


Multidrug resistance-1 (MDR1) acts as a chemotherapeutic drug efflux pump in tumor cells, although its physiological functions remain enigmatic. Using a recently developed MDR1-knockin reporter allele (Abcb1aAME), we found that constitutive MDR1 expression among hematopoietic cells was observed in cytolytic lymphocytes—including CD8+ cytotoxic T lymphocytes (CTLs) and natural killer cells—and regulated by Runt-related (Runx) transcription factors. Whereas MDR1 was dispensable for naive CD8+ T cell development, it was required for both the normal accumulation of effector CTLs following acute viral infection and the protective function of memory CTLs following challenge with an intracellular bacterium. MDR1 acted early after naive CD8+ T cell activation to suppress oxidative stress, enforce survival, and safeguard mitochondrial function in nascent CTLs. These data highlight an important endogenous function of MDR1 in cell-mediated immune responses and suggest that ongoing efforts to intentionally inhibit MDR1 in cancer patients could be counterproductive.

Introduction
Multidrug resistance-1 (MDR1; encoded by ABCB1 in humans and Abcb1a and Abcb1b in mice) is a membrane-associated, ATP-dependent efflux pump recognized and named for removing cytostatic drugs from tumor cells. Accordingly, MDR1 has been viewed as a “dedicated drug handler” in mammalian cells (Borst and Schinkel, 2013; Zhou, 2008), and numerous MDR1 inhibitors have been developed and tested in clinical cancer trials, with little success (Bauer et al., 2005; Kelly et al., 2012; O’Brien et al., 2010; Peck et al., 2001; Seiden et al., 2002). However, MDR1 is also expressed in a number of normal cell types and tissues (Schinkel et al., 1995; Sugawara et al., 1988; Thiebaut et al., 1987), and both the presence of MDR1 orthologues in prokaryotes and a growing body of literature suggest that this transporter has conserved endogenous functions in eukaryotes that extend beyond interacting with synthetic medicines.
In the immune system, MDR1 expression has been reported in skin dendritic cells, CD4+-induced T regulatory and T effector (Teff) cells, CD8+ CTLs, and natural killer (NK) cells (Chaudhary et al., 1992; Chaudhary and Roninson, 1991; Egashira et al., 1999; Randolph et al., 1998). MDR1 has been suggested to regulate egress of skin dendritic cells into lymphatic vessels, promote induced T regulatory development, and protect IFN-γ–producing (T helper [Th]1) and IL-17–secreting (Th17) CD4+ T cells from bile acid–driven oxidative stress in the small intestine (Cao et al., 2017; Randolph et al., 1998; Tanner et al., 2013). By contrast, the function of MDR1 in CTLs and NK cells has remained controversial (Egashira et al., 1999; Gupta et al., 1992), but has important implications in the design and delivery of vaccines and immunotherapies.
A paucity of genetic mouse models and specific antibodies has hampered a more robust understanding of MDR1 expression and function in vivo. Mice lacking one (Abcb1a−/−; Schinkel et al., 1994) or both (Abcb1a−/−Abcb1b−/−; hereafter referred to as Abcb1a/1b−/−; Schinkel et al., 1997) MDR1 genes are only available on the FVB background, in contrast to most C57BL/6J (B6)–derived transgenic and knockout alleles. In addition, no currently available antibodies detect MDR1 proteins in live mouse cells by flow cytometry. Thus, MDR1 expression is most commonly inferred indirectly, based on the efflux of fluorescent transport substrates, such as rhodamine 123 (Rh123; Ludescher et al., 1992). However useful, these dyes are also substrates for related multidrug transporters (e.g., ABCC1, ABCC3, ABCG2) and are, thus, nonspecific (Strouse et al., 2013; Zhou et al., 2001). To begin addressing these limitations, we previously generated an MDR1-reporter allele (Abcb1aAME) on the B6 background, in which the Abcb1a stop codon was replaced with a bicistronic reporter cassette containing a P2A peptide and a fluorescent transgene, ametrine, to reflect endogenous Abcb1a mRNA levels (Cao et al., 2017).
Using Abcb1aAME/+ reporter mice here, we found that cytolytic lymphocytes, including CD8+ CTLs and NK cells, constitutively express Abcb1a. Runt-related (Runx) transcription factors, which instruct CTL and NK cell development (Cruz-Guilloty et al., 2009; Lotem et al., 2013; Rapp et al., 2017; Taniuchi et al., 2002), bound and maintained chromatin accessibility at the MDR1 (Abcb1a-Abcb1b) locus. Most importantly, endogenous MDR1 expression was required for both effector and memory CTL responses to intracellular pathogens in vivo and acted early after CD8+ T cell activation to suppress oxidative stress, promote mitochondrial function, and enforce survival. These results unmasked a previously unrecognized function of MDR1 in cell-mediated immunity, which could be leveraged to identify endogenously generated MDR1 transport substrates and to inform the safe and rationale use of MDR1 inhibitors in human clinical trials.
Results and discussion
Endogenous MDR1 expression across the hematopoietic system
An integrated view of endogenous MDR1 expression throughout the hematopoietic system is lacking and could provide insight into protein function. Therefore, we used heterozygous Abcb1aAME/+ reporter mice to quantify steady-state Abcb1a expression in >100 immune cell types and developmental stages from five major lymphoid and nonlymphoid tissues: bone marrow, thymus, spleen, lung, and small intestine lamina propria (siLP; Fig. 1, A and B; and Table S1). This analysis incorporated 11 high-content (10–13 color) flow cytometry panels and used parallel gating of Abcb1aAME/+ reporter and wild-type B6 subsets (Table S1), to account for variable auto-fluorescence between cell types and to quantify normalized Abcb1a expression (Fig. 1 A).
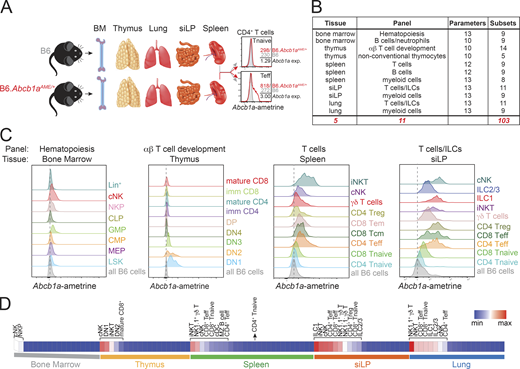
Endogenous Abcb1a expression across the hematopoietic system. (A) Tissues (bone marrow [BM], thymus, spleen, lung, and small intestine lamina propria [siLP]) were harvested from three pairs of 6–8-wk-old female B6 wild-type or heterozygous Abcb1aAME/+ reporter mice to profile endogenous MDR1 (Abcb1a) gene expression (exp) across the mouse hematopoietic compartment. Identically gated control and reporter cell subsets were gated, and normalized Abcb1a expression for each cell type was calculated by dividing ametrine MFI in Abcb1aAME/+ reporter cells by the background MFI in wild-type B6 cells; two examples of this analysis are shown for cells in spleen (top: CD4+ naive [Tnaive]; bottom: CD4+ effector/memory [Teff]). (B) Names and descriptions of the FACS antibody panels used to discriminate hematopoietic cell types in the tissues indicated in A. See also Table S1 for a full list of the cell types and developmental stages analyzed and the gating hierarchies. ILCs, innate lymphoid cells. (C) Representative Abcb1aAME reporter expression, determined by flow cytometry as in A, in cells from (left to right) bone marrow, thymus, spleen, and siLP. CLP, common lymphoid progenitor; CMP, common myeloid progenitor; cNK, conventional NK cells (mix of immature and mature); DN, double negative; GMP, granulocyte/macrophage progenitor; ILC1, group 1 innate lymphoid cells; ILC2/3, group 2/3 innate lymphoid cells; iNKT, invariant NK T cells; LSK, Lin−Sca-1+c-Kit−; MEP, megakaryocyte/erythrocyte progenitor; NKP, NK progenitor; Tcm, central memory T cells; Tem, effector memory T cells; Treg, regulatory T cells. Abcb1aAME expression in each gated population is shown; due to space constraints, gray/shaded peaks show background ametrine expression in all live B6 wild-type cells, gated only on forward/side scatter and viability, from the same tissue. Vertical dotted lines indicate background ametrine MFIs in all B6 wild-type cells. Representative of three pairs of B6 wild-type and Abcb1aAME/+ reporter mice analyzed over two independent experiments. (D) Normalized mean Abcb1aAME reporter expression (n = 3) in all 103 hematopoietic cell types and developmental stages analyzed as in C (see also Fig. S1). Each tissue is presented in rank order from high to low Abcb1aAME/+ reporter expression. Cell types displaying Abcb1aAME expression above background in each tissue are annotated. Spleen CD4+ Tnaive cells are annotated as a negative reference point (i.e., cells that lack biologically significant MDR1 expression; Cao et al., 2017). max, maximum; min, minimum.
Endogenous Abcb1a expression across the hematopoietic system. (A) Tissues (bone marrow [BM], thymus, spleen, lung, and small intestine lamina propria [siLP]) were harvested from three pairs of 6–8-wk-old female B6 wild-type or heterozygous Abcb1aAME/+ reporter mice to profile endogenous MDR1 (Abcb1a) gene expression (exp) across the mouse hematopoietic compartment. Identically gated control and reporter cell subsets were gated, and normalized Abcb1a expression for each cell type was calculated by dividing ametrine MFI in Abcb1aAME/+ reporter cells by the background MFI in wild-type B6 cells; two examples of this analysis are shown for cells in spleen (top: CD4+ naive [Tnaive]; bottom: CD4+ effector/memory [Teff]). (B) Names and descriptions of the FACS antibody panels used to discriminate hematopoietic cell types in the tissues indicated in A. See also Table S1 for a full list of the cell types and developmental stages analyzed and the gating hierarchies. ILCs, innate lymphoid cells. (C) Representative Abcb1aAME reporter expression, determined by flow cytometry as in A, in cells from (left to right) bone marrow, thymus, spleen, and siLP. CLP, common lymphoid progenitor; CMP, common myeloid progenitor; cNK, conventional NK cells (mix of immature and mature); DN, double negative; GMP, granulocyte/macrophage progenitor; ILC1, group 1 innate lymphoid cells; ILC2/3, group 2/3 innate lymphoid cells; iNKT, invariant NK T cells; LSK, Lin−Sca-1+c-Kit−; MEP, megakaryocyte/erythrocyte progenitor; NKP, NK progenitor; Tcm, central memory T cells; Tem, effector memory T cells; Treg, regulatory T cells. Abcb1aAME expression in each gated population is shown; due to space constraints, gray/shaded peaks show background ametrine expression in all live B6 wild-type cells, gated only on forward/side scatter and viability, from the same tissue. Vertical dotted lines indicate background ametrine MFIs in all B6 wild-type cells. Representative of three pairs of B6 wild-type and Abcb1aAME/+ reporter mice analyzed over two independent experiments. (D) Normalized mean Abcb1aAME reporter expression (n = 3) in all 103 hematopoietic cell types and developmental stages analyzed as in C (see also Fig. S1). Each tissue is presented in rank order from high to low Abcb1aAME/+ reporter expression. Cell types displaying Abcb1aAME expression above background in each tissue are annotated. Spleen CD4+ Tnaive cells are annotated as a negative reference point (i.e., cells that lack biologically significant MDR1 expression; Cao et al., 2017). max, maximum; min, minimum.
Abcb1a expression was low or absent throughout most stages of bone marrow hematopoiesis and thymic T cell development, whereas it was expressed in several mature myeloid and lymphoid subsets in the spleen and upregulated in nearly all lymphocytes in siLP (Fig. 1, C and D; and Fig. S1). Elevated Abcb1a expression in the intestine was not a generic feature of all mucosal tissues, as Abcb1a expression in these same cell types was significantly lower in the lung vs. gut (mean Abcb1a expression in lung, 442.8; mean Abcb1a expression in siLP, 844.3; P = 0.0051, paired Student’s t test; Fig. 1, C and D; and Fig. S1 A). Thus, preferential expression of Abcb1a in intestinal lymphocytes is compatible with current models that consider MDR1 a transporter of xenobiotic substances in the gastrointestinal tract (Borst and Schinkel, 2013; Cohen, 1989; Schinkel, 1997).

