


Cytokine release syndrome (CRS) is a frequently observed side effect of chimeric antigen receptor (CAR)-T cell therapy. Here, we report self-regulating T cells that reduce CRS severity by secreting inhibitors of cytokines associated with CRS. With a humanized NSG-SGM3 mouse model, we show reduced CRS-related toxicity in mice treated with CAR-T cells secreting tocilizumab-derived single-chain variable fragment (Toci), yielding a safety profile superior to that of single-dose systemic tocilizumab administration. Unexpectedly, Toci-secreting CD19 CAR-T cells exhibit superior in vivo antitumor efficacy compared with conventional CD19 CAR-T cells. scRNA-seq analysis of immune cells recovered from tumor-bearing humanized mice revealed treatment with Toci-secreting CD19 CAR-T cells enriches for cytotoxic T cells while retaining memory T-cell phenotype, suggesting Toci secretion not only reduces toxicity but also significantly alters the overall T-cell composition. This approach of engineering T cells to self-regulate inflammatory cytokine production is a clinically compatible strategy with the potential to simultaneously enhance safety and efficacy of CAR-T cell therapy for cancer.
Introduction
Chimeric antigen receptor (CAR)-T cell therapy has demonstrated remarkable successes in the treatment of relapsed or refractory hematological malignancies (Majzner and Mackall, 2019). Multiple FDA-approved products are now available as second-line therapy for B-cell lymphoma, reflecting increasing confidence in the efficacy and safety of CAR-T cell therapy for cancer. However, the administration of CAR-T cell therapies remains limited to major medical centers to date, in part due to the potential for severe and acute toxicities that require the availability of comprehensive intensive care units (Gutierrez et al., 2020; Neelapu, 2019). Consequently, CAR-T cell therapy remains out of reach for patients who cannot access major medical institutions.
A common toxicity associated with CAR-T cell therapy is cytokine release syndrome (CRS), characterized by sudden and dramatic elevations of inflammatory cytokines and immunomodulatory proteins (Lee et al., 2019; Davila et al., 2014; Majzner and Mackall, 2019). CRS of varying severity is commonly observed in patients treated with CD19 CAR-T cell therapy (Fowler et al., 2022; Abramson et al., 2020; Neelapu et al., 2022), and it has also been reported for CAR-T cell therapy targeting HER2, BCMA, and mesothelin (Morgan et al., 2010; Berdeja et al., 2021; Tanyi et al., 2017). Severe CRS has been identified as the root cause of multiple patient deaths (Brentjens et al., 2010; Morgan et al., 2010), and the severity of CRS has also been associated with an increased incidence of infectious complications (Hill et al., 2018; Park et al., 2018), highlighting a significant need to develop effective preventive, diagnostic, and therapeutic methods for CRS management.
At present, tocilizumab and corticosteroids are the most common treatments for CRS, with additional agents such as etanercept and infliximab serving as less frequently used alternatives (Brudno and Kochenderfer, 2016; Lee et al., 2014). Corticosteroids have pleiotropic effects on the immune system, which may compromise T-cell treatment efficacy by non-specifically inhibiting the antitumor functions of engineered T cells (Maude et al., 2014a; Davila et al., 2014). Tocilizumab, a neutralizing antibody for interleukin-6 receptor α (IL-6Rα), was first considered as a treatment for CRS based on the observation that patients undergoing CRS after CAR-T cell transfer had dramatically elevated levels of IL-6 (Davila et al., 2014; Grupp et al., 2013; Lee et al., 2015; Maude et al., 2014b; Porter et al., 2015). In 2017, the administration of tocilizumab became an FDA-approved strategy for CRS management in adoptive T-cell therapy (Si and Teachey, 2020).
Although corticosteroids and tocilizumab often show clinical efficacy in ameliorating CRS, current methods for CRS diagnosis rely on close monitoring of clinical signs such as body temperature and blood pressure, which are indirect measurements of CRS that can be confounded by comorbidities and other clinical factors. The clinical community has made a concerted effort to establish a standardized grading system for the diagnosis of CRS (Lee et al., 2019) and to develop algorithms that can quantify a patient’s propensity for CRS based on the expression of predictive biomarkers (Hay et al., 2017; Teachey et al., 2016). However, clinical grading systems developed to date remain reliant on monitoring clinical signs or serum cytokine levels, and predictive algorithms are not yet able to replace precise and timely diagnosis at the bedside.
As a precaution against the possibility of runaway immune responses and fatal CRS symptoms, numerous systems incorporating suicide genes have been proposed (Di Stasi et al., 2011; Wang et al., 2011; Diaconu et al., 2017; Hong et al., 2020). However, such strategies necessitate the termination of T-cell therapy, and it remains unclear whether triggering the suicide process would lead to sufficiently rapid reversal of life-threatening CRS and improve patient outcomes. Finally, the prophylactic administration of tocilizumab is also under evaluation, with preliminary results suggesting a decrease in severe CRS but no statistically significant difference in overall CRS occurrence (Caimi et al., 2021; Locke et al., 2017). Although generally well tolerated, tocilizumab does contain a black-box warning for serious infections (Si and Teachey, 2020; Kroschinsky et al., 2017). These results underscore the challenge of precisely modulating CRS and associated toxicities through systemic, exogenous manipulation of cytokine signaling.
As an alternative, we hypothesized that tumor-reactive T cells engineered to self-modulate cytokine signaling activities could reduce CRS occurrence and severity, consequently reducing the need for external mitigation measures for CRS. Indeed, it has been shown that expression of membrane-bound IL-6 receptor on CAR-T cells allows engineered T cells to neutralize IL-6 signaling, although the system was evaluated in immunodeficient mice and thus unable to formally assess effects on CRS (Tan et al., 2020). Separately, CD19 and BCMA CAR-T cells engineered to express anti-IL-6 scFv and IL-1 receptor antagonist (IL-1Ra) have been tested in the clinic (Xue et al., 2021). Results from this trial indicate expression of anti-IL-6 single-chain variable fragment (scFv) and IL-1Ra did not compromise CAR-T cell efficacy, but the strategy also did not reduce the overall incidence of CRS (18/18 patients experienced CRS) nor the frequency of severe CRS (4/18 patients experienced grade-3 CRS) compared with conventional CD19 and BCMA CAR-T cell therapies for the same indications (Berdeja et al., 2021; Munshi et al., 2021; Shah et al., 2021; Wang et al., 2020). It is noteworthy that the preclinical evaluation of this strategy was similarly limited to the use of immunodeficient mouse models, which could not meaningfully assess the occurrence and thus amelioration of CRS (Xue et al., 2021).
CRS is a systemic immune response that involves not only CAR-T cells but also endogenous immune cells such as monocytes and macrophages. As such, standard tumor xenograft models in NOD/scid/γ−/− (NSG) are inadequate for the investigation of CRS and its modulation. A prior report by Giavridis et al. (2018) demonstrated that intraperitoneal (i.p.) injection of high-dose Raji lymphoma cells followed by i.p. injection of high-dose CD19 CAR-T cells in SCID-beige mice can trigger symptoms resembling CRS. However, little to no human IL-6 was produced in this model, consistent with the fact that T cells are not the major producer of IL-6 (Obstfeld et al., 2017; Singh et al., 2017). Another study by Norelli et al. (2018) made use of NSG-SGM3 mice humanized with hematopoietic stem and progenitor cells (HSPCs) isolated from human cord blood and demonstrated the ability to induce CRS-like symptoms including fever and weight loss, as well as the production of human IL-6. We hypothesize that the use of humanized mouse models can enable the identification of superior CAR-T cell designs with the ability to modulate CRS while retaining robust antitumor efficacy. In particular, we posit that a reliable model can support not only the evaluation of a single construct but also the head-to-head comparison of multiple designs to identify a winning configuration.
Here, we report the engineering of primary human CAR-T cells that secrete scFvs or antagonist proteins against IL-1, IL-6Rα, tumor necrosis factor (TNF)-α, and interferon (IFN)-γ and evaluate each design’s ability to buffer the cytokine production and effector functions of CAR-T cells. We observe that CD19 CAR-T cells engineered to secrete a tocilizumab-based scFv (Toci) reduce the severity of toxicities induced by CAR-T cell administration in tumor-bearing humanized mice, resulting in a more favorable safety profile compared with systemic tocilizumab antibody administration. We further note the unexpected observation that Toci-secreting CAR-T cells exhibit superior in vivo antitumor efficacy compared with conventional CAR-T cells. Single-cell RNA sequencing (scRNA-seq) demonstrates that mice treated with Toci-secreting CAR-T cells benefit from an increased proportion of highly functional T cells enriched in memory phenotype and cytotoxic function while reducing cell-cycle activity. The self-regulating T cells reported here represent a new approach to increasing the safety of adoptive T-cell therapy by providing cell-autonomous means to prevent and control CRS.
Results
Cytokine modulators derived from FDA-approved antibodies neutralize cytokine signaling
CRS is a systemic disorder characterized by the upregulation of a wide variety of cytokines. For example, IFN-γ and TNF-α produced by T cells can lead to the activation of macrophages and endothelial cells, which can in turn release additional proinflammatory cytokines such as IL-6 (Shimabukuro-Vornhagen et al., 2018; Obstfeld et al., 2017; Singh et al., 2017). Furthermore, IL-1 production by monocytes has been shown to increase after CAR-T cell infusion, and IL-1 blockade using anakinra (i.e., the mature IL-1Ra protein with signal sequence removed) was reported to ameliorate CRS severity in mouse models (Norelli et al., 2018). Separately, human CAR-T cells engineered to express murine IL-1Ra were shown to reduce CRS-like symptoms in the SCID-beige mouse model by Giavridis et al. (2018), although it remained unclear whether a fully human system would have similar effects. To evaluate the relative merits of targeting different cytokines, we engineered T cells to secrete soluble inhibitors of IL-6, TNF-α, IFN-γ, and IL-1 signaling and performed head-to-head comparisons of their ability to reduce CRS associated with CAR-T cell therapy (Fig. 1, A and B).
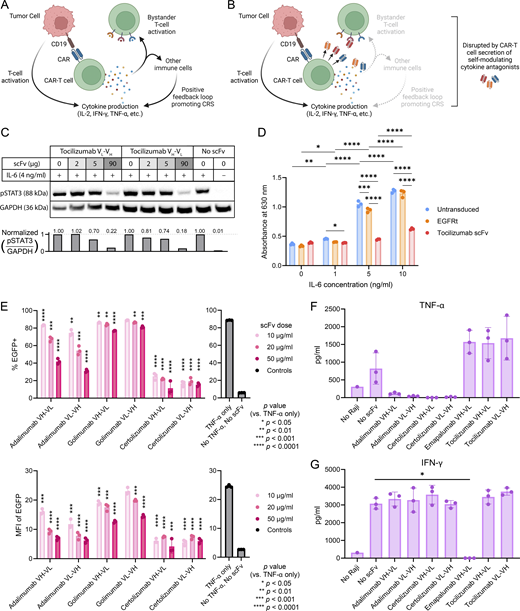
T cells can be engineered to modulate cytokine signaling. (A and B) Schematic of CRS regulation by CAR-T cells engineered to secrete cytokine modulators. Generated with BioRender. (A) Uncontrolled CAR-T cell activation may lead to immune overstimulation and result in CRS. (B) Self-regulating CAR-T cells secrete scFvs or antagonist proteins that block cytokine signaling, thus preventing immune overstimulation. (C–G) Cytokine- and cytokine-receptor–binding scFvs bind their intended targets. (C) Toci blocks IL-6–induced STAT3 signaling in primary human CD4+ T cells. T cells were coincubated with Toci for 3 h and then treated with or without 4 ng/ml human IL-6 protein for 30 min. Cell lysates were analyzed by western blot and probed for phosphorylated STAT3 (pSTAT3) and GAPDH. (D) Toci inhibits IL-6 signaling in IL-6 reporter HEK-Blue cells. IL-6 reporter HEK-Blue cells transfected with plasmids encoding the transduction marker EGFRt orToci were stimulated with the indicated concentration of IL-6 for 24 h. The amount of STAT3-induced SEAP secretion by the HEK-Blue cells was quantified by spectrophotometer. Triplicate wells were separately seeded, transfected, stimulated with IL-6, and quantified. Statistical significance was determined by two-way ANOVA corrected for multiple comparisons using Tukey’s method. (E) Adalimumab- and certolizumab-based scFvs block TNF-α–induced NF-κB signaling. NFκB-EGFP reporter Jurkat cells were incubated with indicated concentrations of TNF-α scFvs overnight before stimulation with 10 ng/ml of TNF-α. EGFP expression induced by NFκB signaling was quantified by flow cytometry, with percent EGFP+ and EGFP MFI shown. Triplicate wells were seeded, treated with the indicated scFvs, and quantified. Statistical significance was determined by two-tailed Student’s t tests comparing each condition against the “TNF-α only” control with correction for multiple comparisons using the two-stage step-up procedure by Benjamini, Krieger, and Yekutieli for false discovery rate < 1.00%. (F and G) Adalimumab- and certolizumab-based scFvs block TNF-α binding (F) and emapalumab-based scFv blocks IFN-γ binding (G). CD19 CAR-T cells were cocultured with Raji cells and donor-matched immune cells (monocytes, macrophages, and dendritic cells) to stimulate cytokine secretion. Addition of scFvs concentrated from HEK293T supernatant binds specific cytokines and blocks their detection by Cytometric Bead Array assay. Triplicate wells were separately seeded, treated with scFvs, and quantified. Pairwise comparisons against the no-scFv control were performed using Welch’s t test, with multiple-comparison correction by the Holm–Sidak method using a significance threshold of 0.05. In panels D–G, means of triplicates are shown with error bars indicating ± 1 standard deviation (SD). Statistical significance levels for all panels: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. The experiments in panels C–E were each performed once. Experiments shown in F and G were performed twice using two different healthy donors’ cells; data from one representative donor are shown. Source data are available for this figure: SourceData F1.
