Hydrolethalus syndrome (HLS) is a lethal, autosomal recessive ciliopathy caused by the mutation of the conserved centriole protein HYLS1. How HYLS1 controls centriole function is poorly understood. Here, we show that mice harboring the HYLS1 disease mutation die shortly after birth and exhibit developmental defects that recapitulate several manifestations of HLS. These phenotypes arise from a loss of centriole integrity that causes tissue-specific defects in cilia assembly and function. We show that HYLS1 is recruited to the centriole by CEP120 and stabilizes the localization of centriole inner scaffold proteins that ensure the integrity of the centriolar microtubule wall. The HLS disease mutation reduced the centriole localization of HYLS1 and caused degeneration of the centriole distal end. We propose that tissue-specific defects in centriole integrity caused by the HYLS1 mutation prevent ciliogenesis and contribute to HLS phenotypes.
Introduction
Centrioles are barrel-shaped cellular organelles composed of a ninefold triplet arrangement of microtubules with three important functions. First, centrioles form the core of the centrosome, a microtubule organizing center that nucleates the interphase microtubule cytoskeleton and guides the assembly of the mitotic spindle apparatus during cell division (Gönczy, 2012; Nigg and Holland, 2018). Second, centrioles function as basal bodies that dock at the plasma membrane and initiate the biogenesis of cilia that have motile or sensory functions (Breslow and Holland, 2019; Nigg and Raff, 2009). Finally, recent work revealed centrioles function as signaling centers that restrict the proliferation of polyploid cells in mammals (Sladky et al., 2020, 2022). Centrioles are remarkably stable structures that persist over multiple cell cycles with minimal turnover of their molecular components. Understanding how centrioles are assembled and maintained remains an important challenge.
In proliferating cells, centriole biogenesis is tightly coupled to cell cycle progression (Holland et al., 2010; Guderian et al., 2010; Cunha-Ferreira et al., 2013). G1 cells contain a pair of parent centrioles that differ in age and structure, with only the oldest parent centriole possessing distal appendages required to nucleate cilia (Nigg and Stearns, 2011; Tanos et al., 2013). Centriole duplication starts at the onset of S-phase when a new procentriole is assembled orthogonally at the base of each parent centriole. Procentrioles elongate during the S and G2 phases but are maintained in an immature state that is unable to duplicate. In mitosis, procentrioles separate from parent centrioles and undergo maturation to become replication-competent parent centrioles (Azimzadeh and Marshall, 2010; Gönczy, 2012; Nigg and Holland, 2018). At the same time, the mitotic spindle evenly partitions centriole pairs into daughter cells to maintain appropriate centriole numbers. Errors in centriole duplication and assembly have been linked to a variety of diseases, including cancer, growth retardation, and neurodevelopmental defects (Basto et al., 2008; Coelho et al., 2015; Serçin et al., 2016; Levine et al., 2017; Levine and Holland, 2018).
Mammalian centrioles can be divided into three main regions: a proximal end, which defines the centriolar ninefold symmetry and is essential for centriole duplication; a central core, required for the cohesion of the centriolar microtubule triplets; and a distal end, required for basal body membrane docking and ciliogenesis (LeGuennec et al., 2021). Centriole duplication begins with the assembly of the cartwheel comprised of the proteins SAS-6, STIL, and CEP135, which link to the centriolar microtubule wall through the protein CPAP (Tang et al., 2009; Schmidt et al., 2009; Lin et al., 2013; Pelletier et al., 2006; Vásquez-Limeta et al., 2022). The establishment of the centriolar microtubule triplet begins with the formation of a single microtubule (the A-tubule) that attaches to the cartwheel, upon which the B- and C-tubules are assembled (Guichard et al, 2010, 2013; Li et al., 2012). Together with CPAP, CEP295 and CEP120 coordinate the recruitment of several proteins required for centriole elongation and structural integrity (Chang et al., 2016; Tsai et al., 2019). Accordingly, the depletion of CPAP leads to short or broken centrioles (Vásquez-Limeta et al., 2022; Zheng et al., 2016), while the depletion of CEP120, CEP295, or their interacting factors leads to short centrioles that lack distal appendages and the ability to ciliate. By contrast, overexpression of CPAP, CEP120, or CEP295 promotes the over-elongation of the centriolar microtubule wall (Lin et al., 2013; Chang et al., 2016; Comartin et al., 2013). Despite a broad understanding of the molecular players, how these proteins cooperate to ensure centriole stability remains unclear.
A large spectrum of disorders, termed ciliopathies, arise from defects in the centriole–cilia apparatus. While complete loss of cilia function leads to developmental arrest and mid-gestation embryonic lethality, some ciliopathies are compatible with life and produce diverse phenotypes such as polydactyly, mental retardation, and infertility (Reiter and Leroux, 2017; Nigg and Raff, 2009). Hydrolethalus syndrome (HLS) is a severe autosomal recessive ciliopathy characterized by developmental defects (polydactyly, hydrocephaly, and cleft lip or palate) that lead to lethality around birth. HLS is caused by a D211G point mutation in the conserved centriole protein HYLS1 and is enriched in the Finnish population (Mee et al., 2005; Honkala et al., 2009). HYLS1 is required to anchor basal bodies at the plasma membrane and assemble the transition zone required for cilia biogenesis and signaling, and recent work has implicated cilia dysfunction as the major cause of HLS defects (Dammermann et al., 2009; Wei et al., 2016; Serwas et al., 2017; Hou et al., 2020; Chen et al., 2021; Takeda et al., 2024). HYLS1 is dispensable for centriole duplication and centrosome function in Caenorhabditis elegans, suggesting a basal body–specific function (Dammermann et al., 2009; Serwas et al., 2017). However, we previously identified HYLS1 in a genome-wide screen for genes required to arrest cell proliferation upon centriole amplification (Evans et al., 2021), indicating that in contrast to C. elegans, HYLS1 may also function in centriole assembly in mammalian cells. Consistent with a centriole-specific role, the depletion of HYLS1 in human cells was recently reported to lead to unstable centrioles with abnormal microtubule triplets (Takeda et al., 2024). Moreover, HYLS1 is required to elongate the unusually long spermatocyte centrioles in flies (Hou et al., 2020), and HYLS1 overexpression promoted the elongation of tubulin-based centriole-like structures in the cytoplasm of human cells (Takeda et al., 2024). Ultimately, how the HYLS1 D211G mutation impacts the role of HYLS1 in centriole integrity to support cilia assembly is incompletely understood.
In this study, we uncover a tissue-specific role of HYLS1 in centriole structural integrity. Depletion of HYLS1 or knock-in of the HLS disease-causing mutation results in short or broken centrioles that replicate and maintain a normal centrosome content. Centriole structural abnormalities arise from reduced recruitment of mutant HYLS1 to the centriole and lead to the impaired localization of inner scaffold centriolar proteins and defective assembly of centriole distal appendages. Together, our data show that ciliogenesis defects in HLS result from a loss of centriole integrity driven by mutant HYLS1.
Results
Hyls1 D226G leads to tissue-specific ciliogenesis defects and perinatal lethality
To probe the underlying cause of HLS, we knocked-in the corresponding human HYLS1 D211G mutation into the murine Hyls1 gene (Hyls1 D226G) (Fig. S1 A). Wildtype and heterozygous Hyls1 D226G mice (hereafter Hyls1+/+ and Hyls1+/DG, respectively) were born at Mendelian ratios with no obvious phenotype, while homozygous Hyls1DG/DG mice died at birth or shortly thereafter (Fig. 1 A). Hyls1DG/DG embryos were similar in size and weight to Hyls1+/+ and Hyls1+/DG embryos at embryonic day 14.5 (E14.5) but were smaller and lighter at E18.5 and at birth (Fig. S1, B–D), suggesting late developmental growth defects. Hyls1DG/DG pups displayed frequent polydactyly (Fig. 1 B), a cleft palate (Fig. 1 C), breathing abnormalities (data not shown), and a modest reduction in cerebral cortex size (Fig. S1, E and F). The kidney at E18.5 and postnatal day 0 (P0) was also smaller in Hyls1DG/DG animals compared with Hyls1+/+ and Hyls1+/DG animals (Fig. 1 D), and histological analysis revealed small cysts, dilated tubules, and increased fibrosis in Hyls1DG/DG kidneys (Fig. S1 G). We next analyzed cilia assembly in several tissues of P0 animals. Compared with Hyls1+/+ and Hyls1+/DG control mice, the number of cilia was substantially reduced in the kidney and trachea, but not in the heart, brain, or thymus of Hyls1DG/DG animals (Fig. 1, E and F). Analysis of cilia in the lungs of Hyls1DG/DG animals showed a modest, but significant decrease in cilia number (Fig. 1, E and F).
