


ERCC1-XPF is a multifunctional endonuclease involved in nucleotide excision repair (NER), interstrand cross-link (ICL) repair, and DNA double-strand break (DSB) repair. Only two patients with bi-allelic ERCC1 mutations have been reported, both of whom had features of Cockayne syndrome and died in infancy. Here, we describe two siblings with bi-allelic ERCC1 mutations in their teenage years. Genomic sequencing identified a deletion and a missense variant (R156W) within ERCC1 that disrupts a salt bridge below the XPA-binding pocket. Patient-derived fibroblasts and knock-in epithelial cells carrying the R156W substitution show dramatically reduced protein levels of ERCC1 and XPF. Moreover, mutant ERCC1 weakly interacts with NER and ICL repair proteins, resulting in diminished recruitment to DNA damage. Consequently, patient cells show strongly reduced NER activity and increased chromosome breakage induced by DNA cross-linkers, while DSB repair was relatively normal. We report a new case of ERCC1 deficiency that severely affects NER and considerably impacts ICL repair, which together result in a unique phenotype combining short stature, photosensitivity, and progressive liver and kidney dysfunction.

Introduction
The six billion base pairs of the human genome are continually exposed to DNA-damaging agents, causing a wide variety of genomic DNA lesions, including UV light–induced photoproducts, intra- and interstrand cross-links (ICLs), and DNA double-strand breaks (DSBs). To ensure genomic integrity, these DNA lesions need to be repaired in a specific and efficient manner. Different DNA repair mechanisms have evolved that each act upon a specific subset of DNA lesions. While many DNA repair proteins are involved in a single pathway, the heterodimeric endonuclease ERCC1-XPF is shared between several mechanistically distinct DNA repair pathways (Manandhar et al., 2015).
XPF is unstable and rapidly degraded, which is prevented by its heterodimerization with ERCC1 (Biggerstaff et al., 1993). The interaction between these two proteins is mediated by a double helix-hairpin helix (HhH)2 motif located in their respective C-termini, as well as through the central domain of ERCC1, and the nuclease domain of XPF (de Laat et al., 1998b; Jones et al., 2020; Tripsianes et al., 2005; Tsodikov et al., 2005). The binding to double-stranded DNA is facilitated by ERCC1, while XPF mediates the association with single-stranded DNA (ssDNA) through its (HhH)2 domain (Tsodikov et al., 2005). Once positioned, XPF employs its catalytic activity through its highly conserved nuclease motif to cleave different DNA substrates at the 5′ junction of bubbles and single-stranded 3′ overhangs extending from a DNA double helix (de Laat et al., 1998a; Enzlin and Schärer, 2002; Matsunaga et al., 1996). A key role of ERCC1-XPF was first demonstrated in nucleotide excision repair (NER), where it cleaves the damaged DNA 5′ to the lesion (Sijbers et al., 1996a).
NER recognizes a wide variety of structurally unrelated DNA lesions, including UV-induced photoproducts, via its two sub-pathways: global genome repair (GGR) and transcription-coupled repair (TCR). The bulk of the NER activity relies on GGR, where the XPC protein recognizes photoproducts throughout the genome and recruits the transcription factor IIH (TFIIH) complex (Sugasawa et al., 1998; Volker et al., 2001). DNA lesions that cause stalling of RNA polymerase II (RNAPII) during transcription trigger the recruitment of TCR-specific proteins, such as CSB, CSA, and UVSSA, which in turn recruit the TFIIH complex (Nakazawa et al., 2020; van der Weegen et al., 2020). Following TFIIH recruitment to the DNA lesion, GGR and TCR funnel into a common molecular mechanism involving the association of XPA and XPG (Marteijn et al., 2014), which stimulate the helicase activity of TFIIH and form a stable preincision complex together with RPA (Kokic et al., 2019; Riedl et al., 2003; Wakasugi and Sancar, 1998). The ERCC1-XPF heterodimer is recruited to the NER complex by XPA through the XPA-binding domain of ERCC1 (Tsodikov et al., 2007; Volker et al., 2001). A dual incision by the coordinated activity of the endonucleases XPF (5′ from the lesion) and XPG (3′ from the lesion) releases a stretch of ssDNA containing the DNA lesion (Li et al., 1995; Matsunaga et al., 1995; O’Donovan et al., 1994; Staresincic et al., 2009), after which the generated gap is filled in and ligated to complete repair.
In addition to NER, it was shown that ERCC1-XPF also plays an essential role in ICL repair (Klein Douwel et al., 2014). This repair pathway is initiated by stalled replication and involves recognition of the ICL-stalled replication fork by the multi-protein core Fanconi anemia (FA) complex, which serves as an E3 ubiquitin ligase complex for FANCD2. The mono-ubiquitylation of FANCD2 triggers the recruitment of SLX4, which in turn targets ERCC1-XPF to ICLs (Abdullah et al., 2017; Klein Douwel et al., 2014; Walden and Deans, 2014; Wood, 2010). Once recruited, ERCC1-XPF mediates unhooking of the ICL by incising the parental DNA strand on either side of the lesion to enable subsequent lesion bypass and repair (Kuraoka et al., 2000). Finally, ERCC1-XPF has also been implicated in DSB repair by homologous recombination (Ahmad et al., 2008), during which it can efficiently cleave different recombination intermediates (Wyatt et al., 2017). Alternatively, cells use the error-prone RAD52-dependent single-strand annealing pathway, which involves annealing of regions with microhomology and subsequent removal of the nonhomologous 3′ ssDNA tails by ERCC1-XPF (Adair et al., 2000; Li et al., 2013, 2019). However, the precise role of ERCC1-XPF in these DSB repair pathways is not fully understood.
The importance of DNA repair pathways is underscored by the clinical phenotype of individuals with inherited DNA repair–deficiency disorders. Inactivation of GGR results in Xeroderma pigmentosum (XP; MIM 278700, 610651, 278720, 278730, 278740, 278760, and 278780; DiGiovanna and Kraemer, 2012), which is characterized by photosensitivity and a 2,000-fold increased risk of skin cancer. Neurodegeneration, growth retardation, and multisystem disease without skin cancer are observed in Cockayne syndrome (CS; MIM 216400 and 133540). CS patients have a selective genetic defect in TCR that prevents the processing of DNA damage–stalled RNAPII (Karikkineth et al., 2017; Nakazawa et al., 2020; Nakazawa et al., 2012; Ribeiro et al., 2018; Tufegdžić Vidaković et al., 2020). Mutations in essential ICL repair genes cause FA (MIM 227650, 300514, 227645, 605724, 227646, 600901, 603467, 614082, 609053, 609054, 614083, 614087, 610832, 613390, and 613951), which is characterized by anemia, skeletal abnormalities, organ malformations, and genomic instability (Auerbach, 2009). The role of ERCC1-XPF in ICL repair is underscored by the existence of mutations in XPF that give rise to the FA phenotype (Bogliolo et al., 2013; Ceccaldi et al., 2016; Kashiyama et al., 2013). Inherited defects in DSB repair cause a wide range of disorders associated with radiosensitivity, cancer predisposition, immunodeficiency, and neurodegeneration (Helfricht et al., 2020; McKinnon and Caldecott, 2007).
DNA repair–deficiency disorders are rare and occur with an estimated frequency of ∼1 per 200,000 live births worldwide for XP, CS, and FA. However, only two reports of individuals with bi-allelic ERCC1 (MIM 126380) mutations have been described to date (Jaspers et al., 2007; Kashiyama et al., 2013). Both individuals displayed features consistent with CS and died at 14 mo and 2.5 yr. The first and most severely affected individual (165TOR) carried a premature stop codon (Q158X) on one allele and an F231L missense variant on the other (Jaspers et al., 2007). The second individual (CS20LO) was homozygous for the F231L missense variant (Kashiyama et al., 2013). Cells from both individuals showed sensitivity to UV-induced DNA damage, and 165TOR in particular was also mildly sensitive to ICL-inducing agents. Both individuals died in early childhood (1–2 yr; Jaspers et al., 2007; Kashiyama et al., 2013). A third individual (XP202DC) with bi-allelic ERCC1 mutations who died at 37 yr of age is cited in a meeting abstract (Imoto, K., et al. 2007. Patients with defects in the interacting nucleotide excision repair proteins ERCC1 or XPF show xeroderma pigmentosum with late onset severe neurological degeneration. [Abstract] J. Invest. Dermatol. 127:S92). This individual was compound heterozygous for a nonsense mutation (K226X) and a splice mutation in ERCC1. A detailed phenotypic description is not available, and the impact of these ERCC1 mutations is unknown.
The phenotype of the two aforementioned individuals is more severe than that expected from NER deficiency alone, consistent with the involvement of ERCC1-XPF in multiple DNA repair pathways. In line with this, either ERCC1 or XPF-KO mice displayed high embryonic lethality and decreased life span and died due to severe liver failure (McWhir et al., 1993; Tian et al., 2004; Weeda et al., 1997), which is not observed in NER-deficient XPA-KO mice (de Vries et al., 1995; Nakane et al., 1995).
In the current study, we describe two siblings with bi-allelic ERCC1 mutations who have a unique phenotype of short stature, photosensitivity, progressive cholestatic liver disease, and renal tubulopathy. Both individuals developed progressive liver impairment and required liver transplantations before the age of 10 yr. Functional studies show that the steady-state protein levels of ERCC1 and XPF were dramatically reduced in patient cells and knock-in epithelial cells carrying the missense variant found in the patients. Additionally, the mutant ERCC1 protein interacted only weakly with other NER and ICL repair proteins. We report a new case of ERCC1 deficiency that strongly affects NER and has a considerable impact on ICL repair, which together result in a unique phenotype combining short stature, photosensitivity, and progressive liver and kidney dysfunction.
Results
Two siblings with photosensitivity, short stature, and progressive liver and kidney dysfunction
Sibling 1 (PV50LD; Fig. 1 A) is 13 yr of age, the eldest of three siblings to healthy, unrelated parents of mixed ethnicity including Indigenous Australian, Maltese, and Anglo-Celtic heritage. During the pregnancy, her mother had a transient dilated cardiomyopathy that resolved postpartum. She was born at 39 wk of gestation, and her birth weight was 1.9 kg (Z = −3.8), length 44 cm (Z = −2.44), and head circumference 29.5 cm (Z = −3.8). She had poor growth in infancy, and at 18 mo of age, she was noted to have liver dysfunction with a predominantly cholestatic pattern. Liver ultrasound and magnetic resonance cholangiopancreatography were normal. Liver function progressively declined, as evident from progressive increases in γ-glutamyltransferase (GGT), alanine transaminase (ALT), and bilirubin levels (Fig. S1, A–C). At a liver biopsy (at age 3.5 yr), the lobular parenchyma showed variation in hepatocyte nuclear morphology, with some much larger nuclei and cells with double nuclei (Fig. S1 D) to areas where the cells have small, unremarkable nuclei. There was mild portal fibrosis and mild fibrous portal expansion and mild focal interface inflammation with no ductopenia or periductal fibrosis (Fig. S1 E).
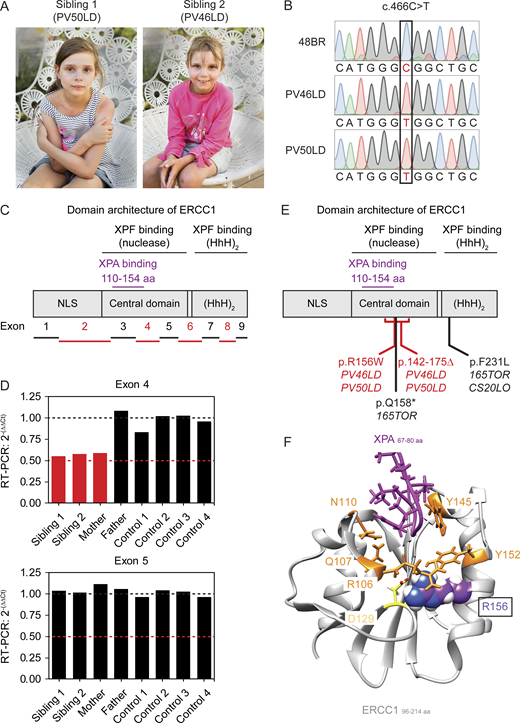
Two siblings with ERCC1 deficiency.(A) Picture of the siblings PV50LD (left) and PV46LD (right). (B) Sanger sequencing of a region in the ERCC1 gene from genomic DNA of the indicated cell lines. See Fig. S2 A for additional NGS data on the ERCC1 missense variant. (C) Schematic representation of the exons in the ERCC1 gene mapped onto the domains in the ERCC1 protein. (D) Multiplex real-time qPCR results on genomic DNA extracted from lymphocytes or epithelial cells from which regions in exon 4 or exon 5 of the ERCC1 gene were amplified (see Table S1). Exon 27 of the CFTR gene was used as an internal standard. The 2-(ΔΔCt) values are shown for the indicated individuals. All values are based on technical triplicates. The black line indicates the expected value when two functional copies of the indicated ERCC1 exon are present, while the red line indicates the expected value when only a single copy is present due to an intragenic deletion on the other allele. See Fig. S2 B for additional NGS data on the intergenic ERCC1 deletion. (E) Schematic representation of domains in the ERCC1 protein with known patient mutations. (F) Structure of the ERCC1 central domain bound to a short XPA peptide. Residues that line the XPA-binding pocket (R106, Q107, N110, Y145, and Y152) or that form a salt bridge just below this pocket (R156 and D129) are indicated. The R156 residue is substituted for W in the siblings.
Two siblings with ERCC1 deficiency.(A) Picture of the siblings PV50LD (left) and PV46LD (right). (B) Sanger sequencing of a region in the ERCC1 gene from genomic DNA of the indicated cell lines. See Fig. S2 A for additional NGS data on the ERCC1 missense variant. (C) Schematic representation of the exons in the ERCC1 gene mapped onto the domains in the ERCC1 protein. (D) Multiplex real-time qPCR results on genomic DNA extracted from lymphocytes or epithelial cells from which regions in exon 4 or exon 5 of the ERCC1 gene were amplified (see Table S1). Exon 27 of the CFTR gene was used as an internal standard. The 2-(ΔΔCt) values are shown for the indicated individuals. All values are based on technical triplicates. The black line indicates the expected value when two functional copies of the indicated ERCC1 exon are present, while the red line indicates the expected value when only a single copy is present due to an intragenic deletion on the other allele. See Fig. S2 B for additional NGS data on the intergenic ERCC1 deletion. (E) Schematic representation of domains in the ERCC1 protein with known patient mutations. (F) Structure of the ERCC1 central domain bound to a short XPA peptide. Residues that line the XPA-binding pocket (R106, Q107, N110, Y145, and Y152) or that form a salt bridge just below this pocket (R156 and D129) are indicated. The R156 residue is substituted for W in the siblings.

