


Autoinflammatory diseases include a number of monogenic systemic inflammatory diseases, as well as acquired autoinflammatory diseases such as gout. Here, we show that the myeloid Src-family kinases Hck, Fgr, and Lyn are critical for experimental models of gout, as well as for genetically determined systemic inflammation in the Ptpn6me-v/me-v (motheaten viable) mouse model. The Hck−/−Fgr−/−Lyn−/− mutation abrogated various monosodium urate (MSU) crystal–induced pro-inflammatory responses of neutrophils, and protected mice from the development of gouty arthritis. The Src-family inhibitor dasatinib abrogated MSU crystal–induced responses of human neutrophils and reduced experimental gouty arthritis in mice. The Hck−/−Fgr−/−Lyn−/− mutation also abrogated spontaneous inflammation and prolonged the survival of the Ptpn6me-v/me-v mice. Spontaneous adhesion and superoxide release of Ptpn6me-v/me-v neutrophils were also abolished by the Hck−/−Fgr−/−Lyn−/− mutation. Excessive activation of tyrosine phosphorylation pathways in myeloid cells may characterize a subset of autoinflammatory diseases.
Introduction
Autoinflammatory diseases such as monogenic autoinflammatory diseases and gout place substantial burden on health-care systems worldwide. Molecular details of their pathomechanism are still poorly understood, hindering their molecular classification and limiting our therapeutic options.
Autoinflammatory diseases comprise a diverse family of diseases characterized by systemic inflammation, primarily caused by excessive activation of the innate immune response, i.e., without a dominant adaptive immune (autoimmune) component (Krainer et al., 2020). A number of autoinflammatory diseases are caused by monogenic gain-of-function mutations in various inflammasome components (de Jesus et al., 2015). However, a large number of other monogenic autoinflammatory diseases, caused by dysregulation (mostly hyperactivation) of other innate immune mechanisms, have also been identified (Savic et al., 2020; de Jesus et al., 2015). In addition, acquired mechanisms, such as hyperuricemia in gout, also lead to unique types of autoinflammation caused by excessive activation of the innate immune response (Szekanecz et al., 2021).
Gout is the most common inflammatory arthritis, affecting more than 40 million people with an alarmingly increasing incidence worldwide (Dalbeth et al., 2016; Dalbeth et al., 2019; Danve and Neogi, 2020; Dehlin et al., 2020). Gout is an autoinflammatory diseases caused by the deposition of monosodium urate (MSU) crystals in the joints, triggering an acute inflammatory response through myeloid cells such as neutrophils (Rock et al., 2013; So and Martinon, 2017). This inflammatory response is manifested in highly painful acute flares (Dalbeth et al., 2019). Repeated flares cause substantial structural joint damage (Chhana and Dalbeth, 2014) and the deterioration of the quality of life of the affected individuals. Together with associated pathologies such as obesity, hypertension, cardiovascular diseases, and chronic kidney disease, gout causes highly significant morbidity and mortality (Danve and Neogi, 2020; Dalbeth et al., 2019). In contrast to the dramatic improvements in other inflammatory joint diseases such as rheumatoid arthritis, very little progress has been made in the understanding and therapy of gout in the last decades (Danve and Neogi, 2020). Indeed, in contrast to rheumatoid arthritis, no improvement in the premature mortality gap in gout has been observed in the last two decades (Fisher et al., 2017). Those issues point to the need for further mechanistic studies on the pathogenesis of gout.
One of the earliest examples of genetically determined severe systemic inflammation models is the so-called motheaten (“me”) phenotype in experimental mice (Green and Shultz, 1975). A similar though less severe phenotype (termed motheaten viable or “me-v”) was later also identified (Shultz et al., 1984). Both phenotypes are caused by spontaneous mutations showing an autosomal recessive inheritance (Green and Shultz, 1975; Shultz et al., 1984). Later studies have revealed that the two phenotypes are caused by loss-of-function mutations in the Ptpn6 gene encoding SH2 domain-containing phosphatase 1 (SHP1), a negative regulator of hematopoietic cell function (Tsui et al., 1993; Shultz et al., 1993; Tsui and Tsui, 1994). While the motheaten (Ptpn6me) mutation caused a complete loss of SHP1 expression, the motheaten viable (Ptpn6me-v) mutation resulted in a hypomorphic mutation, in line with the less severe in vivo phenotype (Tsui et al., 1993; Shultz et al., 1993; Tsui and Tsui, 1994; Kozlowski et al., 1993). Both motheaten (Ptpn6me/me) and motheaten viable (Ptpn6me-v/me-v) mice suffer from severe dermatitis, arthritis, and pneumonitis (Green and Shultz, 1975; Shultz et al., 1984), causing lethality with a mean lifespan of ∼3 wk for the Ptpn6me/me and ∼2 mo for the Ptpn6me-v/me-v mice (Shultz et al., 1984). Since the survival and main pathological findings of the motheaten viable (Ptpn6me-v/me-v) mice were not affected by crossing them to the B and T cell–deficient Prkdcscid/scid or Rag1−/− backgrounds (Dominique and Francis, 1995; Yu et al., 1996), the phenotype is likely caused by hyperactivation of the innate, rather than the adaptive immune response (and therefore should be classified as an autoinflammatory rather than an autoimmune disease model). Lineage-specific conditional deletion of Ptpn6 revealed that early lethality was likely due to loss of SHP1 in neutrophils, whereas additional autoimmune features, emerging at later times, were likely mostly caused by SHP1 deficiency in dendritic cells (Abram et al., 2013; Abram and Lowell, 2017).
Src-family tyrosine kinases are the best-known family of non-receptor tyrosine kinases (Thomas and Brugge, 1997). They are involved in a number of critical cellular functions including proliferation, differentiation, adhesion, and migration, as well as in signaling by a number of cell surface receptors such as growth factor receptors, integrins, and various immunoreceptors (Thomas and Brugge, 1997; Parsons and Parsons, 2004; Espada and Martin-Perez, 2017). Accordingly, Src-family kinases play major roles in diverse physiological and pathological processes such as hematological malignancies, cancer progression, platelet function, and immunity/inflammation (Yeatman, 2004; Zhang and Yu, 2012; Senis et al., 2014). Src-family kinases have also been proposed as potential therapeutic targets in various diseases including hematological malignancies, solid tumors, and other pathologies such as Alzheimer’s disease (Martellucci et al., 2020; Kim et al., 2009; Nygaard et al., 2014).
Within the immune system, Src-family kinases are primarily responsible for leukocyte adhesion, as well as for signal transduction by lymphocyte antigen receptors and Fc-receptors (Lowell, 2004; Futosi et al., 2013; Futosi and Mócsai, 2016). Myeloid cells such as neutrophils and macrophages primarily express the Src-family kinases Hck, Fgr, and Lyn, which are involved in integrin and Fc-receptor signal transduction in vitro (Futosi et al., 2013; Futosi and Mócsai, 2016; Lowell et al., 1996; Mócsai et al., 1999; Mócsai et al., 2006), as well as in integrin- and Fc-receptor-mediated disease processes in vivo (Hirahashi et al., 2006; Kovács et al., 2014). However, several other cellular functions of myeloid cells proceed independently of, or in some cases are even dampened by, Src-family kinases, suggesting that the role of these kinases in myeloid cells is mainly restricted to integrin and Fc-receptor signaling (Mócsai et al., 2002; Zhang et al., 2005; Lowell, 2011; Kovács et al., 2014).
Prior studies revealed that Src-family kinases are also activated by MSU crystals in neutrophils (Gaudry et al., 1995; Tatsiy et al., 2020) and in the THP-1 human monocytic leukemia cell line (Liu et al., 2001). Sporadic experiments showed that the tyrosine kinase inhibitors PP1/PP2 inhibit MSU crystal–induced activation of Syk and Tec in neutrophils (Popa-Nita et al., 2007; Popa-Nita et al., 2008) and of NF-κB in THP-1 cells (Liu et al., 2001), whereas effects on other cellular functions were controversial. No genetic studies or in vivo experiments on the role of Src-family kinases in MSU crystal–induced inflammatory processes have been reported. Therefore, the role of Src-family kinases in the pathogenesis of gout is still unresolved.
There is also substantial evidence for the interaction of Src-family kinases with the SHP1 phosphatase (Somani et al., 1997; Frank et al., 2004; Pereira et al., 2004; Shi et al., 2021). However, the biological relevance of this interaction is still unclear, given the complex interaction between Src-family kinases and SHP-1 and the evidence for both synergistic and antagonistic effects of the two signaling molecules (Somani et al., 1997; Frank et al., 2004). This warrants further studies on the role for Src-family kinases in the autoinflammatory phenotypes caused by loss of SHP1 function in the motheaten or motheaten viable mice.
The above issues prompted us to test in vitro responses and in vivo inflammation caused by MSU crystals or the motheaten viable (Ptpn6me-v/me-v) mutation in mice lacking the myeloid Src-family kinases Hck, Fgr, and Lyn. Our results indicate a critical role for myeloid Src-family kinases in various MSU crystal–induced in vitro functional responses of neutrophils, as well as a major role for those kinases in the inflammatory response in an in vivo gout model. Similarly, myeloid Src-family kinases proved to be critical for in vitro neutrophil functions and in vivo inflammatory reactions in the motheaten viable (Ptpn6me-v/me-v) mice. We also provide evidence for the therapeutic efficacy of a clinically approved Abl/Src-inhibitor on human neutrophils and on our in vivo gout model. Taken together, our results indicate a critical role for Src-family kinases in two distinct models of autoinflammation and point to a potential novel direction of future pharmacological intervention. Together with other recent reports, our results also suggest that excessive activation of tyrosine phosphorylation pathways in myeloid cells may delineate a novel class of autoinflammatory diseases.
Results
Myeloid Src-family kinases in MSU crystal–induced neutrophil activation
As a first approach to test the role of myeloid Src-family kinases in MSU crystal–induced responses, we tested MSU crystal–induced activation of neutrophils isolated from mice lacking Hck, Fgr, and Lyn, the three major Src-family kinases expressed in myeloid cells such as neutrophils and macrophages (Hck−/−Fgr−/−Lyn−/− mice). As shown in Fig. 1 A, activation of WT neutrophils by 1 mg/ml MSU crystals triggered a robust respiratory burst response, whereas no response could be observed in Hck−/−Fgr−/−Lyn−/− neutrophils under identical conditions (P = 9.6 × 10−9). Analysis of increasing MSU doses revealed a parallel increase of ROS production in WT neutrophils, but no such response from Hck−/−Fgr−/−Lyn−/− mutants at any MSU doses used could be observed (Fig. 1 B; P = 2.8 × 10−6). This was not due to abrogation of the intrinsic ability of ROS production since the PKC-activating agent PMA was able to trigger nearly normal superoxide release from Hck−/−Fgr−/−Lyn−/− neutrophils (Fig. S1 A; P = 0.27).
