


SHP2 (PTPN11) acts upstream of SOS1/2 to enable RAS activation. Allosteric SHP2 inhibitors (SHP2i) in the clinic prevent SHP2 activation, block proliferation of RTK- or cycling RAS mutant-driven cancers, and overcome “adaptive resistance.” To identify SHP2i resistance mechanisms, we performed genome-wide CRISPR/Cas9 knockout screens on two SHP2i-sensitive cell lines, recovering genes expected to cause resistance (NF1, PTEN, CDKN1B, LZTR1, and RASA2) and novel targets (INPPL1, MAP4K5, epigenetic modifiers). We screened 14 additional lines with a focused CRISPR library targeting common “hits” from the genome-wide screens. LZTR1 deletion conferred resistance in 12/14 lines, followed by MAP4K5 (8/14), SPRED2/STK40 (6/14), and INPPL1 (5/14). INPPL1, MAP4K5, or LZTR1 deletion reactivated ERK signaling. INPPL1-mediated sensitization to SHP2i required its NPXY motif but not lipid phosphatase activity. MAP4K5 acted upstream of MEK through a kinase-dependent target(s); LZTR1 had cell-dependent effects on RIT and RAS stability. INPPL1, MAP4K5, or LZTR1 deletion also conferred SHP2i resistance in vivo. Defining the SHP2i resistance landscape could suggest effective combination approaches.
Introduction
The RAS/ERK cascade is a key signaling pathway downstream of receptor tyrosine kinases (RTKs), cytokine receptors, and integrins. Hyperactivation of this pathway, caused by gene amplifications, chromosomal abnormalities, or mutations, is among the most common oncogenic events in human cancer (Fernández-Medarde and Santos, 2011; Prior et al., 2012). Multiple inhibitors targeting pathway components, including aberrant RTKs (e.g., EGFR, HER2, and MET) or tyrosine kinase (TK) fusions (e.g., BCR-ABL, FLT3-ITD), KRASG12C or BRAFV600E, and WT MEK or ERK (Drosten and Barbacid, 2020; Lee et al., 2020), have been developed. These drugs can evoke dramatic tumor regressions in some patients and prolong their survival. Inevitably, however, resistance emerges, resulting in patient relapse and, in the absence of other, more durable modalities (e.g., immune therapies), patient demise (Labrie et al., 2022; Nagano et al., 2018; Rosenzweig, 2018; Ryan and Corcoran, 2018).
Mechanisms of resistance to RAS/ERK cascade inhibitors are multiple and complex (Labrie et al., 2022; Nagano et al., 2018; Rosenzweig, 2018; Ryan and Corcoran, 2018). Often, mutations in the target protein disable inhibitor binding. Alternatively, bypass mutations can occur in genes encoding downstream signaling components or components of parallel pathways. In addition, epigenetic alterations can induce a change in cell state (e.g., epithelial to mesenchymal transition, lineage switching) that renders malignant cells agnostic to the targeted oncogene. Some tumor cells treated with RAS/ERK pathway inhibitors exhibit “adaptive resistance,” in which ERK inhibition results in de-repression of MYC targets, including RTKs and their ligands, which drive increased upstream signaling and overcome inhibitor action (Ahmed et al., 2019; Fedele et al., 2021; Fedele et al., 2018; Hallin et al., 2020; Hao et al., 2019; Lou et al., 2019; Mainardi et al., 2018; Misale et al., 2019; Xue et al., 2020).
The protein tyrosine phosphatase SHP2, encoded by PTPN11, is required for full activation of the RAS/ERK pathway (Chan et al., 2008; Grossmann et al., 2010; Neel et al., 2003). SHP2 functions upstream of RAS through as yet unknown substrate(s) to promote the activity of the guanine nucleotide exchange factors SOS1/2, although it might also have a role in RAS-GAP regulation and/or in parallel pathways. For these reasons, SHP2 inhibition has been proposed as a therapeutic strategy for malignancies driven by RTKs or TK fusions, “cycling” KRAS mutants, or, in combination with other pathway inhibitors for combating adaptive resistance (Fedele et al., 2021; Fedele et al., 2018; Lou et al., 2019; Mainardi et al., 2018; Ryan and Corcoran, 2018; Sun et al., 2014). Recently, allosteric inhibitors of SHP2 (SHP2i) were developed, and several have entered clinical trials as monotherapies or in various combinations (Chen et al., 2016; Quintana et al., 2020; Sun et al., 2020; Yuan et al., 2020). Because prospective identification of potential routes to drug resistance could suggest more effective combination therapies, we sought to identify potential mechanisms of SHP2i resistance using genome-wide loss-of-function screens.
Results
Genome-wide loss-of-function screens for SHP099 resistance
To systemically identify key regulators whose loss leads to SHP2i resistance, we performed genome-wide CRISPR KO screens on two FLT3-ITD–driven human acute myeloid leukemia (AML) lines, MOLM13 and MV4-11. These lines were chosen based on their marked sensitivity to SHP2 inhibition. We transduced each line with the TKOv3 lentiviral CRISPR library, which targets 18,053 protein-coding genes with four guide RNAs per gene (Hart et al., 2017; Mair et al., 2019), at 1,000× representation per replicate (n = 2 biological replicate screens per cell line). Cells were then cultured in vehicle (DMSO) or with the tool inhibitor SHP099 at 7× IC50 for 12 doublings (Fig. 1 A). The prolonged culture period and high concentration of inhibitor was designed to enrich for bona fide drivers of drug resistance as opposed to synthetic lethal genes. At the end of the incubation period, genomic DNA (gDNA) was isolated from each sample and sequenced. Enrichment/dropout of individual genes was assessed by consistent effects on single-guide RNAs (sgRNAs) targeting the same gene using robust rank aggregation, as implemented in the MaGeCK (Li et al., 2014b) package (Fig. 1, B and C; and Table S1).

CRISPR screens for SHP099 resistance. (A) Workflow for genome-wide CRISPR KO screens. (B and C) Results of genome-wide CRISPR KO screens of MOLM13 (B) and MV4-11 (C) cells, analyzed by MaGeCK; red circles indicate enriched genes with FDR < 0.05, blue circles label enriched genes with 0.05 < FDR < 0.1, and green circles show genes with 0.1 < FDR < 0.2. (D) Workflow for focused CRISPR mini-screens of 14 cancer lines. (E and F) Results of CRISPR mini-screens of MOLM13 (E) and MV4-11 (F) cells. Points represent log2-fold enrichment (SHP099 vs. DMSO) of each sgRNA present in the library targeting the indicated gene compared with non-targeting (ctrl) sgRNAs; significance was assessed by Student’s t test with FDR correction. (G) Heat map showing significant hits in CRISPR mini-screens across 14 cancer lines. Yellow color indicates FDR < 0.05 enrichment for a given gene in a cell line.
CRISPR screens for SHP099 resistance. (A) Workflow for genome-wide CRISPR KO screens. (B and C) Results of genome-wide CRISPR KO screens of MOLM13 (B) and MV4-11 (C) cells, analyzed by MaGeCK; red circles indicate enriched genes with FDR < 0.05, blue circles label enriched genes with 0.05 < FDR < 0.1, and green circles show genes with 0.1 < FDR < 0.2. (D) Workflow for focused CRISPR mini-screens of 14 cancer lines. (E and F) Results of CRISPR mini-screens of MOLM13 (E) and MV4-11 (F) cells. Points represent log2-fold enrichment (SHP099 vs. DMSO) of each sgRNA present in the library targeting the indicated gene compared with non-targeting (ctrl) sgRNAs; significance was assessed by Student’s t test with FDR correction. (G) Heat map showing significant hits in CRISPR mini-screens across 14 cancer lines. Yellow color indicates FDR < 0.05 enrichment for a given gene in a cell line.
Resistance genes that scored in both cell lines included known negative regulators of the RAS/ERK pathway (e.g., NF1, SPRED2, etc.), negative regulators of parallel pathways (e.g., PTEN), negative regulators of the cell cycle (e.g., CDKN1B, FBXW7, RB1, etc.), and PTPRJ, which encodes a receptor tyrosine phosphatase reported to target FLT3 (Godfrey et al., 2012; Kresinsky et al., 2018). We also found more unexpected genes, including INPPL1, MAP4K5, BIRC6, and the epigenetic regulators SUV39H1 and WHSC1 (Fig. 1, B and C; and Table S1). Gene ontology (GO) analysis of the 50 top-ranked genes from MOLM13 and MV4-11 cells revealed enrichment for genes with the annotations “negative regulation of protein serine/threonine kinase activity,” “negative regulation of protein phosphorylation” (MOLM13 and MV4-11), “negative regulation of cell cycle G1/S phase transition” (MOLM13), “negative regulation of cyclin-dependent protein kinase activity” (MV4-11), and “negative regulation of RAS protein signal transduction” (MV4-11), among others (Fig. S1 A and Table S2). These results comport with known functions of the SHP2 pathway.
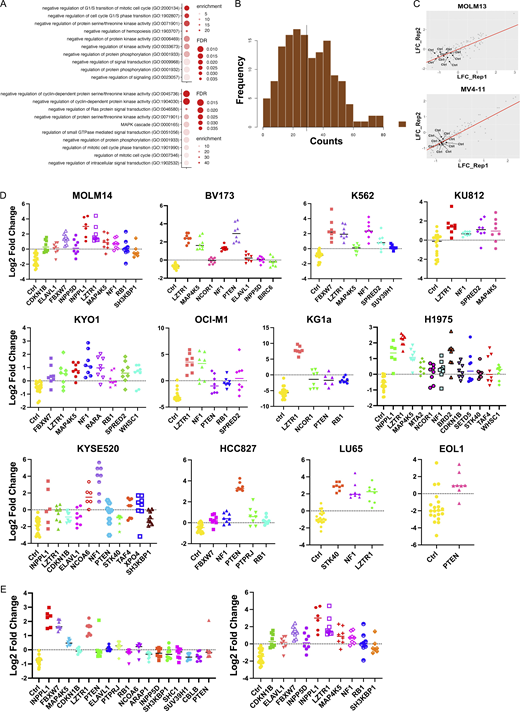
Focused CRISPR mini-screens identify genes conferring SHP099 resistance in 12 additional cell lines. (A) GO analysis of top 50 resistance genes from genome-wide MOLM13 (top panel) and MV4-11 (bottom panel) screens. Select pathways (colored red in Table S2) are shown. (B) sgRNA distribution in cloned CRISPR mini-library: 141/142 sgRNAs were detected. (C) Correlation between replicate CRISPR mini-screens of MOLM13 and MV4-11 cells. Log2-fold enrichment of each sgRNA is shown for treatment (SHP099 at 7× IC50 for each line) vs. vehicle (DMSO) groups at screen termination. All non-targeting sgRNAs (Ctrl) were depleted as expected. (D) Significantly enriched genes (FDR < 0.05) in CRISPR mini-screens of the indicated cancer cell lines. (E) Effect of SHP099 concentration on recovery of resistance genes in MOLM14 cells. Left: 5× IC50 (3 µM). Right: 7× IC50 (5 µM).
