


Enteric helminths form intimate physical connections with the intestinal epithelium, yet their ability to directly alter epithelial stem cell fate has not been resolved. Here we demonstrate that infection of mice with the parasite Heligmosomoides polygyrus bakeri (Hpb) reprograms the intestinal epithelium into a fetal-like state marked by the emergence of Clusterin-expressing revival stem cells (revSCs). Organoid-based studies using parasite-derived excretory-secretory products reveal that Hpb-mediated revSC generation occurs independently of host-derived immune signals and inhibits type 2 cytokine–driven differentiation of secretory epithelial lineages that promote their expulsion. Reciprocally, type 2 cytokine signals limit revSC differentiation and, consequently, Hpb fitness, indicating that helminths compete with their host for control of the intestinal stem cell compartment to promote continuation of their life cycle.

Introduction
Intestinal helminths remain pervasive throughout the animal kingdom by manipulating host defense pathways to prioritize tissue adaptation and repair over parasite expulsion (King and Li, 2018). This host defense strategy begins at the gut epithelium, a monolayer of cells continually replenished by intestinal stem cells (ISCs) residing at the crypt base. As frontline effectors of barrier integrity, a key attribute of the ISC compartment is its ability to undergo extensive transcriptional reprogramming in response to injury (Santos et al., 2018; de Sousa e Melo and de Sauvage, 2019). During homeostasis, ISCs, marked by the expression of leucine rich repeat containing G protein-coupled receptor 5 (Lgr5), reside at the crypt base where they are supported by growth factor–secreting Paneth cells and various stromal cell subsets (Clevers, 2013). In this setting, ISCs are responsible for the perpetual turnover of the entire repertoire of gut epithelial cell lineages (Clevers, 2013). In response to diverse forms of damage, Lgr5+ ISC are depleted but rapidly replenished, at least in part, by dedifferentiation of lineage-committed cells to swiftly regenerate the intestinal barrier (Meyer et al., 2022; Ouladan and Gregorieff, 2021; Larsen and Jensen, 2021). These cell fate changes are often accompanied by upregulation of genes normally restricted to the fetal gut, suggesting that replenishment of stem cells following injury requires the temporary reversion or reprogramming of the adult gut epithelium into an embryonic state (Yui et al., 2018; Ohara et al., 2022). Indeed, we have recently identified a damage-induced ISC population marked by the expression of Clusterin (Clu), which we termed “revival” stem cells (revSCs), that are critical for gut regeneration following irradiation and dextrate sulfate sodium–induced colitis (Ayyaz et al., 2019).
The intestinal epithelium is also critical for host defense against diverse species of parasitic helminths (Coakley and Harris, 2020). By executing the “weep-and-sweep” response, the epithelium increases mucus secretion and cell turnover to actively promote worm expulsion (Anthony et al., 2007; Cliffe et al., 2005; Shea-donohue et al., 2001). The type 2 immune response, orchestrated by IL-4 and IL-13, is fundamental to driving these epithelial responses during helminth infection. For example, IL-4 and IL-13 drive the differentiation and effector functions of secretory epithelial cells such as tuft and goblet cells that produce lipid mediators (McGinty et al., 2020) and anti-helminth effector molecules (e.g., Relmβ), respectively, that can promote worm expulsion (Barner et al., 1998; Horsnell et al., 2007; Herbert et al., 2009). However, early work showed that artificially enhancing the type 2 immune response by administration of long-acting IL-4 complexes was required to expel Heligmosomoides polygyrus bakeri (Hpb), a natural parasitic roundworm of mice, during primary infection (Urban et al., 1995). These results suggested that Hpb actively limits type 2 immunity.
While the ability of helminths to counter the host immune response during infection has been previously demonstrated, these effects have been largely attributed to modulation of the innate and adaptive immune compartments. Only recently have studies investigated whether helminths directly target the intestinal epithelium to regulate host defense (Drurey et al., 2021; Duque-Correa et al., 2020). By interrogating Hpb infection in mice, we demonstrate that these parasites directly reprogram the ISC compartment, independently of host-derived factors. This reprogramming event coincides with adult parasite colonization of the intestinal lumen and is characterized by activation of a fetal-like, regenerative Hippo pathway signature and the emergence of Clu-expressing revSCs. Morphological and single-cell transcriptomic analyses of intestinal organoids exposed to Hpb-conditioned medium (Hpb-CM) revealed direct targeting of the ISC compartment. Hpb-CM also suppressed IL-13–induced secretory cell differentiation, while deletion of type 2 cytokine signaling in vivo led to an epithelial-intrinsic expansion of revSCs and improved parasite fitness. Collectively, our study reveals how a helminth parasite co-opts a tissue development program to counter type 2 immune–mediated expulsion and maintain chronic infection.
Results and discussion
Enhanced fetal reprogramming of the intestinal epithelium at the luminal stage of Hpb infection
A recent report demonstrated that expression of Ly6a (encoding Sca-1) marks epithelial cells undergoing a fetal-like reversion event in granuloma-associated crypts during the tissue-dwelling stage of Hpb infection (6 d post-infection [dpi]; Nusse et al., 2018). We performed a kinetic analysis of duodenal tissue from Hpb-infected adult C57BL/6 mice using RNA sequencing (RNA-seq) that confirmed an increase in several fetal-associated genes (Mustata et al., 2013) including Ly6a expression on day 6 after infection (Fig. 1 A). However, we detected a more robust fetal gene signature during the luminal stage of infection (day 14), a time when adult worms establish residence alongside the intestinal villi (Fig. 1, A and B). To corroborate these results, we extracted intestinal crypts from Hpb-infected mice at different time points and cultured them under standard organoid growth conditions (Fig. 1 C). Adult intestinal organoids normally develop into polarized structures that mimic the crypt/villus architecture and are driven by abundant Lgr5+ ISCs (Date and Sato, 2015). Although crypts extracted at early time points after infection yielded typical budding organoids with abundant Paneth cells (Fig. 1 D), crypts on day 14 after infection developed into hollow spheres resembling fetal organoids (Fig. 1, D and E; Mustata et al., 2013). Furthermore, crypts and epithelial cells isolated on day 14 after infection showed elevated expression of several fetal genes including Clu, Il1rn, and Msln (Figs. 1 F and S1 A). These findings were confirmed using single-molecule RNA in situ hybridization (RNAscope), which showed the loss of Olfm4+ ISC and a transient induction of Ly6a in granuloma-associated crypts on day 6, but sustained induction of Il1rn and Il33 in distinct domains along the crypt–villus axis and the luminal stage-specific induction of Clu (Fig. 1, G and H; and Fig. S1 B).

A fetal-associated transcriptional signature at the luminal stage of Hpb infection. (A) Analysis of RNA-seq dataset (Gentile et al., 2020) showing reads per kilobase million of fetal-associated genes (Mustata et al., 2013) in duodenal tissue 6 and 14 dpi. Specific genes of interest are depicted by name. Heatmap was created using the Heatmapper web tool (Babicki et al., 2016); uninf, uninfected. (B) GSEA of fetal-associated transcripts 14 dpi (in comparison to Mustata et al., 2013). NES, normalized enrichment score. (C–E) C57BL/6 mice were infected with 150 L3 Hpb larvae, and SI crypts were extracted 2, 6, and 14 dpi and plated in Matrigel domes for 24–48 h. Image in C created with Biorender.com. (D) Representative photomicrographs of organoids cultured from Hpb-infected intestines, 24 h after culture time point; arrowheads indicate Paneth cells. (E) Frequency of organoids with spheroid morphology. (F) qPCR analysis of organoids cultured from Hpb-infected intestines. (G) RNAscope of fetal-associated transcripts in Hpb-infected tissue. a and b are enlarged insets from left panels; Gr, granuloma. (H) Colorimetric RNAscope of Clu mRNA and quantification of Clu+ areas. Scale bar, 50 μm (D–G); 100 μm (H). Data shown are representative of one (A and B) or two or more (D–H) independent experiments, n > 3 biological replicates. Statistical tests: nonparametric one-way ANOVA (E), t test (F and H); *, P < 0.05; ***, P < 0.005.
