


NUP188 encodes a scaffold component of the nuclear pore complex (NPC) and has been implicated as a congenital heart disease gene through an ill-defined function at centrioles. Here, we explore the mechanisms that physically and functionally segregate Nup188 between the pericentriolar material (PCM) and NPCs. Pulse-chase fluorescent labeling indicates that Nup188 populates centrosomes with newly synthesized protein that does not exchange with NPCs even after mitotic NPC breakdown. In addition, the steady-state levels of Nup188 are controlled by the sensitivity of the PCM pool, but not the NPC pool, to proteasomal degradation. Proximity-labeling and super-resolution microscopy show that Nup188 is vicinal to the inner core of the interphase centrosome. Consistent with this, we demonstrate direct binding between Nup188 and Cep152. We further show that Nup188 functions in centriole duplication at or upstream of Sas6 loading. Together, our data establish Nup188 as a component of PCM needed to duplicate the centriole with implications for congenital heart disease mechanisms.
Introduction
The enclosure of the genome within the nuclear membranes occurred alongside the evolution of nuclear pore complexes (NPCs), which control all molecular traffic between the nucleus and cytoplasm. There are ∼30 nucleoporins or “nups” that construct modular subcomplex building blocks that come together in multiples of eight to assemble ∼100 megadalton transport channels (Hampoelz et al., 2019). The major architectural units of the NPC scaffold consist of the Nup107-160 complex (also termed the “Y” or “outer ring” complex), in addition to the Nup93 or “inner ring complex” (Amlacher et al., 2011; Bui et al., 2013; Kim et al., 2018; Kosinski et al., 2016; Siniossoglou et al., 2000; von Appen et al., 2015). The latter consists of Nup93, Nup155, Nup35 (Nup53), Nup205, and Nup188 (Amlacher et al., 2011; Vollmer and Antonin, 2014). The ring complexes provide anchor points for Phe-Gly (FG)–rich nups that establish a size-selective diffusion barrier and provide binding sites for shuttling nuclear transport receptors (NTRs/karyopherins/importins/exportins) bound to cargo (Schmidt and Görlich, 2016; Wente and Rout, 2010).
In addition to their well-established roles at NPCs, some nups moonlight in other subcellular locations, such as the nucleus (Capelson et al., 2010; Capitanio et al., 2018; Kalverda et al., 2010; Liang et al., 2013; Vaquerizas et al., 2010), or by binding the mitotic apparatus (Wozniak et al., 2010). For example, a fraction of the Nup107-160 complex is recruited to kinetochores after nuclear envelope and NPC breakdown during mitosis (Belgareh et al., 2001; Loïodice et al., 2004; Zuccolo et al., 2007), where it helps to recruit the γ-Tubulin ring complex (Mishra et al., 2010). This association might also recruit NTRs and components of the Ran GTPase system, which also play a central role in spindle assembly (Clarke and Zhang, 2008; Zhang et al., 2014). Other nups have also been shown to interact with the mitotic spindle (Cross and Powers, 2011) and spindle assembly checkpoint components (Iouk et al., 2002; Lussi et al., 2010; Markossian et al., 2015; Ródenas et al., 2012; Rodriguez-Bravo et al., 2014; Schweizer et al., 2013). Furthermore, there is evidence to support that both Nup62 (Hashizume et al., 2013) and Nup188 (Itoh et al., 2013) localize to centrosomes, the major microtubule organizing centers in mammalian cells. In general, the molecular mechanisms that define nup function in association with the mitotic apparatus remain to be fully determined.
Understanding the full spectrum of nup function is becoming more pressing as increasing evidence supports that disruption of the nuclear transport system is causative of a wide range of neurodegenerative diseases (Sakuma and D’Angelo, 2017) and cancers (Köhler and Hurt, 2010; Rodriguez-Bravo et al., 2018; Simon and Rout, 2014). In addition, modern patient genomics is revealing a remarkable list of nup disease variants associated with, for example, triple A syndrome (Tullio-Pelet et al., 2000), steroid-resistant nephrotic syndrome (Braun et al., 2018; Braun et al., 2016; Miyake et al., 2015), nonprogressive congenital ataxia (Zanni et al., 2019), and heterotaxy (Fakhro et al., 2011; Manheimer et al., 2018). Heterotaxy is a disorder of left-right patterning that can lead to mispositioned hearts and a severe form of congenital heart disease (Sutherland and Ware, 2009). Interestingly, other nups such as Gle1 have also been associated with left-right patterning in zebrafish, suggesting a specific role for nups in development (Jao et al., 2017; Kaneb et al., 2015). Indeed, while it is likely that some disease-causing nup malfunction is linked directly to defects in nuclear transport, our prior investigation of a copy number variant of NUP188 in a heterotaxy patient (Fakhro et al., 2011) suggested a role for Nup188 at the bases of cilia, key organelles essential for left-right patterning in the developing embryo (Del Viso et al., 2016).
Cilia are built atop centrioles, which are ancient organelles that consist of ninefold radially arranged triplets of microtubules (Azimzadeh and Marshall, 2010; Gönczy, 2012; Nigg and Holland, 2018). While their most evolutionarily conserved function is thought to be the formation of cilia, centrioles also form the core of centrosomes by acquiring pericentriolar material (PCM) consisting of γ-Tubulin ring complexes and centrosome proteins (Ceps, among many others; Andersen et al., 2003; Jakobsen et al., 2011). During interphase, PCM is organized into multiple distinct “layers” with unique molecular components in each (Fry et al., 2017; Lawo et al., 2012; Mennella et al., 2012; Sonnen et al., 2012). For example, Cep192 is a major component of the inner layer, with Cep152 and Pericentrin each populating the intermediate and outer layers, respectively (Lawo et al., 2012; Mennella et al., 2012; Sonnen et al., 2012). Interestingly, this distinct radial-layer architecture is altered as cells enter G2/M, when centrosomes “mature” with the addition of more PCM (Haren et al., 2009; Khodjakov and Rieder, 1999; Lawo et al., 2012; Lee and Rhee, 2011; Sonnen et al., 2012; Zhu et al., 2008). In addition, the delivery of PCM components to the centrosome is thought to require, in some cases, an intermediary in the form of centriolar satellites, cytosolic granules that have PCM1 as their foundational molecular component (Gupta et al., 2015; Hori and Toda, 2017; Kubo et al., 1999; Tollenaere et al., 2015). Mature centrosomes contribute to spindle formation and the successful capture and segregation of chromosomes (Prosser and Pelletier, 2017).
After chromosome segregation, daughter cells inherit a single centrosome consisting of two centrioles, only one of which has PCM; the other acquires PCM through a centriole-to-centrosome conversion process in G1 (Fong et al., 2018; Fu et al., 2016; Izquierdo et al., 2014; Kim et al., 2019). To double the centrosome number, each of the centrioles must first be duplicated in S-phase by a nascent “pro” centriole that assembles perpendicularly from the parent centriole outer wall (Nigg and Holland, 2018). This is achieved by a highly coordinated series of yet to be fully defined molecular events triggered by the recruitment of polo-like–kinase 4 (Plk4; Bettencourt-Dias et al., 2005; Habedanck et al., 2005; O’Connell et al., 2001) by Cep152 and Cep192 (Cizmecioglu et al., 2010; Hatch et al., 2010; Kim et al., 2013; Park et al., 2014; Sonnen et al., 2013) and its phosphorylation of a network of proteins including STIL, CPAP, and Sas6 (Dammermann et al., 2004; Delattre et al., 2004; Dzhindzhev et al., 2017; Kemp et al., 2004; Kirkham et al., 2003; Kratz et al., 2015; Leidel et al., 2005; Leidel and Gönczy, 2003; Moyer et al., 2015; Moyer and Holland, 2019; Ohta et al., 2014; Pelletier et al., 2004; Stevens et al., 2010; Tang et al., 2011). Sas6 helps form a supramolecular cartwheel-shaped structure that serves as a scaffold for microtubule nucleation (Gönczy, 2012). Procentrioles acquire additional proteins that help control microtubule growth (Nigg and Holland, 2018); however, they are not considered to be fully mature centrioles until they have acquired distal and subdistal appendages and, with them, the capability to form cilia (Hoyer-Fender, 2010).
Here, we build on our prior work suggesting that Nup188 surrounds the centrioles at cilia bases (Del Viso et al., 2016). We specifically explored the mechanisms that lead to Nup188 recruitment to PCM and determined that newly synthesized Nup188 populates PCM at all stages of the cell cycle. Our data support a model where modulating Nup188 turnover rate ultimately controls its steady-state levels at centrosomes. We further uncovered proximity interactions with established PCM components and show direct binding to Cep152. Lastly, we provide evidence that Nup188 is required for centriole duplication. Thus, Nup188 is a shared component of both NPCs and PCM required for the centriole life cycle.
Results
Nup188 accumulates at centrosomes with cyclin-like behavior
To better determine the mechanism of Nup188 recruitment to PCM, we first examined the steady-state distribution of Nup188 in interphase and mitosis. Immunofluorescence microscopy showed α-Nup188 staining at the nuclear periphery and in the nucleus of interphase cells and a spatially distinct pool that colocalized with γ-Tubulin, which labels centrosomes (α-γ-Tubulin, red; Fig. 1 A). Interestingly, we observed a continuous increase of α-Nup188 signal at centrosomes as cells progressed into mitosis, reaching a peak in metaphase with approximately threefold higher levels of fluorescence relative to interphase levels (mitosis stage assessed with Hoechst staining) before diminishing in telophase (Fig. 1, A and B). These results are consistent with published work (Itoh et al., 2013), and the temporal signature of this accumulation (and loss) is similar to mitotic cyclins and to other PCM components that drive centrosome maturation during mitosis, including γ-Tubulin (Fig. 1 C; Woodruff et al., 2014).

Nup188 levels increase at centrosomes during mitosis. (A) Immunofluorescence micrographs of HeLa-M cells stained with α-Nup188 (green) and α-γ-Tubulin (red) antibodies (with merge) during interphase and mitosis. Mitotic stages were defined by the morphology of the Hoechst-stained chromosomes (blue). Scale bar is 5 µm. Magnifications of the boxed areas encompassing centrosomes (green, red, and merge) are shown at right. Scale bar is 0.5 µm. (B and C) Plot of the total fluorescence (in a.u.) of α-Nup188 (B) and α-γ-Tubulin (C) staining fluorescence of individual centrosomes normalized (norm.) to background (extracellular) fluorescence in interphase (I; n = 100), prophase (P; n = 29), metaphase (Me; n = 41), anaphase (A; n = 39), and telophase (T; n = 40) from three independent experiments. Mean ± SD is indicated. (D) Several nups including Nup188 exhibit cyclin-like behavior. Western blots assessing the indicated nup levels in total protein samples generated from HeLa-M cells synchronized and released from S-phase. Approximate cell cycle stage is indicated at the top and is informed by assessing Cyclin A and B synthesis and degradation. α-β-Actin is used to assess total protein load in each sample. Numbers at left are positions of molecular weight standards (in kilodaltons).
Nup188 levels increase at centrosomes during mitosis. (A) Immunofluorescence micrographs of HeLa-M cells stained with α-Nup188 (green) and α-γ-Tubulin (red) antibodies (with merge) during interphase and mitosis. Mitotic stages were defined by the morphology of the Hoechst-stained chromosomes (blue). Scale bar is 5 µm. Magnifications of the boxed areas encompassing centrosomes (green, red, and merge) are shown at right. Scale bar is 0.5 µm. (B and C) Plot of the total fluorescence (in a.u.) of α-Nup188 (B) and α-γ-Tubulin (C) staining fluorescence of individual centrosomes normalized (norm.) to background (extracellular) fluorescence in interphase (I; n = 100), prophase (P; n = 29), metaphase (Me; n = 41), anaphase (A; n = 39), and telophase (T; n = 40) from three independent experiments. Mean ± SD is indicated. (D) Several nups including Nup188 exhibit cyclin-like behavior. Western blots assessing the indicated nup levels in total protein samples generated from HeLa-M cells synchronized and released from S-phase. Approximate cell cycle stage is indicated at the top and is informed by assessing Cyclin A and B synthesis and degradation. α-β-Actin is used to assess total protein load in each sample. Numbers at left are positions of molecular weight standards (in kilodaltons).
