


Neutrophil infiltration is a hallmark of periodontitis, a prevalent oral inflammatory condition in which Th17-driven mucosal inflammation leads to destruction of tooth-supporting bone. Herein, we document that neutrophil extracellular traps (NETs) are early triggers of pathogenic inflammation in periodontitis. In an established animal model, we demonstrate that neutrophils infiltrate the gingival oral mucosa at early time points after disease induction and expel NETs to trigger mucosal inflammation and bone destruction in vivo. Investigating mechanisms by which NETs drive inflammatory bone loss, we find that extracellular histones, a major component of NETs, trigger upregulation of IL-17/Th17 responses, and bone destruction. Importantly, human findings corroborate our experimental work. We document significantly increased levels of NET complexes and extracellular histones bearing classic NET-associated posttranslational modifications, in blood and local lesions of severe periodontitis patients, in the absence of confounding disease. Our findings suggest a feed-forward loop in which NETs trigger IL-17 immunity to promote immunopathology in a prevalent human inflammatory disease.

Introduction
Neutrophils play key roles in antimicrobial immunity and acute innate immune responsiveness. However, exaggerated neutrophil recruitment and activation are linked to immunopathology in a plethora of disease settings (Burn et al., 2021; Hajishengallis, 2020; Ley et al., 2018; Soehnlein et al., 2017). As such, neutrophils have been implicated in the pathogenesis of diseases of inflammation-triggered bone destruction, such as rheumatoid arthritis (RA) and periodontitis (Dutzan et al., 2018; Hajishengallis, 2020; O’Neil et al., 2020; Wright et al., 2014). Periodontitis is a prevalent human disease that affects the tissues supporting the dentition. In this condition, dysregulated mucosal inflammation triggers destruction of soft tissues and the supporting alveolar bone, leading to tooth loss in severe cases (Hajishengallis, 2015). Beyond local tissue destruction, periodontitis is linked with the occurrence and severity of systemic inflammatory diseases, including cardiovascular disease, diabetes, and RA (Hajishengallis and Chavakis, 2021).
Increased neutrophil recruitment and activation have been thoroughly documented in patients with periodontitis. Neutrophil infiltration is a dominant feature in disease lesions (Dutzan et al., 2016; Hajishengallis and Chavakis, 2021; Landzberg et al., 2015; Williams et al., 2021), and neutrophil hyperactivation has been documented in blood samples of periodontitis patients (Gustafsson et al., 2006; Landzberg et al., 2015). Indeed, neutrophil-mediated immunopathology has been long suspected to be a driving force of disease pathology and demonstrated to be so in animal models of disease (Dutzan et al., 2018). However, the mechanisms by which neutrophils contribute to disease pathology are not fully understood. Recently, neutrophil extracellular traps (NETs) have been proposed as culprits in neutrophil-mediated periodontal immunopathology (Magan-Fernandez et al., 2020; White et al., 2016). NET structures and NET-associated proteins have been documented both in tissue lesions and in circulation of patients with common/chronic forms of periodontal disease (Hirschfeld et al., 2015; Vitkov et al., 2009; White et al., 2016). Furthermore, classic periodontal pathobionts such as Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans have been shown to directly mediate NETs (Bryzek et al., 2019; Hirschfeld et al., 2017).
NETs are extracellular mesh-like structures composed of a backbone of decondensed chromatin (DNA) and decorated with cytosolic and granule neutrophil proteins (Hirschfeld et al., 2017; Papayannopoulos, 2018). Classic NET-associated proteins include histones (bearing distinct posttranslational modifications such as citrullination and carbamylation), neutrophil elastase (NE), and myeloperoxidase. NETs are released by activated neutrophils following stimulation by microbial or sterile inflammatory stimuli, primarily through a cell death process termed NET formation (NETosis; Papayannopoulos, 2018), and can trap and neutralize microbes (Brinkmann et al., 2004), conceivably preventing the spread of bacterial and fungal infections. However, excessive NET formation or impaired NETs clearance is linked to immunopathology in several disorders (Brinkmann et al., 2004; Hidalgo et al., 2022; Papayannopoulos, 2018). Through the process of NETosis, intracellular autoantigens and enzymes are released which can directly damage tissues, thus contributing to the pathogenesis of diseases such as sepsis, thrombosis, atherosclerosis, and autoimmunity (Denning et al., 2019; Kimball et al., 2016; Tsourouktsoglou et al., 2020).
Recently, we have documented fibrin-mediated NETosis in oral tissues in the setting of a genetic defect in fibrinolysis, plasminogen deficiency, which leads to severe periodontitis (Silva et al., 2021). Yet, whether NETs participate in inflammatory bone loss in the absence of a fibrinolytic defect and are relevant to common forms of periodontitis has not been detailed. Furthermore, the mechanisms by which NETs mediate tissue inflammation in periodontitis remain unclear.
Our current work demonstrates a functional role for NETs in the induction of periodontal immunopathology and identifies extracellular histones as a pathogenic factor in triggering disease. Using the established model of ligature-induced periodontitis (LIP), we find that NETs accumulate within the disease lesions and demonstrate that inhibition/removal of NETs and/or inhibition of extracellular histones significantly protects from periodontal bone loss. Intriguingly, our work connects NET-mediated immunopathology with the induction of a well-established pathogenic mechanism in periodontitis, namely IL-17/Th17 inflammation (Gaffen and Moutsopoulos, 2020). While IL-17/Th17 immunity has been established as a pathogenic mechanism in periodontitis (Dutzan et al., 2018; Eskan et al., 2012; Xiao et al., 2017), our current work demonstrates that neutrophils contribute to the initial triggering of Th17 responses, at least partially, through NETosis. Indeed, NETs are detected in periodontitis tissues prior to the accumulation of Th17 cells and are located in proximity to T cells in the early lesions of disease. Critically, removal of NETs and/or inhibition of extracellular histones inhibits IL-17/Th17 cell accumulation. Finally, through studies in a well-curated cohort of severe periodontitis patients, without confounding disease (such as RA), we document that NET-associated histones with distinct posttranslational modifications are present in local tissues and peripheral blood of patients with periodontitis and that their levels correlate with disease severity, revealing clinical relevance of our experimental findings. Collectively, our studies, through multiple complementary approaches, provide evidence for the role of NETs and extracellular histones as early disease triggers of pathogenic inflammation in periodontitis and support crossregulation of neutrophil and IL-17 immunity in disease pathogenesis.
Results
NETosis is evident in experimental periodontitis
Increased neutrophil recruitment is evident in the experimental periodontitis LIP model. Indeed, RNA sequencing (RNA-seq) of oral mucosal tooth-adjacent tissues (gingiva) following LIP revealed distinct transcriptomic signatures in LIP compared with control (CTL). Top upregulated pathways in LIP included pathways related to inflammation, neutrophil-mediated immunity, and neutrophil activation (Fig. 1, A and B; and Fig. S1 A). Consistent with transcriptomic profiles and resembling human disease (Dutzan et al., 2018), flow cytometric analysis of LIP-associated gingival tissues revealed a significant accumulation of neutrophils (CD45+CD11b+CD11clow/medLy6G+ cells) within the affected lesions at early time points (3–18 h) after disease induction, with a second wave of neutrophil infiltration at later time points (Fig. 1 C and Fig. S1, B and C). Most importantly, inflammatory bone loss in LIP required neutrophils as antibody-depletion (1A8) significantly reduced inflammatory bone destruction (Fig. 1 D and Fig. S1, D and E), as previously reported (Dutzan et al., 2018). Having established that neutrophils participate in inflammatory bone destruction in this in vivo model of periodontitis, we next investigated whether NETs were involved in this process. We first sought to determine whether NETs could be identified within the lesions of LIP. For this, we stained gingival tissues with or without LIP for citrullinated histone H3 (citH3), a marker of NETs, as well as Ly6G, a classic neutrophil marker. We detected positive staining for citH3 in LIP tissues in association with “string-like formations,” characteristic of NETs and in proximity to Ly6G+ neutrophils (Fig. 1, E and F). Overall, these results indicate that NETs are present in affected tissues in the LIP model.

Evidence of NETosis in lesions of experimental periodontitis. (A and B) Bulk RNA-seq in mouse gingival tissues from CTL (n = 4) and LIP (n = 5, 5 d). (A) Principal components analysis. (B) Gene Ontology (GO) biological processes in ascending order of P value. (C) Flow cytometry analysis of mouse gingiva CTL/LIP mice at indicated times (0–120 h). Absolute number of neutrophils (CD45+CD11b+CD11clow/medLy6G+) per standardized tissue block (n = 3–14) is shown. (D) Bone loss after LIP with isotype control (clone 2A3) or anti-Ly6G (clone 1A8) treatment. Bar graph depicts bone loss (n = 6–7, 6 d). (E and F) Immunofluorescence for citH3 and Ly6G in gingival tissue (CTL/LIP, 18 h). (E) Representative confocal images; scale bars are 50 µm. (F) Quantification, mean fluorescence intensity of citH3 staining (n = 7–8). (G and H) LIP following treatment with DNase-I or vehicle treatment. (G) Bone loss measurement (n = 11–12, 6 d). (H) RNA-seq of gingival tissues (18 h after LIP) with DNase-I (n = 5) or vehicle (n = 5). Heatmap analysis of select significantly differentially expressed genes. Each column represents a single sample. (I and J) TRAP-stained sections of gingival tissues from DNase-I or vehicle-treated mice with or without LIP (n = 6–7, 6 d). (I) TRAP-positive cells appear purplish to dark red in the sections. Scale bars are 50 µm; representative image. (J) Quantification of osteoclasts per condition, number of osteoclast/bone surface (N/mm). Data are representative of three (D, F, G, and J) or four (C) independent experiments. Graphs show the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. One-way ANOVA with Tukey’s multiple comparison test (B–D, G, and J), unpaired t test (F).
