


Inheritable and de novo variants in the cardiac voltage-gated sodium channel, Nav1.5, are responsible for both long-QT syndrome type 3 (LQT3) and Brugada syndrome type 1 (BrS1). Interestingly, a subset of Nav1.5 variants can cause both LQT3 and BrS1. Many of these variants are found in channel structures that form the channel fast inactivation machinery, altering the rate, voltage dependence, and completeness of the fast inactivation process. We used a series of mutants at position 1784 to show that the most common inheritable Nav1.5 variant, E1784K, alters fast inactivation through two separable mechanisms: (1) a charge-dependent interaction that increases the noninactivating current characteristic of E1784K; and (2) a hyperpolarized voltage dependence and accelerated rate of fast inactivation that decreases the peak sodium current. Using a homology model built on the NavPaS structure, we find that the charge-dependent interaction is between E1784 and K1493 in the DIII–DIV linker of the channel, five residues downstream of the putative inactivation gate. This interaction can be disrupted by a positive charge at position 1784 and rescued with the K1493E/E1784K double mutant that abolishes the noninactivating current. However, the double mutant does not restore either the voltage dependence or rates of fast inactivation. Conversely, a mutant at the bottom of DIVS4, K1641D, causes a hyperpolarizing shift in the voltage dependence of fast inactivation and accelerates the rate of fast inactivation without causing an increase in noninactivating current. These findings provide novel mechanistic insights into how the most common inheritable arrhythmogenic mixed syndrome variant, E1784K, simultaneously decreases transient sodium currents and increases noninactivating currents, leading to both BrS1 and LQT3.
Introduction
Voltage-gated sodium channels are heterotetrameric channels formed by four domains (DI–DIV) each containing six α-helical transmembrane segments (S1–S6; Noda et al., 1984). The first four segments in each domain (S1–S4) form a voltage sensor, whereas the S5, S6, and extracellular S5–S6 linker of each domain form the channel pore. In response to membrane depolarization, the S4s move toward the extracellular membrane surface. This movement is transferred through the S4–S5 linkers to the channel pore, leading to channel opening and an inward sodium current that causes further depolarization.
Sodium currents are transient; after activation, the channel rapidly inactivates (Hodgkin and Huxley, 1952). Fast inactivation of sodium channels occurs in two steps: (1) movement of the fast inactivation voltage sensor, DIVS4; and (2) binding of the putative fast inactivation particle, a conserved IFM motif located in the intracellular DIII–DIV linker (Capes et al., 2013; West et al., 1992). Upon depolarization, DIVS4 moves outward and allows for IFM binding to a hydrophobic pocket at the bottom of DIIIS6 and DIVS5 and S6, occluding the pore and shutting off the sodium current (Jiang et al., 2020). Both the DIII–DIV linker and the DIV voltage sensor are sites for a number of Brugada syndrome type 1 (BrS1) and long-QT syndrome type 3 (LQT3) variants (Jiang et al., 2020). Generally, mutants that alter the movement of DIVS4 change the voltage dependence and rate of fast inactivation (Capes et al., 2013; Peters et al., 2017), whereas mutants in the DIII–DIV linker can both alter the rates of inactivation and cause a noninactivating/persistent sodium current (Patton et al., 1992; West et al., 1992).
A key regulator of channel fast inactivation is the sodium channel C terminus. Mutants in the C terminus alter the voltage dependence, rate, and stability of the fast-inactivated state (Bezzina et al., 1999; Deschênes et al., 2000). In the cardiac sodium channel, Nav1.5, disease-causing variants within the C terminus include the most prevalent BrS1 and LQT3 variant, E1784K (Makita et al., 2008). E1784K hyperpolarizes the voltage dependence of fast inactivation, accelerates fast inactivation onset and recovery, and increases the fraction of noninactivating/persistent sodium current (Wei et al., 1999; Deschênes et al., 2000; Makita et al., 2008; Abdelsayed et al., 2015; Peters et al., 2016; Veltmann et al., 2016; Gade et al., 2020). Structural and biochemical studies have suggested an interaction between the C terminus of the sodium channel and the DIII–DIV linker (Motoike et al., 2004; Wingo et al., 2004; Shah et al., 2006; Sarhan et al., 2012; Shen et al., 2017). This interaction is modified by the presence of calcium, fibroblast growth factors, and calmodulin; however, mutants in the C terminus modify inactivation in the absence of these modulatory factors (Wingo et al., 2004; Wei et al., 1999; Peters et al., 2016; Abdelsayed et al., 2017; Yan et al., 2017; Gade et al., 2020). The first eukaryotic sodium channel structure of the cockroach sodium channel, NavPas, shows extensive interactions between the proximal C terminus and not only the DIII–DIV linker, but also the cytoplasmic side of the DIVS4 voltage sensor (Shen et al., 2017). Interestingly, many of the negatively charged residues in the C terminus, including E1784E (E1423 in NavPas), are conserved between human voltage-gated sodium channels and NavPas, despite only 36–43% overall homology.
Although many studies have been published on the prevalence and biophysical impacts of the arrhythmogenic variant E1784K (Deschênes et al., 2000; Makita et al., 2008; Peters et al., 2016; Veltmann et al., 2016), until recently little was known about the mechanism by which E1784K, and other C-terminal variants associated with BrS1 and LQT3, lead to altered inactivation (Peters et al., 2017; Peters, 2017; Clairfeuille et al., 2019; Gade et al., 2020). We previously showed that, in E1784K, the DIVS4 moves at more hyperpolarized potentials (Peters et al., 2017). The corresponding hyperpolarization and accelerated onset of fast inactivation are sufficient to cause a depolarization of the conductance–voltage (GV) relationship (Peters et al., 2017). We also showed that E1784K is preferentially altered by the presence of extracellular protons but were unable to provide a mechanism. A later structure of a NavPas-Nav1.7 channel chimera bound to the α-scorpion toxin AaH2 confirmed the existence of a series of electrostatic interactions between the C terminus with both the DIII–DIV linker and the DIVS4 and showed that the interactions between the bottom of DIVS4 and the C terminus perform a key role in defining the rates and voltage dependence of fast inactivation (Clairfeuille et al., 2019). Most recently, Gade et al. (2020) found that E1784E interacts with K1493 in the DIII–DIV linker and that disrupting this interaction leads to an increase in persistent current that can be rescued when the interaction is restored in the opposite direction by the K1493E/E1784K double mutant. They also showed that this interaction is necessary for calmodulin-dependent regulation of persistent sodium currents (Gade et al., 2020). These results agree with a previous study showing that, relative to other LQT3 variants, E1784K is relatively insensitive to calcium-dependent reductions in persistent current (Abdelsayed et al., 2017). Together, these data support the model proposed by Clairfeuille et al. (2019) that C-terminal interactions with both the DIVS4 and DIII–DIV linker act as switches to control channel fast inactivation.
These studies leave unanswered questions about this channel region and the mechanism by which it modulates channel inactivation. Thus far, all Nav1.5 variants that cause a mixed phenotype of BrS1 and LQT3 lead to both an increase in persistent sodium current and a hyperpolarized voltage dependence or accelerated rate of fast inactivation in electrophysiological experiments (Bezzina et al., 1999; Zareba et al., 2001; Grant et al., 2002; Chen et al., 2005; Makita et al., 2008; Kanters et al., 2014). Furthermore, the C-terminal mutants that were studied at both interaction sites alter fast-inactivation voltage dependence or rate and the magnitude of persistent current (Peters et al., 2017; Clairfeuille et al., 2019; Gade et al., 2020). Whether these effects are due to separable mechanisms or are inherently linked has yet to be addressed. It is also unknown whether E1784K disrupts the C-terminal interactions with both the DIVS4 and DIII–DIV linker or the DIII–DIV linker alone. Because E1784K alters the movement of DIVS4, it may have a downstream effect of breaking the C-terminal interaction with DIVS4.
To further investigate the role of these two interaction sites in defining channel fast inactivation rates and persistent current, we studied a series of mutants introduced into the C terminus at position 1784, into the DIII–DIV linker at positions 1492, 1493, and 1500, and into the DIV voltage sensor at position 1641. Overall, our results suggest that E1784K alters fast inactivation kinetics and voltage dependence in a manner that is separable from its effects on persistent sodium current and reveals novel interactions between the C terminus and other sodium channel structures associated with fast inactivation.
Materials and methods
DNA constructs
The variants at position 1784 include negatively charged, positively charged, neutral, and aromatic residues. The E1784E and the E1784K constructs in pPol1 were used in a previous study from our laboratory. The E1784A point mutant was made by Ziwei Ding (Simon Fraser University). The other mutants at positions 1784, 1641, and 1493 were introduced into the pPol1 construct using a QuikChange Lightning kit. The K1492E/E1784K and K1500E/E1784K mutants were made by Applied Biological Materials. The C373F mutant was included in all plasmids to increase the tetrodotoxin (TTX) sensitivity of the channels and facilitate gating current recordings (Satin et al., 1992).
