


OTU deubiquitinase with linear linkage specificity (OTULIN) regulates inflammation and cell death by deubiquitinating linear ubiquitin chains generated by the linear ubiquitin chain assembly complex (LUBAC). Biallelic loss-of-function mutations causes OTULIN-related autoinflammatory syndrome (ORAS), while OTULIN haploinsuffiency has not been associated with spontaneous inflammation. However, herein, we identify two patients with the heterozygous mutation p.Cys129Ser in OTULIN. Consistent with ORAS, we observed accumulation of linear ubiquitin chains, increased sensitivity to TNF-induced death, and dysregulation of inflammatory signaling in patient cells. While the C129S mutation did not affect OTULIN protein stability or binding capacity to LUBAC and linear ubiquitin chains, it did ablate OTULIN deubiquitinase activity. Loss of activity facilitated the accumulation of autoubiquitin chains on LUBAC. Altered ubiquitination of LUBAC inhibits its recruitment to the TNF receptor signaling complex, promoting TNF-induced cell death and disease pathology. By reporting the first dominant negative mutation driving ORAS, this study expands our clinical understanding of OTULIN-associated pathology.

Introduction
Ubiquitination is the reversible posttranslational covalent modification in which ubiquitin (Ub) is conjugated to target proteins to modulate their function. Ub itself can be ubiquitinated to generate poly Ub chains by attachment of the incoming Ub to any of seven different lysine residues (K6, K11, K27, K29, K33, K48, K63) or in a linear manner using the N-terminal methionine (M1). Linear or M1-linked Ub chains are particularly important for innate immune and cytokine receptor signaling. This is best studied in the context of tumor necrosis factor (TNF) receptor (TNFR1) signaling, where M1-Ub chains regulate the balance between inflammatory signaling and cell death through nuclear factor-κB (NFκB) and mitogen-activated protein kinases (MAPK) (Swatek and Komander, 2016; Jahan et al., 2021).
Ub modification is catalyzed by the repetitive function of three enzymes: E1 (Ub-activating enzyme), E2 (Ub-conjugating enzyme), and E3 (Ub ligase). To date, the linear Ub chain assembly complex (LUBAC) is the only E3 enzyme complex identified to generate M1-linked Ub chains (Kirisako et al., 2006). LUBAC is composed of three subunits: a large isoform of heme-oxidized iron regulatory protein 2 Ub ligase 1 (HOIL-1L), HOIL-1L interacting protein (HOIP), and SHANK-associated RH domain-interacting protein (SHARPIN) (Kirisako et al., 2006; Gerlach et al., 2011; Ikeda et al., 2011; Tokunaga et al., 2011). LUBAC attaches M1-Ub chains to a variety of substrates, and the full extent of M1-Ub chain regulation of intracellular signaling pathways is an active area of research. TNF stimulation triggers LUBAC and cellular inhibitor of apoptosis proteins recruitment to the TNF receptor signaling complex to modify receptor-interacting protein kinase 1 (RIPK1) and NFκB essential modulator (NEMO) with M1-Ub chains and K63-linked Ub chains, respectively (Haas et al., 2009). This stabilizes the TNFR1 signaling complex (complex I) and allows for sequential recruitment and phosphorylation of TAK1, IKKα, and IKKβ. This leads to IκBα degradation and consequent phosphorylation and activation of the NFκB transcription factors comprised of p65 and p50 subunits, and activation of the MAPKs p38, JNK, and ERK1/2 (Haas et al., 2009; Emmerich et al., 2013; Holbrook et al., 2019). Inappropriate ubiquitination of complex I causes its destabilization and consequent formation of complex IIa, which leads to caspase-8–initiated apoptosis, or complex IIb, which triggers necroptosis (Holbrook et al., 2019). Mutations in LUBAC components, HOIP and HOIL1 in humans and SHARPIN in mice, lead to dysregulation of M1-Ub–stabilized signaling, autoinflammation, and immunodeficiency (Gerlach et al., 2011; Ikeda et al., 2011; Tokunaga et al., 2011; Boisson et al., 2012, 2015; Oda et al., 2019).
Ubiquitination is counteracted by deubiquitinating enzymes (DUBs). In humans, a single DUB selectively and effectively disassembles M1-Ub chains, OTU deubiquitinase with linear linkage specificity (OTULIN) (Keusekotten et al., 2013; Rivkin et al., 2013). A second DUB, the Ub-specific protease CYLD, comprises a M1-selective catalytic domain (Komander et al., 2009; Sato et al., 2015); however, full-length CYLD preferentially cleaves K63 chains (Elliott et al., 2021). Both OTULIN and CYLD bind HOIP in the LUBAC complex, either directly (OTULIN) or indirectly via SPATA2 (CYLD). It appears that CLYD binds to LUBAC when LUBAC is recruited to receptor signaling complexes while OTULIN bound to LUBAC is not observed in this context (Elliott et al., 2016). Notably, LUBAC undergoes autoubiquitination and a key role of OTULIN is to remove these M1-Ub chains to maintain appropriate LUBAC function (Keusekotten et al., 2013; Rivkin et al., 2013; Elliott et al., 2014, 2016; Schaeffer et al., 2014; Kupka et al., 2016; Schlicher et al., 2016).
OTULIN within and outside LUBAC serves to globally downregulate inflammatory M1-Ub chains (Stangl et al., 2019). Consistently, downregulation of OTULIN results in enhanced M1-linked ubiquitination of LUBAC and its substrates (Fiil et al., 2013; Hrdinka et al., 2016). Accumulation of linear Ub chains triggers inflammatory signaling (Damgaard et al., 2016, 2019, 2020) and inhibits LUBAC’s ability to regulate NFκB signaling and protect against cell death (Fuseya and Iwai, 2021). Interestingly, while loss of OTULIN in myeloid cells triggers aberrant inflammation and cytokine release (Damgaard et al., 2016), some cell types, such as fibroblasts, compensate for the loss of OTULIN by downregulating LUBAC, and as a trade-off, these cells are unable to appropriately respond to cytokine signals and undergo apoptosis (Damgaard et al., 2019).
Similar to patients with loss-of-function mutations in LUBAC, patients harboring biallelic loss-of-function mutations in OTULIN develop severe auto-inflammatory disease with skin involvement, termed ORAS (OTULIN-related auto-inflammatory syndrome, also known as Otulipenia, OMIM #617099) (Damgaard et al., 2016; Zhou et al., 2016; Tao et al., 2021; Zinngrebe et al., 2022). Furthermore, mice deficient for OTULIN or expressing a catalytically inactive OTULIN mutant (cysteine to alanine at position 129, C129A) die mid-gestation due to angiogenic deficits and aberrant cell death mediated by TNFR1 signaling and RIPK1 kinase activity (Rivkin et al., 2013; Damgaard et al., 2016). To date, all reported ORAS-associated variants have been biallelic loss of function, while heterozygous loss-of-function variants can compromise patient immune response to certain pathogens such as Staphylococcus aureus (S. aureus) with the potential to result in severe necrotizing disease (Spaan et al., 2022). Herein, we describe the first heterozygous, dominant negative variant in OTULIN to cause ORAS, identified in two unrelated individuals. This unexpected result demonstrates the importance of understanding the functional consequences of specific mutations and expands our understanding of OTULIN biology and mutations causing ORAS.
Results and discussion
Patient 1 (P1) phenotype
The patient is a 22-mo-old male, born to non-consanguineous Caucasian parents. Born at 31 wk of gestation by emergency cesarean section for maternal eclampsia and required mechanical ventilation for 1 day after birth. At 4 days old he developed peri-umbilical erythema and was diagnosed with a presumed abscess, which was refractory to broad-spectrum antibiotics. Surgery was performed and significant inflammation was observed around a urachal remnant, with high numbers of neutrophils seen on histopathological examination, yet no bacteria was cultured from the site. The surgical closure dehisced and he was left with an abdominal wound that failed to close (Fig. 1 A). Across the period of healing of 2 mo, the only positive culture was a light growth of methicillin-sensitive S. aureus.

Patients with the heterozygous C129S variant in OTULIN exhibit clinical features consistent with ORAS. (A) Photographs detailing surgical wound progression of P1. (B) Acute respiratory deterioration in P1. (C) Sterile pustular abscesses at sites of previous trauma, e.g., injection site on ankle from P1. (D) Concentration of CRP and frequencies of platelets, neutrophils, monocytes, and eosinophils in patient blood measured weekly. Gray sections indicate normal range, and therapeutic interventions and key events in P1’s treatment are denoted by dotted lines. (E) Photographs of P2’s tender erythematous plaques and patches over the trunk and bilateral lower limbs and ulcerating wounds with necrotizing fasciitis. (F) Pedigrees of the kindreds presenting ORAS-like disease and carrying C129S heterozygous mutations of OTULIN, P1, P2, and NT (indicates not tested).
Patients with the heterozygous C129S variant in OTULIN exhibit clinical features consistent with ORAS. (A) Photographs detailing surgical wound progression of P1. (B) Acute respiratory deterioration in P1. (C) Sterile pustular abscesses at sites of previous trauma, e.g., injection site on ankle from P1. (D) Concentration of CRP and frequencies of platelets, neutrophils, monocytes, and eosinophils in patient blood measured weekly. Gray sections indicate normal range, and therapeutic interventions and key events in P1’s treatment are denoted by dotted lines. (E) Photographs of P2’s tender erythematous plaques and patches over the trunk and bilateral lower limbs and ulcerating wounds with necrotizing fasciitis. (F) Pedigrees of the kindreds presenting ORAS-like disease and carrying C129S heterozygous mutations of OTULIN, P1, P2, and NT (indicates not tested).
At 8 wk of age, the patient suffered an acute respiratory deterioration (Fig. 1 B) associated with extensive body edema, hypotension, and presumed pulmonary edema. High ventilation pressures were required to maintain lung oxygenation. Again, no organisms were isolated other than a rhinovirus in nasopharyngeal aspirate. Interestingly, corticosteroids were initiated as a supportive measure and the lungs immediately improved. Furthermore, the initiation of corticosteroid treatment correlated with the abdominal wound beginning to heal. This continued, although he developed a spontaneous cutaneous stoma from his duodenum (Fig. 1 A). In addition to steroids, he was treated with TNF-blockade (Adalimumab), which seemed to improve the rate of healing allowing for a reduced steroid dose, and JAK inhibition (Ruxolitinib). Furthermore, the rate of healing was noted to be remarkably better when he was covered with broad-spectrum antibiotic prophylaxis, initially with Timentin, and subsequently intravenous and then nasogastric Co-trimoxazole. During this time, the patient also developed a systolic murmur associated with a 1.2 × 1.6 mm echogenic focus on the tricuspid valve and was treated for infective endocarditis, but again with no causative organism identified on repeated culture.
