


ATP-sensitive potassium (KATP) channels composed of Kir6.x and sulfonylurea receptor (SURs) subunits couple cellular metabolism to electrical activity. Cantú syndrome (CS) is a rare disease caused by mutations in the genes encoding Kir6.1 (KCNJ8) and SUR2A (ABCC9) that produce KATP channel hyperactivity due to a reduced channel block by physiological ATP concentrations. We functionally characterized the p.S1054Y SUR2A mutation identified in two CS carriers, who exhibited a mild phenotype although the mutation was predicted as highly pathogenic. We recorded macroscopic and single-channel currents in CHO and HEK-293 cells and measured the membrane expression of the channel subunits by biotinylation assays in HEK-293 cells. The mutation increased basal whole-cell current density and at the single-channel level, it augmented opening frequency, slope conductance, and open probability (Po), and promoted the appearance of multiple conductance levels. p.S1054Y also reduced Kir6.2 and SUR2A expression specifically at the membrane. Overexpression of ankyrin B (AnkB) prevented these gain- and loss-of-function effects, as well as the p.S1054Y-induced reduction of ATP inhibition of currents measured in inside-out macropatches. Yeast two-hybrid assays suggested that SUR2A WT and AnkB interact, while p.S1054Y interaction with AnkB is decreased. The p.E322K Kir6.2 mutation, which prevents AnkB binding to Kir6.2, produced similar biophysical alterations than p.S1054Y. Our results are the first demonstration of a CS mutation whose functional consequences involve the disruption of AnkB effects on KATP channels providing a novel mechanism by which CS mutations can reduce ATP block. Furthermore, they may help explain the mild phenotype associated with this mutation.
Introduction
ATP-sensitive potassium (KATP) channels are octamers consisting of four pore-forming Kir6.1 and/or Kir6.2 subunits encoded by KCNJ8 and KCNJ11 genes, respectively, and four ATP-binding cassette proteins SUR1 and/or SUR2 (with two main splice variants, SUR2A and SUR2B) subunits encoded by ABCC8 and ABCC9, respectively (Nichols et al., 2013; Shyng and Nichols, 1997; Hibino et al., 2010). KATP channels are widely distributed throughout the body and may exhibit different subunit compositions; e.g., pancreatic β-cell KATP channels are composed of Kir6.2/SUR1, whereas those of cardiac and vascular smooth muscle cells are predominantly formed by Kir6.2/SUR2A and Kir6.1/SUR2B, respectively (Nichols et al., 2013; Hibino et al., 2010). KATP channel activity is balanced by the blockade produced by intracellular ATP interacting with Kir6.x and the activating effects of Mg nucleotides that bind SURs, which allows them to couple electrical activity to cellular metabolism (Nichols et al., 2013; Hibino et al., 2010).
Cantú syndrome (CS; ORPHA: 1517) is a genetically determined rare disease characterized by hypertrichosis, craniofacial dysmorphology, and neurological, intellectual, and cardiovascular alterations including cardiomegaly, systemic hypotension, persistent ductus arteriosus, alterations in the electrocardiogram, and pulmonary hypertension (Nichols et al., 2013; Huang et al., 2018). CS is caused by mutations in ABCC9 and less frequently in KCNJ8, which lead to KATP channel hyperactivity due to a reduced block of the channel by physiological ATP concentrations (Nichols et al., 2013; Grange et al., 2019; Harakalova et al., 2012; Brownstein et al., 2013; McClenaghan and Nichols, 2022). The mutations can increase the activating effects of Mg nucleotides that would eventually override ATP block or modify the ATP-binding site within Kir6.x subunit, thus reducing channel sensitivity for ATP (Nichols et al., 2013; Grange et al., 2019; Harakalova et al., 2012; Cooper et al., 2015; McClenaghan et al., 2018). The severity of the phenotype varies among CS patients carrying different mutations, but it is accepted that the larger the reduction of ATP block the more severe the phenotype (Cooper et al., 2015). Recent evidence indicates that alterations produced by the CS mutations on KATP channels are more complex than initially suspected (Cooper et al., 2015; McClenaghan et al., 2018) and, thus, additional mechanisms can determine phenotypic variability. The p.S1054Y SUR2A variant was found in two CS patients (Scurr et al., 2011; Harakalova et al., 2012; Grange et al., 2019) and accounts for ∼2.8% of the ABCC9 variants identified in a CS systematic study developed in the context of the International Cantú Syndrome Registry (Grange et al., 2019). The p.S1054Y variant affects a conserved serine located in helix 13 of the second transmembrane domain (TMD2) of SUR2A (Rubaiy, 2016). This mutation, which in silico was predicted to be highly pathogenic (Harakalova et al., 2012), leads to one of the strongest reductions in the ATP block among those mutations analyzed thus far (Scurr et al., 2011; Harakalova et al., 2012; Houtman et al., 2019). Strikingly, its phenotypic consequences are milder than expected for a highly pathogenic mutation (Scurr et al., 2011; Harakalova et al., 2012; Grange et al., 2019). This behavior had been observed with some KCNJ11 (Kir6.2) and ABCC8 (SUR1) mutations associated with neonatal diabetes that greatly decrease ATP blockade but, simultaneously, diminished the expression of the KATP channel subunits at the plasma membrane (Zhou et al., 2010; Lin et al., 2013; Kline et al., 2009). More recently, Zhang et al. (2021) have described the first CS mutation, p.R1154Q SUR2A, which in addition to decreasing ATP blockade, reduces the surface expression of SUR2A subunits in mouse ventricular myocytes.
We thus aimed to determine the consequences of the p.S1054Y SUR2A mutation on channel function and on the membrane expression of channel subunits. Our results demonstrate that the p.S1054Y SUR2A CS mutation augmented macroscopic basal current density and single-channel conductance by promoting the appearance of multiple conductance levels, with the most frequently observed levels being those with the highest amplitudes and open probability (Po). Concomitantly, it reduced the membrane expression of Kir6.2 and SUR2A subunits. Importantly, both gain- and loss-of-function actions were corrected by the overexpression of the adapter protein ankyrin B (AnkB), suggesting that p.S1054Y SUR2A mutation impairs the physiological interaction between AnkB and KATP channels (Kline et al., 2009; Li et al., 2010). Therefore, here we describe the novel mechanism that accounts for the mild phenotype associated with p.S1054Y SUR2A CS mutation.
Materials and methods
cDNA constructs and cell culture and transfection
The cDNAs encoding rat wild-type (WT) and p.S1054Y SUR2A (GenBank accession no. NM_013040; 95.35% homologous to human SUR2A) subcloned in pCMV6 were kindly provided by Dr. Van der Heyden (Department of Medical Physiology, University Medical Center Utrecht, Utrecht, The Netherlands; Houtman et al., 2019) and mouse Kir6.2 (GenBank accession no. NM_010602; 96.41% homologous to human Kir6.2) subcloned in pECE was a generous gift from Dr. Colin Nichols (Department of Cell Biology and Physiology, Washington University School of Medicine, St. Louis, MO; Borschel et al., 2017). The p.E322K Kir6.2 mutation was introduced using the QuikChange Site-Directed Mutagenesis kit (Agilent) and confirmed by direct DNA sequencing as previously described (Tinaquero et al., 2020; Nieto-Marín et al., 2022; Rubio-Alarcón et al., 2021). The study has been conducted in Chinese hamster ovary (CHO) cells and human embryonic kidney 293 (HEK-293) cells purchased from American Type Culture Collection. They had been authenticated by the supplier as appropriate. Mycoplasma tests were conducted routinely for both cell lines and showed no mycoplasma contamination.
Macroscopic currents were recorded in CHO cells grown in Ham-F12 medium supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. The cultures were passaged every 4–5 d using a brief trypsin treatment. Subconfluent cultures were transiently transfected with the cDNA encoding Kir6.2 (3 µg) plus WT or mutant SUR2A (3 µg) together with the cDNA encoding the CD8 antigen (0.5 µg) using FUGENE XtremeGENE (Roche Diagnostics) following the manufacturer’s instructions (Caballero et al., 2017; Pérez-Hernández et al., 2018; Tinaquero et al., 2020).
Single-channel currents were recorded in HEK-293 cells that were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% BFS, and penicillin-streptomycin (5 ml, 1%) at 37°C as previously described (Caballero et al., 2010; Amorós et al., 2013; Gómez et al., 2014). The cultures were passaged every 4–5 d using a brief trypsin treatment. Subconfluent cultures were transiently transfected with the cDNA encoding mutated or WT Kir6.2 (0.5 µg) plus mutated or WT SUR2A (0.5 µg) together with the cDNA encoding the CD8 antigen (0.5 µg) using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s instructions. We coexpressed SUR2A with Kir6.2, instead of Kir6.1, to ensure a robust KATP current (IKATP) density, since the conductance of Kir6.2 channels is higher than that of Kir6.1 (Hibino et al., 2010). 48 h after transfection, cells were incubated with polystyrene microbeads precoated with anti-CD8 antibody (Dynabeads M450; Life Technologies). Most of the cells that were beaded also had channel expression (Gómez et al., 2014; Caballero et al., 2010; Amorós et al., 2013; Caballero et al., 2017; Pérez-Hernández et al., 2018). In some experiments, cells were co-transfected with 0.5 µg of the cDNA encoding GFP-tagged AnkB (GenBank accession no. NM_001148; Origene), whose expression was monitored by the green fluorescent signal. On the day of recordings, CHO or HEK-293 cells were removed from the dish using a cell scraper or by trypsinization, respectively, and the cell suspension was stored at room temperature and used within 12 h for electrophysiological experiments. To minimize the influence of the expression variability, each construct was tested in a large number of cells obtained from at least three different cell batches. Moreover, to avoid putative interferences of culture conditions (passage number, cell density, etc.), currents generated by cells expressing WT and p.S1054Y currents were always recorded in parallel.
Recording techniques
Macroscopic current recordings
Whole-cell recordings
A small aliquot of cell suspension was placed in a 0.5 ml chamber mounted on the stage of an inverted microscope (Nikon TMS; Nikon Co.). After settling to the bottom of the chamber, cells were perfused at ≈1 ml/min with external solution. Macroscopic currents were recorded at room temperature (21–23°C) using the whole cell patch-clamp technique using an Axopatch-200B patch clamp amplifier (Molecular Devices; Caballero et al., 2010; Amorós et al., 2013; Caballero et al., 2017; Pérez-Hernández et al., 2018; Tinaquero et al., 2020; Nieto-Marín et al., 2022). Micropipette resistance was kept below 3.5 MΩ when filled with the internal solution and immersed in the external solution. In all experiments, series resistance was compensated manually using the series resistance compensation unit of the Axopatch amplifier and ≥80% compensation was achieved. Thus, under our experimental conditions, no significant voltage errors (<5 mV) due to series resistance were expected with the micropipettes used. Moreover, the low capacitance (10.0 ± 0.6 pF; n = 27) enabled fast clamp control. Cells were perfused with an external solution containing (in mM) 136 NaCl, 4 KCl, 1.8 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose (pH 7.4 with NaOH), and the recording pipettes were filled with an internal solution containing (in mM) 80 K-aspartate, 42 KCl, 10 KH2PO4, 0.8 MgATP, 3 phosphocreatine, 5 HEPES, and 5 EGTA (pH 7.2 with KOH). The protocol to record macroscopic IKATP consisted of 500-ms depolarizing ramps from −100 to +50 mV (holding potential = 80 mV; Gómez et al., 2009; Gómez et al., 2014). In each experiment, current amplitude was normalized to cell capacitance to obtain the current density.