Endogenous Abcb1a expression across the hematopoietic system. (A) Representative Abcb1aAME reporter expression in select cell types from (left to right) bone marrow, spleen, thymus, and lung. cNK, conventional NK cells; FO, follicular B cells; GC, germinal center B cells; imm. B, immature B cells; iNKT, invariant NK T cells; MZ, marginal zone B cells; recirc. mature B, recirculating mature B cells; T1/2 T1 and T2 transitional B cells, T3, transitional T3 B cells; Treg, regulatory T cells; Tnaive, naive T cells. As in Fig. 1, normalized Abcb1aAME expression across subsets was quantified by comparing ametrine MFIs in identically gated Abcb1aAME/+ reporter or wild-type B6 cells but is presented in all Abcb1aAME/+ reporter subsets and all tissues with only a single gray/shaded peak reflecting the distribution of background ametrine expression in all live wild-type B6 cells, gated only on forward/side scatter and viability, from the same tissue. Vertical dotted lines indicate background ametrine MFIs in all wild-type B6 cells. (B) Representative Abcb1aAME reporter expression, determined by ex vivo flow cytometry and presented as in A, in select myeloid cells from (left to right) spleen, siLP, and lung. cDC, conventional dendritic cells; cMF, classical macrophages; ncMF, nonclassical macrophage; pDC, plasmacytoid dendritic cell. Data are representative of three pairs of B6 wild-type and Abcb1aAME/+ reporter mice analyzed over two independent experiments in A and B. (C) Fluorescence microscopy of frozen spleen sections from wild-type (B6; left) or MDR1-reporter (B6.Abcb1aAME/+; right) mice. Native Abcb1aAME/+ reporter fluorescence (green) and anti-CD11c staining (red) is shown; colocalization of ametrine and CD11c fluorescence is indicated by orange coloring. Magnification ×10; scale bar = 200 µm; representative of five mice per genotype analyzed over two independent experiments.
Endogenous Abcb1a expression across the hematopoietic system. (A) Representative Abcb1aAME reporter expression in select cell types from (left to right) bone marrow, spleen, thymus, and lung. cNK, conventional NK cells; FO, follicular B cells; GC, germinal center B cells; imm. B, immature B cells; iNKT, invariant NK T cells; MZ, marginal zone B cells; recirc. mature B, recirculating mature B cells; T1/2 T1 and T2 transitional B cells, T3, transitional T3 B cells; Treg, regulatory T cells; Tnaive, naive T cells. As in Fig. 1, normalized Abcb1aAME expression across subsets was quantified by comparing ametrine MFIs in identically gated Abcb1aAME/+ reporter or wild-type B6 cells but is presented in all Abcb1aAME/+ reporter subsets and all tissues with only a single gray/shaded peak reflecting the distribution of background ametrine expression in all live wild-type B6 cells, gated only on forward/side scatter and viability, from the same tissue. Vertical dotted lines indicate background ametrine MFIs in all wild-type B6 cells. (B) Representative Abcb1aAME reporter expression, determined by ex vivo flow cytometry and presented as in A, in select myeloid cells from (left to right) spleen, siLP, and lung. cDC, conventional dendritic cells; cMF, classical macrophages; ncMF, nonclassical macrophage; pDC, plasmacytoid dendritic cell. Data are representative of three pairs of B6 wild-type and Abcb1aAME/+ reporter mice analyzed over two independent experiments in A and B. (C) Fluorescence microscopy of frozen spleen sections from wild-type (B6; left) or MDR1-reporter (B6.Abcb1aAME/+; right) mice. Native Abcb1aAME/+ reporter fluorescence (green) and anti-CD11c staining (red) is shown; colocalization of ametrine and CD11c fluorescence is indicated by orange coloring. Magnification ×10; scale bar = 200 µm; representative of five mice per genotype analyzed over two independent experiments.
A second set of results was less consistent with the xenobiotic transport model and revealed that Abcb1a was also constitutively expressed in cytotoxic lymphocytes. First, Abcb1a was upregulated in the bone marrow as common lymphoid progenitors committed to the NK lineage (Fig. 1, C and D). Second, whereas Abcb1a expression was high in early double-negative (e.g., DN1, DN2) thymocytes, it was downregulated in later stages of thymic development and only reemerged late in CD8+, but not CD4+, T cell development (Fig. 1, C and D). This same pattern of Abcb1a expression in developing thymocytes was noted on the Immunological Genome Project’s website (available at: http://www.immgen.org), using RNA-sequencing (RNA-seq) as a readout. Third, naive CD8+ T cells displayed constitutive Abcb1a expression in all peripheral tissues, unlike naive CD4+ T cells (Fig. 1, C and D; and Fig. S1 A). Other lymphocytes with cytotoxic potential—invariant NK T cells and NK1.1+ γδT cells—also displayed constitutive Abcb1a expression (Fig. 1, C and D; and Fig. S1 A). Therefore, Abcb1a expression in mice appears to be both a locally acquired trait of lymphocytes in the intestine and a developmentally imprinted feature in cytotoxic immune cells.
Runx transcription factors promote gene expression at the MDR1 (Abcb1a-Abcb1b) locus
Abcb1a expression was higher in polyclonal CD8+ T cells with central memory or effector memory phenotypes compared with naive cells (Fig. 1 C), suggesting that MDR1 may play a role in the generation or maintenance of memory CTLs. To explore the regulation of Abcb1a expression in antigen-specific CD8+ T cells responding to infection, we crossed Abcb1aAME/+ reporter mice with Thy1.1 congenic/P14 TCR transgenic mice (hereafter referred to as P14 mice) that express a TCR specific for the lymphocytic choriomeningitis virus (LCMV) glycoprotein (gp)33-41 peptide presented on MHC class I (H2-Db; Pircher et al., 1989). Thy1.1+CD8+ naive P14 cells with or without the Abcb1aAME reporter allele were transferred into Thy1.2+ wild-type B6 hosts, which were subsequently infected with the Armstrong strain of LCMV (LCMVArm) to produce an acute infection or were left uninfected (Fig. 2 A). Both control and Abcb1aAME/+ reporter P14 cells underwent marked expansion by day 8 after infection, and both differentiated into expected mixtures of Klrg1+IL-7Rαlo terminal effector (TE) and Klrg1−IL-7Rαhi memory precursor (MP) cells (Joshi et al., 2007; Fig. 2 A). Abcb1a expression was maintained in TE and Klrg1−IL-7Rαlo early effector cells, relative to naive cells, but was upregulated in both MP and Klrg1+IL-7Rαhi double-positive (DP) effector cells from LCMV-infected recipients (Fig. 2, B and C). Thus, Abcb1a expression is highest in effector CD8+ T cells that develop the potential to give rise to both effector memory and central memory T cells.

Runx transcription factors regulate MDR1 expression in CTLs. (A) Naive CD8+ T cells from Thy1.1 congenic/P14 TCR transgenic mice with or without a heterozygous MDR1-reporter allele (Abcb1aAME/+) were transferred into wild-type (Thy1.2) B6 hosts. Recipient mice were left uninfected (no virus) or infected with LCMVArm. After 8 d (d8), Abcb1a-ametrine reporter expression was analyzed by flow cytometry in splenic P14 (Thy1.1+) CD8+ T cell subsets based on differential expression of Klrg1 and IL-7Rα; representative of five mice analyzed over two independent experiments. (B) Ex vivo Abcb1aAME/+ reporter expression (exp) in the indicated donor P14 (Thy1.1+) CD8+ T cell subsets, determined by flow cytometry as in A. For each subset, background ametrine MFIs from wild-type P14 CD8+ T cells (gray/shaded peaks) were divided by those of Abcb1aAME/+ reporter P14 CD8+ T cells (red peaks) to generate normalized Abcb1a expression values; representative of five mice analyzed over two independent experiments. (C) Mean normalized Abcb1aAME/+ reporter expression (n = 5), determined by flow cytometry as in B in the indicated donor P14 (Thy1.1+) CD8+ T cell subsets. Individual data points for each animal are shown. *P < 0.05; one-way ANOVA with Tukey’s correction for multiple comparisons. (D) Naive CD8+ T cells from Thy1.1 congenic/P14 TCR transgenic/Abcb1aAME/+ reporter B6 mice were activated and transduced in vitro with GFP-expressing retroviruses containing control (shCD4) or Runx-targeting (shRunx1, shRunx3, shCbfb) shRNAmirs. Transduced T cells were injected into wild-type B6 (Thy1.2) hosts, which were then infected with LCMVArm. 8 d later, the impact of Runx gene depletion on Abcb1aAME/+ reporter expression in transduced (GFP+) or untransduced (GFP−) P14 (Thy1.1+) TE (Klrg1hiIL-7Rαlo) or MP (Klrg1loIL-7Rαhi) cells was evaluated by flow cytometry. Representative gating strategy is shown from an LCMV-infected mouse receiving P14 cells transduced with shCD4-GFP retroviruses. (E)Abcb1aAME/+ reporter expression, determined by flow cytometry as in D, in P14 TE (left) or MP (right) cells expressing control (shCd4) or Runx-targeting shRNAmirs. Ametrine MFIs for each untransduced (GFP−) or transduced (GFP+) cell type is indicated by color-matched text. Vertical dotted lines indicate mean background ametrine fluorescence in shCd4-expressing cells; representative of five mice analyzed over two independent experiments. (F) Mean relative Abcb1aAME/+ reporter expression (n = 5) in untransduced (GFP−) or transduced (GFP+) P14 TE (Left) or MP (Right) cells, determined by flow cytometry as in D and E. Individual data points for each animal shown. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, one-way ANOVA with Tukey’s correction for multiple comparisons. (G) Top: Runx3 (Lotem et al., 2013), Irf4 (Kurachi et al., 2014), or T-bet (Dominguez et al., 2015) binding across the MDR1 (Abcb1a-Abcb1b) locus, determined by ChIP-seq, in in vitro–cultured resting (no stimulation [stim]) or TCR-activated (stim) naive CD8+ T cells. Middle: Chromatin accessibility, determined by ATAC-seq (Wang et al., 2018), in B6 wild-type or Runx3−/− naive CD8+ T cells activated in vitro (anti-CD3/anti-CD28) for 12 h. Bottom: chromatin accessibility, determined by ATAC-seq (Scott-Browne et al., 2016), in naive, TE (day 8), MP (day 8), memory (day 35), or exhausted (day 20) P14 TCR transgenic CD8+ T cells isolated from LCMV-infected mice. The shaded vertical box highlights a region in Abcb1b exon 19 where TCR-induced Runx3 occupancy corresponds with Irf4 and T-bet binding, as well as with Runx3-dependent chromatin accessibility that is increased in LCMV-specific memory and reduced in LCMV-specific exhausted CTLs. *P < 0.00001; statistically significant binding peaks were called in MACS using base settings.
Runx transcription factors regulate MDR1 expression in CTLs. (A) Naive CD8+ T cells from Thy1.1 congenic/P14 TCR transgenic mice with or without a heterozygous MDR1-reporter allele (Abcb1aAME/+) were transferred into wild-type (Thy1.2) B6 hosts. Recipient mice were left uninfected (no virus) or infected with LCMVArm. After 8 d (d8), Abcb1a-ametrine reporter expression was analyzed by flow cytometry in splenic P14 (Thy1.1+) CD8+ T cell subsets based on differential expression of Klrg1 and IL-7Rα; representative of five mice analyzed over two independent experiments. (B) Ex vivo Abcb1aAME/+ reporter expression (exp) in the indicated donor P14 (Thy1.1+) CD8+ T cell subsets, determined by flow cytometry as in A. For each subset, background ametrine MFIs from wild-type P14 CD8+ T cells (gray/shaded peaks) were divided by those of Abcb1aAME/+ reporter P14 CD8+ T cells (red peaks) to generate normalized Abcb1a expression values; representative of five mice analyzed over two independent experiments. (C) Mean normalized Abcb1aAME/+ reporter expression (n = 5), determined by flow cytometry as in B in the indicated donor P14 (Thy1.1+) CD8+ T cell subsets. Individual data points for each animal are shown. *P < 0.05; one-way ANOVA with Tukey’s correction for multiple comparisons. (D) Naive CD8+ T cells from Thy1.1 congenic/P14 TCR transgenic/Abcb1aAME/+ reporter B6 mice were activated and transduced in vitro with GFP-expressing retroviruses containing control (shCD4) or Runx-targeting (shRunx1, shRunx3, shCbfb) shRNAmirs. Transduced T cells were injected into wild-type B6 (Thy1.2) hosts, which were then infected with LCMVArm. 8 d later, the impact of Runx gene depletion on Abcb1aAME/+ reporter expression in transduced (GFP+) or untransduced (GFP−) P14 (Thy1.1+) TE (Klrg1hiIL-7Rαlo) or MP (Klrg1loIL-7Rαhi) cells was evaluated by flow cytometry. Representative gating strategy is shown from an LCMV-infected mouse receiving P14 cells transduced with shCD4-GFP retroviruses. (E)Abcb1aAME/+ reporter expression, determined by flow cytometry as in D, in P14 TE (left) or MP (right) cells expressing control (shCd4) or Runx-targeting shRNAmirs. Ametrine MFIs for each untransduced (GFP−) or transduced (GFP+) cell type is indicated by color-matched text. Vertical dotted lines indicate mean background ametrine fluorescence in shCd4-expressing cells; representative of five mice analyzed over two independent experiments. (F) Mean relative Abcb1aAME/+ reporter expression (n = 5) in untransduced (GFP−) or transduced (GFP+) P14 TE (Left) or MP (Right) cells, determined by flow cytometry as in D and E. Individual data points for each animal shown. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, one-way ANOVA with Tukey’s correction for multiple comparisons. (G) Top: Runx3 (Lotem et al., 2013), Irf4 (Kurachi et al., 2014), or T-bet (Dominguez et al., 2015) binding across the MDR1 (Abcb1a-Abcb1b) locus, determined by ChIP-seq, in in vitro–cultured resting (no stimulation [stim]) or TCR-activated (stim) naive CD8+ T cells. Middle: Chromatin accessibility, determined by ATAC-seq (Wang et al., 2018), in B6 wild-type or Runx3−/− naive CD8+ T cells activated in vitro (anti-CD3/anti-CD28) for 12 h. Bottom: chromatin accessibility, determined by ATAC-seq (Scott-Browne et al., 2016), in naive, TE (day 8), MP (day 8), memory (day 35), or exhausted (day 20) P14 TCR transgenic CD8+ T cells isolated from LCMV-infected mice. The shaded vertical box highlights a region in Abcb1b exon 19 where TCR-induced Runx3 occupancy corresponds with Irf4 and T-bet binding, as well as with Runx3-dependent chromatin accessibility that is increased in LCMV-specific memory and reduced in LCMV-specific exhausted CTLs. *P < 0.00001; statistically significant binding peaks were called in MACS using base settings.