Throughout his intensive care unit admission of many months, he developed repeated areas of swelling and erythema consistent with panniculitis, and additionally developed lesions that were believed to represent pathergy, with sterile pustular abscesses forming at the sight of prior trauma, including injection and cannula sites at the ankle and thigh (Fig. 1 C). Consistent with the abdominal wound healing, these lesions were responsive to corticosteroids.
Investigations demonstrated raised inflammatory markers (C reactive protein [CRP] 100–200 mg/L) and elevated neutrophils (30–50 × 109/L), monocytes (2–10 × 109/L), eosinophilia (1–3 × 109/L), and a persistent thrombocytopaenia (below 100 × 109/L), which continued until inflammation was controlled with corticosteroids (Fig. 1 D). The patient also demonstrated an initial hypogammaglobulinemia (IgG = 0.59 g/L, IgA < 0.07 g/L), although this may have been related to prematurity. In contrast to the elevated myeloid populations, lymphocyte subsets were largely normal (data not shown). With time, he has begun to develop antibodies normally (IgA = 0.31 g/L, IgM = 0.61 g/L) with IgG still replaced. A biopsy of perilesional skin from the abdominal wound showed heavy acute inflammatory exudate with diffuse suppuration and areas of necrosis. Persistent inflammation and evidence of cell death were considered consistent with an autoinflammatory disease caused by an inborn genetic error.
Patient 2 (P2) phenotype
The second patient is a full-term Saudi female born to non-consanguineous parents with insignificant antenatal history. She was asymptomatic until the age of 7 mo when she presented to a tertiary hospital in Riyadh, Saudi Arabia, with multiple tender skin lesions over the upper and lower limbs bilaterally, associated with a fever (38–39°C) and generalized edema (Fig. 1 E). This presentation coincided with the administration of routine 6-mo vaccine injections, including hexavalent (diphtheria, tetanus, and pertussis; hepatitis B; inactivated poliovirus vaccine; Haemophilus influenzae type b), bacille Calmette-Guerin, and pneumococcal conjugate vaccines, which were received 2 wk prior to the presentation.
The patient was admitted and started empirically on broad-spectrum antibiotics. Initial investigations showed significant leukocytosis (45.5 × 109/L) with neutrophilia (40.9 × 109/L), high CRP (264 mg/L), and negative blood and urine cultures. No improvement was noticed after 72 h of intravenous (IV) antibiotics as her fever was persistent, and some of her lesions progressed to a picture of tender cellulitis-like plaques evolving into necrotizing fasciitis with ulceration (Fig. 1 E). Debridement and fasciotomy were performed on multiple sites, and tissue samples were sent for cultures and histopathology. Biopsy results showed mixed infiltration of neutrophils and plasma cells with the area of necrosis indicating acute and chronic inflammation. No pathogen was identified as all her tissue cultures for fungal, bacterial, and mycobacterial organisms were negative. A thorough immunological and autoimmune workup was done with no significant results (Table S1), and, therefore, genetic testing was sent.
Following the surgical debridement, the patient showed slow improvement in terms of fever pattern and trend of laboratory results, with no development of new lesions. She completed a 3-wk course of IV antibiotics and was discharged with close follow-up. At the age of 12 mo, she presented to the hospital complaining of the same manifestations with fever and new multiple tender erythematous plaques and patches over the trunk and bilateral lower limbs. Again, her initial investigations showed leukocytosis (19.9 × 109/L) with neutrophilia (12.9 × 109/L), very high CRP (393 mg/L), and negative cultures. Skin biopsy revealed panniculitis. With these two attacks of hyperinflammatory response in the setting of negative cultures, the patient was managed as a case of an autoinflammatory/immune-dysregulation disorder caused by an inborn genetic error. This was supported by the prompt clinical and biochemical improvement after starting corticosteroids. Notably, no antibiotics were started during this admission.
Given the phenotype, response to steroid, and potentially involved signaling pathways, the patient was started on anti-TNF (Infliximab) with adjunctive Methotrexate, and corticosteroid was successfully weaned off. She was followed during her daycare visits for Infliximab infusion every 4 wk. Initially, she had recurrent simple chest infections, which notably improved after adding prophylactic antibiotics. Otherwise, she remains in clinical and biochemical remission since starting anti-TNF therapy, and she completed her third year of life in June 2023, achieving 2 years of disease remission.
Genetic studies
Trio exome sequencing was performed on P1 and his parents; this identified a de novo variant in OTULIN, ENST00000284274.5:c.386G>C, resulting in a missense change from cysteine to serine at position 129 (p.Cys129Ser) (Fig. 1 F). P2 underwent whole genome sequencing and a variant in OTULIN, ENST00000284274.5:c.385T>A, which results in the same cysteine to serine change at position 129, was identified. Follow-up testing on P2’s parents confirmed the de novo nature of the variant (Fig. 1 F).
The C129S OTULIN variant was previously unreported and absent from databases of normal variation, including gnomAD (v2.1.1 and v3.1.2). In silico pathogenicity prediction tools were equivocal with 11 out of 22 queried by the Varsome website consistent with a deleterious effect, a Combined Annotation Dependent Depletion score of 24.6, and a moderate physiochemical change (Grantham score = 112). Significantly, cysteine 129 is part of the OTULIN catalytic domain and is essential for OTULIN enzymatic activity (Keusekotten et al., 2013; Rivkin et al., 2013). The amino acid change from cysteine to serine is predicted to render the catalytic site inactive, leading to a loss of OTULIN activity. Indeed, a mouse model of a similar variant, C129A OTULIN, has been shown to drive an autoinflammatory disease phenotype characterized by aberrant cell death and dysregulation of TNF and the type I interferon (IFNαβ) signaling pathways (Heger et al., 2018). We hypothesized that the C129S variant likewise drives autoinflammatory disease. However, as all reported ORAS patient variants result in almost complete loss of OTULIN function, this heterozygous C129S variant may act in a dominant negative fashion to drive disease.
Heterozygous C129S variant of OTULIN does not alter protein stability but affects OTULIN deubiquitinase activity
The majority of ORAS-related mutations destabilize OTULIN expression; thus, we began by assessing C129S OTULIN stability. C129S OTULIN exhibited equivalent protein stability with similar melting temperature to wild type (WT) OTULIN in a Tycho NT.6 assay (Fig. 2 A). Consistent with this, we observed similar expression of both OTULIN protein and OTULIN transcript in patient fibroblasts compared with healthy donor (HD) fibroblasts (Fig. 2, B and C; and Fig. S1 A). Destabilization of OTULIN protein has been reported to result in the loss of the LUBAC components HOIP and SHARPIN in certain cell types including fibroblasts (Damgaard et al., 2019). However, we did not observe a consistent decrease in transcription or protein expression of any LUBAC component (SHARPIN, HOIP, or HOIL-1) in patient fibroblasts compared with HD (Fig. 2, B and C; and Fig. S1 A).
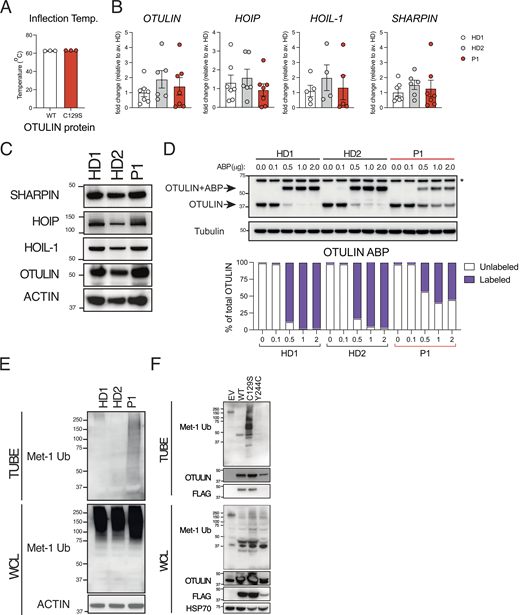
C129S OTULIN mutant protein associates with increased M1-Ub and cell death in patient cells. (A) C129S OTULIN protein thermostability is comparable to WT OTULIN protein, as assessed by Tycho. (B) Transcription of OTULIN, SHARPIN, HOIL-1, and HOIP in HD1, HD2, and patient fibroblasts was assessed by qPCR. (C) Protein levels of OTULIN, SHARPIN, HOIL-1, HOIP, and actin were assessed in HDs (HD1, HD2) and patient fibroblasts by immunoblot. (D) Fibroblast lysates from HD1, HD2, and patient lines were treated with the OTULIN ABP at stated concentrations to assess OTULIN activity. OTULIN and Tubulin (loading control) were assessed by immunoblot. Arrows indicate unlabeled (OTULIN) and labeled (OTULIN+ABP) OTULIN. * indicates non-specific band. Densitometry analysis of labeled and unlabeled OTULIN bands was performed and graphed as a percentage of total signal. (E and F) TUBE reagent was used to pull-down polyubiquitin in cell lysates. Linear Ub chains (M1-Ub) were then specifically probed for in TUBE pull-down and WCL samples. Equal loading was confirmed by blotting for actin in the WCL. This was performed in (E) lysates from HD1, HD2, and P1 fibroblasts and in (F) lysates from HEK 293T cells transiently transfected with EV, WT, C129S, or Y244C OTULIN. (A and B) Data were pooled from at least three experiments, where each dot is representative of a single experimental result and statistical significance was assessed by unpaired t test, means ± SEMs. (C–F) Data representative of at least three experiments; indicated molecular weight values in kilodaltons (kD). Source data are available for this figure: SourceData F2.
C129S OTULIN mutant protein associates with increased M1-Ub and cell death in patient cells. (A) C129S OTULIN protein thermostability is comparable to WT OTULIN protein, as assessed by Tycho. (B) Transcription of OTULIN, SHARPIN, HOIL-1, and HOIP in HD1, HD2, and patient fibroblasts was assessed by qPCR. (C) Protein levels of OTULIN, SHARPIN, HOIL-1, HOIP, and actin were assessed in HDs (HD1, HD2) and patient fibroblasts by immunoblot. (D) Fibroblast lysates from HD1, HD2, and patient lines were treated with the OTULIN ABP at stated concentrations to assess OTULIN activity. OTULIN and Tubulin (loading control) were assessed by immunoblot. Arrows indicate unlabeled (OTULIN) and labeled (OTULIN+ABP) OTULIN. * indicates non-specific band. Densitometry analysis of labeled and unlabeled OTULIN bands was performed and graphed as a percentage of total signal. (E and F) TUBE reagent was used to pull-down polyubiquitin in cell lysates. Linear Ub chains (M1-Ub) were then specifically probed for in TUBE pull-down and WCL samples. Equal loading was confirmed by blotting for actin in the WCL. This was performed in (E) lysates from HD1, HD2, and P1 fibroblasts and in (F) lysates from HEK 293T cells transiently transfected with EV, WT, C129S, or Y244C OTULIN. (A and B) Data were pooled from at least three experiments, where each dot is representative of a single experimental result and statistical significance was assessed by unpaired t test, means ± SEMs. (C–F) Data representative of at least three experiments; indicated molecular weight values in kilodaltons (kD). Source data are available for this figure: SourceData F2.
