


Resistance to KRAS therapy in pancreatic ductal adenocarcinoma (PDAC) involves cellular plasticity, particularly the epithelial-to-mesenchymal transition (EMT), which poses challenges for effective targeting. Chronic pancreatitis, a known risk factor for PDAC, elevates TGFβ levels in the tumor microenvironment (TME), promoting resistance to KRAS therapy. Mechanistically, TGFβ induces the formation of a novel protein complex composed of SMAD3, SMAD4, and the nuclear factor NFAT5, triggering EMT and resistance by activating key mediators such as S100A4. Inhibiting NFAT5 attenuates pancreatitis-induced resistance to KRAS inhibition and extends mouse survival. Additionally, TGFβ stimulates PDAC cells to secrete CCL2, recruiting macrophages that contribute to KRAS bypass through paracrine S100A4. Our findings elucidate the role of TGFβ signaling in EMT-associated KRAS therapy resistance and identify NFAT5 as a druggable target. Targeting NFAT5 could disrupt this regulatory network, offering a potential avenue for preventing resistance in PDAC.

Introduction
Pancreatic ductal adenocarcinoma (PDAC) exhibits addiction to oncogenic KRAS (KRAS*) (Cancer Genome Atlas Research Network, 2017; Ying et al., 2012), with the quasi-mesenchymal (QM) subtype demonstrating the shortest overall survival, the highest epithelial-to-mesenchymal transition (EMT) gene signature, and the least dependency on KRAS signaling across classical and exocrine-like subtypes (Collisson et al., 2011). Despite the significant tumor growth suppression observed with KRAS inhibitors (KRASi) in preclinical models (Hallin et al., 2022; Wang et al., 2022), this antitumor effect is transient (Punekar et al., 2022), and EMT frequently emerges as a phenotype in resistant cells (Adachi et al., 2020; Hou et al., 2020; Hou and Wang, 2022; Peng et al., 2019; Shao et al., 2014). However, targeting TGFβ signaling and EMT is challenging due to the difficulties in developing drugs for transcription factors and the associated toxicities (Massague and Sheppard, 2023). Overcoming EMT-associated therapy resistance remains a primary objective.
Transforming growth factor-beta (TGFβ), a master driver of EMT (Xu et al., 2009), is abundant in the tumor microenvironment (TME), primarily sourced from cancer-associated fibroblasts and macrophages (Hou et al., 2020; Riesle et al., 1997). Chronic pancreatitis, a key risk factor for PDAC, induces fibrosis, recruits macrophages, and elevates TGFβ (Riesle et al., 1997; Slater et al., 1995). This study demonstrates that pancreatitis promotes resistance to KRAS*-targeted therapy through canonical TGFβ signaling, dependent on SMAD3 and SMAD4, but not SMAD2. Despite the direct DNA binding of SMAD3 and SMAD4, their binding strength is weak, necessitating cofactors for the activation of various gene targets (Hill, 2016). Notably, the nuclear factor of activated T cells 5 (NFAT5) is identified as an interactor of SMAD3 and SMAD4 and a critical mediator of TGFβ-driven KRAS* independency in PDAC.
Belonging to the Rel family, NFAT5 possesses a Rel-homology domain (RHD) for DNA binding (Lee et al., 2019). Functional and mechanistic studies reveal that the NFAT5–SMAD3/4 complex binds to the promoter of genes mainly involved in extracellular matrix (ECM) remodeling to activate their transcription. Among them, S100 Calcium Binding Protein A4 (S100A4) is a top target that supports the KRAS* bypass. Additionally, TGFβ pathway activation recruits S100A4-positive macrophages. Inhibition of NFAT5 suppresses S100A4 expression in both tumor cells and macrophages, preventing EMT-associated KRASi resistance and impairing escaper tumor maintenance.
Results
Chronic pancreatitis-induced resistance to KRASi hinges on TGFβ signaling
Pancreatitis is a key risk factor for PDAC (Gandhi et al., 2022; Kirkegard et al., 2018), resulting in TGFβ elevation in the microenvironment (Glaubitz et al., 2023; Ishihara et al., 1998). Our previous findings reveal that TGFβ drives KRAS* therapy resistance (Hou et al., 2020), prompting us to investigate whether chronic pancreatitis fosters the bypass of KRAS* dependency in PDAC. We utilized iKPC (p48-Cre, tetO_LKrasG12D, ROSA_rtTA, Trp53Lox/+) genetically engineered mice on a C57BL/6 background, in which KrasG12D expression was regulated by the tet-ON promoter and induced by doxycycline (dox) treatment (Ying et al., 2012). RNA profiling data comparing KRAS*-expressing (KRAS* on) and KRAS*-depleted (KRAS* off) tumors from iKPC mice revealed an elevation of inflammatory pathways and TGFβ signaling in KRAS* off tumors versus KRAS* on tumors (Fig. 1, A and B), suggesting that these factors may play a crucial role in KRAS* therapy resistance.

Pancreatitis drives KRAS* bypass. (A and B) GSEA analysis of RNA-seq data comparing KRAS* on and off tumors. PDAC cells from iKPC mice were orthotopically transplanted into C57BL/6 mice, followed by dox water administration to maintain KRAS* activation. After 1 wk, four mice continued receiving dox water (ON) while four were switched to normal water to deactivate KRAS* (OFF). 5 days later, tumors were collected for RNA-seq analysis. (A) Top deregulated gene sets in KRAS* OFF or ON tumors by GSEA analysis. (B) Enrichment plots of IFNα response and TGFβ signaling gene sets. (C) Experimental design for inducing chronic pancreatitis in spontaneous PDAC models using CAE at 100 μg/kg. (D) Kaplan–Meier survival analysis comparing mouse groups with KRAS* expression (on dox), KRAS* depletion (off dox), and KRAS* depletion plus CAE treatment (off dox + CAE). (E) Representative histological images illustrating the time-course analysis of malignant lesions and tumors during pancreatitis-induced tumor relapse. H&E, Mason’s Trichrome staining, and immunohistochemistry (IHC) were performed. (F) Quantification of IHC signal-positive cells using ImageJ. The percentage of relative area was calculated as 100*(positive cell area)/(total cell area). (G) Quantification of TGFβ IHC signal intensity using ImageJ. The optical density was calculated as log(max intensity/mean intensity), where the max intensity is 255 for 8-bit images. Log-rank (Mantel-Cox) test was used for D. Statistical analysis was performed using one-way ANOVA for F and G. The P values: ns, not significant; *, P < 0.05; **, P < 0.01, ***, P < 0.001; ****, P < 0.0001. Error bars represent the median ± SEM. All experimental data was verified in at least two independent experiments.
Pancreatitis drives KRAS* bypass. (A and B) GSEA analysis of RNA-seq data comparing KRAS* on and off tumors. PDAC cells from iKPC mice were orthotopically transplanted into C57BL/6 mice, followed by dox water administration to maintain KRAS* activation. After 1 wk, four mice continued receiving dox water (ON) while four were switched to normal water to deactivate KRAS* (OFF). 5 days later, tumors were collected for RNA-seq analysis. (A) Top deregulated gene sets in KRAS* OFF or ON tumors by GSEA analysis. (B) Enrichment plots of IFNα response and TGFβ signaling gene sets. (C) Experimental design for inducing chronic pancreatitis in spontaneous PDAC models using CAE at 100 μg/kg. (D) Kaplan–Meier survival analysis comparing mouse groups with KRAS* expression (on dox), KRAS* depletion (off dox), and KRAS* depletion plus CAE treatment (off dox + CAE). (E) Representative histological images illustrating the time-course analysis of malignant lesions and tumors during pancreatitis-induced tumor relapse. H&E, Mason’s Trichrome staining, and immunohistochemistry (IHC) were performed. (F) Quantification of IHC signal-positive cells using ImageJ. The percentage of relative area was calculated as 100*(positive cell area)/(total cell area). (G) Quantification of TGFβ IHC signal intensity using ImageJ. The optical density was calculated as log(max intensity/mean intensity), where the max intensity is 255 for 8-bit images. Log-rank (Mantel-Cox) test was used for D. Statistical analysis was performed using one-way ANOVA for F and G. The P values: ns, not significant; *, P < 0.05; **, P < 0.01, ***, P < 0.001; ****, P < 0.0001. Error bars represent the median ± SEM. All experimental data was verified in at least two independent experiments.
To investigate the role of pancreatitis in regulating KRAS* bypass, we chose a well-established method for inducing acute or chronic pancreatitis through repetitive injections of caerulein (CAE) (Ferreira et al., 2017; Komar et al., 2017). These iKPC mice were administered dox water starting at 4 wk of age to initiate tumorigenesis. Upon reaching a pancreatic tumor size of ∼1 cm in diameter, dox was discontinued to halt KRAS* expression. One week after dox withdrawal, mice received injections of either vehicle or CAE to induce chronic pancreatitis (Fig. 1 C). While control mice remained tumor-free, at least 36% (10 escapers out of 28 confirmed, 2 undetermined) of the CAE-treated mice developed escaper tumors less than or around a year (Fig. 1 D). These escaper tumors were undifferentiated, expressed high levels of TGFβ, and contained a significant number of M2-like macrophages as reflected by robust F4/80 and ARG1 expression (Fig. 1, E and F).
By histological analysis, we observed increased fibrosis in residual tumor lesions compared with KRAS*-expressing tumors at the onset of treatment regimens (Fig. 1 E). These lesions continued to decrease in size by day 14 after dox withdrawal. In contrast, the CAE-treated group displayed pancreatitis characterized by pancreatic damage with increased fibrosis, elevated TGFB1 expression, and enhanced infiltration of F4/80+ macrophages by day 14 (Fig. 1, E and F). Although we did not observe an increase in the number of TGFβ+ cells, we noted that pancreatitis induction significantly enhanced the TGFβ signal intensity, as reflected by the optical density analysis of IHC staining (Fig. 1 G). Moreover, despite low infiltration levels, we observed a significant increase in CD8+ T cells following KRAS* depletion (Fig. 1, E and F). However, the induction of pancreatitis reduced CD8+ T cell infiltration, and escaper tumors were found to be deprived of CD8+ T cells, similar to KRAS*-expressing primary tumors. In contrast, we did not identify significant changes in immune-suppressive myeloid cells and dendritic cells, as indicated by ARG1 and CD11c staining, respectively (Fig. 1, E and F).
To further corroborate these findings in an alternative genetic model, parallel pharmacological studies were conducted using orthotopically transplanted luciferase-expressing KPC (p48-Cre, lox-stop-lox KrasG12D, Trp53lox/+) (Bardeesy et al., 2006) PDAC cells in C57BL/6 mice. 1 wk after implantation, tumor-bearing mice received treatment with the KRASG12D inhibitor (G12Di) MRTX1133, CAE, and/or an anti-TGFβ neutralizing antibody (Fig. 2 A). Our findings reveal that inducing pancreatitis with CAE promotes KPC PDAC tumor growth compared with the vehicle control while neutralizing TGFβ suppresses this effect (Fig. 2, B–E). Although G12Di significantly reduces tumor growth, pancreatitis induction leads to resistance. However, the acceleration of tumor growth by CAE is mitigated by TGFβ blockade (Fig. 2, B–E). Notably, TGFβ neutralization alone has minimal impact on tumor growth, and we observed no additive or synergistic anti-tumor effects between KRAS inhibition and TGFβ neutralization in this KPC tumor model.

Pancreatitis promotes KRASi resistance through TGFβ. (A) Experimental design for inducing pancreatitis in orthotopically transplanted PDAC models. (B) Bioluminescence imaging (BLI) monitoring of tumor formation across different treatment groups: vehicle control + saline + IgG (V), vehicle control + caerulein (CAE, 100 μg/kg) + IgG (C), vehicle control + saline + α-TGFβ neutralizing antibody (250 μg per dose, T), vehicle control + CAE + α-TGFβ neutralizing antibody (CT), MRTX1133 (10 mg/kg, BID) + saline + IgG (M), MRTX1133 + CAE + IgG (MC), MRTX1133 + saline + α-TGFβ neutralizing antibody (MT), and MRTX1133 + CAE + α-TGFβ neutralizing antibody (MCT). (C) Images of collected tumors at humane endpoints. (D) Comparison of tumor weights. (E) Comparison of tumor volumes. (F and G) Histological analysis using H&E (F) and IHC (G) staining to characterize tumor morphology and TAMs. (H) Quantification of IHC signal-positive cells from G using ImageJ. Statistical analysis was performed using one-way ANOVA for D, E, and H. The P values: ns, not significant; *, P < 0.05; **, P < 0.01, ***, P < 0.001; ****, P < 0.0001. Error bars represent the median ± SEM. All experimental data was verified in at least two independent experiments.