Runx transcription factors, particularly Runx1 and Runx3, are essential for the development and maintenance of CD8+ T cells and NK cells (Naito et al., 2011). Runx3 is upregulated during positive selection of CD8+ thymocytes (Liu et al., 2005), consistent with the emergence of Abcb1a expression late in CD8+ T cell thymic development (Fig. 1, C and D). In the periphery, Runx3 promotes naive CD8+ T cell differentiation into both DP and MP CTLs, as well as tissue-resident memory cells, following acute infections (Milner et al., 2017; Wang et al., 2018). As these Runx3-dependent subsets all displayed elevated Abcb1a expression, relative to either naive CD8+ T cells or to many other hematopoietic cell lineages (Fig. 1, C and D), we used retroviral expression of previously validated short hairpin RNAs expressed in a microRNA context (shRNAmirs; Wang et al., 2018) to test whether Runx transcriptional activity is required to maintain CTL-intrinsic expression of Abcb1a in vivo (Fig. 2 D). Consistent with this possibility, depletion of Runx1, Runx3, or the obligate Runx DNA-binding cofactor Cbfb in P14 cells responding to LCMVArm infection each impaired endogenous Abcb1a expression in TE or MP cells (Fig. 2, D–F), although Runx1 depletion had the strongest effect (Fig. 2, D–F). Because Runx1 expression is increased in CTLs following Runx3 depletion (Wang et al., 2018), it seems less likely that Runx1 is sufficient to sustain Abcb1a expression in the absence of Runx3, and more likely that Runx1 and Runx3 act in concert to promote endogenous Abcb1a expression during CTL responses to infection.
The mouse MDR1 locus encodes two functionally redundant transporter proteins (Abcb1a, Abcb1b), with Abcb1a reflecting the mouse orthologue of human ABCB1. Unlike its stable expression in vivo, Abcb1a was lost upon in vitro TCR stimulation of naive CD8+ T cells that were subsequently cultured in high or low IL-2–containing media (Pipkin et al., 2010; Fig. S2, A and B). However, these cells retained MDR1 transport (Rh123 efflux) activity, apparently because of Abcb1b upregulation (Fig. S2, B and C). Loss of Abcb1a expression occurred within 48 h of TCR stimulation and paralleled Runx1 downregulation (Fig. S2 C), further suggesting that both Runx1 and Runx3 are required to maintain Abcb1a expression in CTLs. By contrast, Runx3 was sufficient in the absence of Runx1 to maintain, and even increase, Abcb1b expression in cultured CTLs; Abcb1b upregulation correlated with increased Runx3 protein expression, despite stable Runx3 mRNA expression (Fig. S2 C; data not shown), and shRNAmir-mediated depletion of either Runx3 or Cbfb reduced MDR1-dependent Rh123 efflux and abolished Abcb1b mRNA upregulation upon in vitro expansion (Fig. S2, D and E). These results suggest that interplay among Runx1, Runx3, and T cell activation signals underlies dynamic regulation of MDR1 (Abcb1a, Abcb1b) gene expression in CTLs.

Regulation of MDR1 expression in T lymphocytes by Runx transcription factors. (A) Top: Abcb1a-ametrine reporter expression (exp) in ex vivo-isolated Abcb1aAME/+ naive CD8+ T cells (left) or Abcb1aAME/+ naive CD8+ T cells activated in vitro with anti-CD3/anti-CD28 antibodies (for 2 d) and then expanded until day 6 (d6) in effector-like (high [hi] IL-2 TE) or memory-like (low [lo] IL-2 TM) CTL differentiation conditions (right). Background ametrine expression is shown in wild-type CD8+ T cells (gray/shaded peaks) for comparison. Color-matched text indicates raw (B6.Abcb1aAME/+; red, B6; gray) and normalized (i.e., background subtracted; black) ametrine MFIs; representative of three independent experiments. Bottom: MDR1-dependent Rh123 efflux, determined by flow cytometry, in naive or in vitro–expanded wild-type (B6) CD8+ T cells as above. Color-matched text indicates raw (no elacridar; red, elacridar; gray) and corrected (i.e., background divided; black) Rh123 MFIs; representative of three independent experiments. (B) Mean Abcb1aAME reporter expression (top) or MDR1-dependent Rh123 efflux (bottom; n = 3) in naive or d6 in vitro–expanded wild-type or Abcb1aAME/+ reporter hi IL-2 TE or lo IL-2 TM cells as in A. Individual data points for each animal are shown analyzed over three independent experiments. *P < 0.05, **P < 0.01; one-way ANOVA with Tukey’s correction for multiple comparisons. (C) Mean relative mRNA abundance + SEM (n = 3), determined by TaqMan qPCR, of Abcb1a (red) or Abcb1b (blue; Top) or Runx1 (red) or Runx3 (blue; Bottom) in naive CD8+ T cells cultured in hi IL-2 TE (closed circles) or lo IL-2 TM (open circles) differentiation conditions as in A and B. (D) Left: MDR1-dependent Rh123 efflux in wild-type (B6) CD8+ T cells cultured for 6 d in lo IL-2 TM differentiation conditions as in A–C. Cells were transduced 24 h after activation with ametrine-expressing retroviruses containing control (shCD4; red), Runx3 (shRunx3; blue), or Cbfβ (shCbfb; green) shRNAmirs. Rh123 efflux is shown in live, ametrine+ T cells. Background Rh123 efflux is shown in elacridar-treated control shCD4-expressing cells (shCD4+E; gray). Color-matched text indicates Rh123 MFIs; representative of five independent experiments. Right: Mean normalized Rh123 efflux (n = 5), determined by flow cytometry as above, in shCD4-, shRunx3-, and shCbfb-expressing CD8+ T cells. Individual data points for each animal are shown. *P < 0.05, ***P < 0.001, one-way ANOVA with Tukey’s correction for multiple comparisons. (E) Mean relative Abcb1a (Left) or Abcb1b (Right) gene expression ± SEM (n = 3), determined by TaqMan qPCR, in ex vivo naive CD8+ T cells or in vitro–generated (day 6-expanded) lo IL-2 TM cells transduced with control (shCD4) or Runx-targeting shRNAmirs (shRunx3, shCbfb) as in D. *P < 0.05, **P < 0.01, one-way ANOVA with Tukey’s correction for multiple comparisons. (F) Top: Naive CD8+ T cells from CD45.1 wild-type (B6; blue) or CD45.2 MDR1-deficient (Abcb1a/1b−/−; B6.Mdr1ko; red) mice were purified, mixed at 1:1 ratios, and then stimulated and expanded for 6 d in hi IL-2 TE or lo IL-2 TM conditions. Ratios and genotypes were confirmed before culture by CD45.1 staining and Rh123 efflux; elacridar was added to some cultures to inhibit MDR1 transport activity. Bottom: Mean relative abundance (Log2 fold-change; ± SEM; n = 7) of congenically cultured MDR1-deficient vs. wild-type hi IL-2 TE (left) or lo IL-2 TM (right) cells in the absence (closed circles) or presence (open circles) of elacridar, as above. Data are presented relative to d0 ratios. *P < 0.05, two-way ANOVA. (G) Top: Kinetics of CD69 expression (Left) or carboxyfluorescein diacetate succinimidyl ester (CFSE) dilution (Right) in resting or stimulated B6 wild-type or MDR1ko lo IL-2 TM cells with or without elacridar. Live (viability dye−) CD8+ T cells are shown; representative of four experiments. Bottom: Kinetics of mean relative CD69 expression (left) or proliferation (CFSE dilution; right; ± SEM; n = 4) in wild-type (B6; blue) or MDR1-deficient (B6.Mdr1ko; red) lo IL-2 TM cells activated and expanded in the absence (closed circles) or presence (open circles) of elacridar as above. Proliferation index reflects the projected number of cells based on CFSE dilution peaks (Cao et al., 2017). (H) Top: PCA of gene expression (n = 3), determined by RNA-seq, in in vitro–activated and expanded wild-type (B6; blue) or MDR1-deficient (B6.Mdr1ko; red) lo IL-2 TM cells with or without elacridar at d6. Bottom: Fold-change/fold-change plots showing differential gene expression (n = 3), determined by RNA-seq as above, in d6-activated wild-type (B6) or MDR1-deficient (B6.Mdr1ko) lo IL-2 TM cells treated with or with or without elacridar. Blue lines = 1.8-fold-change. Genes whose expression increased (up; n = 248) or decreased (down; n = 898) ≥1.8-fold in both elacridar-treated wild-type (B6+E; x axis) and MDR1-deficient CTLs (B6.Mdr1ko; y axis) are highlighted red and blue, respectively. Only transcripts with a minimum expression ≥0.1 and multiclass variance ≤1 and whose expression was changed ≤1.5-fold in elacridar- vs. control-treated B6.Mdr1ko CTLs are shown (n = 22,746). The gene encoding mouse MDR1 (Abcb1a) is highlighted green for reference. Data incorporate three independent experiments.