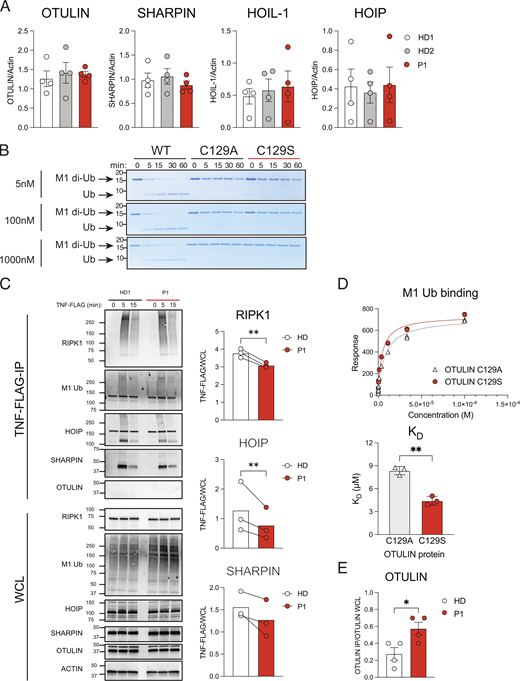
The C129S mutation ablates OTULIN deubiquitinase activty, but not OTULIN protein stability or substrate binding. (A) Fibroblast lysates from HD1, HD2, and P1 lines assessed for protein levels of OTULIN, HOIP, HOIL-1, SHARPIN, and actin (loading control) were assessed by immunoblot. Densitometry analysis of protein expression was performed using Fiji and graphed as a function of actin loading control. Data were pooled from four experiments, where each dot indicates an individual experimental result, means ± SEM, and statistical significance was assessed by one-way ANOVA. (B) Recombinant WT OTULIN and C129A and C129S mutant OTULIN proteins were incubated at stated concentrations with M1-linked di-Ub chains. Cleavage capacity was assessed from 0 to 60 min. Arrows indicate uncleaved M1–di-Ub and cleaved Ub; indicated molecular weight values in kD. Data is representative of three repeats. (C) HD1 and P1 fibroblasts were stimulated with TNF-FLAG for indicated time points; the TNFR1 signaling complex was then assessed by Flag pull-down. Immunoblotting was performed for RIPK1, M1-Ub, HOIP, SHARPIN, OTULIN, and actin as a loading control; indicated molecular weight values in kD. Graphs show densitometry quantification of proteins immunoprecipitated at 5 min with TNF-FLAG relative to the signal observed in WCL. Data pooled from three experiments where HD indicates HD1 or HD4 and lines connect values from the same experiment. Significance assessed by ratio paired t test and ** indicates P < 0.01. (D) C129S and C129A binding capacity to M1-Ub was assessed by SPR; data representative of three repeats and KD values summarized in column graph; significance assessed by paired Student’s t test where ** indicates P < 0.01. (E) Densitometry quantification of OTULIN pulled down in four repeats of endogenous SHARPIN IP (Fig. 5 C); significance assessed by Student’s t test, where * indicates P < 0.05. Source data are available for this figure: SourceData FS1.
The C129S mutation ablates OTULIN deubiquitinase activty, but not OTULIN protein stability or substrate binding. (A) Fibroblast lysates from HD1, HD2, and P1 lines assessed for protein levels of OTULIN, HOIP, HOIL-1, SHARPIN, and actin (loading control) were assessed by immunoblot. Densitometry analysis of protein expression was performed using Fiji and graphed as a function of actin loading control. Data were pooled from four experiments, where each dot indicates an individual experimental result, means ± SEM, and statistical significance was assessed by one-way ANOVA. (B) Recombinant WT OTULIN and C129A and C129S mutant OTULIN proteins were incubated at stated concentrations with M1-linked di-Ub chains. Cleavage capacity was assessed from 0 to 60 min. Arrows indicate uncleaved M1–di-Ub and cleaved Ub; indicated molecular weight values in kD. Data is representative of three repeats. (C) HD1 and P1 fibroblasts were stimulated with TNF-FLAG for indicated time points; the TNFR1 signaling complex was then assessed by Flag pull-down. Immunoblotting was performed for RIPK1, M1-Ub, HOIP, SHARPIN, OTULIN, and actin as a loading control; indicated molecular weight values in kD. Graphs show densitometry quantification of proteins immunoprecipitated at 5 min with TNF-FLAG relative to the signal observed in WCL. Data pooled from three experiments where HD indicates HD1 or HD4 and lines connect values from the same experiment. Significance assessed by ratio paired t test and ** indicates P < 0.01. (D) C129S and C129A binding capacity to M1-Ub was assessed by SPR; data representative of three repeats and KD values summarized in column graph; significance assessed by paired Student’s t test where ** indicates P < 0.01. (E) Densitometry quantification of OTULIN pulled down in four repeats of endogenous SHARPIN IP (Fig. 5 C); significance assessed by Student’s t test, where * indicates P < 0.05. Source data are available for this figure: SourceData FS1.
Whilst the C129S mutation did not alter OTULIN protein stability, this mutation did, as predicted, affect OTULIN deubiquitinase activity. C129S OTULIN, like the previously reported C129A mutation, failed to hydrolyze M1–di-Ub in a cell-free cleavage assay over a range of concentrations (Fig. S1 B). To confirm that C129S OTULIN was being expressed in patient cells and the equivalent levels of OTULIN protein observed were not the result of compensation from the WT allele, we used a chemically enhanced, linear di-Ub activity-based probe (ABP). This ABP covalently binds to catalytically active OTULIN and causes a size shift identifiable by western blot (Weber et al., 2017). Using this OTULIN-specific ABP, we confirmed that in patient fibroblasts, both WT (active) and C129S (inactive) forms of OTULIN are coexpressed in seemingly equal amounts (Fig. 2 D).
C129S mutant OTULIN drives the accumulation of linear Ub chains in the presence of WT OTULIN
The primary function of OTULIN is to negatively regulate the accumulation of M1-Ub chains through deubiquitination of this specific chain type. As we had observed that the catalytically inactive C129S OTULIN is stably expressed in patient cells, we sought to determine the effect on global M1-Ub chain accumulation. Using a GST-NEMO linear tandem Ub binding entity (TUBE) pull-down, we observed significant accumulation of M1-Ub chains in patient cells compared with HD fibroblasts (Fig. 2 E). This accumulation of M1-Ub chains in patient fibroblasts could be attributed to the 50% decrease in OTULIN activity due to the C129S mutant allele. Indeed, fibroblasts generated from OTULIN-haploinsufficent individuals also exhibit increased M1-Ub chain accumulation (Spaan et al., 2022). However, transient transfection of human embryonic kidney (HEK) 293T cells with C129S OTULIN triggered M1-Ub chain accumulation (Fig. 2 F). Transfection with WT OTULIN or, interestingly, Y244C OTULIN, a mutation that decreases OTULIN stability associated with ORAS (Zhou et al., 2016) and OTULIN haploinsuffiency (Spaan et al., 2022), did not cause the same M1-Ub accumulation (Fig. 2 F). Importantly, HEK 293T cells express functional OTULIN endogenously, therefore C129S OTULIN, unlike WT and Y244C OTULIN, directly drives the accumulation of M1-Ub chains, despite the presence of endogenous OTULIN. This also likely explains the difference between OTULIN-haploinsufficent fibroblasts and transfection of a loss of function OTULIN variant such as Y244C. Unlike OTULIN-haploinsufficent cells, HEK 293T cells have sufficient endogenous OTULIN to control M1-Ub chain levels in spite of transfection with a loss-of-function OTULIN variant.
Consistent with increased overall M1-Ub, we observed that transient transfection of C129S OTULIN, but not WT OTULIN or two representative OTULIN loss-of-function mutants, N341D (Spaan et al., 2022) and Y244C (Zhou et al., 2016), triggered NFκB activity as assessed by luciferase assay (Fig. 3 A). NFκB luciferase activity increased in relation to the increasing concentration of C129S OTULIN transfected, demonstrating a causal relationship between C129S OTULIN expression and spontaneous NFκB activity. Furthermore, cotransfection of WT and C129S OTULIN into HEK 293T cells across a range of ratios revealed that C129S OTULIN was able to drive NFκB activity even at lower concentrations than WT OTULIN (Fig. 3 B). Collectively, these data indicate C129S OTULIN has a dominant negative effect over WT OTULIN and may thereby drive autoinflammatory disease, even in a heterozygous context.
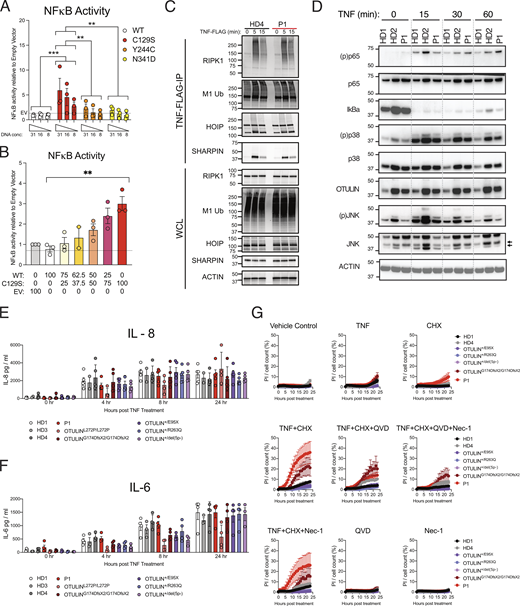
Heterozygous C129S OTULIN fibroblasts exhibit increased sensitivity to TNF-induced cell death. (A and B) NFκB activity was assessed by luciferase reporter assay, where HEK 293T cells were transfected with NFκB luciferase reporter, Renilla control, and (A) stated concentrations of EV, WT, C129S, Y244C, and N341D OTULIN or (B) as a ratio between WT and C129S OTULIN. (C) HD4 and P1 fibroblasts were stimulated with TNF-FLAG for indicated time points; the TNFR1 signaling complex was then assessed by Flag pull-down. Immunoblotting was performed for RIPK1, M1-Ub, HOIP, SHARPIN, and actin as a loading control. (D) HD1, HD2, and P1 fibroblast lines were treated with TNF (100 ng/ml) for indicated time points. Immunoblot was then performed to assess phosphorylated (p) and total levels of p65, p38, JNK as well as total levels IκBa and OTULIN. Actin was used as a loading control. (E–G) Fibroblasts from up to four HDs, P1, two ORAS patients (OTULINL272P/L272P and OTULING174DfsX2/G174DfsX2) and three OTULIN-haploinsufficent patients (OTULIN+/E95X, OTULIN+/R263Q, and OTULIN+/del(5p-)) were used. (E and F) Indicated fibroblast lines were treated with 10 ng/ml TNF for stated time points. Supernatants were then assessed for secretion of (E) IL-8 and (F) IL-6. (G) Stated fibroblasts were treated with TNF (100 ng/ml), CHX (50 μg/ml), QVD-OPh (10 μM), and Nec-1 (10 μM) or a combination thereof, as indicated. Cell death was measured over a period of 24 h by cellular inclusion of propidium iodide dye. (A–G) Data is pooled from at least three experiments, where each dot indicates an individual experimental result, means ± SEM, and statistical significance was assessed by (A, E, and F) two-way ANOVA or (B) one-way ANOVA, where ** indicates P < 0.01 and *** denotes P < 0.001. (C and D) Data representative of at least three experiments, indicated molecular weight values in kD. Source data are available for this figure: SourceData F3.
