, but not in cbs-1(yq357) sams-1(tm3957) double mutants (Fig. 4 E), consistent with the requirement for SAMS-1 in the first step for producing Hcy (Fig. 3 A). Hcy supplementation to HT115 E. coli essentially abolished the lysosome-ameliorating effects in both cbs-1(yq357) single and cbs-1(yq357) sams-1(tm3945) double mutants, as evidenced by the reappearance of enlarged lysosomes (Fig. 4 E). Taken together, these results provided further evidence that Hcy is responsible for the lysosomal defects and growth impairment in cbs-1 mutants.
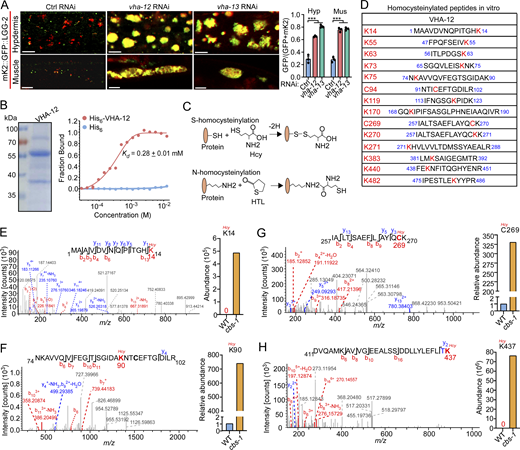
Hcy binds to and homocysteinylates V-ATPase subunits
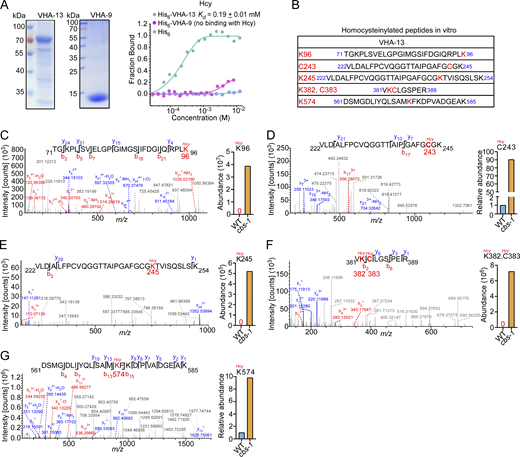
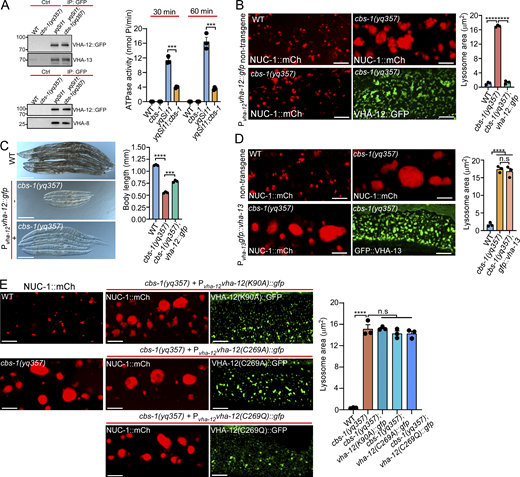
Given that lysosomal pH is elevated in cbs-1 mutants, we next investigated how Hcy disrupts lysosomal acidification. Lysosomal pH is mainly controlled by V-ATPase, a multi-subunit complex that pumps protons into lysosomes (Mindell, 2012; Vasanthakumar and Rubinstein, 2020). We knocked down individual genes encoding V-ATPase subunits by RNAi (Table S1). In WT animals, inactivation of vha-12 and -13, which encode the B and A subunits of the V1 subcomplex of V-ATPase, caused similar lysosomal defects to cbs-1 mutants (Fig. 5 A). We therefore investigated whether Hcy exerted an inhibitory role on V-ATPase so as to impair lysosomal acidification. Using microscale thermophoresis (MST) assays, we found that Hcy bound directly to purified VHA-12 and VHA-13, with Kd values of 0.28 ± 0.01 mM and 0.19 ± 0.01 mM, respectively (Fig. 5 B and Fig. S3 A). In comparison, Hcy had no obvious binding with the V1 F subunit, VHA-9 (Fig. S3 A). These results suggest that Hcy binds to V-ATPase subunits with certain specificities. Because Hcy can covalently link to proteins at cysteine (C-Hcy, S-homocysteinylation) or lysine (K-Hcy, N-homocysteinylation) residues (Fig. 5 C) and impair protein functions (Jakubowski, 2019; Zhang et al., 2018; Zinellu et al., 2017), we further investigated whether Hcy binding caused homocysteinylation of these V-ATPase subunits. Using mass spectrometry, we identified multiple cysteine and lysine residues that were homocysteinylated in VHA-12 and -13, following their incubation with Hcy (Fig. 5 D and Fig. S3 B). We next sought to determine whether these modifications on VHA-12 and -13 occurred in cbs-1 mutants. To do so, we expressed VHA-12::GFP as single-copy insertions in the genome in both WT and cbs-1(yq357) mutants and immunoprecipitated VHA-12::GFP using GFP antibody. Using LC-MS/MS, we analyzed the immunoprecipitated VHA-12::GFP and the coprecipitated endogenous VHA-13. Multiple cysteine and lysine residues were found to be homocysteinylated in VHA-12 in vivo (Fig. 5, E–H), some of which overlapped with the homocysteinylated residues identified in vitro (Fig. 5 D). Several homocysteinylated sites were also identified in the coprecipitated endogenous VHA-13 (Fig. S3, C–G), and these sites were consistent with the sites identified by in vitro homocysteinylation assays (Fig. S3 B). In all cases, the abundance of homocysteinylation at each identified AA residue was significantly higher in cbs-1 mutants than in WT. Collectively, these data suggest that the elevated Hcy led to homocysteinylation of V-ATPase subunits.