We wondered whether the increase in α-Nup188 staining reflected a rise in total levels of Nup188 during mitosis. We therefore arrested cells in S-phase and synchronously released them into the cell cycle while monitoring nup levels by Western blot (Fig. 1 D). To monitor synchronous entry into mitosis, the increase and rapid clearance of Cyclin A and Cyclin B, which demarcate the G2-M and the metaphase-anaphase transitions, respectively (Furuno et al., 1999; Pines and Hunter, 1989), were simultaneously assessed (Fig. 1 D). Nup188 levels began rising at 8 h after S-phase release and peaked at 12 h, which corresponds to a time point just after Cyclin B degradation. Interestingly, this behavior was not mirrored by other nups such as Nup214 and the other components of the inner ring complex, including Nup93 and Nup155, whose levels remained roughly consistent through the time course; however, we observed a modest increase for Nup205, the paralog of Nup188.
We next assessed additional nups that have been functionally linked to cell cycle progression, including the FG-nups, Nup62 (Hashizume et al., 2013; Wu et al., 2016), and Nup98 (Cross and Powers, 2011). Here, we observed an even more striking cyclin-like behavior: both of these proteins increased and then rapidly diminished with kinetics that mirrored those of Cyclin B. We further note that Nup96, but not other components of the outer ring complex, was previously shown to decline in mitosis as well (Chakraborty et al., 2008). Together, these correlations between nup levels and cell cycle stage support that some nups, including Nup188, respond to cell cycle cues.
Centrosomal Nup188 is populated by newly synthesized protein, not from NPCs
We next asked whether the increase of Nup188 at centrosomes during mitosis was derived from NPC breakdown or from new protein synthesis. To test this, we generated a stable HeLa cell line that expresses a SNAP-tagged Nup188 fusion protein behind a doxycycline (dox)-inducible promoter. To reduce the potential for overexpression artifacts, we established conditions where SNAP-Nup188 is produced below endogenous levels (i.e., undetectable by the α-Nup188 antibody; Fig. S1 A). We also ensured that SNAP-Nup188 showed cyclin-like behavior just like the endogenous protein (Fig. 2 A).

Nup188 is stabilized upon proteasome inhibition. (A) SNAP-Nup188 levels are below endogenous Nup188 levels. Western blot (and fluorescence labeling of SNAP-Nup188 with a 647-SiR dye, top panel) of total protein extracts derived from SNAP-Nup188–expressing HeLa cells collected after 48 h in the presence (+) or absence (−) of dox. Note that the putative position of SNAP-Nup188 is indicated, as it is expressed at levels that cannot be detected with the α-Nup188 antibody. α-β-Actin is a protein load reference. (B) Inhibition of the proteasome stabilizes Nup188 levels leaving mitosis. Nocodazole-arrested HeLa-M cells were released in the presence of MG132 or carrier alone (DMSO). Western blots of total protein extracts using the indicated primary antibodies were performed at the indicated time points. α-β-Actin is a protein load reference. (C) Nup188 is ubiquitylated. Western blots of input (5%) and immunoprecipitated (IP) fractions (100%) of FLAG-Nup188 or FLAG alone from cell extracts coexpressing HA-Ubiquitin (HA-Ub) treated with MG132 or DMSO. Use legend at top to interpret whether a given plasmid or treatment has been added (+) or not (−). FLAG-Nup188 was detected with an α-Flag antibody and HA-Ub–conjugates using α-HA antibodies. (D) Western blots examining the levels of BirA*-Nup188 in total protein samples derived from BirA*-Nup188–expressing HeLa cells treated with increasing amounts of dox. Expected position of BirA*-Nup188 is indicated relative to the observed endogenous Nup188; α-α-Tubulin is a reference for total protein loads. Numbers at left of all panels are positions of molecular weight standards (in kilodaltons).
Nup188 is stabilized upon proteasome inhibition. (A) SNAP-Nup188 levels are below endogenous Nup188 levels. Western blot (and fluorescence labeling of SNAP-Nup188 with a 647-SiR dye, top panel) of total protein extracts derived from SNAP-Nup188–expressing HeLa cells collected after 48 h in the presence (+) or absence (−) of dox. Note that the putative position of SNAP-Nup188 is indicated, as it is expressed at levels that cannot be detected with the α-Nup188 antibody. α-β-Actin is a protein load reference. (B) Inhibition of the proteasome stabilizes Nup188 levels leaving mitosis. Nocodazole-arrested HeLa-M cells were released in the presence of MG132 or carrier alone (DMSO). Western blots of total protein extracts using the indicated primary antibodies were performed at the indicated time points. α-β-Actin is a protein load reference. (C) Nup188 is ubiquitylated. Western blots of input (5%) and immunoprecipitated (IP) fractions (100%) of FLAG-Nup188 or FLAG alone from cell extracts coexpressing HA-Ubiquitin (HA-Ub) treated with MG132 or DMSO. Use legend at top to interpret whether a given plasmid or treatment has been added (+) or not (−). FLAG-Nup188 was detected with an α-Flag antibody and HA-Ub–conjugates using α-HA antibodies. (D) Western blots examining the levels of BirA*-Nup188 in total protein samples derived from BirA*-Nup188–expressing HeLa cells treated with increasing amounts of dox. Expected position of BirA*-Nup188 is indicated relative to the observed endogenous Nup188; α-α-Tubulin is a reference for total protein loads. Numbers at left of all panels are positions of molecular weight standards (in kilodaltons).

Centrosomes are populated by newly synthesized Nup188, not exchange from NPCs. (A) Like endogenous Nup188, SNAP-Nup188 undergoes mitotic oscillation. Western blots showing the levels of SNAP-Nup188 in total protein samples derived from synchronized cells. Numbers at left are positions of molecular weight standards (in kilodaltons). Approximate cell cycle stage is indicated at the top, referencing Cyclin B synthesis and degradation; α-β-Actin is used to assess total protein loads. (B) As in A, but SNAP-Nup188 protein is visualized by fluorescence. SNAP-Nup188 is first pulse labeled with a 647-SiR dye (red/old) and then chase labeled at the indicated time points with a TMR-Star dye (yellow/new). (C) Plot of fluorescence from B with two additional experimental replicates. The mean fluorescence from these three experiments is represented ±SD. (D) Fluorescence images of HeLa cells expressing SNAP-Nup188 were synchronized with thymidine at S-phase and labeled with 647-SiR dye (red). Subsequently, these cells were released from thymidine block and then allowed to undergo mitosis. The newly synthesized SNAP-Nup188 was labeled with TMR-star dye (yellow) at each time point shown. α-Pericentrin labeling the centrosome is shown in magenta. DNA was visualized by Hoechst staining (blue). Scale bar is 5 µm. Magnifications of boxed regions encompassing centrosomes in red, yellow, and magenta are shown on the right. Scale bar is 0.5 µm. Bottom panels show background fluorescence in cells not expressing SNAP-Nup188 (-dox). n.a., not applicable.
Centrosomes are populated by newly synthesized Nup188, not exchange from NPCs. (A) Like endogenous Nup188, SNAP-Nup188 undergoes mitotic oscillation. Western blots showing the levels of SNAP-Nup188 in total protein samples derived from synchronized cells. Numbers at left are positions of molecular weight standards (in kilodaltons). Approximate cell cycle stage is indicated at the top, referencing Cyclin B synthesis and degradation; α-β-Actin is used to assess total protein loads. (B) As in A, but SNAP-Nup188 protein is visualized by fluorescence. SNAP-Nup188 is first pulse labeled with a 647-SiR dye (red/old) and then chase labeled at the indicated time points with a TMR-Star dye (yellow/new). (C) Plot of fluorescence from B with two additional experimental replicates. The mean fluorescence from these three experiments is represented ±SD. (D) Fluorescence images of HeLa cells expressing SNAP-Nup188 were synchronized with thymidine at S-phase and labeled with 647-SiR dye (red). Subsequently, these cells were released from thymidine block and then allowed to undergo mitosis. The newly synthesized SNAP-Nup188 was labeled with TMR-star dye (yellow) at each time point shown. α-Pericentrin labeling the centrosome is shown in magenta. DNA was visualized by Hoechst staining (blue). Scale bar is 5 µm. Magnifications of boxed regions encompassing centrosomes in red, yellow, and magenta are shown on the right. Scale bar is 0.5 µm. Bottom panels show background fluorescence in cells not expressing SNAP-Nup188 (-dox). n.a., not applicable.
Knowing that SNAP-Nup188 mirrors the behavior of Nup188, we pulse-labeled S-phase synchronized cells with a SNAP-binding 647-silicon rhodamine (SiR) dye. At each time point after release from S-phase, we chase-labeled SNAP-Nup188 with a tetramethylrhodamine (TMR)-Star dye. Thus, we followed two pools of SNAP-Nup188: an existing “old” pool and one that was derived from new synthesis. Interestingly, by monitoring total levels of SNAP-Nup188 (old) and SNAP-Nup188 (new) at each time point, we observed that the old pool was unchanged as cells progressed through mitosis (Fig. 2 B, fluorescence quantified and plotted in Fig. 2 C, red line). In contrast, the newly synthesized pool appeared to peak and then decline after mitosis. Therefore, there is a fraction of SNAP-Nup188 that is newly synthesized that is subject to mitotic oscillation (Fig. 2, B and C, black line in C).
That there might be two distinct pools of Nup188 correlates with the observation that Nup188 exists at NPCs and at centrosomes. To understand whether the centrosomal Nup188 pool is populated by new synthesis or exchange from intact NPCs (during interphase) or NPC breakdown (during mitosis), we directly examined the localization of old and new SNAP-Nup188 in synchronized cells. Consistent with the steady-state distribution of Nup188, pulse-labeled SNAP-Nup188 (old) can be found at both NPCs and at centrosomes (located by α-Pericentrin labeling; Fig. 2 D). Interestingly, by 4 h after release from the S-phase block, we no longer detected SNAP-Nup188 (old) at centrosomes, whereas the pool at NPCs remained unchanged. In contrast, after just 1 h of release from S-phase arrest, we observed a striking accumulation of SNAP-Nup188 (new) at centrosomes (Fig. 2 D). These data suggest that new Nup188 is targeted to centrosomes, where it likely turns over, or to NPCs, where it is incapable of exchanging with the centrosome pool.
That Nup188 populates the centrosome from a newly synthesized pool during interphase was surprising because most nups that associate with the mitotic apparatus do so after release from the NPC during nuclear envelope breakdown (Belgareh et al., 2001; Cross and Powers, 2011; Joseph et al., 2004; Loïodice et al., 2004; Lussi et al., 2010; Wong et al., 2006). We therefore asked whether the NPC-associated pool of SNAP-Nup188 (old) could reach the centrosome during metaphase when NPCs are disassembled. Remarkably, even under these conditions, we only observed SNAP-Nup188 (new) visibly colocalized with the anti-Pericentrin label (Fig. 2 D; 12 h). While it was formally possible that there was a small pool of SNAP-Nup188 (old) that was masked under these conditions, even after mitosis when SNAP-Nup188 (old) was reassembled into NPCs, we were unable to detect any at centrosomes (Fig. 2 D; 14 and 16 h). These data suggest a model where there may be a mechanism dictating that once assembled into NPCs, Nup188 is incompetent for binding the centrosome, which can only be populated by a virgin pool.