Heterozygous C129S OTULIN fibroblasts exhibit increased sensitivity to TNF-induced cell death. (A and B) NFκB activity was assessed by luciferase reporter assay, where HEK 293T cells were transfected with NFκB luciferase reporter, Renilla control, and (A) stated concentrations of EV, WT, C129S, Y244C, and N341D OTULIN or (B) as a ratio between WT and C129S OTULIN. (C) HD4 and P1 fibroblasts were stimulated with TNF-FLAG for indicated time points; the TNFR1 signaling complex was then assessed by Flag pull-down. Immunoblotting was performed for RIPK1, M1-Ub, HOIP, SHARPIN, and actin as a loading control. (D) HD1, HD2, and P1 fibroblast lines were treated with TNF (100 ng/ml) for indicated time points. Immunoblot was then performed to assess phosphorylated (p) and total levels of p65, p38, JNK as well as total levels IκBa and OTULIN. Actin was used as a loading control. (E–G) Fibroblasts from up to four HDs, P1, two ORAS patients (OTULINL272P/L272P and OTULING174DfsX2/G174DfsX2) and three OTULIN-haploinsufficent patients (OTULIN+/E95X, OTULIN+/R263Q, and OTULIN+/del(5p-)) were used. (E and F) Indicated fibroblast lines were treated with 10 ng/ml TNF for stated time points. Supernatants were then assessed for secretion of (E) IL-8 and (F) IL-6. (G) Stated fibroblasts were treated with TNF (100 ng/ml), CHX (50 μg/ml), QVD-OPh (10 μM), and Nec-1 (10 μM) or a combination thereof, as indicated. Cell death was measured over a period of 24 h by cellular inclusion of propidium iodide dye. (A–G) Data is pooled from at least three experiments, where each dot indicates an individual experimental result, means ± SEM, and statistical significance was assessed by (A, E, and F) two-way ANOVA or (B) one-way ANOVA, where ** indicates P < 0.01 and *** denotes P < 0.001. (C and D) Data representative of at least three experiments, indicated molecular weight values in kD. Source data are available for this figure: SourceData F3.
Heterozygous C129S OTULIN has minimal effect on NFκB signaling in patient fibroblasts
LUBAC-mediated M1-Ub is required for stabilization of the TNFR signaling complex (Haas et al., 2009). TNFR1-mediated autoinflammation has been suggested as a key driver of disease in both ORAS patients and mouse models with OTULIN defects (Damgaard et al., 2016, 2019; Zhou et al., 2016; Heger et al., 2018). All living individuals with biallelic loss-of-function mutations in OTULIN are maintained on an anti-TNF monoclonal antibody (Damgaard et al., 2016, 2019; Zhou et al., 2016; Zinngrebe et al., 2022). Similarly, TNF blocking therapy, Adalimumab, allowed both C129S OTULIN heterozygous patients to be weaned to low doses or off corticosteroids. Blockade of TNF signaling dramatically reduced CRP levels and promoted a decrease in numbers of circulating myeloid cells in P1 (Fig. 1 D). Furthermore, Adalimumab therapy supported abdominal wound healing in P1. P1 is now 2 mo post-op following closure of the wound. A Gore-Tex patch was used to recreate his abdominal wall (Fig. 1 A) and is currently re-established on full feeds. We, therefore, investigated whether C129S OTULIN alters TNF signaling in patient cells.
We performed a Flag-TNF pull-down to assess ubiquitination of the TNFR1 signaling complex in patients and HD fibroblasts. Flag-TNF stimulation resulted in recruitment and modification of RIPK1 at the TNFR1 signaling complex in both HD- and patient-derived fibroblasts; however, we observed consistently less modification of RIPK1 in patient cells (Fig. 3 C, second HD and quantification shown in Fig. S1 C). Consistent with this, we observed reduced recruitment of LUBAC components SHARPIN and HOIP to the TNFR1 signaling complex in patient cells compared with HD and this was associated with reduced M1-Ub (Fig. 3 C and Fig. S1 C). As previously described, OTULIN is not recruited to the TNFR1 signaling complex in either HD or patient cells (Draber et al., 2015; Elliott et al., 2016; Hrdinka et al., 2016) (Fig. S1 C).
We next sought to determine if this altered M1-Ub at the TNFR1 signaling complex altered inflammatory signaling. Comparison of activation of MAPKs and the NFκB response upon TNF stimulation of C129S OTULIN heterozygous and two HD fibroblast lines revealed minimal differences in response (Fig. 3 D). Although we do observe a slight increase in phosphorylation of the NFκB subunit p65 in patient fibroblasts at baseline, in response to TNF stimulation, the degradation of IKBα, and phosphorylation of p65, p38, and JNK were comparable between patient and HD lines over time (Fig. 3 D). Similarly, secretion of the inflammatory cytokines: IL-8 and IL-6 in response to TNF stimulation was comparable between C129S OTULIN heterozygous, OTULIN-haploinsufficent (Spaan et al., 2022), and HD fibroblast lines (Fig. 3, E and F). We also assessed IL-8 and IL-6 secretion from two classical ORAS lines (Zhou et al., 2016) and observed a slightly delayed response in fibroblasts bearing the homozygous L272P loss-of-function mutation but not the G174DfsX2 mutation (Fig. 3, E and F).
Collectively, this data demonstrates that C129S OTULIN heterozygosity does not significantly alter MAPK or NFκB signaling in response to TNF stimulation in patient fibroblasts. In line with this, studies in previously reported ORAS patient fibroblasts with biallelic loss-of-function mutations in OTULIN have a varied effect on NFκB and MAPK activation downstream of TNFR1. Both Zinngrebe et al. (2022) and Zhou et al. (2016) observed enhanced M1-Ub at the TNFR1 signaling complex. However, this only correlated to increased NFκB and MAPK activation in the ORAS patients reported by Zhou et al. (2016), while Zinngrebe et al. (2022) reported a similar signaling response between patient and HD cells (Zinngrebe et al., 2022). In contrast, Damgaard et al. (2019) compared an ORAS fibroblast line to an HD and observed decreased phosphorylation of IκBα, JNK, and p38 as well as decreased IL-8 secretion (Damgaard et al., 2019). These differences could reflect differences in the specific mutation of OTULIN, consequent LUBAC or TNFR1 signaling complex stability, or other experimental variables. Additionally, Damgaard et al. (2016) identified the myeloid cell linage as the main source of inflammatory cytokines such as TNF in a mouse model of loss-of-OTULIN function (Damgaard et al., 2016), and thus, dysregulation of NFκB-driven inflammation may be more easily observed in these cell types or the overexpression systems we employed (Fig. 3, A and B).
Heterozygous C129S OTULIN fibroblasts exhibit increased sensitivity to TNF-induced cell death
Reduced recruitment of LUBAC to the TNFR1 signaling complex observed in heterozygous C129S OTULIN patient cells indicates that while survival and inflammatory signaling pathways are unaffected at early time points (Fig. 3 D), complex I formation in these cells may have decreased stability (Fig. 3 C). Decreased stability of TNFR1 signaling complex I results in the formation of complex II and leads to TNF-induced cell death. One of the key cellular differences between OTULIN haploinsufficiency and ORAS is sensitivity to TNF-induced cell death. OTULIN-haploinsufficient fibroblasts are not sensitive to TNF-induced cell death while fibroblasts from ORAS patients exhibit enhanced cell death, primarily apoptosis, downstream of TNFR1 signaling (Damgaard et al., 2019; Spaan et al., 2022; Zinngrebe et al., 2022). Thus, to determine if the heterozygous C129S mutation was acting in a dominant negative fashion to drive an ORAS-like disease rather than OTULIN haploinsufficiency, we compared viability of primary C129S OTULIN patient fibroblasts to those derived from ORAS and OTULIN-haploinsufficent patients, as well as HDs. Upon treatment with TNF in combination with the protein synthesis inhibitor cycloheximide (CHX), C129S OTULIN patient fibroblasts died at a similar rate to OTULIN biallelic loss-of-function fibroblasts, as assessed by cellular inclusion of propidium iodide dye. C129S heterozygous OTULIN and ORAS fibroblasts exhibited markedly increased sensitivity to TNF-induced cell death compared with HD and OTULIN-haploinsufficent cells (Fig. 3 G). Treatment with the pan caspase inhibitor QVD significantly reduced TNF-CHX–induced cell death in the C129S heterozygous and ORAS lines, while the RIPK1 kinase inhibitor, necrostatin-1 (Nec-1), had minimal effect. Combination treatment of QVD and Nec-1 significantly blocked TNF-CHX–induced cell death in C129S heterozygous OTULIN and ORAS cells (Fig. 3 G). Thus, C129S heterozygous OTULIN patient fibroblasts die primarily by apoptosis and, to a lesser extent, by necroptosis in response to TNF+CHX stimulation. This phenotype is consistent with fibroblasts derived from ORAS patients, despite our patient having different LUBAC/OTULIN levels (Fig. 2 C and Fig. S1 A) and distinguishes these cells from those of OTULIN-haploinsufficient individuals (Damgaard et al., 2019; Spaan et al., 2022).
This increased sensitivity to TNF-induced cell death is likely a strong contributor to C129S heterozygous OTULIN patient pathology, as both P1 and P2 suffered from necrotizing wounds that improved upon TNF blockade. TNF-driven cell death in fibroblasts may increase the presence of danger-associated molecular patterns and thereby enhance inflammatory signaling from myeloid cells. This data emphasizes the importance of controlling not only TNF-driven inflammatory signaling but also TNF-induced cell death in ORAS patients.