Evidence of NETosis in lesions of experimental periodontitis. (A and B) Bulk RNA-seq in mouse gingival tissues from CTL (n = 4) and LIP (n = 5, 5 d). (A) Principal components analysis. (B) Gene Ontology (GO) biological processes in ascending order of P value. (C) Flow cytometry analysis of mouse gingiva CTL/LIP mice at indicated times (0–120 h). Absolute number of neutrophils (CD45+CD11b+CD11clow/medLy6G+) per standardized tissue block (n = 3–14) is shown. (D) Bone loss after LIP with isotype control (clone 2A3) or anti-Ly6G (clone 1A8) treatment. Bar graph depicts bone loss (n = 6–7, 6 d). (E and F) Immunofluorescence for citH3 and Ly6G in gingival tissue (CTL/LIP, 18 h). (E) Representative confocal images; scale bars are 50 µm. (F) Quantification, mean fluorescence intensity of citH3 staining (n = 7–8). (G and H) LIP following treatment with DNase-I or vehicle treatment. (G) Bone loss measurement (n = 11–12, 6 d). (H) RNA-seq of gingival tissues (18 h after LIP) with DNase-I (n = 5) or vehicle (n = 5). Heatmap analysis of select significantly differentially expressed genes. Each column represents a single sample. (I and J) TRAP-stained sections of gingival tissues from DNase-I or vehicle-treated mice with or without LIP (n = 6–7, 6 d). (I) TRAP-positive cells appear purplish to dark red in the sections. Scale bars are 50 µm; representative image. (J) Quantification of osteoclasts per condition, number of osteoclast/bone surface (N/mm). Data are representative of three (D, F, G, and J) or four (C) independent experiments. Graphs show the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. One-way ANOVA with Tukey’s multiple comparison test (B–D, G, and J), unpaired t test (F).
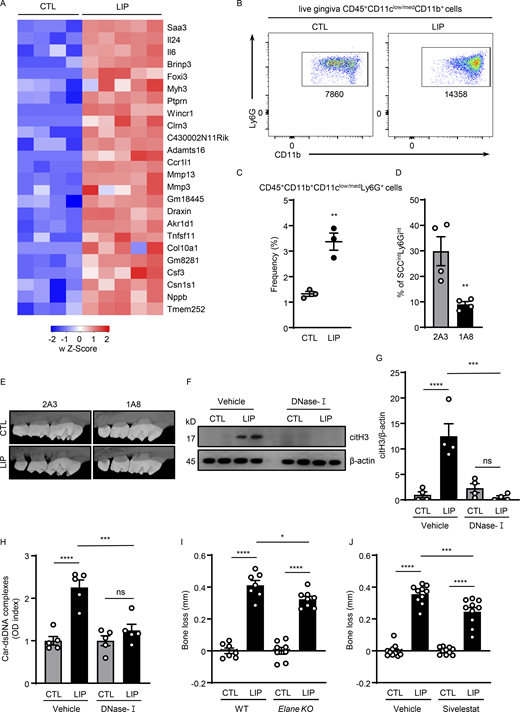
NETs play a pathogenic role in inflammatory bone loss. (A) Mouse gingival tissues of CTL (n = 4) and LIP (n = 5, 5 d) were harvested, and processed for RNA-seq analysis. Heatmap analysis of the RNA-seq data. Significantly differentially expressed genes between CTL and LIP are shown. Each column represents an individual sample. (B and C) Flow cytometry analysis of mouse oral gingival tissues in CTL and LIP (n = 3, 5 d) mice. FACS plot shows changes in neutrophil numbers (B) and graph indicates percentage of CD45+CD11b+ CD11clow/medLy6G+ cells (C). (D) Percentage of neutrophils in gingival tissue after 1A8 (n = 4) or 2A3 (n = 4) treatment. (E) Representative microcomputed tomography images of maxilla from isotype control-treated and anti-Ly6G-treated mice (n = 3). (F–H) Gingival tissues with or without LIP (18 h) in mice treated with DNase-I (n = 4–5) or vehicle (n = 4–5) were harvested, processed, and then subjected to (F and G) immunoblot for citH3 and (H) ELISA for Car–dsDNA complexes. (I and J) Bone loss measurements with or without LIP (6 d) in (I) WT and Elane KO (n = 7–8) mice or (J) mice treated with sivelestat or vehicle (n = 10). Bar graph depicts bone loss. Data are representative of three (C, D, and H) or four (F, G, I, and J) independent experiments. Graphs show the mean ± SEM. * P <0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. One-way ANOVA with Tukey’s multiple comparison test (G–J), unpaired t test (C and D). Source data are available for this figure: SourceData FS1.
NETs play a pathogenic role in inflammatory bone loss. (A) Mouse gingival tissues of CTL (n = 4) and LIP (n = 5, 5 d) were harvested, and processed for RNA-seq analysis. Heatmap analysis of the RNA-seq data. Significantly differentially expressed genes between CTL and LIP are shown. Each column represents an individual sample. (B and C) Flow cytometry analysis of mouse oral gingival tissues in CTL and LIP (n = 3, 5 d) mice. FACS plot shows changes in neutrophil numbers (B) and graph indicates percentage of CD45+CD11b+ CD11clow/medLy6G+ cells (C). (D) Percentage of neutrophils in gingival tissue after 1A8 (n = 4) or 2A3 (n = 4) treatment. (E) Representative microcomputed tomography images of maxilla from isotype control-treated and anti-Ly6G-treated mice (n = 3). (F–H) Gingival tissues with or without LIP (18 h) in mice treated with DNase-I (n = 4–5) or vehicle (n = 4–5) were harvested, processed, and then subjected to (F and G) immunoblot for citH3 and (H) ELISA for Car–dsDNA complexes. (I and J) Bone loss measurements with or without LIP (6 d) in (I) WT and Elane KO (n = 7–8) mice or (J) mice treated with sivelestat or vehicle (n = 10). Bar graph depicts bone loss. Data are representative of three (C, D, and H) or four (F, G, I, and J) independent experiments. Graphs show the mean ± SEM. * P <0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. One-way ANOVA with Tukey’s multiple comparison test (G–J), unpaired t test (C and D). Source data are available for this figure: SourceData FS1.
NET removal protects from inflammation-induced bone destruction
We next evaluated whether timely removal of NETs will protect from periodontal bone destruction. We used DNase-I, a nuclease that dismantles NETs in vivo, as a systemic treatment. Consistent with this function, gingival tissues from DNase-I–treated animals had significantly reduced tissue citrullination and carbamylation (O'Neil et al., 2020), as documented by Western blot and ELISA analysis (Fig. S1, F–H). DNase-I treatment led to significant protection from periodontal bone loss compared with vehicle CTL (Fig. 1 G). Similarly, NE (Elane) KO mice, which have been previously shown to have impaired NETosis (Warnatsch et al., 2015), also displayed reduced periodontal bone loss compared with WT counterparts (Fig. S1 I), although the effect of NE elimination was less pronounced than DNase-I treatment. Pharmacologic inhibition of elastase with Silvelestat also protected from periodontal bone loss in vivo (Fig. S1 J).
To understand the putative mechanisms by which NETs mediate periodontal immunopathology, we performed RNA-seq analysis of gingival tissues with or without LIP in the presence of DNase-I or vehicle treatment. Using this unbiased approach, we aimed to explore the pathways and genes upregulated during experimental periodontitis that were inhibited in the presence of DNase-I treatment. We found that pathways and genes inhibited by DNase-I treatment were related to inflammatory responses (Il1a, Il1b, Ptgs2, and Tnf), myeloid cell activation (Trem1, Clec4e, Cd14, and Adam8), neutrophil recruitment (Csf3r, Cxcl1, Cxcl2, Cxcr2, Cxcl4), and osteoclast differentiation and activation (Fos, Ncr2, Mapk8, Fosl1, and Fosb; Fig. 1 H). These results indicate that NETs participate in the induction of the overall inflammatory milieu in periodontitis. Consistent with the inhibition of inflammation and bone loss, we additionally document reduced numbers of osteoclasts in the lesions of LIP when treated with DNase-I. By staining for tartrate-resistant acid phosphatase (TRAP), a classic method to identify osteoclasts, we show that while osteoclasts become prominent during LIP, they are significantly decreased in numbers in the presence of DNase-I treatment (Fig. 1, I and J). Overall, our data reveal that NETosis drives inflammatory pathways leading to bone destruction in periodontitis.
Inhibition of citrullination protects from periodontal bone loss
NETosis has been associated with unique posttranslational modifications of proteins, most notably, citrullination, which has served as a marker for NETosis (Wang et al., 2009), and has been shown to potentiate the inflammatory functions of NETs (Tsourouktsoglou et al., 2020).