All DNA constructs were cloned in 10β Escherichia coli cells (New England Biolabs), purified using a Qiagen Midi-prep kit, and sequenced by either Eurofin MWG Operon sequencing service or Genewiz sequencing service. We used NotI to linearize all constructs. Transcription was performed using a T7 mMESSAGE mMACHINE high-yield capped RNA transcription kit (Ambion).
Oocyte preparation
The Xenopus laevis oocyte preparation has been published previously (Jones et al., 2011; Peters et al., 2017). All animal surgery and animal care procedures were performed in accordance with the policies of the Simon Fraser Animal Care Committee and the Canadian Council of Animal Care.
Data acquisition
We performed cut-open voltage clamp experiments as described previously (Peters et al., 2017). We used a CA-1B amplifier (Dagan Corp.) in the cut-open mode. Data were lowpass filtered at 10 kHz, digitized at 50 kHz, and recorded using Patchmaster (HEKA Electronik). We permeabilized cells via bottom bath perfusion with intracellular solution supplemented with 0.1% saponin. After a 10–60-s exposure, saponin-free intracellular solution was washed in.
For ionic current recordings, the extracellular solution contained (in mM) 96 NaCl, 4 KCl, 1 MgCl2, 2 CaCl2, and 5 HEPES. For ionic current recordings, the intracellular solution contained (in mM) 9.6 NaCl, 88 KCl, 11 EGTA, and 5 HEPES. For gating current recordings, the extracellular solution at pH 7.4 contained (in mM) 117.7 NMDG, 122.3 Mes, 10 HEPES, and 2 Ca(OH)2, and the extracellular solution at pH 6.0 contained (in mM) 70 NDMG, 170 Mes, 10 HEPES, and 2 Ca(OH)2. The ratio of NMDG to Mes was changed to make solutions at the correct pH of the same osmolarity. For gating current recordings, the intracellular solution contained (in mM) 120 NMDG, 120 Mes, 10 HEPES, and 2 EGTA. Mes was substituted in place of HEPES for ionic solutions at pH 6.0. HCl and NMDG were used to titrate ionic solutions to the desired pH, and Mes and NMDG were used for gating current solutions. Gating currents were recorded after addition of 20 µl of 50 µM TTX to the 0.5-ml top chamber, which yields a final TTX concentration of ≈2 µM. Data at low pH is matched in all cells to data at pH 7.4. In gating current experiments, TTX was applied after each replacement of the extracellular solution. Capacitance was compensated before recordings, and leak was subtracted using either a P/4 (ionic recordings) or P/8 (gating current recordings) protocol.
Voltage protocols
All voltage protocols have been previously described (Peters et al., 2017). To measure the voltage dependence of sodium conductance through the channel pore, we depolarized channels to voltages between −100 and +60 mV for 20 ms from a holding potential of −150 mV. Conductance was determined by dividing the peak current by the voltage minus the experimentally observed reversal potential. For comparisons of proton block, we compared absolute conductance values while our GV relationship comparisons were performed with normalized conductance. GV relationships were fitted by a single Boltzmann function.
To measure steady-state fast inactivation, we conditioned cells to voltages between −150 and −10 mV for 500 ms. After the conditioning pulse, currents were elicited by a depolarizing pulse to −10 mV. The peak currents were normalized and plotted versus conditioning potential. Steady-state fast inactivation curves were fitted by a single Boltzmann function.
We measured noninactivating current as the fraction of current remaining at the end of 100-ms depolarizations to voltages between −30 and 0 mV. To measure the fraction of channels that failed to inactivate, the persistent current was divided by the peak inward current at the same potential to show relative noninactivating current. For noninactivating currents, five traces were averaged for each measurement.
We measured fast inactivation recovery with a double pulse protocol. After a 500-ms depolarization to 0 mV, channels were allowed to recover for varying lengths of time at potentials between −130 and −70 mV, after which currents were measured by a pulse to −10 mV. Recovery time courses were fitted by a double exponential function with the fast time constant being analyzed as the time constant of fast inactivation.
We measured open-state fast inactivation by fitting the decay of macroscopic currents with a single exponential function. Closed-state fast inactivation was measured by depolarizing cells to −70 or −50 mV for varying amounts of time from a holding potential of −150 mV. The amount of current remaining was measured during a test pulse to −10 mV, and the normalized current versus onset time was fitted by a single exponential function at −70 mV and a double exponential equation at −50 mV.
Charge–voltage (QV) relationships were measured by 20-ms depolarizations to potentials between −150 mV and +40 mV from a holding potential of −150 mV. This was followed by a 20-ms hyperpolarization to −150 mV. We measured the activation of the S4 segments by integrating the outward gating currents during the initial depolarization steps to give the amount of charge moved at a given membrane potential. We fitted the resulting QV curve with a Boltzmann function.
We measured the voltage dependence of gating current deactivation by integrating the outward charge during a 20-ms depolarization to +50 mV after a 20-ms repolarization between 20 and −150 mV after a 20-ms depolarization to +50 mV. The amount of charge was plotted versus voltage and fitted with a Boltzmann function. Results were similar if gating charge return was measured as the inward charge during the repolarizing step. We measured ramp currents using a 0.15-mV/ms voltage ramp from −120 to +40 mV. Currents were normalized to the peak inward sodium current measured at −20 mV in the same cell.
Data analysis
All midpoint values are means, and measurements of error listed are SEM. All statistical analysis was performed using JMP statistical software. Comparisons of means were made using a two-factor repeated-measures ANOVA in which the main factors analyzed were mutant (nominal variable) and pH (repeated, scalar variable). A significant interaction between mutant and pH was interpreted as a difference in the proton effect on the dependent variable on at least one mutant relative to the others. Unless stated for a specific variant, P values reported in text are those of the main factors or interaction factor from the full ANOVA. P values from individual variants were compared with E1784E, using a Dunnet’s correction for multiple comparisons. In cases in which a similar gating parameter was measured at multiple membrane potentials (e.g., fast inactivation recovery), only the largest or smallest P value is reported for significant results or nonsignificant results, respectively. For comparisons of time constants, the log of the time constants was compared, as the log values are normally distributed. Tests of the K1493E, K1492E/E1784K, K1493E/E1784K, and K1500E/E1784K mutant against E1784K and E1784E channels at pH 7.4 were performed with a one-factor ANOVA followed by Tukey’s post hoc test. Comparisons of E1784E and K1641D channels were performed using Student’s t test. For measurements of correlation, the Pearson correlation coefficient is given. The test for a significant correlation was against the null hypothesis that the Pearson correlation coefficient is 0. Statistical significance was measured at α < 0.05.
Homology models
Molecular graphics and analyses were performed with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics (RBVI) at the University of California, San Francisco (supported by NIGMS P41-GM103311; Pettersen et al., 2004). The Homology model of the Nav1.5 sequence on the NavPaS structure was constructed using MODELLER software run at RBVI.
Nav1.5 current models
To simulate ionic currents from Nav1.5 channel variants, we used the model proposed in our previous article (Peters et al., 2017). The forward and reverse rates (α and β) for the DIII transition between the deactive (D) and active (A) states for all models were shifted to more hyperpolarized potentials to account for voltage-clamp fluorimetry data from DIII (Varga et al., 2015; Peters, 2017). The transition rates between the DIVD and DIVA states (α and β) and DIVA and DIVI states (FI and FR) were fitted to experimental inactivation voltage dependence, onset, and recovery, as well as the fraction of noninactivating current for each of the simulated channel variants. All other rates are the same as those previously reported (Peters et al., 2017). Rates that differ from those previously reported can be found in Table S9. All models were run in the Spyder Python environment. Graphing was performed in IGOR Pro. Voltage changes were simulated as an exponential time course with time constants from the fitting of capacitive transients in ionic current solutions.
Online supplemental material
Source data for all figures, including means, SEM, and number of independent recordings for each mutant at pH 7.4, pH 7.0, and pH 6.0, are found in Tables S1, Table S2, Table S3, Table S4, Table S5, Table S6, Table S7, and Table S8. Note that the number of independent recordings is smaller for E1784K, as its effects have been reported in many previous publications and our present results are consistent with the published data. Table S9 contains transition rates for the model in Fig. 8. Fig. S1 contains average ramp currents that are discussed in relation to the differences between window current and noninactivating current in Discussion.
Results
For Figs. 1, 2, 3, 4, and 5, position 1784 variants are shown using the following color scheme: the negatively charged amino acids, E1784E and E1784D, are shown as black and gray, respectively; positively charged amino acids, E1784K and E1784R, are shown as warm colors; neutral amino acids, E1784A, E1784Q, and E1784V, are shown as shades of blue; and the aromatic amino acid, E1784W, is shown in green.