It is also notable that the addition of prophylactic antibiotics played a role in wound healing for both patients. Loss of skin barrier function caused by cell death may have perpetuated inflammatory signaling and TNF-induced cell death through the sensing of commensal organisms. Interestingly, OTULIN haploinsufficiency confers a predisposition to sporadic severe necrosis of the skin and lungs, typically, but not exclusively, after infection with S. aureus. This is due to insufficient OTULIN levels in haploinsufficent patient fibroblasts to ensure deubiquitination of the signaling protein, caveolin-1 (Spaan et al., 2022). Data from that study also indicates that ORAS patients should exhibit similar susceptibility to bacterial infection; however, aside from observations made in this study, this is yet to be reported. P1’s phenotype was more pervasive than OTULIN-haploinsufficent patients, but his wound was exacerbated by microbial infection. Along with corticosteroid treatment, antibiotics were essential to wound recovery, consistent with bacterial invasion contributing to his phenotype. Relatedly, P2 experienced recurrent chest infections that have been controlled by the addition of prophylactic antibiotics to her treatment regime. Despite repeated testing, we did not identify a causative pathogen in either patient; however, the improvement of disease burden upon prophylactic antibiotic treatment indicates a microbial contribution to patient pathology, and this should be taken into account when treating ORAS patients.
Heterozygous C129S OTULIN drives IFNαβ-driven inflammation
In contrast to ORAS, patients with heterozygous loss-of-function mutations in OTULIN do not exhibit overt autoinflammatory disease, and thorough investigation into specific patient cell types did not reveal any subclinical inflammatory disturbances (Spaan et al., 2022). While we did not observe differences in NFκB-driven cytokines in C129S heterozygous OTULIN fibroblasts compared with control lines upon TNF stimulation (Fig. 3, E and F), we did identify a marked increase in transcription of proinflammatory cytokines, IFNB1 and IL6, and to a lesser extent TNF in the peripheral blood mononuclear cells (PBMCs) of P1 (Fig. 4 A). Furthermore, a similar inflammatory gene signature was observed in the human monocyte line, THP-1s cells upon stable expression of C129S OTULIN but not WT OTULIN (Fig. 4 B).

C129S OTULIN heterozygosity drives elevated IFNαβ signaling in patient cells. (A–C) Transcription of inflammatory genes including IFNB1, IRF7, IL6, and TNF was assessed by qPCR in (A) patient and HD PBMCs, in (B) THP-1 cells stably expressing WT or C129S OTULIN or non-transduced controls, and in (C) fibroblasts from up to four HDs, P1, two ORAS patients (OTULINL272P/L272P and OTULING174DfsX2/G174DfsX2), and three OTULIN-haploinsufficent patients (OTULIN+/E95X, OTULIN+/R263Q and OTULIN+/del(5p-)). Inflammatory genes of interest are expressed as fold change relative to the (A and C) average of the healthy donors or (B) non-transduced THP-1s. (D) Activation of IFNαβ signaling was assessed in fibroblasts from indicated donors by ISG Score. ISG Score was generated from the assessment of six known ISGs (IRF7, ISG15, MX1, SIGLEC1, IFIT1, and RSAD2). (A) Data representative of one experiment, technical replicates graphed. (B–D) Data are pooled from at least three experiments, where each dot indicates an individual experimental result, means ± SEM, and statistical significance was assessed by one-way ANOVA. * indicates P < 0.05. For C and D, the following comparisons to P1 were performed: HDs, ORAS, and OTULIN-haploinsufficent.
C129S OTULIN heterozygosity drives elevated IFNαβ signaling in patient cells. (A–C) Transcription of inflammatory genes including IFNB1, IRF7, IL6, and TNF was assessed by qPCR in (A) patient and HD PBMCs, in (B) THP-1 cells stably expressing WT or C129S OTULIN or non-transduced controls, and in (C) fibroblasts from up to four HDs, P1, two ORAS patients (OTULINL272P/L272P and OTULING174DfsX2/G174DfsX2), and three OTULIN-haploinsufficent patients (OTULIN+/E95X, OTULIN+/R263Q and OTULIN+/del(5p-)). Inflammatory genes of interest are expressed as fold change relative to the (A and C) average of the healthy donors or (B) non-transduced THP-1s. (D) Activation of IFNαβ signaling was assessed in fibroblasts from indicated donors by ISG Score. ISG Score was generated from the assessment of six known ISGs (IRF7, ISG15, MX1, SIGLEC1, IFIT1, and RSAD2). (A) Data representative of one experiment, technical replicates graphed. (B–D) Data are pooled from at least three experiments, where each dot indicates an individual experimental result, means ± SEM, and statistical significance was assessed by one-way ANOVA. * indicates P < 0.05. For C and D, the following comparisons to P1 were performed: HDs, ORAS, and OTULIN-haploinsufficent.
We observed increased transcription of IFNB1 and IL6 in C129S OTULIN heterozygous fibroblasts; however, this was quite variable and ORAS fibroblasts exhibited a similar trend (Fig. 4 C). OTULIN-haploinsufficent fibroblasts exhibited similar transcription of IFNB1 and IL6 compared with HDs (Fig. 4 C). The elevated transcription of IL6 in C129S OTULIN heterozygous patient fibroblasts was reflected in elevated secretion at baseline identified by ELISA (Fig. 3 F). Interestingly, this was not observed in the biallelic loss-of-function ORAS lines and may reflect the retained presence of LUBAC to support NFκB activation in our patient fibroblasts (Fig. 2 C and Fig. S1 A). Alternatively, IL6 transcription can also be driven by type I IFN signaling (Nan et al., 2018), so increased basal secretion of IL-6 from our patient cells may reflect the notably higher IFNB1 transcription observed in this line.
IFNB1 is a member of the type I IFN family. This master cytokine family triggers transcription of hundreds of genes collectively known as interferon-stimulated genes (ISGs). We assessed the basal transcription of six ISGs (SIGLEC1, IFIT1, ISG15, RSAD2, MX1, and IRF7) and generated an ISG Score as per Rice et al. (2013). Consistent with elevated IFNB1 in patient cells, we also observed an elevated ISG Score compared with HDs and OTULIN-haploinsufficent lines (Fig. 3 E). Transcription of the ISG IRF7 was also elevated in patient PBMCs compared with HDs; however, due to material limitations, we could not generate a full ISG Score for patient PBMCs (Fig. 3 D). This elevated inflammatory gene transcription is consistent with the ORAS phenotype, where C129S OTULIN heterozygous fibroblasts exhibited similar ISG Scores to the two biallelic loss-of-function ORAS lines assessed (Fig. 3 E). This is in line with results reported in the literature to date, as Tao et al. (2021) recently identified an elevated type I IFN gene signature in the blood of ORAS patients due to loss of OTULIN resulting in accumulation of Ub chains on proteasomal subunits inhibiting proteasome function (Tao et al., 2021). In contrast, expression of ISGs in PBMCs from OTULIN-haploinsufficient patients was not elevated (Spaan et al., 2022).
To counter inflammatory signaling driven by elevated type I IFNs observed in patient cells, the JAK inhibitor Ruxolitinib was also trialed in P1. However, P1’s discontinuous bowel wound may impede absorption, so the exact benefit of this drug is currently unknown. Given that ORAS patients have been demonstrated to have an ISG signature (Tao et al., 2021) and the high success of JAK inhibitor treatment in other autoinflammatory diseases that have an ISG signature (Sanchez et al., 2018), it would be of interest to determine if Ruxolitinib could be used as a support therapy for all ORAS patients.
C129S OTULIN mutation allows for accumulation of M1-Ub chains on LUBAC
The data above establish that a heterozygous C129S mutation in OTULIN drives an ORAS-like phenotype. In contrast to other ORAS variants that lead to loss or substantial decrease in OTULIN protein levels, C129S OTULIN has comparable stability with WT OTULIN, and total OTULIN protein levels in patient fibroblasts are similar to the levels observed in HDs (Fig. 2, A and C; and Fig. S1 A). We observed similar levels of OTULIN pulled down in the TUBE assay in HEK cells transiently overexpressing both WT and C129S OTULIN (Fig. 2 F), implying that C129S OTULIN can bind to linear Ub chains; yet as Fig. S1 A shows, C129S OTULIN cannot cleave linear Ub chains. Thus, we hypothesized that the C129S mutant OTULIN is likely to be acting in a dominant negative manner to inhibit WT OTULIN function and driving inflammatory disease.
C129A OTULIN is the highest affinity M1–di-Ub receptor described to date with a binding constant of 112 nM (Keusekotten et al., 2013). Using Surface Plasmon Resonance (SPR), we confirmed C129S OTULIN similarly binds to M1–di-Ub with high affinity; indeed, C129S affinity for M1-Ub is slightly higher than C129A OTULIN (Fig. S1 D). Binding affinity of WT OTULIN cannot be assessed by SPR due to its catalytic activity. We hypothesized that C129S OTULIN was binding to M1-Ub chains in patient cells and protecting these chains from hydrolysis by WT OTULIN. To assess this, we performed a competitive cleavage assay. Increasing concentrations of C129S OTULIN protein were incubated with M1–di-Ub prior to the addition of WT OTULIN. Yet, even at a 5:1 M ratio of OTULIN C129S to WT, all di-Ub was cleaved in this cell-free assay, demonstrating that C129S OTULIN does not directly protect M1-Ub chains from hydrolysis by WT OTULIN (Fig. 5 A). This data also indicates that OTULIN binding to M1-Ub chains is highly dynamic.