To examine centriole integrity, we performed ultrastructure expansion microscopy (U-ExM) in the kidney, trachea, lung, heart, brain, and thymus of control (Hyls1+/+; Hyls1+/DG) and Hyls1DG/DG mice. Centriole integrity was determined using two distinct methods: (1) a centriole asymmetry score (AS) that quantifies the relative asymmetry in the centriole microtubule wall and (2) a length score (L) that measures the longest centriole microtubule length (Fig. S2, A and B). A diminished AS score characterizes broken centrioles, while short centrioles have a normal AS score but a short length. Consistent with the reduced cilia, we found that ∼50–55% of centrioles in the kidneys and trachea from Hyls1DG/DG mice had structural defects, appearing short or broken (Fig. 2, A–D and Fig. S2 C). Analysis of centriole integrity in the lungs and heart showed that ∼35–40% of the centrioles were defective (Fig. 2, E–H and Fig. S2 C). These defects were less evident in the brains and thymus of Hyls1DG/DG animals, where only ∼30% or ∼20% of the centrioles appeared abnormal, respectively (Fig. 2, I–L and Fig. S2 C). By contrast, analysis of centrioles in brain multiciliated cells (MCCs) by U-ExM did not reveal obvious structural issues in Hyls1DG/DG mice, and immunofluorescence analysis showed that multiciliogenesis seems to occur normally in these animals (Fig. S2, D–F). Our data show that the Hyls1DG/DG mouse model recapitulates some key features of the human HLS, which is driven by tissue-specific defects in cilia assembly.
Hyls1 D226G/D211G drives ciliogenesis defects through loss of distal appendages
We generated mouse embryonic fibroblasts (MEFs) from Hyls1+/+, Hyls1+/DG, and Hyls1DG/DG animals and examined their centriole and cilia defects in more detail. Cells were serum-starved to induce ciliation and stained for centrosomes (γ-TUBULIN), distal appendages (CEP164), and cilia (acetylated α-TUBULIN). While centrosome number was maintained for all genotypes, we observed a dramatic reduction in the appearance of centriole distal appendages in Hyls1DG/DG MEFs, and cilia formation was nearly completely abolished (Fig. 3, A and B; and Fig. S3 A). The rare cilia observed in Hyls1DG/DG MEFs were of normal length (Fig. S3, B and D) and capable of recruiting smoothened in response to smoothened agonist (SAG) treatment (Fig. S3, C and D).
Since MCCs appeared unaffected in the brains of Hyls1DG/DG mice, we also examined cilia assembly during the differentiation of ependymal cell progenitors in vitro. Consistent with our observations in vivo, no clear differences were observed in the number of cilia assembled in Hyls1DG/DG MCCs (Fig. S3, E and F). Furthermore, analysis of centriole amplification steps during MCC differentiation in vitro by U-ExM showed that Hyls1 D226G does not impact centriole assembly (Fig. S3 G).
To assess whether distal appendage and cilia defects observed in Hyls1DG/DG MEFs were conserved in humans, we knocked in the D211G mutation into both HYLS1 alleles in human RPE1 cells (hereafter HYLS1DG/DG) (Fig. S4 A). HYLS1DG/DG RPE1 cells were similarly defective in cilia assembly, with only ∼1% of serum-starved HYLS1DG/DG RPE1 cells forming cilia compared with 80% of HYLS1+/+ cells (Fig. 3, C and D). In addition, HYLS1DG/DG RPE1 cells exhibited the same lack of distal appendage formation as Hyls1DG/DG MEFs despite maintaining normal centrosome counts (Fig. 3, C and D; and Fig. S4 B). Cell cycle analysis showed that the D211G mutation did not affect cell cycle progression (Fig. S4 C). To compare the impact of the HYLS1 D211G mutation with the loss of HYLS1, we generated HYLS1 knockout RPE1 cells (hereafter HYLS1−/−) (Fig. S4 D). Like HYLS1 mutant cells, loss of HYLS1 led to a severe defect in distal appendage formation and cilia assembly (Fig. S4, B, E, and F). We conclude that HYLS1DG/DG is a loss-of-function mutation that does not impact centrosome number but impairs centriole distal appendage formation and ciliation.
Finally, we performed U-ExM on serum-starved MEFs and RPE1 cells. In rare cases where cilia were present, they displayed normal structure and localized CEP164 at their distal end (Fig. 3, E and F; and Fig. S4 G). As expected, daughter centrioles from HYLS1DG/DG and HYLS1−/− RPE1 cells showed evidence of short or disintegrating centrioles (Fig. 3 F and Fig. S4 G). Together, these data confirm that HYLS1 mutation or loss leads to defects in centriole distal end formation and decreased cilia formation.
HYLS1 is required for centriole structural integrity
To characterize the centriole integrity defect in more detail, we performed U-ExM analysis in proliferating Hyls1DG/DG MEFs and HYLS1DG/DG and HYLS1−/− RPE1 cells. Centrioles from Hyls1+/+ and Hyls1+/DG MEFs and HYLS1+/+ RPE1 cells had a normal structure with a length of ∼400 nm (Fig. 4, A and B; and Fig. S5, A–C). Consistent with our data in serum-starved cells, ∼60% of centrioles in Hyls1DG/DG MEFs and ∼55% of HYLS1DG/DG RPE1 cells displayed broken centrioles, and most of the remaining centrioles were short (<400 nm in length) (Fig. 4, A and B; and Fig. S5, A and B). HYLS1−/− RPE1 cells showed a similar phenotype to Hyls1DG/DG MEFs and HYLS1DG/DG RPE1 cells (Fig. S5 C). These data show that the HYLS1 disease mutation mirrors the effects of HYLS1 knockout, resulting in defects in the assembly and/or maintenance of the centriole distal end.
Given the short and broken centrioles observed with HYLS1 deficiency, we speculated that HYLS1 may impact the localization of proteins involved in the early steps of centriole assembly and elongation. To better understand at what stage of centriole assembly HYLS1 is required, we analyzed the localization of proximal, central, or distal end centriole proteins in RPE1 cells. Similar to what we observed for CEP135 (Fig. 3 D, Fig. S4 B, and Fig. S5 D), the proximal end cartwheel component SAS-6 was unaffected by HYLS1 D211G mutation (Fig. 4 C) or HYLS1 loss (Fig. S5 E). Analysis of proteins involved in centriole elongation and integrity showed that neither HYLS1 loss nor HYLS1 mutation affected the localization of CPAP, CEP295, CEP120, or CP110 to centrioles in HYLS1DG/DG RPE1 cells and HYLS1−/− RPE1 cells (Fig. 4 C; and Fig. S5, E and F). By contrast, recruitment of the inner scaffold proteins POC5 and CENTRIN2 was diminished in HYLS1DG/DG and HYLS1−/− RPE1 cells (Fig. 4 D; and Fig. S5, E and F). Similarly, recruitment of the centriole distal-end proteins C2CD3 and TALPID3, as well as the distal appendage protein CEP164, was also diminished by HYLS1 D211G mutation or HYLS1 loss (Fig. 4 D; and Fig. S5, E and F). These data suggest that HYLS1 loss or mutation results in defective recruitment of inner scaffold proteins, which may cause the disintegration of the centriole microtubule wall.