T cells can be engineered to modulate cytokine signaling. (A and B) Schematic of CRS regulation by CAR-T cells engineered to secrete cytokine modulators. Generated with BioRender. (A) Uncontrolled CAR-T cell activation may lead to immune overstimulation and result in CRS. (B) Self-regulating CAR-T cells secrete scFvs or antagonist proteins that block cytokine signaling, thus preventing immune overstimulation. (C–G) Cytokine- and cytokine-receptor–binding scFvs bind their intended targets. (C) Toci blocks IL-6–induced STAT3 signaling in primary human CD4+ T cells. T cells were coincubated with Toci for 3 h and then treated with or without 4 ng/ml human IL-6 protein for 30 min. Cell lysates were analyzed by western blot and probed for phosphorylated STAT3 (pSTAT3) and GAPDH. (D) Toci inhibits IL-6 signaling in IL-6 reporter HEK-Blue cells. IL-6 reporter HEK-Blue cells transfected with plasmids encoding the transduction marker EGFRt orToci were stimulated with the indicated concentration of IL-6 for 24 h. The amount of STAT3-induced SEAP secretion by the HEK-Blue cells was quantified by spectrophotometer. Triplicate wells were separately seeded, transfected, stimulated with IL-6, and quantified. Statistical significance was determined by two-way ANOVA corrected for multiple comparisons using Tukey’s method. (E) Adalimumab- and certolizumab-based scFvs block TNF-α–induced NF-κB signaling. NFκB-EGFP reporter Jurkat cells were incubated with indicated concentrations of TNF-α scFvs overnight before stimulation with 10 ng/ml of TNF-α. EGFP expression induced by NFκB signaling was quantified by flow cytometry, with percent EGFP+ and EGFP MFI shown. Triplicate wells were seeded, treated with the indicated scFvs, and quantified. Statistical significance was determined by two-tailed Student’s t tests comparing each condition against the “TNF-α only” control with correction for multiple comparisons using the two-stage step-up procedure by Benjamini, Krieger, and Yekutieli for false discovery rate < 1.00%. (F and G) Adalimumab- and certolizumab-based scFvs block TNF-α binding (F) and emapalumab-based scFv blocks IFN-γ binding (G). CD19 CAR-T cells were cocultured with Raji cells and donor-matched immune cells (monocytes, macrophages, and dendritic cells) to stimulate cytokine secretion. Addition of scFvs concentrated from HEK293T supernatant binds specific cytokines and blocks their detection by Cytometric Bead Array assay. Triplicate wells were separately seeded, treated with scFvs, and quantified. Pairwise comparisons against the no-scFv control were performed using Welch’s t test, with multiple-comparison correction by the Holm–Sidak method using a significance threshold of 0.05. In panels D–G, means of triplicates are shown with error bars indicating ± 1 standard deviation (SD). Statistical significance levels for all panels: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. The experiments in panels C–E were each performed once. Experiments shown in F and G were performed twice using two different healthy donors’ cells; data from one representative donor are shown. Source data are available for this figure: SourceData F1.
To target IL-6, TNF-α, and IFN-γ signaling pathways, we generated scFvs from several FDA-approved antibodies currently in use for treating inflammatory and autoimmune diseases (Fig. S1 A). Both orientations of light and heavy chains (VL-VH or VH-VL) were evaluated to identify scFvs that can be efficiently produced by human cells. Western blot on producer-cell culture supernatant indicated that all scFv variants except the VL-VH version of emapalumab scFv were efficiently secreted (Fig. S1 B).
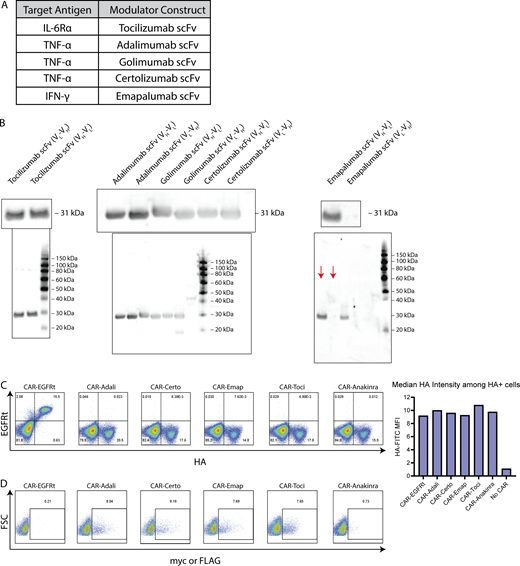
Characterization of cytokine-modulating constructs. (A) scFvs were derived from antibodies reported to bind IL-6Rα, TNF-α, or IFN-γ. (B) The light chain (VL) and heavy chain (VH) was ordered in either VL-VH or VH-VL orientation for each scFv, and fused to the murine kappa chain signal sequence plus an N-terminal FLAG tag (tocilizumab) or c-Myc tag (all other scFvs). HEK293T cells were transfected with plasmids encoding each scFv construct, and culture supernatant was collected 2 days after transfection and analyzed by western blot. The presence of secreted scFv was detected by FLAG (tocilizumab) or c-Myc staining. Full gels are shown below images of zoomed-in bands. Red arrows indicate the relevant lanes in the emapalumab gel; other lanes show samples irrelevant to this study. (C) CAR expression was detected by surface antibody staining for the HA tag. (D) Cytokine modulator expression was detected by intracellular staining for the c-Myc or FLAG tag. Experiments in B–D were each performed once. Source data are available for this figure: SourceData FS1.
Characterization of cytokine-modulating constructs. (A) scFvs were derived from antibodies reported to bind IL-6Rα, TNF-α, or IFN-γ. (B) The light chain (VL) and heavy chain (VH) was ordered in either VL-VH or VH-VL orientation for each scFv, and fused to the murine kappa chain signal sequence plus an N-terminal FLAG tag (tocilizumab) or c-Myc tag (all other scFvs). HEK293T cells were transfected with plasmids encoding each scFv construct, and culture supernatant was collected 2 days after transfection and analyzed by western blot. The presence of secreted scFv was detected by FLAG (tocilizumab) or c-Myc staining. Full gels are shown below images of zoomed-in bands. Red arrows indicate the relevant lanes in the emapalumab gel; other lanes show samples irrelevant to this study. (C) CAR expression was detected by surface antibody staining for the HA tag. (D) Cytokine modulator expression was detected by intracellular staining for the c-Myc or FLAG tag. Experiments in B–D were each performed once. Source data are available for this figure: SourceData FS1.
Next, we evaluated the ability of each scFv to bind its intended target cytokine. Tocilizumab-derived scFvs were shown to inhibit IL-6–induced STAT3 signaling in a dose-dependent manner in primary human CD4+ T cells (Fig. 1 C) and in HEK-Blue IL-6 reporter cells (Fig. 1 D). Similarly, scFvs derived from certolizumab and, to a lesser extent, adalimumab were effective in blocking TNF-α–induced NF-κB signaling in Jurkat cells (Fig. 1 E). Results showed golimumab-derived scFvs were less impactful, thus these designs were eliminated from further consideration. The ability of certolizumab- and adalimumab-derived scFvs to bind TNF-α was further corroborated by cytokine measurements in the supernatant of co-cultures containing CD19+ Raji lymphoma cells and donor-matched CD19 CAR-T cells and immune cells (monocytes, macrophages, and dendritic cells), in which addition of adalimumab- and certolizumab-derived scFvs specifically and efficiently inhibited the detection of TNF-α, albeit without statistical significance due to large intersample variability in the no-scFv control group (Fig. 1 F). In the same assay, IFN-γ detection was specifically and significantly inhibited in samples treated with the emapalumab-derived scFv (Fig. 1 G), confirming its ability to bind IFN-γ.
We next linked a second-generation HA-tagged CD19 CAR containing a 4-1BB costimulatory domain to scFvs derived from tocilizumab, certolizumab, or emapalumab via a self-cleaving T2A sequence (Fig. 2 A). The CD19 CAR was confirmed to have similar surface expression levels regardless of whether an scFv is coexpressed with the CAR (Fig. S1 C). Intracellular staining further confirmed the expression of each scFv in the bicistronic constructs (Fig. S1 D).
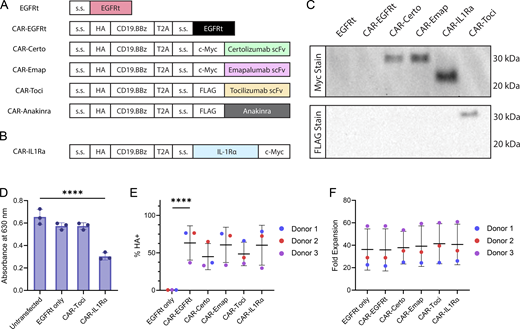
Cytokine-modulating scFvs can be efficiently coexpressed with CD19 CAR in primary human T cells. (A and B) Schematic of CAR constructs coexpressed with cytokine-modulating scFvs and proteins. Each scFv was fused to a murine kappa signal sequence (s.s.) for secretion and an N-terminal c-Myc or FLAG tag to enable detection by western blots. EGFRt and IL-1Ra are fused to the native EGFR and native IL-1Ra s.s., respectively. CARs are fused to the GM-CSF s.s. and an N-terminal HA tag. (C) scFvs and IL-1Ra are efficiently secreted by CAR-T cells. Primary human T cells were retrovirally transduced with constructs depicted in A and B, and culture supernatants were analyzed by western blot. Full gel images are available in online source data. (D) Recombinant IL-1Ra inhibits IL-1 signaling. IL-1 reporter HEK-Blue cells were transfected with plasmids encoding the indicated constructs and stimulated with 10 ng/ml IL-1β for 24 h. The amount of IL-1-induced SEAP secretion by the HEK-Blue cells was quantified by spectrophotometer. Triplicate wells were separately seeded, transfected, treated with IL-1β, and quantified. Statistical significance was determined by one-way ANOVA for comparisons against the untransfected condition using Dunnett’s correction for multiple comparisons (****P < 0.0001). (E and F) Coexpression of cytokine modulator does not compromise CAR expression or CAR-T cell ex vivo expansion. (E) CAR surface expression was measured by anti-HA antibody staining and flow cytometry (****P < 0.0001). (F) CAR-T cell expansion by day 14 of ex vivo cell culture is unaffected by cytokine modulator expression. Data from three different donors are shown. Statistical significance was determined by linear regression analysis with CAR-EGFRt as the reference group, with donor included as covariate to account for donor correlation. There was no statistically significant difference across different CAR-expressing constructs in panels E and F. In panels D–F, means of triplicates are shown with error bars indicating ± 1 SD. The experiments in panels C and D were each performed once. Panels E and F show data from three different donors, each evaluated with triplicate samples once. Source data are available for this figure: SourceData F2.