Inside-out recordings
In some experiments, currents were recorded at room temperature from excised inside-out macropatches from HEK-293 cells (Caballero et al., 2010; Amorós et al., 2013; Gómez et al., 2014). Recordings were made using the same solution on both sides of the patch containing (in mM) 140 KCl, 10 HEPES, 1 MgCl2, and 1 EGTA (pH 7.4 with KOH). Patch pipettes were pulled from 1.0 mm o.d. borosilicate capillary tubes (GD1, Narishige Co., Ltd.). When filled with the high-K+-containing solution, tip resistances were between 1.0 and 1.5 MΩ. Inside-out recordings were performed by applying 3-s ramps from a holding potential of −80 mV to potentials ranging −80 and +60 mV in control conditions and in the presence of increasing concentrations of MgATP (0.01–10 mM). MgATP-induced inhibition at −80 mV was used as an index of block and concentration-response curves were constructed and a Hill equation was fitted to the data to calculate the concentration that produces the 50% of the maximum block (IC50).
Single-channel recordings
Single-channel currents were recorded at room temperature (21–23°C) using the cell-attached patch-clamp configuration (Shindo et al., 1998; Gómez et al., 2009; Caballero et al., 2010; Amorós et al., 2013; Gómez et al., 2014). Using this configuration, the intracellular environment is completely preserved, and channel activity can be measured in an intact cell. Cells were suspended in bath solution containing (in mM) 140 KCl, 1.8 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose; pH 7.4 with KOH. This high-K+ solution was used to achieve a resting membrane potential of zero. Since Po of KATP channels in the cell-attached configuration is close to 0 due to the channel block by endogenous ATP (Babenko et al., 1998), channel activity was almost undetectable under basal conditions. For this reason, all recordings were done in the continuous presence of the K channel opener pinacidil (100 µM) in the bath solution (Shindo et al., 1998). Patch pipettes were pulled from 1.5 mm o.d. borosilicate capillary tubes (Harvard Apparatus Ltd.), coated at the tip with Sylgard (Dow Corning), and fire-polished with a microforge (Mod. MF-830; Narishige). When filled with pipette solution containing (in mM) 140 KCl and 10 HEPES; pH 7.4 with KOH, tip resistances were between 5 and 10 MΩ. The micropipettes were gently lowered onto the cells to get a gigaohm seal after applying suction. After seal formation, the cells were lifted from the bottom of the perfusion bath and current recordings were started.
Single-channel protocols and analysis
Single-channel currents were recorded by applying repetitive 10-s pulses from a holding potential of 0 to −120 mV or to potentials between −120 and +40 mV in 20 mV steps. Current data were sampled at 10 kHz and filtered at 1 kHz. The apparent number of active channels in a patch was determined by visual inspection of the current traces and patches with more than one channel were discarded. The experimental conditions were optimized to reduce the number of active channels on each recording: e.g., the amount of cDNA used for cell transfection was reduced by 85% and the tip resistance of the pipettes was increased approximately threefold compared to whole-cell recordings. Under these conditions, around 10% of cells with active channels had more than one channel. All data analysis was performed using pCLAMP software and opening events were captured using the event detection tool of Clampfit 10 (Gómez et al., 2009; Caballero et al., 2010; Amorós et al., 2013; Gómez et al., 2014). Linear leak and capacity currents were digitally subtracted with the average currents of non-active sweeps. The Po was obtained for each experiment by dividing the time that the channel remains in the open state by the total recording time.
The opening frequency (fo) was calculated for each experiment as the inverse of the closed time between events.
To measure opening and closing kinetics, durations of channel open and closed times from all the experiments in each condition were pooled in a joint events table. The latter was used to generate dwell-time histograms constructed by plotting pooled dwell-time data as a function of the number of events per bin as described by Sigworth and Sine (1987) for the analysis of open-time and closed-time constants.
Open dwell-time histograms were fitted by a monoexponential equation to obtain the τOPEN, while log-binned closed dwell-time histograms were fitted by a double Gaussian that yielded τfCLOSED and τsCLOSED. This biphasic time course has been attributed to intraburst and interbust closures (Trube and Hescheler, 1984). Comparison of gating kinetics was performed with the use of an F statistical test on the sum of squared errors of the functions (see below).
Amplitude of unitary currents was determined for each experiment by direct measurements of fully resolved openings, which allowed the calculation of average values. Unitary amplitude was also calculated from pooled data from all the experiments in each group using a Gaussian distribution fit to amplitude histograms that were constructed by plotting amplitude data as a function of the number of events per bin (bin width = 0.05 pA). The amplitude histogram for currents recorded in cells expressing Kir6.2+SUR2A p.S1054Y was fitted by a triple Gaussian distribution. In this case, pooled opening events were distributed among three different conductance levels according to their amplitude: O1 (smaller than −5 pA); O2 (between −10 and −5 pA); and O3 (greater than −10 pA). Pooled open dwell-time values of each event were distributed among the three levels and Po was calculated for each level as the ratio between the sum of open dwell-times and the total recording time.
Current–voltage relationships were constructed by plotting the single-channel current amplitude as a function of the membrane potential and conductance was calculated from the slope of the fit of a linear function to the data recorded at potentials between −120 and 0 mV. At more positive potentials, a mild inward rectification was produced, in accordance with previous results (Shindo et al., 1998). To quantify putative changes in this process, the rectification index was calculated by dividing the single current amplitude generated by the most depolarized pulse (+40 mV) over the current amplitude generated by the most hyperpolarized pulse (−120 mV), as previously described (Amorós et al., 2013).
Biotinylation assay
A biotinylation assay was conducted using previously described procedures (Ponce-Balbuena et al., 2018) to determine putative changes in the membrane expression of KATP channel subunits. 48 h after co-transfection of Kir6.2 with WT or p.S1054Y SUR2A constructs in the absence or presence of AnkB, HEK-293 cells were washed twice with ice-cold PBS and biotinylated for 25 min at 4°C using PBS containing 0.5 mg/ml of EZ Link Sulfo-NHS-SS-Biotin (Thermo Fisher Scientific). Plates were washed twice with PBS-200 mM glycine (to quench unlinked biotin) and again with PBS only. Cells were collected in RIPA buffer containing 50 mM Tris·HCl (pH = 7.5), 150 mM NaCl, 1% Nonidet P-40, 0.1% SDS, 0.5% sodium deoxycholate, and 1 mM PMSF and protease inhibitor cocktail (Sigma-Aldrich Inc.) for protein extraction. Subsequently, the extract (2 mg) was incubated with Streptavidin Sepharose (50 µl; GE Healthcare) overnight at 4°C. To separate biotinylated fraction, samples were centrifuged at 3,000 rpm for 2 min at 4°C. The fraction of biotinylated proteins was washed by several centrifugations prior to Western blot analysis. Detection of Kir6.2, SUR2A, AnkB, and ezrin proteins was carried out in HEK-293 cells transfected with Kir6.2+SUR2A and co-transfected or not with AnkB by Western blot following previously described procedures (Caballero et al., 2017; Ponce-Balbuena et al., 2018; Pérez-Hernández et al., 2018; Tinaquero et al., 2020; Nieto-Marín et al., 2022). Nuclei and cell debris were removed by centrifugation at 14,000 rpm for 20 min at 4°C. The total protein amount of the extracts was calculated with the bicinchoninic acid method (Pierce). Samples (200 µg) were run on 4–15% Mini-PROTEAN TGX stain-free gels (Bio-Rad) and, afterwards, protein was transferred to nitrocellulose membranes. Nonspecific binding sites were blocked with 5% non-fat dried milk in PBS with Tween-20 (0.05%) for 1 h at room temperature. Membranes were then incubated with rabbit polyclonal anti-Kir6.2 (1:200; APC-020 Alomone), mouse monoclonal anti-SUR2A (1:500; MA5-27637; Thermo Fisher Scientific), mouse monoclonal anti-AnkB (1:400; 33-3700; Invitrogen) and mouse monoclonal anti-ezrin (1:400; ab4069; Abcam) primary antibodies overnight at 4°C. All the antibodies had been validated by the manufacturers. The anti-Kir6.2 antibody is directed against the peptide (C)SVAVAKAKPKFSIS corresponding to amino acid residues 372–385 of rat Kir6.2 (the peptide confirmation was done by amino acid analysis and mass spectrometry). The immunogen of the anti-SUR2A is the peptide SSIVDAGLVLVFSEGILVECDTGPNLLQHKNGLFSTLVMTNK corresponding to amino acids 1505–1546 within the cytoplasmic C-terminus of mouse SUR2A. After the incubation with the primary antibodies, samples were incubated for 1 h with fluorescent-dye conjugated secondary antibodies (anti-mouse HRP 1:10,000, 115-035-003; Jackson). Membranes were washed three times with PBS-Tween after adding primary and secondary antibodies. Protein expression was visualized using the LED imaging modules of the Chemidoc MP System. Expression of the proteins in the biotinylated (membrane) fraction was normalized to the total protein expression (input).
Yeast two-hybrid assay
To analyze the protein–protein interaction between SUR2A and AnkB, we conducted a GAL4-based yeast two hybrid assay using the ProQuest Two-Hybrid System with Gateway Technology (PQ1000101; Invitrogen; Ganguly et al., 2018). WT or p.S1054Y SUR2A were first cloned into bait pDEST32 vector encoding for the GAL4 DNA binding domain (DBD) and AnkB was cloned into prey pDEST22 encoding for the activation domain (AD). Cloning was performed using Gateway recombination cloning technology procedures following manufacturer’s instructions. To assess the interaction between SUR2A and AnkB, both DBD/SUR2A and AD/AnkB were co-transformed into MaV203 competent cells, a modified strain of Saccharomyces cerevisiae in which endogenous GAL4 is deleted. Moreover, this yeast strain was also modified to integrate in its genome reporter genes, such as the auxotrophic markers HIS3 or URA3, whose regulatory regions were engineered to contain the DNA binding sites for the DBD/protein fusion (bait). Therefore, if the two tested proteins interact in the nucleus, DBD and AD of GAL4 will bring together to reconstitute transcriptional activation, leading to the expression of the reporter genes. The protranscriptional effect over the HIS3 reporter allows the cells to synthesize histidine and growth in a selective medium containing 3 amino-1,2,4-triazole (3AT, 50 mM) and lacking Leu-Trp-His (SC-Leu-Trp-His-3AT). The interaction was also tested by cloning SUR2A (WT or p.S1054Y) into prey pDEST22 and AnkB into bait pDEST32 vectors. As a negative control, yeast cells were transformed with empty vectors, whereas positive controls yeast cells were transformed with vectors encoding Krev1 and RalGDS interacting proteins. As an additional positive control, we used Kir6.2 and AnkB whose interaction is known (Li et al., 2010). Protein–protein interaction between SUR2A and AnkB was further determined based on the expression of the URA3 reporter, which encodes the Ura3 enzyme that converts 5-fluoroorotic acid (5FOA) to 5-fluorouracil, which is toxic and inhibits cell growth. Therefore, an interaction is confirmed if growth is inhibited when cells are plated on selective medium lacking Leu-Trp but containing 5FOA (0.2%; SC-Leu-Trp-5FOA).
