


In the human thymus, a CD10+ PD-1+ TCRαβ+ differentiation pathway diverges from the conventional single positive T cell lineages at the early double-positive stage. Here, we identify the progeny of this unconventional lineage in antigen-inexperienced blood. These unconventional T cells (UTCs) in thymus and blood share a transcriptomic profile, characterized by hallmark transcription factors (i.e., ZNF683 and IKZF2), and a polyclonal TCR repertoire with autoreactive features, exhibiting a bias toward early TCRα chain rearrangements. Single-cell RNA sequencing confirms a common developmental trajectory between the thymic and blood UTCs and clearly delineates this unconventional lineage in blood. Besides MME+ recent thymic emigrants, effector-like clusters are identified in this heterogeneous lineage. Expression of Helios and KIR and a decreased CD8β expression are characteristics of this lineage. This UTC lineage could be identified in adult blood and intestinal tissues. In summary, our data provide a comprehensive characterization of the polyclonal unconventional lineage in antigen-inexperienced blood and identify the adult progeny.

Introduction
Conventional CD4+ and CD8+ T cells are the main and best-studied T cell populations generated in the thymus. After the successful rearrangement of the TCRα and β locus in the thymus, each precursor T cell expresses a single, unique TCR. These cells are subsequently selected for MHC binding affinity. Only cells with a moderate binding affinity further differentiate to mature CD4 or CD8 single-positive (SP) conventional T cells (CTCs), a process called positive selection. After stringent selection, a small percentage of these T cells emigrate from the thymus as dormant, stem cell–like cells without effector function. These naive cells recirculate between blood and secondary lymphoid organs and will acquire effector function only after encountering their cognate foreign antigen (Chaplin, 2010).
Concurrently, several minor lineages of so-called unconventional T cell (UTC) populations are generated by different selection mechanisms. In recent years, there has been a growing appreciation of the diverse role of unconventional populations, bridging innate and adaptive immunity. The best-studied unconventional αβ T cell subsets include natural killer T cells (NKT) and mucosal-associated invariant T cells (MAIT). These two distinct populations express semi-invariant TCRs that recognize non-peptide antigens in complex with nonclassical MHC class I–like molecules, CD1d and MHC-related molecule-1 (MR1), respectively (Pellicci et al., 2020).
Besides these semi-invariant TCRαβ populations, a UTC lineage with a polyclonal TCR repertoire has been described in mice. Unconventional TCRαβ+ CD8αα+ intestinal intraepithelial lymphocytes (IELs), in addition to the induced memory TCRαβ+ CTCs and the TCRγδ+ cells, form a prominent thymus-derived T cell population that guards the intestinal epithelium (Cheroutre et al., 2011). Thymic IEL precursors (IELps) divert from conventional T cell development upon high-affinity TCR interaction at the CD4 CD8 double-positive (DP) stage. Although high-affinity TCR interaction usually leads to clonal deletion, some precursor T cells are induced to differentiate into CD4 CD8 double-negative (DN) CD3+ T cells, which acquire an activated phenotype and express intestinal homing receptors (McDonald et al., 2014).
Our research previously identified an unconventional CD10+ PD-1+ subset of mature CD8αβ+ T cells coexpressing CD8αα dimers in the human postnatal thymus (PNT; Verstichel et al., 2017). This innate-like population displays an effector phenotype associated with agonist selection. The TCR repertoire is diverse with a biased usage of TCRα joining (TRAJ)–proximal V gene segments and TCRα variable (TRAV)–proximal J gene segments, indicating that these thymocytes are selected early in the DP stage (Verstichel et al., 2017). This characteristic repertoire was confirmed by Daley et al. (2019), who furthermore observed an increased percentage of TCRs with hydrophobic amino acid (AA) doublets or central cysteines in the complementarity-determining region 3 (CDR3), characteristic of self-reactive TCRs. The same TCR characteristics were identified in type A IELps in mice (Daley et al., 2019; Wirasinha et al., 2018).
The aim of the present study is to identify the progeny of this agonist-selected CD10+ PD-1+ population in the human periphery. Studies were performed on antigen-inexperienced cord blood (CB), as CB CTCs have not yet been activated by foreign antigens and therefore are easily discriminated from innate-like UTCs. In this manuscript, we show evidence that the agonist-selected CD10+ PD-1+ population indeed leaves the human thymus by identifying a corresponding population in CB based on a shared transcriptomic and TCR repertoire profile. Comprehensive analysis of the CB UTC lineage identified an MME+ cell cluster with a similar transcriptome as PNT PD-1+ cells and several effector-like clusters in CB. Finally, we could show that Helios+ KIR+ cells, which are found in adult blood and tissues, are part of this UTC lineage.
Results
The PNT and CB PD-1+ populations share a similar transcriptomic and proteomic profile
The unconventional agonist-selected population was previously defined in human PNT as CD3+ TCRγδ− CD4− CD8α+ CD10+ PD-1+ (Fig. S1 A). The progeny of this PNT PD-1+ population in human CB was tentatively defined as CD3+/low TCRγδ− CD4− CD8α+ PD-1+ (Fig. 1 A; Verstichel et al., 2017). CD10 membrane expression, which is prominent in PNT PD-1+ cells (Fig. S1A), was less prominent in the CB PD-1+ population (Fig. 1 B). However, MME mRNA (encoding CD10) was significantly upregulated in the CB PD-1+ population compared with the conventional CD3+ PD-1− population (Fig. 1 C). To examine the relatedness of PNT and CB PD-1+ populations, both were comprehensively analyzed by means of transcriptome and proteome analyses and compared with the CD3+ PD-1− population. Principal component analysis (PCA) of the sorted CB populations indicated that the PD-1+ populations from the different donors clustered together and, similar to the respective PNT populations, shared more features with the unconventional TCRγδ population than with conventional PD-1− populations (Fig. 1 D and Fig. S1 C). Volcano plots comparing the transcriptomes of the PD1+ and PD1− populations showed significant upregulation in both CB and PNT PD-1+ populations of the hallmark transcription factors (TFs) ZNF683 (Hobit), IKZF2 (Helios), RUNX3, ID3, and TBX21 (T-bet); and downregulation of RORA and FOXP1 that mediates quiescence, and SATB1 that is required for positive and negative selection (Fig. 1 C and Fig. S1 B; Kondo et al., 2016; Wei et al., 2016). Notably, both CB and PNT PD-1+ populations highly expressed the unconventional TCRαβ marker TRGC2 (TCRγ constant 2), a gene progressively silenced in the conventional T cell lineage during passage through the CD4+ CD8+ DP stage in the thymus (Fig. 1 C and Fig. S1 B; Kisielow et al., 2011). Mass spectrometry–based proteomics confirmed upregulation of Helios in the PNT PD-1+ population and, although not reaching the significance threshold, in CB (Fig. 1 C and Fig. S1 B). Flow cytometric analysis validated the upregulation of Helios in both PNT and CB populations. Helios expression was stronger in the PD-1+ population in PNT compared with the CB (Fig. 1 E and Fig. S1 D). In search of additional distinctive markers, membrane proteins identified in the transcriptomic and proteomic profile of the PNT and CB PD-1+ populations were confirmed by flow cytometry: CD3low CD8βlow CCR7− and EVI2B+ (Fig. 1 E and Fig. S1 D). As expected, a correlation between the significantly differentially expressed genes and abundance of the corresponding proteins was observed (Fig. S1 E). When zooming in on the genes or proteins that were differentially expressed between the PD-1+ and PD-1− populations either in PNT or in CB, a highly significant positive correlation was revealed between the respective PNT and CB populations at both the RNA and protein level (Fig. 1 F, Fig. S1 F, and Table S1). Supporting this correlation between the PNT and CB PD-1+ population, gene set enrichment analysis (GSEA) confirmed that the significantly upregulated genes in the PNT PD-1+ population were also significantly enriched in the CB PD-1+ population (Fig. 1 G). Based on the similarities in their transcriptomic and proteomic profile, it is hypothesized here that the CB CD3+/low TCRγδ− CD4− CD8α+ PD-1+ population is the progeny of the PNT PD-1+ population.

RNA and protein expression profile by the PNT CD10 + PD-1 + population. (A) Representative gating strategy for the CD3+ TCRγδ− CD4− CD8α+ CD10+ PD-1+ populations in human PNT. (B) Volcano plots of differentially expressed genes (left) and proteins (right) between the PNT CD10+ PD-1+ and CD10− PD-1− populations. Triangles indicate data points outside the y-axis range. Data points with a |log2 fold change| >0.6 and adjusted P < 0.05 are colored (upregulated in blue, downregulated in orange). (C) PCA of the transcriptome (left, donor corrected) and proteome (right) analysis of the sorted populations from three different PNT donors. (D) Flow cytometric analysis of CD8α, CD8β, CD3, Helios, CCR7, and EVI2B on the CD10− PD-1− and CD10+ PD-1+ population in PNT, representative of at least three PNT donors. (E) Heatmap showing the corresponding relative protein abundance of all significantly differentially expressed genes between the PNT CD10+ PD-1+ and CD10− PD-1− population (left) or between the CB CD3+/low PD-1+ and CD3+ PD-1− population (right). Differentially expressed genes of which the corresponding protein was not detected are not shown. The heatmap shows column- and row-scaled, normalized, mean protein abundance. (F) Bar graph showing the overlap in identified (left) and significantly differentially expressed (right) RNAs and proteins in PNT and CB populations.
RNA and protein expression profile by the PNT CD10 + PD-1 + population. (A) Representative gating strategy for the CD3+ TCRγδ− CD4− CD8α+ CD10+ PD-1+ populations in human PNT. (B) Volcano plots of differentially expressed genes (left) and proteins (right) between the PNT CD10+ PD-1+ and CD10− PD-1− populations. Triangles indicate data points outside the y-axis range. Data points with a |log2 fold change| >0.6 and adjusted P < 0.05 are colored (upregulated in blue, downregulated in orange). (C) PCA of the transcriptome (left, donor corrected) and proteome (right) analysis of the sorted populations from three different PNT donors. (D) Flow cytometric analysis of CD8α, CD8β, CD3, Helios, CCR7, and EVI2B on the CD10− PD-1− and CD10+ PD-1+ population in PNT, representative of at least three PNT donors. (E) Heatmap showing the corresponding relative protein abundance of all significantly differentially expressed genes between the PNT CD10+ PD-1+ and CD10− PD-1− population (left) or between the CB CD3+/low PD-1+ and CD3+ PD-1− population (right). Differentially expressed genes of which the corresponding protein was not detected are not shown. The heatmap shows column- and row-scaled, normalized, mean protein abundance. (F) Bar graph showing the overlap in identified (left) and significantly differentially expressed (right) RNAs and proteins in PNT and CB populations.

RNA and protein expression profile by the CB CD3 +/low PD-1 + population. (A) Representative gating strategy for the CD3+/low TCRγδ− CD4− CD8α+ PD-1+ population in human CB. (B) CD10 expression on the CD3+ PD-1− (orange) and CD3+/low PD-1+ (blue) populations in CB, representative of at least three CB donors. (C) Volcano plots of differentially expressed genes (left) and proteins (right) between the CB CD3+/low PD-1+ and CD3+ PD-1− populations. Triangles indicate data points outside the y-axis range. Data points with a |log2 fold change| >0.6 and adjusted P < 0.05 are colored (upregulated in blue, downregulated in orange). (D) PCA of the transcriptome (left, donor corrected) and proteome (right) analysis of the sorted populations from three different CB donors. (E) Flow cytometric analysis of CD8α, CD8β, CD3, Helios, CCR7, and EVI2B on the CD3+ PD-1− and CD3+/low PD-1+ populations in CB, representative of at least three CB donors. (F) Scatterplots and Spearman correlation coefficient comparing the log2 fold change of the significantly differentially expressed genes (left) and proteins (right) of CB (CD3+/low PD-1+ versus CD3+ PD-1−) and PNT (CD10+ PD-1+ versus CD10− PD-1−). (G) GSEA showing the significantly upregulated gene set from PNT CD10+ PD-1+ versus CD10− PD-1−, on the CB CD3+/low PD-1+ versus CD3+ PD-1− population. NES and FDR q value are shown.
RNA and protein expression profile by the CB CD3 +/low PD-1 + population. (A) Representative gating strategy for the CD3+/low TCRγδ− CD4− CD8α+ PD-1+ population in human CB. (B) CD10 expression on the CD3+ PD-1− (orange) and CD3+/low PD-1+ (blue) populations in CB, representative of at least three CB donors. (C) Volcano plots of differentially expressed genes (left) and proteins (right) between the CB CD3+/low PD-1+ and CD3+ PD-1− populations. Triangles indicate data points outside the y-axis range. Data points with a |log2 fold change| >0.6 and adjusted P < 0.05 are colored (upregulated in blue, downregulated in orange). (D) PCA of the transcriptome (left, donor corrected) and proteome (right) analysis of the sorted populations from three different CB donors. (E) Flow cytometric analysis of CD8α, CD8β, CD3, Helios, CCR7, and EVI2B on the CD3+ PD-1− and CD3+/low PD-1+ populations in CB, representative of at least three CB donors. (F) Scatterplots and Spearman correlation coefficient comparing the log2 fold change of the significantly differentially expressed genes (left) and proteins (right) of CB (CD3+/low PD-1+ versus CD3+ PD-1−) and PNT (CD10+ PD-1+ versus CD10− PD-1−). (G) GSEA showing the significantly upregulated gene set from PNT CD10+ PD-1+ versus CD10− PD-1−, on the CB CD3+/low PD-1+ versus CD3+ PD-1− population. NES and FDR q value are shown.