Both siblings have liver dysfunction and progressive kidney impairment. (A–C) Liver function of patient 1 (PV50LD) and patient 2 (PV46LD) is represented by GGT (A), ALT (B), and bilirubin (C) levels measured until liver transplantation. (D) 40× (high power) hematoxylin and eosin stain of the liver of sibling 1 at age 3.5 yr. The lobular parenchyma shows some variation in hepatocyte nuclear morphology from field to field, with some much larger nuclei (red arrow) and cells with double nuclei (black arrow), to areas where the cells have small, unremarkable nuclei. The significance of this is unclear, but it may represent regeneration following some previous insult. There is a sparse, focal inflammatory infiltrate comprising small lymphocytes abutting the interface with very occasional extension into lobular parenchyma. The infiltrate does not contain significant numbers of plasma cells. No cytoplasmic or canalicular cholestasis is seen, and rosette formation is not seen. Right: magnification from left picture. (E) Trichrome stain of liver, 10× (low power) of the liver of sibling 1 at age 3.5 yr. Most portal tracts contain unremarkable bile ducts, without evidence of ductopenia. Several show mild fibrous expansion with short spurs of collagen extending into periportal parenchyma. The reticulin pattern appears normal without evidence of nodular regenerative hyperplasia. Arteries and veins appear normal. Right: magnification from left picture. (F) Kidney function of sibling 1 (PV50LD) and sibling 2 (PV46LD) is represented by serum creatinine values over time. The green bar represents the normal values for each parameter. Scale bar is 10 µm.
Both siblings have liver dysfunction and progressive kidney impairment. (A–C) Liver function of patient 1 (PV50LD) and patient 2 (PV46LD) is represented by GGT (A), ALT (B), and bilirubin (C) levels measured until liver transplantation. (D) 40× (high power) hematoxylin and eosin stain of the liver of sibling 1 at age 3.5 yr. The lobular parenchyma shows some variation in hepatocyte nuclear morphology from field to field, with some much larger nuclei (red arrow) and cells with double nuclei (black arrow), to areas where the cells have small, unremarkable nuclei. The significance of this is unclear, but it may represent regeneration following some previous insult. There is a sparse, focal inflammatory infiltrate comprising small lymphocytes abutting the interface with very occasional extension into lobular parenchyma. The infiltrate does not contain significant numbers of plasma cells. No cytoplasmic or canalicular cholestasis is seen, and rosette formation is not seen. Right: magnification from left picture. (E) Trichrome stain of liver, 10× (low power) of the liver of sibling 1 at age 3.5 yr. Most portal tracts contain unremarkable bile ducts, without evidence of ductopenia. Several show mild fibrous expansion with short spurs of collagen extending into periportal parenchyma. The reticulin pattern appears normal without evidence of nodular regenerative hyperplasia. Arteries and veins appear normal. Right: magnification from left picture. (F) Kidney function of sibling 1 (PV50LD) and sibling 2 (PV46LD) is represented by serum creatinine values over time. The green bar represents the normal values for each parameter. Scale bar is 10 µm.
She experienced a large number of recurrent infections including tonsillitis, chicken pox, hand-foot-and-mouth disease, pneumonia, bronchitis, and recurrent episodes of fever, abdominal pain, and pale stools with no cause found. Immunological investigations did not identify any immunodeficiency. She developed episodes of ocular and skin photosensitivity. At age 6 yr, renal dysfunction was detected, with features suggestive of proximal tubular dysfunction characterized by albuminuria (sub-nephrotic range) and hypercalciuria. Her renal function fluctuated, with intermittent episodes of acute kidney injury, progressive kidney impairment with increasing creatinine levels (Fig. S1 F), and minimal response to acetyl cholinesterase inhibition. Renal ultrasound showed small kidneys with increased echogenicity and reduced corticomedullary differentiation. Lung function tests showed mild to moderate restrictive lung disease.
Developmental milestones were normal, but some learning difficulties were evident at school age with no sign of regression. Vision and hearing were normal. Growth remained very slow despite supplemental feeding. She was relatively stable until 9.5 yr of age, when she displayed evidence of liver decompensation with a progressively rising bilirubin level (Fig. S1 C) and international normalized ratio. She underwent orthotopic liver transplantation at age 9 yr 10 mo. Following liver transplant, her tubulopathy has stabilized, although her serum creatinine continues to increase at a slower rate (Fig. S1 F). She is clinically stable. At age 12 yr, ovarian insufficiency was diagnosed. Brain magnetic resonance imaging (MRI) at age 12 yr showed mild cerebral atrophy with moderate cerebellar atrophy and mild brainstem atrophy. At last assessment (aged 13.5 yr), growth was slow (weight 22.4 kg [Z = −6.08] and height 134.7 cm [Z = −3.72]). She had a very slim build with poor muscle bulk and a paucity of subcutaneous fat. She had developed freckling on sun-exposed areas (Fig. 1 A). She had mildly deep-set eyes, and her scalp hair was thin. There were no radial ray abnormalities. Neurological examination showed mild weakness, minimal ataxia, and depressed reflexes.
Sibling 2 (PV46LD; Fig. 1 A) is 11 yr of age, a younger sister to sibling 1. The pregnancy was complicated by placenta previa and transient maternal cardiomyopathy, and sibling 2 was born at 35 wk of gestation with a birth weight of 1.79 kg (third centile), a length of 45 cm (50th centile), and a head circumference of 29 cm (less than third centile). She had a paucity of subcutaneous fat and similar facial features to her older sibling. She exhibited failure to thrive in the first year of life and liver impairment from age 2 yr, with significantly increased GGT, ALT, and bilirubin levels (Fig. S1, A–C). Liver biopsy at age 6 yr showed damage to the intrahepatic bile ducts, resulting in periductal fibrosis. As with the liver biopsy from her sister, a moderate number of double-nucleated hepatocytes was seen, some with large nuclei and large nucleoli.
She had episodes of ocular and skin photosensitivity. She had findings of a renal tubulopathy with mild renal impairment, with progressively increasing creatinine levels (Fig. S1 F). Renal ultrasound showed small kidneys with nephrocalcinosis. Developmental milestones were normal, but a mild intellectual disability (IQ 66) was diagnosed at school age. There has been slow forward progress with neurodevelopmental milestones and no definite regression. Vision and hearing were normal. The liver impairment was progressive, and at age 8 yr, she underwent liver transplantation. Brain MRI at age 5 yr was normal, but a repeat MRI at 10 yr showed moderate cerebellar atrophy and mild cerebral atrophy. At age 11 yr, measurement of estrogen, follicle-stimulating hormone, and luteinizing hormone showed a pattern suggestive of ovarian insufficiency. At last assessment at age 11 yr, growth was slow (weight 20 kg [Z = −3.89] and height 120.7 cm [Z = −3.17]). She had a very thin build with minimal subcutaneous fat and poor muscle bulk, familial facial features, mildly deep-set eyes, and freckling on sun-exposed areas (Fig. 1 A). She had no radial ray abnormalities. Neurological examination showed mild weakness with mild ataxia and absent reflexes.
Molecular analysis identifies novel bi-allelic ERCC1 mutations
Exome sequencing revealed that both siblings harbor a novel missense variant in exon 4 of the ERCC1 gene (p.R156W; c.466C>T) on the paternal allele (Fig. S2 A). The presence of the c.466C>T missense variant was confirmed by Sanger sequencing in both affected siblings (Fig. 1 B). Whole-genome sequencing revealed a deletion in exon 4 on the maternal allele (hg19: chr19:45,922,224-45,924,375; Fig. S2 B), which was confirmed by comparative quantitative PCR (qPCR) of exon 4 and exon 5 of ERCC1 in both siblings and the mother (Fig. 1, C and D). The deletion, which is expected to be a null allele, was detected in neither the father nor in four negative control samples (Fig. 1 D).

Identification of the missense variant in ERCC1 by NGS. (A) Genomic sequencing data for both siblings and parents, visualized using the Integrative Genomics Viewer. The location of ERCC1 on chromosome 19q13.32 appears at the top (red line), with genomic coordinates, reference nucleotide sequence, and amino acid sequence below. An apparently homozygous variant, c.466C>T, is seen in both siblings (right panel). This is seen in the heterozygous state in one parent (left panel), while the other is apparently homozygous for the reference sequence. Please note that as ERCC1 is in reverse orientation, this variant appears as the corresponding G>A. A schematic of read depth, with color blocks highlighting departure from the reference sequence, appears above separate contiguous reads for each family member. (B) Raw NGS data of patient viewed with Integrative Genomics Viewer software. Each line represents “one read,” and the gray bars represents DNA fragments that have been reassembled. The ERCC1 gene deletion appears as horizontal red lines.
Identification of the missense variant in ERCC1 by NGS. (A) Genomic sequencing data for both siblings and parents, visualized using the Integrative Genomics Viewer. The location of ERCC1 on chromosome 19q13.32 appears at the top (red line), with genomic coordinates, reference nucleotide sequence, and amino acid sequence below. An apparently homozygous variant, c.466C>T, is seen in both siblings (right panel). This is seen in the heterozygous state in one parent (left panel), while the other is apparently homozygous for the reference sequence. Please note that as ERCC1 is in reverse orientation, this variant appears as the corresponding G>A. A schematic of read depth, with color blocks highlighting departure from the reference sequence, appears above separate contiguous reads for each family member. (B) Raw NGS data of patient viewed with Integrative Genomics Viewer software. Each line represents “one read,” and the gray bars represents DNA fragments that have been reassembled. The ERCC1 gene deletion appears as horizontal red lines.
While a previously described pathogenic ERCC1 missense variant (p.F231L; c.693C>G) is located in the (HhH)2 domain (Fig. 1 E), the missense variant (p.R156W) is located within the central domain of ERCC1 and is in close proximity to the XPA-binding pocket (110–154 aa) of ERCC1 (Fig. 1 E). Structural analysis of the ERCC1 central domain revealed a narrow V-shaped hydrophobic pocket that binds a short motif (67–80 aa) present in XPA (Fig. 1 F; Tsodikov et al., 2007). Amino acids that line the XPA-binding pocket in ERCC1 (Fig. 1 F) are important for NER but dispensable for other ERCC1-dependent repair pathways (Orelli et al., 2010). The R156 residue that is substituted in the patients is located just below the XPA-binding pocket and forms a salt bridge with the opposing amino acid D129 (Fig. 1 F). The R156W substitution therefore likely affects the stability of the XPA-binding pocket. It is possible that this substitution also weakens the interaction between the central domain of ERCC1 and the nuclease domain of XPF (Jones et al., 2020), and we would predict that this could lead to a mild to moderate destabilization of the heterodimer interface, resulting in reduced stability of the protein.
PV46LD and PV50LD fibroblasts display a severe NER defect
A hallmark of NER deficiency is strong sensitivity to UV-C irradiation. To address the impact of the ERCC1 deficiency on NER function, we obtained primary fibroblasts from skin biopsies of both affected siblings. To allow clonogenic survival assays, we immortalized PV50LD cells by introducing hTERT (Fig. S3 A). As a control, we generated full ERCC1-KO cells in RPE1-hTERT cells by CRISPR-Cas9 (Fig. 2 A). Clonogenic UV-C survival assay showed that PV50LD-hTERT cells are hypersensitive to UV-C irradiation, although not to the same extent as full ERCC1-KO cells (Fig. 2 B), suggesting a severe defect in NER.
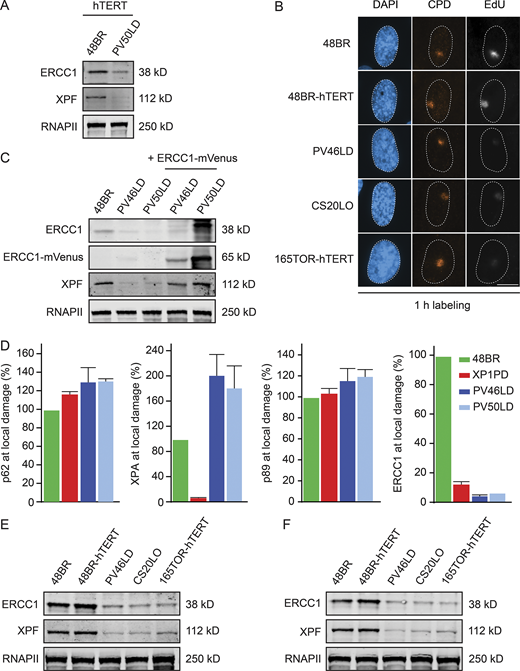
ERCC1 protein expression in patient fibroblasts.(A) Western blot of hTERT-immortalized 48BR and PV50LD cells. (B) Representative microscopy images of UDS in the indicated cell lines. (C) Western blot on 48BR and the patient fibroblasts with or without lentiviral transduction with ERCC1-mVenus. Note that the decreased protein levels of XPF are fully reversed. (D) Quantification of local damage staining shown in Fig. 3 A. Data represent mean ± SEM. (E and F) Western blot of the indicated cell lines stained for ERCC1 and XPF (n = 2). Scale bar is 5 µm. The number of cells used for quantification is shown in Table S6. Uncropped Western blot data are shown in Data S1. CPD, cyclobutane pyrimidine dimer.
ERCC1 protein expression in patient fibroblasts.(A) Western blot of hTERT-immortalized 48BR and PV50LD cells. (B) Representative microscopy images of UDS in the indicated cell lines. (C) Western blot on 48BR and the patient fibroblasts with or without lentiviral transduction with ERCC1-mVenus. Note that the decreased protein levels of XPF are fully reversed. (D) Quantification of local damage staining shown in Fig. 3 A. Data represent mean ± SEM. (E and F) Western blot of the indicated cell lines stained for ERCC1 and XPF (n = 2). Scale bar is 5 µm. The number of cells used for quantification is shown in Table S6. Uncropped Western blot data are shown in Data S1. CPD, cyclobutane pyrimidine dimer.