To characterize the colonies that contain bait and prey proteins, a series of patching and replica plating steps onto selection/screen plates was performed following manufacturer’s instructions. To this end, the yeasts transformed with bait and prey vectors were plated onto a single selective medium lacking Leu-Trp (SC-Leu-Trp) plate at 30°C for 18 h. Afterwards, replicas were sequentially plated (a first incubation at 30°C for 24 h followed by a second incubation at 30°C for 1 d) onto selection plates in the presence of SC-Leu-Trp-His-3AT or SC-Leu-Trp-5FOA. Densitometric analysis of the images was performed using ImageJ to analyze putative changes in the SUR2A–AnkB interaction induced by the p.S1054Y mutation (Chatin et al., 2014).
Statistical analyses
Results are expressed as mean ± SEM. Paired or unpaired t test or one-way ANOVA followed by Tukey’s test were used to assess statistical significance where appropriate. To make comparisons between fits of pooled data or concentration-dependent effects, an F test was used. To take into account repeated sample assessments, data were analyzed with multilevel mixed-effects models. Normality assumption was verified using the Shapiro–Wilk test. Variance was comparable between groups throughout the manuscript. A value of P < 0.05 was considered significant. For the different groups of experiments sample size was chosen empirically according to previous experience in the calculation of experimental variability. No statistical method was used to predetermine sample size and no particular procedure was followed for randomization/allocation of the respective experimental groups. The experiments were not blinded due to the nature of the experimental design and platforms, but all the data were analyzed in an identical manner for all conditions to eliminate possible operator bias.
Results
Effects of p.S1054Y SUR2A mutation on macroscopic IKATP
Fig. 1 A shows representative macroscopic IKATP traces recorded with the whole-cell patch-clamp configuration by applying a voltage-ramp from −100 to +50 mV in two different CHO cells expressing Kir6.2+SUR2A WT and Kir6.2+SUR2A p.S1054Y channels. As can be observed, the amplitude of the IKATP generated by WT channels in basal conditions was very small due to the block of the channels by cytosolic ATP. Indeed, under these conditions the current was almost linear to the voltage at this range of membrane potentials (Fig. 1 B). The presence of p.S1054Y SUR2A significantly increased the density of the basal current at all the potentials tested (Fig. 1, A and B), and slightly changed the morphology of the current traces elicited by the voltage-ramps (Fig. 1 A), exhibiting the weak inward rectification at positive potentials characteristic of this current. These results could be explained by the marked reduction in ATP block induced by the mutation (Scurr et al., 2011; Harakalova et al., 2012; Houtman et al., 2019).

The p.S1054Y SUR2A mutation increases macroscopic basal IKATP. (A) Superimposed representative current traces recorded by applying a 500-ms voltage-ramp from −100 to +50 mV (inset) in two different CHO cells expressing Kir6.2+SUR2A WT (black line) or Kir6.2+SUR2A p.S1054Y (blue line) channels. The dotted line represents the zero current level. (B) Current density–voltage relationships for currents recorded in cells expressing Kir6.2+SUR2A WT or Kir6.2+SUR2A p.S1054Y channels. Data points represent the mean ± SEM of the number of experiments indicated in the figure. *P < 0.05 vs. Kir6.2+SUR2A WT (unpaired t test; from left to right: P value = 0.000026 at −100 mV, 0.0000001 at −90 mV, 0.0002 at −80 mV, 0.04 at −40 mV, 0.01 at −30 mV, 0.009 at −20 mV, 0.006 at −10 mV, 0.004 at 0 mV, 0.004 at +10 mV, 0.002 at +20 mV, 0.001 at +30 mV, 0.0012 at +40 mV, and 0.0006 at +50 mV). (C and D) Single-channel recordings obtained by applying 10-s pulses from 0 to −120 mV in untransfected HEK-293 cells in the absence (C) or presence (D) of 100 µM pinacidil. (E and F) Pinacidil-induced single-channel currents recorded by applying 10-s pulses from 0 to −120 mV in HEK-293 cells expressing Kir6.2+SUR2A WT (E) or Kir6.2+SUR2A p.S1054Y (F) in the absence (control) or presence of 10 µM glibenclamide. (G) Single-channel currents recorded by applying 10-s pulses from 0 to −120 mV in HEK-293 cells expressing Kir6.2+SUR2A p.S1054Y in the absence of pinacidil.
The p.S1054Y SUR2A mutation increases macroscopic basal IKATP. (A) Superimposed representative current traces recorded by applying a 500-ms voltage-ramp from −100 to +50 mV (inset) in two different CHO cells expressing Kir6.2+SUR2A WT (black line) or Kir6.2+SUR2A p.S1054Y (blue line) channels. The dotted line represents the zero current level. (B) Current density–voltage relationships for currents recorded in cells expressing Kir6.2+SUR2A WT or Kir6.2+SUR2A p.S1054Y channels. Data points represent the mean ± SEM of the number of experiments indicated in the figure. *P < 0.05 vs. Kir6.2+SUR2A WT (unpaired t test; from left to right: P value = 0.000026 at −100 mV, 0.0000001 at −90 mV, 0.0002 at −80 mV, 0.04 at −40 mV, 0.01 at −30 mV, 0.009 at −20 mV, 0.006 at −10 mV, 0.004 at 0 mV, 0.004 at +10 mV, 0.002 at +20 mV, 0.001 at +30 mV, 0.0012 at +40 mV, and 0.0006 at +50 mV). (C and D) Single-channel recordings obtained by applying 10-s pulses from 0 to −120 mV in untransfected HEK-293 cells in the absence (C) or presence (D) of 100 µM pinacidil. (E and F) Pinacidil-induced single-channel currents recorded by applying 10-s pulses from 0 to −120 mV in HEK-293 cells expressing Kir6.2+SUR2A WT (E) or Kir6.2+SUR2A p.S1054Y (F) in the absence (control) or presence of 10 µM glibenclamide. (G) Single-channel currents recorded by applying 10-s pulses from 0 to −120 mV in HEK-293 cells expressing Kir6.2+SUR2A p.S1054Y in the absence of pinacidil.
Effects of p.S1054Y SUR2A mutation on single Kir6.2+SUR2A channel currents
To characterize the biophysical mechanisms underlying the effects of the p.S1054Y mutation, single-channel currents were recorded in HEK-293 cells using the cell-attached configuration of the patch-clamp technique. Fig. 1 E shows representative single-channel traces recorded at −120 mV in a HEK-293 cell expressing Kir6.2+SUR2A WT channels in the continuous presence of the KATP channel opener pinacidil (100 µM) and in the absence (top) or the presence (bottom) of the KATP channel blocker glibenclamide (10 µM). According to previous reports (Shindo et al., 1998), single-channel activity recorded in cells expressing Kir6.2+SUR2A WT was characterized by spontaneous openings in bursts followed by periods of channel closure. This current was abolished by glibenclamide confirming that it was generated by KATP channels. Conversely, non-transfected HEK-293 cells did not generate any current both in the absence (Fig. 1 C) or presence of pinacidil (Fig. 1 D), since these cells do not express endogenous KATP channels (Zhong et al., 2014; Shindo et al., 1998). Fig. 2 A shows single-channel traces recorded in two different HEK-293 cells expressing Kir6.2+SUR2A WT (left) or Kir6.2+SUR2A p.S1054Y (right) channels by applying 10-s pulses to −120 mV from a holding potential of 0 mV in the continuous presence of 100 μM pinacidil. Single-channel activity generated by Kir6.2+SUR2A WT channels yielded mean fo (Fig. 2 B) and Po (Fig. 2 C) values of 13.4 ± 2.3 Hz and 0.027 ± 0.004 (n = 26), respectively. In cells expressing Kir6.2+SUR2A p.S1054Y, opening burst periods were longer (Fig. 2 A, right) and thus, fo (Fig. 2 B) and Po (Fig. 2 C) were significantly higher (n = 15) than in cells expressing SUR2A WT. The current recorded in cells expressing Kir6.2+SUR2A p.S1054Y was also abolished by 10 µM glibenclamide, demonstrating that it was generated by KATP channels (Fig. 1 F). In the absence of pinacidil, some cells expressing Kir6.2+SUR2A p.S1054Y channels generated single-channel currents with very low fo and Po (Fig. 1 G). The presence of this current could be the consequence of the reduced block by the endogenous intracellular ATP induced by the mutation and may underlie the p.S1054Y-induced increase in the macroscopic IKATP recorded using the whole-cell configuration (Fig. 1, A and B). Gating kinetics was characterized by means of dwell-time histograms constructed by plotting pooled dwell-time data as a function of the number of events per bin. The fit of a monoexponential function to the open dwell-time histograms yielded a τOPEN of 0.33 ms (Fig. 2 D). The mutation also modified gating kinetics by reducing the τOPEN (P < 0.05; Fig. 2 E). To accurately define the kinetics of the closed states, closed dwell-time histograms were constructed as log-binned histograms and fitted by a double Gaussian function (Sigworth and Sine, 1987), where each peak corresponds to the time constants (τfCLOSED and τsCLOSED; Fig. 3, A and B), which describe intraburst and interbust closures, respectively (Fig. 3 C), as previously described (Trube and Hescheler, 1984). As can be observed, the mutation also reduced the τsCLOSED (Fig. 3 B), without modifying the τfCLOSED (Fig. 3, A and B).

The p.S1054Y SUR2A mutation increases fo and Po. (A) Single-channel recordings obtained by applying 10-s pulses from 0 to −120 mV in HEK-293 cells expressing Kir6.2+SUR2A WT (left) or Kir6.2+SUR2A p.S1054Y (right). (B and C) Mean fo (B) and Po (C) for single-channel currents recorded in cells expressing Kir6.2+SUR2A WT or Kir6.2+SUR2A p.S1054Y. Each bar represents the mean ± SEM of the number of experiments/cells indicated in the figure and each dot represents one experiment/cell. *P = 0.0043 and P = 0.0044 for the comparisons of fo (B) and Po (C), respectively, vs. Kir6.2+SUR2A WT (unpaired t test). (D and E) Open dwell-time histograms for currents recorded in cells expressing Kir6.2+SUR2A WT (D) or Kir6.2+SUR2A p.S1054Y (E; bin width = 0.1 ms). Continuous lines represent the fit of a monoexponential function to the data, which yielded the indicated τOPEN. Histograms were obtained by pooling data from 26 (Kir6.2+SUR2A WT) and 15 (Kir6.2+SUR2A p.S1054Y) experiments. *P = 0.016 vs. Kir6.2+SUR2A WT (F test).
The p.S1054Y SUR2A mutation increases fo and Po. (A) Single-channel recordings obtained by applying 10-s pulses from 0 to −120 mV in HEK-293 cells expressing Kir6.2+SUR2A WT (left) or Kir6.2+SUR2A p.S1054Y (right). (B and C) Mean fo (B) and Po (C) for single-channel currents recorded in cells expressing Kir6.2+SUR2A WT or Kir6.2+SUR2A p.S1054Y. Each bar represents the mean ± SEM of the number of experiments/cells indicated in the figure and each dot represents one experiment/cell. *P = 0.0043 and P = 0.0044 for the comparisons of fo (B) and Po (C), respectively, vs. Kir6.2+SUR2A WT (unpaired t test). (D and E) Open dwell-time histograms for currents recorded in cells expressing Kir6.2+SUR2A WT (D) or Kir6.2+SUR2A p.S1054Y (E; bin width = 0.1 ms). Continuous lines represent the fit of a monoexponential function to the data, which yielded the indicated τOPEN. Histograms were obtained by pooling data from 26 (Kir6.2+SUR2A WT) and 15 (Kir6.2+SUR2A p.S1054Y) experiments. *P = 0.016 vs. Kir6.2+SUR2A WT (F test).