The decrease in V-ATPase activity accounts for lysosomal defects in cbs-1 mutants
To assess the effects of Hcy binding and homocysteinylation on V-ATPase, we measured V-ATPase activity by immunoprecipitating the single-copy-inserted VHA-12::GFP from both WT and cbs-1(yq357) animals. Endogenous VHA-13 and VHA-8 (the E subunit of the V1 subcomplex) were co-immunoprecipitated with VHA-12::GFP in both WT and cbs-1(yq357) animals (Fig. 6 A), suggesting that Hcy did not affect the assembly of the V1 subcomplex. However, compared with WT, the V-ATPase activities in cbs-1(yq357) mutants were significantly decreased at the experimental time points (Fig. 6 A). This suggests that the highly accumulated Hcy inhibited lysosomal V-ATPase activity. To corroborate this conclusion, we reinforced the expression of VHA-12 by introducing a transgenic array expressing VHA-12::GFP under the control of the vha-12 promoter (Pvha-12vha-12::gfp). VHA-12::GFP colocalized well with VHA-5::RFP that marks multivesicular bodies/late endosomes/lysosomes (Shi et al., 2022), but not the early endosome marker 2xFYVE::mK2 (mKate2), suggesting that VHA-12::GFP was correctly targeted to late endosomes/lysosomes (Fig. S4 A). Importantly, overexpressed VHA-12::GFP strongly suppressed the lysosomal enlargement in cbs-1(yq357) mutants (Fig. 6 B) and significantly restored the shortened body length (Fig. 6 C). By comparison, similar overexpression of GFP::VHA-13 had no rescuing effect on lysosomal defects in cbs-1(yq357) animals (Fig. 6 D). Moreover, we overexpressed VHA-12::GFP containing mutations of the homocysteinylated residues detected by in vivo mass spectrometry analysis (Fig. 5, E–G). While overexpression of VHA-12(K90A), VHA-12(C269A), and VHA-12(C269Q) failed to alleviate the lysosomal enlargement in cbs-1(yq357) animals (Fig. 6 E), overexpression of VHA-12(K14A) or VHA-12(K437A) mostly suppressed the lysosomal enlargement (Fig. S4 B). Thus, the homocysteinylation of individual AA residues probably affects VHA-12 with differential effects. Taken together, these findings suggest that the accumulated Hcy in cbs-1 mutants exerts an inhibitory effect on V-ATPase by directly binding to and homocysteinylating V-ATPase subunits. This in turn inhibits the V-ATPase activity and impairs lysosomal acidification and consequently leads to dysfunction of lysosomes.
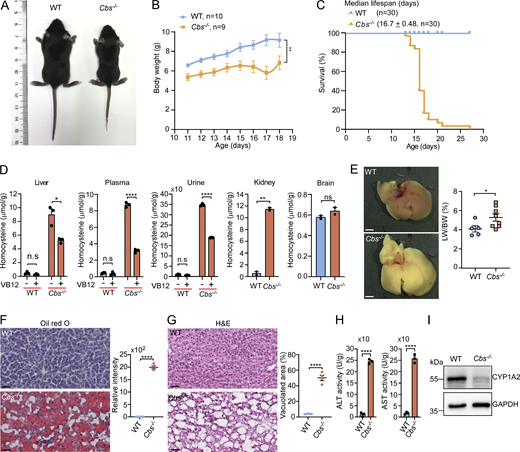
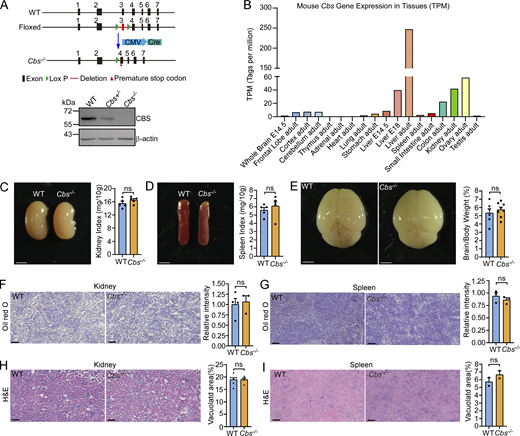
Hcy accumulation leads to developmental defects and liver damage in Cbs knockout mice
To investigate whether Hcy accumulation similarly causes lysosomal dysfunction in mammals, we generated Cbs knockout (Cbs−/−) mice (Fig. 7 A and Fig. S5 A). CBS is predominantly expressed in the liver, kidney, and ovary in adult mice (Fig. S5 B) (Yue et al., 2014). Cbs−/− mice exhibited growth retardation with significantly reduced body weights and premature mortality with 50% of animals dying by ∼17 days (Fig. 7, B and C), consistent with previous reports (Kruger, 2017; Watanabe et al., 1995). The Hcy levels in the liver, kidney, plasma, and urine, but not brain, were markedly elevated in Cbs−/− mice (Fig. 7 D). Administration of VB12 via oral gavage, however, significantly reduced the Hcy levels in the liver, plasma, and urine (Fig. 7 D). Because liver is the primary site for Hcy metabolism, we next investigated the effect of Hcy accumulation on the liver. Compared with the WT, Cbs−/− mice displayed markedly swollen and yellowish hepatic tissue with increased ratio of liver-to-body weight (Fig. 7 E). Oil Red O staining revealed a severe accumulation of lipid droplets, and hematoxylin and eosin (H&E) staining further indicated a widespread vacuolation in Cbs−/− livers (Fig. 7, F and G). In addition, Cbs−/− mouse livers exhibited strongly elevated activities of aspartate aminotransferase and alanine aminotransferase, as well as a marked reduction in CYP1A2 protein levels (Fig. 7, H and I). Altogether, these data suggest that Cbs−/− mice had severe hepatic damage, which is analogous to the liver damage reported in HCU patients (Gaull and Schaffner, 1971; Maclean et al., 2010; Majtan et al., 2017; Robert et al., 2005; Watanabe et al., 1995).
Unlike the liver, the kidney, spleen, and brain in Cbs−/− mice were superficially normal compared with that in WT mice (Fig. S5, C–E). Oil Red O and H&E staining did not reveal obvious abnormality of the kidney and spleen, either (Fig. S5, F–I). Thus, the liver is more severely affected in Hcy-accumulating mice.