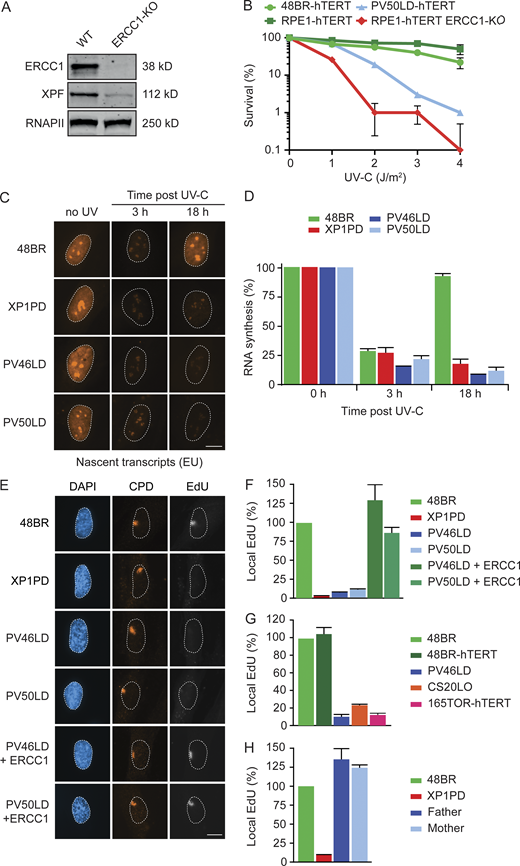
Patient fibroblasts display a strong NER defect.(A) Western blot of RPE1-hTERT WT and ERCC1-KO cells. (B) Clonogenic UV-C survival in the indicated cell lines (n = 2). See Fig. S3 A for Western blot analysis. (C and D) Representative microscopy images of 5-ethynyl-uridine (EU) incorporation into nascent transcripts (C) and quantification of nascent transcripts (D) in unirradiated cells or at 3 h and 18 h after UV-C in the indicated cell lines (n = 2 or 3). (E–H) Representative microscopy images of EdU incorporation at repair sites (E) and quantification of UDS (F–H) in the indicated cell lines (n = 2–4). See Fig. S3 B for additional UDS microscopy images and Fig. S3 C for Western blot analysis. Data represent mean ± SEM. Scale bar is 5 µm. The number of cells used for quantification is shown in Table S6. Uncropped Western blot data are shown in Data S1. CPD, cyclobutane pyrimidine dimer.
Patient fibroblasts display a strong NER defect.(A) Western blot of RPE1-hTERT WT and ERCC1-KO cells. (B) Clonogenic UV-C survival in the indicated cell lines (n = 2). See Fig. S3 A for Western blot analysis. (C and D) Representative microscopy images of 5-ethynyl-uridine (EU) incorporation into nascent transcripts (C) and quantification of nascent transcripts (D) in unirradiated cells or at 3 h and 18 h after UV-C in the indicated cell lines (n = 2 or 3). (E–H) Representative microscopy images of EdU incorporation at repair sites (E) and quantification of UDS (F–H) in the indicated cell lines (n = 2–4). See Fig. S3 B for additional UDS microscopy images and Fig. S3 C for Western blot analysis. Data represent mean ± SEM. Scale bar is 5 µm. The number of cells used for quantification is shown in Table S6. Uncropped Western blot data are shown in Data S1. CPD, cyclobutane pyrimidine dimer.
UV-induced DNA lesions encountered during transcription are repaired by TCR, which can be measured by the recovery of RNA synthesis (RRS) assay (Nakazawa et al., 2010). We measured RRS in 48BR fibroblasts (WT), patient fibroblasts, and included XPA-deficient (XP1PD) primary fibroblasts as a control. Labeling of nascent transcripts by 5-ethynyl-uridine incorporation showed a strong UV-induced decrease in nascent transcripts in all cell lines at 3 h after UV-C. While 48BR fully recovered transcription within 18 h after UV-C irradiation, both patient fibroblasts recovered even less than the NER-deficient XP-A patient cells (Fig. 2, C and D), suggesting that TCR is virtually absent under these conditions.
To examine GGR activity, we measured unscheduled DNA synthesis (UDS) in nondividing cells (Nakazawa et al., 2010). To this end, primary fibroblasts were locally irradiated with UV-C light followed by pulse-labeling with the thymidine analogue 5-ethynyl-deoxyuridine (EdU) to measure repair. Clear EdU incorporation was detected in 48BR cells at sites of local UV-induced DNA damage. However, only very low levels of EdU incorporation were detectable in the patient fibroblasts at similar levels as in XPA-deficient fibroblasts (XP1PD; Fig. 2, E and F). The UDS levels in PV46LD and PV50LD cells were comparable to, or even lower than, the levels measured in the previously described ERCC1-deficient 165TOR (Jaspers et al., 2007) and CS20LO (Kashiyama et al., 2013) cells, which we included in parallel (Fig. 2 G and Fig. S3 B). Importantly, reexpression of mVenus-tagged ERCC1 in both fibroblasts fully rescued EdU incorporation (Fig. 2, E and F; and Fig. S3 C), confirming that the strong UDS defect is due to ERCC1 deficiency. We also obtained primary fibroblasts from the parents, which displayed normal UDS at sites of local UV damage (Fig. 2 H), suggesting that the respective heterozygous ERCC1 defect in the parents is fully compensated by the WT allele.
PV46LD and PV50LD fibroblasts have very low ERCC1 and XPF protein levels
NER involves the highly coordinated and sequential assembly of DNA repair complexes, during which XPA recruitment occurs downstream of TFIIH but upstream of ERCC1-XPF (Volker et al., 2001). To address to which extent the patient fibroblasts still support NER complex assembly, we locally irradiated fibroblasts with UV-C and monitored the recruitment of several core NER proteins by immunofluorescent labeling (Fig. 3 A, quantification, and Fig. S3 D). Clear recruitment of TFIIH, XPA, and ERCC1 at sites of local UV-induced DNA damage was detected in 48BR cells, while XPA-deficient XP1PD cells failed to recruit ERCC1 as previously described (Fig. 3 A and Fig. S3 D; Volker et al., 2001). Both PV46LD and PV50LD cells showed normal recruitment of TFIIH and XPA, while ERCC1 recruitment to sites of local UV-induced DNA damage was undetectable (Fig. 3 A and Fig. S3 D).
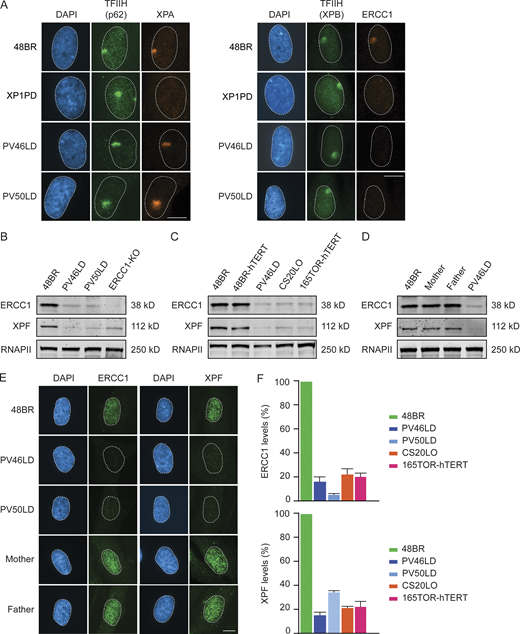
Patient fibroblasts have low ERCC1 and XPF protein levels.(A) Representative microscopy images of different antibody stainings after local UV-C irradiation. Quantification of these data is shown in Fig. S3 D. (B–D) Western blot of the indicated cell lines stained for ERCC1 and XPF. Additional Western blot data are shown in Fig. S3, E and F. (E and F) Microscopy images (E) and quantification of the ERCC1 and XPF protein levels based on Western blot (F) in the indicated cell lines. Data represent mean ± SEM. Scale bar is 5 µm. Uncropped Western blot data are shown in Data S1.
Patient fibroblasts have low ERCC1 and XPF protein levels.(A) Representative microscopy images of different antibody stainings after local UV-C irradiation. Quantification of these data is shown in Fig. S3 D. (B–D) Western blot of the indicated cell lines stained for ERCC1 and XPF. Additional Western blot data are shown in Fig. S3, E and F. (E and F) Microscopy images (E) and quantification of the ERCC1 and XPF protein levels based on Western blot (F) in the indicated cell lines. Data represent mean ± SEM. Scale bar is 5 µm. Uncropped Western blot data are shown in Data S1.
The loss of ERCC1 localization could be due to a failure of ERCC1 recruitment, a general reduction in ERCC1 protein levels, or a combination of the two. Western blot analysis indeed revealed that the steady-state levels of both ERCC1 and XPF were dramatically reduced in PV46LD and PV50LD cells (Fig. 3 B). Residual ERCC1 expression was, however, still detected when compared with full ERCC1-KO cells (Fig. 3 B). The reduction in ERCC1 and XPF protein levels in PV46LD cells was highly similar to the reduced levels detected in either CS20LO or 165TOR cells (Fig. 3 C and Fig. S3, E and F), suggesting that the more severe phenotype in the individual from which these cells were derived is not due to a stronger impact on protein stability. In line with our UDS results, we detected normal ERCC1 and XPF expression in cells from the parents, indicating compensation by the WT allele (Fig. 3 D). Immunofluorescent labeling confirmed that ERCC1 and XPF protein levels were severely reduced, while cells from the parents were indistinguishable from WT cells (Fig. 3 E). Quantification of Western blot data shows that ERCC1 protein levels were reduced to ∼20% in patient fibroblasts, also resulting in similarly reduced levels of XPF levels (∼20%; Fig. 3 F).
Knock-in of the R156W missense variant leads to ERCC1-XPF protein instability and a severe NER defect
It is tempting to speculate that the low steady-state protein levels of ERCC1-XPF and the strong NER defect in the patient fibroblasts are caused by the R156W amino acid substitution in ERCC1. However, considering that the affected individuals are siblings, we cannot exclude that other genetic features shared between them may also contribute to this phenotype.
To directly assess this, we decided to generate knock-in lines (KIs) carrying the R156W amino acid substitution in RPE1-hTERT cells using CRISPR-Cas9 technology. We obtained homozygous KI clones carrying the patient mutation in ERCC1 (p.R156W; c.466C>T), which was confirmed by Sanger sequencing in two individual clones (Fig. 4 A). The missense mutation caused a severe reduction of ERCC1 and XPF protein levels in both KI clones (clone 2–17 and 2–51; Fig. 4 B and Fig. S4, A and B). Quantification of the Western blot data showed that the ERCC1 and XPF protein levels were almost as low as in ERCC1-KO cells (Fig. 4 C). These findings establish that the R156W amino acid substitution has a strong impact on the stability of ERCC1.
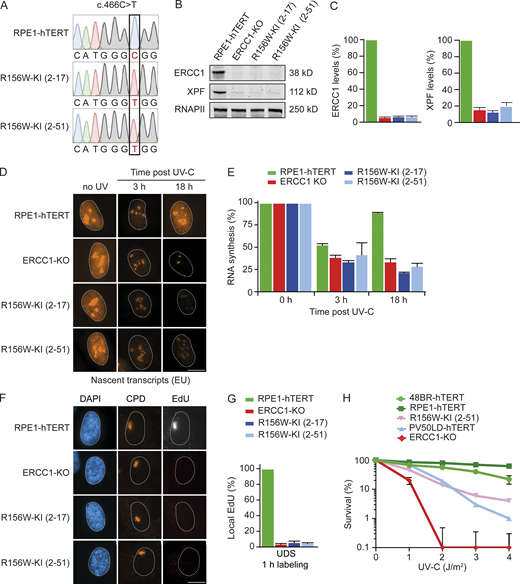
Knock-in of R156W in RPE1 cells causes a strong NER defect.(A) Sanger sequencing of a region in the ERCC1 gene from genomic DNA of the indicated cell lines. (B) Western blot of the indicated cell lines stained for ERCC1 and XPF. See Fig. S4, A and B, for additional Western blot data. (C) Quantification of ERCC1 and XPF protein levels based on Western blot in the indicated cell lines. (D and E) Representative microscopy images (D) and quantification of nascent transcripts (E) in unirradiated cells or at 3 h and 18 h after UV-C in the indicated cell lines (n = 2). (F and G) Representative microscopy images (F) and quantification of UDS (G) in the indicated cell lines (n = 2). (H) Clonogenic UV-C survival in the indicated cell lines (n = 2–4). Data represent mean ± SEM. Scale bar is 5 µm. The number of cells used for quantification is shown in Table S6. Uncropped Western blot data are shown in Data S1. CPD, cyclobutane pyrimidine dimer.
Knock-in of R156W in RPE1 cells causes a strong NER defect.(A) Sanger sequencing of a region in the ERCC1 gene from genomic DNA of the indicated cell lines. (B) Western blot of the indicated cell lines stained for ERCC1 and XPF. See Fig. S4, A and B, for additional Western blot data. (C) Quantification of ERCC1 and XPF protein levels based on Western blot in the indicated cell lines. (D and E) Representative microscopy images (D) and quantification of nascent transcripts (E) in unirradiated cells or at 3 h and 18 h after UV-C in the indicated cell lines (n = 2). (F and G) Representative microscopy images (F) and quantification of UDS (G) in the indicated cell lines (n = 2). (H) Clonogenic UV-C survival in the indicated cell lines (n = 2–4). Data represent mean ± SEM. Scale bar is 5 µm. The number of cells used for quantification is shown in Table S6. Uncropped Western blot data are shown in Data S1. CPD, cyclobutane pyrimidine dimer.
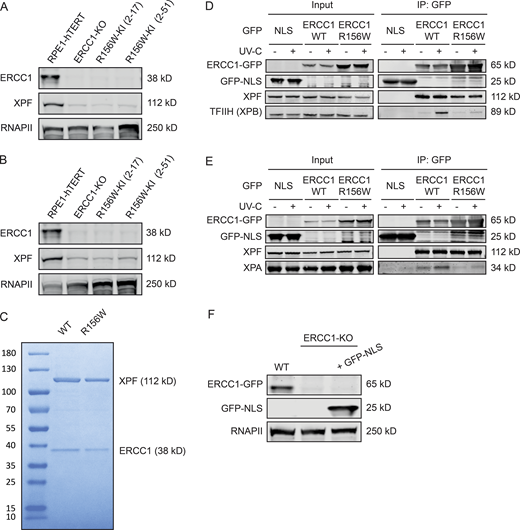
Co-IP of ERCC1WT-GFP and ERCC1R156W-GFP.(A and B) Western blot of the indicated RPE1 cell lines stained for ERCC1 and XPF. (C) Purified recombinant XPF-HA-ERCC1WT-His and XPF-HA-ERCC1R156W-His proteins. (D and E) Co-IP of U2OS GFP-NLS, ERCC1WT-GFP, and ERCC1R156W-GFP in the absence or presence of UV-C showing the UV-induced interaction with TFIIH (D) or XPA (E). The panel in Fig. 6 C is a composite based on these individual Co-IPs. (F) Western blot of the indicated U2OS cell lines stained for ERCC1 and GFP. Uncropped Western blot data are shown in Data S1. CPD, cyclobutane pyrimidine dimer.
Co-IP of ERCC1WT-GFP and ERCC1R156W-GFP.(A and B) Western blot of the indicated RPE1 cell lines stained for ERCC1 and XPF. (C) Purified recombinant XPF-HA-ERCC1WT-His and XPF-HA-ERCC1R156W-His proteins. (D and E) Co-IP of U2OS GFP-NLS, ERCC1WT-GFP, and ERCC1R156W-GFP in the absence or presence of UV-C showing the UV-induced interaction with TFIIH (D) or XPA (E). The panel in Fig. 6 C is a composite based on these individual Co-IPs. (F) Western blot of the indicated U2OS cell lines stained for ERCC1 and GFP. Uncropped Western blot data are shown in Data S1. CPD, cyclobutane pyrimidine dimer.