Despite their reduced overall recruitment, U-ExM analysis showed that CENTRIN2 and CEP164 were still detectable at some broken centrioles in HYLS1DG/DG and HYLS1−/− RPE1 cells (Fig. 4 B and Fig. S5 C). A centriole coverage analysis revealed that CENTRIN2 localized in a normal pattern at the distal end of centrioles in HYLS1DG/DG RPE1 cells (Fig. S6, A–C). We analyzed CENTRIN2, POC5, C2CD3, and TALPID3 localization in RPE1 cells with duplicating centrioles. HYLS1DG/DG RPE1 cells recruited CENTRIN2, C2CD3, and TALPID3 to procentrioles, while structurally intact centrioles had CENTRIN2 and POC5 in their inner lumen and C2CD3 and TALPID3 localized at the distal end (Fig. S6 C). Furthermore, we observed procentrioles assembling orthogonally from some centriole fragments in G2-arrested HYLS1DG/DG and HYLS1−/− RPE1 cells (Fig. S6 D). Together, these data suggest that the defective recruitment of centriole inner scaffold and distal end proteins in HYLS1DG/DG and HYLS1−/− cells arises as a result of the unstable centriole microtubule wall.
HYLS1 overexpression drives assembly of centriolar microtubules
The centriole phenotype caused by HYLS1 D211G is reminiscent of phenotypes observed with loss of the known elongation factors CPAP, CEP120, and CEP295. We therefore tested if overexpression of HYLS1 would lead to abnormally long centrioles. We generated stable cell lines carrying a doxycycline-inducible WT or D211G HYLS1-mNeonGreen transgene (hereafter HYLS1WT-mNG or HYLS1DG-mNG) in HYLS1−/− DLD1 cells (Fig. S7 A). Consistent with what was observed in RPE1 cells, HYLS1 loss led to impaired recruitment of inner scaffold, distal end, and distal appendage proteins in DLD1 cells (Fig. 5, A–D; and Fig. S7, B and C). Overexpression of HYLS1WT-mNG restored the localization of CENTRIN2 and CEP164 at 2 and 4 days after doxycycline addition (Fig. 5, A–D). Furthermore, analysis by U-ExM confirmed that most centrioles elongated normally and were structurally intact at these time points (Fig. 5, E–H and Fig. S7 D). By contrast, overexpression of HYLS1DG-mNG was unable to effectively rescue CENTRIN2 and CEP164 localization (Fig. 5, A–D), and most centrioles remained defective (Fig. 5, E–H and Fig. S7 D). Overexpression of HYLS1WT-mNG drove an increase in centriole length as well as the presence of centrioles harboring an extended microtubule at the distal end (Fig. 5, E–G and Fig. S7 D), a phenotype that was rarely observed in cells overexpressing HYLS1DG-mNG (Fig. 5, E–G and Fig. S7 D). CP110 localized to the distal end of both broken and intact centrioles in all the genotypes analyzed (Fig. 5 E).
To define whether the defects observed in HYLS1DG/DG cells were entirely driven by instability in the centriolar microtubule wall, we attempted to rescue the centriole structure with the microtubule-stabilizing agent Taxol. Previous literature suggested that structurally weak centrioles are prone to further destabilize in mitosis (Wang et al., 2017; Schweizer et al., 2021). Therefore, we taxol-treated G2-phase HYLS1+/+ and HYLS1DG/DG RPE1 cells, arrested them in mitosis overnight, and analyzed centriole integrity in the following G1-phase after mitotic release with the CDK1 inhibitor RO3306 (Fig. S7 E). Taxol treatment decreased the percentage of defective centrioles and induced the over-elongation of non-acetylated centriolar microtubules observed by U-ExM analysis in HYLS1DG/DG cells (Fig. S7, F and G). These data suggest that HYLS1 stabilizes the centriole microtubule wall to maintain the structural integrity of centrioles.
HYLS1 D211G shows defective centriole recruitment
Since HYLS1 D211G behaves similarly to a complete loss of HYLS1 protein, we wondered whether HYLS1 localization to the centriole was affected by the HLS mutation. We knocked in a HA tag onto the C-terminus of HYLS1 in HYLS1+/+ and HYLS1DG/DG RPE1 cells. HYLS1WT-HA was absent on G1 centrioles that lacked the procentriole marker SAS-6 but increased in abundance as cells duplicated centrioles in the S-phase and progressed through the cell cycle (Fig. 6, A and B; and Fig. S8 A). Interestingly, HYLS1 localization was decreased in serum-starved ciliated cells (Fig. 6, C and D). In contrast to HYLS1WT-HA, HYLS1DG-HA was barely detected at centrioles throughout the cell cycle, suggesting that HYLS1 D211 is critical for the localization of HYLS1 to the centriole (Fig. 6, A and B; and Fig. S8 A).
High-resolution analysis of RPE1 HYLS1WT-HA cells with U-ExM showed that HYLS1 was more abundant at younger centrioles compared with older parent centrioles or basal bodies of ciliated cells (Fig. 6 E and Fig. S8 B). In addition, HYLS1DG-HA was undetectable at centrioles, confirming our immunofluorescence analysis (Fig. 6 E and Fig. S8 B). To examine the localization of HYLS1 D211G in cells with normal centrioles, we analyzed HYLS1+/+ DLD1 cells carrying a doxycycline-inducible WT or D211G HYLS1-HA transgene (hereafter HYLS1WT-HA or HYLS1DG-HA). Consistent with what was observed in RPE1 cells, HYLS1WT-HA localized asymmetrically to the two parent centrioles in G1 cells and was recruited to the microtubule wall during early procentriole assembly (Fig. S8 C). In the growing procentrioles, HYLS1 was localized as a cap-like structure around the microtubule wall as the microtubules became acetylated (Fig. S8 C). Endogenous wildtype HYLS1 partially restored HYLS1DG-HA recruitment to the centrioles of DLD1 cells, but the cap-like structure around the microtubule wall was absent (Fig. S8 C). To confirm HYLS1 localization with a different tag, we performed U-ExM in DLD1 cells expressing HYLS1-EGFP. This analysis further supported the asymmetric localization of HYLS1 at younger parent centrioles and its recruitment during the early stages of centriole duplication (Fig. S8 D). Together, these data show that the D211G mutation prevents the effective recruitment of HYLS1 to the centriole microtubule wall during the early stages of procentriole assembly.
Next, we set out to establish how the HYLS1 D211G mutation impacts its recruitment to the centriole. Our data suggests that, like CEP120 (Tsai et al., 2019), HYLS1 is enriched at the younger parent centriole and required for C2CD3 and TALPID3 recruitment. Our immunofluorescence analysis, however, showed that HYLS1 mutation (Fig. 4 C) or HYLS1 depletion (Fig. S5, E and F; and Fig. S7 B) did not impair CEP120 recruitment to the centriole. Therefore, we tested whether CEP120 may be responsible for HYLS1 recruitment to the centriole. CEP120 depletion by siRNA reduced the centriole recruitment of HYLS1WT-mNG to levels similar to HYLS1DG-mNG (Fig. 6, F and G; and Fig. S8, F–H). HYLS1 and CEP120 colocalized at centrioles, showing a similar asymmetric localization at parent centrioles and early recruitment to procentrioles (Fig. 6 H). Analysis of cells at the early stages of centriole duplication showed that CEP120 was recruited to procentrioles before HYLS1 (Fig. S8 I). To test whether HYLS1 acts upstream or downstream of centriole elongation factors, we tested whether HYLS1 overexpression affects CPAP or CEP120 recruitment to the centriole. Consistent with the prior observation that HYLS1 mutation or depletion did not impact the localization of these proteins to the centriole, HYLS1 overexpression did not increase the centriole recruitment of CPAP (Fig. S8 J) or CEP120 (Fig. 6 I). These data suggest that CEP120 recruits HYLS1 to the centriole, which then subsequently leads to the recruitment of TALPID3 and C2CD3.
