


The β-myosin heavy chain expressed in ventricular myocardium and the myosin heavy chain (MyHC) in slow-twitch skeletal Musculus soleus (M. soleus) type-I fibers are both encoded by MYH7. Thus, these myosin molecules are deemed equivalent. However, some reports suggested variations in the light chain composition between M. soleus and ventricular myosin, which could influence functional parameters, such as maximum velocity of shortening. To test for functional differences of the actin gliding velocity on immobilized myosin molecules, we made use of in vitro motility assays. We found that ventricular myosin moved actin filaments with ∼0.9 µm/s significantly faster than M. soleus myosin (0.3 µm/s). Filaments prepared from isolated actin are not the native interaction partner of myosin and are believed to slow down movement. Yet, using native thin filaments purified from M. soleus or ventricular tissue, the gliding velocity of M. soleus and ventricular myosin remained significantly different. When comparing the light chain composition of ventricular and M. soleus β-myosin, a difference became evident. M. soleus myosin contains not only the “ventricular” essential light chain (ELC) MLC1sb/v, but also an additional longer and more positively charged MLC1sa. Moreover, we revealed that on a single muscle fiber level, a higher relative content of MLC1sa was associated with significantly slower actin gliding. We conclude that the ELC MLC1sa decelerates gliding velocity presumably by a decreased dissociation rate from actin associated with a higher actin affinity compared to MLC1sb/v. Such ELC/actin interactions might also be relevant in vivo as differences between M. soleus and ventricular myosin persisted when native thin filaments were used.
Introduction
Myriad of essential cellular processes including cytokinesis, cargo transport, cell migration, and muscle contraction are driven by motor proteins, such as myosins of different classes. The myosin holoenzyme consists of myosin heavy chains (MyHCs) and light chains. Myosin heavy chains are composed of an N-terminal catalytic head or motor domain, an adjacent neck domain, and a C-terminal tail domain of variable design and function. The catalytic motor domains produce movement and force by cyclic interactions with actin filaments powered by the chemical energy derived from ATP hydrolysis. Members of myosin class II dimerize by their coiled-coil tail domains, thus forming hexameric, two-headed protein complexes composed of two heavy chains and two pairs of myosin light chains (MLC), one essential light chain (ELC), and one regulatory light chain (RLC) associated with each MyHC.
MLCs are calmodulin family members, which bind noncovalently to the so-called IQ motifs located in the MyHC neck domain and are needed for the structural integrity of the myosin protein complex (Lowey et al., 1993b; VanBuren et al., 1994). MLCs can have regulatory functions on the motor activity of the myosin holoenzyme by modulating functional properties, such as maximal velocity of shortening of muscle fibers or even processive behavior (Amrute-Nayak et al., 2019; Greaser et al., 1988; Reggiani et al., 1997). Reports on hypertrophic cardiomyopathy (HCM)-associated MLC mutations also suggest functional relevance of the associated MLCs (Guhathakurta et al., 2017; Hernandez et al., 2007; Huang and Szczesna-Cordary, 2015). In class-II myosins, ELC binds the IQ motif next to the motor domain, while the RLC binds to the nearby second IQ motif (Rayment et al., 1993). Muscle tissue ELCs consist of 150–208 amino acids and have molecular weights ranging from ∼17–23 kD. They are of various lengths and can be grouped into shorter (also known as A2-type) ELCs, such as MLC3f expressed in fast muscle fibers and longer (also known as A1-type) isoforms expressed in slow and fast skeletal muscle fibers and cardiomyocytes (Lowey and Risby, 1971).
The finding that myopathies such as HCM are often caused by point mutations in cardiac sarcomeric proteins has generated an increasing interest for these molecules (Rayment et al., 1995). In HCM, mainly the ventricular motor protein β-myosin heavy chain (β-MyHC) or the cardiac myosin-binding protein C are affected. Within β-MyHC, most mutations are located in the motor domain, which is responsible for cyclic interactions with actin filaments and generation of motion and force powered by ATP hydrolysis. Thus, mutations within the myosin motor can result in functional alterations of the actomyosin complex and might lead to organ malfunction. β-MyHC in ventricular myocardium is the product of the same gene (MYH7) on chromosome 14q1 (Epstein et al., 1992; Geisterfer-Lowrance et al., 1990), which is also expressed in slow-twitch type-I muscle fibers found in, e.g., Musculus soleus (M. soleus; Lompré et al., 1984; Schiaffino and Reggiani, 1996). Normal and mutant β-MyHC are incorporated into the sarcomeres of both cardiac and slow-twitch skeletal muscle (Becker et al., 2007; Cuda et al., 1993; Nier et al., 1999; Tripathi et al., 2011; Yu et al., 1993), the latter one being more easily accessible and less subject to the adaptive responses seen in myocardium. Consequently, single M. soleus fibers or myosin extracts from HCM patients have often been used to study functional effects of HCM-related β-myosin mutations compared to wild-type myosin from healthy individuals (Cuda et al., 1997; Kirschner et al., 2005; Köhler et al., 2002; Seebohm et al., 2009; Thedinga et al., 1999). However, besides variations of MyHC isoforms, some reports suggested variations in the MLC composition between individual M. soleus fibers, which could influence functional parameters like maximum velocity of fiber shortening (Greaser et al., 1988; Reiser et al., 1985; Sweeney et al., 1988). Additionally, the isoform composition of the myosin mechanoenzyme regarding MyHCs and MLCs can vary during development and can be shifted by various stimuli, such as thyroid hormones (Biral et al., 1999; Sartore et al., 1981). This suggests that even when myosin molecules share the same heavy chain, their functional properties might differ due to differences in other components of the myosin complex, such as MLCs.
Unlike shorter A2-type ELCs, A1 ELC isoforms contain an 40–45 amino acid long, positively charged N-terminal extension, which can transiently interact with acidic residues on filamentous actin (F-actin), thereby bridging the actin filament and the myosin-II motor domain and likely modulating myosin motor mechanoenzymatic function (Andreev et al., 1999; Milligan et al., 1990; Sutoh, 1982; Sweeney, 1995; Timson et al., 1998). A1 ELC isoforms MLC1v and MLC1sb found in ventricular and slow-twitch skeletal muscle tissue have been shown to be identical and encoded by the same gene in mouse (Barton and Buckingham, 1985) and human (Fodor et al., 1989). In some slow-twitch muscle fibers, however, a second MLC1s isoform has been detected in addition to MLC1sb (Pinter et al., 1981; Sarkar et al., 1971; Weeds, 1976). Like MLC1v or MLC1sb, this isoform called MLC1sa belongs to the A1-type ELCs. MLC1sa contains a positively charged N-terminal extension, which is slightly longer than that observed in MLC1sb. The presence of different slow-type ELC isoforms in slow-twitch skeletal muscle is heterogeneous across species and can vary between slow muscles in the same species or even fibers from the same muscle (Bicer and Reiser, 2004; Biral et al., 1982; Carraro et al., 1981; Staron and Pette, 1987), and is therefore not generally considered.
As several studies reported modulating effects of A1 ELC isoforms on myosin motor mechanoenzymatic function, the presence of an additional A1 ELC such as MLC1sa with a longer N-terminal extension might cause changes in functional properties, such as maximal velocity of shortening. Indeed, in pig diaphragm slow muscle fibers, maximal velocity of shortening was reported to be inversely related to the relative level of MLC1sa (Reiser and Bicer, 2006). However, in the studied muscle fibers, functional effects upon changes in the relative level of MLC1sa were accompanied by changes in the expression of a fast A2 ELC isoform MLC1f and of a slow-type troponin-T isoform. The aims of the present study are, therefore, (1) to test whether differences between ventricular and slow M. soleus myosin regarding the speed of actomyosin chemomechanical interaction persist also in a reduced system, such as the actin gliding assay. Assuming that there are differences regarding the protein isoform composition of myosin holoenzymes in ventricular and slow-twitch M. soleus tissue, we aim to test (2) whether possible differences in functional parameters of myosin holoenzymes can be attributed to the presence of the long ELC isoform MLC1sa expressed in slow-twitch muscle fibers.
We found that M. soleus myosin moved actin filaments significantly slower than ventricular myosin. Slower actin gliding was associated with the presence of MLC1sa in M. soleus myosin; and on a single M. soleus fiber level, a higher relative content of MLC1sa was related with significantly slower actin gliding. To exclude artificial effects of filaments polymerized from isolated actin, we also confirmed differences in actin filament gliding velocity between myosin holoenzymes with or without MLC1sa using native thin filaments extracted from ventricular and M. soleus tissue.
Materials and methods
Animals
Female New Zealand bastard white rabbits, aged between 4 and 7 mo were euthanized following the regulations from the German Animal Welfare Act. The animals used for the collection of M. soleus and ventricular muscle tissue were registered under §4/2004/462 or 504, and §4/10 Kraft, in accordance with the regulations existing at that time. For atrial samples, New Zealand bastard white rabbits with the authorization numbers I2A291 F17478 + F17313 (approved to Dr. Jan Faix, Hannover Medical School) were used.
Protein purification
Myosin
Full-length myosin was purified from native M. soleus or ventricular tissue of three and four New Zealand bastard white rabbits, respectively. Each tissue sample resulted in one myosin preparation. From two animals, both M. soleus and ventricular tissue were used for myosin preparations. To test for possible preparation-to-preparation variation, M. soleus myosin was isolated in two individual preparations from the same animal. First, the tissue was disintegrated using a liquid nitrogen cooled pestle and mortar, followed by myosin extraction for 20 min in extraction buffer (0.3 M KCl, 0.15 M K2HPO4, 0.01 M Na4P2O7, 1 mM MgCl2, and 2 mM DTT, pH 6.8). After centrifugation at 65,000 rpm (TLA 120.2; Beckman Coulter) for 1 h, the supernatant was diluted with pure water containing 2 mM DTT to precipitate myosin. After incubation for 40 min on ice and subsequent centrifugation for 30 min at 27,000 rpm (Ti70; Beckman Coulter), the pellet was resuspended in myosin buffer (0.3 mM KCl, 25 mM HEPES, 4 mM MgCl2, 1 mM EGTA, and 2 mM DTT). Protein concentration was determined using Bradford protein assay (#5000006; Bio-Rad Laboratories, Inc.). Myosin was snap frozen over liquid nitrogen with the addition of 50% glycerol to prevent damage during long-term storage at −80°C.
For the preparation of full-length myosin from chemically skinned single M. soleus fibers, fibers were isolated from small bundles as described before (Kraft et al., 1995; Thedinga et al., 1999). Single fibers were sorted by their resting sarcomere length using laser light diffraction to ensure that only slow-twitch type-I fibers were used for myosin extraction. In brief, after isolation from bundles, muscle fibers were allowed to rest for 20 min at the bottom of a fiber preparation chamber under relaxing conditions at 5°C. Individual fibers were then placed in the center of a 1.2-mW diode laser beam with a wavelength of 650 nm. The resulting diffraction pattern was projected onto a scaled analysis mask in a known distance below the preparation chamber bottom (Fig. S1). From the distance between main laser beam and the first maximum of diffracted laser light, the sarcomere length could be determined. Only fibers of resting sarcomere lengths of 2.0 µm or shorter were identified as slow-twitch type-I fibers and used for subsequent myosin extraction. Myosin from each fiber was extracted by placing the fiber attached to a glass capillary in a tube containing 5 μl extraction buffer for 30 min. Afterwards, the fiber remains were removed by pulling the glass capillary out of the tube. The resulting myosin extracts were used immediately for in vitro motility assays. Remains of the single-fiber myosin extracts were stored at −20°C for subsequent SDS-PAGE analysis of myosin light and heavy chain isoforms.