The TCR repertoires of the PNT PD-1+ and CB PD-1+ populations share highly characteristic features
TCRα rearrangements are known to occur in a non-random manner, starting at the J-proximal V segments and the V-proximal J segments. As multiple sequential rearrangements may occur during the DP thymocyte stage, later rearrangements tend to be biased toward J-distal V segment and V-distal J segment usage. As shown in our previous publication and confirmed by others, the TCRα usage of the PNT PD-1+ population is biased toward early rearrangements similar to early DP thymocytes, in contrast to late DP and conventional thymocytes (Verstichel et al., 2017; Daley et al., 2019; Park et al., 2020). Additionally, the presence of cysteines within two positions of the CDR3 apex (cysteine index) and enrichment of hydrophobic AA doublets at positions 6 and 7 of the CDR3 (hydrophobic index) is reported for the PNT PD-1+ population (Daley et al., 2019; Stadinski et al., 2016). Similar CDR3 properties are also reported to result in strong TCR–ligand interactions (Lagattuta et al., 2022; Košmrlj et al., 2008; Logunova et al., 2020). Finally, in contrast to NKT or MAIT cells, the repertoire of the PNT PD-1+ population was determined to be polyclonal (Fig. S2, A and B). Thus, to obtain additional evidence for a precursor–progeny relationship between the PNT and CB PD-1+ populations, the TCR repertoire of the CB PD-1+ population was analyzed for these characteristics and compared with the PNT PD-1+ TCR repertoire (Fig. S2, A–G for PNT).

Distinctive TCR repertoire of the PNT CD10 + PD-1 + population. (A–G) show the already known TCR characteristics for the PNT CD10+ PD-1+ population (Verstichel et al., 2017; Daley et al., 2019), which were here confirmed and added to allow easy comparison with the CB TCR repertoire shown in Fig. 2. (A) Representative tree maps showing CDR3α clonotype usage in relation to repertoire size for the CD10− PD-1− (top) and CD10+ PD-1+ (bottom) populations. Each rectangle represents one CDR3 clonotype and its size corresponds to its relative frequency in the repertoire (rectangle colors are categorized from J-proximal (pink) to J-distal (white) for the TRAV gene segments). (B) D75 (percentage of clonotypes required to occupy 75% of the total TCR repertoire) analysis for the TCRα chain, comparing the PD-1− and PD-1+ population (individual values and mean ± SEM). Mean difference was not significant. (C) Representative heatmap illustrating the difference in TCR Vα usage between the PD-1− and PD-1+ population. (D) Representative cumulative percentage of TRAV (left) or TRAJ (right) gene segment usage. The x axis represents the location in the TRAV or TRAJ locus. (E) Area under the curve determined from the cumulative plots from each sample (individual values and mean ± SEM, n = 6). Šídák’s multiple comparisons test was used to assess the statistically significant difference. P value < 0.05 (*), P value < 0.001 (***). (F and G) Cysteine index (F; percentage of unique sequences with cysteine within two positions of the CDR3 apex) and hydrophobic index (G; percentage of unique sequences with self-reactive hydrophobic CDR3 position 6 and 7 doublets) of the CDR3α. (H) Percentage of unique sequences containing a TRDV1 segment. Paired t-tests were used to assess statistical significance (B and F–H). Connected values correspond to paired populations of the same biological replicate (n = 6). P value > 0.05 (ns), P value < 0.05 (*), P value < 0.001 (***). (I) Flow cytometric analysis of the Vδ1+ (A13 clone) cells in both CB populations, representative of six CB donors.
Distinctive TCR repertoire of the PNT CD10 + PD-1 + population. (A–G) show the already known TCR characteristics for the PNT CD10+ PD-1+ population (Verstichel et al., 2017; Daley et al., 2019), which were here confirmed and added to allow easy comparison with the CB TCR repertoire shown in Fig. 2. (A) Representative tree maps showing CDR3α clonotype usage in relation to repertoire size for the CD10− PD-1− (top) and CD10+ PD-1+ (bottom) populations. Each rectangle represents one CDR3 clonotype and its size corresponds to its relative frequency in the repertoire (rectangle colors are categorized from J-proximal (pink) to J-distal (white) for the TRAV gene segments). (B) D75 (percentage of clonotypes required to occupy 75% of the total TCR repertoire) analysis for the TCRα chain, comparing the PD-1− and PD-1+ population (individual values and mean ± SEM). Mean difference was not significant. (C) Representative heatmap illustrating the difference in TCR Vα usage between the PD-1− and PD-1+ population. (D) Representative cumulative percentage of TRAV (left) or TRAJ (right) gene segment usage. The x axis represents the location in the TRAV or TRAJ locus. (E) Area under the curve determined from the cumulative plots from each sample (individual values and mean ± SEM, n = 6). Šídák’s multiple comparisons test was used to assess the statistically significant difference. P value < 0.05 (*), P value < 0.001 (***). (F and G) Cysteine index (F; percentage of unique sequences with cysteine within two positions of the CDR3 apex) and hydrophobic index (G; percentage of unique sequences with self-reactive hydrophobic CDR3 position 6 and 7 doublets) of the CDR3α. (H) Percentage of unique sequences containing a TRDV1 segment. Paired t-tests were used to assess statistical significance (B and F–H). Connected values correspond to paired populations of the same biological replicate (n = 6). P value > 0.05 (ns), P value < 0.05 (*), P value < 0.001 (***). (I) Flow cytometric analysis of the Vδ1+ (A13 clone) cells in both CB populations, representative of six CB donors.
Analysis of the CDR3α and CDR3β clonotypes revealed that the CB PD-1+ population was polyclonal and the degree of polyclonality was similar to the conventional PD-1− T cell population (Fig. 2 A). This was quantified by calculating the D75 values, i.e., the percentage of unique clonotypes required to occupy 75% of the total TCR repertoire for both populations. The D75 values were about 30% for both CB populations, indicating that the bulk of the repertoire consists of a wide variety of clonotypes. Moreover, no significant mean difference between the two CB populations could be identified, supporting the notion that the UTC population was equally polyclonal as the conventional T cell population in CB (Fig. 2 B). The CB PD-1+ population exhibited biased usage of early J-proximal TCR Vα rearrangements and early V-proximal TCR Jα rearrangements, and this bias was similar to that found in the PNT PD-1+ population (Fig. 2, C–E; and Fig. S2, C–E). The cysteine index of the TCRβ chain was significantly higher in the CB PD-1+ population compared with the PD-1− population, and the same trend could be observed for the TCRα chain (Fig. 2 F). The hydrophobic index of the TCRβ chain was likewise significantly higher in the CB PD-1+ population compared with the PD-1− population. However, such a trend could not be established for the TCRα chain (Fig. 2 G). In line with this, the CDR3β repertoire exhibited higher interaction strength values (strength and volume parameters) compared with the PD-1− population counterpart (Fig. 2 H; Košmrlj et al., 2008; Lagattuta et al., 2022). In contrast, polarity, a property associated with CTCs, was reduced in the CB PD-1+ population (Fig. 2 H; Bolotin et al., 2017). Finally, TCR sequencing of the CB populations revealed a significantly higher percentage of TRAJ sequences using the TCR δ variable 1 (TRDV1) gene segment (instead of a TRAV gene segment) in the CB PD-1+ population (Fig. 2 I). When subsequently analyzing the PNT populations, an increased TRDV1 usage was also observed in the PNT PD-1+ population compared with their PD-1− counterparts (Fig. S2 H). Vδ1+ cells expressing a hybrid TRDV1-TRAJ-TRAC TCR chain and coexpressing a TCRβ chain rather than a TCRγ chain have been previously reported in human peripheral blood. This population, termed δ/αβ T cells, recognizes antigens presented by both HLA and CD1d (Pellicci et al., 2014). By using an anti-Vδ1 antibody, an enrichment of Vδ1 membrane expression was shown in the CB PD-1+ population (Fig. 2 J and Fig. S2 I).

Distinctive TCR repertoire of the CB PD-1 + population. (A) Representative tree maps showing CDR3α (left) and CDR3β (right) clonotype usage in relation to repertoire size for the CD3+ PD-1− (top) and CD3+/low PD-1+ (bottom) populations. Each rectangle represents one CDR3 clonotype, and its size corresponds to its relative frequency in the repertoire. Rectangle colors for CDR3α are categorized from J-proximal (pink) to J-distal (white) for the TRAV gene segments, and for CDR3β, they are chosen randomly. (B) D75 (percentage of clonotypes required to occupy 75% of the total TCR repertoire) analysis for the TCRα chain, comparing the PD-1− and PD-1+ population (individual values and mean ± SEM). Mean difference was not significant. (C) Representative heatmap illustrating the difference in J-proximal versus J-distal TCR Vα usage between the PD-1− and PD-1+ populations. (D) Cumulative percentage of TRAV (left) or TRAJ (right) gene segment usage by the PD-1− (orange) and PD-1+ (blue) population in a representative donor. The x axis represents the location in the TRAV or TRAJ locus. (E) Area under the curve determined from the cumulative plots from each sample (individual values and mean ± SEM, n = 6). Šídák's multiple comparisons test was used to assess the statistically significant difference. P value < 0.0001 (****). (F and G) Cysteine index (F; percentage of unique sequences with cysteine within two positions of the CDR3 apex) and hydrophobic index (G; percentage of unique sequences with self-reactive hydrophobic CDR3 position 6 and 7 doublets) of the CDR3α (left) and CDR3β (right). (H) Physicochemical properties (strength, volume, and polarity) of the CDR3α (left) and CDR3β (right). (I) Percentage of unique sequences containing a TRDV1 segment. (J) Flow cytometric analysis of the percentage of Vδ1+ (A13 clone) cells in both CB populations. Paired t tests were used to assess statistical significance (B and F–J). Connected values correspond to paired populations of the same biological replicate (n = 6). P value > 0.05 (ns), P value < 0.05 (*), P value < 0.01 (**), P value < 0.0001 (****).
Distinctive TCR repertoire of the CB PD-1 + population. (A) Representative tree maps showing CDR3α (left) and CDR3β (right) clonotype usage in relation to repertoire size for the CD3+ PD-1− (top) and CD3+/low PD-1+ (bottom) populations. Each rectangle represents one CDR3 clonotype, and its size corresponds to its relative frequency in the repertoire. Rectangle colors for CDR3α are categorized from J-proximal (pink) to J-distal (white) for the TRAV gene segments, and for CDR3β, they are chosen randomly. (B) D75 (percentage of clonotypes required to occupy 75% of the total TCR repertoire) analysis for the TCRα chain, comparing the PD-1− and PD-1+ population (individual values and mean ± SEM). Mean difference was not significant. (C) Representative heatmap illustrating the difference in J-proximal versus J-distal TCR Vα usage between the PD-1− and PD-1+ populations. (D) Cumulative percentage of TRAV (left) or TRAJ (right) gene segment usage by the PD-1− (orange) and PD-1+ (blue) population in a representative donor. The x axis represents the location in the TRAV or TRAJ locus. (E) Area under the curve determined from the cumulative plots from each sample (individual values and mean ± SEM, n = 6). Šídák's multiple comparisons test was used to assess the statistically significant difference. P value < 0.0001 (****). (F and G) Cysteine index (F; percentage of unique sequences with cysteine within two positions of the CDR3 apex) and hydrophobic index (G; percentage of unique sequences with self-reactive hydrophobic CDR3 position 6 and 7 doublets) of the CDR3α (left) and CDR3β (right). (H) Physicochemical properties (strength, volume, and polarity) of the CDR3α (left) and CDR3β (right). (I) Percentage of unique sequences containing a TRDV1 segment. (J) Flow cytometric analysis of the percentage of Vδ1+ (A13 clone) cells in both CB populations. Paired t tests were used to assess statistical significance (B and F–J). Connected values correspond to paired populations of the same biological replicate (n = 6). P value > 0.05 (ns), P value < 0.05 (*), P value < 0.01 (**), P value < 0.0001 (****).
To conclude, a series of characteristic features of the TCR repertoire of the PNT PD-1+ population can be tracked within the CB PD-1+ population. This strongly suggests that the CB PD-1+ T cell population is the progeny of the PNT PD-1+ T cell population and that biased TCRα chain usage and self-reactive features of both TCR chains are acquired during early thymic agonist selection and preserved after thymic egress.
The CB UTC population extends beyond the CD3+/low PD-1+ cells
The high level of PD-1 expression by the PNT PD-1+ population is attributed to an elevated and persistent TCR signaling during thymic agonist selection (McDonald et al., 2014; Pobezinsky et al., 2012). It is therefore hypothesized that, after leaving the thymus, PD-1 may be downregulated. Indeed, the PNT PD-1+ population downregulates PD-1 upon in vitro culture with IL-15 (Billiet et al., 2020). Consequently, the CD3+/low PD-1+ CB population may not include the complete human unconventional population but only the more recent thymic emigrants of that population. Therefore, the discriminatory markers identified above (Fig. 1 E) were used in a flow cytometric analysis of the entire CD3+/low TCRγδ− CD4− CD8α+ fraction in CB (Fig. 3 A). In addition to the expected conventional CCR7+ population, the resulting uniform manifold approximation and projection (UMAP) and flow cytometry data analysis using self-organizing maps (FlowSOM) clustering revealed a distinct cluster 2 of CCR7− cells that contained all PD-1+ cells (Fig. 3 A). Within this CCR7− cluster 2, part of the PD-1− cells were Helios+ and EVI2B+, two markers associated with the PNT PD-1+ population (Fig. 3 A and Fig. S1 B). Here, it was hypothesized that these Helios+ and EVI2B+ cells may represent PD-1− progeny of the PNT PD-1+ population. Consequently, the CB UTC population was further studied within the CCR7− EVI2B+ as well as within the CD3+/low PD-1+ population.

scRNA-seq of human CB reveals a heterogeneous unconventional population. (A) UMAP analysis of flow cytometry data gated on all CD3+/low TCRγδ− CD4− CD8α+ cells from a representative CB donor. FlowSOM clustering results are projected onto the left UMAP, identifying three clusters. (B) Schematic workflow of the CB processing. (C) Gating strategy on the CD3+/low TCRγδ− cells for the four different sorts to isolate the different populations of interest. (D) UMAP of 24,727 CB single cells, colored by the 13 identified cell clusters. (E) UMAP representation of the four different sorts, with the cells from the particular sorts colored according to the cell clusters and the remaining cells in gray.
scRNA-seq of human CB reveals a heterogeneous unconventional population. (A) UMAP analysis of flow cytometry data gated on all CD3+/low TCRγδ− CD4− CD8α+ cells from a representative CB donor. FlowSOM clustering results are projected onto the left UMAP, identifying three clusters. (B) Schematic workflow of the CB processing. (C) Gating strategy on the CD3+/low TCRγδ− cells for the four different sorts to isolate the different populations of interest. (D) UMAP of 24,727 CB single cells, colored by the 13 identified cell clusters. (E) UMAP representation of the four different sorts, with the cells from the particular sorts colored according to the cell clusters and the remaining cells in gray.
