


Acid-sensing ion channels (ASICs) are trimeric cation-selective channels activated by extracellular acidification. Amongst many pathological roles, ASICs are an important mediator of ischemic cell death and hence an attractive drug target for stroke treatment as well as other conditions. A peptide called Hi1a, isolated from Australian funnel web spider venom, inhibits ASIC1a and attenuates cell death in a stroke model up to 8 h after stroke induction. Here, we set out to understand the molecular basis for Hi1a’s action. Hi1a is a bivalent toxin with two inhibitory cystine knot domains joined by a short linker. We found that both Hi1a domains modulate human ASIC1a gating with the N-terminal domain impairing channel activation while the C-terminal domain produces a “pro-open” phenotype even at submicromolar concentrations. Interestingly, both domains bind at the same site since a single point mutation, F352A, abolishes functional effects and reduces toxin affinity in surface plasmon resonance measurements. Therefore, the action of Hi1a at ASIC1a appears to arise through a mutually exclusive binding model where either the N or C domain of a single Hi1a binds one ASIC1a subunit. An ASIC1a trimer may bind several inhibitory N domains and one or more pro-open C domains at any one time, accounting for the incomplete inhibition of wild type Hi1a. We also found that the functional differences between these two domains are partially transferred by mutagenesis, affording new insight into the channel function and possible novel avenues of drug design.
Introduction
Acid-sensing ion channels (ASICs) are trimeric cation-selective ion channels activated by extracellular pH stimuli lower than pH 7 (Rook et al., 2021a, 2021b, 2021c). They are expressed throughout the brain and body and are critical players in mediating ischemic-induced cell death, pain, as well as fear conditioning (Bohlen et al., 2011; Du et al., 2017; Mazzuca et al., 2007; Wemmie et al., 2003, 2004; Xiong et al., 2004). Consequently, they are drug targets of high significance. Yet despite much effort and several promising leads, no ASIC-selective small molecule compounds have entered clinical use (Vullo and Kellenberger, 2019). The highest affinity ASIC modulators are toxins derived from snakes, spiders, and sea anemone (Bohlen et al., 2011; Chassagnon et al., 2017; Diochot et al., 2004, 2012; Escoubas et al., 2000). Understanding how these diverse toxins modulate the ASIC family may lead to the development of new therapeutics targeting ASICs.
The best studied ASIC toxin is psalmotoxin1 (PcTx1), a 40 amino acid peptide that adopts an inhibitory cystine knot (ICK) fold stabilized by three disulfide bonds (Escoubas et al., 2000, 2003). Toxins containing ICK folds modulate many ion channels including voltage-gated sodium, calcium, and potassium channels, and are highly stable in vivo (Daly and Craik, 2011; Er et al., 2017). Certain ICK toxins also modulate voltage-insensitive sodium-selective ASIC channels. PcTx1 binds within the acidic pocket (Baconguis and Gouaux, 2012; Dawson et al., 2012), a region of the ASIC extracellular domain important for proper pH gating (Baconguis and Gouaux, 2012; Rook et al., 2021a, 2021b, 2021c, 2023). PcTx1 binding increases the apparent affinity for protons, causing the channel to desensitize at physiological pHs (Chen et al., 2005). A more recently discovered toxin named Hi1a shows great promise as both an ASIC1a inhibitor and a potential lead for ASIC1a-targeted drugs. Hi1a contains two PcTx1-like ICK domains linked together to form a double-knot or bivalent structure (Chassagnon et al., 2017) (Fig. 1 A). Hi1a combines partial inhibition with extremely high potency. Moreover, administration of Hi1a up to 8 h after an ischemic stroke event can reduce infarct damage and functional loss (Chassagnon et al., 2017). Thus, Hi1a is of tremendous clinical interest, given the prevalence of stroke and other pathophysiological conditions ASIC1a is involved in such as cardiac disease (Chassagnon et al., 2017; Redd et al., 2021). Yet, the precise mechanism of how the bivalent Hi1a works remains unclear. A possible model is that one domain binds with high affinity, targeting Hi1a to ASIC1a and enabling the second lower affinity domain to allosterically modulate the channel through a yet-to-be-discovered site. An alternative is a mutually exclusive binding model where each ASIC subunit binds a different Hi1a molecule, by either N- or C-domain, and the pattern of modulation depends on the number and type of each domain bound to the trimer.

Modulation of hASIC1a by Hi1a wild type as well as both N and C domains. (A) Sequence alignment of Hi1a, the isolated N and C domains, Hi1a-CN, and PcTx1. Solid black denotes complete conservation. (B) Structures of PcTx1 (green, from PDB accession no. 4FZ0) or Hi1a (PDB accession no. 2N8F) showing the N domain (blue) and C domain (red). (C) Structure of cASIC1 bound to PcTx1 (PDB accession no. 4FZ0). PcTx1 is colored in green while individual ASIC subunits are colored in black, grey, or white. (D) Outside-out patch recording of hASIC1a responses to pH 6 following incubation with varying concentrations of Hi1a. (E) Example of excised patch recordings as in D but using N domain (left) or C domain (middle) or Hi1a-CN (right). (F) Concentration–response curves of hASIC1a to Hi1a wild type (purple), N domain (blue), C domain (red), or Hi1a-CN (orange). Error bars indicate SEM.
Modulation of hASIC1a by Hi1a wild type as well as both N and C domains. (A) Sequence alignment of Hi1a, the isolated N and C domains, Hi1a-CN, and PcTx1. Solid black denotes complete conservation. (B) Structures of PcTx1 (green, from PDB accession no. 4FZ0) or Hi1a (PDB accession no. 2N8F) showing the N domain (blue) and C domain (red). (C) Structure of cASIC1 bound to PcTx1 (PDB accession no. 4FZ0). PcTx1 is colored in green while individual ASIC subunits are colored in black, grey, or white. (D) Outside-out patch recording of hASIC1a responses to pH 6 following incubation with varying concentrations of Hi1a. (E) Example of excised patch recordings as in D but using N domain (left) or C domain (middle) or Hi1a-CN (right). (F) Concentration–response curves of hASIC1a to Hi1a wild type (purple), N domain (blue), C domain (red), or Hi1a-CN (orange). Error bars indicate SEM.