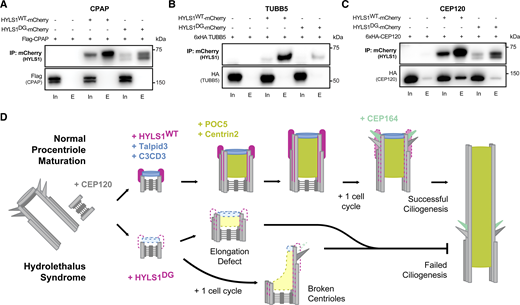
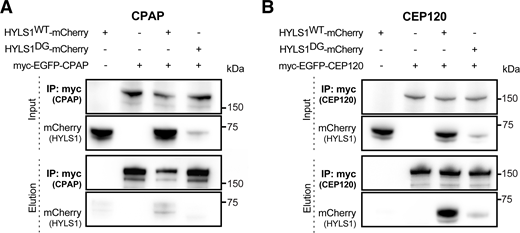
HYLS1 interacts with the elongation factor CEP120
Finally, we tested whether HYLS1 interacts with proteins involved in centriole elongation and/or structural integrity by co-immunoprecipitation analysis in DLD1 HYLS1 add-back cell lines. We began by analyzing whether HYLS1 interacts with CPAP in human cells, similar to what was previously reported in C. elegans (Dammermann et al., 2009). We failed to detect an interaction with CPAP when HYLS1 was used as the bait for immunoprecipitation (Fig. 7 A), but a weak interaction with HYLS1 was observed when CPAP was the immunoprecipitation bait (Fig. S9 A). HYLS1 was recently reported to interact with TUBB5 (Takeda et al., 2024), but we could not confirm this in our hands (Fig. 7 B). Since HYLS1 required CEP120 for recruitment to the centriole, we tested whether HYLS1 and CEP120 interacted. We observed a robust interaction between HYLS1 and CEP120 in reciprocal co-immunoprecipitations (Fig. 7 C and Fig. S9 B). While we observed a clear reduction in the binding of HYLS1 D211G to CEP120, the mutant HYLS1 protein was also less stable, making it challenging to evaluate whether the D211G mutation itself or protein instability was the cause of the reduced binding. Taken together, these data show that HYLS1 interacts with CEP120, which is required for HYLS1 centriolar localization.
Discussion
How HYLS1 disease mutation impairs centriole assembly in humans and leads to HLS remains incompletely understood. Using genetically engineered mice and cell lines, we identify centriole structural instability as a key feature responsible for the ciliogenesis defects caused by HYLS1 mutation. We show that the HYLS1 D211G disease mutation reduces recruitment of HYLS1 to the centriole, which in turn drives degeneration of the centriole distal end that harbors distal appendages required to initiate ciliogenesis. Strikingly, defective centrioles lacking HYLS1 duplicate and maintain a normal centrosome number. Thus, we identify a role of HYLS1 in maintaining centriole integrity, offering mechanistic insight into how HYLS1 mutation leads to defects in ciliogenesis.
Prior work in C. elegans, Xenopus, D. melanogaster, and human cells has revealed that loss of HYLS1 leads to impaired ciliogenesis (Dammermann et al., 2009; Wei et al., 2016; Hou et al., 2020; Chen et al., 2021; Serwas et al., 2017; Takeda et al., 2024). However, the extent of the ciliation defect resulting from the HYLS1 disease mutation varies across species and cell types (Dammermann et al., 2009; Wei et al., 2016; Serwas et al., 2017; Hou et al., 2020; Chen et al., 2021; Takeda et al., 2024). In this study, we developed a mammalian HLS model system where we knock in the D226G or the D211G mutation in mice and RPE1 cells, respectively. U-ExM analysis revealed that HYLS1 mutation or loss leads to centrioles that are short or broken at their distal end, consistent with a weakened microtubule wall. Similar structural integrity defects have been described upon loss of the centriole microtubule wall protein WDR90 and the centriole inner scaffold proteins HAUS6 and CCDC15 (Steib et al., 2020; Schweizer et al., 2021; Arslanhan et al., 2023). Like HYLS1, WDR90 depletion has also been shown to impair the recruitment of the inner scaffold proteins POC5 and CENTRIN2 (Steib et al., 2020). Thus, our data indicate that the ciliogenesis defects underlying the clinical features of HLS result, at least in part, from a structural weakness of the centriole microtube wall driven by dysfunctional HYLS1 protein. Prior work has shown that HYLS1 localizes at the basal bodies and has implicated a direct role of HYLS1 in ciliogenesis and ciliary signaling in human cell lines (Chen et al., 2021). Thus, we do not exclude that some HLS phenotypes may arise from specific abnormalities in cilia assembly or function. Defects in centriole integrity and cilia assembly were tissue-specific in the HLS mouse model and were not evident in the brains or the thymus of Hyls1DG/DG animals. We speculate that a variable dependency on HYLS1 for centriole integrity or the presence of centriolar proteins with overlapping functions could explain this tissue specificity. The complete absence of cilia is lethal embryonically, suggesting that the tissue-specific defects in ciliogenesis caused by HYLS1 mutation lead to more subtle phenotypes that support embryonic development in HLS patients.
CPAP, CEP120, and CEP295 bind microtubules and have also been shown to regulate centriole length and/or structural integrity (Tang et al., 2009; Schmidt et al., 2009; Chang et al., 2016; Sharma et al., 2016, 2018, 2021; Vásquez-Limeta et al., 2022). Although HYLS1 loss, mutation, or overexpression did not affect the recruitment of these proteins, HYLS1 loss or mutation drove a strong decrease in the recruitment of the downstream CEP295 and CEP120 interacting proteins POC5, C2CD3, and TALPID3. In particular, C2CD3 and TALPID3 are recruited by CEP120 to the procentrioles during centriole duplication and stabilize the centriole distal end (Wang et al., 2018). We conclude that HYSL1 regulates the early steps of procentriole assembly and elongation by facilitating the recruitment of proteins required for centriole structural integrity and distal end assembly. In the future, it would be interesting to explore how HYLS1 cooperates with CEP120 and other centriole proteins to promote proper centriole distal end assembly.
The centriole integrity defects observed in HYLS1 mutant cells were partly rescued by microtubule stabilization with Taxol. Moreover, we show that HYLS1 overexpression leads to over-elongated centriolar microtubules and centrioles with an extended microtubule filament at their distal end. Similar phenotypes have been described for CPAP, CEP120, CEP295, or POC5 overexpression (Kohlmaier et al., 2009; Schmidt et al., 2009; Lin et al., 2013; Comartin et al., 2013; Chang et al., 2016; Schweizer et al., 2021), suggesting that HYLS1 might cooperate with these proteins to regulate the early steps of centriole assembly. Consistent with the data presented here, a recent study showed the accumulation of centriole-like microtubule structures in the cytoplasm upon HYLS1 overexpression (Takeda et al., 2024), possibly explaining the requirement of HYLS1 for the elongation of the giant centriole/basal body in fly spermatocytes (Hou et al., 2020). The impaired localization of the distal end proteins and distal appendages observed in HYLS1-deficient cells likely arises because of the structural instability of the centriole and/or defective elongation of centriolar microtubules. This is consistent with the fact that CENTRIN2, POC5, C2CD3, TALPID3, and CEP164 were still observed at broken centrioles despite an overall impairment in their recruitment.