Focused CRISPR mini-screens identify genes conferring SHP099 resistance in 12 additional cell lines. (A) GO analysis of top 50 resistance genes from genome-wide MOLM13 (top panel) and MV4-11 (bottom panel) screens. Select pathways (colored red in Table S2) are shown. (B) sgRNA distribution in cloned CRISPR mini-library: 141/142 sgRNAs were detected. (C) Correlation between replicate CRISPR mini-screens of MOLM13 and MV4-11 cells. Log2-fold enrichment of each sgRNA is shown for treatment (SHP099 at 7× IC50 for each line) vs. vehicle (DMSO) groups at screen termination. All non-targeting sgRNAs (Ctrl) were depleted as expected. (D) Significantly enriched genes (FDR < 0.05) in CRISPR mini-screens of the indicated cancer cell lines. (E) Effect of SHP099 concentration on recovery of resistance genes in MOLM14 cells. Left: 5× IC50 (3 µM). Right: 7× IC50 (5 µM).
Focused CRISPR screens identify genes that confer SHP099 resistance in multiple cancer cell lines
We next asked how frequently resistance genes from MOLM13 and MV4-11 cells were shared across a larger panel of cancer cells bearing different driver mutations. To this end, we constructed a focused CRISPR “mini-library” comprising lentiviruses expressing sgRNAs targeting 33 common hits from the above screens and several control sgRNAs (refer to Materials and methods for details; Fig. S1 B and Table S3). 14 cancer cell lines were transduced with the mini-library in two independent experiments and cultured in vehicle or SHP099 (7× IC50) for 10 doublings (Fig. 1 D). Lines were chosen based on their PTPN11 dependency in DepMap (Dempster et al., 2019; Pacini et al., 2021), their observed sensitivity to SHP099 in our hands, and their diverse driver mutations (Table S3). Most of these were AML (n = 10, driven by FLT3-ITD, BCR-ABL, or JAK2V617F) lines; in addition, a few solid tumor lines driven by mutant EGFR or RASG12C were assessed (n = 4). The distribution of sgRNAs was determined by sequencing of gDNA, and enrichment of each gene at the experimental endpoint was evaluated by one-tailed Student’s t test with false discovery rate (FDR) < 0.05 considered significant. Replicates were well correlated (Fig. S1 C), and non-targeting (control) guides were depleted as expected (Fig. 1, E and F; and Fig. S1 D). Several of the common resistance hits were either tumor suppressor genes or encoded negative regulators of the cell cycle (e.g., PTEN, NF1, FBXW7, CDKN1B, and RB1). Germ line mutations in the negative regulators NF1, SPRED2, and LZTR1 also cause RASopathies (Aoki et al., 2016; Zenker et al., 2022). Others, such as INPPL1 and MAP4K5, have no clear role in RAS/ERK pathway regulation or in cancer.
Across the 14 lines, the largest number of significant genes (FDR < 0.05) were found in MOLM13 and MV4-11 cells, which is to be expected given that the focused CRISPR library was designed based on our genome-wide screens of these cell lines (Fig. 1 G). Deletion of several genes conferred SHP099 resistance in multiple cell lines, including LZTR1 (12/14), NF1 (12/14), PTEN (8/14), MAP4K5 (8/14), STK40 (6/14), FBXW7 (6/14), SPRED2 (6/14), and INPPL1 (5/14). Some hits were common to hematopoietic and solid tumor lines (e.g., LZTR1, NF1, PTEN, STK40, INPPL1, MAP4K5) and might predict frequent mechanisms of resistance in patients treated with SHP2is. The higher the SHP099 dose, the fewer significant hits that were recovered (Fig. S1 E). Moreover, “hits” differed significantly in their relative enrichment compared with control guides. These differences most likely reflect their relative ability to confer resistance, although other factors, including differential sgRNA efficiency and/or RNA/protein half-lives, might also contribute. Regardless, confirmed hits from this screen (performed at 7× IC50) are likely to be genes whose absence causes substantial resistance to SHP099. We decided to further explore the mechanism by which the deletion of LZTR1, a near-universal hit, as well as two unexpected hits, INPPL1 and MAP45K, confer SHP099 resistance.
Structural basis for SHP2i resistance caused by INPPL1 deletion
We first attempted for confirm that INPPL1 deletion with either of two sgRNAs conferred SHP099 resistance in vitro. Parental and INPPL1-deleted (KO) MOLM13, MOLM14, and MV4-11 cells were cultured under standard conditions in the presence or absence of SHP099, and cell number was inferred by PrestoBlue assay. KO cells showed substantially higher readings, consistent with higher cell number (Fig. 2 A, left panel). Importantly, re-expressing WT INPPL1 in KO MOLM13 cells restored their sensitivity to SHP099, ruling out off target effects of the INPPL1 sgRNAs as the cause of SHP099 resistance (Fig. 2 A, right panel). INPPL1 KO also caused resistance to TNO-155 and RMC2550, two other SHP2 inhibitors, confirming that the effects of SHP099 on cell proliferation, and the resistance conferred by INPPL1 deficiency, reflected SHP2 inhibition rather than off-target effects of the tool compound (Fig. S2). DAPI staining revealed that SHP099 causes G1 arrest and cell death (sub-G0 cells), both of which were decreased in KO cells (Fig. S3).
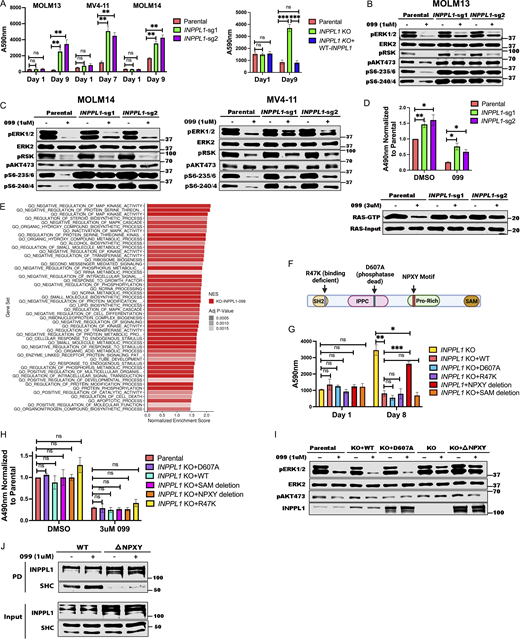
INPPL1 KO acts downstream of RAS to promote ERK reactivation and SHP099 resistance. (A)INPPL1 KO causes resistance to SHP099. Left: PrestoBlue assays of MOLM13, MOLM14, and MV4-11 cells, treated with 1, 2, or 2 µM SHP099, respectively. Right: Proliferation of parental MOLM13 cells, INPPL1 KO MOLM13 cells, and INPPL1 KO MOLM13 cells re-expressing WT INPPL1 in 1 µM SHP099. (B and C) Immunoblots of lysates from parental and INPPL1 KO MOLM13 (B), MOLM14 (C), and MV4-11 (C) cells treated with vehicle or SHP099 (1 µM) for 1 h; note increased activity of ERK pathway (pERK, pS6-235/6) in SHP099-treated INPPL1 KO cells. (D)INPPL1 KO causes increased RAS activation in MOLM13 cells. Cells were treated with vehicle (DMSO) or SHP099 (3 µM) for 1 h, and RAS-GTP was quantified by active RAS ELISA (Cytoskeleton). Luminescence at A490 nm was normalized to parental DMSO values. Lower panel shows RAS RBD-pulldown assays on MOLM13 cell lysates. (E) Top 50 significantly different MSigDB GO biological processes in INPPL1 KO vs. parental MOLM13 cells based on differentially expressed genes in RNAseq. (F) Domain structure of INPPL1 indicating mutants/deletions of functional domains assessed. (G) NPXY motif but not lipid phosphatase activity of INPPL1 is required to restore SHP099 sensitivity. WT INPPL1 and the indicated INPPL1 mutants were re-expressed in INPPL1 KO MOLM13 cells, and cell number after exposure to SHP099 (1 µM) for 8 d was inferred by PrestoBlue assays. (H) NPXY motif is dispensable for INPPL1 effects on RAS activation. WT INPPL1 and the indicated INPPL1 mutants were re-expressed in INPPL1 KO MOLM13 cells. Cells were treated with vehicle (DMSO) or SHP099 (3 µM) for 1 h, and RAS activation was assessed by ELISA. All readings are normalized to vehicle-treated parental MOLM13 cells. (I) NPXY motif is required for ERK regulation by INPPL1. Parental or INPPL1 KO MOLM13 cells reconstituted with the indicated mutants were treated with SHP099 (1 µM) for 1 h, and cell lysates were prepared and analyzed by immunoblotting with the indicated antibodies. (J) NPXY motif is required for SHC binding. INPPL1 KO MOLM13 cells were reconstituted with WT- or NPXY deleted-INPPL1 bearing a C-terminal Strep tag. Cells were treated with vehicle (DMSO) or SHP099 (1 µM) for 1 h, and INPPL1 was recovered on Strep-Tactin beads and analyzed by immunoblotting as indicated. All immunoblotting experiments were independently performed three times except for those in J, which were performed twice. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Source data are available for this figure: SourceData F2.
