Text and Interview by Ruth Williams
Kinases and their targets have been the subject of Hunter's career for almost three decades.
Before 1979, kinases were only thought to stick phosphates on two of the twenty amino acids: serine and threonine. But then Tony Hunter discovered that tyrosine could also be phosphorylated (1, 2), thereby uncovering an entirely new mechanism of protein regulation in cells. Since then, Hunter has worked on all sorts of protein phosphorylation events and the kinases that deliver them. Indeed, he has been instrumental in deducing the human kinome (3, 4).

Protein phosphorylation events have an impact on practically every cellular pathway to some degree, but the main focus of Hunter's work has been their particular relevance in cancer (5). Hunter, who is a fellow of the Royal Society and member of the National Academy of Sciences, is director of the Cancer Center and American Cancer Society professor at the Salk Institute in San Diego.
In a recent interview, Hunter recounted the tale of his tyrosine phosphorylation discovery. And it's a tale with an important lesson: never dismiss an anomalous result—even if you're using out-of-date reagents!
EARLY FOCUS
How did you get started in science? My father was a surgeon in the UK National Health Service. He got me interested in biology fairly early. Then, when I went to public school at the age of 13, I was pushed up a class, and within the first week a decision had to be made whether I should take classics or science as my major subject.
My father and the headmaster had a conversation, the result of which was that I was pretty much specialized in science from then on.
That was very young for such a decision. Were you happy with it? I could have been happy either way, I expect. Clearly, I was good at science. I was not so good at math, which was an issue and still is. But science was easy, so I never really questioned the decision.
Did you ever think about following in your father's footsteps? I did, but he strongly discouraged me. He felt that the National Health Service leveled all doctors, so that the talented ones really weren't given the due they deserved.
I don't think I would have been a very good doctor. I don't have the people skills or the necessary compassion.
So you chose biology when you went off to Cambridge? I read natural sciences at Cambridge and specialized in biochemistry for my final year. But I didn't go to university straight after my A levels. I was still only 16 and I didn't feel ready. Most people going to university are 18, and that's the drinking age, so I think it makes sense to go when you're 18 to enjoy it!
You then stayed on at Cambridge? Someone in the biochemistry department suggested that I apply for an MRC studentship to do graduate work. I thought, “Well, it seems like a reasonable thing to do, and the path of least resistance.”
I opted to join Asher Korner's lab, which was the one lab in the department doing anything resembling molecular biology at the time. He actually left in the middle of my Ph.D. to take up the chair in biology in Sussex, but I decided not to move with him.
Was that rather disrupting? Not at all. He never spent much time talking to us. He let us do what we wanted. We each had our own projects that we developed, so it didn't make a lot of difference. Nevertheless, he was very important in creating a great lab environment and recruiting the best graduate students. Four out of the nine students who were there at the same time as me are now fellows of the Royal Society. It was really a very eminent group of young scientists.
What was it about molecular biology that appealed to you? It was the mid-1960s, and the genetic code was just being solved. The first protein structures were just beginning to emerge. The structure of DNA had been solved. It seemed like if you really wanted to understand how cells or organisms worked, you'd have to understand how the molecules worked inside the cells.
It was a really exciting time, because we were right there at the cutting edge, even as students. In the department in the center of town, we were very much the poor cousins to the Laboratory of Molecular Biology (LMB) up the road, where all the high profile molecular science was going on. But we went up there for seminars, and people from there, like Fred Sanger, Sydney Brenner, and Max Perutz, would come and lecture to us, which was fantastic.
UK TO USA TO UK…
If it was going so well, what made you head off to the States? After my Ph.D., I stayed on as a college fellow for four years and was planning on staying longer, but I married Pippa Marrack, who was a grad student at the LMB. She was a couple of years behind me and wanted to come to UCSD to do a postdoc with Dick Dutton.
Obviously, we had to stick together, so Alan Munro, who had done a one-year sabbatical here at the Salk Institute suggested that I work with Walter Eckhart, a new faculty member at Salk who worked on polyoma virus, a small DNA tumor virus. I thought, “Sounds interesting,” and I met Eckhart at a meeting in London, and he agreed to take me into his lab. So in 1971, Pippa and I came out to San Diego.
I teamed up with a postdoc in Walter Eckhart's lab, and we produced a bunch of papers together, setting up polyoma virus DNA synthesis as an in vitro model for DNA replication. That worked pretty well. But during this time, Pippa and I split up, and I decided I would go back to Cambridge.
Then six months later, back in Cambridge, we burned the lab down. The origin of the fire is still obscure, but it was probably caused by ether. It was pretty bad. We lost most of our stuff, but luckily the liquid nitrogen canister containing all our precious biologicals survived.