Here, we investigated the mechanism of Hi1a’s modulation and distinguished between these two models using a combination of fast perfusion electrophysiology and surface plasmon resonance (SPR). We first clearly define the activities of the individual N- and C-terminal ICK domains upon the channel, providing novel information about how the individual domains can contribute to the effect of the linked wild type bivalent Hi1a. We also find strong evidence that each domain of Hi1a can bind to the acidic pocket with nanomolar affinity but with distinct functional effects. We further identify mutations within the more distinct C-terminal domain that account for the unusual modulatory pattern. Taken together, our data strongly support a mutually exclusive binding model and identify the ICK fold as a highly versatile platform for modulating ASIC function with a large and unexplored space of possible pharmacological effects.
Materials and methods
Cell culture, mutagenesis, and transfection
Human embryonic kidney (HEK) 293T cells with the endogenous ASIC1 gene deleted via CRISPR (Rook et al., 2021a, 2021b, 2021c) were used for all electrophysiology experiments. These HEK293T cells were maintained in MEM supplemented with 10% equifetal bovine serum (Atlas Biologicals) and PenStrep (Gibco) and passaged every 3–4 days but not >25 passages. Cells were plated on 35-mm tissue-cultured treated Petri dishes and transfected 1–2 days later using polyethylenimine 25k (Polysciences, Inc.) with a mass ratio of 1:3 (cDNA:PEI). A pcDNA3.1(+) vector containing the sequence for codon-optimized human ASIC1a (hASIC1a) was used for all experiments unless otherwise indicated. Mutations to this, or toxin expression constructs, were introduced using site-directed mutagenesis PCR and confirmed by Sanger sequencing.
Electrophysiology
Electrophysiology recordings were performed 1–3 days after transfection as described elsewhere. Briefly, outside-out patches or whole-cell recordings were obtained using borosilicate patch pipettes with resistances of 3–6 MΩ when filled with an internal pipette solution. The internal pipette solution was (in mM) 135 CsF, 11 EGTA, 10 HEPES, 10 MES, 2 MgCl2, and 1 CaCl2, and pH was adjusted to 7.4 using CsOH. External solutions were comprised of (in mM) 150 NaCl, 1 CaCl2, 1 MgCl2, and either 10 HEPES (pH 7.45) or 10 MES (pH 6 or less) and adjusted using Tris-base to the indicated pH. All external solutions contained 0.02% bovine serum albumin (BSA) to prevent peptide adsorption. Note that BSA also chelates calcium ions, making direct comparisons of calcium between BSA-containing and BSA-lacking solutions difficult. Unless otherwise stated, toxins were only supplied in the conditioning pH line and not the activation pH line. Data were acquired at 20–50 kHz and filtered online at 10 kHz using Clampex11, an Axopatch 200 B amplifier, and a 1550 converter (all Molecular Devices) at room temperature and with a holding potential of −60 mV. Series resistance was routinely compensated by 90–95% when the peak amplitude exceeded 100 pA. A home-built double or triple barrel perfusion pipette (Vitrocom) attached to a piezo translator (Physik Instrumente) under computer control was used for fast perfusion. Piezo voltage commands were generally filtered between 50 and 100 Hz.
Peptide purification
The pLicC-MBP-APETx2 vector was purchased from AddGene (#72668). The coding sequence of Hi1a was custom synthesized and cloned in place of APETx2 using the NEBuilder system resulting in the pLicC-MBP-Hi1a vector (Fig. S1). The MalEss signal sequence was removed and the resulting vector was transformed into SHuffle cells (C3029J), grown at 30°C until the OD600 reached ∼1.0, and then expression was induced with 0.1 mM IPTG. Bacterial cultures expressing MBP-Hi1a were grown overnight at 18°C and pelleted. Bacterial pellets were resuspended in lysis buffer (350 mM NaCl, 20 mM imidazole, 1 mM PMSF, and 1 mg/ml lysozyme, pH 8.0) and then sonicated. Cell lysates were passed through a gravity Ni-NTA column (HisPur, PI88221; Thermo Fisher Scientific) and then eluted with a 500 mM imidazole elution buffer. The eluate was incubated with TEV protease overnight to liberate the toxin from MBP. The cleaved toxin solution was precipitated overnight at 4°C with 1% trifluoroacetic acid (TFA). The precipitated solution was then filtered and loaded onto an Agilent 1260 Infinity II HPLC system and the peptide was isolated using a C18 column (Zorbax 300, #880995-902; Agilent). Peptides were isolated using a 10–50% Buffer B gradient. Buffer B was composed of 90% acetonitrile, 10% H2O, and 0.1% TFA. Toxin aliquots were collected, concentrated to a stock volume of ∼500 μl, and stored at −80°C. Toxin concentration was assessed using A280 measurements and the corresponding toxin’s calculated molar absorptivity.
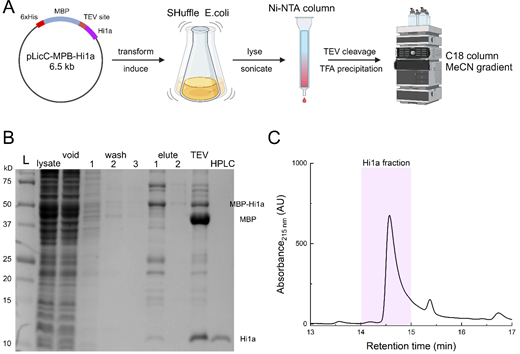
Toxin purification. (A) Workflow of toxin purification using the indicated expression vector transformed into SHuffle cells followed by initial column purification, TEV cleavage, TFA precipitation, and reverse phase purification. Figure prepared using Biorender. (B) Example purification gel showing the ladder, lysate, void, three sequential washes, two sequential elutions, post-TEV cleavage, and final HPLC purification using C18 column. (C) HPLC chromatograph illustration of the Hi1a peak and approximate fraction collected. Source data are available for this figure: SourceData FS1.
Toxin purification. (A) Workflow of toxin purification using the indicated expression vector transformed into SHuffle cells followed by initial column purification, TEV cleavage, TFA precipitation, and reverse phase purification. Figure prepared using Biorender. (B) Example purification gel showing the ladder, lysate, void, three sequential washes, two sequential elutions, post-TEV cleavage, and final HPLC purification using C18 column. (C) HPLC chromatograph illustration of the Hi1a peak and approximate fraction collected. Source data are available for this figure: SourceData FS1.