Cytokine-modulating scFvs can be efficiently coexpressed with CD19 CAR in primary human T cells. (A and B) Schematic of CAR constructs coexpressed with cytokine-modulating scFvs and proteins. Each scFv was fused to a murine kappa signal sequence (s.s.) for secretion and an N-terminal c-Myc or FLAG tag to enable detection by western blots. EGFRt and IL-1Ra are fused to the native EGFR and native IL-1Ra s.s., respectively. CARs are fused to the GM-CSF s.s. and an N-terminal HA tag. (C) scFvs and IL-1Ra are efficiently secreted by CAR-T cells. Primary human T cells were retrovirally transduced with constructs depicted in A and B, and culture supernatants were analyzed by western blot. Full gel images are available in online source data. (D) Recombinant IL-1Ra inhibits IL-1 signaling. IL-1 reporter HEK-Blue cells were transfected with plasmids encoding the indicated constructs and stimulated with 10 ng/ml IL-1β for 24 h. The amount of IL-1-induced SEAP secretion by the HEK-Blue cells was quantified by spectrophotometer. Triplicate wells were separately seeded, transfected, treated with IL-1β, and quantified. Statistical significance was determined by one-way ANOVA for comparisons against the untransfected condition using Dunnett’s correction for multiple comparisons (****P < 0.0001). (E and F) Coexpression of cytokine modulator does not compromise CAR expression or CAR-T cell ex vivo expansion. (E) CAR surface expression was measured by anti-HA antibody staining and flow cytometry (****P < 0.0001). (F) CAR-T cell expansion by day 14 of ex vivo cell culture is unaffected by cytokine modulator expression. Data from three different donors are shown. Statistical significance was determined by linear regression analysis with CAR-EGFRt as the reference group, with donor included as covariate to account for donor correlation. There was no statistically significant difference across different CAR-expressing constructs in panels E and F. In panels D–F, means of triplicates are shown with error bars indicating ± 1 SD. The experiments in panels C and D were each performed once. Panels E and F show data from three different donors, each evaluated with triplicate samples once. Source data are available for this figure: SourceData F2.
For IL-1R targeting, the CD19 CAR was linked to anakinra equipped with murine kappa signal sequence and an N-terminal FLAG tag (Fig. 2 A). However, anakinra was found to be poorly expressed in this format (Fig. S1 D). In response, we reconfigured the construct to express full-length human IL-1Ra with its native signal sequence and labeled the protein with a C-terminal c-Myc tag to minimize disruption of the native sequence (Fig. 2 B). The resulting IL-1Ra protein, similar to the scFv-based constructs, was efficiently secreted by primary human T cells as confirmed by western blot (Fig. 2 C). The ability of secreted IL-1Ra to inhibit NF-κB and AP-1 downstream of IL-1 signaling was further confirmed using HEK-Blue IL-1β reporter cells (Fig. 2 D).
Importantly, coexpression of the cytokine modulators did not compromise CAR expression levels or CAR-T cell expansion during ex vivo culture, indicating compatibility of this engineering strategy with cell manufacturing (Fig. 2, E and F). Since cytokines are a critical part of T-cell mediated antitumor function, we investigated whether the coexpression of cytokine modulators in CAR-T cells would compromise the T cells’ antitumor effector functions. CD19 CAR-T cells with and without secretion of cytokine modulators were subjected to repeated antigen challenge by the addition of fresh CD19+ Raji tumor cells every 2 days. All CAR-T cells showed comparable efficiency in target-cell lysis and T-cell proliferation (Fig. 3 A), demonstrating the in vitro antitumor response of CAR-T cells was not inhibited by the coexpression of cytokine modulators. To further confirm that scFv secretion does not compromise CAR-T cell function, we compared the cytotoxicity of (a) T cells expressing the CD19 CAR plus tocilizumab scFv (CAR-Toci) versus (b) T cells expressing the CD19 CAR plus a non-signaling transduction marker (CAR-EGFRt), each against four different CD19+ target lines across a range of effector-to-target (E:T) ratios. Results show that CAR-Toci cells are largely comparable and occasionally superior in target-cell lysis compared with CAR-EGFRt cells (Fig. 3 B).

Secretion of cytokine-modulating scFv does not compromise on-target cytotoxicity by CAR-T cells. (A) CAR-T cells with and without cytokine modulator secretion exhibit similar cytotoxicity and proliferation response to repeated antigen stimulation. CD19 CAR-T cells or EGFRt-transduced control T cells were stimulated with fresh CD19+, EGFP+ Raji tumor cells every 2 days. Viable EGFP+ tumor cells and viable EGFRt+ or HA+ (CAR+) T cells were quantified by flow cytometry. Two independent experiments using two different donors’ cells were performed with triplicate samples; data from one representative donor are shown. (B) Mock-transduced, CAR-EGFRt, and CAR-Toci cells were coincubated with CD19+, ffLuc-expressing Raji, Ramos, NALM6, and K562 cells at the indicated E:T ratios for 48 h. Viable target cells were quantified by bioluminescence measurement, and percent lysis was calculated by normalizing to the signal from wells treated with mock-transduced T cells. Each coincubation condition was performed in triplicates, with two independent experiments performed using two different donors’ cells. Statistical significance was determined by two-way ANOVA analysis applied to each donor’s CAR-EGFRt and CAR-Toci samples. Significance levels are shown for the effects of CAR construct type, adjusted for E:T ratio in the statistical model: *P < 0.05, ***P < 0.001, ****P < 0.0001. In all figure panels, means of triplicates are shown with error bars indicating ± 1 SD.
Secretion of cytokine-modulating scFv does not compromise on-target cytotoxicity by CAR-T cells. (A) CAR-T cells with and without cytokine modulator secretion exhibit similar cytotoxicity and proliferation response to repeated antigen stimulation. CD19 CAR-T cells or EGFRt-transduced control T cells were stimulated with fresh CD19+, EGFP+ Raji tumor cells every 2 days. Viable EGFP+ tumor cells and viable EGFRt+ or HA+ (CAR+) T cells were quantified by flow cytometry. Two independent experiments using two different donors’ cells were performed with triplicate samples; data from one representative donor are shown. (B) Mock-transduced, CAR-EGFRt, and CAR-Toci cells were coincubated with CD19+, ffLuc-expressing Raji, Ramos, NALM6, and K562 cells at the indicated E:T ratios for 48 h. Viable target cells were quantified by bioluminescence measurement, and percent lysis was calculated by normalizing to the signal from wells treated with mock-transduced T cells. Each coincubation condition was performed in triplicates, with two independent experiments performed using two different donors’ cells. Statistical significance was determined by two-way ANOVA analysis applied to each donor’s CAR-EGFRt and CAR-Toci samples. Significance levels are shown for the effects of CAR construct type, adjusted for E:T ratio in the statistical model: *P < 0.05, ***P < 0.001, ****P < 0.0001. In all figure panels, means of triplicates are shown with error bars indicating ± 1 SD.
CD19 CAR-T cells induce CRS-like toxicities in humanized NSG-SGM3 mice
We next aimed to evaluate the constructs in vivo. The humanized NSG-SGM3 mouse model used in the CRS study by Norelli et al. (2018) represents the most biologically relevant mouse model for CRS reported to date. However, the need to generate CAR-T cells from humanized mice and adoptively transfer them into tumor-bearing mice humanized with the same donor’s HSPCs places a significant constraint on the total number of mice that could be generated, which in turn limits the number of constructs one could simultaneously evaluate in vivo. To support the head-to-head comparison of our panel of constructs, we utilized a modified version of the NSG-SGM3 mouse model, in which NSG-SGM3 mice were first humanized with human fetal liver CD34+ HSPCs and subsequently engrafted with CD19+ Raji cells 3 wk after humanization. 6 days after tumor injection, mice were given tail-vein injections of T cells derived from adult healthy-donor blood (Fig. 4 A). In this system, three potential sources of toxicities could be expected: (a) tumor progression, (b) CRS, and (c) graft-versus-host disease (GvHD) resulting from interactions among adult human CAR-T cells, human immune cells derived from fetal HSPCs, and endogenous murine cells. A pilot study was first performed to determine whether such a mouse model could be used to specifically assess CRS-related toxicity.

Humanized NSG-SGM3 mice exhibit CRS-related toxicity upon CD19 CAR-T cell transfer. (A) Schematic of the humanized NSG-SGM3 mouse model with Raji xenograft and CD19 CAR-T cell therapy. Generated with BioRender. The table summarizes potential sources of toxicity that may be observed for each treatment group. (B–E) Pilot study demonstrates CAR-induced toxicity in NSG-SGM3 mouse model. Weight, temperature, and tumor radiance of Raji tumor-bearing mice without treatment (B; n = 2), with T cells expressing EGFRt only (C; n = 4), with T cells expressing CD19 CAR and EGFRt (D; n = 4), or with T cells expressing CD19 CAR and tocilizumab scFv (E; n = 4). Each trace represents an individual mouse. Weight and temperature were measured at least once daily, and the end of each weight and temperature trace indicates the end point of the mouse. Bioluminescence imaging frequency was limited to no more than once weekly starting 2 days after T-cell injection due to animal welfare concerns for mice experiencing severe toxicity; thus the end of each radiance trace indicates last data point available but does not necessarily correlate with the end point. (F) Mice treated with different T cells exhibit divergent phenotypes at the endpoint, with CAR-EGFRt T cells inducing symptoms consistent with CRS. This study was performed once.
Humanized NSG-SGM3 mice exhibit CRS-related toxicity upon CD19 CAR-T cell transfer. (A) Schematic of the humanized NSG-SGM3 mouse model with Raji xenograft and CD19 CAR-T cell therapy. Generated with BioRender. The table summarizes potential sources of toxicity that may be observed for each treatment group. (B–E) Pilot study demonstrates CAR-induced toxicity in NSG-SGM3 mouse model. Weight, temperature, and tumor radiance of Raji tumor-bearing mice without treatment (B; n = 2), with T cells expressing EGFRt only (C; n = 4), with T cells expressing CD19 CAR and EGFRt (D; n = 4), or with T cells expressing CD19 CAR and tocilizumab scFv (E; n = 4). Each trace represents an individual mouse. Weight and temperature were measured at least once daily, and the end of each weight and temperature trace indicates the end point of the mouse. Bioluminescence imaging frequency was limited to no more than once weekly starting 2 days after T-cell injection due to animal welfare concerns for mice experiencing severe toxicity; thus the end of each radiance trace indicates last data point available but does not necessarily correlate with the end point. (F) Mice treated with different T cells exhibit divergent phenotypes at the endpoint, with CAR-EGFRt T cells inducing symptoms consistent with CRS. This study was performed once.
Four treatment groups were included in this pilot: (1) untreated (PBS only), (2) T cells expressing a transduction marker only (EGFRt), (3) CAR-EGFRt cells, and (4) CAR-Toci cells. In this setup, the untreated group should only experience tumor progression. Mice treated with EGFRt T cells would experience tumor progression and GvHD. Finally, mice treated with CAR-EGFRt or CAR-Toci T cells could experience toxicities related to tumor progression, GvHD, and CRS (Fig. 4 A). However, the CD19 CAR may enable T cells to reduce tumor burden. Furthermore, the CAR-Toci group may experience reduction of CRS symptoms. We hypothesized that CRS-related toxicity could be disentangled from tumor and GvHD-related toxicity by comparing the timing and type of symptoms exhibited by the different treatment groups in this study. In all in vivo experiments in this work, immediately prior to injection into mice, transduced T cells were diluted with untransduced T cells so that every construct has the same percent CAR positivity, and the same number of total T cells as well as the same number of CAR+ T cells were injected into each treatment group.
