


The interception of blood-borne bacteria in the liver defines the outcomes of invasive bacterial infections, but the mechanisms of this antibacterial immunity are not fully understood. This study shows that natural antibodies (nAbs) to capsules enable liver macrophage Kupffer cells (KCs) to rapidly capture and kill blood-borne encapsulated bacteria in mice. Affinity pulldown with serotype-10A capsular polysaccharides (CPS10A) of Streptococcus pneumoniae (Spn10A) led to the identification of CPS10A-binding nAbs in serum. The CPS10A–antibody interaction enabled KCs to capture Spn10A bacteria from the bloodstream, in part through complement receptors on KCs. The nAbs were found to recognize the β1-6-linked galactose branch of CPS10A and similar moieties of serotype-39 S. pneumoniae and serotype-K50 Klebsiella pneumoniae capsules. More importantly, the nAbs empowered KCs to capture serotype-39 S. pneumoniae and serotype-K50 K. pneumoniae in the liver. Collectively, our data have revealed a highly effective immune function of nAb against encapsulated bacteria and emphasize the concept of treating septic encapsulated bacterial diseases with monoclonal antibodies.
Introduction
Invasive infections of encapsulated bacteria in sterile organ/tissue sites are a leading cause of human morbidity and mortality (GBD 2019 Antimicrobial Resistance Collaborators, 2022). Capsules, the outermost cellular structures of encapsulated bacteria, are known as the “slippery” or antiphagocytic coat of encapsulated bacteria due to the resistance to phagocytic killing of host defense (Comstock and Kasper, 2006). Certain physical properties of the capsules (e.g., hyperviscosity and negative charge) hinder the recognition and binding by phagocytes to encapsulated bacteria (Brown and Gresham, 2012; Nahm and Katz, 2012; Taylor and Roberts, 2002). Virtually, all bacterial capsules are composed of capsular polysaccharides (CPSs) (An et al., 2024; Whitfield et al., 2020). There are great inter- and intraspecies variations in chemical structure and antigenicity of CPSs (An et al., 2024; Whitfield et al., 2020). Many bacteria produce a large number of capsule types (An et al., 2024). As an example, there are 103 capsular serotypes in Streptococcus pneumoniae, a leading cause of community-acquired pneumonia, blood infection, and meningitis (Ganaie et al., 2023; Silva-Costa et al., 2024). Capsule types are associated with the virulence level of encapsulated bacteria as exemplified by the dominance of the low-numbered pneumococcal serotypes/serogroups in severe pneumonia in the preantibiotic era (White, 1938) and of Escherichia coli K1 in neonatal meningitis (Robbins et al., 1974).
The liver is regarded as the vascular “firewall” against invading bacteria in the blood circulation (Balmer et al., 2014; Jenne and Kubes, 2013). The importance of the liver in filtering blood bacteria is demonstrated by the dominant role of the organ in trapping blood-borne bacteria (Benacerraf et al., 1959; Brown et al., 1981; Rogers, 1956). More recent investigations have shown that the liver macrophages—Kupffer cells (KCs)—are the major immune cells for the capture of commensal and potentially pathogenic bacteria (An et al., 2022; Broadley et al., 2016; Huang et al., 2022; Zeng et al., 2016). This functional dominance of KCs is accompanied by their exceptional representation in cell number among tissue-resident macrophages since KCs constitute ∼90% of total tissue macrophages in humans and mice (Bilzer et al., 2006).
While KC-mediated antibacterial immunity is vital for host blood sterility and health, it is largely unknown how KCs capture circulating bacteria with such astonishing speed and capacity. Recent studies have highlighted the strict requirement of pattern recognition receptors for KC capture of blood-borne bacteria, likely due to the high shear force in the high-speed hepatic bloodstream (Kubes and Jenne, 2018). However, in contrast to the massive number of microbes that potentially enter the blood circulation, the full receptor arsenal for KC capture of invading bacteria is still a black hole since only a few pathogen receptors on KCs have been identified to date. The complement receptor CRIg is the first known receptor for KC capture of complement C3-opsonized bacteria (Broadley et al., 2016; Helmy et al., 2006; Zeng et al., 2016). Our recent studies have shown that capsule types define bacterial survival in the bloodstream and thereby virulence by structure-dependent variability of capsules in escaping phagocytic capture of KCs in the liver (An et al., 2022; Huang et al., 2022). Encapsulated bacteria differ in evasion of hepatic clearance in a capsule type-dependent manner, which stratifies bacteria into high-virulence (HV) and low-virulence (LV) serotype groups. While the HV serotypes completely circumvent hepatic recognition, the LV counterparts are partially captured by KCs to various extents. The asialoglycoprotein receptor (ASGR) was identified as the first known receptor on KCs for pneumococcal LV serotype-7F and -14 capsules (An et al., 2022). However, it remains unknown how KCs recognize many other LV capsule types.
Natural antibodies (nAbs) are spontaneously produced by B-1 cells and marginal zone B cells without deliberate immunization (Galili et al., 1984; Martin et al., 2001; Springer et al., 1961). The majority of nAbs recognize polysaccharide antigens. Human and mouse plasma contain nAbs against phosphorylcholine of pneumococcal cell wall teichoic acid, which confers a modest level of protection against pneumococcal infections (Briles et al., 1981b; Haas et al., 2005). nAbs against the O127 lipopolysaccharide (LPS) of enteropathogenic E. coli promote pathogen capture by KCs (Zeng et al., 2018). However, the specific functions of many nAbs are largely speculative.
This study sought to understand how KCs capture LV serotypes of S. pneumoniae in the liver and identified plasma nAbs as the binding receptor for the capsules of serotype 10A (CPS10A). We further revealed that this antibody-capsule binding interaction enables KCs to capture and eliminate blood-borne serotype-10A S. pneumoniae (Spn10A) in the liver. The broad function of the anti-CPS10A nAbs in host defense against other encapsulated bacteria was also validated.
Results
Circulating Spn10A are captured by KCs
To identify host receptor(s) recognizing LV capsules, we chose serotype-10A S. pneumoniae (Spn10A), which displays an LV phenotype in mice (An et al., 2022). Spn10A represents one of the prevalent serotypes in invasive pneumococcal disease after the introduction of polysaccharide conjugate vaccines (Plainvert et al., 2023; Yun et al., 2021) and has been added to the recent conjugate vaccines (Kobayashi et al., 2024). We first verified the LV phenotype of Spn10A using natural isolates in mice. In contrast to 100% mortality of mice after intravenous (i.v.) infection with 106 colony forming unit (CFU) of the HV strains D39 (serotype 2) and TIGR4 (serotype 4), all mice survived infection of five selected Spn10A strains (Fig. 1 A). Accordingly, mice infected with Spn10A bacteria became undetectable in the blood at 12 h after infection and remained undetectable ever since, whereas all of the D39- and TIGR4-infected mice showed severe bacteremia before falling to the infection (Fig. 1 B). We further assessed the clearance process of Spn10A strains in the bloodstream in the first 30 min after i.v. infection. Opposite to the stable presence of D39 and TIGR4 in the bloodstream, all the five tested Spn10A strains rapidly disappeared from circulation, which is characterized by an extremely short 50% clearance time (CT50) or the time window during which half of the inoculum is absent from circulation (Fig. 1 C). The serotype-dependent virulence phenotypes were verified using isogenic capsule-switch derivatives of serotype-6A strain TH870. All of the mice infected by the TH870 derivative carrying the serotype-10A CPS gene cluster survived the i.v. infection, whereas infection with the isogenic serotype-2 and -4 counterparts led to 100% mortality with severe bacteriemia (Fig. 1, D and E). In a similar manner, the isogenic strain of serotype 10A was rapidly cleared from the bloodstream as compared with the counterparts of serotypes 2 and 4 (Fig. 1 F). These results showed that Spn10A belongs to the LV category of S. pneumoniae.

Capture of circulating Spn10A by liver macrophages. (A and B) Survival rates (A) and bacteremia levels (B) of mice i.v. infected with 106 CFU of D39, TIGR4, and five serotype-10A pneumococcal strains. The serotype of each strain is denoted with superscripted characters. n = 10. The dashed line represents the detection limit. (C) Bacteremia kinetics of mice in the first 30 min after i.v. infection, as in A. n = 6. (D and E) Survival rates (D) and bacteremia levels (E) of mice i.v. infected with 106 CFU of isogenic capsule-switched serotype-2, -4, and -10A strains. n = 10. (F) Bacteremia kinetics of mice in the first 30 min after i.v. infection, as in D. n = 6. (G) Bacterial distribution in the blood and major organs of mice after i.v. infection with 106 CFU of TH86010A. n = 6. (H) IVM detection of pneumococcal capture in the liver sinusoids of Clec4f-DTR mice treated with (+DT) or without (−DT) DT after i.v. infection with 5 × 107 CFU of TH86010A. Representative images (left) showed S. pneumoniae (green), KCs (red), and microvasculature (cyan) at 15 min after infection. Scale bar, 10 μm. n = 2. Bacteria immobilized on KCs (right panel) were quantified. The clearance processes are demonstrated in Video 1. Data were representative results (H) or pooled from two independent experiments (A–G). Unpaired t test (H) was performed. ****, P < 0.0001.
Capture of circulating Spn10A by liver macrophages. (A and B) Survival rates (A) and bacteremia levels (B) of mice i.v. infected with 106 CFU of D39, TIGR4, and five serotype-10A pneumococcal strains. The serotype of each strain is denoted with superscripted characters. n = 10. The dashed line represents the detection limit. (C) Bacteremia kinetics of mice in the first 30 min after i.v. infection, as in A. n = 6. (D and E) Survival rates (D) and bacteremia levels (E) of mice i.v. infected with 106 CFU of isogenic capsule-switched serotype-2, -4, and -10A strains. n = 10. (F) Bacteremia kinetics of mice in the first 30 min after i.v. infection, as in D. n = 6. (G) Bacterial distribution in the blood and major organs of mice after i.v. infection with 106 CFU of TH86010A. n = 6. (H) IVM detection of pneumococcal capture in the liver sinusoids of Clec4f-DTR mice treated with (+DT) or without (−DT) DT after i.v. infection with 5 × 107 CFU of TH86010A. Representative images (left) showed S. pneumoniae (green), KCs (red), and microvasculature (cyan) at 15 min after infection. Scale bar, 10 μm. n = 2. Bacteria immobilized on KCs (right panel) were quantified. The clearance processes are demonstrated in Video 1. Data were representative results (H) or pooled from two independent experiments (A–G). Unpaired t test (H) was performed. ****, P < 0.0001.
We further characterized bacterial organ distribution at various time points after infection using representative strain TH86010A. With the rapid disappearance from the circulation in the course of blood infection, the pneumococci were mostly trapped in the liver at 5, 10, and 30 min after inoculation (Fig. 1 G). There were marginal levels of bacteria in the spleen, lung, heart, and kidney at the three time points. The liver-trapped bacteria were rapidly killed because the combined CFU values from the blood and five organs were similar to the inoculum at 5 min, but were dramatically reduced to 52% and 16% of the inoculum at 10 and 30 min, respectively. These results showed that Spn10A bacteria are rapidly eliminated in the liver.
Based on the dominant role of liver macrophage KCs in capturing the LV pneumococci (An et al., 2022), we determined the contribution of KCs to the clearance of Spn10A. Spn10A was significantly delayed in clearance in Clec4f-DTR mice treated with diphtheria toxin (DT) to specifically deplete KCs compared with that without DT treatment, no matter with an infection dose of 106 or 5 × 107 CFU (Fig. S1). We used intravital microscopy (IVM) to visualize the KC capture of Spn10A in the liver sinusoids (Fig. 1 H and Video 1). IVM imaging showed rapid tethering of circulating bacteria onto KCs after i.v. inoculation but a dramatic reduction of immobilized bacteria on KCs was observed in KC-deficient mice. The number of KC-immobilized bacteria in each field of view (FOV) in KC-deficient mice was only 15% of that was observed at the vascular wall of the liver sinusoids in control mice. These results demonstrated that KCs are the dominant immune cells responsible for capturing Spn10A in the liver.