INPPL1 KO acts downstream of RAS to promote ERK reactivation and SHP099 resistance. (A)INPPL1 KO causes resistance to SHP099. Left: PrestoBlue assays of MOLM13, MOLM14, and MV4-11 cells, treated with 1, 2, or 2 µM SHP099, respectively. Right: Proliferation of parental MOLM13 cells, INPPL1 KO MOLM13 cells, and INPPL1 KO MOLM13 cells re-expressing WT INPPL1 in 1 µM SHP099. (B and C) Immunoblots of lysates from parental and INPPL1 KO MOLM13 (B), MOLM14 (C), and MV4-11 (C) cells treated with vehicle or SHP099 (1 µM) for 1 h; note increased activity of ERK pathway (pERK, pS6-235/6) in SHP099-treated INPPL1 KO cells. (D)INPPL1 KO causes increased RAS activation in MOLM13 cells. Cells were treated with vehicle (DMSO) or SHP099 (3 µM) for 1 h, and RAS-GTP was quantified by active RAS ELISA (Cytoskeleton). Luminescence at A490 nm was normalized to parental DMSO values. Lower panel shows RAS RBD-pulldown assays on MOLM13 cell lysates. (E) Top 50 significantly different MSigDB GO biological processes in INPPL1 KO vs. parental MOLM13 cells based on differentially expressed genes in RNAseq. (F) Domain structure of INPPL1 indicating mutants/deletions of functional domains assessed. (G) NPXY motif but not lipid phosphatase activity of INPPL1 is required to restore SHP099 sensitivity. WT INPPL1 and the indicated INPPL1 mutants were re-expressed in INPPL1 KO MOLM13 cells, and cell number after exposure to SHP099 (1 µM) for 8 d was inferred by PrestoBlue assays. (H) NPXY motif is dispensable for INPPL1 effects on RAS activation. WT INPPL1 and the indicated INPPL1 mutants were re-expressed in INPPL1 KO MOLM13 cells. Cells were treated with vehicle (DMSO) or SHP099 (3 µM) for 1 h, and RAS activation was assessed by ELISA. All readings are normalized to vehicle-treated parental MOLM13 cells. (I) NPXY motif is required for ERK regulation by INPPL1. Parental or INPPL1 KO MOLM13 cells reconstituted with the indicated mutants were treated with SHP099 (1 µM) for 1 h, and cell lysates were prepared and analyzed by immunoblotting with the indicated antibodies. (J) NPXY motif is required for SHC binding. INPPL1 KO MOLM13 cells were reconstituted with WT- or NPXY deleted-INPPL1 bearing a C-terminal Strep tag. Cells were treated with vehicle (DMSO) or SHP099 (1 µM) for 1 h, and INPPL1 was recovered on Strep-Tactin beads and analyzed by immunoblotting as indicated. All immunoblotting experiments were independently performed three times except for those in J, which were performed twice. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Source data are available for this figure: SourceData F2.

INPPL1, MAP4K5, or LZTR1 KO confer resistance to other SHP2 inhibitors. (A) Top left: PrestoBlue assays on parental, INPPL1 KO, or MAP4K5 KO MOLM13 cells treated with RMC-4550 (60 nM). Top right: PrestoBlue assays on parental or LZTR1 KO cells treated with RMC-4550 (30 nM). Bottom left: PrestoBlue assays on parental, INPPL1 KO, or MAP4K5 KO MV4-11 cells treated with RMC-4550 (200 nM). Bottom right: PrestoBlue assays on parental or LZTR1 KO MV4-11 cells treated with RMC-4550 (300 nM). (B) Same design as in A, but with TNO155. Doses: 60 nM for parental and INPPL1 KO/MAP4K5 KO MOLM13 cells, 30 nM for parental and LZTR1 KO MOLM13 cells, 200 nM for parental and INPPL1 KO/MAP4K5 KO MV4-11 cells, or 300 nM for parental and LZTR1 KO cells. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
INPPL1, MAP4K5, or LZTR1 KO confer resistance to other SHP2 inhibitors. (A) Top left: PrestoBlue assays on parental, INPPL1 KO, or MAP4K5 KO MOLM13 cells treated with RMC-4550 (60 nM). Top right: PrestoBlue assays on parental or LZTR1 KO cells treated with RMC-4550 (30 nM). Bottom left: PrestoBlue assays on parental, INPPL1 KO, or MAP4K5 KO MV4-11 cells treated with RMC-4550 (200 nM). Bottom right: PrestoBlue assays on parental or LZTR1 KO MV4-11 cells treated with RMC-4550 (300 nM). (B) Same design as in A, but with TNO155. Doses: 60 nM for parental and INPPL1 KO/MAP4K5 KO MOLM13 cells, 30 nM for parental and LZTR1 KO MOLM13 cells, 200 nM for parental and INPPL1 KO/MAP4K5 KO MV4-11 cells, or 300 nM for parental and LZTR1 KO cells. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.

Effects of INPPL1, MAP4K5, or LZTR1 deletion on cell cycle response to SHP099 in MV4-11 cells. (A) Cell cycle distribution of parental, INPPL1, MAP4K5, and LZTR1 KO MV4-11 cells treated with vehicle (DMSO) or SHP099 (2 µM) for 6, 24, or 48 h. (B) Statistical analysis (Student’s t test) of cell cycle distribution data from A. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Effects of INPPL1, MAP4K5, or LZTR1 deletion on cell cycle response to SHP099 in MV4-11 cells. (A) Cell cycle distribution of parental, INPPL1, MAP4K5, and LZTR1 KO MV4-11 cells treated with vehicle (DMSO) or SHP099 (2 µM) for 6, 24, or 48 h. (B) Statistical analysis (Student’s t test) of cell cycle distribution data from A. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
INPPL1 encodes a protein also known as SHIP2, an SH2-domain containing 5′ phosphatidyinositol phosphatase expressed broadly in hematopoietic and non-hematopoietic tissues (Elong Edimo et al., 2014; Eramo and Mitchell, 2016; Ramos et al., 2019; Thomas et al., 2017). SHIP2 catalyzes the dephosphorylation of PtdIns(4,5)P2, PtdIns(3,4,5)P3, PtdIns(3,5)P2, and several water-soluble phosphoinositides, and is best known as a negative regulator of insulin-stimulated AKT activation. We explored how INPPL1 deficiency causes SHP2i resistance. As expected, we observed increased levels of pAKT-S473 in INPPL1-KO MOLM13 cells in the presence or absence of SHP099. Activation of the AKT pathway results in increased mTORC1 activity, and ultimately increased activity of p70S6 kinase (S6K). Phosphorylation of S240/244 in ribosomal protein S6 (pS6-240/4), a selective target of S6K (Roux et al., 2007; Saxton and Sabatini, 2017), also was elevated in KO cells, providing additional evidence for AKT/mTORC1 pathway activation (Fig. 2, B and C). Unexpectedly, ERK, and its downstream target RSK showed increased phosphorylation in INPPL1-KO cells, as did S235/236 of S6 (pS6-235/6), which can be phosphorylated by S6K or RSK. Whereas SHP099 treatment strongly suppressed ERK, RSK, and S235/236 phosphorylation, suppression was attenuated in KO cells. SHP099 also resulted in decreased RAS activation (RAS-GTP) in parental cells, and this effect was attenuated by INPPL1 KO. In addition, RAS activation was enhanced in vehicle-treated KO cells, indicating that INPPL1 has an important role in negative regulation of RAS in these cells (Fig. 2 D).
To gain further insight into the predominant pathway(s) that confer SHP099 resistance, we performed RNA sequencing (RNAseq) on parental and SHP099-treated MOLM13 cells (Fig. 2 E and Fig. S4, A and B; and Table S4). In vehicle (DMSO)-treated cells, GO pathways most significantly enriched in INPPL1 KO (compared with parental) cells featured annotations such as “biosynthetic process” and “metabolic process” (Fig. S4 A), pathways that are regulated by the AKT/mTORC1 pathway (Saxton and Sabatini, 2017). By contrast, in the presence of SHP099, INPPL1-KO cells predominantly showed enrichment for MAPK pathway gene sets (e.g., “regulation of MAPK,” “regulation of MAPK cascade,” “MAPK kinase activity”). Moreover, MYC genes, DUSP family, and SPRY family genes are known targets of ERK, and were significantly upregulated in INPPL1-KO, compared with parental, cells (Fig. 2 E and Fig. S4 B). Taken together, these findings suggest that although the AKT/mTORC1 and RAS/ERK pathways both show enhanced activation upon INPPL1 deletion (and therefore are negatively regulated by INPPL1), increased activation of the former contributes predominantly to basal transcription (i.e., in the presence of vehicle), but increased RAS/ERK pathway activation likely is the major reason for SHP2i resistance.

Effect of INPPL1 or MAP4K5 KO on basal gene expression and following SHP099 treatment of MOLM13 cells. (A) RNAseq analysis of INPPL1 KO MOLM13 cells treated with vehicle (DMSO) 2.5 h compared with parental controls. Top: Top 50 most significantly enriched GO biological processes. Bottom: Volcano plot showing significantly up- and down-regulated genes (q < 0.05, red dots). (B) Volcano plot showing significantly up- and down-regulated genes (q < 0.05, red dots) in RNAseq analysis of INPPL1-KO MOLM13 cells treated with SHP099 (1 µM) for 2.5 h, compared with cognate parental controls. (C) Same as A, but for MAP4K5-KO cells vs. parental controls. (D) Same as B, but for SHP099-treated MAP4K5 KO vs. parental MOLM13 cells.
Effect of INPPL1 or MAP4K5 KO on basal gene expression and following SHP099 treatment of MOLM13 cells. (A) RNAseq analysis of INPPL1 KO MOLM13 cells treated with vehicle (DMSO) 2.5 h compared with parental controls. Top: Top 50 most significantly enriched GO biological processes. Bottom: Volcano plot showing significantly up- and down-regulated genes (q < 0.05, red dots). (B) Volcano plot showing significantly up- and down-regulated genes (q < 0.05, red dots) in RNAseq analysis of INPPL1-KO MOLM13 cells treated with SHP099 (1 µM) for 2.5 h, compared with cognate parental controls. (C) Same as A, but for MAP4K5-KO cells vs. parental controls. (D) Same as B, but for SHP099-treated MAP4K5 KO vs. parental MOLM13 cells.