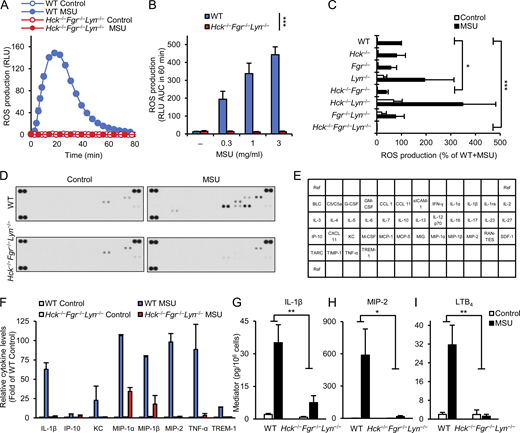
Myeloid Src-family kinases are required for MSU crystal–induced neutrophil activation. (A–I) WT or Hck−/−Fgr−/−Lyn−/− neutrophils (A–I), along with various single and double knockout neutrophils (C) were stimulated with 1 mg/ml (A and C–I) or the indicated concentrations (B) of MSU crystals. (A–C) Luminometric measurement of reactive oxygen production. (D–I) Cells were incubated for 18 h followed by determination of cytokine, chemokine, and lipid mediator levels from cell-free supernatants using a commercial cytokine array (D) or ELISA assays (G–I). (E) Map of the different analytes on the cytokine array. (F) Quantification of cytokine array results by densitometry. Panels A (mean and SD) and D show results from a representative experiment. Bar graph shows mean and SEM from 4 (B), 3–8 (C), 2 (F), and 5–8 (G–I) independent experiments. Panels C and F show data normalized to WT MSU and WT control samples, respectively. Panel F shows analytes with at least fivefold MSU-induced increase in WT samples. RLU, relative luminescence unit; AUC, area under the curve. Two-way ANOVA genotype × stimulus interaction analysis; *, P < 0.05; **, P < 0.01; ***, P < 0.001. See the text for actual P values. Source data are available for this figure: SourceData F1.
Myeloid Src-family kinases are required for MSU crystal–induced neutrophil activation. (A–I) WT or Hck−/−Fgr−/−Lyn−/− neutrophils (A–I), along with various single and double knockout neutrophils (C) were stimulated with 1 mg/ml (A and C–I) or the indicated concentrations (B) of MSU crystals. (A–C) Luminometric measurement of reactive oxygen production. (D–I) Cells were incubated for 18 h followed by determination of cytokine, chemokine, and lipid mediator levels from cell-free supernatants using a commercial cytokine array (D) or ELISA assays (G–I). (E) Map of the different analytes on the cytokine array. (F) Quantification of cytokine array results by densitometry. Panels A (mean and SD) and D show results from a representative experiment. Bar graph shows mean and SEM from 4 (B), 3–8 (C), 2 (F), and 5–8 (G–I) independent experiments. Panels C and F show data normalized to WT MSU and WT control samples, respectively. Panel F shows analytes with at least fivefold MSU-induced increase in WT samples. RLU, relative luminescence unit; AUC, area under the curve. Two-way ANOVA genotype × stimulus interaction analysis; *, P < 0.05; **, P < 0.01; ***, P < 0.001. See the text for actual P values. Source data are available for this figure: SourceData F1.

Analysis of PMA-induced superoxide production. (A and B) WT or Hck−/−Fgr−/−Lyn−/− neutrophils (A) and human neutrophils treated with vehicle or 100 nM dasatinib (B) were stimulated with 100 nM PMA, followed by a spectrophotometric analysis of superoxide release. Kinetic curves (A and B) show mean and SEM of five (A) or three (B) independent experiments. Two-way ANOVA genotype × stimulus (A) or inhibitor × stimulus (B) interaction analysis; n.s., not significant. See the text for actual P values.
Analysis of PMA-induced superoxide production. (A and B) WT or Hck−/−Fgr−/−Lyn−/− neutrophils (A) and human neutrophils treated with vehicle or 100 nM dasatinib (B) were stimulated with 100 nM PMA, followed by a spectrophotometric analysis of superoxide release. Kinetic curves (A and B) show mean and SEM of five (A) or three (B) independent experiments. Two-way ANOVA genotype × stimulus (A) or inhibitor × stimulus (B) interaction analysis; n.s., not significant. See the text for actual P values.
We next tested MSU crystal–induced respiratory burst in single or double Hck-, Fgr- and Lyn-deficient neutrophils (Fig. 1 C). Though the Hck−/− or Fgr−/− single mutants tended to show a partially reduced response and the Lyn−/− cells tended to mount an increased respiratory burst, none of those differences proved to be statistically significant (P = 0.31, 0.34, and 0.81, respectively). Of the various double mutant cells, Hck−/−Fgr−/− neutrophils showed a partially but statistically significantly reduced response (P = 0.030), whereas neither the Hck−/−Lyn−/− nor the Fgr−/−Lyn−/− double mutants showed a statistically significant difference (P = 0.59 and 0.48, respectively), although the Hck−/−Lyn−/− cells tended to mount an augmented respiratory burst response. It should also be noted that all cells carrying the Lyn−/− mutation (Lyn−/− single, as well as Hck−/−Lyn−/− or Fgr−/−Lyn−/− double mutants) showed a higher basal respiratory burst which, together with the tendency for an augmented response in Lyn−/− and Hck−/−Lyn−/− cells, may be related to the partial inhibitory effect of Lyn under certain conditions (Pereira and Lowell, 2003). Importantly, Hck−/−Fgr−/−Lyn−/− triple knockout neutrophils again showed a completely abolished respiratory burst response (P = 1.2 × 10−5). Taken together, Hck, Fgr, and Lyn play a critical, yet substantially overlapping role in the MSU crystal–induced neutrophil respiratory burst.
Neutrophils also release various proinflammatory mediators, thus amplifying their own recruitment and contributing to the organization of the inflammatory response (Mócsai, 2013; Németh and Mócsai, 2016). This was first tested by a cytokine array approach (Fig. 1, D–F). As shown in the cytokine array profiles in Fig. 1 D (see array map in Fig. 1 E), MSU crystals triggered a robust cytokine and chemokine release from WT but not from Hck−/−Fgr−/−Lyn−/− neutrophils. Fig. 1 F depicts the semi-quantitative densitometric analysis of mediators showing at least fivefold increase in WT cells upon administration of MSU crystals. Importantly, all those responses were strongly reduced or even completely abrogated in Hck−/−Fgr−/−Lyn−/− cells (Fig. 1 F). Some of these and other mediators have also been assessed by quantitative ELISA (Fig. 1, G–I). Those experiments indicated that WT but not Hck−/−Fgr−/−Lyn−/− neutrophils release IL-1β (P = 0.0056), MIP-2 (P = 0.026), and the lipid mediator LTB4 (P = 0.0031) upon activation by MSU crystals (Fig. 1, G–I).
Taken together, MSU crystals triggered substantial neutrophil activation which was completely blocked by the Hck−/−Fgr−/−Lyn−/− triple knockout mutation. Analysis of single and double mutants revealed substantial functional overlap between Hck, Fgr, and Lyn.
Src-family kinases in MSU crystal–induced macrophage activation
Although gouty arthritis is dominated by massive neutrophil infiltration and activation, other myeloid cells such as macrophages might also play an important role (So and Martinon, 2017). Therefore, we tested the role of Src-family kinases in MSU crystal–induced macrophage responses. Bone marrow–derived WT macrophages showed well-measurable ROS production upon activation by MSU crystals (Fig. S2 A), and this response was further enhanced after pre-activation (priming) of the cells with bacterial LPS (Fig. S2 B). Hck−/−Fgr−/−Lyn−/− macrophages showed a strongly reduced ROS production both without (P = 0.0014) and with (P = 3.5 × 10−4) LPS priming. We have also tested MSU crystal–induced IL-1β and MIP-2 release from unprimed and LPS-primed macrophages (Fig. S2, C and D). In agreement with the two-step model of IL-1β production (upregulation followed by inflammasome-mediated maturation), MSU crystal–induced IL-1β release was strongly enhanced by LPS priming in WT macrophages. Interestingly, and in contrast to our findings on neutrophils, Hck−/−Fgr−/−Lyn−/− macrophages showed normal (or slightly even enhanced) IL-1β release both without (P = 0.079) and with (P = 0.56) LPS priming (Fig. S2 C). MSU-induced MIP-2 release was less pronounced, especially in LPS-primed cells, but this response also proceeded normally in Hck−/−Fgr−/−Lyn−/− macrophages (Fig. S2 D; P = 0.63–0.68). Taken together, Src-family kinases are required for MSU-induced ROS production but not IL-1β or MIP-2 release of macrophages.

MSU crystal–induced macrophage responses. (A–D) WT or Hck−/−Fgr−/−Lyn−/− bone marrow–derived macrophages were stimulated with 1 mg/ml MSU crystals without (A–D) or with (B–D) overnight priming of the cells with 1 μg/ml ultrapure LPS. (A and B) Luminometric measurement of reactive oxygen production. (C and D) IL-1β (C) and MIP-2 (D) levels determined from cell-free supernatants after incubation for 24 h. Kinetic curves (A) show mean and SD of a representative experiment. Bar graph shows mean and SEM from three (B) or four to five (C and D) independent experiments. RLU, relative luminescence unit. Two-way ANOVA genotype × stimulus interaction analysis; n.s., not significant; **, P < 0.01; ***, P < 0.001. See the text for actual P values.
MSU crystal–induced macrophage responses. (A–D) WT or Hck−/−Fgr−/−Lyn−/− bone marrow–derived macrophages were stimulated with 1 mg/ml MSU crystals without (A–D) or with (B–D) overnight priming of the cells with 1 μg/ml ultrapure LPS. (A and B) Luminometric measurement of reactive oxygen production. (C and D) IL-1β (C) and MIP-2 (D) levels determined from cell-free supernatants after incubation for 24 h. Kinetic curves (A) show mean and SD of a representative experiment. Bar graph shows mean and SEM from three (B) or four to five (C and D) independent experiments. RLU, relative luminescence unit. Two-way ANOVA genotype × stimulus interaction analysis; n.s., not significant; **, P < 0.01; ***, P < 0.001. See the text for actual P values.