CBS deficiency causes lysosomal defects in mouse liver and kidney
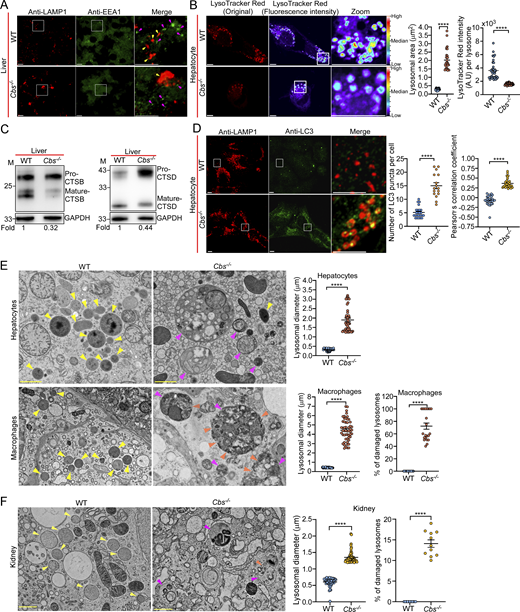
We next analyzed lysosomes in both Cbs−/− and WT mice. Firstly, we performed immunostaining of liver sections using antibodies of EEA1 and LAMP1. The results showed that Cbs−/− livers contained enlarged LAMP1-positive lysosomes, while EEA1-positive endosomes were not obviously affected (Fig. 8 A). We then isolated hepatocytes and performed LysoTracker Red staining, and found that Cbs−/− hepatocytes had enlarged lysosomes with weaker LysoTracker Red staining than WT hepatocytes (Fig. 8 B). Consistent with this, the maturation of lysosomal CTSB) and CTSD was strongly reduced in Cbs−/− hepatocytes (Fig. 8 C). In addition, we found that Cbs−/− hepatocytes accumulated significantly higher numbers of LC3 puncta that were colocalized with LAMP1 (Fig. 8 D). Together, these findings suggest that lysosomes in Cbs−/− mouse livers were dysfunctional.
We next analyzed lysosomes with TEM. In Cbs−/− liver tissues, both hepatocytes and macrophages exhibited markedly enlarged lysosomes (2–14-fold increase in diameter) compared with that in WT (Fig. 8 E). These enlarged abnormal lysosomes accumulated various undigested cargos, including mitochondria and membrane materials (Fig. 8 E). Notably, a large proportion (∼60%) of lysosomes in liver macrophages displayed severe structural damage, characterized by discontinuous or broken limiting membranes (Fig. 8 E). Lysosomes in Cbs−/− mouse kidneys were also greatly enlarged compared with that in the kidneys of WT mice (one to three-fold increase in diameter), which contained diverse undigested materials (Fig. 8 F). Among the lysosomes in kidney cells, ∼15% were found to be damaged (Fig. 8 F). These findings indicate that Cbs deficiency leads to the high level of Hcy, which impairs lysosomal function and consequently accumulation of lysosomal cargos and lysosomal damage in multiple tissues/organs, which recapitulates the findings in C. elegans and suggest that lysosomal damage is probably one of the contributing factors for pathogenesis of HCU and other Hcy-related diseases.
Discussion
Lysosomal function and homeostasis are known to be disrupted under many stress and pathological conditions (Ballabio and Bonifacino, 2020; Gan et al., 2019; Liu et al., 2012; Parenti et al., 2015; Platt et al., 2012). Nevertheless, it is less understood whether lysosomes are directly affected by abnormally accumulated metabolic intermediates, which frequently cause metabolic disorders. In this study, we reveal that Hcy, the HCU-causing metabolite generated in Met-cysteine metabolism, impairs lysosome function and leads to progressive lysosomal pathology in both C. elegans and mice. We demonstrated that, in C. elegans, loss of cbs-1 function leads to a strong accumulation of Hcy, which causes lysosomal dysfunction and enlargement and severely impairs animal development and growth. The lysosomal dysfunction manifests first as impaired degradative capacity, resulting in the accumulation of various cellular cargos, including proteins, lipids, and autophagic substrates. With undegraded materials accumulating, lysosomes become dramatically enlarged; and in severe cases, the accumulating undigested cargos lead to rupture of lysosomal membranes. The progressive deterioration of lysosomes leads to severe developmental delay, growth arrest, and eventual organismal death. Our findings thus highlight the essential role of appropriate Hcy metabolism in maintaining lysosomal function, integrity, and organismal development.
Our findings establish a direct causal link between Hcy accumulation and lysosomal dysfunction. Through unbiased genetic suppressor screening, we revealed that mutations in sams-1, which block SAM synthesis and consequently Hcy production, completely suppress the lysosomal defects in cbs-1 mutants. The causative role of Hcy is further supported by dietary and metabolic interventions. First, bacterial diets with varying Met contents affect lysosomal function through their impact on Hcy levels. HT115 bacteria, which contain 1.5-fold less Met than OP50, reduce Hcy levels in cbs-1 mutants by ∼1.5-fold. Although these mutants still maintain higher Hcy levels than WT animals, this reduction is probably below a critical threshold, which significantly alleviates both developmental and lysosomal defects. Importantly, supplementation of either Met or Hcy to cbs-1 mutants grown on HT115 bacteria is sufficient to restore the severe developmental and lysosomal defects, demonstrating that Hcy accumulation is the primary cause of the phenotype. Additionally, promoting Hcy metabolism through VB12 supplementation effectively rescues the phenotypes, providing further evidence that Hcy levels directly determine the severity of lysosomal dysfunction.