Clearance of Spn10A from the circulation by KC s. Bacteremia kinetics of Clec4f-DTR mice treated with (+DT) or without (−DT) DT before i.v. infection with 106 (left) or 5 × 107 (right) CFU of TH86010A. n = 6. Data were all pooled from two independent experiments. Ordinary two-way ANOVA with Tukey’s multiple comparisons test was performed. ****, P < 0.0001.

Clearance of Spn10A from the circulation by KC s. Bacteremia kinetics of Clec4f-DTR mice treated with (+DT) or without (−DT) DT before i.v. infection with 106 (left) or 5 × 107 (right) CFU of TH86010A. n = 6. Data were all pooled from two independent experiments. Ordinary two-way ANOVA with Tukey’s multiple comparisons test was performed. ****, P < 0.0001.
IVM shows diminished liver capture of Spn10A in KC-deficient mice.S. pneumoniae (green), KCs (red), and microvasculature (cyan) in the liver sinusoids of Clec4f-DTR mice treated with (+DT) or without (−DT) DT in the first 1 min after i.v. infection with 5 × 107 CFU of TH86010A were shown. Graphic analysis is shown in Fig. 1 H. Playback speed is 25 frames per second.
IVM shows diminished liver capture of Spn10A in KC-deficient mice.S. pneumoniae (green), KCs (red), and microvasculature (cyan) in the liver sinusoids of Clec4f-DTR mice treated with (+DT) or without (−DT) DT in the first 1 min after i.v. infection with 5 × 107 CFU of TH86010A were shown. Graphic analysis is shown in Fig. 1 H. Playback speed is 25 frames per second.
KCs capture Spn10A by recognizing CPS
To understand how KCs capture Spn10A, we hypothesized that the macrophage recognizes the serotype-10A CPS (CPS10A) on the basis of our previous work with serotype-14 pneumococci (An et al., 2022). This possibility was tested by i.v. administration of purified CPS10A before i.v. bacterial inoculation. While CPS10A-treated mice showed stable bacteremia at various time points in the first 30 min after TH86010A infection, the same treatment with CPS10A did not show any obvious impact on early clearance of TH291214 (Fig. 2 A). Serotype-specific blocking of serotype-10A but not serotype-14 pneumococcal clearance with CPS10A was also verified by inhibition against Spn10A trapping in the liver. In contrast to the dramatic disappearance of the Spn14 inoculum from the body of CPS10A-treated mice at 30 min after i.v. inoculation with TH291214, CPS10A-treated mice retained 98% Spn10A of the inoculum at 30 min, the majority of which was located in the blood (Fig. 2 B). Due to the extremely short time window between the injection of free CPS10A and retro-orbital bleeding for CFU plating after injection of bacteria, it is unlikely that this blocking effect was due to any regulator effect of the CPS on KCs. These data strongly suggested that KCs capture Spn10A by recognizing the CPS.

Capsular branch-dependent KC capture of circulating Spn10A. (A and B) Bacteremia kinetics (A) and bacterial distribution at 30 min (B) of mice i.v. treated with 400 μg CPS10A or PBS before i.v. infection with 106 CFU of TH86010A and TH291214. n = 6. (C and D) Bacteremia kinetics (C) and bacterial distribution at 30 min (D) of mice i.v. infected with 106 CFU of TH86010A and three serotype-10B pneumococcal strains. n = 6. (E) Biochemical structure of repeating units of CPS10A and CPS10B. (F) Schematic representation of the cps loci of Spn10A and Spn10B. (G) Representative images of TH860∆wcrG (left), TH860 (middle), and TH860∆cps (right) stained with Anthony’s staining method. Scale bar, 2 μm. (H and I) Bacteremia kinetics (G) and bacterial distribution at 30 min (H) of mice i.v. infected with 106 CFU of TH86010A or TH86010A∆wcrG. n = 6. (J and K) Bacteremia kinetics (I) and bacterial distribution at 30 min (J) of mice i.v. treated with 400 μg CPS10A or CPS10A∆wcrG and i.v. infected with 106 CFU of TH86010A. n = 6. (L) Representative IVM images of liver sinusoids (left) and quantitation of bacteria immobilized on KCs (right) of mice i.v. treated with 400 μg CPS10A or CPS10A∆wcrG and i.v. infected with TH86010A. n = 2. The clearance processes are demonstrated in Video 2. Data were representative results (G and L) or pooled from two independent experiments (A–D and H–K). Ordinary two-way ANOVA with Tukey’s multiple comparisons test (A, C, H, and J) and unpaired t test (K) were performed. ****, P < 0.0001; ns, not significant.
Capsular branch-dependent KC capture of circulating Spn10A. (A and B) Bacteremia kinetics (A) and bacterial distribution at 30 min (B) of mice i.v. treated with 400 μg CPS10A or PBS before i.v. infection with 106 CFU of TH86010A and TH291214. n = 6. (C and D) Bacteremia kinetics (C) and bacterial distribution at 30 min (D) of mice i.v. infected with 106 CFU of TH86010A and three serotype-10B pneumococcal strains. n = 6. (E) Biochemical structure of repeating units of CPS10A and CPS10B. (F) Schematic representation of the cps loci of Spn10A and Spn10B. (G) Representative images of TH860∆wcrG (left), TH860 (middle), and TH860∆cps (right) stained with Anthony’s staining method. Scale bar, 2 μm. (H and I) Bacteremia kinetics (G) and bacterial distribution at 30 min (H) of mice i.v. infected with 106 CFU of TH86010A or TH86010A∆wcrG. n = 6. (J and K) Bacteremia kinetics (I) and bacterial distribution at 30 min (J) of mice i.v. treated with 400 μg CPS10A or CPS10A∆wcrG and i.v. infected with 106 CFU of TH86010A. n = 6. (L) Representative IVM images of liver sinusoids (left) and quantitation of bacteria immobilized on KCs (right) of mice i.v. treated with 400 μg CPS10A or CPS10A∆wcrG and i.v. infected with TH86010A. n = 2. The clearance processes are demonstrated in Video 2. Data were representative results (G and L) or pooled from two independent experiments (A–D and H–K). Ordinary two-way ANOVA with Tukey’s multiple comparisons test (A, C, H, and J) and unpaired t test (K) were performed. ****, P < 0.0001; ns, not significant.
Since Spn10A belongs to serogroup 10 of S. pneumoniae, which contains five serotypes with similar capsular structures (Ganaie et al., 2020; Geno et al., 2015), we further assessed early clearance of three serotype-10B strains in our collection (TH16770, TH16771, and TH16772). Surprisingly, all three strains showed much slower clearance than serotype-10A strains (Fig. 2 C). Consistently, the CFUs of the serotype-10B strains recovered from the blood, liver, and spleen at 30 min were significantly higher than that of TH86010A (Fig. 2 D). This result suggested that serotype-10B CPS does not bind to the host receptor(s) that recognizes CPS10A.
Serotypes 10A and 10B are highly similar in chemical structure (Fig. 2 E) and setting of biosynthesis genes (Fig. 2 F). However, CPS10A structurally differs from CPS10B in two aspects: a β1-6-linked galactopyranose (Galp) branch (absent in CPS10B) and an α1-2 linkage between galactose and ribitol-5-phosphate (α1-4 linkage in CPS10B). We thus tested if the Galp branch in CPS10A is responsible for the serotype-specific KC capture of Spn10A by mutating the wcrG gene in the cps10a locus of TH860, encoding the enzyme that adds β1-6-linked Galp branches to GalNAc in the CPS10A repeating unit (Yang et al., 2011). wcrG in the cps10b locus is a pseudogene. We generated a ∆wcrG mutant in the TH860 background and confirmed that the strain still produced a capsule layer comparable with the parent strain (Fig. 2 G). Removing the branch moiety made TH860 much more resistant to hepatic clearance. The CT50 of TH860∆wcrG mutant was elongated to 15.5 from 0.9 min in the parental strain (Fig. 2 H). The total bacteria detected from the blood, liver, and spleen at 30 min after infection were three times higher than that in mice infected with the mutant (Fig. 2 I). Consistently, pretreatment of mice with purified CPS from TH860∆wcrG (CPS10A∆wcrG) did not hinder the shuffling of TH860 from the blood (Fig. 2 J) to the liver (Fig. 2 K) as compared with the intact CPS10A. IVM imaging showed fivefold more KC-immobilized bacteria in the mutant CPS-treated mice as compared with mice pretreated with the normal CPS (Fig. 2 L and Video 2). These data indicated that the β1-6-linked Galp branch on CPS10A is recognized by an uncharacterized receptor(s) for KC capture of serotype-10A pneumococci.
IVM shows inhibition of free CPS10A but not CPS10A without β1-6-linked galactose branch on KC capture of Spn10A. Pneumococcal capture in the liver sinusoids of WT mice i.v. treated with 400 μg CPS10A or CPS10A∆wcrG before i.v. infection with 5 × 107 CFU of TH86010A are shown. Graphic analysis is shown in Fig. 2 L. Playback speed is 25 frames per second.
IVM shows inhibition of free CPS10A but not CPS10A without β1-6-linked galactose branch on KC capture of Spn10A. Pneumococcal capture in the liver sinusoids of WT mice i.v. treated with 400 μg CPS10A or CPS10A∆wcrG before i.v. infection with 5 × 107 CFU of TH86010A are shown. Graphic analysis is shown in Fig. 2 L. Playback speed is 25 frames per second.
Serum antibodies specifically bind to the capsule of Spn10A
KCs may capture pathogens by membrane receptors directly or by soluble pattern recognition molecules indirectly (Kubes and Jenne, 2018). To determine whether KC-mediated capture of Spn10A relies on soluble component(s) in serum, we detected binding interactions between isolated KCs and bacteria in an in vitro system. The visualization of KC binding showed that only by the addition of mouse serum can KCs capture Spn10A, and the inhibition was achieved by CPS10A but not CPS10A∆wcrG (Fig. 3 A). These data indicate that KCs capture Spn10A by recognizing the β1-6-linked Galp branch on CPS10A in the presence of the serum.