The p.S1054Y SUR2A mutation reduces τsCLOSED. (A and B) Log-binned closed dwell-time histograms for currents recorded in cells expressing Kir6.2+SUR2A WT (A) or Kir6.2+SUR2A p.S1054Y (B). Continuous lines represent the fit of a double Gaussian function to the data, which yielded the indicated τfCLOSED and τsCLOSED. Histograms were obtained by pooling data from 26 (Kir6.2+SUR2A WT) and 15 (Kir6.2+SUR2A p.S1054Y) experiments. *P = 0.0023 vs. Kir6.2+SUR2A WT (F test). (C) Expanded view of a single-channel current trace recorded at −120 mV in a cell expressing Kir6.2+SUR2A WT highlighting the intraburst and interburst closures.
The p.S1054Y SUR2A mutation reduces τsCLOSED. (A and B) Log-binned closed dwell-time histograms for currents recorded in cells expressing Kir6.2+SUR2A WT (A) or Kir6.2+SUR2A p.S1054Y (B). Continuous lines represent the fit of a double Gaussian function to the data, which yielded the indicated τfCLOSED and τsCLOSED. Histograms were obtained by pooling data from 26 (Kir6.2+SUR2A WT) and 15 (Kir6.2+SUR2A p.S1054Y) experiments. *P = 0.0023 vs. Kir6.2+SUR2A WT (F test). (C) Expanded view of a single-channel current trace recorded at −120 mV in a cell expressing Kir6.2+SUR2A WT highlighting the intraburst and interburst closures.
Amplitude of unitary currents was calculated from a Gaussian distribution fit to amplitude histograms that were constructed by plotting pooled amplitude data as a function of the number of events per bin. In cells expressing Kir6.2+SUR2A WT, the Gaussian distribution fit yielded discrete peaks at −0.06 pA (closed channels) and −7.68 pA (open channels; Fig. 4 A). Mean open channel amplitude value obtained by averaging the amplitude of the opening events measured for each experiment (n = 26) was −6.7 ± 0.5 pA in cells expressing Kir6.2+SUR2A WT, while it was significantly higher (−10.2 ± 0.6 pA, P < 0.05) in the presence of the p.S1054Y mutation (Fig. 4 C). The fit of the amplitude histograms of closed events recorded in cells expressing the mutation by a single Gaussian distribution yielded a peak at 0.01 pA (Fig. 4 B). Strikingly, close observation of the current traces revealed that at least three levels of conductance (designated as O1, O2, and O3) could be identified. Examples of these conductance levels are shown in an expanded view of a representative trace (inset of Fig. 4 B). Consequently, a triple Gaussian distribution was needed to fit the amplitude histograms of open channels (Fig. 4 B). The most frequently observed level (O2) displayed a similar amplitude (−7.05 pA) to that of Kir6.2+SUR2A WT (P > 0.05), while O1 and O3 peaked at −3.65 and −14.0 pA, respectively (Fig. 4 B). To further characterize each level, we pooled all experiments (n = 15) and opening events were incorporated into one of the three levels according to their amplitude (Fig. 4 D; see Materials and methods). The dwell-times of the opening events were distributed among their corresponding levels and, as can be observed in Fig. 4 E, they were significantly smaller (O1) or higher (O2 and O3) than those of Kir6.2+SUR2A WT (P < 0.05). Po was calculated as the ratio between the sum of open dwell-times of each level and the total recording time (Fig. 4 F) and the results showed that Po of O2 and O3 were much higher than those of O1 and also higher than those of Kir6.2+SUR2A WT, indicating that the most frequent levels were those with the largest amplitudes, open dwell-times, and Po.

The p.S1054Y SUR2A mutation leads to the appearance of multiple conductance levels. (A and B) Amplitude histograms of currents recorded at −120 mV constructed by plotting amplitude data as a function of the number of events per bin (bin width = 0.05 pA) together with the fit of a Gaussian function (solid lines) for single-channel currents recorded in cells expressing Kir6.2+SUR2A WT (A) or Kir6.2+SUR2A p.S1054Y (B). Histograms were obtained by pooling data from 26 to 15 experiments, respectively. The fit to the open channel data recorded in cells expressing the p.S1054Y mutation required a triple Gaussian function. In B, an expanded view of single-channel traces is shown to better appreciate the appearance of multiple conductance levels. (C) Mean single-channel amplitude values averaged from n experiments/cells in each group, and each dot represents one experiment/cell. **P = 0.0002 vs. Kir6.2+SUR2A WT. (D–F) Unitary current amplitudes (D), open-dwell times (E), and Po (F) calculated by pooling all the opening events. In cells expressing SUR2A p.S1054Y data were distributed among the three groups (O1–O3) according to the amplitude of the opening events. In D and E, each bar shows the mean ± SEM of the indicated number of events and in D each dot represents one opening event. *P = 0.0001 for all comparisons vs. Kir6.2+SUR2A WT (one-way ANOVA).
The p.S1054Y SUR2A mutation leads to the appearance of multiple conductance levels. (A and B) Amplitude histograms of currents recorded at −120 mV constructed by plotting amplitude data as a function of the number of events per bin (bin width = 0.05 pA) together with the fit of a Gaussian function (solid lines) for single-channel currents recorded in cells expressing Kir6.2+SUR2A WT (A) or Kir6.2+SUR2A p.S1054Y (B). Histograms were obtained by pooling data from 26 to 15 experiments, respectively. The fit to the open channel data recorded in cells expressing the p.S1054Y mutation required a triple Gaussian function. In B, an expanded view of single-channel traces is shown to better appreciate the appearance of multiple conductance levels. (C) Mean single-channel amplitude values averaged from n experiments/cells in each group, and each dot represents one experiment/cell. **P = 0.0002 vs. Kir6.2+SUR2A WT. (D–F) Unitary current amplitudes (D), open-dwell times (E), and Po (F) calculated by pooling all the opening events. In cells expressing SUR2A p.S1054Y data were distributed among the three groups (O1–O3) according to the amplitude of the opening events. In D and E, each bar shows the mean ± SEM of the indicated number of events and in D each dot represents one opening event. *P = 0.0001 for all comparisons vs. Kir6.2+SUR2A WT (one-way ANOVA).
To determine the effects of the p.S1054Y mutation on the slope conductance, single-channel current–voltage relationships were constructed for currents recorded at potentials between −120 and +40 mV (for simplicity, in Kir6.2+SUR2A p.S1054Y the mean amplitude value was incorporated for each potential; Fig. 5 A). Plotting the mean amplitude as a function of the membrane potential yielded an almost linear relationship at negative potentials, with a reversal potential ∼0 mV (due to symmetrical 150 mM extracellular and intracellular [K+]) and mild inward rectification at more positive potentials, according to previous results (Shindo et al., 1998). In the presence of the p.S1054Y mutation, single-channel amplitudes at potentials between −60 and −120 mV were significantly higher (P < 0.05), while at potentials positive to 0 mV current amplitudes were not significantly different (Fig. 5 A). As a consequence of these effects, the slope conductance (γ) calculated by the linear regression to the current–voltage relationships was increased from 62.4 ± 3.9 to 98.0 ± 4.0 pS (P < 0.05; Fig. 5 A). A close inspection of the current–voltage relationships at potentials positive to 0 mV showed that the linear regression fit remarkably deviates from the experimental data points measured for Kir6.2+SUR2A p.S1054Y channels suggesting a greater inward rectification in the presence of the mutation. To quantify this phenomenon, a rectification index (Amorós et al., 2013) was calculated by dividing the current amplitude generated by the most depolarized pulse by the current amplitude generated by the most hyperpolarized pulse. Fig. 5 B demonstrated that the rectification index was significantly higher for currents recorded in cells expressing the p.S1054Y SUR2A mutation. Overall, the results obtained in these groups of experiments suggest that the p.S1054Y mutation produced a remarkable effect on channel gating characterized by an increase in opening frequency and the appearance of different conductance levels that eventually lead to augmented unitary conductance and Po of the channel.

The p.S1054Y SUR2A mutation increases KATP channel slope conductance. (A) Single-channel current–voltage relationships for currents recorded in cells expressing Kir6.2+SUR2A WT or Kir6.2+SUR2A p.S1054Y. Solid lines represent the fit of a linear function to the data. Each point represents the mean ± SEM of the number of experiments/cells indicated in the figure. *P < 0.05 vs. Kir6.2+SUR2A WT (unpaired t test; from left to right: P value = 0.031 at −120 mV, 0.037 at −100 mV, 0.018 at −80 mV, and 0.039 at −60 mV). (B) Rectification index calculated for each experiment as the ratio between the current recorded at +40 and −120 mV. Each bar represents the mean ± SEM of the number of experiments/cells indicated in the figure and each dot represents one experiment/cell. *P = 0.01 vs. Kir6.2+SUR2A WT (unpaired t test).
The p.S1054Y SUR2A mutation increases KATP channel slope conductance. (A) Single-channel current–voltage relationships for currents recorded in cells expressing Kir6.2+SUR2A WT or Kir6.2+SUR2A p.S1054Y. Solid lines represent the fit of a linear function to the data. Each point represents the mean ± SEM of the number of experiments/cells indicated in the figure. *P < 0.05 vs. Kir6.2+SUR2A WT (unpaired t test; from left to right: P value = 0.031 at −120 mV, 0.037 at −100 mV, 0.018 at −80 mV, and 0.039 at −60 mV). (B) Rectification index calculated for each experiment as the ratio between the current recorded at +40 and −120 mV. Each bar represents the mean ± SEM of the number of experiments/cells indicated in the figure and each dot represents one experiment/cell. *P = 0.01 vs. Kir6.2+SUR2A WT (unpaired t test).
Effects of p.S1054Y SUR2A mutation on the membrane expression of Kir6.2 and SUR2A subunits
To determine a putative effect of p.S1054Y on the expression of Kir6.2 and SUR2A subunits at the plasma membrane, we conducted a cell surface biotinylation assay. To this end, HEK-293 cells were transfected with the cDNA encoding Kir6.2 plus WT or p.S1054Y SUR2A and the levels of expression of total protein (inputs) and membrane Kir6.2 and SUR2A were measured by Western blot (Fig. 6). The cytosolic protein ezrin was used as a negative control. Fig. 6 A shows a representative image of a biotinylation assay depicting the total and membrane expression (biotinylated fractions) of the three proteins in samples run in the same gel. Fig. 6 B shows the densitometry of the total expression of Kir6.2 and SUR2A in cells expressing p.S1054Y SUR2A (n = 7), while Fig. 6 C shows the densitometry of the relative surface expression of Kir6.2 and SUR2A normalized to that measured in cells expressing the WT form. We did not detect significant differences in the expression of total Kir6.2 and SUR2A proteins between cells transfected with WT or p.S1054Y SUR2A (Fig. 6 B). Conversely, the expression of both subunits at the plasma membrane was significantly reduced in cells expressing the mutation (Fig. 6, A and C). As expected, ezrin was not detected in the biotinylated extracts from any experimental group (Fig. 6 A).