Surface plasmon resonance
hASIC1a with a C-terminal twin strep tag (TST) was expressed by transient transfection in HEK293T ASIC1 KO cells. After transfection, the adherent ASIC1 KO cells were allowed to express the channel for ∼3 days. Cells expressing hASIC1a-TST were then isolated by centrifugation and solubilized by end-over-end spinning for 1 h at 4°C in a buffer containing 40 mM n-dodecyl-β-maltoside (DDM), 150 mM NaCl, and 20 mM Tris (pH 8.0). The solubilized hASIC1a-TST was immobilized onto a Biacore series S sensor chip (SA) until a response unit (RU) level of 700 was achieved. SPR kinetic analysis was performed using a Biacore T200 with a flow rate of 50 μl/min. Kinetics were observed in the association phase over 60 s with a wide range of different peptide concentrations. The length of the observed disassociation phase depended on the peptide tested, with lengths ranging from 100 s (Hi1aC) up to 400 s (Hi1a). Kinetic experiments were conducted with a running buffer composed of 150 mM NaCl, 20 mM HEPES, and 1 mM DDM (pH 7.5). Kinetic data were analyzed using Biacore’s T200 evaluation software version 3.2 (Cytiva) with the double referencing method (Myszka, 1999). Fitting to a 1:1 analyte to ligand binding mode yielded the on rate (kon), off rate (koff), and apparent equilibrium binding constant (KD).
Statistics, data analysis, and simulations
Kinetic simulations were performed using Kinetic Model Builder (Goldschen-Ohm et al., 2014) in “Eigen solver mode” using rate constants for association and dissociation from SPR data. The model was run for 300 s with 10 ms time steps. At time 1 s, 100 µM each of N- and C-domain toxins was added and removed at 201 s.
Online supplemental material
Fig. S1 shows the general workflow and quality control for recombinant peptide production.
Results
Preliminary characterization of wild type Hi1a, and N and C domains
Prior work using recombinant Hi1a established several distinct functional properties. First, Hi1a is selective for ASIC1a, does not inhibit ASIC2 or ASIC3, and shows weak potentiation of ASIC1b (Chassagnon et al., 2017). Second, the inhibition of hASIC1a occurs with a subnanomolar IC50 (0.52 ± 0.06 nM) and is incomplete (∼80% inhibition) (Chassagnon et al., 2017). Third, Hi1a slows the rising phase of channel activation (Chassagnon et al., 2017). Given the remarkable protective effect of Hi1a in both neuronal and cardiac stroke models (Chassagnon et al., 2017; Redd et al., 2021), we set out to investigate how Hi1a produces this unique set of features.
Hi1a is a bivalent ICK toxin made of two PcTx1-like domains fused together (Fig. 1, A and B). The N-terminal domain is similar in sequence to PcTx1 (67% sequence identity) while the C domain is more distinct (46% identity, Fig. 1 A). To investigate the functional properties of wild type Hi1a and each individual domain, we purified each recombinant toxin and examined their properties using outside-out patch clamp electrophysiology (see Materials and methods, Fig. S1). Our purified Hi1a behaved as previously described at hASIC1a. Namely, we found high potency and incomplete inhibition (0.3 ± 0.1 nM, n = 6; 77 ± 9% inhibition, Fig. 1, D and F) along with slowing of the rising phase (no toxin: 8.1 ± 2.2 ms, n = 6; 100 nM Hi1a: 170 ± 36 ms, n = 5, P < 1e−6Fig. 1 D and Fig. 2 C). We next turned to studying the individual N and C domains. Here again, our data were consistent with past work (Chassagnon et al., 2017). The N domain produced inhibition but at a much higher concentration than wild type Hi1a (IC50 7 ± 1 µM, n = 5–7, Fig. 1, E and F). This is a lower potency than previously found (1.0 ± 0.1 µM [Chassagnon et al., 2017]), most likely due to the use of higher affinity rat ASIC1a in that study (Chassagnon et al., 2017; Cristofori-Armstrong et al., 2019). The C domain did not produce any detectable inhibition (Fig. 1, E and F, P = 0.67 versus no inhibition). While the C domain potentiates rat ASIC1a at 1 µM (Chassagnon et al., 2017), we did not observe this in our hASIC1a recordings. However, we did note two observations not previously reported. First, the isolated C domain slowed activation times at relatively low concentrations (Fig. 1 E; and Fig. 2, A and C, 10–90% rise time before toxin: 7.7 ± 2.5 ms, 88 ± 26 ms after 100 nM C domain, n = 6; 18 ± 6-fold slower, P = 0.0003 versus before toxin). This was also observed with wild type Hi1a in our experiments (Fig. 1 D and Fig. 2 C) and previously (Chassagnon et al., 2017) but not with the N domain (Fig. 1 E and Fig. 2 C). Second, the deactivation decay time constants of hASIC1a responses were nearly 10-fold slower in the presence of C domain toxin (Fig. 1 E; and Fig. 2, A and D, no toxin: 4.2 ± 0.3 ms; 1 μM C domain: 79 ± 18 ms, 9 ± 1.2-fold slower, n = 7, P = 0.005 versus no toxin). The slowing of deactivation was concentration-dependent and detectable at concentrations as low as 10 nM (Fig. 2 A, 2.6 ± 0.3-fold slowing of deactivation by 10 nM C domain, P < 0.004). This phenomenon was not observed with either the N domain (except at the highest concentration of 10 µM) or wild type Hi1a (Fig. 1, D and E; and Fig. 2 D, 6 ± 2 ms deactivation time constant before toxin, 7 ± 2 ms after 100 nM Hi1a, n = 6, P = 0.65 versus before toxin). These two new observations demonstrate that the isolated C domain can interact with hASIC1a with much higher affinity than expected. Furthermore, the C domain is likely responsible for the slowing of channel opening observed in wild type Hi1a.

