


Compared with naïve B cells, the B cell receptor (BCR) signal in germinal center (GC) B cells is attenuated; however, the significance of this signaling attenuation has not been well defined. Here, to investigate the role of attenuation of BCR signaling, we employed a Csk mutant mouse model in which Csk deficiency in GC B cells resulted in augmentation of net BCR signaling with no apparent effect on antigen presentation. We found that Csk is required for GC maintenance and efficient antibody affinity maturation. Mechanistically, ROS-induced apoptosis was exacerbated concomitantly with mitochondrial dysfunction in Csk-deficient GC B cells. Hence, our data suggest that attenuation of the BCR signal restrains hyper-ROS production, thereby protecting GC B cells from apoptosis and contributing to efficient affinity maturation.

Introduction
Germinal centers (GCs) are the primary sites where B cells undergo affinity maturation, the process whereby the average affinity of antibodies increases with time after immunization. Affinity maturation relies on antigen-driven somatic hypermutation (SHM) of their immunoglobulin genes followed by positive selection. Proliferation and SHM occur in the dark zone (DZ), and then the B cells shuttle to the light zone (LZ), where they exit the cell cycle and undergo positive selection for antigen-binding, allowing them to re-enter the DZ and proliferate (Victora and Nussenzweig, 2012).
Positive selection of GC B cells is based on the affinity of their cell surface B cell receptors (BCRs) for antigen. Two, not necessarily mutually exclusive, models have been proposed for this selection mechanism. First, higher-affinity GC B cells would receive stronger BCR signals favoring their survival compared to low-affinity cells. Alternatively, by acquiring and processing more antigens in a short time window, higher-affinity GC B cells would be more effective in engaging T cells and receiving helper T cell signals (Allen et al., 2007; Cyster and Allen, 2019). Strong support for the latter model comes from experiments providing a small number of GC B cells with antigen in a BCR-independent manner via the cell surface receptor DEC-205 (Victora et al., 2010). In these experiments, BCR-independent antigen delivery through a DEC-205–dependent endocytic pathway produced a burst of GC B cell proliferation. However, recent studies have provided evidence that BCR signaling also contributes to the selection events (Chen et al., 2023; Kräutler et al., 2017; Yada et al., 2024). In contrast to the well-developed insights into roles of T cell help in positive selection, studies of the role of BCR signaling have been hampered, at least partly, by the fact that the BCR has dual functions as a signal transducer and an endocytic receptor, the latter of which captures antigens, and presents peptides from them to cognate T cells that select B cells displaying higher levels of peptide–major histocompatibility (pMHC) complexes.
As for the nature of BCR signaling in GC B cells, initial experiments using in vitro isolated GC B cells showed that they are less sensitive to soluble antigen stimulation (Khalil et al., 2012) compared with naïve B cells. This finding, together with the lower levels of surface BCR expression on GC B cells compared with naïve and memory B cells (Khalil et al., 2012), led to the view that GC BCR signaling is generally attenuated in vivo. More recent in vivo and ex vivo studies supported this view and further demonstrated that BCR signaling pathways in GC B cells are also qualitatively different from those of naïve B cells (Luo et al., 2018, 2019). Nevertheless, whether and how BCR signaling per se and its attenuation in GC B cells play a role in GC selection remains poorly understood.
Genetic manipulation of the signaling components is a powerful approach to address the in vivo role of BCR signaling alone; however, such manipulations may affect both signaling and endocytic functions. For instance, ablation of SHP-1 (a Tyr phosphatase known as a negative regulator of BCR signaling) (Tamir et al., 2000) in GC B cells leads to an increase of both BCR signaling and T cell help (Yam-Puc et al., 2021), therefore making it difficult to interpret the effects of BCR signaling alone. Hence, to circumvent this problem and directly examine the role of the attenuation of GC BCR signaling, here we used a mouse model deficient in a BCR signal attenuator, Csk, since Csk ablation led to augmentation of net BCR signaling with no apparent effect on antigen presentation. Our data suggest that attenuation of BCR signaling, partly executed by Csk, is necessary to restrain ROS-mediated apoptosis, thereby contributing to GC selection and efficient affinity maturation.
Results
A mouse model with altered BCR signaling but normal T cell help
We initially considered which mouse model would be feasible to affect the overall BCR signal but not antigen presentation. BCR signals trigger the phosphorylation of Igα/Igβ immunoreceptor tyrosine activation motifs by the Src-family kinase Lyn. Csk is well known to negatively regulate Lyn by phosphorylating the inhibitory tyrosine residue (Y507) in its C-terminal tail (Cheng et al., 1996; Okada, 2012). Because of the following, we considered that GC-specific deletion of Csk could be a feasible model. First, Csk is the major proximal tyrosine phosphorylation-mediated BCR signal attenuator; it is not directly involved in T cell–mediated signaling (CD40 and cytokine signaling) (Kurosaki, 1997). Second, a recent report showed that ubiquitination of Igα/Igβ mediated by Cbl/Cbl-b affects antigen presentation in naïve, but not in GC B cells (Li et al., 2020). Therefore, in the case of GC B cells, changes in the status of Igα/Igβ potentially induced by Csk deletion are not expected to affect subsequent antigen presentation.
Based on the above considerations, we experimentally verified whether BCR signaling is indeed augmented without affecting antigen presentation activity in Csk-deficient GC B cells. We first examined the signaling status in Csk-deficient GC B cells by conducting adoptive cotransfer experiments using Csk+/+ CreERT2 B1-8hi and Cskf/f CreERT2 B1-8hi B cells for inducible GC B cell–specific deletion of Csk. After transfer and immunization with NP-CGG, tamoxifen was administered on day 8 and 9, when most of the donor B cells had differentiated into GC B cells (Fig. 1 A) (Inoue et al., 2017). Using this protocol, Csk mRNA and protein were efficiently depleted on day 12 (Fig. 1 B). As expected, a significant decrease of C-terminal inhibitory phosphorylation of Lyn (pY507) was observed in the absence of Csk (Fig. 1 C). We also traced the activation status of the protein tyrosine kinase Syk, assessed by Tyr525/526 phosphorylation, demonstrating that Syk in Csk-deficient GC B cells was slightly but significantly more phosphorylated on Tyr525/526, and the difference was more conspicuous upon in vitro BCR stimulation compared with that in control cells (Fig. 1 D). Western blot analysis revealed that activation of tyrosine phosphorylation of Src family kinases (Y416) and phospho-Erk1/2 (T202/Y204) were both enhanced in lysates from purified Csk-deficient GC B cells, even though total Lyn amount was decreased, possibly caused by degradation of activated Lyn as is the case in naïve B cells treated with Csk inhibitor (Lu et al., 2021) (Fig. 1 E). The enhanced BCR signal was also evidenced by the expression of a Nur77-GFP reporter, an indicator of BCR signal strength (Zikherman et al., 2012), in Cskf/f CreERT2 B1-8hi Nur77-GFP LZ GC B cells compared with that in their control Csk+/+ counterparts (Fig. 1 F). When we focused on the small population of the LZ cells that signal through BCR, the magnitude of GFP expression and the frequency of Nur77-GFPhi LZ cells were significantly higher in Cskf/f cells (labeled as LZ [GFPhi]). As expected from the report that, in contrast to LZ cells, DZ GC B cells do not receive significant BCR signals (Mueller et al., 2015), the Nur77-GFPhi cells were rarely detectable in both control and Csk-deficient DZ B cells. Together, we conclude that the net BCR signaling is augmented in Csk-deficient LZ GC B cells.