Our findings reveal that Hcy binds to and homocysteinylates the Α and Β subunits of the V1 subcomplex of V-ATPase, thereby inhibiting V-ATPase activity and consequently impairing lysosomal acidification. We identified high levels of homocysteinylation at multiple lysine and cysteine residues in VHA-12 (Β subunit) and VHA-13 (Α subunit) in cbs-1 mutants, and that endogenous V-ATPase activity was significantly inhibited as a result. Consistent with this, reinforced expression of VHA-12, the catalytic subunit of V-ATPase, mostly rescued the lysosomal defects and the shortened body length of cbs-1 animals. Thus, while highly accumulated Hcy might affect a variety of proteins and disrupt their functions (Jakubowski, 2019; Zinellu et al., 2017), non-covalent binding and covalent modification are probably the major mechanism by which Hcy inhibits V-ATPase to disrupt lysosomal function and lysosome-related cellular processes. In yeast, it was reported that mutation of the CYS4 gene, which encodes the homolog of C. elegans CBS-1 and mammalian CBS, caused inhibition of V-ATPase. It was suggested that CYS4 deficiency caused low levels of reduced glutathione in the cytosol is responsible for the V-ATPase inhibition (Oluwatosin and Kane, 1997). In addition, in bovine V-ATPase, the correct formation of disulfide bonds between three conserved cysteine residues in the ATP-binding region (P-loop) in the V1 A subcomplex is important for its activity. Disruption of the redox state of these residues led to inactivation of V-ATPase (Feng and Forgac, 1992; Feng and Forgac, 1994; Liu et al., 1997). In this study, we revealed that C269 in VHA-12 and C243 in VHA-13 were homocysteinylated in C. elegans cbs-1 mutants. We reason that, as reported for yeast and bovine V-ATPases, homocysteinylation of the cysteine residues probably disrupts the redox states required for formation of appropriate disulfide bonds within the proteins, leading to inhibition of V-ATPase activity. The dual protein-inhibitory mechanism by non-covalent binding and covalent modification is probably very important in Hcy-related pathologies, given that Hcy, as an intermediate in Met-cysteine metabolism, can modify various cellular proteins through homocysteinylation of both cysteine and lysine residues. As a matter of fact, homocysteinylation has been shown to affect numerous proteins crucial for diverse cellular functions. For example, homocysteinylation of collagen and elastin disrupts extracellular matrix structure, potentially contributing to vascular complications (Hubmacher et al., 2010). In the nervous system, homocysteinylation of tau protein enhances its aggregation propensity and may accelerate the formation of neurofibrillary tangles characteristic of Alzheimer’s disease (Zhang et al., 2008). Similarly, modification of α-synuclein by Hcy promotes its oligomerization, potentially exacerbating Parkinson’s disease pathology (Zhou et al., 2023). In addition, elevated Hcy levels are strongly associated with cardiovascular diseases, including atherosclerosis and thrombosis, and various neurodegenerative disorders such as Alzheimer’s disease, Parkinson’s disease, and cognitive decline (Jakubowski, 2019). These pathological effects may be mediated by both non-covalent binding and protein homocysteinylation, although the complete spectrum of protein targets and the functional consequences of their modification remain to be fully elucidated.
Our findings suggest that Hcy-induced lysosomal impairment is one of the cellular mechanisms underlying the pathogenesis of HCU, a life-threatening genetic disorder characterized by developmental delay, intellectual disability, skeletal abnormalities, and vascular complications (Kruger, 2017; Watanabe et al., 1995). Mutations in CBS cause classical HCU, but the cellular mechanisms remain poorly understood. Cbs−/− mice exhibit severe lysosomal dysfunction and accumulation of lysosomal cargos in tissues with high Hcy accumulation, particularly in the liver and kidney, which perfectly recapitulates the lysosomal defects observed in C. elegans cbs-1 mutants. The liver, as the major organ of metabolism, is rich in lysosomes and mitochondria and is thus particularly susceptible to the metabolic stress resulting from CBS deficiency (Evans and Hendriksz, 2017; Mansouri et al., 2018; Matté et al., 2009; Robert et al., 2005; Watanabe et al., 1995). In addition, lysosomes in the kidney of Cbs−/− mice also exhibit enlargement and cargo accumulation. This finding is particularly relevant as the kidney expresses CBS and other metabolic enzymes and is another major site of Hcy metabolism. In rats, impaired CBS function in the kidney leads to local Hcy accumulation (Prathapasinghe et al., 2008). Consistent with these findings, end-stage renal disease patients exhibit elevated Hcy levels, highlighting the dysregulated Hcy metabolism in renal diseases (Brosnan et al., 1995; Kruger, 2017; van Guldener and Stehouwer, 2005). The identification of V-ATPase as a critical target provides a direct molecular link between Hcy accumulation and lysosomal dysfunction. These findings further suggest that Hcy-induced lysosomal impairment is one of the cellular mechanisms underlying the pathogenesis of HCU and above mentioned Hcy-related diseases. Given that Hcy accumulation may cause damage to many different proteins (Jakubowski, 2019), it remains to be investigated whether other intracellular organelles, such as mitochondria, also suffer from Hcy-induced impairment or damage (Gaull and Schaffner, 1971).
Materials and methods
C. elegans strains and genetics
C. elegans cultures and genetics were performed following standard procedures.
The WT strain used in the experiments was the N2 (Bristol) strain. All mutants and strains generated by gene editing, integrated arrays, single-copy arrays, and extrachromosomal arrays are listed in Table S2.
Experimental reagents and expression vectors
EMS mutagenesis and gene cloning
Synchronized L4-stage yqIs205 animals expressing GFP::LGG-2 were treated with 50 mM EMS for 4 h. The resulting F2 progeny were grown at 20°C on NGM plates seeded with OP50 E. coli. F2 adults at 48 h after L4 stage were examined for abnormal GFP::LGG-2 structures with fluorescence microscopy. cbs-1(yq399) mutants with abnormal GFP::LGG-2 structures were isolated from ∼10,000 haploid genomes. sams-1(yq547) and sams-1(yq548) mutations, which alleviated the shortened body length and lysosomal enlargement in cbs-1(yq357) mutants, were isolated by screening the F2 progeny (∼20,000 haploid genomes) of EMS-treated cbs-1(yq357) mutants fed on OP50 E. coli. For mapping of mutation sites, single-nucleotide polymorphism mapping was carried out with the CB4856 (Hawaiian) strain. Identification of mutation sites was performed by using whole-genome sequencing and the MiModD tool on the Galaxy platform (Minevich et al., 2012; Schneeberger, 2014). Transgenic expression of genes was further employed to rescue defects in corresponding mutants for validating the cloned genes.