Conductance
We measured the voltage dependence of channel conductance from macroscopic inward sodium currents at membrane potentials between −100 and +40 mV. The fraction of maximal conductance at pH 6.0 compared with pH 7.4 in the same cell was used to measure proton block of the sodium current. Sample current traces for each position 1784 mutant are shown in Fig. 1 A. There is a significant mutant-dependent effect on the midpoint of the GV relationship (P = 0.001), with both E1784V and E1784R being significantly more depolarized than E1784E at pH 7.4 (P = 0.0004 and P = 0.0118, respectively; Fig. 1 B). Similar to previous data (Peters et al., 2017; Deschênes et al., 2000; Gade et al., 2020; Abdelsayed et al., 2015), E1784K shows a trend toward depolarization compared with E1784E (P = 0.1554; Fig. 1 B). There is also a significant pH-dependent depolarization of the GV midpoint in all position 1784 variants (P < 0.0001; Fig. 1 C). Decreased extracellular pH significantly reduces the maximal conductance in all mutants (P < 0.0001; Fig. 1 D). The proton-dependent block of maximal conductance is significantly different between channel variants (P = 0.0016), ranging from 24% in E1784W to 40% in E1784E and 48% in E1784K at pH 6.0.
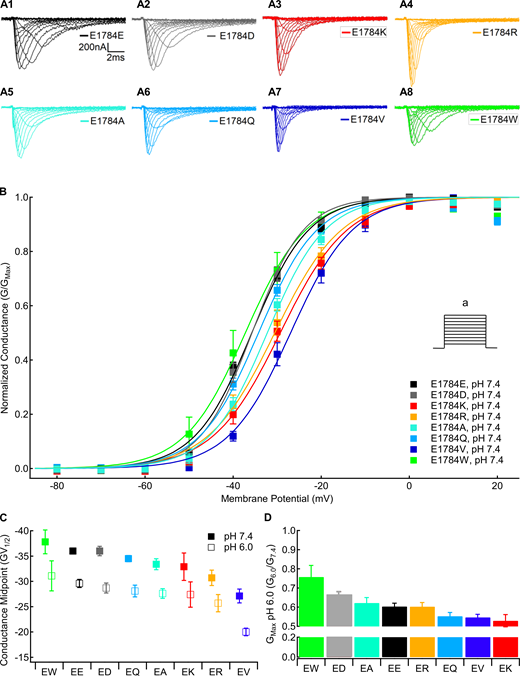
Mutants at residue 1784 in Nav1.5 alter the voltage dependence of conductance and proton block of maximal conductance.(A) Sample macroscopic sodium currents recorded in residue 1784 mutants at extracellular pH 7.4. (B) Normalized GV relationships of residue 1784 mutants at extracellular pH 7.4. Conductance was determined from peak currents measured during depolarizations between −100 and 40 mV (a) from a prepulse potential of −150 mV. (C) Average midpoints of the GV relationships for residue 1784 mutants at extracellular pH 7.4 and pH 6.0 (n = 5–10; Table S1). (D) Fraction of conductance remaining at extracellular pH 6.0 in residue 1784 mutants (n = 5–10). Error bars are SEM.
Mutants at residue 1784 in Nav1.5 alter the voltage dependence of conductance and proton block of maximal conductance.(A) Sample macroscopic sodium currents recorded in residue 1784 mutants at extracellular pH 7.4. (B) Normalized GV relationships of residue 1784 mutants at extracellular pH 7.4. Conductance was determined from peak currents measured during depolarizations between −100 and 40 mV (a) from a prepulse potential of −150 mV. (C) Average midpoints of the GV relationships for residue 1784 mutants at extracellular pH 7.4 and pH 6.0 (n = 5–10; Table S1). (D) Fraction of conductance remaining at extracellular pH 6.0 in residue 1784 mutants (n = 5–10). Error bars are SEM.
Fast inactivation
We measured the voltage dependence of fast inactivation using 500-ms prepulses to membrane potentials between −150 and −10 mV, followed by a test pulse to −10 mV. There is a significant mutant-dependent shift in the midpoint of the fast inactivation voltage dependence (P < 0.0001; Fig. 2 A). At pH 7.4, E1784K, E1784R, and E1784Q are shifted toward significantly more hyperpolarized membrane potentials compared with E1784E (P ≤ 0.0289 in all cases), whereas E1784V shows a nonsignificant trend (P = 0.0772; Fig. 2 A, inset). In contrast, the voltage dependence of fast inactivation in both E1784A and E1784W are shifted toward significantly more depolarized potentials compared with E1784E (P = 0.0163 and P < 0.0001, respectively; Fig. 2 A, inset).
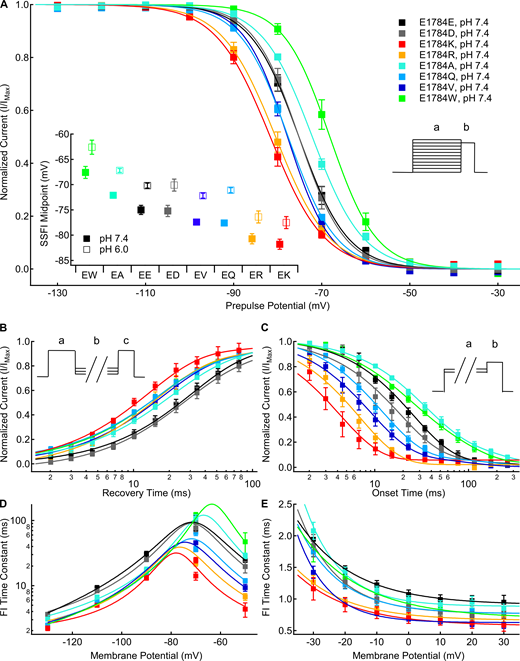
Mutants at residue 1784 in Nav1.5 alter the rates and voltage dependence of fast inactivation.(A) Average voltage dependence of fast inactivation in residue 1784 mutants at extracellular pH 7.4. The voltage dependence of fast inactivation was determined by measuring current remaining during a test pulse to −10 mV (b) following conditioning pulses to voltages between −150 and −10 mV (a). Inset: Average midpoints of the steady-state fast inactivation (SSFI) voltage dependence for residue 1784 mutants at pH 7.4 and pH 6.0 (n = 5–9; Table S2). (B) Average time course of fast inactivation recovery at −90 mV in residue 1784 mutants at extracellular pH 7.4 (n = 3–5; Table S3). Rates of recovery from fast inactivation were determined by measuring current during a test pulse (c) following recovery pulses of varying lengths (b) after an initial depolarizing pulse (a). (C) Average time course of fast inactivation onset at −50 mV in residue 1784 mutants at extracellular pH 7.4 (n = 3–5; Table S4). The rates of closed-state fast inactivation onset were determined by measuring the current remaining in a test pulse (b) following conditioning pulses of varying durations (a). (D) Average time constants of fast inactivation (FI) recovery and closed-state onset plotted versus voltage for residue 1784 mutants at extracellular pH 7.4 (n = 3–5; Tables S3 and Table S4). (E) Average time constants of open-state fast inactivation onset plotted versus voltage for residue 1784 mutants at extracellular pH 7.4 (n = 4–9; Table S5). Error bars are SEM.
Mutants at residue 1784 in Nav1.5 alter the rates and voltage dependence of fast inactivation.(A) Average voltage dependence of fast inactivation in residue 1784 mutants at extracellular pH 7.4. The voltage dependence of fast inactivation was determined by measuring current remaining during a test pulse to −10 mV (b) following conditioning pulses to voltages between −150 and −10 mV (a). Inset: Average midpoints of the steady-state fast inactivation (SSFI) voltage dependence for residue 1784 mutants at pH 7.4 and pH 6.0 (n = 5–9; Table S2). (B) Average time course of fast inactivation recovery at −90 mV in residue 1784 mutants at extracellular pH 7.4 (n = 3–5; Table S3). Rates of recovery from fast inactivation were determined by measuring current during a test pulse (c) following recovery pulses of varying lengths (b) after an initial depolarizing pulse (a). (C) Average time course of fast inactivation onset at −50 mV in residue 1784 mutants at extracellular pH 7.4 (n = 3–5; Table S4). The rates of closed-state fast inactivation onset were determined by measuring the current remaining in a test pulse (b) following conditioning pulses of varying durations (a). (D) Average time constants of fast inactivation (FI) recovery and closed-state onset plotted versus voltage for residue 1784 mutants at extracellular pH 7.4 (n = 3–5; Tables S3 and Table S4). (E) Average time constants of open-state fast inactivation onset plotted versus voltage for residue 1784 mutants at extracellular pH 7.4 (n = 4–9; Table S5). Error bars are SEM.