Pancreatitis promotes KRASi resistance through TGFβ. (A) Experimental design for inducing pancreatitis in orthotopically transplanted PDAC models. (B) Bioluminescence imaging (BLI) monitoring of tumor formation across different treatment groups: vehicle control + saline + IgG (V), vehicle control + caerulein (CAE, 100 μg/kg) + IgG (C), vehicle control + saline + α-TGFβ neutralizing antibody (250 μg per dose, T), vehicle control + CAE + α-TGFβ neutralizing antibody (CT), MRTX1133 (10 mg/kg, BID) + saline + IgG (M), MRTX1133 + CAE + IgG (MC), MRTX1133 + saline + α-TGFβ neutralizing antibody (MT), and MRTX1133 + CAE + α-TGFβ neutralizing antibody (MCT). (C) Images of collected tumors at humane endpoints. (D) Comparison of tumor weights. (E) Comparison of tumor volumes. (F and G) Histological analysis using H&E (F) and IHC (G) staining to characterize tumor morphology and TAMs. (H) Quantification of IHC signal-positive cells from G using ImageJ. Statistical analysis was performed using one-way ANOVA for D, E, and H. The P values: ns, not significant; *, P < 0.05; **, P < 0.01, ***, P < 0.001; ****, P < 0.0001. Error bars represent the median ± SEM. All experimental data was verified in at least two independent experiments.
In another independent study, we observed similar results that chronic pancreatitis induction in the CAE;G12Di-treated group was associated with significantly faster tumor progression compared with vehicle;G12Di-treated controls; this CAE-driven tumor progression was partially attenuated with TGFβ blockade (Fig. S1, A–D). Western blot analysis of collected tumors revealed that CAE treatment induced SMAD3 phosphorylation and the upregulation of EMT transcription factors (TFs) such as SNAI2, ZEB1, and ZEB2 (Fig. S1 E), indicating TGFβ signaling activation. TGFβ blockade downregulated these markers (Fig. S1 E). In addition, by histological examination on days 7 and 21, we found that CAE treatment induced inflammation in the normal pancreas, with minimal impact on immune cell infiltration upon TGFβ blockage (Fig. S1 F). CAE;G12Di-treated tumors lost the ductal-like phenotype and were more undifferentiated relative to vehicle;G12Di controls (Fig. 2 F); TGFβ blockage restored a differentiated ductal-like phenotype and reduced the number of M2-like tumor-associated macrophages (TAMs) in the CAE;G12Di-treated tumors (Fig. 2, F–H). In summary, our data suggest that pancreatitis induction accelerates pancreatic cancer growth and contributes to KRAS* targeting resistance in a TGFβ-dependent manner.

TGFβ pathway activation is required for pancreatitis-induced KRASi resistance. (A) Experimental design for inducing pancreatitis in orthotopically transplanted PDAC models. (B) BLI monitoring tumor formation in comparison groups: MRTX1133 (10 mg/kg, BID) + saline + IgG (M), MRTX1133 + CAE (100 μg/kg) + IgG (MC), and MRTX1133 + CAE + α-TGFβ neutralizing antibody (250 μg per dose, MCT). Collected tumors are shown below. (C and D) Statistical comparison of tumor weight (C) and tumor volume (D) among the three experimental arms. (E) Western blot analysis of canonical TGFβ pathway activation status in PDAC tissues under different treatments. (F) H&E staining of mouse pancreas following various treatments. (G) IHC staining of TGFB1 in KRAS*-expressing tumors and KRAS*-depleted tumors for 5 days from iKPC mice. (H) Quantification of TGFβ signal intensity in G using ImageJ. OD, optical density. (I) Quantification of relative TGFβ-positive area in G using ImageJ. Statistical analysis for C, D, H, and I involved one-way ANOVA. The P values: ns, not significant; *, P < 0.05; **, P < 0.01, ***, P < 0.001; ****, P < 0.0001. Error bars represent the median ± SEM. All experimental data was verified in at least two independent experiments. Source data are available for this figure: SourceData FS1.
TGFβ pathway activation is required for pancreatitis-induced KRASi resistance. (A) Experimental design for inducing pancreatitis in orthotopically transplanted PDAC models. (B) BLI monitoring tumor formation in comparison groups: MRTX1133 (10 mg/kg, BID) + saline + IgG (M), MRTX1133 + CAE (100 μg/kg) + IgG (MC), and MRTX1133 + CAE + α-TGFβ neutralizing antibody (250 μg per dose, MCT). Collected tumors are shown below. (C and D) Statistical comparison of tumor weight (C) and tumor volume (D) among the three experimental arms. (E) Western blot analysis of canonical TGFβ pathway activation status in PDAC tissues under different treatments. (F) H&E staining of mouse pancreas following various treatments. (G) IHC staining of TGFB1 in KRAS*-expressing tumors and KRAS*-depleted tumors for 5 days from iKPC mice. (H) Quantification of TGFβ signal intensity in G using ImageJ. OD, optical density. (I) Quantification of relative TGFβ-positive area in G using ImageJ. Statistical analysis for C, D, H, and I involved one-way ANOVA. The P values: ns, not significant; *, P < 0.05; **, P < 0.01, ***, P < 0.001; ****, P < 0.0001. Error bars represent the median ± SEM. All experimental data was verified in at least two independent experiments. Source data are available for this figure: SourceData FS1.
Cdkn2b hinders canonical TGFβ pathway driven KRAS* bypass
The TGFβ signaling pathway exhibits multifunctionality (Massague, 2012), capable of activating target genes through both SMADs-dependent (canonical) and -independent (non-canonical) mechanisms (Fabregat and Caballero-Diaz, 2018). R-SMADs, such as SMAD2 and SMAD3, undergo phosphorylation by TGFβ receptors (TGFβR) upon ligand binding, forming heterotrimers with SMAD4 to regulate gene expression. The non-canonical TGFβ pathway involves the activation of other signaling pathways, including MAPK, JNK/p38 MAPK, PI3K/Akt cascades, and Rho-like GTPases (Zhang, 2009). Despite the genetic inactivation of SMAD4 in approximately one-third of PDAC cases, our analysis of the TCGA human PDAC dataset indicates a decreased frequency of genetic alterations of SMAD4 in poorly differentiated and undifferentiated tumor subtypes (Fig. 3 A), suggesting a potential requirement for an active canonical TGFβ pathway in these EMT-associated aggressive PDAC subtypes.

TGFβ signaling determines PDAC sensitivity to KRAS* targeted therapy. (A) Genetic mutation rates of KRAS, TP53, and SMAD4 in PDAC subtypes, including well, moderately, and poorly differentiated (diff’d), and undifferentiated (undiff’d) subtypes. The QCMG PDAC dataset from cBioPortal was used for the study. (B) Comparison of cancer spheroid formation using three distinct iKPC PDAC cells. (C) The inability of TGFβ to induce KRAS*-independent cancer spheroid formation in some iKPC cell lines. (D) Assessment of Cdkn2a and Cdkn2b expression in various iKPC cell lines. (E) Determination of knockdown efficiency for Cdkn2a and Cdkn2b via qRT-PCR. (F and G) Comparative analysis of TGFβ-driven, KRAS*-independent cancer spheroid formation following the knockdown of Cdkn2a or Cdkn2b in two distinct KRAS* bypass-deficient iKPC cell lines. (H) Comparison of cancer spheroid formation from KPC PDAC cells upon treatment with G12Di MRTX1133 (0.3 μM), murine recombinant TGFβ (0.5 ng/ml), and TGFβRi SB505124 (3 μM). (I) Cancer spheroid formation from human PDAC MIA PaCa-2 cells upon treatment with KRASG12C inhibitor (G12Ci) ARS-1620 (7.5 μM), human recombinant TGFβ (0.5 ng/ml), and TGFβRi SB505124 (3 μM). (J) Cancer spheroid formation from human PDAC Panc 04.03 upon treatment with G12Di MRTX1133 (0.2 μM), human recombinant TGFβ (0.5 ng/ml), and TGFβRi SB505124 (3 μM). (K) Cancer spheroid formation from human PDAC AsPC-1 upon treatment with G12Di MRTX1133 (0.3 μM), human recombinant TGFβ (0.5 ng/ml), and TGFβRi SB505124 (1 μM). (L) Representative images of spheroids from KRAS*-independent escaper tumor cell lines under treatment with TGFβRi SB505124 (3 μM). The control E5 images in Fig. 3 L and Fig. 7 F were from the same experiment. Statistical analysis was performed using one-way ANOVA for B and E–K. The P values: ns, not significant; *, P < 0.05; **, P < 0.01, ***, P < 0.001; ****, P < 0.0001. Error bars represent the median ± SEM. All experimental data was verified in at least three independent experiments.
TGFβ signaling determines PDAC sensitivity to KRAS* targeted therapy. (A) Genetic mutation rates of KRAS, TP53, and SMAD4 in PDAC subtypes, including well, moderately, and poorly differentiated (diff’d), and undifferentiated (undiff’d) subtypes. The QCMG PDAC dataset from cBioPortal was used for the study. (B) Comparison of cancer spheroid formation using three distinct iKPC PDAC cells. (C) The inability of TGFβ to induce KRAS*-independent cancer spheroid formation in some iKPC cell lines. (D) Assessment of Cdkn2a and Cdkn2b expression in various iKPC cell lines. (E) Determination of knockdown efficiency for Cdkn2a and Cdkn2b via qRT-PCR. (F and G) Comparative analysis of TGFβ-driven, KRAS*-independent cancer spheroid formation following the knockdown of Cdkn2a or Cdkn2b in two distinct KRAS* bypass-deficient iKPC cell lines. (H) Comparison of cancer spheroid formation from KPC PDAC cells upon treatment with G12Di MRTX1133 (0.3 μM), murine recombinant TGFβ (0.5 ng/ml), and TGFβRi SB505124 (3 μM). (I) Cancer spheroid formation from human PDAC MIA PaCa-2 cells upon treatment with KRASG12C inhibitor (G12Ci) ARS-1620 (7.5 μM), human recombinant TGFβ (0.5 ng/ml), and TGFβRi SB505124 (3 μM). (J) Cancer spheroid formation from human PDAC Panc 04.03 upon treatment with G12Di MRTX1133 (0.2 μM), human recombinant TGFβ (0.5 ng/ml), and TGFβRi SB505124 (3 μM). (K) Cancer spheroid formation from human PDAC AsPC-1 upon treatment with G12Di MRTX1133 (0.3 μM), human recombinant TGFβ (0.5 ng/ml), and TGFβRi SB505124 (1 μM). (L) Representative images of spheroids from KRAS*-independent escaper tumor cell lines under treatment with TGFβRi SB505124 (3 μM). The control E5 images in Fig. 3 L and Fig. 7 F were from the same experiment. Statistical analysis was performed using one-way ANOVA for B and E–K. The P values: ns, not significant; *, P < 0.05; **, P < 0.01, ***, P < 0.001; ****, P < 0.0001. Error bars represent the median ± SEM. All experimental data was verified in at least three independent experiments.
TGFβ serves as a major inducer of EMT, a process often positively associated with resistance to targeted, chemo-, and immunotherapies in various cancers (Byers et al., 2013; Shibue and Weinberg, 2017; Zheng et al., 2015). TGFβ is highly expressed in PDAC tissues, regardless of KRAS* targeting (Fig. 1 E and Fig. S1, G–I). In line with our previous findings (Hou et al., 2020), we observed that TGFβ1 efficiently promoted several iKPC PDAC cell lines to bypass KRAS* in spheroid assays (Fig. 3 B). Notably, some iKPC cell lines (251, 276) were unable to bypass KRAS* under TGFβ1 treatment (Fig. 3 C). Gene expression analysis revealed higher expression of tumor suppressors cyclin-dependent kinase inhibitor 2a and 2b (Cdkn2a and Cdkn2b) in iKPC cell lines that failed to bypass KRAS* compared with those that could (Fig. 3 D). Consistent with previous reports indicating that TGFβ can stimulate CDKN2B expression via the SMAD4–SMAD2/3–FOXO complex to induce cell cycle arrest (Seoane et al., 2001; Thillainadesan et al., 2012), knockdown of Cdkn2b, rather than Cdkn2a, prevented TGFβ1-induced cell death and enabled PDAC cells to become KRAS*-independent (Fig. 3, E–G). Thus, we conclude that CDKN2B serves as a key barrier to TGFβ-driven KRAS* bypass.