Myeloid Src-family kinases in MSU crystal–induced in vivo arthritis
To determine the role of Src-family kinases in MSU crystal–induced inflammatory processes in vivo, we injected MSU crystals into the right hind paws of the experimental mice, along with vehicle (PBS) injected into the left hind paws as controls. As shown in the photographs taken 24 h later (Fig. 2 A), MSU crystals triggered inflammation and paw swelling in WT mice, whereas this response was much less pronounced in Hck−/−Fgr−/−Lyn−/− triple knockout mice. The paw volume was then followed for 3 d using a digital plethysmometer. Those measurements revealed a rapid and sustained MSU crystal–induced increase of paw volume in WT mice, whereas a strongly reduced swelling response could be observed in Hck−/−Fgr−/−Lyn−/− animals (Fig. 2 B; P = 1.9 × 10−15). Analysis of the paw volume change in the first 24 h after MSU crystal injection in a larger cohort of mice also revealed a substantially reduced MSU-induced response in Hck−/−Fgr−/−Lyn−/− animals (Fig. 2 C; P = 0.0019).

Myeloid Src-family kinases mediate MSU crystal–induced arthritis in vivo. (A–G) WT or Hck−/−Fgr−/−Lyn−/− mice or various single and double mutants (D and E) were subjected to gouty arthritis by injecting the right hind paws of the mice with 1 mg MSU crystal suspended in 50 μl PBS, along with injecting left hind paws with PBS alone as controls. MSU crystal–induced inflammation was followed by photographing on day 1 (A; scale bar: 10 mm), measuring the paw volume by plethysmometry (B–E), and examining hypernociception (F and G). Panel A shows a representative experiment. Quantitative curves and bar graphs show mean and SEM from 11 to 13 (B), 32–33 (C), 5–13 (D and E; except for a single Hck−/−Lyn−/− mouse), and 11–16 (F and G) mice per genotype from 3 (B), 5 (C), 2–3 (D and E), and 2 (F and G) independent experiments. Two-way ANOVA genotype × stimulus (A–E) or genotype × time (F and G) interaction analysis; *, P < 0.05; **, P < 0.01; ***, P < 0.001. See the text for actual P values.
Myeloid Src-family kinases mediate MSU crystal–induced arthritis in vivo. (A–G) WT or Hck−/−Fgr−/−Lyn−/− mice or various single and double mutants (D and E) were subjected to gouty arthritis by injecting the right hind paws of the mice with 1 mg MSU crystal suspended in 50 μl PBS, along with injecting left hind paws with PBS alone as controls. MSU crystal–induced inflammation was followed by photographing on day 1 (A; scale bar: 10 mm), measuring the paw volume by plethysmometry (B–E), and examining hypernociception (F and G). Panel A shows a representative experiment. Quantitative curves and bar graphs show mean and SEM from 11 to 13 (B), 32–33 (C), 5–13 (D and E; except for a single Hck−/−Lyn−/− mouse), and 11–16 (F and G) mice per genotype from 3 (B), 5 (C), 2–3 (D and E), and 2 (F and G) independent experiments. Two-way ANOVA genotype × stimulus (A–E) or genotype × time (F and G) interaction analysis; *, P < 0.05; **, P < 0.01; ***, P < 0.001. See the text for actual P values.
We also tested the potential overlap between Hck, Fgr, and Lyn in MSU crystal–induced inflammation using single and double mutant mice by following the increase of paw volume for 3 d (Fig. 2 D). Paw volume changes in the first 24 h after MSU crystal injection are shown in Fig. 2 E. Similar to the in vitro findings, Hck−/−, Fgr−/−, or Lyn−/− single knockout mice showed similar paw swelling as WT animals (Fig. 2 D; P = 0.85, 0.63, and 0.79, respectively; and Fig. 2 E; P = 0.88, 0.21, and 0.64, respectively). Of the various double mutants, Hck−/−Fgr−/− mice showed a modest but statistically significant reduction in paw swelling (Fig. 2 D; P = 0.050; and Fig. 2 E; P = 0.040), whereas Fgr−/−Lyn−/− mice were moderately hyper-responsive (Fig. 2 D; P = 0.0023; and Fig. 2 E; P = 0.025). A single Hck−/−Lyn−/− double knockout mouse showed a mostly normal response. Importantly, Hck−/−Fgr−/−Lyn−/− mice again showed a substantially reduced MSU crystal–induced swelling response (Fig. 2 D; P = 2.1 × 10−9; and Fig. 2 E; P = 3.1 × 10−9).
Since the most prominent feature of an acute gout attack is severe pain, we also tested pain sensation in mice after MSU crystal injection. As a first approach, we measured the mechanonociceptive threshold by dynamic plantar esthesiometry (Fig. 2 F). The mechanonociceptive threshold of the MSU crystal–injected limb was strongly reduced 6 and 24 h after MSU crystal injection in WT animals (Fig. 2 F). Importantly, this drop of mechanonociceptive threshold was much less pronounced in Hck−/−Fgr−/−Lyn−/− mice (P = 9.7 × 10−5), indicating a less painful course of MSU crystal–induced joint inflammation in the absence of myeloid Src-family kinases. As another approach, we also tested the distribution of weight bearing between the left (control-injected) and right (MSU crystal–injected) hind limbs. As shown in Fig. 2 G, WT mice placed significantly more weight on their control-treated hind limbs, especially at the 6 h time point. This difference between the two limbs tended to be less prominent in Hck−/−Fgr−/−Lyn−/− mice, although the overall difference between the two genotypes did not reach statistical significance (P = 0.26).
Taken together, Hck, Fgr, and Lyn play an important role in both the clinical signs and certain nociceptive changes during the development of MSU crystal–induced arthritis, with a substantial overlap between the three kinases.
Analysis of the in vivo inflammatory microenvironment
To gain a more detailed insight into the in vivo changes during MSU crystal–induced inflammation, we next tested various aspects of the development of the inflammatory tissue microenvironment. We first performed in vivo imaging of ROS production using two different bioluminescence probes, measuring either the activity of myeloperoxidase (MPO; Gross et al., 2009; Kovács et al., 2014; Fig. 3, A and B) or superoxide generation by the phagocytic NADPH-oxidase enzymes (Fig. 3, C and D). As shown in Fig. 3 A, MSU crystals triggered a robust MPO-derived bioluminescence signal in WT but not Hck−/−Fgr−/−Lyn−/− mice. Quantitative analysis (Fig. 3 B) revealed sustained MPO activity for at least 2 d and complete absence of this response in Hck−/−Fgr−/−Lyn−/− animals at all investigated time points (P = 6.9 × 10−13, 7.1 × 10−9, and 1.8 × 10−4 for the 6, 24, and 48 h time points, respectively). Similarly, MSU crystals triggered robust bioluminescence signals corresponding to the activity of the NADPH-oxidase in WT but not in Hck−/−Fgr−/−Lyn−/− mice (Fig. 3 C). Quantification of this response also revealed complete protection in the mutant mice at all measured time points (Fig. 3 D; P = 0.0027, 4.9 × 10−4, and 2.6 × 10−6 at 6, 24, and 48 h, respectively). Therefore, myeloid Src-family kinases are required for in vivo ROS production at the site of MSU crystal injection.
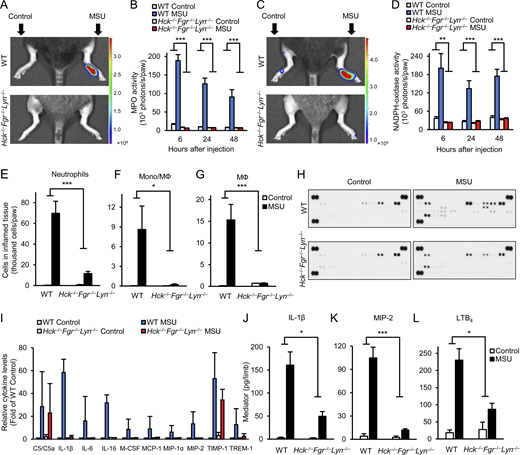
Analysis of the inflammatory microenvironment.(A–L) WT or Hck−/−Fgr−/−Lyn−/− mice were subjected to gouty arthritis as described in Fig. 2. (A–D) In vivo MPO (A and B) and NADPH-oxidase (C and D) activity was determined by chemiluminescence imaging after i.p. injection of luminol or lucigenin, respectively. Color-coded photon flux intensity is superposed on the grayscale photo of the mice after 24 h of MSU injection (A and C) and quantified in defined regions of interest at the indicated time points (B and D). (E–G) The hind paws were flushed after 24 h of MSU injection and the number of neutrophils (E) or monocytes/macrophages (F and G) was determined by flow cytometry. (H–L) The cell-free supernatants of the tissue infiltrates were probed using a commercial cytokine array (H and I) or analyzed by ELISA for the indicated pro-inflammatory mediators (J–L). Panels A, C, and H show representative images. Bar graphs show mean and SEM from 6–12 (B and D), 8–20 (E), 3 (F and G), 2 (I), and 13–27 (J–L) mice per genotype from 2 (B and D), 8 (E), 1 (F and G), 2 (I), and 4–7 (J–L) independent experiments. Two-way ANOVA genotype × stimulus interaction analysis; *, P < 0.05; **, P < 0.01; ***, P < 0.001. See the text for actual P values. Source data are available for this figure: SourceData F3.
Analysis of the inflammatory microenvironment.(A–L) WT or Hck−/−Fgr−/−Lyn−/− mice were subjected to gouty arthritis as described in Fig. 2. (A–D) In vivo MPO (A and B) and NADPH-oxidase (C and D) activity was determined by chemiluminescence imaging after i.p. injection of luminol or lucigenin, respectively. Color-coded photon flux intensity is superposed on the grayscale photo of the mice after 24 h of MSU injection (A and C) and quantified in defined regions of interest at the indicated time points (B and D). (E–G) The hind paws were flushed after 24 h of MSU injection and the number of neutrophils (E) or monocytes/macrophages (F and G) was determined by flow cytometry. (H–L) The cell-free supernatants of the tissue infiltrates were probed using a commercial cytokine array (H and I) or analyzed by ELISA for the indicated pro-inflammatory mediators (J–L). Panels A, C, and H show representative images. Bar graphs show mean and SEM from 6–12 (B and D), 8–20 (E), 3 (F and G), 2 (I), and 13–27 (J–L) mice per genotype from 2 (B and D), 8 (E), 1 (F and G), 2 (I), and 4–7 (J–L) independent experiments. Two-way ANOVA genotype × stimulus interaction analysis; *, P < 0.05; **, P < 0.01; ***, P < 0.001. See the text for actual P values. Source data are available for this figure: SourceData F3.