To further establish that the NER defect in patient fibroblasts is caused by the R156W amino acid substitution in ERCC1, we measured TCR activity by monitoring transcription restart after UV irradiation. While parental RPE1-hTERT cells showed normal restart at 18 h after UV, both R156W-KI clones showed a strong TCR defect comparable to full ERCC1-KO cells (Fig. 4, D and E). Similarly, UDS experiments revealed normal GGR activity in parental RPE1-hTERT cells, which was severely reduced below 10% in both R156W-KI clones and ERCC1-KO cells (Fig. 4, F and G). Finally, we measured UV sensitivity of one R156W-KI clone (2–51) in comparison to the hTERT-immortalized patient fibroblast PV50LD. The R156W-KI cells showed a strong UV-sensitive phenotype, although not as sensitive as full ERCC1-KO cells (Fig. 4 H). Interestingly, the UV-sensitive phenotype of the R156W-KI was very similar to that of the PV50LD-hTERT patient fibroblasts. These findings establish that the low ERCC1-XPF protein levels and the strong NER defect are a direct consequence of the R156W amino acid substitution in ERCC1.
ERCC1R156W substitution causes partial cytoplasmic localization
The patient fibroblasts and the KI cells display dramatically reduced protein levels and strongly reduced NER activity. We next wished to explore whether the NER defect is only caused by reduced ERCC1-XPF protein levels, or whether the ERCC1R156W mutant protein has reduced activity as an endonuclease or in NER.
To address this, we generated ERCC1-KO cells by CRISPR-Cas9 in U2OS cells equipped with the Flp-In/T-Rex system. Using Flp-based site-directed recombination, we targeted cDNAs encoding GFP-tagged ERCC1WT or ERCC1R156W to the genomic Flp recognition target (FRT) site to enable inducible expression from a strong viral promoter. Western blot analysis showed that ERCC1WT and ERCC1R156W were expressed at similar levels and that expression of either protein rescued the reduced protein levels of XPF observed in ERCC1-KO cells (Fig. 5 A). These cells are therefore an excellent model system to disentangle the specific impact of the ERCC1R156W substitution when expressed at WT levels from the impact of lower ERCC1 expression in the patient fibroblasts and KI epithelial cells.
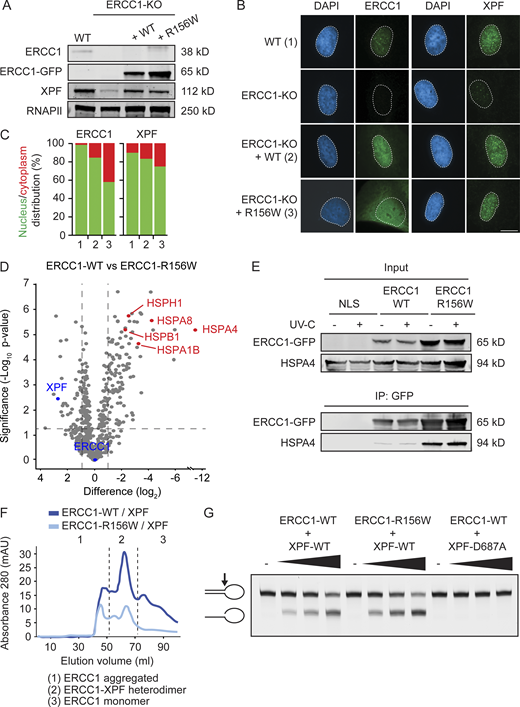
The ERCC1R156W substitution causes partial cytoplasmic localization.(A) Western blot of U2OS WT, ERCC1-KO, and ERCC1-KO complemented with either ERCC1WT or ERCC1R156W. (B and C) Microscopy images (B) and nuclear/cytoplasmic distribution (C) of ERCC1 and XPF in complemented U2OS cells. (D) Volcano plot depicting the differential interactome of ERCC1WT over ERCC1R156W after pull-down and analysis by label-free MS. The enrichment (log2) is plotted on the x axis, and the significance (−log10 P value) is plotted on the y axis. The dashed lines indicate an enrichment of twofold on the x axis (log2 of 1) and a significance of 0.05 (−log10 P value of 1.3) on the y axis. Note that interactors in red bind more strongly to ERCC1R156W than to ERCC1WT. (E) Co-IP of GFP-NLS, ERCC1WT-GFP, and ERCC1R156W-GFP in the absence or presence of UV-C. (F) Gel-filtration profiles of recombinant purified ERCC1WT-XPF or ERCC1R156W-XPF in mega-arbitrary units (mAU). The dashed lines indicate the approximate volumes at which aggregated ERCC1 (<50 ml), ERCC1-XPF heterodimer (50–70 ml), and ERCC1 monomer (>70 ml) elute. See Fig. S4 C for additional data. (G) Nuclease activity assay of ERCC1WT-XPF or ERCC1R156W-XPF on a stem-loop DNA substrate. Scale bar is 5 µm. The number of cells used for quantification is shown in Table S6. Uncropped Western blot data are shown in Data S1.
The ERCC1R156W substitution causes partial cytoplasmic localization.(A) Western blot of U2OS WT, ERCC1-KO, and ERCC1-KO complemented with either ERCC1WT or ERCC1R156W. (B and C) Microscopy images (B) and nuclear/cytoplasmic distribution (C) of ERCC1 and XPF in complemented U2OS cells. (D) Volcano plot depicting the differential interactome of ERCC1WT over ERCC1R156W after pull-down and analysis by label-free MS. The enrichment (log2) is plotted on the x axis, and the significance (−log10 P value) is plotted on the y axis. The dashed lines indicate an enrichment of twofold on the x axis (log2 of 1) and a significance of 0.05 (−log10 P value of 1.3) on the y axis. Note that interactors in red bind more strongly to ERCC1R156W than to ERCC1WT. (E) Co-IP of GFP-NLS, ERCC1WT-GFP, and ERCC1R156W-GFP in the absence or presence of UV-C. (F) Gel-filtration profiles of recombinant purified ERCC1WT-XPF or ERCC1R156W-XPF in mega-arbitrary units (mAU). The dashed lines indicate the approximate volumes at which aggregated ERCC1 (<50 ml), ERCC1-XPF heterodimer (50–70 ml), and ERCC1 monomer (>70 ml) elute. See Fig. S4 C for additional data. (G) Nuclease activity assay of ERCC1WT-XPF or ERCC1R156W-XPF on a stem-loop DNA substrate. Scale bar is 5 µm. The number of cells used for quantification is shown in Table S6. Uncropped Western blot data are shown in Data S1.
Previous studies have shown that missense mutations in XPF can cause mislocalization of ERCC1-XPF in the cytoplasm (Ahmad et al., 2010). In line with this, we also detected that ∼40% of the ERCC1R156W protein pool mislocalized to the cytoplasm, which was only ∼15% in ERCC1WT-expressing cells (Fig. 5, B and C). This mislocalization of ERCC1 also increased the fraction of cytoplasmic XPF (Fig. 5, B and C), indicating that ERCC1R156W may be partially misfolded.
To test this possibility, we performed immunoprecipitation experiments on ERCC1WT-GFP and ERCC1R156W-GFP followed by quantitative label-free mass spectrometry (MS) to map differential interactors of the two ERCC1 proteins. Importantly, quantitative MS confirmed that equal amounts of ERCC1WT-GFP and ERCC1R156W-GFP protein were pulled down (Fig. 5 D). Strikingly, the interactome of ERCC1R156W was enriched for heat-shock proteins (HSPs), in particular of the HSPA family, while the interaction with XPF was quantitatively reduced compared with ERCC1WT (Fig. 5 D). The HSPA proteins are protein-folding chaperones that prevent aggregation of misfolded proteins (Stetler et al., 2010). Co-immunoprecipitation (Co-IP) experiments indeed confirmed that ERCC1R156W-GFP strongly interacted with HSPA4 compared with ERCC1WT (Fig. 5 E), suggesting partial misfolding of ERCC1R156W.
To address to which extent ERCC1R156W-XPF is still active as an endonuclease, we purified the recombinant complex from Sf9 insect cells (Fig. S4 C). Our previously established procedure involves a gel-filtration step that allows us to assess whether ERCC1-XPF is in a dimeric or aggregated state (Fig. 5 F, fractions 2 and 1, respectively). The yield of the recombinant ERCC1R156W-XPF complex was lower and showed a higher level of aggregated protein. Moreover, we also noticed a reduction in the fraction of heterodimeric protein compared with ERCC1WT-XPF (Fig. 5 F). Nonetheless, the heterodimeric recombinant ERCC1R156W-XPF in fraction 2 was fully active when incubated with a stem-loop model DNA substrate, while the catalytically inactive ERCC1-XPFD687A, included as a negative control, was devoid of incision activity (Fig. 5 G). These findings suggest that ERCC1R156W-XPF is partially misfolded and mislocalized, but the protein fraction that folds properly is still active as an endonuclease.
The ERCC1R156W mutant protein fails to efficiently interact with core NER factors
We next addressed how the ERCC1R156W substitution affected NER using the reconstituted ERCC1-KO cells. Because the R156W substitution is located just underneath the XPA-binding pocket in ERCC1 (Fig. 1 F), we asked whether ERCC1R156W could still interact with NER proteins in response to UV irradiation. To test this, we immunoprecipitated ERCC1WT-GFP, ERCC1R156W-GFP, or GFP fused to a nuclear localization signal (GFP-NLS) as a control from UV-irradiated cells and subsequently performed label-free MS. By comparing ERCC1-GFP with GPF-NLS, we identified constitutive as well as UV-induced interactors of ERCC1. Our analysis revealed that both ERCC1WT and ERCC1R156W interacted with XPF and the ICL repair-specific scaffold proteins SLX4 and SLX4IP, albeit quantitatively reduced in ERCC1R156W-expressing cells (Fig. 6, A and B). Strikingly, the UV-induced interaction with the TFIIH subunit XPB in ERCC1WT cells was completely lost in ERCC1R156W cells (Fig. 6, A and B). Co-IP experiments confirmed that ERCC1R156W failed to associate with either TFIIH or XPA in response to UV irradiation, while robust interactions were detected after pull-down of ERCC1WT (Fig. 6 C and Fig. S4, D and E).
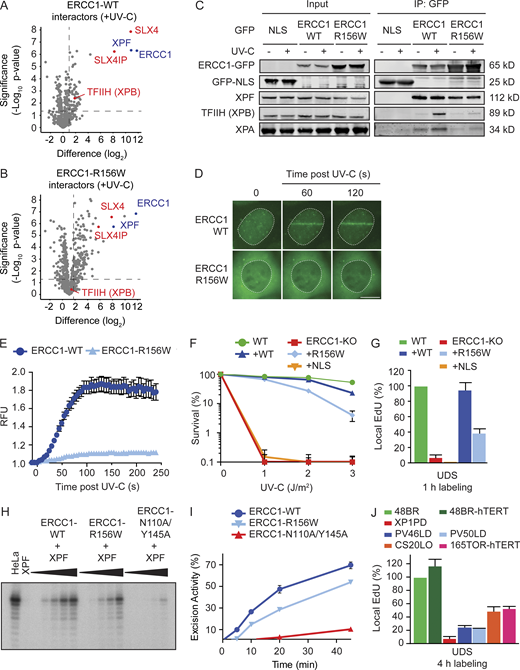
ERCC1R156W supports residual repair by NER.(A and B) Volcano plots depicting the interactome of ERCC1WT-GFP (A) or ERCC1R156W-GFP (B) after pull-down from UV-irradiated cells and analysis by label-free MS. The dashed lines indicate an enrichment of twofold on the x axis (log2 of 1) and a significance of 0.05 (−log10 P value of 1.3) on the y axis. (C) Co-IP of GFP-NLS, ERCC1WT-GFP, and ERCC1R156W-GFP in the absence or presence of UV-C. This panel is a composite of two representative Co-IPs. See Fig. S4, D and E, for the two individual Co-IPs. (D and E) Microscopy images (D) and quantification (E) of the recruitment of ERCC1WT-GFP and ERCC1R156W-GFP to sites of local UV-C laser irradiation (n = 2) in relative fluorescence units (RFUs). (F) Clonogenic UV-C survival in U2OS ERCC1-KO cells complemented with ERCC1WT-GFP, ERCC1R156W-GFP, or GFP-NLS (n = 3–6). See Fig. S4 F for Western blot analysis. (G) UDS quantification of U2OS ERCC1-KO cells complemented with ERCC1WT-GFP, ERCC1R156W-GFP, or GFP-NLS (n = 2–4). (H) NER assay with recombinant ERCC1-XPF dimer on a plasmid containing a dG-AAF lesion at different time points (0, 5, 10, 20, and 45 min) after incubation. (I) Quantification of the in vitro NER activity of ERCC1WT and ERCC1R156W (n = 1). (J) Quantification of UDS with 4-h EdU labeling in the indicated cell lines (n = 2–4). Data represent mean ± SEM. Scale bar is 5 µm. See Fig. S5 A for UDS microscopy images. The number of cells used for quantification is shown in Table S6.
ERCC1R156W supports residual repair by NER.(A and B) Volcano plots depicting the interactome of ERCC1WT-GFP (A) or ERCC1R156W-GFP (B) after pull-down from UV-irradiated cells and analysis by label-free MS. The dashed lines indicate an enrichment of twofold on the x axis (log2 of 1) and a significance of 0.05 (−log10 P value of 1.3) on the y axis. (C) Co-IP of GFP-NLS, ERCC1WT-GFP, and ERCC1R156W-GFP in the absence or presence of UV-C. This panel is a composite of two representative Co-IPs. See Fig. S4, D and E, for the two individual Co-IPs. (D and E) Microscopy images (D) and quantification (E) of the recruitment of ERCC1WT-GFP and ERCC1R156W-GFP to sites of local UV-C laser irradiation (n = 2) in relative fluorescence units (RFUs). (F) Clonogenic UV-C survival in U2OS ERCC1-KO cells complemented with ERCC1WT-GFP, ERCC1R156W-GFP, or GFP-NLS (n = 3–6). See Fig. S4 F for Western blot analysis. (G) UDS quantification of U2OS ERCC1-KO cells complemented with ERCC1WT-GFP, ERCC1R156W-GFP, or GFP-NLS (n = 2–4). (H) NER assay with recombinant ERCC1-XPF dimer on a plasmid containing a dG-AAF lesion at different time points (0, 5, 10, 20, and 45 min) after incubation. (I) Quantification of the in vitro NER activity of ERCC1WT and ERCC1R156W (n = 1). (J) Quantification of UDS with 4-h EdU labeling in the indicated cell lines (n = 2–4). Data represent mean ± SEM. Scale bar is 5 µm. See Fig. S5 A for UDS microscopy images. The number of cells used for quantification is shown in Table S6.