To comprehensively study the heterogeneity of the UTCs in CB, single-cell RNA sequencing (scRNA-seq) was performed. CB of two different donors was depleted of CD4+, CD14+, CD19+, and CD235+ cells (Fig. 3 B). Of the first donor, the UTCs were sorted as CD8α+ CD3+/low PD-1+ (sort 1) or CD8α+ CCR7− EVI2B+ (sort 2) within the CD3+/low TCRγδ− CD4− window. Both fractions were labeled with different hashtags before they were further processed, enabling subsequent assignment of the single cells to their corresponding sorting strategy. For the second donor, the sorting strategy was expanded to include CD4− CD8α− DN cells (sort 3 and 4). In sorts 3 and 4, the respective conventional populations were also sorted and added in equal portions before further analysis (Fig. 3 C). Sort 4 was combined with cellular indexing of transcriptomes and epitopes sequencing (CITE-seq) to capture the expression of 277 membrane proteins. Using a droplet-based single-cell platform, 3′ gene expression libraries were constructed. Reciprocal PCA (RPCA) was used to integrate the Seurat objects resulting from the separate sorts. This approach resulted in 24,727 cells included in the scRNA-seq analysis after quality control and filtering. Leiden clustering applied to this filtered and integrated Seurat object defined 13 distinct clusters (Fig. 3 D). Both CD8α+ CD3+/low PD-1+ (sort 1) and CD8α+ CCR7− EVI2B+ (sort 2) consisted mainly of clusters 1–5, suggesting that these clusters represent the UTCs. Focusing on these five clusters, cluster 1 is relatively overrepresented in the CD3+/low PD-1+ sorts 1 and 3, and cluster 4 is relatively enriched in the CCR7− EVI2B+ sorts 2 and 4, highlighting that indeed the two different sorting strategies captured slightly different UTC subpopulations (Fig. 3 E).
Defining UTC clusters using transcriptomics
This heterogeneity consisting of 13 different clusters has not been reported before for CB CD8+ T cells. Therefore, the clusters were manually annotated based on prominently upregulated genes (Fig. 4, A and B; and Tables S2 and S3). One non-T cell cluster was detected, which was annotated as NK cells based on high expression of NK-associated genes (GZMB, TYROBP) and absence of membrane CD3 and TCRαβ (Fig. 4 A and Fig. S4). The NK cluster was assumed to be a contaminant due to lenient gating for CD3. Two minor T cell clusters were annotated. A cycling T cell cluster was annotated based on upregulated effector (i.e., GZMA, GZMK) and cycling genes (i.e., PCNA, MKI67, CDC6; Fig. 4 A and Fig. S3 A). In addition, NKT/MAIT cells were identified based on coexpression of KLRB1, SLC4A10, DPP4, and IL7R (Fig. 4, A, C, and F; and Fig. S3 A; Domínguez Conde et al., 2022; Parrot et al., 2020). The NKT/MAIT cells were the main “contaminant” in the CCR7− EVI2B+ sorts (Fig. S3 B). From here on, the NKT and MAIT cells will be referred to as the NKT/MAIT population and the novel polyclonal population identified in this study as the UTC population.
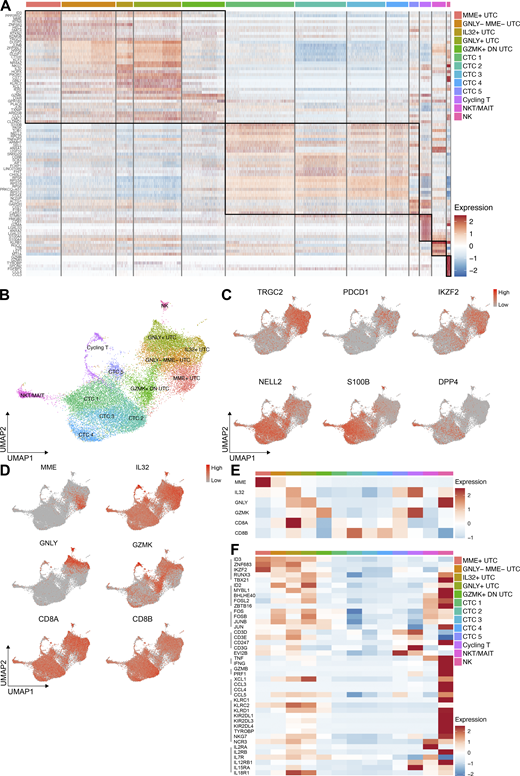
Annotation of the UTC clusters in CB. (A) Heatmap showing the expression of the top 10 differentially expressed genes per defined cluster in CB. Recurrent genes are not repeated. The genes are listed in Table S3. (B) UMAP visualization of the 13 identified cell clusters in CB. (C) UMAP feature plots representing discriminating genes between the UTC and CTC clusters. (D) UMAP feature plots representing differentially expressed genes used to annotate the different UTC clusters. (E) Heatmap showing the mean expression of the UTC signature genes in the different clusters in CB. (F) Heatmap showing the mean expression of characteristic UTC genes in the different clusters in CB.
Annotation of the UTC clusters in CB. (A) Heatmap showing the expression of the top 10 differentially expressed genes per defined cluster in CB. Recurrent genes are not repeated. The genes are listed in Table S3. (B) UMAP visualization of the 13 identified cell clusters in CB. (C) UMAP feature plots representing discriminating genes between the UTC and CTC clusters. (D) UMAP feature plots representing differentially expressed genes used to annotate the different UTC clusters. (E) Heatmap showing the mean expression of the UTC signature genes in the different clusters in CB. (F) Heatmap showing the mean expression of characteristic UTC genes in the different clusters in CB.

Defining the T cell clusters in CB. (A) UMAP feature plots of characteristic genes for the cycling T cells (top) and MAIT cells (bottom). (B) Stacked bar chart showing the composition of the 13 identified clusters (left) or the five different UTC clusters (right) in the different sorts. (C) Integration of the CB scRNA-seq dataset with previously published fetal CD45+ and postnatal CD3+ thymic scRNA-seq datasets (Park et al., 2020). UMAP visualization of the annotated thymic clusters (left) and CB clusters (right) after integration.
Defining the T cell clusters in CB. (A) UMAP feature plots of characteristic genes for the cycling T cells (top) and MAIT cells (bottom). (B) Stacked bar chart showing the composition of the 13 identified clusters (left) or the five different UTC clusters (right) in the different sorts. (C) Integration of the CB scRNA-seq dataset with previously published fetal CD45+ and postnatal CD3+ thymic scRNA-seq datasets (Park et al., 2020). UMAP visualization of the annotated thymic clusters (left) and CB clusters (right) after integration.
Based on their upregulation of hallmark thymic UTC genes, five UTC clusters were annotated (Fig. 4, A and B). The unconventional marker genes TRGC2, PDCD1, and IKZF2 were expressed homogeneously in these UTC clusters, with some expression in the cycling T and MAIT/NKT cells (Fig. 4 C). The remaining clusters were considered CTC clusters based on the expression of typical conventional naive T cell markers (FOXP1 and SELL; Fig. 4, A and B). Furthermore, markers (i.e., NELL2 and S100B) that were strongly overexpressed in the bulk transcriptome of the conventional PD-1− populations in both PNT and CB (Fig. 1 C and Fig. S1 B) were homogeneously expressed in the CTC clusters and largely absent in the UTC clusters, except for the GZMK+ DN UTC cluster. Likewise, DPP4 (encoding CD26) was expressed homogeneously and exclusively by the CTC clusters and strongly in the NKT/MAIT cluster (Fig. 4 C).
Based on the differentially expressed genes, unique distinctive annotations were provided for the individual UTC clusters (Fig. 4 B and Table S3). MME and GNLY (encoding granulysin) are solely expressed by the UTC clusters. IL32 is highly expressed by IL32+ UTCs but is also expressed at lower levels by different CTC and UTC clusters. The cluster that bridges the bulk of the UTCs and the CTCs had a characteristically high expression of GZMK without overexpression of other cytolytic effector genes and a low expression of CD8A and CD8B and was annotated as the GZMK+ DN UTC cluster (Fig. 4, D and E).
The CB UTC clusters expressed the typical TFs of the PNT UTC lineage, ID3, ZNF683, IKZF2, RUNX3, and TBX21, although very limited in the GZMK+ DN UTCs. Importantly, expression of these TFs was absent in the other T cell clusters (Fig. 4 F). ID3, ZNF683, and IKZF2 were expressed highest in the MME+ UTCs. As expected, the CD3+/low PD-1+ population (sort 1 and 3) was enriched for these MME+ UTCs, the recent thymic emigrants (Fig. S3 B). Previously described innateness-associated TFs (i.e., ID2, MYBL1, BHLHE40, and FOSL2) were also expressed by the UTC clusters. These TFs are known to be enriched in NKT, MAIT, and NK cells (Gutierrez-Arcelus et al., 2019). Of note, the UTC clusters did not express ZBTB16, clearly differentiating them from the NKT/MAIT cells (Fig. 4 F). The activator protein 1 (AP-1) TFs (i.e., FOS, JUN) were constitutively expressed in both the UTC and NKT/MAIT clusters (Fig. 4 F). With regard to effector function, low constitutive expression of cytokines (i.e., TNF and IFNG) and genes involved in cytolysis (i.e., GNLY and granzymes) were observed in the GNLY+ UTCs, IL32+ UTCs, and GZMK+ DN UTCs (Fig. 4, E and F). These three clusters were considered effector UTC clusters. These effector clusters showed expression of multiple NK receptors (i.e., KLRC2, KLRD1, NCR3, and KIRs), which are not expressed by CTCs. Finally, UTCs expressed components of the IL-2, IL-7, and IL-15 receptors, as well as IL-12 and IL-18 receptor components, which are required for inflammation-induced cytokine responses. Expression of the genes discussed above was mostly absent in CTCs and cycling T cells (Fig. 4 F).
Transcriptomic similarities of ZNF683+ CD8αα+ thymocytes and CB UTCs suggest a lineage relationship
It was established above that the TCR repertoires of the PNT PD-1+ and CB PD-1+ populations are similar, suggesting that the thymic population is the precursor of the CB UTCs. To further explore this developmental pathway, the CB scRNA-seq dataset was integrated with previously published fetal CD45+ and postnatal CD3+ thymic scRNA-seq datasets (Fig. S3 C; Park et al., 2020). As published, the CD3+ DP PNT cells diverged into two main pathways: the unconventional pathway consisting of GNG4+CD8αα+ T(I), ZNF683+CD8αα+ T(II), and TCRγδ cells, and the conventional pathway of CD4+ and CD8+ SP T cells (Fig. 5 A). Integration with the CB populations showed that the CB UTC clusters partially overlapped with the PNT UTCs, whereas the CB CTC partially overlapped with the PNT SP T cells (Fig. 5, A and B). When focusing on the UTC pathway, the CB MME+ UTCs overlapped with the PNT CD8αα+ T(I) as well as CD8αα+ T(II) cells (Fig. 5 B). Based on the expression of hallmark differentially expressed genes (i.e., GNG4, MME, and ZNF683), the CB MME+ UTCs seemed to originate from the PNT ZNF683+CD8αα+ T(II) rather than from the GNG4+CD8αα+ T(I) (Fig. 5, C and D). Therefore, a TSCAN trajectory analysis was performed with the ZNF683+CD8αα+ T(II) as the population of origin. This analysis revealed a common pathway passing through PNT ZNF683+CD8αα+ T(II) and CB MME+ UTCs, leading to a branching point at the GNLY− MME− UTC cluster. The GNLY− MME− UTC cluster gave rise to three distinct lineages: the GNLY+ UTCs (lineage 1), the GZMK+ DN UTC (lineage 2), and the IL32+ UTCs (lineage 3; Fig. 5, E and F). The common pathway included ZNF683+ cells, which upregulated ID3 and differentiated in TBX21 (T-bet) positive cells (Fig. 5, G and H). During terminal differentiation, all three lineages expressed high levels of AP-1 TFs and gradually upregulated different effector markers (Fig. 5 G). When analyzing the data in regulons, the transcriptional regulation of the UTCs and the CTCs was significantly different. As expected, the CTCs were mainly regulated by the conventional TFs FOXP1 and RORA, while in the UTCs, KLF4, which negatively regulates TCR-mediated proliferation in CD8+ T cells, and RUNX3 were prominent (Fig. 5 I; Mamonkin et al., 2013; Hao et al., 2017). In conclusion, analysis of transcriptome similarities suggests that the PNT ZNF683+CD8αα+ T(II) thymocytes gave rise to three effector UTC lineages in CB.

UTC pathway analysis reveals three effector lineages. (A) Integration of the CB scRNA-seq dataset with a previously published PNT CD3+ scRNA-seq dataset (Park et al., 2020). UMAP visualization of the annotated PNT clusters (left) and CB clusters (right) after integration. (B) UMAP visualization highlighting the PNT and CB UTCs and showing the overlay of the integrated UTCs derived from PNT or CB. (C) UMAP feature plots of GNG4, MME, ZNF683, and IKZF2 expression by UTCs from PNT (red) or CB (blue). Only cells with a relatively high expression are colored. (D) Heatmap showing the mean expression for hallmark differentially expressed genes per UTC population. (E) UMAP visualization of the TSCAN trajectory analysis of the UTC populations with the PNT ZNF683+CD8αα+ T(II) as the original population. (F) Dendrogram of the predicted UTC lineages. (G) Heatmap showing the varying gene expression in the pseudotime for the different lineages. (H) For each lineage, ID3 or TBX21 expression is shown per single cell and summarized as the mean (gray line). (I) The RSS is plotted per cell type for the most prominent UTC (blue) and CTC (orange) pathways. The cell populations on the x axis are ordered according to pseudotime.