Mice in the untreated group maintained stable weight and temperature for 7 days before declining due to high tumor burden (Fig. 4 B). Mice treated with T cells expressing only EGFRt reached the humane end point at approximately the same pace as untreated mice, on days 9–11 after T-cell injection. However, unlike untreated mice, animals treated with EGFRt-only T cells experienced a marked decline in weight and a gradual decline in temperature starting 2 days after T-cell injection, consistent with onset of GvHD that compounded the negative health effects of tumor progression (Fig. 4 C). In contrast, animals treated with T cells expressing CAR-EGFRt experienced dramatic and irreversible weight loss within 24 h of T-cell injection, and the weight loss was accompanied by a significant but temporary dip in body temperature (Fig. 4 D). All mice in the CAR-EGFRt group reached the humane end point within 3–4 days after T-cell injection, with CRS-associated symptoms including seizure and uncontrolled bleeding (Fig. 4, D and F). These results indicate that CRS-related toxicities develop substantially faster than GvHD- and tumor-induced fatalities.
Among all mice tested, the only one to survive long-term was in the CAR-Toci group (Fig. 4 E). Animals in this group experienced early and rapid weight decline similar to that exhibited by mice in the CAR-EGFRt group. However, three out of four mice in this group either stabilized or recovered in weight and survived for at least as long as mice in the EGFRt-only and PBS control groups. One mouse fully recovered in weight, eradicated the tumor, and remained healthy and tumor-free when the experiment was terminated at 30 days after T-cell injection. Taken together, these results suggest that the humanized mouse model can induce CRS-associated symptoms subsequent to on-target CAR-T cell therapy and can serve as a test system for evaluating different CRS-modulating strategies.
CD19 CAR-T cells coexpressing tocilizumab scFv reduce CRS-related toxicity in vivo
We next applied the humanized NSG-SGM3 mouse model to compare the performance of CD19 CAR-T cells coexpressing EGFRt or various cytokine modulators, including IL-1Ra and scFvs derived from certolizumab, emapalumab, or tocilizumab. Consistent with prior observations, tumor-bearing humanized mice showed a rapid decline in weight and temperature after receiving T cells expressing CAR-EGFRt, and a similar pattern was observed with CAR-T cells that secrete certolizumab- and emapalumab-based scFvs (Fig. 5, A and B). By comparison, mice treated with CAR-T cells expressing tocilizumab scFv or, to a lesser extent, IL-1Ra remained relatively stable in weight and temperature (Fig. 5, A and B). In this experiment, all animals in both the CAR-Toci and CAR-IL1Ra treatment groups survived the initial toxicity, eradicated the tumor xenograft, and achieved long-term survival (Fig. 5, C and D). However, two mice in the CAR-IL1Ra group, as well as two mice in the CAR-EGFRt group and one mouse in the CAR-emapalumab group, showed ataxic gait in the absence of tumor signal, suggesting neurological toxicity. In contrast, no mice in the CAR-Toci group exhibited those symptoms. These results indicate that engineering CAR-T cells to secrete cytokine modulators, particularly tocilizumab scFv, is a promising strategy to ameliorate CRS without compromising antitumor efficacy.

CAR-T cells secreting IL-6Rα and IL-1R inhibitors reduce CRS-related toxicity and extend survival. NSG-SGM3 mice were humanized as described in Fig. 4 A, engrafted with 10 × 106 Raji lymphoma cells, and treated with 8 × 106 T cells expressing the indicated construct (n = 3 for untreated; n = 6 for all other treatment groups). (A and B) Mice treated with CD19 CAR-expressing T cells experienced rapid (A) weight loss and (B) temperature drop after T-cell injection, but secretion of Toci or IL-1Ra reduced weight loss and almost completely prevented temperature drop. Each trace represents an individual mouse. Green dashed lines represent the baseline level (100% initial weight or 38°C body temperature). Red dashed lines indicate substantial weight loss (85% initial weight) or temperature drop (36°C). (C) Tumor progression was quantified by bioluminescence imaging. Each trace represents an individual mouse. Weight, temperature, and tumor radiance were measured as described in Fig. 3. (D) Kaplan–Meier curve for overall survival. Statistical significance was determined by log-rank (Mantel-Cox) test with the Holm–Sidak correction for multiple comparisons against the CAR-EGFRt group (*P < 0.05). This study was performed once.
CAR-T cells secreting IL-6Rα and IL-1R inhibitors reduce CRS-related toxicity and extend survival. NSG-SGM3 mice were humanized as described in Fig. 4 A, engrafted with 10 × 106 Raji lymphoma cells, and treated with 8 × 106 T cells expressing the indicated construct (n = 3 for untreated; n = 6 for all other treatment groups). (A and B) Mice treated with CD19 CAR-expressing T cells experienced rapid (A) weight loss and (B) temperature drop after T-cell injection, but secretion of Toci or IL-1Ra reduced weight loss and almost completely prevented temperature drop. Each trace represents an individual mouse. Green dashed lines represent the baseline level (100% initial weight or 38°C body temperature). Red dashed lines indicate substantial weight loss (85% initial weight) or temperature drop (36°C). (C) Tumor progression was quantified by bioluminescence imaging. Each trace represents an individual mouse. Weight, temperature, and tumor radiance were measured as described in Fig. 3. (D) Kaplan–Meier curve for overall survival. Statistical significance was determined by log-rank (Mantel-Cox) test with the Holm–Sidak correction for multiple comparisons against the CAR-EGFRt group (*P < 0.05). This study was performed once.
The results above led us to focus on CAR-Toci as the lead candidate in the cytokine-modulator design. However, we noted some discrepancies in the results of the two humanized mouse studies reported thus far. Specifically, the severity of CRS in the CAR-EGFRt group was sufficient to kill all mice within 4 days in the first study (Fig. 4 D), whereas the majority of mice in the CAR-EGFRt group survived for more than 1 month after T-cell infusion in the second study (Fig. 5 D). Similarly, mice in the CAR-Toci group had much higher survival rates in this second study compared with the pilot, although it should be noted that the second study had to be terminated 39 days after T-cell injection due to COVID-related shutdown, and symptoms such as fur loss, jaundice, and increased respiratory effort indicated many of the mice likely would have declined due to progressing GvHD if given enough time.
Humanized mouse models are inherently highly variable, and donor-to-donor variations in both the CD34+ cells used for humanization and the T cells used for CAR-T cell generation could further contribute to variability in experimental outcomes. To ascertain reproducibility of results, we performed three additional humanized mouse studies using different donors’ naïve/memory T (Tnm) cells. Results across the three independent experiments again displayed variability in median survival, but each consistently showed that mice treated with T cells expressing CAR-Toci outperformed the CAR-EGFRt group in survival (Fig. 6), confirming the benefit of tocilizumab scFv secretion in reducing CAR-T cell–induced toxicities. In the final repetition, we further included CAR-T cells engineered to coexpress Toci and IL-1Ra. However, this combination did not improve but instead reduced overall survival (Fig. 6 C), with the majority of animals reaching humane end point with symptoms such as anemia and head tilt, consistent with CRS-like symptoms and neurological toxicity. The construct encoding both Toci and IL-1Ra is larger than the construct encoding Toci or IL-1Ra alone, and this increase in payload size corresponded to a decrease in both CAR positivity and the level of transgene expression among CAR+ cells (Fig. 6 D). As previously noted, in all of our studies, transduced T cells are diluted with donor-matched untransduced T cells to reach the same percent CAR positivity prior to injection into mice. This normalization ensured every treatment group received the same number of total T cells as well as the same number of CAR+ T cells. However, the normalization step could not compensate for lower gene-expression levels on a per-cell basis (i.e., each CAR+ cell in the CAR-Toci-IL1Ra group still expressed lower transgene levels, as quantified by median fluorescence intensity (MFI), than each CAR+ cell in the CAR-Toci or CAR-IL1Ra groups). Reduced gene expression due to a large transgene footprint has been shown to reduce the antitumor efficacy of CAR-T cells (Zah et al., 2020). It is plausible that the reduced gene expression level compromised both the antitumor efficacy and the cytokine-modulating capability of the CAR-Toci-IL1Ra T cells.

CD19 CAR-T cells that co-express Toci consistently result in extended survival in humanized NSG-SGM3 model for CRS. The humanized mouse study was repeated three additional times to compare T cells expressing CD19 CAR (CAR-EGFRt) or CD19 CAR plus Toci (CAR-Toci). Each experiment used fetal HSPCs from a different donor, as well as CAR-T cells from a different adult healthy donor. (A–C) Several Raji tumor doses were tested: (A) 4.5 × 106, (B) 5.0 × 106, and (C) 10 × 106. Mice were treated with 8 × 106 CAR-T cells in every study. Weight, temperature, and radiance data are shown for each study. Each trace represents an individual mouse. Survival data are shown in Kaplan–Meier curves, with statistical significance determined by log-rank (Mantel-Cox) test against the CAR-EGFRt group (P values shown in figure). Holm–Sidak correction for multiple comparisons was applied in C, which did not yield statistical significance. (D) CD19 CAR expression level of cells used in the study shown in C, as detected by surface staining of pre-infusion T-cell suspensions for the CAR’s N-terminal HA tag. Prior to injection, T-cell cultures were diluted with donor-matched untransduced T cells so that each treatment group had the same percent CAR positivity as measured by HA staining. For A, n = 9 mice in CAR-EGFGRt group and n = 10 mice in CAR-Toci group. For B, n = 10 in both groups. For C, n = 4 in CAR-EGFGRt and n = 5 in both CAR-Toci and CAR-Toci-IL1Ra groups. Experiments in this figure were each performed once.
CD19 CAR-T cells that co-express Toci consistently result in extended survival in humanized NSG-SGM3 model for CRS. The humanized mouse study was repeated three additional times to compare T cells expressing CD19 CAR (CAR-EGFRt) or CD19 CAR plus Toci (CAR-Toci). Each experiment used fetal HSPCs from a different donor, as well as CAR-T cells from a different adult healthy donor. (A–C) Several Raji tumor doses were tested: (A) 4.5 × 106, (B) 5.0 × 106, and (C) 10 × 106. Mice were treated with 8 × 106 CAR-T cells in every study. Weight, temperature, and radiance data are shown for each study. Each trace represents an individual mouse. Survival data are shown in Kaplan–Meier curves, with statistical significance determined by log-rank (Mantel-Cox) test against the CAR-EGFRt group (P values shown in figure). Holm–Sidak correction for multiple comparisons was applied in C, which did not yield statistical significance. (D) CD19 CAR expression level of cells used in the study shown in C, as detected by surface staining of pre-infusion T-cell suspensions for the CAR’s N-terminal HA tag. Prior to injection, T-cell cultures were diluted with donor-matched untransduced T cells so that each treatment group had the same percent CAR positivity as measured by HA staining. For A, n = 9 mice in CAR-EGFGRt group and n = 10 mice in CAR-Toci group. For B, n = 10 in both groups. For C, n = 4 in CAR-EGFGRt and n = 5 in both CAR-Toci and CAR-Toci-IL1Ra groups. Experiments in this figure were each performed once.