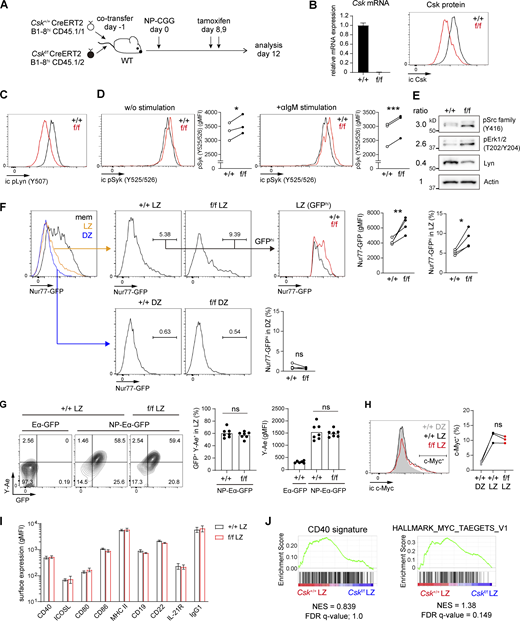
Augmented BCR signaling with normal T cell help in Csk-deficient GC B cells. (A) Experimental design. Congenically marked Csk+/+ and Cskf/f CreERT2 B1-8hi naïve B cells were cotransferred as a 1:1 mixture into wild-type hosts, which were immunized with NP-CGG/alum, treated with tamoxifen (p.o.) on days 8 and 9, and analyzed on day 12. (B) Real-time quantitative PCR analysis of Csk mRNA expression (left, n = 3) and flow cytometry of intracellular Csk protein expression (right) in Csk+/+ and Cskf/f GC B cells. (C) Flow cytometry of intracellular pLyn Y507 expression in Csk+/+ and Cskf/f GC B cells. (D) Flow cytometry of intracellular pSyk Y525/526 expression in Csk+/+ and Cskf/f GC B cells without anti-IgM stimulation (left), or after stimulation by anti-IgM for 1 h in vitro (right). Graphs showing cumulative data of geometric mean fluorescence intensity (gMFI). n = 3. (E) Western blot analysis. Lysates from purified Csk+/+ and Cskf/f GC B cells were blotted with antibodies to phosho-Src family (Y416), phosho-Erk1/2 (T202/Y204), total Lyn, and actin. Values on the left indicate relative signal intensity (Cskf/f/Csk+/+) after normalized by actin signal. (F) Left, flow cytometry of Nur77-GFP expression in GC and memory B cells. Donor LZ (CD45.1+NP+CD38−CD86hiCXCR4lo) or DZ (CD45.1+NP+CD38−CD86loCXCR4hi) GC B cells were separated as Csk+/+ (CD45.1+CD45.2−) and Cskf/f (CD45.1+CD45.2+), and the GFPhi populations were gated. Right, cumulative data of gMFI and frequency of the GFPhi population. n = 4. (G) In vitro antigen presentation assay. Mice were prepared as described in A. Purified splenic B cells were incubated with NP-Eα-GFP or Eα-GFP for 1 h at 37°C. Antigen-presenting LZ GC B cells were detected by staining the LZ GC B cells with the Y-Ae antibody. Left, flow cytometry of LZ GC B cells; middle, cumulative data of the percentage of GFP+ Y-Ae+ LZ GC B cells; right, cumulative data of gMFI of Y-Ae in LZ GC B cells. n = 6–7, pooled from two independent experiments. (H) Left, flow cytometry of intracellular c-Myc expression in Csk+/+ and Cskf/f GC B cells. Right, cumulative data of the c-Myc+ frequency in each population. (I) Flow cytometry and quantification of gMFI of surface BCR and molecules relevant to GC T–B cell interactions in Csk+/+ and Cskf/f GC B cells. Error bars, ± SD (n = 3). (J) GSEA showing the enrichment of genes upregulated after ligation of CD40 (left) and Myc target genes (right), comparing Cskf/f and Csk+/+ LZ B cells. *, P < 0.05; **, P < 0.01; ***, P < 0.001 by paired t test. ns, not significant. Representative of two (B, C, D, G, H, and I) or three (E and F) independent experiments. Source data are available for this figure: SourceData F1.
Augmented BCR signaling with normal T cell help in Csk-deficient GC B cells. (A) Experimental design. Congenically marked Csk+/+ and Cskf/f CreERT2 B1-8hi naïve B cells were cotransferred as a 1:1 mixture into wild-type hosts, which were immunized with NP-CGG/alum, treated with tamoxifen (p.o.) on days 8 and 9, and analyzed on day 12. (B) Real-time quantitative PCR analysis of Csk mRNA expression (left, n = 3) and flow cytometry of intracellular Csk protein expression (right) in Csk+/+ and Cskf/f GC B cells. (C) Flow cytometry of intracellular pLyn Y507 expression in Csk+/+ and Cskf/f GC B cells. (D) Flow cytometry of intracellular pSyk Y525/526 expression in Csk+/+ and Cskf/f GC B cells without anti-IgM stimulation (left), or after stimulation by anti-IgM for 1 h in vitro (right). Graphs showing cumulative data of geometric mean fluorescence intensity (gMFI). n = 3. (E) Western blot analysis. Lysates from purified Csk+/+ and Cskf/f GC B cells were blotted with antibodies to phosho-Src family (Y416), phosho-Erk1/2 (T202/Y204), total Lyn, and actin. Values on the left indicate relative signal intensity (Cskf/f/Csk+/+) after normalized by actin signal. (F) Left, flow cytometry of Nur77-GFP expression in GC and memory B cells. Donor LZ (CD45.1+NP+CD38−CD86hiCXCR4lo) or DZ (CD45.1+NP+CD38−CD86loCXCR4hi) GC B cells were separated as Csk+/+ (CD45.1+CD45.2−) and Cskf/f (CD45.1+CD45.2+), and the GFPhi populations were gated. Right, cumulative data of gMFI and frequency of the GFPhi population. n = 4. (G) In vitro antigen presentation assay. Mice were prepared as described in A. Purified splenic B cells were incubated with NP-Eα-GFP or Eα-GFP for 1 h at 37°C. Antigen-presenting LZ GC B cells were detected by staining the LZ GC B cells with the Y-Ae antibody. Left, flow cytometry of LZ GC B cells; middle, cumulative data of the percentage of GFP+ Y-Ae+ LZ GC B cells; right, cumulative data of gMFI of Y-Ae in LZ GC B cells. n = 6–7, pooled from two independent experiments. (H) Left, flow cytometry of intracellular c-Myc expression in Csk+/+ and Cskf/f GC B cells. Right, cumulative data of the c-Myc+ frequency in each population. (I) Flow cytometry and quantification of gMFI of surface BCR and molecules relevant to GC T–B cell interactions in Csk+/+ and Cskf/f GC B cells. Error bars, ± SD (n = 3). (J) GSEA showing the enrichment of genes upregulated after ligation of CD40 (left) and Myc target genes (right), comparing Cskf/f and Csk+/+ LZ B cells. *, P < 0.05; **, P < 0.01; ***, P < 0.001 by paired t test. ns, not significant. Representative of two (B, C, D, G, H, and I) or three (E and F) independent experiments. Source data are available for this figure: SourceData F1.
We next determined whether the absence of Csk alters the amount of antigen presentation to T cells. We used a conjugate of hapten NP and recombinant Eα-GFP, which contains pEα (specific for the Eα peptide from the I-E molecule) fused to GFP (Pape et al., 2007). Then we quantitated pMHC-II using a specific antibody Y-Ae (Murphy et al., 1992). As seen in Fig. 1 G, both control and Csk-deficient LZ GC B cells had a comparable frequency of pMHC-II presenting GFP+ Y-Ae+ cells with similar intensity of Y-Ae staining after incubation with NP-Eα-GFP molecules ex vivo. Hence, antigen presentation to T cells by LZ B cells is not significantly affected by Csk deficiency.
This conclusion was further substantiated by the observation that c-Myc expression by LZ B cells, which is known to be mainly regulated by the strength of T cell help (Finkin et al., 2019), was not altered due to ablation of Csk (Fig. 1 H). Expression of surface BCR and molecules relevant to T–B cell interaction on GC B cells, including CD40, ICOSL, CD80, CD86, MHC-II, CD19, CD22, and IL-21R, were comparable between Csk-deficient and control GC B cells (Fig. 1 I). Moreover, we did not detect any significant enrichment of genes upregulated by CD40 stimulation as well as c-Myc target genes in Csk-deficient LZ B cells by RNA sequencing (RNA-seq) and subsequent gene set enrichment analysis (GSEA) (Fig. 1 J). Together, these results demonstrate that antigen presentation to T cells and subsequent receipt of T cell–mediated help are not significantly affected by the loss of Csk. Hence, the Csk-deficient mouse model can allow us to test the contribution of augmented BCR signaling alone to GC selection.