UTC pathway analysis reveals three effector lineages. (A) Integration of the CB scRNA-seq dataset with a previously published PNT CD3+ scRNA-seq dataset (Park et al., 2020). UMAP visualization of the annotated PNT clusters (left) and CB clusters (right) after integration. (B) UMAP visualization highlighting the PNT and CB UTCs and showing the overlay of the integrated UTCs derived from PNT or CB. (C) UMAP feature plots of GNG4, MME, ZNF683, and IKZF2 expression by UTCs from PNT (red) or CB (blue). Only cells with a relatively high expression are colored. (D) Heatmap showing the mean expression for hallmark differentially expressed genes per UTC population. (E) UMAP visualization of the TSCAN trajectory analysis of the UTC populations with the PNT ZNF683+CD8αα+ T(II) as the original population. (F) Dendrogram of the predicted UTC lineages. (G) Heatmap showing the varying gene expression in the pseudotime for the different lineages. (H) For each lineage, ID3 or TBX21 expression is shown per single cell and summarized as the mean (gray line). (I) The RSS is plotted per cell type for the most prominent UTC (blue) and CTC (orange) pathways. The cell populations on the x axis are ordered according to pseudotime.
TCRγδ− CCR7− CD26− cells represent the polyclonal UTC population in CB
To identify phenotypical differences between the different clusters, CITE-seq was included in sort 4 of our scRNA-seq experimental setup (Fig. 3 C and Table S4). Membrane protein data were acquired for 3,615 single cells across all clusters (Fig. 6 A). CD10 (encoded by MME) was expressed exclusively in the MME+ UTC cluster. A small fraction of the MME+ UTC cluster expressed the thymic T cell immaturity marker CD1a, suggesting that this cluster included the recent thymic emigrants (Fig. 6 B). In support of this hypothesis, CD10+ PD-1+ and CD1a+ PD-1+ cells were absent in adult blood, in line with reduced thymic output in adults (Fig. S4, A–C).

CCR7 − CD26 − constitutes the largest unconventional population in CB. (A) UMAP visualization of the single cells in the scRNA-seq analysis from which RNA data (gray) or combined RNA and protein data (blue) was determined. (B) Protein-based UMAP visualizations showing the expression of the indicated cell surface protein markers, only visualizing the single cells from which protein data was collected. (C) Flow cytometric analysis of a CD4-depleted CB. The CD3+/low TCRγδ− cells were further gated for CCR7− CD26− cells, for which UMAP visualizations are shown. Representative of at least three CB donors. (D) Percentage of the CD3+ CD4− cells in CB which are TCRγδ+, CCR7− CD26−, or CD161high (individual values and mean ± SEM, n = 10). Holm-Šídák’s multiple comparisons test was used to assess the statistically significant difference. P value < 0.05 (*), P value < 0.01 (**). Gating strategy is shown in Fig. S4 E. (E) Heatmap depicting the mean expression of cell surface protein markers used to differentiate the UTC and CTC clusters from the NKT/MAIT and NK clusters.
CCR7 − CD26 − constitutes the largest unconventional population in CB. (A) UMAP visualization of the single cells in the scRNA-seq analysis from which RNA data (gray) or combined RNA and protein data (blue) was determined. (B) Protein-based UMAP visualizations showing the expression of the indicated cell surface protein markers, only visualizing the single cells from which protein data was collected. (C) Flow cytometric analysis of a CD4-depleted CB. The CD3+/low TCRγδ− cells were further gated for CCR7− CD26− cells, for which UMAP visualizations are shown. Representative of at least three CB donors. (D) Percentage of the CD3+ CD4− cells in CB which are TCRγδ+, CCR7− CD26−, or CD161high (individual values and mean ± SEM, n = 10). Holm-Šídák’s multiple comparisons test was used to assess the statistically significant difference. P value < 0.05 (*), P value < 0.01 (**). Gating strategy is shown in Fig. S4 E. (E) Heatmap depicting the mean expression of cell surface protein markers used to differentiate the UTC and CTC clusters from the NKT/MAIT and NK clusters.

Phenotyping the unconventional populations in CB. (A) Flow cytometric analysis of surface CD10 and CD1a expression by the CD3+/low TCRγδ− CD4− PD-1+ populations in CB and adult blood. Representative of each four donors. (B) Flow cytometric assessment of the percentage of CD10+ cells in the CD3+ TCRγδ− CD4− CD8α+ PD-1− population (orange) and CD3+/low TCRγδ− CD4− PD-1+ population (blue) in CB (top) and adult blood (bottom; individual values and mean ± SEM, n = 5 for CB, n = 6 for adult blood). (C) Dunn’s multiple comparisons test was used to assess the statistically significant difference. P value > 0.05 (ns), P value < 0.01 (**). RT-qPCR analysis for MME transcripts in the populations as defined in B, expression fold change determined via ΔΔCt analysis with JY cells as control condition (individual values and mean ± SEM, n = 3 for CB, n = 4 for adult blood). Dunn’s multiple comparisons test showed no statistically significant differences. (D) Protein-based UMAP visualizations showing the expression of the indicated cell surface protein markers, only visualizing the single cells from which protein data was collected. (E) Flow cytometric gating strategy for the unconventional populations in CB. Gating strategy used to obtain the percentages in Fig. 6 D. (F) Flow cytometric analysis of NKT (left) and MAIT (right) cells in adult blood (top) and CB (bottom). The CD3+/low TCRγδ− CD4− population was analyzed for both adult blood and CB. Representative of each two donors. (G) Cell lysis of anti-HLA (W6/32 hybridoma) or anti-CD3 (OKT3 hybridoma) target cells after 4 h of co-incubation with CTCs or UTCs in different effector–target ratios (E:T). The CTCs and UTCs were sorted from CB and incubated overnight with IL-15 before the assay (mean ± SEM, n = 3). (H) Screening of cytokine secretion of 44 soluble cytokines (Table S5) was measured using a multiplex immunoassay. Log10-value of the secreted concentration by freshly sorted UTCs and UTC clones after 24 h of stimulation with PMA + ionomycin. Mean secretion of three UTC clones is shown. (I) Secreted IFN-γ, granzyme B, GM-CSF, MIP-1α, MIP-1β, and IL-2 measured in the supernatant of CTCs and UTCs after stimulation with PMA + ionomycin for 24 h. Wilcoxon matched-pairs signed rank test was used to assess statistically significant differences in cytokine secretion between the populations (mean ± SEM, n = 7). P value < 0.05 (*).
Phenotyping the unconventional populations in CB. (A) Flow cytometric analysis of surface CD10 and CD1a expression by the CD3+/low TCRγδ− CD4− PD-1+ populations in CB and adult blood. Representative of each four donors. (B) Flow cytometric assessment of the percentage of CD10+ cells in the CD3+ TCRγδ− CD4− CD8α+ PD-1− population (orange) and CD3+/low TCRγδ− CD4− PD-1+ population (blue) in CB (top) and adult blood (bottom; individual values and mean ± SEM, n = 5 for CB, n = 6 for adult blood). (C) Dunn’s multiple comparisons test was used to assess the statistically significant difference. P value > 0.05 (ns), P value < 0.01 (**). RT-qPCR analysis for MME transcripts in the populations as defined in B, expression fold change determined via ΔΔCt analysis with JY cells as control condition (individual values and mean ± SEM, n = 3 for CB, n = 4 for adult blood). Dunn’s multiple comparisons test showed no statistically significant differences. (D) Protein-based UMAP visualizations showing the expression of the indicated cell surface protein markers, only visualizing the single cells from which protein data was collected. (E) Flow cytometric gating strategy for the unconventional populations in CB. Gating strategy used to obtain the percentages in Fig. 6 D. (F) Flow cytometric analysis of NKT (left) and MAIT (right) cells in adult blood (top) and CB (bottom). The CD3+/low TCRγδ− CD4− population was analyzed for both adult blood and CB. Representative of each two donors. (G) Cell lysis of anti-HLA (W6/32 hybridoma) or anti-CD3 (OKT3 hybridoma) target cells after 4 h of co-incubation with CTCs or UTCs in different effector–target ratios (E:T). The CTCs and UTCs were sorted from CB and incubated overnight with IL-15 before the assay (mean ± SEM, n = 3). (H) Screening of cytokine secretion of 44 soluble cytokines (Table S5) was measured using a multiplex immunoassay. Log10-value of the secreted concentration by freshly sorted UTCs and UTC clones after 24 h of stimulation with PMA + ionomycin. Mean secretion of three UTC clones is shown. (I) Secreted IFN-γ, granzyme B, GM-CSF, MIP-1α, MIP-1β, and IL-2 measured in the supernatant of CTCs and UTCs after stimulation with PMA + ionomycin for 24 h. Wilcoxon matched-pairs signed rank test was used to assess statistically significant differences in cytokine secretion between the populations (mean ± SEM, n = 7). P value < 0.05 (*).
CD26 (DPP4) was strongly expressed by the CTCs and NKT/MAIT cells. The latter also highly expressed CD161 (KLRB1), similar to the NK cells. CD54 (ICAM1) and CD244 (2B4) are known to be induced in many immune cell types during inflammatory responses (Bui et al., 2020; Speiser et al., 2001). CD54 and CD244 were highly expressed by the effector type UTCs but not by the earliest CD1a+ CD10+ cells and the GZMK+ DN UTC cluster. Expression of NK receptors such as CD158b (KIR2DL2/DL3), CD244, and CD94 was mainly observed in the GNLY+ UTC and IL32+ UTC clusters and was absent in CTC clusters (Fig. 6 B and Fig. S4 D).
Based on these CITE-seq results, a set of cell surface markers was defined to quantify the polyclonal UTC lineage including the effector clusters in CB (Fig. 6 C). Flow cytometric analysis of the CD3+ TCRγδ− cells of a CD4-depleted CB showed a distinct CCR7− CD26− population. The UMAP visualization of this CCR7− CD26− population clearly showed a gradient of Helios expression with the CD10+ recent thymic emigrants having the highest expression. Whereas CD10 stained the immature UTCs, CD54 preferentially stained the effector UTCs. Finally, Eomes+ Granzyme K+ cells are included of which a minority is ultimately CD8α−. CD161+ NKT/MAIT cells are indeed not included in the CCR7−CD26− population (Fig. 6 C).
Next, the size of this CD3+ TCRγδ− CD4− CCR7− CD26− population, expressed as a percentage of the CD3+ CD4− T cells, was determined (Fig. 6 D and Fig. S4 E). Although the percentages in the different donors varied substantially, the UTC population is generally significantly larger than the TCRγδ+ or CD161high NKT/MAIT population in CB. It constitutes the largest UTC population in CB (Fig. 6 D).
Because of their scarcity in human CB, the unconventional semi-invariant NKT/MAIT populations have only been studied to a limited extent (Koay et al., 2016). When analyzing NKT precursors as Vα24-Jα18+ CD1d–PBS-57-tetramer+ and MAIT precursors as Vα7.2+ MR1–5-OP-RU-tetramers+ in CB, these cells (although very limited) were predominantly CCR7− CD26+ and were therefore included in our NKT/MAIT cluster (Fig. S4 F). Furthermore, they can easily be discriminated from the polyclonal UTCs in CB using the surface markers CD161, CD26, CD117 (KIT), CD194 (CCR4), CD103 (ITGAE), and CD196 (CCR6; Fig. 6 E and Fig. S4 D). Our scRNA-seq and CITE-seq data did not incorporate TCR sequencing. Therefore, no further distinction was made between NKT or MAIT cells in this cluster.
Functional testing of the CD3+ TCRγδ− CCR7− CD26− CB UTCs revealed ex vivo CD3-induced killing activity (Fig. S4 G). Upon activation, a spectrum of chemokines including IL-8, MIP-1α (CCL3), MIP-1β (CCL4), and fractalkine (CX3CL1), and the cytokines IL-2, FLT-3L, PDGF-AA, GM-CSF, IL-10, IFN-γ, and TNFα was produced (Fig. S4, H and I).
The UTC lineage is discriminated from CTCs by the markers Helios, KIR, and CD8β
Many characteristic markers of the UTCs (i.e., PD-1, Helios) are related to activation by autoantigens in the thymus. Therefore, the stability and specificity of these hallmarks were tested in vitro and in vivo. Both CB populations were culture-expanded with interleukins only as an in vitro equivalent for steady-state persistence of the cells in tissues. Similar to the PNT PD-1+ population, the CB UTCs extensively proliferated in the presence of IL-15 (Fig. S5, A and B; Billiet et al., 2020). CD26 expression changed during culture, while Helios proved to be a stable feature distinguishing between the CTCs and UTCs, even after proliferation (Fig. 7 A; and Fig. S5, C and D). CD158b (KIR2DL2/DL3) was expressed in a minority of the UTCs, and this expression remained stable during culture. Moreover, no KIR expression could be observed in culture-expanded CTCs (Fig. 7 A). Therefore, it was hypothesized that Helios+ KIR+ T cells detected in vivo belong to the UTC lineage.

Stability of the UTC phenotype in vitro and in vivo. (A) Proliferation assessed by CellTrace Violet dye dilution for the CD8α+ CCR7− CTCs (orange) and CCR7− CD26− UTCs (blue) isolated from CB, after 5 d of incubation in the presence of IL-7 (10 ng/ml) or IL-15 (10 ng/ml). Representative of three experiments. (B) Cell count in CTC and UTC cultures after 5 d of proliferation with IL-7 or IL-15, started at day 0 with 10,000 cells per population (dotted line; mean ± SEM, n = 2). (C) Representative flow cytometric analysis of Helios expression by the (remaining) CTCs and UTCs, after 5 d of proliferation with IL-15. (D) Representative dot plots of CD26 expression by the CTCs or UTCs after 5 d of incubation with IL-7 or IL-15. Proliferation assessed by CellTrace Violet dye dilution. (E) Flow cytometric analysis of CD8αα homodimer expression by the CB CD8α+ CCR7− CTCs (orange) and CCR7− CD26− UTCs (blue), assessed by TL-tetramer binding. Note that DN cells are included in the gating strategy. Representative of three CB donors. (F) Mean fluorescence intensity (MFI) of Helios and CD26 expression by CTC or UTC clones. Individual values and mean ± SEM of 8 or 16 clones per population are shown. Unpaired t tests were used to assess statistical significance. P value < 0.001 (***), P value < 0.0001 (****). (G) Schematic overview of the lineage tracing experiments in NSG-huIL-15 mice. Equal numbers of CB-derived CTCs or UTCs were intrahepatically injected into sublethally irradiated NSG-huIL-15 pups. After 4–5 wk, the presence of the human CD45+ CD3+ population was determined in the bone marrow, thymus, lungs, spleen, bone marrow, small intestine (IEL), and liver. (H) Flow cytometric analysis of the retrieved CD45+ CD3+ cells from NSG-huIL-15 mice injected with either sorted CTCs (orange) or UTCs (blue) from the same CB donor, 4–5 wk after injection. Representative of three experiments. (I) UMAP visualization of the adult tissue clusters in the integrated dataset, as annotated by Domínguez Conde et al. (2022). (J) Flow cytometric analysis of CD8β expression on the CD45+ CD3+ TCRγδ− CD4−Helios− cells (green) and the CD45+ CD3+ TCRγδ− CD4− CCR7− Helios+ KIR+ cells (blue) isolated from the small intestine (IELs) of humanized NSG-huIL-15 mice, 6 mo after humanization. Representative of two mice.