To evaluate the role of citrullination in our model, we performed LIP in peptidylarginine deiminase-4 (Padi4) KO mice. Padi4 KO mice displayed significant protection from periodontal bone destruction compared with their WT counterparts (Fig. 2 A and Fig. S2, A and B). Consistent with the results obtained by genetic inhibition of PAD4, in vivo pharmacologic inhibition of PAD-mediated citrullination using the compound Cl-amidine (Knight et al., 2014) also conferred substantial protection from periodontal bone loss (Fig. 2 B). Interestingly, inhibition of pathology in the Padi4 KO mice occurred despite comparable accumulation of immune cells, including neutrophils during LIP. Flow cytometry for total hematopoietic CD45+ cells, CD11b+CD11clow/medLy6G+ neutrophils, and other myeloid cells revealed a comparable accumulation of myeloid cells during LIP in Padi4 KO mice and WT counterparts (Fig. 2 C and Fig. S2, C–H). Similarly, immunostaining in gingival tissues of CTL and LIP in Padi4 KO mice and WT counterparts revealed comparable Ly6G and myeloperoxidase (MPO) in Padi4 KO mice and WT counterparts, with minimal citH3 in Padi4 KO mouse gingiva (Fig. 2, D–H). Of note, overall staining area for MPO was comparably diffuse in Padi4 KO with WT mice, consistent with MPO being present in the extracellular space.

Inhibition of citrullination protects from periodontal bone loss. (A and B) Bone loss measurements with or without LIP (6 d) in (A) WT and Padi4 KO (n = 5–6) mice or (B) mice treated with CI-amidine or vehicle (n = 10). (C) Flow cytometry analysis of mouse oral gingival tissues after LIP (18 h) in WT and Padi4 KO mice (n = 4). Graph indicating absolute number of CD45+ or neutrophils (CD45+CD11b+CD11clow/medLy6G+) per standardized tissue block. (D–H) Immunofluorescence for citH3, Ly6G, MPO in gingival tissues from Padi4 KO and WT mice, with or without LIP (n = 5, 18 h). (D and G) Representative images and conditions are indicated. Scale bars are 50 µm. (E, F, and H) Quantification, mean fluorescence intensity for citH3, Ly6G, and MPO staining (n = 5). Data are representative of three (A, C, E, F, and H) or four (B) independent experiments. Graphs show the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. One-way ANOVA with Tukey’s multiple comparison test (A, B, E, F, and H), unpaired t test (C).
Inhibition of citrullination protects from periodontal bone loss. (A and B) Bone loss measurements with or without LIP (6 d) in (A) WT and Padi4 KO (n = 5–6) mice or (B) mice treated with CI-amidine or vehicle (n = 10). (C) Flow cytometry analysis of mouse oral gingival tissues after LIP (18 h) in WT and Padi4 KO mice (n = 4). Graph indicating absolute number of CD45+ or neutrophils (CD45+CD11b+CD11clow/medLy6G+) per standardized tissue block. (D–H) Immunofluorescence for citH3, Ly6G, MPO in gingival tissues from Padi4 KO and WT mice, with or without LIP (n = 5, 18 h). (D and G) Representative images and conditions are indicated. Scale bars are 50 µm. (E, F, and H) Quantification, mean fluorescence intensity for citH3, Ly6G, and MPO staining (n = 5). Data are representative of three (A, C, E, F, and H) or four (B) independent experiments. Graphs show the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. One-way ANOVA with Tukey’s multiple comparison test (A, B, E, F, and H), unpaired t test (C).

Citrullination of NETs potentiates inflammatory bone destruction. (A and B) Representative TRAP-stained sections of gingival tissues with or without LIP (6 d) in WT (n = 5) and Padi4 KO (n = 5) mice. (A) TRAP-positive cells appear as purplish to dark red in the sections. Scale bars are 50 µm; representative image. (B) Quantification of osteoclasts per condition, number of osteoclast/bone surface (N/mm). (C–H) Flow cytometry analysis of mouse oral gingival tissues with or without LIP (18 h) in WT (n = 4) and Padi4 KO (n = 4) mice. Graph indicating absolute number of CD45+ (C), neutrophils (CD45+CD11b+CD11clow/medLy6G+; D), lymphoid DCs (CD11b−CD11c+; E), myeloid DCs (CD11b+ CD11clow-med; F), MNPs (CD11b+CD11clow-medLy6G−Ly6Clow-highSSClow; G), and eosinophils (CD11b+CD11clow-medLy6G−Ly6Clow-neg SSChigh; H) per standardized tissue block. Data are representative of three (B–H) independent experiments. Graphs show the mean ± SEM. *P < 0.05, **P < 0.01, ****P < 0.0001. One-way ANOVA with Tukey’s multiple comparison test (B), unpaired t test (C–H).
Citrullination of NETs potentiates inflammatory bone destruction. (A and B) Representative TRAP-stained sections of gingival tissues with or without LIP (6 d) in WT (n = 5) and Padi4 KO (n = 5) mice. (A) TRAP-positive cells appear as purplish to dark red in the sections. Scale bars are 50 µm; representative image. (B) Quantification of osteoclasts per condition, number of osteoclast/bone surface (N/mm). (C–H) Flow cytometry analysis of mouse oral gingival tissues with or without LIP (18 h) in WT (n = 4) and Padi4 KO (n = 4) mice. Graph indicating absolute number of CD45+ (C), neutrophils (CD45+CD11b+CD11clow/medLy6G+; D), lymphoid DCs (CD11b−CD11c+; E), myeloid DCs (CD11b+ CD11clow-med; F), MNPs (CD11b+CD11clow-medLy6G−Ly6Clow-highSSClow; G), and eosinophils (CD11b+CD11clow-medLy6G−Ly6Clow-neg SSChigh; H) per standardized tissue block. Data are representative of three (B–H) independent experiments. Graphs show the mean ± SEM. *P < 0.05, **P < 0.01, ****P < 0.0001. One-way ANOVA with Tukey’s multiple comparison test (B), unpaired t test (C–H).
Histones trigger inflammatory bone loss in periodontitis
We next sought to further dissect mechanisms by which NETs mediate inflammatory bone destruction in periodontitis. We hypothesized that histones, one of the major components of NETs, may have a role in triggering local immunopathology. We, therefore, aimed to deplete extracellular histones with antibody treatments and evaluated disease severity following histone depletion. For our studies, we used anti-histone H3 or H4 antibodies as well as antibodies for citH3 and appropriate isotype control IgG treatment. To evaluate antibody activity, we measured histone protein levels within the gingiva tissues after antibody blockade. Western blot analysis confirmed that systemic antibody treatment reduced but did not totally deplete histone levels within the tissue lesions. Indeed, all antibodies utilized were able to significantly reduce levels of respective histones (range of depletion 50–85%; Fig. 3, A, C, and E; and Fig. S3, A–C). Inhibition of either H3, H4, or citH3 led to significant protection from periodontal bone loss, revealing a pathogenic role for extracellular histones in disease pathology (Fig. 3, B, D, and F). To further define the contribution of extracellular histones in disease immunopathology, we examined whether histone inhibition would affect inflammatory pathways previously linked to immunopathology in the LIP model, namely the IL-17–mediated inflammatory response (Dutzan et al., 2018). Histones have been shown to trigger differentiation of the IL-17–secreting T cell directly and indirectly subset Th17 (Wilson et al., 2022). In this regard, we observed that upregulation of IL-17–related genes (Il17a, S100a9, and Il6) was substantially reduced in the presence of anti-citH3 inhibition (Fig. 3 G). Most importantly, we document significantly reduced accumulation of IL-17–positive cells (CD45+IL-17eYFP+; Fig. 3, H–J) and Th17 cells (CD4+IL-17eYFP+; Fig. 3, K–M) with reduced γδT+IL-17eYFP+ cell numbers (albeit not to a level of significance; Fig. S3, D–F). Collectively, our data suggest that extracellular histones contribute to the induction of IL-17–mediated inflammation and periodontal bone destruction.

Histones trigger periodontal inflammation and bone destruction. Mice with or without LIP were treated with isotype IgG, anti-H3, anti-H4, or anti-citH3. (A, C, and E) Western blot analysis for H3, H4, citH3, and β-actin in gingival tissues with or without LIP (6 d) in isotype IgG (n = 5), (A) anti-H3 (n = 5), (C) anti-H4 (n = 5), or (E) citH3 (n = 5) treated mice. Densitometric values for H3, H4, and citH3 were normalized against those of β-actin. (B, D, and F) Bone loss measurements in mice treated with isotype IgG (n = 4–10), (B) anti-H3 (n = 5), (D) anti-H4 (n = 10), or (F) citH3 (n = 4–5) treated mice, with or without LIP. (G) qPCR analysis for Il17a, S100a9, and Il6 in gingival tissues in mice treated with isotype IgG (n = 7) or anti-citH3 (n = 5, fold change 4 d after LIP shown for each treatment). (H–M) Flow cytometry analysis of gingival in IL-17acreR26ReYFP mice treated with isotype IgG (n = 5) or anti-citH3 (n = 5), with or without LIP. (H and K) FACS plot and graph indicating (I and L) numbers and (J and M) percentage of CD45+eYFP+ or CD4+eYFP+ cells. Data are representative of three (A–C, E, F, I, J, L, and M) or four (G) independent experiments. Graphs show the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. One-way ANOVA with Tukey’s multiple comparison test (B, D, F, I, and L) or unpaired t test (A, C, E, G, J, and M).