Deletion of Csk decreases GC B cell competitiveness and impairs affinity maturation
The effect of enhancing BCR signal on GC B cell competitiveness was examined by an adoptive cotransfer experiment as described in Fig. 1 A. 3 days after the last tamoxifen treatment (day 12), we found a significant competitive disadvantage of Csk-deficient GC B cells over their control counterparts; the mutant cells were then almost completely competed out on day 18 (Fig. 2, A and B). Csk deficiency also resulted in an impaired DZ/LZ compartmentalization; the Csk-deficient GC B cells were skewed to CXCR4hi CD86lo DZ phenotype (Fig. 2, A and B).

Competitive disadvantage and impaired affinity maturation of Csk-deficient GC B cells. (A) Flow cytometry analysis of Csk+/+ and Cskf/f GC B cells (NP+ GL7+ CD38−) of day 12 or day 18 splenocytes in mice prepared as in Fig. 1 A. (B) Cumulative data of the donor GC B cell frequency among the total GC B cell in each condition, NP+ GC B cell number (**, P < 0.01; ***, P < 0.001 by paired t test.), the Cskf/f:Csk+/+ ratio of NP+ GC B cell number (**, P < 0.01; ***, P < 0.001 by unpaired Student’s t test), and the DZ:LZ ratio (*, P < 0.05 by paired t test). Pooled from two independent experiments (A and B). (C) Experimental design for W33L mutation analysis. Lethally irradiated wild-type CD45.2+ mice were reconstituted with Csk+/+ CreERT2 CD45.1+ or Cskf/f CreERT2 CD45.1+ BM cells mixed with μMT BM cells. Mixed BM chimeric mice were immunized with NP-CGG/alum, administered tamoxifen on day 8 and 9, and then NP+ IgG1+ GC B cells were single-cell sorted on day 11.5 for W33L mutation analysis and the serum were analyzed by ELISA. (D) Left, gating strategy. Right, cumulative data of NP+ IgG1+ GC B cell numbers. Cells for single-cell sorting are gated in red. **, P < 0.01 by unpaired t test. (E) Left, pie charts showing the frequency of GC B cell clones containing the W33L mutation. The numbers in the center indicate the total clones analyzed in each mouse. Right, cumulative data of the frequency of W33L mutation. n = 8 for Csk+/+, n = 7 for Cskf/f. (F) Anti-NP IgG1 serum titers on day 11.5 were measured by ELISA. Total and high-affinity anti-NP titers were measured using NP29-BSA (left) and NP1-BSA (middle), respectively. Right, cumulative data of NP1:NP29 titer ratio. n = 8 for each genotype (D and F). *, P < 0.05; **, P < 0.01 by unpaired Mann–Whitney test (E and F). Pooled from three independent experiments (D, E, and F).
Competitive disadvantage and impaired affinity maturation of Csk-deficient GC B cells. (A) Flow cytometry analysis of Csk+/+ and Cskf/f GC B cells (NP+ GL7+ CD38−) of day 12 or day 18 splenocytes in mice prepared as in Fig. 1 A. (B) Cumulative data of the donor GC B cell frequency among the total GC B cell in each condition, NP+ GC B cell number (**, P < 0.01; ***, P < 0.001 by paired t test.), the Cskf/f:Csk+/+ ratio of NP+ GC B cell number (**, P < 0.01; ***, P < 0.001 by unpaired Student’s t test), and the DZ:LZ ratio (*, P < 0.05 by paired t test). Pooled from two independent experiments (A and B). (C) Experimental design for W33L mutation analysis. Lethally irradiated wild-type CD45.2+ mice were reconstituted with Csk+/+ CreERT2 CD45.1+ or Cskf/f CreERT2 CD45.1+ BM cells mixed with μMT BM cells. Mixed BM chimeric mice were immunized with NP-CGG/alum, administered tamoxifen on day 8 and 9, and then NP+ IgG1+ GC B cells were single-cell sorted on day 11.5 for W33L mutation analysis and the serum were analyzed by ELISA. (D) Left, gating strategy. Right, cumulative data of NP+ IgG1+ GC B cell numbers. Cells for single-cell sorting are gated in red. **, P < 0.01 by unpaired t test. (E) Left, pie charts showing the frequency of GC B cell clones containing the W33L mutation. The numbers in the center indicate the total clones analyzed in each mouse. Right, cumulative data of the frequency of W33L mutation. n = 8 for Csk+/+, n = 7 for Cskf/f. (F) Anti-NP IgG1 serum titers on day 11.5 were measured by ELISA. Total and high-affinity anti-NP titers were measured using NP29-BSA (left) and NP1-BSA (middle), respectively. Right, cumulative data of NP1:NP29 titer ratio. n = 8 for each genotype (D and F). *, P < 0.05; **, P < 0.01 by unpaired Mann–Whitney test (E and F). Pooled from three independent experiments (D, E, and F).
To further investigate whether an increase in net BCR signaling affects the ability of GC B cells to undergo affinity maturation, we prepared a B cell–specific inducible Csk knockout in a non-BCR transgenic background by generating a bone marrow (BM) mixed chimera reconstituted with BM cells from μMT and Cskf/f CreERT2 mice. As a control, we used mice reconstituted with μMT and Csk+/+ CreERT2 BM cells. As expected by our experimental strategy, >95% of splenic B cells were derived from donor CD45.1+ B cells while donor-derived non-B cells remained only <10% in reconstituted BM chimeric mice (Fig. S1 A). These chimeric mice were immunized with NP-CGG and tamoxifen was administered on days 8 and 9 (Fig. 2 C). Similar to the results of the above competitive experiments using B1-8hi BCR knock-in B cells, we also detected a decrease in the numbers of NP+ GC B cells but not in those of non-GC B cells in the absence of Csk on day 11.5 (Fig. 2 D and Fig. S1 B). The NP+ IgG1+ GC B cells were then single-cell sorted for the analysis of a canonical affinity-improving Trp33 to Leu (W33L) mutation in the IgHV186.2 gene (Allen et al., 1988). While the total number of SHM in the VH region and the percentage of unproductive mutations were comparable (Fig. S1, C and D), suggesting that the machinery required for SHM event is largely intact, the frequency of GC B cells harboring the W33L mutation was significantly lower in μMT/Cskf/f CreERT2 mice (Fig. 2 E). Accordingly, a relative high-affinity anti-NP serum IgG1 antibody response, as measured by the ratio of anti-NP1-BSA over anti-NP29-BSA IgG1 titers, was also significantly lower in μMT/Cskf/f CreERT2 mice, while the total anti-NP29-BSA titer was barely decreased (Fig. 2 F). Collectively, an increase of BCR signaling alone induced by deletion of Csk led to defective selection in the GC, leading to impaired antibody affinity maturation.

Analysis of μMT and Csk f/f CreERT2 mixed BM chimera. Mixed BM chimera was generated as described in Fig. 2 C and Materials and methods. (A) Left, flow cytometry analysis of live splenocytes. Donor (Csk+/+ CreERT2 or Cskf/f CreERT2)-derived B cells or non-B cells were gated as CD45.1+CD45.2− population. Right, cumulative data of the frequency of donor-derived cells in each population. Representative of two independent experiments. (B) Plots in Fig. 2 D were additionally gated for non-GC B cell population (left), and the cell numbers were quantified (right). (C) BCR repertoire sequences from single-cell sorted GC B cells (Fig. 2, C and E) were selected for clones whose VDJ sequences had been completely obtained for mutation analysis (n = 399 for Csk+/+, n = 230 for Cskf/f GC B cells). The graph shows the number of nucleotide mutations in VH region. Each circle represents a single cell. (D) Pie charts showing the frequency of clones harboring mutations with unproductive VDJ segments. Pooled from three independent experiments (B, C, and D).