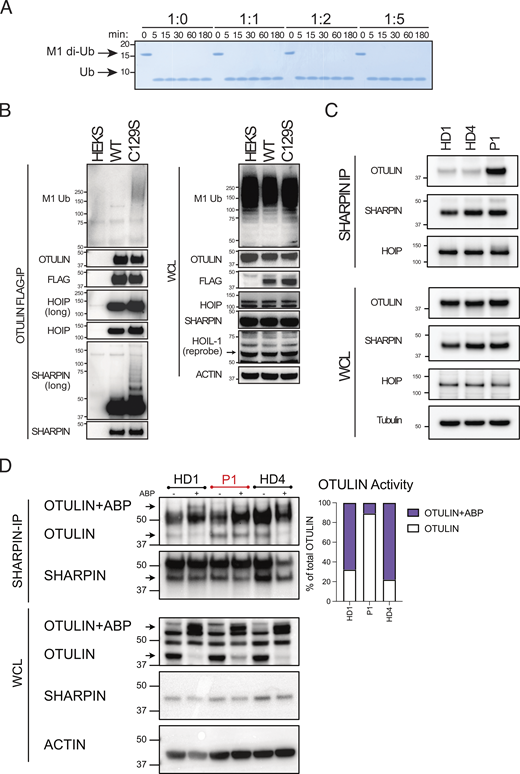
C129S OTULIN dominantly promotes LUBAC autoubiquitination. (A) Cleavage of M1-linked di-Ub was assessed in competitive settings. Increasing concentrations of C129S OTULIN were incubated with WT OTULIN M1-linked di-Ub for 0–180 min. (B) FLAG-tagged WT and C129S OTULIN were stably expressed in HEK 293T cells. Flag IP and WCL lysates from WT OTULIN, C129S OTULIN, and parental HEK 293T cells were immunoblotted for M1-Ub, OTULIN, FLAG, HOIP, SHARPIN, HOIL-1, and actin (loading control). (C and D) Endogenous SHARPIN IP was performed on two HDs and P1 fibroblasts. (C) SHARPIN IP and WCL lysates were immunoblotted for OTULIN, HOIP, SHAPRIN, and actin (loading control). (D) SHARPIN-IP and WCL lysates were treated with the OTULIN ABP probe to assess OTULIN activity when bound to LUBAC. OTULIN, SHARPIN, and actin (loading control) were assessed by immunoblot; arrows indicate unlabeled (OTULIN) and labeled (OTULIN+ABP) OTULIN, and this is quantified by densitometry. (A–D) Data is representative of three repeats and indicated molecular weight values in kD. Source data are available for this figure: SourceData F5.
C129S OTULIN dominantly promotes LUBAC autoubiquitination. (A) Cleavage of M1-linked di-Ub was assessed in competitive settings. Increasing concentrations of C129S OTULIN were incubated with WT OTULIN M1-linked di-Ub for 0–180 min. (B) FLAG-tagged WT and C129S OTULIN were stably expressed in HEK 293T cells. Flag IP and WCL lysates from WT OTULIN, C129S OTULIN, and parental HEK 293T cells were immunoblotted for M1-Ub, OTULIN, FLAG, HOIP, SHARPIN, HOIL-1, and actin (loading control). (C and D) Endogenous SHARPIN IP was performed on two HDs and P1 fibroblasts. (C) SHARPIN IP and WCL lysates were immunoblotted for OTULIN, HOIP, SHAPRIN, and actin (loading control). (D) SHARPIN-IP and WCL lysates were treated with the OTULIN ABP probe to assess OTULIN activity when bound to LUBAC. OTULIN, SHARPIN, and actin (loading control) were assessed by immunoblot; arrows indicate unlabeled (OTULIN) and labeled (OTULIN+ABP) OTULIN, and this is quantified by densitometry. (A–D) Data is representative of three repeats and indicated molecular weight values in kD. Source data are available for this figure: SourceData F5.
Though informative, results attained in these cell-free assays contrast with conditions in patient cells where M1-Ub chains do accumulate in the heterozygous 1:1 setting. To understand the mechanism driving accumulation of M1-Ub chains, we investigated some of the known OTULIN substrates including LUBAC. OTULIN binds to HOIP and thereby controls autoubiquitination of the LUBAC complex (Elliott et al., 2014; Schaeffer et al., 2014; Kelsall et al., 2019; Fuseya et al., 2020). In cells derived from C129A OTULIN mutant mice, ubiquitinated LUBAC components are readily observed (Heger et al., 2018). To test whether C129S OTULIN also facilitates ubiquitination of LUBAC, even in the presence of WT OTULIN, we stably expressed FLAG-tagged WT or C129S OTULIN in HEK 293T cells. Immunoprecipitation (IP) of FLAG-tagged OTULIN revealed that SHARPIN and, to a lesser extent, HOIP immunoprecipitated with C129S OTULIN were more heavily ubiquitinated compared with the HOIP and SHARPIN pulled down by WT OTULIN (Fig. 5 B). Additionally, the overall amount of M1-Ub chains immunoprecipitated by C129S OTULIN was elevated compared with WT OTULIN (Fig. 5 B). Thus, in cells, C129S OTULIN can bind to LUBAC and facilitate autoubiquitination of the complex as well as driving the accumulation of M1-Ub chains on other substrates. Notably, in this system of stable overexpression of mutant C129S OTULIN, endogenous WT OTULIN is unable to deubiquitinate SHARPIN.
Due to variability of protein expression, over-expression of C129S OTULIN in HEK 293T cells does not entirely recapitulate the heterozygous setting. We, therefore, assessed OTULIN binding to the LUBAC complex in patient and HD fibroblasts by IP of endogenous SHARPIN. We observed a marked increased interaction of OTULIN and LUBAC in patient fibroblasts compared with HD (Fig. 5 C, repeats quantified in Fig. S1 E). Furthermore, using the OTULIN ABP to determine catalytic activity, we confirmed that in C129S OTULIN patient cells, the majority of OTULIN bound to the LUBAC complex is catalytically inactive (Fig. 5 D). Thus, C129S OTULIN heterozygosity leads to accumulation of M1-Ub chains on LUBAC complexes, which reduces LUBAC recruitment to the TNFR1 signaling complex. Decreased LUBAC recruitment promotes TNF-induced cell death, which contributes to the key feature of the ORAS-like disease, necrotizing tissue.
Conclusion
We have demonstrated the mechanism by which a heterozygous variant in OTULIN can act in a dominant negative manner to cause an ORAS-like disease. Considering the recently published phenotype of OTULIN-haploinsufficient patients, it is surprising that these patients suffer from an autoinflammatory disease. The C129S OTULIN heterozygous patient phenotype is likely due to the unique mutation in the catalytic domain of OTULIN, which changes OTULIN into an inactive, M1-linkage specific Ub receptor without affecting its stability, cellular levels, or the OTULIN/LUBAC interaction. Thus, the compensatory mechanisms that limit inflammation in ORAS patients, specifically the downregulation of LUBAC components in fibroblasts and lymphocytes, do not occur in these patients. It is interesting to note that individuals with a heterozygous N341D OTULIN mutation do not present with an ORAS-like disease, instead exhibit OTULIN haploinsufficiency-related complications or no phenotype (Spaan et al., 2022). N341 is part of the OTULIN catalytic triad, but unlike C129S OTULIN, N341D OTULIN has decreased affinity to negatively charged proteins such as Ub (Keusekotten et al., 2013). It is likely that this decreased affinity results in a loss-of-function OTULIN rather than a dominant negative OTULIN, as observed with the C129S OTULIN mutation.
Study of our C129S heterozygous patients has also increased our understanding of the spectrum of the ORAS phenotype (Table S2), particularly in terms of bacterial invasion. Decreased or complete loss of OTULIN renders individuals at higher risk of severe necrotizing disease upon bacterial infection (Spaan et al., 2022). The increased sensitivity to TNF-induced cell death and increased inflammatory gene transcription in C129S OTULIN patient cells, as well as the transfer of these features into OTULIN-sufficient cell lines by overexpression of the C129S OTULIN mutant (Fig. 4 B) demonstrates that the necrotizing wounds exhibited by these patients may be enhanced by microbes, and addition of antibiotics to the treatment regime may prove beneficial, particularly in the context of treatment with therapeutics which block the key antimicrobial cytokine, TNF.
This is the first report of ORAS-like disease caused by a heterozygous variant. Importantly, understanding the disease-causing effects of this mutation resulted in patient diagnosis and effective, lifesaving treatment. Our study demonstrates the importance of understanding the functional implications of a variant for clinical diagnosis and expands our understanding of ORAS and OTULIN biology.
Materials and methods
Human subjects
Informed consent was obtained from each patient, in accordance with local regulations, and a protocol for research on human subjects was approved by the institutional review boards (IRB) of Walter and Eliza Hall Institute of Medical Research (WEHI), Human ethics number: HREC ID 18/07. All participants, or their legal representatives, consented to take part in this study and to have the results of this research published.
Patient cells
Patient PBMC samples and fibroblast lines were generated from a peripheral blood sample and skin biopsy from the patient at Sydney Children’s Hospital after obtaining consent HREC/15/MonH/31 and HREC/15/SCHN/346. The HD1, OTULINL272P/L272P, and OTULING174DfsX2/G174DfsX2 fibroblast lines (Zhou et al., 2016) were generated from skin biopsies taken at the National Institutes of Health (NIH), NIH Institutional Review Board–approved natural human history study (https://www.clinicaltrials.gov/ct2/show/NCT02974595) NIH #17-I-0016 “Studies of the natural history, pathogenesis, and outcome of autoinflammatory diseases.” HD2 and HD3 fibroblast lines were purchased from the American Type Culture Collection and Lonza, respectively. HD4 and all OTULIN-haploinsufficent lines (Spaan et al., 2022) were generated as per IRBs of Institut National de la Santé et de la Recherche Médicale (protocol C10-16) and The Rockefeller University (protocols JCA-0698 and JCA-0695). HD5 and HD6 PBMC samples were generated from blood draws conducted on adults participating in the Volunteer Blood Donor Registry at WEHI, Human ethics number: HREC ID 18/07. PBMCs were isolated from whole blood using Ficoll (GE Healthcare), frozen in fetal calf serum (FCS) (Sigma-Aldrich) with 10% DMSO (Sigma-Aldrich), and stored in liquid nitrogen (long term) or −80°C (short term) until use. Fibroblasts were cultured in 10% FCS Dulbecco’s modified Eagle’s medium (DMEM) (Gibco), 40 mM NaHCO3, penicillin (100 U/ml; Sigma-Aldrich), and streptomycin (100 μg/ml; Sigma-Aldrich) supplemented with 10% FCS at 37°C in a humidified atmosphere with 5% CO2.
Cell culture
Human THP-1 cells were obtained from the American Type Culture Collection (catalog no. TIB-202) and cultured in RPMI 1640 (prepared in-house, RPMI 1640 powder [Life Technologies], 23.8 mM sodium bicarbonate [NaHCO3] [Merck], 1 mM sodium pyruvate [C3H3NaO3] [Sigma-Aldrich], penicillin [100 U/ml; Sigma-Aldrich], and streptomycin [100 μg/ml; Sigma-Aldrich]) supplemented with 10% FCS. THP-1s were cultured at 37°C in a humidified atmosphere with 10% CO2. HEK 293T cells were maintained in DMEM, 40 mM NaHCO3, penicillin (100 U/ml; Sigma-Aldrich), and streptomycin (100 μg/ml; Sigma-Aldrich) supplemented with 10% FCS and cultured at 37°C in a humidified atmosphere with 5% CO2.