RNAi
RNAi experiments were performed with RNAi-competent OP50 (Neve et al., 2020) or HT115 E. coli. Briefly, 3–5 L4-stage animals were picked on NGM plates seeded with OP50 or HT115 bacteria expressing specific dsRNA and cultured at 20°C to produce progeny. ≥30 L4-stage progeny animals were transferred to a fresh RNAi plate for a further 48 h of culture, and then the phenotypes were analyzed.
Generation of extrachromosomal and integrated arrays
Transgenic animals with extrachromosomal arrays were obtained by microinjecting plasmids of interest into hermaphrodites at 12 h after L4 stage. Animals carrying extrachromosomal transgenic arrays were isolated from the F1 progeny. To obtain integrated arrays, worms carrying extrachromosomal arrays were treated with TMP and UV irradiation (350 J/m2) by UVP Crosslinker (Analytik Jena). The resulting F2 progeny animals were screened for stable expression of the transgenes of interests. Worms were then outcrossed six times with the N2 strain to get rid of background mutations.
Generation of deletion mutants and knock-in strains
sgRNAs were cloned into the pDD162 vector that also expresses Cas9 (pDD162-Peft-3::Cas9-Pu6::sgRNA). Gene-specific single-guide RNAs and single-stranded oligodeoxynucleotides used for repair templates were designed by using the CRISPR design program (https://crispor.tefor.net/) (Table S5). The dpy-10 mutation was used as a co-injection selection marker in F1 progeny. Dumpy (dpy) or roller (rol) F1 worms were screened for successful recombination via restriction enzyme digestion followed by sequencing to confirm the mutations.
Knock-in of the Plgg-2mK2::gfp::3×flag::lgg-2 and Pcbs-1cbs-1::gfp::3×flag reporters into the genome was performed as reported previously (Dickinson et al., 2015). Briefly, the 638-bp homologous arms of lgg-2 and 600-bp homologous arms of cbs-1 were cloned into the anti-hygromycin gene-expressing pDD282 vector. sgRNAs of lgg-2 and cbs-1 were cloned into pDD162 (Table S5). A mix containing the plasmid expressing Cas9-sgRNA (pDD162), the plasmid carrying the repair template (pDD282), and the plasmid expressing the Pmyo-2mCh::unc-54utr (pCFJ90) co-injection marker was injected into hermaphrodites at 12 h after L4 stage. Following injection, worms were maintained on hygromycin-containing NGM plates, and F2 progeny were screened to exclude the external chromosome array. Positive candidates were further sequenced to confirm the correct knock in of reporters.
Generation of single-copy insertion worms
Single-copy insertion of Pvha-12vha-12::gfp transgenes into the genome was performed as reported previously (Frøkjaer-Jensen et al., 2008). In brief, the Pvha-12vha-12::gfp DNA cassette was cloned into pCZGY2729 containing LG IV–targeted homology arms and a hygromycin resistance gene used for the repair template. sgRNA of cxTi10882 Mos1 sites is listed in Table S5. A mix containing the Cas9-sgRNA–expressing plasmid (pCZGY2750), the repair template-expressing plasmid (pCZGY2729), and the plasmid expressing the Podr-1gfp co-injection marker was injected into hermaphrodites at 12 h after L4 stage. After injection, worms were screened on hygromycin-NGM plates to exclude the external chromosome array. Positive candidates were further sequenced to determine the correct insertion.
Microscopy and imaging
All microscopy and imaging were performed at 20°C. Differential interference contrast and fluorescence images were acquired using an AxioImager M2 microscope (Carl Zeiss) equipped with an AxioCam monochrome digital camera (Carl Zeiss). Confocal images were captured with an inverted confocal microscope (LSM 880 with Airyscan model; Carl Zeiss) with a 100× 1.46-NA oil objective lens and lasers of 488 nm or 543 nm. Image processing and analysis were performed with LSM Image Browser, ZEN software (Carl Zeiss, Inc), and ImageJ. Lysosome sizes were quantified by measuring the area of individual lysosomes in C. elegans by ZEN software (Carl Zeiss, Inc) and the diameters of individual lysosomes in TEM images of mice by ImageJ. Body lengths were measured in adult worms anesthetized with 2.5 mM levamisole. To quantify whole-animal fluorescence intensity, worms synchronized at the same developmental stage were anesthetized and imaged with identical settings. Fluorescence intensity was measured in fixed regions with equal area for individual worms.
Assessing lysosome maturation
C. elegans animals expressing Phsp-16nuc-1::sfgfp::Cherry were synchronized to 24 h after L4 stage and incubated at 33°C for 30 min to induce expression of NUC-1::sfGFP::Cherry. NUC-1::sfGFP::Cherry signals were evaluated at 4, 8, 12, and 24 h after heat shock at 20°C. Because GFP is quenched in the acidic lumen of lysosomes, lysosomes with Cherry-only signals were scored as mature lysosomes, while lysosomes with dual labeling of GFP and Cherry were scored as immature lysosomes.
LysoTracker Red staining
To examine lysosomes in worms, adult animals synchronized at L4 after 48 h were soaked in M9 buffer containing LysoTracker Red DND-99 (1:50 dilution) for 1 h at 20°C in the dark. After a recovery for 2 h on fresh OP50-seeded NGM plates at 20°C in the dark, the worms were imaged with confocal microscopy. To examine lysosomes in mouse hepatocytes, primary hepatocytes cultured at 37°C with 5% CO2 supplement were incubated with LysoTracker Red DND-99 (1:3,000 dilution) for 5 min and imaged with confocal microscopy.
Lifespan assays
10 synchronized L4-stage worms were grown on an NGM plate seeded with E. coli OP50 or HT115 at 20°C and recorded as day 1. Animals were transferred to a new plate every day to separate the offspring until egg-laying stopped. The animals were then transferred to a fresh plate every 2 days. Animals that failed to respond to touch were considered dead. In total, 100 animals were assessed in each biological repeat.