In this study, we report the interaction of HYLS1 with the centriole elongation factor CEP120 and show that CEP120 is required for HYLS1 recruitment to the centriole (Fig. 7 D). Our data suggest that the reduced stability of HYLS1 D211G, coupled with impaired binding of the mutant protein to CEP120, leads to defects in HYLS1 D211G recruitment to the centriole. We propose that reduced HYLS1 D211G recruitment impairs the stability of the centriole microtubule wall and decreases the abundance of proteins required for the cohesion of centriolar microtubules (POC5 and CENTRIN2) and centriole distal end assembly (TALPID3 and C2CD3). Disruption of centriole elongation and centriole integrity creates a fragile microtubule wall that breaks to produce centrioles that lack distal appendages and cannot ciliate (Fig. 7 D). We conclude that tissue-specific defects in centriole structural integrity are a key driver of HLS syndrome.
Materials and methods
Animals
Mice were housed and cared for in an AAALAC-accredited facility. All animal experiments were approved by the Johns Hopkins University Animal Care and Use Committee (MO21M300).
Mouse model
Hyls1+/DG mice were generated using CRISPR/Cas9 technology as previously described (Phan et al., 2022; Sladky et al., 2022). Briefly, one sgRNA (5′-CTACTCGGTCTATCTTGCCC-3′) was co-injected with an single-stranded DNA (ssDNA) repair template (5′-CAGTCTCTTTTGTACTCAAAATATCTGGCTACTCGACCCATTTTTCCCCGGTTTCGGCTTAACTGATCCAGTCTGGGGAG-3′) containing the GAC > GGT modification coding for the amino acid 211 as well as a restriction cut site for Taq1 at the site of the edit. Johns Hopkins University Transgenic Core performed pronuclear injection of one-cell B6SJL/F2 embryos (The Jackson Laboratory). Injected embryos were transferred into the oviducts of pseudopregnant females. Offspring resulting from embryo injections were genotyped and sequenced to check for the presence of the edit. The following primers were used for PCR amplification: forward (5′-GAACGAATGTTAGCTGCTGC-3′) and reverse (5′-GCGGGAAGGGAAGCTTCTTG-3′).
B6.Cg-Hyls1em1Ahol/J (Stock No. 039570; Jax) mice are available from The Jackson Laboratory.
Mouse tissue collection and sectioning
Kidneys, trachea, lungs, heart, and thymus of P0 mice were harvested and embedded fresh in O.C.T. compound (VWR Scientific). Brains of P0 mice were harvested, fixed in 4% paraformaldehyde (PFA) for 2 h, and embedded in 30% sucrose for 3 days at 4°C. This was followed by mounting the brains in OCT compound. Tissues in OCT were frozen for cryosectioning (CM1950 cryostat; Leica), and 20-μm sections were collected on Superfrost Plus microscope slides (Thermo Fisher Scientific).
Multiciliated cells
Mouse ependymal cell cultures were prepared as previously described (Delgehyr et al., 2015; LoMastro et al., 2022). Briefly, brains were dissected from P0 mice in Hank’s solution (10% HBSS, 5% HEPES, 5% sodium bicarbonate, and 1% penicillin/streptomycin [P/S] in pure water) and the telencephalon was cut into pieces. After enzymatic digestion (DMEM GlutaMAX, 1% P/S, 3% papain [3126; Worthington], 1.5% 10 mg/ml DNAse, and 2.4% 12 mg/ml cysteine) for 45 min at 37°C, the digestion was stopped by a solution of trypsin inhibitors (Leibovitz’s L15 medium, 10% 1 mg/ml trypsin inhibitor [Worthington], and 2% 10 mg/ml DNAse) and cells were washed with L15 medium. Cells were resuspended in DMEM GlutaMAX supplemented with 10% fetal bovine serum (FBS) and 1% P/S and grown for 4–5 days on poly-L-lysine-coated flasks. Once cells were confluent, they were incubated overnight with vigorous shaking (250 rpm) and then replated at a density of 2 × 105 cells/cm2 on poly-L-lysine–coated coverslips in DMEM GlutaMAX, 10% FBS, and 1% P/S. The following day, the medium was replaced with serum-free DMEM GlutaMAX and 1% P/S to trigger ependymal differentiation in vitro (differentiation day 0). For immunostaining, cells were fixed on differentiation day 7 with 4% PFA for 20 min at RT or with 100% ice-cold methanol for 10 min at −20°C.
Limb staining
Limbs were stained as previously described (Rigueur and Lyons, 2014) with the modification that only bone was stained. Briefly, limbs were fixed in 95% ethanol. The samples were transferred to acetone overnight at RT. Samples were then placed in 80% ethanol and 20% (glacial) acetic acid overnight. After washing two times with 70% ethanol, limbs were incubated in 95% ethanol overnight. To preclear the tissue, 95% ethanol was removed and 1% potassium hydroxide (KOH) solution was added for 1 h at RT. The KOH solution was replaced by Alizarin red solution (0.005% [wt/vol] in 1% [wt/vol] KOH) for 3–4 h at RT and then replaced with a 50/50 glycerol/KOH (1%) solution for 30 min. The glycerol/KOH solution was changed and samples were incubated at RT for 3–4 days. If excess dye persisted, the glycerol/KOH was changed again and maintained until imaging. After imaging, samples were transferred to 100% glycerol for long-term storage.
Cell culture
MEFs were cultured in Dulbecco’s modified Eagle’s medium (DMEM-1X; Corning) containing 4.5 g/liter glucose and L-glutamine without sodium pyruvate, supplemented with 10% FBS, 1% P/S, and 0.1 mM β-mercaptoethanol. MEFs were maintained at 37°C in a 5% CO2 and 3% O2 incubator. MEFs were frozen down in 60% complete DMEM, 30% FBS, and 10% DMSO. RPE1 cells were cultured in Dulbecco’s modified Eagle’s medium/Hams F-12 50/50 Mix (DMEM/F-12 50/50 1X; Corning) without L-glutamine, supplemented with 10% fetal bovine essence (FBE), 1% P/S, 1% L-glutamine, and 0.5% sodium bicarbonate. RPE1 cells were frozen down in 60% complete DMEM/F-12 media, 30% FBE, and 10% DMSO. DLD1 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM-1X; Corning) containing 4.5 g/liter glucose and L-glutamine without sodium pyruvate, supplemented with 10% FBE, 1% P/S, and 1% L-glutamine. DLD1 cells were frozen down in complete DMEM (95%) and DMSO (5%). Both RPE1 and DLD1 cells were maintained at 37°C in 5% CO2 incubators.
Cloning
All DNA constructs were cloned into a pcDNA5/FRT/TO vector backbone (Life Technologies) and expressed from a CMV promoter under the control of two tetracycline operator sites. All constructs were full-length proteins unless otherwise noted.
Cell line generation
Wildtype (Hyls1+/+), heterozygous (Hyls1+/DG), and homozygous (Hyls1DG/DG) embryos from a CRISPR-Cas9 mediated Hyls1 D226G knock-in mouse model were harvested at E14.5 and transferred to a dish containing sterile 1X PBS. After the heart, liver, and brain were removed, each embryo was transferred to a new dish containing 0.25% trypsin-EDTA (Thermo Fisher Scientific) and cut into fine pieces. The processed embryo was transferred to a 15-ml tube, the volume was filled to 3 ml with 0.25% trypsin-EDTA and incubated overnight at 4°C. The tubes containing embryos were then incubated for 30 min at 37°C. MEF culture media was added to a final volume of 8 ml and the solution was pipetted vigorously to break down the digested tissue into a cell suspension. The supernatant was transferred to a new tube and the previous step was repeated to collect more supernatant. The mixed solution was then cultured into a 10-cm tissue culture dish. CRISPR-Cas9–mediated technology was used to generate HYLS1−/− DLD1 and RPE1 cells as previously described (Gliech et al., 2024). Briefly, HYLS1 sgRNA (5′-GCTGCGCAGAAAGCCAGATG-3′) was cloned into the Lenti-CRISPR-V2 backbone (a gift from Feng Zhang; plasmid #52961; Addgene) and transfected with the pCMV-VSV-G (a gift from Bob Weinberg, plasmid #8454; Addgene) and the psPAX2 (a gift from Didier Trono, plasmid #12260; Addgene) in HEK 293T cells using calcium phosphate. 48 h later, viral supernatant was harvested, filtered, mixed with polybrene (10 μg/ml; Sigma-Aldrich), and administered to cells for 24 h. Transduced cells were then selected with puromycin (2.5 μg/ml) for 72 h. HYLS1 mutation and HYLS1 knockout cells were analyzed by sequencing for D226G/D211G knock-in or INDELs introduced by CRISPR, respectively. Unfortunately, we were unable to confirm HYLS1 knockout by western blot.