Tycho measurement for inflection temperature
The proteins were diluted to 1.0 μM in Tris buffer (20 mM Tris-HCl [pH 7.4], 150 mM NaCl, and 1 mM dithiothreitol [DTT]). Thermal shift assays were performed using Tycho NT.6 (NanoTemper Technologies). The inflection temperatures of each protein were calculated by the Tycho NT.6 software.
Real-time quantitative PCR (qPCR) for cell lines
5 × 106 PBMCs, 0.5–1 × 106 THP-1 cells, or confluent wells of fibroblasts were lysed using TRIzol reagent (Invitrogen) for 5 min at room temperature. Samples were either stored at −80°C or immediately processed. RNA extraction was performed as per the manufacturers’ instructions. cDNA was generated from 0.3 to 1 μg of total RNA using the Invitrogen SuperScript III First-Strand Synthesis System for reverse transcription PCR (Invitrogen). qPCR was performed using SYBR Green/ROX qPCR Master Mix (Thermo Fisher Scientific) on a ViiA 7 Real-Time PCR system (Thermo Fisher Scientific). Primers for genes of interest and housekeeping genes were used as described previously (Davidson et al., 2022) or listed in Table S3. Each sample was run in duplicate, and samples were normalized using the housekeeping gene ACTIN. Results were analyzed using the ΔΔCt method and represented as fold change of the average of HD samples from the same experiment.
Immunoblotting
0.3 × 105 fibroblasts were seeded in 6-well plates (Corning) and cultured for 18–24 h prior to lysis or stimulation for stated time points with 10 ng/ml TNF (kind gift from Prof. John Silke, WEHI, Parkville, Australia). Fibroblasts were lysed in 1× radioimmunoprecipitation assay buffer (1% Triton X-100, 20 mM tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 0.5% sodium deoxycholate, 3.5 mM SDS, 10% glycerol, 10 mM NaPPi, 5 mM NaF, and 1 mM Na3VO4) supplemented with 1 mM phenylmethylsulfonyl fluoride and cOmplete protease inhibitors (Roche Biochemicals) for 30 min at 4°C. Samples were processed through Pierce centrifuge columns (Thermo Fisher Scientific) to remove DNA. After addition of reducing SDS–polyacrylamide gel electrophoresis (SDS-PAGE) sample loading buffer (1.25% SDS, 12.5% glycerol, 62.5 mM tris-HCl [pH 6.8], 0.005% bromophenol blue, and 50 mM DTT) and denaturation at 95°C for 5–10 min, samples were separated on Novex 4–12% precast SDS-PAGE gels (Thermo Fisher Scientific) with MES running buffer (Thermo Fisher Scientific) and subsequently transferred onto polyvinylidene difluoride membrane (Millipore). Membranes were blocked in 5% skim milk in tris-buffered saline (TBS) containing 0.1% Tween 20 (Sigma-Aldrich) (TBST) before overnight incubation with specific primary antibodies in 5% bovine serum albumin (Sigma-Aldrich) or 5% skim milk in TBST at 4°C: anti-OTULIN raised in rabbit (#14127; Cell Signaling Technology [CST]) or anti-OTULIN raised in sheep (University of Dundee, DU43487), anti-SHARPIN (#4444; CST), anti-HOIP (#99633; CST), HOIL-1 (clone 2E2; Sigma-Aldrich), anti–M1 (#4560; CST), (p)p65 (#3033; CST), p65 (#4764S; CST), IkBα (#9242; CST), (p)p38 (#ab195049; Abcam), p38 (#ab31828; Abcam), (p)JNK (#4668; CST), JNK (#9252; CST), RIPK1 (#3493; CST), anti-Flag–horseradish peroxidase (HRP) (1:10,000 A8592; Sigma-Aldrich), or anti-actin–HRP (1:10,000, sc-47778; Santa Cruz Biotechnology). All listed primary antibodies were used at 1:1,000 unless otherwise stated. Membranes were washed three times with TBST and incubated with appropriate HRP-conjugated secondary antibodies and washed again three times. Finally, membranes were developed (Chemiluminescent HRP Substrate; Millipore) and imaged using the ChemiDoc Touch Imaging System (Bio-Rad) or film (GE Healthcare or Thermo Fisher Scientific). Band densitometry was assessed using ImageJ2, version 2.9.0/1.53t software.
Generation of recombinant OTULIN proteins
The full-length OTULIN construct (WT and C129A mutant) has been described previously (Keusekotten et al., 2013). OTULIN mutant (C129A and C129S) was generated in pOPINB by site-directed mutagenesis using In-Fusion HD Cloning system with inverse PCR. OTULIN constructs were expressed in Rosetta2 (DE3) pLacI cells. Cells were grown at 37°C in 2xYT medium with 50 μg/ml kanamycin and 34 μg/ml chroramphenicol to an OD600 of 0.6. The cultures were cooled to 18°C before overnight induction with 200 μM of IPTG. Cells were resuspended in lysis buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 2 mM β-mercaptoethanol, DNase I, lysozyme, and protease inhibitor cocktail) and lysed by sonication. Lysates were clarified by centrifugation at 19,500 rpm for 30 min and applied to cobalt resin for gravity flow purification. The His6 tag was cleaved overnight with His-3C protease during dialysis (20 mM Tris-HCl [pH 8.0], 50 mM NaCl, and 2 mM β-mercaptoethanol). Cleaved proteins were reapplied to cobalt resin, and flow-through was collected. The proteins were further purified by anion exchange chromatography (Resource Q; GE Healthcare). Eluted proteins were subjected to size exclusion chromatography (HiLoad 16/600 Superdex 75; GE Healthcare) in SEC buffer (20 mM Tris-HCl [pH 7.4], 150 mM NaCl, 4 mM DTT). Purified proteins were concentrated and flash-frozen prior to storage at −80°C.
Ub chain cleavage assay
Qualitative DUB assays were performed as previously described (Keusekotten et al., 2013). Briefly, OTULIN WT and mutants (C129A and C129S) were diluted to a 2× stock concentration in 20 mM Tris-HCl (pH 7.4), 150 mM NaCl, and 10 mM DTT, and incubated with 1 μM of M1–di-Ub in the buffer (50 mM Tris-HCl [pH 7.4], 50 mM NaCl, and 5 mM DTT) at 37°C. The samples were taken at different time points and mixed with 4× lithium dodecyl sulfate (LDS) sample buffer to stop the reaction. The samples were resolved by SDS-PAGE and visualized by Coomassie staining. For competitive cleavage assays, the indicated concentration of OTULIN (C129A) was preincubated with 5 μM of M1–di-Ub for 5 min on ice, and 0.5 nM of WT OTULIN was added to the reaction.
Assessment of OTULIN activity using ABP
Fibroblast samples were lysed in lysis buffer (25 mM HEPES, pH 7.5, 150 mM NaCl, 0.2% NP-40, 10% glycerol, 1 mM DTT) supplemented with PhosSTOP phosphatase inhibitor (Roche) and without protease inhibitor cocktail. Lysates were divided into aliquots (20–50 μl). 20 μg/ml or stated concentrations of OTULIN ABP were added to the samples and incubated at room temperature for 30 min. Reactions were stopped by boiling in reducing sample buffer and analyzed as per the Immunoblotting section. Band densitometry was assessed using ImageJ2, version 2.9.0/1.53t software.
TUBE pull-down
Endogenous polyUb conjugates were assessed in fibroblast lines using TUBE affinity reagents. Briefly, TUBE lysis buffer (20 mM Na2HPO4, 20 mM NaH2PO4, 1% NP-40, 2 mM EDTA) was supplemented with 1 mM DTT, cOmplete protease inhibitors (Roche Biochemicals), and 50 μg/ml of GST-TUBE1 (Lifesensors). HD and patient fibroblasts were plated in 15-cm2 dishes at 4 × 106 cells/dish and cultured for 48 h. HEK293T cells were transfected or transduced to express OTULIN or OTULIN mutants as described in later sections. Cells were lysed in 1 ml of ice-cold TUBE lysis buffer and processed as described previously (Hjerpe et al., 2009). After isolation, whole cell lysate (WCL) and TUBE pull-down samples were boiled in reducing sample buffer and analyzed as described in the Immunoblotting section.
Plasmid mutagenesis
We purchased a custom pLV plasmid expressing N terminal FLAG tagged OTULIN and mCherry reporter from Vector Builder. C129S, Y244C, and N341D OTULIN plasmids were obtained via site-directed mutagenesis of the pLV FLAG-OTULIN plasmid using the QuikChange Lightning Kit (Agilent Technologies). Mutagenesis primers were designed using the Agilent Primer Design Program and listed in Table S4. Primers were synthesized by Integrated DNA Technologies. Successful mutagenesis was confirmed via Sanger sequencing.
Transient transfection of HEK 293T cells
For Fig. 2 F, 4 × 106 HEK 293T cells were seeded into 15-cm2 dishes (Corning) and cultured overnight. The next morning 12 μg of plasmid C129S OTULIN, Y244C OTULIN (see Plasmid mutagenesis), WT OTULIN expressing plasmids, or an empty vector (EV) plasmid (VectorBuilder) DNA was transfected into HEK 293T cells using FugeneHD (Promega) diluted in OptiMEM (Thermo Fisher Scientific), according to manufacturer’s instructions (3 μl of FugeneHD/1 μg of DNA). 24 h later, transfection was removed and replaced with fresh 10% FCS DMEM. Transfection efficacy was confirmed by assessing mCherry fluorescence using ZOE Fluorescent Imager (Biorad). 24 h later (i.e., 48 h after transfection) cells were washed with PBS and lysed for TUBE pull-down.
Luciferase assay for NFκB activity
HEK 293T cells were plated in a 24-well plate (Corning), 1.5 × 105 cells per well, and left to adhere overnight. Cells were then transfected with 80 ng/well NFκB Luciferase reporter plasmid (pGL4.32[luc2P/NF-κB-RE/Hygro]) (#E8491; Promega) and 40 ng/well Renilla control (pGL4.74-hRluc/TK) (#E6921; Promega), as well as stated concentrations of plasmid C129S OTULIN, Y244C OTULIN, N341D OTULIN (see Plasmid mutagenesis), WT OTULIN expressing plasmids, or EV plasmid control (VectorBuilder). DNA was transfected into HEK 293T cells using FugeneHD diluted in OptiMEM according to the manufacturer’s instructions (3 μl of FugeneHD/1 μg of DNA). 18 h later, transfection media was removed and cells were collected by pipetting with 300 μl of PBS. 100 μl was replated into a 96-well white OptiPlate (PerkinElmer); cells were then centrifuged (1,500 rpm, 5 min, room temperature) and PBS was replaced with 10% FCS DMEM. Cells were rested for 5 h, after which transfection efficacy was confirmed by assessing mCherry fluorescence using ZOE Fluorescent Imager (Biorad). Upon confirmation of comparable transfection, efficacy media was removed and cells were processed as per Dual-Glo Luciferase Assay System (Promega) to assess luminescence of NFκB Luciferase activity and Renilla. For each sample, NFκB activity relative to Renilla was determined and then normalized to relevant EV control.