Stability of the UTC phenotype in vitro and in vivo. (A) Proliferation assessed by CellTrace Violet dye dilution for the CD8α+ CCR7− CTCs (orange) and CCR7− CD26− UTCs (blue) isolated from CB, after 5 d of incubation in the presence of IL-7 (10 ng/ml) or IL-15 (10 ng/ml). Representative of three experiments. (B) Cell count in CTC and UTC cultures after 5 d of proliferation with IL-7 or IL-15, started at day 0 with 10,000 cells per population (dotted line; mean ± SEM, n = 2). (C) Representative flow cytometric analysis of Helios expression by the (remaining) CTCs and UTCs, after 5 d of proliferation with IL-15. (D) Representative dot plots of CD26 expression by the CTCs or UTCs after 5 d of incubation with IL-7 or IL-15. Proliferation assessed by CellTrace Violet dye dilution. (E) Flow cytometric analysis of CD8αα homodimer expression by the CB CD8α+ CCR7− CTCs (orange) and CCR7− CD26− UTCs (blue), assessed by TL-tetramer binding. Note that DN cells are included in the gating strategy. Representative of three CB donors. (F) Mean fluorescence intensity (MFI) of Helios and CD26 expression by CTC or UTC clones. Individual values and mean ± SEM of 8 or 16 clones per population are shown. Unpaired t tests were used to assess statistical significance. P value < 0.001 (***), P value < 0.0001 (****). (G) Schematic overview of the lineage tracing experiments in NSG-huIL-15 mice. Equal numbers of CB-derived CTCs or UTCs were intrahepatically injected into sublethally irradiated NSG-huIL-15 pups. After 4–5 wk, the presence of the human CD45+ CD3+ population was determined in the bone marrow, thymus, lungs, spleen, bone marrow, small intestine (IEL), and liver. (H) Flow cytometric analysis of the retrieved CD45+ CD3+ cells from NSG-huIL-15 mice injected with either sorted CTCs (orange) or UTCs (blue) from the same CB donor, 4–5 wk after injection. Representative of three experiments. (I) UMAP visualization of the adult tissue clusters in the integrated dataset, as annotated by Domínguez Conde et al. (2022). (J) Flow cytometric analysis of CD8β expression on the CD45+ CD3+ TCRγδ− CD4−Helios− cells (green) and the CD45+ CD3+ TCRγδ− CD4− CCR7− Helios+ KIR+ cells (blue) isolated from the small intestine (IELs) of humanized NSG-huIL-15 mice, 6 mo after humanization. Representative of two mice.

In vitro and in vivo UTC lineage markers. (A) Dot plots of flow cytometric markers expressed by the CTCs (upper row) or UTCs (bottom row) after 5 d of incubation with IL-7 (10 ng/ml, left) or IL-15 (10 ng/ml, right). Proliferation assessed by CellTrace Violet dye dilution. Representative of three experiments. (B) Both the CTCs and UTCs were sorted from CB as strictly CD4− CD8α+ (left). Assessment of CD8α and CD8β expression by the CTC and UTC populations after 2 wk of proliferation with IL-15. Representative of three experiments. (C) Representative examples of CD8α and CD8β expression by CTC- or UTC-derived clones. (D) Percentage of CD8αα+ or DN cells (gated as shown in Fig. 7 C) in CTC- or UTC-derived clones after at least 2 wk of expansion. Individual values and mean ± SEM of 60 clones per population are shown. Mann-Whitney was used to assess statistical significance. P value < 0.001 (***), P value < 0.0001 (****). (E) Flow cytometric analysis of the retrieved CD45+ CD3+ cells from NSG-huIL-15 mice injected with either sorted CTCs (orange) or UTCs (blue) from the same CB donor, 4–5 wk after injection. Antibodies against CD158a/h, CD158b1/b2, and CD158e1/e2 were combined as KIR staining. The human cell number retrieved from the liver was too low to perform intranuclear Helios staining. Representative of three experiments.
In vitro and in vivo UTC lineage markers. (A) Dot plots of flow cytometric markers expressed by the CTCs (upper row) or UTCs (bottom row) after 5 d of incubation with IL-7 (10 ng/ml, left) or IL-15 (10 ng/ml, right). Proliferation assessed by CellTrace Violet dye dilution. Representative of three experiments. (B) Both the CTCs and UTCs were sorted from CB as strictly CD4− CD8α+ (left). Assessment of CD8α and CD8β expression by the CTC and UTC populations after 2 wk of proliferation with IL-15. Representative of three experiments. (C) Representative examples of CD8α and CD8β expression by CTC- or UTC-derived clones. (D) Percentage of CD8αα+ or DN cells (gated as shown in Fig. 7 C) in CTC- or UTC-derived clones after at least 2 wk of expansion. Individual values and mean ± SEM of 60 clones per population are shown. Mann-Whitney was used to assess statistical significance. P value < 0.001 (***), P value < 0.0001 (****). (E) Flow cytometric analysis of the retrieved CD45+ CD3+ cells from NSG-huIL-15 mice injected with either sorted CTCs (orange) or UTCs (blue) from the same CB donor, 4–5 wk after injection. Antibodies against CD158a/h, CD158b1/b2, and CD158e1/e2 were combined as KIR staining. The human cell number retrieved from the liver was too low to perform intranuclear Helios staining. Representative of three experiments.
CD8β expression is characteristically lower than CD8α in the PNT PD-1+ population, resulting in CD8αα homodimers that can be visualized using fluorochrome-labeled thymic leukemia (TL) antigen-tetramers (Verstichel et al., 2017). Here, the homogeneously increased expression of CD8αα homodimers was evident also for the CCR7− CD26− CB UTC lineage (Fig. S5 E). During culture with IL-15, CD8β expression further decreased on UTCs, whereas expression on CTCs was stable (Fig. 7 B).
To investigate the long-term stability of the CB phenotype, single-cell clones of both lineages were culture-expanded and the phenotype of the resulting clones was analyzed. CD8αα+ and DN clones were frequently observed in UTC-derived clones, whereas CTC-derived clones remained predominantly CD8αβ+ (Fig. 7 C and D). This further confirms that downregulation of CD8β, and therefore the enrichment of CD8αα homodimers, is an exclusive characteristic of UTCs in CB. Although Helios expression was induced in CTC-derived clones, expression remained significantly higher in UTC-derived clones. Similarly, CD26 expression was induced on UTC-derived clones but remained significantly higher in CTC-derived clones (Fig. S5 F).
To determine whether these UTC markers were also preserved in vivo, equal numbers of CB-derived CTCs or UTCs were injected into immune-deficient NOD SCID gamma (NSG)-huIL-15 pups (Fig. S5 G). NSG-huIL-15 mice are transgenic for human IL-15, the growth and survival factor for UTCs (Fig. S5, A and B). After 4–5 wk, human CD45+ CD3+ cells were observed in the bone marrow, spleen, lungs, and a few cells in the liver of mice injected with either sorted CTCs or sorted UTCs. The CTCs partly remained CCR7+ and partly were activated and became CCR7− and PD-1+, possibly due to xenoreactivity, which is known to occur after injection of human T cells in NSG mice (Fig. S5 H). Note that KIR was not expressed by the activated CTCs. In contrast, PD-1 expression was not prominent in the UTC-injected mice, while a prominent KIR+ fraction was observed (Fig. 7 E). The UTCs remained predominantly CCR7−, Helioshigh, and CD8βlow compared with the CTCs (Fig 7 E and Fig. S5 H).
In conclusion, based on in vitro and in vivo data, UTCs differ from CTCs by a high expression of Helios and KIR and a decreased CD8β expression.
UTCs home to the intestine as well as to other tissues
In the blood and intestinal IELs of UTC-injected mice, no human cells could be observed (data not shown). To assess the tissue homing or tissue-resident UTCs, the CB scRNA-seq dataset was integrated with a previously published scRNA-seq dataset of human adult tissues (Fig. 8, A and B; and Fig. S5 I; Domínguez Conde et al., 2022). The interposition of the different tissues was in accordance with the published data, with the jejunum samples from the epithelial and lamina propria rather distancing themselves from the other tissues (Fig. 8 A). CB NK cells colocalized with NK cells derived from bone marrow, lungs, and spleen, and CB NKT/MAIT cells mostly colocalized with intestinal MAIT cells (Fig. 8, A and B; and Fig. S5 I). The CB CTCs clustered together and overlapped with the cells derived from blood and lymph nodes. With regard to the UTC lineage, the effector GNLY+ UTC and IL32+ UTC clusters integrated with cells derived from multiple organs: bone marrow, lungs, liver, spleen, and jejunum (Fig. 8, A and B). Of note, IKZF2+ and KIR2DL3+ cells of CB as well as of the adult tissues were present in these overlapping clusters (Fig. 8 C). These data further strengthen the hypothesis that the Helios+ KIR+ cells present in these tissues could be part of the same UTC lineage as the CB UTCs.

Homing characteristics of the UTC lineage. (A–C) Integration of the CB scRNA-seq dataset with a previously published human adult scRNA-seq dataset (Domínguez Conde et al., 2022). (A) UMAP visualization colored by tissue. BLD, blood; BMA, bone marrow; JEJEPI, jejunum samples from the epithelium; JEJLP, jejunum samples from the lamina propria; LIV, liver; LLN and MLN, lymph nodes; LNG, lung; SPL, spleen. (B) UMAP visualization of the 13 CB clusters in the integrated dataset. (C) UMAP feature plots of IKZF2 and KIR2DL3 expression in adult tissues (blue) and CB (red). Only cells with a relatively high expression are colored. (D) Flow cytometric data showing the presence of CD4 and CD8 SP cells in the thymus of humanized NSG-huIL-15 mice and the presence of CD3+ TCRγδ− CD4− CD8α+ CD10+ PD-1+ cells. Representative of two mice. (E) Heatmap showing the expression of hallmark TFs in the human PNT CD10− PD-1− and CD10+ PD-1+ populations (sorted from three different donors) and the corresponding expression in the CD3+/low PD-1+ population sorted from the thymus of humanized immune system (HIS) NSG-huIL-15 mice (sorted from nine mice). (F) Flow cytometric analysis of the CD45+ CD3+ TCRγδ− CD4− cells isolated from humanized NSG-huIL-15 mice, 6 mo after humanization. Representative of two mice. (G) Flow cytometric analysis of all CD3+ TCRγδ− CD4− cells from human CB, adult peripheral blood and IELs isolated from the small intestine. Each representative of at least three donors.
Homing characteristics of the UTC lineage. (A–C) Integration of the CB scRNA-seq dataset with a previously published human adult scRNA-seq dataset (Domínguez Conde et al., 2022). (A) UMAP visualization colored by tissue. BLD, blood; BMA, bone marrow; JEJEPI, jejunum samples from the epithelium; JEJLP, jejunum samples from the lamina propria; LIV, liver; LLN and MLN, lymph nodes; LNG, lung; SPL, spleen. (B) UMAP visualization of the 13 CB clusters in the integrated dataset. (C) UMAP feature plots of IKZF2 and KIR2DL3 expression in adult tissues (blue) and CB (red). Only cells with a relatively high expression are colored. (D) Flow cytometric data showing the presence of CD4 and CD8 SP cells in the thymus of humanized NSG-huIL-15 mice and the presence of CD3+ TCRγδ− CD4− CD8α+ CD10+ PD-1+ cells. Representative of two mice. (E) Heatmap showing the expression of hallmark TFs in the human PNT CD10− PD-1− and CD10+ PD-1+ populations (sorted from three different donors) and the corresponding expression in the CD3+/low PD-1+ population sorted from the thymus of humanized immune system (HIS) NSG-huIL-15 mice (sorted from nine mice). (F) Flow cytometric analysis of the CD45+ CD3+ TCRγδ− CD4− cells isolated from humanized NSG-huIL-15 mice, 6 mo after humanization. Representative of two mice. (G) Flow cytometric analysis of all CD3+ TCRγδ− CD4− cells from human CB, adult peripheral blood and IELs isolated from the small intestine. Each representative of at least three donors.
To further study these tissues, a humanized mouse model was generated by injecting human CD34+ hematopoietic progenitor cells (HPCs) into NSG-huIL-15 pups. These cells differentiated in the murine thymus to conventional CD4 and CD8 SP T cells (Fig. 8 D). Similar to human PNT, a PD-1+ population was present expressing the hallmark TFs of the UTCs described above. This population was prominent 10–12 wk after transplantation (Fig. 8, D and E). To allow sufficient time for the cells to expand on human IL-15, the tissues were harvested after 6 mo. A prominent CD3+ TCRγδ− CD4− CCR7− Helios+ KIR+ fraction could be observed in all investigated tissues: bone marrow, spleen, lung, liver, blood, and small intestine (IEL; Fig. 8 F). Again, CD8β expression was reduced compared with the Helios− CD3+ TCRγδ− CD4− cells (Fig. S5 J).
Next, human adult peripheral blood was assessed. A CCR7− Helios+ population could also be observed within the CD3+ TCRγδ− CD4− population. This population consisted of a KIR+ population, a CD161+ population, and a KIR− CD161− population (Fig. 8 G). Contrary to CB CD161+ NKT/MAIT cells, the CD161+ cells in peripheral blood expressed Helios (Fig. 8 G; Leeansyah et al., 2015). However, these were easily distinguished from the UTCs by the intermediate expression of Helios, the absence of KIR expression, and the presence of CD161. A corresponding CD161− CCR7− Helios+ population could be observed in the human IELs isolated from the small intestine. This population likewise included the KIR+ IELs (Fig. 8 G).