Next, we determined infiltration of leukocytes at the site of inflammation. As shown in Fig. 3, E–G, injection of MSU crystal–induced robust accumulation of neutrophils and monocytes/macrophages in the affected limbs of WT mice, whereas both neutrophil and monocyte/macrophage infiltration was strongly reduced in Hck−/−Fgr−/−Lyn−/− animals (P = 0.0012, 0.034, and 6.3 × 10−4 for Fig. 3, E–G, respectively).
We have also tested the accumulation of inflammatory mediators at the site of MSU crystal injection. As a first approach, a cytokine array showed MSU crystal–induced accumulation of a number of mediators in WT but not Hck−/−Fgr−/−Lyn−/− animals (Fig. 3 H; see array map in Fig. 1 E). Fig. 3 I shows the semi-quantitative densitometric analysis of those mediators that showed at least fivefold increase upon MSU crystal injection in WT mice. Most of those responses (except for C5/C5a and TIMP-1) were practically completely abrogated in Hck−/−Fgr−/−Lyn−/− animals (Fig. 3 I). Further quantitative ELISA assays (Fig. 3, J–L) confirmed MSU crystal–induced accumulation of IL-1β, MIP-2, and LTB4 in WT mice but strongly reduced responses in Hck−/−Fgr−/−Lyn−/− animals (P = 0.021, 2.6 × 10−4, and 0.013, respectively).
Taken together, myeloid Src-family kinases play an important role in diverse aspects of the MSU crystal–induced inflammation process, including ROS production, leukocyte infiltration, and accumulation of various proinflammatory mediators.
Mechanistic studies on MSU crystal–induced neutrophil activation
Having established the role of Src-family kinases in MSU crystal–induced activation of neutrophils and macrophages, as well as in the development of experimental gout, we next aimed to understand how Src-family kinases contribute to MSU crystal–induced inflammation.
Since MSU crystals are actively phagocytosed by myeloid cells, we first tested how deficiency of Src-family kinases affects this process, using a flow cytometry–based approach. As shown in the flow cytometric histograms in Fig. 4 A and their quantification in Fig. 4 B, MSU crystals caused a substantial shift in the side-scatter characteristics of WT mouse neutrophils, likely reflecting increased intracellular complexity due to phagocytosis of MSU crystals. This response was strongly diminished by pretreating WT neutrophils with the actin polymerization inhibitor cytochalasin D (P = 4.2 × 10−4), confirming the active uptake of the MSU crystals. Importantly, this phagocytic response was also strongly reduced by the Hck−/−Fgr−/−Lyn−/− mutation (Fig. 4, A and B; P = 0.0021). The effect of the Hck−/−Fgr−/−Lyn−/− mutation was similar to that of cytochalasin D treatment of WT neutrophils (P = 0.24) and cytochalasin D treatment could not further diminish the response of Hck−/−Fgr−/−Lyn−/− cells (P = 0.52).
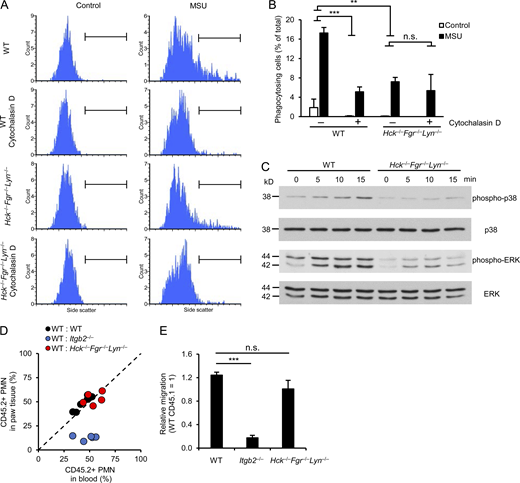
Impaired phagocytic activity and intracellular signaling but normal migratory ability of Hck−/−Fgr−/−Lyn−/−neutrophils. (A–C) WT or Hck−/−Fgr−/−Lyn−/− neutrophils were stimulated with 1 mg/ml MSU crystals followed by assessment of phagocytosis of the MSU crystals (A and B), intracellular phosphorylation (C). Where indicated, the cells were pretreated with 10 μM cytochalasin D. (D and E) Mixed bone marrow chimeras carrying CD45.1-expressing WT and CD45.2-expressing WT, Itgb2−/−, or Hck−/−Fgr−/−Lyn−/− hematopoietic cells were subjected to MSU crystal–induced arthritis as described above. 24 h later, the hind paws were flushed, the ratio of CD45.1- and CD45.2-expressing neutrophils in the paw infiltrate was determined by flow cytometry and compared to the ratio in the peripheral blood. Panel D shows the results for each individual mouse, whereas panel E shows the accumulation of the CD45.2-expressing WT, Itgb2−/−, and Hck−/−Fgr−/−Lyn−/− neutrophils relative to the CD45.1-expressing WT cells. Panels A and C show representative images from two to three independent experiments. Panel B shows mean and SEM from two to six independent experiments. Panel E shows data mean and SEM of five to six chimeras per group from three independent experiments. Two-way ANOVA inhibitor × stimulus or genotype × stimulus interaction analysis (B) and one-way ANOVA (E); n.s., not significant; **, P < 0.01; ***, P < 0.001. See the text for actual P values. Source data are available for this figure: SourceData F4.
Impaired phagocytic activity and intracellular signaling but normal migratory ability of Hck−/−Fgr−/−Lyn−/−neutrophils. (A–C) WT or Hck−/−Fgr−/−Lyn−/− neutrophils were stimulated with 1 mg/ml MSU crystals followed by assessment of phagocytosis of the MSU crystals (A and B), intracellular phosphorylation (C). Where indicated, the cells were pretreated with 10 μM cytochalasin D. (D and E) Mixed bone marrow chimeras carrying CD45.1-expressing WT and CD45.2-expressing WT, Itgb2−/−, or Hck−/−Fgr−/−Lyn−/− hematopoietic cells were subjected to MSU crystal–induced arthritis as described above. 24 h later, the hind paws were flushed, the ratio of CD45.1- and CD45.2-expressing neutrophils in the paw infiltrate was determined by flow cytometry and compared to the ratio in the peripheral blood. Panel D shows the results for each individual mouse, whereas panel E shows the accumulation of the CD45.2-expressing WT, Itgb2−/−, and Hck−/−Fgr−/−Lyn−/− neutrophils relative to the CD45.1-expressing WT cells. Panels A and C show representative images from two to three independent experiments. Panel B shows mean and SEM from two to six independent experiments. Panel E shows data mean and SEM of five to six chimeras per group from three independent experiments. Two-way ANOVA inhibitor × stimulus or genotype × stimulus interaction analysis (B) and one-way ANOVA (E); n.s., not significant; **, P < 0.01; ***, P < 0.001. See the text for actual P values. Source data are available for this figure: SourceData F4.
We also tested the activation of MSU-induced downstream signaling pathways. As shown in Fig. 4 C, MSU crystals triggered phosphorylation of the ERK and p38 MAPKs, both of which was strongly inhibited by the Hck−/−Fgr−/−Lyn−/− mutation.
A possible explanation for our findings could be a defective intrinsic migratory capacity of Hck−/−Fgr−/−Lyn−/− neutrophils. To test this in vivo in a manner not biased by the defective inflammatory environment in the Hck−/−Fgr−/−Lyn−/− mice, we performed a competitive in vivo migration experiment in mixed bone marrow chimeras carrying CD45.1-expressing WT and CD45.2-expessing WT or Hck−/−Fgr−/−Lyn−/− hematopoietic cells (WT:WT and WT:Hck−/−Fgr−/−Lyn−/− chimeras, respectively). As controls, we have also generated and used mixed bone marrow chimeras carrying CD45.1-expressing WT and CD45.2-expressing CD18-deficient (Itgb2−/−) hematopoietic cells (WT:Itgb2−/− chimeras). The mixed bone marrow chimeras were subjected to MSU crystal–induced arthritis and the percentage of CD45.1-expressing and CD45.2-expressing neutrophils in the blood and the paw tissue infiltrate was determined by flow cytometry. Results of those experiments are shown in the representative histograms in Fig. S3 and are summarized in Fig. 4, D and E. As expected, there was no difference between the percentage of CD45.2-expressing (WT) neutrophils between the blood and paw tissue in WT:WT chimeras (Fig. S3 A and Fig. 4 D), indicating that the WT neutrophils from the two different (CD45.1 and CD45.2) origins were equally capable of accumulating at the site of inflammation. In contrast, the percentage of CD45.2-expressing (Itgb2−/−) neutrophils was dramatically reduced in the paw tissue of WT:Itgb2−/− chimeras compared to that in the blood of the same mice (Fig. S3 B and Fig. 4 D), likely reflecting strongly reduced ability of Itgb2−/− neutrophils to accumulate in the inflamed paw. Importantly, the percentage of CD45.2-expressing (Hck−/−Fgr−/−Lyn−/−) neutrophils was similar between the blood and paw tissue in WT:Hck−/−Fgr−/−Lyn−/− chimeras (Fig. S3 C and Fig. 4 D), indicating that Hck−/−Fgr−/−Lyn−/− neutrophils are capable of accumulating at the site of MSU-induced inflammation as well as WT cells. Those results are also summarized in Fig. 4 E, depicting the migration of the CD45.2-expressing WT, Itgb2−/−, and Hck−/−Fgr−/−Lyn−/− neutrophils relative to the CD45.1-expressing WT cells within the same animals. As expected from Fig. S3 and Fig. 4 D, Itgb2−/− neutrophils showed a strongly reduced relative migratory capacity (P = 7.7 × 10−8), whereas Hck−/−Fgr−/−Lyn−/− neutrophils showed practically normal migration (P = 0.14). Those results suggest that, in contrast to the CD18-deficient Itgb2−/− neutrophils, Hck−/−Fgr−/−Lyn−/− neutrophils are intrinsically capable of accumulating at the site of MSU crystal–induced inflammation.

Competitive in vivo migration assays. (A–C) Representative flow cytometric histograms of competitive in vivo migration experiments in mixed bone marrow chimeras with CD45.1-expressing WT and CD45.2-expressing WT (A), Itgb2−/− (B), or Hck−/−Fgr−/−Lyn−/− (C) neutrophils, shown in Fig. 4, D and E (see Fig. 4 for more detail).
Competitive in vivo migration assays. (A–C) Representative flow cytometric histograms of competitive in vivo migration experiments in mixed bone marrow chimeras with CD45.1-expressing WT and CD45.2-expressing WT (A), Itgb2−/− (B), or Hck−/−Fgr−/−Lyn−/− (C) neutrophils, shown in Fig. 4, D and E (see Fig. 4 for more detail).