The recruitment of ERCC1-XPF to NER complexes is fully dependent on XPA (Volker et al., 2001), raising the question of whether ERCC1R156W is still recruited to sites of UV-induced DNA damage. To address this, we monitored the recruitment of ERCC1-GFP to UV-induced DNA lesions using live-cell imaging. To this end, cells were irradiated with a UV-C (266-nm) laser, which triggered the rapid recruitment of ERCC1WT-GFP within the first 60 s following irradiation, reaching a plateau around 120 s (Fig. 6, D and E). In contrast, only very weak recruitment of ERCC1R156W-GFP could be detected upon UV-C irradiation using identical conditions (Fig. 6, D and E). These findings suggest that the inability of ERCC1R156W to interact with XPA severely limits its association with the NER complex.
We next asked how the limited association of ERCC1R156W with NER complexes affects DNA repair activity. Surprisingly, expression of ERCC1R156W protected ERCC1-KO cells from UV irradiation, although not to the same extent as ERCC1WT (Fig. 6 F). Expression of GFP-NLS did not rescue the UV-sensitive phenotype of ERCC1-KO cells, showing this is a specific effect of the reexpression of ERCC1 (Fig. 6 F and Fig. S4 F). Furthermore, ERCC1R156W still showed ∼40% of UDS compared with ERCC1WT (Fig. 6 G), suggesting that ERCC1R156W has residual repair activity. To corroborate this result, we measured the in vitro NER activity of recombinant heterodimeric ERCC1R156W-XPF protein on a plasmid containing an N-(deoxyguanosin-8-yl)-2-acetylaminofluorene (dG-AAF) adduct together with an ERCC1-XPF–deficient cell extract containing all other NER proteins (Fig. 6, H and I). ERCC1R156W-XPF retained significant NER activity, although at a significantly reduced level compared with ERCC1WT-XPF. This in vitro activity was substantially higher than that of ERCC1N110A/Y145A-XPF (Fig. 6, H and I), which was shown to be fully impaired in its interaction with XPA through mutations in the XPA-binding pocket of ERCC1 (Orelli et al., 2010).
Our findings in reconstituted ERCC1-KO cells suggest that ERCC1R156W still supports residual (∼40%) repair activity when expressed at similar levels as ERCC1WT. To attempt to measure residual repair in the PV46LD and PV50LD primary fibroblasts, which showed severely reduced expression of the ERCC1R156W mutant protein, we performed UDS experiments in which we allowed cells to incorporate EdU for extended periods of time (4 h) to capture residual repair. While UDS was still below ∼10% in XPA-deficient XP1PD cells, in PV46LD and PV50LD cells UDS increased from ∼10% to ∼20% UDS under these conditions (Fig. 6 J and Fig. S5 A). These findings show that the ERCC1R156W mutant protein, expressed at low levels in patient cells, does support residual NER activity, likely explaining the mild XP-like clinical features seen in both siblings. Under these conditions, we also detected residual UDS in previously described ERCC1-deficient 165TOR (Jaspers et al., 2007) and CS20LO (Kashiyama et al., 2013) cells at even higher levels than detected in PV46LD and PV50LD cells (Fig. 6 J and Fig. S5 A).

Microscopy images of NER assays. (A) Representative microscopy images of UDS with 4-h EdU labeling in the indicated cell lines. (B) Microscopy images of U2OS ERCC1WT-GFP after BrdU sensitization, followed by local UV-A laser irradiation. Cells are stained for DAPI and FANCD2. (C) Microscopy images of U2OS ERCC1WT-GFP after trioxsalen sensitization followed by local UV-A laser irradiation. Cells are stained for DAPI and XRCC4. (D) Clonogenic IR survival of RPE1-hTERT WT, ERCC1-KO, and R156W-KI cells (n = 2). (E) Microscopy images of γH2AX foci in the indicated cell lines. 48BR was treated with 2 µM DNA-PK inhibitor (NU7441) for 24 h. A selection of time points of the microscopic images is shown in Fig. 8 E. Scale bar is 5 µm. (F) Location of select patient mutations in ERCC1-XPF. Structures of XPF (HLH: white; nuclease domain: blue; HhH domain: light blue), ERCC1 (central domain: beige; HhH domain: yellow), and XPA67–80 (magenta). The location of patient mutations mentioned in the discussion are shown as spheres: XPFR799W (red: located at the interface of the XPF-nuclease and ERCC1 central domains); ERCC1F231L (orange: located at the interface of the XPF and ERCC1 HhH domains); and ERCC1R156W (atom color: mutation disrupting the ERCC1 central domain). The figure was made using the Chimera extensible molecular modeling system located at University of California San Francisco (http://www.cgl.ucsf.edu/chimera) using an overlay of coordinates 6SXA (ERCC1-XPF) and 2JNW (ERCC1-XPA). CPD, cyclobutane pyrimidine dimer. DNA-PKi, DNA-PK inhibitor.
Microscopy images of NER assays. (A) Representative microscopy images of UDS with 4-h EdU labeling in the indicated cell lines. (B) Microscopy images of U2OS ERCC1WT-GFP after BrdU sensitization, followed by local UV-A laser irradiation. Cells are stained for DAPI and FANCD2. (C) Microscopy images of U2OS ERCC1WT-GFP after trioxsalen sensitization followed by local UV-A laser irradiation. Cells are stained for DAPI and XRCC4. (D) Clonogenic IR survival of RPE1-hTERT WT, ERCC1-KO, and R156W-KI cells (n = 2). (E) Microscopy images of γH2AX foci in the indicated cell lines. 48BR was treated with 2 µM DNA-PK inhibitor (NU7441) for 24 h. A selection of time points of the microscopic images is shown in Fig. 8 E. Scale bar is 5 µm. (F) Location of select patient mutations in ERCC1-XPF. Structures of XPF (HLH: white; nuclease domain: blue; HhH domain: light blue), ERCC1 (central domain: beige; HhH domain: yellow), and XPA67–80 (magenta). The location of patient mutations mentioned in the discussion are shown as spheres: XPFR799W (red: located at the interface of the XPF-nuclease and ERCC1 central domains); ERCC1F231L (orange: located at the interface of the XPF and ERCC1 HhH domains); and ERCC1R156W (atom color: mutation disrupting the ERCC1 central domain). The figure was made using the Chimera extensible molecular modeling system located at University of California San Francisco (http://www.cgl.ucsf.edu/chimera) using an overlay of coordinates 6SXA (ERCC1-XPF) and 2JNW (ERCC1-XPA). CPD, cyclobutane pyrimidine dimer. DNA-PKi, DNA-PK inhibitor.
The impact of ERCC1R156W in ICL repair
During ICL repair, the SLX4 scaffold protein recruits ERCC1-XPF to perform the unhooking incisions that enable subsequent repair. Our MS analysis revealed that ERCC1R156W still interacted with SLX4, albeit at reduced levels compared with ERCC1WT (Fig. 6, A and B). Co-IP experiments indeed confirmed a strong interaction of ERCC1WT with SLX4, which was not affected by UV irradiation, while the interaction with ERCC1R156W was strongly reduced (Fig. 7 A). To address if this reduced interaction affected the recruitment of ERCC1 to ICLs, we locally irradiated cells with a UV-A laser (365 nm) in the presence of trioxsalen, which is a psoralen derivative that forms ICLs upon UV irradiation (Velimezi et al., 2018). Local irradiation with the UV-A laser triggered the recruitment of endogenous FANCD2 only in cells sensitized with trioxsalen, demonstrating that ICLs were induced under our conditions (Fig. S5 B). We could detect strong recruitment of GFP-ERCC1WT-GFP to local ICLs, while the recruitment of ERCC1R156W was much weaker (Fig. 7, B and C), suggesting that the decreased interaction with SLX4 also lowers the efficiency with which ERCC1R156W is recruited to ICLs. However, clonogenic survival assays after exposure to the ICL-inducing agent mitomycin C (MMC) showed that ERCC1R156W rescued the hypersensitivity of ERCC1-KO cells to MMC, although slightly less than ERCC1WT (Fig. 7 D). These experiments show that the ERCC1R156W mutant protein supports ICL repair close to WT levels when the mutant protein is expressed near normal levels.
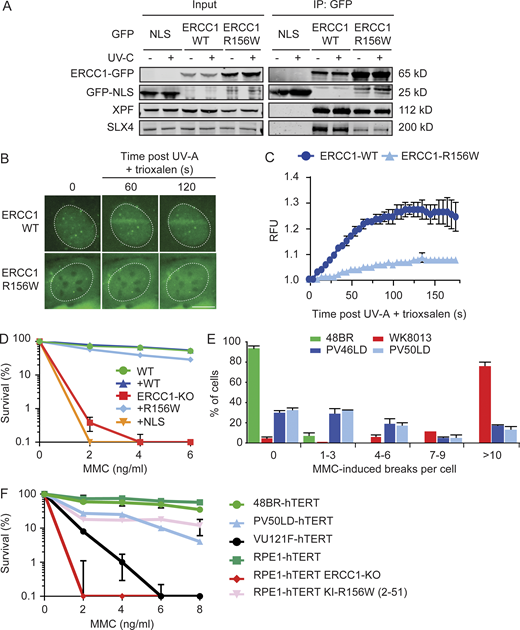
ERCC1R156W has a mild impact on ICL repair.(A) Co-IP of U2OS GFP-NLS, ERCC1WT-GFP, and ERCC1R156W-GFP in the absence or presence of UV-C. (B and C) Microscopy images (B) and quantification (C) of the recruitment of ERCC1WT-GFP and ERCC1R156W-GFP to sites of local UV-A laser irradiation in the presence of trioxsalen (n = 2). See Fig. S5 B for additional controls. (D) Clonogenic MMC survival of U2OS ERCC1-KO cells complemented with either ERCC1WT-GFP or ERCC1R156W-GFP (n = 2). (E) MMC-induced chromosome breakage assay of the indicated cell lines. WK8013 is a cell line derived from an FA patient (n = 2). (F) Clonogenic MMC survival of the indicated cell lines. VU121-F is a cell line derived from an FA patient (n = 2–4). Data represent mean ± SEM. Scale bar is 5 µm. The number of cells used for quantification is shown in Table S6. Uncropped Western blot data are shown in Data S1. RFU, relative fluorescence unit.
ERCC1R156W has a mild impact on ICL repair.(A) Co-IP of U2OS GFP-NLS, ERCC1WT-GFP, and ERCC1R156W-GFP in the absence or presence of UV-C. (B and C) Microscopy images (B) and quantification (C) of the recruitment of ERCC1WT-GFP and ERCC1R156W-GFP to sites of local UV-A laser irradiation in the presence of trioxsalen (n = 2). See Fig. S5 B for additional controls. (D) Clonogenic MMC survival of U2OS ERCC1-KO cells complemented with either ERCC1WT-GFP or ERCC1R156W-GFP (n = 2). (E) MMC-induced chromosome breakage assay of the indicated cell lines. WK8013 is a cell line derived from an FA patient (n = 2). (F) Clonogenic MMC survival of the indicated cell lines. VU121-F is a cell line derived from an FA patient (n = 2–4). Data represent mean ± SEM. Scale bar is 5 µm. The number of cells used for quantification is shown in Table S6. Uncropped Western blot data are shown in Data S1. RFU, relative fluorescence unit.
To address whether PV46LD and PV50LD patient fibroblasts, which showed substantially reduced expression of ERCC1R156W, still supported efficient ICL repair, we performed an MMC-induced chromosome breakage assay. This method measures MMC-induced chromosome breaks in metaphase cells caused by a deficiency in ICL repair. While 48BR (WT) cells accumulated very few breaks after MMC, we measured significant MMC-induced chromosome breakage in Fanconi patient–derived WK8103 cells (Poll et al., 1984; Fig. 7 E). Both PV46LD and PV50LD showed an intermediate phenotype with increased break formation in response to MMC, although not to the same extent as Fanconi cells (Fig. 7 E). In line with this, PV50LD-hTERT cells and RPE1-hTERT R156W-KI cells were sensitive to MMC in clonogenic survival assays, while FANCF-deficient VU121F-hTERT cells (Joenje et al., 1997) and especially ERCC1-KO cells were hypersensitive to MMC (Fig. 7 F). In conclusion, the lower expression of ERCC1R156W and the decreased interaction with SLX4 has a considerable impact on ICL repair, although patient cells are not nearly as sensitive as Fanconi or full ERCC1-KO cells. This likely explains why no FA-like features are apparent in the two siblings.
The impact of ERCC1R156W in DSB repair
Given the role of ERCC1-XPF in DSB repair, we also addressed to what extent ERCC1R156W still supports this repair process. To this end, we performed live-cell imaging after UV-A laser irradiation in cells sensitized with BrdU to generate local DSBs (Lukas et al., 2003). Recruitment of endogenous XRCC4 could be detected at sites of local UV-A laser irradiation only after BrdU sensitization (Fig. S5 C), showing that DSBs were induced under our conditions. Recruitment of ERCC1WT-GFP to sites of DSBs was clearly detected, whereas this was much weaker for ERCC1R156W (Fig. 8, A and B). However, clonogenic survival assays after ionizing radiation (IR) showed no difference between ERCC1-KO cells reconstituted with either ERCC1WT or ERCC1R156W (Fig. 8 C), suggesting that the mutant ERCC1 protein supports DSB repair.
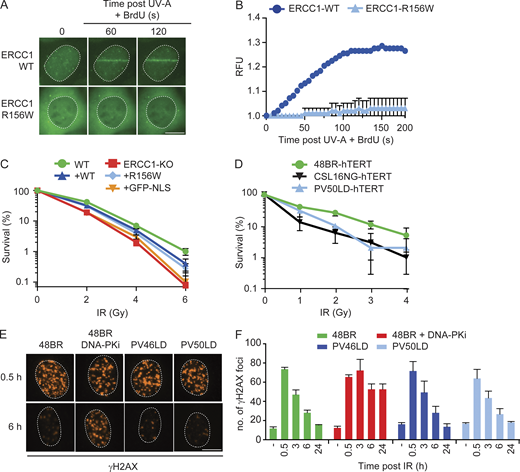
ERCC1R156W has no impact on DSB repair.(A and B) Microscopy images (A) and quantification (B) of the recruitment of ERCC1WT-GFP and ERCC1R156W-GFP to local UV-A laser irradiation after BrdU sensitization (n = 2). See Fig. S5 C for additional controls. (C) Clonogenic IR survival of U2OS ERCC1-KO cells complemented with either ERCC1WT-GFP or ERCC1R156W-GFP (n = 3–6). (D) Clonogenic IR survival of the indicated cell lines. CSL16NG is an XRCC4-deficient patient. See Fig. S5 D for additional IR survivals in RPE1 cells (n = 2). (E and F) Microscopy images (E) and quantification (F) of γH2AX foci in the indicated cell lines. 48BR was treated with 2 µM DNA-PK inhibitor (DNA-PKi; NU7441) for 24 h. Microscopy images of all time points are shown in Fig. S5 E (n = 2). Data represent mean ± SEM. Scale bar is 5 µm. The number of cells used for quantification is shown in Table S6. RFU, relative fluorescence unit.