Determination of resting sarcomere length by laser diffraction. (A) Schematic drawing. (B) Photograph of the setup.

Determination of resting sarcomere length by laser diffraction. (A) Schematic drawing. (B) Photograph of the setup.
Isolated actin and polymerization of filaments
Actin was prepared from chicken M. pectoralis major or rabbit back muscle as described previously (Pardee and Spudich, 1982), and fluorescently labeled actin filaments were polymerized from tissue purified chicken pectoralis muscle G-actin as reported previously for rabbit back muscle actin (Scholz and Brenner, 2003). 0.12 nM chicken or rabbit G-actin was mixed with actin polymerisation buffer containing 10 mM HEPES, pH 8, 100 mM KCl, and 0.25 nM rhodamine phalloidin (P1951; Sigma-Aldrich). Unlabeled F-actin was polymerized accordingly, with the difference that 0.25 µM unlabeled phalloidin (P2141; Sigma-Aldrich) was used. Actin filaments were used for up to 3 d.
Native thin filaments
Native thin filaments were prepared from M. soleus or ventricular muscle tissue as described previously (Tobacman and Sawyer, 1990). Mechanically isolated M. soleus or ventricular myofibrils were washed in 10 mM KH2PO4, pH 7.0, 0.1 M NaCl, 5 mM MgCl2, 0.5 mM EGTA, 1 mM DTT, and 0.1 mM AEBSF. After low-speed centrifugation at 5,000 rpm (TLA 110, 4°C; Beckman Coulter) for 7 min, the pellets were resuspended for 5 min in 1.5 vol of 10 mM KH2PO4, pH 7.0, 0.1 M KCl, 5 mM ATP, 5 mM MgCl2, 10 mM DTT, and 0.1 mM AEBSF to extract thin filaments. From the remaining pellet of a low-speed centrifugation at 5,000 rpm (TLA 110, 4°C, 7 min; Beckman Coulter), thin filaments were extracted once again with 1.5 vol of 10 mM KH2PO4, pH 7.0, 0.1 M KCl, 5 mM ATP, 5 mM MgCl2, 10 mM DTT, and 0.1 mM AEBSF before the sample was spun down at 5,000 rpm (TLA 110, 4°C; Beckman Coulter) for 15 min. The pooled extracts were clarified by a 15-min centrifugation at 46,000 × g (TLA 110, 29,000 rpm, 4°C; Beckman Coulter). The thin filaments were then collected by a 2-h centrifugation at 150,000 × g (TLA 110, 53,000 rpm, 4°C; Beckman Coulter). The pellets were homogenized in 20 mM KH2PO4, pH 6.0, 0.1 M KCl, 5 mM MgCl2, 1 mM ATP, and 0.1 mM AEBSF by three strokes with a Dounce homogenizer. After lowering the pH to 5.75, contaminating actomyosin was eliminated by a 5-min centrifugation at 32,000 rpm (TLA 110, 4°C; Beckman Coulter). The supernatant was dialyzed overnight against a buffer containing 20 mM imidazole, pH 6.5, 50 mM NaCl, 5 mM MgCl2, 1 mM DTT, and 0.1 mM AEBSF. The dialyzed sample was centrifuged for 5 min at 32,100 rpm (Beckman Coulter, TLA 120.2, 4°C), the pellets were discarded, and the purified thin filaments were collected by a 2-h centrifugation at 150,000 × g (TLA 120.2, 53,000 rpm, 4°C; Beckman Coulter). The thin filaments were stored on ice until use for up to 12 d. For the use in in vitro motility assays, thin filaments were resuspended in actin polymerization buffer and fluorescently labeled by the addition of 0.2 nM rhodamine phalloidine.
Total internal reflection fluorescence (TIRF) microscopy
Microscope image acquisition
Movement of fluorescently labeled actin or native thin filaments was recorded using a custom-made TIRF microscope with single-fluorophore sensitivity (Amrute-Nayak et al., 2008; Rump et al., 2011) and modifications as described below. In our inverted objective-type TIRF microscope, rhodamine phalloidin–labeled actin or thin filaments were excited by light of 532 nm wavelength produced by a Nd:YAG laser (Compass 315M-150 SL; Coherent). Both exciting laser light and emitted fluorescence light passed through a high numerical aperture 60× oil immersion objective lens (Plan Apo 60×, NA 1.45, oil; Olympus). Via a QV2 four-channel simultaneous-imaging system (Photometrics) with a respective dichroic mirror (T585 LPXR; Chroma), rhodamine fluorescence signals were projected onto a back-illuminated EMCCD camera (iXon DV887; Andor Technology, cooled to −80°C) and recorded with the software Andor SOLIS for imaging (version 4.15.30000.0). The videos were recorded at a constant sample temperature of 23°C with a frame rate of 5 Hz and converted by Andor SOLIS to 16-bit grayscale. TIFF-stacks for analysis with the computer program ImageJ (W.S. Rasband, National Institutes of Health) as described below.
In vitro motility
For all experiments with a 0.1% nitrocellulose-coated surface, first full-length myosin at a concentration of 1 mg/ml was immobilized on the surface for 2 min. After rinsing with motility extraction buffer (500 mM NaCl, 10 mM HEPES, pH 7.0, 5 mM MgCl2, and 2.5 mM MgATP), the surface was blocked with Pluronic F-127 (Sigma-Aldrich) for 1 min. Assay buffer AB (25 mM imidazole, pH 7.4, 25 mM NaCl, 4 mM MgCl2, 1 mM EGTA, 10 mg/ml D-glucose, and 10 mM DTT) was used to rinse the chamber before blocking of inactive myosin heads occurred for 1 min by addition of 6 pM unlabeled F-actin. AB containing additional 10 U/ml glucose oxidase, 800 U/ml catalase, and 2 mM ATP (A++) was flown into the chamber to release unlabeled F-actin from intact myosin heads. After a subsequent wash step with AB free of ATP (A+), 0.6 pM fluorescently labeled F-actin was added to the chamber and incubated for 1 min. Unbound F-actin was rinsed out using A+. Filament gliding was initiated by adding A++ to the chamber, and in vitro motility was immediately recorded. Temperature during recording was kept at 23°C. For experiments using native thin filament preparations, A+ and A++ contained additional 1 mM CaCl2. Under the used experimental conditions, addition of 1 mM CaCl2 in the presence of 1 mM EGTA results in a concentration of free calcium of approximately pCa 5. Thin filament gliding velocity in in vitro motility assays has been reported to be maximal at pCa levels exceeding 5–6 (Homsher et al., 1996; Sata et al., 1995). Using short native thin filaments, filament gliding on myosin molecules immobilized to nitrocellulose-coated coverslips was more robust (i.e., longer runs before detachment) compared to BSA-coated coverslips. Therefore, velocities of native thin filaments were measured on nitrocellulose-coated coverslips.
F-actin gliding on myosin extracted from single muscle fibers is generally measured on BSA-coated surfaces (Thedinga et al., 1999). As an alternative to nitrocellulose 0.5 mg/ml, BSA (A6003; Sigma-Aldrich) was used to coat the surface of the flow chamber for 5 min before full-length myosin at a concentration of 1 mg/ml was immobilized for 2 min. The chamber was rinsed with AB before inactive myosin was blocked using unlabeled F-actin for 1 min. A++ was used to release F-actin from active myosin heads. The chamber was once again washed with A+ before rhodamine phalloidin–labeled F-actin or native thin filaments were introduced for 1 min. Filament gliding and subsequent data recording were initiated using A++. Temperature during recording was kept at 23°C.
Gel electrophoresis
MyHC isoforms
For the separation of MyHC isoforms, a large format SDS-PAGE setup with integrated cooling core was used (Protean II xi; Bio-Rad Laboratories, Inc.). A 5% stacking gel, containing 5% glycerol and a 6.5% separating gel containing 10% glycerol were cast using an acrylamide/bisacrylamide ratio of 30:0.3 (ROTIPHORESE Gel 30, 3029.1; ROTIPHORESE Gel A, 3037.1; Roth). The running buffer contained 0.0625 M Tris, pH 8.3, 0.48 M glycine, and 0.25% SDS, and was precooled to 4°C. Isoforms were separated at 10–25 mA for 32 h at 4°C and subsequently stained using SYPRO Ruby Protein Gel Stain (S4942; Supelco).