MSU crystals are able to induce neutrophil extracellular trap (NET) formation. To test the role of Src-family kinases in this response, MSU crystal–induced DNA externalization by WT and Hck−/−Fgr−/−Lyn−/− neutrophils was followed by real-time fluorescence imaging using Sytox Green, which labels externalized DNA, along with staining all DNA by a Hoechst dye. NET-like structures were observed within 1 h after neutrophil activation and further accumulated by the end of the 150-min observation period (Fig. S4 A). Importantly, no substantial difference could be observed in this response between WT and Hck−/−Fgr−/−Lyn−/− cells (Fig. S4 A). Quantification based on the determination of the area covered by NETs also failed to show a statistically significant difference between the two genotypes (Fig. S4 B; P = 0.50).

Analysis of MSU crystal–induced DNA externalization. (A–D) Nuclei of WT and Hck−/−Fgr−/−Lyn−/− mouse neutrophils (A and B), or of WT neutrophils treated with vehicle or 100 nM dasatinib (C and D) were stained with 0.5 μg/ml Hoechst dye followed by addition of 0.2 mg/ml MSU crystals along with 50 nM Sytox-Green DNA to visualize extracellular DNA. Panels A and C are representative images (scale bar: 200 µm), and panels B and D show the quantification of the externalized DNA area as mean and SEM from four (A and B) or three (C and D) independent experiments. Two-way ANOVA genotype × stimulus or inhibitor × stimulus interaction analysis; n.s., not significant. See the text for actual P values.
Analysis of MSU crystal–induced DNA externalization. (A–D) Nuclei of WT and Hck−/−Fgr−/−Lyn−/− mouse neutrophils (A and B), or of WT neutrophils treated with vehicle or 100 nM dasatinib (C and D) were stained with 0.5 μg/ml Hoechst dye followed by addition of 0.2 mg/ml MSU crystals along with 50 nM Sytox-Green DNA to visualize extracellular DNA. Panels A and C are representative images (scale bar: 200 µm), and panels B and D show the quantification of the externalized DNA area as mean and SEM from four (A and B) or three (C and D) independent experiments. Two-way ANOVA genotype × stimulus or inhibitor × stimulus interaction analysis; n.s., not significant. See the text for actual P values.
Dasatinib inhibits MSU crystal–induced activation of human neutrophils
We also attempted to test the role of Src-family kinases in human neutrophils by a pharmacological approach. To this end, we tested the effect of dasatinib, an inhibitor of Abl- and Src-family kinases used in the clinical therapy of various hematological malignancies (Hehlmann, 2020; Keating, 2017).
As shown in Fig. 5 A, MSU crystals evoked robust ROS production from vehicle-treated human neutrophils, and this response was strongly inhibited by dasatinib in a concentration-dependent manner, reaching practically complete inhibition (P = 0.014) at 100 nM inhibitor concentration. This was not due to a general abrogation of ROS production since the PKC agonist PMA was able to trigger ROS production of human neutrophils treated with 100 nM dasatinib (Fig. S1 B; P = 0.36). Human neutrophils showed modest LTB4 release in the presence of MSU crystals, which was more pronounced in TNF-primed neutrophils (Fig. 5, B and C). 100 nM dasatinib tended to reduce the moderate MSU crystal–induced lipid mediator release of unprimed neutrophils (Fig. 5 B; P = 0.11) and strongly inhibited the robust response of TNF-primed neutrophils (Fig. 5 C; P = 0.021). Dasatinib also dramatically reduced MSU crystal–induced phosphorylation of the ERK and p38 MAPKs (Fig. 5 D). Finally, phagocytosis of the MSU crystals was greatly inhibited by dasatinib or the actin polymerization inhibitor cytochalasin D, both when tested by flow cytometry (Fig. 5, E and G; P = 0.0019 and 8.3 × 10−4 for dasatinib and cytochalasin D, respectively), or by phase contrast microscopy (Fig. 5, F and H; note phagocytosed crystals marked by arrows; P = 4.2 × 10−6 and 2.9 × 10−6 for dasatinib and cytochalasin D, respectively).
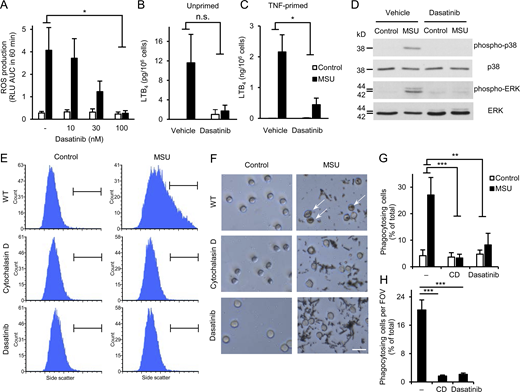
Dasatinib inhibits MSU crystal–induced proinflammatory functions of human neutrophils. Human neutrophils were pretreated with 100 nM or the indicated concentrations of dasatinib or 10 μM cytochalasin D, followed by stimulation with 1 mg/ml MSU crystals. (A) Luminometric measurement of reactive oxygen production. (B and C) LTB4 release determined from cell-free supernatants after 18 h incubation with the crystals of unprimed (B) or TNF-primed (10 ng/ml TNF; C) neutrophils. (D) Analysis of intracellular p38/ERK phosphorylation. (E–H) Phagocytosis of the MSU crystals assessed by flow cytometry (E and G) or phase contrast microscopy (F and H; scale bar: 20 µm). Panels D, E, and F show representative images. Bar graph shows mean and SEM from four to six (A), two to three (B and C), and three (G and H) independent experiments. RLU, relative luminescence unit; AUC, area under the curve. Two-way ANOVA inhibitor × stimulus interaction analysis (A–C and G) and one-way ANOVA (H); n.s., not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001. See the text for actual P values. Source data are available for this figure: SourceData F5.
Dasatinib inhibits MSU crystal–induced proinflammatory functions of human neutrophils. Human neutrophils were pretreated with 100 nM or the indicated concentrations of dasatinib or 10 μM cytochalasin D, followed by stimulation with 1 mg/ml MSU crystals. (A) Luminometric measurement of reactive oxygen production. (B and C) LTB4 release determined from cell-free supernatants after 18 h incubation with the crystals of unprimed (B) or TNF-primed (10 ng/ml TNF; C) neutrophils. (D) Analysis of intracellular p38/ERK phosphorylation. (E–H) Phagocytosis of the MSU crystals assessed by flow cytometry (E and G) or phase contrast microscopy (F and H; scale bar: 20 µm). Panels D, E, and F show representative images. Bar graph shows mean and SEM from four to six (A), two to three (B and C), and three (G and H) independent experiments. RLU, relative luminescence unit; AUC, area under the curve. Two-way ANOVA inhibitor × stimulus interaction analysis (A–C and G) and one-way ANOVA (H); n.s., not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001. See the text for actual P values. Source data are available for this figure: SourceData F5.
We have also tested the effect of dasatinib on MSU crystal–induced NET formation by mouse neutrophils in the assay system described above. As shown in Fig. S4, C and D, MSU crystal–induced NET formation was not affected by 100 nM dasatinib (P = 0.99).
Taken together, dasatinib potently inhibited MSU crystal–induced inflammatory responses and crystal phagocytosis by human neutrophils, but did not substantially affect the intrinsic ability of the cells to produce superoxide or to form NETs upon activation by MSU crystals.
Dasatinib inhibits MSU crystal–induced in vivo arthritis
We have also tested the effect of dasatinib on MSU crystal–induced in vivo arthritis. Mice were treated with 50 mg/kg dasatinib or vehicle every 12 h by oral gavage. In a prophylactic setting (Fig. 6, A–H), dasatinib administration began 1 h before MSU crystal injection. Kinetic analysis of the paw volume measured by plethysmometry (Fig. 6 A) revealed greatly reduced paw swelling in dasatinib-treated mice compared to vehicle-treated animals (P = 1.4 × 10−7). This was confirmed at the 24 h time point in a larger cohort of mice (Fig. 6 B; P = 4.5 × 10−6). We have also tested MSU crystal–induced in vivo ROS production by bioluminescence imaging 24 h after MSU crystal injection. As shown in Fig. 6, C and D, MSU crystals caused dramatic increase of in vivo MPO activity in the absence of dasatinib, and this response was completely abolished by dasatinib treatment (P = 8.9 × 10−5). As shown in Fig. 6 E, MSU-induced neutrophil infiltration was also greatly reduced by dasatinib treatment (P = 1.9 × 10−6). Dasatinib administration also reduced IL-1β (Fig. 6 F; P = 0.016), MIP-2 (Fig. 6 G; P = 0.0021), and LTB4 (Fig. 6 H) levels at the site of MSU crystal injection, indicating defective accumulation of pro-inflammatory mediators at the inflammatory sites.

Dasatinib inhibits MSU crystal–induced arthritis in vivo. (A– I ) Mice were treated with or without 50 mg/kg dasatinib every 12 h, starting either 1 h prior (A–H) or 4 h after (I) injection of MSU crystals into the hind paws of the experimental animals as described in Figs. 2 and 3. (A, B, and I) Arthritis was followed by measuring paw swelling using a plethysmometer. (C and D) In vivo MPO activity was determined by chemiluminescence imaging. Representative color-coded photon flux intensity superposed on the grayscale photo of the mice (C) and quantified in defined regions of interest (D). (E–H) Analysis of the inflammatory microenvironment by flow cytometry (E) or ELISA (F–H). Panels B–H show results obtained 24 h after MSU crystal injection. Panels C and D show representative color-coded photon flux intensity superposed on the grayscale photo of the mice (C) and quantified in defined regions of interest (D). Kinetic curves and bar graphs show mean and SEM from 9 to 14 (A), 14–24 (B), 7 (D), 4–15 (E–H), and 14–24 (I) mice per group from 3 (A), 2 (D), 3–5 (E–H), and 3–5 (I) independent experiments. Two-way ANOVA inhibitor × stimulus interaction analysis; n.s., not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001. See the text for actual P values.
Dasatinib inhibits MSU crystal–induced arthritis in vivo. (A– I ) Mice were treated with or without 50 mg/kg dasatinib every 12 h, starting either 1 h prior (A–H) or 4 h after (I) injection of MSU crystals into the hind paws of the experimental animals as described in Figs. 2 and 3. (A, B, and I) Arthritis was followed by measuring paw swelling using a plethysmometer. (C and D) In vivo MPO activity was determined by chemiluminescence imaging. Representative color-coded photon flux intensity superposed on the grayscale photo of the mice (C) and quantified in defined regions of interest (D). (E–H) Analysis of the inflammatory microenvironment by flow cytometry (E) or ELISA (F–H). Panels B–H show results obtained 24 h after MSU crystal injection. Panels C and D show representative color-coded photon flux intensity superposed on the grayscale photo of the mice (C) and quantified in defined regions of interest (D). Kinetic curves and bar graphs show mean and SEM from 9 to 14 (A), 14–24 (B), 7 (D), 4–15 (E–H), and 14–24 (I) mice per group from 3 (A), 2 (D), 3–5 (E–H), and 3–5 (I) independent experiments. Two-way ANOVA inhibitor × stimulus interaction analysis; n.s., not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001. See the text for actual P values.