Specific binding of serum antibodies to the capsule of Spn10A. (A) Representative confocal images (left) and quantitative analysis (right) showing mouse primary KCs binding with Spn10A. The KCs were incubated with TH86010A supplemented with or without 10% serum, and 100 μg/ml CPS10A or CPS10A∆wcrG. Scale bar, 10 μm. n = 6. (B) Schematic illustration of the strategy for screening CPS10A-binding proteins. (C) Plot of proteins significantly enriched by CPS10A-coated beads. The top six enriched proteins compared with that of CPS10A∆wcrG-coated beads are labeled in red. n = 3. The detailed results are listed in Table S1. (D) ELISA detection of IgG and IgM to CPS10A or CPS10A∆wcrG in WT mouse serum. n = 3. (E) Flow cytometry detection of the IgG and IgM deposition on the surface of TH86010A or TH86010A∆wcrG incubated with mouse serum. (F) ELISA detection of IgG and IgM to CPS10A or CPS10A∆wcrG in sera of 2-mo-old children. n = 18. Data were representative results (A and D–F) or pooled from three independent experiments (C). Ordinary two-way ANOVA with Sidak’s multiple comparisons test (A and D) and paired t test (F) were performed. ***, P < 0.001; ****, P < 0.0001; ns, not significant.
Specific binding of serum antibodies to the capsule of Spn10A. (A) Representative confocal images (left) and quantitative analysis (right) showing mouse primary KCs binding with Spn10A. The KCs were incubated with TH86010A supplemented with or without 10% serum, and 100 μg/ml CPS10A or CPS10A∆wcrG. Scale bar, 10 μm. n = 6. (B) Schematic illustration of the strategy for screening CPS10A-binding proteins. (C) Plot of proteins significantly enriched by CPS10A-coated beads. The top six enriched proteins compared with that of CPS10A∆wcrG-coated beads are labeled in red. n = 3. The detailed results are listed in Table S1. (D) ELISA detection of IgG and IgM to CPS10A or CPS10A∆wcrG in WT mouse serum. n = 3. (E) Flow cytometry detection of the IgG and IgM deposition on the surface of TH86010A or TH86010A∆wcrG incubated with mouse serum. (F) ELISA detection of IgG and IgM to CPS10A or CPS10A∆wcrG in sera of 2-mo-old children. n = 18. Data were representative results (A and D–F) or pooled from three independent experiments (C). Ordinary two-way ANOVA with Sidak’s multiple comparisons test (A and D) and paired t test (F) were performed. ***, P < 0.001; ****, P < 0.0001; ns, not significant.
To identify host factor(s) that promotes KC capture of Spn10A, we performed an affinity pull-down of CPS10A-binding protein(s) in mouse serum and membrane proteins extracted from mouse nonparenchymal cells using CPS10A- and CPS10A∆wcrG-coated beads (Fig. 3 B). Proteomics analysis of the resulting proteins revealed that 37 proteins were at least twofold enriched by CPS10A-coated beads (Fig. 3 C and Table S1). With the consideration of protein abundance and enrichment fold, immunoglobulin (Ig) heavy constant mu (IGHM), Ig gamma-3 chain C region (IGHG3), complement C4-B (CO4B), Ig kappa chain V-VI region XRPC 24 (KV6A2), Ig heavy chain V region X44 (HVM37), and Ig kappa constant (IGKC) were among the most enriched proteins (Fig. 3 C). Collectively, these hits represented various fragments of antibodies. In agreement, the ELISA test revealed more CPS10A-reactive IgG and IgM in mouse serum than antibodies bound to CPS10A∆wcrG (Fig. 3 D). In addition, the CPS10A-reactive IgG almost exclusively belonged to the IgG3 subtype (Fig. S2 A), which is a coincidence with our mass spectrometry data. We further determined the capsule-reactive antibodies in mouse serum with purified CPSs from 13 additional serotypes of S. pneumoniae that were covered by the 13-valent pneumococcal polysaccharide conjugate vaccine (PCV13) (Briles et al., 2019). The binding interaction was not detected in any of these serotypes beyond CPS10A (Fig. S2 B). This result excluded the possibility that the CPS10A-binding antibodies might have resulted from the well-known binding of pneumococcal cell wall phosphocholine to nAbs (Briles et al., 1981b).

Specific binding of serum antibodies to the capsule of Spn10A. (A) ELISA detection of IgG subtypes of anti-CPS10A IgG in mouse serum. n = 3. (B) ELISA detection of IgG and IgM to different pneumococcal CPSs in mouse serum. n = 3. (C) ELISA detection of IgG and IgM to CPS10A in serum of male and female C57BL/6 mice. n = 5. (D) ELISA detection of serum IgG and IgM to CPS10A in SPF and GF mice. n = 3. (E) ELISA detection of serum IgG and IgM to CPS10A in C57BL/6, BALB/c or CD1 mice. n = 3. (F and G) Representative confocal images showing the deposition of IgG (F) or IgM (G) on TH86010A or TH86010A∆wcrG with percentages of deposition in the right. Scale bar, 10 μm. n = 5. Data were all representative results. Ordinary two-way ANOVA with Sidak’s multiple comparisons test (C and D) and unpaired t test (F and G) were performed. ****, P < 0.0001; ns, not significant.

Specific binding of serum antibodies to the capsule of Spn10A. (A) ELISA detection of IgG subtypes of anti-CPS10A IgG in mouse serum. n = 3. (B) ELISA detection of IgG and IgM to different pneumococcal CPSs in mouse serum. n = 3. (C) ELISA detection of IgG and IgM to CPS10A in serum of male and female C57BL/6 mice. n = 5. (D) ELISA detection of serum IgG and IgM to CPS10A in SPF and GF mice. n = 3. (E) ELISA detection of serum IgG and IgM to CPS10A in C57BL/6, BALB/c or CD1 mice. n = 3. (F and G) Representative confocal images showing the deposition of IgG (F) or IgM (G) on TH86010A or TH86010A∆wcrG with percentages of deposition in the right. Scale bar, 10 μm. n = 5. Data were all representative results. Ordinary two-way ANOVA with Sidak’s multiple comparisons test (C and D) and unpaired t test (F and G) were performed. ****, P < 0.0001; ns, not significant.
Zeng et al. have shown that nAbs to E. coli LPS are more abundantly produced in female mice and drive a sex-dependent but microbiota-independent hepatic clearance of enteropathogenic E. coli (Zeng et al., 2018). To test the potential sex-dependent production of anti-CPS10A antibodies, we compared the antibody titers between male and female C57BL mice. There was a similar level of IgG and IgM antibodies to CPS10A in both female and male mice (Fig. S2 C). We next tested the possibility that microbiota induced the anti-CPS10A antibodies using germ-free (GF) mice. The mice raised in GF facilities produced a comparable level of anti-CPS10A antibodies as specific pathogen–free (SPF) mice (Fig. S2 D). We also detected abundant anti-CPS10A antibodies in mice with different genetic backgrounds (Fig. S2 E). Through flow cytometry, we detected IgG and IgM in serum deposited on Spn10A while no positive signals on the surface of Spn10A∆wcrG were observed (Fig. 3 E). This was further confirmed by microscopy that >95% of Spn10A but not Spn10A∆wcrG were covered by serum IgG and IgM (Fig. S2, F and G). To assess the production of anti-CSP10A nAbs in humans, we tested CPS10A-reactive IgG and IgM antibodies in the serum samples that had been previously collected from 2-mo-old children (Wang et al., 2022). Using the capsule from Spn10A∆wcrG as a negative control, we detected significant levels of anti-CSP10A IgG antibodies in all of the 18 tested individuals (Fig. 3 F), some of which might be maternal antibodies. Likewise, there were substantial levels of anti-CSP10A IgM antibodies in some of the infants. Consistent with the previous finding that humans produce nAbs to pneumococcal polysaccharides (Bornstein et al., 1968; Gray et al., 1983), this result strongly suggests the production of anti-CPS10A nAbs in humans.
Anti-capsule antibodies enable KCs to capture Spn10A
To determine whether anti-CPS10A antibodies contribute to hepatic trapping of serotype-10A S. pneumoniae, we assessed early clearance of TH86010A in μMT mice, which lack mature B cells and antibodies. The antibody-deficient mice normally cleared Spn14 as wild-type (WT) control in the first 30 min after infection but completely failed to shuffle TH86010A from the bloodstream (Fig. 4 A) to the liver (Fig. S3 A). Similar results were obtained with the other four Spn10A strains (Fig. S3, B and C). To verify the specific immunity of antibodies against Spn10A, we tested if mouse serum and purified immunoglobulins could rescue the immune deficiency of μMT mice in clearing Spn10A. Instillation of mouse serum in μMT mice 5 min prior to i.v. bacterial inoculation led to a dose-dependent enhancement in early clearance of Spn10A (Fig. 4 B). In a similar manner, the clearance of TH86010A in μMT mice was restored by the administration of purified serum IgG (Fig. 4 C) or IgM (Fig. 4 D) in advance. While the previous study has shown that anti-LPS nAbs drive a more effective clearance of enteropathogenic E. coli in female mice (Zeng et al., 2018), Spn10A bacteria were cleared from the bloodstream of male and female mice at similar levels (Fig. 4 E), which was in line with the equivalent levels of the IgG and IgM antibodies to CPS10A between male and female mice (Fig. S2 C). GF and SPF mice showed comparable efficacy in clearing Spn10A bacteria (Fig. S3 D) as the similar antibody levels of these mice (Fig. S2 D). These results showed that serotype-specific plasma antibodies are essential for hepatic clearance of blood-borne serotype-10A S. pneumoniae, which is independent of gender or microbiota.

Clearance of Spn10A mediated by anti-CPS10A nAb s. (A) Bacteremia kinetics of WT and μMT mice i.v. infected with 106 CFU of TH86010A or TH291214. n = 6. (B–D) Bacteremia kinetics of antibody-deficient mice i.v. treated with various levels of mouse serum (B), purified natural IgG (C), or purified natural IgM (D) before i.v. infection, as in A. n = 6. (E) Bacteremia kinetics of male and female mice infected as in A. n = 6. (F) Representative IVM images of liver sinusoids (left) and quantitation of bacteria immobilized on KCs (right) of antibody-deficient mice i.v. infected with TH86010A or TH291214. Scale bar, 10 μm. n = 2. The clearance processes are demonstrated in Videos 3 and 4. (G) Representative confocal images showing TH86010A or TH291214 binding to primary mouse KCs supplemented with 20% serum of normal or antibody-deficient mice. KC-tethered bacteria are quantified in the right panel. Scale bar, 10 μm. n = 5. (H and I) Survival rate of antibody-deficient mice i.v. (H) or intranasally (i.n.) (I) infected with 107 CFU of TH86010A or TH291214. n = 10. (J) ELISA detection of IgG and IgM to CPS10A in serum of WT and Cd19−/− mice. n = 3. (K) Bacteremia kinetics of WT or Cd19−/− mice i.v. infected with 106 CFU of TH86010A or TH291214. n = 6. (L) Representative IVM images of liver sinusoids (left) and quantitation of bacteria immobilized on KCs (right) of Cd19−/− mice i.v. infected with 5 × 107 CFU of TH86010A or TH291214. Scale bar, 10 μm. n = 2. The processes of bacterial capture are demonstrated in Videos 4 and 5. Data were representative results (F, G, and L) or pooled from two independent experiments (A–E and H–K). Ordinary two-way ANOVA with Tukey’s (A–E and K) or Sidak’s (J) multiple comparisons test, unpaired t test (F, G, and L) and log-rank test (H and I) were performed. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant.
Clearance of Spn10A mediated by anti-CPS10A nAb s. (A) Bacteremia kinetics of WT and μMT mice i.v. infected with 106 CFU of TH86010A or TH291214. n = 6. (B–D) Bacteremia kinetics of antibody-deficient mice i.v. treated with various levels of mouse serum (B), purified natural IgG (C), or purified natural IgM (D) before i.v. infection, as in A. n = 6. (E) Bacteremia kinetics of male and female mice infected as in A. n = 6. (F) Representative IVM images of liver sinusoids (left) and quantitation of bacteria immobilized on KCs (right) of antibody-deficient mice i.v. infected with TH86010A or TH291214. Scale bar, 10 μm. n = 2. The clearance processes are demonstrated in Videos 3 and 4. (G) Representative confocal images showing TH86010A or TH291214 binding to primary mouse KCs supplemented with 20% serum of normal or antibody-deficient mice. KC-tethered bacteria are quantified in the right panel. Scale bar, 10 μm. n = 5. (H and I) Survival rate of antibody-deficient mice i.v. (H) or intranasally (i.n.) (I) infected with 107 CFU of TH86010A or TH291214. n = 10. (J) ELISA detection of IgG and IgM to CPS10A in serum of WT and Cd19−/− mice. n = 3. (K) Bacteremia kinetics of WT or Cd19−/− mice i.v. infected with 106 CFU of TH86010A or TH291214. n = 6. (L) Representative IVM images of liver sinusoids (left) and quantitation of bacteria immobilized on KCs (right) of Cd19−/− mice i.v. infected with 5 × 107 CFU of TH86010A or TH291214. Scale bar, 10 μm. n = 2. The processes of bacterial capture are demonstrated in Videos 4 and 5. Data were representative results (F, G, and L) or pooled from two independent experiments (A–E and H–K). Ordinary two-way ANOVA with Tukey’s (A–E and K) or Sidak’s (J) multiple comparisons test, unpaired t test (F, G, and L) and log-rank test (H and I) were performed. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant.

