


Few studies have assessed hypomorphic variants in ZAP70, leading to profound, late-onset combined immunodeficiency.
A previously healthy 21-year-old man presented with primary EBV B cell lymphoproliferative disease (LPD). He developed hemophagocytic lymphohistiocytosis treated with rituximab, although EBV LPD rapidly relapsed. He subsequently underwent matched, unrelated donor hematopoietic cell transplantation complicated by fatal hepatic sinusoidal obstructive syndrome.
The family history included profound immune dysregulation in his younger sister, characterized by juvenile systemic lupus erythematosus/juvenile idiopathic arthritis overlap diagnosed at 7 years of age, HPV+ warts, and EBV viremia. She failed therapy with methotrexate, leflunomide, sulfasalazine, hydroxychloroquine, rituximab, tofacitinib, abatacept, tocilizumab, and adalimumab. After NIH evaluation, she was confirmed to be HLA:B27-positive and had a marked clinical response to ustekinumab without loss of viral control. Maternal history included epidermodysplasia verruciformis, recurrent infectious rectovaginal fistulas, and lymphocytic colitis.
Whole-exome sequencing (WES) revealed novel homozygous, splice-site variants in ZAP70 (c.1623+5G>A) in all 3 affected family members. WES homology was consistent with founder effect without consanguinity, although the mother and father were fourth cousins. cDNA analysis of patient cells confirmed aberrant splicing between exons 12 and 13 (Figure 1).
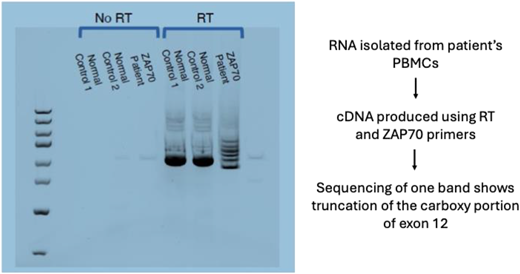
cDNA generated from RNA of the proband, his affected sister, and his affected mother confirmed aberrant splicing at Exon 12 and Exon 13 of ZAP70, consistent with the homozygous, splice-site variant found on whole-exome sequencing.
cDNA generated from RNA of the proband, his affected sister, and his affected mother confirmed aberrant splicing at Exon 12 and Exon 13 of ZAP70, consistent with the homozygous, splice-site variant found on whole-exome sequencing.
All affected family members had CD4 lymphopenia, although CD8 central and effector memory T cells were relatively preserved. Naïve CD4+ and CD8+ T cells were nearly absent in the proband and his sister. Assays in cells from all affected individuals showed reduced ZAP70-dependent T cell receptor stimulation (Figure 2) and decreased T cell proliferation to mitogens. Maternal lymphocyte phenotyping showed increased NK cells with strong cytolytic function, which may have contributed to her relatively mild phenotype. Markedly increased phospho-S6 and PD-1 expression in CD8 cells as well as increased CD4+ and CD8+ TIGIT and PD-1 double-positive cells were consistent with immune dysregulation and T cell exhaustion.
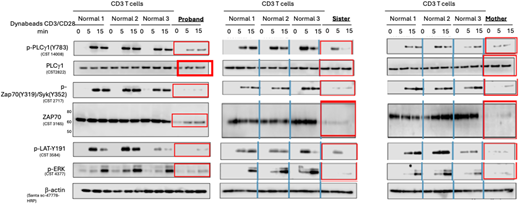
T cells from the proband, his affected sister, and his affected mother showed decreased total ZAP70 protein as well as decreased ZAP70 signaling (phospho-ZAP70, phospho-LAT, and phospho-ERK).
T cells from the proband, his affected sister, and his affected mother showed decreased total ZAP70 protein as well as decreased ZAP70 signaling (phospho-ZAP70, phospho-LAT, and phospho-ERK).
Here we present a 3-member kindred with variable expressivity of a novel homozygous, hypomorphic, splice-site variant in ZAP70. While all family members suffered from severe viral infections, they also presented with multiple features not typically associated with ZAP70 deficiency, including late-onset CID, profound immune dysregulation, and relative preservation of total and memory CD8+ T cell populations.