Nup188 distribution at steady state is controlled by differential turnover
That new synthesis and turnover could control Nup188 localization at centrosomes prompted us to examine both of these processes in isolation. To examine transcription, we performed quantitative PCR on cDNA generated from mRNA derived from cells arrested in S-phase and released into mitosis (Fig. 3 A). As expected, we observed approximately fourfold higher CYCLIN B mRNA levels when M-phase (12 h after S-phase release) and S-phase samples were directly related (Fig. 3 A). We performed a similar analysis of nup messages. Surprisingly, as shown in Fig. 3 A, we observed little change in nup transcripts with only NUP35/53 mRNA and NUP62 mRNA showing approximately twofold increases in mitotic extracts. NUP188 mRNA, by contrast, was unchanged. Thus, it is likely that the observed increase in Nup188 levels during mitosis was due to an inhibition of its degradation, not transcriptional regulation. This idea would also explain the observation that the SNAP-Nup188 protein was similarly regulated despite being controlled by another promoter (Fig. 2 A).

Nup188 turns over rapidly at centrosomes but is stable at NPCs. (A) NUP188 mRNA levels are unchanged between S- and M-phase. Plot shows transcript levels at M-phase relative to S-phase levels as determined by RT-qPCR of the indicated nup messages. Shown are mean transcript levels (± SD) from three independent experiments. Dotted line reflects a ratio of 1, which indicates no change in relative levels between these cell cycle stages. Red, blue, and orange are inner ring, outer ring, and a mixture of cytoplasmic filament/nuclear basket nup genes, respectively. (B) Asynchronous HeLa cells producing SNAP-Nup188 were treated with DMSO (carrier-only control) and then labeled with 647-SiR dye (red) at indicated time points after drug addition, before imaging by fluorescence microscopy. Centrosomes were labeled with α-Pericentrin (green). Bar is 5 µm. Magnifications of boxed regions encompassing centrosomes in red, green, and merge are shown on the right. Scale bar is 0.5 µm. (C) A representative plot (one of three independent replicates) of the mean intensity (± SD) of SNAP-Nup188 fluorescence at centrosomes in individual cells from the experiment in B over time. (D) As in C but plotting nuclear envelope fluorescence. (E–G) As in B–D but cells were treated with cycloheximide. (H and I) As in B and C but cells were treated with MG132. (J) Sample fluorescence images of cells expressing SNAP-Nup188 treated with MG132 for 0 or 4 h. Line profiles of the fluorescence along the white lines shown in images bisecting the nucleus are shown in right panels. Note the y axis scale and intranuclear fluorescence in bottom panels. Arrows denote location of nuclear rim along line profile plot.
Nup188 turns over rapidly at centrosomes but is stable at NPCs. (A) NUP188 mRNA levels are unchanged between S- and M-phase. Plot shows transcript levels at M-phase relative to S-phase levels as determined by RT-qPCR of the indicated nup messages. Shown are mean transcript levels (± SD) from three independent experiments. Dotted line reflects a ratio of 1, which indicates no change in relative levels between these cell cycle stages. Red, blue, and orange are inner ring, outer ring, and a mixture of cytoplasmic filament/nuclear basket nup genes, respectively. (B) Asynchronous HeLa cells producing SNAP-Nup188 were treated with DMSO (carrier-only control) and then labeled with 647-SiR dye (red) at indicated time points after drug addition, before imaging by fluorescence microscopy. Centrosomes were labeled with α-Pericentrin (green). Bar is 5 µm. Magnifications of boxed regions encompassing centrosomes in red, green, and merge are shown on the right. Scale bar is 0.5 µm. (C) A representative plot (one of three independent replicates) of the mean intensity (± SD) of SNAP-Nup188 fluorescence at centrosomes in individual cells from the experiment in B over time. (D) As in C but plotting nuclear envelope fluorescence. (E–G) As in B–D but cells were treated with cycloheximide. (H and I) As in B and C but cells were treated with MG132. (J) Sample fluorescence images of cells expressing SNAP-Nup188 treated with MG132 for 0 or 4 h. Line profiles of the fluorescence along the white lines shown in images bisecting the nucleus are shown in right panels. Note the y axis scale and intranuclear fluorescence in bottom panels. Arrows denote location of nuclear rim along line profile plot.
Consistent with the idea that Nup188 levels at centrosomes are controlled by degradation, we treated cells with the proteasome inhibitor MG132 and monitored Nup188 by Western blot in cells synchronized and released from mitosis. As shown in Fig. S1 B, MG132 treatment led to a stabilization of Nup188 for several hours after release from M-phase arrest compared with carrier-alone (DMSO) samples, which declined as expected. Further, we detected ubiquitin-conjugated species associated with immunoprecipitations of FLAG-Nup188 (when coexpressed with HA-Ubiquitin), suggesting it could be directly targeted by the proteasome (Fig. S1 C). Thus, it is likely that at least a portion of Nup188 is degraded at the end of mitosis. However, as Cyclin B is also stabilized under these conditions, we could not fully rule out that MG132-mediated mitotic arrest was simply upstream of Nup188 turnover.
We therefore more directly examined SNAP-Nup188 turnover at both NPCs and centrosomes outside of any mitotic regulation. Asynchronous cells were treated with cycloheximide to inhibit all protein synthesis, and the levels of SNAP-Nup188 fluorescence were monitored over 4 h by labeling with a 647-SiR dye at each time point. As a control, we first measured the fluorescence of SNAP-Nup188 at the nuclear envelope and at centrosomes in carrier (DMSO) treated cells, which remained unchanged in both locations over this time period (Fig. 3, B–D). In cycloheximide-treated cells, fluorescence of SNAP-Nup188 at the nuclear envelope also remained constant over 4 h, which is consistent with the idea that Nup188 does not appreciably turn over once integrated into NPCs (Fig. 3, E and G). In contrast, we observed the complete loss of SNAP-Nup188 fluorescence at centrosomes between 1 and 2 h (t1/2 of 51 min; Fig. 3, E and F). Furthermore, to confirm that this loss was due to proteasome-mediated degradation, we also treated cells with MG132 (Fig. 3 H). Here, we observed a striking increase in SNAP-Nup188 fluorescence at centrosomes to levels approximately five times higher than those of DMSO-treated samples (Fig. 3 I, note change in y axis scale), alongside a concomitant increase in α-Pericentrin staining. Interestingly, we also observed an increase in SNAP-Nup188 fluorescence within the nucleus, which overshadowed and precluded our ability to directly assess specific nuclear envelope fluorescence under these conditions (Fig. 3 J, compare line profiles of 0 and 4 h images). Nonetheless, we interpreted these data in a model in which Nup188 is a potential substrate of the proteasome, which is a major determinant of Nup188 levels at centrosomes.
Nup188 is found proximal to the inner layer of the PCM
To further substantiate that Nup188 is a bona fide component of centrosomes, we turned to super-resolution microscopy, which has revealed that the interphase centrosome is organized in sequential concentric layers that extend radially out to ∼200 nm from the centriole core (Fry et al., 2017; Lawo et al., 2012; Mennella et al., 2012; Sonnen et al., 2012). We labeled SNAP-Nup188 with a TMR-Star dye and performed 3D structured illumination microscopy (3D-SIM) while colabeling the inner (α-Cep192), intermediate (α-Cep152), and outer (α-Pericentrin) centrosome layers (Fig. 4 D; Lawo et al., 2012; Mennella et al., 2012; Sonnen et al., 2012). As shown in Fig. 4 A, SNAP-Nup188 fluorescence appeared in a circular pattern typical of PCM components when visualized down the long axis of the centriole (Fig. 4 D, top view of centrosome). All three established PCM labels exhibited a qualitatively similar distribution, although unlike Cep152, which is only found at the proximal ends of centrioles (Cizmecioglu et al., 2010; Lukinavičius et al., 2013; Sir et al., 2011; Sonnen et al., 2013), SNAP-Nup188 coated the entire centriole length (see side view, Fig. 4 B).

Nup188 is found proximal to the inner PCM layer. (A) Micrographs generated using 3D-SIM show the localization of SNAP-Nup188 (labeled with TMR-Star; red) colabeled (in green) with α-Cep192, α-Cep152, and α-Pericentrin marking inner, intermediate, and outer radial layers of the interphase PCM, respectively. Scale bar is 1 µm. Boxed regions encompassing the centrosomes showing green, red, and merge panels are magnified at right. Arrowheads point to SNAP-Nup188 that extends off circular core. Scale bar is 0.25 µm. (B) 3D-SIM image of a side view of SNAP-Nup188 and α-Cep152 labeling of centrosome. Scale bar is 0.25 µm. (C) Plot showing the quantification of the radius (R) of the centrosomal ring for the indicated proteins based on a ring structure fitting to individual images (n indicates number of images from three independent replicates). Mean (solid circle) and SD (bars) are shown. (D) Approximate-scale schematic of centrosome showing centriole in center (white circles are microtubules) and location of Nup188 within the inner layer.
Nup188 is found proximal to the inner PCM layer. (A) Micrographs generated using 3D-SIM show the localization of SNAP-Nup188 (labeled with TMR-Star; red) colabeled (in green) with α-Cep192, α-Cep152, and α-Pericentrin marking inner, intermediate, and outer radial layers of the interphase PCM, respectively. Scale bar is 1 µm. Boxed regions encompassing the centrosomes showing green, red, and merge panels are magnified at right. Arrowheads point to SNAP-Nup188 that extends off circular core. Scale bar is 0.25 µm. (B) 3D-SIM image of a side view of SNAP-Nup188 and α-Cep152 labeling of centrosome. Scale bar is 0.25 µm. (C) Plot showing the quantification of the radius (R) of the centrosomal ring for the indicated proteins based on a ring structure fitting to individual images (n indicates number of images from three independent replicates). Mean (solid circle) and SD (bars) are shown. (D) Approximate-scale schematic of centrosome showing centriole in center (white circles are microtubules) and location of Nup188 within the inner layer.
To gain quantitative insight into SNAP-Nup188’s radial distribution, we fitted a theoretical circle to individual SNAP-Nup188 images and used these results to generate an average image (Fig. 4 C). This analysis showed that the average radius of SNAP-Nup188 fluorescence was 129 nm, which placed it between the inner layer demarked by Cep192 (R of 125 nm; Fig. 4 C) and the intermediate layer (R of 146 nm; Fig. 4 C). Thus, Nup188 is found proximal to the inner layer of the interphase centrosome. This observation predicts that Nup188 may physically interact with inner- or intermediate-layer components such as Cep192 or Cep152.
Interestingly, unlike Cep192 and Cep152, SNAP-Nup188 was also found in extensions that emanate outward from its circular core that often intercalated into Pericentrin (Fig. 4 A, arrowheads). These extensions are particularly obvious when comparing montages of SNAP-Nup188 images to those of either α-Cep192 or α-Cep152 (Fig. S2, A–D). We wondered whether these might reflect interactions with microtubules. We therefore de-polymerized microtubules using nocodazole but found little change to these structures (Fig. S2 E). Nonetheless, these data further support the conclusion that Nup188 is a component of PCM associated with the inner/intermediate centrosome layer and likely other, yet to be defined structures.

Nup188 centrosome labeling has extensions that emanate from its circular core and is unperturbed by microtubule depolymerization. (A–D) 3D-SIM image montages of centrosomes labeled with TMR-Star (SNAP-Nup188) and indicated Ceps. Scale bar is 0.10 µm. (E) Neither Nup188 extensions nor α-PCM1 labeling at centrosomes is perturbed by microtubule disruption. 3D-SIM images of SNAP-Nup188 (TMR-Star dye, red) and α-PCM1 (green) at centrosomes in cells treated with nocodazole or DMSO. Arrowheads point to extensions of SNAP-Nup188, which sometimes colocalize with the α-PCM1 label. Scale bar is 0.25 µm.
Nup188 centrosome labeling has extensions that emanate from its circular core and is unperturbed by microtubule depolymerization. (A–D) 3D-SIM image montages of centrosomes labeled with TMR-Star (SNAP-Nup188) and indicated Ceps. Scale bar is 0.10 µm. (E) Neither Nup188 extensions nor α-PCM1 labeling at centrosomes is perturbed by microtubule disruption. 3D-SIM images of SNAP-Nup188 (TMR-Star dye, red) and α-PCM1 (green) at centrosomes in cells treated with nocodazole or DMSO. Arrowheads point to extensions of SNAP-Nup188, which sometimes colocalize with the α-PCM1 label. Scale bar is 0.25 µm.