MLC isoforms
Myosin samples were separated using a small format 15% SDS-PAGE gel with an acrylamide/bisacrylamide ratio of 37.5:1 (ROTIPHORESE Gel 30, 3029.1; Roth). Proteins were stained using Imperial Protein Stain (Thermo Fisher Scientific); and subsequent densitometric analysis was carried out using an argus X1 gel documentation system (version 7.14.22; Biostep). Images were recorded as 16-bit grayscale .TIFF images; and proteins were quantified using Image Quant TL 1D (v.8.2.0.0; GE) software.
Western blot analyses of MLC isoforms were performed after separation of proteins on a small format 12.5% SDS-PAGE gel. For analysis of total protein, the nitrocellulose blotting membrane was stained with SYPRO Ruby protein gel stain (Invitrogen) prior to immunolabeling. Atrial and ventricular essential light chains MLC1a and MLC1s/v were immunolabeled using anti-MYL4 (PA5-49205; Thermo Fisher Scientific). The atrial RLC (MLC2a) was labeled with an MLC2a-specific antibody (311011; Synaptic Systems GmbH).
Phospho-SDS-PAGE
For detection of phosphorylated light chains, extracted myosin samples were precipitated using 10 times the volume of ultrapure water containing 2 mM DTT and 0.5 mM AEBSF. After incubation for 30 min on ice and centrifugation at 40,000 rpm (TLA 120.2; Beckman Coulter), the pellet was resuspended in 1D sample buffer (62.5 mM Tris, pH 6.8, 15% Glycerol, 1% SDS, and 0.002% bromophenol blue) containing one tablet of PhosSTOP (4906837001; Roche) per 1 ml. Proteins were separated using 12% Criterion TGX Precast Midi Protein Gel (5671043; Bio-Rad Laboratories, Inc.). The gel was first stained for phosphorylated proteins using Pro-Q Diamond Phosphoprotein Gel Stain (P33301; Molecular Probes) with PeppermintStick Phosphoprotein Molecular Weight Standards (P27167; Molecular Probes) as a positive control. Subsequently, total protein was detected using SYPRO Ruby Protein Gel Stain (S4942; Supelco). After densitometric analysis of the Pro-Q Diamond signal (D) and the SYPRO Ruby signal (S), the D/S ratios were calculated as a measure of phosphorylation (as described by manufacturer’s instructions; Molecular Probes, Pro-Q Diamond Phosphoprotein Gel Stain).
Data analysis and statistics
The velocity of fluorescently labeled actin or native thin filaments was manually analyzed without any further image processing using the plug-in MTrackJ (Meijering et al., 2012) for the computer program ImageJ.
Velocity data was checked for normal distribution using Origin version 2020 (OriginLab Corporation), which was also used to produce data plots. Mean values were calculated from the mean filament velocities of individual assay chambers as individual experiments. T tests for significant differences between data sets were performed using the two samples, unequal variance TTEST function of Microsoft Excel 2016 (Microsoft Corporation).
Online supplemental material
Fig. S1 shows determination of resting sarcomere length by laser diffraction. Fig. S2 shows gliding velocities of actin filaments on ventricular and M. soleus β-/slow myosin purified from the same animal or the same M. soleus tissue. Fig. S3 shows SDS-PAGE analyses of actin and muscle thin filament preparations. Fig. S4 shows sequence comparison of MLC1s isoforms. Fig. S5 shows electrophoretic analysis of MLC composition and phosphorylation. Fig. S6 shows MLC composition and actin filament gliding velocity on nitrocellulose-coated coverslips of myosin extracted from individual type-I M. soleus fibers as a function of the ELC MLC1sa/MLC1sb/v ratio. Table S1 shows mass spectrometry identification of MyHC isoforms. Table S2 shows mass spectrometry identification of MLC isoforms. Table S3 shows phosphorylation levels of M. soleus and ventricular myosin RLCs. Video 1 shows actin filament gliding on M. soleus (left) and ventricular (right) myosin preparations on a BSA-coated chamber surface.
Results
Gliding of actin filaments on ventricular myosin is significantly faster than on slow skeletal muscle myosin
Full-length ventricular β-/slow myosin molecules were prepared from native muscle tissue and immobilized on a BSA-coated assay chamber surface. Tissue-derived ventricular β-/slow myosin moved fluorescently labeled actin filaments with 0.872 ± 0.047 µm/s (Fig. 1 A). This was significantly faster than actin filament gliding on tissue purified full-length β-/slow M. soleus myosin (0.279 ± 0.011 µm/s, P = 3.33*10−11; Fig. 1 A and Video 1). An additional, hierarchical statistical analysis using the number of animals as N confirmed a significantly higher actin gliding velocity on ventricular myosin compared to movement on M. soleus myosin (n = 4 and 3 animals, respectively; P = 0.00345). Actin filament gliding velocities were independent on the myosin preparation from muscle tissue; and a significant difference remained when M. soleus and ventricular myosin purified from the same animal were compared (Fig. S2).

Gliding velocities of actin filaments on ventricular and M. soleus β-/slow myosin. (A) On a BSA-coated assay chamber surface M. soleus β-/slow myosin molecules purified from native tissue moved actin filaments with 0.279 µm/s (±0.011 µm/s SE, n = 19 assay chambers), while native tissue derived ventricular β-/slow myosin moved actin filaments with 0.872 µm/s (±0.047 µm/s SE, n = 21). (B) On a nitrocellulose-coated assay chamber surface immobilized M. soleus β-/slow myosin molecules moved actin filaments with 0.101 µm/s (±0.007 µm/s SE, n = 7, 2 mM MgATP, T = 23°C). Under the same conditions, ventricular β-/slow myosin moved actin filaments with 0.200 µm/s (±0.010 µm/s SE, n = 6). Data points represent mean gliding velocities of individual assay chambers, while bars represent mean values ± SE. Data could be described by normal distributions (intrinsic curves).
Gliding velocities of actin filaments on ventricular and M. soleus β-/slow myosin. (A) On a BSA-coated assay chamber surface M. soleus β-/slow myosin molecules purified from native tissue moved actin filaments with 0.279 µm/s (±0.011 µm/s SE, n = 19 assay chambers), while native tissue derived ventricular β-/slow myosin moved actin filaments with 0.872 µm/s (±0.047 µm/s SE, n = 21). (B) On a nitrocellulose-coated assay chamber surface immobilized M. soleus β-/slow myosin molecules moved actin filaments with 0.101 µm/s (±0.007 µm/s SE, n = 7, 2 mM MgATP, T = 23°C). Under the same conditions, ventricular β-/slow myosin moved actin filaments with 0.200 µm/s (±0.010 µm/s SE, n = 6). Data points represent mean gliding velocities of individual assay chambers, while bars represent mean values ± SE. Data could be described by normal distributions (intrinsic curves).
Actin filament gliding on M. soleus (left) and ventricular (right) myosin preparations on a BSA-coated chamber surface. The average actin filament velocity on M. soleus myosin was 0.292 and 0.829 µm/s on ventricular myosin (T = 23°C, 2 mM MgATP, played at speed of acquisition).
Actin filament gliding on M. soleus (left) and ventricular (right) myosin preparations on a BSA-coated chamber surface. The average actin filament velocity on M. soleus myosin was 0.292 and 0.829 µm/s on ventricular myosin (T = 23°C, 2 mM MgATP, played at speed of acquisition).