Regulation of MDR1 expression in T lymphocytes by Runx transcription factors. (A) Top: Abcb1a-ametrine reporter expression (exp) in ex vivo-isolated Abcb1aAME/+ naive CD8+ T cells (left) or Abcb1aAME/+ naive CD8+ T cells activated in vitro with anti-CD3/anti-CD28 antibodies (for 2 d) and then expanded until day 6 (d6) in effector-like (high [hi] IL-2 TE) or memory-like (low [lo] IL-2 TM) CTL differentiation conditions (right). Background ametrine expression is shown in wild-type CD8+ T cells (gray/shaded peaks) for comparison. Color-matched text indicates raw (B6.Abcb1aAME/+; red, B6; gray) and normalized (i.e., background subtracted; black) ametrine MFIs; representative of three independent experiments. Bottom: MDR1-dependent Rh123 efflux, determined by flow cytometry, in naive or in vitro–expanded wild-type (B6) CD8+ T cells as above. Color-matched text indicates raw (no elacridar; red, elacridar; gray) and corrected (i.e., background divided; black) Rh123 MFIs; representative of three independent experiments. (B) Mean Abcb1aAME reporter expression (top) or MDR1-dependent Rh123 efflux (bottom; n = 3) in naive or d6 in vitro–expanded wild-type or Abcb1aAME/+ reporter hi IL-2 TE or lo IL-2 TM cells as in A. Individual data points for each animal are shown analyzed over three independent experiments. *P < 0.05, **P < 0.01; one-way ANOVA with Tukey’s correction for multiple comparisons. (C) Mean relative mRNA abundance + SEM (n = 3), determined by TaqMan qPCR, of Abcb1a (red) or Abcb1b (blue; Top) or Runx1 (red) or Runx3 (blue; Bottom) in naive CD8+ T cells cultured in hi IL-2 TE (closed circles) or lo IL-2 TM (open circles) differentiation conditions as in A and B. (D) Left: MDR1-dependent Rh123 efflux in wild-type (B6) CD8+ T cells cultured for 6 d in lo IL-2 TM differentiation conditions as in A–C. Cells were transduced 24 h after activation with ametrine-expressing retroviruses containing control (shCD4; red), Runx3 (shRunx3; blue), or Cbfβ (shCbfb; green) shRNAmirs. Rh123 efflux is shown in live, ametrine+ T cells. Background Rh123 efflux is shown in elacridar-treated control shCD4-expressing cells (shCD4+E; gray). Color-matched text indicates Rh123 MFIs; representative of five independent experiments. Right: Mean normalized Rh123 efflux (n = 5), determined by flow cytometry as above, in shCD4-, shRunx3-, and shCbfb-expressing CD8+ T cells. Individual data points for each animal are shown. *P < 0.05, ***P < 0.001, one-way ANOVA with Tukey’s correction for multiple comparisons. (E) Mean relative Abcb1a (Left) or Abcb1b (Right) gene expression ± SEM (n = 3), determined by TaqMan qPCR, in ex vivo naive CD8+ T cells or in vitro–generated (day 6-expanded) lo IL-2 TM cells transduced with control (shCD4) or Runx-targeting shRNAmirs (shRunx3, shCbfb) as in D. *P < 0.05, **P < 0.01, one-way ANOVA with Tukey’s correction for multiple comparisons. (F) Top: Naive CD8+ T cells from CD45.1 wild-type (B6; blue) or CD45.2 MDR1-deficient (Abcb1a/1b−/−; B6.Mdr1ko; red) mice were purified, mixed at 1:1 ratios, and then stimulated and expanded for 6 d in hi IL-2 TE or lo IL-2 TM conditions. Ratios and genotypes were confirmed before culture by CD45.1 staining and Rh123 efflux; elacridar was added to some cultures to inhibit MDR1 transport activity. Bottom: Mean relative abundance (Log2 fold-change; ± SEM; n = 7) of congenically cultured MDR1-deficient vs. wild-type hi IL-2 TE (left) or lo IL-2 TM (right) cells in the absence (closed circles) or presence (open circles) of elacridar, as above. Data are presented relative to d0 ratios. *P < 0.05, two-way ANOVA. (G) Top: Kinetics of CD69 expression (Left) or carboxyfluorescein diacetate succinimidyl ester (CFSE) dilution (Right) in resting or stimulated B6 wild-type or MDR1ko lo IL-2 TM cells with or without elacridar. Live (viability dye−) CD8+ T cells are shown; representative of four experiments. Bottom: Kinetics of mean relative CD69 expression (left) or proliferation (CFSE dilution; right; ± SEM; n = 4) in wild-type (B6; blue) or MDR1-deficient (B6.Mdr1ko; red) lo IL-2 TM cells activated and expanded in the absence (closed circles) or presence (open circles) of elacridar as above. Proliferation index reflects the projected number of cells based on CFSE dilution peaks (Cao et al., 2017). (H) Top: PCA of gene expression (n = 3), determined by RNA-seq, in in vitro–activated and expanded wild-type (B6; blue) or MDR1-deficient (B6.Mdr1ko; red) lo IL-2 TM cells with or without elacridar at d6. Bottom: Fold-change/fold-change plots showing differential gene expression (n = 3), determined by RNA-seq as above, in d6-activated wild-type (B6) or MDR1-deficient (B6.Mdr1ko) lo IL-2 TM cells treated with or with or without elacridar. Blue lines = 1.8-fold-change. Genes whose expression increased (up; n = 248) or decreased (down; n = 898) ≥1.8-fold in both elacridar-treated wild-type (B6+E; x axis) and MDR1-deficient CTLs (B6.Mdr1ko; y axis) are highlighted red and blue, respectively. Only transcripts with a minimum expression ≥0.1 and multiclass variance ≤1 and whose expression was changed ≤1.5-fold in elacridar- vs. control-treated B6.Mdr1ko CTLs are shown (n = 22,746). The gene encoding mouse MDR1 (Abcb1a) is highlighted green for reference. Data incorporate three independent experiments.
To gain molecular insight into MDR1 gene regulation, we used the published assay for transposable-accessible chromatin (ATAC)-seq and chromatin immunoprecipitation (ChIP)–seq data sets (Kurachi et al., 2014; Lotem et al., 2013; Scott-Browne et al., 2016; Wang et al., 2018) to examine regions of chromatin accessibility and transcription factor binding that develop upon naive CD8 T cell activation in vitro and that are present in mature LCMV-specific CTL subsets in vivo. Multiple regions of chromatin accessibility were observed across the MDR1 (Abcb1a-Abcb1b) locus in naive CD8+ T cells; these were most accessible in memory CTLs (isolated 35 d after LCMVArm infection), much less accessible in “exhausted” CTLs (isolated 20 d after LCMV-clone 13 infection), and reduced in Runx3−/− naive CD8+ T cells after 12 h of TCR stimulation in vitro (Fig. 2 G). Runx3 occupied an accessible region in Abcb1b intron 19 in stimulated, but not resting, CD8+ T cells, together with IRF4 and T-bet—transcription factors that act concertedly with Runx3 to program effector and memory CTL differentiation. IRF4 and T-bet also bound additional regions of Runx3-dependent accessible chromatin in Abcb1a intron 3 (Fig. 2 G). Thus, Runx3 binds and maintains chromatin accessibility at multiple cis-regulatory regions across the MDR1 locus in activated CTLs, which is consistent with the preferential expression of Abcb1a in cytotoxic lymphocytes in vivo and may inform broader mechanisms of MDR1 gene regulation in other normal and malignant cell types.
MDR1 is required for CTL function in vivo
To decipher the function of MDR1 during CTL responses to infection, we backcrossed FVB mice lacking both MDR1 genes (Abcb1a/1b−/−) to pure B6 (12 generations) and then bred B6-derived Abcb1a/1b−/− mice with P14 mice to obtain animals in which gp33-specific CD8+ T cells are null for MDR1 function. We produced 1:1 congenic mixtures of Thy1.1+Thy1.2+ MDR1-sufficient and Thy1.1+Thy1.2− MDR1-deficient naive P14 cells, transferred 5000 mixed cells into wild-type B6 (Thy1.1−Thy1.2+) mice, infected recipients with LCMVArm to induce an acute infection, and monitored the accumulation and differentiation of virus-specific CTLs over time (Fig. 3 A). P14 CTLs lacking MDR1 failed to accumulate to wild-type levels in the blood of LCMV-infected mice, with the relative abundance of MDR1-deficient vs. MDR1-sufficient P14 cells decreasing markedly within the first 1–2 weeks of LCMV infection before stabilizing after day 35 (Fig. 3 B). Thus, MDR1 function may be particularly important during the early response of antigen-primed CTLs. Frequencies and absolute numbers of MDR1-deficient P14 cells were also reduced in spleens of infected mice, relative to MDR1-sufficient counterparts; this was evident at the peak of the effector response (day 8), as well as at both early (day 35) and late (day 90) memory time points (Fig. 3, C and D). Loss of MDR1 impacted both Klrg1+IL-7Rαlo and Klrg1−IL-7Rαhi CTLs equally (Fig. 3, C and D), further suggesting that MDR1 regulates early events in the activation or expansion of primed CTLs, as opposed to selectively regulating effector or memory CTL differentiation per se.

MDR1 is required for CTL responses to intracellular pathogens in vivo. (A) Naive CD8+ T cells from Thy1.1+Thy1.2+ MDR1-sufficient (P14; blue) or Thy1.1+Thy1.2−Abcb1a/1b−/− (P14.Mdr1ko; red) mice were purified, mixed at 1:1 ratios, and cotransferred into wild-type B6 (Thy1.2+) hosts, which were subsequently infected with LCMVArm; responses of virus-specific P14 cells with or without MDR1 were followed in blood and spleens over time. (B) Top: Frequencies of MDR1-sufficient (P14; blue) or MDR1-deficient (P14.Mdrko; red) P14 cells in peripheral blood of LCMV-infected mice over time. Live (viability dye−) CD8+Thy1.1+CD44hi P14 cells are shown; representative of ≤33 mice analyzed over three independent experiments. Bottom left: Mean percentages ± SEM of MDR1-sufficient (P14; blue) or MDR1-deficeint (P14.Mdr1ko; red) P14 cells in peripheral blood of LCMV-infected mice, determined by flow cytometry as above, at days 8 (n = 33), 15 (n = 18), 35 (n = 23), 70 (n = 15), and 90 (n = 15) after infection. Percentages shown are of total live CD8+ T cells in blood. ***P ≤ 0.001, ****P ≤ 0.0001; paired two-tailed Student’s t test. Bottom right: Mean relative abundance (Log2 fold-change; ± SEM) of congenically transferred MDR1-deficient vs. MDR1-sufficient P14 cells at days 8 (n = 33), 15 (n = 18), 35 (n = 23), 70 (n = 15), and 90 (n = 15) after infection. Dotted horizontal line indicates the starting 1:1 ratio (Log2 fold-change = 0). (C) Abundance and phenotypes (TE; Klrg1hiIL-7Rαlo; MP; Klrg1loIL-7Rαhi) of MDR1-sufficient (P14; blue) or MDR1-deficient (P14.Mdr1ko; red) P14 CTLs in spleens of LCMV-infected mice at days 8, 35, and 90 after LCMV infection as above. Data are representative of ≤14 mice per time point analyzed over two independent experiments. (D) Top: Percentages (left) or absolute numbers (right) of MDR1-sufficient (P14; blue) or MDR1-deficient (P14.Mdr1ko; red) P14 CTLs in spleens of LCMV-infected mice, determined by flow cytometry as above, at days 8 (n = 8), 35 (n = 9), or 90 (n = 14) after infection. Individual data points for each animal are shown; black lines connect data from each mouse analyzed over two independent experiments. Bottom: Mean percentages ± SEM of Klrg1+IL-7Rα− (left) or Klrg1−IL-7Rα+ (right) cells among MDR1-sufficeint (P14; blue) or MDR1-deficient (P14.Mdr1ko; red) P14 CTLs from spleens of LCMV-infected mice at days 8 (n = 8), 35 (n = 9), or 90 (n = 14), determined by flow cytometry as in C. *P ≤ 0.05, **P ≤ 0.01, ****P ≤ 0.0001; paired two-tailed Student’s t test. (E) Thy1.1+Thy1.2+ MDR1-sufficient (P14; blue) or Thy1.1+Thy1.2− MDR1-deficient (P14.Mdr1ko; red) long-lived memory P14 cells were FACS purified from congenically transferred and LCMV-infected wild-type B6 mice (as in A) on day 90 after infection. Memory cells were transferred into separate groups of naive B6 recipients, which were then challenged with recombinant LM-gp33. A third group of unmanipulated (no P14 memory cells) naive B6 mice were infected with an equal dose of LM-gp33. (F) Kaplan-Meier survival curves (left) or mean weight loss (right; ± SEM) in LM-gp33 infected mice as in E. Both graphs incorporate data from 20 mice per group infected over two independent experiments. *P ≤ 0.05, ****P ≤ 0.0001; two-way ANOVA. NS, not significant. (G) Mean CFU (c.f.u.) ± SEM of LM-gp33 in spleens of mice 5 d after LM-gp33 infection (as in F) for no P14 transfer recipients (n = 6), MDR1-sufficient P14 recipients (n = 10), and MDR1-deficient P14 recipients (n = 7). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001; one-way ANOVA with Tukey’s correction for multiple comparisons. (H) Frequencies of MDR1-sufficient (P14; blue) or MDR1-deficient (P14.Mdr1ko; red) memory P14 cells in spleens of LM-gp33–infected animals at day 5 after infection (as in E–G). Live (viability dye−) CD8+ T cells are shown; representative of ≤10 mice per group infected over two independent experiments. (I) Mean absolute numbers ± SEM of MDR1-sufficient (P14; blue) or MDR1-deficient (P14.Mdr1ko; red) P14 cells in spleens of LM-gp33–infected animals at day 5, determined by flow cytometry as in H for no P14 transfer recipients (n = 6), MDR1-sufficient P14 recipients (n = 10), and MDR1-deficient P14 recipients (n = 7). *P < 0.05; paired two-tailed Student’s t test.