Proximity-labeling uncovers PCM-specific interactors
Having more clearly established that Nup188 has the characteristics of a component of PCM, we sought to define the molecular interactions that contribute to this association. We therefore turned to a biotin-ligase proximity-labeling approach (Roux et al., 2012) to avoid the challenges with extracting Nup188 from two relatively insoluble protein assemblies (i.e., NPCs and centrosomes). Moreover, as Nup188 turns over rapidly at centrosomes, we wanted to exploit that this approach reflects a “history” of proximity associations, thus increasing the likelihood that we would find specific Ceps. To reduce background biotinylation, we generated a stable HeLa cell line that expresses the promiscuous biotin ligase (BirA*; Roux et al., 2012) both alone and fused to Nup188, expressed behind a dox-inducible promoter (Fig. 5 A). We then titrated dox concentrations to produce below-endogenous Nup188 levels (Fig. S1 D). To ensure the specificity of BirA*-Nup188–labeling, we stained cells with fluorescently labeled (Alexa Flour 488) streptavidin. Gratifyingly, we detected streptavidin-labeling at centrosomes (indicated by α-γ-Tubulin, red) in interphase and mitosis and at the nuclear periphery (in interphase; Fig. 5 B, upper panels, maximum intensity projections shown). We did not detect any streptavidin-labeling of either centrosomes or NPCs when BirA* was expressed on its own (Fig. 5 B, lower panels). Consistent with these observations, probing total proteins separated by SDS-PAGE and transferred onto nitrocellulose with streptavidin coupled to HRP showed a distinct subset of biotinylated proteins between BirA* and BirA*-Nup188 cells (Fig. 5 C).
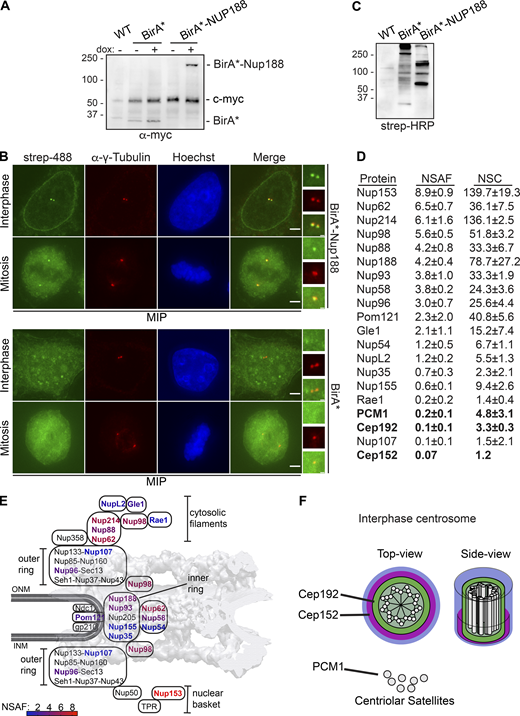
Proximity-labeling predicts physical interactions between Nup188 and PCM. (A) BirA* and BirA*-Nup188 are stably and inducibly expressed at similar levels. Western blots of whole cell extracts from BirA*- and BirA*-Nup188–expressing cell lines. Both BirA* and BirA*-Nup188 are under the control of a dox-inducible promoter and produced with a myc epitope to allow detection by Western blot. Note that endogenous c-myc is also detected in all lanes and serves as a total protein load reference. Numbers at left are positions of molecular weight standards (in kD); − and + denote absence or presence of dox, respectively. (B) Centrosomes and the nuclear envelope are specifically biotinylated in cell lines expressing BirA*-Nup188. Maximum-intensity projections (MIPs) of a Z-series of fluorescence micrographs of BirA*-Nup188 (top panels) and BirA* alone (bottom panels) expressing cells. DNA is stained with Hoechst (blue) to help identify metaphase cells and visualize the nucleus. Biotinylated proteins are labeled with streptavidin (strep) coupled to Alexa Fluor 488 (green). Centrosomes are labeled with α-γ-Tubulin (red). Scale bar is 5 µm. Boxed regions of centrosomes in green, red, and merge are magnified at right. Scale bar is 0.5 µm. (C) Total protein from HeLa cells (WT) and those producing BirA* and BirA*-Nup188 were separated by SDS-PAGE; biotinylated proteins detected by strep coupled to HRP. Numbers at left are positions of molecular weight standards (in kD). (D) Table of biotinylated proteins identified in BirA*-Nup188–expressing cells with NSAF and normalized spectral count (NSC) values (mean ± SD) from three independent replicates. See Table S1 for all proteins identified. (E) Schematic of the relative location of nups within a diagram of the NPC superimposed on a cryo-EM map of the NPC structure (von Appen et al., 2015). Biotinylated nups are color coded based on the heat map scale of NSAF values indicated at bottom. INM and ONM are inner and outer nuclear membrane, respectively. (F) Approximate scale diagram of centrosome showing position of Cep192 and also PCM1 as a component of centriolar satellites.
Proximity-labeling predicts physical interactions between Nup188 and PCM. (A) BirA* and BirA*-Nup188 are stably and inducibly expressed at similar levels. Western blots of whole cell extracts from BirA*- and BirA*-Nup188–expressing cell lines. Both BirA* and BirA*-Nup188 are under the control of a dox-inducible promoter and produced with a myc epitope to allow detection by Western blot. Note that endogenous c-myc is also detected in all lanes and serves as a total protein load reference. Numbers at left are positions of molecular weight standards (in kD); − and + denote absence or presence of dox, respectively. (B) Centrosomes and the nuclear envelope are specifically biotinylated in cell lines expressing BirA*-Nup188. Maximum-intensity projections (MIPs) of a Z-series of fluorescence micrographs of BirA*-Nup188 (top panels) and BirA* alone (bottom panels) expressing cells. DNA is stained with Hoechst (blue) to help identify metaphase cells and visualize the nucleus. Biotinylated proteins are labeled with streptavidin (strep) coupled to Alexa Fluor 488 (green). Centrosomes are labeled with α-γ-Tubulin (red). Scale bar is 5 µm. Boxed regions of centrosomes in green, red, and merge are magnified at right. Scale bar is 0.5 µm. (C) Total protein from HeLa cells (WT) and those producing BirA* and BirA*-Nup188 were separated by SDS-PAGE; biotinylated proteins detected by strep coupled to HRP. Numbers at left are positions of molecular weight standards (in kD). (D) Table of biotinylated proteins identified in BirA*-Nup188–expressing cells with NSAF and normalized spectral count (NSC) values (mean ± SD) from three independent replicates. See Table S1 for all proteins identified. (E) Schematic of the relative location of nups within a diagram of the NPC superimposed on a cryo-EM map of the NPC structure (von Appen et al., 2015). Biotinylated nups are color coded based on the heat map scale of NSAF values indicated at bottom. INM and ONM are inner and outer nuclear membrane, respectively. (F) Approximate scale diagram of centrosome showing position of Cep192 and also PCM1 as a component of centriolar satellites.
Confident in the specificity of the biotinylation in our cell lines, we affinity purified biotinylated proteins using streptavidin magnetic beads from both BirA*-Nup188 and BirA* alone cell extracts. Protein eluates were subjected to trypsin digestion followed by liquid chromatography tandem-mass spectrometry (LC-MS/MS) peptide identification (Table S1). As the majority of Nup188 is associated with NPCs, most identified peptides corresponded to nups. We normalized specific peptide abundance to the total peptides identified in a given experiment and to the length (in amino acids) of the protein, which we present as a normalized spectral abundance factor (NSAF; Fig. 5 D). We also color coded nup names in a heat map, which we superimposed on the NPC structure to give a sense of the location of the biotinylation hot spots in the NPC (Fig. 5 E). As expected, based on Nup188’s established location within the inner ring complex, we detected Nup93 as one of the top interactors. Consistent with this, Nup62 and Nup58 were also identified, suggesting that the BirA* enzyme is likely proximal to the N-terminus of Nup93 where these FG-nups connect to the inner ring (Stuwe et al., 2015). Interestingly, Nup98 has a high NSAF value consistent with other data supporting that Nup188 binds to this FG-nup (Fischer et al., 2015; Onischenko et al., 2017). Furthermore, the highest NSAF values were found for Nup153 and Nup214, components of the nuclear basket and cytoplasmic filaments, respectively. These data suggest that Nup188 may function in several locations in the NPC and/or associate with these nups in other subcellular locations.
Consistent with the idea that Nup188 may function outside of the NPC, among the ∼80 proteins identified (see complete lists, Table S1) we found many nuclear components including RNPs and chromatin-binding proteins. Critically, we identified two components of PCM, Cep152 and Cep192, and one, PCM1, that is a component of both centriolar satellites and PCM (Fig. 5, D and F; and Table S1). Of these, Cep192 and PCM1 were found in all three biological replicates with mean NSAF values of 0.1 and 0.2, respectively. While this is expectedly low (due to the overall low abundance and rapid turnover of Nup188 at centrosomes versus NPCs), they were remarkably specific, as no other centrosome components were detected. Thus, these data further support that Nup188 is found proximal to the inner layer of the PCM. To help explain the interaction with PCM1, we tested whether SNAP-Nup188 came into proximity to PCM1 at centriolar satellites or centrosomes. As shown in Fig. S2 E, we observed a few instances where PCM1 foci at centrosomes were adjacent to or colocalized with SNAP-Nup188 by 3D-SIM in a microtubule-independent fashion. These data at least provide a rationale for why PCM1 was detected by Biotin Identification (BioID).
Nup188 directly interacts with Cep152
To assess whether proximity-labeling reflected biochemical interactions between Nup188 and Ceps, we immunoprecipitated GFP-Cep152, GFP-PCM1, and GFP-Cep192 from whole cell extracts and tested whether they copurified with FLAG-Nup188. As shown in Fig. 6 A, there was a clear enrichment of FLAG-Nup188 found in immunoprecipitated fractions of GFP-Cep152 and GFP-Cep192 compared with affinity purifications of GFP alone (Fig. 6 A). We also observed a modest specificity to GFP-PCM1. Thus, Nup188 can specifically physically interact with all three of these Ceps, suggesting that the proximity-labeling reflected biochemical interactions with centrosome components.

Nup188 directly interacts with Cep152. (A) Anti-GFP nanobody immunoprecipitations (IPs) were performed on cell extracts derived from HEK293T cells coexpressing FLAG-Nup188 and either GFP, GFP-Cep192, GFP-Cep152, or GFP-PCM1. Western blots using the indicated primary antibodies were performed to detect proteins in input (2%) and IP (100%) fractions. (B)35S-labeled Nup188 was generated in a reticulocyte vitro transcription/translation mix (IVT; left lane) and used to probe a nitrocellulose membrane with the indicated immobilized proteins generated from IPs (see Ponceau panel for total protein loads). Specific binding of 35S-Nup188 was detected by autoradiography (AR). Numbers at left of all panels are position of molecular weight markers (in kilodaltons).
Nup188 directly interacts with Cep152. (A) Anti-GFP nanobody immunoprecipitations (IPs) were performed on cell extracts derived from HEK293T cells coexpressing FLAG-Nup188 and either GFP, GFP-Cep192, GFP-Cep152, or GFP-PCM1. Western blots using the indicated primary antibodies were performed to detect proteins in input (2%) and IP (100%) fractions. (B)35S-labeled Nup188 was generated in a reticulocyte vitro transcription/translation mix (IVT; left lane) and used to probe a nitrocellulose membrane with the indicated immobilized proteins generated from IPs (see Ponceau panel for total protein loads). Specific binding of 35S-Nup188 was detected by autoradiography (AR). Numbers at left of all panels are position of molecular weight markers (in kilodaltons).