To investigate the impact of TGFβ on the development of resistance to KRASi, we utilized a KPC PDAC cell line and three human KRAS* PDAC cell lines for spheroid formation analysis. TGFβ1 actively promoted the formation of cancer spheroids resistant to KRASi from both mouse and human PDAC cells with the intact TGFβ pathway (KPC [1860], Mia PaCa-2, and Panc 04.03, respectively, Fig. 3, H–J). Conversely, the combined inhibition of the TGFβ receptor (TGFβRi) with KRASi led to the eradication of cancer spheroids (Fig. 3, H–J). However, SMAD4-deficient human PDAC cells AsPC-1 did not exhibit a response to TGFβ1 or TGFβRi (Fig. 3 K), indicating the necessity of canonical TGFβ pathway activation for KRASi resistance. Furthermore, de novo generated KRAS*-independent escaper tumors from iKPC mice, which exhibit a hybrid or QM-like phenotype (Kapoor et al., 2014), displayed hypersensitivity to TGFβRi (Fig. 3 L). These observations underscore the indispensable role of TGFβ pathway activation in the survival of KRASi-resistant cells.
NFAT5 interacts with SMAD3 and SMAD4
To determine whether the canonical or non-canonical TGFβ pathway is crucial for the development of KRAS* independency, we conducted knockdown experiments targeting SMADs to block the canonical pathway (Fig. 4, A and B). The results indicated that canonical TGFβ pathway factors, Smad4 and Smad3, but not Smad2, were essential for KRAS* bypass in PDAC (Fig. 4 C). Notably, SMAD3 and SMAD4, along with their downstream EMT transcription factors, have been considered chemically undruggable. Attempts to target upstream elements of the TGFβ signaling cascade, such as TGFβRi, have led to cardiovascular toxicities and chronic inflammation (Huang et al., 2021). To address these challenges, we hypothesized that targeting specific interacting factor(s) of SMAD3 and SMAD4 essential for KRAS* bypass might enhance KRASi efficacy while minimizing adverse effects.
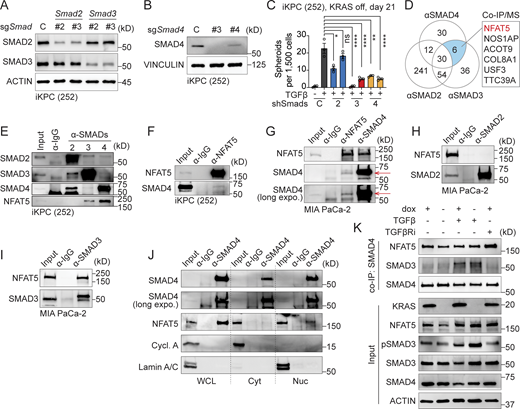
NFAT5 interacts with SMAD3 and SMAD4. (A and B) Western blot analysis to determine the knockdown efficacy of Smad2, Smad3, and Smad4. (C) Examination of TGFβ-driven, KRAS*-independent iKPC cancer spheroid formation after SMADs knockdown compared to the scramble control. (D) Venn diagram illustrating the IP/MS results. Endogenous SMAD2, SMAD3, and SMAD4 were used as baits to pull down proteins in iKPC PDAC cells, with an IgG antibody serving as the negative control. Positive hits are defined as those with an abundance ratio >10 compared with IgG. (E and F) Validation of protein interactions through co-IP/western blot analysis in mouse iKPC PDAC cells using SMADs and NFAT5 as baits. (G–I) Validation of NFAT5–SMADs protein interactions through co-IP/western blot analysis in human MIA PaCa-2 PDAC cells. (J) Cell fractionation followed by pulldown of NFAT5 using α-IgG or α-SMAD4 antibody. WCL: whole cell lysate; Cyt: cytosol fraction; Nuc: nuclear fraction. (K) Analysis of NFAT5 and SMADs interaction under different treatments by co-IP/western blots. All experimental data was verified in at least two independent experiments. Source data are available for this figure: SourceData F4.
NFAT5 interacts with SMAD3 and SMAD4. (A and B) Western blot analysis to determine the knockdown efficacy of Smad2, Smad3, and Smad4. (C) Examination of TGFβ-driven, KRAS*-independent iKPC cancer spheroid formation after SMADs knockdown compared to the scramble control. (D) Venn diagram illustrating the IP/MS results. Endogenous SMAD2, SMAD3, and SMAD4 were used as baits to pull down proteins in iKPC PDAC cells, with an IgG antibody serving as the negative control. Positive hits are defined as those with an abundance ratio >10 compared with IgG. (E and F) Validation of protein interactions through co-IP/western blot analysis in mouse iKPC PDAC cells using SMADs and NFAT5 as baits. (G–I) Validation of NFAT5–SMADs protein interactions through co-IP/western blot analysis in human MIA PaCa-2 PDAC cells. (J) Cell fractionation followed by pulldown of NFAT5 using α-IgG or α-SMAD4 antibody. WCL: whole cell lysate; Cyt: cytosol fraction; Nuc: nuclear fraction. (K) Analysis of NFAT5 and SMADs interaction under different treatments by co-IP/western blots. All experimental data was verified in at least two independent experiments. Source data are available for this figure: SourceData F4.
SMAD3 and SMAD4 exhibit weak DNA binding affinity through the MH1 domain (Hill, 2016). Instead, other TFs or transcriptional regulators may cooperatively bind with them or act as pioneer factors, facilitating chromatin opening and enabling SMADs to access their binding sites (Hill, 2016). This distinctive feature prompted us to explore chemically druggable SMAD3 and SMAD4 interactors that mediate TGFβ-driven KRAS* bypass. Through optimized co-immunoprecipitation (co-IP)/mass spectrometry (MS) analysis using endogenous SMAD proteins as baits, we successfully identified several TFs or transcriptional regulators. Among them, nuclear factor NFAT5 emerged as the sole interactor that was bound with both SMAD3 and SMAD4, excluding IgG and SMAD2 (Fig. 4 D). We validated protein interactions of endogenous SMAD3, SMAD4, and NFAT5 through co-IP/western blot, demonstrating the conservation of complex formation in both human and mouse PDAC cells (Fig. 4, E–I). Moreover, this protein–protein interaction was observed exclusively in the nucleus, not in the cytoplasm (Fig. 4 J), suggesting the potential involvement of DNA in the formation of the NFAT5–SMADs complex.
To demonstrate whether the formation of NFAT5–SMADs depends on KRAS* and TGFβ signaling pathways, we performed co-IP and western blot analysis to examine the interaction between NFAT5 and SMAD4 in iKPC cells following modulation of KRAS* or TGFβ signaling. The input control data indicate that both KRAS* and TGFβ signaling pathways upregulate NFAT5 expression, with KRAS* having a dominant regulatory effect compared with TGFβ (Fig. 4 K). Our co-IP data reveal that SMAD4 and NFAT5 interact constitutively, regardless of KRAS* or TGFβ signaling activation (Fig. 4 K). However, the interaction between SMAD3 and SMAD4 is TGFβ-dependent, and SMAD3 is required for TGFβ-driven KRAS* bypass (Fig. 4, C and K). In KRAS*-depleted cells, the enhanced interaction between NFAT5 and SMAD4 induced by TGFβ may be attributed to increased NFAT5 expression.
Therefore, the formation of the NFAT5–SMADs complex is TGFβ-dependent, supporting the biochemical changes observed with CAE treatment.
NFAT5 is upregulated in PDAC
In contrast to other members of the Rel family, NFAT5 is insensitive to calcium/calcineurin signaling, typically exists in a dimerized state, and does not synergize with FOS or JUN (Lopez-Rodriguez et al., 1999, 2001; Stroud et al., 2002). NFAT5 plays a regulatory role in ambient hypertonicity (Miyakawa et al., 1999) and is involved in the development and activation of immune cells (Lee et al., 2019). A tissue microarray (TMA) study revealed a positive correlation between NFAT5 expression and pancreatic tumorigenesis (Fig. 5, A and B). Correspondingly, elevated NFAT5 expression is associated with poor overall survival in the TCGA PDAC dataset (Fig. 5 C). Patient stratification indicates that patients with high NFAT5 expression have significantly shorter overall survival compared with those with low NFAT5 expression in the SMAD4 wildtype (SMAD4 wt) cohort (Fig. 5 D). However, in patients with SMAD4 mutations or deletions (SMAD4 mut/del), overall survival is similar regardless of NFAT5 expression levels (Fig. 5 E). These findings suggest that NFAT5 may play a key role in patients with intact SMAD4 by interacting with the canonical TGFβ pathway.

NFAT5 is upregulated in PDAC. (A) Human tissue microarray (TMA) analysis of NFAT5 during pancreatic disease progression. (B) Quantification of histological scores in chronic pancreatitis (CP), PanIN, and PDAC. (C–E) Kaplan-Meier survival analysis of PDAC patients with high or low NFAT5 expression in the TCGA PAAD dataset, including overall survival (OS) analysis in the entire cohort (C), the SMAD4 wildtype (wt) cohort (D), and the SMAD4 mutation or deletion (mut/del) cohort (E). (F) IHC staining of NFAT5 in spontaneous tumors from iKPC mice. (G) Quantification of nuclear NFAT5 staining signal intensity in F using ImageJ. (H) IHC staining of NFAT5 in tumor tissues collected from iKPC mice during pancreatitis-induced tumor relapse. (I) Quantification of nuclear NFAT5 staining signal intensity in H using ImageJ. (J) IHC staining of NFAT5 in transplanted tumors under treatments with KRASi, CAE, and α-TGFβ neutralizing antibody. (K) Quantification of nuclear NFAT5 staining signal intensity in J using ImageJ. One-way ANOVA was used for statistical analysis for G, I, and K; the Chi-square test was performed for B; the Log-rank (Mantel-Cox) test was used for C–E. The P values: ns, not significant; *, P < 0.05; **, P < 0.01, ***, P < 0.001; ****, P < 0.0001. Error bars represent the median ± SEM. All experimental data was verified in at least two independent experiments.
NFAT5 is upregulated in PDAC. (A) Human tissue microarray (TMA) analysis of NFAT5 during pancreatic disease progression. (B) Quantification of histological scores in chronic pancreatitis (CP), PanIN, and PDAC. (C–E) Kaplan-Meier survival analysis of PDAC patients with high or low NFAT5 expression in the TCGA PAAD dataset, including overall survival (OS) analysis in the entire cohort (C), the SMAD4 wildtype (wt) cohort (D), and the SMAD4 mutation or deletion (mut/del) cohort (E). (F) IHC staining of NFAT5 in spontaneous tumors from iKPC mice. (G) Quantification of nuclear NFAT5 staining signal intensity in F using ImageJ. (H) IHC staining of NFAT5 in tumor tissues collected from iKPC mice during pancreatitis-induced tumor relapse. (I) Quantification of nuclear NFAT5 staining signal intensity in H using ImageJ. (J) IHC staining of NFAT5 in transplanted tumors under treatments with KRASi, CAE, and α-TGFβ neutralizing antibody. (K) Quantification of nuclear NFAT5 staining signal intensity in J using ImageJ. One-way ANOVA was used for statistical analysis for G, I, and K; the Chi-square test was performed for B; the Log-rank (Mantel-Cox) test was used for C–E. The P values: ns, not significant; *, P < 0.05; **, P < 0.01, ***, P < 0.001; ****, P < 0.0001. Error bars represent the median ± SEM. All experimental data was verified in at least two independent experiments.
Histological analysis of mouse tumors revealed a significant increase of nuclear NFAT5 following KRAS* depletion (Fig. 5, F and G), particularly after chronic pancreatitis induction and in escaper tumors (Fig. 5, H and I). Neutralization of TGFβ suppressed nuclear NFAT5 expression (Fig. 5, J and K), suggesting NFAT5 as a potential downstream target of TGFβ. Additionally, de novo–generated iKPC escaper tumors, especially KRAS*-independent ones, exhibited a significant upregulation of Nfat5 expression compared with primary KRAS*-expressing tumors (Fig. 6 A). Subtype analysis indicated that Nfat5 expression is highest in QM-like escapers, followed by hybrid ones (Fig. 6 A). Collectively, our data point to Nfat5 as a potential regulator of EMT-associated KRAS* bypass.