INPPL1 is composed of several well-characterized domains (Fig. 2 F), including an N-terminal SH2 domain, a lipid phosphatase domain, a proline-rich region containing a NPXY motif that binds phosphotyrosine-binding domain–containing proteins, and a C-terminal sterile alpha motif domain, which reportedly mediates protein–protein interactions (Elong Edimo et al., 2014; Eramo and Mitchell, 2016; Ramos et al., 2019; Thomas et al., 2017). We asked which domain(s) is/are critical for INPPL1 to confer SHP2i sensitivity by restoring expression of WT INPPL1 and various INPPL1 mutants to INPPL1 KO MOLM13 cells and assessing proliferation in the presence of SHP099 (Fig. 2 G). All constructs were overexpressed, but to comparable levels (Fig. 2 I). Only the NPXY motif deletion phenocopied the effects of INPPL1 KO; remarkably, the ability of INPPL1 to confer sensitivity to SHP099 was independent of its lipid phosphatase activity (Le Coq et al., 2017), but instead appears to require its ability to bind (an)other protein(s) through its NPXY motif. Notably, expression of NPXY motif–deleted INPPL1 restored activated RAS to levels comparable to WT INPPL1-expressing cells treated with SHP099, but ERK phosphorylation remained elevated (Fig. 2, H and I). These findings suggest that although INPPL1 regulates RAS in MOLM13 cells, its effects on SHP2 sensitivity are mediated downstream of RAS but upstream of ERK through an NPXY-binding protein(s). Src homology and collagen (SHC) binds to the NPXY motif (Habib et al., 1998; Pesesse et al., 2001; Wisniewski et al., 1999), and, as expected, NPXY deletion greatly compromised SHC/INPPL1 co-immunoprecipitation (Fig. 2 J). Notably, SHC also scored as a resistance “hit” (FDR < 0.05) in our genomic and mini-screens of MOLM13 and MV4-11 cells (Fig. 1, B, C, E, and G; and Table S1). These results suggest that a SHC/INPPL1 complex is required to confer SHP099 sensitivity in these cells, presumably by recruiting one or more additional proteins to limit ERK activation downstream of RAS.
MAP4K5 KO promotes resistance downstream of RAS through its kinase activity
MAP4K5 (a.k.a. KHS, GCKR) encodes a member of the MAP4K family best known for its positive role in JNK activation (Chuang et al., 2016; Tung and Blenis, 1997). Several MAP4K family members also can phosphorylate LATS1/2, resulting in HIPPO pathway activation (Meng et al., 2015). However, the function of MAP4K5 is less clear. Using two independent sgRNAs, we first confirmed that MAP4K5 deletion caused resistance to SHP099 (and other SHP2is) in MOLM13, MV4-11, KU812, KYO1 cells (Fig. 3 A and Fig. S2). Similar to the effects of INPPL1 deletion, SHP099-treated MAP4K5 KO cells showed reduced G1 arrest and decreased sub-G0 populations compared to parental controls (Fig. S3). The effect of MAP4K5 deletion was unique among the MAP4Ks, even though all members of the family were expressed in these lines (Fig. 3 B). Re-expressing WT MAP4K5, but not a kinase-defective mutant (Chuang et al., 2016), in KO MOLM13 cells restored SHP099 sensitivity (Fig. 3, A and C).
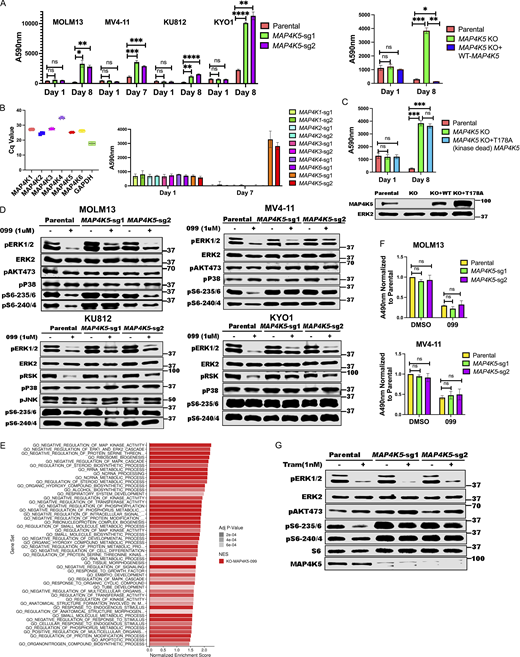
MAP4K5 KO promotes SHP099 resistance downstream of RAS in a kinase-dependent manner. (A)MAP4K5 KO causes SHP099 resistance. Left: PrestoBlue assays on MOLM13, MV4-11, KU812, or KYO1 cells treated with 1, 2, 5, or 10 µM SHP099, respectively. Right: PrestoBlue assays of parental MOLM13 cells, MAP4K5 KO MOLM13 cells, and MAP4K5 KO MOLM13 cells reconstituted with WT MAP4K5 in the presence of SHP099 (1 µM). (B) Deletion of MAP4K5, but not other MAP4K family members, causes resistance to SHP099 in MOLM13 cells. Left: All MAP4K family members are expressed in MOLM13 cells as detected by qRT-PCR. Right: Only MAP4K5 KO causes resistance to SHP099. MOLM13 cells with deletion of the indicated MAP4K family member gene were treated with SHP099 (1 µM) and assessed by PrestoBlue assay on day 7. (C) MAP4K5 requires kinase activity to restore SHP099 sensitivity. Top: Proliferation assays on parental MOLM13 cells, MAP4K5 KO MOLM13 cells, or MAP4K5 KO MOLM13 cells reconstituted with kinase-dead MAP4K5 (T178A) in the presence of 1 µM SHP099. Bottom: Immunoblot showing reconstitution of MAP4K5 KO MOLM13 cells with WT or kinase-dead (T178A) MAP4K5. The immunoblotting experiment was performed once. (D) Increased ERK pathway activity in SHP099-treated MOLM13, MV4-11, KU812, and KYO1 cells lacking MAP4K5. Cells were treated with SHP099 (1 µM) for 1 h, lysed, and analyzed by immunoblotting with the indicated antibodies. Experiments for each cell line were independently performed twice. (E) Top 50 significantly different MSigDB GO biological processes in SHP099-treated MAP4K5 KO vs. parental MOLM13 cells, based on differentially expressed genes from RNAseq. (F)MAP4K5 KO does not affect RAS activation in MOLM13 or MV4-11 cells. MOLM13 and MV4-11 cells were treated with vehicle (DMSO) or SHP099 (3 µM MOLM13, 5 µM MV4-11) for 1 h, lysed, and assessed for RAS activation by ELISA. (G) MAP4K5 regulates ERK activation upstream of MEK. Parental or MAP4K5 KO MOLM13 cells were treated with vehicle (DMSO) or the MEK inhibitor trametinib (1 nM) for 1 h, lysed, and analyzed by immunoblotting. Experiments were independently performed three times. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Source data are available for this figure: SourceData F3.
MAP4K5 KO promotes SHP099 resistance downstream of RAS in a kinase-dependent manner. (A)MAP4K5 KO causes SHP099 resistance. Left: PrestoBlue assays on MOLM13, MV4-11, KU812, or KYO1 cells treated with 1, 2, 5, or 10 µM SHP099, respectively. Right: PrestoBlue assays of parental MOLM13 cells, MAP4K5 KO MOLM13 cells, and MAP4K5 KO MOLM13 cells reconstituted with WT MAP4K5 in the presence of SHP099 (1 µM). (B) Deletion of MAP4K5, but not other MAP4K family members, causes resistance to SHP099 in MOLM13 cells. Left: All MAP4K family members are expressed in MOLM13 cells as detected by qRT-PCR. Right: Only MAP4K5 KO causes resistance to SHP099. MOLM13 cells with deletion of the indicated MAP4K family member gene were treated with SHP099 (1 µM) and assessed by PrestoBlue assay on day 7. (C) MAP4K5 requires kinase activity to restore SHP099 sensitivity. Top: Proliferation assays on parental MOLM13 cells, MAP4K5 KO MOLM13 cells, or MAP4K5 KO MOLM13 cells reconstituted with kinase-dead MAP4K5 (T178A) in the presence of 1 µM SHP099. Bottom: Immunoblot showing reconstitution of MAP4K5 KO MOLM13 cells with WT or kinase-dead (T178A) MAP4K5. The immunoblotting experiment was performed once. (D) Increased ERK pathway activity in SHP099-treated MOLM13, MV4-11, KU812, and KYO1 cells lacking MAP4K5. Cells were treated with SHP099 (1 µM) for 1 h, lysed, and analyzed by immunoblotting with the indicated antibodies. Experiments for each cell line were independently performed twice. (E) Top 50 significantly different MSigDB GO biological processes in SHP099-treated MAP4K5 KO vs. parental MOLM13 cells, based on differentially expressed genes from RNAseq. (F)MAP4K5 KO does not affect RAS activation in MOLM13 or MV4-11 cells. MOLM13 and MV4-11 cells were treated with vehicle (DMSO) or SHP099 (3 µM MOLM13, 5 µM MV4-11) for 1 h, lysed, and assessed for RAS activation by ELISA. (G) MAP4K5 regulates ERK activation upstream of MEK. Parental or MAP4K5 KO MOLM13 cells were treated with vehicle (DMSO) or the MEK inhibitor trametinib (1 nM) for 1 h, lysed, and analyzed by immunoblotting. Experiments were independently performed three times. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Source data are available for this figure: SourceData F3.
In some cell lines tested, we were unable to detect activated JNK (pJNK) in the presence or absence of SHP099. However, even in a line in which pJNK was detectable (e.g., KU812), we observed no difference upon MAP4K5 deletion, nor was p38 activation affected (Fig. 3 D). By contrast, SHP099-treated MAP4K5 KO cells bearing different RTK driver mutations, e.g., FLT3-ITD (MOLM13, MV4-11), and BCR-ABL (KU812, KYO1), showed increased pERK levels compared with cognate parental controls. Consistent with functional consequences of this increase in ERK activation, RNAseq revealed that the top enriched GO pathways in SHP099-treated MAP4K5 KO cells (but not in vehicle-treated cells; Fig. S4 C) had annotations including “negative regulation of MAP kinase activity” and “negative regulation of ERK1 and ERK2 cascade,” and known ERK-responsive genes were upregulated (Fig. 3 E, Fig. S4 D, and Table S5). In contrast to the effects of INPPL1 deficiency, RAS activation was unaltered in vehicle- or SHP099-treated MAP4K5 KO cells (Fig. 3 F). Taken together, these results indicate that MAP4K family members have redundant functions in JNK and p38 activation in MOLM13, MV4-11, KU812, and KYO1 cells, but MAP4K5 has a specific role in regulating ERK activation downstream of RAS. Consistent with this notion, MAP4K5 KO cells are sensitive to MEKi treatment (Fig. 3 G).