A fetal-associated transcriptional signature at the luminal stage of Hpb infection. (A) Analysis of RNA-seq dataset (Gentile et al., 2020) showing reads per kilobase million of fetal-associated genes (Mustata et al., 2013) in duodenal tissue 6 and 14 dpi. Specific genes of interest are depicted by name. Heatmap was created using the Heatmapper web tool (Babicki et al., 2016); uninf, uninfected. (B) GSEA of fetal-associated transcripts 14 dpi (in comparison to Mustata et al., 2013). NES, normalized enrichment score. (C–E) C57BL/6 mice were infected with 150 L3 Hpb larvae, and SI crypts were extracted 2, 6, and 14 dpi and plated in Matrigel domes for 24–48 h. Image in C created with Biorender.com. (D) Representative photomicrographs of organoids cultured from Hpb-infected intestines, 24 h after culture time point; arrowheads indicate Paneth cells. (E) Frequency of organoids with spheroid morphology. (F) qPCR analysis of organoids cultured from Hpb-infected intestines. (G) RNAscope of fetal-associated transcripts in Hpb-infected tissue. a and b are enlarged insets from left panels; Gr, granuloma. (H) Colorimetric RNAscope of Clu mRNA and quantification of Clu+ areas. Scale bar, 50 μm (D–G); 100 μm (H). Data shown are representative of one (A and B) or two or more (D–H) independent experiments, n > 3 biological replicates. Statistical tests: nonparametric one-way ANOVA (E), t test (F and H); *, P < 0.05; ***, P < 0.005.

Fetal reversion of the intestinal epithelium at the luminal stage of Hpb infection and following Hpb-CM stimulation of SI organoids. Related to Figs. 1 and 2. (A) Epithelial cells were extracted from the SI of uninfected mice on day 14 after Hpb infection, and expression of the indicated fetal genes was assessed by qPCR. (B) RNAscope of Il33, Il1rn, and Clu on days 6 and 14 after Hpb infection of SI tissues. (C) Heatmap of reads per kilobase million was created using the Heatmapper web tool (Babicki et al., 2016). (D) GSEA of transcripts associated with mature enterocytes, goblet cells, and Paneth cells following Hpb-CM stimulation of organoids. To exclude the possibility of endotoxin or host protein contamination in Hpb-CM, the following three experiments were performed: (E)Hpb adult worms were incubated in organoid basal medium. 24 h later, CM was collected (Hpb-CM), and the worms were moved into fresh organoid basal medium for an additional 24 h. At the 48-h time point, CM was collected again (Hpb-CM 48H). SI organoids were stimulated on day 0 of culture with Hpb-CM, Hpb-CM 48H, or control ENR medium. qPCR analysis of selected fetal-associated genes is shown. (F)Hpb-CM was passed through a 3-kD filter, and the flowthrough was collected. SI organoids were stimulated on day 0 of culture with crude Hpb-CM, flowthrough fraction of Hpb-CM (Hpb-CM <3 kD), or control ENR medium. (G) SI organoids were stimulated on day 0 of culture with Hpb-CM, 1 ng/ml LPS, 10 ng/ml LPS, or control ENR medium. qPCR analysis of selected fetal-associated genes as well as Il6 and Tnfa is shown. Scale bar, 50 μm. Data shown are representative of two independent experiments (A, B, and E–G) or one independent experiment (C and D), n > 3 biological replicates. Statistical tests: t test (A–G); *, P < 0.05; **, P < 0.01; ***, P < 0.005.
Fetal reversion of the intestinal epithelium at the luminal stage of Hpb infection and following Hpb-CM stimulation of SI organoids. Related to Figs. 1 and 2. (A) Epithelial cells were extracted from the SI of uninfected mice on day 14 after Hpb infection, and expression of the indicated fetal genes was assessed by qPCR. (B) RNAscope of Il33, Il1rn, and Clu on days 6 and 14 after Hpb infection of SI tissues. (C) Heatmap of reads per kilobase million was created using the Heatmapper web tool (Babicki et al., 2016). (D) GSEA of transcripts associated with mature enterocytes, goblet cells, and Paneth cells following Hpb-CM stimulation of organoids. To exclude the possibility of endotoxin or host protein contamination in Hpb-CM, the following three experiments were performed: (E)Hpb adult worms were incubated in organoid basal medium. 24 h later, CM was collected (Hpb-CM), and the worms were moved into fresh organoid basal medium for an additional 24 h. At the 48-h time point, CM was collected again (Hpb-CM 48H). SI organoids were stimulated on day 0 of culture with Hpb-CM, Hpb-CM 48H, or control ENR medium. qPCR analysis of selected fetal-associated genes is shown. (F)Hpb-CM was passed through a 3-kD filter, and the flowthrough was collected. SI organoids were stimulated on day 0 of culture with crude Hpb-CM, flowthrough fraction of Hpb-CM (Hpb-CM <3 kD), or control ENR medium. (G) SI organoids were stimulated on day 0 of culture with Hpb-CM, 1 ng/ml LPS, 10 ng/ml LPS, or control ENR medium. qPCR analysis of selected fetal-associated genes as well as Il6 and Tnfa is shown. Scale bar, 50 μm. Data shown are representative of two independent experiments (A, B, and E–G) or one independent experiment (C and D), n > 3 biological replicates. Statistical tests: t test (A–G); *, P < 0.05; **, P < 0.01; ***, P < 0.005.
Direct reprogramming of the ISC compartment by Hpb-CM
One interpretation of our findings is that luminal adult worms trigger fetal reprogramming because of the host immune response to parasite-induced breaches in the epithelium, as previously suggested for the larval stages of infection (Nusse et al., 2018). An alternative, but not mutually exclusive, possibility is that fetal reprogramming is driven by recognition of helminth-derived products by intestinal epithelial cells without a priori damage. To test the latter possibility, we stimulated organoids from the duodenum of uninfected mice with Hpb-CM collected from adult Hpb parasites. Strikingly, Hpb-CM–stimulated organoids assumed a spheroid morphology with robust induction of Clu and other fetal markers, while also downregulating the homeostatic ISC markers Lgr5 and Olfm4 (Fig. 2, A–D). RNA-seq followed by fast gene set enrichment analysis (fGSEA) confirmed the direct fetal-like reprogramming of organoids by Hpb-CM (Fig. 2, E and F; Mustata et al., 2013). Further analysis revealed that this reprogramming event was accompanied by the suppression of diverse lineages of differentiated epithelial cells (Fig. 2 G; and Fig. S1, C and D; Haber et al., 2017) including goblet and Paneth cells, similar to findings reported by Drurey et al. (2021). We also excluded the possibility that this fetal-like reversion of the epithelium is mediated by endotoxin contamination of Hpb-CM (Fig. S1, E–G), in agreement with previous studies establishing the inability of small intestinal organoids to respond to LPS due to lack of TLR4 expression (Kayisoglu et al., 2021). Importantly, helminth-induced fetal-like reprogramming of organoids was not observed in response to excretory-secretory products prepared from Nippostrongylus brasiliensis (Nippo-CM), a rat parasite that briefly transits through the small intestine (SI) of infected mice (Fig. 2, H–J). Collectively, these results suggest that fetal reprogramming is associated with parasites that chronically infect the SI.
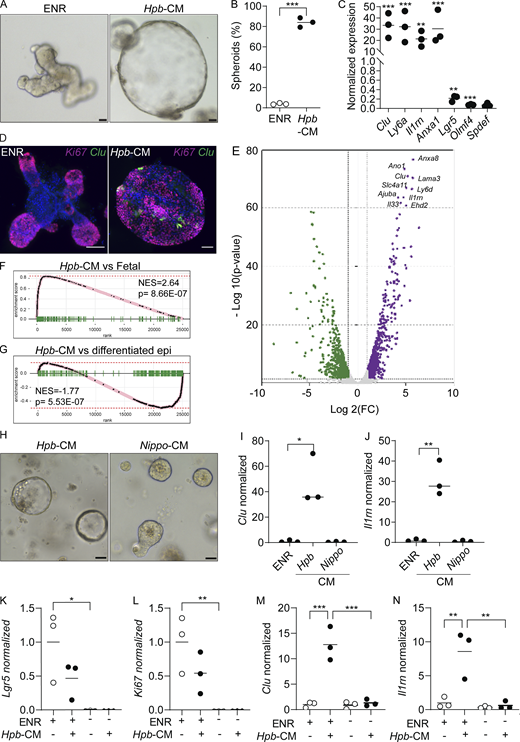
Hpb-CM directly induces fetal reversion of intestinal organoids. (A) SI organoids were stimulated with ENR (control) or Hpb-CM medium for 24 h after 3 d in culture. (B) Frequency of organoids with a spheroid morphology. (C) qPCR analysis of SI organoids stimulated with Hpb-CM for 24 h from the plating of fresh crypts. (D) Confocal photomicrographs of SI organoids from CluGFP crypts (Ayyaz et al., 2019) stimulated with Hpb-CM for 24 h on day 3 of culture. (E) Volcano plot of differentially expressed genes from RNA-seq analysis comparing Hpb-CM–treated organoids to control organoids, false discovery rate < 0.05. For a complete list of differentially expressed genes, see Table S1. (F and G) GSEA of fetal-associated transcripts (F) and differentiated epithelial cell markers (G). NES, normalized enrichment score. (H–J) Representative photomicrographs (H) and qPCR analysis (I and J) of SI organoids stimulated with Nippo-CM for 24 h. (K–N) qPCR analysis of SI established organoids stimulated with Hpb-CM for 24 h in the presence or absence of ENR. Scale bar, 50 μm. Data shown are representative of two or more independent experiments, n = 3 biological replicates; statistical tests: t test (B, C, I, and J), two-way ANOVA (K–N); *, P < 0.05; **, P < 0.01; ***, P < 0.005.