We made use of the Flp-In System to express HYLS1WT or HYLS1DG tagged with either 1x-mNeonGreen, 1x-mCherry, or 1x-HA (cloned into the pcDNA5 FRT/TO plasmid) in DLD1 HYLS1+/+ or HYLS1−/− cells. Cell lines were generated by transfecting the pcDNA5 FRT/TO plasmids with the POG44 Flp-recombinase plasmid into DLD1 Flp-In T-Rex cells. Cells were then selected with 400 µg/ml hygromycin B for at least 2 wk. Expression of reporters was induced with 1 µg/ml of doxycycline. Knock-in of an endogenous 1xHA tag on the C-terminus of the HYLS1 gene (HYLS1WT-HA) or an endogenous knock-in of the HYLS1 D211G mutation in HYLS1WT-HA RPE1 cells (HYLS1DG-HA) was generated as previously described (Ghetti et al., 2021). Briefly, an Alt-R™ crRNA targeting the HYLS1 translational stop codon (IDT - 5′-AltR1-AGAUUUAAGAAGGAGAAAGA-AltR2-3′) or HYLS1 D211 (IDT - 5′-AltR1-GAAACCGGGGCAAGACAGAC-AltR2-3′) was annealed with a tracrRNA (#1072532; IDT) and combined with recombinant S.p. Cas9 nuclease (#1081058; IDT) and the HDR donor DNA (HYLS1WT-HA: 5′-GTTGTGACCTTGCAAATGGTGTCATACCCAGGAAGCTTCCCTTTCCTCTTAGTCCTAGTGGCAGCTACCCATACGACGTACCAGATTACGCTTAATCTCCTTCTTAAATCTTTTTAAACTTCTTTCACAGGATTGTTTGAGATAA-3′; HYLS1DG-HA: 5′-TTCTCCCAAAGCTGGACCAGTTAAGCCGAAACCGGGGCAAGACAGGTCGAGTAGCCCGGTATTTTGAGTACAAACGGGACTGGGACTC-3′). The prepared RNP was nucleofected into cells using the Lonza 4D-Nucleofector system with the P3 Primary Cell 4D-NucleofectorTM X KitS (#V4XP-3032; Lonza) and by following the protocols. HYLS1WT-HA and HYLS1DG-HA were analyzed by sequencing for 1xHA tag or D211G knock-in, respectively.
Treatments in cultured cells
To rescue centriole integrity with Taxol treatment, RPE1 cells were arrested in late G1 with Palbociclib (PB – 200 nM; Sigma-Aldrich), a CDK4/6 inhibitor, for 24 h. Cells were then released into fresh media containing Dimethylenastron (2 μM; Sigma-Aldrich) to arrest the cells in mitosis. 12 h after the initial release from PB (cells in G2-phase), DMSO or Taxol (10 μM; Sigma-Aldrich) were added and the cells were kept overnight in mitosis. The following morning, RO3306 (10 μM; Sigma-Aldrich), a Cdk1 inhibitor, was added to the cells to force them to exit mitosis. Cells (in G1-phase) were fixed 5 h later followed by immunofluorescence or U-ExM. To induce ciliogenesis in MEFs and RPE1 cells, cells were seeded on coverslips and serum-starved by adding the corresponding culture media without FBS/FBE for 48 h. Cells were fixed in 4% PFA and treated accordingly for immunofluorescence or U-ExM. Hedgehog signaling response was analyzed by treating serum-starved MEFs with 500 nM of SAG (#566660; Sigma-Aldrich) 24 h before the cells were fixed.
CEP120 siRNA-mediated gene depletion
DLD1 cells were transfected with 100 nM CEP120 siRNA (#s45768; Thermo Fisher Scientific) using Lipofectamine RNAiMAX Transfection Reagent (Life Technologies) according to the manufacturer’s guidelines. Cells were fixed for immunofluorescence analysis 48 h after transfection.
Immunofluorescence staining in cultured cells
Cells were grown on glass coverslips and fixed for 10 min in ice-cold MeOH at −20°C or 20 min in 4% PFA at RT. MEFs, RPE1, and DLD1 cells were blocked in a blocking solution containing 2.5% FBS, 200 mM glycine, and 0.1% Triton X-100 in 1X PBS for 1 h, and MCCs were blocked for 1–2 h in the blocking buffer containing 1X PBS, 0.1% Triton X-100, and 10% normal donkey serum at RT. Cells were then incubated in primary antibody diluted in the blocking solution for 1 h (MEFs, RPE1, and DLD1 cells) or overnight (MCCs) at RT and subsequently washed with PBST (0.1% Triton X-100 in 1X PBS) three times for 10 min each. Secondary antibodies were added for 1–2 h at RT. DNA was stained with DAPI for 5 min and cells were washed with PBST three times for 10 min each. Coverslips were then mounted on microscope slides using ProLong Gold antifade mountant (Invitrogen).
Immunofluorescence staining in tissue sections
Slides containing tissue sections were washed in 1X PBS before antigen retrieval was performed using L.A.B. solution (#24310-500; Polysciences) for 10 min at RT. After washing with 1X PBS, the sections were blocked for 1 h at RT in a blocking solution (10% goat serum, 0.2% Triton-X, and 1X PBS). Primary antibodies were incubated for 3 h at RT or overnight at 4°C in the blocking buffer. After washing the sections three times with 1X PBS, secondary antibodies were incubated for 3 h at RT. After washing the slides three times with 1X PBS, the tissue sections were mounted using ProLong Gold Antifade Mountant (Invitrogen).
U-ExM
U-ExM was performed based on a previously published protocol (Gambarotto et al., 2019, 2021). Briefly, cells were seeded on a 25-mm (MEFs, RPE1, and DLD1) or a 12-mm (MCCs at differentiation day 6) glass coverslip in a 6-well plate. Cells were rinsed with 1X PBS, and 2 ml of the anchor solution (1X PBS, 1.4% formaldehyde, and 2% acrylamide mixture prepared fresh) was added. The plate was incubated at 37°C for 5 h (MEFs, RPE1, and DLD1) or overnight (mouse tissues and MCCs). A coverslip was then placed on top of an 850 µl (MEFS, RPE1, and DLD1) or 50 µl (MCCs) drop of precooled monomer solution (1X PBS, 23% sodium acrylate [wt/vol], 10% acrylamide, 0.1% N,N′-methylenbisacrylamide, 0.5% tetramethylenediamide (TEMED), and 0.5% ammonium persulfate) sitting on top of parafilm in a humid chamber. For tissue sections, the monomer solution was added on top of the slide containing the tissue section and a coverslip was placed on top. The humidified chamber was incubated on ice for 30 min (MEFs, RPE1, DLD1, and MCCs) or for 60 min (mouse tissues) and then moved to 37°C for 1 h (MEFs, RPE1, DLD1, and MCCs) or for 2 h (mouse tissues). Following gel polymerization, a 4-mm biopsy punch was used to create several punches from each coverslip. Punches were transferred to a 50 ml Falcon tube (MEFs, RPE1, DLD1, and tissue sections) or into a 1.5 ml Eppendorf tube containing denaturation buffer (200 mM SDS, 200 mM NaCl, and 50 mM Tris) and incubated for 15 min at RT with gentle agitation. The 50-ml Falcon tubes were transferred to a water bath at 95°C for 1.5 h with gentle agitation every 20 min. The Eppendorf tubes containing the MCC punches were incubated at 95°C for 1 h. Punches from mouse tissues were kept in a denaturation buffer with gentle agitation for an additional 1 h at RT. Punches were washed in water three times for 20 min each and kept overnight at RT with gentle agitation for the first round of gel expansion. Expanded punches were transferred to 1X PBS for 1 h at RT and blocked with 2% BSA in PBS at 37°C for 30 min–1 h. This step reduced the gel size to ∼2× the original 4 mm width. Primary antibodies were diluted in 2% BSA in PBS and stained for 3 h (MEFs, RPE1, DLD1, and tissue sections) or overnight (MCCs) at 37°C with gentle agitation. After washing the punches three times for 10 min at RT in PBST (1X PBS, 0.1% Tween-20), secondary antibodies and DAPI were added for 3 h at 37°C. This was followed by another round of washes with PBST (three times for 10 min at RT) and three washes with water for 20 min each. Gels were incubated in water overnight at room temperature with gentle agitation for the second round of gel expansion. Expansion factors were calculated by measuring the expanded punches with calipers and dividing the value measured by four (initial size of the gel after using the 4 mm-biopsy punch). The gel expansion was consistently 3.9–4.1×.