LZTR1 KO promotes SHP099 resistance through cell context–dependent effects on RIT1 and/or RAS family members
Deletion of LZTR1 using individual sgRNAs also caused resistance to SHP099 and other SHP2is in multiple cell lines (Fig. 4 A; and Figs. S2 and S3). LZTR1 was reported by different groups to promote the degradation of RAS family members or RIT1 through recruitment of a CUL3 ubiquitin ligase complex (Bigenzahn et al., 2018; Castel et al., 2019). Germ-line LZTR1 mutations also are responsible for some cases of the RASopathy Noonan syndrome (Aoki et al., 2016; Zenker et al., 2022). Given the controversy over the target(s) of the LZTR1-containing ubiquitin ligase complex, we assessed the levels of RIT1 and RAS family members across multiple cell lines from our panel. Consistent with the observations of Castel et al. (2019), RIT1 levels were increased in all LZTR1 KO lines tested. Elevated muscle RAS oncogene homologue (MRAS) levels were also seen in nearly all LZTR1-deficient lines. By contrast, increased levels of other RAS proteins were cell dependent (Fig. 4 B). For example, in MV4-11, the levels of Ki-ras2 Kirsten rat sarcoma viral oncogene homologue (KRAS), neuroblastoma RAS viral oncogene homologue (NRAS), and Harvey rat sarcoma viral oncogene homologue (HRAS) were unaffected by LZTR1 KO. By contrast, in K562 cells, LZTR1 deficiency led to increased levels of KRAS, HRAS, NRAS, and RAS-related (RRAS), in addition to the regularly seen elevation of RIT1 and MRAS (Fig. 4 B and Fig. S5, A and B). K562 cells also showed increased levels of RAS-GTP (Fig. 4 C). The cognate transcripts for RAS family members were unaffected by LZTR1 deficiency, consistent with differential post-transcriptional regulation of RAS/RIT expression levels (Fig. S5 C).
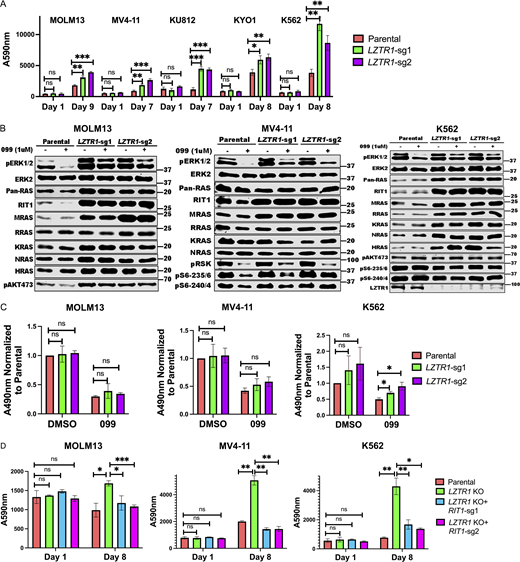
LZTR1 KO has cell context–dependent effects on RIT1 and/or RAS family members but promotes SHP099 resistance through RIT1. (A)LZTR1 KO cells are resistant to SHP099. PrestoBlue assays on parental and LZTR1 KO MOLM13, MV4-11, KU812, KYO1, or K562 cells in 0.5, 2, 5, 5 or 5 µM SHP099, respectively. (B) Cell context–dependent regulation of RIT and RAS family proteins by LZTR1. Parental and LZTR1 KO cells were treated with SHP099 (1 µM) for 1 h, lysed, and analyzed by immunoblotting with the indicated antibodies. Experiments for each line were independently performed for twice. (C)LZTR1 KO increases active RAS levels in K562 cells. MOLM13, MV4-11, and K562 cells were treated with vehicle (DMSO) or 3 µM (MOLM13) or 5 µM (other lines) SHP099 for 1 h, and RAS-GTP levels were assessed by ELISA. All readings were normalized to those of vehicle-treated parental cells for each line. (D)RIT1 KO restores sensitivity of LZTR1 KO cells to SHP099. Parental, LZTR1-KO, and LZTR1-KO plus RIT1-KO MOLM13, MV4-11, and K562 cells were treated with 0.5, 2, and 5 µM SHP099, respectively, for 8 d, and cell number was analyzed by PrestoBlue assay; *, P < 0.05; **, P < 0.01; ***, P < 0.001. Source data are available for this figure: SourceData F4.
LZTR1 KO has cell context–dependent effects on RIT1 and/or RAS family members but promotes SHP099 resistance through RIT1. (A)LZTR1 KO cells are resistant to SHP099. PrestoBlue assays on parental and LZTR1 KO MOLM13, MV4-11, KU812, KYO1, or K562 cells in 0.5, 2, 5, 5 or 5 µM SHP099, respectively. (B) Cell context–dependent regulation of RIT and RAS family proteins by LZTR1. Parental and LZTR1 KO cells were treated with SHP099 (1 µM) for 1 h, lysed, and analyzed by immunoblotting with the indicated antibodies. Experiments for each line were independently performed for twice. (C)LZTR1 KO increases active RAS levels in K562 cells. MOLM13, MV4-11, and K562 cells were treated with vehicle (DMSO) or 3 µM (MOLM13) or 5 µM (other lines) SHP099 for 1 h, and RAS-GTP levels were assessed by ELISA. All readings were normalized to those of vehicle-treated parental cells for each line. (D)RIT1 KO restores sensitivity of LZTR1 KO cells to SHP099. Parental, LZTR1-KO, and LZTR1-KO plus RIT1-KO MOLM13, MV4-11, and K562 cells were treated with 0.5, 2, and 5 µM SHP099, respectively, for 8 d, and cell number was analyzed by PrestoBlue assay; *, P < 0.05; **, P < 0.01; ***, P < 0.001. Source data are available for this figure: SourceData F4.
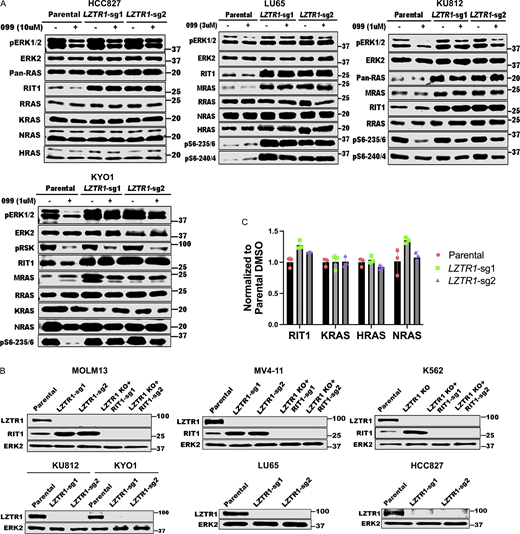
LZTR1 regulation of RAS family proteins is cell context dependent. (A)LZTR1 KO differentially affects RAS family protein levels in HCC827, LU65, KU812, and KYO1 cells. Experiments for each line were independently performed twice. (B) Immunoblots showing LZTR1 and RIT1 levels in the indicated cell lines used in Fig. 5, B and D. Experiments were independently performed once. (C) RIT1, KRAS, HRAS, and NRAS mRNA levels in K562 cells are not affected by LZTR1 KO. Source data are available for this figure: SourceData FS5.
LZTR1 regulation of RAS family proteins is cell context dependent. (A)LZTR1 KO differentially affects RAS family protein levels in HCC827, LU65, KU812, and KYO1 cells. Experiments for each line were independently performed twice. (B) Immunoblots showing LZTR1 and RIT1 levels in the indicated cell lines used in Fig. 5, B and D. Experiments were independently performed once. (C) RIT1, KRAS, HRAS, and NRAS mRNA levels in K562 cells are not affected by LZTR1 KO. Source data are available for this figure: SourceData FS5.
These results indicate that RIT1 is a universal substrate for LZTR1 (followed by MRAS), whereas other RAS family members are conditional substrates based on the cell context (and potentially, differences in composition of LZTR1-containing ubiquitin ligase complexes; see Discussion). The suppression of ERK activation upon SHP099 treatment was attenuated in all LZTR1 KO lines (Fig. 4 B and Fig. S5 A). Notably, although the effect of LZTR1 deficiency on the levels of RAS family members differed in various cell lines, RIT1 KO restored SHP099 sensitivity to all the LZTR1 KO cell lines tested, including K562 (Fig. 4 D and Fig. S5 C). The latter finding indicates that, despite the upregulation of multiple RAS family members in LZTR1 KO K562 cells, RIT1 is the major effector of SHP099 resistance.
INPPL1, MAP4K5, or LZTR1 KO confer cross-resistance to TK inhibitors
Although SHP2 inhibitors are only in early phase trials, the FLT3 inhibitor gilteritinib is used clinically for the treatment of FLT3-ITD+ AML (Perl et al., 2022; Tarver et al., 2020), and the BCR-ABL inhibitor dasatinib is used in BCR-ABL+ AML. MOLM13 and MV4-11 express FLT3-ITD, whereas KU812, KYO1, and K562 cells express BCR-ABL, and our results indicate that INPPL1, MAP4K5, and LZTR1 act downstream of these TK fusions. Therefore, we asked whether deletion of one or more of these SHP2i resistance hits also caused gilteritinib or dasatinib resistance. Indeed, deletion of any of these genes caused resistance to gilteritinib in both FLT3-ITD–expressing lines tested. In addition, deletion of MAP4K5 or LZTR1 caused resistance to dasatinib in BCR-ABL expressing lines (Fig. 5, A and B). We did not test the effects of INPPL1 deficiency because it does not confer SHP099 resistance in BCR-ABL–driven lines.