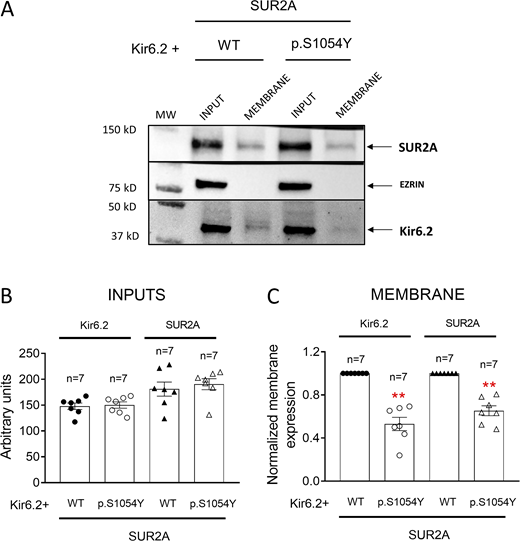
The p.S1054Y SUR2A mutation reduces Kir6.2 and SUR2A membrane expression. (A–C) Representative Western blot images (A) and densitometric analyses (B and C) of biotinylation assays showing the total (Input, B) or surface (Membrane, C) expression of Kir6.2 (37 kD) and SUR2A (120 kD) in cells expressing Kir6.2+SUR2A WT or Kir6.2+SUR2A p.S1054Y. The cytosolic protein ezrin (81 kD) was used as a negative control. In B and C, each bar shows the mean ± SEM of the number of independent experiments indicated in the figure, and each dot represents one experiment. **P = 0.001 for all comparisons vs. Kir6.2+SUR2A WT (unpaired t test). Source data are available for this figure: SourceData F6.
The p.S1054Y SUR2A mutation reduces Kir6.2 and SUR2A membrane expression. (A–C) Representative Western blot images (A) and densitometric analyses (B and C) of biotinylation assays showing the total (Input, B) or surface (Membrane, C) expression of Kir6.2 (37 kD) and SUR2A (120 kD) in cells expressing Kir6.2+SUR2A WT or Kir6.2+SUR2A p.S1054Y. The cytosolic protein ezrin (81 kD) was used as a negative control. In B and C, each bar shows the mean ± SEM of the number of independent experiments indicated in the figure, and each dot represents one experiment. **P = 0.001 for all comparisons vs. Kir6.2+SUR2A WT (unpaired t test). Source data are available for this figure: SourceData F6.
The effects of p.S1054Y SUR2A mutation are abolished by AnkB
The results obtained thus far indicate that the p.S1054Y SUR2A mutation leads to a combination of gain- and loss-of-function effects. As mentioned above, this dual mechanism has been described for KCNJ11 and ABCC8 mutations associated with neonatal diabetes (Zhou et al., 2010; Lin et al., 2013; Kline et al., 2009). These mutations prevent the interaction between the cytoskeletal adapter protein AnkB and KATP channels leading to a strong decrease ATP blockade and, simultaneously, to a diminished expression of the channel subunits at the plasma membrane (Zhou et al., 2010; Lin et al., 2013; Kline et al., 2009). Thus, we surmized that the effects of p.S1054Y SUR2A could be mediated by the disruption of the KATP channel interaction with AnkB. To test this hypothesis, we recorded single-channel currents in cells expressing Kir6.2 plus WT or p.S1054Y SUR2A and co-transfected with the cDNA encoding AnkB. HEK-293 cells endogenously express AnkB and, as expected, its expression level increased in AnkB-transfected cells (Fig. 7, A and B). Moreover, AnkB was not detected in the biotinylated extracts both in cells transfected or not with AnkB (Fig. 7 A) and the presence of the p.S1054Y SUR2A mutation did not modify AnkB expression compared to the WT SUR2A form, either in AnkB-transfected or non-transfected cells (Fig. 7 C). In AnkB-transfected cells, Kir6.2+SUR2A WT single-channel properties such as fo, Po, or open (−7.8 pA) and closed (−0.03 pA) amplitude values were not significantly different (n = 19; P > 0.05; Fig. 8, A–D and F) from those measured in AnkB-non transfected cells (Fig. 2). Importantly, in the presence of AnkB, the length of the opening burst periods (Fig. 8 A, right), fo (Fig. 8 B), and Po (Fig. 8 C) were similar in cells expressing WT or p.S1054Y SUR2A. Furthermore, as can be observed in the amplitude histogram, a single Gaussian was used to fit both closed and open channel amplitudes (Fig. 8 E), indicating that overexpression of AnkB prevented the appearance of multiple conductance levels induced by the mutation. Mean single-channel amplitude was not modified by the mutation either (Fig. 8 F). Moreover, overexpression of AnkB also abolished the increase in the slope conductance values produced by p.S1054Y SUR2A (γ = 59.8 ± 2.8 and 58.5 ± 1.9 pS for WT and p.S1054Y SUR2A, respectively; P > 0.05; Fig. 8 G) and the increase of the outward rectification of the current (rectification index = 0.41 ± 0.03 and 0.48 ± 0.06 for WT and p.S1054Y SUR2A, respectively; P > 0.05). Dwell-time histograms also showed that opening and closing kinetics were not affected by the p.S1054Y mutation in the presence of AnkB (Fig. 9, A–E).
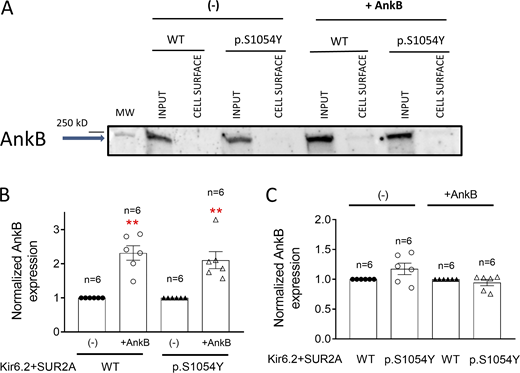
The p.S1054Y SUR2A mutation did not modify AnkB expression. (A) Representative Western blot images of biotinylation assays showing the total (Input) or surface (Membrane) expression of AnkB (220 kD) in cells expressing Kir6.2+SUR2A WT or Kir6.2+SUR2A p.S1054Y and co-transfected or not with AnkB. (B and C) Densitometric analysis of biotinylation assays showing the total expression of AnkB in cells expressing Kir6.2+SUR2A WT or Kir6.2+SUR2A p.S1054Y co-transfected or not with AnkB. In B, the AnkB expression measured in cells co-transfected with AnkB was normalized to that measured in AnkB non-transfected cells. In C, the AnkB expression measured in cells expressing p.S1054Y was normalized to the AnkB expression measured in cells expressing WT SUR2A. In B and C, each bar shows the mean ± SEM of the number of independent experiments indicated in the figure, and each dot represents one experiment. **P = 0.0001 and P = 0.0013 for cells expressing WT and p.S1054Y, respectively, together with AnkB vs. AnkB non-transfected cells (unpaired t test). Source data are available for this figure: SourceData F7.
The p.S1054Y SUR2A mutation did not modify AnkB expression. (A) Representative Western blot images of biotinylation assays showing the total (Input) or surface (Membrane) expression of AnkB (220 kD) in cells expressing Kir6.2+SUR2A WT or Kir6.2+SUR2A p.S1054Y and co-transfected or not with AnkB. (B and C) Densitometric analysis of biotinylation assays showing the total expression of AnkB in cells expressing Kir6.2+SUR2A WT or Kir6.2+SUR2A p.S1054Y co-transfected or not with AnkB. In B, the AnkB expression measured in cells co-transfected with AnkB was normalized to that measured in AnkB non-transfected cells. In C, the AnkB expression measured in cells expressing p.S1054Y was normalized to the AnkB expression measured in cells expressing WT SUR2A. In B and C, each bar shows the mean ± SEM of the number of independent experiments indicated in the figure, and each dot represents one experiment. **P = 0.0001 and P = 0.0013 for cells expressing WT and p.S1054Y, respectively, together with AnkB vs. AnkB non-transfected cells (unpaired t test). Source data are available for this figure: SourceData F7.

AnkB corrects the effects of the p.S1054Y SUR2A mutation on single-channel properties. (A) Single-channel recordings obtained by applying 10-s pulses from 0 to −120 mV in cells expressing Kir6.2+SUR2A WT (left) or Kir6.2+SUR2A p.S1054Y (right) co-transfected with AnkB. (B and C) Mean fo (B) and Po (C) for single-channel currents recorded in cells expressing Kir6.2+SUR2A WT or Kir6.2+SUR2A p.S1054Y and co-transfected with AnkB. (D and E) Amplitude histograms for currents recorded in cells expressing Kir6.2+SUR2A WT (D) or Kir6.2+SUR2A p.S1054Y (E; bin width = 0.05 pA) co-transfected with AnkB. Continuous lines represent the fit of a Gaussian function to the data. Histograms were obtained by pooling data from 19 (Kir6.2+SUR2A WT) and 21 (Kir6.2+SUR2A p.S1054Y) experiments. (F) Mean unitary current amplitude measured in both experimental groups. In B, C, and F, each bar represents the mean ± SEM of the number of experiments/cells indicated in the figure and each dot represents one experiment/cell (unpaired t test). (G) Single-channel current–voltage relationships constructed for currents recorded in cells expressing Kir6.2+SUR2A WT or Kir6.2+SUR2A p.S1054Y co-transfected with AnkB. Solid lines represent the fit of a linear function to the data. Each point represents the mean ± SEM of the number of experiments/cells indicated in the figure (unpaired t test).
AnkB corrects the effects of the p.S1054Y SUR2A mutation on single-channel properties. (A) Single-channel recordings obtained by applying 10-s pulses from 0 to −120 mV in cells expressing Kir6.2+SUR2A WT (left) or Kir6.2+SUR2A p.S1054Y (right) co-transfected with AnkB. (B and C) Mean fo (B) and Po (C) for single-channel currents recorded in cells expressing Kir6.2+SUR2A WT or Kir6.2+SUR2A p.S1054Y and co-transfected with AnkB. (D and E) Amplitude histograms for currents recorded in cells expressing Kir6.2+SUR2A WT (D) or Kir6.2+SUR2A p.S1054Y (E; bin width = 0.05 pA) co-transfected with AnkB. Continuous lines represent the fit of a Gaussian function to the data. Histograms were obtained by pooling data from 19 (Kir6.2+SUR2A WT) and 21 (Kir6.2+SUR2A p.S1054Y) experiments. (F) Mean unitary current amplitude measured in both experimental groups. In B, C, and F, each bar represents the mean ± SEM of the number of experiments/cells indicated in the figure and each dot represents one experiment/cell (unpaired t test). (G) Single-channel current–voltage relationships constructed for currents recorded in cells expressing Kir6.2+SUR2A WT or Kir6.2+SUR2A p.S1054Y co-transfected with AnkB. Solid lines represent the fit of a linear function to the data. Each point represents the mean ± SEM of the number of experiments/cells indicated in the figure (unpaired t test).