Hpb-CM directly induces fetal reversion of intestinal organoids. (A) SI organoids were stimulated with ENR (control) or Hpb-CM medium for 24 h after 3 d in culture. (B) Frequency of organoids with a spheroid morphology. (C) qPCR analysis of SI organoids stimulated with Hpb-CM for 24 h from the plating of fresh crypts. (D) Confocal photomicrographs of SI organoids from CluGFP crypts (Ayyaz et al., 2019) stimulated with Hpb-CM for 24 h on day 3 of culture. (E) Volcano plot of differentially expressed genes from RNA-seq analysis comparing Hpb-CM–treated organoids to control organoids, false discovery rate < 0.05. For a complete list of differentially expressed genes, see Table S1. (F and G) GSEA of fetal-associated transcripts (F) and differentiated epithelial cell markers (G). NES, normalized enrichment score. (H–J) Representative photomicrographs (H) and qPCR analysis (I and J) of SI organoids stimulated with Nippo-CM for 24 h. (K–N) qPCR analysis of SI established organoids stimulated with Hpb-CM for 24 h in the presence or absence of ENR. Scale bar, 50 μm. Data shown are representative of two or more independent experiments, n = 3 biological replicates; statistical tests: t test (B, C, I, and J), two-way ANOVA (K–N); *, P < 0.05; **, P < 0.01; ***, P < 0.005.
The addition of EGF, Noggin, and R-spondin-1 (ENR) is an integral part of the organoid culture medium, reflecting the importance of morphogens such as WNTs and bone morphogenetic proteins in the regulation of ISC self-renewal and lineage commitment (Santos et al., 2018). To assess whether organoids depend on niche factors for helminth-driven reprogramming, established organoids were replated in the absence of ENR supplementation and stimulated with Hpb-CM. While the removal of niche factors resulted in an expected decrease in proliferation and loss of Lgr5 expression, Hpb-CM was unable to initiate a fetal transcriptional response (Fig. 2, K–N), indicating that reprogramming requires niche factors and likely targets a mitotic progenitor cell.
Hpb-CM reprogramming of the ISC compartment is partially dependent on the Hippo signaling pathway
The Hippo signaling pathway is a well-established regulator of tissue regeneration in response to damage and linked to fetal reprogramming (Yui et al., 2018; Gregorieff and Wrana, 2017). Consistently, Hpb infection induced nuclear translocation of YAP, a transcriptional effector of Hippo signaling, in the intestinal epithelium at the luminal stage of infection (Fig. S2 A). Although many of the fetal-associated genes, including Clu, are YAP target genes, Hpb-CM–induced fetal-like reprogramming of organoids and in vivo Clu induction were only partially Yap dependent (Fig. S2, B–D; and data not shown). Furthermore, treatment of organoids with recombinant CLU was not sufficient to induce spheroids, and stimulation of Clu-deficient organoids with Hpb-CM did not prevent organoid reprogramming (Fig. S2, E–G; and not depicted). Finally, animals lacking YAP in the intestinal epithelium (VillinCreYapfl/fl mice) had worm burdens equivalent to those of littermate controls, indicating that this transcriptional activator is dispensable for Hpb persistence (Fig. S2 H). Thus, Clu expression represents a broader transcriptional network mediating fetal-like reprogramming that does not exclusively rely on YAP-dependent signaling.

Fetal-like reprogramming is partially Yap dependent and is not amplified by IFNγ. Related to Figs. 1, 2, and 3. (A) Immunohistochemistry for active YAP; squares, area in bottom panel; open arrowheads, nuclear YAP expression; black arrowheads, worm presence. (B–D) qPCR analysis of the indicated genes from Yap−/− and littermate control SI organoids stimulated with Hpb-CM for 24 h. (E–G) qPCR analysis of Clu−/− and littermate control SI organoids stimulated with Hpb-CM for 24 h. (H–K)Vilcre;Yapfl/fl and littermate control Vilcre;Yapfl/+ mice were infected with 150 L3 Hpb larvae, and adult worms were counted 28 dpi. (I–K) qPCR analysis of the indicated genes from SI organoids stimulated, on day 0 of culture, with ENR (control) or 10 ng/ml IFNγ with or without Hpb-CM for 24 h. (M) Monocle 3 direct exports of the trajectory analysis using CBCs as the pseudotime source variable, showing the distribution of the different cell clusters along the pseudotime variable. Scale bar, 100 μm. Data shown are representative of two or more independent experiments, n > 3 biological replicates; statistical tests: two-way ANOVA (B–L); *, P < 0.05; **, P < 0.01; ***, P < 0.005; ****, P < 0.001.
Fetal-like reprogramming is partially Yap dependent and is not amplified by IFNγ. Related to Figs. 1, 2, and 3. (A) Immunohistochemistry for active YAP; squares, area in bottom panel; open arrowheads, nuclear YAP expression; black arrowheads, worm presence. (B–D) qPCR analysis of the indicated genes from Yap−/− and littermate control SI organoids stimulated with Hpb-CM for 24 h. (E–G) qPCR analysis of Clu−/− and littermate control SI organoids stimulated with Hpb-CM for 24 h. (H–K)Vilcre;Yapfl/fl and littermate control Vilcre;Yapfl/+ mice were infected with 150 L3 Hpb larvae, and adult worms were counted 28 dpi. (I–K) qPCR analysis of the indicated genes from SI organoids stimulated, on day 0 of culture, with ENR (control) or 10 ng/ml IFNγ with or without Hpb-CM for 24 h. (M) Monocle 3 direct exports of the trajectory analysis using CBCs as the pseudotime source variable, showing the distribution of the different cell clusters along the pseudotime variable. Scale bar, 100 μm. Data shown are representative of two or more independent experiments, n > 3 biological replicates; statistical tests: two-way ANOVA (B–L); *, P < 0.05; **, P < 0.01; ***, P < 0.005; ****, P < 0.001.
Hpb-CM–induced epithelial reprogramming is not regulated by IFNγ signals
A previous study demonstrated that the induction of Ly6a+ ISCs in granuloma-associated crypts was IFNγ dependent (Nusse et al., 2018). Although our RNA-seq and RNAscope data (Fig. 1, A and G) confirmed the induction of Ly6a mRNA on day 6 after Hpb infection, expression of this gene was decreased by day 14 after infection. Nevertheless, we tested the possibility that the fetal reversion induced by Hpb-CM is regulated by IFNγ. As expected for an IFNγ-inducible gene (Khan et al., 1990), Ly6a was induced following organoid stimulation with IFNγ. However, IFNγ did not induce other fetal-associated genes we tested, and its addition to Hpb-CM–stimulated organoids had no effect or decreased induction of other fetal genes (Fig. S2, I–L). Therefore, IFNγ signals do not support Hpb-CM–induced epithelial reprogramming.