Histones trigger periodontal inflammation and bone destruction. Mice with or without LIP were treated with isotype IgG, anti-H3, anti-H4, or anti-citH3. (A, C, and E) Western blot analysis for H3, H4, citH3, and β-actin in gingival tissues with or without LIP (6 d) in isotype IgG (n = 5), (A) anti-H3 (n = 5), (C) anti-H4 (n = 5), or (E) citH3 (n = 5) treated mice. Densitometric values for H3, H4, and citH3 were normalized against those of β-actin. (B, D, and F) Bone loss measurements in mice treated with isotype IgG (n = 4–10), (B) anti-H3 (n = 5), (D) anti-H4 (n = 10), or (F) citH3 (n = 4–5) treated mice, with or without LIP. (G) qPCR analysis for Il17a, S100a9, and Il6 in gingival tissues in mice treated with isotype IgG (n = 7) or anti-citH3 (n = 5, fold change 4 d after LIP shown for each treatment). (H–M) Flow cytometry analysis of gingival in IL-17acreR26ReYFP mice treated with isotype IgG (n = 5) or anti-citH3 (n = 5), with or without LIP. (H and K) FACS plot and graph indicating (I and L) numbers and (J and M) percentage of CD45+eYFP+ or CD4+eYFP+ cells. Data are representative of three (A–C, E, F, I, J, L, and M) or four (G) independent experiments. Graphs show the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. One-way ANOVA with Tukey’s multiple comparison test (B, D, F, I, and L) or unpaired t test (A, C, E, G, J, and M).
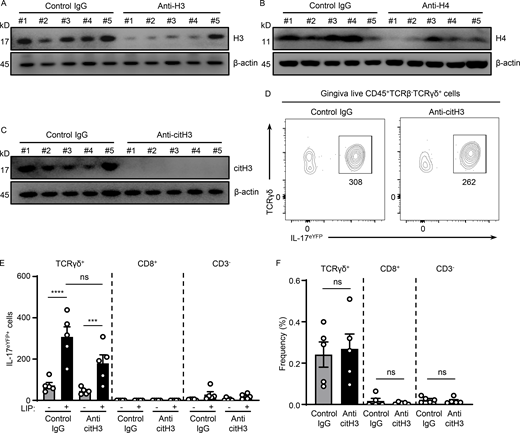
Histones meditate IL-17 cell accumulation in periodontitis. (A–C) Western blot analysis for H3, H4, citH3, and β-actin in mouse oral gingival tissues with or without LIP (6 d) in isotype IgG H3 (n = 5), (A) anti-H3 (n = 5), (B) anti-H4 (n = 5), or (C) anti-citH3 treated mice (n = 5). (D–F) Flow cytometry analysis of mouse oral gingival tissues with or without LIP (4 d) in isotype IgG (n = 5) and anti-citH3 (n = 5) treated IL-17acreR26ReYFP mice. (D) FACS plot and graphs indicating (E) numbers and (F) percentage of γδT+eYFP+, CD8eYFP+, as well as CD3−eYFP+ cells. Data are representative of three (A–C, E, and F) independent experiments. Graphs show the mean ± SEM. ***P < 0.001, ****P < 0.0001. One-way ANOVA with Tukey’s multiple comparison test (E and F). Source data are available for this figure: SourceData FS3.
Histones meditate IL-17 cell accumulation in periodontitis. (A–C) Western blot analysis for H3, H4, citH3, and β-actin in mouse oral gingival tissues with or without LIP (6 d) in isotype IgG H3 (n = 5), (A) anti-H3 (n = 5), (B) anti-H4 (n = 5), or (C) anti-citH3 treated mice (n = 5). (D–F) Flow cytometry analysis of mouse oral gingival tissues with or without LIP (4 d) in isotype IgG (n = 5) and anti-citH3 (n = 5) treated IL-17acreR26ReYFP mice. (D) FACS plot and graphs indicating (E) numbers and (F) percentage of γδT+eYFP+, CD8eYFP+, as well as CD3−eYFP+ cells. Data are representative of three (A–C, E, and F) independent experiments. Graphs show the mean ± SEM. ***P < 0.001, ****P < 0.0001. One-way ANOVA with Tukey’s multiple comparison test (E and F). Source data are available for this figure: SourceData FS3.
NETs potentiate the induction of IL-17 inflammation in periodontitis
We next explored whether NETs have a role in triggering the accumulation of IL-17/Th17 cells in periodontitis. Consistent with results from anti-histone treatment, treatment with DNase-I also led to a significant reduction of CD45+ IL17+ cells, as well as CD4+IL-17+ (Th17) cells during LIP (Fig. 4, A–C; and Fig. S4, A and B), further supporting a role for NETs in the induction of IL-17/Th17 responses.

NETs potentiate IL-17/Th17 inflammation in periodontitis. (A–C) Flow cytometry analysis of IL-17acreR26ReYFP mice oral gingival tissues with or without LIP (4 d) treated with DNase-I (n = 4) or vehicle (n = 4) control. (A) FACS plot of CD45eYFP+ and (B and C) quantification of CD45eYFP+ and CD4eYFP+ cells (LIP, 4 d, numbers of cells/per standardized block). (D–F) Flow cytometry analysis in WT mice with or without LIP (n = 4, 4 d). (D) FACS plot and quantification of (E) CD45+TLR2+ or (F) CD4+TLR2+ cells. (G) Immunofluorescence for citH3 and CD3 in mice without/with LIP (n = 3, 18 h). Scale bars, 50 µm; representative images shown. (H) Total oral microbial biomass in mice treated with DNase-I (n = 8) or vehicle (n = 7) treated mice at 18 h and 6 d after LIP. (I and J) 16S rRNA sequencing of LIP microbial communities from DNase-I and vehicle-treated mice at 18 h and 6 d (n = 7–8). (I) Plots show alpha-diversity based on the Shannon index. (J) Principal Coordinate analysis (PCoA) of beta-diversity based on Bray-Curtis dissimilarity. Vehicle and DNase-I are not significantly different regarding beta-diversity at either 18 h or 6 d. Data are representative of three (B, C, E, F, and H) independent experiments. Graphs show the mean ± SEM. *P < 0.05, **P < 0.01. Unpaired t test (B, C, E, F, H, and I) or permutational multivariate ANOVA (J).
NETs potentiate IL-17/Th17 inflammation in periodontitis. (A–C) Flow cytometry analysis of IL-17acreR26ReYFP mice oral gingival tissues with or without LIP (4 d) treated with DNase-I (n = 4) or vehicle (n = 4) control. (A) FACS plot of CD45eYFP+ and (B and C) quantification of CD45eYFP+ and CD4eYFP+ cells (LIP, 4 d, numbers of cells/per standardized block). (D–F) Flow cytometry analysis in WT mice with or without LIP (n = 4, 4 d). (D) FACS plot and quantification of (E) CD45+TLR2+ or (F) CD4+TLR2+ cells. (G) Immunofluorescence for citH3 and CD3 in mice without/with LIP (n = 3, 18 h). Scale bars, 50 µm; representative images shown. (H) Total oral microbial biomass in mice treated with DNase-I (n = 8) or vehicle (n = 7) treated mice at 18 h and 6 d after LIP. (I and J) 16S rRNA sequencing of LIP microbial communities from DNase-I and vehicle-treated mice at 18 h and 6 d (n = 7–8). (I) Plots show alpha-diversity based on the Shannon index. (J) Principal Coordinate analysis (PCoA) of beta-diversity based on Bray-Curtis dissimilarity. Vehicle and DNase-I are not significantly different regarding beta-diversity at either 18 h or 6 d. Data are representative of three (B, C, E, F, and H) independent experiments. Graphs show the mean ± SEM. *P < 0.05, **P < 0.01. Unpaired t test (B, C, E, F, H, and I) or permutational multivariate ANOVA (J).

NETs support IL-17 cell accumulation in periodontitis. (A and B) Flow cytometry analysis of IL-17acreR26ReYFP mice oral gingival tissues after LIP (4 d) treated with DNase-I (n = 4) or vehicle (n = 4). Graphs indicating the percentage of (A) CD45+eYFP+ or (B) CD4+eYFP+ cells. (C and D) Flow cytometry analysis of mouse oral gingival tissues in CTL/LIP mice at the indicated times (n = 3, 0–120 h). Graphs indicating numbers of (C) CD45+ or (D) CD4+IL-17+ cells. (E and F) Flow cytometry analysis in WT mice with or without LIP (n = 4, 4 d). Graph indicating percentage of (E) CD45+ or (F) CD4+TLR2+ cells. (G) 16S rRNA sequencing of LIP microbial communities from DNase-I and vehicle-treated mice at 18 h and 6 d (n = 7–8). Relative abundance of most abundant bacteria, OTU level. (H and I) Total oral microbial biomass in WT (n = 4) and Padi4 KO (n = 4) mice at (H) 18 h and (I) 6 d after LIP. Data are representative of three (A–F, H, and I) independent experiments. Graphs show the mean ± SEM. *P < 0.05. ns, not significant. One-way ANOVA with Tukey’s multiple comparison test (C and D), Unpaired t test (A, B, E, F, H, and I).