INPPL1, MAP4K5, or LZTR1 KO confer cross resistance to gilteritinib and dasatinib. (A) Parental MOLM13 and MV4-11 cells and the indicated KO derivatives were treated with 2 nM (MOLM13) or 0.6 nM gilteritinib (MV4-11) for 8 d and analyzed by PrestoBlue assays. (B) Parental KU812, KYO1, and K562 and the indicated KO cells were treated with 120, 60, and 100 pM dasatinib, respectively, for 8 d and analyzed by PrestoBlue assays. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
INPPL1, MAP4K5, or LZTR1 KO confer cross resistance to gilteritinib and dasatinib. (A) Parental MOLM13 and MV4-11 cells and the indicated KO derivatives were treated with 2 nM (MOLM13) or 0.6 nM gilteritinib (MV4-11) for 8 d and analyzed by PrestoBlue assays. (B) Parental KU812, KYO1, and K562 and the indicated KO cells were treated with 120, 60, and 100 pM dasatinib, respectively, for 8 d and analyzed by PrestoBlue assays. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
INPPL1-, MAP4K5-, or LZTR1-KO confers SHP099 resistance in vivo
Finally, we tested whether the hits identified in our in vitro screen confer SHP099 resistance in vivo. To this end, MV4-11 cells were transduced with a lentivirus expressing luciferase to enable monitoring of tumor burden by bioluminescence imaging. We then generated INPPL1-, MAP4K5-, or LZTR1-deleted, luciferase-expressing lines by transduction with lenti-Cas9–expressing viruses co-expressing non-targeting or specific sgRNAs. MV4-11 cells and their derivatives (1.5 × 106) were injected through tail veins into NSG mice. 10 d later, treatment with SHP099 (50 mg/kg/d) was initiated, and tumor burden was evaluated weekly by total body luminescence (Fig. 6 A). By week 3, mice bearing INPPL1-, MAP4K5-, or LZTR1 KO MV4-11 cells all showed significantly higher tumor burdens than mice injected with parental cells (Fig. 6, B and C). Consistent with these data, INPPL1, MAP4K5, or LZTR1 deletion also significantly shortened survival of injected mice (Fig. 6 D).

INPPL1-, MAP4K5-, or LZTR1-KO MV4-11 cells are resistant to SHP099 in vivo. (A) Workflow for mouse experiments. (B) Bioluminescence imaging of mice injected with parental and INPPL1-, MAP4K5-, LZTR1-KO MV4-11 cells on day 0 and monitored for 21 d. Note increased tumor burden caused by each KO line. (C) Quantification of data in B by Student’s t test. (D) Deletion of INPPL1, MAP4K5, or LZTR1 in MV4-11 cells significantly shortens survival of mice. **, P < 0.01; ***, P < 0.001; Mantel log-rank test.
INPPL1-, MAP4K5-, or LZTR1-KO MV4-11 cells are resistant to SHP099 in vivo. (A) Workflow for mouse experiments. (B) Bioluminescence imaging of mice injected with parental and INPPL1-, MAP4K5-, LZTR1-KO MV4-11 cells on day 0 and monitored for 21 d. Note increased tumor burden caused by each KO line. (C) Quantification of data in B by Student’s t test. (D) Deletion of INPPL1, MAP4K5, or LZTR1 in MV4-11 cells significantly shortens survival of mice. **, P < 0.01; ***, P < 0.001; Mantel log-rank test.
Discussion
The essential role of SHP2 in RTK/RAS/ERK pathway signaling and the frequent activation of this pathway in multiple malignancies prompted the development of SHP2is for cancer therapy. Such inhibitors, alone or in combination with drugs targeting other cascade components or parallel pathways, are in clinical trials for RTK- or cycling KRAS mutant–dependent cancers. To anticipate SHP2i resistance mechanisms mimicking gene downregulation, silencing, or inactivation, we performed CRISPR/Cas9 genome-scale KO screens in two FLT3-ITD–driven human AML lines that are highly sensitive to SHP2 inhibition, followed by validation of common hits in 12 additional cancer lines. Our studies identified common and bespoke resistance mechanisms across these lines, including both “expected” and “novel” genes. The former included genes encoding known negative regulators of the RAS/ERK pathway (e.g., NF1, SPRED2), the parallel PI3K/AKT pathway (PTEN), and negative regulators of cell cycle (e.g., CDKN1B, RB1). Several of these genes were also found in previous genome-wide CRISPR loss-of-function screens for BRAF inhibitor resistance and metastasis (Chen et al., 2015; Shalem et al., 2014). However, more detailed analysis of three of the unexpected hits, INPPL1 (SHIP2), MAP4K5, and LZTR1, reveal new insights into RAS/ERK pathway regulation (Fig. 7). Deletion of each of these genes also results in SHP2i resistance in mice and cross-resistance to the FLT3-ITD inhibitor gilteritinib.

Model showing sites of action of INPPL1, MAP4K5, and LZTR1. SHP2 plays a positive role for RAS activation upstream of SOS. INPPL1 works both upstream and downstream of RAS, negatively regulating ERK activity. However, INPPL1 deletion confers SHP099 resistance mainly by its actions downstream of RAS. MAP4K5 acts downstream of RAS, but upstream of MEK. LZTR1 can act on RAS proteins or RIT1 but confers SHP099 resistance mainly through the latter.
Model showing sites of action of INPPL1, MAP4K5, and LZTR1. SHP2 plays a positive role for RAS activation upstream of SOS. INPPL1 works both upstream and downstream of RAS, negatively regulating ERK activity. However, INPPL1 deletion confers SHP099 resistance mainly by its actions downstream of RAS. MAP4K5 acts downstream of RAS, but upstream of MEK. LZTR1 can act on RAS proteins or RIT1 but confers SHP099 resistance mainly through the latter.
We chose to screen for SHP2i resistance in hematopoietic cell lines because of their high degree of sensitivity. Notably, clinical SHP2is are being tested predominantly in solid tumor cell lines. Although we did test our “hits” in several such lines, the overall landscape of resistance will likely differ in solid tumors. For this reason, parallel studies are underway in our laboratory to address SHP2i resistance alone and in combination with other targeted therapies in non-small cell lung cancer lines. Nevertheless, blood malignancies driven by TK fusion genes also develop adaptive and stable resistance, and SHP2is might be clinically relevant in these settings as well (Melgar et al., 2019).
INPPL1 is typically regarded as a negative regulator of the PI3K/AKT/mTORC1 pathway, but substantial evidence suggests additional, if not alternative, roles (Blero et al., 2001; Clément et al., 2001; Habib et al., 1998; Jurynec and Grunwald, 2010; Pesesse et al., 2001; Sleeman et al., 2005; Wang et al., 2004; Wisniewski et al., 1999). Two Inppl1 KO mouse models exhibit increased activation of AKT and S6K, although the phenotypes of these mice differ, most likely reflecting the distinct parts of the Inppl1 locus removed and/or removal of the adjacent Phox2a gene in one model (Clément et al., 2001; Sleeman et al., 2005). Consistent with these findings, we observed increased pAKT and pS6-240/4 in INPPL1-KO cells. However, we also found that INPPL1 deficiency increased RAS/ERK activation, and RNAseq suggested that ERK pathway activation is important, if not essential, in driving SHP2i resistance. Notably, INPPL1 overexpression impairs ERK and/or AKT activation in response to multiple growth factors in various cell systems (Blero et al., 2001; Ishihara et al., 1999; Pesesse et al., 2001; Sasaoka et al., 2003; Wang et al., 2004). Decreased ERK activation in these experiments reflects sequestration of SHC from GRB2 by its binding to INPPL1 and thus might be an artifactual consequence of over-expression. More compellingly, zebrafish Ship2 morphants show dorsal-ventral patterning defects characteristic of increased FGF signaling along with enhanced ERK phosphorylation and ERK-dependent gene expression (Jurynec and Grunwald, 2010). Moreover, the “pure” Inppl1 KO model (i.e., not including Phox2a) exhibits craniofacial abnormalities and runting, phenotypes that could reflect altered RAS/ERK activation (Sleeman et al., 2005). Likewise, a variety of homozygous inactivating mutations of human INPPL1 cause opsismodysplasia, a rare skeletal dysplasia syndrome, rather than an insulin hypersensitivity syndrome (Below et al., 2013; Fradet and Fitzgerald, 2017; Huber et al., 2013; Li et al., 2014a). Taken together, it seems clear that INPPL1 can and does regulate the MEK/ERK pathway, at least in some cell contexts. Our results clearly reveal much more widespread, albeit non-ubiquitous, regulation of ERK activation by INPPL1.
The physiological function of INPPL1 lipid phosphatase activity also remains unresolved. Overexpression of catalytically inactive INPPL1 in smooth muscle cells suppresses growth factor–induced MEK/ERK activation, while enhancing AKT activation (Sasaoka et al., 2003). As noted above, such results could reflect artifactual sequestration of SHC. By contrast, knock-in mice with global expression of catalytically impaired INPPL1 are runted and show craniofacial defects similar to those caused by global INPPL1 deficiency, as well as increased IGF1 (but not FGF2)-stimulated MEK/ERK activation. However, INPPL1 levels are significantly reduced in these mice, making it difficult to attribute the observed effects to catalytic impairment alone. Missense mutants affecting the phosphatase domain have been identified in opsismodysplasia cohorts (Below et al., 2013; Fradet and Fitzgerald, 2017; Huber et al., 2013; Li et al., 2014a), but again, whether the mutant proteins are expressed at normal levels has not been established.
Our structure-function analyses show that phosphatase-inactive INPPL1 restores SHP099 sensitivity and decreases pAKT and pERK to the same extent as the WT protein. The sterile alpha motif and pro-rich domains are similarly dispensable. Instead, mutation of the NPXY motif, which enables binding to phosphotyrosine-binding domain–containing proteins, disables the ability of INPPL1 to restore SHP2i sensitivity to INPPL1-KO lines. SHC binds to this motif and is the most likely mediator of the observed effects on ERK activation. Remarkably, however, the NPXY mutant restores the elevated RAS levels seen in KO cells to those of WT-reconstituted or parental cells yet fails to normalize ERK phosphorylation. These results suggest that INPPL1 has a dual negative role in ERK regulation, acting upstream and downstream of RAS. Importantly, although the INPPL1 mutants were over-expressed compared with parental INPPL1 in our reconstitution experiments, expression of the phosphatase-dead D607A mutant (rescue-competent) and the NPXY deletion mutant (rescue-incompetent) were comparable.