MDR1 is required for CTL responses to intracellular pathogens in vivo. (A) Naive CD8+ T cells from Thy1.1+Thy1.2+ MDR1-sufficient (P14; blue) or Thy1.1+Thy1.2−Abcb1a/1b−/− (P14.Mdr1ko; red) mice were purified, mixed at 1:1 ratios, and cotransferred into wild-type B6 (Thy1.2+) hosts, which were subsequently infected with LCMVArm; responses of virus-specific P14 cells with or without MDR1 were followed in blood and spleens over time. (B) Top: Frequencies of MDR1-sufficient (P14; blue) or MDR1-deficient (P14.Mdrko; red) P14 cells in peripheral blood of LCMV-infected mice over time. Live (viability dye−) CD8+Thy1.1+CD44hi P14 cells are shown; representative of ≤33 mice analyzed over three independent experiments. Bottom left: Mean percentages ± SEM of MDR1-sufficient (P14; blue) or MDR1-deficeint (P14.Mdr1ko; red) P14 cells in peripheral blood of LCMV-infected mice, determined by flow cytometry as above, at days 8 (n = 33), 15 (n = 18), 35 (n = 23), 70 (n = 15), and 90 (n = 15) after infection. Percentages shown are of total live CD8+ T cells in blood. ***P ≤ 0.001, ****P ≤ 0.0001; paired two-tailed Student’s t test. Bottom right: Mean relative abundance (Log2 fold-change; ± SEM) of congenically transferred MDR1-deficient vs. MDR1-sufficient P14 cells at days 8 (n = 33), 15 (n = 18), 35 (n = 23), 70 (n = 15), and 90 (n = 15) after infection. Dotted horizontal line indicates the starting 1:1 ratio (Log2 fold-change = 0). (C) Abundance and phenotypes (TE; Klrg1hiIL-7Rαlo; MP; Klrg1loIL-7Rαhi) of MDR1-sufficient (P14; blue) or MDR1-deficient (P14.Mdr1ko; red) P14 CTLs in spleens of LCMV-infected mice at days 8, 35, and 90 after LCMV infection as above. Data are representative of ≤14 mice per time point analyzed over two independent experiments. (D) Top: Percentages (left) or absolute numbers (right) of MDR1-sufficient (P14; blue) or MDR1-deficient (P14.Mdr1ko; red) P14 CTLs in spleens of LCMV-infected mice, determined by flow cytometry as above, at days 8 (n = 8), 35 (n = 9), or 90 (n = 14) after infection. Individual data points for each animal are shown; black lines connect data from each mouse analyzed over two independent experiments. Bottom: Mean percentages ± SEM of Klrg1+IL-7Rα− (left) or Klrg1−IL-7Rα+ (right) cells among MDR1-sufficeint (P14; blue) or MDR1-deficient (P14.Mdr1ko; red) P14 CTLs from spleens of LCMV-infected mice at days 8 (n = 8), 35 (n = 9), or 90 (n = 14), determined by flow cytometry as in C. *P ≤ 0.05, **P ≤ 0.01, ****P ≤ 0.0001; paired two-tailed Student’s t test. (E) Thy1.1+Thy1.2+ MDR1-sufficient (P14; blue) or Thy1.1+Thy1.2− MDR1-deficient (P14.Mdr1ko; red) long-lived memory P14 cells were FACS purified from congenically transferred and LCMV-infected wild-type B6 mice (as in A) on day 90 after infection. Memory cells were transferred into separate groups of naive B6 recipients, which were then challenged with recombinant LM-gp33. A third group of unmanipulated (no P14 memory cells) naive B6 mice were infected with an equal dose of LM-gp33. (F) Kaplan-Meier survival curves (left) or mean weight loss (right; ± SEM) in LM-gp33 infected mice as in E. Both graphs incorporate data from 20 mice per group infected over two independent experiments. *P ≤ 0.05, ****P ≤ 0.0001; two-way ANOVA. NS, not significant. (G) Mean CFU (c.f.u.) ± SEM of LM-gp33 in spleens of mice 5 d after LM-gp33 infection (as in F) for no P14 transfer recipients (n = 6), MDR1-sufficient P14 recipients (n = 10), and MDR1-deficient P14 recipients (n = 7). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001; one-way ANOVA with Tukey’s correction for multiple comparisons. (H) Frequencies of MDR1-sufficient (P14; blue) or MDR1-deficient (P14.Mdr1ko; red) memory P14 cells in spleens of LM-gp33–infected animals at day 5 after infection (as in E–G). Live (viability dye−) CD8+ T cells are shown; representative of ≤10 mice per group infected over two independent experiments. (I) Mean absolute numbers ± SEM of MDR1-sufficient (P14; blue) or MDR1-deficient (P14.Mdr1ko; red) P14 cells in spleens of LM-gp33–infected animals at day 5, determined by flow cytometry as in H for no P14 transfer recipients (n = 6), MDR1-sufficient P14 recipients (n = 10), and MDR1-deficient P14 recipients (n = 7). *P < 0.05; paired two-tailed Student’s t test.
We also asked whether long-lived memory CTLs that develop in the absence of MDR1 are functional. For this, we FACS-purified Thy1.1+Thy1.2+ MDR1-sufficient or Thy1.1+Thy1.2− MDR1-deficient memory P14 cells from spleens of congenically transferred mice 90 d after LCMV infection, transferred 30,000 cells of each genotype into separate groups of naive B6 recipients, and then challenged these animals with a high dose (80,000 CFUs) of recombinant Listeria monocytogenes expressing gp33 (LM-gp33), which requires exogenous P14 memory cells for protection (Olson et al., 2013); unmanipulated naive B6 mice (receiving no P14 cells) were infected in parallel to confirm memory CTL-dependent protection (Fig. 3 E). Indeed, memory CTL-dependent protection against LM-gp33 challenge also required MDR1, as both naive B6 mice and mice receiving MDR1-deficient P14 memory cells displayed increased morbidity (weight loss) and mortality compared with recipients of MDR1-sufficient P14 memory cells (Fig. 3 F). Among animals surviving until day 5, significantly more L.monocytogenes was recovered from spleens of MDR1-deficient vs. MDR1-sufficient P14 recipients, whereas both the frequency and absolute numbers of MDR1-deficient P14 cells were reduced relative to MDR1-sufficient controls (Fig. 3, G–I). These data suggest that both primary and secondary CTL responses to intracellular pathogens require endogenous MDR1 function. By contrast, we found no evidence that MDR1 regulates the development or homeostasis of naive CD8+ T cells as (1) neither FVB- nor B6-derived MDR1-null mice showed altered frequencies or numbers of naive CD8+ T cells, relative to wild-type littermates; and (2) loss of MDR1 affected neither the engraftment nor the persistence (over 8 d) of naive P14 cells transferred into uninfected recipient mice (data not shown).
MDR1 enforces survival and mitochondrial fitness in early mitotic CTLs
To explore the timing and mechanisms by which endogenous MDR1 function might promote CTL responses to infection, we performed single-cell RNA-seq (scRNA-seq) analysis on congenically cotransferred control and MDR1-deficient P14 cells 8 d after LCMVArm infection. After normalizing gene expression and deconvoluting cell genotypes (based on antibody hashtags), we reduced the dimensionality of the data with Uniform Manifold Approximation and Projection (UMAP; Becht et al., 2018) and performed Louvain clustering. Both control and MDR1-deficient P14 cells segregated into five cell clusters, including early mitotic cells, Klrg1hi TE cells, and Il7rhi MP cells (Fig. 4 A). The expected distribution of control and MDR1-deficient CTLs was observed in all but one cluster (cluster 3), which corresponded to early mitotic CTLs (expressing high levels of Cdk1, Pcna, and Mki67) and which contained significantly fewer than expected MDR1-null cells (chi-square P = 0.396E-06; Fig. 4, B and C). Among this actively dividing CTL cluster, cells lacking MDR1 displayed increased gene expression associated with autophagy and apoptosis (Fig. 4 D); many of these genes were also increased in all MDR1-deficient vs. control CTLs, irrespective of cluster (data not shown). As autophagy is a major pathway through which damaged mitochondria are cleared (Um and Yun, 2017) and cell-intrinsic apoptosis is initiated by the release of cytochrome c from depolarized mitochondrial membranes (Green, 2019), these transcriptional signatures suggest that a primary function of MDR1 may be to promote mitochondrial fitness in recently activated CTLs. Consistent with this possibility, day 8 LCMV-specific CTLs produced more mitochondrial superoxide (mitoSOX) and less ATP in the absence of MDR1 (Fig. 4, E–G).

MDR1 enforces survival and mitochondrial fitness in early mitotic CTLs. (A) scRNA-seq analysis of congenically transferred wild-type and MDR1-deficient (Abcb1a/1b−/−; Mdr1ko) P14 CTLs from LCMV-infected mice at day 8 after infection (as in Fig. 3 A). UMAP was used to visualize dimensionally reduced transcriptomics data. Left: Louvain clustering identified five cell clusters (color-coded); Right: CTL distribution by genotype (P14; blue, P14.Mdr1ko; orange) is shown. (B) Distribution of control (P14; black) or MDR1-deficient (P14.Mdr1ko; gray) CTLs across the five clusters identified in A. Percentages of total cells (per genotype) is shown; exact chi-square P values are indicated. (C) Normalized expression (TPM) of cluster-defining genes as in A. (D) Top: Volcano plot showing differential gene expression between cluster 3 (mitotic) control (P14) and MDR1-deficient (P14.Mdr1ko) CTLs (cluster 3 identified as in A and C). Gene expression is based on the mean expression among all cells (per genotype) within the cluster; genes significantly increased or decreased (P < 0.05, DESeq2) in MDR1-deficient vs. control CTLs are highlighted red or blue, respectively. Bottom: GO terms significantly enriched (P < 0.05, ClusterProfiler) among genes identified above that are increased in MDR1-deficient vs. control P14 cells. All scRNA-seq data are from P14 cells sorted from five pooled spleens of LCMV-infected mice; data shown are analyzed from one independent experiment and representative of two independent experiments. TOR, target of rapamycin. (E) Mitochondrial function (fxn) was assessed in congenically transferred MDR1-sufficient (P14; blue) and MDR1-deficient (P14.Mdr1ko; red) P14 CTLs 8 d after LCMV infection. (F) Left: Ex vivo analysis of control (Thy1.1+Thy1.2+; blue) or MDR1-deficient (Thy1.1+Thy1.2−; red) P14 CTLs or endogenous (Endog.; gray) CD8+ T cells from spleens of LCMV-infected or uninfected B6 mice. Middle: Mitochondrial superoxide production, determined by mitoSOX staining and flow cytometry, in MDR1-sufficient (P14; blue) or MDR1-deficient (P14.Mdr1ko; red) P14 CTLs or in endogenous (Endog.; gray) CD8+ T cells from congenically transferred and LCMV-infected wild-type B6 mice (Top; as in E). Parallel mitoSOX staining was performed on endogenous CD8+ T cells from spleens of uninfected wild-type B6 mice; CD8+ T cells from uninfected animals were treated ex vivo with or without the mitochondrial complex III inhibitor, Antimycin A, to validate staining (Bottom). Color-matched text indicates mitoSOX MFIs; representative of five mice analyzed over two independent experiments. Right: Mean relative mitoSOX MFIs (n = 5) in MDR1-sufficient (P14; blue) or MDR1-deficient (P14.Mdr1ko; red) P14 CTLs or in endogenous (Endog.; gray) CD8+ T cells, determined by flow cytometry as above. Individual data points for each animal are shown. **P < 0.01, paired two-tailed Student’s t test. (G) Mean ATP concentration (n = 5), determined by ex vivo CellTiter-Glo Luminescent Viability Assay at day 8, in MDR1-sufficient (P14; blue) or MDR1-deficient (P14.Mdr1ko; red) P14 CTLs or in endogenous (Endog.; gray) CD8+ T cells, from congenically transferred and LCMV-infected mice as in E and F. Individual data points for each animal are shown analyzed over three independent experiments. **P < 0.01, paired two-tailed Student’s t test.