Hi1a C domain modulates hASIC1a kinetics at nanomolar concentrations. (A) Left: Excised patch response of wild type hASIC1a preincubated with isolated C domain toxin at indicated concentrations. Same patch as in Fig. 1 E (middle). Right: Expanded time base of the boxed region in A (left). (B) Same experiment as in panel A but for hASIC1a F352A mutation. (C) Rise times (10–90%) of pH 6 evoked responses in excised patches for wild type (open circles) or F352A (stars) in the indicated concentration of Hi1a (purple), N domain (blue), C domain (red), or Hi1a-CN (orange). (D) Deactivation time constants for wild type (open symbols) or F352A (filled symbols) for Hi1a (100 nM, purple), N domain (1 µM, blue), C domain (1 µM, red), or Hi1a-CN (100 nM, orange). For all plots, error bars indicate SEM.
Hi1a C domain modulates hASIC1a kinetics at nanomolar concentrations. (A) Left: Excised patch response of wild type hASIC1a preincubated with isolated C domain toxin at indicated concentrations. Same patch as in Fig. 1 E (middle). Right: Expanded time base of the boxed region in A (left). (B) Same experiment as in panel A but for hASIC1a F352A mutation. (C) Rise times (10–90%) of pH 6 evoked responses in excised patches for wild type (open circles) or F352A (stars) in the indicated concentration of Hi1a (purple), N domain (blue), C domain (red), or Hi1a-CN (orange). (D) Deactivation time constants for wild type (open symbols) or F352A (filled symbols) for Hi1a (100 nM, purple), N domain (1 µM, blue), C domain (1 µM, red), or Hi1a-CN (100 nM, orange). For all plots, error bars indicate SEM.
The bivalent nature of Hi1a allows for an intriguing possible two-site avidity mechanism of action. It is possible the N domain binds to the acidic pocket with high affinity just as PcTx1 while the C domain binds to a distinct yet-to-be-identified modulatory site. The binding of the N domain in the acidic pocket would increase the local concentration of the C domain near its putative modulatory site, enabling the C domain to interact and modify gating. This avidity model would also account for the slow wash-off of Hi1a in functional measurements. However, it is also possible that the N and C domain both bind within the acidic pocket in a mutually exclusive fashion. In this model, each of the three ASIC subunits would be bound by a different Hi1a molecule with either an N or C domain at the acidic pocket. To distinguish between these possibilities, we mutated the Phe352 position found at the outer edge of the acidic pocket to alanine. Mutation of Phe352 markedly attenuates PcTx1 inhibition and binding (Heusser et al., 2022; Saez et al., 2015; Sherwood et al., 2009). And as expected, the F352A mutation essentially abolishes Hi1a inhibition (Chassagnon et al., 2017). If the C domain binds within the acidic pocket, it should also be sensitive to the F352A mutation. Consistent with this prediction, the slowing of rise-times induced by either Hi1a or the C domain was eliminated in the F352A mutation (Fig. 2, B and C, F352A rise times before Hi1a: 19 ± 3 ms, after 100 nM Hi1a: 20 ± 3 ms, 1.0 ± 0.1-fold change, n = 5, P = 0.843 versus no toxin; before C domain 17 ± 5 ms, after 1 μM C domain 13 ± 4 ms, 0.8 ± 0.2-fold change, n = 5, P = 0.5476 versus no toxin). Similarly, the slowing of hASIC1a wild type deactivation by the C domain was eliminated by the F352A mutation (Fig. 2, B and D, deactivation before C domain: 1.8 ± 0.7 ms, after 1 μM C domain: 2 ± 0.7 ms, 1.0 ± 0.1-fold change, n = 5, P = 0.935 versus no toxin). These F352A data indicate that the C domain modulates through binding to the acidic pocket, supporting a mutually exclusive binding model.
To further distinguish between a mutually exclusive binding model and a two-site avidity model, we reversed the N and C domains and placed the C domain upstream of the N domain in a construct called Hi1a-CN (see Fig. 1 A for sequence). If Hi1a works through a two-site avidity mechanism, then the C domain of Hi1a-CN should be poorly positioned to interact with any putative modulatory site. The N domain should bind the acidic pocket as before, leaving the C domain to essentially float above the channel without an interaction site. However, if Hi1a works through a mutually exclusive binding model, then Hi1a-CN should still modulate just as Hi1a wild type. Hi1a-CN inhibited channel activation with an IC50 of 16 ± 6 nM (Fig. 1, E and F, n = 5). Based on the shallow slope of the inhibition curve, Hi1a-CN appears to produce partial inhibition but this is difficult to determine based on the concentrations we probed (Fig. 1 F). Consistent with a mutually exclusive model, Hi1a-CN slowed hASIC1a rise times (rise times before Hi1a-CN: 9 ± 4 ms, after 100 nM Hi1a-CN: 35 ± 11 ms, 5.2 ± 1.1-fold change, n = 5, P = 0.024 versus no toxin, Fig. 1 E and Fig. 2 C). Hi1a-CN slowed channel deactivation as well (deactivation before Hi1a-CN: 13 ± 9 ms, after 100 nM Hi1a-CN: 49 ± 16 ms, 6.6 ± 2.8-fold change, n = 5, P < 1e−6, Fig. 1 E and Fig. 2 C), just as the C domain alone did. Thus, Hi1a-CN possesses the general features of Hi1a wild type with inhibition and slowing of rise-time but has a stronger “C domain fingerprint” as evidenced by slow deactivation (Fig. 1 F). These data, combined with the F352A results, strongly favor the mutually exclusive binding model.