Complex I purification
6 × 106 fibroblasts were plated in 15-cm dishes and treated at indicated time points with 1 μg/ml Flag-TNF (kind gift from Rebecca Feltham, WEHI, Parkville, Australia). Following treatments, cells were lysed in 1 ml DISC lysis buffer (150 mM sodium chloride, 2 mM EDTA, 1% Triton X-100, 10% glycerol, 20 mM Tris, pH 7.5, Roche complete protease inhibitor cocktail, Roche phosSTOP phosphatase inhibitor) at 4°C for 30 min. The lysates were centrifuged at 15,000 × g for 10 min. Complex I was immunoprecipitated with 20 μl anti-Flag beads (Sigma-Aldrich) overnight at 4°C. The beads were washed four times with 1 ml DISC lysis buffer and eluted with 2× LDS buffer (NuPAGE; Invitrogen). Proteins were separated by SDS-PAGE (NuPAGE) and analyzed as per the Immunoblotting section.
Cytokine analysis
To measure secretion of IL-6 and IL-8 upon TNF stimulation, fibroblast cell lines were seeded at 5 × 103 cells per well in a 96-well flat-bottomed plate in DMEM-FCS 10% media. 18–24 h later, the cells were washed once with PBS and the culture media was replaced. Cells were treated with 10 ng/ml of TNF or vehicle control (ddH2O) at 4-, 8-, and 24-h time points. The plate was centrifuged at 1,500 rpm for 5 min at room temperature and supernatants were collected and stored at −20°C for ELISA. Concentrations of IL-6 and IL-8 were measured using the Human IL-6 Quantikine ELISA Kit and Human IL-8/CXCL8 Quantikine kits (R&D Systems) as per the manufacturer’s instructions.
Analysis of cell death
To measure cell death levels, fibroblast cell lines were seeded at 2 × 104 cells per well in a 48-well flat-bottomed plate. 18–24 h later, cells were treated with either one single treatment or one combination treatment. Single treatments were 100 ng/ml TNF, 50 μg/ml CHX, 20 μM QVD, 20 μM DMSO, and 10 μM Nec-1. Combination treatments were TNF+CHX, TNF+CHX+QVD, TNF+CHX+QVD+Nec-1, and TNF+CHX+Nec-1. Propidium iodide (PI) was added to cell media and cells were imaged using an Incucyte, every hour over 24 h. Cell death was graphed as PI Object Count/Phase Object Count (%).
ISG Score calculation
ISG Score was calculated from SYBR green qPCR results for the following ISGs: IRF7, ISG15, SIGLEC1, MX1, IFIT1, and RSAD2, as per Pescarmona et al. (2019). Expression of each ISG was calculated using the formula E−ΔCt then normalized to the geometric mean of ACTIN expression. For each ISG, individual samples were then normalized to the average of all HDs run in the experiment. Finally, the median of the relative expression of these six ISGs were used to calculate the final IFN score.
Stable expression of OTULIN and C129S OTULIN in cell lines
Third-generation lentiviral constructs (pLV) for WT OTULIN (VectorBuilder) and C129S OTULIN (see Plasmid mutagenesis) were used to generate lentivirus as described previously (Davidson et al., 2022). Briefly, 1.5 × 106 HEK 293T cells were seeded into a 10-cm2 dish (Corning); after confirmation of cell adherence, HEK 293T cells were transiently transfected with pLV OTULIN plasmids, pMDL (packaging), RSV-REV (packaging), and VSVg (envelope) using LipoFectMax (ABP biosciences), diluted in OptiMEM, to generate lentiviral particles. The cell culture supernatant was replaced 24 h later with appropriate media (i.e., 10% FCS RPMI for THP-1 cells or 10% FCS DMEM for HEK 293T cells). Supernatant was collected a further 24 h later and filtered through a 0.45-μm filter prior to transduction of cell lines. For transduction of THP-1 and HEK 293T cells, 0.5-3 × 106 cells were centrifuged with the lentivirus in the presence of polybrene (Sigma-Aldrich) at 839 × g for 3 h at 32°C and cultured at 37°C overnight. Cells were washed ×3 with media to remove all lentivirus. Effectively transduced cells were subsequently selected for by fluorescence-activated sorting of mCherry-positive cells to generate stable cell lines carrying FLAG-tagged OTULIN or FLAG-tagged C129S OTULIN.
SPR
All SPR binding assays were performed on a Biacore S200 (Cytiva) at 25°C. M1–di-Ub was immobilized by amine coupling to a single flow cell of a Series S CM5 sensor chip (Cytiva) in immobilization buffer (20 mM HEPES, pH 7.4, 150 mM NaCl). The chip surface was preconditioned with a 1:1 mix of 11.5 mg/ml N-hydroxysuccinimide and 75.0 mg/ml 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride. M1–di-Ub was diluted to 0.2 mg/ml in 10 mM sodium acetate, pH 4.5, and immobilized to around 800 response units. The first flow cell was left empty and used as the reference cell. All flow cells were then blocked with 1 M ethanolamine, pH 8.0. A dilution series of full-length OTULIN C129A and C129S was prepared in duplicate from a starting concentration of 100 μM in SPR running buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5 mM Tris (2-Carboxyethyl) phosphine hydrochloride, 0.005% Tween 20). Both OTULIN mutants were injected as analytes over the chip at 30 μl/min for 60 s followed by 120 s dissociation. The KD (dissociation constant) was calculated using the steady-state affinity model in the Biacore S200 Evaluation software 1.1 (Cytiva), and the resulting binding curves were plotted in Prism 10 (GraphPad Software).
IP
For endogenous SHARPIN IP, HD and patient fibroblasts were plated in 15-cm2 dishes at 4 × 106 cells/dish and cultured for 48 h. 5 μg of polyclonal SHARPIN antibody (14626-1-AP; Proteintech) was bound to 50 μl Dynabeads (Thermo Fisher Scientific) per sample by incubating for 2 h at 4°C on a rotator. Beads were washed ×3 with PBS and resuspended in 500 μl PBS. To crosslink antibody to beads, 5 mM of BS3 (Thermo Fisher Scientific) was added and incubated for 30 min at room temperature on a rotator. Crosslink reaction was halted by addition of 1 M Tris-HCl and beads were incubated for a further 15 min at room temp on rotator. Beads were washed ×3 with DISC buffer (20 mM Tris-HCl, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, and 10% Glycerol) supplemented with protease inhibitor cocktail tablet (Roche) and Phos-STOP phosphatase inhibitor tablet (Roche). Cells were lysed in DISC lysis buffer for 30 min on ice. Insoluble material was removed by centrifugation at 13,000 rpm at 4°C for 30 min. 50 μl of each sample was kept for WCL and processed as per immunoblot samples. SHARPIN antibody–conjugated beads were added per 1 ml of lysate and incubated at 4°C overnight on a rotator. Samples were washed 3× with DISC buffer and resuspended in 30 μl of sample buffer. Samples were then boiled for 5 min and subjected to immunoblotting (as per the Immunoblotting section). The following modifications to the endogenous SHARPIN IP protocol were made to assess the activity of LUBAC bound-OTULIN using the OTULIN ABP. Antibody/bead crosslinking was not performed. Cells were lysed in a modified DISC buffer (20 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 10% glycerol, and 1 mM DTT) supplemented with Phos-STOP phosphatase inhibitor tablet for 30 min on ice. Note, this IP must be performed without protease inhibitors for OTULIN ABP to bind efficiently. Insoluble material was removed by centrifugation at 13,000 rpm at 4°C for 30 min. Each sample was collected and split into two replicate tubes, one of which was treated with OTULIN ABP (10 μg/ml). 50 μl/sample was collected for WCL and processed as per immunoblot samples. For SHARPIN IP, 5 μg/ml of polyclonal SHARPIN antibody was added to each tube and incubated for 2 h at 4°C on a rotator. 50 μl Dynabeads/sample were washed in modified DISC buffer and added to the samples for a further 3 h. Samples were then washed with modified DISC buffer and resuspended in 30 μl of sample buffer. The next day, samples were boiled for 5 min and immunoblotting was performed. Band densitometry was assessed using ImageJ2, version 2.9.0/1.53t software.
Online supplemental material
Fig. S1 shows quantification of OTULIN and LUBAC protein levels and formation of the TNFR1 signaling complex in HD and P1 fibroblasts, as well as assessment of C129S OTULIN deubiquitinase activity and substrate binding capacity. Table S1 shows details hematological and immunological parameters of P2. Table S2 provides a pathophysiological comparison between ORAS, OTULIN haploinsufficiency, and OTULIN+/C129S P1 and P2. Table S3 lists OTULIN and LUBAC component qPCR primer sequences. Table S4 shows details of mutagenesis primers used to generate OTULIN variant plasmids.
Data availability
The data in the figures are available in the published article and the online supplemental material.
Acknowledgments
The authors thank Saskia Vadder (University of Bonn) and Daniel Simpson (WEHI) for technical assistance.
This work was supported by grants from the Australian National Health and Medical Research Council (2003159, 2003756; S.L. Masters, GNT1178122; D. Komander, 2017929; N. Lalaoui), fellowships from the Victorian Endowment for Science Knowledge and Innovation (S.L. Masters), the Howard Hughes Medical Institute-Wellcome International Research Scholarship (S.L. Masters), and the Sylvia and Charles Viertel Foundation (S.L. Masters). S. Davidson acknowledges funding from National Health and Medical Research Council (NHMRC) grants (GNT1143412 and GNT2003756). Y. Shibata acknowledges funding from NHMRC (GNT1182757) and the Naito Foundation.
Author contributions: E.P. Kirk and R. Walsh identified the variant in P1. C. Russell, A. van Beek, and M.W.Y. Li were involved in P1 treatment, and together with P.E. Gray, J.M. Alqanatish, A. Almojali, A. Alrasheed, and W. Alsuwairi were involved in identification of variant and treatment of P2. S. Davidson, Y. Shibata, S. Collard, H. Zheng, K. Kong, J.M. Sun, P. Laohamonthonku, A. Cerra, T. Kratina, and N. Lalaoui were involved in functional validation studies. S. Davidson, Y. Shibata, J. Alqanatish, A. Almojali, P.E. Gray, D. Komander, and S.L. Masters wrote the manuscript with input from all authors. P.E. Gray, D. Komander, and S.L. Masters were involved in the supervision and conception of the study together with S. Davidson and Y. Shibata.