MDR1 enforces survival and mitochondrial fitness in early mitotic CTLs. (A) scRNA-seq analysis of congenically transferred wild-type and MDR1-deficient (Abcb1a/1b−/−; Mdr1ko) P14 CTLs from LCMV-infected mice at day 8 after infection (as in Fig. 3 A). UMAP was used to visualize dimensionally reduced transcriptomics data. Left: Louvain clustering identified five cell clusters (color-coded); Right: CTL distribution by genotype (P14; blue, P14.Mdr1ko; orange) is shown. (B) Distribution of control (P14; black) or MDR1-deficient (P14.Mdr1ko; gray) CTLs across the five clusters identified in A. Percentages of total cells (per genotype) is shown; exact chi-square P values are indicated. (C) Normalized expression (TPM) of cluster-defining genes as in A. (D) Top: Volcano plot showing differential gene expression between cluster 3 (mitotic) control (P14) and MDR1-deficient (P14.Mdr1ko) CTLs (cluster 3 identified as in A and C). Gene expression is based on the mean expression among all cells (per genotype) within the cluster; genes significantly increased or decreased (P < 0.05, DESeq2) in MDR1-deficient vs. control CTLs are highlighted red or blue, respectively. Bottom: GO terms significantly enriched (P < 0.05, ClusterProfiler) among genes identified above that are increased in MDR1-deficient vs. control P14 cells. All scRNA-seq data are from P14 cells sorted from five pooled spleens of LCMV-infected mice; data shown are analyzed from one independent experiment and representative of two independent experiments. TOR, target of rapamycin. (E) Mitochondrial function (fxn) was assessed in congenically transferred MDR1-sufficient (P14; blue) and MDR1-deficient (P14.Mdr1ko; red) P14 CTLs 8 d after LCMV infection. (F) Left: Ex vivo analysis of control (Thy1.1+Thy1.2+; blue) or MDR1-deficient (Thy1.1+Thy1.2−; red) P14 CTLs or endogenous (Endog.; gray) CD8+ T cells from spleens of LCMV-infected or uninfected B6 mice. Middle: Mitochondrial superoxide production, determined by mitoSOX staining and flow cytometry, in MDR1-sufficient (P14; blue) or MDR1-deficient (P14.Mdr1ko; red) P14 CTLs or in endogenous (Endog.; gray) CD8+ T cells from congenically transferred and LCMV-infected wild-type B6 mice (Top; as in E). Parallel mitoSOX staining was performed on endogenous CD8+ T cells from spleens of uninfected wild-type B6 mice; CD8+ T cells from uninfected animals were treated ex vivo with or without the mitochondrial complex III inhibitor, Antimycin A, to validate staining (Bottom). Color-matched text indicates mitoSOX MFIs; representative of five mice analyzed over two independent experiments. Right: Mean relative mitoSOX MFIs (n = 5) in MDR1-sufficient (P14; blue) or MDR1-deficient (P14.Mdr1ko; red) P14 CTLs or in endogenous (Endog.; gray) CD8+ T cells, determined by flow cytometry as above. Individual data points for each animal are shown. **P < 0.01, paired two-tailed Student’s t test. (G) Mean ATP concentration (n = 5), determined by ex vivo CellTiter-Glo Luminescent Viability Assay at day 8, in MDR1-sufficient (P14; blue) or MDR1-deficient (P14.Mdr1ko; red) P14 CTLs or in endogenous (Endog.; gray) CD8+ T cells, from congenically transferred and LCMV-infected mice as in E and F. Individual data points for each animal are shown analyzed over three independent experiments. **P < 0.01, paired two-tailed Student’s t test.
MDR1 mitigates activation-induced oxidative stress
Elucidating early activation events in small/physiological numbers of antigen-specific CD8+ T cells in vivo is not feasible. Therefore, we used in vitro cell culture to more precisely map the early events in CD8+ T cell activation and proliferation that may be impacted by MDR1 deficiency. Consistent with our in vivo results, naive CD8+ T cells lacking MDR1 displayed reduced accumulation vs. wild-type counterparts upon in vitro differentiation into either effector- or memory-like CTLs (Fig. 5 A). Blocking MDR1 transport activity with elacridar (Hyafil et al., 1993) also reduced the accumulation of wild-type CTLs—to a similar extent as Abcb1a/1b ablation—but had no impact on the already blunted expansion of MDR1-null CTLs (Fig. 5 A). In addition, MDR1 promoted CTL accumulation in a cell-intrinsic manner, which we confirmed using congenic coculture experiments where CD45.1 wild-type and CD45.2 MDR1-deficient CTLs were activated and expanded together in the same tissue culture wells (Fig. S2 F). Reduced accumulation of MDR1-deficient CTLs was associated with increased cell death (Fig. 5 B), whereas MDR1-deficient cells that remained viable displayed wild-type level activation (CD69 upregulation) and proliferation (carboxyfluorescein diacetate succinimidyl ester dilution) kinetics (Fig. S2 G). Also in line with our in vivo results, loss or inhibition of MDR1 in cultured CTLs led to (1) increased gene expression associated with autophagy and apoptosis (as well as hypoxia; Fig. 5 C and Fig. S2 H); and (2) increased mitoSOX and decreased ATP production (Fig. 5, D–F). These same metabolic parameters were unaffected by MDR1 deficiency in resting naive CD8+ T cells (Fig. 5, D–F). These in vitro results corroborate our in vivo findings and reinforce a model in which MDR1 acts early and selectively in activated CTL to enforce metabolic fitness.

MDR1 suppresses oxidative stress in recently activated CTLs. (A) Expansion of wild-type (B6; blue) or MDR1-deficient (Abcb1a/1b−/−; B6.Mdr1ko; red) CD8+ T cells activated (anti-CD3/anti-CD28) and expanded in vitro in effector-like (high [hi] IL-2 TE, 100 U/ml; top) or memory-like (low [lo] IL-2 TM, 10 U/ml; bottom) conditions in the presence (+E; open circles) or absence (-E; closed circles) of the selective MDR1 antagonist, elacridar. Data are shown as mean absolute numbers ± SEM (n = 8) analyzed over four independent experiments. ***P < 0.001, ****P < 0.0001, two-way ANOVA. (B) Top: Cell death, determined by fixable viability dye staining and flow cytometry, in cocultured CD45.1 wild-type (B6; blue) or CD45.2 MDR1-deficient (B6.Mdr1ko; red) memory-like CTLs expanded for 6 d in the presence or absence of elacridar (lo IL-2 TM day [d] 6; as in A). Color-matched text indicates dead cell percentages; representative of four independent experiments. Bottom: Relative cell death (n = 4) of congenically cultured CD45.1 wild-type (B6; blue) or CD45.2 MDR1-deficient (B6.Mdr1ko; red) d6 lo IL-2 TM cells, determined by flow cytometry as above. Individual data points for each animal are shown; horizontal lines indicate mean values. *P < 0.05; one-way ANOVA with Tukey’s correction for multiple comparisons. (C) Heatmap of representative genes whose expression is increased (Up) or decreased (Down) by loss or inhibition of MDR1 in d6 lo IL-2 TM cells, determined by RNA-seq. Genes were selected for presentation based on their inclusion in significantly enriched GO pathways, determined by the R script package ClusterProfiler. Data incorporate three independent experiments. max., maximum; min., minimum. (D) Mitochondrial superoxide production, determined by mitoSOX staining and flow cytometry, in ex vivo–isolated B6 wild-type (B6; black) or MDR1-null (B6.Mdr1ko; gray) naive CD8+ T cells or in d6-expanded B6 wild-type (B6; blue) or MDR1-deficient (B6.Mdr1ko; red) lo IL-2 TM cells treated with or without elacridar as above. The mitochondrial complex III inhibitor, Antimycin A, was added to resting (naive) wild-type CD8+ T cells to induce mitoSOX production as a positive staining control. Color-matched text indicates mitoSOX MFIs; representative of five independent experiments. (E) Relative mitoSOX MFIs (n = 5), determined by flow cytometry as in D, in resting or d6-expanded B6 wild-type or MDR1ko CD8+ T cells. Individual data points for each animal are shown; horizontal lines indicate mean values. *P < 0.05, **P < 0.01, one-way ANOVA with Tukey’s correction for multiple comparisons. ko, knockout; NS, not significant; wt, wild-type. (F) Relative ATP concentrations (n = 5), determined by CellTiter-Glo, in resting or d6-expanded B6 wild-type or MDR1ko CD8+ T cells, as in D and E. Individual data points for each animal are shown; horizontal lines indicate mean values. **P < 0.01, one-way ANOVA with Tukey’s correction for multiple comparisons. NS, not significant. (G) Left: Ratio of reduced (GSH) to oxidized (GSSG) glutathione (± SEM; n = 4) in wild-type (B6; blue) or MDR1-deficient (B6.Mdr1ko; red) naive CD8+ T cells before or after in vitro activation (anti-CD3/anti-CD28). Absolute concentration of oxidized GSSG (middle) or total GSH (right; ± SEM; n = 4) in wild-type (B6; blue) or MDR1-deficient (B6.Mdr1ko; red) naive CD8+ T cells before or after in vitro activation as above. Data are representative of four mice per group analyzed over three independent experiments. *P ≤ 0.05, **P ≤ 0.01; paired two-tailed Student’s t test. (H) Mean ATP concentration (± SEM; n = 4), determined by ex vivo CellTiter-Glo Luminescent Viability Assay in MDR1-sufficient (B6; blue) or MDR1-deficient (B6.Mdr1ko; red) naive CD8+ T cells before or after in vitro activation as in G. **P ≤ 0.01; paired two-tailed Student’s t test.
MDR1 suppresses oxidative stress in recently activated CTLs. (A) Expansion of wild-type (B6; blue) or MDR1-deficient (Abcb1a/1b−/−; B6.Mdr1ko; red) CD8+ T cells activated (anti-CD3/anti-CD28) and expanded in vitro in effector-like (high [hi] IL-2 TE, 100 U/ml; top) or memory-like (low [lo] IL-2 TM, 10 U/ml; bottom) conditions in the presence (+E; open circles) or absence (-E; closed circles) of the selective MDR1 antagonist, elacridar. Data are shown as mean absolute numbers ± SEM (n = 8) analyzed over four independent experiments. ***P < 0.001, ****P < 0.0001, two-way ANOVA. (B) Top: Cell death, determined by fixable viability dye staining and flow cytometry, in cocultured CD45.1 wild-type (B6; blue) or CD45.2 MDR1-deficient (B6.Mdr1ko; red) memory-like CTLs expanded for 6 d in the presence or absence of elacridar (lo IL-2 TM day [d] 6; as in A). Color-matched text indicates dead cell percentages; representative of four independent experiments. Bottom: Relative cell death (n = 4) of congenically cultured CD45.1 wild-type (B6; blue) or CD45.2 MDR1-deficient (B6.Mdr1ko; red) d6 lo IL-2 TM cells, determined by flow cytometry as above. Individual data points for each animal are shown; horizontal lines indicate mean values. *P < 0.05; one-way ANOVA with Tukey’s correction for multiple comparisons. (C) Heatmap of representative genes whose expression is increased (Up) or decreased (Down) by loss or inhibition of MDR1 in d6 lo IL-2 TM cells, determined by RNA-seq. Genes were selected for presentation based on their inclusion in significantly enriched GO pathways, determined by the R script package ClusterProfiler. Data incorporate three independent experiments. max., maximum; min., minimum. (D) Mitochondrial superoxide production, determined by mitoSOX staining and flow cytometry, in ex vivo–isolated B6 wild-type (B6; black) or MDR1-null (B6.Mdr1ko; gray) naive CD8+ T cells or in d6-expanded B6 wild-type (B6; blue) or MDR1-deficient (B6.Mdr1ko; red) lo IL-2 TM cells treated with or without elacridar as above. The mitochondrial complex III inhibitor, Antimycin A, was added to resting (naive) wild-type CD8+ T cells to induce mitoSOX production as a positive staining control. Color-matched text indicates mitoSOX MFIs; representative of five independent experiments. (E) Relative mitoSOX MFIs (n = 5), determined by flow cytometry as in D, in resting or d6-expanded B6 wild-type or MDR1ko CD8+ T cells. Individual data points for each animal are shown; horizontal lines indicate mean values. *P < 0.05, **P < 0.01, one-way ANOVA with Tukey’s correction for multiple comparisons. ko, knockout; NS, not significant; wt, wild-type. (F) Relative ATP concentrations (n = 5), determined by CellTiter-Glo, in resting or d6-expanded B6 wild-type or MDR1ko CD8+ T cells, as in D and E. Individual data points for each animal are shown; horizontal lines indicate mean values. **P < 0.01, one-way ANOVA with Tukey’s correction for multiple comparisons. NS, not significant. (G) Left: Ratio of reduced (GSH) to oxidized (GSSG) glutathione (± SEM; n = 4) in wild-type (B6; blue) or MDR1-deficient (B6.Mdr1ko; red) naive CD8+ T cells before or after in vitro activation (anti-CD3/anti-CD28). Absolute concentration of oxidized GSSG (middle) or total GSH (right; ± SEM; n = 4) in wild-type (B6; blue) or MDR1-deficient (B6.Mdr1ko; red) naive CD8+ T cells before or after in vitro activation as above. Data are representative of four mice per group analyzed over three independent experiments. *P ≤ 0.05, **P ≤ 0.01; paired two-tailed Student’s t test. (H) Mean ATP concentration (± SEM; n = 4), determined by ex vivo CellTiter-Glo Luminescent Viability Assay in MDR1-sufficient (B6; blue) or MDR1-deficient (B6.Mdr1ko; red) naive CD8+ T cells before or after in vitro activation as in G. **P ≤ 0.01; paired two-tailed Student’s t test.