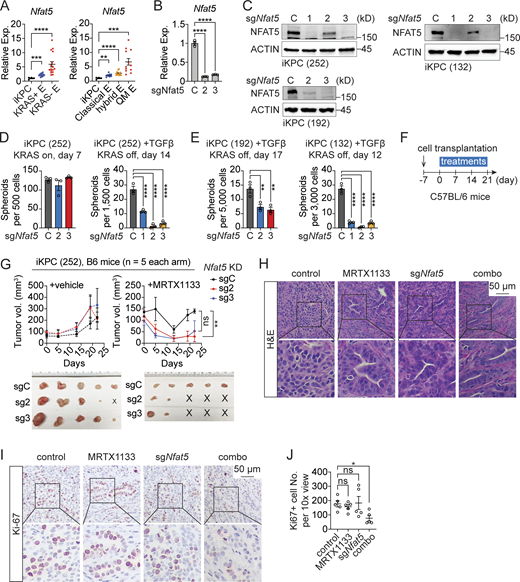
NFAT5 is essential for TGFβ-driven KRAS* bypass. (A)Nfat5 expression in primary KRAS*-expressing PDAC tumors (iKPC), KRAS*-reactivated escaper tumors (KRAS+ E), and KRAS*-independent escaper tumors (KRAS− E) from iKPC mice (left). The same dataset was reanalyzed to indicate Nfat5 expression in different subtypes of escaper tumors from iKPC mice (right), including classical, hybrid, and QM escapers. (B and C) Knockdown efficiency of Nfat5 in iKPC cells assessed by RT-PCR (B) and western blot (C) analysis. (D and E) Cancer spheroid formation assay comparing Nfat5 knockdown to the vehicle control in KRAS*-expressing iKPC PDAC cells and TGFβ-driven KRAS* bypass. Three different iKPC cell lines were used for the study. (F) Experimental design to assess the anti-tumor effect of Nfat5 knockdown in combination with G12Di in vivo. (G) Comparison of tumor growth between the scramble control and Nfat5 knockdown under treatment with G12Di MRTX1133 (10 mg/kg, QD) or vehicle control. Tumors were collected on day 21. (H) Tumor characterization by H&E staining. (I) Characterization of tumors from G by IHC staining. (J) Quantification of Ki67+ cell number per 10× view from I by ImageJ. One-way ANOVA was used for statistical analysis for A, B, D, E, and J; the unpaired, two-tailed t test was used for G at the time point of tumor collection. The P values: ns, not significant; *, P < 0.05; **, P < 0.01, ***, P < 0.001; ****, P < 0.0001. Error bars represent the median ± SEM. All experimental data was verified in at least two independent experiments. Source data are available for this figure: SourceData F6.
NFAT5 is essential for TGFβ-driven KRAS* bypass. (A)Nfat5 expression in primary KRAS*-expressing PDAC tumors (iKPC), KRAS*-reactivated escaper tumors (KRAS+ E), and KRAS*-independent escaper tumors (KRAS− E) from iKPC mice (left). The same dataset was reanalyzed to indicate Nfat5 expression in different subtypes of escaper tumors from iKPC mice (right), including classical, hybrid, and QM escapers. (B and C) Knockdown efficiency of Nfat5 in iKPC cells assessed by RT-PCR (B) and western blot (C) analysis. (D and E) Cancer spheroid formation assay comparing Nfat5 knockdown to the vehicle control in KRAS*-expressing iKPC PDAC cells and TGFβ-driven KRAS* bypass. Three different iKPC cell lines were used for the study. (F) Experimental design to assess the anti-tumor effect of Nfat5 knockdown in combination with G12Di in vivo. (G) Comparison of tumor growth between the scramble control and Nfat5 knockdown under treatment with G12Di MRTX1133 (10 mg/kg, QD) or vehicle control. Tumors were collected on day 21. (H) Tumor characterization by H&E staining. (I) Characterization of tumors from G by IHC staining. (J) Quantification of Ki67+ cell number per 10× view from I by ImageJ. One-way ANOVA was used for statistical analysis for A, B, D, E, and J; the unpaired, two-tailed t test was used for G at the time point of tumor collection. The P values: ns, not significant; *, P < 0.05; **, P < 0.01, ***, P < 0.001; ****, P < 0.0001. Error bars represent the median ± SEM. All experimental data was verified in at least two independent experiments. Source data are available for this figure: SourceData F6.
NFAT5 is essential for TGFβ driven KRAS* targeting resistance
To elucidate the role of NFAT5 in regulating tumor responses to KRAS* targeted therapy, we conducted Nfat5 knockdown in iKPC PDAC cells (Fig. 6, B and C). While proving dispensable for the growth of KRAS*-expressing cancer spheroids, Nfat5 emerged as an essential factor for TGFβ-driven, KRAS*-independent spheroid formation in three distinct iKPC PDAC cell lines (Fig. 6, D and E). Consistently, the knockdown of Nfat5 attenuated tumor growth in vivo under treatment with G12Di, in contrast to the vehicle control (Fig. 6, F and G). Compared with untreated tumors, Nfat5 knockdown induced tumor differentiation and a decrease in Ki67-positive cancer cells (Fig. 6, H–J), signifying the suppression of EMT and cancer cell proliferation.
NFAT5 can be inhibited by a small molecule compound, KRN2, which specifically disrupts the binding of NF-κB p65 to the NFAT5 promoter region (Han et al., 2017). We confirmed that KRN2 suppressed Nfat5 expression in PDAC cells in a dose-dependent manner, with >50% inhibition observed at concentrations >0.3 μM (Fig. 7 A). Consistent with genetic results, KRN2 demonstrated the following inhibitory effects: (1) impairment of TGFβ-driven KRAS* bypass (Fig. 7 B), (2) attenuation of KRASi persistence in cancer spheroid formation from KPC and Mia PaCa-2 cells (Fig. 7, C and D), excluding SMAD4-deficient cancer spheroid formation (Fig. 7 E), (3) suppression of QM-like or hybrid KRAS*-independent escaper spheroid growth as comparable to TGFβRi (Fig. 7, F and G), (4) inhibition of mouse KPC allograft tumor growth in combination with KRASi (Fig. 7, H–L), (5) reduction of human Mia PaCa-2 xenograft tumor growth cooperatively with KRASi (Fig. 7, M–O), (6) restriction of mouse escaper tumor growth in vivo (Fig. 7 P), and (7) blockage of chronic pancreatitis-induced KRASi resistance (Fig. 7, Q–S). KRN2 exhibited minimal systemic toxicities, reflected by stable mouse weights during treatment in both allograft and xenograft models (Fig. 7, K and N). Additionally, KRN2 induced mouse tumor differentiation compared to the vehicle control in the KPC transplanted model (Fig. 7 L). In contrast to Nfat5 knockdown (Fig. 6 G), KRN2 monotherapy prolonged mouse survival in immune-competent mice (Fig. 7 J), suggesting that NFAT5 may have non-cancer cell-autonomous functions in regulating tumor maintenance. Alternatively, other targets of KRN2 may also play a role in PDAC. Thus, NFAT5 forms a transcriptional regulatory complex with SMAD3 and SMAD4 to mediate TGFβ-driven KRAS* bypass and sustain EMT-associated escaper tumor growth.

NFAT5 inhibition mitigates KRAS* targeted therapy resistance. (A) Western blot analysis to determine the dose-dependent inhibition of NFAT5 expression by chemical compound KRN2. (B) Comparison of cancer spheroid formation under the treatment of different combinations of dox, TGFβ (0.5 ng/ml), and KRN2 (1 μM) in three distinct iKPC PDAC cell lines. (C) Cancer spheroid formation assay to assess the combination effect of G12Di MRTX1133 (0.03 μM) and KRN2 (1 μM) in KPC PDAC cells. (D) Cancer spheroid formation assay to determine the combination effect of G12Ci ARS-1620 (5 μM) and KRN2 (0.3 μM) in human PDAC MIA PaCa-2 cells. (E) Cancer spheroid formation assay to determine the combination effect of G12Di MRTX1133 (0.3 μM) and KRN2 (0.3 μM) in human PDAC AsPC-1 cells. (F) Comparison of cancer spheroid formation under treatment of DMSO control, TGFβRi SB505124 (3 μM), and KRN2 (1 μM) in three KRAS*-independent escaper tumor cell lines from iKPC mice without CAE treatment. The control E5 images in Fig. 3 L and Fig. 7 F were from the same experiment. (G) Comparison of cancer spheroid formation under treatment of DMSO control, TGFβRi SB505124 (3 μM), and KRN2 (1 μM) in three KRAS*-independent escaper tumor cell lines from CAE-treated iKPC mice. (H) Experimental design to evaluate the anti-tumor effect of KRN2 (3 mg/kg, QD) monotherapy and its combination with G12Di MRTX1133 (10 mg/kg, QD) in vivo. (I) BLI imaging to monitor tumor formation. (J) Kaplan–Meier survival analysis. OS, overall survival. (K) Measurement of mouse body weight along treatments. (L) Tumor characterization by H&E staining. (M) Schematic of the experimental design to assess the combined inhibition of KRAS and NFAT5 in the MIA PaCa-2 orthotopic xenograft model. Tumor-bearing mice were under treatment of vehicle control, G12Ci MRTX849 (100 mg/kg, QD), KRN2 (3 mg/kg, QD), and the combination (combo). (N) Measurement of mouse body weight along treatments. (O) Comparison of tumor weight and size on day 28. (P) Analysis of escaper tumor growth comparing treatment of vehicle control and KRN2 (3 mg/kg, QD). KRAS*-independent escaper tumor cells E725 were transplanted into nude mice subcutaneously. Tumors were collected and imaged on day 28. (Q) Comparison of tumor growth by BLI under treatments: MRTX1133 (10 mg/kg, BID) + saline + vehicle (M), MRTX1133 + CAE (100 μg/kg) + vehicle (MC), and MRTX1133 + CAE + KRN2 (3 mg/kg, QD, MCK). The KPC PDAC cells (1860) were orthotopically transplanted in immunocompetent mice. Tumors were collected on day 21. (R and S) Statistical comparison of tumor volume (R) and tumor weight (S) among the three experimental arms. One-way ANOVA was used for statistical analysis for B–G, R, and S; the unpaired, two-tailed t test was used for O and P at the time point of tumor collection. The P values: ns, not significant; *, P < 0.05; **, P < 0.01, ***, P < 0.001; ****, P < 0.0001. Error bars represent the median ± SEM. All experimental data was verified in at least two independent experiments. Source data are available for this figure: SourceData F7.
NFAT5 inhibition mitigates KRAS* targeted therapy resistance. (A) Western blot analysis to determine the dose-dependent inhibition of NFAT5 expression by chemical compound KRN2. (B) Comparison of cancer spheroid formation under the treatment of different combinations of dox, TGFβ (0.5 ng/ml), and KRN2 (1 μM) in three distinct iKPC PDAC cell lines. (C) Cancer spheroid formation assay to assess the combination effect of G12Di MRTX1133 (0.03 μM) and KRN2 (1 μM) in KPC PDAC cells. (D) Cancer spheroid formation assay to determine the combination effect of G12Ci ARS-1620 (5 μM) and KRN2 (0.3 μM) in human PDAC MIA PaCa-2 cells. (E) Cancer spheroid formation assay to determine the combination effect of G12Di MRTX1133 (0.3 μM) and KRN2 (0.3 μM) in human PDAC AsPC-1 cells. (F) Comparison of cancer spheroid formation under treatment of DMSO control, TGFβRi SB505124 (3 μM), and KRN2 (1 μM) in three KRAS*-independent escaper tumor cell lines from iKPC mice without CAE treatment. The control E5 images in Fig. 3 L and Fig. 7 F were from the same experiment. (G) Comparison of cancer spheroid formation under treatment of DMSO control, TGFβRi SB505124 (3 μM), and KRN2 (1 μM) in three KRAS*-independent escaper tumor cell lines from CAE-treated iKPC mice. (H) Experimental design to evaluate the anti-tumor effect of KRN2 (3 mg/kg, QD) monotherapy and its combination with G12Di MRTX1133 (10 mg/kg, QD) in vivo. (I) BLI imaging to monitor tumor formation. (J) Kaplan–Meier survival analysis. OS, overall survival. (K) Measurement of mouse body weight along treatments. (L) Tumor characterization by H&E staining. (M) Schematic of the experimental design to assess the combined inhibition of KRAS and NFAT5 in the MIA PaCa-2 orthotopic xenograft model. Tumor-bearing mice were under treatment of vehicle control, G12Ci MRTX849 (100 mg/kg, QD), KRN2 (3 mg/kg, QD), and the combination (combo). (N) Measurement of mouse body weight along treatments. (O) Comparison of tumor weight and size on day 28. (P) Analysis of escaper tumor growth comparing treatment of vehicle control and KRN2 (3 mg/kg, QD). KRAS*-independent escaper tumor cells E725 were transplanted into nude mice subcutaneously. Tumors were collected and imaged on day 28. (Q) Comparison of tumor growth by BLI under treatments: MRTX1133 (10 mg/kg, BID) + saline + vehicle (M), MRTX1133 + CAE (100 μg/kg) + vehicle (MC), and MRTX1133 + CAE + KRN2 (3 mg/kg, QD, MCK). The KPC PDAC cells (1860) were orthotopically transplanted in immunocompetent mice. Tumors were collected on day 21. (R and S) Statistical comparison of tumor volume (R) and tumor weight (S) among the three experimental arms. One-way ANOVA was used for statistical analysis for B–G, R, and S; the unpaired, two-tailed t test was used for O and P at the time point of tumor collection. The P values: ns, not significant; *, P < 0.05; **, P < 0.01, ***, P < 0.001; ****, P < 0.0001. Error bars represent the median ± SEM. All experimental data was verified in at least two independent experiments. Source data are available for this figure: SourceData F7.