Tocilizumab scFv secretion provides superior toxicity control than single-dose systemic tocilizumab antibody injection
We next examined whether engineering CAR-T cells to secrete Toci scFv provides an advantage over direct administration of tocilizumab, which is commonly given to patients upon diagnosis of CRS. Compared with antibody injection, scFv-secreting CAR-T cells have the potential advantage of being intrinsically linked to the time and place where cytokine modulation is required—i.e., the scFv is produced whenever and wherever the CAR-T cells are present, capitalizing on CAR-T cells’ ability to locally expand and contract in response to tumor burden. To assess this hypothesis, we compared CAR-Toci cells against CAR-EGFRt cells with or without tocilizumab antibody administration in the humanized mouse model. Raji-bearing humanized mice were treated with CAR-Toci cells or CAR-EGFRt cells. In one CAR-EGFRt group, mice were also given one intravenous injection of 8 mg/kg of tocilizumab 2 days after T-cell injection. The antibody dose matches that used in human patients, and the injection time was chosen based on the typical timeline of weight loss observed in this humanized mouse model (Fig. 4 A and Fig. 5). Consistent with expectations, cases of ≥10% weight loss began to appear by 2 days after T-cell injection (Fig. 7 A). Results indicate that tocilizumab administration failed to halt the acute weight loss and temperature drop induced by CAR-EGFRt cell injection, whereas mice treated with CAR-Toci cells avoided this acute toxicity and achieved the longest median survival (Fig. 7, A–D). Cytokine measurements in peripheral blood collected before and after T-cell injection reveal that scFv secretion prevented an acute spike in IFN-γ and TNF-α levels immediately after T-cell injection (Fig. 7 E). Furthermore, scFv secretion resulted in lower levels of MCP-1, IL-6, IL-8, and IL-18 inflammatory cytokines that increased significantly in the CAR-EGFRt treatment groups by day 16 after T-cell injection and could only be partially tempered by tocilizumab antibody therapy (Fig. 7 E). While it remains possible that further fine-tuning of the timing, frequency, and dosage of tocilizumab administration may improve toxicity control, these results underscore the benefit of a self-regulated cell therapy that can modulate cytokine signaling without the need for external intervention.

Tocilizumab scFv secretion enables superior toxicity control compared to single-dose systemic tocilizumab antibody administration. NSG-SGM3 mice were humanized as described in Fig. 4 A, engrafted with 10 × 106 Raji lymphoma cells, and treated with 8 × 106 T cells expressing either CAR-EGFRt (n = 12) or CAR-Toci (n = 6). Half (n = 6) of the mice treated with CAR-EGFRt cells were given a single 8 mg/kg dose of tocilizumab via tail-vein injection 2 days after T-cell injection. (A) Weight of individual mice in each treatment group normalized to weight on the day of T-cell injection. Weights were measured twice daily, with jaggedness corresponding to natural daily weight fluctuations. Red arrow indicates time of tocilizumab injection. (B) Rectal temperature of individual mice. (C) Tumor radiance as quantified by bioluminescence imaging. Each trace in A–C corresponds to an individual mouse. (D) Kaplan–Meier curve for overall survival. Statistical significance was determined by log-rank (Mantel-Cox) test with the Holm–Sidak correction for multiple comparisons against the CAR-Toci group (**P < 0.01). (E) Cytokine levels in peripheral blood collected on days −4, 1, 6, and 16 relative to the time of T-cell injection. Red dotted lines indicate the limit of detection (LOD), and data points with values below the LOD are plotted on the LOD line. Means are shown with error bars indicating ± 1 SD. Statistical significance was determined by Welch’s ANOVA with Dunnett’s T3 multiple comparisons test. Statistical significance levels for all panels: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. This study was performed twice with two different donors’ cells; data from one donor are shown.
Tocilizumab scFv secretion enables superior toxicity control compared to single-dose systemic tocilizumab antibody administration. NSG-SGM3 mice were humanized as described in Fig. 4 A, engrafted with 10 × 106 Raji lymphoma cells, and treated with 8 × 106 T cells expressing either CAR-EGFRt (n = 12) or CAR-Toci (n = 6). Half (n = 6) of the mice treated with CAR-EGFRt cells were given a single 8 mg/kg dose of tocilizumab via tail-vein injection 2 days after T-cell injection. (A) Weight of individual mice in each treatment group normalized to weight on the day of T-cell injection. Weights were measured twice daily, with jaggedness corresponding to natural daily weight fluctuations. Red arrow indicates time of tocilizumab injection. (B) Rectal temperature of individual mice. (C) Tumor radiance as quantified by bioluminescence imaging. Each trace in A–C corresponds to an individual mouse. (D) Kaplan–Meier curve for overall survival. Statistical significance was determined by log-rank (Mantel-Cox) test with the Holm–Sidak correction for multiple comparisons against the CAR-Toci group (**P < 0.01). (E) Cytokine levels in peripheral blood collected on days −4, 1, 6, and 16 relative to the time of T-cell injection. Red dotted lines indicate the limit of detection (LOD), and data points with values below the LOD are plotted on the LOD line. Means are shown with error bars indicating ± 1 SD. Statistical significance was determined by Welch’s ANOVA with Dunnett’s T3 multiple comparisons test. Statistical significance levels for all panels: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. This study was performed twice with two different donors’ cells; data from one donor are shown.
CD19 CAR-T cells equipped with tocilizumab scFv show superior in vivo antitumor efficacy compared with conventional CD19 CAR-T cells
Although the CAR-Toci design emerged as the lead candidate from the humanized mouse model, IL-1Ra was an additional target of interest as highlighted by multiple prior studies (Giavridis et al., 2018; Norelli et al., 2018; Xue et al., 2021) and its performance in our humanized mouse model. To further evaluate the relative merit of the CAR-Toci and CAR-IL1Ra designs, we next compared their antitumor efficacy in a lymphoma xenograft model. In the humanized mouse studies, animals that survived toxicity associated with CRS and GvHD uniformly eradicated the lymphoma burden (Figs. 3, 4, 5, and 6). However, a high dose (8 × 106) of CAR+ T cells was used in the humanized mouse studies to induce CRS symptoms, and the reconstituted immune system and overall inflammatory state of the mice may have also contributed to tumor rejection. To more stringently assess whether coexpression of the tocilizumab scFv would compromise antitumor efficacy, we utilized the standard Raji lymphoma model in non-humanized NSG mice commonly used for preclinical evaluation of CAR-T cell therapies (Zah et al., 2016a, 2016b; Alizadeh et al., 2019; Baeuerle et al., 2019; Brentjens et al., 2007) (Fig. 8 A). Tumor-bearing mice were treated with 2 × 106 T cells expressing CAR-EGFRt, CAR-Toci, CAR-IL1Ra, or EGFRt only. At this dose, the CAR-EGFRt T cells had little efficacy, resulting in only a slight delay in tumor progression and no significant improvement in survival compared with the EGFRt-only control (Fig. 8, B and C). Animals treated with CAR-IL1Ra T cells showed intermediate tumor control but did not extend median survival relative to CAR-EGFRt T cells. In contrast, animals treated with CAR-Toci T cells exhibited superior tumor control, resulting in a significant increase in survival (Fig. 8, B and C). We confirmed that expression of either CAR-Toci or CAR-IL1Ra did not significantly impact the CD4:CD8 ratios of the resulting T-cell products; thus, the difference in antitumor activity was not due to changes in the frequency of cytotoxic T cells (Fig. 8 D). These results indicate cytokine modulation does not compromise antitumor efficacy, and the secretion of Toci may enhance therapeutic efficacy in vivo.

CAR-T cells secreting tocilizumab scFvs show superior anti-tumor efficacy in in vivo stress test against lymphoma xenograft. (A) Schematic of the Raji lymphoma xenograft model in non-humanized NSG mice. Generated with BioRender. (B) Kaplan–Meier curve for overall survival (n = 6 in each treatment group). Statistical significance was determined by log-rank (Mantel-Cox) test with the Holm–Sidak correction for multiple comparisons against the CAR-EGFRt group (**P < 0.01). (C) Tumor progression was quantified by bioluminescence imaging. Each trace represents an individual mouse. This study was performed once. (D) CD4 and CD8 expression distributions among T cells engineered to express CAR with and without cytokine modulators showed little variation based on construct identity. Thawed cell stocks were surface stained with anti-CD4 and anti-CD8 antibodies and analyzed by flow cytometry. Donor 246’s Tnm cells were used on two separate occasions to generate CAR-T cells (batch 1 and batch 2). No cryopreserved stocks were available for CAR-Toci cells from batch 2 for this analysis. Data from three independent experiments are shown; each test condition was performed with triplicate samples.
CAR-T cells secreting tocilizumab scFvs show superior anti-tumor efficacy in in vivo stress test against lymphoma xenograft. (A) Schematic of the Raji lymphoma xenograft model in non-humanized NSG mice. Generated with BioRender. (B) Kaplan–Meier curve for overall survival (n = 6 in each treatment group). Statistical significance was determined by log-rank (Mantel-Cox) test with the Holm–Sidak correction for multiple comparisons against the CAR-EGFRt group (**P < 0.01). (C) Tumor progression was quantified by bioluminescence imaging. Each trace represents an individual mouse. This study was performed once. (D) CD4 and CD8 expression distributions among T cells engineered to express CAR with and without cytokine modulators showed little variation based on construct identity. Thawed cell stocks were surface stained with anti-CD4 and anti-CD8 antibodies and analyzed by flow cytometry. Donor 246’s Tnm cells were used on two separate occasions to generate CAR-T cells (batch 1 and batch 2). No cryopreserved stocks were available for CAR-Toci cells from batch 2 for this analysis. Data from three independent experiments are shown; each test condition was performed with triplicate samples.
Co-expression of CD19 CAR and tocilizumab scFv enriches for T cells with robust but balanced effector functions in vivo
To better understand how secretion of tocilizumab scFv by CD19 CAR-T cells could both reduce toxicity and enhance antitumor efficacy, we performed scRNA-seq on cells recovered from NSG-SGM3 mice to investigate cell-type and transcriptomic differences mediated by tocilizumab scFv secretion. NSG-SGM3 mice were humanized, engrafted with Raji lymphoma cells, and treated with T cells expressing CAR-EGFRt or CAR-Toci. 8 days after T-cell injection, the liver and spleen of each mouse were harvested, and cells expressing human CD45 were isolated by magnetic bead–based cell sorting prior to sample preparation for scRNA-seq.
The vast majority of detected events were T cells, with only a single cluster of non-T cells comprising neutrophils and macrophages (cluster 7; Fig. 9 A). Aside from this neutrophil/macrophage cluster and a small cluster of CD8+ cytotoxic T cells with chemokine signatures (cluster 8; Fig. 9 A), phenotype distribution is more strongly correlated with whether Toci is expressed than with tissue location (Fig. 9 B). Among the eight T-cell clusters, four were dominated by cell-cycle genes (clusters 1, 2, 4, and 5; Fig. 9 A and Fig. S2). Notably, CAR-EGFRt samples were substantially more enriched in these cell-cycle-dominated clusters compared with CAR-Toci samples (Fig. 9 B). In contrast, CAR-Toci samples were enriched in T-cell clusters associated with cytotoxic and memory phenotypes, characterized by high levels of granzymes, perforin, and granulysin (clusters 0 and 8; Fig. 9 B; and Fig. 2, A and B) as well as markers of both central and effector memory T cells in cluster 0 (Fig. S3). CAR-Toci samples were also enriched for CD4+ and CD8+ T cell clusters with naïve-like phenotypes characterized by TCF7, LEF1, and CCR7 expression (clusters 3 and 6; Fig. S2, B and C).

CAR-T cells secreting tocilizumab scFvs promote the enrichment of cytotoxic T cells with memory signatures in vivo. (A) Pooled scRNA-seq data from the livers and spleens of mice treated with either CAR-EGFRt or CAR-Toci T cells were visualized via UMAP. (B) UMAP split by sample (left) and quantification of cluster frequencies (right) reveal CAR-EGFRt samples are dominated by T cells actively undergoing cell cycling, whereas CAR-Toci samples are enriched in cytotoxic T cells that exhibit signatures of memory phenotypes. (C) Reclustering of scRNA-seq data after regression of cell-cycle features. (D) Reclustered UMAP split by sample. (E) Sample composition by cluster. This study was performed once.
CAR-T cells secreting tocilizumab scFvs promote the enrichment of cytotoxic T cells with memory signatures in vivo. (A) Pooled scRNA-seq data from the livers and spleens of mice treated with either CAR-EGFRt or CAR-Toci T cells were visualized via UMAP. (B) UMAP split by sample (left) and quantification of cluster frequencies (right) reveal CAR-EGFRt samples are dominated by T cells actively undergoing cell cycling, whereas CAR-Toci samples are enriched in cytotoxic T cells that exhibit signatures of memory phenotypes. (C) Reclustering of scRNA-seq data after regression of cell-cycle features. (D) Reclustered UMAP split by sample. (E) Sample composition by cluster. This study was performed once.