Analysis of μMT and Csk f/f CreERT2 mixed BM chimera. Mixed BM chimera was generated as described in Fig. 2 C and Materials and methods. (A) Left, flow cytometry analysis of live splenocytes. Donor (Csk+/+ CreERT2 or Cskf/f CreERT2)-derived B cells or non-B cells were gated as CD45.1+CD45.2− population. Right, cumulative data of the frequency of donor-derived cells in each population. Representative of two independent experiments. (B) Plots in Fig. 2 D were additionally gated for non-GC B cell population (left), and the cell numbers were quantified (right). (C) BCR repertoire sequences from single-cell sorted GC B cells (Fig. 2, C and E) were selected for clones whose VDJ sequences had been completely obtained for mutation analysis (n = 399 for Csk+/+, n = 230 for Cskf/f GC B cells). The graph shows the number of nucleotide mutations in VH region. Each circle represents a single cell. (D) Pie charts showing the frequency of clones harboring mutations with unproductive VDJ segments. Pooled from three independent experiments (B, C, and D).
DZ skewing, but not GC competitiveness, in Csk-deficient GC B cells was rescued by normalization of mTORC1 activity
To address the underlying mechanism by which the augmentation of the BCR signal affects GC positive selection, we first focused on mTORC1 signaling because of the following. In GC B cells, the mTORC1 pathway is regulated, at least to some extent, by in vitro BCR stimulation (Luo et al., 2018); indeed, we observed reproducibly hyper-mTORC1 activity, assessed by phosphorylation of ribosomal protein S6 (pS6) on serine 235 and 236 (targets of S6 kinase, itself a target of mTORC1), in Csk-deficient LZ B cells (Fig. 3 A, “−rapamycin”). Given a previous report showing that enhanced mTORC1 activity promotes the DZ phenotype and leads to the loss of GC competitiveness (Ersching et al., 2017), we reasoned that the phenotypes in Csk-deficient GC B cells could be simply accounted for by hyper-mTORC1 activity.

DZ skewing, but not the competitive disadvantage, is rescued by rapamycin. (A) Left, flow cytometry of intracellular pS6 protein expression in Csk+/+ and Cskf/f LZ and DZ GC B cells. “−rapamycin” samples were prepared as in Fig. 1 A. “+rapamycin” samples were prepared as in Fig. 3 B. Right, cumulative data of pS6 gMFI. n = 3 for −rapamycin, n = 4 for +rapamycin. *, P < 0.05 by paired t test. (B) Experimental design. Congenically marked Csk+/+ and Cskf/f CreERT2 B1-8hi naïve B cells were co-transferred as a 1:1 mixture into MtorF2108L/F2108L hosts, which were immunized with NP-CGG/alum, administered tamoxifen on day 8 and 9, injected with rapamycin daily during days 9–12, and analyzed on day 12. (C) Left, flow cytometry analysis of Csk+/+ and Cskf/f GC B cells (donor NP+ CD38−) in mice prepared as in Fig. 3 B. Right, cumulative data of the DZ:LZ ratio. The ratio of Csk+/+ was ∼1.8-fold, and that of Cskf/f was ∼2.2-fold decreased upon rapamycin treatment, respectively. (D) Cumulative data of the Cskf/f:Csk+/+ ratio of NP+ GC B cell numbers. n = 4–6, representative of two independent experiments. †, P < 0.05 by paired t test; *, P < 0.05; **, P < 0.01 by unpaired Mann–Whitney test.
DZ skewing, but not the competitive disadvantage, is rescued by rapamycin. (A) Left, flow cytometry of intracellular pS6 protein expression in Csk+/+ and Cskf/f LZ and DZ GC B cells. “−rapamycin” samples were prepared as in Fig. 1 A. “+rapamycin” samples were prepared as in Fig. 3 B. Right, cumulative data of pS6 gMFI. n = 3 for −rapamycin, n = 4 for +rapamycin. *, P < 0.05 by paired t test. (B) Experimental design. Congenically marked Csk+/+ and Cskf/f CreERT2 B1-8hi naïve B cells were co-transferred as a 1:1 mixture into MtorF2108L/F2108L hosts, which were immunized with NP-CGG/alum, administered tamoxifen on day 8 and 9, injected with rapamycin daily during days 9–12, and analyzed on day 12. (C) Left, flow cytometry analysis of Csk+/+ and Cskf/f GC B cells (donor NP+ CD38−) in mice prepared as in Fig. 3 B. Right, cumulative data of the DZ:LZ ratio. The ratio of Csk+/+ was ∼1.8-fold, and that of Cskf/f was ∼2.2-fold decreased upon rapamycin treatment, respectively. (D) Cumulative data of the Cskf/f:Csk+/+ ratio of NP+ GC B cell numbers. n = 4–6, representative of two independent experiments. †, P < 0.05 by paired t test; *, P < 0.05; **, P < 0.01 by unpaired Mann–Whitney test.
To test this idea, we conducted an adoptive transfer experiment, the same as in Fig. 1 A, except that MtorF2108L/F2108L rapamycin-resistant mutant mice were used as recipients and were treated with rapamycin to decrease mTORC1 activity in a donor B cell–intrinsic manner (Ersching et al., 2017; Inoue et al., 2021) (Fig. 3 B). We carefully titrated the rapamycin concentration so that it gave rise to a similar level of mTORC1 activity in both donor-derived control and Csk-deficient GC B cells (Fig. 3 A, “+rapamycin”). The rapamycin treatment partially but significantly corrected the DZ skewing phenotype more conspicuously in Csk-deficient GC B cells compared with control GC B cells (Fig. 3 C), but the loss of GC competitiveness was not corrected (Fig. 3 D), suggesting the involvement of the other factor(s) than the enhanced mTORC1 activity in the decreased GC competitiveness of mutant GC cells.
Augmented apoptosis due to enhanced mitochondrial ROS in the absence of Csk
As a first step toward identifying factor(s) that cause the decreased competitiveness in the absence of Csk, we determined the proliferation and apoptotic status in GC B cells. Under the experimental conditions as Fig. 1 A, we found that Csk-deficient GC B cells exhibited somewhat less proliferation than control cells as assessed by the incorporation of the nucleotide analog 5-ethynyl-2′-deoxyuridine (EdU) over a 0.5-h period (Fig. 4 A). Next, we reasoned that a GC B cell survival might be also affected by the absence of Csk. Csk-deficient GC B cells had a higher frequency of active Caspase 3+ (aCasp3+) apoptotic cells compared with control cells (Fig. 4 B). This difference was largely attributed to increased apoptosis in the DZ in Csk-deficient GC cells (Fig. S2 A).

Mitochondrial dysfunction and enhanced ROS production lead to apoptotic cell death. (A) Proliferation status of Csk+/+ and Cskf/f GC B cells was assessed by EdU incorporation 30 min after an EdU injection. Mice were prepared as in Fig. 1 A. Left, flow cytometry analysis. Right, cumulative data of the EdU+ frequency in GC B cells. n = 7. (B) Left, flow cytometry analysis of active caspase-3 (aCasp3) staining in Csk+/+ and Cskf/f GC B cells prepared as in Fig. 1 A. Right, cumulative data of the aCasp3+ frequency in GC B cells. n = 6. (C) Mitochondrial status of Csk+/+ and Cskf/f GC B cells prepared as in Fig. 1 A. Flow cytometry and cumulative gMFI of GC B cells stained with MitoTracker Green FM (left), MitoTracker CMTMRos (middle), and MitoSOX (right). n = 4. (D) Experimental scheme. (E) Left, flow cytometry analysis of Csk+/+ and Cskf/f GC B cells stained with MitoSOX after incubation with NAC for 1 h. Right, cumulative data of gMFI. n = 4. (F) Left, flow cytometry analysis of aCasp3 staining in Csk+/+ and Cskf/f GC B cells after incubation with NAC for 1 h. Right, cumulative data of aCasp3+ frequency in GC B cells. *, P < 0.05 by paired t test. (G) Left, flow cytometry analysis of purified wild-type naïve B cells and IgG1+ GC B cells incubated with anti-IgM and anti-IgG, respectively, with or without NAC for 18 h and stained with MitoSOX. Right, cumulative data of gMFI. (H) Left, flow cytometry analysis of purified Csk+/+ or Cskf/f IgG+ GC B cells incubated with anti-IgG. Right, cumulative data of gMFI. n = 3. *, P < 0.05; **, P < 0.01; ***, P < 0.001 by paired t test (A–C, E, and F). *, P < 0.05; ***, P < 0.001 by unpaired t test (G and H). Representative of two independent experiments.