Emergence of revSCs at the luminal stage of Hpb infection
For a more in-depth analysis into the effect Hpb-CM exerts on the ISC compartment, we performed single-cell RNA-seq (scRNA-seq) on control and Hpb-CM–stimulated organoids. Clustering of cells was performed using previously published epithelial lineage-defining markers (Haber et al., 2017; Ayyaz et al., 2019). Our analysis yielded eight cell clusters, identifying stem (Lgr5+ crypt base columnar cells [CBCs]) and progenitor cells as well as mature secretory populations (Fig. 3, A and B). In accordance with our bulk RNA-seq results (Fig. 2), Hpb-CM expanded a distinct cell cluster enriched in fetal genes, which was rare in control-treated organoids, that we previously identified as revSCs following irradiation (Ayyaz et al., 2019). By contrast, goblet and Paneth cell clusters were significantly reduced in Hpb-CM–stimulated organoids, whereas secretory progenitor cells (marked by Atoh1 and Spdef expression) were enriched (Fig. 3, A and C). Under these culture conditions, we were not able to detect a sufficient number of tuft cells for statistical analysis. Next, we performed trajectory analyses in which the pseudotime scale indicates gene expression changes that inform the potential differentiation paths from a chosen source population (i.e., CBCs). This analysis demonstrated that transit amplifying (TA) cells are displaced within the Uniform Manifold Approximation and Projection (UMAP) space by revSC expansion in Hpb-CM–stimulated organoids (Figs. 3 D and S2 M), implying a central role for revSCs in driving renewal of the gut epithelium during Hpb infection in vivo. To validate this possibility, we performed lineage-tracing studies using tamoxifen-inducible Clu fate-mapping mice (Ayyaz et al., 2019). As shown in Fig. 3, E–G, labeling Clu-expressing cells on day 5–7 after infection and assessing the tissue 12 d later yielded rare Tomato (Tom)+ cells. By contrast, labeling Clu-expressing cells on day 12 after infection resulted in robust generation of Tom+ ribbons along the crypt-villus axis 12 d later, a pattern indicative of progeny derived from Clu-expressing stem cells. Consistent with their stem cell capacity, revSCs gave rise to Tom+ cells that also expressed DCLK or UEA1, representing tuft and goblet cells, respectively. Remarkably, however, these secretory lineages were less abundant in Tom+ ribbons compared with Tom− ribbons (Fig. 3, H and I). Together, these data suggest that Hpb profoundly rewires the differentiation trajectories of ISCs and TA cells in favor of revSCs, which in turn, display a reduced capacity to generate secretory cell lineages with direct anti-helminth activity.
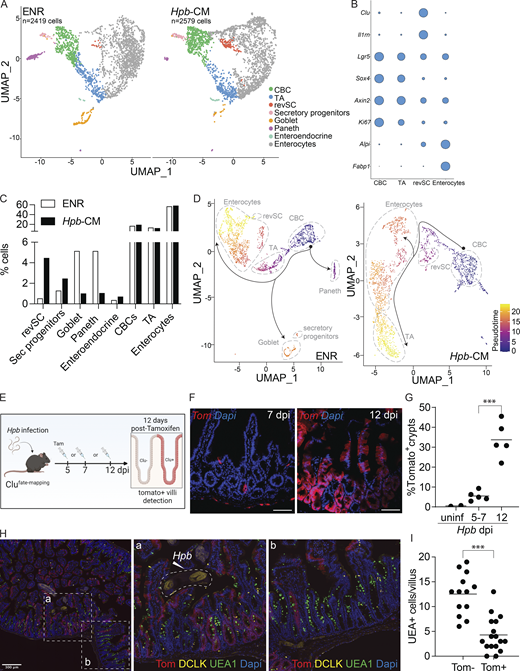
Hpb-CM expands revSCs and limits secretory cell differentiation. SI organoids were stimulated with Hpb-CM for 24 h from the plating of fresh crypts, and scRNA-seq analysis was performed. (A) UMAP projection plots; colors represent cells clustered together based on gene expression similarity. (B) Representative cluster identifying markers; circle size represents average expression of indicated transcripts (complete gene expression list can be found in Table S2). (C) The proportion of each cluster within each sample is presented. (D) Trajectory analysis using CBCs as the pseudotime source variable (pseudotime = 0). (E–G)Clu fate-mapping (Clu-CreERT; Rosa26-LSL-tdTomato; Ayyaz et al., 2019) mice were infected with Hpb, and Clu+ Tom+ cells were labeled by tamoxifen injection 5, 7, or 12 dpi and imaged 12 d after each injection; scale bar, 100 μm; image created with Biorender.com. (H and I) Immunofluorescent staining for DCLK (yellow), UEA1 (green), and DAPI (blue) as well as detection of Tom (red) in Clu fate-mapping mice on 24 dpi (mice were injected with tamoxifen on 12 dpi). Data shown are representative of one (A–D) or three or more (E–I) independent experiments, n = 3–5 biological replicates. Statistical tests: two-way ANOVA (G), t test (I); ***, P < 0.005.
Hpb-CM expands revSCs and limits secretory cell differentiation. SI organoids were stimulated with Hpb-CM for 24 h from the plating of fresh crypts, and scRNA-seq analysis was performed. (A) UMAP projection plots; colors represent cells clustered together based on gene expression similarity. (B) Representative cluster identifying markers; circle size represents average expression of indicated transcripts (complete gene expression list can be found in Table S2). (C) The proportion of each cluster within each sample is presented. (D) Trajectory analysis using CBCs as the pseudotime source variable (pseudotime = 0). (E–G)Clu fate-mapping (Clu-CreERT; Rosa26-LSL-tdTomato; Ayyaz et al., 2019) mice were infected with Hpb, and Clu+ Tom+ cells were labeled by tamoxifen injection 5, 7, or 12 dpi and imaged 12 d after each injection; scale bar, 100 μm; image created with Biorender.com. (H and I) Immunofluorescent staining for DCLK (yellow), UEA1 (green), and DAPI (blue) as well as detection of Tom (red) in Clu fate-mapping mice on 24 dpi (mice were injected with tamoxifen on 12 dpi). Data shown are representative of one (A–D) or three or more (E–I) independent experiments, n = 3–5 biological replicates. Statistical tests: two-way ANOVA (G), t test (I); ***, P < 0.005.
An oxidative stress response mediates Hpb-induced fetal reversion of intestinal organoids
To gain mechanistic insight into how Hpb-CM reprograms the ISC compartment, we next assessed the transcriptional changes within each cluster identified in our scRNA-seq experiment. The number of differentially expressed genes (DEGs) was highest in enterocytes and TA cells following Hpb-CM stimulation, and the most significant pathways identified were seen in the TA cell compartment (Fig. 4, A and B). Specifically, pathway analysis indicated a metabolic change in organoids following Hpb-CM stimulation as well as a striking oxidative stress response (Fig. 4 B). To exclude the possibility that nutrient deprivation was contributing to fetal reprogramming, we used purified HES (Valanparambil et al., 2014). To this end, Hpb-CM was concentrated over a 3-kD filter and used to stimulate organoids at a 10-fold dilution. As shown in Fig. 4, C–E, diluting concentrated HES in fresh ENR resulted in induction of spheroid formation as well as the fetal-associated genes Clu and Il1rn to the same extent as Hpb-CM, indicating that nutrient deprivation is not likely the cause of cell reprogramming. In additional agreement with the induction of an oxidative stress response, Hmox1, a well-known indicator of oxidative stress (Poss and Tonegawa, 1997), was induced following Hpb-CM stimulation (Fig. 4 F). We next directly tested the role of oxidative stress by stimulating organoids with Hpb-CM in the presence of elevated concentrations of the antioxidant N-acetylcysteine (NAC; Ezeriņa et al., 2018). The addition of NAC suppressed spheroid formation and the induction of fetal-associated genes (Fig. 4, G–J), establishing the oxidative stress response as an important mediator of Hpb-driven fetal-like reversion of the ISC compartment.

An oxidative stress response drives Hpb-CM–induced fetal reversion of intestinal organoids. (A) Number of DEGs in each cell cluster identified by scRNA-seq analysis described in Fig. 3. (B) Pathway analysis based on DEGs was performed using Metascape (Zhou et al., 2019). (C–E) SI organoids were stimulated at the start of culture with ENR (control), Hpb-CM, or a 10-fold dilution of HES for 24 h. (C) Frequency of organoids with a spheroid morphology. (D and E) qPCR analysis of the indicated genes normalized to Hprt expression. (F–J) SI organoids were stimulated at the start of culture with ENR (control) or Hpb-CM. qPCR analysis is shown (G–J). SI organoids were stimulated with ENR (control) or Hpb-CM in the presence of increasing concentrations of NAC for 24 h. qPCR analysis of the indicated genes is shown. Scale bar, 50 μm; data shown are representative of one (A and B) or three or more (C–H) independent experiments, n = 3–5 biological replicates. Statistical tests: one-way ANOVA (C–E), t test (F), two-way ANOVA (H–J); *, P < 0.05; *, P < 0.01; ***, P < 0.005; ****, P < 0.001.