Gliding velocities of actin filaments on ventricular and M. soleus β-/slow myosin purified from the same animal or the same M. soleus tissue. (A) On BSA-coated coverslips M. soleus myosin molecules moved actin filaments with 0.263 µm/s (±0.005 µm/s SE, n = 5 assay chambers, left) or 0.273 µm/s (±0.005 µm/s SE, n = 3 assay chambers, right) significantly slower than ventricular myosin purified from the same animals (1.004 ± 0.062 µm/s SE, n = 6 assay chambers and 1.077 ± 0.016 µm/s SE, n = 3 assay chambers, respectively). No significant differences were detected between M. soleus and ventricular myosin preparations from both animals (P = 0.3036 and P = 0.3379, respectively). (B) From one animal, M. soleus myosin was extracted in two preparations, and no significant difference regarding gliding velocity was observed (0.282 ± 0.019 µm/s SE, n = 10 assay chambers; 0.269 ± 0.011 µm/s SE, n = 4 assay chambers; P = 0.5926). Data points represent mean gliding velocities of individual assay chambers, while bars represent mean values ± SE.

Gliding velocities of actin filaments on ventricular and M. soleus β-/slow myosin purified from the same animal or the same M. soleus tissue. (A) On BSA-coated coverslips M. soleus myosin molecules moved actin filaments with 0.263 µm/s (±0.005 µm/s SE, n = 5 assay chambers, left) or 0.273 µm/s (±0.005 µm/s SE, n = 3 assay chambers, right) significantly slower than ventricular myosin purified from the same animals (1.004 ± 0.062 µm/s SE, n = 6 assay chambers and 1.077 ± 0.016 µm/s SE, n = 3 assay chambers, respectively). No significant differences were detected between M. soleus and ventricular myosin preparations from both animals (P = 0.3036 and P = 0.3379, respectively). (B) From one animal, M. soleus myosin was extracted in two preparations, and no significant difference regarding gliding velocity was observed (0.282 ± 0.019 µm/s SE, n = 10 assay chambers; 0.269 ± 0.011 µm/s SE, n = 4 assay chambers; P = 0.5926). Data points represent mean gliding velocities of individual assay chambers, while bars represent mean values ± SE.
The difference in gliding velocity was independent of the assay chamber coating and persisted on nitrocellulose-coated assay chamber surface (Fig. 1 B). Overall, actin filament gliding on a nitrocellulose-coated surface was significantly slower compared to BSA-coated flow chambers (ventricular β-myosin P = 3.213*10−12; M. soleus β-myosin P = 1.493*10−12; Fig. 1). Yet, ventricular myosin remained to move actin filaments significantly faster (0.200 ± 0.010 µm/s, P = 4.24*10−5) than M. soleus myosin (0.101 ± 0.007 µm/s; Fig. 1 B).
Using native muscle thin filaments, the differences in gliding speed between ventricular and M. soleus β-/slow myosin remains
Filaments polymerized from isolated actin are not the native interaction partner of striated muscle myosin, as they lack regulatory proteins, such as tropomyosin and the troponin complex. Consequently, the absence of actin-associated proteins might influence actomyosin interactions, and such actin filaments are generally believed to slow down movement. However, compared to F-actin, tissue purified M. soleus thin filaments decelerated the movement on M. soleus myosin (0.082 ± 0.004 µm/s, P = 0.041; Fig. 2 A). In contrast, thin filaments prepared from ventricular muscle moved significantly faster on ventricular β-myosin molecules (0.604 ± 0.029 µm/s, P = 6.65*10−14) than F-actin (Fig. 2 B).

Gliding velocities of actin filaments and native ventricular and M. soleus thin filaments on native tissue purified ventricular and M. soleus β-/slow myosin on a nitrocellulose-coated assay chamber surface. (A) Movement of F-actin (0.101 ± 0.007 µm/s SE, n = 7 assay chambers) and of M. soleus native thin filaments (0.082 ± 0.004 µm/s SE, n = 21) on M. soleus myosin. (B) Movement of F-actin on ventricular β-myosin molecules (0.200 ± 0.010 µm/s SE, n = 6) was significantly slower than that of thin filaments prepared from ventricular myofibrils (0.604 ± 0.029 µm/s SE, n = 28). (C) Native thin filaments purified from M. soleus were translocated significantly slower by M. soleus myosin (0.082 ± 0.004 µm/s SE, n = 21, T = 23°C) than by ventricular myosin (0.247 ± 0.016 µm/s SE, n = 21). (D) M. soleus myosin molecules moved cardiac thin filaments significantly slower (0.247 ± 0.009 µm/s SE, n = 19) than ventricular myosin molecules (0.604 ± 0.029 µm/s SE, n = 28). Data points represent mean gliding velocities of individual assay chambers, while bars represent mean values ± SE. Data could be described by normal distributions (intrinsic curves).
Gliding velocities of actin filaments and native ventricular and M. soleus thin filaments on native tissue purified ventricular and M. soleus β-/slow myosin on a nitrocellulose-coated assay chamber surface. (A) Movement of F-actin (0.101 ± 0.007 µm/s SE, n = 7 assay chambers) and of M. soleus native thin filaments (0.082 ± 0.004 µm/s SE, n = 21) on M. soleus myosin. (B) Movement of F-actin on ventricular β-myosin molecules (0.200 ± 0.010 µm/s SE, n = 6) was significantly slower than that of thin filaments prepared from ventricular myofibrils (0.604 ± 0.029 µm/s SE, n = 28). (C) Native thin filaments purified from M. soleus were translocated significantly slower by M. soleus myosin (0.082 ± 0.004 µm/s SE, n = 21, T = 23°C) than by ventricular myosin (0.247 ± 0.016 µm/s SE, n = 21). (D) M. soleus myosin molecules moved cardiac thin filaments significantly slower (0.247 ± 0.009 µm/s SE, n = 19) than ventricular myosin molecules (0.604 ± 0.029 µm/s SE, n = 28). Data points represent mean gliding velocities of individual assay chambers, while bars represent mean values ± SE. Data could be described by normal distributions (intrinsic curves).
When using native thin filaments purified from M. soleus or ventricular myofibrils, the gliding velocities on tissue purified M. soleus and ventricular β-/slow myosin on a nitrocellulose-coated surface remained significantly different. Movement of native thin filaments purified from M. soleus muscle on M. soleus myosin was significantly slower (0.082 ± 0.004 µm/s, P = 9.065*10−10) than on ventricular myosin (0.247 ± 0.016 µm/s; Fig. 2 C). A similar effect was observed when cardiac native thin filaments were used. M. soleus β-myosin translocated cardiac thin filaments with 0.247 ± 0.009 µm/s significantly slower compared to movement on ventricular β-myosin molecules (0.604 ± 0.029 µm/s, P = 8.16*10−13; Fig. 2 D).
Differences in gliding speed between native thin filaments from ventricular or slow muscle tissue might arise from diverse muscle-type specific compositions of protein isoforms (Fig. S3). Thin filaments prepared from M. soleus myofibrils had a decelerating effect on M. soleus myosin (Fig. 2 A), while they had an accelerating effect on ventricular myosin (0.247 ± 0.016 µm/s SE, n = 21 assay chambers compared to 0.200 ± 0.010 µm/s for F-actin). However, with all tested actin or native thin filament types, gliding velocities of M. soleus and ventricular β-/slow myosin molecules remained significantly different.
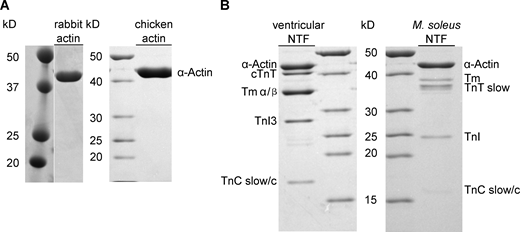
SDS-PAGE analyses of actin and muscle thin filament preparations. (A) Rabbit back muscle actin (#1610374; Colormarker Biorad, Precision Plus Protein Dual Color Standard) and chicken actin preparations (#26614; Thermo Fisher Scientific, PageRuler Unstained Protein Ladder). (B) Ventricular (left) and M. soleus (right) muscle native thin filament composition. Source data are available for this figure: SourceData FS3.