Mitochondrial dysfunction and enhanced ROS production lead to apoptotic cell death. (A) Proliferation status of Csk+/+ and Cskf/f GC B cells was assessed by EdU incorporation 30 min after an EdU injection. Mice were prepared as in Fig. 1 A. Left, flow cytometry analysis. Right, cumulative data of the EdU+ frequency in GC B cells. n = 7. (B) Left, flow cytometry analysis of active caspase-3 (aCasp3) staining in Csk+/+ and Cskf/f GC B cells prepared as in Fig. 1 A. Right, cumulative data of the aCasp3+ frequency in GC B cells. n = 6. (C) Mitochondrial status of Csk+/+ and Cskf/f GC B cells prepared as in Fig. 1 A. Flow cytometry and cumulative gMFI of GC B cells stained with MitoTracker Green FM (left), MitoTracker CMTMRos (middle), and MitoSOX (right). n = 4. (D) Experimental scheme. (E) Left, flow cytometry analysis of Csk+/+ and Cskf/f GC B cells stained with MitoSOX after incubation with NAC for 1 h. Right, cumulative data of gMFI. n = 4. (F) Left, flow cytometry analysis of aCasp3 staining in Csk+/+ and Cskf/f GC B cells after incubation with NAC for 1 h. Right, cumulative data of aCasp3+ frequency in GC B cells. *, P < 0.05 by paired t test. (G) Left, flow cytometry analysis of purified wild-type naïve B cells and IgG1+ GC B cells incubated with anti-IgM and anti-IgG, respectively, with or without NAC for 18 h and stained with MitoSOX. Right, cumulative data of gMFI. (H) Left, flow cytometry analysis of purified Csk+/+ or Cskf/f IgG+ GC B cells incubated with anti-IgG. Right, cumulative data of gMFI. n = 3. *, P < 0.05; **, P < 0.01; ***, P < 0.001 by paired t test (A–C, E, and F). *, P < 0.05; ***, P < 0.001 by unpaired t test (G and H). Representative of two independent experiments.

Apoptosis and differentially expressed genes in Csk-deficient GC B cells. (A) Cumulative data of flow cytometry analysis of aCasp3 staining in Csk+/+ and Cskf/f DZ and LZ GC B cells in mice prepared as in Fig. 1 A. n = 4. *, P < 0.05 by unpaired Mann–Whitney test. ns, not significant. Representative of two independent experiments. (B) Volcano plots showing the differentially expressed genes in LZ and DZ GC B cells (FDR cutoff = 0.1, minimal fold-change = 2). (C) GSEA of RNA-seq data. Hallmark gene sets marginally enriched in Csk+/+ LZ and Csk+/+ DZ GC B cells (FDR q-val < 0.5) were listed with normalized enrichment score (NES) and nominal (NOM) P value (p-val). (D) Expression of selected genes including Bcl2-family genes, Nr4a1, Casp3, Ccnd3, and Cdk4 in Csk+/+ and Cskf/f DZ and LZ GC B cells based on RNA-seq analysis. Fragments per kilobase of exon per million reads (FPKM) values are shown. Error bars, ± SD (n = 2 or 3). (E) Relative expression (Cskf/f/Csk+/+) of selected genes in D. n = 2 for Csk+/+ DZ, Cskf/f DZ, Cskf/f LZ and n = 3 for Csk+/+ LZ from single RNA-seq experiment.
Apoptosis and differentially expressed genes in Csk-deficient GC B cells. (A) Cumulative data of flow cytometry analysis of aCasp3 staining in Csk+/+ and Cskf/f DZ and LZ GC B cells in mice prepared as in Fig. 1 A. n = 4. *, P < 0.05 by unpaired Mann–Whitney test. ns, not significant. Representative of two independent experiments. (B) Volcano plots showing the differentially expressed genes in LZ and DZ GC B cells (FDR cutoff = 0.1, minimal fold-change = 2). (C) GSEA of RNA-seq data. Hallmark gene sets marginally enriched in Csk+/+ LZ and Csk+/+ DZ GC B cells (FDR q-val < 0.5) were listed with normalized enrichment score (NES) and nominal (NOM) P value (p-val). (D) Expression of selected genes including Bcl2-family genes, Nr4a1, Casp3, Ccnd3, and Cdk4 in Csk+/+ and Cskf/f DZ and LZ GC B cells based on RNA-seq analysis. Fragments per kilobase of exon per million reads (FPKM) values are shown. Error bars, ± SD (n = 2 or 3). (E) Relative expression (Cskf/f/Csk+/+) of selected genes in D. n = 2 for Csk+/+ DZ, Cskf/f DZ, Cskf/f LZ and n = 3 for Csk+/+ LZ from single RNA-seq experiment.
So, next, we focused on our RNA-seq dataset (Fig. 1 J) to dissect the molecular basis for the observed phenotypes by identifying differentially expressed genes in LZ and DZ GC B cells upon Csk depletion (Table S1); six genes upregulated and four genes downregulated in Cskf/f LZ, and 271 genes upregulated and 34 genes downregulated in Cskf/f DZ GC B cells (false discovery rate [FDR] cutoff = 0.1, minimal fold-change = 2). One of the genes upregulated in Csk-deficient LZ B cells is Inppl1 encoding SHIP2 protein, which is associated with FcγRIIB and may be related to the regulation of apoptosis (Fig. S2 B) (March and Ravichandran, 2002; Muraille et al., 2000). Overall, GSEA did not identify significantly enriched (FDR q-val < 0.25) gene sets. Marginally (FDR q-val < 0.5) enriched gene sets are shown in Fig. S2 C. From the analysis of the mRNA expression levels of Bcl2-family member genes, we found that Csk-deficient DZ GC B cells tended to express lower levels of anti-apoptotic Bcl2a1 gene (Fig. S2, D and E).
Given the importance of c-Myc levels in positively selected LZ GC cells in subsequent DZ proliferation (Finkin et al., 2019), the Fig. 1 H data showing comparable c-Myc levels suggest that the cell division program is initiated normally in Csk-deficient LZ GC B cells. Considering that apoptosis occurs predominantly in Csk-deficient DZ cells, it is likely that the enhanced cell death in the mutant DZ cells eventually results in a decrease in overall EdU incorporation. This interpretation is supported by the following two lines of evidence: (i) rescue of the decreased numbers of mutant GC cells by haploinsufficiency of Bim, one of the proapoptotic molecules, as discussed below (Fig. 5); (ii) comparable expression levels of transcripts of cyclin D3, a crucial cell cycle regulator of DZ GC B cells (Pae et al., 2021; Ramezani-Rad et al., 2020), and cyclin-dependent kinase 4, a major partner of cyclin D3 in murine B cells (Tanguay et al., 2001), between control and mutant GC B cells (Fig. S2, D and E). However, we cannot completely exclude the possibility that the proliferation program is also affected by augmented BCR signaling, thereby contributing to the decreased numbers of mutant GC cells.

Rescue of the Csk KO GC phenotype by Bim haploinsufficiency. (A) Experimental design. Congenically marked Csk+/+, Csk+/+Bcl2l11f/+, Cskf/f, or Cskf/fBcl2l11f/+ × CreERT2 B1-8hi naïve B cells were transferred independently into wild-type hosts, which were immunized with NP-CGG/alum, treated with tamoxifen on day 8 and 9, and analyzed on day 12 or 13. (B) Flow cytometry analysis of donor NP+ (CD45.1+ NP+) cells. GC B cells were also stained with aCasp3. (C) Cumulative data of aCasp3+ frequency in GC B cells (left), GC B cell number (middle), and the DZ:LZ ratio (right). n = 6–16, pooled from three independent experiments (B and C). (D) Experimental design for W33L mutation analysis. Lethally irradiated wild-type CD45.2+ mice were reconstituted with Cskf/f CreERT2 CD45.1+ or Cskf/fBcl2l11f/+ CreERT2 CD45.1+ BM cells mixed with μMT BM cells. Mixed BM chimeric mice were immunized with NP-CGG/alum, administered tamoxifen on day 8 and 9, and then NP+ IgG1+ GC B cells were single-cell sorted on day 11.5 for W33L mutation analysis. (E) Left, gating strategy. Right, cumulative data of NP+ IgG1+ GC B cell numbers. Cells for single-cell sorting are gated in red. (F) Left, pie charts showing the frequency of GC B cell clones containing the W33L mutation. The numbers in the center indicate total clones analyzed in each mouse. Right, cumulative data of the frequency of W33L mutation. (G) Anti-NP IgG1 serum titers on day 11.5 measured by ELISA. Total and high affinity anti-NP titers were measured using NP29-BSA (left) and NP1-BSA (middle), respectively. Right, cumulative data of NP1:NP29 titer ratio. n = 5–7 (D–G). *, P < 0.05; **, P < 0.01; ***, P < 0.001 by unpaired Mann–Whitney test. Pooled from two independent experiments (E, F, and G).