An oxidative stress response drives Hpb-CM–induced fetal reversion of intestinal organoids. (A) Number of DEGs in each cell cluster identified by scRNA-seq analysis described in Fig. 3. (B) Pathway analysis based on DEGs was performed using Metascape (Zhou et al., 2019). (C–E) SI organoids were stimulated at the start of culture with ENR (control), Hpb-CM, or a 10-fold dilution of HES for 24 h. (C) Frequency of organoids with a spheroid morphology. (D and E) qPCR analysis of the indicated genes normalized to Hprt expression. (F–J) SI organoids were stimulated at the start of culture with ENR (control) or Hpb-CM. qPCR analysis is shown (G–J). SI organoids were stimulated with ENR (control) or Hpb-CM in the presence of increasing concentrations of NAC for 24 h. qPCR analysis of the indicated genes is shown. Scale bar, 50 μm; data shown are representative of one (A and B) or three or more (C–H) independent experiments, n = 3–5 biological replicates. Statistical tests: one-way ANOVA (C–E), t test (F), two-way ANOVA (H–J); *, P < 0.05; *, P < 0.01; ***, P < 0.005; ****, P < 0.001.
Counter-regulation of the ISC compartment by Hpb and type 2 cytokines
Tuft and goblet cells are secretory epithelial lineages that orchestrate the initiation and effector functions, respectively, of the type 2 immune response that is responsible for parasitic helminth expulsion (Coakley and Harris, 2020; von Moltke et al., 2016; Gerbe et al., 2016). Therefore, building on our scRNA-seq and imaging results (Fig. 3), we tested whether Hpb-CM was able to directly inhibit type 2 cytokine–induced differentiation of secretory cells. While IL-13 stimulation of organoids resulted in robust Spdef, Clca1 (encoding Gob5), Muc2, and Dclk1 mRNA expression, the addition of Hpb-CM prevented induction of these lineage-specific transcripts (Fig. 5, A–D; Drurey et al., 2021). Suppression of IL-13–induced differentiation of tuft and goblet cells was also confirmed by immunofluorescence microscopy (Fig. 5, E–G). By contrast, pretreatment of organoids with IL-13 to promote secretory cell differentiation limited fetal gene expression upon subsequent exposure to Hpb-CM (Fig. 5, H and I). Therefore, Hpb-CM and type 2 cytokines act in a counter-regulatory manner on the ISC compartment.

Counter-regulation of the ISC compartment by helminths and type 2 cytokine signaling. (A–D) SI organoids were stimulated at the start of culture with Hpb-CM ± IL-13, and qPCR analysis was performed for the indicated genes 48 h later. (E) Photomicrographs of organoids stimulated with Hpb-CM ± IL-13 for 48 h. (F and G) DCLK+ (F) and MUC2+ (G) cells were counted in 20 individual organoids from two mice. (H and I) SI organoids were stimulated with IL-13 for 10 d (over three passages) before Hpb-CM stimulation, and qPCR analysis for the indicated genes was performed 24 h after Hpb-CM stimulation. (J and K) C57BL/6 and Stat6−/− mice were infected with Hpb, and crypts were extracted 14 dpi and plated in Matrigel domes for 24 h. Representative photomicrographs (J) and quantification of ex vivo spheroid generation (K) are shown. (L and M)Clu expression by in situ hybridization of WT and Stat6−/− tissues 14 dpi. Representative photomicrographs (L) and quantification (M) of Clu+ tissue area. (N) Quantification of Clu+ intestinal tissue from Il4raWT and Il4raΔIEC mice 14 dpi. (O and P) Fecal Hpb egg counts from WT and Stat6−/− (O) and Il4raWT and Il4raΔIEC (P) mice 14 dpi. (Q) Quantification of Clu+ intestinal area from Clu fate-mapping mice injected with tamoxifen 12 dpi and PBS or IL-4C 10, 12, and 14 dpi. Tissues were harvested on day 24 after infection. Scale bar, 50 μm (E and J); 1 mm (L). All data shown are representative of two or more independent experiments, n > 3 biological replicates. Statistical tests: two-way ANOVA (A–K and O), t test (M, N, and Q); *, P < 0.05; **, P < 0.01; ***, P < 0.005.
Counter-regulation of the ISC compartment by helminths and type 2 cytokine signaling. (A–D) SI organoids were stimulated at the start of culture with Hpb-CM ± IL-13, and qPCR analysis was performed for the indicated genes 48 h later. (E) Photomicrographs of organoids stimulated with Hpb-CM ± IL-13 for 48 h. (F and G) DCLK+ (F) and MUC2+ (G) cells were counted in 20 individual organoids from two mice. (H and I) SI organoids were stimulated with IL-13 for 10 d (over three passages) before Hpb-CM stimulation, and qPCR analysis for the indicated genes was performed 24 h after Hpb-CM stimulation. (J and K) C57BL/6 and Stat6−/− mice were infected with Hpb, and crypts were extracted 14 dpi and plated in Matrigel domes for 24 h. Representative photomicrographs (J) and quantification of ex vivo spheroid generation (K) are shown. (L and M)Clu expression by in situ hybridization of WT and Stat6−/− tissues 14 dpi. Representative photomicrographs (L) and quantification (M) of Clu+ tissue area. (N) Quantification of Clu+ intestinal tissue from Il4raWT and Il4raΔIEC mice 14 dpi. (O and P) Fecal Hpb egg counts from WT and Stat6−/− (O) and Il4raWT and Il4raΔIEC (P) mice 14 dpi. (Q) Quantification of Clu+ intestinal area from Clu fate-mapping mice injected with tamoxifen 12 dpi and PBS or IL-4C 10, 12, and 14 dpi. Tissues were harvested on day 24 after infection. Scale bar, 50 μm (E and J); 1 mm (L). All data shown are representative of two or more independent experiments, n > 3 biological replicates. Statistical tests: two-way ANOVA (A–K and O), t test (M, N, and Q); *, P < 0.05; **, P < 0.01; ***, P < 0.005.
The main driver of secretory cell differentiation is STAT6-dependent type 2 cytokine signaling (via IL-4 and IL-13; Finkelman et al., 2004). To test the involvement of type 2 immune signaling in the regulation of Hpb-induced fetal-like reprogramming in vivo, we infected Stat6-deficient mice with Hpb. Remarkably, loss of STAT6 resulted in increased ex vivo fetal-associated spheroid formation and in situ intestinal Clu expression (Fig. 5, J–M). To specifically test the role of epithelial-intrinsic type 2 immune signaling in the induction of Clu+ revSCs, VillinCreIl4rafl/fl mice (Il4raΔIEC) were infected with Hpb, and a trend toward increased in situ Clu expression was detected (Figs. 5 N and S3 A). We next assessed Hpb fitness and persistence by evaluating adult worm burden in Stat6−/− mice (Fig. S3 C) and fecal egg counts in Stat6−/− and Il4raΔIEC mice (Fig. 5, O and P). In association with enhanced Clu expression, Stat6−/− and Il4raΔIEC mice had elevated worm and egg burdens on day 14 after infection, suggesting that epithelial-intrinsic type 2 immune signaling is needed to limit the induction of revSCs and worm persistence. To control for differences in parasite fitness, we took a gain-of-function approach and treated Clu-fate mapping mice with low-dose IL-4 complexes (IL4C) on days 10, 12, and 14 after Hpb infection. This transient amplification of type 2 cytokine signals led to a reduction of Hpb-induced Tom+ ribbons without a difference in Hpb egg production (Fig. 5 Q and Fig. S3, B and D). Finally, to assess the immune landscape in Stat6−/− and Il4raΔIEC mice following Hpb infection, we performed immunophenotyping of the SI, Peyer’s patches (PPs), and mesenteric lymph nodes (MLNs) by flow cytometry (Fig. S3, E–J). As expected, accumulation of GATA3+ Th2 cells in the MLNs, PPs, and SI of Hpb-infected animals was largely eliminated in Stat6−/− mice. By contrast, infected Il4raΔIEC mice showed numbers of Th2 cells similar to littermate controls, indicating that direct type 2 cytokine signaling, not immune deviation per se, is responsible for driving epithelial reprogramming. Taken together, our data establish a balanced regulation of the ISC compartment by helminths and type 2 immune signaling that supports durable infection.