ERCC1R156W has no impact on DSB repair.(A and B) Microscopy images (A) and quantification (B) of the recruitment of ERCC1WT-GFP and ERCC1R156W-GFP to local UV-A laser irradiation after BrdU sensitization (n = 2). See Fig. S5 C for additional controls. (C) Clonogenic IR survival of U2OS ERCC1-KO cells complemented with either ERCC1WT-GFP or ERCC1R156W-GFP (n = 3–6). (D) Clonogenic IR survival of the indicated cell lines. CSL16NG is an XRCC4-deficient patient. See Fig. S5 D for additional IR survivals in RPE1 cells (n = 2). (E and F) Microscopy images (E) and quantification (F) of γH2AX foci in the indicated cell lines. 48BR was treated with 2 µM DNA-PK inhibitor (DNA-PKi; NU7441) for 24 h. Microscopy images of all time points are shown in Fig. S5 E (n = 2). Data represent mean ± SEM. Scale bar is 5 µm. The number of cells used for quantification is shown in Table S6. RFU, relative fluorescence unit.
To also address this in the patient cells, we performed clonogenic survival assays in hTERT-immortalized fibroblasts after exposure to increasing doses of IR. Fibroblasts deficient in XRCC4 (CS16NG-hTERT; Guo et al., 2015) were clearly sensitive to IR already at the lowest dose (2 Gy). However, PV50LD-hTERT cells were not very sensitive at 2 Gy and only showed an increasing sensitivity at higher doses (Fig. 8 D). In fact, full ERCC1-KO cells and R156W-KI cells only displayed marginal sensitivity to IR (Fig. S5 D), suggesting a minor contribution of ERCC1-XPF to protect cells against IR. Another method to assess DSB repair capacity is monitoring the resolution of IR-induced γH2AX foci in time. Both 48BR and patient fibroblasts showed a normal clearance of γH2AX foci within 24 h after irradiation with a physiological dose of 2 Gy (Fig. 8, E and F; and Fig. S5 E). However, treatment of 48BR cells with DNA-dependent protein kinase (DNA-PK) inhibitor fully suppressed γH2AX foci resolution at all time points analyzed (Fig. 8, E and F). These findings suggest that PV46LD and PV50LD cells are fully proficient in DSB repair under low damage load.
Discussion
A new case of bi-allelic ERCC1 mutations
Only two individuals with bi-allelic ERCC1 mutations have been reported to date, both of whom displayed CS-like features. The most severely affected individual (165TOR) had a nonsense variant (Q158X) and an F231L missense variant in the (HhH)2 motif of ERCC1 (Fig. S5 F), displayed growth retardation, developmental failure, and contractures, and died after the first year of life due to pneumonia (Jaspers et al., 2007). The second affected individual (CS20LO) had contractures, microcephaly, and hypertonia and was homozygous for the F231L missense variant and died in the second year of life (Kashiyama et al., 2013).
We report two siblings with bi-allelic ERCC1 mutations—a paternally inherited missense variant (p.R156W; c.466C>T) and a null-allele due to a maternally inherited intragenic deletion. All the remaining ERCC1 protein expressed in cells from these patients carries the R156W amino acid substitution, which disrupts a salt bridge between the positively charged R156 residue and the opposing negatively charged D129 residue located just below the XPA-binding pocket in the central domain of ERCC1. The impact of this substitution is twofold: (1) it strongly diminishes the overall stability of ERCC1 and its binding partner XPF, and (2) it specifically affects the interaction with XPA (Fig. 9).

Model for the dual impact of the ERCC1 missense variant. Left: healthy individuals have a stable pool of ERCC1-XPF heterodimer available for DNA repair. From this pool, there is robust recruitment of ERCC1WT during NER and ICL repair by XPA and SLX4, respectively. Right: cells from ERCC1 patients show strongly reduced levels of ERCC1-XPF heterodimer due to a destabilizing missense variant. In addition, the ERCC1R156W mutant protein that is still present in lower amounts has decreased ability to interact with XPA and SLX4, resulting in weak recruitment during NER and ICL repair. This leads to a strong NER defect accompanied by a milder ICL repair defect. We propose that this combination, possibly together with an impact in another ERCC1-dependent DNA repair pathway not studied by us, results in a unique phenotype combining short stature, photosensitivity, and progressive liver and kidney dysfunction.
Model for the dual impact of the ERCC1 missense variant. Left: healthy individuals have a stable pool of ERCC1-XPF heterodimer available for DNA repair. From this pool, there is robust recruitment of ERCC1WT during NER and ICL repair by XPA and SLX4, respectively. Right: cells from ERCC1 patients show strongly reduced levels of ERCC1-XPF heterodimer due to a destabilizing missense variant. In addition, the ERCC1R156W mutant protein that is still present in lower amounts has decreased ability to interact with XPA and SLX4, resulting in weak recruitment during NER and ICL repair. This leads to a strong NER defect accompanied by a milder ICL repair defect. We propose that this combination, possibly together with an impact in another ERCC1-dependent DNA repair pathway not studied by us, results in a unique phenotype combining short stature, photosensitivity, and progressive liver and kidney dysfunction.
Reduced nuclear protein levels of ERCC1 due to partial misfolding
Western blot and immunofluorescence analyses revealed that the overall ERCC1 and XPF protein levels are dramatically reduced to below 20% of WT levels in the fibroblasts from both siblings. This effect is also recapitulated by the bi-allelic KI of the missense mutation (p.R156W; c.466C>T) in the endogenous ERCC1 locus of RPE1-hTERT cells. Interestingly, complete loss of ERCC1 in mice led to death within 4 wk, while increasing protein levels to around 15% of the levels in WT mice increased the life span fivefold (Weeda et al., 1997), suggesting that even low levels of ERCC1 considerably increase the potential for viability. Consistent with our findings, previous studies have shown that missense mutations in the central domain of ERCC1 generally cause destabilization (Sijbers et al., 1996b), indicating that this region is important for protein stability.
The recently reported cryogenic electron microscopy structure of the full-length ERCC1-XPF heterodimer (Jones et al., 2020) reveals that the ERCC1R156 residue is located on the very edge of the heterodimer. We speculate that the increased bulkiness and altered electronic structure of the substitution of an arginine by tryptophan not only disturbs the XPA-binding pocket but also the dimerization interface between the central domain of ERCC1 and the nuclease domain of XPF, causing general destabilization (Fig. S5 F). Of interest, the missense variant XPFR799W is located at the same interface on the XPF side (Fig. S5 F) and also results in destabilization and severely reduced XPF protein levels (Mori et al., 2018). In one affected individual (CALIF1010) who presented with CS-like and segmental progeroid features, the XPFR799W variant was inherited together with an intergenic deletion in the other XPF allele, resulting in a strong NER defect combined with a mild ICL repair defect (Mori et al., 2018), similar to the siblings reported in this study.
Ectopic expression of an inducible version of ERCC1R156W for 24 h enabled us to reach WT levels of this mutant protein, which resulted in mislocalization in the cytoplasm, a reduced interaction with XPF, and an increased interaction with protein-folding chaperones as detected by MS. These findings all lend support to the notion that ERCC1R156W is partially misfolded and degraded in patient cells, resulting in an ∼80% reduction in ERCC1 nuclear protein levels. KI of ERCC1R156W in RPE1-hTERT cells supports this conclusion.
When comparing the ERCC1 and XPF protein levels in PV46LD and PV50LD cells with those in cells from more severely affected individuals (165TOR and CS20LO), we noted that these levels were comparable, or perhaps even lower, in cells from the siblings described in this study (Fig. 3 C and Fig. S3, E and F; Jaspers et al., 2007; Kashiyama et al., 2013). Note that CS20LO is homozygous for ERCC1F231L, while 165TOR is heterozygous and carries an additional premature stop on the other ERCC1 allele (Q158X). These findings suggest that in addition to reduced protein levels, the functionality of the mutant protein that is still expressed and the combination of the two mutated alleles need to be taken into account as a potential modulator of phenotypic severity and expressivity.
ERCC1R156W: Differential impact on ERCC1-dependent DNA repair pathways
Expression of ERCC1R156W-GFP in ERCC1-KO cells at levels similar to ERCC1WT enabled us to disentangle the impact of the amino acid substitution on protein stability from the impact on protein functionality. While the mutant ERCC1R156W protein was severely impaired in its UV-induced interaction with XPA and TFIIH and failed to efficiently localize to sites of UV-induced DNA lesions, we still detected considerable (∼40%) repair activity in UDS assays, clonogenic survivals, and in vitro NER assays. These findings suggest that a mutant ERCC1 protein that only interacts very weakly with the NER complex can still support considerable levels of NER (∼40%) activity inside cells. Together, these findings suggest that the low expression level of ERCC1 (∼20%) combined with the residual GGR activity of the mutant proteins that is still expressed provides sufficient protection to prevent PV46LD and PV50LD from developing full-blown XP. Indeed, residual GGR activity was still detected under more sensitive conditions in UDS assays when compared with fibroblasts from a severely affected and cancer-prone XP-A patient. Nonetheless, both patients could still be at risk for developing skin cancer, and protection from sunlight is therefore advised. The residual activity in PV46LD and PV50LD cells was either similar to or lower than the repair activity we detected in 165TOR or CS20LO cells (Fig. 6 J; Jaspers et al., 2007; Kashiyama et al., 2013). The clinical severity therefore does not correlate with NER activity but is likely due to a role of ERCC1 outside NER that is important during development.
Despite our findings that ERCC1R156W showed a reduced interaction with SLX4 and was also recruited less efficiently to sites of local ICL induction, further analysis revealed that the ERCC1R156W mutant protein was only mildly affected in supporting ICL repair when ectopically expressed in ERCC1-KO cells. However, the impact on ICL repair was much milder than the impact of the R156W substitution on NER, which, combined with earlier findings that the repair of ICLs requires much less ERCC1 protein than the repair of NER-specific DNA lesions (Jaspers et al., 2007; Sijbers et al., 1996b), may very well explain the mild impact on ICL repair. In patient fibroblasts, which have strongly reduced levels of ERCC1, we did detect moderate sensitivity, as well as increased chromosome breakage upon exposure to the ICL-inducing compound MMC, albeit much milder than cells from an FA patient that were included in parallel. Importantly, neither PV46LD nor PV50LD have displayed any overt signs of FA-like clinical features, suggesting that the ERCC1R156W mutant protein provides sufficient protection against the low endogenous ICL load in these patients.
ERCC1 deficiency causes liver and kidney impairment
Mice that are KO for either ERCC1 or XPF show severe runting (i.e., smaller and weaker than WT mice) and die before the first 4 wk of life due to severe liver failure (McWhir et al., 1993; Sijbers et al., 1996b; Tian et al., 2004; Weeda et al., 1997). Liver-specific expression of ERCC1 in these mice partially corrected the smaller size and extended life span up to 12 wk but also unmasked severe kidney dysfunction and renal failure as a secondary cause of death (Selfridge et al., 2001). KO mice specific for XP or FA do not display liver disease, suggesting that this phenotype is not caused by a NER or ICL repair defect. It is possible that partial redundancy between these two pathways, which is lost in ERCC1-KO mice deficient in both pathways, could explain this phenotype. In line with this idea, hepatocyte and kidney-proximal tubule cells became polyploid in ERCC1-KO mice and accumulated high levels of p53 (McWhir et al., 1993; Selfridge et al., 2001; Weeda et al., 1997), suggesting that accumulation of endogenous DNA damage in these organs may occur. The precise nature of this endogenous DNA lesion, however, remains unclear.
Interestingly, mice with joint inactivation of both NER and ICL repair do not display liver dysfunction, suggesting that redundancy between these DNA repair pathways is not the sole explanation and that an additional function of ERCC1-XPF outside these DNA repair pathways contributes to the liver impairment (Mulderrig and Garaycoechea, 2020). It is possible that ERCC1-XPF is involved in additional DNA repair pathways that deal with endogenous DNA damage. For instance, ERCC1-XPF was recently shown to act upon alternative DNA structures, such as Z-DNA, during which it cooperates with mismatch repair proteins rather than NER or ICL repair factors (McKinney et al., 2020). In addition, ERCC1-XPF was shown to be involved in a sub-pathway of base excision repair together with the RECQ1 helicase (Woodrick et al., 2017).
The two siblings (PV46LD and PV50LD) in this study exhibited a failure to thrive, short stature, and a lack of subcutaneous fat, reminiscent of the small size observed in ERCC1-KO mice (McWhir et al., 1993; Selfridge et al., 2001; Weeda et al., 1997). Moreover, the siblings developed liver impairments with a predominantly cholestatic picture that led to orthotopic liver transplantation before the age of 9 yr. This intervention clearly prevented early death but did not improve the failure to thrive, as evident from post-transplant growth of weight, height, and head circumference well below the third centile.
Of interest, cholestatic liver disease was reported as a common feature in Egyptian CS patients, suggesting that this should be monitored more closely in CS patients from other ethnic backgrounds to see if this is a more common feature than previously recognized (Abdel Ghaffar et al., 2011). Although no liver abnormalities have been described in XP patients, there are some cases of hepatic cytolysis (cell breakdown or bursting) in FA patients (Masserot-Lureau et al., 2012), which may be distinct from the liver abnormalities observed in the ERCC1-deficient siblings. Histological examination of a liver biopsy from the older sibling (PV50LD) revealed changes in hepatocyte morphology with nuclear enlargement and variability, as noted in the ERCC1-deficient mice (Fig. S1 D; Núñez et al., 2000).
The second cause of death in ERCC1-KO mice with liver-specific expression of ERCC1 was severe kidney failure (Selfridge et al., 2001). Both siblings also display renal dysfunction, with features suggestive of proximal tubular dysfunction leading to progressive kidney impairment. However, tubular function stabilized following the liver transplant, but kidney function requires ongoing monitoring. The renal phenotype of the ERCC1-KO mice—with dilated tubules containing leaked proteinaceous material consistent with a tubulopathy (McWhir et al., 1993; Selfridge et al., 2001; Weeda et al., 1997)—is similar to that of the patients described in our study. Moreover, renal impairment and, in some instances, failure has been reported in combined XP/CS (Ben Chehida et al., 2017; Kondo et al., 2016; Kralund et al., 2013) as well as CS (Funaki et al., 2006; Reiss et al., 1996; Sato et al., 1988), while an association between FA and structural anomalies of the kidney has also been reported (Sathyanarayana et al., 2018). The phenotype in these siblings is distinct from prior reports of individuals with bi-allelic ERCC1 mutations and also distinct from the known phenotypic entities, CS and XP. Neither individual had features suggestive of FA. There may be parallels between the phenotype of these siblings and a previously reported individual with bi-allelic ERCC1 mutations (XP2020DC; p.K266X; IVS6-26G>A) who died at the age of 37 (Gregg et al., 2011; Imoto, K., et al. 2007. Patients with defects in the interacting nucleotide excision repair proteins ERCC1 or XPF show xeroderma pigmentosum with late onset severe neurological degeneration. [Abstract] J. Invest. Dermatol. 127:S92). Although no liver impairment was reported, this patient developed severe brain atrophy at age 15, which developed into progressive neurodegeneration with dementia. Mild brain atrophy has also been noted in both siblings at age 13 and 11, indicating that the neurological development of both siblings should be monitored carefully.