NFAT5–SMADs complex binds to and transcriptionally activates S100a4
To unravel the regulatory role of the NFAT5–SMADs complex in KRAS* bypass, we conducted transcriptomic analysis in TGFβ-treated, KRAS*-depleted iKPC spheroids (Fig. 8 A). Notably, the knockdown of Nfat5 and Smad3 as well as the inhibition of NFAT5 by KRN2 resulted in the top downregulation of gene sets related to EMT and NFκB while upregulating genes associated with fatty acid metabolism and mTORC1 signaling (Fig. 8 B; and Fig. S2, A and B). The overlapping genes downregulated by Nfat5 and Smad3 knockdown were enriched in ECM and collagen biosynthesis pathways, while upregulated genes were enriched mainly in GTPase regulation and lipid metabolism (Fig. S2 C).
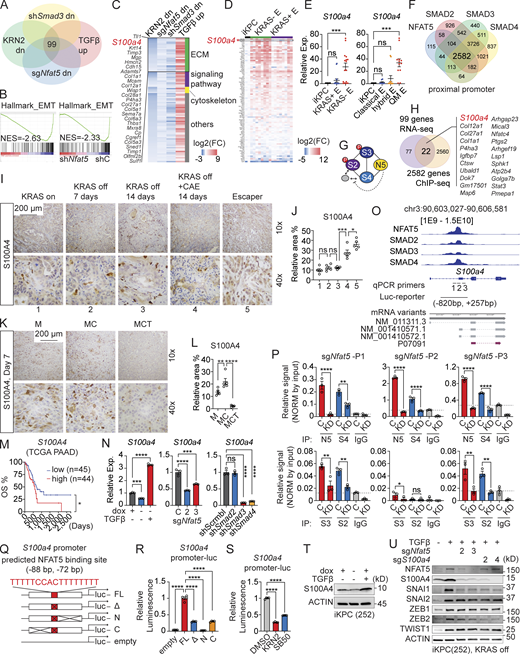
S100A4 is a direct target of the NFAT5–SMADs complex. (A) Summary of RNA-seq analysis to identify candidate targets of the NFAT5–SMADs complex. (B) GSEA analysis to identify the loss of the EMT gene signature after the inhibition or knockdown of NFAT5. (C) Intersection of RNA-seq datasets to identify 99 candidate genes potentially activated by the NFAT5–SMADs complex. (D) Expression profile of gene candidates in primary and escaper PDAC tumor cells from iKPC mice. (E) Comparison of S100a4 expression in primary and escaper PDAC tumors based on KRAS reactivation status (left) and tumor subtypes (right). (F) Summary of ChIP-seq data revealing genes with proximal promoters bound by NFAT5 and SMADs. (G) Schematic representation of the NFAT5–SMADs interaction. (H) Overlapping genes between the 99 candidates from RNA-seq and 2,582 genes from ChIP-seq. (I) IHC staining of S100A4 in tumors during pancreatitis-driven KRAS* bypass and escaper tumors. (J) Quantification of relative S100A4 signal-positive area in I using ImageJ. (K) IHC staining of S100A4 in transplanted tumors under treatments with KRASi, CAE, and α-TGFβ neutralizing antibody. (L) Quantification of relative S100A4 signal-positive area in K using ImageJ. (M) Kaplan-Meier survival analysis of PDAC patients based on high or low S100A4 expression in TCGA PAAD dataset. (N) Expression changes of S100a4 after treatments with dox or TGFβ, following knockdown of Nfat5 or Smad2/3/4. (O) Binding of NFAT5 and SMADs at the S100a4 promoter. (P) NFAT5–SMADs binding comparison at the S100a4 promoter in Nfat5 wildtype and knockdown iKPC cells. (Q and R) Comparison of luciferase activity driven by full length (FL) of or truncated S100a4 promoter. (S) Comparison of S100a4 activation under treatment of NFAT5i KRN2 (1 μM) or TGFβRi SB505124 (3 μM) by luciferase reporter assay under the control of the S100a4 promoter (FL). (T) Western blot analysis of S100A4 expression regulated by TGFβ in iKPC spheroids. (U) Western blot analysis of EMT TF expression after Nfat5 or S100a4 knockdown. Statistical analysis for E, J, L, N, P, R, and S involved one-way ANOVA; the Log-rank (Mantel-Cox) test was used for M. The P values: ns, not significant; *, P < 0.05; **, P < 0.01, ***, P < 0.001; ****, P < 0.0001. Error bars represent the median ± SEM. All experimental data was verified in at least two independent experiments. Source data are available for this figure: SourceData F8.
S100A4 is a direct target of the NFAT5–SMADs complex. (A) Summary of RNA-seq analysis to identify candidate targets of the NFAT5–SMADs complex. (B) GSEA analysis to identify the loss of the EMT gene signature after the inhibition or knockdown of NFAT5. (C) Intersection of RNA-seq datasets to identify 99 candidate genes potentially activated by the NFAT5–SMADs complex. (D) Expression profile of gene candidates in primary and escaper PDAC tumor cells from iKPC mice. (E) Comparison of S100a4 expression in primary and escaper PDAC tumors based on KRAS reactivation status (left) and tumor subtypes (right). (F) Summary of ChIP-seq data revealing genes with proximal promoters bound by NFAT5 and SMADs. (G) Schematic representation of the NFAT5–SMADs interaction. (H) Overlapping genes between the 99 candidates from RNA-seq and 2,582 genes from ChIP-seq. (I) IHC staining of S100A4 in tumors during pancreatitis-driven KRAS* bypass and escaper tumors. (J) Quantification of relative S100A4 signal-positive area in I using ImageJ. (K) IHC staining of S100A4 in transplanted tumors under treatments with KRASi, CAE, and α-TGFβ neutralizing antibody. (L) Quantification of relative S100A4 signal-positive area in K using ImageJ. (M) Kaplan-Meier survival analysis of PDAC patients based on high or low S100A4 expression in TCGA PAAD dataset. (N) Expression changes of S100a4 after treatments with dox or TGFβ, following knockdown of Nfat5 or Smad2/3/4. (O) Binding of NFAT5 and SMADs at the S100a4 promoter. (P) NFAT5–SMADs binding comparison at the S100a4 promoter in Nfat5 wildtype and knockdown iKPC cells. (Q and R) Comparison of luciferase activity driven by full length (FL) of or truncated S100a4 promoter. (S) Comparison of S100a4 activation under treatment of NFAT5i KRN2 (1 μM) or TGFβRi SB505124 (3 μM) by luciferase reporter assay under the control of the S100a4 promoter (FL). (T) Western blot analysis of S100A4 expression regulated by TGFβ in iKPC spheroids. (U) Western blot analysis of EMT TF expression after Nfat5 or S100a4 knockdown. Statistical analysis for E, J, L, N, P, R, and S involved one-way ANOVA; the Log-rank (Mantel-Cox) test was used for M. The P values: ns, not significant; *, P < 0.05; **, P < 0.01, ***, P < 0.001; ****, P < 0.0001. Error bars represent the median ± SEM. All experimental data was verified in at least two independent experiments. Source data are available for this figure: SourceData F8.

Discovery of S100a4 as a direct target of the NFAT5–SMADs complex. (A and B) GSEA to unveil deregulated gene sets by Smad3 and Nfat5 knockdown (A) and KRN2 (B). (C) GSEA to show the shared deregulated genes following Smad3 and Nfat5 knockdown. (D) Overview of DNA binding regions for NFAT5, SMAD2, SMAD3, and SMAD4 in the mouse genome as determined by ChIP-seq. (E) GSEA to indicate genes bound by the NFAT5–SMADs complex (left) and genes exclusively bound by SMADs (right).
Discovery of S100a4 as a direct target of the NFAT5–SMADs complex. (A and B) GSEA to unveil deregulated gene sets by Smad3 and Nfat5 knockdown (A) and KRN2 (B). (C) GSEA to show the shared deregulated genes following Smad3 and Nfat5 knockdown. (D) Overview of DNA binding regions for NFAT5, SMAD2, SMAD3, and SMAD4 in the mouse genome as determined by ChIP-seq. (E) GSEA to indicate genes bound by the NFAT5–SMADs complex (left) and genes exclusively bound by SMADs (right).
Further analysis, intersecting the three datasets with TGFβ-upregulated genes in KRASG12D-depleted iKPC cells, revealed 99 overlapping genes, with approximately one-third of them associated with the ECM (Fig. 8, A and C). S100 calcium-binding protein A4 (S100a4), a key regulator of ECM and EMT (Boye and Maelandsmo, 2010), emerged as one of the top genes upregulated by TGFβ but downregulated following interference with Nfat5 and Smad3 (Fig. 8 C). Comparative analysis with other candidate genes highlighted elevated expression of S100a4 in KRAS*-independent escaper tumors, particularly in the QM subtype (Fig. 8, D and E).
To identify DNA bound by the NFAT5–SMADs complex, we conducted Chromatin IP followed by next-generation sequencing (ChIP-seq) using antibodies binding to NFAT5, SMAD2, SMAD3, and SMAD4 (Fig. 8 F and Fig. S2 D). Our analysis revealed that 2,582 genes were bound by NFAT5 and SMAD2/3/4 at the proximal promoter (±1 kb, P < 0.01), while only 113 genes were bound by NFAT5 and SMAD3/4 (Fig. 8 F). Additionally, 3,726 genes were still bound by the SMAD2/3/4 complex. This data implies that NFAT5 may not interfere with the formation of heterotrimers of SMADs. Instead, the NFAT5-containing complex may block the interaction between SMAD2 and its partners, regulating a unique group of genes (Fig. 8 G). Accordingly, genes bound by the SMAD2/3/4–NFAT5 complex were enriched in oxidative phosphorylation and non-canonical NF-κB signaling, while genes bound by the SMAD2/3/4 complex were mainly enriched in cell adhesion and development (Fig. S2 E). Overlapping the 2,582 genes bound by the NFAT5–SMADs complex with the 99 genes transcriptionally upregulated by the NFAT5–SMADs complex identified 22 genes, most of which are ECM-related genes, including S100a4 (Fig. 8 H). Additionally, we predicted TF binding motifs using TFmotifView (Leporcq et al., 2020) and discovered several NFAT5 and SMADs binding sites in the human S100A4 and mouse S100a4 gene loci (Fig. S3 A).

The NFAT5–SMADs complex regulates canonical TGFβ pathway targets. (A) Predicted DNA binding motifs for NFAT5 and SMADs, with predicted binding sites on the S100a4 (S100A4) gene region in the mouse (human) genome. (B) Expression levels of EMT TFs following Nfat5 knockdown or inhibition, and the binding of NFAT5 and SMADs to the DNA regions of EMT TFs. (C) The binding of NFAT5 and SMADs at the Nfat5 promoter.
The NFAT5–SMADs complex regulates canonical TGFβ pathway targets. (A) Predicted DNA binding motifs for NFAT5 and SMADs, with predicted binding sites on the S100a4 (S100A4) gene region in the mouse (human) genome. (B) Expression levels of EMT TFs following Nfat5 knockdown or inhibition, and the binding of NFAT5 and SMADs to the DNA regions of EMT TFs. (C) The binding of NFAT5 and SMADs at the Nfat5 promoter.
S100A4 belongs to the S100 protein family and is localized in the cytoplasm, nuclei, and the ECM (Boye and Maelandsmo, 2010). Upon calcium binding, it undergoes a conformational change to recognize its target proteins. S100A4 plays a multifaceted role in cancers, particularly in PDAC, where it is reported to regulate tumor growth, metastasis, and angiogenesis via activating Src and focal adhesion kinase signaling pathways (Che et al., 2015). Our observations indicate that S100A4 is expressed in both cancer cells and stromal cells in PDAC, with elevated expression noted after pancreatitis induction and in escaper tumors compared with primary tumors (Fig. 8, I and J). The neutralization of TGFβ suppressed pancreatitis-induced S100A4 expression in the TME (Fig. 8, K and L), indicating the TGFβ–NFAT5 axis as the major upstream regulator of S100a4. Moreover, S100A4 emerges as a potential prognostic marker for human PDAC (Ai et al., 2008), with high expression positively correlated with poor patient survival (Fig. 8 M).
NFAT5 was previously identified as a downstream effector of integrin α6β4 signaling, activating S100A4 expression in breast and colon cancer cells (Chen et al., 2009, 2011). We revealed that S100a4 was upregulated by TGFβ in KRAS*-depleted iKPC spheroids, a process dependent on NFAT5 and SMAD3/4 but not SMAD2 (Fig. 8 N). Interestingly, KRAS* signaling also upregulated S100a4 expression (Fig. 8 N), which may be related to its function in driving EMT. ChIP-seq data revealed enriched binding peaks of NFAT5 and SMADs around the second exon of S100a4 (Fig. 8 O), where the transcription start site (TSS) of mRNA variants GenBank NM_001410571 and GenBank NM_001410572 is located. Through ChIP-qPCR analysis comparing Nfat5 knockdown to wildtype control, we demonstrated that the binding of the NFAT5–SMADs complex to the promoter region of S100a4 depended on NFAT5 (Fig. 8 P). The signal intensity of NFAT5 and SMAD4 was notably higher than that of SMAD3 and SMAD2, suggesting that the complex may bind DNA via NFAT5 and SMAD4.