Clearance of Spn10A in μMT mice and GF mice. (A) Bacterial distribution at 30 min of WT or μMT mice i.v. infected with 106 CFU of TH86010A or TH291214. n = 6. (B and C) Bacteremia kinetics (B) and bacterial distribution at 30 min (C) of μMT mice i.v. infected with 106 CFU of four serotype-10A pneumococcal strains. n = 3. (D) Bacteremia kinetics of GF and SPF mice i.v. infected with 106 CFU of TH86010A. n = 3. Ordinary two-way ANOVA with Tukey’s multiple comparisons test was performed. ns, not significant.

Clearance of Spn10A in μMT mice and GF mice. (A) Bacterial distribution at 30 min of WT or μMT mice i.v. infected with 106 CFU of TH86010A or TH291214. n = 6. (B and C) Bacteremia kinetics (B) and bacterial distribution at 30 min (C) of μMT mice i.v. infected with 106 CFU of four serotype-10A pneumococcal strains. n = 3. (D) Bacteremia kinetics of GF and SPF mice i.v. infected with 106 CFU of TH86010A. n = 3. Ordinary two-way ANOVA with Tukey’s multiple comparisons test was performed. ns, not significant.
The immune function of anti-CPS10A antibodies was further manifested by antibody-dependent hepatic capture of TH86010A via IVM imaging of the liver sinusoids. In contrast to the effective capture of TH86010A by KCs of WT mice, μMT mice displayed a significant reduction in KC-associated pneumococci (Fig. 4 F and Video 3). However, Spn14 bacteria were similarly captured by KCs between μMT and WT mice. We also verified the antibody-driven host–pathogen interaction under the in vitro conditions. Primary mouse KCs abundantly bound to TH86010A bacteria in the presence of WT mouse serum but KCs exhibited a marginal level of bacterial binding when being co-incubated with that of μMT mouse serum (Fig. 4 G). In sharp contrast, Spn14 bacteria were abundantly tethered to KCs regardless of the serum source, which agrees with the ASGR-mediated capture of Spn14 by KCs (An et al., 2022). Finally, septic infection experiments showed that antibody-mediated bacterial capture in the liver is essential for host survival against serotype-10A S. pneumoniae. While all the tested WT mice survived infection with TH86010A pneumococci that were inoculated via either i.v. route (Fig. 4 H) or intranasal route (Fig. 4 I), both the infection regiments resulted in 100% mortality of μMT mice. However, the antibody-deficient mice fully survived against infection with Spn14 pneumococci. These experiments demonstrated the essential role of antibody-mediated capsule recognition in serotype-specific host defense against serotype-10A S. pneumoniae.
IVM shows diminished KC capture of Spn10A in μMT mice. Pneumococcal capture in the liver sinusoids of WT or μMT mice i.v. infected with 5 × 107 CFU of TH86010A were shown. Graphic analysis is shown in Fig. 4 F. Playback speed is 25 frames per second.
IVM shows diminished KC capture of Spn10A in μMT mice. Pneumococcal capture in the liver sinusoids of WT or μMT mice i.v. infected with 5 × 107 CFU of TH86010A were shown. Graphic analysis is shown in Fig. 4 F. Playback speed is 25 frames per second.
The presence of relatively high levels of anti-CPS10A antibodies in the serum of naive mice strongly suggested the nature of nAb (Kearney et al., 2015). This possibility was tested using Cd19−/− mice that lack B-1a cells, the major source of nAbs (Baumgarth et al., 2005; Rickert et al., 1995). The anti-CPS10A antibodies were barely detected in the serum of Cd19−/− mice (Fig. 4 J). Accordingly, Cd19−/− mice were found to display severe impairment in the clearance of Spn10A but not Spn14 bacteria (Fig. 4 K). Specific impairment in hepatic capture of Spn10A but not Spn14 bacteria was also observed in Cd19−/− mice by IVM imaging (Fig. 4 L and Videos 4 and 5). These data allowed us to conclude that the anti-CPS10A antibodies belong to nAbs.
IVM shows unaffected KC capture of Spn14 in nAb-deficient mice. Pneumococcal capture in the liver sinusoids of WT, μMT, or Cd19−/− mice i.v. infected with 5 × 107 CFU of TH291214 were shown. Graphic analysis is shown in Fig. 4, F and L. Playback speed is 25 frames per second.
IVM shows unaffected KC capture of Spn14 in nAb-deficient mice. Pneumococcal capture in the liver sinusoids of WT, μMT, or Cd19−/− mice i.v. infected with 5 × 107 CFU of TH291214 were shown. Graphic analysis is shown in Fig. 4, F and L. Playback speed is 25 frames per second.
IVM shows diminished KC capture of Spn10A in Cd19−/−mice. Pneumococcal capture in the liver sinusoids of WT or Cd19−/− mice i.v. infected with 5 × 107 CFU of TH86010A are shown. Graphic analysis is shown in Fig. 4 L. Playback speed is 25 frames per second.
IVM shows diminished KC capture of Spn10A in Cd19−/−mice. Pneumococcal capture in the liver sinusoids of WT or Cd19−/− mice i.v. infected with 5 × 107 CFU of TH86010A are shown. Graphic analysis is shown in Fig. 4 L. Playback speed is 25 frames per second.
The complement system is required for nAb-mediated hepatic clearance of Spn10A
To understand the mechanism of nAb-mediated clearance of Spn10A, we assessed the potential involvement of antibody receptors using mice lacking the known IgM receptors (FCAMR–Fcamr−/− and FCMR–Fcmr−/−) or the four phagocytosis-associated IgG receptors (FcγRI, FcγRIIB, FcγRIII, and FcγRIV–FcRα null). None of these mice showed obvious deficiency in early clearance of Spn10A as compared with WT mice (Fig. 5 A), indicating that KCs do not use these known Fc receptors for binding to nAb-opsonized Spn10A. In light of our earlier observation that complement components C1q, C1s, C1r, C4b, and C3 were abundantly enriched by CPS10A-coated beads (Fig. S4 A and Table S1), we hypothesized that KCs rely on the complement system to capture nAb-opsonized Spn10A. Because the classical pathway of the complement system is known to mediate antibody-driven phagocytosis (Zipfel and Skerka, 2009), we tested nAb-induced complement C3 deposition on bacteria. C3 coated on Spn10A when sera from WT mice were added while sera from μMT mice could not induce C3 deposition due to the lack of nAbs (Fig. 5 B). We then determined the potential contribution of complement proteins C1q and C3, two key components of the classical pathway. Clearance of Spn10A was significantly delayed in both C1qa−/− and C3−/− mice, although the former displayed a more severe phenotype (CT50, 11.4 min) than C3−/− mice (CT50, 2.7 min) as compared with that in WT mice (CT50, 1.3 min) (Fig. 5 C). In the same fashion, C1qa−/− and C3−/− mice showed lower levels of liver-trapped bacteria (Fig. S4 B). The defect of C1qa−/− and C3−/− mice in bacterial clearance was not due to the potential difference in nAb production because these mice showed similar levels of anti-CPS10A nAbs as WT mice (Fig. S4 C). These experiments indicated that anti-CPS10A nAbs enable KCs to capture Spn10A, at least in part by activating complement-mediated phagocytosis.

The importance of C3 and C3 receptors in nAb -mediated pneumococcal capture. (A) Bacteremia kinetics of antibody receptor-deficient mice i.v. infected with 106 CFU of TH86010A. n = 6. (B) Flow cytometry detection of C3 deposition on TH86010A surface after incubation with serum from μMT or WT mice. (C) Bacteremia kinetics of C1qa−/− and C3−/− mice infected as in A. n = 6. (D) Representative IVM images of liver sinusoids (left) and quantitation of bacteria immobilized on KCs (right) of C1qa−/− and C3−/− mice i.v. infected with 5 × 107 CFU of TH86010A. n = 2. The clearance processes are demonstrated in Video 6. (E) Survival rates of WT, C1qa−/−, or C3−/− mice i.v. infected with 108 CFU of TH86010A. n = 10. (F) Bacteremia kinetics of C3-receptor-deficient mice infected as in A. n = 6. (G and H) Representative IVM images of liver sinusoids (G) and quantitation of bacteria immobilized on KCs (H) of C3-receptor-deficient mice i.v. infected with 5 × 107 CFU of TH86010A. Scale bar, 10 μm. n = 2. The clearance processes are demonstrated in Video 7. (I) Survival of C3- and C3-receptor-deficient mice i.v. infected with 108 CFU of TH86010A. n = 10. (J and K) Bacteremia kinetics (J) and bacterial distribution at 30 min (K) of C5−/− and C9−/− mice infected as in (A). n = 6. (L) Working model of nAbs in driving hepatic clearance of encapsulated bacteria. Invading bacteria in the blood are bound by nAbs, which subsequently activates the complement system. C3 opsonzid bacteria are captured by C3 receptors CR3 and CRIg on KCs. Data were representative results (B, D, G, and H) or pooled from two independent experiments (A, C, E, F, and I–K). Ordinary two-way ANOVA with Tukey’s multiple comparisons test (A, C, F, and J), one-way ANOVA with Tukey’s multiple comparisons test (D and H), and log-rank test (E and I) were performed. *, P < 0.05; **, P < 0.01; ****, P < 0.0001; ns, not significant.
The importance of C3 and C3 receptors in nAb -mediated pneumococcal capture. (A) Bacteremia kinetics of antibody receptor-deficient mice i.v. infected with 106 CFU of TH86010A. n = 6. (B) Flow cytometry detection of C3 deposition on TH86010A surface after incubation with serum from μMT or WT mice. (C) Bacteremia kinetics of C1qa−/− and C3−/− mice infected as in A. n = 6. (D) Representative IVM images of liver sinusoids (left) and quantitation of bacteria immobilized on KCs (right) of C1qa−/− and C3−/− mice i.v. infected with 5 × 107 CFU of TH86010A. n = 2. The clearance processes are demonstrated in Video 6. (E) Survival rates of WT, C1qa−/−, or C3−/− mice i.v. infected with 108 CFU of TH86010A. n = 10. (F) Bacteremia kinetics of C3-receptor-deficient mice infected as in A. n = 6. (G and H) Representative IVM images of liver sinusoids (G) and quantitation of bacteria immobilized on KCs (H) of C3-receptor-deficient mice i.v. infected with 5 × 107 CFU of TH86010A. Scale bar, 10 μm. n = 2. The clearance processes are demonstrated in Video 7. (I) Survival of C3- and C3-receptor-deficient mice i.v. infected with 108 CFU of TH86010A. n = 10. (J and K) Bacteremia kinetics (J) and bacterial distribution at 30 min (K) of C5−/− and C9−/− mice infected as in (A). n = 6. (L) Working model of nAbs in driving hepatic clearance of encapsulated bacteria. Invading bacteria in the blood are bound by nAbs, which subsequently activates the complement system. C3 opsonzid bacteria are captured by C3 receptors CR3 and CRIg on KCs. Data were representative results (B, D, G, and H) or pooled from two independent experiments (A, C, E, F, and I–K). Ordinary two-way ANOVA with Tukey’s multiple comparisons test (A, C, F, and J), one-way ANOVA with Tukey’s multiple comparisons test (D and H), and log-rank test (E and I) were performed. *, P < 0.05; **, P < 0.01; ****, P < 0.0001; ns, not significant.
Indispensable role of the complement system in nAb-mediated Spn10A capture. (A) Mass spectrometry results showing enriched complement components by CPS10A-beads compared with that of CPS10A∆wcrG-beads. n = 3. The detailed results are listed in Table S1. (B) Bacterial distribution at 5 min of WT, C1qa−/−, or C3−/− mice i.v. infected with 106 CFU of TH86010A. n = 6. (C) ELISA detection of IgG and IgM to CPS10A in serum of complement- and C3-receptor-deficient mice. n = 5. (D) TH86010A binding to primary mouse KCs with incubation of 10% WT, C1qa−/−, C3−/−, or μMT serum. n = 6. (E) Bacterial distribution at 5 min of C3-receptor-deficient mice i.v. infected with 106 CFU of TH86010A. n = 6. (F) TH86010A binding to primary mouse KCs from C3-receptor-deficient mice with incubation of 10% WT serum. n = 6. Data are representative results (C) or pooled from two independent experiments (A, B, and D–F). Ordinary one-way ANOVA with Tukey’s multiple comparisons test (D and F) was performed. **, P < 0.01; ****, P < 0.0001.