The rates of fast inactivation recovery and closed-state fast inactivation onset were measured using double pulse protocols (Fig. 2, B and C, insets). Time courses of fast inactivation recovery at −90 mV are shown for all position 1784 mutants in Fig. 2 B. Time courses of closed state fast inactivation onset are shown for all position 1784 mutants at −50 mV in Fig. 2 C. The time constants of fast inactivation are plotted versus voltage in Fig. 2 D. There was a significant mutant effect on the time constant of fast inactivation recovery and onset at all membrane potentials (P < 0.0001). Except for E1784D (P = 0.1054), at pH 7.4 all the position 1784 variants significantly speed the rate of fast inactivation recovery at −90 mV compared with E1784E (P < 0.0001 in all cases), with E1784K being the most rapid (Fig. 2 B). At −50 mV and pH 7.4, E1784K, E1784Q, E1784R, and E1784V speed the rate of inactivation onset compared with E1784E (P ≤ 0.0029 in all cases; Fig. 2 C). Typically, onset and recovery from fast inactivation are most rapid for E1784K, whereas the recovery from inactivation is slowest in E1784E and E1784D, and the onset of inactivation is slowest in E1784A and E1784W (Fig. 2 D).
We measured the rate of open-state fast inactivation by fitting a single exponential function to macroscopic current decay at membrane potentials between −30 and +30 mV. The time constants of open-state fast inactivation are plotted versus voltage for all position 1784 mutants in Fig. 2 E. There is a significant mutant-dependent effect (P ≤ 0.0091) on the time constant of open-state fast inactivation at all membrane potentials. At pH 7.4, E1784K, E1784R, and E1784V inactivate significantly faster than E1784E at all potentials from −30 to +20 mV (P ≤ 0.013 in all cases). These three variants are the fastest to inactivate across the voltage range, whereas E1784E and E1784A typically show the slowest inactivation.
Gating charge activation and deactivation
We measured the voltage dependence of gating charge activation (QV) in position 1784 mutants by integrating the outward gating current during 20-ms depolarizations from a holding potential of −150 mV to membrane potentials between −150 and +40 mV. This was followed by a 20-ms repolarization to −150 mV. Sample gating currents are shown in Fig. 3 A1. We measured the voltage dependence of gating current deactivation in position 1784 mutants using 20-ms repolarizations between 20 and −150 mV after a depolarization to +50 mV. The amount of returning charge was assessed by integrating the outward gating currents during subsequent depolarizing pulses to +50 mV. Sample deactivating gating currents are shown in Fig. 3 A2. There are significant mutant-dependent shifts in the midpoint of the activation QV relationship (Fig. 3, B and D) and deactivation-voltage relationship (P < 0.0001 in both cases; Fig. 3, C and D). E1784K, E1784Q, E1784R, and E1784V shift the QV relationship to significantly more hyperpolarized potentials compared with E1784E (P ≤ 0.0308 in all cases), whereas E1784W shows a nonsignificant trend toward more depolarized potentials (P = 0.1832; Fig. 3 D).
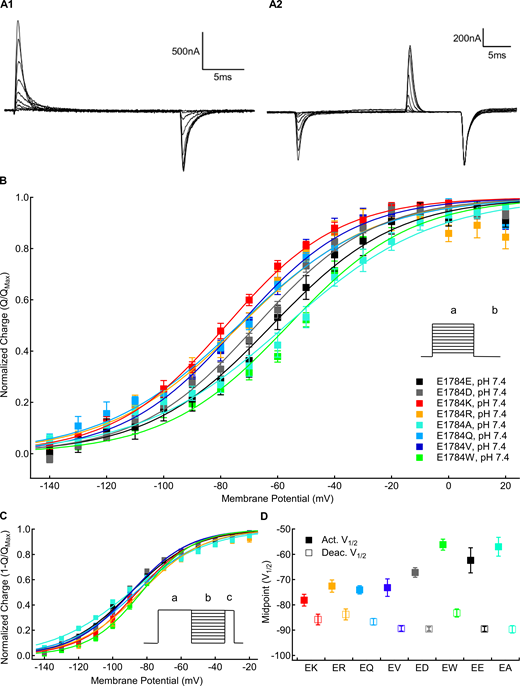
Mutants at residue 1784 alter gating current voltage dependence.(A1) Sample gating currents recorded with the activation protocol described in the text. (A2) Sample gating currents recorded with the deactivation protocol described in the text. (B) Average voltage dependence of gating charge activation for residue 1784 mutants recorded at extracellular pH 7.4 (n = 4–6). Outward QVs were determined by integrating the charge moved during depolarizations between −150 and +60 mV (a) from a holding potential of −150 mV. (C) Average voltage dependence of gating charge deactivation for residue 1784 mutants recorded at extracellular pH 7.4 (n = 4–5). To measure the voltage dependence of gating current deactivation, cells were hyperpolarized to membrane potentials between −150 and +20 mV (b) following a depolarization pulse to +50 mV (a). This was followed by depolarization to +50 mV (c). (D) Average midpoints of gating charge activation and deactivation for residue 1784 mutants at extracellular pH 7.4 (n = 4–6; Tables S6 and Table S7). Error bars are SEM.
Mutants at residue 1784 alter gating current voltage dependence.(A1) Sample gating currents recorded with the activation protocol described in the text. (A2) Sample gating currents recorded with the deactivation protocol described in the text. (B) Average voltage dependence of gating charge activation for residue 1784 mutants recorded at extracellular pH 7.4 (n = 4–6). Outward QVs were determined by integrating the charge moved during depolarizations between −150 and +60 mV (a) from a holding potential of −150 mV. (C) Average voltage dependence of gating charge deactivation for residue 1784 mutants recorded at extracellular pH 7.4 (n = 4–5). To measure the voltage dependence of gating current deactivation, cells were hyperpolarized to membrane potentials between −150 and +20 mV (b) following a depolarization pulse to +50 mV (a). This was followed by depolarization to +50 mV (c). (D) Average midpoints of gating charge activation and deactivation for residue 1784 mutants at extracellular pH 7.4 (n = 4–6; Tables S6 and Table S7). Error bars are SEM.
Notably, the ordering of mutants differs between gating current activation and deactivation (Fig. 3 D). E1784K activates at the most hyperpolarized potential but has the third most depolarized voltage dependence of deactivation. In contrast, E1784A has the second most depolarized voltage dependence of gating current activation and the most hyperpolarized gating charge deactivation. Thus, whereas E1784K and E1784R have ∼7 mV of hysteresis between gating charge activation and deactivation, E1784E, E1784A, and E1784W approach 30 mV of hysteresis.
Noninactivating current
To test for mutant-dependent changes in the completeness of fast inactivation, we measured persistent sodium current at the end of 100-ms depolarizations to membrane potentials between −30 and 0 mV (Fig. 4 A, inset). Five traces were averaged for each recording. Currents were normalized to the peak inward current to compare the fraction of noninactivating current (Fig. 4 A). There is a significant mutant-dependent effect on the normalized persistent current at all measured membrane potentials (P < 0.0001 in all cases; Fig. 4 B). At all membrane potentials, E1784K and E1784R have significantly larger fractions of noninactivating current compared with E1784E at pH 7.4 (P ≤ 0.0086 in all cases). The fraction of noninactivating sodium current is similar to E1784E in all other mutants. E1784Q had no discernable noninactivating sodium current in many recordings. There is a significant interaction between decreasing extracellular pH and mutant at all membrane potentials (P ≤ 0.0160). The fraction of noninactivating current is significantly increased in E1784K and E1784R by decreasing extracellular pH at all membrane potentials (P ≤ 0.0005 in all cases; Fig. 4 C). This is not the case for the other mutants.
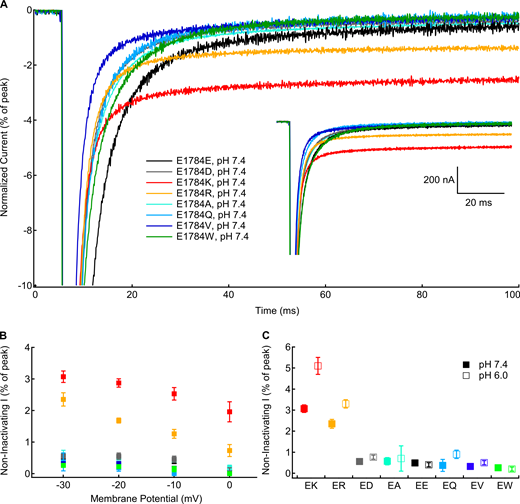
Positively charged mutants at residue 1784 increase noninactivating current.(A) Sample noninactivating currents recorded at −10 mV from residue 1784 mutants at pH 7.4 normalized to peak current. Inset: Unnormalized currents presented in A. (B) Average noninactivating current as a percentage of peak current for residue 1784 mutants recorded between −30 and 0 mV at extracellular pH 7.4 (n = 4–5; Table S8). (C) Average noninactivating current at −30 mV as a percentage of peak current for residue 1784 mutants at extracellular pH 7.4 and pH 6.0 (n = 4–5; Table S8). Error bars are SEM.