SDS-PAGE analyses of actin and muscle thin filament preparations. (A) Rabbit back muscle actin (#1610374; Colormarker Biorad, Precision Plus Protein Dual Color Standard) and chicken actin preparations (#26614; Thermo Fisher Scientific, PageRuler Unstained Protein Ladder). (B) Ventricular (left) and M. soleus (right) muscle native thin filament composition. Source data are available for this figure: SourceData FS3.
Faster actin gliding on ventricular myosin cannot be explained by contaminations with faster atrial myosin
Both M. soleus and ventricular myosin contain the same myosin heavy chain (β-/slow-MyHC), encoded by gene MYH7 (Table S1). However, we found that myosin prepared from these two sources moved actin or native thin filaments with significantly different velocities; and the question remained whether M. soleus β-MyHC samples were decelerated compared to ventricular myosin or ventricular β-MyHC was accelerated by factors other than β-MyHC.
Since it has been reported that in some species ventricles might also express the faster atrial MyHC isoform α-MyHC in addition to β-MyHC to some extent (Sartore et al., 1981) or in response to increased levels of thyroid hormones (Lompré et al., 1984), we tested for possible contaminations with atrial myosin heavy or light chains in our myosin preparations. For rabbit ventricular myosin, no visible contamination with α-MyHC could be detected in MyHC isoform SDS-PAGE gels (Fig. 3 A). While atrial myosin samples resolved in a double band representing the larger α-MyHC and slightly smaller β-MyHC isoform (Fig. 3 A, lower panel), ventricular myosin showed only one band corresponding to β-MyHC (Fig. 3 A, upper and lower panel). A mass spectrometric analysis of ventricular myosin identified predominantly β-MyHC and traces of α-MyHC, which were below the detection limit of ∼5% of our SDS-PAGE analysis (Table S1). Contamination with atrial MLCs was also not detectable by SDS-PAGE (Fig. 3 B) or by Western blot analysis (Fig. 3 C). Neither antibodies raised against the atrial RLC MLC2a did detect any respective protein in ventricular myosin samples (Fig. 3 C, lower panel), nor antibodies detecting both atrial and ventricular essential light chains MLC1a and MLC1v did suggest the presence of MLC1a in ventricular myosin samples (Fig. 3 C, upper panel).

SDS-PAGE and Western blot analyses of native tissue purified ventricular and M. soleus myosin heavy and light chain composition. (A) 8% (top) and 6.5% (bottom) SDS-PAGE analyses of MyHCs in ventricular and M. soleus myosin. Rabbit M. psoas myosin containing mainly MyHC-IIb and atrial myosin containing both α- and β-MyHC served as references. No α-MyHC could be detected in myosin purified from ventricular tissue, while M. soleus myosin contained traces of MyHC-IIa (faint upper band). (B) 15% SDS-PAGE analysis of MLCs in atrial, ventricular, and M. soleus myosin preparations. Atrial myosin contained the ELC MLC1a and RLC MLC2a, while ventricular myosin contained ELC MLC1sb/v and RLC MLC2s/v. In addition to the latter two light chains, M. soleus myosin contained the ∼0.7 kD larger ELC MLC1sa. (C) Western blot analysis of MLCs in atrial and ventricular myosin preparations. Both atrial and ventricular ELCs MLC1a and MLC1s/v were immunolabeled by the antibody anti-MYL4 (top), and no signal of MLC1a was detected in ventricular myosin preparations. The atrial RLC (MLC2a) was labeled with an MLC2a specific antibody (bottom) and was absent in ventricular myosin samples. Source data are available for this figure: SourceData F3.
SDS-PAGE and Western blot analyses of native tissue purified ventricular and M. soleus myosin heavy and light chain composition. (A) 8% (top) and 6.5% (bottom) SDS-PAGE analyses of MyHCs in ventricular and M. soleus myosin. Rabbit M. psoas myosin containing mainly MyHC-IIb and atrial myosin containing both α- and β-MyHC served as references. No α-MyHC could be detected in myosin purified from ventricular tissue, while M. soleus myosin contained traces of MyHC-IIa (faint upper band). (B) 15% SDS-PAGE analysis of MLCs in atrial, ventricular, and M. soleus myosin preparations. Atrial myosin contained the ELC MLC1a and RLC MLC2a, while ventricular myosin contained ELC MLC1sb/v and RLC MLC2s/v. In addition to the latter two light chains, M. soleus myosin contained the ∼0.7 kD larger ELC MLC1sa. (C) Western blot analysis of MLCs in atrial and ventricular myosin preparations. Both atrial and ventricular ELCs MLC1a and MLC1s/v were immunolabeled by the antibody anti-MYL4 (top), and no signal of MLC1a was detected in ventricular myosin preparations. The atrial RLC (MLC2a) was labeled with an MLC2a specific antibody (bottom) and was absent in ventricular myosin samples. Source data are available for this figure: SourceData F3.
Although slow skeletal muscle myosin and ventricular myosin share the same MyHC, they differ in their light-chain composition
Immobilized M. soleus muscle β-/slow myosin molecules drove gliding of F-actin and native thin filaments with a lower velocity compared to ventricular β-/slow myosin, yet we could not detect any differences regarding their MyHCs or relevant contaminations with faster heavy or light chain isoforms in ventricular myosin preparations. However, from Fig. 3 B, it became obvious that they differed in their light chain compositions. M. soleus myosin contained, besides β-/slow-MyHC, slow/ventricular essential light chain MLC1sb/v, and slow/ventricular RLC MLC2s/v, one additional, myosin-associated protein migrating slightly slower than MLC2s/v. This protein was identified by mass spectrometry as the essential myosin light chain (ELC) isoform MLC1sa (Table S2), which was absent in ventricular myosin (Fig. 3 B). Protein bands representing identical MLC1sb and MLC1v from M. soleus and ventricular myosin, respectively, have been identified by the same sequence with mass spectrometry (Table S2). Compared to MLC1sb/v, MLC1sa has a 13–amino acid–larger N-terminal region (Fig. S4). This N-terminal region of long ELC isoforms has been reported to interact transiently with actin filaments (Milligan et al., 1990), thus serving as a second actin binding site of myosin molecules (Miyanishi et al., 2002; Nieznańska et al., 2002) and possibly modifying actomyosin interaction kinetics (Petzhold et al., 2014; Sweeney, 1995; VanBuren et al., 1994). Myosin preparations from M. soleus tissue contained significant amounts of both ELC isoforms MLC1sa and MLC1sb with a higher relative content of MLC1sa (Fig. 3 B).

Sequence comparison of MLC1s isoforms. RefSeq accession nos. for MYL3 (MLC1sb/v) NP_000249 and for MYL6B (MLC1sa) NP_002466 were taken from NCBI. MYL3 (MLC1sb/v): 21,932.05 D, 195 amino acids; MYL6B (MLC1sa): 22,763.98 D, 208 amino acids. Red dashes indicate charge relevant changes and additional lysine residues within the N-terminal extensions.

Sequence comparison of MLC1s isoforms. RefSeq accession nos. for MYL3 (MLC1sb/v) NP_000249 and for MYL6B (MLC1sa) NP_002466 were taken from NCBI. MYL3 (MLC1sb/v): 21,932.05 D, 195 amino acids; MYL6B (MLC1sa): 22,763.98 D, 208 amino acids. Red dashes indicate charge relevant changes and additional lysine residues within the N-terminal extensions.
Besides differences in myosin heavy or light chain isoform compositions, also post-translational modifications such as phosphorylation might affect actomyosin interactions, thus leading to differences in actin gliding velocities. Standard SDS-PAGE and Western blot analysis do not exclude differences between M. soleus and ventricular myosin regarding the degree of phosphorylation, e.g., of myosin RLCs. Unlike in smooth muscle, RLC phosphorylation does not act as an activation switch in striated muscle but rather modulates actomyosin interactions during striated muscle contraction. In skeletal muscle fibers, RLC phosphorylation accelerates actomyosin crossbridge cycling kinetics by increasing the rate of non–force-generating to force-generating state transition (Sweeney and Stull, 1990). In contrast, in vitro motility assays showed that actin filament gliding slows down upon RLC phosphorylation due to an increase in the duty cycle of skeletal muscle myosin (Greenberg et al., 2009). Thus, to test for possible differences in the degree of RLC phosphorylation between our M. soleus and ventricular samples, which might influence actin gliding velocity, we analyzed M. soleus and ventricle muscle-derived myosin samples accordingly. To perform an electrophoresis-based ratiometric analysis, we measured the intensity of the Pro-Q Diamond phosphorylation signal (D) and the SYPRO Ruby protein signal (S) of the respective protein bands and calculated the D/S ratios of the fluorescence. We found that in M. soleus and ventricular myosin, RLCs were, if at all, phosphorylated to very little and comparable degrees (Fig. 4). Compared to a ratio of the positive control (45 kD band of the PeppermintStick phosphoprotein standard) of 3.00 ± 0.362 (SE, n = 4) phosphorylation to protein fluorescence, RLCs of M. soleus and ventricular myosin showed much lower but comparable ratios of 0.23 ± 0.017 (SE, n = 5) and 0.21 ± 0.018 (SE, n = 5), respectively (Table S3). The unphosphorylated control protein (116 kD band of the PeppermintStick phosphoprotein standard) had a D/S fluorescence ratio of 0.28 ± 0.094 (SE, n = 4). A picture of the respective gel is shown as Fig. S5.