The lack of obvious specificity between the binding of Nup188 to the tested centrosome components (Fig. 6 A) prompted us to more thoroughly investigate which, if any, of these proteins directly interacted with Nup188. As these proteins are all extremely challenging to produce recombinantly, we turned to a far Western approach. We first generated 35S-labeled Nup188 using an in vitro transcription and translation reaction mix, which produced several protein products (likely derived from full-length and prematurely terminated Nup188 transcripts) around the molecular weight of Nup188 (Fig. 6 B). We used this mixture to probe a nitrocellulose membrane with near-equivalent amounts of immobilized GFP, GFP-Cep192, GFP-Cep152, and GFP-PCM1 immunoprecipitated from whole cell extracts (Fig. 6 B; Ponceau). After washing, 35S-Nup188 binding was detected by autoradiography (Fig. 6 B). Gratifyingly, GFP-Cep152 was specifically radioactive. Indeed, there was hardly any detectable radioactivity at positions with the considerably more abundant GFP or the other GFP-fusions. These results support the conclusion that Nup188 can directly interact with Cep152.
Specific knockdown of Nup188 impacts centriolar satellite distribution
That Nup188 directly interacts with Cep152 raised the possibility that it may be required for its recruitment to centrosomes. We therefore used siRNA to knock down Cep152 (and Cep192 and PCM1) and examined any impact on SNAP-Nup188 localization (Fig. S3 A). Importantly, all three of these proteins were depleted with the siRNA treatments (Fig. S3 B). However, despite these clear reductions in protein levels, we were unable to convincingly observe any changes to SNAP-Nup188 localization, with mean values of SNAP-Nup188 fluorescence at centrosomes remaining unchanged (Fig. S3 A). Likewise, depletion of Nup188 did not impact the recruitment of either Cep192 or Cep152 to centrosomes (Fig. S3, C–E) despite the fact that we could clear SNAP-Nup188 from centrosomes with siRNA treatments as short as 12 h due to its rapid turnover (Fig. S4, A–C). Taken together, these data suggest that a single interaction interface with Cep152 cannot explain the association of Nup188 with centrosomes, and there are likely other binding partners that remain to be uncovered.

Centrosome associations of Nup188 and PCM proteins are independent of each other. (A) Plot of the mean fluorescence at centrosomes of SNAP-Nup188 labeled with 647-SiR dye in cells treated with indicated siRNAs. More than 30 cells were quantified with mean ± SD shown. (B) Western blots (or fluorescence image/top panel) of total protein from SNAP-NUP188–expressing HeLa cell extracts prepared from cells treated with the indicated siRNAs. α-β-Actin is a protein load reference. Numbers at left are positions of molecular weight standards (in kilodaltons). (C). Representative plot of α-Cep192 mean fluorescence ± SD at centrosomes (n > 30). (D) Representative plot of α-Cep152 mean fluorescence ± SD at centrosomes (n > 30). (E) Western blots of total protein from HeLa cell extracts prepared from cells treated with NUP188 siRNA. α-β-Actin is a protein load reference. n.s., not significant as calculated from two-way ANOVA. Numbers at left are positions of molecular weight standards (in kilodaltons).
Centrosome associations of Nup188 and PCM proteins are independent of each other. (A) Plot of the mean fluorescence at centrosomes of SNAP-Nup188 labeled with 647-SiR dye in cells treated with indicated siRNAs. More than 30 cells were quantified with mean ± SD shown. (B) Western blots (or fluorescence image/top panel) of total protein from SNAP-NUP188–expressing HeLa cell extracts prepared from cells treated with the indicated siRNAs. α-β-Actin is a protein load reference. Numbers at left are positions of molecular weight standards (in kilodaltons). (C). Representative plot of α-Cep192 mean fluorescence ± SD at centrosomes (n > 30). (D) Representative plot of α-Cep152 mean fluorescence ± SD at centrosomes (n > 30). (E) Western blots of total protein from HeLa cell extracts prepared from cells treated with NUP188 siRNA. α-β-Actin is a protein load reference. n.s., not significant as calculated from two-way ANOVA. Numbers at left are positions of molecular weight standards (in kilodaltons).

The centrosomal pool of Nup188 is sensitive to siRNA knockdown. (A) Western blots (or fluorescence image/top panel) of total protein from SNAP-NUP188–expressing HeLa cell extracts prepared from cells treated with the indicated siRNAs for 12 or 24 h. α-β-Actin is a protein load reference. Numbers at left are positions of molecular weight standards (in kilodaltons). (B) Fluorescence micrographs of SNAP-Nup188 (labeled with 647-SiR, red) in HeLa cells treated with NUP188 or scrambled siRNA for 12 or 24 h. Centrosomes are labeled with α-Pericentrin (green). Scale bar is 5 µm. Magnifications of boxed region are shown at right. Scale bar is 0.5 µm. (C and D) Plots of mean SNAP-Nup188 fluorescence (647-SiR) ± SD at centrosomes and the nuclear envelope in the indicated conditions where >30 cells were quantified.
The centrosomal pool of Nup188 is sensitive to siRNA knockdown. (A) Western blots (or fluorescence image/top panel) of total protein from SNAP-NUP188–expressing HeLa cell extracts prepared from cells treated with the indicated siRNAs for 12 or 24 h. α-β-Actin is a protein load reference. Numbers at left are positions of molecular weight standards (in kilodaltons). (B) Fluorescence micrographs of SNAP-Nup188 (labeled with 647-SiR, red) in HeLa cells treated with NUP188 or scrambled siRNA for 12 or 24 h. Centrosomes are labeled with α-Pericentrin (green). Scale bar is 5 µm. Magnifications of boxed region are shown at right. Scale bar is 0.5 µm. (C and D) Plots of mean SNAP-Nup188 fluorescence (647-SiR) ± SD at centrosomes and the nuclear envelope in the indicated conditions where >30 cells were quantified.
As knockdown of Cep152 was insufficient to upset Nup188 PCM accumulation, we assessed Nup188 centrosome function in the context of the established roles for all three proximity interactors: Cep152, Cep192, and PCM1. First, as PCM1 is a major component of centriolar satellites, we tested the distribution of PCM1 in the context of siRNA knockdown of NUP188. Strikingly, PCM1-puncta were no longer distributed in a close-radial association with centrosomes but instead were dispersed throughout the cytosol (Fig. 7 A). To quantitatively assess PCM1 distribution around centrosomes, we plotted the total fluorescence of α-PCM1 staining as a function of its radial distribution from the centrosome axis (Fig. 7 B). Cells treated with NUP188 siRNA had on average ∼30% less fluorescence than control cells treated with scrambled siRNA at radii within 5 µm of the centrosome (Fig. 7, A and B). Importantly, the reduction of α-PCM1 fluorescence reflected the dispersal of PCM1, not its disappearance, as shown in Fig. 7 C, where we have plotted the cumulative distribution in the same cell areas: PCM1 foci are more likely to populate cytosol further from the centrosome axis in cells treated specifically with NUP188 siRNA. Concerned that this effect on satellites might be due to a disruption of nuclear transport, we also tested knockdown of several additional nups including Nup93, Nup62, and Nup214. Over the time course of this experiment, we observed similar levels of knockdown for each of these proteins (Fig. S5 A); yet, only the knockdown of Nup188 had any significant impact on PCM1 distribution. Indeed, even reduction of Cep192 levels did not influence PCM1 localization (Fig. 7, B and C). Thus, when considered in the context of evidence for a putative, albeit indirect, physical interaction, we suggest that this effect may be a direct consequence of perturbing a functional relationship between Nup188 and PCM1.

Knockdown of NUP188 specifically impacts centriolar satellite distribution. (A) Fluorescence images of U2OS cells labeled with α-PCM1 (fluorescence inverted, red in magnification at right) and α-Centrin (green) to localize centrioles, with merged image showing Hoechst-stained DNA (blue). Cells were treated for 48 h with siRNAs specific to indicated nup transcripts with scrambled siRNA control before imaging. Scale bar is 1 µm. Boxed regions of centrosomes in green, red, and merge are magnified at right. Scale bar is 0.5 µm. (B) Plot of the total fluorescence of α-PCM1–labeled centriolar satellites within an annulus of indicated radii around an axis of α-Centrin fluorescence in 10 cells, each treated with the indicated siRNA. Error bars indicate ±SD. (C) As in B but plotting the cumulative distribution of α-PCM1 labeling throughout the cell (n = 10). Error bars indicate mean ± SD.
Knockdown of NUP188 specifically impacts centriolar satellite distribution. (A) Fluorescence images of U2OS cells labeled with α-PCM1 (fluorescence inverted, red in magnification at right) and α-Centrin (green) to localize centrioles, with merged image showing Hoechst-stained DNA (blue). Cells were treated for 48 h with siRNAs specific to indicated nup transcripts with scrambled siRNA control before imaging. Scale bar is 1 µm. Boxed regions of centrosomes in green, red, and merge are magnified at right. Scale bar is 0.5 µm. (B) Plot of the total fluorescence of α-PCM1–labeled centriolar satellites within an annulus of indicated radii around an axis of α-Centrin fluorescence in 10 cells, each treated with the indicated siRNA. Error bars indicate ±SD. (C) As in B but plotting the cumulative distribution of α-PCM1 labeling throughout the cell (n = 10). Error bars indicate mean ± SD.

FACS analysis confirms S-phase arrest in NUP188 siRNA–treated cells. (A) Western blots of total protein from U2OS extracts prepared from cells treated with the indicated siRNAs. α-β-Actin is a protein load reference. Numbers at left are positions of molecular weight standards (in kilodaltons). (B) FACS profiles of DNA content (from DAPI fluorescence in a.u.) in asynchronous (red lines) and S-phase synchronized (aphidicolin, blue lines) U2OS cells treated with the indicated siRNAs.
FACS analysis confirms S-phase arrest in NUP188 siRNA–treated cells. (A) Western blots of total protein from U2OS extracts prepared from cells treated with the indicated siRNAs. α-β-Actin is a protein load reference. Numbers at left are positions of molecular weight standards (in kilodaltons). (B) FACS profiles of DNA content (from DAPI fluorescence in a.u.) in asynchronous (red lines) and S-phase synchronized (aphidicolin, blue lines) U2OS cells treated with the indicated siRNAs.
Nup188 is required for centriole duplication through Sas6 loading
We next explored how Nup188 might impact the function of Cep152 and Cep192, which have been ascribed diverse roles in the centrosome life cycle including in centriole duplication (Kim et al., 2013; Sonnen et al., 2013; Zhu et al., 2008) and centrosome maturation (Gomez-Ferreria et al., 2007; Joukov et al., 2014; O’Rourke et al., 2014; Zhu et al., 2008). We therefore began by testing the impact of knockdown of Nup188 and other nups, including Nup93, Nup62, and Nup214, on centriole duplication. Centriole number was assessed by immunostaining with either α-Centrin or α-CP110 antibodies, which recognize the distal ends of centrioles. Here, the results were again remarkably specific for Nup188 (Fig. 8, A and B). For example, knockdown of Nup62 and Nup93 did not lead to statistically different changes in centriole number, with ∼50% of cells having between two and four centrioles, like the scrambled siRNA-treated control. Small fractions of each of these cell populations also had supernumerary centrioles, which were particularly striking in Nup214-depleted cells (Fig. 8, A and B). In contrast, only 20% of NUP188 siRNA–treated cells had between two and four centrioles, with the other 80% containing zero to two. Thus, knockdown of Nup188 leads to a specific abrogation of centriole duplication. Importantly, FACS analysis of DNA content confirmed that these effects were not likely due to any marked delay of the cell cycle, as the proportions of cells in G1 and G2 were similar in Nup188 and Nup93 knockdown populations, but only Nup188 knockdown had any significant impact on centriole number (Fig. S5 B).