Indispensable role of the complement system in nAb-mediated Spn10A capture. (A) Mass spectrometry results showing enriched complement components by CPS10A-beads compared with that of CPS10A∆wcrG-beads. n = 3. The detailed results are listed in Table S1. (B) Bacterial distribution at 5 min of WT, C1qa−/−, or C3−/− mice i.v. infected with 106 CFU of TH86010A. n = 6. (C) ELISA detection of IgG and IgM to CPS10A in serum of complement- and C3-receptor-deficient mice. n = 5. (D) TH86010A binding to primary mouse KCs with incubation of 10% WT, C1qa−/−, C3−/−, or μMT serum. n = 6. (E) Bacterial distribution at 5 min of C3-receptor-deficient mice i.v. infected with 106 CFU of TH86010A. n = 6. (F) TH86010A binding to primary mouse KCs from C3-receptor-deficient mice with incubation of 10% WT serum. n = 6. Data are representative results (C) or pooled from two independent experiments (A, B, and D–F). Ordinary one-way ANOVA with Tukey’s multiple comparisons test (D and F) was performed. **, P < 0.01; ****, P < 0.0001.
We further validated the contribution of the complement system to KC capture of Spn10A under in vitro conditions using serum from C1qa−/− or C3−/− mice. Both sera from C1qa−/− mice and C3−/− mice were less able to promote KC binding to Spn10A (Fig. S4 D). However, these sera displayed relatively milder phenotypes than that from μMT mice, suggesting that nAbs promote bacterial clearance in the liver by an uncharacterized complement-independent mechanism(s). In a similar manner, IVM imaging showed that KCs of C1qa−/− and C3−/− mice were significantly defective in capturing Spn10A in the liver sinusoids (Fig. 5 D and Video 6). Consistently, C1qa−/− and C3−/− mice succumbed to otherwise nonlethal infection of Spn10A (Fig. 5 E). These results fully demonstrated that the complement classical pathway is vital for the nAb-mediated capture of Spn10A by KCs.
IVM shows diminished KC capture of Spn10A in complement-deficient mice. Pneumococcal capture in the liver sinusoids of WT or Cd19−/− mice i.v. infected with 5 × 107 CFU of TH86010A were shown. Graphic analysis us shown in Fig. 5 D. Playback speed is 25 frames per second.
IVM shows diminished KC capture of Spn10A in complement-deficient mice. Pneumococcal capture in the liver sinusoids of WT or Cd19−/− mice i.v. infected with 5 × 107 CFU of TH86010A were shown. Graphic analysis us shown in Fig. 5 D. Playback speed is 25 frames per second.
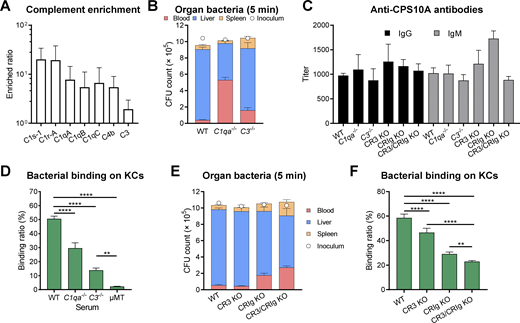
To determine how KCs capture nAb/C3-opsonized pneumococci, we tested the role of CR3 and CRIg, two complement receptors that are significantly expressed on KCs (Helmy et al., 2006). While CR3-deficient (Itgam−/−) mice did not show any obvious defect in early clearance of Spn10A, CRIg-deficient (Vsig4−/−) mice displayed a modest phenotype (Fig. 5 F). However, the simultaneous loss of both receptors (CR3/CRIg KO) yielded a more significant impact on bacterial clearance. In a similar manner, mice with the combined absence of CR3 and CRIg (CR3/CRIg KO) showed the most severe impairment in hepatic capture of Spn10A than the mice lacking either receptor (Fig. S4 E). The functional redundancy of CR3 and CRIg in mediating KC capture of nAb/C3-opsonized pneumococci was also verified with primary KCs. KCs from CR3/CRIg KO mice displayed a lower capacity for bacterial binding than those from the single receptor-deficient mice (Fig. S4 F). Likewise, IVM imaging also revealed more severe deficiency with CR3/CRIg KO mice than CR3- or CRIg-deficient mice in KC capture of Spn10A in the liver sinusoids (Fig. 5, G and H; and Video 7). Accordingly, CR3/CRIg KO mice were more susceptible to infection with Spn10A (Fig. 5 I). The complement activation can also lead to the formation of a membrane attack complex (C5b-9) on target cells, which results in cellular lysis (Densen and Ram, 2015). We thus assessed the contribution of the membrane attack complex to the nAb-driven clearance of Spn10A using C5- and C9-deficient mice since C5 and C9 are essential factors in the formation of membrane attack complex (Densen and Ram, 2015). Both C5−/− and C9−/− mice showed normal clearance of serotype-10A pneumococci (Fig. 5 J) and hepatic capture/killing (Fig. 5 K). This result agrees with the current dogma that the thick peptidoglycan layer of Gram-positive bacteria prevents the complement system from full access to the bacterial cell membrane and thus the formation of the membrane attack complex (Densen and Ram, 2015). These lines of evidence allowed us to propose a working model to explain how anti-capsule antibodies drive hepatic clearance of blood-borne bacteria by engaging the complement system and multiple C3 receptors (Fig. 5 L).
IVM shows diminished KC capture of Spn10A in complement receptor-deficient mice. Pneumococcal capture in the liver sinusoids of WT, CR3 KO, CRIg KO, or CR3/CRIg KO mice i.v. infected with 5 × 107 CFU of TH86010A were shown. Graphic analysis is shown in Fig. 5 G. Playback speed is 25 frames per second.
IVM shows diminished KC capture of Spn10A in complement receptor-deficient mice. Pneumococcal capture in the liver sinusoids of WT, CR3 KO, CRIg KO, or CR3/CRIg KO mice i.v. infected with 5 × 107 CFU of TH86010A were shown. Graphic analysis is shown in Fig. 5 G. Playback speed is 25 frames per second.
Anti-CPS10A nAbs recognize other Galp branch-containing capsules
Our mass spectrometry data (Fig. 3 B and Table S1) revealed significant enrichment of variable regions of multiple monoclonal antibodies that have been previously reported, including XRPC 24 (X24) and XRPC 44 (X44) that are known to recognize β1-6 galactan (Rudikoff et al., 1973). Since CPS10A contains β1-6 galactose, we tested whether these monoclonal antibodies react with CPS10A by generating recombinant IgM and IgG3 forms of X24 and X44 based on the additional sequence information available in the UniProt databases. ELISA results showed specific binding to CPS10A by the IgM and IgG3 forms of X24 and X44 (Fig. 6 A). IgM form of both antibodies had relatively higher levels of binding affinity to CPS10A than the IgG3 counterparts, which might be due to different valences of IgM (pentamer) and IgG3 (monomer).

Recognition of other capsule types by anti-CPS10A antibodies. (A) ELISA detection of X24-IgM, -IgG3 (left) and X44-IgM, -IgG3 (right) binding to CPS10A. n = 3. (B and C) Bacteremia kinetics of μMT mice i.v. treated with 0, 2.5, and 5 μg X24 or X44-IgM (B) and -IgG3 (C) before i.v. infection with 106 CFU of TH86010A. n = 6. (D) ELISA detection of antibodies to CPSs with galactose branch in WT mouse serum. n = 6. (E) Bacteremia kinetics of mice i.v. treated with 400 μg CPS39 before infection, as in B. n = 6. (F) The repeating unit structures of CPSs for serotypes 10A and 39 of S. pneumoniae and serotype K50 of K. pneumoniae. (G) ELISA detection of IgG and IgM to K. pneumoniae K50 and K10 capsules in mouse serum. n = 6. (H) Bacteremia kinetics of mice i.v. inoculated with 800 μg CPS of K. pneumoniae K50 (CPSK50) before i.v. infection with 106 CFU of K. pneumoniae K50. n = 6. Data were all pooled from two independent experiments. Ordinary two-way ANOVA with Tukey’s multiple comparisons test (B, C, E, and H) and one-way ANOVA with Tukey’s multiple comparisons test (G) were performed. ***, P < 0.001; ****, P < 0.0001.
Recognition of other capsule types by anti-CPS10A antibodies. (A) ELISA detection of X24-IgM, -IgG3 (left) and X44-IgM, -IgG3 (right) binding to CPS10A. n = 3. (B and C) Bacteremia kinetics of μMT mice i.v. treated with 0, 2.5, and 5 μg X24 or X44-IgM (B) and -IgG3 (C) before i.v. infection with 106 CFU of TH86010A. n = 6. (D) ELISA detection of antibodies to CPSs with galactose branch in WT mouse serum. n = 6. (E) Bacteremia kinetics of mice i.v. treated with 400 μg CPS39 before infection, as in B. n = 6. (F) The repeating unit structures of CPSs for serotypes 10A and 39 of S. pneumoniae and serotype K50 of K. pneumoniae. (G) ELISA detection of IgG and IgM to K. pneumoniae K50 and K10 capsules in mouse serum. n = 6. (H) Bacteremia kinetics of mice i.v. inoculated with 800 μg CPS of K. pneumoniae K50 (CPSK50) before i.v. infection with 106 CFU of K. pneumoniae K50. n = 6. Data were all pooled from two independent experiments. Ordinary two-way ANOVA with Tukey’s multiple comparisons test (B, C, E, and H) and one-way ANOVA with Tukey’s multiple comparisons test (G) were performed. ***, P < 0.001; ****, P < 0.0001.
Importantly, both the IgM form (Fig. 6 B) and IgG3 form (Fig. 6 C) of the antibodies significantly accelerated early clearance of serotype-10A pneumococci in antibody-deficient μMT mice in a dose-dependent manner when they were i.v. inoculated 2 min prior to bacterial infection. However, the IgM antibodies were in general more effective than the IgG3 counterparts when both antibody forms were used at the same concentrations. While the IgM antibodies were still effective at the dose of 2.5 µg (Fig. 6 B), the IgG3 proteins showed only marginal impact on pneumococcal clearance at this dose (Fig. 6 C). In the context of the functional dependence of nAbs on the complement system (Fig. 5 C), this functional difference could be caused by the higher potency of IgM antibodies in C3 activation (Hajishengallis et al., 2017). These results indicated that anti-CPS10A nAbs and previously described anti-β1-6 galactan antibodies belong to the same pool of anti-polysaccharide nAbs in mice.
Many pneumococcal serotypes contain galactose branches in the capsular repeating units (Geno et al., 2015), including α1-2 galactopyranose (Galp) in serotypes 15B, 15C and 33F, α1-3 Galp in serotype 12F, α1-4 Galp in serotype 17F, β1-4 galactofuranose (Galf) in serotype 20B, and β1-6 Galp in serotype 39 (Fig. S5). We thus determined if anti-CPS10A nAbs react with any of these galactose branch-containing capsules. ELISA test revealed significant binding between purified serum IgG or IgM and serotype-39 CPS (CPS39), but no obvious antibody binding was detected with the other six serotypes (Fig. 6 D). To determine whether the same nAbs bind to both CPS10A and CPS39, we performed in vivo competitive blocking of Spn10A clearance with free CPS39. Pretreatment with CPS39 remarkably inhibited early clearance of Spn10A (Fig. 6 E), which was to a similar extent to what was observed with CPS10A (Fig. 2 A). This finding is consistent with the presence of a β1-6-linked Galp branch and an identical glycan composition in both CPS10A and CPS39, although there are significant differences between the two serotypes in glycan linkage and acetylation (Petersen et al., 2014) (Fig. 6 F). Through an additional search, we also realized that the capsule of serotype-K50 Klebsiella pneumoniae (CPSK50) also contained the terminal β1-6-linked Galp (Altman and Dutton, 1983) (Fig. 6 F). ELISA revealed significant binding of CPSK50 to purified serum IgG and IgM (Fig. 6 G). Consistently, pretreatment of mice with CPSK50 significantly blocked early clearance of Spn10A (Fig. 6 H). These results uncovered broad recognition of bacterial capsules by circulating nAbs.