The p.S1054Y mutation did not modify open and closed time constants in the presence of AnkB. (A and B) Open dwell-time histograms for currents recorded in cells expressing Kir6.2+SUR2A WT (A) or Kir6.2+SUR2A p.S1054Y (B) co-transfected with AnkB (bin width = 0.1 ms). Continuous lines represent the fit of a monoexponential function to the data, which yielded the indicated τOPEN (F test). (C and D) Log-binned closed dwell-time histograms for currents recorded in cells expressing Kir6.2+SUR2A WT (C) or Kir6.2+SUR2A p.S1054Y (D) co-transfected with AnkB (bin width = 0.5 ms). Continuous lines represent the fit of double Gaussian function to the data, which yielded the indicated τfCLOSED and τsCLOSED. Histograms were obtained by pooling data from 19 (Kir6.2+SUR2A WT) and 21 (Kir6.2+SUR2A p.S1054Y) experiments (F test). (E) Mean open-dwell times for currents recorded in cells expressing Kir6.2+SUR2A WT or Kir6.2+SUR2A p.S1054Y co-transfected with AnkB. Each bar represents the mean ± SEM of the number of experiments/cells indicated in the figure and each dot represents one experiment/cell (unpaired t test).
The p.S1054Y mutation did not modify open and closed time constants in the presence of AnkB. (A and B) Open dwell-time histograms for currents recorded in cells expressing Kir6.2+SUR2A WT (A) or Kir6.2+SUR2A p.S1054Y (B) co-transfected with AnkB (bin width = 0.1 ms). Continuous lines represent the fit of a monoexponential function to the data, which yielded the indicated τOPEN (F test). (C and D) Log-binned closed dwell-time histograms for currents recorded in cells expressing Kir6.2+SUR2A WT (C) or Kir6.2+SUR2A p.S1054Y (D) co-transfected with AnkB (bin width = 0.5 ms). Continuous lines represent the fit of double Gaussian function to the data, which yielded the indicated τfCLOSED and τsCLOSED. Histograms were obtained by pooling data from 19 (Kir6.2+SUR2A WT) and 21 (Kir6.2+SUR2A p.S1054Y) experiments (F test). (E) Mean open-dwell times for currents recorded in cells expressing Kir6.2+SUR2A WT or Kir6.2+SUR2A p.S1054Y co-transfected with AnkB. Each bar represents the mean ± SEM of the number of experiments/cells indicated in the figure and each dot represents one experiment/cell (unpaired t test).
To determine the consequences of the overexpression of AnkB over the membrane expression of Kir6.2 and SUR2A, we conducted a biotinylation assay in HEK-293 cells transfected with Kir6.2 plus WT or p.S1054Y SUR2A. Fig. 10 shows the detection of Kir6.2, SUR2A, and ezrin proteins in the inputs and membrane fractions from samples run in the same gel. As shown in the representative images (Fig. 10 A) and confirmed by the densitometric analysis (n = 7; Fig. 10 B), in AnkB-transfected cells membrane expressions of Kir6.2 and SUR2A were not different in cells transfected with WT or p.S1054Y SUR2A subunits.
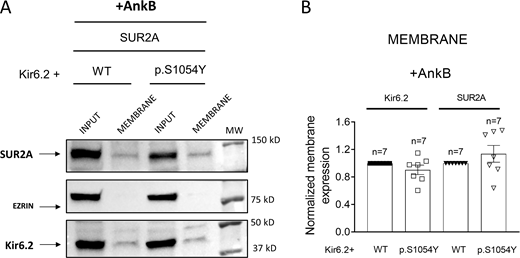
AnkB corrects the reduction of Kir6.2 and SUR2A membrane expression induced by the p.S1054Y SUR2A mutation. (A and B) Representative Western blot images (A) and densitometric analysis (B) of biotinylation assays showing the total (Input) or surface (Membrane) expression of Kir6.2 (37 kD) and SUR2A (120 kD) in cells expressing Kir6.2+SUR2A WT or Kir6.2+SUR2A p.S1054Y and co-transfected with AnkB. The cytosolic protein ezrin (81 kD) was used as a negative control. In B, each bar shows the mean ± SEM of the number of independent experiments indicated in the figure (unpaired t test). Source data are available for this figure: SourceData F10.
AnkB corrects the reduction of Kir6.2 and SUR2A membrane expression induced by the p.S1054Y SUR2A mutation. (A and B) Representative Western blot images (A) and densitometric analysis (B) of biotinylation assays showing the total (Input) or surface (Membrane) expression of Kir6.2 (37 kD) and SUR2A (120 kD) in cells expressing Kir6.2+SUR2A WT or Kir6.2+SUR2A p.S1054Y and co-transfected with AnkB. The cytosolic protein ezrin (81 kD) was used as a negative control. In B, each bar shows the mean ± SEM of the number of independent experiments indicated in the figure (unpaired t test). Source data are available for this figure: SourceData F10.
AnkB overexpression corrects the reduction in ATP block induced by the p.S1054Y SUR2A mutation
To test the functional relevance of our findings regarding the role of AnkB in the p.S1054Y SUR2A effects, we assessed the concentration dependence of the MgATP inhibition on IKATP recorded in excised inside-out macropatches from HEK-293 cells expressing Kir6.2+SUR2A WT, Kir6.2+SUR2A p.S1054Y, or Kir6.2+SUR2A p.S1054Y+AnkB. Fig. 11 A shows representative current traces generated by cells expressing Kir6.2+SUR2A p.S1054Y (upper panel) or Kir6.2+SUR2A p.S1054Y+AnkB (lower panel) in control conditions and in the presence of 0.1 mM MgATP by applying 3-s voltage ramps from −80 to +60 mV (Fig. 11 A, top inset). The MgATP-inhibition measured at −80 mV was used as an index of block and a Hill equation was fitted to the respective concentration-response data (Fig. 11 B). As expected, the presence of p.S1054Y shifted the curve to the right and significantly increased the IC50 (from 0.037 ± 0.015 to 0.34 ± 0.05 mM) compared to Kir6.2+SUR2A WT (P < 0.05, n ≥ 3 for each concentration). Interestingly, the concentration-response curve for currents recorded in cells coexpressing AnkB and Kir6.2+SUR2A p.S1054Y overlapped (IC50 = 0.044 ± 0.019 mM) with that obtained for cells expressing Kir6.2+SUR2A WT (P > 0.05 vs. Kir6.2+SUR2A WT), suggesting that the presence of AnkB rescued the KATP channel affinity for MgATP that had been reduced by the p.S1054Y mutation.

AnkB corrects the reduced KATP channel affinity for MgATP block induced by the p.S1054Y mutation. (A) Superimposed current traces elicited by 3-s ramps from −80 to +60 mV (see a scheme of the protocol in the inset at the top) in inside-out macropatches from HEK-293 cells expressing Kir6.2+SUR2A p.S1054Y or Kir6.2+SUR2A p.S1054Y+AnkB in the absence and presence of 0.1 mM MgATP. Dashed lines represent the zero current level. (B) Concentration–response curves constructed by plotting ATP inhibition (expressed as remaining current) of currents recorded at −80 mV in inside-out macropatches from HEK-293 cells expressing Kir6.2+SUR2A WT, Kir6.2+SUR2A p.S1054Y, or Kir6.2+SUR2A p.S1054Y and AnkB. Solid lines represent the fit of a Hill equation to the data. An F test was used to demonstrate differences in the IC50. Each point represents the mean ± SEM of n ≥ 3 experiments.
AnkB corrects the reduced KATP channel affinity for MgATP block induced by the p.S1054Y mutation. (A) Superimposed current traces elicited by 3-s ramps from −80 to +60 mV (see a scheme of the protocol in the inset at the top) in inside-out macropatches from HEK-293 cells expressing Kir6.2+SUR2A p.S1054Y or Kir6.2+SUR2A p.S1054Y+AnkB in the absence and presence of 0.1 mM MgATP. Dashed lines represent the zero current level. (B) Concentration–response curves constructed by plotting ATP inhibition (expressed as remaining current) of currents recorded at −80 mV in inside-out macropatches from HEK-293 cells expressing Kir6.2+SUR2A WT, Kir6.2+SUR2A p.S1054Y, or Kir6.2+SUR2A p.S1054Y and AnkB. Solid lines represent the fit of a Hill equation to the data. An F test was used to demonstrate differences in the IC50. Each point represents the mean ± SEM of n ≥ 3 experiments.
The p.S1054Y mutation reduces the interaction of SUR2A with AnkB
The results obtained so far suggest that the presence of the p.S1054Y SUR2A mutation reduces the interaction of AnkB with the KATP channel. There are data demonstrating that Kir6.2 and AnkB do interact (Li et al., 2010). To explore whether AnkB can also interact with SUR2A, we conducted a yeast-two hybrid assay that is an accepted method to detect protein–protein interactions (Paiano et al., 2019). We cloned WT or p.S1054Y SUR2A into the bait pDEST32 vector encoding the GAL4 DBD and AnkB was cloned into the prey pDEST22 vector encoding the GAL4 AD (see Materials and methods). The principle of this assay is that if the tested proteins do interact, then the DBD and AD of the transcription factor GAL4 will be indirectly connected and transcription of the reporters (HIS3 and URA3) gene will occur (Paiano et al., 2019). DBD/SUR2A and AD/AnkB were co-transformed into MaV203 competent cells. As can be observed in Fig. 12 A, yeasts transformed with SUR2A WT and AnkB were able to growth in SC-Leu-Trp-His-3AT selective medium. This result indicated that the transformation of both proteins allowed the expression of HIS3 leading to the synthesis of histidine, and, in turn, to growth of transformed yeasts in a medium lacking histidine. Conversely, the presence of p.S1054Y mutation significantly reduced yeast growth compared to WT SUR2A (Fig. 12, B and L). These results were confirmed by cloning SUR2A (WT or p.S1054Y) into prey pDEST22 and AnkB into bait pDEST32 vectors (Fig. 12 C). The use of negative (yeast cells transfected with empty vectors) and positive (cloning of Krev1 and RalGDS interacting proteins) controls demonstrated the reliability of the system (Fig. 12, E and F). As an additional positive control, we determined that yeasts transformed with DBD/Kir6.2 and AD/AnkB were able to growth in SC-Leu-Trp-His-3AT selective medium (Fig. 12 G). The interaction between SUR2A and AnkB was also determined based on the GAL4-induced protranscriptional effect over the promoter of the URA3 reporter, enabling the yeast to express the Ura3 enzyme that converts 5FOA into the toxic derivative 5-fluorouracil. To this end, growth was determined plating the cells onto SC-Leu-Trp-5FOA medium that contains 5FOA (0.2%). The interaction of SUR2A WT with AnkB was suggested by a small yeast growth (Fig. 12 H), similar to that produced by positive controls (Fig. 12 J), while the negative control remarkably increased yeast growth (Fig. 12 K). The presence of p.S1054Y SUR2A mutation slightly increased growth compared to WT SUR2A, suggesting that it was reducing the SUR2A-AnkB interaction (Fig. 12, I and L).