A balanced regulation of the ISC niche by helminths and type 2 immune signaling supports durable infection. Related to Fig. 5. (A) Representative photomicrographs of Clu RNAscope of Il4raWT and Il4raΔIEC tissue sections on day 14 after Hpb infection. (B) Representative photomicrographs of dtTomato expression in Clu fate-mapping mice treated with PBS or IL4C as described in Fig. 5 Q and Materials and methods. (C)Hpb worm burden 28 dpi in WT and Stat6−/− mice. (D)Hpb egg burden following IL4C treatment. (E–J) Immune phenotyping on day 14 after Hpb infection. SI (E), PPs (F), and MLNs (G) were harvested from WT and Stat6−/− mice. Single-cell suspensions were made, and flow cytometry was performed. Cells were gated for viable CD45+CD3+B220−CD4+CD62L−CD44high cells, and Gata3+ cells were identified as Th2 cells. Percentage of Gata3+ cells of the CD62L−CD44+ population. (H–J) SI (H), PPs (I), and MLN (J) were harvested from Il4raWT and Il4raΔIEC mice. Single-cell suspensions were made, and flow cytometry was performed. Cells were gated for viable CD45+CD3+B220−CD4+CD62L−CD44high cells, and Gata3+ cells were identified as Th2 cells. Percentage of Gata3+ cells of the CD62L−CD44+ population. Scale bar, 500 μm. Data shown are representative of two or more independent experiments, n = 6 biological replicates; statistical tests: t test (C, D, and H–J); two-way ANOVA (E–G); *, P < 0.05; ***, P < 0.005.
A balanced regulation of the ISC niche by helminths and type 2 immune signaling supports durable infection. Related to Fig. 5. (A) Representative photomicrographs of Clu RNAscope of Il4raWT and Il4raΔIEC tissue sections on day 14 after Hpb infection. (B) Representative photomicrographs of dtTomato expression in Clu fate-mapping mice treated with PBS or IL4C as described in Fig. 5 Q and Materials and methods. (C)Hpb worm burden 28 dpi in WT and Stat6−/− mice. (D)Hpb egg burden following IL4C treatment. (E–J) Immune phenotyping on day 14 after Hpb infection. SI (E), PPs (F), and MLNs (G) were harvested from WT and Stat6−/− mice. Single-cell suspensions were made, and flow cytometry was performed. Cells were gated for viable CD45+CD3+B220−CD4+CD62L−CD44high cells, and Gata3+ cells were identified as Th2 cells. Percentage of Gata3+ cells of the CD62L−CD44+ population. (H–J) SI (H), PPs (I), and MLN (J) were harvested from Il4raWT and Il4raΔIEC mice. Single-cell suspensions were made, and flow cytometry was performed. Cells were gated for viable CD45+CD3+B220−CD4+CD62L−CD44high cells, and Gata3+ cells were identified as Th2 cells. Percentage of Gata3+ cells of the CD62L−CD44+ population. Scale bar, 500 μm. Data shown are representative of two or more independent experiments, n = 6 biological replicates; statistical tests: t test (C, D, and H–J); two-way ANOVA (E–G); *, P < 0.05; ***, P < 0.005.
Expulsion of multicellular helminths from the intestinal lumen requires production of type 2 cytokines that direct the epithelial weep-and-sweep response (Anthony et al., 2007). It is well documented that helminths secrete immunomodulatory factors that target type 2 cytokine–producing cells to limit anti-helminth immunity (Hewitson et al., 2009). Here we demonstrate that helminths also evade expulsion through direct reprogramming of the intestinal epithelium into a fetal-like state. Interestingly, similar to our findings using adult Hpb-CM, Drurey et al. (2021) showed that Hpb infective L3 larvae were also capable of suppressing IL-4/IL-13–induced differentiation of goblet and tuft cells. However, they did not test the ability of L3 larvae to induce fetal-like reversion of the epithelium, an event we found to be dominant during the luminal stage of infection. This result raises the possibility that helminth-induced fetal reversion and inhibition of secretory cell formation might be two mutually exclusive events, an avenue of great interest for future studies.
Taken together, our results suggest that, although fetal reversion of the intestine is critical for regeneration upon injury, parasites such as Hpb that have evolved with their hosts coopt this restorative pathway to persist and continue their life cycle. Our study provides a new conceptual framework that not only may lead to improved anthelmintics that interfere with parasite-epithelium signaling networks, but also spur helminth-based therapies to rejuvenate the intestinal barrier following acute injury.
Materials and methods
Animals
All experiments were performed in accordance with the McGill University Health Centre Research Institute Animal Resource Division with approved animal use protocol #7977. WT C57BL/6, Stat6−/−, and Yapfloxed mice were obtained from The Jackson Laboratory. VillinCRE mice were kindly provided by Nicole Beauchemin (McGill University). Il4rafl/fl mice were kindly provided by Frank Brombacher (International Center for Genetic Engineering and Biotechnology, Cape Town, South Africa). VillinCREIl4rafl/fl and VillinCREYapfl/fl were bred in-house. Clu-CreERT2; Rosa26-LSL-tdTomato, CluGFP, and ClucreERT2/creERT2 were generated as previously described (Ayyaz et al., 2019). Homozygote Clu-CreERT2 are effectively Clu deficient (Clu−/−; Ayyaz et al., 2019). All mice were bred and maintained under specific pathogen–free conditions. All experiments were performed using littermates with the exception of Stat6−/− mice. Female and male mice 6–10 wk of age were used.
Helminth infection
For Hpb infection, mice were infected by gavage with 150 L3 stage larvae diluted in sterile water. Mice were euthanized at the indicated time points, and tissues were harvested for analysis. For histology, the proximal 5 cm of the SI was harvested on the indicated days, flushed, and fixed in 10% formalin for 24 h before standard formalin-fixed, paraffin-embedded (FFPE) processing. For Nippo infection, infectious third-stage larvae (L3) were maintained by passaging through Lewis rats (Envigo), as previously described (Liang et al., 2012). For histology of Nippo-infected intestines, mice were infected s.c. with 500 L3 larvae. The proximal 10–12 cm of the SI was harvested after 7 d, flushed, and fixed in 10% formalin for 2 h. Intestines were then rolled into “Swiss rolls,” placed in a tissue cassette, and fixed for an additional 18–24 h before standard FFPE processing.
SI organoid culture
The SI was harvested, flushed with PBS, cut into three 10-cm pieces, and opened longitudinally. Intestine pieces were rinsed with PBS and then incubated in 2 mM EDTA at 4°C for 30 min with gentle agitation. Crypts were further released from the SI by vigorous shaking in 10 ml PBS, three times, and passed through a 70-μm cell strainer. Isolated crypts were resuspended in 10 ml basal organoid medium (Advanced DMEM/F12 [Gibco], 10 mM Hepes [Gibco], 1× GlutaMAX [Gibco], and 1% penicillin/streptomycin [Gibco]). Crypts were plated in 30 μl Matrigel (Corning) domes in 24-well plates, and 500 μl of culture medium was added to each well. Organoids culture medium (ENR): basal organoid medium with the addition of N2 Supplement (1:100; Gibco), B27 Supplement (1:50; Gibco), 1 mM NAC, 50 ng/ml recombinant EGF, in-house made R-spondin 1, and Noggin CM (1:50). Organoids were maintained for the indicated times and passaged every 3–4 d by physical dissociation of the Matrigel and replating in fresh ENR medium. For Hpb-CM and/or Nippo-CM stimulation, Hpb-CM and/or Nippo-CM were supplemented with N2 (1:100; Gibco), B27 (1:50; Gibco), 1 mM NAC, 50 ng/ml recombinant EGF, in-house made R-spondin 1, and Noggin CM (1:50). For IL-13 stimulation, organoids were treated with 5 ng/ml of IL-13 simultaneously with Hpb-CM for 24 h or pretreated with 5 ng/ml IL-13 for 10 d (over three passages) before stimulation with Hpb-CM for 18 h.