Phosphorylation levels of native tissue-purified M. soleus and ventricular myosin RLCs. Compared to a phosphorylation to protein fluorescence (Pro-Q/SyproRuby) ratio of the positive control (pPS; 45 kD band of the PeppermintStick phosphoprotein standard) of 3.00 ± 0.362 (SE, n = 4), RLCs of M. soleus (MLC2s) and ventricular (MLC2v) myosin showed much lower and comparable ratios of 0.23 ± 0.017 (SE, n = 5) and 0.21 ± 0.018 (SE, n = 5), respectively. The unphosphorylated control protein (upPS; 116 kD band of the PeppermintStick phosphoprotein standard) had a Pro-Q/SyproRuby fluorescence ratio of 0.28 ± 0.094 (SE, n = 4). Neither D/S ratios of M. soleus and ventricular RLCs were significantly different nor ratios of RLCs and of the unphosphorytated control protein. Bars represent mean values ± SE.
Phosphorylation levels of native tissue-purified M. soleus and ventricular myosin RLCs. Compared to a phosphorylation to protein fluorescence (Pro-Q/SyproRuby) ratio of the positive control (pPS; 45 kD band of the PeppermintStick phosphoprotein standard) of 3.00 ± 0.362 (SE, n = 4), RLCs of M. soleus (MLC2s) and ventricular (MLC2v) myosin showed much lower and comparable ratios of 0.23 ± 0.017 (SE, n = 5) and 0.21 ± 0.018 (SE, n = 5), respectively. The unphosphorylated control protein (upPS; 116 kD band of the PeppermintStick phosphoprotein standard) had a Pro-Q/SyproRuby fluorescence ratio of 0.28 ± 0.094 (SE, n = 4). Neither D/S ratios of M. soleus and ventricular RLCs were significantly different nor ratios of RLCs and of the unphosphorytated control protein. Bars represent mean values ± SE.
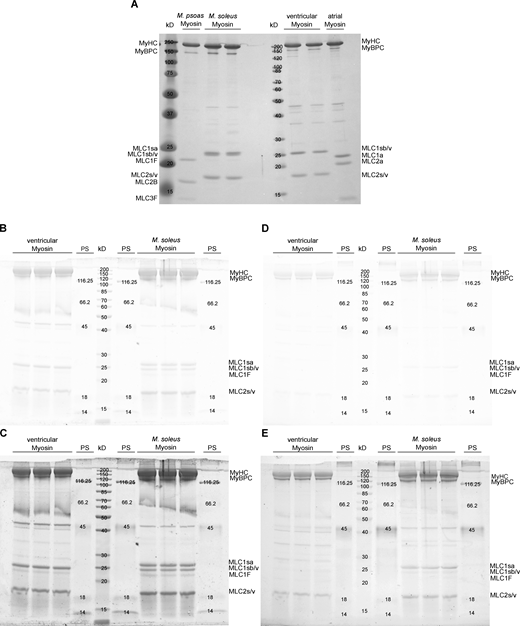
Electrophoretic analysis of MLC composition and phosphorylation. (A–C) Imperial protein stain (Thermo Fisher Scientific) SDS-PAGE of the MLCs of skeletal M. psoas and M. soleus, as well as cardiac ventricular and atrial tissue, (B) SyproRuby, PS = PeppermintStick, and (C) SyproRuby color corrected (−25 contrast, 0.22 gamma correction), (D) ProQ, and (E) ProQ color corrected (−25 contrast, 0.22 gamma correction). Protein bands representing the RLCs are found at a molecular weight of ∼19 kD. Source data are available for this figure: SourceData FS5.

Electrophoretic analysis of MLC composition and phosphorylation. (A–C) Imperial protein stain (Thermo Fisher Scientific) SDS-PAGE of the MLCs of skeletal M. psoas and M. soleus, as well as cardiac ventricular and atrial tissue, (B) SyproRuby, PS = PeppermintStick, and (C) SyproRuby color corrected (−25 contrast, 0.22 gamma correction), (D) ProQ, and (E) ProQ color corrected (−25 contrast, 0.22 gamma correction). Protein bands representing the RLCs are found at a molecular weight of ∼19 kD. Source data are available for this figure: SourceData FS5.
A higher content of the essential MLC1sa is associated with a reduced actin gliding velocity
Since myosin preparations from native M. soleus tissue contained significant amounts of both ELC isoforms MLC1sa and MLC1sb, we extracted myosin freshly from single M. soleus fibers (see Materials and methods) and tested for possible fiber-to-fiber variations regarding the MLC1sa/MLC1sb ratio and respective actin gliding velocities on a BSA-coated assay chamber. All single fibers tested contained both ELC isoforms, most of them MLC1sa at a higher content (Fig. 5 A). A higher relative content of MLC1sa compared to the ventricular ELC MLC1sb/v was associated with significantly slower actin gliding (grouping cut-off >60%: 0.314 ± 0.008 µm/s and <40% MLC1sa: 0.387 ± 0.014 µm/s, respectively. P = 0.002; Fig. 5 B, black and light grey squares, respectively, and inset). This suggests MLC1sa as the factor slowing down M. soleus compared to ventricular β-/slow-MyHC. In Fig. 5 A, fibers 6 and 7 represent exemplary fibers containing >60 or <40% MLC1sa, respectively. Fibers containing MLC1sa and MLC1sb/v to almost equal amounts, exemplary represented by fibers 1 and 3 in Fig. 5 A, translocated actin filaments at intermediate speed (Fig. 5 B, grey squares). A plot of the actin gliding velocity versus ELC MLC1sa/MLC1sb/v ratio derived from 19 individual M. soleus fibers shows a decrease of actin filament velocity with increasing MLC1sa/MLC1sb/v ratio (Fig. 5 B). A linear regression to the velocity data had a slope of −0.0383 ± 0.007. Myosin extracted freshly from six single M. soleus fibers moved F-actin with 0.124–0.202 µm/s on a nitrocellulose-coated surface (Fig. S6). A linear regression to the respective velocity versus MLC1sa/MLC1sb/v ratio plot had a slope of −0.0392 ± 0.008, which was very close to the MLC1sa-related deceleration of actin filament movement observed on BSA.