Structure of repeating unit of pneumococcal CPSs with galactose branch. Structure of repeating units of serotype-10A, -12F, -15B, -15C, -17F, -20B, -33F, and -39 capsule are shown in which galactose branches are labeled.

Structure of repeating unit of pneumococcal CPSs with galactose branch. Structure of repeating units of serotype-10A, -12F, -15B, -15C, -17F, -20B, -33F, and -39 capsule are shown in which galactose branches are labeled.
Anti-capsule nAbs enhance hepatic clearance of multiple encapsulated bacteria
Since anti-CPS10A nAbs also recognized CPS39, we determined whether the nAbs played a role in the early clearance of serotype-39 pneumococci (Spn39). In contrast to the severe impairment of antibody-deficient μMT mice in clearing Spn10A (Fig. 4 A), Spn39 bacteria were similarly cleared between the mutant and WT mice (Fig. 7 A). This result suggested that Spn39 bacteria are cleared by nAb-mediated and additional redundant mechanisms or that the nAb-mediated in vitro CPS39 binding does not operate in vivo. A previous study has shown that nAbs significantly enhance hepatic clearance of enteropathogenic E. coli in the absence of the complement system (C3−/− mice) (Zeng et al., 2018). We thus tested early clearance of Spn39 in C3−/− μMT mice lacking both antibodies and C3. The double knockout mice displayed severe deficiency in shuffling Spn39 bacteria from the blood (Fig. 7 A) to the liver (Fig. 7 B), indicating the anti-capsule nAbs and complement system represent two unique immune mechanisms against Spn39. In line with this conclusion, C3−/− mice exhibited a significant but partial defect in the early clearance of this bacterium. The impact of these dual immune mechanisms on hepatic clearance of Spn39 was also observed by IVM imaging of the liver sinusoids. Spn39 bacteria were abundantly captured by KCs of C3−/− and μMT mice but completely bypassed the hepatic trapping in C3−/− μMT mice (Fig. 7, C and D; and Video 8).