Increased mitoSOX production (i.e., oxidative stress) can be both a cause and a consequence of defective ATP synthesis (Martínez-Reyes and Cuezva, 2014; Zorov et al., 2014). Thus, we finally sought to understand whether endogenous MDR1 function more directly regulates oxidation reduction (redox) or bioenergetic pathways. For this, we analyzed the kinetics of oxidative stress and ATP synthesis in recently activated naive CD8+ T cells that have or lack MDR1, and we focused on the synthesis and redox status of glutathione—an endogenous antioxidant cofactor produced by activated T cells to buffer oxidized free radicals (Wang et al., 2011)—to obtain a more comprehensive view of oxidative stress. As expected, TCR stimulation diminished reduced glutathione (GSH) stores in both wild-type and MDR1-deficient CD8+ T cells and within 12 h of TCR stimulation (Fig. 5 G). By contrast, only wild-type cells were capable of replenishing reduced GSH levels over the next 12–36 h (Fig. 5 G). Decreased levels of reduced GSH in activated MDR1-null CD8+ T cells were associated specifically with accumulation of oxidized glutathione dipeptides (GSSG), whereas both total and reduced GSH synthesis remained intact in the absence of MDR1 (Fig. 5 G, data not shown). In addition, alterations in the GSH redox balance preceded deficits in ATP synthesis in MDR1-deficient CTLs, the latter of which did not manifest until 48 h after stimulation (Fig. 5 H). Together, these data suggest that the primary endogenous function of MDR1 in CTLs may be to limit activation-induced oxidative stress.
General conclusions
By establishing contemporary reporter and loss-of-function alleles in mice, we have shown that the MDR1 transporter is highly expressed in CTLs, where it is regulated by interplay between Runx transcription factors and T cell activation signals and necessary for both primary and secondary responses to infection. These data highlight a previously unrecognized endogenous function of MDR1 in cell-mediated immunity and raise questions about the safety and utility of systemic MDR1 inhibitors in cancer therapy. At a molecular level, both in vivo scRNA-seq and in vitro mechanistic studies support a model in which MDR1 function is summoned rapidly after antigen-dependent CTL priming to buffer TCR-driven oxidized free radical production. It is interesting in this regard that production of reactive oxygen/nitrogen species is considered a general response of adaptive lymphocytes to antigen receptor signaling, whereas most noncytotoxic lymphocytes lack constitutive MDR1 expression (Fig. 1). Therefore, it is possible that the metabolic programs of activated CTLs are unique among adaptive lymphocytes in their generation of oxidized free radicals or endogenous MDR1 transport substrates. Finally, decreased mitochondrial metabolism and increased oxidative stress shown here in MDR1-deficient CTLs resembles that previously described in MDR1-deficient enterocytes, tumor cells, and CD4+ T cells exposed to bile acids in the intestine (Cao et al., 2017; Ho et al., 2018; Hwang et al., 2019). Thus, it remains possible that a unifying molecular function of MDR1 may eventually be traced to the transport of a common class of endogenous metabolites that is produced by all cells in a manner commensurate with responses to oxidative stress.
Materials and methods
Mice
C57BL/6J (B6)–derived Abcb1aAME/+ reporter mice were generated as previously described (Cao et al., 2017). B6-derived wild-type (stock no. 000664) and Rag1−/− (stock no. 002216) mice were purchased from The Jackson Laboratory. FVB/N (FVB)-derived Abcb1a/1b−/− mice (model no. 1487) were purchased from Taconic and backcrossed for 10 generations under a breeding license to obtain B6-derived Abcb1a/1b−/− mice. B6-derived Abcb1aAME/+ reporter and Abcb1a/1b−/− mice were bred to Thy1.1 congenic/P14 TCR transgenic mice (Wang et al., 2018). All mice were bred and used in experiments in accordance with protocols approved by the Scripps Florida institutional animal care and use committee.
T cell isolation and culture
Naive CD8+ T cells were isolated from spleen and peripheral lymph nodes by magnetic negative isolation (Stem Cell Technologies, Inc.). Purified CD8+ T cells were cultured in DMEM supplemented with 10% heat-inactivated FBS, essential amino acids, nonessential amino acids, sodium pyruvate, arginine/asparagine/folic acid, 10 mM Hepes, 2 mM l-glutamine, 50 µM 2-mercaptoethanol, penicillin-streptomycin, and gentamycin (all from Thermo Fisher Scientific). For in vitro CD8+ T cell activation, magnetically isolated CD8+ T cells were activated at 4 × 105 cells/cm2 and 106 cells/ml in 24-well flat bottom plates coated with anti-CD3 (1 µg/ml) and anti-CD28 (1 µg/ml; BioLegend) after precoating with 200 µg/ml goat anti-hamster IgG (Thermo Fisher Scientific). Activated T cells were removed from antibody-coated wells after 48 h and recultured at 5 × 105 cells/ml in the indicated concentrations of recombinant human IL-2 (rhIL-2; National Institutes of Health Biorepository). For in vitro coculture experiments, 1:1 mixtures of magnetically enriched CD45.1 wild-type and CD45.2 Abcb1a/1b−/− naive CD8+ T cells were activated and cultured as above. In some experiments, MDR1 was blocked by adding elacridar (100 nM; Sigma-Aldrich) immediately before T cell activation and again every 36–48 h.
Retroviral plasmids and transductions
GFP- or ametrine-expressing murine retroviral vectors containing shRNAmirs against Cd4, Runx1, Runx3, and Cbfb have been described and validated previously (Wang et al., 2018). Generation of retroviral particles and T cell transductions were performed as previously described (Cao et al., 2017) using TransIT-LT1 transfection reagent (Mirus). Briefly, CD8+ T cells were transduced 24 h after activation; culture media was replaced with viral supernatant containing 10 µg/ml polybrene and centrifuged at 2,000 rpm for 1 h at room temperature. For in vitro culture experiments, transduced cells were kept in a 37°C incubator until 48 h after activation, after which time retroviral supernatants were replaced with fresh media containing 10 or 100 U/ml of rhIL-2, as indicated. For adoptive transfer of transduced P14 cells, T cell cultures were incubated for 3 h in a 37°C CO2 incubator after retroviral infections and centrifugation, and 5 × 105 cells were transferred into naive B6 mice for LCMV infection as above.
Flow cytometry
All surface and intracellular FACS stains were performed at 4°C for 30 min and then washed with PBS before data acquisition. For Rh123 efflux, cells were labeled with 0.1 µM Rh123 (Sigma-Aldrich) for 30 min at 4°C in ice-cold complete media. Labeled cells were then washed with ice-cold fresh media, resuspended in fresh room temperature media, and incubated for 30–60 min in a 37°C incubator. An aliquot of labeled cells was treated with elacridar (100 nM) to monitor background efflux activity. Any surface stains performed after Rh123 efflux were conducted at 4°C as above. Mitochondrial superoxide was quantified via FACS analysis of mitoSOX Red staining (Thermo Fisher Scientific; excitation/emission = 510/580 nm). Briefly, cells were labeled concomitantly with mitoSOX Red (5 µM) and cell surface FACS antibodies in PBS and incubated for 15 min in a 37°C incubator protected from light. To confirm specificity, an aliquot of cells was treated for 15 min with the mitochondrial complex III inhibitor, Antimycin A (1 µM; Abcam), at 37°C before mitoSOX staining. Labeled cells were washed twice and resuspended with warm PBS before acquisition on the cytometer. Anti-mouse antibodies used for FACS analysis included Alexa700-CD34, BV605-CD11b, BV711-CD127, PE-CF594-CD135, BV711-CD138, FITC-CD1d, Percp-Cy5.5-CD21/CD35, PE-CF594-CD25, PE-Cy7-CD27, BV421-CD5, Alexa700-CD8a, BUV395-CD8a, PE-CF594-CD93, PE-Cy7-CD95, BV421-Gr-1, Percp-Cy5.5-IgD, BV650-IgM, BUV395-Ly-6G, BV650-MHCII, BUV395-NK1.1, BV605-Sca1, PE-CF594-Siglec-F (from BD Biosciences); Alexa700-B220, FITC-B220, BV421-c-Kit, PE-c-Kit, BV421-CD103, Percp-Cy5.5-CD115, FITC-CD11b, BV605-CD11b, BV650-CD150, BUV395-CD19, FITC-CD19, FITC-CD25, BV650-CD3, FITC-CD3, BV711-CD4, Percp-Cy5.5-CD4, PE-Cy7-CD44, BV650-CD44, Percp-Cy5.5-CD48, Percp-Cy5.5-CD62L, PE-CD8A, PE-CX3CR1, FITC-Gr-1, BV421-Lin Cocktail, BV711-Ly-6C, Percp-Cy5.5-TCRβ, PE-Cy7-TCRβ, BV605-TCRβ, FITC-CD69, BV421-CD90.1, APC-KLRG1 (from BioLegend); and PE-Cy7-CD11c, PE-Cy7-CD122, FITC-CD16/CD32, FITC-CD24, and PE-CD38 (from eBioscience). Vital dyes used in this study included fixable viability eFluor 506 and eFluor 660 (eBioscience). All FACS data were acquired on LSRII or FACSCanto II instruments (BD Biosciences), and analyzed using FlowJo 9 or 10 software (TreeStar, Inc.). Primary FACS data of endogenous Abcb1aAme reporter expression are available at ImmPort (available at: https://www.immport.org) under the accession no. SDY1595.
Fluorescent microscopy
Spleens from B6 or Abcb1aAME/+ reporter mice were washed with PBS, embedded in Tissue-Tek O.C.T. Compound (Thermo Fisher Scientific) and flash frozen in liquid nitrogen. Tissues were cut on a CM1950 cryostat (Leica) at 5 µm thickness. Slides were postfixed in cold acetone (Sigma-Aldrich), blocked with 5% normal donkey serum (Sigma-Aldrich), and stained with anti-CD11c BOND RTU Primary (Leica Biosystems) with Alexa594 F(ab′)2 and donkey anti-rabbit IgG (H+L) secondary (1:500; Jackson Immunoresearch). Sections were mounted with ProLong Diamond Mountant (Thermo Fisher Scientific). Images were captured on an Eclipse microscope (Nikon).
LCMV/L.monocytogenes infections
5 × 103 naive CD8+ T cells isolated from spleens of wild-type P14, P14.Abcb1aAME/+, or P14.Abcb1a/1b−/− mice were injected (i.v.) into 6–8-wk-old wild-type B6 recipients. The next day, recipient animals were either left uninfected or infected (i.p.) with 2 × 105 PFU LCMVArm. In some experiments, naive CD8+ T cells from spleens of P14.Abcb1aAME/+ reporter mice were activated and transduced in vitro (as above) with GFP-expressing retroviral vectors containing control (shCD4), or Runx-targeting (shRunx1, shRunx3, shCbfb) shRNAmirs before in vivo transfer. 5 × 105 transduced P14 cells were injected (i.v.) into 6–8-wk-old wild-type B6 recipients, which were infected (i.p.) 1 h later with 2 × 105 PFU LCMVArm. For congenic T cell transfer and infection experiments, naive CD8+ T cells isolated from spleens of wild-type P14 (Thy1.1/Th1.2 heterozygous) or P14.Abcb1a/1b−/− (Thy1.1 homozygous) mice were mixed in a 1:1: ratio, and 5 × 103 mixed cells were coadoptively transferred (i.v.) into 6–8-wk-old wild-type B6 recipients before infection (i.p.) with 2 × 105 PFU LCMVArm 24 h later. For LM-gp33 challenge experiments, viable CD8+CD44hi P14 cells with (Thy1.1/Th1.2 heterozygous) or without (Thy1.1 homozygous) MDR1 were FACS-purified from spleens of LCMV-infected mice on day 90 after infection. 3 × 104 cells of each genotype were injected (i.v.) into separate groups of naive (6–8-wk-old) wild-type B6 recipients, which were then infected (i.v.) with 8 × 104 CFU of LM-gp33 24 h later. Mice were sacrificed 5 d after infection. Ex vivo T cell analysis was performed by FACS; bacterial titers were quantified by plating serial dilutions of spleen lysates on brain heart infusion agar as described previously (Olson et al., 2013).
ATP assays
ATP concentrations were quantified using the CellTiter-Glo Luminescent Cell Viability kit (Promega), in accordance with manufacturer’s instructions. Briefly, cells were resuspended in complete media, and 25 µl of titrating cells numbers (104, 5 × 103, 2.5 × 103, and 1.25 × 103) were pipetted, in triplicate, into standard white opaque 384-well microplates (Thermo Fisher Scientific). Control wells were prepared containing complete media without cells to determine background luminescence. Tenfold serial dilutions of ATP (100 mM–0.1 mM) were prepared fresh in complete media to generate ATP standard curves for each plate. An equal volume of 25 µl CellTiter-Glo reagent was added to each sample and shaken for 2 min on an orbital shaker to induce cell lysis. The plate was incubated for an additional 10 min at room temperature to stabilize the luminescent signal. Luminescence was recorded, in duplicate, using a SpectraMax M5 microplate reader (Molecular Devices) with an integration time of 0.5 s per well. ATP concentrations (per 5 × 103 cells) were calculated using the mean corrected luminescence signals, after background subtraction and normalization for cell viability (determined by flow cytometry).