Positively charged mutants at residue 1784 increase noninactivating current.(A) Sample noninactivating currents recorded at −10 mV from residue 1784 mutants at pH 7.4 normalized to peak current. Inset: Unnormalized currents presented in A. (B) Average noninactivating current as a percentage of peak current for residue 1784 mutants recorded between −30 and 0 mV at extracellular pH 7.4 (n = 4–5; Table S8). (C) Average noninactivating current at −30 mV as a percentage of peak current for residue 1784 mutants at extracellular pH 7.4 and pH 6.0 (n = 4–5; Table S8). Error bars are SEM.
Correlations
We previously showed that the hyperpolarization of the gating charge activation voltage dependence in E1784K is due to the hyperpolarized movement of the DIVS4 voltage sensor (Peters et al., 2017). Because the DIVS4 acts as the voltage sensor for fast inactivation, we predicted that the midpoint of our activation QV curve would correlate to the midpoint of fast inactivation voltage dependence if the shifts in the DI–DIII voltage sensors are relatively minimal. From most hyperpolarized to most depolarized, the midpoints of fast inactivation voltage dependence and gating charge activation at pH 7.4 were K < R < Q < V < D < E < A < W and K < Q < V < R < D < E < A < W, respectively (Fig. 5 B). There is a significant correlation between the midpoint of the fast inactivation voltage dependence curve and the QV curve at pH 7.4 (R = 0.92, P = 0.0011). Notably, the correlation between the midpoints of the GV and QV curves is not significant, and the correlation coefficient is negative, indicating that depolarized conductance occurred with hyperpolarized charge movement (R = −0.5658, P = 0.1438; Fig. 5 A).

The voltage dependence of gating charge movement correlates to the voltage dependence of fast inactivation but not conductance. (A) Midpoint of the GV relationship for residue 1784 mutants plotted versus the midpoint of the QV relationship (n = 4–10). (B) Midpoint of the voltage dependence of fast inactivation for residue 1784 mutants plotted versus the midpoint of the QV relationship (n = 4–9). (C) Midpoint of the GV relationship for residue 1784 mutants plotted versus the time constant of open state fast inactivation at −10 mV (n = 4–10). (D) Fraction of maximal conductance blocked by decreasing extracellular pH to pH 6.0 for residue 1784 mutants plotted versus the time constant of closed-state inactivation onset at −50 mV (n = 3–10).
The voltage dependence of gating charge movement correlates to the voltage dependence of fast inactivation but not conductance. (A) Midpoint of the GV relationship for residue 1784 mutants plotted versus the midpoint of the QV relationship (n = 4–10). (B) Midpoint of the voltage dependence of fast inactivation for residue 1784 mutants plotted versus the midpoint of the QV relationship (n = 4–9). (C) Midpoint of the GV relationship for residue 1784 mutants plotted versus the time constant of open state fast inactivation at −10 mV (n = 4–10). (D) Fraction of maximal conductance blocked by decreasing extracellular pH to pH 6.0 for residue 1784 mutants plotted versus the time constant of closed-state inactivation onset at −50 mV (n = 3–10).
We also showed that the depolarized midpoint of the GV curve in E1784K is due to the altered fast inactivation (Peters et al., 2017). Thus, we predicted that the midpoint of the GV curve would correlate to fast inactivation parameters. At pH 7.4, there is a significant correlation between the midpoint of the GV curve and the time constants of open-state fast inactivation at −20, −10, 0, and 10 mV (R ≥ 0.71, P ≤ 0.0468; shown at −10 mV in Fig. 5 C). Similarly, the proton block of maximal conductance at pH 6.0 is correlated with the rate of closed-state fast inactivation at −70 and −50 mV (R = 0.79 and P = 0.0201 at −70 mV; R = 0.73 and P = 0.0398 at −50 mV; Fig. 5 D).
Interactions between the C terminus and the DIII–DIV linker and DIVS4
Similar to the previous study on K1493E (Gade et al., 2020), our homology model of Nav1.5 built on the American cockroach structure in the closed, noninactivated state (Shen et al., 2017) shows that residue E1784E comes within 5 Å of residue K1493 in the DIII–DIV linker (Fig. 6, A and C). To confirm that noninactivating currents in E1784K are due to the disruption of an interaction between E1784E and K1493 (Gade et al., 2020), we made the K1493E/E1784K double mutant to restore the potential electrostatic interaction in the opposite direction. Sample traces of E1784E, E1784K, K1493E, and K1493E/E1784K noninactivating currents at −20 mV are shown in Fig. 7 A. Unlike previous data, K1493E does not increase the fraction of noninactivating current compared with E1784E (P ≥ 0.5263 between −20 and 0 mV; Fig. 7 B; Gade et al., 2020). The K1493E/E1784K double mutant has significantly smaller noninactivating currents than E1784K at −30, −20, and −10 mV (P < 0.0001 in all cases; Fig. 7 B). K1493E/E1783K noninactivating currents are not significantly different from E1784E channels at any voltage (P > 0.1812). We also measured noninactivating current for two other mutants in the DIII–DIV linker, K1492E/E1784K and K1500E/E1784K, at −20 mV. K1500E/E1784K has significantly larger noninactivating currents than E1784E (P < 0.0001; Fig. 7 B). In contrast to previous results, the fraction of noninactivating current in K1492E/E1784K does not significantly differ from either E1784E or K1493E/E1784K (P = 0.6834 and P = 0.9767, respectively) but has significantly less noninactivating current at −20 mV compared with E1784K (P < 0.0001; Fig. 7 B; Gade et al., 2020).

A homology model of Nav1.5 suggests that E1784E associates with K1493 in the DIII–DIV linker. (A) Homology model of the Nav1.5 sequence on the NavPaS structure. The C terminus, DIII–DIV linker, and DIVS4–S5 linker are colored pink, blue, and green, respectively. Black and red boxes show the channel region shown in B and C, respectively. (B) In the homology model, the C terminus of the channel (in pink) is folded back toward the underside of the DIV voltage sensor (in green). (C) In the homology model of Nav1.5, residue E1784E comes within 5 Å of residue K1493 in the DIII–DIV linker. All residues with atoms with 5 Å of E1784E are shown as sticks (PDB accession no. 5X0M; Shen et al., 2017).
A homology model of Nav1.5 suggests that E1784E associates with K1493 in the DIII–DIV linker. (A) Homology model of the Nav1.5 sequence on the NavPaS structure. The C terminus, DIII–DIV linker, and DIVS4–S5 linker are colored pink, blue, and green, respectively. Black and red boxes show the channel region shown in B and C, respectively. (B) In the homology model, the C terminus of the channel (in pink) is folded back toward the underside of the DIV voltage sensor (in green). (C) In the homology model of Nav1.5, residue E1784E comes within 5 Å of residue K1493 in the DIII–DIV linker. All residues with atoms with 5 Å of E1784E are shown as sticks (PDB accession no. 5X0M; Shen et al., 2017).
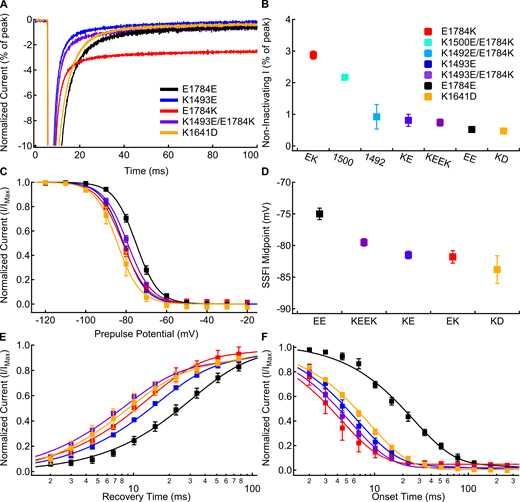
The K1493E/E1784K double mutant abolishes noninactivating current, but not the changes to fast inactivation voltage dependence or rates.(A) Sample currents recorded at −20 mV from E1784E (black), K1493E (blue), E1784K (red), K1493E/E1784K (purple), and K1641D (orange) channels at extracellular pH 7.4 normalized to the peak inward current. Sample currents shown were from cells with similar peak sodium currents. (B) Average noninactivating current as a percent of peak current for E1784E (EE), K1493E (KE), E1784K (EK), K1492E/E1784K (1492), K1500E/E1784K (1500), K1493E/E1784K (KEEK), and K1641D (KD) channels recorded at −20 mV at pH 7.4 (n = 3–7; Table S8). (C) Average voltage dependence of fast inactivation in E1784E, K1493E, E1784K, K1493E/E1784K, and K1641D channels at extracellular pH 7.4. (D) Average midpoints of the fast inactivation voltage dependence for E1784E, K1493E, E1784K, K1493E/E1784K, and K1641D at pH 7.4 (n = 5–9; Table S2). (E) Average time course of fast inactivation recovery at −90 mV in E1784E, K1493E, E1784K, K1493E/E1784K, and K1641D channels at extracellular pH 7.4 (n = 3–7; Table S3). (F) Average time course of fast inactivation onset at −50 mV in E1784E, K1493E, E1784K, K1493E/E1784K, and K1641D channels at extracellular pH 7.4 (n = 3–7; Table S4). Error bars are SEM.