Additionally, we utilized a luciferase reporter under the control of the S100a4 promoter to demonstrate that TGFβ activated S100a4 expression in KRAS*-depleted iKPC PDAC cells, while depletion of the predicted NFAT5 binding site in the S100a4 promoter completely diminished the activity of the luciferase reporter (Fig. 8, Q and R). Furthermore, chemical inhibition of NFAT5 or TGFβR suppressed S100a4 activation (Fig. 8 S).
We also validated the upregulation of S100A4 by TGFβ at the protein level in cancer spheroids (Fig. 8 T). Together, these data support the role of NFAT5 as a pioneer factor in priming SMADs to bind to the S100a4 promoter and activate its expression.
The canonical TGFβ signaling pathway activates EMT TFs at both transcriptional and posttranslational levels (Nieto et al., 2016). Despite not observing consistent transcriptional regulation of EMT TFs by Nfat5 or direct binding of NFAT5 onto their promoters (Fig. S3 B), we identified that knockdown of Nfat5 led to a decrease in the protein levels of SNAI1 and ZEB2 (Fig. 8 U). This suggests that NFAT5 may regulate their posttranslational modification or degradation. Furthermore, S100A4 protein expression was also downregulated after depleting Nfat5 (Fig. 8 U). Consistently, SNAI1 and ZEB2 among EMT TFs were downregulated after the knockdown of S100a4 (Fig. 8 U). These findings suggest that the TGFβ–NFAT5–S100A4 axis may regulate EMT through SNAI1 and ZEB2. Additionally, TGFβ upregulates NFAT5 (Fig. 8 U), and SMADs bind to the NFAT5 promoter (Fig. S3 C), providing further support for NFAT5 as a downstream target of canonical TGFβ signaling.
S100A4 mediates therapy resistance driven by the NFAT5–SMADs complex
To assess the essential role of S100A4 in TGFβ-driven KRAS* bypass, we conducted S100a4 knockdown in iKPC cells (Fig. 9 A). We observed the suppression of the MAPK and AKT pathways after S100a4 depletion (Fig. 9 B). These pathways are the main downstream signaling of KRAS and critical for cancer cell proliferation and survival (Hou and Wang, 2022; Punekar et al., 2022), indicating a potential mechanism by the TGFβ–NFAT5–S100A4 axis to induce KRAS* independency. However, the modest changes in these pathways suggest the presence of other regulators of the MAPK and AKT pathways, as well as additional effectors of S100A4 in PDAC that may contribute to therapy resistance.
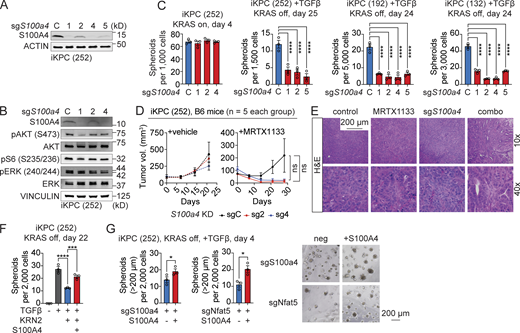
S100A4 is required for KRAS* bypass driven by the TGFβ–NFAT5 axis. (A) Knockdown efficiency of S100A4 in iKPC cells by western blot. (B) Examination of pathway activation after knockdown of S100a4 in iKPC cells by western blot. (C) TGFβ-driven, KRAS*-independent cancer spheroid formation comparison between S100a4 wildtype and knockdown in three distinct iKPC cell lines. (D) Tumor growth analysis of subcutaneously transplanted S100a4 wildtype and knockdown (KD) iKPC cells under treatment of vehicle control or G12Di MRTX1133 (10 mg/kg, QD). (E) Tumor characterization by H&E staining. (F and G) Rescue of TGFβ-driven, KRAS*-independent cancer spheroid formation by S100A4 after NFAT5 inhibition (KRN2, 1 μM) (F) and after knockdown of S100a4 and Nfat5 (G). Statistical analysis for C and F involved one-way ANOVA; the unpaired, two-tailed t test was used for G and for D at the time point of tumor collection. The P values: ns, not significant; *, P < 0.05; **, P < 0.01, ***, P < 0.001; ****, P < 0.0001. Error bars represent the median ± SEM. All experimental data was verified in at least two independent experiments. Source data are available for this figure: SourceData F9.
S100A4 is required for KRAS* bypass driven by the TGFβ–NFAT5 axis. (A) Knockdown efficiency of S100A4 in iKPC cells by western blot. (B) Examination of pathway activation after knockdown of S100a4 in iKPC cells by western blot. (C) TGFβ-driven, KRAS*-independent cancer spheroid formation comparison between S100a4 wildtype and knockdown in three distinct iKPC cell lines. (D) Tumor growth analysis of subcutaneously transplanted S100a4 wildtype and knockdown (KD) iKPC cells under treatment of vehicle control or G12Di MRTX1133 (10 mg/kg, QD). (E) Tumor characterization by H&E staining. (F and G) Rescue of TGFβ-driven, KRAS*-independent cancer spheroid formation by S100A4 after NFAT5 inhibition (KRN2, 1 μM) (F) and after knockdown of S100a4 and Nfat5 (G). Statistical analysis for C and F involved one-way ANOVA; the unpaired, two-tailed t test was used for G and for D at the time point of tumor collection. The P values: ns, not significant; *, P < 0.05; **, P < 0.01, ***, P < 0.001; ****, P < 0.0001. Error bars represent the median ± SEM. All experimental data was verified in at least two independent experiments. Source data are available for this figure: SourceData F9.
The results of functional assays demonstrated significant impairment in TGFβ-driven, KRAS*-independent cancer spheroid formation after S100a4 knockdown from three independent iKPC cell lines, while no suppression was observed in KRAS*-expressing cancer spheroids (Fig. 9 C). Furthermore, in vivo studies indicated that the loss of S100a4 significantly attenuated tumor growth and led to tumor differentiation in combination with G12Di treatment, particularly when compared with the vehicle control group (Fig. 9, D and E).
S100A4 is a chaperone protein that forms homodimers or heterodimers with its target proteins. These complexes either bind to specific cell surface receptors, activating signaling pathways in the ECM, or modulate the functions of their binding partners in the cytoplasm (Boye and Maelandsmo, 2010). We observed that the addition of recombinant S100A4 protein to the culture media rescued TGFβ-induced, KRAS*-independent cancer spheroid growth following NFAT5 inhibition and after the knockdown of Nfat5 or S100a4 (Fig. 9, F and G). The data indicate that signaling through extracellular S100A4 is required for TGFβ–NFAT5-driven KRAS* bypass, although intracellular S100A4 may also play a role in the process. Taken together, these findings strongly support the notion that S100A4 acts as a key downstream effector in the TGFβ–SMAD3/4–NFAT5 cascade.
Tumor-associated macrophages feed cancer cells by paracrine S100A4
S100A4 is expressed in various cell types, including fibroblasts, immune cells, and endothelial cells (Boye and Maelandsmo, 2010). We observed robust S100A4 expression in stromal cells of both primary and escaper PDAC tumors (Fig. 8 I), prompting a comprehensive examination of S100A4 expression in the TME. Utilizing single-cell RNA-sequencing (scRNA-seq) analysis on mouse autochthonous PDAC tumors from both KPC and iKPC models, we found that TAMs expressed significantly higher levels of S100a4 compared with other cell types, irrespective of KRAS* depletion or inhibition (Fig. 10 A). This observation was consistent with elevated S100a4 expression in bone marrow–derived macrophages (mBMDMs) from wildtype C57BL/6 mice compared with iKPC PDAC cells (Fig. 10 B). Further analysis revealed predominant expression of S100a4 in M0 and M2 polarized macrophages, with minimal expression in the M1 subtype (Fig. 10 C). Conditioned media derived from both KRAS*-expressing and -inhibited PDAC cells exhibited a similar effect on inducing S100a4 in macrophages (Fig. 10 C). Though TGFβ is also highly expressed in mBMDMs and tumor-educated macrophages (Fig. 10, B and C), the inhibition of NFAT5 suppressed S100a4 expression, while modulation of the TGFβ pathway by treating BMDMs with recombinant TGFβ or TGFβRi had no significant impact (Fig. 10 D). It highlights that the regulation of S100a4 in macrophages is NFAT5-dependent, TGFβ pathway-independent.

Macrophages promote KRAS* bypass by providing paracrine S100A4. (A) Single-cell RNA-seq analysis to reveal S100a4 expression in tumors collected from KPC and iKPC mice, treated with MRTX1133 (Ki, 10 mg/kg, BID) or with KRAS off for 5 days. (B) Differential expression of Tgfb1 and S100a4 in mBMDMs compared to iKPC cells. (C) Assessment of Tgfb1 and S100a4 expression in mBMDMs post-treatment with M0 inducer (M-CSF), M1 inducer (LPS + IFNγ), M2 inducer (IL-4), tumor-conditioned medium collected from KPC cells (CM), and tumor-conditioned medium collected from KRAS-inhibited KPC cells (+KRASi CM). (D)S100a4 expression in mBMDMs under treatment with TGFβ (0.5 ng/ml), NFAT5i KRN2 (1 μM), or TGFβRi SB505124 (3 μM). (E) IHC staining of F4/80 and S100A4 in transplanted tumors with wildtype or Nfat5 knockdown after MRTX1133 treatment. (F) Quantification of relative F4/80 signal-positive area in E using ImageJ. (G) Quantification of S100A4 high stroma cell number in E using ImageJ. (H) IHC staining of F4/80 in transplanted tumors post MRTX1133 and KRN2 treatment. (I) Quantification of relative F4/80 signal-positive area in H using ImageJ. (J) TGFβ-driven, KRAS*-independent cancer spheroid formation with or without co-culture of mBMDMs (Mφs, 30,000 cells/well) after S100a4 knockout. (K) KRAS*-independent cancer spheroid formation in co-culture with mBMDMs. (L) KRAS*-independent, Nfat5-knockdown cancer spheroid formation in co-culture with mBMDMs. (M) KRAS*-independent cancer spheroid formation in co-culture with mBMDMs under treatment of S100A4 and TGFβ neutralizing antibodies. The concentrations for IgG isotype control, α-S100A4 antibody and α-TGFβ antibody were 10, 5, and 10 μg/ml, respectively. (N) Overlapping genes between RNA-seq datasets and secretome database. (O) Expression changes of Ccl2 in iKPC cells post TGFβ treatment, after Smad2/3/4 knockdown, and Nfat5 knockdown. (P) NFAT5 and SMADs binding at the Ccl2 promoter. (Q)Ccl2 expression in primary and escaper PDAC tumors based on KRAS reactivation status (left) and tumor subtypes (right). Statistical analysis for C, D, F, G, I, K–M, O, and Q involved one-way ANOVA; the unpaired, two-tailed t test was used for B and J. The P values: ns, not significant; *, P < 0.05; **, P < 0.01, ***, P < 0.001; ****, P < 0.0001. Error bars represent the median ± SEM. All experimental data was verified in at least two independent experiments.