GSH assays
Total, reduced, and oxidized (GSSG) GSH levels were quantified using the GSH/GSSG Ratio Detection Assay Kit (Abcam) following the manufacturer’s instructions. Briefly, 104 cells were lysed and deproteinized using a Deproteinizing (TCA) Sample Preparation Kit (Abcam). 25 µl of the deproteinized lysates were pipetted, in duplicate, into black side/clear bottom 384-well microplates (Greiner Bio-One). Twofold serial dilutions of reduced GSH (10 µM–0.1563 µM) and GSSG (5 µM–0.0781 µM) were prepared fresh for standard curves. For detection of reduced GSH, 25 µl of GSH assay mixture was added into each reduced GSH standard and sample wells. For detection of total GSH, 25 µl of total GSH assay mixture was added into each oxidized GSSG standard and sample wells. Plates were incubated for 30 min in the dark, and fluorescence was recorded at excitation/emission = 490/520 nm on the Molecular Devices SpectraMax M5 microplate reader. Reduced GSH and total GSH concentrations were calculated by fitting corrected (background subtracted) mean fluorescence intensities (MFIs) to standard curves; oxidized GSSG concentrations were calculated based on the difference between total GSH and reduced GSH.
Quantitative PCR (qPCR)
RNA was isolated from cultured or ex vivo isolated cells using RNeasy columns (Qiagen) as per the manufacturer’s instructions using on-column DNase treatment. RNA was used to synthesize cDNA via a high capacity cDNA reverse transcription kit (Life Technologies). Taqman qPCR was performed on a StepOnePlus real time PCR instrument (Life Technologies) using the following Taqman primer/probe sets (Life Technologies): Abcb1a (Mm00440761_m1), Abcb1b (Mm00440736_m1), Runx1 (Mm01213404_m1), Runx3 (Mm01213404_m1), and Gapdh (mm99999915_g1).
Bioinformatics analyses
ChIP-seq
Raw sequencing reads for Runx3 (GSE50131; Lotem et al., 2013), Tbx21 (GSE72408; Dominguez et al., 2015), and Irf4 (GSE54191; Kurachi et al., 2014) were downloaded from the Sequence Read Archive database. Reads were aligned to UCSC mm10 with Bowtie2 (Langmead and Salzberg, 2012), and peaks were called in MACS (Zhang et al., 2008) using base settings. Bigwig files of binding peaks were generated and visualized on the UCSC genome browser. Peaks were filtered in R-studio (using GenomeInfoDb and ChIP blacklist GenomicRanges packages) to remove reads with alternative annotations, mitochondrial DNA, or blacklist sites (Carroll et al., 2014).
ATAC-seq
ATAC-seq data were downloaded from Gene Expression Omnibus (GEO; GSE111149, GSE88987), aligned to UCSC mm10 with Bowtie2 (Langmead and Salzberg, 2012) and visualized on the UCSC genome browser.
RNA-Seq
Next-generation RNA-seq was performed on three biologically independent replicates of B6 wild-type or Abcb1a/1b−/− CD8+ T cells activated and expanded for 6 d in 10 U/ml of rhIL-2–containing media with or without elacridar. High quality total RNA (RNA integrity number >9 by Bioanalyzer; Agilent Technologies) was isolated using RNeasy columns with on-column DNase treatment (Qiagen) and processed with the NEBNext Ultra II ribosomal RNA-depletion module human/mouse/rat (New England BioLabs Inc.) using 400 ng total RNA as input. The ribosomal RNA-depleted RNA was processed using the NEBNext Ultra II directional RNA kit for Illumina (New England BioLabs Inc.) to generate strand-specific libraries. Final libraries were amplified by PCR (10 cycles), size selected using beads to enrich for fragments >400 bp, and purified using Ampure XP beads (Beckman Coulter). Final libraries were validated on the Agilent 2100 Bioanalyzer DNA chips and quantified on the Qubit (Thermo Fisher Scientific). Libraries were pooled at equimolar ratios and loaded onto the Illumina NextSeq500 flow cell and sequenced using 40-bp paired-end chemistry. Approximately 25 million reads pass filter (base quality score >Q30; suggesting <1 error in 1,000 bp) were generated per library. Raw sequencing reads (fastq files) were mapped to the mm10 genome, and normalized transcript abundance, displayed as transcripts per million (TPM), were quantified using Salmon (Patro et al., 2017). Differential gene expression was analyzed using DESeq2 and visualized using the MultiPlot studio module in GenePattern (https://cloud.genepattern.org/gp). Principle component analysis(PCA)of normalized TPM data were computed in R script. Gene ontology (GO) term enrichment analysis (using differentially expressed gene sets defined by DESeq2) was performed using the R package ClusterProfiler (Yu et al., 2012); heatmaps of representative genes within significantly enriched GO pathways were visualized using Morpheus (https://software.broadinstitute.org/morpheus/).
scRNA-seq
scRNA-seq analyses was performed on FACS-purified Thy1.1+Thy1.2+ MDR1-sufficient and Thy1.1+Thy1.2− MDR1-deficient P14 CD8+ T cells from 10 congenically transferred and LCMV-infected wild-type B6 mice at day 8 after infection. P14 cells with or without MDR1 were barcoded with TotalSeq-A anti-mouse hashtag antibodies (BioLegend) to enable sample multiplexing and then counted on the Countess II FL automated cell counter (Thermo Fisher Scientific) using Trypan blue to determine viability and cell number (Stoeckius et al., 2018). Samples were pooled equally and adjusted to a final concentration between 700–1,200 cells/µl; 35,000 total cells were loaded into one lane of the single-cell A chip kit (P/N 1000009). Single-cell gel beads in emulsion (GEM) were generated using the 10X Chromium Controller (10X Genomics). Single-cell GEM and sequencing libraries were generated using the Single-Cell 3′ Library and Gel Bead Kit V2 (P/N 120267) according to the manufacturer’s recommendations. Briefly, GEM-RT was performed on the recovered GEM in a C1000 Touch Thermal Cycler with the following program: 53°C for 45 min, 85°C for 5 min, and held at 4°C. After RT, single-strand cDNA was recovered with the DynaBeads MyOne Silane Beads (Thermo Fisher Scientific; P/N 2000048). The cDNA was amplified with the hashtag oligonucleotide (HTO) additive primer 5′-GTGACTGGAGTTCAGACGTGTGC*T*C-3′ (asterisk indicates a phosphorothioate bond) added at 0.2 µM using the C1000 Touch Thermal Cycler with the 96-Deep Well Reaction Module: 98°C for 3 min; cycled 8×: 98°C for 15 s, 67°C for 20 s, and 72°C for 1 min; 72°C for 1 min; and held at 4°C. Amplified cDNA products were subjected to cleanup, using the SPRIselect Reagent Kit with 0.6× SPRI (Beckman Coulter; P/N B23317) to recover full-length cDNA, and supernatant was purified with 2× SPRI to recover the HTO product. cDNA products were then quantified by Qubit (Thermo Fisher Scientific) and visualized on the Agilent 2100 Bioanalyzer DNA high sensitivity chip. The HTO fraction was amplified with 2× Kapa HiFi HotStart ready mix (Kapa Biosystems, P/N KK2601) and the following oligos: P5 5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGC*T*C-3′ and P7 index primer 5′-CAAGCAGAAGACGGCATACGAGATCGAGTAATGTGACTGGAGTTCAGACGTGT*G*C-3′. The following cycling conditions were used in the C1000 Touch Thermal Cycler to amplify the hashtag library: 95°C for 3 min; cycled 12×: 95°C for 20 s, 64°C for 30 s, and 72°C for 20 s; 72°C for 5 min; and held at 4°C. HTO library was purified with 1.6× SPRI beads. The cDNA was converted to indexed sequencing libraries according to the protocol in the Single-Cell 3′ Reagent Kit v2 User Guide (10X Genomics; CG00052 Rev E) that included the following steps: (1) enzymatic fragmentation, end-repair, and A-tailing; (2) double-sided size selection (0.6× SPRI and 0.8× SPRI to supernatant); (3) adaptor ligation; (4) postligation cleanup (0.8X SPRI); (5) index PCR with the Chromium i7 multiplex kit (P/N 120262) on the C1000 Touch Thermal cycler with the 96-Deep Well Reaction Module: 98°C for 45 s; cycled 15×: 98°C for 20 s, 54°C for 30 s, and 72°C for 20 s; 72°C for 1 min; and held at 4°C; and (6) post-PCR cleanup with double-sided SPRI: 0.6× and 0.8×. The final libraries were run on the Bioanalyzer and quantified with the NEBNext library quantification kit (P/N E7630). The cDNA and HTO libraries were pooled at 10:1 and sequenced for a sequencing depth of 50,000 reads per cell for the cDNA and 2000 reads per cell for the HTO library. Libraries were loaded (1.8 pM) on the Illumina NextSeq500 and sequenced with Read 1 25-bp, index i7 8-bp, and Read 2 98-bp. The Cell Ranger Software Suite pipeline was used to demultiplex samples, align reads to the mm10 reference genome, and generate barcoded data matrices (https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger). Single-cell transcriptomic data were analyzed using the Python-based SCANPY framework (Wolf et al., 2018). Cells expressing <500 genes and genes detected in <3 cells were removed from analysis. Normalization of each cell by total counts over all genes was performed so that every cell had the same total of 10,000 reads, allowing counts to be comparable among cells yielding a total of 4,201 P14 CD8+ T cells—1,503 MDR1-sufficient and 2,698 MDR1-deficient—averaging 1,682 gene counts/cell. Normalized counts were then log-transformed and used for UMAP to visualize phenotypic clusters identified by the Louvain algorithm (Becht et al., 2018). Differential gene expression was analyzed using DESeq2 with the ZINBWAVE R package to account for the excess of zero read counts and better fit the data to a zero inflated negative binomial distribution (Van den Berge et al., 2018) and visualized with ggplot2. GO term enrichment analysis was performed using ClusterProfiler (Yu et al., 2012), as above. The scRNA-seq and bulk RNA-seq data sets presented in this paper are publicly available in the GEO database under accession no. GSE143141.
Quantification and statistical analyses
Statistical analyses were performed using Prism (GraphPad). P values were determined by paired or unpaired Student’s t tests or one-way or two-way ANOVA, as appropriate, and indicated throughout the figure legends. Differences were considered significant when P ≤ 0.05 (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001). Significance levels are specified throughout the figure legends. Data are shown as mean values ± SEM throughout.
Online supplemental material
Fig. S1 shows endogenous Abcb1aAme reporter expression (related to Fig. 1) in hematopoietic cells from different tissues (bone marrow, spleen, thymus, lung, siLP). Fig. S2 shows (1) regulation of MDR1 (Abcb1a, Abcb1b) gene expression in cultured CTL by Runx transcription factors (related to Fig. 2); (2) activation, proliferation, and accumulation of CD45.1 MDR1-sufficient and CD45.2 MDR1-deficient CTL in congenic coculture experiments (related to Fig. 5); and (3) PCA and differential gene expression, determined by RNA-seq in day 6 in vitro–expanded or MDR1-deficient memory-like CTL treated with or without elacridar (related to Fig. 5). Table S1 describes the FACS staining panels used to analyze Abcb1aAme reporter expression, including the specific cell types analyzed in each panel and associated gating strategies/phenotypes (related to Fig. 1 and Fig. S1).
Acknowledgments
The authors are grateful for technical support from the The Scripps Research Institute (TSRI)–Florida Flow Cytometry and Genomics Core Facilities, and for the employees of the TSRI-Florida Animal Resource Center for assistance with animal care. We thank Drs. Ananda Goldrath, Gustavo Martinez, and Joseph G. Crompton for critical discussions and review of the manuscript.
This work was supported by TSRI-Florida via the State of Florida, the National Institutes of Health (grants R01AI118931-01 and R21AI119728 to M.S. Sundrud; grants 5R01AI095634-06 and 5U19 AI109976-05 to M.E. Pipkin; grant R01HL125816 to S.B. Koralov), and the Crohn’s and Colitis Foundation (Senior Research Award 422515 to M.S. Sundrud). A. Sun received medical scientist training grants from the National Institutes of Health (5T32GM007308-36 and 5T32CA009161-39).
Author contributions: M.L. Chen, A. Eliason, K.M. Mendez, A.J. Getzler, C. Mukori, and S. Tsuda performed in vitro and in vivo experiments, analyzed data, and revised the manuscript. H. Diao performed bioinformatic analysis of ChIP-seq and ATAC-seq data. A. Sun, W. Cao and S.Y. Kim generated Abcb1aAME/+ reporter mice. N.E. Bruno provided critical discussions and revised the manuscript. M.E. Pipkin provided key reagents and discussions, supervised in vivo infection experiments, analyzed data, and revised the manuscript. S.B. Koralov supervised the generation of Abcb1aAME/+ reporter mice, provided critical discussions, and revised the manuscript. M.S. Sundrud conceived and supervised the study, designed experiments, analyzed data, and wrote the manuscript.