Nup188 is required for centriole duplication through Sas6 loading. (A) Fluorescence micrographs of U2OS cells treated with the indicated siRNAs. Centrioles are labeled with both α-Centrin (green) and α-CP110 (red). Scale bar is 1 µm. (B) Plot of the quantification of centriole number (α-Centrin foci) in U2OS cells treated with the indicated siRNAs. Three independent replicates of >30 cells per replicate were quantified. Error bars indicate mean ± SD. P values are calculated from two-way ANOVA with Dunnett’s multiple comparison test. (C) Fluorescence micrographs of S-phase–synchronized U2OS cells stained with α-Sas6 (green) and α-Cep192 (red) antibodies. Scale bar is 1 µm. (D) Plot of the percentage of cells with the indicated number of Sas6 foci in S-phase–synchronized U2OS cells treated with the indicated siRNAs. Three independent replicates of >40 cells per replicate were quantified. Error bars indicate mean ± SD. P values are calculated from two-way ANOVA with Dunnett’s multiple comparison test. (E) Plot of mean fluorescence (normalized to intracellular background) of individual α-Sas6 foci. Three independent replicates of >35 cells per replicate were quantified. Error bars indicate mean ± SD. P values are calculated from one-way ANOVA with Tukey’s multiple comparison test. *, P ≤ 0.05; ***, P ≤ 0.001; ****, P ≤ 0.0001.
Nup188 is required for centriole duplication through Sas6 loading. (A) Fluorescence micrographs of U2OS cells treated with the indicated siRNAs. Centrioles are labeled with both α-Centrin (green) and α-CP110 (red). Scale bar is 1 µm. (B) Plot of the quantification of centriole number (α-Centrin foci) in U2OS cells treated with the indicated siRNAs. Three independent replicates of >30 cells per replicate were quantified. Error bars indicate mean ± SD. P values are calculated from two-way ANOVA with Dunnett’s multiple comparison test. (C) Fluorescence micrographs of S-phase–synchronized U2OS cells stained with α-Sas6 (green) and α-Cep192 (red) antibodies. Scale bar is 1 µm. (D) Plot of the percentage of cells with the indicated number of Sas6 foci in S-phase–synchronized U2OS cells treated with the indicated siRNAs. Three independent replicates of >40 cells per replicate were quantified. Error bars indicate mean ± SD. P values are calculated from two-way ANOVA with Dunnett’s multiple comparison test. (E) Plot of mean fluorescence (normalized to intracellular background) of individual α-Sas6 foci. Three independent replicates of >35 cells per replicate were quantified. Error bars indicate mean ± SD. P values are calculated from one-way ANOVA with Tukey’s multiple comparison test. *, P ≤ 0.05; ***, P ≤ 0.001; ****, P ≤ 0.0001.
Finally, to gain further insight into the mechanism of Nup188 knockdown–specific centriole loss, we tested whether Nup188 helped control the recruitment of Sas6, which is thought to be critical for formation of the cartwheel. Because Sas6 is recruited during a short temporal window in S-phase (Leidel et al., 2005; Strnad et al., 2007), we arrested cells in S-phase using aphidicolin. As shown in Fig. S5 B, WT, Nup188-, and Nup93-knockdown cell populations had DNA contents consistent with an S-phase arrest. Under these conditions, reducing levels of Nup188 but not Nup93 led to a marked loss of Sas6 recruitment to centrosomes (α-Cep192, red) with ∼50% of cells showing aberrant number (zero and one) of α-Sas6 foci on centrosomes, thus providing a rationale for the centriole duplication defect (Fig. 8, C and D). Indeed, even in cases where we observed Sas6 at centrosomes under conditions of Nup188 depletion, the α-Sas6–labeling fluorescence was lower, suggesting that fewer Sas6 molecules were recruited in the absence of Nup188 (Fig. 8 E). Thus, Nup188 is specifically required for centriole duplication, likely through a role contributing to Sas6 recruitment and/or stability.
Discussion
Here, we argue that we have made a definitive case for Nup188 being a component of the centrosome. There are several reasons for this assertion: (1) Endogenous Nup188, but also SNAP- and BirA*-fusions of Nup188, reproducibly and unambiguously localize to centrosomes; this localization is not dependent on microtubules, as it is stable under conditions of microtubule depolymerization. (2) We established that Nup188 directly interacts with Cep152. (3) We placed Nup188 within the PCM in a physical context that is consistent with it interacting with Cep152 (i.e., between the centrosome inner and intermediate layers) and likely other, yet to be identified factors. (4) Like other PCM components, Nup188 is further enriched at centrosomes during its maturation in mitosis. (5) We demonstrated the specific disruption of centriole duplication upon Nup188, but not other nup, knockdown.
One of the more interesting aspects of Nup188’s association with centrosomes is that it is recruited at all stages of the cell cycle (Fig. 1). For example, other nups, such as the Nup107-160 complex, are recruited to kinetochores solely during mitosis, presumably from a pool liberated during NPC breakdown (Belgareh et al., 2001; Loïodice et al., 2004). In contrast, mechanisms must be in place to sort newly synthesized Nup188 to at least two different destinations throughout interphase. Once there, the levels of Nup188 accumulation are ultimately controlled by differential turnover, with the NPC pool being extremely stable and the centrosome pool sensitive to proteasomal degradation (Fig. 3). It will be interesting to explore the functional implications of these observations. For example, one prediction is that the centrosomal pool would be much more sensitive to siRNA knockdown (which it is; Fig. S4) and perhaps to overexpression. Thus, this might help to explain why disruption of the PCM function of Nup188 is more sensitive to partial knockdown in our heterotaxy Xenopustropicalis model over its function at NPCs (Del Viso et al., 2016). It might also predict that putative Nup188 disease variants that destabilize Nup188 would be more likely to affect its PCM function as well.
Thus, it seems reasonable to conclude that the modulation of the Nup188 turnover rate is the major determinant of its accumulation at centrosomes during mitosis. The expectation is that Nup188’s turnover is inhibited during G2 and early M-phase to elevate its levels and then stimulated at the end of mitosis to trigger its degradation. While we have not yet identified the molecular players that control this behavior, clear candidates are E3 ubiquitin ligases such as the anaphase-promoting complex or the SKP-CUL1-F Box complex, which have both been implicated in centrosome function by impacting the degradation of PCM components (Arquint et al., 2018; Cunha-Ferreira et al., 2009; D’Angiolella et al., 2010; Drosopoulos et al., 2014; Freed et al., 1999; Fung et al., 2018; Holland et al., 2010; Piva et al., 2002; Prosser et al., 2012). Other E3 ligases have also been implicated as functioning at centrosomes including Mib1 (Čajánek et al., 2015). Regardless, these data all point to proteasomal turnover as a major determinant of centrosome architecture and size. This idea is best illustrated by the observation that inhibiting proteasomes during interphase can lead to over a fivefold increase in the levels of Nup188 at centrosomes (Fig. 3). Indeed, this appears to be a general rule for at least a subset of PCM components (Wigley et al., 1999) and reinforces that Nup188 is a component of PCM.
Further illustrating the central importance of controlling the degradation of centrosomal proteins in time and space, even Plk4 and Sas6 levels at centrosomes, and thus the centriole duplication mechanism, is under the control of the ubiquitin-proteasome system (Arquint and Nigg, 2016; Cunha-Ferreira et al., 2009; Holland et al., 2010; Puklowski et al., 2011). Therefore, a priority going forward is to fully elucidate how Nup188 functions during centriole duplication and whether there is a role for Nup188 turnover in this process as well. Our data support a model in which Nup188 impacts events at or upstream of Sas6 recruitment to give rise to the procentriole (Fig. 8). While we did not identify Sas6 by BioID, this is not surprising as it is only recruited to centrioles for a short time period during G1-S before being degraded (Leidel et al., 2005; Strnad et al., 2007). Nonetheless, even if we cannot be conclusive as to whether Nup188 biochemically interacts with Sas6, we favor models in which it acts through a mechanism analogous to Cep192 and Cep152, which provide platforms for Plk4 responsible for Sas6 recruitment (Arquint and Nigg, 2016; Kim et al., 2013; Sonnen et al., 2013). Certainly, as with many nups, Nup188 is a target of kinases with several predicted polo kinase sites (Gouw et al., 2018), suggesting this as a likely possibility for future investigation. That Nup188 also directly binds to Cep152 (Fig. 6) lends further support to this model, and it will be interesting to investigate how this interaction ultimately modulates Plk4 function to support Sas6 loading.
Centrosomal Nup188 likely makes several direct connections with proteins beyond Cep152, as Cep152 depletion did not impact Nup188 recruitment to this organelle (Fig. S3 A). These results illustrate the persistent challenge of delimiting a function for Nup188 at centrosomes in a context in which the perturbation of NPC function can be unequivocally ruled out. We nonetheless argue that Nup188 at centrosomes directly contributes to mechanisms that control both centriolar satellite distribution (Fig. 7) and centriole duplication (Fig. 8). Such an assertion is based in part on prior data suggesting that Nup188 is in fact dispensable for NPC assembly, likely due to redundancy provided by its paralog Nup205 (Theerthagiri et al., 2010). Most critically, however, we demonstrated a direct binding interaction between Nup188 and Cep152 that almost certainly underlies the remarkable specificity of Nup188 knockdown in causing the observed centrosomal-related defects over even knockdown of its nearest neighbor binding partner (at the NPC), Nup93. These results further predict that Nup93 is not a component of centrosomes and provides one of a handful of examples where one nup might separate from its nup subcomplex to perform a unique function. Another example is Nup96, which interestingly is also specifically degraded during mitosis while the rest of the outer ring complex remains stable (Chakraborty et al., 2008). A model in which Nup188 is disengaged from its binding partner Nup93 at centrosomes could also help to explain why the NPC-assembled pool of Nup188 (which would be bound to Nup93) does not seem able to populate centrosomes even after “solubilization” by NPC breakdown during mitosis (Fig. 2). Such a model needs to be reconciled, however, with our prior data suggesting that both Nup93 and Nup188 can localize to cilia bases (Del Viso et al., 2016).
Further work remains to define the catalog of nups at centrosomes (and at cilia bases; Breslow et al., 2013; Del Viso et al., 2016; Endicott et al., 2015; Kee et al., 2012), with key candidates being the FG-nups Nup62 and Nup98, both of which show analogous, if more striking, cyclin-like behavior during mitosis (Fig. 1). Furthermore, each has been implicated in some form of mitotic regulation: Nup62 has a potential pool at centrosomes (Hashizume et al., 2013), and Nup98 potentially directly impacts spindle assembly (Cross and Powers, 2011) while also localizing to cilia bases (Endicott et al., 2015; Endicott and Brueckner, 2018). Could Nup188 interface with these FG-nups in the PCM, and if so, how? Answers may come from recent work showing that Nup188 and its paralog Nup205 have atomic structures that resemble the karyopherin/importin β–family of NTRs (Andersen et al., 2013; Flemming et al., 2012; Sampathkumar et al., 2013; Stuwe et al., 2014). In fact, evidence indicates that these interactions may be critical for helping to hold the NPC structure together (Onischenko et al., 2017), perhaps by “ordering” the inherently disordered FG-repeats, which themselves have been shown to display a spectrum of collective behaviors and “phases” (Lemke, 2016), some of which are impacted by NTR-binding (Hofweber et al., 2018; Yoshizawa et al., 2018). Could an NTR-like Nup188 also contribute to an ordering or disordering of centrosome organization? Such a hypothesis becomes compelling as the Caenorhabditis elegans SPD-5 is capable of undergoing a phase change to a gel/condensate-like state required for microtubule polymerization (Woodruff et al., 2017). That the more structured layers of the interphase centrosome are lost as the centrosome matures with the addition of more PCM (including Nup188) before mitosis (Haren et al., 2009; Khodjakov and Rieder, 1999; Lawo et al., 2012; Lee and Rhee, 2011; Sonnen et al., 2012; Zhu et al., 2008) suggests that similar changes may occur to the human centrosome as well, however challenging to definitively establish in vivo (Raff, 2019). We look forward to directly examining whether Nup188 impacts this process in future work.