Macrophages promote KRAS* bypass by providing paracrine S100A4. (A) Single-cell RNA-seq analysis to reveal S100a4 expression in tumors collected from KPC and iKPC mice, treated with MRTX1133 (Ki, 10 mg/kg, BID) or with KRAS off for 5 days. (B) Differential expression of Tgfb1 and S100a4 in mBMDMs compared to iKPC cells. (C) Assessment of Tgfb1 and S100a4 expression in mBMDMs post-treatment with M0 inducer (M-CSF), M1 inducer (LPS + IFNγ), M2 inducer (IL-4), tumor-conditioned medium collected from KPC cells (CM), and tumor-conditioned medium collected from KRAS-inhibited KPC cells (+KRASi CM). (D)S100a4 expression in mBMDMs under treatment with TGFβ (0.5 ng/ml), NFAT5i KRN2 (1 μM), or TGFβRi SB505124 (3 μM). (E) IHC staining of F4/80 and S100A4 in transplanted tumors with wildtype or Nfat5 knockdown after MRTX1133 treatment. (F) Quantification of relative F4/80 signal-positive area in E using ImageJ. (G) Quantification of S100A4 high stroma cell number in E using ImageJ. (H) IHC staining of F4/80 in transplanted tumors post MRTX1133 and KRN2 treatment. (I) Quantification of relative F4/80 signal-positive area in H using ImageJ. (J) TGFβ-driven, KRAS*-independent cancer spheroid formation with or without co-culture of mBMDMs (Mφs, 30,000 cells/well) after S100a4 knockout. (K) KRAS*-independent cancer spheroid formation in co-culture with mBMDMs. (L) KRAS*-independent, Nfat5-knockdown cancer spheroid formation in co-culture with mBMDMs. (M) KRAS*-independent cancer spheroid formation in co-culture with mBMDMs under treatment of S100A4 and TGFβ neutralizing antibodies. The concentrations for IgG isotype control, α-S100A4 antibody and α-TGFβ antibody were 10, 5, and 10 μg/ml, respectively. (N) Overlapping genes between RNA-seq datasets and secretome database. (O) Expression changes of Ccl2 in iKPC cells post TGFβ treatment, after Smad2/3/4 knockdown, and Nfat5 knockdown. (P) NFAT5 and SMADs binding at the Ccl2 promoter. (Q)Ccl2 expression in primary and escaper PDAC tumors based on KRAS reactivation status (left) and tumor subtypes (right). Statistical analysis for C, D, F, G, I, K–M, O, and Q involved one-way ANOVA; the unpaired, two-tailed t test was used for B and J. The P values: ns, not significant; *, P < 0.05; **, P < 0.01, ***, P < 0.001; ****, P < 0.0001. Error bars represent the median ± SEM. All experimental data was verified in at least two independent experiments.
In Nfat5-knocked down tumors, we observed a reduction in S100A4 expression in tumor cells; however, abundant macrophages and S100A4-positive stroma cells were still present adjacent to tumor cells (Fig. 10, E–G). Additionally, dual inhibition of KRAS* and NFAT5 did not prevent macrophage infiltration (Fig. 10, H and I). We hypothesized that TAMs might promote KRAS* bypass via paracrine S100A4. To verify this, we co-cultured mBMDMs with TGFβ-treated, S100a4-deficient iKPC cancer spheroids following KRAS* ablation. As predicted, the co-culture rescued S100a4 knockdown (Fig. 10 J). Moreover, mBMDMs were sufficient to promote KRAS*-independent cancer spheroid growth without the supplement of TGFβ and rescue NFAT5 knockdown (Fig. 10, K and L). Considering that TAMs are a key source of TGFβ to support KRASi resistance (Hou et al., 2020), we countered S100A4 and TGFβ by neutralizing antibodies in the co-culture system of iKPC PDAC cells and mBMDMs. The observation revealed that the blockade of S100A4 or TGFβ significantly reduced the KRAS*-independent cancer spheroid formation (Fig. 10 M). Strikingly, the combination of S100A4 and TGFβ antibodies showed an additive effect that almost completely prevented the KRAS* bypass driven by macrophages (Fig. 10 M). We conclude that S100A4 may also have intracellular functions that cannot be blocked by S100A4 neutralization but can be compromised by the blockade of the paracrine TGFβ signaling, which inhibits cancer cell-intrinsic elevation of S100a4. Additionally, while the data suggest that environmental S100A4 from non-cancer cells can contribute to KRAS therapy resistance, S100A4 from cancer cells appears to play the dominant role, as evidenced by the strong tumor ablation following S100a4 knockdown (Fig. 9, C and D).
To elucidate the mechanism underlying the recruitment of macrophages by TGFβ-induced, KRAS*-independent PDAC cells, we analyzed the secretome database from ProteinAtlas and intersected it with our five RNA-seq datasets examining genes upregulated by TGFβ, SMAD3, and NFAT5, as well as genes elevated in escapers (Fig. 10 N). Ccl2 emerged as one of the 24 overlapped gene candidates, a well-established chemokine known to recruit CCR2-positive macrophages (Hou et al., 2020). While KRAS* depletion upregulated Ccl2 expression in iKPC PDAC cells by about 12-fold, TGFβ treatment dramatically elevated Ccl2 to >700-fold (Fig. 10 O). The upregulation of Ccl2 by TGFβ depended on SMAD3 and SMAD4, not SMAD2 or NFAT5 (Fig. 10 O). Correspondingly, we observed binding peaks of SMAD3 and SMAD4 in the promoter region of Ccl2 (Fig. 10 P). The expression of Ccl2 was significantly upregulated in KRAS*-independent escaper tumors, especially in the QM subtype, compared with KRAS*-expressing and -reactivated escaper tumors (Fig. 10 Q).
Discussion
Cancer cells exhibit heterogeneity and hyperplasticity, with EMT serving as a pivotal driver of metastasis and a common adaptive resistance mechanism to various cancer therapies. In this study, we unveil the molecular and cellular mechanisms through which TGFβ, a master regulator of EMT abundant in the TME, fosters resistance to KRAS* targeted therapy (Fig. S4). Our findings elucidate that the nuclear factor NFAT5 interacts with canonical TGFβ pathway key players—SMAD3 and SMAD4—to transcriptionally activate S100a4, a crucial factor for the development of KRAS* independence. While conventional Nfat5 knockout mice exhibit high perinatal lethality due to impaired renal and heart functions, the knockout of Nfat5 in adult mice has minimal impact on viability and fertility (Kuper et al., 2015). Importantly, we demonstrate that NFAT5 is chemically druggable, and mice tolerate the therapy well. Inhibiting NFAT5 not only prevents KRASi resistance but also hampers the growth of QM-like escaper tumors in preclinical models. These findings establish a molecular biological foundation for cellular plasticity-associated therapy resistance and propose a strategy to impede this process.

Schematic representation of intercellular crosstalk promoting KRAS* bypass.
The well-established association between chronic pancreatitis and PDAC is characterized by progressive inflammation and fibrosis (Zheng et al., 2013). Macrophages and TGFβ play pivotal roles in exacerbating this disease (Xue et al., 2015). Our study reveals that repetitive induction of pancreas injury, mimicking chronic pancreatitis, accelerates resistance to KRAS* targeted therapy in PDAC through the activation of the TGFβ pathway. TGFβ from the TME induces Ccl2 expression in cancer cells, initiating a positive feedback loop that further recruits TGFβ-positive macrophages, contributing to cancer cell non-autonomous resistance mechanisms. The upregulation of Ccl2 by TGFβ is dependent on SMAD3 and SMAD4 but independent of NFAT5. Consequently, NFAT5 inhibition alone cannot disrupt macrophage infiltration. Combining an NFAT5 inhibitor with therapies that either block macrophage infiltration (e.g., CCR2 inhibitor or anti-CSF1R antagonistic antibody) or re-polarize TAMs to stimulate anti-tumor immunity (e.g., anti-CD40 agonistic antibody) may offer a synergistic approach. Further investigation is warranted to determine whether NFAT5 also regulates immune cell responses.
The canonical TGFβ pathway exhibits a paradoxical role in cancer, restraining early tumorigenesis while facilitating disease progression and metastasis. SMAD4 is recognized as a prevalent tumor suppressor in PDAC, with its inactivation noted in ∼30% of patients, correlating with poorer overall survival rates. EMT is evident in SMAD4-deficient patient samples, suggesting a dual regulation of EMT by both canonical and non-canonical TGFβ pathways. Our study underscores the significance of NFAT5 in SMAD4-dependent EMT and KRASi resistance. However, the causal relationship between EMT TFs and KRASi resistance warrants further investigation using loss-of-function methods in genetically engineered mouse PDAC models. The upregulation of EMT TFs through SMAD4-independent mechanisms might override NFAT5 inhibition, thus fostering tumor relapse.
We observed that NFAT5 interacts with SMAD4 only in the nuclei, suggesting that DNA might be involved in this interaction. It has been reported that NFAT5 can form a homodimer to clamp DNA and stabilize the interaction (Stroud et al., 2002). Due to the low DNA-binding affinity of SMAD3 and SMAD4 (Hill, 2016), nuclear NFAT5 might serve as a crucial pioneer factor for the DNA binding of SMAD3 and SMAD4. Under hypertonic conditions, NFAT5 can be phosphorylated by kinases such as p38 MAPK, ERK, and CDK5, leading to its activation and translocation into the nucleus (Tong et al., 2006; Zhao et al., 2021). Additionally, methylation of NFAT5 at K668 by the EGFR-EZH2 axis in glioblastoma multiforme has recently been shown to be important for NFAT5 stability, activation, and nuclear accumulation (Li et al., 2023). Thus, the canonical TGFβ pathway and other signaling cascades, including KRAS signaling pathway and the non-canonical TGFβ pathway, may cooperatively regulate the downstream effectors of the NFAT5–SMADs complex via posttranslational modification.
Our mechanistic investigations uncover S100A4 as a pivotal downstream effector of the NFAT5–SMADs complex, orchestrating TGFβ-induced resistance to KRAS* targeted therapy. As a chaperone protein devoid of catalytic activity, S100A4 operates through interactions with binding partners. Intracellular S100A4 engages with proteins involved in cell migration, such as actin, while extracellular multimeric forms can bind to receptors like RAGE. The identification of factors interacting with S100A4 to mediate KRAS* targeted therapy resistance necessitates further exploration. Notably, we observed elevated expression of S100A4 in both cancer cells and TAMs. The ability of TAMs to drive KRAS* bypass and rescue the NFAT5–S100A4 axis knockdown underscores their significance as a major contributor to therapy resistance, providing paracrine TGFβ and S100A4.
Collectively, our findings offer insights into the molecular underpinnings of cellular plasticity-associated therapy resistance and present novel strategies to target this seemingly “undruggable” process. Moreover, our pre-clinical studies suggest that chronic pancreatitis may pose a potential risk factor for KRAS* targeted therapy resistance, warranting evaluation in ongoing clinical trials.
Materials and methods
Study design
The objectives of the research were to identify and validate NFAT5 as a therapeutic target to prevent TGFβ/EMT-driven KRAS* targeted therapy resistance in pancreatic cancer. PDAC mouse models were employed to demonstrate the sufficiency and necessity of TGFβ/EMT in regulating tumor responses to KRAS* targeting. By co-IP/MS studies, NFAT5 was identified to interact with SMAD3 and SMAD4 proteins. Subsequent spheroid assays and mouse studies revealed that the NFAT5–SMADs complex mediated TGFβ induced KRAS* targeted therapy resistance, and inhibition or depletion of NFAT5 prevented EMT and resistance. Followed by RNA profiling and ChIP sequencing, S100A4 was discovered as a downstream gene bound and transcriptionally activated by the NFAT5–SMADs complex. Functional assays in vitro and in vivo validated that S100A4 is essential for the TGFβ–NFAT5 axis to drive KRAS* bypass, maybe through reactivation of major KRAS*downstream pathways-MAPK and AKT. Single-cell RNA sequencing analysis revealed that tumor-associated macrophages expressed high S100a4. Co-culture assays suggested that macrophages supported KRAS* bypass by providing paracrine S100A4. At least five mice were randomly allocated to different treatment groups. Both mouse survival and tumor growth were analyzed depending on downstream applications. In vitro studies were performed and repeated at least three times in two or three distinct cell lines. At least three biological replicates were used.
PDAC mouse models
The iKPC and KPC PDAC mouse models were established and described previously (Bardeesy et al., 2006; Ying et al., 2012). They were bred in pure C57BL/6 background. Dox water (2 mg/ml, ad libitum) was administrated at 4–6 wk of mouse age to activate transgenic KRASG12D expression in iKPC mice. The C57BL/6 mice and nude mice of both sexes were purchased from the Jackson Laboratory and employed to ensure matching sexes of the cell lines.
Mouse experiments
All animal experiments were approved by Rutgers’ Institutional Animal Care and Use Committee (IACUC). All mice were maintained in pathogen-free conditions and received care in compliance with the regulations and certification of the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC International).
For the induction of spontaneous tumor relapse in iKPC mice, the withdrawal of dox water was performed to cease KRAS* expression when the tumor diameter reached ∼1 cm. Chronic pancreatitis was induced by intraperitoneal injection of caerulein starting on day 7 after KRAS* ablation, administered at a dose of 100 μg/kg, eight times a day (every hour) over two consecutive days every month. A saline solution (0.9% NaCl) was used to dissolve caerulein and served as the vehicle control.
For allograft or xenograft tumor models, human or mouse PDAC cells were transplanted orthotopically or subcutaneously into recipient C57BL/6 or nude mice at a concentration of 500,000 cells per injection, as specified in the figures. The cells were resuspended in Opti-MEM and combined with growth factor-reduced Matrigel (Corning) at a 1:1 ratio. Chronic pancreatitis induction in the transplanted tumor model involved intraperitoneal injection of caerulein starting from day 7 after tumor cell inoculation. The caerulein was administered at a dose of 100 μg/kg, eight times a day (every hour) over two consecutive days for the first week, followed by 100 μg/kg, three times a day (every hour) over three consecutive days for the subsequent weeks.