The p.S1054Y SUR2A mutation reduces the interaction between AnkB and SUR2A. (A–G) Representative images of yeast-two hybrid assays showing the growth of yeast cells in selective medium containing 3AT (50 mM) and lacking Leu-Trp-His (SC-Leu-Trp-His-3AT) when SUR2A (WT or p.S1054Y) and AnkB were fused to the prey and bait vectors (A and B) or vice versa (C and D). As a negative control (F), yeast cells were transfected with empty vectors and as positive controls (E and G), yeast cells were transfected with vectors encoding native Krev1 and RalGDS interacting proteins (E) and with Kir6.2 and AnkB (G). (H–K) Representative images showing growth of yeast cells in selective medium lacking Leu-Trp but containing 5FOA (0.2%; SC-Leu-Trp-5FOA) when SUR2A (WT or p.S1054Y) and AnkB were fused to the prey and bait vectors (H and I). In this case positive control (J) inhibited yeast growth, whereas negative control remarkably increased it (K). (L) Densitometric analysis of the images corresponding to yeasts expressing SUR2A (WT or p.S1054Y) and AnkB-encoding vectors. In L, each bar shows the mean ± SEM of the number of independent experiments indicated in the figure. *P = 0.044 vs. DBD/SUR2A WT+AD/AnkB (unpaired t test).
The p.S1054Y SUR2A mutation reduces the interaction between AnkB and SUR2A. (A–G) Representative images of yeast-two hybrid assays showing the growth of yeast cells in selective medium containing 3AT (50 mM) and lacking Leu-Trp-His (SC-Leu-Trp-His-3AT) when SUR2A (WT or p.S1054Y) and AnkB were fused to the prey and bait vectors (A and B) or vice versa (C and D). As a negative control (F), yeast cells were transfected with empty vectors and as positive controls (E and G), yeast cells were transfected with vectors encoding native Krev1 and RalGDS interacting proteins (E) and with Kir6.2 and AnkB (G). (H–K) Representative images showing growth of yeast cells in selective medium lacking Leu-Trp but containing 5FOA (0.2%; SC-Leu-Trp-5FOA) when SUR2A (WT or p.S1054Y) and AnkB were fused to the prey and bait vectors (H and I). In this case positive control (J) inhibited yeast growth, whereas negative control remarkably increased it (K). (L) Densitometric analysis of the images corresponding to yeasts expressing SUR2A (WT or p.S1054Y) and AnkB-encoding vectors. In L, each bar shows the mean ± SEM of the number of independent experiments indicated in the figure. *P = 0.044 vs. DBD/SUR2A WT+AD/AnkB (unpaired t test).
The biophysical alterations over KATP channels induced by p.E322K Kir6.2 mutation are similar than those produced by the presence of p.S1054Y SUR2A
Previous data showed that the p.E322K Kir6.2 mutation prevents the binding of AnkB to Kir6.2 (Kline et al., 2009). We questioned whether unitary currents generated by p.E322K Kir6.2 channels display similar characteristics to those generated by Kir6.2 WT+p.S1054Y SUR2A. Thus, in the next group of experiments, single Kir6.2 p.E322K+SUR2A WT currents were recorded in HEK-293 cells overexpressing or not AnkB. Fig. 13 A shows representative traces recorded at −120 mV in cells not overexpressing AnkB and in the continuous presence of 100 µM pinacidil. Mean fo (Fig. 13 B), Po (Fig. 13 C), and slope conductance (Fig. 13 D) were 27.4 ± 3.3 Hz, 0.16 ± 0.01, and 83.1 ± 2.5 pS, respectively (P < 0.05 vs. Kir6.2 WT+SUR2A WT). Strikingly, as can be observed in the traces shown in Fig. 13 A, multiple conductance states are also apparent. Indeed, a triple Gaussian was needed to fit the amplitude histogram for currents generated by Kir6.2 p.E322K+SUR2A WT channels that yielded peak amplitudes at −3.6, −9.4, and −15.5 pA (Fig. 13 E). Fitting of a monoexponential function to the open dwell-time histogram yielded a τOPEN of 0.15 ms, while the biexponential fit to the closed dwell-time histogram yielded τfCLOSED and τsCLOSED of 1.54 and 28.1 ms, respectively. These results closely resembled most of the changes produced by p.S1054Y, i.e., increase of unitary conductance, opening frequency, and open probability and reduction of τOPEN and τsCLOSED compared to Kir6.2 WT, as well as the appearance of multiple open states. Importantly, currents generated by cells expressing Kir6.2 p.E322K+SUR2A WT and AnkB exhibited significantly smaller fo (Fig. 13 B), Po (Fig. 13 C), and slope conductance (Fig. 13 D) values, and significantly larger τOPEN (0.30 ms) and τsCLOSED (42.1 ms) compared to Kir6.2 p.E322K+SUR2A WT alone (P < 0.05). As demonstrated by the amplitude histograms, the presence of AnkB also suppressed the appearance of multiple open states (Fig. 13 F).

The p.E322K Kir6.2 mutation produced alterations in the function of KATP channel similar to those induced by the presence of p.S1054Y SUR2A. (A) Single-channel recordings obtained by applying 10-s pulses from 0 to −120 mV in HEK-293 cells expressing Kir6.2 p.E322K+SUR2A WT (left) or Kir6.2 p.E322K+SUR2A WT + AnkB. (B and C) Mean fo (B) and Po (C) for single-channel currents recorded in cells expressing Kir6.2 p.E322K+ SUR2A WT co-transfected or not with AnkB. Each bar represents the mean ± SEM of the number of experiments/cells indicated in the figure and each dot represents one experiment/cell. *P = 0.0383 and P = 0.0124 for comparisons of fo (B) and Po (C), respectively, vs. p.E322K (unpaired t test). (D) Current density–voltage relationships for currents recorded in cells expressing Kir6.2 p.E322K+SUR2A WT co-transfected or not with AnkB. The linear regression fit yielded conductance values of 83.1 ± 0.2 and 64.5 ± 1.4 pS for each experimental group, respectively (P < 0.05). Data points represent the mean ± SEM of 11 experiments. *P < 0.05 vs. p.E322K (unpaired t test; from left to right: P value = 0.018 at −120 mV, 0.016 at −100 mV, 0.019 at −80 mV, and 0.032 at +40 mV). (E and F) Amplitude histograms of currents recorded at −120 mV constructed by plotting amplitude data as a function of the number of events per bin (bin width = 0.05 pA) together with the fit of a Gaussian function (solid lines) for single-channel currents recorded in cells expressing Kir6.2 p.E322K+SUR2A WT co-transfected (E) or not (D) with AnkB. Histograms were obtained by pooling data from 16 to 14 experiments, respectively. The fit to the open channel data recorded in cells non–co-transfected with AnkB required a triple Gaussian function.
The p.E322K Kir6.2 mutation produced alterations in the function of KATP channel similar to those induced by the presence of p.S1054Y SUR2A. (A) Single-channel recordings obtained by applying 10-s pulses from 0 to −120 mV in HEK-293 cells expressing Kir6.2 p.E322K+SUR2A WT (left) or Kir6.2 p.E322K+SUR2A WT + AnkB. (B and C) Mean fo (B) and Po (C) for single-channel currents recorded in cells expressing Kir6.2 p.E322K+ SUR2A WT co-transfected or not with AnkB. Each bar represents the mean ± SEM of the number of experiments/cells indicated in the figure and each dot represents one experiment/cell. *P = 0.0383 and P = 0.0124 for comparisons of fo (B) and Po (C), respectively, vs. p.E322K (unpaired t test). (D) Current density–voltage relationships for currents recorded in cells expressing Kir6.2 p.E322K+SUR2A WT co-transfected or not with AnkB. The linear regression fit yielded conductance values of 83.1 ± 0.2 and 64.5 ± 1.4 pS for each experimental group, respectively (P < 0.05). Data points represent the mean ± SEM of 11 experiments. *P < 0.05 vs. p.E322K (unpaired t test; from left to right: P value = 0.018 at −120 mV, 0.016 at −100 mV, 0.019 at −80 mV, and 0.032 at +40 mV). (E and F) Amplitude histograms of currents recorded at −120 mV constructed by plotting amplitude data as a function of the number of events per bin (bin width = 0.05 pA) together with the fit of a Gaussian function (solid lines) for single-channel currents recorded in cells expressing Kir6.2 p.E322K+SUR2A WT co-transfected (E) or not (D) with AnkB. Histograms were obtained by pooling data from 16 to 14 experiments, respectively. The fit to the open channel data recorded in cells non–co-transfected with AnkB required a triple Gaussian function.
Discussion
The results here presented show that the p.S1054Y SUR2A mutation stabilized KATP channels in their open state and reduced the sarcolemmal expression of Kir6.2 and SUR2A subunits without modifying their total expression. Interestingly, all the p.S1054Y-induced effects were completely suppressed when AnkB was overexpressed. Our results would explain the mild phenotype exhibited by the patient despite carrying a mutation that produces a strong reduction in ATP block and had been predicted as highly pathogenic. Furthermore, they provide the first evidence of a CS-associated mutation that combines gain- and loss-of-function effects over KATP channels by disrupting the modulatory actions of the adapter protein AnkB.
We demonstrate that the Ser-to-Tyr substitution at position 1054 of SUR2A increased the density of basal macroscopic IKATP recorded under the whole-cell configuration, an effect that was attributed to the reduction of the block of the channels by cytosolic ATP produced by this mutation (Scurr et al., 2011; Harakalova et al., 2012; Houtman et al., 2019). At the single-channel level, the presence of the mutation accelerated channel activation and increased fo, Po, and slope conductance. Moreover, it also led to the appearance of multiple conductance levels, including one level (O3) whose amplitude was almost twice that of channels composed of WT subunits. An individualized analysis of each conductance level demonstrated that those with the highest amplitudes (O2 and O3) also had the longest dwell-open times and Po. The biophysical mechanism underlying the appearance of multiple conductance levels is unknown, but it could be possible that, as proposed for voltage-gated K channels (Chapman and VanDongen, 2005), it is the consequence of a gating change in the pore-forming subunits that promotes the presence of individual transitions of each subunit from the closed to the active state, instead of an orchestrated transition of the four subunits at a time. Further studies would be needed to determine the ultimate mechanism underlying the appearance of these levels in the presence of p.S1054Y SUR2A. Overall, the effects at the single-channel level suggested that p.S1054Y SUR2A stabilizes the open state and explain the remarkable decrease in the ATP block caused by this mutation (Houtman et al., 2019), since in these channels ATP sensitivity depends on the open-state stability (Enkvetchakul et al., 2000).
The vast majority of the CS mutations identified so far have been characterized using inside-out recordings to determine their consequences only on the channel response to ATP, Mg-nucleotides, or pharmacological blockers such as sulfonylureas. Conversely, functional analyses at the single-channel level to identify the possible biophysical alterations produced by CS mutations are, to the best of our knowledge, absent. For example, it has been suggested that the p.V65M Kir6.1 CS-associated mutation also increased Po, although it was not confirmed at the single-channel level (Cooper et al., 2017). Thus, we do not know whether other CS-associated mutations produce similar effects on the single-channel properties.
Concomitantly, the p.S1054Y mutation reduced the expression of Kir6.2 and SUR2A subunits at the plasma membrane, as shown by biotinylation assays. As mentioned above, it has been shown that the expression of the CS-associated mutation p.R1154Q SUR2A in mouse ventricles leads to a complete loss of SUR2 proteins as a result of an aberrant splicing (Zhang et al., 2021). Since we showed that the total levels of expression of Kir6.2 and SUR2A proteins were unaffected, we ruled out a putative effect at transcriptional or post-transcriptional levels.