Antibody-mediated hepatic clearance of serotype-39 S. pneumoniae and serotype-K50 K. pneumoniae. (A and B) Bacteremia kinetics (A) and bacterial distribution at 30 min (B) of WT, μMT, C3−/−, or C3−/− μMT mice i.v. infected with 106 CFU of TH1682739. n = 6. (C and D) Quantitation of bacteria immobilized on KCs (C) and representative IVM images of liver sinusoids (D) of WT, μMT, C3−/−, or C3−/− μMT mice i.v. infected with 5 × 107 CFU of TH1682739. Scale bar, 10 μm. n = 2. The clearance processes are demonstrated in Video 8. (E and F) Bacteremia kinetics (E) and bacterial distribution at 30 min (F) of WT, μMT, C3−/−, or C3−/− μMT mice i.v. infected with 106 CFU of TH17033K50. n = 6. Data were representative results (C and D) or pooled from two independent experiments (A, B, E, and F). Ordinary two-way ANOVA with Tukey’s multiple comparisons test (A and E) and one-way ANOVA with Tukey’s multiple comparisons test (C) were performed. ***, P < 0.001; ****, P < 0.0001; ns, not significant.
Antibody-mediated hepatic clearance of serotype-39 S. pneumoniae and serotype-K50 K. pneumoniae. (A and B) Bacteremia kinetics (A) and bacterial distribution at 30 min (B) of WT, μMT, C3−/−, or C3−/− μMT mice i.v. infected with 106 CFU of TH1682739. n = 6. (C and D) Quantitation of bacteria immobilized on KCs (C) and representative IVM images of liver sinusoids (D) of WT, μMT, C3−/−, or C3−/− μMT mice i.v. infected with 5 × 107 CFU of TH1682739. Scale bar, 10 μm. n = 2. The clearance processes are demonstrated in Video 8. (E and F) Bacteremia kinetics (E) and bacterial distribution at 30 min (F) of WT, μMT, C3−/−, or C3−/− μMT mice i.v. infected with 106 CFU of TH17033K50. n = 6. Data were representative results (C and D) or pooled from two independent experiments (A, B, E, and F). Ordinary two-way ANOVA with Tukey’s multiple comparisons test (A and E) and one-way ANOVA with Tukey’s multiple comparisons test (C) were performed. ***, P < 0.001; ****, P < 0.0001; ns, not significant.
IVM shows diminished KC capture of Spn39 in nAb-deficient mice. Pneumococcal capture in the liver sinusoids of WT, μMT, C3−/−, or C3−/− μMT mice i.v. infected with 5 × 107 CFU of TH1682739 were shown. Graphic analysis is shown in Fig. 7 D. Playback speed is 25 frames per second.
IVM shows diminished KC capture of Spn39 in nAb-deficient mice. Pneumococcal capture in the liver sinusoids of WT, μMT, C3−/−, or C3−/− μMT mice i.v. infected with 5 × 107 CFU of TH1682739 were shown. Graphic analysis is shown in Fig. 7 D. Playback speed is 25 frames per second.
We finally determined whether anti-capsule nAbs contribute to host clearance of serotype-K50 K. pneumoniae, a major Gram-negative pathogen in hospital-acquired infections (Paczosa and Mecsas, 2016). As observed with Spn39 (Fig. 7 A), the early clearance of K. pneumoniae was marginally affected by the loss of either antibody (in μMT mice) or C3 protein (in C3−/− mice) (Fig. 7 E). However, the simultaneous absence of antibody production and C3 protein in C3−/− μMT mice led to a more pronounced defect in clearing blood-borne serotype-K50 K. pneumoniae. Consistently, the liver of C3−/− μMT mice showed a relatively lower level of bacterial burden as compared with those of C3−/− or μMT mice (Fig. 7 F). Therefore, anti-CPS10A nAbs contribute to the clearance of bacteria with β1-6-linked Galp branch in the capsule. Taken together, these data revealed that anti-capsule nAbs can confer broad protection against multiple encapsulated bacteria.
Discussion
While receptor-mediated bacterial capture by KCs in the liver is vital for host blood sterility and health, only a few such receptors are known. As a matter of fact, the scavenger receptor ASGR is the only capsule receptor that mediates KC capture of encapsulated bacteria, which are frequently associated with bloodstream infections and septic deaths (GBD 2019 Antimicrobial Resistance Collaborators, 2022). With the lead of our recent findings that KCs are able to recognize the LV capsule types (An et al., 2022; Huang et al., 2022), this study has shown that the anti-β1-6 galactan nAbs recognize the capsules of serotype-10A and -39 S. pneumoniae and serotype-K50 K. pneumoniae. More importantly, the nAb–capsule interactions enable KCs to capture these pathogens in the liver. Our data have highlighted that nAbs serve as potent capsule receptors for KC capture of potentially virulent encapsulated bacteria.
nAbs are the serotype-specific receptors for multiple bacterial capsules
nAbs are known to fulfil various immune functions, such as clearance of apoptotic cells, regulation of B cell immune responses and broad defense against invading pathogens (New et al., 2016). nAbs are known to recognize broad spectrum of bacteria, fungi, viruses, and parasites (Choi and Baumgarth, 2008; Sarden et al., 2022; Subramaniam et al., 2010; Yilmaz et al., 2014; Zeng et al., 2018). The nAbs recognizing pneumococcal cell wall phosphocholine is the prototype of anti-microbial nAbs, mainly owing to the early availability of anti-phosphocholine hybridoma antibodies (Andres et al., 1981; Gearhart et al., 1981). More importantly, the anti-phosphocholine antibodies are protective against the bloodstream infections of S. pneumoniae (Arai et al., 2011; Briles et al., 1981a, 1981b, 1992). Zeng et al. have recently reported that nAbs against O127 LPS of enteropathogenic E. coli drive pathogen capture by KCs (Zeng et al., 2018). This study revealed the abundant presence of IgM and IgG3 antibodies toward the capsules of important human pathogens: S. pneumoniae and K. pneumoniae.
Multiple lines of evidence indicate that the anti-capsule antibodies belong to the category of nAbs. First, the anti-capsule antibodies are IgM and IgG3, which is in line with the dominance of these two antibody subtypes in nAbs (Palma et al., 2018). Second, the antibodies found in this work recognize CPSs. This functional property agrees with the fact that most of nAbs in human and mouse recognize glycan antigens (New et al., 2016). Third, we found that the anti-capsule antibodies were barely detectable in Cd19−/− mice that lack B-1a cells. nAbs are mainly produced by B-1 cells and marginal zone B cells, which distinguishes antigen-induced antibodies produced by B-2 cells (Martin et al., 2001). Finally, there are similar levels of the anti-capsule antibodies between the GF and SPF mice, which exclude the possibility of microbiota-induction as described for the nAbs against E. coli LPS (Zeng et al., 2018).
nAbs to CPSs enable KCs to capture circulating bacteria
KCs are extremely capable of capturing the LV capsule types of encapsulated bacteria (An et al., 2022; Huang et al., 2022), but ASGR is the only characterized capsule receptor. In this study, we first verified the LV phenotype of Spn10A isolates and their rapid capture by KCs in the liver. We further demonstrated CPS10A as a specific ligand for KC recognition by competitive blocking of bacterial clearance with purified CPS10A. These lines of information led to the discovery of nAbs as CPS10A-binding proteins by affinity pulldown and protein mass spectrometry. The immune function of anti-CPS10A nAbs was defined by significant impairment of antibody-deficient mice (μMT and Cd19−/− mice) in early blood clearance and hepatic interception of Spn10A. IVM imaging allowed us to visualize and quantify the antibody-dependent immune action of KCs in capturing fast-flowing bacteria in the liver sinusoids. This nAb-mediated KC capture of Spn10A is reminiscent of our recent finding that vaccine-induced antibodies enable KCs to capture the otherwise “uncatchable” HV capsule types of S. pneumoniae (Wang et al., 2023). Zeng et al. have recently shown that nAbs make KCs able to capture enteropathogenic E. coli by recognizing O127 LPS (Zeng et al., 2018). While the authors did not explicitly characterize the contribution of potential nAb–capsule interactions to the phenotype, it appears that the capsule-mediate binding is, at least partially, responsible for the KC capture of Xen-14 E. coli since the strain utilizes the same polysaccharide repeating unit for LPS and capsule biosynthesis. To this end, it is reasonable to believe that nAbs are important for the hepatic clearance of invading bacteria.
Anti-β1-6 galactan nAbs broadly recognize β1-6-linked Galp on multiple CPSs
nAbs are long known to confer protective immunity against microbial infections, but the precise mechanisms of nAb-mediated protection are mostly speculative at the molecular level. A major hurdle against comprehensively studying nAb-mediated antibacterial immunity is the lack of pathogen-specific antigens recognized by nAbs and appropriate model systems. Built on our recent finding that liver macrophages effectively capture serotype 10A and other LV capsule types of S. pneumoniae in a receptor-dependent manner in a mouse blood infection model (An et al., 2022; Huang et al., 2022), we identified CPS10A-binding nAbs by the affinity pulldown approach and subsequently verified the functional importance of these antibodies in activating KC capture of Spn10A in the liver sinusoids. Extended experiments also revealed a rather broad immune recognition of the capsules from additional encapsulated Gram-positive (serotype-39 S. pneumoniae) and Gram-negative (serotype-K50 K. pneumoniae) bacteria. More importantly, nAb-mediated capsule recognition activates pathogen capture and killing by KCs in the liver. Our mutagenesis work in the capsule biosynthesis gene locus of Spn10A initially demonstrates the functional importance of the β1-6-linked Galp on CPS10A in KC capture of the bacteria. Our later affinity pulldown with CPS10A-coated beads dramatically enriched nAbs that resemble the variable regions of the β1-6-linked galactan-binding nAbs, particularly XRPC-24 and XRPC-44. Consistent with the nature of nAbs, the purified preparations of these antibodies from mouse serum were identified to be the IgM and IgG3 forms. Interestingly, both the IgM and IgG3 forms of the nAbs rescued the functional defect of antibody-deficient mice in clearing Spn10A.
The XRPC-24 and XRPC-44 monoclonal antibodies were originally identified as the IgA form from myeloma in BALB/c mice following intraperitoneal injection of mineral oil and were found to bind with β1-6-linked galactotetraose (Potter et al., 1972; Rudikoff et al., 1973). In the following two decades, the amino acid sequences of these antibodies and other β1-6-linked galactan-binding nAbs were determined (Rao et al., 1979; Rudikoff et al., 1980, 1983, 1984). This information served as an important clue for the functional characterization of the CPS10A-binding nAbs in this work. We generated recombinant forms of XRPC-24 and XRPC-44 in both the IgM and IgG3 antibody types and demonstrated that these antibodies behave in the same way as the CPS10A-binding nAbs isolated from mouse serum. This and other lines of information have provided convincing evidence that the CPS10A-binding nAbs represent the same antibody repertoire as XRPC-24 and XRPC-44, which recognize the β1-6-linked Galp epitope.
The realization of the antigenic nature of the CPS10A-binding nAbs made it possible to search for other encapsulated bacteria with the β1-6-linked Galp moiety on their capsule polysaccharide repeats. This led to our subsequent investigation of serotype-39 S. pneumoniae and serotype-K50 K. pneumoniae, which were found to contain the β1-6-linked Galp moiety in the repeating units of the capsules in the Carbohydrate Structure Database (Toukach and Egorova, 2016). On another level, subsequent experiments confirmed the functional contribution of the CPS10A-binding nAbs to hepatic clearance of serotype-39 S. pneumoniae and serotype-K50 K. pneumoniae. While it remains to be determined if the CPS10A-binding nAbs recognize additional polysaccharides, the β1-6-linked Galp moiety is a common component of E. coli LPS O antigens (e.g., O46, O64, O124, O131, O134, O164, and O171) (Liu et al., 2020). In the context of the previous finding that nAbs to the O127 antigen of enteropathogenic E. coli activate KC capture of the circulating bacteria (Zeng et al., 2018), it is reasonable to believe that the CPS10A-binding nAbs represent a broad type of pattern-recognition receptors that enable KCs to capture invading bacteria and perhaps other microbes bearing the β1-6-linked Galp moiety.
nAbs mediate KCs capture of encapsulated bacteria by complement-dependent and -independent mechanisms
Antibodies typically mediate microbial elimination through binding interactions with Fc receptors on immune cells once their variable regions bind to specific antigens on the surface of microbes (West and Kemper, 2022). This principle is manifested by our recent finding that vaccine-induced IgG antibodies immobilize the otherwise liver-resistant capsule types of S. pneumoniae to the surface of liver sinusoidal endothelial cells, leading to effective bacterial capture and killing (Wang et al., 2023). However, our limited investigation has not identified specific Fc receptors that convey the nAb-opsonized bacteria to KCs. The IgG3-mediated KC capture of serotype-10A S. pneumoniae fully operated in mice lacking all of the four known phagocytic Fc receptors for IgG (FcγRI-IV). Likewise, the deletion of two known IgM receptors in mice did not compromise the IgM-mediated KC capture of the bacterium. While it remains to be determined if other uncharacterized Fc receptor(s) on KCs is responsible for physically engaging the nAb-opsonized pneumococci, the available data support an important role of the complement system in executing the nAb-initiated bacterial capture by KCs. This is manifested by delayed bacterial clearance in C3-deficient mice.
Antigen binding of IgG3 and IgM has been shown to activate complement C3 via the C1q-dependent classical pathway and thereby mediates internalization of the immune complex through complement receptors (Azeredo da Silveira et al., 2002; Han et al., 2001; Ogden et al., 2005; Saylor et al., 2010; Weinstein et al., 2015). The current data support the important role of complement receptors CRIg and CR3 on KCs in capturing antibody/C3-opsonized S. pneumoniae. Based on the more pronounced phenotype in CRIg KO mice than CR3-deficient animals, it appears that CRIg plays a more prominent role in immobilizing nAb/C3-opsonized S. pneumoniae. C1q is known to activate C3 via the classical pathway by binding to the Fc region of antibodies once antibody-antigen complexes form (West and Kemper, 2022). The functional deficiency of C1qa KO mice in the nAb-mediated bacterial capture in the liver indicates that the complement classical pathway is involved in the nAb-initiated C3 activation on CPS10A and other nAb-binding capsules. This conclusion is also supported by the CPS10A-mediated pulldown of various components of the classical pathway (e.g., C1q, C1s, C1r, C4b, and C3). However, we noticed that C1q-deficient mice displayed a more severe impairment than C3-deficient mice in the nAb-mediated Spn10A clearance. This information points to an uncharacterized C3-independent mechanism behind the nAb-mediated hepatic antibacterial immunity. This notion is supported by the phenotypic differences among mouse lines lacking C3 and antibodies. The clearance of Spn10A was partially retarded in C3-deficient mice but completely lost in μMT mice.
In this context, we envision that natural IgM and IgG3 antibodies activate KCs via distinct pathways. On one hand, IgM nAb-mediated bacterial capture by KCs more relies on the complement system since pneumococcal polysaccharide vaccine-induced IgM antibodies fully depend on complement receptors CRIg and CR3 to mediate KC capture of HV S. pneumoniae (Wang et al., 2023). It is difficult to imagine that natural IgM antibodies would take on a completely different path to initiate bacterial capture by KCs. On the other hand, IgG3 may functionally engage an uncharacterized Fc receptor(s) on KCs. The early study reported the existence of a mouse IgG3-specific receptor other than known FcγRs on J774A.1 cell line (Diamond and Yelton, 1981). Although mouse FcγRI was reported to bind with mouse IgG3-coated sheep erythrocytes with low affinity (Gavin et al., 1998), mouse IgG3-mediated phagocytosis of Cryptococcus neoformans was not affected in the absence of FcγRs (Saylor et al., 2010). Integrin β1 was regarded as part of a mouse IgG3 receptor, despite no evidence of a direct interaction (Hawk et al., 2019). The nAb-mediated KC capture of Spn10A may provide a new model for identifying novel IgG3 Fc receptor(s) since the nAbs alone confer a strong functional phenotype—shuffling Spn10A from the bloodstream to the liver sinusoids.
The anti-capsule nAbs may have profound implications for the control of encapsulated bacterial diseases