MLC composition and actin filament gliding velocity of myosin extracted from individual type-I M. soleus fibers. (A) Example of MLC SDS-PAGE analysis of individual type-I M. soleus fibers. For each fiber, the respective relative content of MLC1sa (upper band) versus MLC1sb/v (lower band) was determined by densitometric analysis of the SyproRuby-stained 15% SDS-PAGE gel. Note that all tested M. soleus fibers contained both ELC isoforms; the majority showed a higher content of MLC1sa, which was not detected in the control ventricular myosin. (B) Mean actin gliding velocity of 19 individual M. soleus fibers as a function of the ELC MLC1sa/MLC1sb/v ratio. A decrease of actin gliding velocity with increasing MLC1sa/MLC1sb/v ratio is reflected by a linear regression to the velocity data with a slope of −0.0383 ± 0.007 (black line). Inset: On a BSA-coated assay chamber surface myosin extracted from individual type-I M. soleus fibers with a lower MLC1sa/MLC1sb ratio (<40% MLC1sa) moved actin filaments with 0.387 µm/s (±0. 014 µm/s SE, n = 3 muscle fibers representing light grey squares in large plot). Actin gliding on myosin prepared from M. soleus fibers containing >60% MLC1sa was significantly slower (0.314 ± 0.008 µm/s SE, n = 7 representing black squares in large plot, T = 23°C). Data points represent mean gliding velocities of individual muscle fibers, while bars represent mean values ± SE. Source data are available for this figure: SourceData F5.
MLC composition and actin filament gliding velocity of myosin extracted from individual type-I M. soleus fibers. (A) Example of MLC SDS-PAGE analysis of individual type-I M. soleus fibers. For each fiber, the respective relative content of MLC1sa (upper band) versus MLC1sb/v (lower band) was determined by densitometric analysis of the SyproRuby-stained 15% SDS-PAGE gel. Note that all tested M. soleus fibers contained both ELC isoforms; the majority showed a higher content of MLC1sa, which was not detected in the control ventricular myosin. (B) Mean actin gliding velocity of 19 individual M. soleus fibers as a function of the ELC MLC1sa/MLC1sb/v ratio. A decrease of actin gliding velocity with increasing MLC1sa/MLC1sb/v ratio is reflected by a linear regression to the velocity data with a slope of −0.0383 ± 0.007 (black line). Inset: On a BSA-coated assay chamber surface myosin extracted from individual type-I M. soleus fibers with a lower MLC1sa/MLC1sb ratio (<40% MLC1sa) moved actin filaments with 0.387 µm/s (±0. 014 µm/s SE, n = 3 muscle fibers representing light grey squares in large plot). Actin gliding on myosin prepared from M. soleus fibers containing >60% MLC1sa was significantly slower (0.314 ± 0.008 µm/s SE, n = 7 representing black squares in large plot, T = 23°C). Data points represent mean gliding velocities of individual muscle fibers, while bars represent mean values ± SE. Source data are available for this figure: SourceData F5.
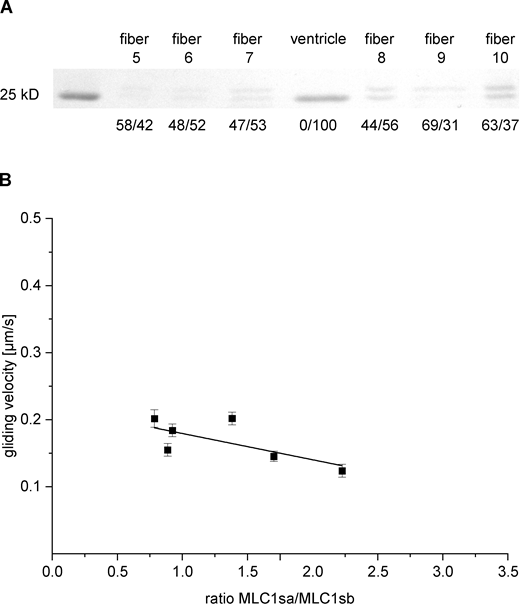
MLC composition and actin filament gliding velocity on nitrocellulose-coated coverslips of myosin extracted from individual type-I M. soleus fibers as a function of the ELC [MLC1sa]/[MLC1sb/v] ratio. (A) MLC SDS-PAGE analysis of individual type-I M. soleus fibers. For each fiber, the respective relative content of MLC1sa (upper band) versus MLC1sb/v (lower band) was determined by densitometric analysis of the Coomassie-stained 15% SDS-PAGE gel. (B) Mean actin gliding velocity of six individual M. soleus fibers as a function of the ELC MLC1sa/MLC1sb/v ratio. A decrease of actin gliding velocity with increasing MLC1sa/MLC1sb/v ratio is reflected by a linear regression to the velocity data with a slope of −0.0392 ± 0.008 (black line). Data points represent mean gliding velocities of individual muscle fibers ± SE. Source data are available for this figure: SourceData FS6.