Conditioned medium and HES preparation
To generate Hpb-CM, WT mice were infected by gavage with 400 L3 Hpb larvae, and adult worms were manually harvested from the SI after 21 d as previously described (Moreno et al., 2011). Sterile adult worms were then cultured overnight in organoid basal medium at ∼130 worms/ml. The following day, culture medium containing helminth secretory products was removed, filtered through a 0.2-μm filter, and kept at −80°C until use. For Nippo-CM preparation, rats were infected s.c. with 3000 L3 Nippo, and adult worms were manually harvested from the SI after 7 d and cultured as described for Hpb-CM. For HES preparation, Hpb-CM was concentrated using a 3-kD Amicon filter tube and used at 10-fold dilution in fresh ENR medium.
ENR withdrawal
SI organoids were grown in ENR-supplemented medium. After 10 d of culture, organoids were passaged and replated in the presence or absence of growth factors for 24 h before stimulation with Hpb-CM for an additional 18 h.
Organoid whole-mounting and confocal microscopy
Established organoids were passaged and replated in a 1:1 ENR:Matrigel solution; 30-μl domes were plated on Nunc Lab-Tek II Chamber Slides and incubated overnight in 200 μl medium at 37°C. Organoids were than treated with 5 ng/ml of IL-13 in ENR or simultaneously with Hpb-CM for 48 h. After stimulation, domes were fixed in 10% formalin for 30 min at room temperature (RT), and permeabilized with PBS-T (PBS + Triton 0.5%) for 15 min. The samples were blocked with 200 μl blocking solution (3% BSA in PBS) for 1 h at RT. Organoids were incubated with primary antibodies overnight at 4 °C, followed by incubation with secondary antibodies at RT for 1 h. The nuclei were stained with DAPI (1 µg/ml). Specific antibodies include anti-mouse/rat Ki67 eFluor 660 (Invitrogen), anti-mouse CD326 (EpCAM) Alexa Fluor 488 (BioLegend), goat anti-GFP (Invitrogen), anti-goat IgG Alexa Fluor 555 (Invitrogen), rabbit anti-mouse Dclk (Abcam), rabbit anti-mouse Muc2 (Abcam), and goat anti-rabbit IgG Alex Fluor 555. All fluorescent images were taken on a Zeiss Confocal LSM700, using a 20×/0.8 M27 Plan-Apochromat objective, after mounting with Invitrogen Prolong Glass Antifade Mounting solution. Tile scanning (5 × 5 tiles) was performed as well as Z-stacking to generate images analyzed using Fiji software (Schindelin et al., 2012).
RNAscope
RNAscope was performed according to manufacturer instructions (ACD Bio). RNAscope probes: Clu (cat# 427891); Il1rn (cat# 495101); Il33 (cat# 400591-C2); and Ly6a (cat# 427571-C2). Images were taken on a Leica Aperio AT Turbo digital pathology scanner, using a 20×/0.75 Plan Apo objective, after mounting with with Cytoseal 60 (Thermo Fisher Scientific). Quantification of Clu expression by colorimetric RNAscope was performed after 20× magnification scanning of entire 5-cm tissue rolls, using the pixel count function of Qupath, a digital pathology image analysis software (Bankhead et al., 2017).
RNA extraction and quantitative PCR (qPCR)
RNA was extracted using Tri reagent (Sigma-Aldrich) or PureLink RNA Mini kit (Invitrogen) according to manufacturer instructions. Primers used: Clu forward, 5′-AAACCAACGCAGAGCGCAAG-3′, and reverse, 5′-CTCCAGAGCATCCTCTTTCTTCTT-3′; Il1rn forward, 5′-AGGCATGTGCTCTACCATCATGCT-3′, and reverse, 5′-GCCGACATGGAATAAGGCTGGC-3′; Msln forward, 5′-TCTCCAAACAGTGGTGTGGG-3′, and reverse, 5′-AAGCAGTAGGAAGCTTCGGC-3′; Lgr5 forward, 5′-AGAGCCTGATACCATCTGCAAAC-3′, and reverse, 5′-TGAAGGTCGTCCACACTGTTGC-3′; Olfm4 forward, 5′-GCTCCTGGAAGCTGTAGTCA-3′, and reverse, 5′-GGCCCCAGGCACCATATTTA-3′; Gob5 forward, 5′-CATCGCCATAGACCACGACG-3′, and reverse, 5′-TTCCAGCTCTCGGGAATCAAA-3′; Muc2 forward, 5′-CTGACCAAGAGCGAACACAA-3′, and reverse, 5′-CATGACTGGAAGCAACTGGA-3′; Spdef forward, 5′-TCCTCTCTGCTCACTCTGAA-3′, and reverse, 5′-AGAGCTCATGTGTATCCCTAGA-3′; Dclk forward, 5′-AACGTCAAGACCACCTCAGC-3′, and reverse, 5′-GAGCCGTCTTCTTGTTCAGC-3′; Il33 forward, 5′-GACACATTGAGCATCCAAGG-3′, and reverse, 5′-TGATTGACTTGCAGGACAGG-3′; Ly6a forward, 5′-GGAGGCAGCAGTTATTGTGG-3′, and reverse, 5′-GCTACATTGCAGAGGTCTTCC-3′; Anxa1 forward, 5′-CAACCATCGTGAAGTGTGCC-3′, and reverse, 5′-ATGCCTTATGGCGAGTTCCG-3′; and Ki67 forward, 5′-CCTGGTCACCATCAAGCGG-3′, and reverse, 5′-ATTCAATACTCCTTCCAAACAGGC-3′.
Bulk RNA-seq
SI organoids were stimulated with ENR (control) or ENR + Hpb-CM medium for 24 h from the start of culture. RNA was extracted using Trizol reagent (Sigma-Aldrich), and quality was checked with the Bioanalyzer RNA 6000 kit. Libraries were prepared using the NEB mRNA stranded library prep Kit (New England Biolabs) and paired-end sequenced on the NovaSeq 6000 at 25 million reads per sample. RNA-seq reads were aligned to the mm10 reference genome using STAR (Dobin et al., 2013). Read counts were calculated using the strand-specific exonic reads of each gene, and duplicate genes were merged using HOMER (Heinz et al., 2010). Transcripts per million were used to evaluate the correlation among replicates for quality control. Differential gene expression was calculated from the raw read counts using edgeR (Robinson et al., 2009). Data sets presented in the manuscript can be found at GEO, accession no. GSE199227.
GSEA
For pathway analyses, custom gene sets were built using genes differentially (up- or down-regulated) expressed in previous cell type-specific transcriptomic studies. A fetal-associated gene list was built using Table S1 in Mustata et al. (2013). A differentiated epithelial cell marker list was built using Table S4 in Haber et al. (2017). Transcriptomes from this work were analyzed for enrichment in differentially expressed genes in those gene sets using the “fgsea” R package (https://github.com/ctlab/fgsea; Korotkevich et al., 2021).
scRNA-seq
SI organoids were stimulated with ENR (control) or ENR + Hpb-CM medium for 24 h on day 0 of culture. Single-cell suspensions were prepared by manual disturbance of Matrigel domes followed by incubation in TrypLE (Gibco) for 40 min at 37°C and loaded on a Next GEM Chip G (PN-1000120) together with Next GEM Single Cell 3ʹ GEM Kit v3.1 (PN-1000121). Single cells were captured on a 10× Genomics Chromium controller with a recovery target of 5,000 cells per sample. cDNAs were generated following the 10X Genomics protocol. cDNAs were size selected using SPRIselect beads from Beckman Coulter (B23318), and their quality was checked with a Bioanalyzer High Sensitivity DNA Kit from Agilent (5067-4626). One quarter of the total cDNA was used to generate libraries using Next GEM Single Cell Library Kit (PN-1000121) and barcoded using the Single Index Kit Set A (PN-111213) following the 10X protocol. Libraries were size-selected using SPRIselect beads, and their quality was checked with the Bioanalyzer High Sensitivity DNA Kit. Libraries’ sizes were centered at 440 bp and paired-end sequenced on a NovaSeq 6000 at 125 M reads per sample. Single-cell matrices were generated using Cellranger (v4.0.0) provided by 10X Genomics. Sequences were aligned with the mm10 mouse transcriptome. Matrices were imported and analyzed with the R package “Seurat” (v4.0.3; Butler et al., 2018). We filtered out genes expressed in fewer than three cells and low-quality cells expressing <200 genes. Cells with >100,000 transcripts were removed, as they likely represent cell doublets (Butler et al., 2018). In addition, immune cells (marked by Cd3g) were removed from the analysis. Each matrix was normalized with Seurat’s “NormalizeData” function using “LogNormalize” as the normalization method and a scale factor equal to 10,000 (default value). Matrices were integrated using the “IntegrateData” function. Linear dimensionality reduction was performed using the “RunPCA” function. Nonlinear dimensionality reduction was then performed using the “RunUMAP” function, considering the Euclidean distances of the first 30 PCA (principal component analysis) components. k-Nearest neighbors graph construction was performed using the “FindNeighbors” function, which also considered the first 30 PCA components. Clustering (Louvain method) was performed using the “FindClusters” function, with a resolution equal to 1.5. Differential expression analysis was performed using the “FindAllMarkers” function to detect markers considering positive log-transformed fold-change values >0.25. Finally, trajectory analysis was performed with the R package “Monocle3” (v0.2.3.0; Trapnell et al., 2014) using CBCs as the source of the pseudotime variable (pseudotime = 0). To display trajectory analysis results, UMAP was performed on each matrix using the “reduce_dimension” function found in the Monocle3 package (McInnes et al., 2018). Data sets presented in the manuscript can be found at GEO, accession no. GSE199227.