NETs support IL-17 cell accumulation in periodontitis. (A and B) Flow cytometry analysis of IL-17acreR26ReYFP mice oral gingival tissues after LIP (4 d) treated with DNase-I (n = 4) or vehicle (n = 4). Graphs indicating the percentage of (A) CD45+eYFP+ or (B) CD4+eYFP+ cells. (C and D) Flow cytometry analysis of mouse oral gingival tissues in CTL/LIP mice at the indicated times (n = 3, 0–120 h). Graphs indicating numbers of (C) CD45+ or (D) CD4+IL-17+ cells. (E and F) Flow cytometry analysis in WT mice with or without LIP (n = 4, 4 d). Graph indicating percentage of (E) CD45+ or (F) CD4+TLR2+ cells. (G) 16S rRNA sequencing of LIP microbial communities from DNase-I and vehicle-treated mice at 18 h and 6 d (n = 7–8). Relative abundance of most abundant bacteria, OTU level. (H and I) Total oral microbial biomass in WT (n = 4) and Padi4 KO (n = 4) mice at (H) 18 h and (I) 6 d after LIP. Data are representative of three (A–F, H, and I) independent experiments. Graphs show the mean ± SEM. *P < 0.05. ns, not significant. One-way ANOVA with Tukey’s multiple comparison test (C and D), Unpaired t test (A, B, E, F, H, and I).
However, the mechanisms by which NETs/histones trigger IL-17/Th17 inflammation in vivo remained unclear. Previous studies have demonstrated that histones can engage TLR2 to promote Th17 differentiation in vitro (Wilson et al., 2022) or can indirectly promote inflammatory responses linked to Th17 differentiation (Warnatsch et al., 2015). NETs could also alter the local microbiome and indirectly affect Th17 differentiation. Th17 accumulation in this model is indeed microbiome-dependent (Dutzan et al., 2018). To explore mechanisms by which NETs may induce IL-17/Th17 accumulation, we first investigated the time course of neutrophil accumulation/NETosis relevant to the accumulation of Th17 cells. We find that initial neutrophil recruitment (3–18 h) and NETosis (18 h) precede the accumulation of Th17 cells in LIP (Fig. 1 and Fig. S4, C and D), supporting a potential role for NETs as initial triggers of immunopathology. Second, we questioned whether T cells in gingiva express TLR2 that could potentially be engaged by histones to directly promote Th17 differentiation. We documented positive TLR2 expression in CD4+ T cells in gingiva with increased expression after disease induction (Fig. 4, D–F; and Fig. S4, E and F). Additionally, we explored whether neutrophil NETs and T cells are in close proximity in gingival tissues, allowing for potential interactions at early time points. By staining for citH3 (a NET marker) and CD3 for T cells, we document NETs in close proximity to T cells at early time points of disease induction (18 h; Fig. 4 G). Finally, we inquired whether NETosis affects the local microbiome during LIP. To assess this, we removed ligatures from mice treated with vehicle or DNase-I during LIP at early and later time points of the model (18 h, 6 d). We evaluated total microbial biomass as well as microbial community diversity, community composition, and relative abundance of microbial species. Strikingly, DNase-I treatment did not affect any of these parameters. Indeed, ligature microbiome had no significant differences in biomass, community diversity, or composition with or without DNase-I treatment at early and late time points (Fig. 4, H–J; and Fig. S4 G). Similar to DNase-I treatment, microbial load/biomass was comparable in Padi4 KO and WT mice (Fig. S4, H and I). Collectively, these studies demonstrate that NETs and extracellular histones promote IL-17/Th17 cell accumulation in vivo in LIP without affecting the local microbiome.
NET complexes, citrullinated and carbamylated histones are markers of disease severity in periodontal patients
We next sought to evaluate the presence of NET complexes in patients with periodontitis. For these studies, we assembled a curated cohort of patients with severe periodontitis in the absence of any known confounders and/or comorbidities (smoking, diabetes, systemic inflammatory disease, history of cancer, use of immunomodulatory or antibiotic treatment or other significant medical history including RA, all exclusionary), and age-gender-matched healthy control (HC). We quantified NET complexes, citH3-double-stranded DNA (dsDNA), carbamylated protein–dsDNA (Car–dsDNA) complexes (previously described as a marker of carbamylated NETs; O'Neil et al., 2020; O'Neil et al., 2021), as well as carbamylated histone H3/H4 (Car-H3/H4), both at the site of disease (gingival crevicular fluid, GCF) and in serum of patients and controls. We further quantified levels of NE as a surrogate for neutrophil presence in GCF and serum as well as absolute numbers of neutrophils in circulation.
At the site of disease, in the GCF, we documented significantly increased levels of Car–dsDNA complexes and Car-H4, as well as increased NE in patients compared with controls (Fig. 5 A). CitH3–DNA complexes were also detected, albeit marginally elevated. In fact, GCF levels of Car-H4, NE, and citH3–DNA complexes significantly correlated with the severity of bone destruction (measured in mm, clinical attachment loss [CAL]) at the specific area where the clinical sample was collected (Fig. 5 C; Car-H4 [r2 = 0.7430, P < 0.0001], NE [r2 = 0.2100, P = 0.0421], and citH3–DNA complexes [r2 = 0.2734, P = 0.0180]).

Levels of NET complexes in GCF and serum correlate with severity of periodontitis. (A) Human GCF samples were analyzed for the levels of Car–dsDNA complexes, Car-H4, citH3–dsDNA complex, and NE activity in HC (n = 10) and periodontitis (perio) patients (n = 10). (B) Serum samples were analyzed to quantify the levels of Car–dsDNA, Car-H3, citH3–dsDNA complex, NE activity, and absolute neutrophils number in HC and perio patients. (C) Correlation analysis of CAL (in mm) with GCF levels of Car-H4 and elastase activity or serum levels of Car-H3 measured in periodontitis patients. (D) Correlation between plasma levels of Car–dsDNA complexes and Car-H3 measured in periodontitis patients. Results are expressed as OD index. Data are representative of three (A and B) independent experiments. Graphs show the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001****P < 0.0001. Unpaired t test (A and B).
Levels of NET complexes in GCF and serum correlate with severity of periodontitis. (A) Human GCF samples were analyzed for the levels of Car–dsDNA complexes, Car-H4, citH3–dsDNA complex, and NE activity in HC (n = 10) and periodontitis (perio) patients (n = 10). (B) Serum samples were analyzed to quantify the levels of Car–dsDNA, Car-H3, citH3–dsDNA complex, NE activity, and absolute neutrophils number in HC and perio patients. (C) Correlation analysis of CAL (in mm) with GCF levels of Car-H4 and elastase activity or serum levels of Car-H3 measured in periodontitis patients. (D) Correlation between plasma levels of Car–dsDNA complexes and Car-H3 measured in periodontitis patients. Results are expressed as OD index. Data are representative of three (A and B) independent experiments. Graphs show the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001****P < 0.0001. Unpaired t test (A and B).
In the serum of patients with periodontitis, we documented significantly increased levels of Car–dsDNA, Car-H3, as well as increased citH3–dsDNA (Fig. 5 B), despite the lack of increase in neutrophils in the circulation as measured both by NE levels and absolute neutrophil numbers (Fig. 5 B).
Furthermore, there was a significant correlation between circulating levels of Car–dsDNA complexes (r2 = 0.6675, P = 0.0013), Car-H3 (r2 = 0.7223, P < 0.0003), and citH3–DNA complexes (r2 = 0.2867, P = 0.0150) with overall disease severity in each subject measured as average loss of attachment throughout the entire dentition (full-mouth CAL in mm, Fig. 5 D). These results in patients with periodontitis reveal that elevated NET complexes including both citrullinated or carbamylated DNA and histones are features of clinical disease severity.
Discussion
Inflammatory diseases linked to bone destruction such as RA and periodontitis are associated with local neutrophil infiltration in disease lesions (Cascao et al., 2010; Landzberg et al., 2015). While neutrophils have been implicated in these conditions, their role in mediating inflammatory bone destruction has not been fully determined. Here, we demonstrate a causative link between NETs and inflammatory bone destruction in vivo in a well-established model of periodontitis. Furthermore, while NETs represent a massive source of inflammatory molecules, and it is very likely that multiple NET components participate in shaping the inflammatory milieu in periodontitis, we determine that specific NET components, primarily extracellular histones, are contributing to disease pathology.
Our studies utilize multiple complementary approaches aimed at genetic or pharmacological inhibition of NETosis and timely removal of NETs to demonstrate a pathogenic role for NETs in the induction of periodontal inflammation and subsequent bone destruction. Indeed, when NETs are removed through DNase-I treatment, inflammatory responses linked to periodontal immunopathology, such as the IL-17/Th17 response, are blunted, accumulation of osteoclasts is reduced, and consequent bone loss is significantly prevented.
Investigating the properties of NETs that are important in mediating periodontal pathology, we determine that citrullination, a classical posttranslational modification typically observed in NETs and shown to potentiate NET-associated inflammation (Tsourouktsoglou et al., 2020), is important for disease immunopathology. As such, mice with a genetic defect in citrullination (Padi4 KO) display significant protection from inflammatory bone loss. Interestingly, protection from periodontal bone loss in Padi4 KO mice occurred, despite comparable numbers of neutrophils within the tissue microenvironment.