Cluster characterization by top genes and lineage T-cell markers. (A) Heatmap of top genes in each cluster stratified by average log2(fold change). (B) Average and percent expression of manually curated lineage T-cell markers within each cluster. (C) Clusters 3 and 6 are enriched for CAR-Toci samples.
Cluster characterization by top genes and lineage T-cell markers. (A) Heatmap of top genes in each cluster stratified by average log2(fold change). (B) Average and percent expression of manually curated lineage T-cell markers within each cluster. (C) Clusters 3 and 6 are enriched for CAR-Toci samples.

Heterogeneous expression of memory and activation markers in CD8 + clusters. Density plots generated using the Nebulosa R package reveal pockets of memory and activation marker expression within CD8+ T-cell clusters.
Heterogeneous expression of memory and activation markers in CD8 + clusters. Density plots generated using the Nebulosa R package reveal pockets of memory and activation marker expression within CD8+ T-cell clusters.
To better characterize the different T-cell subtypes, we further regressed out cell-cycle genes identified by Tirosh et al. (2016) using Seurat’s native CellCycleScoring and ScaleData functions (Fig. 9, C and D; and Fig. S4). Updated clustering confirms the CAR-Toci samples were highly enriched in cytotoxic CD8+ T cells (cluster 2; Fig. 9 E and Fig. S4). Furthermore, CAR-Toci samples were also enriched in cluster 1 (Fig. 9 E), comprising CD4+ T cells with high IL-7R and CD40L expression and strong activation phenotype as indicated by gene ontology (GO) analysis (Fig. 10 A). In contrast, CAR-EGFRt samples were most highly enriched in cluster 0 (Fig. 9 E), comprising CD8+ T cells that exhibit high HLA-DR expression (Fig. S4). HLA-DR+ T cells are capable of antigen presentation provided that co-stimulatory molecules are also expressed (Holling et al., 2004). However, T cells in cluster 0 are devoid of CD80 and CD86 expression (Fig. 10 B), thus antigen presentation by such T cells is likely to trigger anergy rather than productive activation.

Cluster characterization by top genes and lineage T-cell markers following regression of cell-cycle features. (A) Heatmap of top genes in each cluster stratified by average log2(fold change). (B) Average and percent expression of manually curated lineage T-cell markers within each cluster.
Cluster characterization by top genes and lineage T-cell markers following regression of cell-cycle features. (A) Heatmap of top genes in each cluster stratified by average log2(fold change). (B) Average and percent expression of manually curated lineage T-cell markers within each cluster.

Analysis of cell-cycle-regressed scRNA-seq data. (A) Biological Process GO analysis for cluster 1 reveals strong T-cell activation profile. (B) Expression level for activation, exhaustion, and antigen-presentation genes in cluster 0. Expression levels are log-transformed and normalized to total cell transcript content. (C) Pathway network analysis on genes conserved across all clusters for CAR-EGFRt samples. (D) Significance levels for Biological Process GO pathways for CAR-EGFRt samples. (E) Pathway network analysis on genes conserved across all clusters for CAR-Toci samples. (F) Significance levels for Biological Process GO pathways for CAR-Toci samples. The pathways shown in D and F are represented as nodes in CAR-EGFRt and CAR-Toci pathway networks visualized in C and E, respectively. Significance values in D and F were adjusted via g:Profiler’s g:SCS algorithm and thresholded for padj < 10−10. This study was performed once.
Analysis of cell-cycle-regressed scRNA-seq data. (A) Biological Process GO analysis for cluster 1 reveals strong T-cell activation profile. (B) Expression level for activation, exhaustion, and antigen-presentation genes in cluster 0. Expression levels are log-transformed and normalized to total cell transcript content. (C) Pathway network analysis on genes conserved across all clusters for CAR-EGFRt samples. (D) Significance levels for Biological Process GO pathways for CAR-EGFRt samples. (E) Pathway network analysis on genes conserved across all clusters for CAR-Toci samples. (F) Significance levels for Biological Process GO pathways for CAR-Toci samples. The pathways shown in D and F are represented as nodes in CAR-EGFRt and CAR-Toci pathway networks visualized in C and E, respectively. Significance values in D and F were adjusted via g:Profiler’s g:SCS algorithm and thresholded for padj < 10−10. This study was performed once.
Finally, top genes conserved in CAR-EGFRt or CAR-Toci samples were analyzed with g:Profiler (Raudvere et al., 2019) to generate gene enrichment maps (GEMs) for pathway analysis. These networks were visualized in Cytoscape (Shannon et al., 2003) and revealed that T cells harvested from animals treated with CAR-EGFRt T cells were enriched in homeostatic pathways (Fig. 10, C and D). In contrast, T cells from mice treated with CAR-Toci T cells were enriched in T-cell activation and immune response pathways balanced by regulatory mechanisms (Fig. 10, E and F).
The scRNA-seq data yielded substantially more reads for the CAR-EGFRt samples compared with CAR-Toci samples (Fig. 9 B). Coupled with the prominent cell-cycling signatures seen in the CAR-EGFRt group, we examined whether CAR-EGFRt cells in fact expanded better than CAR-Toci cells. Upon inspection, we noted that the scRNA-seq read count did not correspond to the number of huCD45+ cells recovered from the liver and spleen samples; instead, the CAR-EGFRt group emerged with higher counts only after low-quality data points were filtered out during analysis (Fig. S5 A). We had previously collected retro-orbital blood 1 day after T-cell injection into humanized mice from the study shown in Fig. 6 C, and complete blood count analysis showed no difference between the CAR-EGFRt and CAR-Toci groups (Fig. S5 B). Therefore, the increased cell-cycling activity among CAR-EGFRt cells relative to CAR-Toci cells did not lead to a clear increase in the net T-cell count, suggesting the possibility of increased cell death balancing out increased cell division.

CAR-EGFRt and CAR-Toci expression did not significantly alter T-cell count in vivo. (A) Cell counts in scRNA-seq samples. The first filter was applied automatically by the CellRanger alignment software to remove noisy datapoints (i.e., those likely to represent background signal rather than biologically meaningful cell data). The second filter was applied manually according to Seurat vignette instructions to remove low-quality data points (i.e., those with aberrant total or mitochondrial transcriptional content). (B) Complete blood count analysis on retro-orbital blood obtained from humanized mice. Blood samples were collected from mice in the experiment shown in Fig. 6 C, 1 day after T-cell injection. RBC: red blood cell count; HCT: hematocrit or relative volume of erythrocytes; HB: hemoglobin concentration; PLT: platelet count; PCT: platelet hematocrit; RETIC: reticulocyte percentage; MCV: mean corpuscular (erythrocyte) volume; MCHC: mean corpuscular hemoglobin concentration; WBC: white blood cell count; LY: lymphocyte percentage; MO: monocyte percentage; BA: basophil percentage; EO: eosinophil percentage; NE: neutrophil percentage; RDW: red blood (erythrocyte volume) distribution width; PDW: platelet distribution width; RSD: red blood (erythrocyte volume) standard distribution; MPV: mean platelet volume; and MCH: mean corpuscular hemoglobin.
CAR-EGFRt and CAR-Toci expression did not significantly alter T-cell count in vivo. (A) Cell counts in scRNA-seq samples. The first filter was applied automatically by the CellRanger alignment software to remove noisy datapoints (i.e., those likely to represent background signal rather than biologically meaningful cell data). The second filter was applied manually according to Seurat vignette instructions to remove low-quality data points (i.e., those with aberrant total or mitochondrial transcriptional content). (B) Complete blood count analysis on retro-orbital blood obtained from humanized mice. Blood samples were collected from mice in the experiment shown in Fig. 6 C, 1 day after T-cell injection. RBC: red blood cell count; HCT: hematocrit or relative volume of erythrocytes; HB: hemoglobin concentration; PLT: platelet count; PCT: platelet hematocrit; RETIC: reticulocyte percentage; MCV: mean corpuscular (erythrocyte) volume; MCHC: mean corpuscular hemoglobin concentration; WBC: white blood cell count; LY: lymphocyte percentage; MO: monocyte percentage; BA: basophil percentage; EO: eosinophil percentage; NE: neutrophil percentage; RDW: red blood (erythrocyte volume) distribution width; PDW: platelet distribution width; RSD: red blood (erythrocyte volume) standard distribution; MPV: mean platelet volume; and MCH: mean corpuscular hemoglobin.
Taken together, scRNA-seq analysis indicates the overall T-cell population in mice treated with CAR-Toci T cells experienced less cell cycling compared with mice treated with CAR-EGFRt T cells. However, the T cells that were present in CAR-Toci–treated mice were enriched in highly functional phenotypes that combine strong cytotoxicity with memory signatures. This combination is consistent with CAR-Toci T cells’ ability to effectively eradicate tumors while simultaneously reducing toxicity.
Discussion
CRS is a potentially fatal side effect of immunotherapy that has been widely observed in patients treated with CD19 CAR-T cells. Tocilizumab has shown clinical efficacy in the treatment of CRS, but CRS diagnosis and the timing of CRS treatment remains an imprecise process. In this work, we aimed to develop an alternative approach to CRS management by engineering T cells that can lower cytokine signaling without compromising antitumor effector functions, with the goal of generating a therapeutic cell product with significantly reduced risk of CRS.
Results from this study demonstrate that tocilizumab can be converted into an scFv and retain potent inhibitory function against IL-6 signaling. Primary human T cells that express tocilizumab scFv show substantially reduced STAT3 phosphorylation upon IL-6 exposure, indicating effective self-regulation of IL-6 signaling. In a humanized NSG-SGM3 mouse model bearing Raji lymphoma, CD19 CAR-T cells that secrete tocilizumab scFv (CAR-Toci) outperformed T cells expressing CD19 CAR alone or coexpressing CD19 CAR with scFvs targeting TNF-α (certolizumab) or IFN-γ (emapalumab). Consistent with previous reports that IL-1 blockade with anakinra decreased CRS toxicity (Giavridis et al., 2018; Norelli et al., 2018), we found that coexpressing the CD19 CAR with IL-1Ra also resulted in reduced animal mortality. Interestingly, expressing anakinra (i.e., the mature form of IL-1Ra, without its native signal peptide) with a murine kappa signal sequence did not result in appreciable protein production. Instead, full-length IL-1Ra containing its native signal sequence was necessary to yield efficient protein secretion. In this study, we did not observe a substantial reduction in CRS-related toxicity when CD19 CAR-T cells were engineered to secrete scFvs targeting IFN-γ or TNF-α, suggesting IL-6 and IL-1 modulation may be a more effective means of countering CRS symptoms.
IL-6 has been identified as a critical modulator of CRS in patients treated with adoptive T-cell therapy (Lee et al., 2015; Porter et al., 2015; Turtle et al., 2016; Maude et al., 2014a), and it has been shown that IL-6 does not impact the transcriptional profiles or cytotoxicity of CAR-T cells (Tan et al., 2020). Based on these findings, we chose to focus on in-depth characterization of CAR-Toci T cells and demonstrated their ability to exert effective control over CRS without compromising CAR-T cell functions that are essential for antitumor efficacy. In humanized mice bearing Raji tumors, CAR-Toci T cells significantly and reproducibly reduced acute toxicity and extended survival compared with conventional CAR-T cells that did not secrete any cytokine-modulator proteins. Furthermore, in the standard Raji lymphoma model in NSG mice, CAR-Toci cells showed superior tumor control compared with conventional CAR-T cells. Improved antitumor efficacy was not a pre-programmed feature of our design, but this observation echoes the recent finding that IL-6 blockade not only reduces toxicity but also increases the efficacy of immune checkpoint blockade therapy by altering the immune-cell composition in vivo (Hailemichael et al., 2022). Indeed, our scRNA-seq results indicate animals treated with CAR-Toci T cells have a dominant population of cytotoxic T cells that simultaneously exhibit memory phenotypes, leading to the ability to both execute and regulate T-cell effector functions. In contrast, animals treated with conventional CAR-T cells showed strong enrichment of HLA-DR+CD80−CD86− T cells, which can potentially trigger anergy by presenting antigens without co-stimulatory signals. This dichotomy may partially account for the increased anti-tumor efficacy of CAR-Toci T cells compared to conventional CAR-T cells. It is also notable that the presence of HLA-DR+ CD8+ T cells has been shown to correlate with increased severity and therapy resistance of inflammatory diseases such as systemic lupus erythematosus (Viallard et al., 2001) and Kawasaki disease (Wakiguchi et al., 2015). However, formal demonstration of the contribution of HLA-DR+ CD8+ T cells to CRS observed in animals treated with conventional CAR-T cells would require additional analyses, including quantification of their cytokine production capacity and interaction with endogenous immune cells.