Metascape analysis
All differentially expressed genes, in a specific cluster, between ENR and Hpb-CM samples were used in the online Metascape tool (Zhou et al., 2019), and the default “express analysis” option was chosen.
Clu lineage-tracing studies
Tamoxifen-inducible Clu fate-mapping mice (Clu-CreERT/+; Rosa26-LSL-tdTomato; Ayyaz et al., 2019) were infected with 150 Hpb larvae. 2.5 mg tamoxifen (Sigma-Aldrich) was injected i.p. 5, 7, and 12 dpi. To allow the proliferation and differentiation of potential Clu+ stem cells, SIs were harvested 12 d after tamoxifen injection for histological analysis. Specifically, the proximal 5 cm of the duodenum was fixed overnight in 10% formalin, transferred to 30% sucrose in PBS for 24 h, frozen in Optimal Cutting Temperature, and kept at −80° until analysis by confocal microscopy. All fluorescent images were taken on a Zeiss Confocal LSM700, using a 20×/0.8 M27 Plan-Apochromat objective, after mounting with Invitrogen Prolong Glass Antifade Mounting solution. Tile scanning was performed as well as Z-stacking for image analysis using the Fiji software (Schindelin et al., 2012). Tom+ crypts were defined as crypts composed entirely of Tom-labeled cells.
Immunofluorescence staining for DCLK and UEA1
Optimal Cutting Temperature–embedded sections were dried, washed in PBS, and fixed for 10 min in 10% formalin. Permeabilization in 0.05% Triton X-100 in PBS was done for 20 min followed by 1 h of blocking in 3% BSA in PBS. Staining was done overnight at 4°C, followed by washes in 0.05% Triton X-100 in PBS, staining with a secondary antibody for 1 h at RT, and staining with DAPI (1 μg/ml) and mounted. All fluorescent images were taken at 20× using a Zeiss Confocal LSM700. DCLK (rabbit polyclonal, ab31704; Abcam), UEA1 (L9006; Sigma-Aldrich). Secondary antibody: donkey anti-rabbit AF650 (SA5-10041; Invitrogen). Tissue sections were scanned at 20× magnification and Z stacked, and 7 × 7 tiling was performed to image a large area of tissue. Tom+ crypts were defined as crypts composed entirely of Tom-labeled cells. Thereafter, 10–15 Tom+ and 10–15 Tom− villi were identified, and the number of UEA1+ cells was counted.
Immunophenotyping
The distal 10 cm of the ileum was used to extract lamina propria cells as previously described (Gentile et al., 2020). For the PPs and MLNs, tissue was crushed in cold HBSS buffer (HBSS supplemented with 2% FBS and 15 mM Hepes), passed through a 100-μm filter, and centrifuged at 1,800 rpm for 5 min at 4°C. Cell suspensions were incubated with a fixable Viability dye (eFluor 506; eBioscience) for 30 min at 4°C. Cells were then incubated with Fc block (10 min at 4°C), followed by staining (for 30 min at 4°C) with the following antibodies in appropriate combinations of fluorophores. From Invitrogen: B220 (RA3–6B2), CD127 (A7R34), CD62L (MEL-14), and GATA-3 (TWAJ); from BioLegend: CD45 (30F11); and from BD: CD44 (IM7), CD3 (145-2C11), and CD4 (GK1.5). For staining of intracellular proteins, cells were fixed and permeabilized with the FoxP3 Fix/Perm kit (eBioscience) according to manufacturer’s instructions. Data were acquired with a LSR Fortessa (BD Biosciences) and analyzed using FlowJo software (TreeStar).
IL4C treatment
A long-acting form of IL-4 was produced by mixing 10 μg recombinant murine IL-4 (PeproTech) with 50 μg of neutralizing monoclonal antibody (clone 11.B11; BioXcell). Clu fate-mapping mice were injected i.v. with IL4C on days 10, 12, and 14 after Hpb infection and with a single 3-mg tamoxifen i.p. injection on day 12 after infection. The SI tissue as well as feces for Hpb egg counts were harvested on day 12 after tamoxifen injection (day 24 after Hpb infection).
Parasite burden and fecundity
For worm burden assessment, mice were infected with Hpb, and the SI was harvested 28 dpi. Adult worms were individually removed from the intestine and enumerated. To measure parasite fecundity, feces was collected from infected mice 14 dpi, weighed, and placed in 1 ml saturated NaCl solution. The samples were vortexed and left at room temperature for 24 h and then placed at 4°C. The eggs were enumerated and normalized to the volume of the supernatant and weight of feces.
YAP immunohistochemistry
SI tissues for immunohistochemistry were fixed overnight in formalin at room temperature. Samples were then dehydrated, embedded in paraffin, and sectioned at 4 μM. After antigen retrieval (10 mM sodium citrate buffer, pH 6) and blockage of peroxidase activity in 3% H2O2 solution, sections were stained with an active Yap antibody (1:300; ab205270; Abcam). Detection of primary antibodies was achieved using the Dako Envision plus system.
Statistical analysis
GraphPad Prism 9 software was used to perform statistical analyses. Student’s t test or one- or two-way ANOVA were used as appropriate. Nonparametric tests were used when the results did not fit a normal distribution.
Online supplemental material
Fig. S1 refers to Figs. 1 and 2 and shows fetal reversion of the intestinal epithelium at the luminal stage of Hpb infection and after Hpb-CM stimulation of SI organoids. Fig. S2 refers to Figs. 1, 2, and 3 and shows partial dependence of the Hpb-induced fetal program on the YAP-signaling pathway and its relation to IFNγ signaling. Fig. S3 refers to Fig. 5 and shows Clu in situ expression and Hpb egg burden in Stat6−/−, Il4raΔIEC mice and in WT mice after IL4C treatment. It also shows immune-phenotyping of Stat6−/− and Il4raΔIEC mice 14 dpi. Table S1 is a complete list of differentially expressed genes. Related to RNAseq data set in Fig. 2. Table S2 is the complete list of cluster defining markers. Related to scRNAseq data set in Fig. 3.
Data and materials availability
Data sets presented can be found at GEO, accession no. GSE199227.
Acknowledgments
We thank all members of the King and Gregorieff laboratories for their support and feedback in preparing this manuscript. We greatly appreciate the technical support of Marianna Orlova (McGill), Pauline Cassart (McGill), and Sarah Boissel (Montreal Clinical Research Institute) for the scRNA-seq experiments and the McGill University Health Centre Research Institute Animal Resource Division and the Histology platform. Images throughout were created using Biorender.com.
This work was supported by the Canadian Institutes of Health Research (PJT-362757 and FRN-156038). I.L. King holds a Canada Research Chair in Barrier Immunity. M. Malleshaiah holds a Fonds de recherche du Québec—Santé, Junior 1 Scholarship.
Author contributions: Conceptualization: I.L. King, A. Gregorieff, D. Karo-Atar; Methodology: I.L. King, A. Gregorieff, D. Karo-Atar, M. Malleshaiah, L. Diatchenko, J. von Moltke; Investigation: D. Karo-Atar, T. Javkar, S. Ouladan, S. Merritt, G. Fontes, A. Rochette, L. Joumier, M. Parisien, S. Westfall, M.E. Gentile, M.K. Matheson, G.J. Fonseca; Funding acquisition: I.L. King, A. Gregorieff; Writing—original draft: I.L. King, A. Gregorieff, D. Karo-Atar; Writing—review & editing: All authors.