MAP4K5 is one of six related STE-20–like kinases best known for their positive roles in JNK activation (Chuang et al., 2016; Tung and Blenis, 1997). However, other functions have also been attributed to the MAP4K family. For example, MAP4K5 reportedly acts as a positive transducer of canonical and non-canonical WNT signaling in B cells (Shi et al., 2006). Other MAP4K family kinases can phosphorylate and activate LATS1/2 in HEK293 cells, acting in parallel to MST1/2 in the HIPPO pathway, although MAP4K5 was unable to perform this function (Meng et al., 2015). We found that MAP4K5, but not other MAP4K family members, negatively regulates the RAS/ERK pathway in a kinase-dependent manner, acting downstream of RAS but upstream of MEK; notably, we saw no difference in JNK activation in MAP4K5-KO cells. Although the mechanism by which MAP4K5 controls ERK activation remains unclear, recent biochemical studies found that MAP4K5 can phosphorylate all AMPK-related kinases, including MARK1-3 (Liu et al., 2022). MARK1 (a.k.a. C-TAK1) phosphorylates KSR1 and impedes RAF/MEK/ERK activation (Müller et al., 2001). Studies are underway to test this interesting possibility.
Of the three resistance hits we characterized, LZTR1 KO conferred SHP2i resistance the most widely (12/14 lines in the mini-screen). We did not directly test the remaining two lines individually, so we cannot exclude technical failure of LZTR1 to validate in the mini-screen of these lines. Our results comport with previous studies demonstrating a role for LZTR1 in the RAS/MEK/ERK pathway, but also provide new insights into its mechanism of action. As an adaptor for CUL3 E3 ligase complexes, LZTR1 promotes the degradation of target proteins. However, the precise target(s) of the LZTR1/CUL3 complex have been a subject of controversy. RAS family members were suggested initially (Bigenzahn et al., 2018), but subsequent work argued that RIT1 is the primary substrate (Castel et al., 2019). We found that LZTR1 KO invariably upregulated RIT1 and MRAS but not canonical RAS proteins in seven cell lines screened. Nevertheless, in other cell lines, various RAS proteins also were upregulated, with K562 cells showing stabilization of KRAS, HRAS, NRAS, and RRAS. The simplest explanation for these findings is that LZTR1/CUL3 complexes have additional components that await identification. For example, an inhibitor of RAS protein binding could be present, or a RAS-specific adaptor could be missing in lines where only RIT1 is stabilized. A very recent study of leukemogenesis in Lztr1−/− mice also noted altered levels of conventional RAS proteins along with RIT1 and MRAS (Chen et al., 2022). Nevertheless, RIT1 deletion in LZTR1-KO K562 cells restores SHP2 sensitvity, indicating that although LZTR1 deficiency can affect other RAS family proteins in certain cells, RIT1 stabilization is necessary to confer SHP2i resistance.
Although we characterized three targets in detail in this work, several other novel hits warrant further study. STK40, which encodes a poorly understood pseudokinase, was a frequent hit (6/14 lines), whose deletion causes SHP099 resistance in hematologic (FLT3-ITD and BCR-ABL–driven) and solid tumor (EGFR-driven) cells. We also recovered genes encoding epigenetic regulators, including WHSC1, SETD5, and SUV39H1, as well as transcriptional coactivators/repressors (NCOR1, NCOA6). While our screens were designed with the primary goal of identifying mechanisms of SHP099 resistance, we did observe some synthetic lethal genes. For example, KDM5A, which encodes a Jumanji-type histone demethylase, was depleted significantly in SHP099-treated MV4-11 and MOLM13 cells, and several additional genes showed synthetic lethal behavior in MV4-11 cells in particular.
Because SHP2is are in early-stage trials, clinical samples of resistance are not available. Nevertheless, several lines of evidence indicate that genes identified here are relevant to therapeutic outcome in hematologic and other malignancies. Deletion of INPPL1, MAP4K5, or LZTR1 confers gilteritinib resistance (Fig. 5 A); similar effects of LZTR1 deficiency were reported recently (Chen et al., 2022). Moreover, deletion of MAP4K5 or LZTR1 confers dasatinib resistance (Fig. 5 B). High levels of MAP4K5 RNA are associated with favorable prognosis in the TGCA AML cohort, both in univariate analyses and when corrected for age, cytogenetics, French-American-British stage, and RUNX and TP53 status (Bai et al., 2019). Conversely, low MAP4K5 levels correlate with particularly poor outcome in pancreatic ductal adenocarcinoma (Wang et al., 2016). Future studies will be required to assess the generality of those relationships and their utility for combination therapy with SHP2is, as well as to determine whether deletion or mutation of the resistance hits observed in these studies are found in patients treated with SHP2is and other targeted therapies.
Materials and methods
Cell lines and reagents
Cells were maintained in 5% CO2 at 37°C in media conditions described by the vendor or the source laboratory and were tested monthly for mycoplasma by PCR (Young et al., 2010). KO cells were generated by infection with lentiviruses constitutively expressing Cas9 and appropriate sgRNAs. All experiments were performed with early passage lines within 3 mo of defrosting. MOLM13, MV4-11, BV173, K562, H1975, LU65, HCC827 cells were from lab stocks. KYSE520 was obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (German Collection of Microorganisms and Cell Cultures GmbH). KG1a was obtained from Dr. Christopher Park (NYU Grossman School of Medicine, New York, NY, USA) in June 2020; MOLM14 was obtained from Dr. Iannis Aifantis (NYU Grossman School of Medicine, New York, NY, USA) in July 2020; KU812, KYO1, EOL1, OCI-M1 were provided from Dr. Ross L. Levine (Memorial Sloan Kettering Cancer Center, New York, NY, USA) in March 2021. MOLM13, MV4-11, BV173, K562, H1975, LU65, HCC827, KYSE520, KG1a, KU812, KYO1, and EOL1 were cultured in RPMI supplemented with 10% FBS and 1% penicillin/streptomycin. OCI-M1 was maintained in IMDM supplemented with 10% FBS and 1% penicillin/streptomycin.
sgRNAs for non-targeting control and the target genes INPPL1, MAP4K5, LZTR1, and RIT1 were as follows (target sequence): non-targeting: 5′-AACCGGCTGCGCGTTTGCAA-3′; INPPL1-sg1: 5′-GCAGGGCGCACACAAGGCCC-3′; INPPL1-sg2: 5′-CCTGGATATCCATGTCCAGG-3′; MAP4K5-sg1: 5′-AGGACTACGAACTCGTCCAG-3′; MAP4K5-sg2: 5′-TAGGCCAGAAATGTACACAC-3′; LZTR1-sg1: 5′-TATGGTCGAAGTCCACGCTC-3′; LZTR1-sg2: 5′-CGGCCGAGTGGTGGTAACGG-3′; RIT1-sg1: 5′-ACGTACTGACGATACACCTG-3′; RIT1-sg2: 5′-TCGGTGGCTGATGAACTGCA-3′.
Parental and KO MV4-11 cells for mouse experiments were generated with non-targeting, INPPL1-sg1, MAP4K5-sg1, and LZTR1-sg1. sgRNA resistant constructs for INPPL1 and MAP4K5 for re-expression were generated through making silent mutations in PAM sequence for INPPL1-sg1 and MAP4K5-sg1.
Plasmids and lentiviruses
The open reading frames of INPPL1 (GenScript: OHu19348), MAP4K5 (GenScript: OHu02673), and various mutants of these genes were cloned into pLV-EF1a-IRES-Neo (85139; Addgene) using a Gibson Assembly kit (E5510S; NEB). All sgRNAs were cloned into lentiCRISPRv2 (52961; Addgene) by following a published protocol (Sanjana et al., 2014; Shalem et al., 2014). Lentiviruses were produced by co-transfecting HEK293T cells with viral construct and packaging vector DNAs (Hart et al., 2017; Mair et al., 2019). Cells were spin-infected with concentrated viruses and infected cells were recovered by selection with the appropriate antibiotics.
Cell assays
For proliferation and dose-response assays, cell number was inferred by PrestoBlue assay (A13262; Thermo Fisher Scientific). Briefly, cells were seeded in 96-well plates and then incubated with media containing DMSO/various inhibitors. For proliferation assays, viability was assessed, and media (including inhibitors) were refreshed, every 2 d. For dose-response studies, media (including inhibitors) were refreshed once on day 2, and data were collected on day 4. Dose response curves (and IC50s) were generated with R package “drc.” To assess cell cycle distribution, cells were fixed with 70% ethanol overnight at −20°C, followed by DAPI staining the next day, and analyzed by flow cytometry using FlowJo software.
CRISPR mini-library construction
A CRISPR mini-library was constructed based on overlapping hits from the genome wide screens of MV4-11 and MOLM13 cells. The library comprised 142 sgRNAs with 4sgRNA/gene against 33 target genes and 10 non-targeting controls. For the target genes, two sgRNAs were derived from the TKOv3 library and the other two were from the Doench CRISPR KO library and had the highest “Rule Set 2 Scores” (Doench et al., 2016). Mini-library sgRNAs were cloned into the lentiCRISPRv2 backbone according to previously published protocol (Chen et al., 2015) and sequenced to assess library quality (Fig. S1 A).
CRISPR/Cas9 screens
Two independent replicas of each cell line were spin-infected with TKOv3 Cas9-sgRNA or CRISPR mini-library viruses at a low multiplicity of infection (∼0.2) and at 1,000× representation for each sgRNA in the library. On day 2 after infection, puromycin was added to the media, and cells were selected for 8 d. Live cells (for hematopoietic cell lines) after selection were isolated using Ficoll-Paque PLUS Media according to the manufacturer’s instructions, and cultured without antibiotics for additional 2 d. After recovery, a 1,000× library representation of cells for each replica was cultured with DMSO or SHP099 at 7× IC50 for each line for 12 doublings (genomic screens) or 10 doublings (mini-screens). For mini-screens, if the IC50 for the cell line was >1.5 μM, 10 μM SHP099 was used. At screen termination, gDNA was extracted and PCR amplified according to published protocol (Shalem et al., 2014). Final PCR products were sequenced with Illumina NovaSeq 6000 (SP 100 Cycle Flow Cell v1.5). Results were decomplexed with Bowtie (Shalem et al., 2014) to generate count tables and subsequently analyzed with MaGeCK for genomic screens or t test with FDR correction for mini-screens.
gDNA extraction
Briefly, cells (3–5 × 107) were suspended in 6 ml lysis buffer (50 mM Tris, 50 mM EDTA, 1% SDS, pH 8), supplemented with 30 µl 20 mg/ml Proteinase K (19131; Qiagen), and incubated at 55°C overnight. For higher cell numbers, volumes were adjusted appropriately. The next day, 30 µl of 10 mg/ml RNAse A (19101; Qiagen) were added to the sample, which was then incubated at 37°C for 30 min. Then, proteins in the sample were precipitated with 2 ml 7.5 M cold ammonium acetate and centrifuged at >4,000 g for 10 min. Supernatants containing DNA were transferred to a new tube, isopropanol (6 ml) was added, and the samples were centrifuged at >4,000 g for 10 min. Precipitated DNA was washed with 6 ml 70% ethanol, air dried at room temperature, and suspended in nuclease-free water.