Recombinant proteins
The cDNAs of vha-9, vha-12 and vha-13 were cloned into the pET-28a(+) vector with an N-terminal His6-tag and transformed into the E. coli Rosetta (DE3) strain for expression. Recombinant proteins were purified using Chelating Sepharose Fast Flow (17057501; Cytiva) following the supplier’s protocol. Proteins were concentrated and desalted by using Amicon Ultra-4 30K centrifugal filters.
MST experiments
The binding of Hcy to purified proteins was assessed by using the Monolith NT.115 instrument (NanoTemper Technologies). Briefly, proteins or His6 peptides (control) at a concentration of 200 nM were incubated with varying concentrations of Hcy in a 10 μl mixture and loaded into NT.115 standard-coated capillaries (Nanotemper Technologies). MST measurements were carried out at 25°C, with 100% excitation power and high MST power. Data were analyzed with NanoTemper software.
Lysosomal cathepsin activity assay
The lysosomal membrane protein LMP-1 tandemly fused with RFP and 3×HA (LMP-1::RFP:: 3×HA ) was expressed in the hypodermis under the control of the col-12 promoter. Lysosomes were then purified using anti-HA magnetic beads as described previously (Zhang et al., 2023). Briefly, 100–300 mg (wet weight) of adult animals (48 h after L4 stage) expressing LMP-1::RFP::3xHA were collected for each strain and extensively washed in M9 buffer followed by washing in KPBS buffer (136 mM KCl, 10 mM KH2PO4, pH 7.2, 10% glycerol, protease inhibitor cocktail, and 1 mM PMSF) to get rid of sodium. Animals were then homogenized on ice with a Dounce homogenizer. The lysates were centrifuged at 1,000 g for 3 min at 4°C. The supernatants were incubated with 5 μl of anti-HA magnetic beads for 1 h at 4°C and transferred to a magnetic stand to collect the beads. The lysosome-bound magnetic beads were washed with cold KPBS at least four times. The amount of lysosomes bound to magnetic beads from individual strains was evaluated by examining LMP-1 protein levels with western blotting. To measure cathepsin activities, lysosomal enzymes were released from lysosome-bound magnetic beads with 0.1% Triton X-100 in KPBS. After centrifugation, the cathepsin-containing supernatants (referred to as lysosomal samples here) were used to assess cathepsin (B, B/L, and D) activities. To measure CTSB and B/L activities, 20 μl of lysosomal samples was added to a reaction buffer containing 0.1 M sodium citrate (pH 4 for B and pH 5.3 for B/L), 1 mM DTT, and 10 μM nonfluorescent cathepsin substrates in a total volume of 200 μl. For measurement of CTSD activities, DTT was omitted, and the pH of sodium citrate was adjusted to 5.3. All reactions were carried out in a 96-well plate at 37°C for 2 h. The conversion of the nonfluorescent substrates to fluorescent products by cathepsins was measured with a fluorescence plate reader (Molecular Devices) at 460 nm for CTSBB and cathepsin B/L and 425 nm for CTSD.
V-ATPase assay
100–200 mg (wet weight) of C. elegans adults (48 h after L4 stage) carrying single-copy insertion of vha-12::gfp were homogenized in worm protein lysis buffer (50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 0.5% sodium deoxycholate, 10% glycerol, 1% NP-40, and 1 mM PMSF) on ice. After centrifugation, the supernatants were incubated with GFP-Nanoab-Agarose at 4°C for 3 h. The beads were washed extensively with protein lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% NP-40, and 1 mM PMSF), and the bound VHA-12::GFP and associated endogenous VHA-13 were examined by western blotting. The GFP beads with VHA-12::GFP/VHA13 were assayed for ATPase activity following a procedure described previously (Rule et al., 2016). In brief, 10 μl GFP-Nanoab-Agarose were added to HNG buffer (20 mM HEPES, pH 8.5, 13 mM NaCl, and 1% glycerol) containing 5 mM ATP and 5 mM MgCl2 to a total volume of 30 μl for reaction at 37°C. The reaction samples were collected at time points 0, 30, and 60 min. Meanwhile, 1 mM phosphate standards were serially diluted in HNG buffer to make a standard curve. Malachite green/Molybdate work reagent was added to the samples and standards and incubated at 25°C for 25 min. V-ATPase activity was determined by measuring the OD650 on a fluorescence plate reader (Molecular Devices).
Determining Met levels in E. coli strains
OP50 and HT115 E. coli were grown in LB medium to OD600 of 0.6. Bacterial cells were collected and lysed in protein lysis buffer. The lysates were centrifuged at 10,000 rpm for 30 min at 4°C to obtain the supernatant. The concentration of Met in the supernatant was measured with a Methionine Assay Kit following the protocols provided by the supplier.
Hcy measurement
50–110 mg (wet weight) of adult worms (48 h after L4 stage) or mouse tissues (50–500 mg of live, 150–250 mg of kidney, 150–250 mg of heart, 50–300 mg of plasma, and 50–300 mg of urine) were collected and frozen in liquid nitrogen. Whole worms or mouse tissues were homogenized in 500 μl of 80% ice-cold methanol and vortexed at 4°C for 30 min. Samples were then centrifuged at 12,000 g at 4°C for 15 min to collect the supernatants. The pellets were resuspended in 100 μl of freshly prepared 0.5 M DTT in water and vortexed at 4°C for 30 min and centrifuged to collect the supernatants. All supernatants were combined and passed through a 0.2-μm filter. Filtered samples were dried by evaporation and resuspended in a solution containing 50% acetonitrile and 0.1% formic acid in water. Samples were vortexed for 30 min and centrifuged at 12,000 g for 15 min at 4°C. The supernatants were collected for measurement of Hcy levels by liquid chromatography–mass spectrometry on an Orbitrap Exploris 120 (Thermo Fisher Scientific) equipped with an HILIC column (L× I.D. 2.1 × 100 mm, 2.6 μm particle size) flowing at 0.3 ml min−1. The chromatographic temperature and injected volume were 40°C and 5.0 μl. The mobile phase consisted of buffer A (0.1% formic acid) and buffer B (acetonitrile plus 0.1% formic acid) in H2O with positive mode.