Phew! Yeah. We were homeless, but luckily a new university building had just been built right opposite the LMB, and we were offered space there on an empty floor. We actually had a functioning lab again in less than six weeks.
Max Perutz, director of the LMB, generously offered us dining rights in the LMB canteen—the famous canteen where everyone was supposed to sit at tables that did not have any of your lab mates, in order to promote scientific discussion. As a result, we hooked up with a group working on tobacco mosaic virus and had a very fruitful collaboration.
During this time, I applied for a couple of faculty positions in the UK but had no luck. So I wrote to Walter Eckhart, who had offered me a position before I left the Salk. He told me it was still open, so I moved back to San Diego in February, 1975.
…AND BACK
A few years later, you discovered that tyrosines could be phosphorylated. What led up to that? When I got back, I had begun working on polyoma virus again and by this time we knew that a protein called middle T antigen, which gets expressed immediately after the virus infects cells, could by itself transform fibroblasts. We wanted to know how but were at a bit of a loss.
A postdoc then joined my lab and started trying to identify the transforming protein for a different tumor virus: Rous sarcoma virus. We were beaten to the punch by Ray Erikson's group, who identified the Src protein. They also discovered that Src was a kinase.
That led us to test whether the polyoma virus middle T antigen was also a kinase. And it was. Erikson had reported that Src phosphorylates threonine. So I started routine hydrolysis experiments to identify the amino acid target of the polyoma kinase. One evening I ran a hydrolyzed sample of labeled middle T antigen from polyoma-infected cells together with markers for phosphoserine and phosphothreonine—the only known phosphoamino acids at the time. The next day, it was clear that the target amino acid was neither phosphoserine nor phosphothreonine.
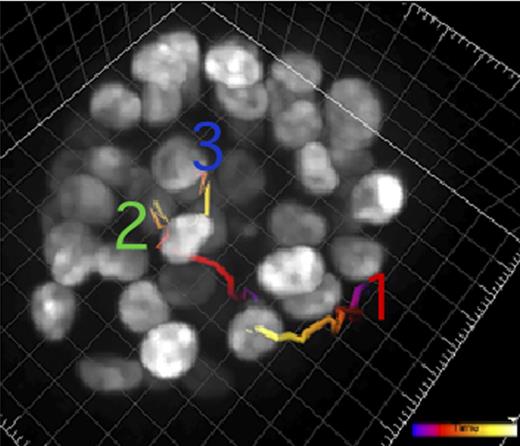
A recent study from Hunter's lab reveals how the action of kinases affects cell movement (colored lines) in a model of cancer.
My biochemistry training came in useful, because I knew that there was a third hydroxyl amino acid, tyrosine, that could potentially be phosphorylated. I crudely synthesized some phosphotyrosine, ran it against the polyoma sample, and found that indeed tyrosine was the polyoma kinase target.
To run the thin layer plates, I had been using an old bottle of pH 1.9 buffer. Then, rather foolishly, I made up some fresh buffer to repeat the experiment. To my horror, I discovered that phosphotyrosine and phosphothreonine migrated together! I spent some time aging the buffer, and it turns out that this causes its pH to drop slightly, allowing the two phosphoamino acids to run separately.
I later ran a sample of the Src kinase product as a control, and much to my amazement, this turned out also to phosphorylate tyrosine. Erikson had been misled by of the comigration of phosphotyrosine and phosphothreonine when he reported that Src is a threonine kinase.
Ah, he should have been using old buffer! Yes! When the story broke, the word spread incredibly fast. I spoke about it in December 1979 in Basel, and soon everyone knew about it and started testing their favorite transforming proteins. Within a year, we knew that tyrosine phosphorylation was important for normal cells, and within three or four years, it was clear this was a major regulatory system. Then in the early '80s, the first mutations in tyrosine kinases and its link with cancer began to be reported.
Since then, you've worked on all sorts of kinases. What's the next big question in kinase biology? Several published studies say that there are thousands of different phosphorylation events in a typical cell. So, the key questions are, What do they all do? And how many of them are noise? For some proteins, we know they're phosphorylated under particular conditions, but it's proved difficult to figure out how that changes their function. Also, for proteins with multiple sites of phosphorylation, do different combinations of phosphates mean different things?
Then, of course, there's the other side of the coin: the phosphatases. There are over 500 kinases and maybe 150 or so phosphatases. So there's a lot of interest in trying to build networks of phosphorylation events—the systems biology approach. I think that's certainly going to be a very important area.