The K1493E/E1784K double mutant abolishes noninactivating current, but not the changes to fast inactivation voltage dependence or rates.(A) Sample currents recorded at −20 mV from E1784E (black), K1493E (blue), E1784K (red), K1493E/E1784K (purple), and K1641D (orange) channels at extracellular pH 7.4 normalized to the peak inward current. Sample currents shown were from cells with similar peak sodium currents. (B) Average noninactivating current as a percent of peak current for E1784E (EE), K1493E (KE), E1784K (EK), K1492E/E1784K (1492), K1500E/E1784K (1500), K1493E/E1784K (KEEK), and K1641D (KD) channels recorded at −20 mV at pH 7.4 (n = 3–7; Table S8). (C) Average voltage dependence of fast inactivation in E1784E, K1493E, E1784K, K1493E/E1784K, and K1641D channels at extracellular pH 7.4. (D) Average midpoints of the fast inactivation voltage dependence for E1784E, K1493E, E1784K, K1493E/E1784K, and K1641D at pH 7.4 (n = 5–9; Table S2). (E) Average time course of fast inactivation recovery at −90 mV in E1784E, K1493E, E1784K, K1493E/E1784K, and K1641D channels at extracellular pH 7.4 (n = 3–7; Table S3). (F) Average time course of fast inactivation onset at −50 mV in E1784E, K1493E, E1784K, K1493E/E1784K, and K1641D channels at extracellular pH 7.4 (n = 3–7; Table S4). Error bars are SEM.
Similar to previous data (Gade et al., 2020), K1493E shifts the voltage dependence of fast inactivation (Fig. 7, C and D) to significantly more hyperpolarized potentials compared with E1784E (P < 0.0001). K1493E also significantly accelerates the rates of fast inactivation recovery (Fig. 7 E) and onset (Fig. 7 F) compared with E1784E (P < 0.0001 and P < 0.0001, respectively). Although K1493E/E1784K rescues noninactivating currents to WT levels, the voltage dependence and rates of fast inactivation are still shifted to similar levels as in E1784K. The midpoint of fast inactivation voltage dependence is shifted to significantly more hyperpolarized potentials in K1493E/E1784K compared with E1784E (Fig. 7, C and D; P = 0.0005). Similarly, the rates of fast inactivation recovery at −90 mV (Fig. 7 E) and fast inactivation onset at −50 mV (Fig. 7 F) are significantly accelerated in K1493E/E1784K compared with those in E1784E (P < 0.0001 and P < 0.0001, respectively).
Our homology model also suggests electrostatic interactions between the α1 helix of the C terminus and the cytoplasmic end of DIVS4, as previously reported (Fig. 6 B,; Clairfeuille et al., 2019). The previous study showed that mutants in the C terminus at this interaction site lead to increases in persistent current in addition to their role in hyperpolarizing the voltage dependence and accelerating the rate of fast inactivation (Clairfeuille et al., 2019). However, this may be due to the proximity of this region of the C terminus to the DIII–DIV linker as opposed to just a disruption of the interaction with DIVS4. We therefore tested the effects of mutating the corresponding site at the cytoplasmic end of DIVS4, K1641D. We also attempted to restore this interaction with the K1641D/D1790K mutant, but found that it does not produce measurable currents. As expected, K1641D causes a significant depolarization of the GV dependence (P < 0.0001; Table S1), a significant hyperpolarization of the voltage dependence of fast inactivation (P = 0.0008; Fig. 7, C and D), a significant acceleration of fast inactivation recovery at −90 mV (P = 0.0005; Fig. 7 E), and a significant acceleration of fast inactivation onset at −50 mV (P < 0.0001; Fig. 7 F); however, K1641D does not significantly increase the noninactivating sodium current at −20 mV (P = 0.6612; Fig. 7, A and B).
Nav1.5 variant models suggest that noninactivating current in E1784K is mechanistically distinct from the altered voltage dependence and rates of fast inactivation
In sodium channels, the fast inactivation process incorporates at least two distinct steps: the voltage-dependent activation of the DIVS4 voltage sensor (Capes et al., 2013) and the voltage-independent binding of the IFM motif in the DIII–DIV linker that occludes the channel pore (Fig. 8 A; Aldrich et al., 1983; West et al., 1992; Jiang et al., 2020). To further elucidate the mechanisms by which channel variants cause noninactivating currents, we modified our previously described model of Nav1.5 gating to simulate the fast inactivation process of E1784E, E1784K, E1784V, E1784W, K1492E/E1784K, and K1641D channels (Peters et al., 2017). This model explicitly simulates each of the four voltage sensors as four-state models with the pore conducting current when the first three voltage sensors are in their active state and the DIV voltage sensor is not in the inactive state.

E1784K alters voltage-dependent and voltage-independent rates of the sodium channel fast inactivation process. (A) Schematic model of two-step sodium channel fast inactivation. The first step is the voltage-dependent activation (white to black) of the DIV voltage sensor. The activation and deactivation rates of this first step determine the voltage dependence, recovery rate, and onset rate of fast inactivation. The second step is the voltage-independent binding of the IFM motif to occlude the channel pore. The ratio of forward and reverse binding rates for IFM determines the fraction of noninactivating current in E1784K. In addition, the forward binding rate determines the maximal fast-inactivation rate at highly depolarized potentials. (B) Simulated macroscopic currents for E1784E (black), E1784K (red), E1784V (blue), E1784W (green), K1493E/E1784K (purple), and K1641D (orange) models in response to depolarizing steps between −100 and +40 mV from a holding potential of −130 mV. (C) Simulated (lines; Sim.) and experimental (symbols; Exp.) voltage dependence of fast inactivation for Nav1.5 variants. (D) Simulated noninactivating currents for Nav1.5 variants at −20 mV normalized to peak inward current. (E) Simulated (lines) and experimental (symbols) recovery time courses from fast inactivation for Nav1.5 variants. (F) Simulated (lines) and experimental (symbols) onset time courses of fast inactivation for Nav1.5 variants.
E1784K alters voltage-dependent and voltage-independent rates of the sodium channel fast inactivation process. (A) Schematic model of two-step sodium channel fast inactivation. The first step is the voltage-dependent activation (white to black) of the DIV voltage sensor. The activation and deactivation rates of this first step determine the voltage dependence, recovery rate, and onset rate of fast inactivation. The second step is the voltage-independent binding of the IFM motif to occlude the channel pore. The ratio of forward and reverse binding rates for IFM determines the fraction of noninactivating current in E1784K. In addition, the forward binding rate determines the maximal fast-inactivation rate at highly depolarized potentials. (B) Simulated macroscopic currents for E1784E (black), E1784K (red), E1784V (blue), E1784W (green), K1493E/E1784K (purple), and K1641D (orange) models in response to depolarizing steps between −100 and +40 mV from a holding potential of −130 mV. (C) Simulated (lines; Sim.) and experimental (symbols; Exp.) voltage dependence of fast inactivation for Nav1.5 variants. (D) Simulated noninactivating currents for Nav1.5 variants at −20 mV normalized to peak inward current. (E) Simulated (lines) and experimental (symbols) recovery time courses from fast inactivation for Nav1.5 variants. (F) Simulated (lines) and experimental (symbols) onset time courses of fast inactivation for Nav1.5 variants.
To model position 1784 variants and K1641D, the transition rates of the DIV voltage sensor were modified to fit experimental fast inactivation data (Table S9; see Materials and methods). Simulated current–voltage relationships for E1784E, E1784K, E1784V, E1784W, K1493E/E1784K, and K1641D are shown in Fig. 8 B. The experimentally observed voltage dependence (Fig. 8 C), recovery (Fig. 8 E), and onset (Fig. 8 F) of fast inactivation in E1784V, E1784W, and K1641D are simulated with changes to the voltage-dependent activation and deactivation rates of the DIV voltage sensor and the voltage-independent forward rate of IFM binding; however, modification of these rates alone is not sufficient to induce noninactivating current in these models (Fig. 8 D).