Cell cycle analysis
Cell cycle profile analysis was performed by staining fixed cells (70% ethanol at −20°C for at least 30 min) with a propidium iodide solution. After fixation, the cells were washed three times with 1X PBS and stained with propidium iodide (50 μg/ml) containing RNaseA (100 μg/ml) in the dark at 37°C for 30 min. Subsequently, the cells were analyzed by flow cytometry using the BD FACSCalibur Flow Cytometer (BD Biosciences). Quantification analysis of cell cycle profiles was generated using the FlowJo software package (FlowJo, LLC.).
Antibodies used for image analysis
The following primary antibodies were used for immunofluorescence or U-ExM in MEFs, RPE1, or DLD1 cells: anti-HA (Rat, #ROAHAHA, 1:500; Roche), anti-SAS-6 (Mouse, #sc-81431, 1:1,000; Santa Cruz Biotechnology), anti-CEP135 (Rabbit, a gift from A. Hyman, Max Planck Institute for Molecular Cell Biology and Genetics, Dresden, Germany, 1:1,000), anti-CPAP (Rabbit, #11517-1-AP, 1:1,000; Proteintech), anti- CEP120 (Rabbit, a gift from M. Mahjoub, Washington University in St. Louis, MO, USA, 1:1,000), anti-CENTRIN2 (Rabbit, Homemade [Moyer and Holland, 2019], 1:1,000), anti- POC5 (Rabbit, #a303-341, 1:1,000; Bethyl Laboratories), anti-CP110 (Rabbit, #12780, 1:1,000; Proteintech), anti-TALPID3 (Rabbit, #24432-1-AP, 1:1,000; Proteintech), anti-C2CD3 (Rabbit, #HPA038552, 1:1,000; Sigma-Aldrich), anti-CEP164 (Rabbit, #ABE2621, 1:1,000; EMD Millipore), anti-ANKRD26 (Rabbit, #GTX128255, 1:1,000; GeneTex), anti-Acetylated α-tubulin (Mouse, #sc-23950, 1:1,000; Santa Cruz Biotechnology), anti-Smoothened (Mouse, #sc-166685, 1:500; Santa Cruz Biotechnology), anti-α-Tubulin (#AA344, 1:1,000; Guinea Pig, Geneva Antibody Facility), anti-β-Tubulin (#AA345, 1:1,000; Guinea Pig, Geneva Antibody Facility), anti-Arl13b (Mouse, #75-287, 1:1,000; NeuroMab/Antibodies Incorporated), and γ-Tubulin (Goat, 1:1,000; Homemade). The following primary antibodies were used for MCC staining: anti-Acetylated-α-Tubulin (Lys40) (Mouse, #12152, 1:1,000; Cell Signaling), anti-SAS-6 (Mouse, #sc-81431, 1:250; Santa Cruz Biotechnology), anti-Deup1 (Rabbit, 1:1,000; homemade), anti-β-Tubulin (#AA345, 1:1,000; Guinea Pig, Geneva Antibody Facility), and anti-CENTRIN2 (Rabbit, 1:1,000; homemade). The following secondary antibodies were used for immunofluorescence or U-ExM: Alexa Fluor conjugated donkey anti-mouse A488 (#A21202; Thermo Fisher Scientific), donkey anti-rat A488 (#A21208; Thermo Fisher Scientific), goat anti-guinea pig A488 (#A11073; Thermo Fisher Scientific), donkey anti-mouse A555 (#A31570; Thermo Fisher Scientific), donkey anti-rabbit A555 (#A31572; Thermo Fisher Scientific), goat anti-guinea pig A555 (#A21435; Thermo Fisher Scientific), donkey anti-rabbit A647 (#A31573; Thermo Fisher Scientific), and donkey anti-rat A647 (#A48272; Thermo Fisher Scientific).
Protein co-immunoprecipitation
HYLS1−/− and HYLS1−/− with HYLS1-mCherry (WT or DG) add-back DLD1 cells were induced with doxycycline (1 μg/ml) and 48 h later transiently transfected with the construct of interest cloned into the pcDNA5 FRT/TO plasmid (Life Technologies). 48 h after the transient transfection, the cells were washed with 1X PBS and then 2 ml of PBS was added to the dish. Using a cell scraper, cells were scraped from the plate into the small amount of remaining PBS and transferred to a 15-ml conical tube on ice. After centrifugation for 5 min at 1,500 rpm at 4°C, the supernatant was removed and cells were snap-frozen. The pellet was resuspended in supplemented lysis buffer (10 mM Tris pH 7.5, 250 mM NaCl, 1 mM EDTA, 50 mM NaF, 50 mM β-glycerophosphate, 0.1%Triton X-100, supplemented with fresh 1% PMSF, 1 mM DTT, and cOmplete mini EDTA-free protease inhibitors [Sigma-Aldrich]) and transferred to a 2-ml Eppendorf tube. After vortexing briefly, cells were kept on ice for 30 min. Each tube was sonicated three times for 6 s at a 30% duty cycle and put on ice between the sonication cycles. Cells were centrifuged for 15 min at 15,000 rpm at 4°C. For each sample, 5% of the lysate was saved as “input,” and the remaining clarified lysate was added to the prewashed beads coupled to mCherry or GFP-binding protein (Rothbauer et al., 2008) and gently tumbled at 4°C for 3–4 h. Beads were then spun down at 4°C at 1,000 rpm for 1 min and the supernatant was discarded. Beads were washed three times with 1 ml of lysis buffer and, the immunopurified protein was analyzed by immunoblot.
Immunoblotting analysis
Cell lysates were loaded on denaturing SDS-polyacrylamide gels and separated by gel electrophoresis under constant volt (100 V) on a Mini Gel Tank (Thermo Fisher Scientific). The separated proteins were transferred to a nitrocellulose membrane using a Mini Trans-blot Module (Bio-Rad) at constant 100 V for 60–90 min, depending on the size of the protein of interest. Membranes were then incubated in blocking buffer (5% dried milk powder with 0.1% Tween-20 in 1X PBS) for 1 h at RT, and primary antibodies (diluted in 3% BSA in 1X PBS) were subsequently incubated overnight at 4°C. Membranes were then washed three times (10 min each) with washing buffer (0.1% Tween-20 in 1X PBS) and incubated with the secondary antibodies (diluted in 3% BSA in 1X PBS) for 1 h at RT. This step was followed by three, 10-min washing steps with a washing buffer before the membrane was incubated with WesternBright ECL (Advansta) for 3 min. The chemoluminescent signal was visualized using a Genesys G:Box Chemi-XX6 system (Syngene). The following primary antibodies were used: anti-HA (Rat, #ROAHAHA, 1:500; Roche), anti-RFP (Rabbit, #ab167453, 1:500; Abcam), anti-FLAG M2 (Mouse, #F3165, 1:500; Sigma-Aldrich), and anti-MYC 4A6 (Rabbit, 05-724, 1:500; EMD Millipore). The following secondary antibodies were used: HRP-conjugated anti-rat (Goat, #7077, 1:5,000; Cell Signaling) or HRP-conjugated anti-rabbit (Goat, #7074, 1:5,000; Cell Signaling).