MLC composition and actin filament gliding velocity on nitrocellulose-coated coverslips of myosin extracted from individual type-I M. soleus fibers as a function of the ELC [MLC1sa]/[MLC1sb/v] ratio. (A) MLC SDS-PAGE analysis of individual type-I M. soleus fibers. For each fiber, the respective relative content of MLC1sa (upper band) versus MLC1sb/v (lower band) was determined by densitometric analysis of the Coomassie-stained 15% SDS-PAGE gel. (B) Mean actin gliding velocity of six individual M. soleus fibers as a function of the ELC MLC1sa/MLC1sb/v ratio. A decrease of actin gliding velocity with increasing MLC1sa/MLC1sb/v ratio is reflected by a linear regression to the velocity data with a slope of −0.0392 ± 0.008 (black line). Data points represent mean gliding velocities of individual muscle fibers ± SE. Source data are available for this figure: SourceData FS6.
Discussion
In the present study, we found that actin gliding velocity on ventricular myosin was significantly faster than on M. soleus myosin, although ventricular and M. soleus myosin share identical heavy chains, RLCs, and one A1 ELC (MLC1sb/v). MLC1sb and MLC1v have been reported to be identical for species from rodents to human (UniProtKB—P16409 (MYL3_rat); P09542 (MYL3_mouse); OMIM 160790 (MYL3, MLC 3, alkali, ventricular, skeletal, slow; human). However, M. soleus myosin can contain also a second A1 essential light chain, MLC1sa, which is different from MLC1sb/v. Slower actin gliding was related to the presence and relative content of MLC1sa to MLC1sb/v in M. soleus myosin.
At saturating ATP concentrations, unloaded muscle shortening and actin gliding velocities, V, are thought to be limited by the detachment of actin-bound myosin heads that resist shortening (Huxley, 1957). Filament gliding velocities are therefore also related to the fraction of the time that a myosin head spends in its strongly-bound attached phase, the so-called duty ratio r [r = τon/(τon + τoff)]. Thus, according to this simple model, actin gliding velocities are determined by V = d*τon−1 (Harada et al., 1990; Uyeda et al., 1990), where d is the displacement per actomyosin interaction (the myosin step size) and τon is the lifetime of the attached states of the myosin heads. The rate of detachment of myosin from actin is limited by the rate of ADP release from actomyosin (Siemankowski and White, 1984; Warshaw et al., 1991; Yamashita et al., 1994) or the rate of attachment of ATP to actomyosin crossbridges (Nyitrai et al., 2006). However, besides speed limitations of unloaded muscle shortening by kinetics of myosin detachment from actin, it also correlates with the maximal ATPase rate (Barany, 1967). Several studies also reported that unloaded velocity of fiber shortening exceeds the detachment limit (Baker et al., 2002; Haldeman et al., 2014; Hooft et al., 2007) and that factors that inhibit attachment kinetics slow down fiber shortening, too. Hence, it has been suggested that also attachment kinetics to actin might limit filament gliding velocity at saturating myosin densities (Brizendine et al., 2015; Stewart et al., 2021). Consequently, possible explanations of a decreased actin gliding velocity in the present study might include (1) an increased duty ratio due to an increased actin affinity of myosin heads and lower detachment rates, (2) slower attachment kinetics of myosin heads to actin, and/or (3) a reduced myosin step size. Slower detachment kinetics would increase the time fraction of strongly bound myosin states and thus the duty ratio of myosin, and might be the result of slower ATP binding to actomyosin or decelerated ADP release from actomyosin crossbridges.
How could the presence of the, compared to ELC MLC1sb/v, longer and more positively charged ELC MLC1sa (Fig. S4) lead to one or more of the above-mentioned decelerating causes? In early studies, it was shown that the unloaded shortening velocity of skinned skeletal muscle fibers as well as the velocity of actin filament gliding measured in in vitro motility assays depended on the ELC isoform composition of myosin. Fibers and extracted myosin with a high content of A1-type ELC LC1 shortened or moved actin filaments with a significantly lower velocity than those containing a high amount of short A2-type ELC LC3 (Greaser et al., 1988; Lowey et al., 1993a, b; Sweeney et al., 1988). Subsequently, many studies reported interactions of the “sticky” N-terminus of ventricular ELC (A1, MYL3) with actin. In an early cryo-EM and traditional helical reconstruction study, the decoration of actin filaments with myosin subfragment 1 (S1) either containing A1-type ELCs or and the shorter A2-type ELCs was compared. The resulting difference map showed an additional density peak at the C-terminal region of actin, which was assigned to the extra N-terminal residues in A1 (Milligan et al., 1990). This finding was consistent with a biochemical study showing that the N-terminal amino acids 1–15 of ventricular A1 ELCs could be crosslinked to acidic residues 361–364 of actin (Sutoh, 1982). It was also reported using myosin-induced actin polymerization experiments that myosin S1 containing N-terminal truncated A1-type ELCs lost the ability to interact with actin and to facilitate actin polymerization (Lowey et al., 2007). In striated muscle, A1-type ELCs can transiently interact with the negatively charged C-terminus of actin, resulting in strongly actin-bound myosin heads with slower kinetics compared to A2 ELCs (Hernandez et al., 2007; Morano, 1999; Sweeney, 1995). In vitro experiments such as peptide and antibody studies in skinned muscle fibers or N-terminal truncated A1-type ELCs were used to show that weakening the A1/actin interaction increased myosin motor activity. The observed increased actin-activated myosin ATPase activity, faster in vitro actin motility speed, and higher maximal shortening velocity of skinned muscle fibers were interpreted as the result of an increased detachment rate of myosin from actin filaments (Bottinelli et al., 1994; Greaser et al., 1988; Hayashibara and Miyanishi, 1994; Lowey et al., 2007; Morano et al., 1995; Petzhold et al., 2014; Rarick et al., 1996; Sweeney, 1995; Timson et al., 1999; Wagner and Weeds, 1977).
The A1 ELC interaction with actin mainly resides in a cluster of conserved lysines K3, K4, K8, and K9 within its 41–43 residue N-terminal extension (Fodor et al., 1989; Seharaseyon et al., 1990) followed by a repeating sequence of proline/alanine residues. The overall 13–amino acid–longer MLC1sa (MYL6B) contains K3, K4, and K9, too, plus two additional positive charges in very close vicinity at position 10 (Fig. S4). Compared to MLC1sb/v, MLC1sa is charged +2 within its first 13 N-terminal residues, which could enhance electrostatic interactions, thus increasing the attachment rate and affinity to actin and decreasing the detachment rate from actin. Longer and more positively charged ELCs such as MLC1sa might, therefore, increase the duty ratio of myosin and therefore the load during filament gliding and lead to a lower velocity of actin filament gliding.
Besides interaction of long A1-type myosin ELCs with actin, intramolecular interactions within the myosin motor domain have been reported. These intramolecular interactions involve the N-terminal extension of A1 ELC and the SH3 domain close to the nucleotide-binding site of the MyHC head domain; and it has been proposed that this might modulate ATPase kinetics, possibly slowing down ADP release or reducing the rate of ATP hydrolysis (Lowey et al., 2007). Hence, a longer and more positively charged N-terminal extension present in MLC1sa could slow down the release of ADP or decrease the rate of ATP hydrolysis even more, thus slowing down actin filament gliding. A transient A1/SH3 domain interaction could also bridge the gap between the ELC binding site at the lever arm of myosin and actin, thus facilitating binding of the N-terminal extension to actin (Lowey et al., 2007).
The additional myosin light chain MLC1sa found in M. soleus fibers comprises an N-terminal extension, which is ∼13 amino acids longer and more positively charged than MLC1sb/v found in ventricular myosin molecules. Following the idea that this might strengthen the interaction to actin and/or the SH3 domain of the myosin motor domain, what could be functional effects? Increased ELC/actin interactions could lead to (1) an altered step size, (2) faster attachment kinetics, and (3) an increased actin affinity with a reduced actin-activated ATPase and slower detachment kinetics. Skeletal muscle myosin has a step size of ∼5–6 nm (Steffen et al., 2001). For cardiac myosin, it has been suggested that an apparent step along actin might consist of 3 and 5 nm substeps of varying frequencies, which eventually add up to a step size of 8 nm (Wang et al., 2016). It has also been proposed that the presence of the long N-terminal extension of ventricular A1-type MLCs can cause changes in the frequency of these subsequent substeps, and therefore of the step size. According to the proposed mechanism, binding of the N-terminal extension to the actin filament leads to an increased frequency of 5 nm steps followed by the additional 3 nm substep thus increasing step size. Yet, a larger step size alone would lead to faster actin gliding. Indeed, a higher frequency of total 8 nm steps was accompanied by lower ATPase rates and lower actin gliding velocities (Wang et al., 2016), which suggests a larger total step size was overcompensated by slower kinetics, such as myosin detachment from actin. Compared to ventricular MLC1sb/v, the longer N-terminal extension of MLC1sa could lead to an even higher frequency of larger steps. Our observation that myosin containing higher amounts of MLC1sa moves actin and native thin filaments at decreased speeds, however suggests that either step size is reduced instead of increased, or an increased step size is overcompensated by other decelerating processes, such as slower crossbridge kinetics. Faster attachment kinetics of myosin to actin resulting from facilitated MLC1sa/actin interaction would lead to increased actin filament speeds, which does not fit our observations. Slower detachment kinetics could be the result of slower ATP binding to actomyosin, stronger and therefore prolonged binding of myosin heads to actin, or decelerated ADP release from actomyosin crossbridges. A decelerated release of ADP could be caused, e.g., by intermolecular strain within the myosin ensemble or by intramolecular ELC/MyHC motor domain interactions. In experiments using myosin extracts from single M. soleus fibers, an increasing relative content of MLC1sa to MLC1sb/v led to deceleration of actin filament gliding. This suggests that in an ensemble of asynchronously working myosin molecules, like in the in vitro motility assay, the presence of MLC1sa-containing myosin heads with decelerated actin detachment might lead to increased load on the ensemble crossbridges. Thus, load-dependent processes such as ADP release could be prolonged also in the primarily unaffected myosin heads, and actin gliding velocity would be reduced.
In a parallel optical trapping study (Wang et al., 2022), we investigated the chemomechanical properties of M. soleus and ventricular β-myosin at the single molecule level and found that the stroke sizes d of ventricular and M. soleus myosin were very comparable (5.32 ± 0.16 and 6 ± 0.19 nm, respectively). The lifetimes of actomyosin complexes, however, were increased significantly for M. soleus myosin molecules compared to ventricular β-myosin. Maximal detachment rates of actomyosin crossbridges at saturating ATP were determined with 88.16 ± 10.41 s−1 for ventricular myosin and with 29.31 ± 1.51 s−1 for M. soleus myosin. This decreased maximal detachment rate of M. soleus myosin was only one-third of the rate determined for ventricular myosin, which agrees well with the presented actin gliding velocity decelerated to one-third. The maximal actomyosin detachment rate most likely represents ADP release rate as the rate-limiting step. Both maximal detachment rate and gliding velocity were reduced by a comparable factor, which suggests that the actin-activated ATPase activity was not changed significantly. A threefold increase in the duration of strongly bound states of the actomyosin crossbridges (τon, measured as lifetimes of the actomyosin complex) found for M. soleus suggests that the duty ratio of M. soleus myosin was increased accordingly. Actin-activated ATPase rates of rabbit ventricular myosin and of Ca2+-activated rabbit M. soleus myofibrils have been measured with 3.08 and 5.1 s−1, respectively (Candau et al., 2003; VanBuren et al., 1995). In combination with the above-described detachment rates of 88.16 s−1 for ventricular myosin and 29.31 s−1 for M. soleus myosin, M. soleus myosin is characterized by an increased duty ratio of ∼0.17 compared to a duty ratio of ∼0.04 calculated for ventricular β-myosin. Thus, slower detachment from actin and an increased duty ratio compared to ventricular myosin might explain decelerated actin and thin filament gliding on M. soleus myosin. However, we cannot exclude the possibility of an additionally increased frictional load induced by interactions of the ELC N-terminus of MLC1sa with actin, but we consider its contribution to deceleration of actin gliding rather small, compared to the kinetic effects described above.
We propose that the longer and more positively charged MLC1sa decelerates actin gliding either due to a higher affinity to actin associated with a decreased dissociation rate from actin compared to MLC1sb/v or by decelerated actomyosin cycling kinetics. Such ELC/actin interactions might also be relevant in vivo, as differences between M. soleus and ventricular myosin remain when native thin filaments were used. While the current study as well as the parallel optical trapping study on the single myosin molecule level primarily refer to rabbit M. soleus and ventricular myosin, it is possible that some effects of HCM-associated mutations in human M. soleus myosin may vary from those in ventricular myosin. Although values of, e.g., shortening velocity and crossbridge kinetics might vary between M. soleus and ventricular myosin, we could show in our recent single molecule study that other functional parameters such as stroke size and myosin rigor stiffness remained unchanged between ventricular and M. soleus myosin (Wang et al., 2022). In previous studies, M. soleus fibers from HCM patients carrying point mutations in their MYH7 gene were compared to M. soleus fibers from healthy individuals. Absolute values of shortening velocity and kinetics determined using M. soleus fibers might be different from ventricular myosin and should not be extrapolated to ventricular muscle without further ado, while other functional parameters are expected to be comparable. However, possible differences between HCM patient and healthy control samples are likely to reflect mutation effects when samples from the same muscle tissue are compared. Nevertheless, tissue-specific differences of some functional parameters suggest the use of, e.g., ventricular cardiomyocytes, ventricular myofibrils, or ventricular myosin in future studies on HCM-associated mutations in ventricular myosin.
Acknowledgments
Henk L. Granzier served as editor.
We are grateful to Bernhard Brenner (deceased) for helpful discussion in early stages of the study. The authors thank Andreas Pich of MHH Core Facility Proteomics for mass spectrometric analysis and identification of ventricular and M. soleus myosin heavy and light chains. The authors also thank Judith Montag for alignment of light chain sequences and Torsten Beier for technical assistance. We thank Jan Faix for sharing muscle tissue (Hannover Medical School, Hannover, Germany).
This work was supported by grants from the Young faculty program of Hannover Medical School (to T. Scholz). T. Wang is supported by a research grant from Fritz Thyssen Stiftung (10.19.1.009MN to M. Amrute-Nayak).
The authors declare no competing financial interests.
Author contributions: T. Scholz conceived the study, which was supervised by T. Scholz, M. Amrute-Nayak, and T. Kraft. J. Osten and T. Scholz designed experiments. M. Mohebbi, J. Osten, P. Uta, T. Wang, and T. Scholz performed experiments. Proteins were purified by J. Osten, F. Matinmehr, and P. Uta. J. Osten, M. Mohebbi, P. Uta, M. Amrute-Nayak, T. Wang, and T. Scholz analysed data. J. Osten and T. Scholz wrote the manuscript with contributions by F. Matinmehr and revisions from J. Osten, M. Amrute-Nayak, T. Kraft, and T. Scholz.