Invasive infections by encapsulated bacteria represent a major threat to public health because many encapsulated bacteria are resistant to many or nearly all of the clinical antimicrobials, such as Acinetobacter baumannii, E. coli, and K. pneumoniae. As manifested by the broad recognition of the β1-6-linked Galp moiety on multiple capsules by the anti-CPS10A antibodies, broad-spectrum monoclonal antibodies against certain common antigenic epitopes of CPSs may be developed as an alternative to antibacterials for future treatment of drug-resistant encapsulated pathogens when the routine treatment practice become obsolete.
Materials and methods
Bacterial strains, cultivation, and genetic manipulation
The bacterial strains used in this research are listed in Table S2. Pneumococci were grown in Todd-Hewitt broth supplemented with 0.5% yeast extract (THY) or tryptic soy agar plates with 5% defibrated sheep blood at 37°C with 5% CO2 as described (Lu et al., 2006). K. pneumoniae strains were grown in Luria-Bertani (LB) broth or on LB agar plates. For animal infection, bacteria were grown to OD620 nm of 0.5–0.55, stored as frozen stocks, and diluted to desirable concentrations in Ringer’s solutions as inoculum. The precise concentration of each inoculum was determined by plating the inoculum immediately before infection.
Pneumococcal ∆wcrG and ∆cps mutants were generated in serotype-10A strain TH860 by natural transformation as described (Liu et al., 2019). TH860 was transformed with PCR fragments of the rpsL1 allele to gain streptomycin resistance (Sung et al., 2001). The wcrG of TH860 was then replaced with Janus Cassette segments containing a rpsL+ allele by using homologous recombination, followed by counterselection with the 1,500-bp homologous flanking regions of wcrG. The cps locus of TH860 was replaced by the chloramphenicol resistance gene from plasmid pIB166 (Biswas et al., 2008). The relevant primers (Pr18745-Pr18750 for ∆wcrG and Pr10489, Pr10492, Pr19929-Pr19932 for ∆cps) are listed in Table S3. Pneumococcal capsule-switch strains were constructed as described (An et al., 2022).
Mouse infection
All infections were performed in 6–8-wk-old female C57/BL6 or ICR mice obtained from Vital River unless otherwise specified. GF mice and SPF mice that were used in the same controlled experiment were purchased from GemPharmatech. All experiments on mice were performed according to the animal protocols approved by the Institutional Animal Care and Use Committees at Tsinghua University. All genetically modified mice were maintained in a C57/BL6 background. C3−/− and μMT mice were obtained from the Jackson Laboratory. CR3 KO (Itgam−/−) mice were generated by CRISPR/Cas9 as described (Wang et al., 2023). CRIg KO (Vsig4−/−) mice were acquired from Genentech. C1qa−/−, Cd19−/−, and Fcamr−/− mice were obtained from GemPharmatech. Fcmr−/− mice were purchased from the Model Organisms Center. Mice that lack all mouse FcγR α-chain genes (FcRα null mice) were generally provided by professor Jeffrey V. Ravetch (Smith et al., 2012). Clec4f-DTR mice were generated and generally provided by Professor Martin Guilliams (Scott et al., 2016). Depletion of KCs in Clec4f-DTR mice was achieved by i.v. injection of 25 ng/g DT to mice 24 h prior to infection. The other mice with multiple genetic modifications were obtained by cross-breeding.
Animal survival was monitored for 7 days or at a humane endpoint (body weight loss >20%). Bacteremia level was assessed by retroorbital bleeding and CFU plating. For bacterial early clearance, bacteremia level was assessed at various time points after i.v. infection. The CT50 values were calculated using the formula , in which plateau and K were generated through one-phase association nonlinear regression analysis of clearance ratio of each strain using GraphPad Prism as described previously (An et al., 2022). Bacteria in different organs were enumerated by plating homogenates of the liver, spleen, lung, kidney, and heart. To determine the factors that affect bacterial clearance, CPSs, serum, and purified antibodies in a volume of 100 μl were i.v. injected to mice 2 min before i.v. infection if necessary. The volumes of intraperitoneal, i.v. and intranasal infection were 200, 100, and 30 μl, respectively.
IVM
IVM imaging of the mouse liver was performed as described (An et al., 2022). Briefly, 108 CFU of pneumococci were stained in 100 μl PBS containing 10 μg FITC (Sigma-Aldrich) in the dark for 30 min. The hepatic microvasculature and KCs were stained by i.v. injection of 2.5 μg AF594 anti-CD31 and AF647 anti-F4/80 antibodies, respectively, for 30 min before i.v. inoculation with 5 × 107 CFU of FITC-labeled S. pneumoniae. This high infection dose was used in IVM imaging because lower doses yielded fewer bacteria passing through the liver blood vessels under the lens of the microscope, likely due to rapid phagocytosis of captured bacteria by KCs. A similar high infection dose was used in the previous IVM study (Zeng et al., 2018).
Images were acquired with a Leica TCS SP8 confocal microscope using 10×/0.45 NA and 20×/0.80 NA HC PL APO objectives. The microscope was equipped with Acousto Optics without filters. Fluorescence signals were detected by photomultiplier tubes and hybrid photodetectors (600 × 600 pixels for time-lapse series and 1,024 × 1,024 pixels for photographs). Three laser excitation wavelengths (488, 585, and 635 nm) were employed by white light laser (1.5 mw, Laser kit WLL2, 470–670 nm). Real-time imaging was monitored for 1 min after infection. Five to ten random fields of view at 10–15 min were selected to calculate the bacteria number per FOV.
Capsule staining
Pneumococcal capsules were stained using Anthony’s staining method (Anthony, 1931). Briefly, S. pneumoniae was suspended in 10% milk and made a smear on the slide. The smear was air-dried and stained with 1% crystal violet for 2 min. The slide was then rinsed with 20% copper sulfate and air-dried. The slide was examined under the oil immersion objective of a light microscope.
CPS purification
Purification of pneumococcal CPSs was described with modification (Lee et al., 2020). Briefly, pneumococci were grown in THY medium and lysed with sodium deoxycholate solution. The pH of the lysate was adjusted to 5.0 to precipitate most soluble proteins. After centrifugation, the supernatant containing CPS was subjected to ultrafiltration against 25 mM sodium acetate to remove cell wall polysaccharide. The CPS was then precipitated by 3% hexadecyl trimethyl ammonium bromide (CTAB) and resuspended in 1 M NaCl solution. Ethanol was sequentially added to the solution to a final concentration of 33% to remove impurities and 80% to precipitate pneumococcal CPSs. CPS of K. pneumoniae was extracted as described (Huang et al., 2022).
Screening of CPS10A-binding proteins
The identification of CPS-binding proteins by affinity pulldown of CPS-coated beads to serum proteins was performed as described with modifications (An et al., 2023). Briefly, CPSs were incubated with 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM; Sigma-Aldrich). The mixture was then ultrafiltered to remove extra DMTMM and mixed with carboxyl latex beads (2 μm; Invitrogen). The beads were blocked in PBS with 1% bovine serum albumin. The membrane proteins of liver non-parenchymal cells (NPCs) were purified using the Mem-PER Plus Membrane Protein Extraction Kit (Thermo Fisher Scientific).
To isolate CPS-binding proteins, 1 × 108 CPS-coated beads were mixed with 100 μl freshly isolated mouse serum and 100 μg purified membrane proteins of liver NPCs in a total of 500 μl PBS at 37°C for 1 h. The beads were washed and then heated at 96°C for 5 min to dissociate CPS-binding proteins. The dissociated proteins were then separated on SDS-PAGE gels to excise protein bands for in-gel digestion. The protein mass spectrometry and analysis were performed as described (An et al., 2022). Protein abundance was compared between the CPS10A- and CPS10A∆wcrG-coated beads and proteins with more than twofold preference for CPS10A beads were regarded as CPS10A-binding candidates.
In vitro bacterial binding to KCs
KCs were isolated by a collagenase–DNase digestion procedure as described (Li et al., 2014). Briefly, mouse liver was perfused from the portal vein with digestion buffer containing HBSS with 0.5 mg/ml collagenase IV (Sigma-Aldrich), 20 μg/ml DNase I (Roche), and 0.5 mM CaCl2 and then digested in digestion buffer for 30 min at 37°C. The homogenate of the liver was filtered through 70-μm strainers and washed. The residue red blood cells were lysed by RBC lysis buffer (BioLegend) and hepatocytes were removed by centrifugation at 50 g for 1 min. The liver NPCs in the supernatant were collected.
Bacterial binding to mouse KCs was performed as described (An et al., 2022). The liver NPCs were seeded into a 48-well plate in RPMI 1640 and incubated for 30 min at 37°C to enrich KCs. The adherent cells (KCs) were then incubated with 1 × 105 CFU of bacteria (MOI = 1), 10% mouse serum, and 100 μg/ml CPS (if necessary) in each well. The plate was then centrifugated at 500 g for 5 min and incubated for 30 min at 37°C with 5% CO2. The number of unbound bacteria in the culture supernatant was determined by plating. The KCs were lysed with sterile water to determine the number of bound bacteria. The bacterial binding ratio was equal to the ratio of the bound bacteria number to the total bacteria number after the incubation.
To visualize in vitro bacterial binding to KCs, the NPCs were blocked by 1% anti-mouse CD16/CD32 for 10 min and then stained with AF647 anti-F4/80 (1:200; BioLegend) for 20 min. The 8 × 105 cells/chamber stained cells were seeded into four-chamber 35-mm glass bottom dishes with a 20-mm microwell (Cellvis) and cultured as mentioned in 48-well plates. After non-adherent cells were removed, 2 × 106 CFU of bacteria (MOI = 10), 20% mouse serum, and 100 μg/ml CPS (if necessary) were added to each chamber in a total volume of 500 μl. The dishes were then centrifugated at 500 g for 5 min and incubated for 30 min at 37°C with 5% CO2. The images were acquired using the Leica TCS SP8 confocal microscope randomly and pictures with >100 bacteria were chosen for quantification.
Enzyme-linked immunosorbent assay (ELISA)
CPS-specific antibody was quantified by ELISA as described (Tian et al., 2024). Briefly, 96-well plates were coated with CPSs and blocked with 5% non-fat dry milk (BD Difco). The mouse serum was serially diluted and added into CPS-coated wells for a 2-h incubation. The detection of anti-CPS antibodies in mouse serum was performed by incubation with HRP-conjugated goat anti-mouse IgG (1:2,000, BE0102-100; EasyBio) or HRP-conjugated goat anti-mouse IgM (1:2,000, E-AB-1008; EasyBio). To determine the subtypes of mouse anti-CPS10A IgG, HRP-conjugated IgG1-, IgG2b-, IgG2c-, and IgG3-specific antibodies (1:2,000, RRID: AB_2890964, AB_2890966, AB_2890967, AB_2890968; Proteintech) were used. Unimmunized children’s sera have been described (Wang et al., 2022). The detection of anti-CPS10A IgM in human serum was performed by incubation with HRP-conjugated goat anti-human IgM (1:2,000, E-AB-1009; Elabscience). The absorbance at a wavelength of 450 nm was detected after incubation with TMB substrate (TIANGEN) and termination. The titer was calculated as the highest dilution degree with a positive/negative ratio >2.
Fluorescence imaging of antibody deposition
Briefly, 108 CFU of pneumococci were stained in 100 μl PBS containing 10 μg AF647-NHS ester (Invitrogen) for 30 min at room temperature (RT). The bacteria were washed three times with PBS, followed by incubation with 50 μl serum for 30 min at RT. After a three-time wash with PBS, the bacteria were incubated with FITC-conjugated goat anti-mouse IgG (1:2,000, BE0111-100; EasyBio) or FITC-conjugated goat anti-mouse IgM (1:2,000, E-AB-1067; Elabscience) for 1 h. Fluorescence images were acquired using the Leica TCS SP8 confocal microscope.
Flow cytometry
Briefly, 108 CFU of pneumococci were incubated in 100 μl PBS containing 20% serum from WT or μMT mice. For detection of antibody deposition, the bacteria were incubated with FITC-conjugated goat anti-mouse IgG (BE0111-100; EasyBio) or FITC-conjugated goat anti-mouse IgM (E-AB-1067; Elabscience) for 20 min. For detection of complement C3 deposition, the bacteria were incubated with HRP-conjugated goat IgG anti-mouse Complement C3 (1:1,000, 0855557; MP Biomedicals) and FITC-conjugated rabbit anti-goat IgG (1:2,000, BE0120-100; EasyBio), sequentially. FITC signals on bacteria were detected by flow cytometry.
Antibody production and purification
The total natural IgG was purified from murine serum using Protein G resin (GenScript) according to the manufacturer’s instructions. The total natural IgM was purified from the flow through serum from Protein G resin using Protein L resin (GenScript).
Recombinant monoclonal antibodies were generated as described (Wang et al., 2023). The coding sequences of light chains and heavy chains of X24 and X44 were synthesized based on amino acid sequences (Uniprot: KV6A1_MOUSE, HVM37_MOUSE, KV6A2_MOUSE, HVM39_MOUSE) and cloned into vectors containing constant region for light chain (pTH16770) or constant region for heavy chain of murine IgG3 (pTH16771) as described (Wang et al., 2023). The coding sequences of heavy chains were fused with PCR-amplified J-chain (GenBank: AB644392.1) and the constant region of the heavy chain of murine IgM (CDS of GenBank: BC096667.1) and cloned into the vector. The related primers and plasmids are listed in Tables S3 and S4. The antibodies were produced by cotransfection of the vectors containing light chains and heavy chains to human HEK293 suspension culture cells (Expi293F) and purified with Protein G resin (IgG) and Protein L (IgM) resin, respectively.
Statistical analysis
All experiments were performed with at least two biological replicates. Statistical analyses were performed using GraphPad Prism software (8.3.0). The error bars depict the standard error of mean (SEM). The significance was determined as described in the figure legend. A P value of <0.05 was considered significant.
Online supplemental material
The online supplemental material consists of five figures, four tables, and eight videos. Fig. S1 shows the dominant role of KCs in the clearance of Spn10A, relating to Fig. 1 H. Fig. S2 shows specificity and wide expression of anti-CPS10A antibody, relating to Fig. 3. Fig. S3 shows supporting data to emphasize the essential role of microbiota-independent anti-CPS10A antibodies in the clearance of Spn10A, relating to Fig. 4. Fig. S4 shows data to support complement system involved in nAb-mediated bacterial clearance, relating to Fig. 5. Fig. S5 shows the chemical structure of capsules used in Fig. 6 D. Table S1 shows the screening result of CPS10A-binding proteins by mass spectrometry. Tables S2, S3, and S4 summarize bacterial strains, primers, and plasmids used in this study, respectively. Video 1 shows the dominant role of KCs in the capture of Spn10A, relating to Fig. 1 H. Video 2 illustrates the inhibition of CPS10A and CPS10A∆wcrG to KC capture of Spn10A, relating to Fig. 2 L. Videos 3, 4, and 5 show the essential and specific role of nAbs in clearance of Spn10A, relating to Fig. 4. Videos 6 and 7 support complement system involved in nAb-mediated bacterial clearance, relating to Fig. 5. Video 8 shows the essential role of nAbs in clearance of Spn39, relating to Fig. 7.
Data availability
All data associated with this study are available in the paper or the online supplemental material.
Acknowledgments
We thank M. Guilliams (Ghent University, Ghent, Belgium) for providing Clec4f-DTR mice, J.V. Ravetch (The Rockefellor University, New York, NY, USA) for providing FcRα null mice, and the Tsinghua University Microbiology Core for flow cytometry, Center for Proteomics for protein mass spectrometry, and Laboratory Animal Research Center for animal experimentation.
This work was supported by grants to J.-R. Zhang from the National Key Research and Development Program of China (2023YFc2306301), the National Natural Science Foundation of China (82330071), and the Tsinghua University Initiative Scientific Research Program (20243080033).
Author contributions: X. Tian: Conceptualization, Investigation, Methodology, Project administration, Validation, Visualization, Writing - original draft, Writing - review & editing, Y. Liu: Formal analysis, Project administration, K. Zhu: Methodology, H. An: Formal analysis, Methodology, Validation, J. Feng: Resources, L. Zhang: Conceptualization, Formal analysis, Investigation, Resources, Supervision, Writing - review & editing, J.-R. Zhang: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing - original draft, Writing - review & editing.
References