Each domain differentially alters proton sensitivity
Since the isolated C domain slows channel deactivation (Figs. 1 and 2), the C domain might also left-shift pH response curves. We constructed activation and SSD curves of hASIC1a alone or in the presence of wild type Hi1a, N, or C domain. Consistent with past work, 1 nM wild type Hi1a slightly right-shifted activation curves by ∼0.1 pH units and left-shifted SSD curves by 0.15 pH units (Control pH50act 6.52 ± 0.02, n = 7–8; + Hi1a pH50act 6.41 ± 0.04, n = 6, P = 0.019; Control pH50SSD 7.08 ± 0.02, n = 7–10; + Hi1a pH50SSD 7.26 ± 0.04, n = 7–9, P = 0.0023, Fig. 3, A and C). As with past experiments (Chassagnon et al., 2017), we also found that Hi1a considerably shallowed the slope of the SSD curves (control slope 8.1 ± 2.0; + Hi1a slope 1.6 ± 0.06, P = 0.029). The N domain produced a substantial left shift in the SSD curve, effectively facilitating SSD at much higher pH values (+1 μM N domain pH50SSD 7.36 ± 0.04, n = 5, P < 1e−6 versus control, Fig. 3). This is somewhat expected given the N domain’s higher sequence identity with PcTx1 (Chen et al., 2005). However, unlike PcTx1 (Heusser et al., 2022), the N domain did not induce a corresponding left shift in the activation curve (+3 μM N domain pH50Act 6.58 ± 0.05, n = 4, P = 0.19 versus control, Fig. 3). The C domain did induce a small but statistically significant left shift in activation curves (+100 nM C domain pH50act 6.65 ± 0.02, n = 4, P = 0.004 versus control, Fig. 3), as well as a right shift in SSD curves (+100 nM C domain pH50SSD 6.97 ± 0.04, n = 4, P = 0.043 versus control). Essentially, the C domain brings the activation and SSD curves closer together in an “inward shift,” potentially resulting in a greater “window current” in the pH 7 to 6.8 range. This surprising effect indicates the C domain has a broadly “pro-open” phenotype, requiring a weaker stimulus to open the pore and a stronger stimulus to inactivate the channel.

Domains of Hi1a differentially influence acid sensitivity . (A–C) Example outside out patch steady-state desensitization experiments for hASIC1a in the presence of 1 nM Hi1a (A), 1 μM N domain (B), or 100 nM C domain (C). (D) Steady-state desensitization and activation curves for hASIC1a alone or in the presence of the indicated toxin. Note either 1 or 3 μM N domain was used for SSD or activation curves, respectively. (E) Summary of pH50’s for steady-state desensitization (downward arrow) and activation (filled circle) across toxin conditions. Symbols denote means for D and individual patches for E. All error bars are SEM.
Domains of Hi1a differentially influence acid sensitivity . (A–C) Example outside out patch steady-state desensitization experiments for hASIC1a in the presence of 1 nM Hi1a (A), 1 μM N domain (B), or 100 nM C domain (C). (D) Steady-state desensitization and activation curves for hASIC1a alone or in the presence of the indicated toxin. Note either 1 or 3 μM N domain was used for SSD or activation curves, respectively. (E) Summary of pH50’s for steady-state desensitization (downward arrow) and activation (filled circle) across toxin conditions. Symbols denote means for D and individual patches for E. All error bars are SEM.
Both N and C domains bind the acidic pocket with high affinity
The functional experiments corroborate prior work showing that Hi1a inhibits the channel with higher potency relative to the N domain (Chassagnon et al., 2017). We also uncovered a surprising capacity for the C domain to modulate channel gating with an apparently higher potency than the N domain (Fig. 1). To directly measure toxin binding, we conducted SPR measurements using hASIC1a with a C-terminal twin-strep tag, solubilized and bound to a streptavidin sensor chip. Initial control experiments with PcTx1 revealed rapid binding with very slow dissociation kinetics, yielding a KD of ∼7 pM (Fig. 4, B and F). Because of the unexpectedly strong binding of PcTx1 in our SPR measurements, we repeated our experiments using the hASIC1a F352A mutation that is expected to reduce the affinity of PcTx1 (Fig, 4, A and B). Consistent with this, the affinity of PcTx1 on the F352A channel was reduced by three orders of magnitude to 14 nM (Fig. 4, C and F). We next tested Hi1a as well as the individual N and C domains and the reversed construct Hi1a-CN. Of these, Hi1a had the highest affinity with a KD of 0.2 nM, followed by Hi1a-CN with a KD of ∼0.8 nM, the N domain with a KD of 1.4 nM, and the C domain with a KD of 9 nM. The estimated association rates ranged from 4.7 × 106 to 6.88 × 107 M−1 s−1 while the dissociation constants spanned a wider range, from 0.18 s−1–2.7 × 10−4 s−1 (Table 1), indicating that dissociation is the main driver of differences between the toxin affinities. Thus, the rank order of toxin affinities is Hi1a > Hi1a-CN > N domain > C domain, which contrasts with the IC50s from functional measurements, where the C domain showed a modulatory effect at much lower concentrations than the N domain (Figs. 1 and 5). The most straightforward explanation is that the functional effects of the C domain require fewer toxins to bind than the N domain. For example, inhibition by the N domain may require all three subunits to be occupied or even just two subunits, as PcTx1 does (Heusser et al., 2022). However, the C domain-induced changes to rise-time and deactivation might need only one subunit bound to be measurable.

All toxins bind to the acidic pocket but with varying affinities. (A) Structure of cASIC1 bound to PcTx1 (PDB accession no. 4FZ0) and zoomed into the acidic pocket. (B) Example multicycle SPR sensorgrams of various concentrations of PcTx1 (left) or Hi1a (right) binding and dissociation. (C) Same as in B but using hASIC1a F352A mutation. Note the compressed time scale due to much faster dissociation. (D and E) SPR sensorgrams for either N domain (D) or C domain (E) over the indicated concentration range. (F) Summary of equilibrium constants for all toxins tested in wild type or F352A channels. N/D stands for not determinable.
All toxins bind to the acidic pocket but with varying affinities. (A) Structure of cASIC1 bound to PcTx1 (PDB accession no. 4FZ0) and zoomed into the acidic pocket. (B) Example multicycle SPR sensorgrams of various concentrations of PcTx1 (left) or Hi1a (right) binding and dissociation. (C) Same as in B but using hASIC1a F352A mutation. Note the compressed time scale due to much faster dissociation. (D and E) SPR sensorgrams for either N domain (D) or C domain (E) over the indicated concentration range. (F) Summary of equilibrium constants for all toxins tested in wild type or F352A channels. N/D stands for not determinable.