Discussion
In the present study, the peripheral progeny of the human thymic unconventional CD10+ PD-1+ population was identified in CB as a separate population distinct from conventional CD8+ T cells and NKT/MAIT cells. This population was remarkably heterogeneous. Five clusters were identified of which the MME+ UTCs and the GNLY− MME− UTCs are the closest related progeny of the thymic CD10+ PD-1+ population and are themselves the precursor clusters of the three effector clusters. Indeed, in adulthood, when thymic output diminishes, the MME+ UTCs are no longer detectable and possibly have differentiated into effector cells. Of the effector clusters, the GNLY+ UTC and IL32+ UTC both contain cells expressing NK receptors, including KIRs. KIR+ cells were not detected in the CTC clusters, ex vivo, nor after manipulation, suggesting that KIR expression is confined to the UTC lineage cells. A fifth UTC cluster, adjacent to the CTCs, was notable for high GZMK expression and low expression of CD8A and CB8B. In the trajectory analysis, this cluster came out as a third effector population arising from the common GNLY− MME− UTC cluster. A distinctive set of membrane markers for this GZMK+ DN UTC cluster was not found. Therefore, the GZMK+ DN UTC cluster could not be isolated and subjected to a TCR analysis to put in evidence the characteristic TCR features of the UTC lineage. Despite the transcriptomic similarity, it, therefore, remains uncertain whether this cluster originates from the PNT CD10+ PD-1+ population.
The UTC lineage is well defined in CB CD8+ TCRαβ+ cells by the absence of CD26 and CCR7 expression. However, the UTC lineage is heterogeneous with regard to the expression of hallmark protein markers such as PD-1, Helios, CD8β, and KIR receptors. Our data suggest that PD-1 expression on the peripheral UTCs is a remnant of TCR stimulation during agonist selection in the thymus and that the peripheral UTCs gradually lose expression of PD-1. Possibly, NK receptors expressed on the effector clusters may inhibit the re-expression of PD-1 upon TCR stimulation (Sottile et al., 2021). In contrast to PD-1, Helios expression seemed more stable in the peripheral tissues. KIR expression was limited in CB and confined to the effector-like GNLY+ UTC and IL32+ UTC clusters. Surprisingly, 5 wk after injection of UTCs in immune-deficient mice, a large percentage of the retrieved cells expressed KIRs. This suggests that UTC clusters may further differentiate into KIR+ effector cells. Alternatively, KIR+ cells may preferentially survive in the tissues.
It is known already for some time that a subpopulation of CD8+ TCRαβ+ cells expresses KIRs (Young et al., 2001; Björkström et al., 2012). This subpopulation has an effector and effector memory phenotype: CD28− CCR7− perforin+ IFN-γ+ (Björkström et al., 2012; Anfossi et al., 2004). As KIR expression is not observed in naive T cells, it is generally accepted that the expression of KIRs is induced at some stage after T cell activation and memory formation, although this has never been shown in vitro nor in vivo (Xu et al., 2005; Arlettaz et al., 2004). When the memory repertoire for CMV or HIV-1 was analyzed, no specific memory T cells were observed in the CD8+ KIR+ population, suggesting that the KIR+ effector memory-phenotype cells may not be part of the virus-specific adaptive immune response (Anfossi et al., 2004). Here, several lines of evidence showed that KIRs may be a lineage marker for UTCs rather than an effector/memory marker. In the CB TCRαβ+ population, KIR expression is limited to the UTCs described here. Neither incubation with interleukins nor activation in vitro was able to induce KIR expression on CTCs. In addition, in vivo activation (probably by xeno-antigens) of CTCs after injection in immune-deficient mice, strongly induced PD-1 expression and a CCR7− effector memory phenotype. However, none of these T cells expressed KIRs. In contrast, the progeny of CB UTCs expressed KIRs in high percentages. Based on these findings, we hypothesize that the TCRαβ+ CD8+ KIR+ population belongs to the UTC lineage described here. Further evidence for this hypothesis was recently published by Schattgen et al. (2022): CD8+ KIR+ T cells were isolated from human healthy adult blood using pMHC multimers that also bind KIRs (Schattgen et al., 2022). These cells express KLRC2, KIR2DL3, and NCR3, all of which are expressed by cells within the GNLY+ UTC and IL32+ UTC clusters. These cells are characterized by prominent expression of IKZF2 and ZNF683, two hallmark TFs of the CB UTC lineage (Schattgen et al., 2022). Importantly, the TCR features of the thymic and CB UTC lineage that we described here, namely preferential use of TRAJ-proximal V gene segments and TRAV-proximal J gene segments, and high frequency of hydrophobic residues and cysteine in the apex of the CDR3s, were all present in the CD8+ KIR+ cells of adult donors (Schattgen et al., 2022). Together, this strongly suggests that the adult KIR-expressing population belongs to the same lineage as the thymic CD10+ PD-1+ and CB CCR7− CD26− UTC lineage. It also suggests that the autoreactive characteristics (hydrophobicity index, cysteine index) of the TCR repertoire are not lost during postnatal peripheral expansion.
TCRαβ+ CD8+ KIR+ cells expressing IKZF2 are found to be enriched at autoimmune inflammatory sites such as the intestine in patients with celiac disease, the kidneys in lupus, and the joints affected by rheumatoid arthritis (Li et al., 2022). The concept of these innate-like T cells originating in the thymus by agonist selection as a consequence of the special characteristics of their TCR may shed new light on the association of KIR+ T cells with autoimmune diseases.
The human thymic CD10+ PD-1+ population was previously described by us and others as the putative human counterpart of murine IELps: the population is activated in the human thymus by high-affinity ligands evidenced by the expression of PD-1 and Helios; is generated from early DP blasts and has a characteristic TCR repertoire with autoreactive features (Verstichel et al., 2017; Wirasinha et al., 2018; Daley et al., 2019). Here, by identifying the progeny of this PD-1+ population in CB, strong evidence was shown that these cells exit the human thymus. In addition, indirect evidence was provided that this lineage may be home to different tissues including the intestine. Using scRNA-seq, Park et al. (2020) recently reported that the PD-1+ population in the human thymus actually consists of two discrete populations: GNG4+CD8αα+ T(I) and ZNF683+CD8αα+ T(II) (Park et al., 2020). Both unconventional CD8αα+ populations express PDCD1 (encoding for PD-1). They present evidence that the human GNG4+CD8αα+ T(I) cells are thymic resident (Park et al., 2020). Here, we showed that CB UTCs express ZNF683, the hallmark gene of human CD8αα+(II) T cells, while virtually no CB UTCs express GNG4, the hallmark gene of human CD8αα+(I) T cells. By integrating the scRNA-seq datasets of the CB UTCs and the thymic populations, it was demonstrated that the CB MME+ UTCs overlap with the thymic ZNF683+CD8αα+ T(II) population, indicating that these cells have similar transcriptomes. Therefore, our data suggest that only the ZNF683+CD8αα+ T(II) cells leave the thymus as the CB UTC population and support the notion that the GNG4+CD8αα+ T(I) population may be thymic resident.
In mice, two types of IELps are distinguished in the thymus: type A IELps, which are PD-1+ and reside in the cortex, and type B IELps, which are T-bet+ and reside in the medulla (Golec et al., 2017; Ruscher et al., 2017). Both IELp types can seed the murine intestine where they express CD8αα; however, type B IELps only seed the intestine in a restricted time period early in life. With progressing age, type B IELps seem to become thymus-resident and type A IELps eventually become the predominant type in the adult murine intestine (Ruscher et al., 2020). Alternatively, Hummel et al. propose that the IELp subsets represent distinct maturation stages rather than distinct lineages, with the T-bet+ IELps progressing through a PD-1+ stage before upregulating T-bet, all arising from one common thymic precursor population (Hummel et al., 2020). In the human thymus, GNG4+CD8αα+ T(I) and ZNF683+CD8αα+ T(II) populations were distinguished. Park et al. (2020) suggests that the GNG4+CD8αα+ T(I) population is the human equivalent of the type A IELps and the ZNF683+CD8αα+ T(II) population is the human equivalent of type B IELps based on transcriptome integration of human and mouse thymus (Park et al., 2020). However, the cortex–medulla location within the thymus (human GNG4+CD8αα+ T(I) are located in the medulla), the emigration out of the thymus (human ZNF683+ cells emigrate from the thymus to CB), as well as the expression of some hallmark TFs (i.e., GNG4, XCL1, ZEB2, and CLDN10) do not fully support this model (Park et al., 2020). Taking all these data into account, it is suggested that the phenotype and emigration of UTC subsets may be different in humans versus mice.
Materials and methods
Sample processing
Human CB was obtained from the Cord Blood Bank UZ Gent. Mononuclear cells (MNCs) were isolated using density gradient centrifugation (LymphoPrep; Axis-Shield, 1114547) and were enriched by magnetically activated cell sorting (MACS) through negative selection using anti-CD4-biotin, anti-CD14-biotin, anti-CD19-biotin, anti-CD235-biotin (homemade), and anti-biotin microbeads (130-090-485; Miltenyi Biotec). Human postnatal thymus and peripheral blood from healthy adult blood donors were processed as previously described (Billiet et al., 2020). All human samples were processed according to the guidelines approved by the Medical Ethical Committee of Ghent University Hospital (CG20171208A, December 8, 2017) after informed consent had been obtained in accordance with the Declaration of Helsinki.
Flow cytometry and antibodies
Staining of surface markers was performed in DPBS (17-512F; Lonza) with 1% FCS (S1810; Biowest) using the antibody-to-cell ratio recommended by the supplier. Intracellular and intranuclear stainings were performed following the supplier’s protocol using BD Cytofix&Cytoperm (554714; BD Biosciences) and the eBioscience Foxp3/Transcription Factor Staining Buffer Set (00-5523-00; eBioscience), respectively. Flow cytometric analysis was performed on the LSR II and cell sorting on the FacsARIA Fusion (both BD Biosciences). Flow cytometry data were analyzed using FACS DIVA software (BD Biosciences) and FlowJo software (TreeStar Inc). The FlowSOM Plugin (version 2.9) was used to cluster flow cytometry data (Van Gassen et al., 2015). Viable cells were gated based on propidium iodide negativity or Fixable Viability Dye (eFluor 506; 65-0866-18; Thermo Fisher Scientific) negativity for surface and intracellular stainings, respectively. The following list of anti-human monoclonal antibodies was used. Allophycocyanin (APC)/AF647-conjugated: CD4 (130-113-250; Miltenyi), CD158b (KIR2DL2/DL3, 130-092-617; Miltenyi), Helios (137221; BioLegend), Granzyme K (370503; BioLegend), NKG2A (CD159a, 130-114-089; Miltenyi), PD-1 (CD279, 367420; BioLegend), and TCRγδ (130-113-500; Miltenyi); APC Cy7/APC Fire750–conjugated: CD8α (344746; BioLegend), CCR7 (CD197, 353246; BioLegend), and TCR Vα7.2 (351713; BioLegend); Brilliant Violet 421–conjugated: CD3 (317344; BioLegend) and CD54 (353131; BioLegend); Brilliant Violet 605–conjugated: CD161 (339915; BioLegend); Brilliant Violet 650–conjugated: CD3 (317323; BioLegend); Brilliant Violet 711–conjugated: CCR7 (CD197, 353227; BioLegend); Brilliant Violet 785–conjugated: CD10 (312237; BioLegend); fluorescein isothiocyanate–conjugated: CD8α (homemade), CD161 (130-114-118; Miltenyi), IFN-γ (554551; BD Biosciences), and TCRγδ (347903; BD Biosciences); phycoerythrin (PE)-conjugated: CD158a/h (KIR2DL1/DS1, 130-116-975; Miltenyi), CD158b1/b2 (KIR2DL2/DL3, IM2278U; Beckman Coulter), CD158e1/e2 (KIR3DL/DS1, IM3292; Beckman Coulter), EVI2B (CD361, A15806; Thermo Fisher Scientific), granzyme B (12-8899-42; eBioscience), granzyme K (370511; BioLegend), PD-1 (CD279, 367404; BioLegend), perforin (12-9994-42; eBioscience), and TCR Vα24-Jα18 (342903; BioLegend); PE Cy7–conjugated: CD8β (25-5273-42; eBioscience), CD10 (312214; BioLegend), Eomes (25-4877-41; eBioscience), and TCRγδ (331222; BioLegend); peridinin chlorophyll protein complex Cy5.5–conjugated: CD4 (344608; BioLegend) and CD26 (302715; BioLegend). CB MNCs were stained with Vδ1 (clone A13 supernatant, which can bind to Vδ1 when incorporated in hybrid Vδ1-Jα-Cα TCR chains, a kind gift from Prof. Dr. Lorenzo Moretta’s laboratory, Bambino Gesù Children's Hospital, Rome, Italy), anti–mouse Ig light chain κ (409506; BioLegend), 5% normal mouse serum (10410; Invitrogen), followed by the appropriate antibodies above to isolate the populations. The CD1d and MR1 tetramer technology was developed jointly by Dr. James McCluskey, Dr. Jamie Rossjohn, and Dr. David Fairlie, and the material was produced by the National Institutes of Health Tetramer Core Facility as permitted to be distributed by the University of Melbourne. CD8αα expression was assessed by staining with PE-conjugated TL-tetramer after previous staining with anti-CD8β antibodies. The TL-tetramer was kindly provided by Prof. Dr. Hilde Cheroutre (Center for Autoimmunity and Inflammation, La Jolla Institute for Immunology, La Jolla, CA, USA).