Noninactivating currents in E1784K are induced only by increasing the ratio of the voltage-independent reverse rates for IFM unbinding compared with the forward rate. This is further illustrated by the K1493E/E1784K model that uses the same rates as the E1784K model for all transitions except the unbinding rate of the IFM motif, for which it uses the WT rate. The K1493E/E1784K model replicates the experimentally observed hyperpolarized voltage dependence (Fig. 8 C), accelerated recovery (Fig. 8 E), and accelerated onset (Fig. 8 F) of fast inactivation, but does not induce noninactivating current (Fig. 8 D).
Discussion
Inheritable and de novo variants in Nav1.5 are typically associated with either LQT3 or BrS1. Whereas LQT3 is caused by gain-of-function variants that increase the noninactivating sodium current, BrS1 is caused by loss-of-function variants that decrease the peak sodium transient current (Wang et al., 1995, 1996; Chen et al., 1998). Interestingly, a subset of sodium channel variants cause both LQT3 and BrS1. To date, six such variants have been characterized in electrophysiology experiments: the DIII–DIV linker variants ΔK1500 and ΔKPQ1505-1507; ΔF1617 in the DIV voltage sensor; and E1784K, L1786Q, and 1795insD in the C terminus of Nav1.5 (Bezzina et al., 1999; Zareba et al., 2001; Grant et al., 2002; Chen et al., 2005; Makita et al., 2008; Kanters et al., 2014). Of these mixed syndrome variants, E1784K is the most common. It is notable that all these variants cause noninactivating sodium currents and hyperpolarize the voltage dependence of fast inactivation or accelerate the rate of fast inactivation. Furthermore, each of these variants is located within regions of the channel previously associated with fast inactivation.
The proximal C terminus is a key regulator of sodium channel fast inactivation and expression. Approximately one third of the initial 30 amino acids of the C terminus are negatively charged glutamates or aspartates. Changing these negative charges disrupts the voltage dependence, rate, and stability of fast inactivation (Bezzina et al., 1999; Deschênes et al., 2000; Baroudi and Chahine, 2000; Veldkamp et al., 2000; Makita et al., 2008; Clairfeuille et al., 2019; Gade et al., 2020). However, it is not known if altered inactivation voltage dependence and level of noninactivating current are due to a single impairment of the channel structure or multiple disrupted interactions. We previously showed that E1784K shifts the movement of DIVS4 to more hyperpolarized potentials and that the subsequent hyperpolarization and acceleration of fast inactivation is sufficient to decrease the transient sodium current (Peters et al., 2017). In addition, we showed that decreasing extracellular pH preferentially decreases the transient sodium current and increases the fraction of noninactivating channels in the E1784K mutant (Peters et al., 2016, 2017). More recently, structural and biophysical studies have proposed a series of interactions between the C terminus of the channel and the DIVS4 and DIII–DIV linker (Shen et al., 2017; Clairfeuille et al., 2019; Gade et al., 2020).
Here, we use a series of amino acid substitutions to probe the underlying pathophysiological mechanism by which E1784K disrupts channel function. The data presented here further support prior results showing a charge-dependent interaction between a C terminus residue, E1784E, and a DIII–DIV residue, K1493 (Gade et al., 2020). However, restoration of this interaction in the opposite direction does not rescue the hyperpolarized voltage dependence or accelerated rates of inactivation seen in E1784K. Furthermore, many of the mutants at position 1784 alter the voltage dependence or rates of inactivation without altering the fraction of persistent current. In addition, models of different channel variants suggest that changes to the voltage dependence of DIVS4 do not induce noninactivating current. Combined, these data suggest that E1784K-dependent increases in persistent current and altered rates and voltage dependence of fast inactivation likely occur through separate structural interactions.
Fast inactivation, gating charge movement, and channel conductance in E1784K
Our prior experiments showed that, despite the depolarizing shift in the GV curve of E1784K, the QV curve is shifted to more hyperpolarized membrane potentials (Peters et al., 2017). The QV relationship, reflecting the combined membrane translocation of all four S4s, is typically thought of as a reflection of channel activation; however, in contrast to potassium channels, the activation of sodium channels primarily results from movement of only three of the S4 segments (DI–DIII), whereas the movement of DIVS4 is the rate-limiting step for Nav fast inactivation (Chanda and Bezanilla, 2002; Capes et al., 2013); thus, shifts in the voltage dependence of fast inactivation should also be reflected by shifts in the QV relationship. Indeed, we find that there is a strong positive correlation (R = 0.92) between the midpoint of the QV curve and the midpoint of channel fast inactivation. Thus, a hyperpolarized channel fast inactivation caused by movement of DIVS4 at more negative potentials leads to a hyperpolarization of the QV relationship. In contrast, the correlation between the midpoint of the GV and QV relationships is not significant and is negative. Although both mutant and structural studies suggest significant coupling between voltage sensors (Chanda et al., 2004; Jiang et al., 2020) and we previously found that E1784K induces a small shift in the DIIIS4 activation (Peters et al., 2017), our results suggest that changes to DIVS4 voltage-sensor movement and, consequently, fast inactivation play a large role in determining the gating of position 1784 variants.
Our data also agree with our prior hypothesis that the altered rates of fast inactivation cause the shifts in the channel GV relationship of the E1784K mutant. When the putative inactivation motif is removed, the E1784K mutant no longer alters the sodium channel conductance (Peters et al., 2017). Here we show that the midpoint of the GV relationship in position 1784 mutants is negatively correlated to the rate of fast inactivation between −20 and +10 mV. Although channel opening is thought to allow for faster channel inactivation, such a relationship could be expected to appear as a positive correlation between the GV and the time constant of inactivation. Instead, the negative correlation we observed suggests that more rapid inactivation decreases the conduction of sodium in E1784K at the membrane potentials when activation is slowest, causing an apparent depolarization of the GV relationship that is not dependent on a depolarized QV. This hypothesis is supported by single-channel recordings showing that a significant number of channels activate after the fast inactivation process has begun (Aldrich et al., 1983).
Interestingly, we found that the rate of channel inactivation is also correlated with the extent to which protons decrease channel conductance. Constructs with the fastest inactivation rates also showed greater proton block at pH 6.0. Protons slow channel activation by depolarizing the voltage-sensor movement (Jones et al., 2013; Peters, 2017). Mutants that accelerate fast inactivation may cause larger decreases in peak current when activation is slowed by protons, similarly to how accelerated fast inactivation depolarizes the GV curve. Because hyperpolarization and acceleration of channel inactivation are shared by mixed syndrome variants including 1795insD and ΔKPQ (Chandra et al., 1998; Veldkamp et al., 2000), the fast inactivation–dependent depolarization of activation and increased effect of protons may also be shared.
The positive charge at position 1784 confers noninactivating current
As in previous studies, we find that E1784K causes a large fraction of noninactivating sodium current. Notably, despite inducing shifts in the voltage dependence and rates of fast inactivation, none of E1784V, E1784Q, or E1784W change the fraction of noninactivating sodium current. E1784V, in particular, exerts a similar effect on the fast inactivation rates and voltage dependence as E1784R, yet does not increase the fraction of noninactivating current. We also note that E1784R confers a significantly smaller fraction of noninactivating current compared with E1784K. The decreased noninactivating current in E1784R is likely due to the positive charge on arginine being split between three nitrogens, in contrast to lysine, in which the positive charge is localized to a single nitrogen. This effect is similar to data from the sodium channel selectivity filter, in which the weaker charge on the K1419R mutant abolishes selectivity for sodium over potassium (Favre et al., 1996). In sum, these data suggest that noninactivating current in the E1784K mutant is due to the positive charge at position 1784.