Kinetic parameters of toxin association and dissociation from SPR fits
| Toxin | KON (M−1 s−1) | KOFF (s−1) | KD (M) |
|---|---|---|---|
| PCTX1 | 3.81e7 ± 3.5e5 (1.03e7± 5.1e4)a | 2.73e−4 ± 1.10e−6 (0.146 ± 3.60e−4)a | 7.12e−12 (1.42e−8)a |
| HI1A | 6.88e7 ± 8.5e5 (7.49e5± 5.2e3)a | 0.014 ± 1.70e−4 (0.157 ± 6.00e−4)a | 1.98e−10 (2.09e−7)a |
| N | 4.68e6 ± 1.3e4 (2.06e6± 2.7e3)a | 0.007 ± 1.50e−5 (0.320 ± 1.50e−4)a | 1.42e−9 (1.5e−7)a |
| C | 2.10e7 ± 1.8e5 | 0.184 ± 0.0017 | 8.75e−9 |
| HI1A-CN | 3.61e7 ± 5.2e5 | 0.030 ± 4.4e−4 | 8.19e−10 |
| S27R | 4.79e7 ± 2.9e5 | 0.001 ± 8.10e−6 | 2.77e−11 |
| G28R | 2.29e7 ± 4.0e5 | 0.048 ± 9.10e−4 | 2.11e−9 |
| N29S | 1.53e7 ± 2.5e5 | 0.182 ± 0.003 | 1.27e−8 |
Smaller italicized values are using F352A hASIC1a mutation.

Relation between toxin potency and affinity. Plot of the affinity measured by SPR (KD) on the ordinate axis versus the potency as measured by functional changes in patch clamp (IC or EC50) on the abscissa. The dotted line denotes a slope of 1.
Relation between toxin potency and affinity. Plot of the affinity measured by SPR (KD) on the ordinate axis versus the potency as measured by functional changes in patch clamp (IC or EC50) on the abscissa. The dotted line denotes a slope of 1.
Is the C domain binding to a distinct site? The F352A mutation abolishes C domain modulation of kinetics, suggesting that the C domain exerts a functional effect via the acidic pocket (Fig. 2). However, this experiment is complicated since the F352A mutation itself affects channel gating (Heusser et al., 2022). To directly measure binding, we repeated the SPR measurements using the F352A channel. Importantly, the affinity of all Hi1a-based toxins was markedly reduced by the F352A mutation (Fig. 4 F). Indeed, the reduction in C domain binding was so great that a KD could not be determined using the F352A hASIC1a due to a large amount of C domain required for such low-affinity binding (Fig. 4 F and Table 1). The reduction in affinity imparted by F352A is solid evidence that all three Hi1a derivatives share a common binding region within the acidic pocket, likely with some overlapping contacts. Combined with the functional data from F352A and Hi1a-CN (Figs. 1 and 2), there is strong evidence that the C domain binds and modulates gating through interactions within the acidic pocket.
Given that the N and C domains bind to the same site, we constructed a kinetic model of a trimer with one toxin binding site per subunit using the association and dissociation constants from SPR (Fig. 6). At equilibrium with saturating concentrations of both toxins, >99% of channels have at least one inhibitory N domain bound and 62% have three N domains. Thus, 38% of channels have at least one C domain bound with 32% having only one C domain and 5.5% having two C domains bound (Fig. 6). This simulation relies on SPR measures of isolated domains and thus cannot reflect the binding and unbinding of those domains in the full Hi1a due to the influence of the linker and adjacent domains, possibly contributing steric effects. Nonetheless, the broad pattern of occupancy is likely to hold with the wild type toxin. With saturating Hi1a, roughly 60% of channels would be exclusively bound by N domains and completely inhibited. The remaining 40% of channels would each have one or more of the pro-open C domains and thus contribute current with a slower rise time.

Simulations of co-applied N and C domain binding and unbinding. (A) Schematic of trimer model where each subunit has a single binding site for either the N or C domain. N domain binding occurs left to right and C domain binding occurs lower to upper. Percentages give the occupancy at time 200 s from panel B. Rate constants taken from SPR data in Table 1. (B) Time-dependent occupancy following application of 100 µM each of N and C domains at time 1 s and removal at time 201 s.
Simulations of co-applied N and C domain binding and unbinding. (A) Schematic of trimer model where each subunit has a single binding site for either the N or C domain. N domain binding occurs left to right and C domain binding occurs lower to upper. Percentages give the occupancy at time 200 s from panel B. Rate constants taken from SPR data in Table 1. (B) Time-dependent occupancy following application of 100 µM each of N and C domains at time 1 s and removal at time 201 s.
Structure activity of the C domain
PcTx1, N, and C domain toxins are all single-domain ICK toxins with high structural similarity. Yet, they produce a wide range of functional effects. PcTx1 binds very tightly and left-shifts both activation and SSD curves (Chen et al., 2005; Heusser et al., 2022). In contrast, the C domain binds with lower affinity and, instead of producing parallel left (or right) shifts of both activation and SSD curves, induces an “inward shift,” bringing the SSD and activation curves closer together resulting in a pro-open phenotype (Fig. 3). The N domain binds with an affinity between PcTx1 and the C domain, yet, does not exert a measurable functional effect until relatively high occupancies (i.e., big difference between KD and IC50, Fig. 5). Furthermore, the N domain seems to produce an “outward shift” in pH response curves, left-shifting the SSD curve. The activation curve was not statistically different, although it was slightly right-shifted (Fig. 3, C and D); therefore the N domain either does not change or possibly right-shifts the activation curve. Given the array of functional phenotypes, we set out to probe the structure–activity relationships by mutating individual residues in the large loop of the C domain to the corresponding amino acids found in PcTx1 and subsequently assessing both binding and functional modulation (Fig. 7 A).

Arg27 is critical for high affinity. (A) Structures of PcTx1 bound to cASIC1 (PDB accession no. 4FZ0) (left) or C domain structure (PDB accession no. 2N8F) modeled in place of PcTx1 using ChimeraX’s matchmaker tool. The divergent β hairpin loop residues are depicted. (B) Inhibition curves of hASIC1a by the indicated C domain mutant. (C) SPR sensorgrams for S27R (left) and G28R (right). (D) Equilibrium constants from SPR fit for C domain mutants. Values for PcTx1 and wild type C domain are shown as dashed lines. (E) hASIC1a outside-out patch responses in varying concentrations of S27R (left) or G28R (right). (F) Summary of fold slowing of rise times (left) and deactivation (right) for hASIC1a responses in the indicated C domain mutation. All error bars are SEM.