Patients with severe CRS symptoms often also experience immune effector cell-associated neurotoxicity syndrome (ICANS), and tocilizumab has not shown efficacy in preventing or treating neurotoxicity (Si and Teachey, 2020). While cytokines such as IL-6 can cross the blood–brain barrier (BBB), antibodies such as tocilizumab have poor distribution in the central nervous system (CNS) (Si and Teachey, 2020). In contrast, CAR-T cells can reach the CNS from peripheral circulation in human patients (Alcantara et al., 2022; Frigault et al., 2019), thus engineering CAR-T cells to locally secrete tocilizumab scFv may enable wider reach of this therapeutic strategy and potentially provide a means to counteract ICANS in addition to CRS. Cases of seizure and ataxia were observed in our humanized NSG-SGM3 mice treated with CD19 CAR-T cells with and without coexpression of cytokine modulators, including IL-1Ra, but no case of neurologic toxicity was observed in mice treated with CAR-Toci cells. Nevertheless, additional experimental systems will be required to fully investigate the potential of cytokine-modulator secretion in reducing CAR-T cell–mediated ICANS. A recent publication reported interim data from a phase-2 trial examining prophylactic anakinra administration for patients receiving commercial CD19 CAR-T cell products, with results indicating that anakinra could cross the BBB and potentially reduce ICANS incidence (Park et al., 2023). The study also shows evidence that the precise timing and dose of anakinra administration plays a key role in clinical outcome and can be dependent on individual patients’ disease and response to therapy. By engineering CAR-T cells to autonomously produce cytokine modulators, whose level waxes and wanes with T-cell expansion—which is itself dynamically responsive to changes in tumor burden and antigen load—one could potentially sidestep the need to precisely monitor and optimize treatment timing for individual patients. The superior toxicity control exhibited by CAR-Toci T cells in our head-to-head comparison against the administration of exogenous tocilizumab upon CRS symptom onset underscores this potential advantage. Of note, in our study, tocilizumab was administered 2 days after T-cell infusion, corresponding to the time when significant weight loss and temperature drop first began to manifest in the CAR-EGFRt treatment groups. This timing decision was based on the fact that tocilizumab is most commonly administered in the clinic after CRS symptoms have been observed. However, IFN-γ and TNF-α levels were already markedly elevated by 1 day after T-cell infusion, illustrating the challenge of determining the optimal timing of intervention based on clinical symptoms such as subject weight and temperature, and highlighting the advantage of a cell-intrinsic cytokine-modulation design.
In addition to Toci, IL1Ra also presented promising activity in reducing toxicity in the humanized mouse model, consistent with prior reports (Giavridis et al., 2018; Norelli et al., 2018). Besides the phase-2 study on prophylactic anakinra administration noted above, preliminary readout on an ongoing clinical trial suggests that administering anakinra to patients experiencing grade ≥2 CRS after CAR-T cell infusion may be effective in preventing severe ICANS (Oliai et al., 2021). However, T cells expressing CAR-IL1Ra did not exhibit the clearly superior anti-tumor efficacy observed with those expressing CAR-Toci in the Raji lymphoma model. This is consistent with clinical data noting no significant impact of anakinra on the anti-tumor efficacy of CAR-T cell therapy (Park et al., 2023). Interestingly, combining Toci and IL1Ra secretion by CAR-T cells resulted in decreased animal survival in the Raji tumor xenograft model, underscoring the fact that additive effects are not always easily achievable in the design of cell-based therapies. To this end, the preclinical models presented in this study may serve as a useful tool in the evaluation of additional designs. For example, it would be of interest to directly compare Toci secretion with the expression of membrane-bound IL-6 receptor (Tan et al., 2020) and evaluate whether the two strategies would have differential impact on both the engineered T cells and surrounding immune cells as well as the tumor microenvironment. Further combinations, such as the clinically evaluated strategy of combining expression of anti–IL-6 scFv and IL-1Ra with genetic knockout of GM-CSF (Yi et al., 2021), could also be assessed to gain an in-depth understanding of each component’s effects. No single preclinical model can fully recapitulate human physiology, and multifaceted evaluations are needed to increase confidence in the translational value of new therapeutic strategies. The combination of human xenograft studies for antitumor efficacy, humanized mouse models for CRS modulation, and transcriptomic studies in humanized mice for overall immune composition and alteration as a consequence of CAR-T cell therapy could provide useful insight into the therapeutic potential of new treatment proposals prior to clinical translation.
Taken together, our study demonstrates that engineering CAR-T cells to self-regulate cytokine signaling through the secretion of cytokine modulators such as Toci can substantively reduce CAR-T cell–induced toxicities. This design strategy has the potential to increase the safety profile of CAR-T cell therapy, potentially paving a path to making this treatment modality more widely available to patients across a wide range of clinical settings.
Materials and methods
Plasmid construction
The heavy and light chain sequences of monoclonal antibodies, besides IL-1Ra, were connected with an 18–amino acid linker to generate cytokine antagonist scFv sequences. A second-generation CD19 CAR containing 4-1BB costimulatory domain was constructed as previously described (Zah et al., 2016b) and connected to the cytokine antagonist scFvs or IL-1Ra with a T2A sequence. All constructs were cloned in MSCV vector, which was a generous gift from Dr. Steven Feldman (National Cancer Institute, Bethesda, MD, USA).
Cell line maintenance
Raji cells (ATCC) were cultured in RPMI-1640 (Lonza) with 10% heat-inactivated FBS (HI-FBS; Life Technologies). Human embryonic kidney 293T (HEK293T) cells (ATCC) were cultured in DMEM (HyClone) with 10% HI-FBS. IL-1β and IL-6 reporter HEK-Blue cells (InvivoGen) were cultured in DMEM with 10% HI-FBS. NFκB-EGFP reporter Jurkat cells were a generous gift from Dr. Xin Lin (MD Anderson, Houston, TX, USA) and were cultured in RPMI-1640 with 10% HI-FBS.
Retrovirus production
HEK293T cells were seeded in 10-cm dishes at 3.5 × 106 cells in 9 ml of DMEM with 10% HI-FBS per dish and transfected by linear polyethylenimine (PEI). 16 h after transfection, cells were washed with 3 ml of PBS (Lonza) without magnesium and calcium and supplemented with 8 ml of DMEM with 10% HI-FBS, 20 mM HEPES, and 10 mM sodium butyrate (Sigma-Aldrich). 8 h later, cells were washed with PBS and supplemented with 7 ml of DMEM with 10% HI-FBS and 20 mM HEPES (DMEM-HEPES). Viral supernatant was collected the following day and filtered through a 0.45-μm filter (VWR). 7 ml of fresh DMEM-HEPES was added to the cells after the supernatant collection. The next day, another viral supernatant was collected, filtered, and combined with the first harvest. The final viral supernatant batch was stored at −80°C until further use.
Generation of CAR-expressing primary human T cells
Primary human T cells (CD3+, CD4+, or CD8+) were isolated from deidentified healthy donor blood obtained from the University of California, Los Angeles (UCLA) Blood and Platelet Center using RosetteSep (Stemcell Technologies) following the manufacturer’s protocol. To obtain CD14−/CD25−/CD62L+ Tnm cells, peripheral mononuclear blood cells (PBMCs) were isolated from healthy donor blood using Ficoll density-gradient separation and subsequently depleted of CD14+ and CD25+ cells before enriching for CD62+ cells by magnetism-activated cell sorting (MACS; Miltenyi). Isolated Tnm cells were stimulated with CD3/CD28 T-cell activation Dynabeads (Life Technologies) at a 1:3 bead-to-cell ratio and transduced with retrovirus 48 and 72 h after stimulation. Dynabeads were removed 7 days after stimulation. T cells were cultured in RPMI-1640 with 10% HI-FBS, 50 U/ml IL-2 (Life Technologies), and 1 ng/ml IL-15 (Miltenyi). Both IL-2 and IL-15 were supplemented every 2–3 days.
Western blots
To generate Toci, HEK293T cells were seeded in 10-cm2 dishes and transiently transfected with plasmids encoding different constructs using PEI. 24 h after transfection, a media change was performed and cells were cultured in serum-free media for another 24 h. Culture supernatant was harvested and concentrated using Amicon Centrifugal filter units (10,000 nominal molecular weight limit; EMD Millipore). For pSTAT3 western blots, primary human T cells were seeded in 12-well plates and incubated with indicated amounts of Toci for 3 h. Indicated wells were subsequently treated with 4 ng/ml of human recombinant IL-6 (BioLegend) for another 30 min before cell harvest. Cell pellets were lysed in 1× radioimmunoprecipitation assay buffer (1% Igepal CA-630, 0.1% SDS, and 0.5% sodium deoxycholate) supplemented with protease and phosphatase inhibitor cocktail tablets (Thermo Fisher Scientific). Samples were run on Bolt 4–12% Bis-Tris Plus gels (Life Technologies) and then transferred to nitrocellulose membranes (Life Technologies). Membranes were blocked with 5% BSA (Amresco) in TBS-T buffer (50 mM Tris, 150 mM NaCl, 0.05% Tween 20) for 1 h at room temperature, then incubated in TBS-T buffer containing pSTAT3 (Y705) antibody (cat: 651002; BioLegend) and 5% BSA overnight at 4°C. GAPDH (cat: G9292; Sigma-Aldrich) was stained as the loading control for all the samples.
For cytokine-modulating construct expression verification, secreted scFvs were concentrated from supernatant of transfected HEK as described above. The presence of secreted scFvs was detected by FLAG (tocilizumab) (cat: F1804-200UG; Sigma-Aldrich) or c-Myc (cat: 130-099-616; Miltenyi Biotec) staining with SNAP i.d. protein detection system (EMD Millipore) following manufacturer’s protocol. Anti-mouse secondary antibody conjugated to horseradish peroxidase (cat: 115-035-062; Jackson ImmunoResearch) was used for both western blots. Images of western blots were visualized using SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific).
Flow cytometry
All flow cytometry experiments were performed with a MACSQuant VYB flow cytometer (Miltenyi). Flow data were analyzed and gated using FlowJo (TreeStar). To verify the expression of different scFvs and anakinra in HEK293T cells, 5 × 104 cells/500 μl/well were seeded in a 24-well plate and transfected with plasmid encoding the scFv or anakinra using PEI. Cells were cultured for 8 h after transfection and then supplemented with 5 µg/ml of Brefeldin A (BioLegend) and 2 µg/ml of monensin (BioLegend) for 20 h. Cells were fixed with 1.5% formaldehyde, permeabilized with ice-cold methanol, stained with antibody binding to FLAG (cat: 130-101-571; Miltenyi Biotec) or myc epitope tag (cat: 130-099-616; Miltenyi Biotec), and assessed by flow cytometry.
Tocilizumab scFv and IL-1Ra functionality assays
IL-1β and IL-6 reporter HEK-Blue cells were seeded in a 24-well plate at 5 × 104 cells/500 μl/well and transfected by linear PEI. 24 h after transfection, 10 ng/ml IL-1β (BioLegend) was added to the IL-1β reporter cells and 1–10 ng/ml IL-6 (BioLegend) was added to the IL-6 reporter cells. 24 h after IL-1β or IL-6 stimulation, levels of secreted embryonic alkaline phosphatase (SEAP) were monitored using QUANTI-Blue (InvivoGen).