A previously reported 15-yr-old boy with bi-allelic XPF mutations (XP51RO; p.R153P; c.458G>C; Jaspers et al., 2007; Niedernhofer et al., 2006) presented with a phenotype with some overlap with the siblings reported here, including photosensitivity without skin cancer, short stature, lack of subcutaneous fat, developmental delay, and renal insufficiency (Niedernhofer et al., 2006). Distinct from the siblings reported here, this boy showed pronounced segmental progeroid features, but a much milder liver picture without cholestasis. The siblings reported here demonstrate that bi-allelic ERCC1 mutations can cause a spectrum of phenotypes, from that typically seen in CS through to a phenotype comprising milder short stature, photosensitivity, and severe liver and kidney impairment, potentially because of a combined strong impact on NER and a simultaneous impact on ICL repair, which may particularly affect these organs.
Materials and methods
All procedures performed in this study are in accordance with the ethical standards of and were approved by the Human Ethics Committee of the Royal Children’s Hospital, Melbourne, Victoria, Australia (HREC36291C) and the UK National Research Ethics Service Committee North East–Newcastle and North Tyneside 2. Patients were recruited into Undiagnosed Diseases Programs for pediatric patients with presumed “orphan” Mendelian disorders. Saliva or blood samples from the patients and their family members were collected for genomic DNA extraction after written informed consent was given. The parents have granted permission to show unredacted photographs of the affected siblings in Fig. 1 A.
Next-generation sequencing (NGS)
DNA extracted from blood of both affected siblings and unaffected parents was subjected to exome capture and sequencing through Oxford Gene Technologies Ltd. Genomic data were analyzed using a custom-made in-house pipeline using an autosomal recessive disease model. A paternally inherited missense variant in NM_202001.2 (ERCC1), g.45922415G>A, c.466C>T was identified in both affected individuals in exon 4 of 8 of the ERCC1 gene. On initial interrogation of the exome data in both affected individuals, this variant appeared to be in a homozygous state but was present in a heterozygous state in the father and absent in the mother, suggesting a possible deletion on the maternal allele. The missense variant is present in the gnomAD database at a frequency of 0.01% (22 heterozygotes, no homozygotes) and has not been previously reported in affected individuals. The missense variant has been deposited in the LOVD database (http://www.LOVD.nl/ERCC1_000020 and http://www.LOVD.nl/ERCC1_000021) and the ClinVar database (variation ID: 978472).
Detection of the deletion by qPCR
To detect the deletion, genomic DNA was extracted from lymphocytes/epithelial cells, and the relevant region of the ERCC1 gene was amplified in a qPCR. qPCR was performed using probes and specific primers (listed in Table S1) designed using the Universal ProbeLibrary Assay Design Center (Roche) and synthesized by Sigma-Aldrich. The primers were designed to include the deleted region in exon 4 and a region within the gene that is not deleted as control (exon 5). A multiplex qPCR using the LightCycler R 480 Probes Master reaction mix (Roche) was performed according to the manufacturer’s protocol to amplify and quantify the ERCC1 gene region, using the CFTR gene (exon 27) as an internal standard (Table S1). The qPCR was performed on the LightCycler R 480 instrument (Roche), and data analysis was performed using the LightCycler R 480 software version 1.5.0 (Roche). American College of Medical Genetics guidelines were followed for interpretation of sequence variation (http://www.acmg.net).
Detection of the missense variant by Sanger sequencing
Genomic DNA was isolated by resuspending cell pellets in whole-cell lysate buffer (50 mM KCL, 10 mM Tris, pH 8.0, 25 mM MgCl2, 0.1 mg/ml gelatin, 0.45% Tween-20, and 0.45% NP-40) containing 0.1 mg/ml Proteinase K (EO0491;Thermo Fisher Scientific) and incubating for 1 h at 56°C followed by a 10-min heat inactivation of Proteinase K at 96°C. Fragments of ∼1 kb spanning the missense mutations were PCR amplified (sequencing primers are listed in Table S1) followed by Sanger sequencing using either the forward or the reverse primer.
Cell lines
Fibroblast cell lines were established from skin biopsies from both affected individuals and parents. All cell lines (listed in Table S2) were cultured at 37°C in an atmosphere of 5% CO2 in DMEM (Thermo Fisher Scientific) supplemented with penicillin/streptomycin (Sigma-Aldrich) and 10% FBS (Bodinco BV). All cell lines were routinely tested for mycoplasma infection.
Fibroblast immortalization with hTERT
The WT (48BR) and PV50LD fibroblasts were immortalized by nucleofection of pBabe-Neo-TERT (Addgene Plasmid #1774) using the Amaxa Nucleofector (program U23). Each electroporation contained 400,000 fibroblasts and 2.5 µg plasmid DNA. After electroporation, cells were seeded in 25-cm2 tissue culture flasks containing 5 ml DMEM and 10% FBS. After 2 d, culture medium was replaced with medium containing 20 µg/ml neomycin.
Plasmid constructs
All plasmids used in this study are listed in Table S3. The GFP gene in pERCC1-GFP-N1 (Houtsmuller et al., 1999) was replaced with mVenus (a gift of Joachim Goedhart, Amsterdam, Netherlands) using AgeI and BsrGI. The GFP-NLS gene (Luijsterburg et al., 2017) was inserted into pcDNA5-FRT-TO-Puro. The ERCC1-mVenus cassette was amplified by PCR (see Table S1 for primers) and inserted as an NheI and BspEI fragment into pLenti-CW57-TO-GFP digested with NheI and AgeI to generate pLenti-CW57-TO-ERCC1WT-mVenus. Overlap PCR was used to introduce the c.466C>T mutations in ERCC1. The overlap PCR product was inserted as an NheI and AgeI fragment to generate pLenti-CW57-TO-ERCC1R156W-mVenus. ERCC1WT and ERCC1R156W were inserted as NheI and AgeI fragments into pcDNA5-FRT-TO-Puro-EGFP-N1 to generate pcDNA5-FRT-TO-Puro-ERCC1WT-EGFP and pcDNA5-FRT-TO-Puro-ERCC1R156W-EGFP. Site-directed mutagenesis was used to introduce point mutations in pMacroBac-XPF-ERCC1WT using the KOD-plus mutagenesis kit (Toyobo) as described in the manufacturer’s protocol, using the primers listed in Table S1. All sequences were verified by Sanger sequencing.
Generation of KO cell lines
To generate single KOs, U2OS(FRT) and RPE1-hTERT (PuroR/TP53-dKO) cells were cotransfected with pLV-U6g-PPB encoding a guide RNA from the Leiden University Medical Center/Sigma-Aldrich single-guide RNA (sgRNA) library (see Table S3 for plasmids and Table S4 for sgRNA sequences) targeting the ERCC1 gene together with an expression vector encoding Cas9-2A-GFP (pX458; Addgene #48138) using Lipofectamine 2000 (Invitrogen). Transfected U2OS(FRT) cells were selected on puromycin (1 µg/ml) for 3 d and plated at low density, after which individual clones were isolated. Transfected RPE1-hTERT cells were FACS sorted on BFP/GFP and plated at low density, after which individual clones were isolated. KO clones were verified by Western blot analysis. The absence of Cas9 integration/stable expression was confirmed by Western blot.
Generation of stable cell lines
A single ERCC1 KO clone in the U2OS(FRT) background (see Table S2) was used to stably express ERCC1WT-GFP, ERCC1R156W-GFP, or GFP-NLS by cotransfection of pcDNA5-FRT-TO-Puro plasmid encoding these cDNAs (2 µg), together with pOG44 plasmid encoding the Flp recombinase (0.5 µg). A polyclonal cell population was obtained after selection on 1 µg/ml puromycin and 4 µg/ml blasticidin S. Expression of the GFP-tagged ERCC1 proteins or GFP-NLS was induced by the addition of 2 µg/ml doxycycline for 24 h.
Generation of KI cell lines
To generate homozygous KIs, RPE1-hTERT cells were first treated for 30 min with 1 µM DNA-PK inhibitor (NU7441) to suppress error-prone DSB repair and increase the use of homology-dependent repair. Subsequently, 350,000 cells were resuspended in 20 µl nucleofector buffer (V4XP-3032; Lonza) in the presence of 4.5 µg plasmid containing Cas9 and sgRNA targeting the ERCC1 gene (see Table S3 for plasmids and Table S4 for sgRNA sequences) and 100 µM single-stranded donor DNA containing the patient mutation as well as silent mutations to destroy the PAM site to prevent recutting by Cas9 and silent mutations to introduce a HindIII restriction site to facilitate screening of KI clones. Cells were electroporated using an Amaxa 4D-X Nucleofector Unit (Lonza) using the EA-104 program. Following electroporation, cells were plated at low density in McCoy medium with 10% FBS. The next day, the medium was replaced with DMEM (10% FBS and 1% penicillin/streptomycin), and cells were allowed to form colonies. Approximately 400 colonies were isolated and expanded. Genomic DNA was isolated, and a fragment including the introduced mutation and the introduced restriction site (HindIII) was amplified by nested PCR (see Table S1 for primers). PCR fragments were digested with HindIII (NEB) for 3 h at 37°C and then separated by gel electrophoresis. The clones that had the introduced HindIII restriction site were analyzed by Sanger sequencing (see Table S1 for primers). We obtained two homozygous KI clones among 400 isolated clones, which corresponds to a KI efficiency of ∼0.5%.
Lentiviral transduction
For lentiviral transduction, mVenus-ERCC1WT or mVenus-ERCC1R156W was inserted into lentiviral vector pLenti-CW57-TO. HEK293T were transfected with vectors encoding these ERCC1 fusions, VSV-G, RRE, and REV using JetPEI (Sigma-Aldrich) to produce virus. The virus-containing supernatant was collected after 24 h and filtered with a 0.44-µm filter. Primary fibroblasts PV46LD and PV50LD were lentivirally transduced in the presence of polybrene (Sigma-Aldrich). Cells were selected with 15 µg/ml puromycin. Expression of the mVenus-tagged ERCC1 proteins was induced by the addition of 2 µg/ml doxycycline for 24 h.
Clonogenic survival assays
Cells were trypsinized, seeded at low density, and allowed to attach. The next day, cells were either mock treated or exposed to an increasing dose of UV light (1, 2, 3, and 4 J/m2 of UV-C 266 nm), an increasing dose of IR (2, 4, 6, and 8 Gy), or increasing concentrations of MMC (2, 4, 6, and 8 ng/ml). After 7–9 d, the cells were washed with 0.9% NaCl and stained with methylene blue. Colonies of >20 cells were scored. Survival experiments were performed in duplicate and repeated at least twice.
Immunoprecipitation for Co-IP
Cells were mock treated or UV-C irradiated (20 J/m2) and harvested after 1 h. Cell pellets were lysed for 20 min on ice in EBC-150 buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 0.5% NP-40, 2 mM MgCl2, and protease inhibitor cocktail [Roche]) supplemented with 500 U/ml Benzonase Nuclease (Novagen). Cell lysates were incubated for 1.5 h at 4°C with GFP-Trap A beads (Chromotek). The beads were then washed six times with EBC-150 buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 0.5% NP-40, 1 mM EDTA, and protease inhibitor cocktail) and boiled in Laemmli-SDS sample buffer.
Western blot
Cells were spun down, washed with PBS, and boiled for 10 min in Laemmli buffer (40 mM Tris, pH 6.8, 3.35% SDS, 16.5% glycerol, 0.0005% Bromophenol Blue, and 0.05 M dithiothreitol). Proteins were separated on 4%–12% Criterion XT Bis-Tris gels (#3450124; Bio-Rad) in NuPAGE MOPSrunning buffer (NP0001-02; Thermo Fisher Scientific) and blotted onto polyvinylidene fluoride membranes (IPFL00010; EMD Millipore). The membrane was blocked with blocking buffer (MB-070-003; Rockland) for 1 h at room temperature. The membrane was then probed with antibodies (listed in Table S5) as indicated. An Odyssey CLx system (LI-COR Biosciences) was used for detection.
RNA recovery assay (RRS)
30,000 cells were seeded on 12-mm glass coverslips in 24-wells plates in DMEM with 1% FBS. After 24 h, cells were irradiated with UV-C at a dose of 6 J/m2 and incubated in conditioned medium for different time periods (0 h, 3 h, and 18 h). Following incubation, nascent transcripts were labeled by incubating the cells with 400 µM 5-ethynyl-uridine (CLK-N002-10; Jena Bioscience), which was then visualized with a Click-iT mix consisting of 50 mM Tris buffer, pH 8.0, 60 µM Atto Azide (647N-101; ATTO-TEC), 4 mM CuSO4•5H2O, 10 mM L-ascorbic acid (A0278; Sigma-Aldrich), and 0.1 µg/ml DAPI (D1306; Thermo Fisher Scientific) for 1 h. Cells were washed three times for 5 min with PBS and mounted on microscope slides (Thermo Fisher Scientific) using Aqua Polymount (Brunschwig).
Immunofluorescence
For immunofluorescent staining, 250,000 cells were seeded on 18-mm glass coverslips. The next day, cells were fixed with 4% paraformaldehyde, followed by permeabilization with 0.5% Triton X-100 for 10 min. Cells were treated with 100 mM glycine in PBS for 10 min to block unreacted aldehyde groups, rinsed with PBS, and equilibrated in wash buffer (PBS containing 0.5% BSA and 0.05% Tween-20; Sigma-Aldrich) for 10 min. Antibody steps and washes were in wash buffer. The primary antibodies were incubated for 2 h at room temperature, followed by secondary antibodies for 1 h and DAPI for 5 min. Primary and secondary antibodies are listed in Table S5. Cells were incubated with 0.1 µg/ml DAPI and mounted using Aqua Polymount.
γH2AX staining
200,000 cells were seeded on 18-mm glass coverslips in 24-well plates with DMEM (10% FBS and 1% P/S). The next day, the cells were exposed to IR by the YXlon International x-ray generator (200 KV, 4 mA; dose rate 2 Gy/min). At the indicated time points, cells were fixed with 3% paraformaldehyde in PBS for 10 min and stained for γH2AX for 1 h (see above). Images were quantified using a custom-built macro in ImageJ that enabled automatic and objective analysis of the number of foci per cell, as described previously (Typas et al., 2015).