We have also tested the effect of dasatinib in a therapeutic setting, administering the first oral dasatinib dose 4 h after MSU crystal injection, allowing the initial inflammation to occur in the absence of dasatinib. As shown in Fig. 6 I, such therapeutic oral dasatinib treatment rapidly attenuated paw swelling (note immediate decline between the 4.5 h peak and the next, 6 h measurement) and it significantly reduced overall paw volume increase during the observed period (P = 0.0098), reaching a level of inhibition comparable to that triggered by the prophylactic administration protocol toward the end of the experiment (compare Fig. 6, A and I).
These results indicate that prophylactic or therapeutic treatment with dasatinib attenuates MSU crystal–induced in vivo inflammatory reactions, also pointing to potential novel therapeutic strategies.
The role of myeloid Src-family kinases in the motheaten viable phenotype
To test the role of myeloid Src-family kinases in an entirely different, genetically determined autoinflammatory reaction, we have crossed the Hck−/−Fgr−/−Lyn−/− and the Ptpn6me-v/me-v (motheaten viable) mice. As shown in Fig. 7 A, Ptpn6me-v/me-v mice showed severe spontaneous inflammatory arthritis of the fingers of their hind limb typical for the motheaten viable mutation. Remarkably, this inflammatory reaction was completely abrogated by crossing the Ptpn6me-v/me-v mutation to the Hck−/−Fgr−/−Lyn−/− background (Fig. 7 A). The Ptpn6me-v/me-v mice also developed severe lung inflammation characterized by a massive infiltration of leukocytes (supposedly mainly neutrophils) into the lung tissue (Fig. 7 B) and substantial increase of the lung weight (Fig. 7 C). Importantly, both the leukocytic infiltration (Fig. 7 B) and the increase of the lung weight (Fig. 7 C; P = 7.7 × 10−5) was dramatically reduced by the Hck−/−Fgr−/−Lyn−/− mutation. The Ptpn6me-v/me-v mutation also caused early lethality of the affected mice with a median survival of 7.0 wk (Fig. 7 D). Crossing the Ptpn6me-v/me-v mutation to the Hck−/−Fgr−/−Lyn−/− background nearly doubled the median survival of the mice to 13.5 wk (Fig. 7 D; P = 0.00001). Therefore, myeloid Src-family kinases play a critical role in the spontaneous inflammation and early lethality of motheaten viable mice.

The Hck −/− Fgr −/− Lyn −/− mutation attenuates inflammation in the motheaten viable model. (A) Representative images of the hind paws of WT, Ptpn6me-v/me-v, and Ptpn6me-v/me-vHck−/−Fgr−/−Lyn−/− mice. (B and C) H&E-stained sections (B; upper panels: 10× magnification, scale bar, 100 µm; lower panels: 40× magnification, scale bar, 25 µm) and weight (C) of lungs from Ptpn6me-v/me-v and Ptpn6me-v/me-vHck−/−Fgr−/−Lyn−/− mice. (D) Survival of the mice of the indicated genotypes. (E–G) neutrophils isolated from mice of the indicated genotypes were stimulated on fibrinogen surface (E and G) or by using 1 nM PMA (F), and superoxide release (E and F) or the percentage of cellular adhesion (G) was determined. Panels A and B show representative images from four independent experiments. Bar graphs show mean and SEM of 15–19 mice per genotype (C) and mean and SEM from three independent experiments (G). Survival curves show the data of 10–29 animals per genotype. Kinetic curves on panels E and F show representative experiments performed in triplicate (mean and SEM) from four independent experiments. Cells were prepared from three to four mice per genotype and pooled for each experiment. One-way ANOVA (C and G), log-rank test (D); n.s., not significant; ***, P < 0.001. See the text for actual P values.
The Hck −/− Fgr −/− Lyn −/− mutation attenuates inflammation in the motheaten viable model. (A) Representative images of the hind paws of WT, Ptpn6me-v/me-v, and Ptpn6me-v/me-vHck−/−Fgr−/−Lyn−/− mice. (B and C) H&E-stained sections (B; upper panels: 10× magnification, scale bar, 100 µm; lower panels: 40× magnification, scale bar, 25 µm) and weight (C) of lungs from Ptpn6me-v/me-v and Ptpn6me-v/me-vHck−/−Fgr−/−Lyn−/− mice. (D) Survival of the mice of the indicated genotypes. (E–G) neutrophils isolated from mice of the indicated genotypes were stimulated on fibrinogen surface (E and G) or by using 1 nM PMA (F), and superoxide release (E and F) or the percentage of cellular adhesion (G) was determined. Panels A and B show representative images from four independent experiments. Bar graphs show mean and SEM of 15–19 mice per genotype (C) and mean and SEM from three independent experiments (G). Survival curves show the data of 10–29 animals per genotype. Kinetic curves on panels E and F show representative experiments performed in triplicate (mean and SEM) from four independent experiments. Cells were prepared from three to four mice per genotype and pooled for each experiment. One-way ANOVA (C and G), log-rank test (D); n.s., not significant; ***, P < 0.001. See the text for actual P values.
Myeloid Src-family kinases in spontaneous activation of Ptpn6me-v/me-v neutrophils
Finally, we have tested the effect of the Hck−/−Fgr−/−Lyn−/− mutation on the spontaneous activation of neutrophils isolated from motheaten viable mice. As shown in Fig. 7 E, Ptpn6me-v/me-v neutrophils showed substantially increased superoxide release when plated on a fibrinogen surface in the absence of any additional stimulus. This spontaneous activation was completely abrogated by the Hck−/−Fgr−/−Lyn−/− mutation (Fig. 7 E). In contrast, superoxide production triggered by the PKC-activating agent PMA was not affected (Fig. 7 F), arguing against an intrinsic defect of the NADPH-oxidase complex. Ptpn6me-v/me-v neutrophils also showed increased spontaneous adhesion to the adherent surface (Fig. 7 G). This phenotype was also completely abrogated by the Hck−/−Fgr−/−Lyn−/− mutation (Fig. 7 G; P = 0.0001). Therefore, myeloid Src-family kinases are also critical for the spontaneous activation of neutrophils from motheaten viable mice.
Discussion
Autoinflammatory diseases are a group of diverse diseases characterized by severe inflammatory reactions without a substantial adaptive immune (autoimmune) component. They include a large number of diverse monogenic autoinflammatory diseases, as well as acquired autoinflammatory diseases such as gout. Using a combined genetic and pharmacological approach on human and mouse cells and in vivo models, here we identify the Src-family kinases Hck, Fgr, and Lyn as critical components of MSU crystal–induced inflammation, as well as of the severe wasting phenotype in the motheaten viable mice. Given the autoinflammatory characteristics of both of those models, our results suggest a major role for myeloid Src-family kinases in certain autoinflammatory disease processes.
Current classification of monogenic autoimmune diseases includes three well-defined groups characterized by augmented inflammasome activation (inflammasomopathies), enhanced interferon signaling (interferonopathies), and enhanced NF-κB pathway activation (relopathiles), with a fourth group comprised of a list of highly diverse other diseases. We have previously shown that mice lacking Src-family kinases, Syk, or PLCγ2 show very similar in vitro neutrophil (Mócsai et al., 2002; Mócsai et al., 1999; Jakus et al., 2009; Kovács et al., 2014; Németh et al., 2016) and in vivo inflammatory (Jakus et al., 2009; Jakus et al., 2010; Kovács et al., 2014; Németh et al., 2017; Németh et al., 2018; Futosi et al., 2021; Szilveszter et al., 2022) phenotypes, and that Syk and PLCγ2 are likely downstream components of signaling by Src-family kinases in neutrophils (Mócsai et al., 2002; Jakus et al., 2009; Futosi and Mócsai, 2016). Those results point to the existence of a Src-family–Syk–PLCγ2 axis (Berton et al., 2005; Mócsai et al., 2010; Németh and Mócsai, 2012; Futosi et al., 2013; Futosi and Mócsai, 2016; Szilveszter et al., 2019; Németh et al., 2020). Importantly, a number of prior reports indicate that certain gain-of-function mutations of PLCγ2 cause spontaneous systemic inflammation (autoinflammation) in humans (APLAID; autoinflammation with PLCγ2-associated antibody deficiency and immune dysregulation syndrome) and mice (Zhou et al., 2012; Jackson et al., 2021; Yu et al., 2005; Abe et al., 2011). A recent paper has also identified gain-of-function mutations in Syk as the likely causative agent of systemic inflammation in human patients and in mice carrying the most severe human mutation (Wang et al., 2021). In addition, gain-of function mutations in Hck (Kanderova et al., 2022; Ernst et al., 2002), Fgr (Abe et al., 2019), or Lyn (Louvrier et al., 2023; de Jesus et al., 2023; Hibbs et al., 2002) cause spontaneous inflammation in pediatric patients or experimental mice. We have also previously shown that the severe early inflammatory reactions of the motheaten mice are caused by SHP-1 deletion in the neutrophil lineage, likely due to excessive activation of tyrosine phosphorylation pathways in those cells (Abram et al., 2013). Our current results, together with those prior reports, delineate a putative novel class of autoinflammatory diseases characterized by excessive activation of tyrosine kinase signaling in neutrophils, either due to increased activity of Src-family kinases, Syk, or PLCγ2, or to reduced activity of the negative regulator SHP-1.
The current therapy of gout relies on urate lowering therapies and treatment of acute gouty attacks using anti-inflammatory drugs. Our results raise the possibility that tyrosine kinase inhibitors acting on Src-family kinases may also have therapeutic benefit in acute gouty attacks. The apparent lack of effect of the Hck−/−Fgr−/−Lyn−/− mutation and of dasatinib on NET formation may possibly provide a unique additional benefit, given the proposed anti-inflammatory role of NET formation during MSU crystal–induced inflammation (Reinwald et al., 2016; Schauer et al., 2014). In addition, inhibitors of Src-family kinases may also have therapeutic benefit in other autoinflammatory diseases with a significant role of enhanced tyrosine kinase signaling in neutrophils.