Image and data analysis
Immunofluorescence images were acquired using a DeltaVision Elite system (GE Healthcare) controlling a scientific CMOS camera (pco.edge 5.5). Images were acquired using an Olympus 60×/1.42 NA or an Olympus 100×/1.4 NA oil objective with Applied Precision immersion oil (n = 1.516) with 0.2 µm z-sections. Acquisition parameters were controlled by SoftWoRx suite (GE Healthcare). Immunofluorescence images of tissue sections were acquired using a Leica SP8 confocal microscope controlling a Leica DFC9000 GTC camera with a 40×/1.30 NA oil immersion objective with 0.2 μm z-step size. U-ExM images of tissue sections, MEFs, RPE1, and DLD1 cells were acquired using a Leica SP8 confocal microscope with a 63×/1.4 NA oil immersion objective with 0.2 μm z-step size and a pixel size of 22.55 nm. Immunofluorescence and U-ExM MCC images were acquired using a Leica SP8 confocal microscope with a 40×/1.30 NA oil immersion objective, or a Zeiss Axio Observer 7 inverted microscope with Slidebook 2023 software (3i—Intelligent; Imaging Innovations, Inc.), CSU-W1 (Yokogawa) T1 Super-Resolution SoRa Spinning Disk, and Prime 95B CMOS camera (Teledyne Photometrics) with a 40×/1.30 NA. Images were deconvolved with Leica’s lightning process software or Microvolution software built into Slidebook. All images presented in the figures are Z-stack maximum intensity projections. Imaging analysis was performed blinded using ImageJ (v2.1.0/1.53c; National Institutes of Health, http://imagej.net).
Quantification of centriole length and relative CENTRIN2 coverage was analyzed using a custom FIJI (Schindelin et al., 2012) macro on lightning-deconvoluted confocal 3D image z-stacks. Centrioles parallel to the XY plane were analyzed and were maximum-intensity projected over the Z axis prior to measurement. Centriole length data were analyzed by acquiring a linear signal intensity profile along the axis of the centriole (getProfile tool). Distal and proximal signal peaks were identified from intensity profiles as the first and last peaks greater than a minimum signal threshold (>10% of max intensity profile signal), and length was measured as the distance between the 50% intensity location for the proximal and distal peaks. Overall centriole length was measured using an acetylated-TUBULIN profile encompassing the total centriole width. Long and short centriole wall lengths were measured as above using narrow intensity profiles encompassing only the width of each centriole wall. Intensity profiles of CENTRIN2 were collected in tandem and plotted relative to centriole length. Centriole defects were defined as described in Fig. S2, A and B. U-ExM analysis was performed using acetylated-TUBULIN and one or two additional centriole markers (CEP135, CENTRIN2, POC5, TALPID3, C2CD3, CP110, and/or CEP164). In duplicating centrioles, the proximal end was further identified by the presence of the procentriole. For the CENTRIN2 coverage analysis, quantifications were performed using CEP135 as a proximal marker and CENTRIN2 as a distal end marker. Centriole length analysis by U-ExM was normalized to the calculated expansion factor for each experiment. Intensity analysis of centriolar proteins was quantified as described in LoMastro et al. (2022). For the HYLS1 abundance analysis in different cell cycle phases, cells in interphase and mitosis were identified by their DAPI pattern. Cells were considered SAS-6 positive when their SAS-6 intensity at the centriole was >10% of the maximum SAS-6 peak of the respective control.
Statistical analysis
Statistical analysis was performed using GraphPad Prism software. For comparisons, we performed unpaired two-tailed Student’s t test with Welch’s correction, and one-way or two-way ANOVA with post-hoc analysis. Data distribution was assumed to be normal, but this was not formally tested. Only statistically significant and relevant comparisons are shown in the figures. More detailed statistical analyses are summarized in supplementary material. For all experiments, averages and standard error of the mean (SEM) are plotted. Information regarding statistics of an individual experiment is provided in the figure legends.
Online supplemental material
Fig. S1 contains the strategy used to generate the Hyls1 D226G mouse model, analysis of the size and weight of Hyls1 D226G embryos and P0 animals, as well as analysis of brain size and kidney histological analysis of Hyls1 D226G animals. Fig. S2 details the approach used to quantify centriole length and integrity defects, quantifications of centriole length in different tissues of the Hyls1 D226G mice, and investigation of Hyls1 D226G mutation in MCC differentiation. Fig. S3 extends the analysis of the effects of Hyls1 D226G in centrosome and distal appendages markers, cilia length, and ciliary recruitment of smoothened, as well as MCC differentiation in vitro. Fig. S4 details the strategy used to generate the HYLS1DG/DG and HYLS1−/− RPE1 cell lines and extends the analysis of the effects of HYLS1DG/DG and HYLS1−/− in centrosome and distal appendages markers, as well as the effect of HYLS1−/− in cilia assembly. Fig. S5 contains the analysis of centriole length in Hyls1DG/DG MEFs and HYLS1DG/DG RPE1 cells and extends the quantification of different centriole markers in HYLS1−/− RPE1 cells. Fig. S6 details the effects of HYLS1 D211G on CENTRIN2 coverage analysis along the centriole, as well as the recruitment of different centriole proteins and the duplicating capacity of centriole fragments in G2-phase arrested HYLS1DG/DG RPE1 cells. Fig. S7 details the strategy used to generate the HYLS1−/− DLD1 cell lines, extends the quantification of different centriole markers in HYLS1−/− DLD1 cells, and contains the analysis of centriole length in HYLS1−/− DLD1 cells with HYLS1-mNG (WT or D211G) add-back and the analysis of centriole defects and centriole length in HYLS1DG/DG RPE1 cells treated with Taxol. Fig. S8 expands the analysis of HYLS1 centriole localization throughout the cell cycle and in serum-starved cells, contains the quantification of centriole levels of HYLS1, CEP120, and CEP135 upon depletion of CEP120, co-localization analysis of HYLS1 and CEP120 in duplicating centrioles, and the effect of HYLS1 overexpression in the recruitment of CPAP to the centriole. Fig. S9 contains reciprocal immunoprecipitation of CPAP and CEP120 with HYLS1. Data S1 shows extended statistical analysis.
Data availability
Correspondence and requests for data and materials should be addressed to A.J. Holland ([email protected]).
Acknowledgments
We thank the Holland Lab for fruitful discussions.
A.J. Holland acknowledges funding by the National Institutes of Health (R01 GM133897, R01 GM114119, R01 CA266199). C.E. Jewett is a Merck Fellow of the Damon Runyon Cancer Research Foundation, DRG-2478-22.
Author contributions: A. Curinha: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing - original draft, Writing - review & editing, Z. Huang: Data curation, Formal analysis, Investigation, Validation, T. Anglen: Conceptualization, Investigation, Methodology, Writing - review & editing, M.A. Strong: Investigation, Resources, C.R. Gliech: Formal analysis, Software, Visualization, Writing - review & editing, C.E. Jewett: Formal analysis, Investigation, Visualization, A. Friskes: Investigation, T.P. Phan: Investigation, Methodology, Z. Nicholas: Conceptualization, Investigation, A.J. Holland: Conceptualization, Formal analysis, Project administration, Resources, Supervision, Writing - original draft, Writing - review & editing.
References
Author notes
Z. Huang and T. Anglen contributed equally to this paper.
Disclosures: The authors declare no competing interests exist.