CD4/CD14/CD19/CD235-depleted CB MNCs were sorted into CD3+ TCRγδ− CD4− CD8α+ PD-1− (PD-1− population) and CD3+/low TCRγδ− CD4− CD8α+ PD-1+ (PD-1+ population) for the transcriptome, TCR and proteome analyses, and quantitative reverse transcription PCR (RT-qPCR). The PNT populations were sorted as previously described (Billiet et al., 2020). CD4/CD14/CD19/CD235-depleted peripheral blood MNCs were sorted into CD3+ TCRγδ− CD4− CD8α+ PD-1− (PD-1− population) and CD3+/low TCRγδ− CD4− CD8α+ PD-1+ (PD-1+ population) for the RT-qPCR. For the single-cell assay, CD4/CD14/CD19/CD235-depleted CB MNCs were sorted into CD3+/low TCRγδ− CD4− CD8α+ PD-1+ (sort 1), CD3+/low TCRγδ− CD4− CD8α+ CCR7− EVI2B+ (sort 2), CD3+ TCRγδ− CD4− PD-1− and CD3+/low TCRγδ− CD4− PD-1+ (sort 3), and CD3+ TCRγδ− CD4− CCR7+ EVI2B− and CD3+/low TCRγδ− CD4− CCR7− EVI2B+ (sort 4). For the in vitro proliferation and functionality experiments and the in vivo lineage tracing study in NSG-huIL-15 mice, CD4/CD14/CD19/CD235-depleted CB MNCs were sorted into CD3+ TCRγδ− CD4− CD8α+ CCR7+ (CTC) and CD3+/low TCRγδ− CD4− CCR7− CD26− (UTC).
RNA sequencing
The populations of interest were each time sorted from three CB and PNT donors. The CD3+ TCRγδ+ population was also sorted and only two TCRγδ+ samples were analyzed for CB. The CD3+/low TCRγδ− CD4− CD8α+ PD-1+ (PD-1+ population) was sorted from the thymi of nine NSG-huIL-15 mice 12 wk after injection with CD34+ HPCs. The PNT and CB populations were sorted in IMDM (12440053; Thermo Fisher Scientific) supplemented with 10% FCS, 2 mM L-glutamine (25030-081; Thermo Fisher Scientific), 100 IU/ml penicillin, and 100 IU/ml streptomycin (15140-122; Thermo Fisher Scientific; complete IMDM, cIMDM) and washed three times in PBS. RNA extraction was performed using the miRNeasy Mini Kit (217004; Qiagen). For poly(A) RNA-seq, the QuantSeq 3′ mRNA FWD kit (Lexogen) was used, followed by single-ended sequencing on the NextSeq500 Sequencing System (Illumina) with a read length of 75 bp. RNA-seq reads were aligned to hg38-noalt using STAR v2.6.0c and quantified on Ensembl v93.
TCR sequencing
For the TCR analysis, the populations of interest were sorted from six CB donors and two new PNT donors. Previously published PNT samples were also reanalyzed (Verstichel et al., 2017). RNA extraction was performed using the miRNeasy Micro Kit (217084; Qiagen) followed by template-switch anchored RT-PCR. High-throughput sequencing of TRA and TRB loci was performed as previously described (Van Caeneghem et al., 2017). Raw sequencing reads from FASTQ files were aligned to reference V, D, and J genes from the GenBank database specifically for “TRA” or “TRB” to build CDR3 sequences using the MiXCR software version 3.0.12 (Bolotin et al., 2015). Following this, the CDR3 sequences were analyzed using VDJtools software version 1.2.1 (Shugay et al., 2015). Out-of-frame sequences were excluded from the analysis, as well as non-functional TRA and TRB segments using IMGT (the international ImMunoGeneTics information system) annotation. TRDV gene segment–containing sequences were filtered as well, except for the analysis where the amount of TRDV1-containing sequences was assessed. Calcbasicstats default function was used to calculate the number of CDR3 N additions. Cumulative gene segment plots were generated using the output from CalcSegmentUsage function. Treemaps were generated using the Treemap Package on RStudio, grouping TRAV and TRAJ segments according to their locus position. D75 repertoire diversity metrics were calculated for the TCRα chain by measuring the percentage of clonotypes required to occupy 75% of the total TCR repertoire. Determination of the CDR3α and CDR3β apex region and cysteine usage was performed following previously described indications (Wirasinha et al., 2018). Hydrophobic CDR3α and CDR3β doublet containing sequences were determined by calculating the percentage of sequences using any of the 175 AA doublets previously identified as promoting self-reactivity (Daley et al., 2019). Physicochemical characteristics (strength, volume, and polarity) of the CDR3β were analyzed using VDJtools software version 1.2.
Liquid chromatography–tandem mass spectrometry (LC-MS/MS) proteomic analysis
Sample preparation
The populations of interest were each time sorted from three CB and PNT donors. Cell pellets (± 1.106 cells per pellet) were resuspended in lysis buffer (8 M urea; 20 mM HEPES, pH 8.0). Samples were sonicated by three pulses of 15 s, interspaced by 1 min pauses on ice at an intensity output of 15 W, and centrifuged for 15 min at 20,000 g at room temperature to remove insoluble components. Proteins were reduced with 5 mM dithiothreitol (Sigma-Aldrich) for 30 min at 55°C and then alkylated by the addition of 10 mM iodoacetamide (Sigma-Aldrich) for 15 min at room temperature in the dark. Samples were further diluted with 20 mM HEPES, pH 8.0, to a final urea concentration of 4 M and proteins were digested with LysC (Wako; 1/100, wt/wt) for 4 h at 37°C. Samples were again diluted to 2 M urea and digested with trypsin (1/100, wt/wt; Promega) overnight at 37°C. The resulting peptide mixture was acidified by the addition of 1% trifluoroacetic acid. Peptides were then purified on a SampliQ SPE C18 cartridge (Agilent), vacuum-dried, and kept at −20°C until measured by LC-MS/MS.
LC-MS/MS analysis
Immediately before injection, purified peptides were redissolved in 15 μl loading solvent (0.1% trifluoroacetic acid/water/acetonitrile (0.1:98:2, vol/vol/vol) and the peptide concentration was determined by measuring on a Lunatic spectrophotometer (Unchained Labs). 2 μg of peptide material of each sample was injected for LC-MS/MS analysis on an Ultimate 3000 RSLC nano-LC (Thermo Fisher Scientific) in-line connected to a Q Exactive HF mass spectrometer (Thermo Fisher Scientific) equipped with a nanospray flex ion source (Thermo Fisher Scientific). Trapping was performed at 10 μl/min for 4 min in loading solvent A on a 20-mm trapping column (made in-house, 100-μm internal diameter, 5-μm beads, C18 Reprosil-HD, Dr. Maisch, Germany). Peptide separation after trapping was performed on a 200-cm-long micropillar array column (PharmaFluidics) with C18-endcapped functionality. The Ultimate 3000’s column oven was set to 50°C. For proper ionization, a fused silica PicoTip emitter (10-μm inner diameter; New Objective) was connected to the μPAC outlet union, and a grounded connection was provided to this union. Peptides were eluted by a nonlinear gradient from 1 to 55% MS solvent B (0.1% formic acid in water/acetonitrile [2:8, vol/vol]) over 145 min, starting at a flow rate of 750 nl/min and switching to 300 nl/min after 15 min, followed by a 15-min washing phase plateauing at 99% MS solvent B. Re-equilibration with 99% MS solvent A (0.1% formic acid in water) was performed at 300 nl/min for 45 min followed by 5 min at 750 nl/min, adding up to a total run length of 210 min. The mass spectrometer was operated in a data-dependent, positive ionization mode, automatically switching between MS and MS/MS acquisition for the 16 most abundant peaks in each MS spectrum. The source voltage was 2.2 kV and the capillary temperature was 275°C. One MS1 scan (m/z 375–1,500, automatic gain control target 3 × 106 ions, maximum ion injection time 60 ms), acquired at a resolution of 60,000 (at 200 m/z), was followed by up to 16 tandem MS scans (resolution 15,000 at 200 m/z) of the most intense ions fulfilling predefined selection criteria (automatic gain control target 1 × 105 ions, maximum ion injection time 80 ms, isolation window 1.5 daltons, fixed first mass 145 m/z, spectrum data type: centroid, intensity threshold 1.3 × 104, exclusion of unassigned, 1, 7, 8, >8 positively charged precursors, peptide match preferred, exclude isotopes on, dynamic exclusion time 12 s). The higher-energy collisional dissociation was set to 28% normalized collision energy, and the polydimethylcyclosiloxane background ion at 445.12003 daltons was used for internal calibration (lock mass).
Data analysis
Data analysis was performed with MaxQuant (version 1.6.2.6) using the Andromeda search engine with default search settings including a false discovery rate (FDR) set at 1% on both the peptide and protein levels. Spectra were searched against the human Swiss-Prot database (from November 2018 with 20,424 entries) separately for PNT and CB T cells. The mass tolerance for precursor and fragment ions was set to 4.5 and 20 ppm, respectively, during the main search. Enzyme specificity was set to the C-terminal of arginine and lysine, also allowing cleavage next to prolines with a maximum of two missed cleavages. Variable modifications were set to oxidation of methionine residues and acetylation of protein N-termini. Matching between runs was enabled with a matching time window of 1.5 min and an alignment time window of 20 min. Only proteins with at least one unique or razor peptide were retained, leading to the identification of 4,539 and 3,584 proteins for PNT and CB, respectively. Proteins were quantified by the MaxLFQ algorithm integrated into the MaxQuant software. A minimum ratio count of two unique or razor peptides was required for quantification. Further data analysis was performed with the Perseus software (version 1.6.2.1) separately for the PNT and CB data set after uploading the protein groups file from MaxQuant. Reverse database hits, potential contaminants, and proteins that are only identified by peptides carrying at least one modified AA were removed. Replicate samples were grouped and proteins with less than three valid values in at least one group were removed, and missing values were imputed from a normal distribution around the detection limit resulting in 3,001 and 2,024 quantified proteins for PNT and CB, respectively.
Gene set enrichment analysis
GSEA was performed using the GSEA software version 4.1.0., a joint project of UC San Diego (San Diego, CA, USA) and Broad Institute (Cambridge, MA, USA; Subramanian et al., 2005; Mootha et al., 2003). The GSEAPreranked tool was run using standard parameters and 1,000 permutations. The gene set contained significantly upregulated genes when comparing the CD10+ PD-1+ population to the CD10− PD-1− cells from human PNT. The gene list was ranked by comparing the PD-1+ population to the PD-1− cells from human CB, ranked from the upregulated genes (left) to the downregulated genes (right). The normalized enrichment score (NES) reflects the degree to which the gene set is overrepresented in the upregulated genes (positive value) or downregulated genes (negative value). The FDR q value is the estimated probability that a gene set with a given NES represents a false-positive finding.
scRNA-seq analysis
Single-cell library preparation and sequencing
The populations of interest were sorted from CD4/CD14/CD19/CD235-depleted CB from two different donors. From the first donor, CD3+/low TCRγδ− CD4− CD8α+ and CD3+/low PD-1+ (sort 1) or CCR7− EVI2B+ (sort 2) were sorted separately. Both fractions were labeled with different TotalSeq anti-human Hashtag antibodies (BioLegend) before being pooled in equal portions and processed together. From the second donor, CD3+ TCRγδ− CD4− PD-1− and CD3+/low TCRγδ− CD4− PD-1+ (sort 3), CD3+ TCRγδ− CD4− CCR7+ EVI2B− and CD3+/low TCRγδ− CD4− CCR7− EVI2B+ (sort 4) were sorted. Considering the smaller percentage of UTCs, the conventional cells were sorted separately and later added in equal portions. Sort 4 was combined with CITE-seq labeling. Cells were incubated for 30 min on ice with 50 µl of staining mix in PBS containing 0.04% BSA, Fc receptor block (PN 422301, TruStain FcX; BioLegend), and a human cell surface protein antibody panel containing 277 oligo-conjugated antibodies (TotalSeq-A, BioLegend), including six TotalSeq-A isotype controls (Table S4). TotalSeq antibodies were diluted in concentrations as recommended by the manufacturer. Sorted single-cell suspensions were resuspended at an estimated final concentration of 1,200 cells/µl and loaded on a Chromium GemCode Single Cell Instrument (10× Genomics) to generate single-cell gel beads-in-emulsion (GEM) at the VIB Single Cell Core. The scRNA-seq libraries were prepared using the GemCode Single Cell 3′ Gel Bead and Library kit, version NextGEM 3.1 (10x Genomics) according to the manufacturer’s instructions with the addition of amplification primers (3 nM), 5′-CCTTGGCACCCGAGAATT*C*C-3′ and 5′-GTGACTGGAGTTCAGACGTGTGC*T*C-3′ during complementary DNA (cDNA) amplification to enrich the TotalSeq-A cell surface and hashtag protein oligos. Library construction was performed according to the manufacturer’s instructions. Sequencing libraries were loaded on an Illumina NovaSeq flow cell at VIB Nucleomics core with sequencing settings according to the recommendations of 10x Genomics, pooled in a 80:25 ratio for the combined 3′ gene expression and cell surface protein libraries, respectively.
Preprocessing of the scRNA-seq and CITE-seq data
The Cell Ranger pipeline (10x Genomics, version 3.1.0) was used to perform sample demultiplexing and to generate FASTQ files for read 1, read 2, and the i7 sample index for the gene expression and cell surface protein libraries. Read 2 of the gene expression libraries was mapped to the reference genome (GRCh38.99) using STAR. The resulting count matrices were subsequently loaded into R for further processing using Seurat version 4.0.5 (Hao et al., 2021). Empty and/or damaged cells were removed from the datasets by filtering on the following three parameters: (i) the number of genes per cell (nFeature > 600), (ii) the number of UMI counts per cell (nCount > 1,150), and (iii) percentage mitochondrial genes per cell (percent.mt < 15). The remaining cells were normalized with the built-in normalization function from the Seurat package using the log normalization method and a scale factor of 10,000. Highly variable features/genes were identified with FindVariableFeatures with the selection method set to vst and the nfeatures parameter to 4,000. Differential gene expression analysis between cell clusters and conditions was performed using the Wilcoxon Rank Sum test through the Seurat function “FindMarkers.” P value adjustment was performed using Bonferroni correction.
CITE-seq antibody reads were quantified using the feature-barcoding functionality within the Seurat package. Antibodies with low expression were filtered out based on inspection of the feature plots for each antibody. After processing, the CITE-Seq and scRNA-seq data of sort 4 were merged into the same Seurat object.