Analysis of protein homocysteinylation
To examine in vitro homocysteinylation of VHA proteins, 50 mM Hcy was incubated with purified recombinant proteins in PBS containing 100 mM HEPES and 50 mM NaCl for 12 h. The samples were resolved by SDS-PAGE as described previously (Olsen et al., 2004). In brief, excised gel bands containing VHA proteins were sequentially treated with 20 mM NH4CO3, dehydrated with acetonitrile, rehydrated in reduction buffer (10 mM DTT and 20 mM NH4CO3) at 56°C, alkylated with 55 mM iodoacetamide (IAA) and 20 mM NH4HCO3, and digested with 4 ng/μl trypsin and 20 mM NH4CO3. The resultant peptide samples were extracted with 1% trifluoroacetic acid and subjected to mass-spectrometry analysis. To examine homocysteinylation of VHA proteins in C. elegans, VHA-12::GFP and GFP::VHA-13 proteins were first immunoprecipitated from worm lysates using GFP-Nanoab-Agarose. The beads were then treated with UTU buffer (6 M urea and 2 M thiourea, pH 8.0) for 30 min, followed by in-solution digestion and desalting to yield peptide samples (Wu et al., 2017). All peptide samples were analyzed by Easy-nLC 1200 (Thermo Fisher Scientific) and mass spectrometry (Orbitrap Exploris 480, Thermo Fisher Scientific). Peptide mixtures were loaded onto a home-made self-pack analytical C18 integrated column (35 cm long with 75 µm inner diameter). Peptides were separated with a binary buffer system of buffer A (0.1% formic acid) and buffer B (80% acetonitrile plus 0.1% formic acid) in H2O. Peptides were eluted using a linear gradient running from 8% to 98% solution B over 60 min. Proteins were identified based on the information-dependent acquisition of fragmentation spectra of multiple charged peptides. Up to 20 data-dependent MS/MS spectra were acquired in the linear ion trap for each full-scan spectrum acquired at 60,000 full-width half-maximum resolution.
Identification of homocysteinylated sites and label-free quantification were performed by Thermo Proteome Discoverer 2.4.0.305. The parameters of Proteome Discoverer were set as follows. “Label-free quantitation” was marked for peptide identification and quantification. Maximum missed cleavage was set to 2, and 6 and 144 were chosen for minimum and maximum peptide length. Precursor and fragment mass tolerance were set to 10 ppm and 0.02 Da, respectively. N-homocysteinylation was identified by characteristic mass shifts +174.04600 Da (Marczak et al., 2011) (with IAA modified in Hcy-SH) or +117.1700 Da (without IAA) at lysine residues. S-homocysteinylation was identified by characteristic mass shifts of +133.19000 Da at cysteine residues.
Knockout mice
Cbs knockout mice were generated by CRISPR-Cas9–mediated insertion of loxP sites flanking exon 3 of the Cbs gene. The service was provided by Cyagen Biosciences. Cbs-floxed mice were then crossed with Cag-Cre mice (provided by Cyagen Biosciences) to generate whole-body Cbs knockout mice. The knockout efficiency was confirmed by western blotting with a CBS polyclonal antibody. All mice were housed in chambers with a light/dark cycle of 12 h at the Animal Core Facility of Yunnan University. All animal experiments were approved by Yunnan University Institutional Animal Care and Use Committee.
Survival analysis in mice
Survival rates of WT and Cbs−/− mice were monitored throughout the postnatal period. The number of animals in each group was as follows: WT (n = 30) and Cbs−/− (n = 30). Survival data were collected at different postnatal days. At each time point, the number of surviving mice was recorded, and the survival rate was calculated as the percentage of mice alive relative to the initial number of mice in each group. Statistical significance was determined using the log-rank test. P ≤ 0.05 was considered statistically significant.
Oil Red O staining
P14 mice were euthanized, and fresh liver tissues were harvested. Tissues were dehydrated in 30% sucrose at 4°C and equilibrated in a 1:1 mixture of 30% sucrose and optimal cutting temperature (OCT) compound. Liver tissues were embedded in dry ice and sectioned using a cryostat (Thermo Fisher Scientific) at a thickness of 8 μm. For Oil Red O staining, liver sections were immersed in 60% isopropanol for 5 min, stained with filtered Oil Red O working solution for 15 min, and immersed in 60% isopropanol for 10 min. Sections were washed with water for 1 min (three times) and stained with hematoxylin for 10 s. Slides were rinsed under running water, mounted with 80% glycerin jelly, and scanned using a slide scanner (#SQS-100; Shengqiang Technology).
H&E staining
Euthanized P14 mice were perfused with 10 ml PBS followed by 10 ml 4% PFA to harvest liver tissues. Liver tissues were dehydrated in 30% sucrose at 4°C and equilibrated in a 1:1 mixture of 30% sucrose and OCT. Liver tissues were then embedded in dry ice and sectioned using a freezing microtome at a thickness of 8 μm. For H&E staining, liver sections were sequentially treated as follows: immersion in PBST (PBS + 0.05% Tween-20) for 10 min, staining with hematoxylin (#G1005-1; ServiceBio) for 30 s, rinsing with running water for 1 min, immersion in hydrochloric acid ethanol differentiation solution for 10 s (twice), rinsing with running water for 1 min, immersion in bluing solution for 15 s, rinsing with running water for 1 min, staining with eosin for 30 s, immersion in 95% ethanol for 10 s, and immersion in absolute ethanol for 30 s. The sections were mounted and scanned using a slide scanner (#SQS-100; Shengqiang Technology).
Mouse treatments
VB12 was administered orally using a 6-gauge gavage needle, starting at postnatal day 5, with a dose of 100 mg/kg per animal.