Arg27 is critical for high affinity. (A) Structures of PcTx1 bound to cASIC1 (PDB accession no. 4FZ0) (left) or C domain structure (PDB accession no. 2N8F) modeled in place of PcTx1 using ChimeraX’s matchmaker tool. The divergent β hairpin loop residues are depicted. (B) Inhibition curves of hASIC1a by the indicated C domain mutant. (C) SPR sensorgrams for S27R (left) and G28R (right). (D) Equilibrium constants from SPR fit for C domain mutants. Values for PcTx1 and wild type C domain are shown as dashed lines. (E) hASIC1a outside-out patch responses in varying concentrations of S27R (left) or G28R (right). (F) Summary of fold slowing of rise times (left) and deactivation (right) for hASIC1a responses in the indicated C domain mutation. All error bars are SEM.
The potency of PcTx1 arises from two sets of interactions: the conserved Trp residues that interact with F352 of the channel (Fig. 4 A) and the polybasic loop that binds within the acidic pocket (Fig. 7 A) (Saez et al., 2011, 2015). The C domain already contains the Trp residues and has a similar initial KR sequence within the polybasic loop (Fig. 1 A). Where PcTx1 contains a KRRRSF and the N domain has KRRHSF, the C domain contains KRSGNKS. We hypothesized that the divergence of the loop sequence led to the lower affinity and distinct characteristics of the C domain compared with PcTx1. To test this, we mutated each side chain in the C domain loop to the corresponding amino acid in PcTx1 (i.e., S27R, G28R, N29S, and K30F) and conducted SPR measurements. The K30F mutation was of particular interest given that an F30A mutation to PcTx1 imparts potentiating activity (Saez et al., 2015). Unfortunately, the yields of the K30F were too low for further experiments. However, all other mutations gave sufficient product. Consistent with the critical role of the polybasic loop, both S27R and G28R C domain variants had increased affinity compared with wild type C domain but not as high as PcTx1. Interestingly, the S27R mutation increased the affinity 300-fold from ∼9 to 0.03 nM while the G28R only increased affinity 4-fold, to 2 nM (Fig. 7, C and D; and Table 1). The N29S mutation did not alter affinity much (Fig. 7, C and D; and Table 1), as expected, based on past studies with an S29A mutation in PcTx1 (Saez et al., 2015).
Having established all three mutated C domain toxins bind, we turned to functional measurements in excised patches. We found that G28R had a largely similar phenotype as the C domain itself, slowing the rising phase of activation (fold change of rise time: 100 nM G28R 15 ± 2, n = 6, p 0.002; Fig. 7, E and F) but not strongly inhibiting the peak response (86 ± 3% peak response in 1 µM G28R; Fig. 7 B). However, neither G28R nor N29S slowed deactivation, in contrast to the C domain alone (deactivation fold change: 100 nM G28R 0.91 ± 0.27, n = 5, P = 0.5; 1 µM N29S 2.1 ± 0.4, n = 5, P = 0.15; Fig. 7 F). These results suggest that the C domain’s deactivation slowing property largely arises from either Gly28 or Asn29 (or both) interacting within the acidic pocket. Consistent with this, the S27R mutation retained the slow deactivation phenotype. Indeed, the S27R mutation slowed deactivation and rise time far more than the C domain itself (Fig. 7). Specifically, 100 nM S27R slowed the rising phase by >60-fold (63 ± 17-fold, n = 11). The effect on deactivation was even more pronounced. Deactivation of hASIC1a into pH 7.4 is multiexponential (MacLean and Jayaraman, 2017) but in the presence of S27R, the kinetics became more complicated with a very prominent slow component (Fig. 7 E). The deactivations were approximated by a two-component exponential decay where the slow component amplitude and time constants increased with increasing S27R concentration (no toxin: area slow 0.1 ± 0.02, tau slow 73 ± 7 ms, n = 12; 100 nM S27R: area slow 0.61 ± 0.02, tau slow 1,830 ± 430 ms, n = 10). The fast time constant also increased in duration with higher S27R concentration (no toxin tau fast 3.0 ± 0.3 ms, n = 12; 100 nm S27R tau fast 50 ± 14 ms, n = 10). Moreover, the S27R mutation led to strong inhibition of the ASIC1a peak response. Based on these data, we propose that the S27R residue imparts higher affinity binding to the C domain scaffold while preserving and even enhancing the slowing of rise times and deactivation that depend on the remaining residues.
Discussion
Our work demonstrates that both the N and C lobs of Hi1a modulate channel gating, with the C domain responsible for the slowing of both the activation and deactivation time courses (Figs. 1 and 2). The slowing of deactivation suggests the C domain might increase the apparent affinity for protons. Consistent with this, the C domain left-shifts activation curves (Fig. 3). However, the C domain also right-shifts SSD curves (Fig. 3), requiring a stronger stimulus to evoke desensitization but a weaker stimulus to provoke activation. Thus, the C domain alone appears to stabilize the open state of the channel. We also observed that both domains bind within the acidic pocket of ASIC1a with submicromolar affinity (Fig. 4). The concentration dependence of Hi1a and the C domain’s functional effects align closely with the binding affinities (Fig. 5). In contrast, PcTx1 and the N domain exhibit a greater shift between the binding affinity and functional efficacy, suggesting a higher degree of occupancy is needed to produce functional effects for these toxins (Fig. 5). Interestingly, the double knot Hi1a would appear to not have this requirement of higher occupancy, which contrasts with the activity of the isolated N domain. Further structure–activity examination of the isolated C domain revealed that Gly28 and Asn29 are important in mediating the slowing of deactivation (Fig. 7). The S27R mutation, however, increases the binding affinity of the C domain greatly, and functional data imply the position may be important for inhibition in PcTx1 and the N domain (Fig. 7). These data reveal that Hi1a works by both domains binding to the same site but with different affinities. Incorporating the rate constants from our SPR data into a kinetic model (Goldschen-Ohm et al., 2014) of a trimer with one binding site per subunit, we calculated that at saturating levels of N and C toxins, 32% of receptors will have one C domain bound and 5.5% will have two C domains bound (Fig. 6). Therefore, this smaller fraction of C domain binding, coupled with the C domains stabilization of the open state, likely accounts for the partial inhibition of saturating Hi1a (Chassagnon et al., 2017) (Fig. 1).