It is still mostly unclear how Src-family kinases contribute to neutrophil activation during a gouty attack and the inflammatory changes in motheaten viable mice. Neutrophils fail to accumulate at the site of gouty inflammation, and this is also likely the case in various organs of motheaten viable mice. A plausible explanation could be the inability of Hck−/−Fgr−/−Lyn−/− neutrophils to migrate to the site of inflammation. However, our MSU-induced arthritis experiments in mixed bone marrow chimeras, along with our previous studies (Mócsai et al., 2002; Kovács et al., 2014), argue against that possibility. An alternative explanation could be that Src-family kinases are required for the release of inflammatory mediators from neutrophils, thus contributing to the inflammation process. Our in vitro and in vivo experiments on MSU crystal–induced changes in inflammatory mediators are in line with that possibility, and such a scenario may also be relevant to the motheaten viable phenotype.
Some of our results raise further questions that may necessitate additional experiments. The role of Src-family kinases in macrophages during MSU crystal–induced inflammation is still unclear, given the differential effect of the Hck−/−Fgr−/−Lyn−/− mutation on the various macrophage responses and the potential difference between bone marrow–derived and true tissue macrophages. It would be important to know whether the remaining MSU crystal–induced inflammatory response in Hck−/−Fgr−/−Lyn−/− mutants is caused by partially normal macrophage responses. The individual contribution of the three kinases also requires further assessment, especially given the tendency of Lyn−/− mutants for augmented responses to MSU crystals, despite complete defect in Hck−/−Fgr−/−Lyn−/− mutants. A possible scenario is a dual (positive and negative) role for one or more Src-family kinases during MSU-induced inflammation, with differential “penetrance” of the lack of the positive and the negative signaling role in the respective single or combined knockouts.
There are a number of further limitations of our study. Additional lineage-specific experiments will be required to better define the lineage(s) utilizing Hck, Fgr, and Lyn during the pathogenesis of gout. Similarly, the receptors recognizing MSU crystals through Src-family kinase signaling need to be identified, also considering prior studies on the role of β2-integrins and Fc-receptors (Popa-Nita and Naccache, 2010), as well as the possibility of a direct cell activation (Zhang et al., 2009). The interaction between Src-family kinases and the NLRP3 or other inflammasomes needs to be addressed in more detail, particularly since our preliminary experiments suggest a rather complex picture. Besides neutrophil-mediated early inflammation, the motheaten mice also develop chronic autoimmunity, likely due to SHP-1 deletion in dendritic cells (Abram et al., 2013). Dendritic cell–specific SHP-1 deletion in Hck−/−Fgr−/−Lyn−/− mice will need to be performed to test the role of Src-family kinases in the autoimmune disease component. Our current experiments cannot exclude the possibility that the in vitro or in vivo effects of dasatinib are at least in part due to inhibition of Abl-family tyrosine kinases. This should be tested using single and combined mutants lacking the Abl-family tyrosine kinases Abl (Abl1) and/or Arg (Abl2). Finally, more detailed experiments will need to be performed to test the role of Src-family kinases in NET formation.
Taken together, we have identified a novel crucial role for the myeloid Src-family kinases Hck, Fgr, and Lyn in two widely used models of autoinflammatory diseases. Together with other very recent reports, our results suggest that excessive activation of tyrosine phosphorylation pathways may delineate a novel class of autoinflammatory diseases.
Materials and methods
Animals
Mice homozygous for the Hcktm1Hev, Fgrtm1Hev, or Lyntm1Sor mutations (referred to as Hck−/−, Fgr−/−, and Lyn−/−, respectively; Lowell, 2004; Chan et al., 1997) were used to generate all possible double and triple knockout combinations (Kovács et al., 2014). All mice were apparently healthy and bred normally in homozygous form, except for the Hck−/−Lyn−/− mutants which showed poor viability, as reported before (Kovács et al., 2014; Xiao et al., 2008). CD18-deficient (Itgb2tm2Bay/tm2Bay, referred to as Itgb2−/−) mice were obtained from Arthur Beaudet (Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA; Scharffetter-Kochanek et al., 1998) and were maintained by crossing Itgb2−/− and Itgb2+/− mice. Ptpn6me-v/me-v (also known as motheaten viable) mice were described previously (Shultz et al., 1984) and used to generate Ptpn6me-v/me-vHck−/−Fgr−/−Lyn−/− quadruple mutant mice. Genotyping was performed by allele-specific PCR. All mice were on the C57BL/6 genetic background. Despite extensive (∼15 generations) backcrosses to the C57BL/6 background, all mice carrying the Hck– allele retained an agouti coat color from the 129 background, likely due to the close genetic linkage of the Hck and the “a” (nonagouti) genes on mouse chromosome 2. WT C57BL/6 animals were used as controls. Both males and females were used for experiments. Mice were mostly used at 8–15 wk of age, except for the Hck−/−Lyn−/− mutants, which were used at 6–7 wk of age because of their poor health status. The mice were kept in individually sterile ventilated cages (Tecniplast). Breeding was performed in a specific pathogen–free facility, whereas experiments were done in a conventional facility.
All animal experiments were approved by the Animal Experimentation Review Boards of Semmelweis University, the University of Pécs, or the University of California, San Francisco.
Isolation of neutrophils and macrophages
Mouse neutrophils were isolated from bone marrow of the femurs and tibias of the mice by hypotonic lysis of red blood cells followed by Percoll (GE Healthcare) gradient centrifugation (Mócsai et al., 2003; Németh et al., 2016). Human neutrophils were isolated from the venous blood of healthy volunteers by Ficoll gradient centrifugation followed by hypotonic lysis (Futosi et al., 2012; Mócsai et al., 2000). Cells were resuspended in Ca2+- and Mg2+-free HBSS (Lonza) supplemented with 20 mM Hepes (Sigma-Aldrich), pH 7.4 (referred to as assay medium), and kept at room temperature until use (up to 30 min).
To obtain mouse bone marrow–derived macrophages, bone marrow cells were cultured in α-MEM (Sigma-Aldrich) complemented with 10% FCS (Invitrogen), 2 mM L-glutamine, antibiotics, 10 mM Hepes (all from Sigma-Aldrich), and 10% conditioned medium from CMG 14-12 cells (Kertész et al., 2012) as a source of recombinant murine M-CSF for 8–10 d on bacterial dishes with changing the media every 2–3 d. Cells were suspended by 5 mM EDTA, washed, and serum-starved for 2 h before the experiments.
Cells were diluted in assay medium, supplemented with 0.5 mM CaCl2 (Sigma-Aldrich) and then prewarmed to 37°C for 10 min prior to stimulation. Unless otherwise stated, 1 mM MgCl2 (Sigma-Aldrich) was added immediately before cell activation. Sterile or endotoxin-free reagents were used throughout the experiments.
In pharmacological studies, cells were pretreated with the tyrosine kinase inhibitor dasatinib (Selleck Chemicals) or DMSO (Sigma-Aldrich) at the indicated concentrations for 30 min.
Preparation of MSU crystals and cell activation
MSU crystals were prepared under pyrogen-free conditions as previously described (Vieira et al., 2015). Briefly, uric acid (Sigma-Aldrich) was dissolved in 0.01 M NaOH (Sigma-Aldrich) at pH 7.1. The solution was filtered using a 0.22 µm filter and kept at room temperature on a rocking shaker for ∼3 d. The crystals formed were washed with 100% ethanol, reduced in size by sonication, and dried under sterile condition. The crystals were stored at room temperature and resuspended in PBS before use.
Plate-based activation of neutrophils and macrophages was performed in white (Greiner Bio-One) or clear (Nunc MaxiSorp) microplates, or tissue culture-treated Corning plates. Cells were plated on 10% FCS (Invitrogen) pre-coated surface, and stimulated with the indicated concentration of MSU crystals (1 mg/ml, unless otherwise stated).
Neutrophil activation in suspension was performed in polypropylene tubes or in FCS-coated wells in the absence of MgCl2 by stimulating the cells with 1 mg/ml MSU crystals.
Functional assays and biochemical studies
For MSU crystal–induced respiratory burst assays, cells were supplemented with 50 µg/ml lucigenin (Sigma-Aldrich), 4 × 105/well neutrophils or macrophages or 105/well human neutrophils were plated on 96-well plates, stimulated with MSU crystals, and their chemiluminescence was followed by a Thermo Fisher Scientific Varioskan Flash multimode plate reader. Alternatively, cell suspensions were supplemented with 100 nM ferricytochrome-c (Sigma-Aldrich), 4 × 105/well mouse or 105/well human neutrophils were plated on 96-well plates, stimulated with 100 nM PMA (Sigma-Aldrich), and their superoxide release was monitored spectrophotometrically using a cytochrome c reduction assay (Mócsai et al., 2002; Jakus et al., 2008; Futosi et al., 2012). For analysis of cytokine release, 500-µl aliquots of 4 × 106/ml neutrophils, 3 × 106/ml macrophages, or 106/ml vehicle- or dasatinib-treated human neutrophils were suspended in DMEM (Sigma-Aldrich), plated on 24-well tissue culture plate (Corning) and incubated for 18 or 24 h, respectively, with 1 mg/ml MSU crystals. Macrophages were primed by 1 µg/ml ultrapure LPS (InvivoGen) overnight, whereas vehicle- or dasatinib-treated human neutrophils were primed by 10 ng/ml recombinant human TNF (PeproTech) for 30 min prior to MSU crystal stimulation. The analysis of inflammatory mediators from the cell-free supernatant was performed as described below.
For phagocytosis assays, mouse or human neutrophils were suspended in Mg2+-free assay medium, preincubated with the indicated inhibitors (100 nM dasatinib or 10 μM cytochalasin D) or vehicle, and then further incubated with 1 mg/ml MSU crystals for 10 min at 37°C. The reaction was then stopped on ice. Mouse neutrophils were then stained for Ly6G antibody (clone 1A8; BD Biosciences) while human neutrophils were stained for biotinylated CD11b antibody (clone M1/70) followed by streptavidin-PerCP-Cy5.5 (both from BD Biosciences) and analyzed by flow cytometry, using a BD Biosciences FACSCalibur and the CellQuest software. Phagocytosis of MSU crystals by human neutrophils was also visible under phase-contrast microscopy and was therefore also analyzed on a Leica DMI 6000B inverted microscope with a 20× phase-contrast objective.