Materials and methods
Plasmids
Plasmids used in the study are listed in Table S2. To construct plasmids pMC1 and pMC2, the GFP ORF from GFP-NUP188 plasmid (gift from Dr. K. Tanaka, Tohoku University, Sendai, Japan; Itoh et al., 2013) was replaced with either SNAP or BirA* coding sequences excised from pSNAPf (New England Biolabs) and pcDNA3.1 mycBioID plasmid (Addgene), respectively. To generate pNV2 and pNV3, SNAP-Nup188 and BirA*-NUP188 were subcloned into pcDNA5/FRT/TO. To construct pNV1, NUP188 sequence was excised from pNV2 leaving only the BirA* ORF. To construct pKD1 and pKD2, the NUP188 coding sequence was amplified by PCR and subcloned into p3XFLAG-CMV-10 vector (Millipore Sigma) or pcDNA3.1/3× myc-A, respectively, using the Gibson Assembly Master Mix (NEB).
Cell culture, transfection, and stable cell line generation
The following human cell lines were used in this study: HeLa-M (gift from Dr. P. De Camilli, Yale School of Medicine, New Haven, CT), HeLa (American Type Culture Collection [ATCC]), HeLa T-Rex-Flp-In (Thermo Fisher Scientific), and U2OS (ATCC). HeLa, HeLa-M, and HeLa T-Rex-Flp-In were cultured in DMEM with 4.5 g/liter glucose (Thermo Fisher Scientific), and U2OS cells (ATCC) were cultured in McCoy’s 5A Medium Modified (Thermo Fisher Scientific). The medium was supplemented with 10% FBS (Millipore Sigma) and 100 U/ml penicillin and 100 µg/ml streptomycin (Thermo Fisher Scientific). The HeLa T-Rex-Flp-In cell line was also supplemented with 100 µg/ml Zeocin (InvivoGen) and 15 µg/ml of Blasticidin (InvivoGen) to maintain the stably integrated FRT site and tetracycline repressor, respectively. All cells were maintained in a humidified incubator at 37°C in 5% CO2.
Transfection of siRNA was performed using RNAiMax (Thermo Fisher Scientific) per the manufacturer’s instructions. Briefly, siRNAs were diluted in 500 µl of optiMEM (Thermo Fisher Scientific) to a final concentration of 30–50 nM (optimized by Western blot) in a six-well plate. After mixing, 8 µl of RNAiMax was added and incubated for 20 min. Cells were trypsinized and diluted in 2 ml antibiotic-free medium and then added to the siRNA mix and incubated for 48 h, with the exception of Sas6 localization experiments, which were assessed after 72 h. Knockdown efficiency was assessed by Western blot. The siRNA duplex oligonucleotides are listed in Table S3.
To generate cell lines stably expressing BirA*, BirA*-NUP188, and SNAP-Nup188, a 9:1 molar ratio of pOG44 and pNV1, pNV2, and pNV3, respectively, was cotransfected into the HeLa T-Rex-Flp-In cell line. Two days after transfection, cells were selected for Hygromycin resistance by growing them in medium containing DMEM-high glucose, 10% Tet system approved FBS (Takara), 100 U/ml penicillin and 100 µg/ml streptomycin, 15 µg/ml of Blasticidin, and 200 µg/ml Hygromycin (Thermo Fisher Scientific). Culture medium was changed every 3 d. Clonal colonies were visible by 14 d, which were then isolated using 8-mm-diameter cloning cylinders (Thermo Fisher Scientific), trypsinized, expanded, and verified by Western blotting.
Cell cycle synchronization and drug treatment
To establish S-phase–synchronized cell populations, HeLa-M and SNAP-NUP188 cell lines were grown to 30% confluency and treated with 2 mM thymidine (Millipore Sigma) for 16 h. Cells were then washed twice with PBS and incubated in medium without thymidine for 8 h. After this incubation, 2 mM thymidine was added to the medium for the next 16 h to block all cells in early S-phase. To enrich for cells in mitosis, S-phase–synchronized cells were released in the presence of 100 nM nocodazole (Millipore Sigma) for 12 h. Mitotic cells were then harvested by gently tapping the culture dish and collecting the medium. To investigate Nup188 turnover, HeLa cells expressing SNAP-NUP188 were treated with 10 µg/ml cycloheximide or 20 µg/ml MG132 or DMSO for up to 4 h at 37°C. To examine the recruitment of Sas6 to centrosomes, U2OS cells were arrested in S-phase by treatment with 1.6 µg/ml aphidicolin (Millipore Sigma) for 24 h. For microtubule-depolymerization experiments, cells were treated with 5 µg/ml nocodazole or DMSO for 3 h at 37°C.
RNA extraction, cDNA synthesis, and RT-quantitative (q)PCR
Comparison of expression levels of nup transcripts in synchronized cells was performed by RT-qPCR. Briefly, cells were synchronized in S-phase and released; cells were then harvested every hour. Total RNA was isolated using the RNeasy Plus Mini kit (Qiagen) per the manufacturer’s protocol. Superscript III RT kit (Thermo Fisher Scientific) was used to synthesize cDNA using Oligo-dT primers (Thermo Fisher Scientific). qPCR of the cDNA template was performed by the iTaq Universal SYBR Green Supermix (Bio-Rad) with 0.2 pmol/µl of gene-specific primers (Integrated DNA Technologies; Table S4). A region of the GAPDH cDNA was amplified as an internal control. The specificity of each amplicon was verified by a melt curve analysis.
Western blots
To extract total protein from whole cell extracts, cells were washed twice with PBS and then lysed in RIPA buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.5% deoxycholate, and 0.1% SDS) supplemented with both protease and phosphatase inhibitor cocktails (Thermo Fisher Scientific) on ice for 10 min. Lysates were harvested using a cell scraper and then clarified by centrifugation for 10 min at 14,000 rpm at 4°C, and supernatants were collected. Protein concentrations were determined by a Bradford assay (Thermo Fisher Scientific). 15–50 µg of protein was separated on a 4%–20% gradient gel (Bio-Rad) and transferred onto 0.2 µm nitrocellulose (Bio-Rad) using the Mini Trans-Blot Cell (Bio-Rad) at 100 V for 60 min. Blocking and antibody incubations were performed with 5% milk (American Bio) in Tris-buffered saline, 0.1% Tween 20 (TBST). Nitrocellulose was blocked with 5% milk in TBST for 1 h and then incubated with primary antibodies (listed in Table S5) for 1 h. Blots were then extensively washed in TBST and then incubated with HRP-conjugated secondary antibodies for 1 h. After washing, proteins were detected using SuperSignal West Femto kit (Thermo Fisher Scientific), and images were acquired with a ChemiDoc XRS+ Imaging system (Bio-Rad) according to the manufacturer’s instructions.
Immunofluorescence microscopy
To determine the localization of proteins in fixed cells by immunofluorescence microscopy, cells were first grown on coverslips, fixed in either −20°C methanol for 5 min or 4% PFA for 10 min at RT and washed twice with PBS. Cells were then blocked with either 5% FBS or 1% BSA in PBS with 0.1% Triton-X 100 (PBST). Cells were then incubated in primary antibody diluted in 1% FBS or 1% BSA in PBST for 1 h. After incubation, cells were washed with PBS and then incubated with secondary antibody diluted in 1% FBS or 1% BSA in PBST for 1 h. To image biotinylated proteins, streptavidin coupled to Alexa Fluor 488 (Thermo Fisher Scientific) was used. Lastly, cells were incubated with Hoechst (Thermo Fisher Scientific) to stain the DNA, and coverslips were then mounted using Fluoromount-G (Electron Microscopy Sciences) before imaging.
Immunostained cells were imaged on a Deltavision wide-field deconvolution microscope (Applied Precision/GE Healthcare Life Sciences) equipped with a 100×/1.40 (Olympus) or PLANAPO 60×/1.42 oil objective (Olympus), a CoolSnap HQ2 CCD camera (Photometrics), and a Xenon arc lamp. Image stacks were acquired in 0.2-µm increments along the z axis.
SNAP-Nup188 labeling
To label SNAP-Nup188 with fluorophores, we used either a 647-SiR dye (SNAP-Cell 647-SiR; NEB) or TMR-Star dye (SNAP-Cell TMR-Star; NEB). Briefly, expression of SNAP-NUP188 was induced by adding doxycyclin (Millipore Sigma) to the culture medium at a final concentration of 100 ng/ml for 48 h. Labeling was performed in living cells by addition of SNAP-Cell 647-SiR or SNAP-Cell TMR-Star directly to the culture medium at a final concentration of 2 µM. Labeling was performed for 30 min at 37°C. Cells were then washed three times in prewarmed medium and further incubated for 30 min at 37°C to remove excess unincorporated dye. After being washed three times with PBS, cells were fixed and processed for immunofluorescence as described above.
To determine whether centrosomes were populated by newly synthesized Nup188 or from exchange from NPCs, SNAP-NUP188–expressing cells were synchronized in S-phase and pulse labeled with SNAP-Cell 647-SiR. Subsequently, the cells were released from the S-phase block and chase labeled every hour with SNAP-Cell TMR-Star before processing for fluorescence microscopy.
To investigate the proteasomal-mediated turnover of SNAP-Nup188 at centrosomes, asynchronous cells were treated with either DMSO, cycloheximide, or MG132 as described above for up to 4 h. Samples were labeled with SNAP-Cell 647-SiR at hourly intervals before being prepared for immunofluorescence.
3D-SIM
To localize SNAP-Nup188 at centrosomes at subdiffraction resolution, we employed super-resolution 3D-SIM. Cells producing SNAP-Nup188 were labeled with the TMR-Star dye, fixed with methanol at −20°C for 5 min, and then immunostained with antibodies recognizing centrosome components. 3D-SIM imaging was performed on a DeltaVision OMX V3 system (GE Healthcare Life Sciences) equipped with a U-PLANAPO 60×/1.42 numerical aperture oil immersion objective lens (Olympus), CoolSNAP HQ2 CCD camera with a pixel size of 0.080 µm (Photometrics), and 488-nm, 561-nm, and 642-nm solid-state lasers (Coherent and MPB Communications). Image stacks were acquired in 0.125-µm increments in the z axis in sequential imaging mode. Samples were illuminated by a coherent scrambled laser light source first passed through a diffraction grating to generate the structured illumination by interference of light orders in the image plane to create a 3D sinusoidal pattern, with lateral stripes ∼0.270 µm apart. The pattern was shifted laterally through five phases and through three angular rotations of 60° for each Z-section, separated by 0.125 µm. Exposure times were typically between 25 and 150 ms, and the power of each laser was adjusted to achieve optimal fluorescence intensities between 2,000 and 4,000 in a raw image of 16-bit dynamic range, at the lowest possible laser power to minimize photo bleaching. Color channels were carefully aligned using an alignment parameter from control measurements with 0.5-µm-diameter multispectral fluorescent beads (Thermo Fisher Scientific).
Image analysis
All wide-field images presented in the figures were subjected to a constrained iterative deconvolution using softWoRx (GE Healthcare Life Sciences). All image analysis/quantification of fluorescence was performed using Fiji (Schindelin et al., 2012) on raw (i.e., nondeconvolved) images.
To quantify total fluorescence intensity of α-Nup188 and α-γ-Tubulin immunostaining at the centrosomes, cells were staged in mitosis by the morphology of the chromosomes stained with Hoechst. The integrated density of a region of interest that encompassed the centrosome was measured and divided by mean background (between cells) fluorescence.
To quantify SNAP-Nup188 fluorescence at centrosomes and the nuclear envelope during turnover experiments, a region of interest across the nuclear envelope and within the centrosome was drawn. The mean fluorescence intensity of these regions of interest were measured and subtracted from the mean fluorescence of intracellular cellular background on an individual cell basis.
To quantify changes in fluorescence intensity of α-PCM1 labeling around centrosomes, the radial profile ImageJ plugin was used. The total fluorescence intensity of an annulus of a given radius with the centrosome as its center was measured. To plot the cumulative distribution of α-PCM1 labeling throughout the cell, the radial fluorescence intensity distribution for each individual cell was normalized to the total fluorescence in each cell, averaged across all cells, and normalized to the maximum value.