Dataset integration and batch correction
To verify that our different sorting strategies/definitions of the UTCs in CB included the same or different cell types, a data integration of our three CB samples was performed using the Seurat package. Therefore, the integration anchor strategy was followed. Integration anchors were identified following the RPCA method instead of the CCA method due to the speed of the former method and the recommendation of the developers that RPCA is more conservative and thus better equipped to handle cell populations that have no matching type between samples. For this integration, integration features were first selected with the “SelectIntegrationFeatures” function and subsequently the samples were scaled (“ScaleData”) and a PCA analysis (“RunPCA”) was executed for each sample separately. Afterward, the integration anchors were identified with the “FindIntegrationAnchors” function. After integration with the “IntegrateData” function, the data was scaled once again and a new PCA analysis was performed for visualization and data exploration.
Subsequently, to identify the progeny of the thymic UTC lineage in CB, a second data integration was performed with CB and PNT samples. Therefore, three fetal (FCAImmP7528283, FCAImmP7555851, and FCAImmP7579218) and four PNT samples (TTA9, TTA10, TTA12, and TTA14) were selected from Park et al. (2020). The FASTQ-files of these thymic samples were acquired and processed as described under Preprocessing of the scRNA-seq and CITE-seq data. By processing the samples of both organs in the same way, the variation that needed to be corrected in the subsequent data integration and batch correction step could be limited. For the latter, the R package Harmony was used (Korsunsky et al., 2019). Prior to the data integration with Harmony, all samples were merged into a combined Seurat object after the following steps were performed: (i) identification of highly variable genes/features (“FindVariableFeatures”), (ii) data scaling (“ScaleData”), and (iii) PCA analysis (“RunPCA”). The data integration was executed with the function “RunHarmony,” taking into account three sources of potential batch effects, namely donor, sort, and chemistry. The latter was added since the PNT samples were processed with the version 2 version of the 10x Genomics scRNA-seq kit, while for the CB samples, the version 3 version was used. Afterward, the integrated object was processed for visualization and data exploration.
A third and final integration was performed to screen for potential tissue homing of the described UTC population. Therefore, scRNA-seq data was downloaded from Domínguez Conde et al. (2022). Three adult tissue donors were selected based on total cell count, namely 637C, D496, and D503. All available data from these three donors were downloaded and processed as described under Preprocessing of the scRNA-seq and CITE-seq data. From the remaining cells after processing, only T cells and NK cells were retained for the data integration with CB. T cells and NK cells were selected based on the manual annotation provided by Domínguez Conde et al. (2022). Subsequently, all adult tissue samples were combined with our CB samples and the same processing steps were followed as for the previous integration between CB and PNT. The data integration was performed with Harmony, taking into account different sources of batch effects, namely donor, sort, and method. Method references the sequencing method of the 10x Genomics kit, namely 5′ or 3′. The latter differed between donor 637C and the other samples. Afterward, the integrated object was processed for visualization and data exploration.
Trajectory analysis and SCENIC
Based on the data integration of the PNT (TTA9, TTA10, TTA12, and TTA14) and CB samples, a trajectory analysis could be performed to identify the progeny of the UTCs. For this trajectory analysis, the TSCAN algorithm (version 1.28.0) was used (Ji and Ji, 2016). To simplify the problem for the algorithm, the populations of interest were selected prior to the actual analysis. For the unconventional populations, these selected cell types were CD8αα(II), MME+ UTC, GNLY− MME− UTC, GNLY+ UTC, GZMK+ DN UTC, and IL32+ UTC. While for the conventional population these were CD8+T, CTC1, CTC2, CTC3, CTC4, and CTC5. The first step in the TSCAN analysis was the calculation of the centroid of each cluster with the “reducedDim” function. These centroids were then connected with a minimum spanning tree (MST) in the subsequent step using the “createClusterMST” function. The cells were subsequently mapped and ordered along this MST to determine their pseudo-time point. This was done with the functions “mapCellsToEdges” and “orderCells.” To identify the genes that might play a role in this differentiation process along the pseudo-time, a differential expression analysis with the tradeSeq package (version 1.4.0) was performed (Van den Berge et al., 2020). Therefore, a generalized additive model was fitted to the data along the pseudotime, and subsequently the associationTest was performed to identify the differentially expressed genes.
To better identify which TFs are important or active in the different cell populations, the data were analyzed with the SCENIC algorithm (Aibar et al., 2017). Note that for the actual analysis, the python implementation of this package, pySCENIC, was used. In the first step, gene regulatory networks and co-expression modules are generated by the GRNBoost 2 algorithm based on the correlation between TFs and other genes. Subsequently, regulons are predicted based on known binding motifs provided by the Aerts lab, and in the final step, the cellular enrichment for the different regulons was calculated using the Aucell algorithm. The output of the different steps was loaded into python to calculate the regulon specificity scores (RSS) for the different cell populations through the pySCENIC package.
The RSS was calculated from the regulon activity score and lies between 0 and 1, with a higher value for RSS indicating a higher specificity of that regulon in the cell type compared with others. The UTC- and CTC-specific regulons were selected according to the following strategy: a regulon that appeared in the top five of one of the UTC or CTC populations and the top 10 of one or more of the remaining UTC or CTC populations was selected for inclusion in the plot. The regulon activity score was calculated based on the rank of the expression value in the cell of all genes involved in the regulon (Ma et al., 2020).
RT-qPCR
Total RNA was prepared from 200,000 cells per FACS-sorted sample using the miRNeasy Micro Kit (217684; Qiagen). cDNA synthesis was performed with the iScript Advanced cDNA Synthesis Kit (1725037; Bio-Rad) according to the manufacturer’s instructions. The mRNA transcript levels were determined via SYBR Green I technology using the LightCycler 480 SYBR Green I Master kit (04707516001) on a LightCycler 480 II (both from Roche) according to the manufacturer’s protocol. The primer pairs (Integrated DNA Technologies) included the following: MME: 5′-AGTCTTCCCAGCCGGCATTC-3′ (forward), 5′-AGCCATGGGTGATTTCGTGTCC-3′ (reverse); TBP: 5′-CACGAACCACGGCACTGATT-3′ (forward), 5′-TTTTCTTGCTGCCAGTCTGGAC-3′ (reverse). MME expression fold change was calculated via ΔΔCt analysis with TBP as housekeeping gene and JY cells as control condition.
T cell expansion
The CellTrace proliferation assays were performed as previously described (Billiet et al., 2020). CTC and UTC clones were generated by FACS sorting single cells and expanding them on irradiated allogeneic feeder cells consisting of a mixture of 40 Gy irradiated peripheral blood MNCs and 50 Gy irradiated JY cells. Cells were cultured in cIMDM, supplemented with 1 μg/ml phytohemagglutinin (Sigma-Aldrich). IL-2 (5 ng/ml; 130-097-748; Miltenyi) was added on day 5 and day 10. Cells were restimulated every 7–14 d. After 14–28 d, grown clones were harvested and assessed via flow cytometry.
51Chromium release assay
Target cells (W6/32 or OKT3 hybridoma) were labeled with 51Chromium (Perkin Elmer) for 90 min at 37°C, washed, and added at 103 cells per well to various ratios of effector T cells (CTC or UTC population after overnight incubation with IL-15) in 96-well V-bottomed plates (NUNC, Thermo Fisher Scientific). After 4 h of coincubation, the supernatant was harvested and measured in a 1450 LSC & Luminescence Counter (Perkin Elmer). Specific lysis was calculated as follows: (experimental release − spontaneous release)/(maximal release − spontaneous release) × 100%.
Cytokine production
To explore the secreted cytokine profile of the CB populations, Luminex High Performance Assays (R&D Systems) were performed. The supernatant of both freshly sorted UTCs and UTC-derived clones after 24 h of stimulation with phorbol myristate acetate (PMA; 1 ng/ml; 16561-29-8; Sigma-Aldrich) + ionomycin (0.5 µg/ml; 56092-82-1; Sigma-Aldrich) was assessed with the Luminex Performance Human XL Cytokine Magnetic Panel 44-plex Fixed Panel (LKTM014; Bio-Techne). Following this, a custom mixed multiplex was used to determine the IFN-γ, granzyme B, GM-CSF, MIP-1α, MIP-1β, and IL-2 concentrations in the supernatant of both freshly sorted CTCs and UTCs after 24 h of stimulation with PMA + ionomycin. All assays were performed conforming to the manufacturer’s protocol, measured with the Bio-Plex 200 system (Bio-Rad), and analyzed with the Bio-Plex Manager software version 6.2. Levels below or above the detection level were set as the lower or upper detection level, respectively.
Mice experiments
All mice were maintained at the animal facility at the UZ Ghent. Animal care and experiments were carried out using protocols approved by the Ghent University Animal Ethics Committee. All animal experiments were performed after approval of the Ethical Committee for Experimental Animals at the Faculty of Medicine and Health Sciences of Ghent University (ECD20-20, Ghent, Belgium). 200,000 CD3+ TCRγδ− CD4− CD8α+ CCR7+ (CTC) cells and 200,000 CD3+/low TCRγδ− CD4− CCR7− CD26− (UTC) cells were freshly sorted from the same CD4/CD14/CD19/CD235-depleted CB donor. Next, these cells were intrahepatically injected into sublethally irradiated (100 cGy) NSG-huIL-15 pups (age 1–3 d). Pups injected with either UTCs or CTCs from the same CB donor were littermates. Male or female in-house bred NOD.Cg-PrkdcscidIl2rgtm1Wjl Tg(IL15)1Sz/SzJ (NSG-huIL-15) mice (The Jackson Laboratory) were housed in cages of up to five mice under pathogen-free conditions. Animals were housed at temperatures of 21.1–24.5°C, 30–70% humidity, and 12:12 light–dark cycles. At 4–5 wk after injection, the mice were sacrificed by cervical dislocation and single-cell suspensions were generated, from the bone marrow, liver, thymus, blood, spleen (Filtjens et al., 2013), and small intestine (IELs; Van Acker et al., 2014) as previously described. Lungs were cut into 2–4 mm2 pieces, followed by digestion using a human Tumor Dissociation Kit (130-095-929; Miltenyi Biotec) and red blood cells lysis using ammonium-chloride-potassium lysis buffer (A1049201; Life Technologies).
The humanized mouse model was generated by intrahepatically injecting CB-derived HPCs into sublethally irradiated NSG-huIL-15 pups (age 1–3 d). The CD34+ HPCs were first isolated by MACS using the CD34 MicroBead kit (130-046-703; Miltenyi Biotec). After 12 wk, nine humanized mice were sacrificed, and the PD-1+ population was sorted from the thymus for RNA analysis, as described above. After 6 mo, the humanized mice were sacrificed, and their organs were processed as described above.
Isolation of IELs from the human small intestine
Human ileum was obtained from patients who underwent right hemicolectomy. Healthy ileum was dissected from the colon, followed by isolation of the mucosal layer. The IELs were further isolated as previously described (Verstichel et al., 2017).
Statistical analysis
Statistical analyses were performed in Prism version 9.3.1. (GraphPad Software) using statistical tests as indicated in figure legends. Results were considered statistically significant when the P value was <0.05.
Online supplemental material
Fig. S1 shows the RNA and protein expression profile of the PNT CD10+ PD-1+ population. Fig. S2 shows the distinctive TCR repertoire of the PNT CD10+ PD-1+ population. Fig. S3 defines the T cell clusters in CB. Fig. S4 phenotypes the unconventional populations in CB. Fig. S5 shows the stability of the UTC phenotype in vitro and in vivo. Table S1 contains the significantly differentially expressed genes and proteins between the PD-1+ and PD-1− population in both PNT and CB. Table S2 lists the number of cells in each identified CB cluster. Table S3 contains the top 10 differentially expressed genes for each identified cluster. Table S4 contains the used CITE-seq cell surface protein antibody panel. Table S5 lists the Luminex Performance Human XL Cytokine Magnetic Panel 44-plex Fixed Panel used.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2022) partner repository with the dataset identifier PXD033392. The sequencing data discussed in this publication have been deposited in the National Center for Biotechnology Information’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through accession number GSE201811.
Acknowledgments
The authors would like to thank Dr. Conny Matthys of the Cord Blood Bank UZ Ghent, Dr. Jo Van Dorpe of Pathology UZ Ghent, Sergi Rodà Llordés for his help and advice to optimize data filtering for analysis of the CDR3 sequences, Gabriële Holtappels for her expertise in Luminex High Performance Assays, Juliette Roels for her tips and tricks regarding R, and the UGent Core Flow Cytometry for their help with flow cytometry and cell sorting. We thank Hilde Cheroutre, Derk Amsen, and Greet Verstichel for critically reading the manuscript and their helpful suggestions.
This work is funded by Research Foundation Flanders (grants 1198422N and 1S58622N), Stichting tegen Kanker (grant FAF-F/2016/756), GOA (grant 2021.0008), Fonds de la Recherche Scientifique CDR (grant CDR_J.0225.20), and the TCR. G.S. Sanchez is supported by Télévie–Fonds de la Recherche Scientifique (grant 7.4586.19 and 7.6529.21).
Author contributions: L. Billiet: Conceptualization, data curation, formal analysis, investigation, methodology, project administration, visualization, writing—original draft. L. De Cock: Conceptualization, data curation, formal analysis, investigation, methodology, software, visualization, writing—review & editing. G.S. Sanchez: Formal analysis, investigation, visualization, writing—review & editing. R.L. Mayer: Formal analysis, investigation, visualization, writing—review & editing. G. Goetgeluk: Investigation. S. De Munter: Writing—review & editing. M. Pille: Writing—review & editing. J. Ingels: Writing—review & editing. H. Jansen: Writing—review & editing. K. Weening: Writing—review & editing. E. Pascal: Writing—review & editing. K. Raes: Writing—review & editing. S. Bonte: Writing—review & editing. T. Kerre: Writing—review & editing. N. Vandamme: Investigation, software, writing—review & editing. R. Seurinck: Software, writing—review & editing. J. Roels: Software, writing—review & editing. M. Lavaert: Software, writing—review & editing. F. Van Nieuwerburgh: Formal analysis, writing—review & editing. G. Leclercq: Supervision, writing—review & editing. T. Taghon: Supervision, writing—review & editing. F. Impens: Funding acquisition, supervision, writing—review & editing. B. Menten: Funding acquisition, supervision, writing—review & editing. D. Vermijlen: Funding acquisition, supervision, writing—review & editing. B. Vandekerckhove: Conceptualization, data curation, funding acquisition, methodology, project administration, supervision, validation, writing—original draft.