Microscopic analysis of fixed cells
Images of fixed samples were acquired on a Zeiss AxioImager M2 or D2 wide-field fluorescence microscope equipped with 63× PLAN APO (1.4 numerical aperture [NA]) oil-immersion objectives (Zeiss) and an HXP 120 metal-halide lamp used for excitation. Fluorescent probes were detected using the following filters for DAPI (excitation filter: 350/50 nm; dichroic mirror: 400 nm; emission filter: 460/50 nm), Alexa 555 (excitation filter: 545/25 nm; dichroic mirror: 565 nm; emission filter: 605/70 nm), or Alexa 647 (excitation filter: 640/30 nm; dichroic mirror: 660 nm; emission filter: 690/50 nm). Images were recorded using ZEN 2012 software and analyzed in ImageJ.
UV laser micro-irradiation
Cells were grown on 18-mm quartz (UV-C) or glass (UV-A) coverslips and placed in a Chamlide CMB magnetic chamber in which growth medium was replaced by CO2-independent Leibovitz L-15 medium (Thermo Fisher Scientific). UV-C laser tracks were made using a diode-pumped solid-state 266-nm Yttrium Aluminum Garnet laser (average power 5 mW, repetition rate up to 10 kHz, and pulse length 1 ns). Prior to UV-A micro-irradiation, cells were either sensitized with 6 µM trioxsalen for 1 h to generate ICLs or with 15 µM BrdU for 24 h to generate DSBs. UV-A laser tracks were made by a diode-pumped solid-state 355-nm Yttrium Aluminum Garnet laser (average power 14 mW and repetition rate up to 200 Hz). Both lasers were integrated in a UGA-42-Caliburn/2L Spot Illumination system (Rapp OptoElectronic).
Micro-irradiation was combined with live-cell imaging in an environmental chamber set to 37°C on an all-quartz wide-field fluorescence Zeiss Axio Observer 7 microscope, using a 100× (1.2 NA) ultrafluar glycerol-immersion objective (UV-C) or a Plan-Neofluar 63× (1.25 NA) oil-immersion objective (UV-A). The laser system is coupled to the microscope via a TriggerBox, and a neutral density (ND-1) filter blocks 90% of the laser light. An HXP 120-V metal-halide lamp was used for excitation. Images were acquired in Zeiss ZEN and quantified in ImageJ.
Chromosome breakage assay
2 × 106 fibroblasts were seeded in 175-cm2 tissue culture flasks with DMEM (10% FBS) and cultured at 37°C in the presence or absence of 50 nM MMC. After 48 h, 600 µl of demecolcine (10 µg/µl) was added to each culture flask, and cells were incubated for an additional 30 min at 37°C to enrich for metaphases. Cells were then trypsinized and resuspended in 75 mM KCL and incubated for 20 min at room temperature. The cells were spun down, resuspended in 10 ml fixative (75% methanol and 25% acetic acid), incubated for 30 min at room temperature, and centrifuged. Pellets were resuspended in 10 ml of fixative and incubated for 5 min at room temperature. This step was repeated, and finally the pellet was resuspended in 0.5–1.0 ml fixative. The cell suspension was dropped on a slide and allowed to dry. Slides were stained for 5 min in a 3% Giemsa solution, rinsed in tap water, and allowed to dry. Slides were coded, and from each coded culture 50 metaphases were examined for chromosomal damage. After scoring, the slides were decoded, and the results were analyzed as presented (Stoepker et al., 2011).
UDS
180,000 cells were seeded on 18-mm glass coverslips in 12-well plates in DMEM with 1% FBS. After 24 h, cells were locally irradiated through a 5-µm filter with 30 J/m2 UV-C. Cells were subsequently pulse labeled with 20 µM EdU (VWR) and 1 µM 5-fluoro-deoxyuridine (Sigma-Aldrich) for either 1 h or 4 h. After labeling, cells were medium chased with 10 µM thymidine in DMEM without supplements for 30 min and fixed for 15 min with 3.7% formaldehyde in PBS. Cells were permeabilized for 20 min in PBS with 0.5% Triton-X100 and blocked in 3% BSA (Thermo Fisher Scientific) in PBS. The incorporated EdU was coupled to Attoazide Alexa Fluor 647 using Click-iT chemistry according to the manufacturer’s instructions (Invitrogen). After coupling, the cells were postfixed with 2% formaldehyde for 10 min and subsequently blocked with 100 mM glycine. DNA was denatured with 0.5 M NaOH for 5 min, followed by blocking with 10% BSA for 15 min. Next, the cells were incubated with an antibody against cyclobutane pyrimidine dimers (see Table S5) for 2 h, followed by secondary antibodies for 1 h, and DAPI for 5 min. Cells were mounted in Polymount (Brunschwig).
MS data acquisition
MS was performed essentially as previously described (Salas-Lloret et al., 2019). All the experiments were performed on an EASY-nLC 1000 system (Proxeon) connected to a Q-Exactive Orbitrap (Thermo Fisher Scientific) through a nano-electrospray ion source. The Q-Exactive was coupled to a 25-cm silica emitter (FS360-75-15-N-5-C25; NewObjective) in-house packed with 1.9 µm C18-AQ beads (Reprospher-DE; Pur; Dr. Manish, Ammerbuch-Entringen, Germany). Samples were run in a 40-min chromatography gradient from 0% to 30% acetonitrile and then increased to 95% acetonitrile before column reequilibration with a flow rate of 200 nl/min. The mass spectrometer was operated in a data-dependent acquisition mode with a top-seven method and a scan range of 300–1,600 m/z. Full-scan MS spectra were acquired at a target value of 3 × 106 and a resolution of 70,000, and the higher-collisional dissociation tandem mass spectra (MS/MS) were recorded at a target value of 105 and with a resolution of 35,000, an isolation window of 2.2 m/z, and a normalized collision energy of 25%. The minimum automatic gain control target was 104. The maximum MS1 and MS2 injection times were 250 and 120 ms, respectively. The precursor ion masses of scanned ions were dynamically excluded from MS/MS analysis for 30 s. Ions with charge 1 and higher than 6 were excluded from triggering MS2 analysis.
MS data analysis
All raw data were analyzed using MaxQuant (version 1.6.6.0) as described previously (Tyanova et al., 2016a). We performed the search against an in silico–digested UniProt reference proteome for Homo sapiens, including canonical and isoform sequences (May 27, 2019). Database searches were performed according to standard settings with the following modifications. Digestion with Trypsin/P was used, allowing four missed cleavages. Oxidation (M) and acetyl (protein N-term) were allowed as variable modifications with a maximum number of three. Carbamidomethyl (C) was disabled as a fixed modification. Label-free quantification (LFQ) was enabled, not allowing Fast LFQ. MaxQuant output data were further processed using the Perseus computational platform (v 1.6.6.0; Tyanova et al., 2016b). LFQ intensity values were log2 transformed, and potential contaminants and proteins identified by site only or reverse peptide were removed. Samples were grouped in experimental categories, and proteins not identified in four out of four replicates in at least one group were also removed. Missing values were imputed using normally distributed values with a 1.8 downshift (log2) and a randomized 0.3 width (log2) considering whole matrix values. Statistical analysis (t tests) was performed to determine which proteins were significantly enriched. Volcano plots were generated, and statistical analysis output tables were further processed in Microsoft Excel. The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD017940.
Purification of recombinant ERCC1-XPF
Baculovirus production was performed as described using the pMacroBac-His-ERCC1/XPF-HA constructs (Enzlin and Schärer, 2002). ERCC1WT and ERCC1R156W were coexpressed with XPFWT from single baculovirus in Sf9 insect cells. The heterodimers were purified over nickel affinity, size exclusion, and heparin chromatography as described (Enzlin and Schärer, 2002). ERCC1-XPF was eluted from the heparin column at around 600 mM NaCl. Protein concentrations ranged from 0.1 to 0.2 mg/ml.
Nuclease assay
A stem-loop substrate (GCCAGCGCTCGG(T)22CCGAGCGCTGGC) containing fluorescent dye Cy5 (50 pmol) at 3′ terminus (IDT) was annealed in a 100-µl annealing buffer (10 mM Tris, pH 7.5, and 50 mM NaCl) by heating at 95°C for 5 min and slowly cooling down over a period of 2 h. 200 fmol of annealed substrate was used in 20-µl reactions containing 25 mM Hepes, pH 8.0, 2 mM MgCl2, 10% glycerol, 0.5 mM β-mercaptoethanol, 0.1 mg/ml BSA, 40 mM NaCl, and 0–40 nM of protein. The reactions were incubated at 30°C for 30 min and stopped by adding 10 µl 90% formamide/10 mM EDTA. After heating at 95°C for 5 min and cooling on ice, 15 µl of each sample was loaded on a 15% denaturing polyacrylamide gel. Gels were run at 30 mA for 30 min and bands visualized by fluorescence by Typhoon RGB (Amersham Biosciences).
In vitro NER assay
XPF-deficient (XP2YO) cell extracts and the plasmid containing site-specific dG-AAF lesion were generated as previously described (Gillet et al., 2005; Shivji et al., 1999). For each reaction, 2 µl of repair buffer (200 mM Hepes-KOH, 25 mM MgCl2, 2.5 mM DTT, 10 mM ATP, 110 mM phosphocreatine, and 1.8 mg/ml BSA, final pH 7.8), 0.2 µl of creatine phosphokinase buffer (2.5 mg/ml creatine phosphokinase from rabbit muscle [Sigma-Aldrich], 10 mM glycine, pH 9.0, and 50% glycerol), 3 µl of XPF-deficient cell extract, NaCl (to a final concentration of 70 mM), and purified ERCC1-XPF proteins in a total volume of 9 µl were prewarmed at 30°C for 10 min. 1 µl plasmid containing dG-AAF (50 ng/µl) was added to each reaction, and the reactions were incubated at 30°C for 45 min. The reaction mixture was then cooled on ice for 5 min, followed by the addition of 0.5 µl of 1 µM complementary strand d(GGGGCATGTGGCGCCGGTAATAGCTACGTAGCTC), and the reaction mixture was denatured by heating at 95°C for 5 min. Following 15 min of annealing at room temperature, 1 µl sequenase mix (containing 0.13 U of sequenase and 2.0 µCi [α-32P] dCTP for each reaction) was added. After preincubation at 37°C for 3 min, 1.2 µl deoxyribonucleotide triphosphate mixture (50 µM dCTP, 100 µM dTTP, 100 µM dATP, and 100 µM dGTP) was added. The reaction mixture was incubated at 37°C for 12 min, and the reaction was stopped by adding 8 µl loading dye (80% formamide and 10 mM EDTA). Samples were heated at 95°C for 5 min, cooled on ice, and loaded onto a 14% denaturing polyacrylamide gel. Gels were run at 45 W for 2.5 h and bands visualized using a PhosphorImager (Typhoon RGB).
Online supplemental material
Fig. S1 shows laboratory values of liver and kidney functions in sibling 1 and sibling 2. Fig. S2 shows genomic sequencing data confirming the missense mutation and the intragenic deletion in the ERCC1 gene. Fig. S3 shows the protein expression of ERCC1 and XPF in patient fibroblasts. Fig. S4 shows purified recombinant ERCC1 and XPF proteins and Co-IP of ERCC1WT and ERCC1R156W. Fig. S5 shows microscopy images of UDS, the γH2AX foci, and the location of patient mutations in ERCC1-XPF. Table S1 contains the sequences of all primers and probes used in this study. Table S2 lists all cell lines tested in this study. Table S3 lists all plasmids involved in this study. Table S4 provides the study’s sgRNA sequences and Table S5 shows the antibodies. Table S6 provides the number of cells used for quantification in the figures. Data S1 displays all the uncropped Western blot files.
Acknowledgments
The authors acknowledge both siblings and their parents for contributing to this paper, Sylvie Noordermeer (Leiden University Medical Center [LUMC], Leiden, Netherlands) for the pLenti-CW57-TO-GFP lentiviral expression vector and advice on generating KI cells, Joachim Goedhart (University of Amsterdam, Amsterdam, Netherlands) for the mVenus cDNA, Bert van der Kooij (LUMC, Leiden, Netherlands) for the DNA-PK inhibitor, Wim Vermeulen and Anja Raams (Erasmus MC, Rotterdam, Netherlands) for providing 165TOR cells, Tomoo Ogi (University of Nagoya, Japan) for providing CS20LO cells, Alan Lehman (University of Sussex, UK) for providing CSL16NG and CSL16NG-hTERT cells, and Joost Schimmel (LUMC, Leiden, Netherlands) for advice on generating KI cells.
This work was funded by a Leiden University Medical Center Research Fellowship and a Nederlandse Organisatie voor Wetenschappelijk Onderzoek VIDI grant (ALW.016.161.320) to M.S. Luijsterburg, a European Research Council starting grant (310913) to A.C.O. Vertegaal, a KWF Kankerbestrijding Young Investigator grant (11367) to R. González-Prieto, a KWF Kankerbestrijding grant (VU 2013-5983) to M.A. Rooimans, and grants from the Korean Institute for Basic Science (IBS-R022-A1) and the National Cancer Institute (P01CA092584) to O.D. Schärer.
Author contributions: K. Apelt generated KO cells, KI cells, and stable cell lines; performed Western blot and immunostainings for ERCC1 and XPF expression, lentiviral transductions, clonogenic survivals (UV, MMC, IR), Co-IP experiments for Western blot analysis, UDS experiments, Co-IP experiments for MS, and γH2AX stainings; and wrote the paper. A. Kragten performed UDS, RRS, and immunostainings after local UV. A.P. Wondergem generated KO and KI cells and performed UDS experiments, Western blot, and Sanger sequencing to confirm the missense mutation. S.M. White provided counseling for the patients, analyzed exome data, performed biopsies, generated primary fibroblasts, and wrote the clinical patient description. S. Lunke and B.T. Wilson generated NGS data. S. Pantaleo and D. Flanagan performed qPCR to validate the deletion. C. Quinlan wrote the clinical description of the kidney. W. Hardikar wrote the clinical description of the liver. M.A. Rooimans and R.M.F. Wolthuis generated hTERT-immortalized 48BR and PV50LD fibroblasts and performed the MMC-induced chromosome breakage assays. R. González-Prieto and A.C.O. Vertegaal analyzed all MS experiments. W.W. Wiegant and H. van Attikum performed clonogenic survivals in U2OS cells (IR). J.-E. Yeo and H.S. Kim purified recombinant proteins and performed the nuclease assay and the in vitro NER assays. O.D. Schärer supervised in vitro NER work, contributed the structural interpretation of the R156W allele, and edited the paper. M.S. Luijsterburg supervised the project and wrote the paper.