Rescue of the Csk KO GC phenotype by Bim haploinsufficiency. (A) Experimental design. Congenically marked Csk+/+, Csk+/+Bcl2l11f/+, Cskf/f, or Cskf/fBcl2l11f/+ × CreERT2 B1-8hi naïve B cells were transferred independently into wild-type hosts, which were immunized with NP-CGG/alum, treated with tamoxifen on day 8 and 9, and analyzed on day 12 or 13. (B) Flow cytometry analysis of donor NP+ (CD45.1+ NP+) cells. GC B cells were also stained with aCasp3. (C) Cumulative data of aCasp3+ frequency in GC B cells (left), GC B cell number (middle), and the DZ:LZ ratio (right). n = 6–16, pooled from three independent experiments (B and C). (D) Experimental design for W33L mutation analysis. Lethally irradiated wild-type CD45.2+ mice were reconstituted with Cskf/f CreERT2 CD45.1+ or Cskf/fBcl2l11f/+ CreERT2 CD45.1+ BM cells mixed with μMT BM cells. Mixed BM chimeric mice were immunized with NP-CGG/alum, administered tamoxifen on day 8 and 9, and then NP+ IgG1+ GC B cells were single-cell sorted on day 11.5 for W33L mutation analysis. (E) Left, gating strategy. Right, cumulative data of NP+ IgG1+ GC B cell numbers. Cells for single-cell sorting are gated in red. (F) Left, pie charts showing the frequency of GC B cell clones containing the W33L mutation. The numbers in the center indicate total clones analyzed in each mouse. Right, cumulative data of the frequency of W33L mutation. (G) Anti-NP IgG1 serum titers on day 11.5 measured by ELISA. Total and high affinity anti-NP titers were measured using NP29-BSA (left) and NP1-BSA (middle), respectively. Right, cumulative data of NP1:NP29 titer ratio. n = 5–7 (D–G). *, P < 0.05; **, P < 0.01; ***, P < 0.001 by unpaired Mann–Whitney test. Pooled from two independent experiments (E, F, and G).
It has been well documented in multiple biological systems that dysregulation of reactive oxygen species (ROS) and resulting mitochondrial dysfunction leads to cell death. Indeed, upon binding to the antigen, naïve B cells were shown to require a second activation signal to avert mitochondrial dysfunction and apoptosis (Akkaya et al., 2018). In Csk-deficient GC B cells, we found decreased staining by MitoTracker Green FM, reflecting mitochondrial mass, and MitoTracker CMTMRos, reflecting mitochondrial membrane potential, both suggestive of mitochondrial dysfunction, with an increase in mitochondrial ROS generation as evidenced by elevated MitoSOX staining (Fig. 4 C). Production of mitochondrial ROS was reported to induce release of mitochondrial intermembrane space proteins into the cytosol, where they activate caspase-dependent and -independent cell death mechanisms (Green et al., 2014). To determine if the enhanced mitochondrial ROS indeed contributed to GC B cell apoptosis, we treated GC B cells with a ROS inhibitor N-acetylcysteine (NAC), a scavenger of oxidant species, and analyzed its effect on cell survival (Fig. 4 D). As expected, NAC treatment successfully normalized the MitoSOX fluorescence in Csk-deficient GC B cells (Fig. 4 E), which resulted in a decrease in the frequency of aCasp3+ apoptotic cells (Fig. 4 F), suggesting that the ROS accumulation contributes to the enhanced apoptosis in Csk-deficient GC B cells. Although the hyper-ROS activity is well correlated with apoptosis, it requires more extensive experiments to make the in vivo cause–effect relationship more solid. Since ROS production in B cells was increased by in vitro BCR stimulation (Fig. 4 G), the augmented BCR signaling is likely to induce ROS accumulation and mitochondrial dysfunction, thereby causing cell death. In fact, Csk-deficient GC B cells showed higher intensity of MitoSOX in this in vitro culture system even without BCR stimulation compared with control cells treated with anti-IgG, which was further increased upon BCR stimulation (Fig. 4 H).
Mitochondrial outer membrane permeabilization results from the interaction of Bcl-2 family proteins that protect or disrupt the outer mitochondrial membrane integrity (Green et al., 2014). Since Bim, a pro-apoptotic member of the Bcl-2 family, is one of the disrupting proteins, the above data predict that decreasing Bim expression would rescue the cell death observed in Csk-deficient GC B cells and subsequent defective selection even though Bim expression itself is not altered upon Csk deletion (Fig. S3). To examine this prediction, we adoptively transferred Csk+/+Bcl2l11+/+ CreERT2 B1-8hi, Csk+/+Bcl2l11f/+ CreERT2 B1-8hi, Cskf/fBcl2l11+/+ CreERT2 B1-8hi, or Cskf/fBcl2l11f/+ CreERT2 B1-8hi B cells independently into congenically marked mice and immunized them with NP-CGG (Fig. 5 A). GC B cell–specific deletion of Csk and Bim was induced by tamoxifen administration. As demonstrated in Fig. 5, B and C, the introduction of Bim haploinsufficiency alone did not largely impact GC B cell phenotype in terms of cell number, apoptosis, and DZ/LZ polarization. In contrast, the elevated GC B cell apoptosis and the decreased GC B cell number in Csk-deficient mice were largely normalized by the introduction of haploinsufficiency of Bim into the Csk knockout background, suggesting that the provision of survival could rescue, at least in part, the abnormality of Csk-deficient GC B cells. Somewhat unexpectedly, the DZ skewing in the absence of Csk was also, to some extent, rescued. This result suggests that, in addition to PI3K/Akt/Foxo1-mediated CXCR4 induction, survival activity also participates in proper LZ/DZ polarization.

Expression of Bim protein in Csk f/f GC B cells. Left, flow cytometry of intracellular (ic) Bim expression in Csk+/+ and Cskf/f GC B cells. Right, cumulative data of gMFI. n = 3. ns, not significant by paired t test. Representative of two independent experiments.
Expression of Bim protein in Csk f/f GC B cells. Left, flow cytometry of intracellular (ic) Bim expression in Csk+/+ and Cskf/f GC B cells. Right, cumulative data of gMFI. n = 3. ns, not significant by paired t test. Representative of two independent experiments.
Furthermore, we examined whether the impaired affinity maturation in Csk-deficient GC B cells could be also rescued by Bim haploinsufficiency. Same as Fig. 2 C, we prepared mixed BM chimera reconstituted with BM cells from μMT and Cskf/fBcl2l11f/+ CreERT2 CD45.1+ mice, immunized them with NP-CGG and administered tamoxifen on day 8 and 9 (Fig. 5 D). Similar to the results from adoptive transfer experiments using B1-8hi B cells (Fig. 5, B and C), NP+ IgG1+ GC B cell number was normalized by decreasing Bim expression (Fig. 5 E). Notably, the frequency of the W33L mutation was also normalized in Cskf/fBcl2l11f/+ CreERT2 GC B cells (Fig. 5 F). Consistently, a relative high affinity anti-NP serum IgG1 antibody response was also significantly increased in μMT/Cskf/fBcl2l11f/+ CreERT2 mice (Fig. 5 G). Collectively, these results demonstrate that augmented BCR signaling in GC B cells induces hyper ROS–mediated apoptosis, thereby constraining normal GC selection and affinity maturation.