Models of Hi1a modulation
We considered two models of Hi1a’s action. First, a two-site avidity model where one domain binds to the acidic pocket and leaves the second linked domain free to bind to another site nearby. This additional site may be relatively low affinity but the linkage of the two domains increases the local concentration to enable occupancy of low affinity sites. When one domain unbinds, it remains in forced proximity through linkage to the still-bound ligand, facilitating rebinding and prolonging the dissociation of the full Hi1a. Next, consider a mutually exclusive binding model where either domain can bind the acidic pocket at any one time but not both at the same time. Five distinct lines of evidence argue for the mutually exclusive binding model over the avidity model. First, an avidity model would be supported if the C domain could not bind the channel without the tethered N domain to guide it, or if the C domain binds but not to the acidic pocket. However, we found that both the N and C domains can bind the acidic pocket with nanomolar KD’s since the F352A mutation attenuates the binding of each domain and functional modulation of the C domain. Second, Hi1a and the N domain have comparable dissociation times (0.014 s−1 for Hi1a and 0.07 s−1 for the N domain, Table 1). This would not be expected of an avidity model where the dissociation should be considerably slowed. Third, the association rates of Hi1a are roughly twice that of the fastest associating single-domain toxins, PcTx1 or the C domain. PcTx1 and the C domain have kons of 4e7 and 2e7 M−1*s−1, respectively, while the kon of Hi1a is 7e7 M−1*s−1. This is consistent with two ligands on each Hi1a molecule with a comparable association constant. Fourth, Hi1a shallows the slope of the SSD curve that appears as a blend of the N-domain left-shifted SSD and C-domain right-shifted SSD phenotypes (Fig. 3 C). This curve is consistent with two functionally distinct domains acting with slightly different potencies. Such a shallowed SSD curve is less likely to arise from a simple avidity model where the N domain is the main driver of binding. Fifth and finally, Hi1a-CN acts very similarly to Hi1a wild type in that it slows the rising phase and deactivation, hallmarks of C domain modulation. An avidity model where the N domain drives the binding is difficult to reconcile with modulation by Hi1a-CN as the short linker length would preclude the C domain of Hi1a-CN from reaching the same modulatory site of the C domain in wild type Hi1a. However, modulation by Hi1a-CN is completely expected of a mutually exclusive binding site model. Importantly, our data do not necessarily preclude the existence of a second site. But if a second site does exist, it likely has minimal functional effect.
The simple mutually exclusive binding model is complicated by the slow wash-off of Hi1a. In our experience, cells exposed to Hi1a do not recover functional responses (see also 12). In contrast, functional responses return quite readily following N domain wash-off (data not shown). Yet, the two toxins have comparable dissociation constants (Fig. 4 F and Table 1). Reconciling similar binding data with disparate functional data is difficult. It is possible that during SPR experiments, hASIC1a trimers dissociated or aggregated in some way that destroyed or occluded a possible C domain binding site. We cannot exclude this possibility, even though the F352A effects suggest the channel is intact. Another possible reconciliation is the different concentration dependencies of binding and functional modulation. PcTx1 dissociates very slowly in our SPR measurements. Similarly, in voltage clamp fluorometry experiments, the PcTx1-bound fluorescence signal dissipates slowly (Heusser et al., 2022), far slower than the functional modulation of PcTx1 washes away. Since the N domain is closely related in sequence to PcTx1, N domain functional modulation fades quickly during wash-off while complete dissociation measured in SPR takes longer. In contrast, Hi1a’s functional wash-off may more closely reflect its actual unbinding process.
It is presently unclear what the precise structural mechanism is for the unusual functional modulation of Hi1a or the C domain. An intriguing aspect of the C domain is that the β hairpin turn contains an “extra” amino acid. In the PcTx1 structures, the β hairpin is wedged into the acidic pocket and “secured” by contacts formed by W24, K25, and R26 on the one side and S29 and F30 on the other. In between these two “anchor points” are the two Arg residues at the apex of the β hairpin. The N domain of Hi1a has a similar assembly but has a KRRH polybasic loop instead of the KRRR seen in PcTx1. However, the C domain has the sequence of KRSGNK, thus containing one additional amino acid. We speculate that the Gly allows some flexibility of the Asn side chain and adjacent residues to interact with new contacts to exert the novel effects of the C domain. But how these all “fit” into the acidic pocket requires detailed structural investigation. Furthermore, this particular combination of amino acids is seemingly able to modify the acidic pocket conformation or distribution of conformations in such a way as to bias the channel toward the open state and away from the desensitized state (Fig. 3, C and D), perhaps by influencing the propensity of the β11–12 linker to isomerize and enable desensitization (Rook et al., 2021a, 2021b, 2021c; Rook et al., 2020).
Conclusions
The double knot ICK configuration offers tremendous therapeutic potential by allowing custom engineering of either domain to produce desired affinity or modulation. Our measurements demonstrate the plasticity of the ICK fold, thus making it an ideal vehicle for channel modulation. However, since these toxins are unlikely to pass the blood–brain barrier, delivery is a challenge. One solution is using intranasal administration which bypasses the barrier as has been done with PcTx1 previously (Pignataro et al., 2007; Hanson and Frey, 2008). Another option is to use Hi1a, or derivatives thereof, outside the blood–brain barrier such as in the peripheral nervous system or in a transplant setting. Indeed, work has already been undertaken to probe the utility of Hi1a in protecting hearts during transplants (Redd et al., 2021). Continued investigation of ASIC-toxin interactions may yield exciting new clinical opportunities as well as valuable research tools.
Data availability
The data are available in the article itself. Additional data are available from the corresponding author upon reasonable request via email.
Acknowledgments
Christopher J. Lingle served as editor.
We thank Dr. Jermaine Jenkins for assistance with SPR as well as Juliana Wagner, Lauren Bainbridge, and especially Dante Hovancik for technical assistance. We also thank members of the MacLean lab for helpful discussion and comments.
This work was supported by National Institutes of Health grants R21MH125135 (to D.M. MacLean) and T90DE021985 (to K.D. Berger).
Author contributions: K.D. Berger: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Validation, Visualization, Writing - review & editing, D.M. MacLean: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Project administration, Supervision, Visualization, Writing - original draft, Writing - review & editing.