Centriole number and Sas6 foci were counted manually in each cell. To quantify changes in Sas6 levels at centrosomes, the mean fluorescence intensity of α-Sas6–labeling at the centrosome was measured and the mean fluorescence of the cellular background was subtracted.
The 3D-SIM images were subjected to SIM reconstruction and image processing using the SoftWoRx 3.7 imaging software package. The channels were then aligned in x, in y, and rotationally using predetermined shifts as measured using a target lens and the SoftWoRx alignment tool.
Input experimental images were chosen manually and cropped to regions that fully encompassed the structure of interest. These images were fitted automatically and used to generate plots and average images. To generate an average image of a structure formed by centrosome-associated proteins, all input images for fitting were aligned by using the fitting output (described above) to define a center of the structure and by cropping a square region of interest around the center. Subsequently, all independent images of the structure from different individual cells were averaged by calculating a mean pixel value for a given pixel position through all cropped images.
Note that the average image (described above), while decreasing image noise, provides information at pixel resolution. Since the center of a ring structure can be at any position within the pixel, the experimental images of multiple independent rings contain information about the intensity distribution with subpixel resolution. To leverage this information, we calculated a radial distribution of intensity around the ring center. For each input image, distance from the ring center (defined by fitting) and the corresponding pixel intensity were calculated for all pixels in the image. These distance–intensity pairs were combined together and binned by the distance from the center to include n = 60 points in each bin. For each bin, a mean pixel value was calculated.
BioID proximity labeling
To determine the nearest neighbor interaction partners of BirA*-Nup188 and BirA* alone, we performed proximity labeling using BioID (Roux et al., 2012). Following the protocol outlined in Firat-Karalar and Stearns (2015), Biotin (Millipore Sigma) was added at a final concentration of 50 µM for 24 h to catalyze proximity-biotinylation. To affinity purify biotinylated proteins, cells were washed with PBS and then lysed in BioID lysis buffer (50 mM Tris, pH 7.4, 500 mM NaCl, 0.4% SDS, 5 mM EDTA, 1 mM DTT, 2% Triton X-100, and protease inhibitors; Thermo Fisher Scientific) for 10 min on ice. To ensure lysis and to break up chromatin, the mixtures were sonicated with a Bioruptor UCD-2000 (Diagenode). The resulting lysate was clarified by centrifugation at 14,000 rpm for 10 min at 4°C. The supernatant was collected and diluted with an equal volume of 50 mM Tris, pH 7.4, to promote binding to streptavidin beads. Streptavidin-coupled magnetic beads (Dynabeads MyOne Streptavidin C1; Thermo Fisher Scientific) were added to the lysate and incubated at 4°C overnight. The beads were collected on a magnet and subjected to sequential washes with the following buffers: (1) 2% SDS; (2) 0.2% deoxycholate, 1% Triton X-100, 500 mM NaCl, 1 mM EDTA, and 50 mM Hepes, pH 7.5; (3) 10 mM Tris, pH 8.1, 250 mM LiCl, 5% NP-40, 0.5% deoxycholate, 1% Triton X-100, 500 mM NaCl, and 1 mM EDTA; and (4) 50 mM Tris, pH 7.4, and 50 mM NaCl.
Mass spectrometry (MS)
Affinity-purified biotinylated proteins were subjected to on-bead trypsin digestion. Briefly, beads were washed twice with 600 µl of 50 mM ammonium bicarbonate, pH 8.0, and then trypsin (Promega) was added (200 ng in 200 µl of 25 mM ammonium bicarbonate, pH 8) to carry out in-bead digestion overnight at 37°C. After the supernatant was transferred to an Eppendorf tube, the beads were rinsed with 100 µl of water and the supernatants were combined. The solution was acidified by adding 5 µl of 20% trifluoroacetic acid and then dried in a SpeedVac (Thermo Fisher Scientific). Peptides were dissolved in 30 µl MS loading buffer (2% acetonitrile, 0.2% trifluoroacetic acid), and protein concentration was determined by a Nanodrop (Thermo Fisher Scientific) A260/A280 measurement. An aliquot of each sample was then further diluted with loading buffer to 0.1 µg/µl, and 5 µl (0.5 µg) was injected for LC-MS/MS analysis.
LC-MS/MS analysis was performed on a Thermo Fisher Scientific Orbitrap Elite mass spectrometer equipped with a Waters nanoAcquity ultra performance liquid chromatograph system using a binary solvent system (A: 100% water, 0.1% formic acid; B: 100% acetonitrile, 0.1% formic acid). Trapping was performed at 5 µl/min, 97% solvent A for 3 min using a Waters Symmetry C18 180 µm × 20 mm trap column. Peptides were separated using an ACQUITY UPLC peptide separation technology ethylene bridged hybrid C18 nanoACQUITY Column 1.7 µm, 75 µm × 250 mm (37°C) and eluted at 300 nl/min with the following gradient: 3% solvent B at initial conditions; 5% B at 3 min; 35% B at 140 min; 50% B at 155 min; 85% B at 160–165 min; and return to initial conditions at 166 min. MS was acquired in the Orbitrap in profile mode over the 300–1,700 m/z range using 1 microscan, 30,000 resolution, automatic gain control target of 1E6, and a full max ion time of 50 ms. Up to 15 tandem mass spectrometry (MS/MS) was collected per MS scan using collision-induced dissociation on species with an intensity threshold of 5,000 and charge states 2 and above. Data-dependent MS/MS was acquired in centroid mode in the ion trap using 1 microscan, AGC target of 2E4, full maximum injection time of 100 ms, 2.0 m/z isolation window, and normalized collision energy of 35. Dynamic exclusion was enabled with a repeat count of 1, repeat duration of 30 s, exclusion list size of 500, and exclusion duration of 60 s.
All MS/MS spectra were searched in-house using the Mascot algorithm (version 2.6.0) for uninterpreted MS/MS spectra after using the Mascot Distiller program to generate Mascot compatible files. The data were searched against the Swiss Protein database with taxonomy restricted to Homo sapiens, allowing for methionine oxidation as a variable modification. Peptide mass tolerance was set to 10 ppm and MS/MS fragment tolerance to 0.5 D. Normal and decoy database searches were run to determine the false discovery rates, and the confidence level was set to 95% within the MASCOT search engine for protein hits based on randomness.
Scaffold (version Scaffold_4.8.2; Proteome Software Inc.) was used to validate MS/MS–based peptide and protein identifications. Peptide identifications were accepted if they could be established at >95.0% probability by the Scaffold Local false discovery rate algorithm. Protein identifications were accepted if they could be established at >95.0% probability and contained at least one identified peptide.
To compare data across different runs and assess the abundance of each proximity partner, normalized spectral counts were determined for BirA* and BirA*-Nup188 samples. A spectral abundance factor (SAF) was calculated by normalizing the total spectral counts for a given protein to its length. To account for variability between independent runs, individual SAF values were then normalized against the sum of all SAFs for a particular run, resulting in an NSAF. Only proteins that were (1) specific to Bir*-Nup188; (2) had NSAF values 2.5-fold greater than found in the BirA-control; and (3) were detected in at least two biological replicates were selected.
Co-immunoprecipitation
To test whether Nup188 biochemically interacted with Cep192, Cep152, and PCM1, we cotransfected HEK293T cells with pFLAG-NUP188 and pEGFP-C2, pGFP-Cep192 (pKM3105), pGFP-Cep152, or pGFP-PCM1. After 24 h, cells were washed with PBS and lysed in 50 mM Tris, pH 7.4, 250 mM NaCl, 1% Igepal, 1 mM PMSF, and protease and phosphatase inhibitors for 20 min on ice. The insoluble material was pelleted by centrifugation at 14,000 rpm for 10 min at 4°C, and the supernatant was incubated with anti-GFP nanobody-conjugated agarose beads (GFP-Trap_A; Chromotek) for 2 h at 4°C. Beads were then washed in lysis buffer three times and eluted with Laemmli sample buffer. Proteins in eluates were separated by SDS-PAGE gels and processed for Western blotting.
Far Western
To assess if Nup188 directly interacts with Ceps, we performed a far Western using a protocol outlined in Einarson et al. (2007). Specifically, we generated radio-labeled Nup188 with TNT Quick Coupled Transcription/Translation System (Promega) in the presence of pKD2 and 35S-methionine (Perkin Elmer). This mixture containing 35S-labeled Nup188 was treated with RNase A (2.5 mg/ml) for 10 min, diluted in 1% nonfat milk, 5% glycerol, 20 mM Hepes (pH 7.5), 50 mM KCl, 10 mM MgCl2, 1 mM DTT, and 0.1% Igepal and incubated for 4 h at 4°C with a nitrocellulose membrane containing GFP, GFP-Cep192, GFP-Cep152, and GFP-PCM1 purified as described above. Blots were washed twice with PBS with 0.2% Triton X-100 and twice in PBS with 0.2% Triton X-100 and 100 mM KCl for 10 min each. The membrane was wrapped in plastic film, and radioactivity was detected with autoradiography.
Statistical analysis
All experiments were performed at least three times. Graphs and statistical analyses were generated using Prism (GraphPad 7.0). P values in all graphs were generated with tests as indicated in figure legends and are represented as follows: ns, P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; and ****, P ≤ 0.0001. All error bars represent the SD from the mean.
Online supplemental material
Fig. S1 shows that SNAP-Nup188 and BirA*-Nup188 are produced below endogenous Nup188 levels and Nup188 is ubiquitylated and stabilized upon proteasome-inhibition. Fig. S2 includes 3D-SIM micrographs showing that SNAP-Nup188 labels extensions that emanate from its circular core and sometimes colocalize with PCM1. Fig. S3 demonstrates that the localization of Nup188 and other PCM components at centrosomes are not interdependent. Fig. S4 shows that the centrosomal pool of Nup188 is more sensitive to siRNA knockdown than its NPC pool. Fig. S5 shows FACS analysis that confirms S-phase arrests in NUP188 siRNA–treated cells in support of Fig. 8. Table S1 lists total proteins identified by BioID. Tables S2, S3, S4, and S5 list plasmids, siRNAs, qPCR primers, and antibodies, respectively, used in this study.
Acknowledgments
We thank J. Kanyo and the Yale Keck Biotechnology Resource Laboratory for help with MS analysis and F.E. Rivera-Molina for expertise with 3D-SIM. We are also grateful to E. Nigg (University of Basel, Basel, Switzerland), T. Stearns (Stanford University, Stanford, CA), K. Tanaka (Tohoku University, Sendai, Japan), L. Pelletier (University of Toronto, Toronto, Canada), K.S. Lee (National Cancer Institute, Bethesda, MD), C. Schlieker (Yale University, New Haven, CT), V. D’Angiolella (University of Oxford, Oxford, UK), T. Melia (Yale School of Medicine, New Haven, CT), and M. King (Yale School of Medicine, New Haven, CT) for advice and reagents.
This work was generously supported by grants from the National Institutes of Health: RO1 HL124402 to C.P. Lusk and M.K. Khokha, which supports N. Vishnoi and K. Dhanasekeran. M. Chalfant is funded by NIH-5F31HL134272 and NIH-5T32GM007223.
The authors declare no competing financial interests.
Author contributions: Conceptualization and experimental design: C.P. Lusk, N. Vishnoi, K. Dhanasekeran, and M.K. Khokha; investigation, formal analysis, and visualization: N. Vishnoi, K. Dhanasekeran, I. Surovstev, M. Chalfant, and C.P. Lusk; writing of the original draft: C.P. Lusk, N. Vishnoi, and K. Dhanasekeran; writing of the review and editing: C.P. Lusk, N. Vishnoi, K. Dhanasekeran, and I. Surovstev; supervision: C.P. Lusk; funding acquisition: C.P. Lusk and M.K. Khokha.