TEM analysis
Adult C. elegans and mouse tissues were fixed using a high-pressure freezer (EM ICE; Leica). Freeze substitution was performed in anhydrous acetone containing 1% osmium tetroxide and 0.1% uranyl acetate dihydrate. The samples were incubated sequentially at −90°C for 72 h, −60°C for 15 h, −30°C for 15 h, and finally at 0°C for 5 h in a freeze-substitution unit (EM AFS2; Leica). After three washes (10 min each) with fresh cold anhydrous acetone, the samples were infiltrated with Embed-812 resin in the following steps: 1:3 (resin) for 3 h, 1:1 for 5 h, 3:1 overnight, and 100% resin for 4 h. The samples were then left in resin overnight and embedded at 60°C for 48 h. Sections were cut to 70 nm using a microtome (EM UC7; Leica), then electron-stained with 2% uranyl acetate for 10 min and lead citrate for 5 min. Images were captured with a Hitachi HT7800 at 80 kV.
Immunostaining and western blotting
Liver cryosections (10 μm) were fixed in pre-chilled acetone (−20°C, 15 min) and washed three times with PBS. The sections were blocked with blocking buffer (2% goat serum and 1% BSA) for 1 h and then incubated with primary antibodies overnight at 4°C. After extensive washing with PBS, the section samples were incubated with fluorophore-conjugated secondary antibodies for 1 h at room temperature. After extensive washing, the samples were mounted with an anti-fade reagent and imaged on a Zeiss LSM 880 Airyscan Confocal Microscope.
For western blotting, liver tissues were homogenized in ice-cold lysis buffer (50 mM Tris-HCl, 1 mM EDTA, 150 mM NaCl, and 1% Triton Χ-100). The lysates were cleared by centrifugation at 10,000 g for 20 min at 4°C. The supernatants were collected and applied to western blotting with standard protocols. All antibodies used for immunostaining and western blotting are listed in Table S3. Western blotting images were captured with SmartChem 610 (SINSAGE Inc). Intensities of protein bands were quantified using ImageJ software. The ratios of individual mature cathepsins, (mature CPL-1/CTSB/CTSD)/(total CPL-1/CTSB/CTSD), in C. elegans cbs-1 mutants or Cbs−/− mice were normalized to that in WT C. elegans or mice to yield the relative fold of maturation of individual cathepsins.
Isolation of mouse primary hepatocyte
Primary mouse hepatocytes were isolated as described before (Charni-Natan and Goldstein, 2020). Briefly, P14 mice were euthanized and intracardially perfused first with HBSS-HEPES-EDTA buffer (25 mM HEPES and 0.5 mM EDTA) and then with digestion buffer (HBSS-HEPES, 4 mM MgSO4, 1.25 mM CaCl2, and 0.4 mg/ml collagenase IV). Following digestion at 37°C for 15 min, the samples were filtered (100 μm) and centrifuged (50 × g, 20 min, 4°C) to collect pelleted cells. After washing twice in PBS (300 × g, 5 min, 4°C), the cells were resuspended in DMEM with 10% FBS and 1% penicillin–streptomycin. Viable hepatocytes (0.3 × 106 cells/cm2) were plated on collagen I–coated dishes and incubated for 4 h. The medium was then replaced, and cells were cultured overnight for further analysis.
Statistical analysis
Statistical analyses were performed using the unpaired two-sided Student’s t test for comparisons between two experimental groups, except for body weight and survival analyses. Body weight data were analyzed using the two-way mixed-effects model followed by Sidak’s multiple comparison test. Survival curves were analyzed using the Kaplan–Meier method and the log-rank (Mantel–Cox) test. Data are presented as mean ± SEM as indicated in the figure legends. All statistical analyses were performed using GraphPad Prism 9. Statistical significance was defined as *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; n.s, not significant.
Online supplemental material
Fig. S1 (related to Fig. 1) characterizes the CBS-1 mutations and expression pattern. Fig. S2 (related to Figs 2 and 3) shows the identification of sams-1 mutants and TEM images of lysosomes in WT and cbs-1(yq357) animals expressing SCAV-3::APEX. Fig. S3 (related to Fig. 5) shows that Hcy binds to and homocysteinylates VHA-13. Fig. S4 (related to Fig. 6) analyzes VHA-12 localization and VHA-12 mutations affecting the homocysteinylated residues. Fig. S5 (related to Fig. 7) characterizes the Cbs−/− mice. Table S1 shows the list of V-ATPase subunits screened by RNAi. Table S2 shows the C. elegans strains used in this study. Table S3 shows the antibodies and reagents used in this study. Table S4 shows the expression vectors used in this study. Table S5 shows the oligonucleotides used for CRISPR/Cas9-mediated gene editing and site-directed mutagenesis.
Data availability
All data are available in the manuscript or the supplementary materials.
Acknowledgments
We thank Dr. I. Hanson for proofreading the manuscript and the Caenorhabditis Genetics Center for C. elegans strains used in this study.
This research was supported by grants 2021YFA1300302 from the National Basic Research Program of China; 32293201 and 92354303 from the National Science Foundation of China. Chonglin Yang is supported by the Program of Yunnan Province Leading Talents in Science and Technology (202105AB160003). Mei Yang is supported by Yunnan Basic Research Project (202401BF070001-015) and the Xingdian Talents Program (C619300A142).
Author contributions: Yang Yang: conceptualization, data curation, formal analysis, investigation, methodology, project administration, resources, validation, visualization, and writing—original draft, review, and editing. Qianjin Kong: conceptualization, data curation, formal analysis, investigation, methodology, project administration, resources, validation, and visualization. Chaolian Liu: formal analysis, investigation, methodology, resources, and validation. Fengyang Wang: conceptualization, data curation, formal analysis, investigation, methodology, project administration, validation, and visualization. Meijiao Li: investigation. Shalan Li: data curation. Yuehui Shi: resources. Leonard Krall: formal analysis and resources. Xin Wang: investigation. Shan He: resources. Kai Jiang: resources. Xuna Wu: formal analysis and resources. Mei Yang: funding acquisition, supervision, and writing—original draft, review, and editing. Chonglin Yang: conceptualization, funding acquisition, project administration, supervision, visualization, and writing—original draft, review, and editing.
References
Author notes
Y. Yang, Q. Kong, and C. Liu contributed equally to this paper.
Disclosures: The authors declare no competing interests exist.