Biochemical assays
RAS activation was assessed by ELISA using a commercially available kit (Cytoskeleton; BK131) according to the manufacturer’s instructions, or by GST-RBD “pull-down” assays. Specifically, 2 million cells from each line were lysed in 100 μl lysis buffer, each sample was adjusted to same protein concentration, and the same amount of total protein was used in each assay. For active RAS pulldown (PD) assays, 1 × 107 MOLM13 cells were lysed in 500 μll RAS PD buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM MgCl2, 5% glycerol, 1% NP40) supplemented with protease and phosphatase inhibitors on ice and centrifuged immediately at 14,000 rpm for 10 min at 4°C. For PD assays, 800 μg of clarified lysats at a concentration of 2 μg/µl were incubated with GST-RBD glutathione agarose beads for 45 min with constant rocking. Beads were washed three times with PD buffer and suspended in 2× SDS-PAGE sample buffer.
For monitoring additional effects on cell signaling pathways, 2 × 106 cells/well were plated in 6-well plates, and the next day the indicated inhibitor(s) were added for 1 h. Cells were harvested and lysed in RIPA buffer (50 mM Tris-HCl pH 8, 150 mM NaCl, 2 mM EDTA, 1% NP-40 and 0.1% SDS), supplemented with protease (40 µg/ml PMSF, 2 µg/ml antipain, 2 µg/ml pepstatin A, 20 µg/ml leupeptin, and 20 µg/ml aprotinin) and phosphatase (10 mM NaF, 1 mM Na3VO4, 10 mM β-glycerophosphate, and 10 mM sodium pyrophosphate) inhibitors. Protein concentration was determined by Coomassie assay according to the manufacturer’s instructions (#ab119211; Abcam). For immunoblotting, total lysate protein (15 μg) was resolved by SDS-PAGE and transferred to nylon membranes in 1× transfer buffer supplemented with 10% methanol. Membranes were incubated with primary antibody (in 5% BSA in TBST) at 4°C overnight, and then with secondary antibodies labeled with IRDye (in 3% skim milk in TBS with 0.1% SDS/0.5% Triton) at room temperature for 1 h, followed by visualization of bands with a LICOR Odyssey CLx apparatus.
Antibodies against p-p42/44 MAPK (#9101; 1:1,000), p-AKT (Ser473; #9271; 1:1,000), p-S6 (Ser240/244; #5364; 1:1,000), p-S6 (Ser235/236; #2211; 1:1,000), total S6 (#2317; 1:1,000), p-SAPK/JNK (Thr183/Tyr185; #4668; 1:1,000), p-p38 (Thr180/Tyr182; #4511), p-RSK (359/363; #9344, 1:1,000), SHIP2 (#2839, 1:1,000), and RRAS (#8446, 1:1,000) were obtained from Cell Signaling. Antibodies against LZTR1 (sc-390166 X, 1:1,000) and ERK2 (sc-1647; 1:1,000) were obtained from Santa Cruz Biotechnology. Antibodies against MAP4K5 (#ab96551, 1:1,000) and MRAS (#ab176570, 1:1,000) were from Abcam. Pan-RAS antibody (Ab-3, 1:1,000) was from Millipore. Antibodies against KRAS (12063-1-AP, 1:5,000), NRAS (10724-1-AP, 1:1,000), and HRAS (18295-1-AP, 1:1,000), were from Proteintech.
A short peptide sequence (Strep-tag: ASWSHPQFEK) was cloned onto the C-terminus of WT and NPXY-deleted INPPL1 and re-expressed in endogenous INPPL1 KO MOLM13 cells. For pulldown of strep-tagged INPPL1, 1 × 107 MOLM13 cells were lysed in 500 μl NP40 buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1%NP40, 5 mM EDTA). After clarification, 800 μg lysate, at a concentration of 2 μg/ul for each sample, were incubated with MagStrep “type3” XT beads (#2-4090-010; IBA) overnight at 4°C. The next day, beads were washed three times with NP40 buffer, then suspended with 2× SDS-PAGE sample buffer, boiled at 95°C for 10 min, and analyzed by immunoblotting.
RNA extraction and sequencing
For RNAseq sample preparation, parental, INPPL1 KO, and MAP4K5 KO MOLM13 cells (2 million cells/well) were plated in 6-well plates. The next day, cells were treated with DMSO or 1 μM SHP099 for 2.5 h, and RNA was extracted immediately afterwards with a Qiagen RNeasy Plus Mini Kit (74136; Qiagen).
RNA levels were quantified using RNA Nano Chips (#5067-1511; Agilent) on an Agilent 2100 BioAnalyzer. RNA-Seq library preps were constructed with the Illumina TruSeq Stranded mRNA Library Prep kit (#20020595; Illumina) using 1,000 ng of total RNA as input, amplified by 10 cycles of PCR, and paired-end sequenced for 50 cycles on an Illumina NovaSeq6000 SP flowcell with a 2% PhiX spike-in.
Sequencing results were demultiplexed and converted to FASTQ format using Illumina bcl2fastq software. The sequencing reads were adapter- and quality-trimmed with Trimmomatic and then aligned to the human genome (build hg38/GRCh38) using the splice-aware STAR aligner. The featureCounts (Liao et al., 2014) program was utilized to generate counts for each gene based on how many aligned reads overlap its exons. These counts were then normalized and used to test for differential expression using negative binomial generalized linear models implemented by the DESeq2 R package. Pathway enrichment analysis was performed for the pre-ranked gene lists based on the differential expression using the fgsea R package and MSigDB gene sets. All sequence data (RNAseq, genome-wide, and focused CRISPR screens) are available in the Gene Expression Omnibus database under accession no. GSE218491.
Quantitative PCR (qPCR)
Total RNA was isolated using the Qiagen RNeasy kit (74136; Qiagen). Complementary DNAs (cDNAs) were generated using the SuperScript IV First Strand Synthesis System (18091050; Invitrogen). qRT-PCR was performed with Fast SYBR Green Master Mix (4385618; Thermo Fisher Scientific), following the manufacturer’s protocol, in 96-well format in C1000 Touch Thermal Cycler (Bio-Rad). Differential gene expression analysis was performed with CFX Manager (Bio-Rad) and normalized to GAPDH expression. Primers used are listed in Table S6.
Mouse experiments
All the animal experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committees at New York University Grossman School of Medicine. To assess the effects of resistance gene depletion on leukemogenesis, Mv4-11 cells were transduced with a luciferase-expressing lentivirus, and then infected with lentiviruses co-expressing Cas9 and sgRNAs for INPPL1, MAP4K5, LZTR1 or a non-targeting control and selected with puromycin. Each cell line (1.5 million cells) was injected to NSG mice through tail vein, and 10 d later, SHP099 (50 mg/kg) through oral gavage daily was initiated. Whole-body bioluminescence imaging using an IVIS imager was performed at the indicated times immediately after retro-orbital injection of 150 mg/kg D-luciferin Firefly (#122799; Perkin-Elmer). Bioluminescence signals were quantified using Living Imaging software with standard regions of interest rectangles.
Statistical analysis
Results are expressed as mean ± SD, as indicated. Statistical significance was assessed by Student’s t test for proliferation assays and active RAS ELISAs. Survival curves were plotted using the Kaplan–Meier algorithm and Prism software, and significance was assessed using the log-rank (Mantel–Cox) test. P < 0.05 was considered statistically significant.
Online supplemental material
Count tables and MaGeCK analysis of genomic CRISPR screens in MOLM13 and MV4-11 are found in Table S1. GO pathway analysis for MOLM13 and MV4-11 genomic screens is found in Table S2 and Fig. S1 A. Information for focused CRISPR screens can be found in Table S3 and Fig. S1. Fig. S2 and Fig. S3, respectively, show that INPPL1, MAP4K5, or LZTR1 KO causes resistance to structurally distinct SHP2 inhibitors (RMC-4550 and TNO155) and their effects on cell cycle regulation. RNAseq data for INPPL1/MAP4K5-KO compared with parental MOLM13 cells are in Tables S4 and S5 and Fig. S4. The effects of LZTR1 KO on RAS and RIT1 protein levels are in Fig. S5. Table S6 provides the sequences of qPCR primers used in this manuscript.
Acknowledgments
We thank Drs. Yi Ban and Xufeng Chen for help with the mouse leukemogenesis experiments and Drs. Hao Ran and Carmine Fedele for technical advice and helpful discussions.
We also thank the PCC Genome Technology Center, Applied Bioinformatics Laboratory, and Immune Monitoring Laboratory shared resources, funded by National Institutes of Health (NIH) National Cancer Institute (NCI) grant P30 CA016087. This work was supported by NIH-NCI grants CA49152 and CA244896 to B.G. Neel and NIH National Human Genome Research Institute grant HG010099 to N.E. Sanjana.
Author contributions: W. Wei: Experimental design, experiments execution, and writing of the manuscript. M.J. Geer: Generating data for in vitro and in vivo experiments, data analysis, and editing of the manuscript. X. Guo: Design, construction, and evaluation of the CRISPR mini-library. I. Dolgalev: RNAseq data analysis and writing of the manuscript. N.E. Sanjana: Advice in genome-wide CRISPR screens, construction of the mini-library. B.G. Neel: Overall supervision of the project, data analysis, writing of the manuscript.