A relevant question that can arise is how an amino acid substitution of a residue located out of the permeation pathway can produce such important biophysical effects. Recent molecular modelling of SUR2A CS-associated mutations predicted that the bulky tyrosine side-chain of the p.S1054Y substitution would produce steric clashes that may lead to local conformational changes, most likely followed by unpredictable global changes affecting the structure of the channel complex (Houtman et al., 2019). However, our results demonstrating the involvement of AnkB could be the actual answer to this question. Members of the Ankyrin family have been involved in targeting KATP channels at the sarcolemma (Li et al., 2010; Cunha and Mohler, 2006; Yang et al., 2020; Sucharski et al., 2020). Indeed, AnkB regulates Kir6.2 and SUR membrane expression and function in cardiac (Li et al., 2010) and β-pancreatic cells (Kline et al., 2013). When the AnkB role is disturbed, such as in the presence of neonatal diabetes-associated KCNJ11 mutants (e.g., p.E322K) that prevent the interaction between Kir6.2 and AnkB or in cardiomyocytes from AnkB-deficient mice (AnkB+/−), membrane expression of KATP channel subunits is reduced (Kline et al., 2009; Li et al., 2010). Interestingly, diminishing AnkB expression also decreases channel sensitivity to ATP block and increases Po (Kline et al., 2009; Li et al., 2010), as p.S1054Y does. Therefore, we surmized that the p.S1054Y mutation decreases the AnkB modulation over KATP channels. A strong support to this contention came from the fact that overexpression of AnkB corrected the changes induced by the p.S1054Y mutation on KATP channel gating and Kir6.2/SUR2A membrane expression. Since the presence of the mutation did not modify AnkB expression, we propose that it leads to a conformational change that reduces the affinity of the channel complex for the scaffolding protein. AnkB has a membrane-binding domain (MBD) that consists of 24 consecutive ANK repeats and is mainly responsible for AnkB-dependent interactions with ion channels (Li et al., 2010). Kline and coworkers elegantly described that AnkB, Kir6.2, and SUR1/SUR2A form a ternary complex based on the interaction of the MBD of AnkB with an eight amino-acid motif in the C-terminal of Kir6.2, where an acidic triplet (EED) including the glutamic acid at position 322, plays a key role (Kline et al., 2009). Indeed, they suggested that AnkB can bind directly to Kir6.2 without requiring SUR2A (Kline et al., 2009). Our results obtained with yeast-two hybrid assays strongly suggested that AnkB not only interacted with Kir6.2, but also with SUR2A and that the presence of p.S1054Y SUR2A mutation reduces this interaction. Furthermore, the p.E322K Kir6.2 mutation, which prevents AnkB binding to Kir6.2 subunits (Kline et al., 2009), increases the unitary conductance, the fo, and Po, and induces the appearance of multiple open states. Thus, single-channel properties of p.E322K Kir6.2 channels resembled most of those produced by p.S1054Y. We also demonstrated that transfection with AnkB reversed all the p.E322K-induced changes, suggesting that the reduced AnkB-binding to the KATP channel was overcome by the overexpression of the scaffolding protein. In the same line, the results obtained by the inside-out recordings demonstrating that AnkB overexpression rescued the reduced ATP sensitivity to ATP block induced by the p.S1054Y mutation further support the critical role of AnkB in the molecular mechanism responsible for the alterations of this CS-associated mutation. All these results would mean that when AnkB cannot properly interact with the KATP channel either through an impaired binding to Kir6.2 (as in the case of the p.E222K mutant) or to SUR2A (as occurs with p.S1054Y), the consequences on channel gating, ATP sensitivity, expression, are almost identical. This leads us to the proposal that simultaneous interaction of the pore-forming and SUR2 subunits with AnkB is necessary for the proper KATP channel function.
Finally, it seems that some of the p.S1054Y-induced changes (increase in slope conductance and appearance of multiple conductance levels) are beyond those that have been described for the reduction of AnkB binding per se. Co-expression of SUR2A subunits with Kir6.2 increases slope conductance and open probability of KATP channels when compared to Kir6.2 channels alone (Fang et al., 2006). Co-expression of the GluA1 subunits of the AMPA receptor (AMPAR) of glutamate with some transmembrane AMPAR-regulatory proteins (TARP) may lead to the appearance of multiple conductance states (Shelley et al., 2012). Therefore, the multiple conductance states of p.S1054Y CS mutation may be the consequence of a decreased AnkB modulation of the channels formed by the association of Kir6.2 and the mutated form of SUR2A.
Physiopathological implications
Pathogenicity of CS mutations has been mainly attributed to their ability to reduce KATP channel block by physiological concentrations of ATP (Cooper et al., 2015; McClenaghan et al., 2018). Recent evidence, including ours, suggest that other functional effects may modulate their phenotypic consequences. The p.S1054Y mutation strongly reduced ATP block (IC50 in our experiments was increased by 10-fold) and was predicted as highly pathogenic. However, in the carriers the presence of the mutation was associated with much milder phenotypes than expected. The first described carrier presented mixed respiratory and metabolic acidosis and generalized hypertrichosis, hepatomegaly, and patent ductus arteriosus at 9 d of age (Scurr et al., 2011). Interestingly, she did not develop pulmonary hypertension, speech delay, learning difficulties or intellectual disability and at the end of the follow up (up to 19 yr old; Scurr et al., 2011), most of her manifestations had disappeared except hypertrichosis, cardiomyopathy, that remained stable, and reflux esophagitis (Scurr et al., 2011). More recently, a systematic study developed in the context of the International Cantú Syndrome Registry identified a second p.S1054Y SUR2A mutation carrier who is a 16-yr-old male that exhibited hypertrichosis, skin and joint abnormalities, arterial hypertension, umbilical hernia, and headache. In his clinical summary it was also stated that he presented developmental delay although no further information was provided (Grange et al., 2019).
Our results demonstrated that the mutation increased slope conductance and Po, which would underlie the marked reduction of the ATP block (gain-of-function). Simultaneously, the presence of the p.S1054Y mutation reduced membrane expression of the channel subunits (loss-of-function). Thus, decrease of the membrane KATP channel expression could counterbalance the hyperactivity of the channel, reducing the severity of the phenotype. Nonetheless, it is possible that the combination of these effects can also lead to unexpected outcomes, as described for some neonatal diabetes-associated mutations producing this dual mechanism that lead to unusual phenotypic consequences including the development of epilepsy (Lin et al., 2013). Our results could also be relevant regarding the pharmacological treatment of CS patients. Although there is no specific treatment for the disease, KATP blockers like glibenclamide could be valuable (Ma et al., 2019). In this context, it seems reasonable to propose that the consequences of the pharmacological treatment with KATP blockers would be different for pure gain-of-function mutations and for mixed gain- and loss-of-function mutations such as p.S1054Y, since an excessive reduction of channel activity with unpredictable consequences could be produced in the latter.
Study limitations
We conducted our study in expression systems, which allowed us to perform a detailed analysis of the consequences of the p.S1054Y SUR2A mutation on the IKATP without the interference of other ion currents. Moreover, we co-expressed WT or mutant SUR2A subunits with Kir6.2, but not with Kir6.1. It has been described that Kir6.1 expression is not affected in cardiomyocytes from AnkB knockout mice and that AnkB and Kir6.1 do not directly interact (Li et al., 2010). Therefore, the effects here reported would be manifested preferentially in those cells expressing KATP channels mostly composed of Kir6.2+SUR2A subunits as in atrial and ventricular myocytes although channels composed by Kir6.2+SUR2A subunits can be also found in other cell types (Nichols et al., 2013). Recent evidence suggests that many of the CS manifestations due to ABCC9 mutations are preferentially due to altered KATP channels containing Kir6.1 rather than Kir6.2 subunits (McClenaghan and Nichols, 2022). However, there are several CS symptoms (e.g., at the cardiovascular or neurological level) that support the importance of the mutation consequences on Kir6.2-containing channels. Interestingly, some gain-of-function KCNJ11 mutations linked to permanent neonatal diabetes (DEND Syndrome) have been associated with neurological disorders (including development delay, and speech and learning disabilities; Gloyn et al., 2006) that may be similar with those appear in CS patients (Roessler et al., 2021). Nevertheless, it has to be considered that the subunit composition of KATP channels throughout the different tissues of the body is complex and still matter of debate. Furthermore, heterotetrameric combination of Kir6.1 and Kir6.2 subunits (Hibino et al., 2010) to form KATP channels expressed in tissues affected by CS cannot be dismissed either. Furthermore, the demonstration that SUR2A and AnkB may interact and that the p.S1054Y mutation reduces the intensity of the interaction also indicates that the consequences of the mutation will be apparent irrespective of the Kir6.x pore-forming subunit. Growing evidence suggests that CS manifestations predominate in tissues where SUR2B subunits are more expressed than SUR2A (Davis et al., 2020; McClenaghan and Nichols, 2022). SUR2A and SUR2B are two splice variants encoded by the ABCC9 gene that vary only in the last 42 amino acids (Hibino et al., 2010). Therefore, since the p.S1054Y mutation is within the core of the SUR2 protein, the changes here described will be common for mutant SUR2A- and SUR2B-containing channels (Cooper et al., 2015).
On the other hand, we did not conduct experiments co-expressing mutated and WT SUR2A subunits to simulate the heterozygous condition of the carriers. It can be predicted that the effects could be partially attenuated by the presence of WT subunits, unless the mutation causes a dominant negative effect (Pérez-Hernández et al., 2018). It is also possible that the gating effects of the mutation depend on the K channel opener used to activate the channels (pinacidil). Interestingly, a previous paper showed identical single-channel properties of Kir6.2+SUR2A when the channels were activated by pinacidil or nicorandil (Shindo et al., 1998).
Conclusions
We demonstrated that the p.S1054Y SUR2A mutation produces gain- and loss-of-function effects that were corrected when AnkB was overexpressed. Our results provide a novel mechanism by which CS mutations can reduce ATP block and help explain the mild phenotype associated with this mutation. Furthermore, this is the first demonstration of a CS mutation whose functional consequences involve the disruption of AnkB effects on KATP channels.
Data availability
The data underlying this article will be shared upon reasonable request to the corresponding author.
Acknowledgments
Jeanne M. Nerbonne served as editor.
We thank Dr. Marcel Van der Heyden and Dr. Colin Nichols for providing us with the cDNA encoding the constructs used in the study.
This work was funded by Ministerio de Ciencia e Innovación [PID2020-118694RB-I00], Comunidad Autónoma de Madrid (S2017/BMD-3738; 2018-T2/BMD-10724 [J. Cebrián]; European Structural and Investment Funds), Universidad Complutense de Madrid and Comunidad Autónoma de Madrid (PR65/19-22358), and ERA-Net for Research on Rare Diseases.
The authors declare no competing financial interests.
Author contributions: T. Crespo-García, M. Rubio-Alarcón, A. Cámara-Checa, M. Dago, J. Rapún, P. Nieto-Marín, M. Marín, J. Cebrián: development of experiments, analysis of the data, preparation of the figures, and critical revision of the MS. J. Tamargo, E. Delpón, and R. Caballero: Conception, design and interpretation of the data, drafting and final approval of the MS.