Discussion
A role of T cell help in GC positive selection has been well established (Victora and Nussenzweig, 2012). T helper cells exert an essential role in this process by engaging with cognate LZ GC B cells through T–B cell interactions and secretion of cytokines. However, it has been less clear whether BCR signaling per se plays a direct role in GC selection, and if it does, how it exerts its function. Particularly, since it is well known that BCR signaling is attenuated in GC B cells, compared with naïve B cells, the significance of this attenuation remained to be determined.
Here, to address this question, we employed a mouse model deficient in a BCR signal attenuator, Csk. As expected from previous reports (Luo et al., 2019), Csk-deficient GC B cells manifested augmented net BCR signaling, while exhibiting apparently normal antigen presentation and receiving a similar magnitude of T cell help. By using this model, we showed that Csk is required for selection in the GC and subsequent efficient antibody affinity maturation. Furthermore, we found that, in mutant GC B cells, ROS-induced apoptosis was exacerbated, likely leading to the defect in GC selection. Together, our data suggest that attenuation of GC BCR signal is required for minimizing ROS-mediated detrimental cellular responses such as apoptosis, thereby contributing to normal GC selection and affinity maturation. In ROS biology, it should be mentioned that an appropriate ROS activity plays a positive role in BCR signaling (Capasso et al., 2010; Wheeler and Defranco, 2012). In this regard, ROS appears to be able to exert both positive and negative effects on BCR signaling, probably depending upon its quantity.
A substantial fraction of recently arrived LZ B cells show low or undetectable affinity for antigen (Mayer et al., 2017; Stewart et al., 2018), demonstrating that an affinity-dependent checkpoint for DZ to LZ entry does not exist. However, it should be noted that the DZ cells with compromised BCR cannot enter the LZ, suggesting the requirement of a BCR tonic signal for the transition of DZ to the LZ (Mayer et al., 2017; Stewart et al., 2018). After moving to the LZ, B cells expressing low-affinity BCRs are prone to die by apoptosis (Victora et al., 2010). Therefore, antigen-derived BCR signaling has been for a long time thought to be required to prevent apoptosis of LZ GC B cells for positive selection. Direct experimental evidence for this concept has been recently obtained: decreasing the BCR signal with a Btk blocker or with deletion of Stim1/2 impacted B cell survival (Chen et al., 2023; Yada et al., 2024).
Here, we demonstrate that augmenting the BCR signal by loss of Csk also impacts GC B cell survival. However, the underlying mechanism differs from a decrease in BCR signal. While apoptosis mainly takes place in the LZ GC cells in the case of loss of BCR signal (treatment with a Btk blocker), apoptosis by Csk ablation predominantly occurs in the DZ, where GC B cells undergo proliferation. Hence, the phenotype induced by a decrease of the BCR signal appears to be quite consistent with the classical direct antigen selection model that, during a vulnerable time before the LZ B cells acquire T cell help, clones that signal through a high-affinity BCR are rescued from apoptosis (Victora and Nussenzweig, 2012). In contrast, apoptosis induced by an augmented BCR signal is more consistent with the previous observations that strong BCR crosslinking alone results in GC B cell death in vivo (Pulendran et al., 1995; Shokat and Goodnow, 1995). These previous in vivo studies were aimed to test whether or not auto-reactive B cells are tolerated during GC reactions. Assuming that BCR affinity is well correlated with its signaling strength, we speculate that a similar negative selection of GC B cells with extraordinarily high-affinity BCRs probably takes place in foreign antigen immunization contexts, which, in turn, might provide one of the potential mechanisms of why an upper-affinity ceiling exists during normal immune responses (Foote and Eisen, 1995).
The overall attenuation of the BCR signal in GC B cells is thought to be mediated by the existence of the negative signaling feedback loop; in GC B cells, activated Akt redirects the targets toward Csk, SHP-1, and HPK1, negative regulators of BCR signaling, thereby dampening signals (Luo et al., 2019). In addition, Lyn-mediated recruitment of SHP-1/SHIP1 to inhibitory receptors is also reported to contribute to negative signaling feedback in the case of naïve B cells (Ingley, 2012). In contrast to our results of GC B cells, a previous study reported that the acute chemical inhibition of Csk in naïve B cells resulted in a decrease in calcium mobilization and Erk activity upon BCR engagement, despite enhanced Src family kinase activation, which is due to an impairment of BCR-mediated phosphatidylinositol 3,4,5-triphosphate (PIP3) production (Lu et al., 2021). The apparent discordance of the BCR signal between our Csk-deficient GC B cells and naïve B cells treated with Csk inhibitor might be caused by differential PIP3 generation. Mechanistically, in naïve B cells with hyperactivated Lyn, hyperphosphorylation of ITIMs in inhibitory receptors takes place, thereby exaggerating SHIP1 activity and subsequent suppression of PIP3. In contrast, in Csk-deficient GC B cells, such PIP3 suppression is not likely to dominate, rather somewhat more PIP3 is generated, as demonstrated by hyper-mTORC1 activity.
The attenuation of the GC BCR signal might result in a very narrow range of signaling breadth for discriminating antigen affinity. So, the question arises of why, despite such strong attenuation, GC B cells still sense affinity differences. We speculate that the following two mechanisms may complement the narrow range of BCR signaling breadth. First, in T cell–mediated signal, it is known that T cells can engage in positive feedback loops in which signals delivered by CD40L on T cells induce GC B cells to upregulate surface and soluble mediators that attract further T cell help (Liu et al., 2015, 2021). Such T cell–mediated loops might explain why relatively modest gains in BCR affinity can elicit disproportionately large clonal bursts (Victora and Nussenzweig, 2022). Second, in addition to the above signaling layer, recent microscopic analysis of immunological synapses indicates that, whereas naïve B cells form an extended, planar interaction with the antigen-bearing surface, GC B cells develop extended, pod-like structures recognizing the antigen-bearing surfaces (Nowosad et al., 2016). Thus, it is also possible that GC B cells possess a greater ability than naïve B cells to discriminate even modest affinity differences at the recognition level.
One limitation of our study is that we could not detect clear changes in gene expression, which might be responsible for the phenotypic changes seen by Western blot or flow cytometry analysis in Csk-deficient GC B cells. One obvious possibility is that these changes may become evident only when focusing on a LZ small population of cells that signal through the BCR (defined as Nur77-GFPhi).
Materials and methods
Mice
Cskf/f (Schmedt et al., 1998; Yagi et al., 2007), B1-8hi (Shih et al., 2002), Nur77-GFP (Zikherman et al., 2012), MtorF2108L (Ersching et al., 2017), μMT (Kitamura et al., 1991), and Bcl2l11f/f (Inoue et al., 2021) mice were described previously. Rosa26-CreERT2 mice were purchased from Taconic Biosciences. C57BL/6 mice were purchased from CLEA Japan and SLC Japan. For the generation of mixed BM chimeric mice, C57BL/6 mice were lethally irradiated by X-rays (8.5 Gy) and then reconstituted by intravenous injection of BM cells from μMT CD45.2+ (80%) mice mixed with those from Csk+/+ CreERT2KI/+ CD45.1+ (20%), Cskf/f CreERT2KI/+ CD45.1+ (20%), or Cskf/fBcl2l11f/+ CreERT2KI/+ CD45.1+ (20%) mice, respectively. Chimeric mice were rested for at least 8 wk before immunization. Mice were bred and maintained under specific pathogen–free conditions, and all animal experiments were performed under the institutional guidelines of Osaka University.