Recently, it was shown that persistent currents in E1784K may be restored by the K1493E mutant in the DIII–DIV linker (Gade et al., 2020). This interaction appears to be one of many possible electrostatic interactions, as the proximal C terminus contains ∼30% negative glutamate and aspartate residues, whereas the DIII–DIV linker contains 12 positively charged residues. Neutralizing positive charges in the DIII–DIV linker, or negative residues in the proximal C terminus, increases the fraction of noninactivating sodium current (Patton et al., 1992; Wei et al., 1999; Shen et al., 2017). We constructed a homology model of the Nav1.5 sequence on the NavPaS structure using the MODELLER program run at the University of California San Francisco RBVI (Sali and Blundell, 1993). As shown previously, this model places residue E1784E within 5 Å of residue K1493 in the DIII–DIV linker (Gade et al., 2020). A model constructed on the SWISS-MODEL server (not depicted) showed a similar result (Bordoli et al., 2009; Arnold et al., 2006; Biasini et al., 2014). To confirm an interaction between E1784E and K1493, we made a double mutant, K1493E/E1784K, that reverses the charge at both sites. The double mutant restores the noninactivating current of E1784K to normal levels, suggesting that a potential interaction between the channel C terminus and the DIII–DIV linker was restored. However, the interaction between K1493 and E1784E may not be specific and may be stabilized by the surrounding residues. Neutralization of either side of this interaction by E1784Q and E1784A or K1493A and K1493I does not increase the fraction of persistent current (Gade et al., 2020), whereas neutralization of E1784E and three other residues (E1773, E1780, and E1781) is sufficient to increase persistent current (Wei et al., 1999). Even the charge reversal of K1493E led to only a mild increase in persistent current in the study by Gade et al. (2020) and a nonsignificant trend toward an increase in our dataset. In addition, a mutant with a less-focused positive charge, E1784R, does not increase persistent current to the same extent as E1784K. In contrast to previously reported results, we also show that the persistent current in E1784K can be largely attenuated by the K1492E mutant, but not the more distant K1500E mutant (Gade et al., 2020). In total, these data indicate that the interactions between the C terminus and DIII–DIV linker in this region may involve multiple residues that allow for several possible electrostatic interactions.
E1784K disrupts multiple interactions
The NavPaS structure, work by Clairfeuille et al. (2019), and our homology model show C-terminal interactions with the DIVS4-S5 linker just downstream of the position homologous to residue 1784 (Shen et al., 2017; Fig. 6 B). These structures also show that the C terminus kinks back upwards to form these interactions at P1785, the residue adjacent to E1784E. In their article, Clairfeuille et al. (2019) proposed that the interaction between the C terminus and the bottom of DIVS4 acts as a switch that maintains the DIVS4 in its downward conformation. When these interactions are broken, DIVS4 may move more easily to its upward confirmation, leading to a hyperpolarizing shift in voltage dependence of fast inactivation. Although the previous data on mutants in this region suggested that the single C-terminal mutants also increased persistent current, this may be due to their proximity to the DIII–DIV linker (Clairfeuille et al., 2019). Disrupting the interaction between the C terminus and DIVS4 in the opposite direction, via the K1641D mutant, hyperpolarizes the voltage dependence and accelerates the rate of inactivation without increasing persistent current.
In contrast to increases in noninactivating current, the rate and voltage dependence of fast inactivation are disrupted by many mutants at position 1784. We also show that the K1493E/E1784K double mutant retains a similar hyperpolarization of fast inactivation and acceleration of fast inactivation as E1784K, despite attenuating persistent current. Thus, the effects of E1784K on the voltage dependence and rates of fast inactivation are likely not due to the disrupted interaction with the DIII–DIV linker. Instead, mutants at position 1784 might disrupt the structure of this region and alter the downstream interactions between the C terminus and DIVS4. Our data show that E1784V, E1784Q, and E1784W alter the voltage dependence and/or rates of fast inactivation in the absence of a change to the persistent sodium current. An interesting aspect of our data is that rather than disrupting interactions in this region, the bulkiest mutant we studied, E1784W, stabilizes the deactive conformation of DIVS4 by depolarizing the voltage dependence and slowing the onset of fast inactivation. We believe this effect, in particular, argues that position 1784 mutants alter fast inactivation voltage dependence through a downstream effect at the C-terminal–DIV interface. Tryptophan is rich in pi electrons, allowing it to form stable interactions with cations and adjacent prolines (Gallivan and Dougherty, 1999; Brandl et al., 2001; Schwans et al., 2013; Zondlo, 2013). Therefore, E1784W could form an interaction with the cations in the DIII–DIV linker, leading to little persistent current, while also stabilizing the kink in the C terminus at P1785, in turn stabilizing the interactions between the C terminus and DIVS4.
Mechanisms of altered fast inactivation and noninactivating current in E1784K
The data and modeling presented in this and prior articles ultimately suggests that the E1784K mutant acts through changes to both DIVS4 movement and binding of the IFM motif to the channel pore to alter fast inactivation and produce noninactivating current, respectively. Sodium channel fast inactivation occurs in two distinct steps (Fig. 8 A). The rates and voltage dependence of fast inactivation are largely determined by the voltage-dependent movements of DIVS4 (Capes et al., 2013); however, channel inactivation also requires voltage-independent binding of the IFM motif in the DIII–DIV linker to occlude the channel pore (Aldrich et al., 1983; West et al., 1992; Jiang et al., 2020).
Our previous work showed that E1784K changes DIVS4 movement, which in turn alters the fast inactivation voltage dependence and rates (Peters et al., 2017). Our experiments and modeling now suggest that changes to these rates alone do not induce noninactivating current. E1784V, K1641D, and K1493E/E1784K cause hyperpolarization of the voltage dependence and acceleration of the recovery and onset rates of inactivation, whereas E1784W depolarizes the voltage dependence of fast inactivation; however, none of these mutants induces noninactivating current. Models with altered DIVS4 movement for each of these variants also lack noninactivating current. This is best illustrated by the K1493E/E1784K variant, for which we were able to use the E1784K model with only the unbinding rate of IFM set to the value from the E1784E model.
In our models, noninactivating currents were induced in E1784K only by accelerating the voltage-independent unbinding of the IFM motif. This is reasonable because the noninactivating current in E1784K is, itself, relatively voltage independent. In contrast, window current is a voltage-dependent current driven by the probability that, in the window of overlap between the fast-inactivation and activation curves, there are some channels that activate but do not inactivate (Stafstrom, 2007; Chadda et al., 2017). Of the variants tested, E1784W is predicted to have the largest window current (depolarized fast inactivation), and E1784V is predicted to have the smallest (depolarized activation and hyperpolarized inactivation). This can be seen in the average current produced by these variants during a voltage ramp protocol (Fig. S1); however, this current occurs in a small voltage range and differs from the voltage-independent current in E1784K that decays linearly toward the equilibrium potential.

E1784 variants differentially affect window currents evoked by ramp depolarizations. Average currents in response to a 0.15 mV/ms ramp from −120 to +40 mV in cells expressing E1784E (n = 3), E1784K (n = 4), E1784V (n = 4), and E1784W (n = 4). All currents were normalized to the peak current at −20 mV from the same cell before averaging. Error bars are SEM.
E1784 variants differentially affect window currents evoked by ramp depolarizations. Average currents in response to a 0.15 mV/ms ramp from −120 to +40 mV in cells expressing E1784E (n = 3), E1784K (n = 4), E1784V (n = 4), and E1784W (n = 4). All currents were normalized to the peak current at −20 mV from the same cell before averaging. Error bars are SEM.
Ultimately, our data support a model in which E1784K induces changes to the voltage dependence and rates of fast inactivation by stabilizing DIVS4 in the active state. In addition, E1784K induces noninactivating currents by destabilizing the IFM bound state of the channel. This difference may be important for drug design; for example, drugs such as anionic aryl sulfonamides designed to target and modify the movements of DIVS4. Our data suggest that although these drugs may restore the fast inactivation voltage dependence of E1784K, they may not be able to reduce the noninactivating current.
Conclusion
We have shown that E1784K-induced increases in noninactivating current are dependent on a positive charge at position 1784, and that mutants at position 1784 can alter fast inactivation voltage dependence and rates without affecting the fraction of noninactivating current. These data, in conjunction with other recent studies on this region, confirm that the C terminus forms multiple interactions with the sodium channel inactivation machinery (Clairfeuille et al., 2019; Gade et al., 2020). We confirm that an interaction occurs between residue E1784E of the C terminus and the DIII–DIV linker at sites K1492 and K1493. Disrupting this interaction causes the characteristic noninactivating sodium current of the most prevalent LQT3 mutant, E1784K; however, restoring this interaction does not restore the rates or voltage dependence of fast inactivation back to their normal levels. Ultimately, E1784K alters both the voltage-dependent transitions in DIVS4 and voltage-independent unbinding of the IFM motif.
Acknowledgments
Christopher J. Lingle served as editor.
Data in this manuscript has been previously incorporated in C.H. Peters’s doctoral thesis (Peters, 2017).
The authors acknowledge Mariam Aziz-Hanna for her assistance with experimentation and Mena Abdelsayed, Madeline Angus, and Mohammad-Reza Ghovanloo for their input on data analysis.
This work was supported by the Natural Sciences and Engineering Research Council of Canada (grants RGPIN/03920-2018 to P.C. Ruben and GXSO117 to C.H. Peters).
The authors declare no competing financial interests.
Author contributions: C.H. Peters and P.C. Ruben designed experiments. C.H. Peters, O.L. Poirier, and A.R. Watkins performed experiments and analyzed data. C.H. Peters and P.C. Ruben interpreted the results of experiments. C.H. Peters prepared figures. C.H. Peters and P.C. Ruben wrote the initial manuscript. All authors edited and approved the final manuscript.