PBMC-derived monocytes, macrophages, and dendritic cells
Different immune cell types were prepared to achieve a more diverse cytokine production profile in an in vitro setting. PBMCs from one healthy donor were thawed and recovered for 1 h in RPMI with 10 % FBS, 1× MEM non-essential amino acid (cat: 11140050; Thermo Fisher Scientific), and 1× pen/strep (cat: 15140122; Life Technologies). After the 1-h recovery, suspension cells were gently removed from the flask and adherent cells were kept in culture for monocyte culture. Cells from the suspension fraction were used for either dendritic cell or macrophage differentiation. Dendritic cell culture was supplemented with 0.2 µg/ml of recombinant human IL-4 (cat: 200-04; Peprotech) and 0.2 µg/ml of recombinant human GM-CSF (cat: 300-03; Peprotech). Cells for macrophage differentiation were seeded in 4.0 × 104 cells/200 μl per well in a 96-well plate and used for experimental setup directly. Cell differentiation was confirmed by morphological change and lineage marker expression by flow cytometry.
Anti-TNF-α and IFN-γ scFv functionality assays
To evaluate the effect of anti-TNF-α scFv on TNF-α signaling, NFκB-EGFP reporter Jurkat cells were seeded in 96-well plate at 3 × 104 cells/200 μl/well and incubated with designated concentrations of TNF-α scFvs overnight at 37°C. The following day, 10 ng/ml of TNF-α (Miltenyi) was added to each well. After 24 h, EGFP reporter signal was evaluated by flow cytometry. To evaluate the cytokine-blocking function of anti-TNF-α and anti-IFN-γ scFvs, secreted scFvs were concentrated from supernatant of transfected HEK as described in the Tocilizumab scFv and IL-1Ra functionality assays section above. Macrophages generated as described in the previous section were seeded in 96-well plates, to which 1.5 × 106 WT Raji lymphoma cells, 1.5 × 106 primary human T cells transduced with CD19 CAR (0.75 × 106 cells each of CD4+ and CD8+ T cells), 3,000 dendritic cells, and 3,000 monocytes were added per well. After the cell coculture was set up, scFv equivalent to 180 µg/ml of tocilizumab (calculated based on molar ratio) was added into each well in a 96-well plate. Supernatants was collected after a 12-h incubation and cytokine concentration was measured with the Cytometric Bead Array assay (cat: 551809; BD Biosciences).
Repeated antigen challenge
Primary human T cells expressing CD19 CAR with or without cytokine antagonists were seeded in a 24-well plate at 5 × 105 CAR+ T cells/1 ml/well and coincubated with Raji cells at a 1:1 E:T ratio. The total number of T cells for each construct was normalized with untransduced T cells. Every 2 days, the number of T cells and remaining target cells were evaluated by flow cytometry. Also, 5 × 105 Raji cells were replenished to each well every 2 days after counting.
Luciferase-based cytotoxicity assay
CD19+ Raji, Ramos, NALM6, and K562 cell lines expressing firefly luciferase were seeded in 96-well U-bottom plates at a concentration of 3.0 × 104 cells/50 μl/well. CAR-EGFRt, CAR-Toci, untransduced, and EGFRt-only T cells from two donors were evaluated for percent CAR positivity by flow cytometry. For each donor, the different T cell groups were normalized to contain the same number of total T cells at a concentration of 3.0 × 105 CAR+ T cells/50 μl. Subsequent dilutions were carried out to obtain the appropriate concentrations for the designated E:T ratio before adding 50 μl of T cells to each well of the 96-well plates. After a 48-h coincubation at 37°C, cells were pelleted by centrifugation, washed with PBS, and resuspended in 25 μl of RPMI + 10% HI-FBS before being transferred to an opaque 96-well flat-bottom plate containing 25 μl of Bio-Glo luciferase assay reagent. The samples were incubated at room temperature for 10 min before evaluating luminescence by BioTek Gen5 plate reader.
Isolation of CD34+ HSPCs from fetal liver
Deidentified fetal liver was obtained from the UCLA Gene and Cellular Therapy Core. The tissue was washed with PBS three to four times and dissected into 3-mm3 pieces in IMDM (Thermo Fisher Scientific). The dissected tissue solution was gently resuspended with a 10-ml syringe connected to a 16G-needle five to seven times to homogenize the tissue completely. Next, 250 U/ml collagenase (MP Biomedicals), 1,200 U/ml hyaluronidase (Sigma-Aldrich), 150 U/ml DNase (Sigma-Aldrich), 1X pen/strep (Life Technologies), and 1X amphotericin (HyClone) were added to the homogenized tissue solution, and the solution was incubated in 37°C for 90 min. The tissue solution was filtered through a 100-μm cell strainer. Mononuclear cell isolation was performed using Ficoll density-gradient separation. After the isolation, CD34+ HSPCs were enriched using MACS (Miltenyi).
Humanized NSG-SGM3 mouse model with allogeneic HSPCs and adoptively transferred T cells
All in vivo experiments were approved by the UCLA Animal Research Committee. 6–8-wk-old NSG-SGM3 (NSGTgCMV-IL3, CSF2, KITLG1Eav/MloySzJ) mice (bred by the UCLA Center for AIDS Research Humanized Mouse Core Laboratory) were sublethally irradiated (200 rads) with a Cesium-137 irradiator and injected 3 × 105 CD34+ HSPCs via retro-orbital injection. 3 or 4 wk after humanization, peripheral blood was collected via facial bleeding, and the cells in peripheral blood were stained for CD3, CD14, CD19, and CD45 and analyzed on a MACSQuant VYB flow cytometer (Miltenyi). Mice with more than 20% human cells were considered successfully humanized. Upon confirming humanization, 4.5–10 × 106 firefly luciferase (ffLuc)–expressing Raji cells were injected via tail-vein injection. 7 days after tumor injection, 8 × 106 T cells expressing the indicated transgene were injected via tail-vein injection. (Note: all T cell lines were normalized to the same percent transgene positivity, such that each animal received the same number of EGFRt+ or CAR+ T cells, as well as the same number of total T cells.) Where noted, a single injection of tocilizumab (Genentech) in saline was delivered via tail vein 2 days after T-cell injection at a dose of 8 mg/kg. Peripheral blood was collected via retro-orbital bleeding on the indicated days. Weight and rectal temperature were assessed once or twice daily after T-cell injection. Tumor progression was monitored by bioluminescence imaging once or twice a week after tumor injection.
Raji lymphoma model in NSG mice
7-wk-old NSG mice were obtained from Jackson Laboratories. The protocol was approved by UCLA Institutional Animal Care and Use Committee. Mice were injected with 5 × 105 EGFP+ ffLuc-expressing Raji lymphoma cells by tail-vein injection, randomized into treatment groups based on radiance levels, and subsequently treated with 2 × 106 CAR-T cells or cells expressing EGFRt only (negative control) via tail-vein injection. Tumor progression/regression was monitored with an IVIS Illumina III LT Imaging System (PerkinElmer). Mice were euthanized at the humane end point.
Plasma collection and cytokine measurements
Peripheral blood was collected in EDTA-coated blood collection tubes and centrifuged at 2,000 × g for 10 min at 4°C. Plasma was aliquoted in microcentrifuge tubes and was immediately stored at −80°C until further use. Plasma cytokines were quantified with the BD Cytometric Bead Array Human Th1/Th2 Cytokine Kit II, BD CBA Flex Sets, and LEGENDplex Human Inflammation Panel 1 (13-plex) assay kit (cat: 740809; BioLegend) following the manufacturer’s protocols.
scRNA-seq and data analysis
Spleens and livers were harvested from Raji tumor-bearing humanized NSG-SGM3 mice 8 days after treatment with T cells. Tissues were homogenized to isolate single-cell suspensions, and CD45+ cells were isolated by magnetic bead–based cell sorting. For each treatment group, tissues from two mice were pooled. scRNA-seq libraries were prepared using a 10× Genomics Chromium Controller, and libraries were sequenced on the Illumina NovaSeq SP platform with 50-bp paired-end reads at the UCLA Technology Center for Genomics & Bioinformatics. For each sample, sequencing was performed with the aim of analyzing 10,000 cells at 20,000 reads per cell.
Analysis of sequencing data was guided by Seurat tutorials and vignettes using the Seurat v4.1.1 R package (Hao et al., 2021). Libraries were filtered to exclude events with low total transcript counts, low number of unique transcripts, and high mitochondrial content, each of which can suggest dead/fragmented cells. Up to 2,000 variable features were identified with Seurat’s FindVariableFeatures function using the vst method and were subsequently used to perform integration of liver CAR-EGFRt, spleen CAR-EGFRt, liver CAR-Toci, and spleen CAR-Toci samples. The resultant pooled dataset was then scaled before running principal component analysis (PCA). The top 50 principal components were selected for performing clustering using the default resolution parameter (0.5), and the resultant clustering scheme, either pooled or split by original sample identity, was visualized via uniform manifold approximation and projection (UMAP). Top genes weighted by average log2(fold change), and canonical T-cell phenotypic/lineage markers were visualized for each cluster by heatmap and dot plot, respectively. The Nebulosa v1.6.0 R package (Alquicira-Hernandez and Powell, 2021) was also employed for gene density visualization of memory and lineage markers. Cell cycle features were then regressed out of the pooled dataset using Seurat’s CellCycleScoring and ScaleData functions using previously identified cell-cycle phase gene signatures reported by Tirosh et al. (2016). Identification of variable features, PCA, clustering, and visualization were repeated as described above. Genes positively enriched in each cluster were subjected to GO analysis via g:Profiler (Raudvere et al., 2019) using the GO: Biological Processes GO term set to identify distinguishing pathways. To assess underlying CAR-EGFRt and CAR-Toci pathway patterns, cells were merged across clusters by treatment (liver CAR-EGFRt + spleen CAR-EGFRt and liver CAR-Toci + spleen CAR-Toci) and analyzed for conserved genes. Positively enriched genes (padj < 10−10) for each treatment group were submitted to g:Profiler for pathway analysis to generate GEMs that were subsequently imported into Cytoscape (Shannon et al., 2003) for visualization.
Online supplemental material
Fig. S1 contains characterization data on the expression of cytokine modulators and their coexpression with CD19 CAR. Figs. S2, S3, and S4 show extended details in clustering and cell-cycle analysis of scRNA-seq data. Fig. S5 A contains cell count information of the samples used in scRNA-seq. Fig. S5 B shows a complete blood cell count analysis of samples collected from the experiment shown in Fig. 6 C.
Data availability
The full western blot gels for Fig. 1 C and Fig. 2 C are available as online source files. The scRNA-seq data are available in the NCBI BioProject database with the accession number PRJNA1084011 (individual BioSample accession numbers are SAMN40268892, SAMN40268893, SAMN40268894, and SAMN40268895). The DNA and protein sequences of self-modulators are available in GenBank, with the following accession numbers: cMyc-adalimumab_scFv: PP478640; cMyc-certolizumab_scFv: PP478641; cMyc-wmapalumab_scFv: PP478642; cMyc-tocilizumab_scFv: PP478643; and IL1Ra-cMyc: PP478644. Other datasets associated with the current study are available from the corresponding author upon reasonable request.
Acknowledgments
We thank Dr. Margherita Norelli and Dr. Monica Casucci for their generous input on the NSG-SGM3 humanized mouse model. We thank the UCLA Humanized Mouse Core and Valerie Rezek for generating the humanized NSG-SGM3 mice. We thank Dr. Mobina Khericha Gandhi for technical assistance. We thank Dr. Willy Hugo and Lu Sun for assistance with bioinformatics analyses.
This work was supported by the Hellman Fellows Fund (grant to Y.Y. Chen) and the Parker Institute for Cancer Immunotherapy (grant to Y.Y. Chen). R.M. Shih is supported by the UCLA-Caltech Medical Scientist Training Program (NIGMS T32 GM008042) and the Whitcome Fellowship (fellowship to R.M. Shih).
Author contributions: M.-Y. Lin and Y.Y. Chen designed the project. M.-Y. Lin, E. Nam, A. Shafer, A. Bouren, M. Ayala Ceja, C. Harris, M. Khericha, K.H. Vo, and M. Kim performed experiments. M.-Y. Lin, E. Nam, R.M. Shih, A. Bouren, M. Ayala Ceja, C. Harris, M. Khericha, C.-H. Tseng, and Y.Y. Chen performed data analysis. Y.Y. Chen, M.-Y. Lin, and R.M. Shih wrote and revised the manuscript.