We further determine that extracellular histone, a key component of NETs, has a direct role in triggering the inflammatory pathology of periodontitis. Histones are released passively during cell injury or actively in the process of NETosis and are recognized for their broad antimicrobial activity, mediating lysis of bacteria, fungi, and viruses (Szatmary et al., 2018). Yet, extracellular histones have well-documented proinflammatory functions beyond their antimicrobial effects. Histones are highly cytotoxic to mammalian cells, promoting cell lysis and necrosis, activating platelets, promoting thrombosis, and activating immune cells through TLR ligation (Hoeksema et al., 2016; Tsourouktsoglou et al., 2020). As such, histone H4 was shown to mediate membrane lysis of smooth muscle cells triggering arterial tissue damage and inflammation and thus contributing to atherosclerosis (Silvestre-Roig et al., 2019). Recently, histones have also been shown to promote osteoclast activation (O'Neil et al., 2023) and to trigger differentiation of T helper cells into the Th17 phenotype via TLR2 ligation on T cells (Wilson et al., 2022). The potential of histones to stimulate Th17 immunity directly or indirectly is particularly relevant for periodontal disease, where Th17 cells have been shown to accumulate in disease lesions (Ikeuchi and Moutsopoulos, 2022) and play a significant role in driving pathogenesis. Yet, while the contribution of NETs and extracellular histones to disease pathology is significant, it is not complete, reflecting that additional NET-independent mechanisms are also operational in disease. As such, remaining IL-17/Th17 inflammation can potentially mediate pathology in a neutrophil-independent manner (Moutsopoulos et al., 2014; Moutsopoulos et al., 2017).
Furthermore, it is extremely difficult to determine whether NETs have a direct or indirect role in the induction of Th17 responses in periodontitis. NETs contain a plethora of pleiotropic inflammatory agents with broad functionalities. Even when considering solely the role of histones, their functions are pleiotropic and their known receptors (TLR2/4) are not selective. Therefore, it is plausible that more than one factor/mechanism is involved in NET-associated induction of IL-17/Th17 in periodontitis. Our current data support that NETs and extracellular histones promote IL-17/Th17 inflammation in vivo in LIP. Additionally, we find that factors that would allow for a direct function of NETs/histones on Th17 are available within the mucosal lesion. NETs and T cells come into close proximity within the mucosal tissues, potentially allowing for intimate contact-dependent interactions. NET formation occurs at early time points in the lesion prior to the accumulation of Th17 cells. Additionally, TLR2, the receptor associated with histone-triggered Th17 differentiation is expressed in T cells and becomes upregulated with disease. Moreover, while NETs have been previously shown to promote Th17 differentiation via upregulation of IL-1 responses (Warnatsch et al., 2015), this critical mechanism is likely not relevant in our model as previous data from our lab has demonstrated that Th17 expansion in periodontitis is independent of IL1 signaling (Dutzan et al., 2018). Finally, we determine that NETosis is not a driver of microbial dysbiosis in this model, which could also indirectly affect Th17 differentiation. Collectively, these studies support a pathogenic role for NETs and extracellular histones in the early induction of IL-17/Th17 immunopathology in periodontitis and reveal a feed-forward loop by which early neutrophil recruitment and NETosis trigger IL-17/Th17 immunity to further amplify neutrophil recruitment and immunopathology.
Importantly, our data provide potential insights relevant to human disease characterization and therapeutic targeting. We find that NET complexes as well as histones H3 and H4 bearing classic NET-associated posttranslational modifications (citrullination and carbamylation) are significantly elevated in local lesions (GCF) and serum of patients with severe periodontitis, and their levels correlate with local bone destruction and overall disease severity.
We believe that important concepts arise from these human findings: First, the significant increase in levels of NET complexes in disease lesions and circulation and their association with the severity of bone destruction suggest a pathogenic role for NETs in the progression of human disease. Furthermore, the significant correlation of levels of carbamylated NETs with the severity of bone destruction suggests considering the utility of NETs as biomarkers in periodontitis, a disease that currently lacks biomarkers that can predict disease initiation and progression. Previously, anti-carbamylated protein antibodies have been associated with erosive bone disease in RA (O'Neil et al., 2020) and their presence in circulation has been demonstrated in patients suffering from both RA and periodontitis (Bright et al., 2018; Magan-Fernandez et al., 2020). We further document significantly increased levels of carbamylated DNA complexes and carbamylated histones in the disease lesions and circulation of patients with periodontitis even in the absence of known inflammatory and autoimmune comorbidities such as RA, history of other autoimmunity, or smoking, suggesting a specific association with the periodontitis process. Yet, further work in larger cohorts of periodontitis subjects with longitudinal assessments of disease progression will be necessary to validate carbamylated DNA/histones as disease biomarkers. Such biomarkers can be particularly important for early disease detection and for identification of disease progression and will ideally facilitate prevention and/or timely targeted therapeutic intervention.
Second, our combined experimental work and human findings suggest considering therapeutic targeting of neutrophil NETosis in the treatment of common forms of periodontitis. One such treatment could be recombinant human DNase-I, a simple and low-cost treatment that has been widely used as a safe therapy in cystic fibrosis, where NET-associated inflammation is well established (Fuchs et al., 1994; Lazarus and Wagener, 2019).
Finally, we propose that our findings may suggest a shared mechanistic link for diseases of inflammatory bone loss such as arthritis and periodontitis. Periodontitis has been strongly epidemiologically associated with RA (Bright et al., 2015; Kaneko et al., 2018; Oliveira et al., 2021). Indeed, RA patients present with a significantly higher incidence and severity of periodontal disease even at the time of diagnosis (Potempa et al., 2017; Scher et al., 2012). The fact that new disease onset RA patients present with higher incidence and severity of periodontitis has promoted the concept that periodontitis may be a predisposing factor for RA. However, whether periodontitis is a trigger for distal disease or if common genetic predispositions and shared mechanisms underlie susceptibility to periodontitis and RA is not conclusively determined (Hajishengallis and Chavakis, 2022; Li et al., 2022). While NETs and their posttranslational modifications, citrullination and carbamylation, are well-described features of RA and periodontitis (Bright et al., 2015; Kaneko et al., 2018; Konig et al., 2016; Oliveira et al., 2021), our current work reveals a pathogenic role for NETs in the process of inflammatory bone loss and may reflect mechanisms shared between RA and periodontitis. Collectively, this work uncovers insights and potential therapeutic avenues for the disease periodontitis and proposes a role for NETosis-driven IL-17/Th17 immunopathology as a pathway that could be broadly relevant for diseases of neutrophil-triggered inflammatory bone loss.
Materials and methods
Human study population
All patients were enrolled in an Institutional Review Board–approved clinical protocol at the National Institutes of Health clinical center (ClinicalTrials.gov Identifier: NCT01568697) and provided written informed consent for participation in this study. All participants were deemed systemically healthy based on detailed medical history and select laboratory bloodwork. Inclusion criteria for this study were as follows: ≥18 yr of age, a minimum of 20 natural teeth, and good general health. Exclusion criteria were as follows: history of hepatitis B or C, history of HIV, prior radiation therapy to the head or neck, active malignancy except localized basal or squamous cell carcinoma of the skin, treatment with systemic chemotherapeutics or radiation therapy within 5 yr, pregnant or lactating, diagnosis of diabetes and/or HbA1C level >6%, less than three hospitalizations in the last 3 yr, and autoimmune disorders such as systemic lupus erythematosus and RA. Additional exclusion criteria included the use of any of the following in the 3 mo before study enrollment: systemic (intravenous, intramuscular, or oral) antibiotics, oral, intravenous, intramuscular, intranasal, or inhaled corticosteroids or other immunosuppressants, cytokine therapy, methotrexate or immunosuppressive chemotherapeutic agents, and large doses of commercial probiotics. Use of tobacco products (including e-cigarettes) within 1 yr of screening was also exclusionary. In addition to systemic screening, oral health was assessed and only participants with no soft tissue lesions, no signs/symptoms of oral/dental infection, and no/minimal gingival inflammation were considered for the health group. Inclusion criteria for the periodontal disease group were moderate to severe periodontitis (≥5 mm probing depth in more than four interproximal sites) and visible signs of tissue inflammation, including erythema/edema, and bleeding upon probing. GCF and serum samples were obtained from healthy and periodontitis subjects according to study procedures. GCF was collected by periopaper GCF collection strips (from Oraflow item #593520) and stored −80°C until extraction. 25 µl of PBS was applied to the periopaper and then centrifuged at 2,000 rpm for 5 min. This process was repeated for a total of 100 µl collection (4 × 25 µl). The extracted GCF was stored at −80°C until use.
Mice
C57BL/6,Padi4−/− (B6.Cg-Padi4Tm1.1Kmow/J, 030315), Elane−/− (B6.129X1-Elanetm1Sds/J, 006112), and R26R-EYFP (B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J) mice were purchased from the Jackson Laboratory. IL-17aCre and R26R-EYFP were crossed to generate IL-17acreR26ReYFP mice. All the mice were maintained under specific pathogen–free conditions. Mice were 6–8 wk of age at the beginning of the experiments and were sex-matched for each experiment. Mice experimental protocols were approved by the National Institute of Dental and Craniofacial Research (NIDCR) Animal Care and Use Committee and performed according to their guidelines.