For bioluminescence imaging, each mouse received a 100 μl injection of D-Luciferin (15 mg/ml, i.p.; Perkin Elmer). After a 10-min interval, mice were imaged using the IVIS Spectrum Imaging System, and images were acquired and analyzed using Living Image 4.3 software.
The following reagents were employed for in vivo studies: MRTX1133 (10 mg/kg, i.p., BID; WuXi AppTec), KRN2 (3 mg/kg, i.p., QD; MCE), MRTX849 (100 mg/kg, oral, QD; MCE), and α-TGFβ neutralizing antibody (Clone: 1D11.16.8, 250 μg per mouse, i.p., twice per week; BioXCell) or IgG isotype control (Clone: MOPC-21; BioXCell).
PDAC cell lines and cancer spheroid culture
Human cell lines MIA PaCa-2 and Panc 04.03 were purchased from the American Type Culture Collection (ATCC) and cultured in DMEM supplemented with 10% FBS and RPMI supplemented with 15% FBS and 20 U/ml human recombinant insulin, respectively. Mouse PDAC cells were isolated from spontaneous tumors developed in iKPC or KPC mice. KPC cell lines were sustained in RPMI (Gibco) with 10% FBS (Gibco), while iKPC cell lines were cultured in RPMI with 10% Tet-approved FBS (Gibco) and doxycycline (1 μg/ml; VWR). Escaper tumor cell lines were maintained in RPMI with 10% Tet-approved FBS (Gibco). For cancer spheroid culture, cells were mixed with 50 μl growth factor-reduced Matrigel (Corning) and plated in 24-well low-attachment cell culture plates (Nunc). Medium was added on top of the solidified Matrigel. We monitored both spheroid size and number for spheroid growth assays. For counting spheroid numbers, we used a cut-off of 100 μm in diameter or specified, depending on the time and cell type. For measuring spheroid size, ImageJ was used. Monthly mycoplasma detection (Lonza) was performed to ensure the absence of contamination.
Reagents used for in vitro cell culture included ARS-1620 (MCE), MRTX849 (MCE), caerulein (MCE), MRTX1133 (WuXi AppTec), KRN2 (MCE), SB505124 (MCE), recombinant human and mouse TGFβ1 (Peprotech), recombinant mouse S100A4 (R&D), recombinant mouse M-CSF (Peprotech), α-TGFβ neutralizing antibody (BioXCell), α-S100A4 neutralizing antibody (R&D), and IgG isotype control (BioXCell). Dosage information is provided in the figure legends.
Plasmid information
All shRNAs targeting Smad2, Smad3, and Smad4 were purchased from Sigma-Aldrich. The sgRNAs targeting Cdkn2a, Cdkn2b, Nfat5, and S100a4 were cloned into the CRISPR/Cas9 All-in-One vector, and viruses were packaged using a second-generation lentiviral system. Puromycin (2–6 μg/ml) was employed to select guide RNA- or shRNA-infected cells. For BLI imaging, PDAC cells were infected with the luciferase-mCherry reporter vector. The primer sequences for cloning are provided in Table S1.
Luciferase reporter assay
Mouse S100a4 promoter was cloned into pGL4.12[luc2CP] vector (Promega). Renilla luciferase vector (Promega) was used as the internal control. pGL4.12 vector and Renilla luciferase vector were co-transfected into iKPC PDAC cells using Lipofectamine 2000 (Invitrogen). After 24 h, the medium was replaced, and cells were subjected to various treatments for an additional 24 h. Subsequently, cells were collected for luciferase reporter assay using the Dual-Luciferase Reporter Assay Kit (Promega) according to the manufacturer’s instructions. The firefly luciferase signal was normalized to Renilla luciferase prior to comparison among different treatment groups.
RNA extraction, qRT-PCR, mRNA sequencing, and GSEA
RNA extraction from 2-D or 3-D cultured cell samples was conducted using the RNA Extraction Kit (Qiagen). Matrigel-based 3-D cells were isolated using Cell Recovery Solution (Corning). RNA concentration was assessed using NanoDrop 2000. The RNA samples were either sent for RNA-seq analysis to the Genomic Center at Rutgers or Genewiz (Azenta) or reverse transcribed for qRT-PCR analysis. The preparation of cDNA utilized 5× All-In-One RT MasterMix (Applied Biological Materials) and the PCR reactions were prepared with SYBR Green PCR Master Mix (Applied Biosystems or Bio-Rad). qRT-PCR was executed on the CFX Opus 96 (Bio-Rad), with statistical analysis conducted using GraphPad Prism. For mRNA sequencing, the parameters were NGS-75 nt Paired End, utilizing the Illumina Next Generation Sequencing instrument.
In the case of scRNA-seq, tumors were dissociated into single cells using the Tumor Dissociation Kit from Miltenyi Biotec, followed by the removal of dead cells using the Dead Cell Removal Kit (Miltenyi Biotec). Cells were resuspended in PBS + 0.05% BSA at a concentration of 500–600 cells/μl for library preparation (10x Genomics). At least 100,000 cells were recovered per sample, ensuring a minimum of 20,000 reads per cell for NGS sequencing. NGS sequencing and bioinformatic analysis were conducted at the Genomic Center at Rutgers New Jersey Medical School. Raw reads were first subjected to barcode deconvolution and aligned to the mm10 reference genome using cellranger (v7.1.0). Subsequent data processing was conducted with the Seurat package (v4.3) in R. Cells deemed of low quality—defined by a percentage of reads of mitochondrial origin exceeding 10%, a percentage of reads of ribosomal origin surpassing 45%, <1,000 feature counts, or exceeding 7,000 feature counts—were filtered from the dataset. Correction for ambient nucleotides was executed utilizing SoupX (v1.6.2), and read counts underwent normalization employing the scTransform method as previously described (Hafemeister and Satija, 2019). Sample integration was accomplished using the Seurat integrate function (Stuart et al., 2019), and subsequent clustering via UMAP was performed based on nearest neighbors, using 40 principal components.
GSEA was performed using GSEA software (4.3.2). The qRT-PCR primer sequences are listed in Table S1.
Antibodies, western blot, IP, co-IP/MS, IHC, and Trichrome stain
Antibody information can be found in Table S2. Western blot analysis, IP, co-IP, and IHC staining followed standard protocols. Human TMA slides were purchased from Biomax. Cell fractionation and Masson’s Trichrome Staining were carried out using commercial kits, adhering to manufacturers’ protocols. MS analysis of proteins pulled down by endogenous SMAD2, SMAD3, SMAD4, NFAT5, and IgG was performed by the Proteomics Core Facility at Rutgers New Jersey Medical School, Specifically, IP samples were subjected to SDS-PAGE separation. Each sample’s gel lane was excised for in-gel trypsin digestion, and the resulting peptides were analyzed using LC-MS/MS on an Orbitrap Fusion Lumos Tribrid mass spectrometer coupled with the Ultimate 3000 nano-LC system (Thermo Fisher Scientific). MS/MS spectra were searched against the UniProt mouse database (55,336 sequences, downloaded 8/30/2021) using the Sequest search engine through the Proteome Discoverer platform (version 2.4) with a false discovery rate <1% for both proteins and peptides. Protein abundance ratios were determined using the Label-Free Quantitation (LFQ) method.
Human tumor tissues were evaluated using two criteria: the percentage of stained area (0% for no staining, 1–10% scored as 1, 11–50% as 2, 51–80% as 3, and 81–100% as 4) and the intensity of staining in the nuclei or cytoplasm (no staining as 0, weak staining as 1, moderate staining as 2, and strong staining as 3). The overall scores were determined by multiplying the assigned scores for the percentage of stained area and staining intensity.
Isolation and culture of bone marrow–derived macrophages
Mouse bone marrow–derived macrophages were isolated as previously described (Hou et al., 2020). Immature macrophages (M0) were induced by recombinant mouse M-CSF (20 ng/ml; BioLegend) for 7 days. Murine IFNγ (10 ng/ml; Peprotech) and LPS (100 ng/ml; Peprotech) were employed for M1 polarization, while murine IL-4 (20 ng/ml; Peprotech) was utilized for M2 polarization. Tumor conditional media were generated by adding fresh complete cell culture medium when tumor cells reached 70–80% confluence. After 24 h, the medium was collected, filtered through a 0.45 μm filter, and stored at −80°C. The working solution consisted of a 1:1 ratio of the collected medium mixed with fresh medium.
ChIP-seq and ChIP-qPCR
ChIP was conducted following the protocol of the SimpleChIP Plus Enzymatic Chromatin IP Kit (#9005; Cell Signaling Tech., Inc.). Briefly, cells were crosslinked with 1% paraformaldehyde and then quenched with 0.125 mol/l glycine. Subsequently, cells were lysed on ice for 30 min using lysis buffer. Chromatin DNA was fragmented to around 200–500 bp through Micrococcal Nuclease digestion, followed by sonication using a sonicator with a 102C probe (Branson Sonifier 450) for five cycles of 20 s on and 20 s off at an output of 15%. The lysate was then incubated overnight with anti-SMAD2, SMAD3, SMAD4, NFAT5, or IgG antibodies at 4°C. Immune complexes were washed and separated using ChIP-Grade Protein G Magnetic Beads. Chromatin was eluted from the antibody/protein G magnetic beads at 65°C for 30 min with gentle vortexing (1,200 rpm). DNA was subsequently reverse-crosslinked at 65°C for 4 h, followed by DNA purification through column elution. The purified DNA samples were sequenced at Azenta Life Sci using the Illumina NovaSeq 6000 Sequencing System (2 × 150 bp). For ChIP-qPCR validation, DNA samples were prepared as described above, and primers were designed based on S100a4-binding peaks from the ChIP-seq data, detailed in Table S1.
Statistical analysis
Statistical analysis was conducted using the unpaired two-tailed Student t test or one-way ANOVA to generate P values. Kaplan–Meier survival curves were generated using GraphPad Prism, and the Log-rank (Mantel-Cox) test was employed for statistical analysis in survival analysis. The P values: ns, not significant; *, P < 0.05; **, P < 0.01, ***, P < 0.001; ****, P < 0.0001.
Online supplemental material
Fig. S1 provides additional data confirming that activation of the TGFβ pathway is essential for pancreatitis-induced resistance to KRASi. Fig. S2 presents RNA-seq and ChIP-seq data analyses identifying S100a4 as a direct target of the NFAT5–SMADs complex. Fig. S3 demonstrates that the NFAT5–SMADs complex regulates canonical TGFβ pathway targets. Fig. S4 illustrates a schematic of the intercellular crosstalk promoting KRAS* bypass. Table S1 lists the primers used, and Table S2 provides information on the antibodies.
Data availability
The sequencing data have been deposited at the Gene Expression Omnibus under the SuperSeries accession number GSE252839. This SuperSeries includes the following SubSeries: single-cell RNA-seq data (GSE252834), RNA-seq data (GSE252836), and ChIP-seq data (GSE252838). All data are available in the main text or the supplementary materials.
Acknowledgments
We especially thank Drs. Ronald A. DePinho, Raymond Birge, and Teresa Wood for the critical reading and editing of the manuscript and thank Dr. Yibin Kang for instructive discussions. We express gratitude to E. McCaffrey and S. Bhatt for their assistance in maintaining the mouse colony, to S. Singh for contributing to flow cytometry analysis, and to L. Fritzky for assisting with IHC and imaging. Special thanks to all members of the Hou laboratory for valuable discussions and support.
This work was funded by National Institutes of Health (NIH) grant NIH K22 CA251491 (P. Hou), AACR-Lustgarten Foundation grant 22-20-67-HOU (P. Hou), Rutgers Cancer Institute of New Jersey New Investigator Award (P. Hou), NIH grant NS046593 (H. Li, Rutgers Center for Advanced Proteomics Research [CAPR]), and NIH grant 1S10OD025047 (H. Li, Rutgers Center for Advanced Proteomics Research [CAPR]). The Flow Cytometry and Immunology Core Laboratory, the Genomics Center, and Cellular Imaging and Histology Core were supported by the Office of the Dean at Rutgers New Jersey Medical School.
Author contributions: D. Deng: Investigation, Methodology, Visualization, Writing - original draft, Writing - review & editing, H. Begum: Investigation, T. Liu: Formal analysis, Investigation, Methodology, Resources, J. Zhang: Investigation, Q. Zhang: Formal analysis, Writing - review & editing, T.-y. Chu: Investigation, H. Li: Formal analysis, Supervision, A. Lemenze: Data curation, Formal analysis, Software, M. Hoque: Conceptualization, Methodology, P. Soteropoulos: Data curation, Project administration, Resources, Supervision, Writing - review & editing, P. Hou: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Visualization, Writing - original draft, Writing - review & editing.