Immunization, adoptive transfer, and treatments
Immunization of mice and adoptive transfer experiments were performed as described previously (Inoue et al., 2021). In brief, mice were immunized by intraperitoneal (i.p.) injection with 30 μg of 4-hydroxy-3-nitrophenylacetyl (NP) conjugated to chicken gamma globulin (NP-CGG) precipitated with 10% aluminum sulfate (Wako). Splenic B cells were purified by magnetic cell depletion using anti-CD43 microbeads and the AutoMACS system (Miltenyi Biotec). Purified B1-8hi B cells containing 5 × 104 NP-binders (a 1:1 mixture of 2.5 × 104 NP-binders for co-transfer experiments) were transferred i.v. into recipient mice. Deletion of the loxp-flanked alleles in Rosa26-CreERT2 mice was induced by oral administration (p.o.) of 2 mg tamoxifen (Sigma-Aldrich) in sunflower oil once per day for 1–3 days. Rapamycin (Tokyo Chemical Industry) was injected i.p. at 1.6 mg/kg in 5% PEG 400 (Sigma-Aldrich)/5% Tween 80 (Sigma-Aldrich) once per day at the indicated time points. EdU (Thermo Fisher Scientific) (1 mg/200 μl in PBS) was injected i.p., and the mice were further treated with 0.4 mg/ml EdU in the drinking water for 3 days when necessary.
Flow cytometry and cell sorting
Single-cell suspensions of splenocytes were analyzed and sorted on FACSCanto II, FACSymphony or FACSAria II (BD). V500 anti-mouse B220, BV786 anti-mouse IgM, BV786 anti-mouse CD38, BV786 anti-mouse CD138, BV510 anti-mouse IgG1, and anti-Csk were purchased from BD Biosciences. APC-Cy7 anti-mouse/human B220, BV421 anti-mouse CD138, PE anti-mouse CD40, PE anti-mouse CD275, PE anti-mouse IL-21R, PE-Cy7 anti-mouse CD38, PerCP/Cy5.5 anti-mouse CD86, FITC anti-mouse CD22, Pacific Blue anti-mouse CD45.2, BV421 anti-mouse CD184, and BV510 anti-mouse CD45.2 were purchased from BioLegend. PE anti-mouse CD80, FITC anti-mouse MHC-II, FITC anti-mouse CD19, FITC anti-mouse CD45.1, eFluor450 GL-7, and APC-eFluor780 anti-mouse CD45.1 was purchased from Thermo Fisher Scientific. Pacific Blue anti-phospho-S6 ribosomal protein (Ser235/236), anti-phospho-Lyn (Tyr507), and anti-phospho-Syk (Tyr525/526) were purchased from Cell Signaling Technology. Anti-c-Myc was purchased from Abcam. Alexa Fluor 647 anti-active caspase-3 was purchased from BD. CaspGLOW fluorescein active caspase-3 staining kit was purchased from Thermo Fisher Scientific. Propidium iodide (as a viability dye) was purchased from Sigma-Aldrich. Data were analyzed using FlowJo software v9.9 and v10.8 (BD).
Single B cell sorting and VH186.2 sequence analysis
NP-specific IgG1+ GC B cells (Live B220+ CD45.1+ IgG1+ NP+ GL7+ CD38−) were single-cell sorted into 96-well plates containing 4 μl/well of ice-cold 0.5× PBS with 10 mM DTT, 1.6 U RNasin Plus RNase Inhibitor (Promega), and 0.1 U SUPERase-In RNase Inhibitor (Thermo Fisher Scientific). cDNA was synthesized in a total volume of 10 μl/well in the original 96-well plates containing 100 ng random primer (pd(N)6, Sigma-Aldrich), 0.5 μl of 10 mM dNTPs (QIAGEN), 0.33% (vol/vol) NP-40, 5 U SuperScript IV reverse transcriptase, and 1× RT buffer (Thermo Fisher Scientific) by incubating at 23°C for 10 min, 50°C for 10 min, and then 80°C for 10 min. VH186.2 gene transcripts were amplified by KOD plus ver.2 DNA polymerase (Toyobo) and primers (5′-TTCTTGGCAGCAACAGCTACA-3′ and 5′-GGATCCAGAGTTCCAGGTCACT-3′) (Kaji et al., 2012) for 40 cycles of 94°C 15 s, 54°C 30 s, and 68°C 55 s reactions, and the PCR products were purified and directly sequenced. SHM analysis was performed by IMGT/V-QUEST program (Brochet et al., 2008).
Western blot analysis
GC B cells purified by cell sorting (FACSAria II) were directly lysed in 1× Laemmli sample buffer supplemented with β-mercaptoethanol and dithiothreitol and then boiled for 5 min at 98°C. SDS-PAGE, transfer, and immunoblotting were performed as described previously (Inoue et al., 2015). After blocking (Blocking One; Nacalai Tesque), membranes were incubated with the following antibodies diluted in Can Get Signal Immunoreaction Enhancer Solution (Toyobo): phospho-Src family (Y416) (clone D49G4; Cell Signaling Technology), phospho-Erk1/2 (T202/Y204) (clone D13.14.4E; Cell Signaling Technology), Lyn (clone C13F9; Cell Signaling Technology), and actin (C-11; Santa Cruz Biotechnology). Images were acquired and quantified with a FUSION Solo S image analyzer (Vilber). Uncropped immunoblot images are available in SourceDataF1.
RNA-seq and GSEA
RNA-seq data used for GSEA were derived from three biological replicates. Cskf/f CreERT2 B1-8hi CD45.1+ or Csk+/+ CreERT2 B1-8hi CD45.1+ B cells were transferred into wild-type C57BL/6J mice. After being immunized i.p. with NP-CGG/alum (day 0), mice were administered tamoxifen on days 8 and 9, and NP-specific IgG1+ donor LZ (CD45.1+ NP+ IgG1+ GL7+ CD38− CD86hi CXCR4lo) and DZ (CD45.1+ NP+ IgG1+ GL7+ CD38− CD86lo CXCR4hi) GC B cells were sorted from splenocytes on day 11. Construction of the DNA library for RNA-seq and sequencing were performed as described previously (Inoue et al., 2017). Briefly, the DNA library was constructed using the NEBNext Ultra RNA Library Prep Kit for Illumina (NEB) from total RNA purified from ∼104 sorted cells. RNA-seq was performed on a HiSeq 1500 sequencer (Illumina) in a 49-bp single-end read mode. The raw data were processed with CASAVA 1.8.2 (Illumina) to generate fastq files. The RNA-seq data were deposited in the Gene Expression Omnibus database under the accession number GSE244802. Differentially expressed genes were identified using DESeq2 implemented in iDEP (Ge et al., 2018) with an FDR cut-off of 0.1 and a minimum fold-change of 2. GSEA was performed with GSEA software from the Broad Institute (Subramanian et al., 2005).
Statistical analysis
Statistical analysis was performed using Prism 8 and 9 (Graphpad).
Online supplemental material
Fig. S1 shows the analysis of μMT and Cskf/f CreERT2 mixed BM chimera (related to Fig. 2, C–F). Fig. S2 shows the apoptosis analysis and the expression of selected genes in Csk-deficient GC B cells (related to Fig. 1 J and Table S1). Fig. S3 shows the expression of Bim protein in Cskf/f GC B cells (related to Fig. 5). Table S1 shows the gene expression (FPKM) from the RNA-seq analysis and the list of differentially expressed genes.
Data availability
The RNA-seq data were deposited in the Gene Expression Omnibus database under accession number GSE244802. All other data supporting the findings of this study are available within the article, supplemental material, and source data file.
Acknowledgments
We thank Michel C. Nussenzweig (The Rockefeller University, New York, NY) for B1-8hi mice, Gabriel D. Victora (The Rockefeller University) for MtorF2108L mice, and Peter D. Burrows for critical reading of the manuscript.
This work was partially supported by the Japan Society for the Promotion of Science KAKENHI (JP21H02749 to T. Inoue and JP22H00450 to T. Kurosaki), the Japan Agency for Medical Research and Development (JP233fa627001 and JP243fa627001), the SENSHIN Medical Research Foundation, the Mochida Memorial Foundation for Medical and Pharmaceutical Research, the Takeda Science Foundation, the Naito Foundation, and the Sumitomo Foundation (to T. Inoue).
Author contributions: T. Inoue: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing—original draft, Writing—review and editing; Y. Matsumoto: Investigation, C. Kawai: Investigation, Resources, Validation; M. Ito: Investigation, S. Nada: Conceptualization, Resources, Writing—review and editing; M. Okada: Conceptualization, Resources, Supervision, Validation; T. Kurosaki: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing—original draft, Writing—review and editing.