LIP
Mice were anesthetized with ketamine/xylazine (100 mg/kg i.p. and 10 mg/kg i.p.) and received a silk ligature around the maxillary left second molar tooth to induce periodontitis. Ligature was removed 6 d after placement and periodontal bone heights were assessed after defleshing and staining with 0.5% eosin and 1% methylene blue (Ricca Chemical Company). Bone loss was measured as the distance from the cemento-enamel junction to the alveolar bone crest as previously described (Dutzan et al., 2018).
Single-cell suspension
For mice, gingiva was dissected and digested for 45 min at 37°C with Collagenase IV (Gibco) and DNase-I (Sigma-Aldrich) as previously described (Dutzan et al., 2018). Mouse gingiva from two ligature areas was pooled for flow cytometry or Western blot analysis.
Flow cytometry
Single-cell suspensions from gingiva were stained with the Live/Dead Cell Viability assay (Invitrogen), and cells were incubated with antiFcγIII/I receptor and fluorochrome-conjugated antibodies against surface markers in PBS with 2% FBS for 20 min at 4°C degrees in the dark. Specific anti-mouse antibodies used in this study are as follows: CD45 (30-F11; BioLegend), CD11b (M1/70; eBioscience), CD11c (N418; eBioscience), Ly6G (1A8; BioLegend), Ly6C (HK1.4; eBioscience), MHCII (M5/11.4.15.2; eBioscience), CD4 (RM4-5; eBioscience), CD8 (H35–17.2; eBioscience), TCRβ (H57-597; eBioscience), TCRγδ (GL3; eBioscience), and TLR2 (6C2; eBioscience). The cells were washed in PBS with 2% FBS, resuspended in FACS buffer (0.5% BSA and 0.1% NaN3 in PBS), and analyzed immediately. The samples were examined using a FACS Fortessa cytometer (BD Biosciences). Data analysis was performed using FlowJo software (Tree Star).
Western blot
For Western blot analysis, gingiva was lysed in a buffer containing 10 mM Tris-HCl at pH 8.0, 1 mM EDTA, 140 mM NaCl, 0.1% SDS, 0.1% sodium deoxycholate, 1% Triton X-100, and a protease inhibitor cocktail (Roche). The lysate was incubated at 4°C for 15 min and then centrifuged at 14,000 g for 15 min at 4°C. The supernatant was collected and the protein concentration was measured by Bradford assay (Thermo Fisher Scientific). Then 15 μg of gingival tissue protein extracts were solubilized in Laemmli’s buffer 4× (Bio-Rad) and denatured by boiling and separated by SDS-PAGE on 12% or 15% gels. Proteins were transferred to polyvinyl difluoride membranes (Millipore). The membranes were blocked in 5% nonfat milk (Bio-Rad) in TBS with tween 20 (Thermo Fisher Scientific) for 1 h and incubated overnight with primary antibodies anti–histone H3 (citrulline R2+R8+R17; ab5103; Abcam), anti–histone H3 (ab1791; Abcam), anti–histone H4 (ab10158; Abcam), and anti–β-actin (4970; Cell Signaling) diluted 1:1,000. The membranes were incubated with HRP-conjugated secondary antibody (7074; Cell Signaling) diluted at a ratio of 1:2,000. For the detection of target proteins, the membranes were developed using a chemiluminescent reagent (WBKLS0500; Millipore) and were subsequently quantified using the Amersham BioSciences Imager 600 (GE Healthcare).
Histology and immunofluorescence analysis
The maxillae of the mice were fixed in 4% paraformaldehyde in PBS at 4°C. The maxillae were decalcified in 10% EDTA for 2 wk at 4°C. The decalcification solution was changed weekly. Decalcified maxillae were sent to Histoserv Inc. and processed for paraffin embedding. For TRAP staining, a mouse maxillae paraffin section (5 µm) was cut and stained using an acid phosphatase kit (378A; Sigma-Aldrich) and then counterstained with aniline blue. The section was scanned with a Hamamatsu NDP slide scanner (Hamamatsu Nanozoomer S60) and imaged using an NDPviewer. Immunofluorescence analysis of NETs was performed as described previously (Silva et al., 2021). Mouse maxillae paraffin sections were cut and immunostained with primary antibodies at 1:1,000 dilution (anti-Ly6G, 551459; BD Pharmingen; anti–histone H3 [citrulline R2+R8+R17], ab5103; Abcam; anti-MPO, ab65871; Abcam) O/N at 4°C. Secondary staining was performed with Rhodamine Red-X anti-rat (712-296-150) or Alexa Fluor 647–conjugated AffiniPure F(ab′)2 anti-rabbit (711-606-152; 1:1,000; Jackson Immuno Research Labs) and 4′,6-diamidino-2-phenylindole (DAPI; 1:1,000) for 1 h at room temperature. Inverted Nikon A1R + confocal microscope (20×-air or 60×-oil immersion) using NIS-Elements software (Nikon) was used to capture images. To quantify the citH3/Ly6G-stained area in the gingiva, a statistical threshold (set at two SDs above the mean of the image histogram) was set. An investigator-drawn region of interest determined the appropriate area of the sample and the square area was reported. The analysis was performed with MetaMorph and results were displayed as means ± SEM. For citH3 and CD3 immunostaining, paraformaldehyde-fixed and EDTA-decalcified maxillae were immersed O/N in 30% sucrose in PBS and frozen in OCT. Frozen tissues sections (10 µm) were stained with FITC-conjugated anti-CD3 primary antibody (1:200 dilution, 11-0031-82; Invitrogen) with Hoechst as nuclear counterstain (1:5,000 dilution, 40046; Biotium), while images were obtained with Leica SP8 confocal microscope (40× oil immersion) using LAS X software.
Microcomputed tomography
Whole maxillae were scanned with a voxel size of 7.4 μm and a 1.0-mm aluminum filter at 70 kVp and 114 μA (μCT 50; Scanco Medical AG). Reconstructed images were converted to DICOM format and used for bone loss analysis.
Mouse treatment protocols
1A8-mediated neutrophil depletion
For in vivo depletion of neutrophils, mice were treated with 0.4 mg anti-Ly6G Ab (clone 1A8; BP0075-1; BioXcell) or anti-IgG2a Ab (isotype control, clone 2A3, BE0085; BioXcell) via i.p. injection on days 1, 2, and 4 (total of three times) and harvested on day 6. Efficacy of polymorphonuclear depletion was confirmed by loss of neutrophil populations in blood and gingiva by flow cytometry.
DNase-I treatment
Mice were injected i.p. with either 1,000 units of DNase-I (4716728001; Millipore Sigma) in 0.9% NaCl or 0.9% NaCl alone. Mice were treated with DNase-I on days 1, 2, and 4 (total of three times) and harvested on day 6.
Cl-amidine treatment in LIP mice
Mice received 50 mg/kg of CI-amidine (506282; EMD Millipore) dissolved in 10% DMSO (Sigma-Aldrich) i.p. in PBS or DMSO alone in PBS as a vehicle control. Mice were treated with CI-amidine on days 1, 2, and 4 (total of three times) and harvested on day 6.
Histone depletion in LIP mice
To deplete histones H3, H4, or citH3, mice were treated with anti–histone H3 (50 μg per mouse, LG2-1; Creative Biolabs), anti–histone H4 (50 μg per mouse, orb225483; Biorbyt), anti–histone H3 (citrulline R2+R8+R17; 50 μg per mouse, ab5103; Abcam) or control IgG (BE0085 or BE0095; BioXcell) via i.p. injection on days 1, 2, and 4 (total of three times) and harvested on day 6.
Sivelestat treatment
For in vivo NE inhibition, mice were administered 50 mg/kg of sivelestat (HY-17443, MedChemExpress) dissolved in vehicle control (10% DMSO + 40% PEG300 + 5% Tween 80 + 45% saline) or vehicle control alone by i.p. route. Mice were treated with sivelestat on days 1, 2, and 4 (total of three times) and harvested on day 6.
RNA-seq analysis and data acquisition
Total RNA was extracted from experimental tissue samples and sequencing libraries were prepared by the Illumina Nextera XT method following the manufacturer’s recommendations (Illumina). The multiplexed libraries were sequenced in a NextSeq500 instrument in 150 bp paired-end mode. Demultiplexed samples were mapped and transcripts were quantified to the GRCm38.v11 mouse genome using the STAR v2.5.2 aligner. Gene-level counts were filtered to remove low-expression genes (keeping genes with >5 counts in at least one sample). The filtered gene expression matrix was analyzed by principal component analysis to identify outlier samples (none detected). Differential gene expression was evaluated by three independent statistical methods (DESeq2, edgeR, and Limma-Voom). Differentially expressed genes (false discovery rate < 0.05) were considered for further analyses based on results from DESeq2.
Quantification of total bacteria
The determination of the total bacterial load was performed via quantitative PCR (qPCR) using 16S rRNA gene primers (Barman et al., 2008). Total genomic DNA from P. gingivalis ATCC 33277 was used to construct standard curves with a known number of 16S rRNA gene copies (ranging from 102–108), which are required to determine the number of 16S rRNA gene copies in each sample. The qPCR reactions had a final volume of 20 μl and included 10 μl of SYBR Green PCR Master Mix (4309155; Applied Biosystems), 2.5 uM of reverse and forward primer, 1 μl of DNA from each sample, and PCR water (Qiagen). Thermal cycle conditions were 50°C for 2 min, 95°C for 2 min, and 40 cycles of 95°C for 15 s and 60°C for 1 min. All PCR reactions were performed in duplicate and were carried out in a 7500 Real-Time PCR system (Thermo Fisher Scientific).