For the analysis of intracellular protein phosphorylation, cells were incubated with 1 mg/ml MSU crystals for the indicated time, followed by lysis using a Triton X-100-based lysis buffer containing 100 mM NaCl, 30 mM Na-Hepes (pH 7.4), 20 mM NaF, 1 mM Na-EGTA, 1% Triton X-100, and 1 mM benzamidine, freshly supplemented with protease and phosphatase inhibitors (0.1 U/ml Aprotinin, 1:100 Mammalian Protease Inhibitor Cocktail, 1:100 Phosphatase Inhibitor Cocktail 2, 1 mM phenylmethylsulphonyl fluoride, and 1 mM Na3VO4; all from Sigma-Aldrich; Jakus et al., 2009). 4× Laemmli’s sample buffer was added to the lysate supernatants and boiled for 10 min. Lysates were then run on SDS-PAGE, blotted to nitrocellulose membranes and probed with phospho-specific (Cell Signaling) or non-phospho-specific (Santa Cruz) antibodies against the ERK and p38 MAPKs, followed by peroxidase-labeled secondary antibodies (GE Healthcare) as described (Jakus et al., 2009; Mócsai et al., 2002; Németh et al., 2016). Signal was developed using the ECL system (GE Healthcare) and exposed to x-ray film.
For the analysis of NET formation, neutrophils were first stained with 0.5 μg/ml Hoechst 33342 dye (Invitrogen) in PBS for 20 min (nuclear staining), followed by addition of 0.2 mg/ml MSU crystals along with 50 nM Sytox-Green DNA (Invitrogen) to visualize extracellular DNA. The cells were added to 96-well plates (Greiner Bio-One) and were imaged using an ImageXpress Micro Confocal High Content Imaging System (Molecular Devices) with images taken every 10 or 15 min for 3 h at 37°C. Fluorescence images were taken using excitation and emission filter pairs of 377/50 and 447/60 nm (Hoechst) or 474/27 and 525/45 nm (Sytox Green). Quantitative analysis was performed using the ImageXpress device’s built-in analysis software.
For experiments with dasatinib, human or mouse neutrophils were preincubated with 100 nM or the indicated concentration of dasatinib, or 0.1% DMSO for vehicle control for 30 min at 37°C prior to the assay procedure.
MSU-induced arthritis
MSU-induced gouty arthritis was induced by injection of 1 mg MSU crystals in 50 µl PBS into the hind paw tissue at the level of the metatarsal bones of experimental mice, with PBS alone injected into the other hind paw as control. Paw swelling was then followed by a digital plethysmometer (Borbely et al., 2015; Botz et al., 2014; Ugo Basile). For analyzing the number of infiltrating neutrophils and the accumulation of inflammatory mediators, the experimental animals were sacrificed 24 h after MSU crystal injection and their hind paws were flushed with 1 ml PBS supplemented with 5 mM EDTA (pH 7.5) and 20 mM Hepes (pH 7.4). The samples were centrifuged and the pellets and supernatants were processed separately. The pellets were resuspended in PBS containing 5% FCS and stained with antibodies against Ly6G (clone 1A8), CD11b (M1/70) or F4/80 (T45-2342; all from BD Biosciences) and the number of neutrophils and macrophages was determined by flow cytometry. The cell-free supernatants were used for the analysis of inflammatory mediator release (see below).
In our pharmacological experiments, 50 mg/kg dasatinib (suspended in mucilago:water mixture) or vehicle was administered to the experimental mice per os every 12 h. Treatment was initiated 1 h before (prophylactic setting) or 4 h after (therapeutic setting) MSU crystal injection. In a few cases of prophylactic administration, another dasatinib dose was administered 13 h before MSU crystal injection; this prior treatment did not affect the effect of dasatinib.
Analysis of inflammatory mediators
The release of inflammatory mediators was determined from the cell-free supernatants of tissue infiltrates collected on day 1 or of in vitro activated neutrophils and macrophages. Qualitative analysis of the inflammatory chemokines and cytokines was performed using a mouse cytokine antibody array kit (Panel A from R&D Systems) according to the manufacturer’s instructions. The levels of IL-1β, MIP-2 and LTB4 were tested by commercial ELISA kits (R&D Systems) according to the manufacturer’s instructions.
In vivo imaging of ROS production
Luminol and lucigenin chemiluminescence assays were used to detect in vivo myeloperoxidase and NADPH-oxidase activity, respectively, as described previously (Gross et al., 2009; Tseng and Kung, 2012). Briefly, mice were anesthetized with 120 mg/kg ketamine (Gedeon Richter) and 6 mg/kg xylazine (Eurovet Animal Health) i.p. and injected i.p. with 150 mg/kg luminol sodium salt (Gold Biotechnology) dissolved in PBS or with 25 mg/kg lucigenin (Tokyo Chemical Industry) dissolved in saline at the indicated time points after MSU crystal administration. Bioluminescence images were captured 10 min after injection using IVIS Lumina III (PerkinElmer) and processed by Living Image software (PerkinElmer). Total radiance values (total photon flux/s) from standardized region of interests of hind paws were used for quantitative analysis (Kovács et al., 2014; Botz et al., 2014).
In vivo functional tests
Mechanonociceptive thresholds of the hind paws were determined by measuring withdrawal using dynamic plantar esthesiometry (Ugo Basile). Increasing force (maximal value of 10 g) was applied to the plantar surfaces until the mice withdraw their affected paw before the induction of the gouty arthritis (baseline) and then 6 and 24 h later, as described previously (Szabó et al., 2005). The threshold of each paw was measured three times at each indicated time point. Mechanical hypersensitivity was expressed as a percentage compared to the initial control thresholds.
Dynamic weight bearing (Bioseb) on the hind limbs was measured before the induction of gouty arthritis and then 6 and 24 h later. Mice were placed into the dynamic weight bearing enclosure on a pressure-sensitive floor where they moved freely for 5 min during video monitoring. Weight bearing on the hind limb was calculated as a percentage of the total weight bearing on the hind limbs, as described previously (Tékus et al., 2018).
Competitive in vivo migration experiments
CD45.1-expressing WT bone marrow cells were mixed with CD45.2-expressing WT, CD18-deficient (Itgb2−/−), or Hck−/−Fgr−/−Lyn−/− bone marrow cells at varying (2:3, 1:1, or 3:2) ratios and injected into lethally irradiated CD45.1-expressing WT recipients to generate mixed bone marrow chimeras as described before (Kovács et al., 2014). 5–8 wk later, the mixed bone marrow chimeras were subjected to MSU crystal–induced arthritis (day 0). Peripheral blood samples were taken on days −1, 0, and 1. Experimental mice were sacrificed 24 h after MSU crystal injection and their hind paws were flushed as described above. The cells in the paw tissue infiltrates were analyzed by flow cytometry as described above using antibodies against Ly6G (clone 1A8) and CD45.2 (clone 104, both from BD BioSciences). Relative migration of CD45.2-expressing neutrophils (relative to CD45.1-expressing WT cells) was calculated as described (Kovács et al., 2014).
Analysis of motheaten viable mice
The survival and disease state of WT, Ptpn6me-v/me-v single, Hck−/−Fgr−/−Lyn−/− triple, and Ptpn6me-v/me-vHck−/−Fgr−/−Lyn−/− quadruple mutant mice were followed. To test lung inflammation, the lungs of the mice were weighed, followed by inflation, formalin fixation, embedding in paraffin, sectioning, and staining with H&E. Neutrophils were isolated, plated on a fibrinogen-coated surface or stimulated with 1 nM PMA, and superoxide production was measured spectrophotometrically using a cytochrome c reduction assay, as described (Abram et al., 2013). For the analysis of cellular adhesion, neutrophils were isolated and labeled with Cell-Tracker Green (Molecular Probes), then plated onto fibrinogen-coated plates for 30 min at 37°C. The plates were then washed and cell adhesion was quantified using a fluorescence microscope.
Presentation of data and statistical analysis
The number of experiments are indicated in the figure legends for each panel. Unless otherwise stated, quantitative graphs and kinetic curves show mean and SEM from all independent in vitro experiments and from all individual mice from the indicated number of in vivo experiments. Unless otherwise stated, statistical analyses were carried out using two-way (factorial) ANOVA, with stimulation and genotype/inhibitor treatment being the two independent variables. Unless otherwise stated, P values mentioned in the text refer to the difference between WT vs. Hck−/−Fgr−/−Lyn−/− or Itgb2−/−, or vehicle- vs. dasatinib-treated samples. In case of kinetic assays, area under the curve was used for statistical analysis. Survival studies were analyzed by the Kaplan-Meier approach and log-rank test. P values below 0.05 were considered statistically significant.
Ethics statement
All animal experiments were approved by the Animal Experimentation Review Board of Semmelweis University, the University of Pécs, or the University of California, San Francisco.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Online supplemental material
Fig. S1 shows PMA-induced superoxide production from WT or Hck−/−Fgr−/−Lyn−/− neutrophils and from vehicle- or dasatinib-treated human neutrophils. Fig. S2 shows MSU crystal–induced superoxide production, IL-1β and MIP-2 release from WT or Hck−/−Fgr−/−Lyn−/− bone marrow–derived macrophages. Fig. S3 shows representative flow cytometric histograms of competitive in vivo migration experiments. Fig. S4 shows MSU crystal–induced DNA externalization by WT or Hck−/−Fgr−/−Lyn−/− and by vehicle- or dasatinib-treated mouse neutrophils.
Acknowledgments
We thank Edina Simon, Nikolett Szénási, Dorottya Deli, and Tamás Kiss for expert technical assistance; Miklós Vértes for help with the initial experiments; and Arthur Beaudet for the Itgb2−/− mice. In vivo imaging was performed in collaboration with the Animal Imaging Core Facility at the Szentágothai Research Centre of the University of Pécs.
This work was supported by the Hungarian Ministry of National Economy (VEKOP-2.3.2-16-2016-00002 to A. Mócsai), the Hungarian National Research, Development and Innovation Fund (KKP-129954 and TKP2021-EGA-24 to A. Mócsai, FK132251 and TKP2021-EGA-29 to T. Németh, K-138046 and TKP2021-EGA-16 to Z. Helyes), the European Social Fund (EFOP-3.6.1.-16-2016-00004), the European Union H2020 IMI2 program (RTCure project No. 777357), the János Bolyai Research Scholarship of the Hungarian Academy of Sciences (to K. Futosi and T. Németh), the New National Excellence Program of the Ministry for Innovation and Technology (UNKP-20-5-SE-4 and UNKP-21-5-SE-2 to T. Németh), and the National Brain Research Program (2017-1.2.1-NKP-2017-00002 to Z. Helyes). S. Tusnády was supported by the Hungarian National Academy of Scientist Education.
Author contributions: K. Futosi and A. Mócsai conceived the study, designed the experiments, analyzed and interpreted the data, and wrote the manuscript. K. Futosi performed most of the experiments. T. Németh provided intellectual advice and contributed to in vivo and phagocytosis experiments. S. Tusnády performed the in vitro NET assays. Á.I. Horváth and Z. Helyes participated in the design, execution, and analysis of the imaging and in vivo functional measurements. C.L. Abram and C.A. Lowell designed and performed the experiments on the motheaten mice. A. Mócsai supervised the project.