Oral microbiome characterization using 16S rRNA gene sequencing
For microbiome analysis, oral murine microbial samples were obtained as previously described (Abusleme et al., 2017). DNA was extracted using a modified version of DNeasy Blood and Tissue kit (Qiagen) that includes a chemical/enzymatic lysis step (Abusleme et al., 2017). The library preparation for sequencing of the 16S rRNA V4 region was performed by PCR using the primers 515F and 806R, which included the adapter for MiSeq sequencing (Illumina). Next, these amplicon libraries were pooled and sequenced following the 2 × 250 bp paired-end protocol (Illumina) obtaining paired-end reads as described previously. The raw sequences were processed according to the Mothur version 1.44.3 pipeline (Schloss et al., 2009). Specifically, paired-end sequences were first joined into contigs. Sequences that contained ambiguous bases, had lengths shorter than 150 bp, or had more than 10 homopolymers, were excluded. Chimeric sequences were removed using UCHIME (Edgar et al., 2011). Thus, a total of 903,843 sequences (mean ± SD per sample = 30,128 ± 4,128) were retained for further analysis. Individual sequences were classified using the Ribosomal Database Project classifier in Mothur and a database combining Human Oral Microbiome Database V15.22 (Escapa et al., 2018) and the Genome Taxonomy Database V202 as reference. Reads were clustered at 97% similarity into operational taxonomic units (OTUs). The taxonomy of each OTU was assigned using the classify.otu command in Mothur with a consensus confidence threshold of 80%.
Quantification of NETs
CitH3 or Car–dsDNA complexes were quantified by ELISA as previously described. A 96-well plate was coated with rabbit polyclonal anti-citH3 (Abcam) or carbamylated-lysine (Cell Biolabs) antibodies at 1:400 in PBS overnight at 4°C. Wells were washed and blocked with 1% BSA at room temperature for 1 h. Diluted sera/plasma (1:10) was added to the wells in (1% BSA) blocking buffer and incubated overnight at 4°C. The wells were washed three times and incubated with mouse monoclonal anti-double stranded DNA antibody (EMD Millipore) at 1:100 in blocking buffer. After washing three times, goat anti-mouse conjugated HRP antibody (Bio-Rad) was added to the wells in blocking buffer at 1:10,000. Wells were washed five times followed by the addition of 3,3′,5,5′-Tetramethylbenzidine substrate (Sigma-Aldrich) and stop solution (Sigma-Aldrich). The absorbance was measured at 450 nm and values were calculated as an OD index. Assay was performed in duplicate. To quantify serum/plasma Car-H3/H4, a similar technique was used; 96-well plates were coated with rabbit polyclonal carbamylated-lysine antibody (Cell Biolabs) at 1:400 and a secondary antibody, either HRP-conjugated mouse anti-H3 or H4 (Abcam) diluted in blocking buffer, was used to develop the plate.
Real-time PCR
Total RNA was isolated from mouse gingival tissues using Trizol reagent (15596018; Life Technologies) and Lysing Matrix D (6913500; MP Biomedicals) tubes and quantified by NanoDrop spectrometer at 260 and 280 nm. For real-time PCR, 1 µg of total RNA was reverse transcribed using an oligo deoxythymidylic acid primer (Invitrogen), and the resulting cDNA was amplified by real-time PCR using an ABI Prism 7500 Sequence Detector (Applied Biosystems/Life Technologies), according to the manufacturer’s protocol. Amplification of murine genes was performed using the Taqman expression assay for Il17a (Mm00434214_m1), S100a9 (Mm00656925_m1), Il6 (Mm00446190_m1), and Hprt (Mm01545399_m1) as a normalization control (Thermo Fisher Scientific). We performed relative quantification using the 2−ΔΔCt method with mouse hprt as an internal control gene; data were expressed as relative fold changes.
Statistical analysis
Statistical analysis was performed in Prism (GraphPad Software, v8). Mean ± SEM of at least three independent experiments was calculated for all experiments. The unpaired two-tailed Student’s t test was used for the comparison of means between two independent groups, and one-way ANOVA was used for the comparison of means between two or more groups. Microbiome alpha-diversity was determined via the non-parametric Shannon Index, and beta-diversity was analyzed via the Bray-Curtis distance. Clustering of microbiome communities according to experimental groups was evaluated via principal coordinate analysis and differences were tested via analysis of permutational multivariate ANOVA. Specific P values are detailed in the figure legends.
Online supplemental material
Fig. S1 A is a heatmap representation of top differentially expressed genes in gingival tissues with/without LIP periodontitis. Fig. S1, B and C, show flow plots and graphs depicting percentage of neutrophils within gingival tissues with or without LIP. Fig. S1, D and E, show that depletion of neutrophils by antibody treatment (1A8) significantly reduces neutrophil accumulation in gingival tissues and inhibits periodontal bone destruction. Fig. S1, F–H, demonstrate a significant reduction of citrullination and carbamylation in gingival tissues with DNase-I treatment, and Fig. S1, I and J, show that genetic/pharmacological inhibition of neutrophil elastase (Elane, silvelastat) protects from inflammatory bone destruction. Fig. S2, A and B, demonstrate a significant reduction in osteoclast numbers in Padi4 KO mice compared with WT mice. Fig. S2, C–H, show a comparable accumulation of immune cells, including neutrophils, lymphoid DCs, myeloid DCs, MNPs, and eosinophils in gingival tissues in both Padi4 KO and WT mice. Fig. S3, A–C, show depletion/reduction of extracellular histones following antibody treatment (anti–histone H3, H4, or citH3) within gingival tissues. Fig. S3, D–F, demonstrate numbers and percentages of γδT+IL-17eYFP+, CD8+IL-17eYFP+, as well as CD3-IL-17eYFP+ cells in the gingival tissues of IL-17acreR26ReYFP mice treated with anti-citH3 compared to the isotype IgG control. Fig. S4, A and B, demonstrate percentages of CD45+IL-17eYFP+ or CD4+IL-17eYFP+ cells in the gingival tissues of IL-17acreR26ReYFP mice after LIP (4 d) treated with DNase-I or vehicle control. Fig. S4, C and D, demonstrate flow cytometric analysis IL-17+/Th17 cells within the gingival tissues at indicated times during LIP induction. Fig. S4, E and F, show percentage of CD45+TLR2+ or CD4+TLR2+ cells in gingival tissues, 4 d following LIP induction. Fig. S4 G shows most abundant OTUs present in the ligature microbiome with or without DNase-I treatment at 18 h and 6 d of LIP periodontitis. Fig. S4, H and I, show shows that oral microbial biomass is comparable between Padi4 KO and WT mice at 18 h and 6 d.
Data availability
Data related to this study are available within the main text and main figures and/or its supplementary materials. RNA-seq datasets have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus database under accession code GSE118166 and GSE228021. Microbiome sequencing data have been deposited to the Sequence Read Archive with the BioProjectID PRJNA949160 (http://www.ncbi.nlm.nih.gov/bioproject/949160). All other data are available from the corresponding author upon reasonable request.
Acknowledgments
We thank all the members of the Moutsopoulos lab for their technical assistance and helpful discussions. We thank the NIDCR Veterinary Resources Core (ZIC DE000740-05); NIDCR/NIDCD Genomics and Computational Biology Core (ZIC DC000086); NIDCR Combined Technical Research Core (ZIC DE000729-09); Genomics Core and the Microbiome and Genetics Core (Center for Cancer Research, National Cancer Institute, National Institutes of Health).
This work was supported by the NIDCR and National Institute of Arthritis and Musculoskeletal and Skin Diseases Intramural Research Programs (ZIADE000736 to N.M. Moutsopoulos, Z01DE0699 to T.H. Bugge, 1K99DE030124-01A1 to L.M. Silva, and ZIAAR041199 for M.J. Kaplan).
Author contributions: Conceptualization: T.S. Kim and N.M. Moutsopoulos. Experimentation: T.S. Kim, L.M. Silva, V.I. Theofilou, T. Greenwell-Wild, D.W. Williams, T. Ikeuchi, L. Brenchley, NIDCD/NIDCR Genomics and Computational Biology Core, and C. Carmona-Rivera. Data analysis: T.S. Kim, L.M. Silva, T. Greenwell-Wild, C. Carmona-Rivera, and L. Li. Writing the manuscript: T.S. Kim and N.M. Moutsopoulos. Manuscript review/edits: L.M. Silva, D.W. Williams, T. Greenwell-Wild, L. Brenchley, C. Carmona-Rivera, T.H. Bugge, P.I. Diaz, and M.J. Kaplan. Funding acquisition and supervision: T.H. Bugge, L.M. Silva, M.J. Kaplan, and N.M. Moutsopoulos. Overall supervision: N.M. Moutsopoulos.
References
The lead contact for NIDCD/NIDCR Genomics and Computational Biology Core is Robert Morell, [email protected].
Data availability
Data related to this study are available within the main text and main figures and/or its supplementary materials. RNA-seq datasets have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus database under accession code GSE118166 and GSE228021. Microbiome sequencing data have been deposited to the Sequence Read Archive with the BioProjectID PRJNA949160 (http://www.ncbi.nlm.nih.gov/bioproject/949160). All other data are available from the corresponding author upon reasonable request.

